
Le dynamisme du phénoptype microglial dans la ...
261
© Louis-Charles Béland, 2021 Le dynamisme du phénoptype microglial dans la pathogénèse de la sclérose latérale amyotrophique Thèse Louis-Charles Béland Doctorat en neurobiologie Philosophiæ doctor (Ph. D.) Québec, Canada
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of Le dynamisme du phénoptype microglial dans la ...

© Louis-Charles Béland, 2021
Le dynamisme du phénoptype microglial dans la pathogénèse de la sclérose latérale amyotrophique
Thèse
Louis-Charles Béland
Doctorat en neurobiologie
Philosophiæ doctor (Ph. D.)
Québec, Canada

i
LE DYNAMISME DU PHÉNOPTYPE MICROGLIAL DANS LA PATHOGÉNÈSE DE
LA SCLÉROSE LATÉRALE AMYOTROPHIQUE
Thèse
Louis-Charles Béland
Sous la direction de :
Jasna Kriz, directrice de recherche

ii
Résumé
La sclérose latérale amyotrophique (SLA) est une maladie neurodégénérative qui cause la
mort des neurones moteurs, induisant une paralysie progressive et menant à la mort quelques
années après l’apparition des premiers symptômes. Comme dans toutes les maladies
neurodégénératives, l’inflammation est une composante principale de la pathogénèse de la
SLA. Une des cellules effectrices de cette inflammation, la cellule microgliale est le sujet
central de cette thèse. Avec l’apparition des symptômes, la microglie passe d’un phénotype
anti-inflammatoire et neuroprotecteur à un phénotype aberrant et neurotoxique. Il est
nécessaire d’élucider les mécanismes cellulaires responsable du changement de phénotype et
l’identification des facteurs maintenant ces phénotypes pour permettre le développement de
thérapies efficaces ciblant l’inflammation. Ainsi dans cette thèse, nous tâchons d’identifier,
en utilisant divers outils moléculaires et modèles animaux, la signature microgliale à
différents stades de la SLA.
Durant le stade présymptomatique, nous identifions, par l’étude du sécrétome microglial, la
cytokine immunosuppressive interleukine-10 comme responsable du maintien du phénotype
anti-inflammatoire. Cette cytokine est à la fois produite par les cellules microgliales et
reconnue par celles-ci, grâce au récepteur IL-10R. Le blocage d’IL-10R a pour conséquence
d’accélérer la progression de la maladie et de diminuer la survie dans un modèle murin.
Aussi, l’augmentation de l’expression de cette cytokine à l’aide d’un vecteur viral mène à
une diminution de la vitesse de la progression de la maladie et allonge la durée de vie chez
ce même modèle murin.
Dans le but de comprendre les mécanismes cellulaires impliqués dans le changement
phénotypique de la microglie avec l’apparition des symptômes de la SLA, nous utilisons un
système-modèle pour le profilage moléculaire spécifique à la microglie. Cet outil, EDTA-
TRAP, consiste en l’immunopurification des ribosomes microgliaux et l’identification
subséquente des ARNm et des protéines séquestrés sur ces ribosomes. Il a été possible
d’identifier une forte disparité dans la nature des ARNm et des protéines les plus régulés. Le
profil transcriptomique de la microglie symptomatique dénote une fonction phagocytaire et
possiblement neuroprotectrice pour la microglie alors que son profil protéomique est
proliférateur et potentiellement neurotoxique. La disparité de ces deux profils s’explique par

iii
la répression de la traduction d’ARNm issus de gènes immunitaires par un mécanisme
impliquant l’interaction de leur 3’UTR et de la protéine liant l’ARN SRSF3. La forme
phosphorylée de cette protéine s’accumule spécifiquement du cytoplasme des cellules
microgliales proportionnellement avec la progression de la maladie. La diminution de
l’expression de SRSF3 à l’aide d’un oligonucléotide interférent rétablit la traduction des
ARNm immunitaires et la fonction phagocytaire de la microglie associée à ces ARNm. Cela
mène à une progression plus lente de la maladie et une plus longue survie dans un modèle
murin. Ainsi, l’utilisation d’un oligonucléotide interférant contre SRSF3 pourrait être une
nouvelle avenue thérapeutique dans le traitement de la SLA.
La compréhension des divers mécanismes cellulaires impliquant la protéine SRSF3 est
importante pour comprendre son dynamisme dans la SLA. SRSF3 est la plus petite protéine
de la famille SR. Cette famille se distingue par leur structure soit un ou deux motifs de
reconnaissance de l’ARN en N-terminus et un domaine riche en arginine et en sérine
hautement phosphorylable en C-terminus. SRSF3 peut être phosphorylé dans le noyau par la
kinase CLK1 ou dans le noyau et dans le cytoplasme par les kinases SRPK. SRSF3 est
impliqué dans l’épissage, le transport nucléo-cytoplasmique et la traduction, entre autres. La
dérégulation du niveau d’expression ou de la phosphorylation de SRSF3 peut drastiquement
changer son rôle dans la cellule. Ainsi, SRSF3 est impliqué dans diverses maladies comme
le cancer, les infections virales ou la SLA. Les mécanismes cellulaires régissant le
dynamisme de SRSF3 doivent ainsi être bien compris dans le but de faire de l’oligonucléotide
interférent contre SRSF3 une thérapie efficace contre la SLA.

iv
Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder that leads to
motoneuron loss, progressive paralysis and death a few years after the first symptoms. As
every other neurodegenerative disorder, inflammation is a main component of ALS
pathogenesis. One of the cell type implicated in this inflammation, the microglia, is the
central subject of this thesis. With the emergence of the symptoms, microglia transforms
from an anti-inflammatory and neuroprotective phenotype to an aberrant and neurotoxic
phenotype. It is necessary to elucidate the cellular mechanisms underlying this phenotypic
change and the factors maintaining these phenotypes to allow the development of effective
inflammation-targeting therapies. In this thesis we seek to identify, using multiple molecular
tools and animal models, the microglial signature at the different stage of the disease.
During the symptomatic stage, we identified the immunosuppressive cytokine IL-10 to be
responsible for maintaining the anti-inflammatory phenotype. This cytokine is at the same
time produced and recognized by microglial cells. Blocking IL-10 receptor accelerated
disease progression and shortened survival in a mouse model. Moreover, the augmentation
of the expression of IL-10, using a viral vector, slowed disease progression and lengthened
lifespan in the same mouse model.
In order to understand the cellular mechanisms implicated in the microglial phenotypic
change in ALS, we used a system-model for the microglia-specific molecular profiling. This
tool, EDTA-TRAP, consists in the microglial poly-ribosome immunopurification and the
subsequent identification of in-translation mRNAs and proteins sequestered on these
ribosomes. It was possible to identify a strong mismatch in the nature of the most regulated
mRNAs and proteins. The transcriptomic profile of symptomatic microglia denotes a
phagocytic and possible neuroprotective function for microglia while its proteomic profile is
proliferative and potentially neurotoxic. The discrepancy between these two profiles is
explained by the repression of immune gene mRNA translation by a mechanism implicating
the interaction of their 3’UTR and the RNA-binding protein SRSF3. The phosphorylated
form of SRSF3 specifically accumulates in microglia cytoplasm proportionally with disease
progression. The decrease of SRSF3 expression by an interfering oligonucleotide restores
immune mRNA translation and the microglial phagocytic function. This leads to and slower

v
disease progression and a longer survival in a mouse model. Hence, the use of an interfering
oligonucleotide against SRSF3 could be a new therapeutic avenue in ALS treatment.
The understanding of the diverse cellular mechanisms implicating SRSF3 is important in
elucidating its dynamic function in ALS. SRSF3 is the shortest protein from the SR family.
This family can be distinguished by their structure : one or two N-terminal RNA recognition
motif and a C-terminal highly phosphorylatable arginine and serine rich domain. SRSF3 can
be phosphorylated in the nucleus by the CLK1 kinase or in the nucleus and the cytoplasm by
the SRPK kinases. SRSF3 is implicated in splicing, nucleocytoplasmic transport and
translation, among others. The deregulation of the expression level or the phosphorylation of
SRSF3 can drastically change its role in the cell. Hence, SRSF3 is implicated in many
diseases such as cancer, viral infection and ALS. The cellular mechanisms governing SRSF3
dynamism should then by well understood in order to make the interfering oligonucleotide
against SRSF3 an efficient therapy to treat ALS.

vi
Table des matières
Résumé ....................................................................................................................................................... ii
Abstract ..................................................................................................................................................... iv
Table des matières .................................................................................................................................... vi
Liste des figures ........................................................................................................................................ xii
Liste des tableaux .................................................................................................................................... xiii
Liste des acronymes ................................................................................................................................ xiv
Remerciements ........................................................................................................................................ xix
Avant-propos ........................................................................................................................................... xx
Introduction ................................................................................................................................................ 1
1.1. Sclérose latérale amyotrophique ................................................................................................... 1
1.1.1. Aspects cliniques et anatomiques de la SLA ........................................................................ 3
1.1.2. Aspects génétiques de la SLA ................................................................................................ 5
1.1.2.1. Superoxyde dismutase Cu/Zn ......................................................................................... 6
1.1.2.2. TAR DNA-binding protein 43 ........................................................................................ 9
1.1.2.3. Protéine liant l’ARN FUS ............................................................................................. 11
1.1.2.4. C9orf72 .......................................................................................................................... 13
1.1.3. Mécanismes de cytotoxicité impliquées dans la pathogénèse de la SLA ......................... 14
1.1.3.1. Dérégulation du cytosquelette, du transport et de la survie axonale .......................... 15
1.1.3.2. Dérégulation de la protéostasie..................................................................................... 18
1.1.3.3. Stress oxydatif et dysfonction mitochondriale ............................................................ 20
1.1.3.4. Sécrétion des protéines agrégées .................................................................................. 24
1.2. Pathogénèse cellulaire non-autonome ........................................................................................ 26
1.2.1. Cellules microgliales ............................................................................................................ 27
1.2.2. Astrocytes .............................................................................................................................. 35
1.2.3. Oligodendrocytes et cellules de Schwann ........................................................................... 39
1.2.4. Macrophages et monocytes .................................................................................................. 41
1.2.5. Lymphocytes ......................................................................................................................... 43
1.2.6. Autres cellules immunitaires ................................................................................................ 44
1.3. Inflammation et pathogénèse dans la SLA ................................................................................. 45
1.3.1. Gènes associés à la SLA et leur impact sur l’inflammation............................................... 45
1.3.2. L’inflammation excessive et la réponse immunitaire inefficace ....................................... 47
1.3.3. Le profilage moléculaire à spécificité cellulaire pour l’étude de l’inflammation............. 51
1.4. Hypothèses et objectifs de la thèse ............................................................................................. 53

vii
Chapitre 2 - IL-10 Controls Early Microglial Phenotypes and Disease Onset in ALS Caused by
Misfolded Superoxide Dismutase 1 ....................................................................................................... 55
2.1. Résumé ......................................................................................................................................... 55
2.2. Abstract ......................................................................................................................................... 56
2.3. Introduction .................................................................................................................................. 57
2.4. Materials and Methods ................................................................................................................ 58
2.4.1. Generation of TLR2-Fluc-AcGFP;SOD1G93A and TLR2-Fluc-AcGFP;SOD1G37R
transgenic mice ................................................................................................................................ 58
2.4.2. Virus construction and preparation ...................................................................................... 59
2.4.3. In vivo bioluminescence imaging ........................................................................................ 59
2.4.4. Lippolysaccharide treatment ................................................................................................ 60
2.4.5. Surgical procedures .............................................................................................................. 60
2.4.5.1. Stereotaxic brain injection ............................................................................................ 60
2.4.5.2. Intracerebroventricular brain infusion.......................................................................... 60
2.4.5.3. Intrathecal injection of AAV-CD11b-IL10 ................................................................. 61
2.4.5.4. Tissue collection ............................................................................................................ 61
2.4.6. Immunofluorescence ............................................................................................................ 62
2.4.7. Western blots......................................................................................................................... 62
2.4.8 Primary adult microglia cultures ........................................................................................... 63
2.4.8.1. Primary culture: IL-10 treatments ................................................................................ 63
2.4.9. Reverse transcriptase PCR ................................................................................................... 64
2.4.10. Cytokine array ..................................................................................................................... 65
2.4.11. Flow cytometry ................................................................................................................... 65
2.5. Results ........................................................................................................................................... 66
2.5.1. TLR2-luc-GFP;SOD1G93A mice, a mouse model for live analysis of early microglia
phenotypes in ALS .......................................................................................................................... 66
2.5.2. Preonset ALS is associated with marked downregulation of innate immune response and
anti-inflammatory microglia profiles ............................................................................................. 66
2.5.3. Enhanced IL-10 production by presymptomatic resident SOD1G93A microglia ............... 70
2.5.4. Deregulation of TLR2 is IL-10 dependent .......................................................................... 73
2.5.5. Chronic infusion with IL-10R blocking antibody precipitates disease onset in
the SOD1G93A mouse model ............................................................................................................ 75
2.6. Discussion ..................................................................................................................................... 77
2.7. Footnotes ...................................................................................................................................... 81
2.8. References .................................................................................................................................... 82

viii
2.9. Figures .......................................................................................................................................... 87
2.9.1. Figure 1: In vivo biophotonic/bioluminescence imaging of TLR2-SOD1 mice. .............. 87
2.9.2. Figure 2: Immunofluorescence and in situ hybridization analysis. ................................... 89
2.9.3. Figure 3: IL-10 overexpression in in vitro adult microglia primary cultures. .................. 91
2.9.4. Figure 4: Presymptomatic microglial phenotype is not mediated by peripheral immune
cells. ................................................................................................................................................. 93
2.9.5. Figure 5: Modulating the microglial response by blocking the IL-10 pathway. .............. 95
2.9.6. Figure 6: Treatment with blocking IL-10R antibodies accelerates disease onset
in SOD1G93A mice, whereas AAV-CD11b-cMyc-IL10 virus intrathecal injection delays onset
and extends survival. ....................................................................................................................... 97
2.9.7. Figure 7: Intracerebroventricular infusion of blocking IL-10R antibodies increases
inflammation in the lumbar spinal cord, whereas IL-10 increases anti-inflammatory markers. 99
Chapitre 3 - SRSF3 acts as a ribosome-based checkpoint and regulates translation of immune
mRNAs in ALS microglia..................................................................................................................... 101
3.1. Résumé ....................................................................................................................................... 101
3.2. Abstract ....................................................................................................................................... 102
3.3. Introduction ................................................................................................................................ 103
3.4. Results ......................................................................................................................................... 104
3.4.1. Creation of the CD11b-Flag-EGFP-mRPL10a /hSOD1G93A Mouse Model ................ 104
3.4.2. Chronic Dissociation of mRNA and Protein Molecular Signatures in ALS Microglia . 105
3.4.3. Highly Upregulated Transcripts in ALS Microglia Are Not Translated ......................... 108
3.4.5. SRSF3 is Responsible for the Translational Repression of Immune mRNAs by Binding
to their 3’UTR ............................................................................................................................... 109
3.4.6. Upregulation of pSRSF3 in Lumbar Spinal Cord Microglia Correlates with Clinical
Onset of Disease ............................................................................................................................ 110
3.4.7. Disease-Associated Increase in pSRSF3 is Conserved Across Species .......................... 112
3.4.8. Targeting Srsf3 Restores Immune Gene Translation and Microglia Phagocytic Function
........................................................................................................................................................ 112
3.5. Discussion ................................................................................................................................... 114
3.6. Acknowledgments ...................................................................................................................... 117
3.7. Author Contributions ................................................................................................................. 117
3.8. Declaration of interests .............................................................................................................. 117
3.9. Figures titles and legends .......................................................................................................... 118
3.9.1. Figure 1. Creation of the CD11b-Flag-EGFP-mRPL10a /hSOD1G93A Mouse Model 118
3.9.2. Figure 2. Chronic Dissociation of mRNA and Protein Molecular Signatures in ALS
Microglia........................................................................................................................................ 120

ix
3.9.3. Figure 3. Highly Upregulated Transcripts in ALS Microglia Are Translationally
Arrested by SRSF3 ........................................................................................................................ 122
3.9.4. Figure 4. Upregulation of pSRSF3 in Lumbar Spinal Cord Microglia Correlates With
Clinical Onset of Disease in Rodent and Human Tissue. ........................................................... 124
3.9.5. Figure 5. Targeting Srsf3 Restores Immune Gene Translation and Microglia Phagocytic
Function ......................................................................................................................................... 126
3.9.6. Figure 6. Proposed Mechanisms of Microglial Rescue After Srsf3 Knockdown ........... 128
3.10. Tables titles and legends .......................................................................................................... 129
3.10.1. Table 1. Top regulated transcripts ................................................................................... 129
3.11. STAR Methods......................................................................................................................... 130
3.11.1 Generation and characterization of CD11b Flag-EGFP-RPL10a x G93A SOD1 mouse
........................................................................................................................................................ 130
3.11.2. EDTA-TRAP from Flag-EGFP-RPL10a x hSOD G93A mice. .................................... 131
3.11.3. Immunofluorescence ........................................................................................................ 131
3.11.4. mRNA isolation ................................................................................................................ 132
3.11.5. Affymetrix microarray ..................................................................................................... 132
3.11.6. Peptide isolation ................................................................................................................ 133
3.11.7. Label-free quantification .................................................................................................. 133
3.11.8. EDTA-TRAP data analysis .............................................................................................. 133
3.11.9. Western Blot ..................................................................................................................... 134
3.11.10. Cross-linked immunoprecipitation ................................................................................ 135
3.11.11. qRT-PCR ......................................................................................................................... 135
3.11.12. 3’UTR analysis ............................................................................................................... 136
3.11.13. Human sections immunochemistry ............................................................................... 137
3.11.14. Phagocytosis assay ......................................................................................................... 137
3.11.15. Morpholino treatment ..................................................................................................... 138
3.11.16. SRSF3 ELISA ................................................................................................................. 138
3.11.17. STAR Methods References ............................................................................................ 139
3.12. Supplemental Information Titles and legends ........................................................................ 140
3.12.1. Table S1. Supplementary data to Figure 2 ...................................................................... 140
3.12.2. Figure S1 Supplementary data to Figure 2 Proteomic Validation Through
Immunoblotting ............................................................................................................................. 140
3.12.3. Table S2 Supplementary data to Figure 3G .................................................................... 141
3.12.4. Table S3 Supplementary data to figure 3H ..................................................................... 142

x
3.12.5. Table S4 Primer list .......................................................................................................... 142
3.13. References ................................................................................................................................ 143
Chapitre 4 – The molecular and biological functions of SRSF3 and its implication in diseases ..... 149
4.1. Résumé ....................................................................................................................................... 149
4.2. Abstract ....................................................................................................................................... 150
4.3. Introduction ................................................................................................................................ 151
4.4. SR protein family ....................................................................................................................... 152
4.4.1. SR protein conservation ..................................................................................................... 152
4.4.2. SR protein structure ............................................................................................................ 154
4.4.3. SR protein phosphorylation................................................................................................ 156
4.5. SRSF3 controls multiple molecular functions in the nucleus and the cytoplasm. ................. 157
4.5.1. Alternative splicing ............................................................................................................. 159
4.5.2. Transcription ....................................................................................................................... 161
4.5.3. miRNA maturation ............................................................................................................. 162
4.5.4. mRNA nuclear export......................................................................................................... 162
4.5.5. Translation ........................................................................................................................... 164
4.6. SRSF3 regulates many biological pathways in health and disease......................................... 164
4.6.1. Cellular proliferation and cancer ....................................................................................... 165
4.6.2. Viral infection ..................................................................................................................... 167
4.6.3. Gametogenesis and development....................................................................................... 168
4.6.4. Metabolism .......................................................................................................................... 170
4.6.5. Inflammation ....................................................................................................................... 170
4.6.6. Central nervous system homeostasis and disorders .......................................................... 171
4.7. Conclusion .................................................................................................................................. 174
Discussion générale ............................................................................................................................... 176
5.1. Résumé des chapitres précédents .............................................................................................. 176
5.2. Profilage du sécrétome de la microglie présymptomatique .................................................... 178
5.3. Profilage moléculaire de la microglie dans la neuroinflammation ......................................... 180
5.3.1. L’étude du profil transcriptomique de la microglie symptomatique ............................... 180
5.3.2. Le phénotype DAM dans les maladies neurodégénératives ............................................ 181
5.3.3. Le dynamisme du profil protéomique de la microglie dans la pathogénèse de la SLA . 186
5.4. La protéine SRSF3 dans la pathogénèse de la SLA ................................................................. 192
5.4.1. La phosphorylation de SRSF3 dans la SLA ...................................................................... 193

xi
5.4.2. La distribution cytoplasmique de SRSF3 dans la microglie symptomatique ................. 195
5.4.3. Les avenues thérapeutiques ciblant SRSF3....................................................................... 197
Conclusion ............................................................................................................................................. 200
Bibliographie ......................................................................................................................................... 203

xii
Liste des figures
Figure 1: La découverte des gènes associés à la SLA ............................................................................. 6
Figure 2: La structure de la protéine SOD1 et ses mutations liées à la SLA ......................................... 8
Figure 3: La structure de la protéine TDP43 et ses mutations liées à la SLA ..................................... 11
Figure 4: Les mécanismes de cytotoxicité participant à la pathogénèse de la SLA ............................ 15
Figure 5: La dérégulation du cytosquelette menant à la perte des jonctions neuromusculaires ......... 17
Figure 6: La dérégulation de la protéostasie .......................................................................................... 20
Figure 7: Le stress oxydatif et la dysfonction mitochondriale.............................................................. 24
Figure 8: La propagation prionique dans le SNC .................................................................................. 26
Figure 9: L’activation microgliale dans la neuroinflammation aigue .................................................. 31
Figure 10: L'activation chronique de la microglie dans la SLA ........................................................... 35
Figure 11: Le dialogue neurone-glie et la mort neuronale .................................................................... 39
Figure 12: L'activation du système immunitaire dans la SLA .............................................................. 49
Figure 13: La dualité de l'inflammation dans la SLA: hyperinflammation et immunodéficience ..... 51
Figure 14: SR protein family structure ................................................................................................. 155
Figure 15: SRSF3 molecular functions in the cell............................................................................... 158
Figure 16: Gene targets of SRSF3 in biological pathways ................................................................. 174
Figure 17:La régulation de la voie de signalisation NF-κB par G3BP2 et HSPB1........................... 189
Figure 18: La régulation des kinases de SRSF3 dans la SLA ............................................................ 194

xiii
Liste des tableaux
Table 1:Evolutional conservation of the genes from the SR protein family...................................... 154
Tableau 2:Comparaison des profils transcriptomiques du phénotype DAM ..................................... 183
Tableau 3: La régulation des protéines microgliales impliquées dans la voie de signalisation NF-κB
................................................................................................................................................................ 188

xiv
Liste des acronymes
AAV: Adeno-associated virus
AGE: Advanced glycation end-product
ALS: Amyotrophic lateral sclerosis
ATP: Adenosine triphosphate
BDNF: Brain-derived neurotrophic factor
BHE: Barrière hémato-encéphalique
CCS: Copper chaperone for SOD1
CD: Cluster of differentiation
cDNA: Complementary deoxyribonucleic acid
CMH: Complexe majeur d’histocompatibilité
CMV: Cytomegalovirus
CSF: Cerebrospinal fluid
CSP: Cellule de Schwann périsynaptique
CTD : C-terminal domain
CX3CL1: Fractalkine
DAM: Disease associated microglia
DAMP: Danger associated molecular pattern
DFT: Démence fronto-temporale
DMEM: Dulbecco’s modified eagle medium
EBSS: Earle’s balanced salt solution
EDTA: Ethylenediaminetetraacetic acid
ERAD: Endoplasmic reticulum associated degradation
fALS: Familial amyotrophic lateral sclerosis
FACS: Fluorescent activated cell sorting
FBS: Fetal bovine serum
FoxP3: Forkhead box protein 3

xv
G-CSF: Granulocyte colony stimulating factor
GDNF: Glia-derived neurotrophic factor
GFP: Green fluorescent protein
GPX: Glutathion peroxydase
GR: Glutathion réductase
GSH: Glutathion
GSS: Glutathion synthétase
HPRT1: Hypoxanthine-guanine phosphoribotransferase
hSOD1: human SOD1
HSP: Heat shock protein
IGF-1 : Insulin-like growth factor
IFN: Interféron
IL: Interleukin
IL-10R: Interleukin-10 receptor
IRES: Internal ribosome entry site
ITR: Inverted terminal repeat
JNM: Jonction neuromusculaire
KIR3DL2: Killer cell immunoglobulin-like cell receptor 3 DL 2
LPS: Lipopolysaccharide
Luc: luciferase
MGnD: Microglial neurodegenerative phenotype
miRNA: MicroRNA
MPTP: Methylphényltétrahydropyridine
mRNA: messenger RNA
mSOD1: misfolded superoxide dismutase Cu/Zn
NF-κB: Nuclear factor κB
NLS: Nuclear localisation signal

xvi
OB: Olfactory bulb
PAMP: Pathogen associated molecular pattern
PBS: Phosphate buffered saline
PCR: Polymerase chain reaction
PN: post-natal
pri-miRNA: Primary micro-RNA
pSRSF3 : Phosphorylated SRSF3
qRT-PCR: Quantitive reverse transcriptase polymerase chain reaction
RNA: Ribonucleic acid
RNS: Reactive nitrogen species
ROI: Region of interest
ROS: Reactive oxygen species
RRM: RNA recognition motif
scAAV: self-complementary adeno-associated virus
SEM: Standard error of the mean
siRNA: Small interfering RNA
SLA: Sclérose latérale amyotrophique
SOD1: Superoxyde dismutase Cu/Zn
SNC: Système nerveux central
SNP: Système nerveux périphérique
SDS: Sodium dodecylsulfate
SDS-PAGE: Sodium dodecylsulfate – polyacrylamide gel electrophoresis
SRSF3 : Serine-arginine rich splicing factor 3
SSC: Saline sodium citrate buffer
TDP43: TAR DNA-binding protein 43kDa
TEA: Triethanolamine
TGF: Tumor growth factor

xvii
TLR: Toll-like receptor
TNF: Tumor necrosis factor
TRAP: Translating ribosome immunopurification
Treg: Lymphocyte T régulateur
Tweak: TNF-like weak inducer of apoptosis
UPR: Unfolded protein response
UPS: Ubiquitin-proteasome system
UTR: Untranslated region
VEGF: Vascular endothelium growth factor
YM1: Chitinase-like protein 3

xviii
à Maude, qui a été à mes côtés durant toutes
ces années

xix
Remerciements
Je tiens à remercier, pour commencer, mes amis, les membres de ma famille et ma conjointe,
Maude Langelier, pour leur support émotionnel durant les périodes plus éprouvantes de ce
doctorat. Votre soutien m’a donné le courage de continuer jusqu’au bout.
Je remercie aussi mes collègues que j’ai cotoyé tous les jours : Kallol Dutta, Priyanka Patel,
Silvia Pozzi, Amélie Poulin-Brière, Sunny Kumar, Yuan Chen Weng, Mathieu Gravel,
Senthil Krishnasamy, Sai Sampath Thammisetty, Romina Barreto, Reza Rahimian et Pierre
Cordeau, Tereza Ljutić et Andrea Markovinović. Merci pour vos conseils durant mon
cheminement.
Je souhaite remercier Dr. Jean-Pierre Julien pour m’avoir engagé dans son laboratoire en
2009 et pour m’avoir donné la piqûre pour la recherche en neurosciences, Dre Ivana Munitić
de l’Université de Rijeka pour m’avoir accueilli dans son laboratoire pendant 3 mois dans
lequel j’ai pu enseigner mon savoir, et Dre. Jasna Križ pour m’avoir pris sous son aile pendant
ma maîtrise et mon doctorat. Merci pour toutes les opportunités que vous m’avez donné de
faire rayonner mes travaux à travers les divers congrès, stages et symposiums auxquels j’ai
participé.
Je veux aussi remercier mes mentores : Christine Bareil, Geneviève Soucy et Mélanie
Lalancette-Hébert pour avoir pu répondre à mes questions et mes inquiétudes à tous
moments. Votre aide a été considérable à ma réussite. Je souhaite remercier tout spécialement
Hejer Boutej qui m’a guidé à travers ces années ardues et m’a poussé à réussir. Aucun aspect
de mon travail n’aurait été possible sans ton aide.
Finalement, je veux remercier mes deux acolytes, Mme Laurence Renaud et Dr. Vincent
Picher-Martel. Les moments passé ensemble tant au laboratoire qu’ailleurs ont été parmi les
meilleurs de ces dernières années. Merci pour vos conseils, merci pour votre camaraderie

xx
Avant-propos
Dans le Chapitre 2, l’article « IL-10 Controls Early Microglial Phenotypes and Disease Onset
in ALS Caused by Misfolded Superoxide Dismutase 1 » a été publié dans le journal « Journal
of Neurosciences » le 20 janvier 2016. J’y suis co-premier auteur avec M. Mathieu Gravel.
Auteurs : Mathieu Gravel, Louis-Charles Béland, Geneviève Soucy, Essam Abdelhamid,
Reza Rahimian, Claude Gravel, et Jasna Kriz.
Contributions des auteurs : Mathieu Gravel et Louis-Charles Béland ont planifié et réalisé
des expériences et ont participé à la rédaction de l’article. Geneviève Soucy a les injections
intrathécales. Essam Abdelhamid et Reza Rahimian ont réalisé des expériences. Jasna Kriz a
planifié des expériences et a participé à la rédaction de l’article.
Dans le Chapitre 3, l’article « SRSF3 acts as a ribosome-based checkpoint and regulates
translation of immune mRNAs in ALS microglia » a été soumis dans le journal « Neuron »
le 9 décembre 2020. J’y suis premier auteur.
Auteurs : Louis-Charles Béland, Hejer Boutej, Romina Barreto, Emiliano Trias, Sai Sampath
Thammisetty, Charles Joly-Beauparlant, Arnaud Droit, Vincent Picher-Martel, Nicolas
Dupré, Luis Barbeito, Jasna Kriz
Contribution des auteurs : Louis-Charles Béland a planifié et réalisé des expériences et a écrit
l’article. Hejer Boutej a planifié et réalisé des expériences et a écrit l’article. Romina Nunez-
Barreto, Sai Sampath Thammisetty et Emiliano Trias ont réalisé des expériences. Charles
Joly-Beauparlant et Arnaud Droit ont réalisé les études de bioinformatique. Vincent Picher-
Martel et Nicolas Dupré ont fourni des tissus. Luis Barbeito et Jasna Kriz ont planifié le
projet.

1
Introduction
1.1. Sclérose latérale amyotrophique
La sclérose latérale amyotrophique (SLA) est une maladie neurodégénérative caractérisée
par la dénervation des jonctions neuromusculaires et la mort des motoneurones supérieurs et
inférieurs causant une paralysie tardive (diagnostiquée en moyenne vers 55 ans) mais
progressive menant à la mort par insuffisance respiratoire 3 à 5 après le diagnostic (Taylor et
al. 2016). On compte environ 10% de cas familiaux de SLA, causés par des mutations dans
une panoplies gènes, et 90% de cas sporadiques. Dans les deux types de cas, l’agrégation de
protéines mal repliées est responsable de la dégénérescence des neurones moteurs
(Robberecht et Philips 2013). Cette mort neuronale est accompagnée par une réponse
inflammatoire chronique qui corrèle avec la propagation de la maladie dans les tissus affectés
(Robberecht et Philips 2013). Le pronostic de la SLA est plutôt difficile à établir et vient du
fait que cette maladie est, en soi, très hétérogène entre les patients. L’hétérogénéité de la SLA
vient de la variabilité de plusieurs facteurs tels que l’âge à l’apparition des symptômes, le site
d’apparition des symptômes, l’implication de gènes mutants ou l’association avec d’autres
conditions comme la démence fronto-temporale (DFT) (Hardiman et al. 2017). Bien que la
SLA a été décrite pour la première fois à la fin du 19e siècle, la science n’a toujours pas
découvert de remède définitif pour traiter cette maladie. Avec un taux d’incidence annuel
global de 1,9 par 100 000 individus, plus de 220 000 personnes sont atteintes de la SLA dans
le monde (Chiò et al. 2013, Arthur et al. 2016). Les prévisions estiment qu’environ 375 000
personnes en souffriront en 2040 avec le vieillissement de la population mondiale (Arthur et
al. 2016). Étant donné la perte rapide de la qualité de vie des personnes atteintes et de leur
famille, l’absence actuelle de traitement efficace et l’augmentation prévue des cas dans les
années à venir fait en sorte que la recherche sur la SLA soit d’une importance capitale.
C’est entre les années 1865 et 1869 que le docteur Jean-Martin Charcot, aussi connu comme
le père de la neurologie, a décrit la sclérose latérale amyotrophique à la suite d’une série
d’étude sur des tissus post-mortem (Kumar et al. 2011). Ainsi, dans ses œuvres complètes,
Charcot corrèle les symptômes cliniques de ses patients (atrophie progressive des muscles,
contractions fibrillaires, faiblesse motrice précédant ou accompagnant l’atrophie et rigidité

2
au repos due à des contractures spasmodiques) à des observations anatomiques. Charcot y
parle d’une détérioration de la colonne latérale de la moelle épinière associée aux
contractions fibrillaires (fasciculation) et à la spasticité, ainsi qu’une détérioration des
cellules de la corne antérieure de la matière grise de la moelle épinière associée à l’atrophie
musculaire (Goetz 2000). En 1874, Charcot propose le terme sclérose latérale amyotrophique
pour désigner cette maladie, un terme qui est d’abord basé sur la description anatomique de
la maladie au lieu de sa description clinique, contrairement à l’habitude de l’époque (Goetz
2000).
À la fin des années 1980, des agrégats cytoplasmiques de neurofilaments ubiquitinés dans le
soma des motoneurones de patients atteints de la SLA furent détectés (Murayama et al. 1989).
Il fut découvert plus tard que ces agrégats cytoplasmiques contiennent la protéine TAR DNA-
binding protein 43 (TDP43), délocalisée dans le cytoplasme et, elle aussi, ubiquitinée
(Neumann et al. 2006). Cette protéine semble aussi impliquée dans la DFT, où elle est, encore
une fois, agrégée dans le cytoplasme (Arai et al. 2006). La DFT est une maladie cognitive
parfois retrouvée chez les patients atteint de SLA. Depuis la découverte de la délocalisation
et l’agrégation de TDP43 dans la SLA, la présence de TDP43 dans le cytoplasme des
neurones est reconnue comme une des caractéristiques principales de la SLA, permettant,
entre autres, la confirmation de son diagnostic (Feneberg et al. 2018).
À la même période, la présence de gliose dans la moelle épinière de patients atteint de la SLA
fut découverte (McGeer et al. 1988). Ainsi, en utilisant le marqueur du complexe majeur
d’histocompatibilité (HLA-DR) présent sur les cellules microgliales et le marqueur GFAP
exprimé par les astrocytes, il fut possible d’identifier la présence d’une microglie activée de
et d’astrocytes réactifs, tous deux indicatifs d’une neuroinflammation accompagnant la
neurodégénération. Si à ce moment le rôle des différents éléments de la glie n’était pas
évident, il est maintenant possible d’affirmer que les astrocytes, tout comme les cellules
microgliales, contribuent à la pathogénèse de la SLA et exacerbent la progression de la
maladie (Robberecht et Philips 2013). L’aspect de non-autonomie cellulaire de la
pathogénèse de la SLA et la présence de neuroinflammation mit beaucoup d’espoir dans
l’utilisation de médicaments anti-inflammatoires pour le traitement de cette maladie.

3
Malheureusement, à ce jour, tous les traitements anti-inflammatoires testés en phase
cliniques ont échoué (Benatar 2007, DeLoach et al. 2015).
Ainsi, plus de 150 ans après la découverte de la SLA par Jean-Martin Charcot, seuls deux
traitements sont reconnus mondialement et utilisés pour le traitement de cette maladie, soit
le riluzole et l’edaravone qui ne peuvent malheureusement que ralentir la progression de la
maladie sans toutefois la guérir complètement (Miller et al. 2012, Rothstein 2017). Le
riluzole, un inhibiteur de la neurotransmission glutamatergique, a été approuvé en 1995 et
augmente l’espérance de vie de 1 à 3 mois (Miller et al. 2012). Plus récemment, en 2017,
l’antioxydant edaravone (Rothstein 2017) fut à son tour approuvé. Toutefois, alors que
l’utilisation de l’edaravone ralentit la progression de la maladie, elle n’augmente pas
l’espérance de vie des patients (Sawada 2017). La situation actuelle démontre l’urgence
critique pour le développement de nouveaux traitements.
1.1.1. Aspects cliniques et anatomiques de la SLA
La SLA peut se déclarer de diverses façons selon la région du SNC premièrement affectée.
Différents sous-groupes de la maladie peuvent justement se distinguer selon la première
région atteinte. Ainsi, avec la maladie débutant dans la moelle épinière, les premiers
symptômes sont des fasciculations et une faiblesse musculaire dans un membre, souvent
unilatérale au début. Avec la maladie débutant dans le bulbe rachidien, on assiste à des
difficultés dans l’élocution, la mastication et la déglutition (Renton et al. 2014, Taylor et al.
2016). De plus, l’hétérogénéité dans l’apparition des symptômes est un des aspects de
l’hétérogénéité générale du pronostic de la SLA. Par exemple, les patients qui développent
des symptômes bulbaires en premier ont un pronostic généralement plus mauvais que les
patients développant la maladie dans leurs membres, tandis que la maladie débutant dans les
neurones et les muscles régissant la respiration donne le pire pronostic (Ragagnin et al. 2019).
L’apparition de ces symptômes est expliquée par la perte des jonctions neuromusculaires
(JNM). Bien que la perte des JNM pourrait être précédée par la mort du corps cellulaire
neuronal, l’hypothèse la plus répandue voudrait plutôt que la SLA soit une axonopathie

4
distale où la neurodégénération commence à la plaque motrice et affecte subséquemment le
reste du motoneurone (Cappello et Francolini 2017). En effet, un modèle murin confirme
qu’à l’apparition des symptômes près de 60% des plaques motrices sont déjà dénervées.
Chronologiquement, la dénervation puis la perte des axones moteurs commencent bien avant
l’apparition des symptômes et la mort neuronale en tant que telle (Fischer et al. 2004). À la
suite de l’apparition des symptômes, les corps cellulaires des neurones deviennent vacuolés,
précédant leur mort (Fischer et al. 2004). Toutefois, ce ne sont pas tous les neurones moteurs
qui ont la même susceptibilité à la neurodégénération dans la SLA. Bien que les
motoneurones alpha du tract corticospinal dégénèrent, les neurones oculomoteurs et les
motoneurones du noyau d’Onuf sont résistants à la neurodégénération (Ragagnin et al. 2019).
Toutefois, les inclusions cytoplasmiques de TDP43 sont détectables dans les neurones du
cortex moteur, du tronc cérébral et de la moelle épinière chez les patients et les modèles
murins (Arai et al. 2006, Tan et al. 2007, Shan et al. 2009, Okamoto et al. 2011), ainsi que
dans les neurones d’une panoplie de régions cérébrales comme le lobe occipital, l’amygdale
ou l’hippocampe (Geser et al. 2008, Ragagnin et al. 2019). La présence d’agrégats toxiques
et la nature elle-même des neurones moteurs ne sont donc pas suffisant pour expliquer la
susceptibilité de certains motoneurones à la dégénération. La taille, la sensibilité à
l’excitotoxicité liée au glutamate et au Ca2+ ainsi que l’innervation d’un certains types de
fibres musculaires et la stimulation excitatrice par les motoneurones gamma sont quelques
hypothèses qui tentent d’expliquer la susceptibilité des motoneurones alpha à la
neurodégénération (Lalancette-Hebert et al. 2016, Ragagnin et al. 2019).
Accompagnant la mort des neurones moteurs du tract corticospinal, vient une atrophie
musculaire, comme le nom de la maladie l’indique (Ragagnin et al. 2019). Comme mentionné
plus haut, à la suite de la perte des JNM, les patients atteint de la SLA développent de
l’hyperréflexie, de la spasticité, des fasciculations, une faiblesse et une atrophie musculaire
(Tandan et Bradley 1985, Goetz 2000). Évidemment, la progression de ces symptômes dans
les muscles suit la propagation et le degré de mort neuronale dans la moelle épinière. Durant
les stades avancés de la maladie, la qualité de vie des patients diminue grandement lors de
l’atteinte des muscles régissant la respiration (Hazenberg et al. 2016). Ainsi, l’utilisation de
ventilation non-invasive est utilisé pour pallier l’insuffisance respiratoire grandissante. En

5
fin de vie, l’insuffisance respiratoire, l’impuissance à excréter les sécrétions trachéales et les
pneumonies subséquentes ont raison des patients atteints de la SLA (Braun et al. 2018).
1.1.2. Aspects génétiques de la SLA
Dans la SLA, la majorité des cas, soit 90%, sont de forme sporadique (Robberecht et Philips
2013). Plusieurs hypothèses ont été émises pour expliquer cette apparition sporadique tels
que l’exposition aux métaux lourds ou au paraformaldéhyde, ou encore les traumatismes
crâniens (Talbott et al. 2016). Toutes ces hypothèses n’ont jamais été formellement
confirmées, laissant l’âge comme seul facteur de risque pour la SLA. La forme sporadique
ne diffère pas cliniquement de la forme familiale, mais certaines mutations peuvent causer
l’apparition de la maladie à un âge moins avancé ou bien une progression plus rapide.
Toutefois, étant donnée l’hétérogénéité de la SLA, certains cas sporadiques peuvent avoir le
même pronostic qu’un cas familial agressif.
Environ 10% des cas de SLA sont de forme familiale, c’est-à-dire, d’une forme génétique
qui se transmet verticalement. Depuis l’association de la SLA avec des mutations dans le
gène superoxyde dismutase Cu/Zn (SOD1) en 1993 (Rosen et al. 1993), plusieurs dizaines
de gènes ont été associés avec le développement de cette maladie (Fig.1). À ce jour, les
découvertes de gènes impliqués dans la forme familiale de SLA peuvent expliquer environ
75 à 80% des cas familiaux. Des gènes mutés les plus communs, on retrouve C9orf72, (40%
des cas familiaux), SOD1 (20%), TDP43 et FUS (5% chacun) (Robberecht et Philips 2013,
Taylor et al. 2016). Beaucoup des gènes impliqués dans la SLA ont normalement un rôle
dans le métabolisme de l’ARN, ou dans la stabilité des protéines. Une fois mutées, les
protéines issues de ces gènes se retrouvent mal repliées et peuvent, non-seulement perdre
leurs fonctions homéostatiques, mais aussi gagner des fonctions toxiques. Les protéines mal
repliées s’oligomérisent et s’agrègent, empêchant le fonctionnement normal de la cellule. De
plus, le mauvais repliement de certaines protéines peut se propager de façon prionique, vers
les protéines d’autres cellules encore non affectées (Ayers et Cashman 2018). Dans cette
section, seront énumérés les principaux gènes associés à la SLA ainsi que leurs mécanismes
uniques les impliquant à la pathogénèse de cette maladie.

6
Figure 1: La découverte des gènes associés à la SLA
Depuis la découverte de l’implication du gène SOD1 dans la SLA, une panoplie de gènes a
été lié au le développement de la maladie. Figure adaptée de «Genetics of Amyotrophic
lateral sclerosis» (Gregory et al. 2020)
1.1.2.1. Superoxyde dismutase Cu/Zn
SOD1 est une métalloenzyme responsable de la détoxification des espèces réactives de
l’oxygène (reactive oxygen species, ROS). SOD1 peut fixer un atome de cuivre ou de zinc,
nécessaire à sa fonction enzymatique, à la suite de la formation d’un homodimère (Broom et
al. 2015). La majeure partie de SOD1 est retrouvée dans le cytosol, mais une petite quantité
(5%) est retrouvée dans l’espace intermembranaire mitochondrial. SOD1 est recrutée dans
l’espace intermembranaire mitochondrial dans sa forme d’apoenzyme (sans l’atome
métallique) avec l’aide du récepteur de translocation membranaire TOM (Greco et al. 2019).
Dans un environnement hypoxique où le régime de la chaîne respiratoire mitochondrial est
plus élevé, la chaperonne du cuivre pour SOD1 (copper chaperone for SOD1, CCS) est
recrutée depuis le cytosol vers l’espace intermembranaire mitochondrial pour compléter la
maturation de SOD1 (Tafuri et al. 2015).

7
Dans la SLA, on dénombre plus de 150 mutations dans le gène codant la protéine SOD1
(Fig.2) causant une maladie généralement d’origine lombaire et uniquement motrice (Picher-
Martel et al. 2016). Cependant, la SLA causée par la SOD1 mutée peut être, dans de rares
cas, accompagnée par la DFT (Robberecht et Philips 2013). Les mutations de SOD1 causent
son mauvais repliement et induisent son oligomérisation et son agrégation (Ayers et Cashman
2018). Lorsque mal repliée, SOD1 peut induire sa forme aberrante aux unités SOD1
correctement repliées, à la manière d’un prion. Ce mécanisme pourrait aussi être partagé par
la forme oxydée de SOD1, retrouvée dans les tissus SLA à la suite du stress oxydatif associé
à la maladie (Ezzi et al. 2007). La plupart des mutations de ce gène influencent la structure
quaternaire de la protéine SOD1, c’est-à-dire sa dimérisation et sa fixation d’atome
métallique (Broom et al. 2015). Les régions de SOD1 responsables de la dimérisation sont
relativement petites et composées principalement de résidus hydrophobes (Fig.2). Ainsi, la
moindre mutation affecte grandement la structure tertiaire de SOD1 et son potentiel à former
un dimère (Broom et al. 2015). De plus, bien que les mutations du gène SOD1 induisent tous
un mauvais repliement de la protéine, certaines mutations (A4V) causent une forme plus
agressive de la maladie (Cudkowicz et al. 1997) alors que d’autres (D90A) sont liées à une
progression plus lente (Andersen et al. 1996). Cela est possiblement dû à des différences dans
les potentiels d’oligomérisation, d’agrégation et de diffusion prionique (Broom et al. 2014).
La propagation prionique de la protéine SOD1 semble être dépendant de son tryptophane à
la position 32 (Fig.2) (McAlary et al. 2019), acide aminé conservé uniquement chez les
primates (DuVal et al. 2019). Toutefois, bien que les mutations de SOD1 affectent sa
structure quaternaire, il semble que la toxicité de la protéine mutée vienne uniquement d’un
gain de fonction toxique. La perte de fonction de SOD1 mutante est moins claire, mais
pourrait causer, du moins en partie, le stress oxydatif dans les neurones moteurs (Saccon et
al. 2013).

8
Figure 2: La structure de la protéine SOD1 et ses mutations liées à la SLA
La protéine issue du gène SOD1, lorsque mal repliée est impliquée dans la pathogénèse de la
SLA. Plus de 150 mutations, listées en partie sur cette figure, peuvent induire un mauvais
repliement de la protéine. La structure mal repliée de SOD1 l’empêche de se dimériser
correctement en masquant les résidus de l’interface du dimère, ces résidus représentant moins
de 10% de la séquence totale. Figure adaptée de «Is SOD1 loss of function involved in
amyotrophic lateral sclerosis ?» (Saccon et al. 2013).
Alors que la délocalisation et l’agrégation de TDP43 sont communes à tous les cas de SLA,
comme mentionné précédemment (Arai et al. 2006, Tan et al. 2007, Shan et al. 2009,
Okamoto et al. 2011), il existe un débat à savoir si l’agrégation de la protéine SOD1 est une
caractéristique générale de la SLA. Plusieurs anticorps contre la forme repliée de SOD1 ont
été produits et testés sur des sections de moelle épinière de patient avec des résultats
contradictoires selon les études (Liu et al. 2009, Bosco et al. 2010, Kerman et al. 2010, Paré
et al. 2018). Il n’y a donc pas de consensus sur l’importance de SOD1 dans les cas de SLA
sporadique ou les modèles animaux non SOD1.
Tout de suite après la découverte de l’implication de SOD1 dans la SLA en 1993 (Rosen et
al. 1993), des modèles de souris transgéniques ont été produits afin de mimer le
développement de la maladie. Le premier modèle murin, développé en 1994, exprime 34
copies du gène humain SOD1 avec la mutation G93A (Gurney et al. 1994) et reste encore un
des modèles les plus utilisés encore à ce jour. Cette souris développe la maladie dans les
membres postérieurs, unilatéralement pour commencer. La maladie se propage ensuite dans
le reste du corps et la souris en meurt, quelques mois après l’apparition des symptômes. Dans

9
ce modèle, la dénervation et la mort neuronale sont accompagnées par une gliose importante
(Picher-Martel et al. 2016, Spiller et al. 2018). L’astrogliose et la microgliose suivent la
propagation de la maladie dans la moelle épinière et exacerbe sa progression (Béland et al.
2020). Généralement, ce modèle murin reproduit bien les caractéristiques anatomiques et
cliniques de la SLA chez l’humain (Picher-Martel et al. 2016). Des modèles murins
exprimant d’autres mutation pour SOD1 (G37R, G85R, D90A, pour en nommer quelques-
uns) existent aussi mais sont moins utilisés que le modèle hSOD1G93A (Wong et al. 1995,
Bruijn et al. 1997, Jonsson et al. 2006). Malgré l’utilisation répandue des modèles pour SOD1
mutante, plusieurs critiques pourraient être faites concernant ces modèles. Premièrement, une
récente étude semble montrer que la microgliose chez les souris hSOD1G93A serait excessive,
ce qui pourrait être dû au grand nombre de copies du gène humain, exprimé à une proportion
de 17 :1 contre le gène murin (Gurney et al. 1994, Spiller et al. 2018, Ragagnin et al. 2019).
De plus, étant donné que les mutations de SOD1 ne représentent que 20% des formes
familiales et moins de 1% des formes sporadiques de la SLA (moins de 3% des cas totaux)
(Robberecht et Philips 2013), l’utilisation de ce modèle ne représente que très peu la diversité
génétique de la maladie. Toutefois, les modèles murins exprimant SOD1 mutées restent très
importants pour l’étude des mécanismes de toxicité cellulaire en aval de la toxicité
directement liée au gène mutant. Ces mécanismes cellulaires dans les motoneurones et dans
les autres cellules du système nerveux central (SNC) des modèles de SOD1 mutante, ainsi
que les caractéristiques anatomiques de la maladie chez ces modèles sont, généralement, les
mêmes que chez l’humain (Picher-Martel et al. 2016).
1.1.2.2. TAR DNA-binding protein 43
TDP43 est une protéine liant l’ADN et l’ARN. Elle est codée par le gène TARDBP et
impliquée à différents niveaux dans le métabolisme de l’ARN comme la transcription,
l’épissage, le transport nucléo-cytoplasmique des ARN et la traduction (Weskamp et
Barmada 2018). Ainsi, la protéine TDP43 peut réguler l’expression d’une panoplie de gènes,
incluant sa propre expression. L’autorégulation des protéines liants l’ARN est un mécanisme
commun, impliquant la production d’isoformes non-fonctionnelles d’ARNm par épissage
alternatif (Polymenidou et al. 2011). Durant un stress cellulaire, TDP43 est surexprimée et
est exportée dans le cytoplasme de façon transitoire (Johnson et al. 2011, Swarup et al. 2012).

10
Lors du stress cellulaire chronique associé à la SLA, cette surexpression et cette
délocalisation sont permanentes. Dans les modèles murins, une expression de TDP43 deux à
quatre fois supérieure à la normale est suffisante pour causer de la neurodégénération
(Wegorzewska et Baloh 2011). Cette surproduction observée chez les patients atteints de la
SLA correspond à un niveau d’expression 2,5 fois supérieur à la normale (Swarup et al.
2012). Dans les cas familiaux de SLA causés par une mutation dans le gène TARDBP, la
dérégulation de l’expression de TDP43 s’explique par la présence de la majorité des
mutations dans la région C-terminale du gène (Fig.3) (Barmada et Finkbeiner 2010). Les
mutations au niveau de de cette région augmentent le potentiel de délocalisation et
d’agrégation de TDP43 en stabilisant la protéine. Traduite dans le cytoplasme, la rétention
subséquente de TDP43 mutée ou mal repliée hors du noyau l’empêcherait de s’autoréguler,
favorisant sa surexpression (Weskamp et Barmada 2018). Dans la SLA, la délocalisation et
l’agrégation de TDP43 ont pour effet à la fois une perte de fonctions normales et un gain de
fonctions toxiques. Ainsi, comme perte de fonction normale, on retrouve une baisse dans la
stabilité des ARNm ou encore un épissage alternatif aberrant suivit d’une augmentation de
la dégradation des ARNm non-sens (Weskamp et Barmada 2018). TDP43 gagne les mêmes
fonctions toxiques que les autres gènes impliqués dans la SLA (tel qu’il sera vu dans la
section 1.1.3) mais est aussi impliqué dans l’activation de la voie de signalisation NF-κB
dans les neurones moteurs (Swarup et al. 2011) et dans l’augmentation de la formation des
granules de stress, affectant leur nombre et leur taille (Khalfallah et al. 2018). La
délocalisation chronique de TDP43 dans le cytoplasme affecte le dynamisme des granules de
stress et contribue à la séquestration pathologique d’ARNm, participant à la dérégulation de
l’homéostasie cellulaire (Dewey et al. 2011, Khalfallah et al. 2018).

11
Figure 3: La structure de la protéine TDP43 et ses mutations liées à la SLA
L’agrégation et la délocalisation de la protéine TDP43 sont des caractéristiques de la SLA
retrouvées à la fois dans les cas sporadiques et les cas familiaux. La majorité des mutations
du gène TARDBP causant la maladie (dont une partie est représentée sur la figure ci-haut) se
situent dans la région C-terminale du gène, soit le domaine riche en glycine. Figure adaptée
de «TAR DNA-binding protein 43 in Neurodegenerative Disease» (Chen-Plotkin et al. 2010).
Les modèles murins exprimant TDP43 mutée ou TDP43 sauvage à des niveaux semblables
à ceux retrouvés chez les patients atteints de la SLA ne récapitulent que rarement toutes les
caractéristiques de la SLA. Les neurones moteurs de ces modèles possèdent bel et bien des
inclusions cytoplasmiques de TDP43, mais la neurodégénération n’est pas aussi intense que
chez l’humain ou chez les modèles murins exprimant la protéine SOD1 mutante (Swarup et
al. 2011, Wegorzewska et Baloh 2011, Mitchell et al. 2015, Picher-Martel et al. 2016). La
maladie induite par TDP43 est accompagnée dans certains modèles par des troubles cognitifs,
rappelant la DFT chez l’humain. Cependant, contrairement aux modèles pour SOD1 mutante,
l’utilisation de TARDBP comme gène cible représente mieux la diversité des cas de SLA.
1.1.2.3. Protéine liant l’ARN FUS
Tout comme TDP43, la protéine liant l’ARN FUS (Fused in sarcoma) est impliquée dans le
métabolisme de l’ARN. Ainsi, des mutations dans ce gène mènent eux aussi à un gain de
fonction toxique et une perte de fonctions normales associées à la stabilité des ARNm
(Ishigaki et Sobue 2018). Une bonne partie des mutations de FUS se retrouve dans son signal
de localisation nucléaire, favorisant donc son accumulation cytoplasmique, son absence dans

12
le noyau et son autorégulation aberrante. Dans le noyau, FUS est normalement responsable
de l’épissage alternatif (Zhou et al. 2013). Ainsi, dans les cellules exprimant la protéine FUS
mutante, l’épissage alternatif est altéré provoquant un déséquilibre des différentes isoformes
des gènes cibles de FUS et une augmentation de la dégradation des ARNm non-sens (Lagier-
Tourenne et al. 2012, Fujioka et al. 2013, Zhou et al. 2013, Ishigaki et Sobue 2018). De plus,
dans les neurones, FUS est impliquée dans le transport d’ARNm le long des axones en
formant des organites non-membraneux pour acheminer ces ARNm vers les dendrites. Les
mutations dans FUS diminuent son potentiel de séparation de phase liquide-liquide
nécessaire pour à la formation de ces organites, altérant ainsi les fonctions pré-synaptiques
des neurones (Murakami et al. 2015). Ne pouvant plus passer de la phase soluble à ces
organites non-membraneux, la protéine FUS mutante forme des agrégats cytoplasmiques
toxiques (Chen et al. 2019). Non seulement FUS est impliquée dans l’épissage alternatif et
le transport des ARN, mais cette protéine joue aussi un rôle dans la traduction lorsque qu’elle
est retrouvée dans le cytoplasme. (Sévigny et al. 2020). En effet, FUS réprime la traduction
en interagissant avec les polyribosomes, une répression plus forte lorsque le gène FUS
possède des mutations liées à la SLA. Cette répression traductionnelle contribue donc à la
cytotoxicité de cette protéine et à la pathogénèse de la SLA (Sévigny et al. 2020).
Beaucoup d’études se sont consacrées aux mécanismes de toxicité cellulaire de FUS mutante
in vivo et in vitro (Fushimi et al. 2011, Kryndushkin et al. 2011, Sharma et al. 2016, López-
Erauskin et al. 2018, Sévigny et al. 2020). Ces études sont essentielles à la compréhension
de la pathogénèse de la SLA et de la toxicité cellulaire en aval de la mort neuronale. Les
modèles animaux exprimant la protéine FUS mutante et représentatifs de l’évolution de la
maladie chez l’humain sont peu nombreux (Mitchell et al. 2013, Sephton et al. 2014, Picher-
Martel et al. 2016, Stephenson et Amor 2017). Tous comme pour TDP43, l’agrégation
cytoplasmique de FUS dans les neurones moteurs constitue un aspect commun de la
pathogénèse de la maladie dans les cas sporadiques et les cas familiaux de la SLA (Deng et
al. 2010). Ainsi, l’utilisation de modèles animaux dans lesquels la protéine FUS mutante
forme des agrégats insolubles est représentative de la diversité de la maladie retrouvée chez
l’humain.

13
1.1.2.4. C9orf72
Contrairement aux gènes mentionnés plus haut, la mutation du gène C9orf72, causant environ
40% des cas de SLA familiale (Robberecht et Philips 2013), n’est pas une délétion ou un
polymorphisme d’un seul nucléotide, mais bien par une expansion répétée hexanucléotidique
de GGGGCC (Gendron et Petrucelli 2018). Le nombre de répétition de ce motif de six
nucléotides va de plusieurs dizaines à quelques milliers de répétitions (Gendron et Petrucelli
2018). Le nombre de répétitions corrèle négativement avec l’âge à l’apparition des
symptômes, mais n’a pas de corrélation avec la vitesse de progression de la maladie chez
l’humain (Benussi et al. 2014, Nordin et al. 2015). De plus, la toxicité cellulaire induite par
l’expansion hexanucléotidique dans le gène C9orf72 se distingue de la toxicité cellulaire
induite par les autres gènes ci-dessous parce qu’elle n’est pas seulement causée par
l’agrégation cytoplasmique de protéines mal repliées, mais aussi par la présence de foyers
d’ARN dans le noyau (Gendron et Petrucelli 2018). Les répétitions GGGGCC forment des
structures secondaires dans l’ARN appelées G-quadruplexes et seraient possiblement
responsables de la perte de fonction de ce gène en réduisant son expression (Belzil et al.
2013). De plus, l’expansion nucléotidique dans C9orf72 est responsable d’un gain de
fonction toxique promouvant la neurodégénérescence possiblement en affectant le
métabolisme de l’ARN, son exportation vers le cytoplasme et en stimulant la formation de
P-bodies (Gendron et Petrucelli 2018). Mieux encore, les répétitions d’ARN peuvent être
traduites en six différentes répétitions de dipeptides, selon le cadre de lecture et le brin lu.
Ces dernières semblent aussi participer aux mécanismes toxiques dans les neurones moteurs,
mais à un niveau moindre comparativement aux foyers d’ARN (Gendron et Petrucelli 2018)
probablement en favorisant l’accumulation cytoplasmique de TDP43. Finalement,
l’expansion hexanucléotidique dans le gène C9orf72 semble être responsable d’une altération
dans le transport nucléo-cytoplasmique en affectant la composition et la fonction normale
des pores nucléaires (Zhang et al. 2016). Ce mécanisme semble précéder et même causer
l’accumulation cytoplasmique de TDP43 (Zhang et al. 2015).
Les mécanismes toxiques liant l’expansion hexanucléotidique de C9orf72 à la pathogénèse
de la SLA ne sont pas, à ce jour, très bien compris, d’autant plus que les rares modèles
animaux utilisant ce gène n’ont été développés que relativement récemment (Hukema et al.

14
2014, Chew et al. 2015, Picher-Martel et al. 2016). Ces modèles reproduisent bien les aspects
moléculaires et cellulaires de la maladie, mais, ne vont qu’exceptionnellement mimer les
symptômes de la SLA ou de la DFT (Liu et al. 2016, Picher-Martel et al. 2016).
1.1.3. Mécanismes de cytotoxicité impliquées dans la pathogénèse de la SLA
Dans les formes sporadique et familiale de la SLA, l’accumulation cytoplasmique
d’oligomères et d’agrégats toxiques de protéines mal repliées est en amont de plusieurs
mécanismes de toxicité cellulaire menant à la destruction des JNM et la mort des neurones
moteurs. Bien que la nature de la protéine agrégée varie selon la nature du gène mutant dans
les cas familiaux, celle-ci mène toutefois à la délocalisation cytoplasmique de TDP43,
présente dans la majorité des cas de SLA et des modèles animaux (Fig.4A) (Neumann et al.
2006, Taylor et al. 2016). De plus, les différents mécanismes toxiques des protéines mal
repliées (mentionnés dans la section précédente) convergent vers les mêmes mécanismes
généraux de toxicité cellulaire tels que la dérégulation du cytosquelette et du transport axonal,
la dérégulation de la protéostasie (par un stress du réticulum endoplasmique et une
dysfonction des voies de dégradation des protéines), la dysfonction mitochondriale
accompagnée du stress oxydatif ou encore la sécrétion des protéines mal repliées (Fig.4B-E)
(Robberecht et Philips 2013, Taylor et al. 2016), Il est à noter que ce dernier mécanisme
influence l’inflammation (Henkel et al. 2004, Zhao et al. 2015) et la propagation prionique
de la SLA (Urushitani et al. 2006, Iguchi et al. 2016). À fin de comprendre comment les
motoneurones dégénèrent dans la SLA, il est nécessaire d’explorer les mécanismes sous-
jacents à la perte des JNM, à la rétraction de l’axone et puis à la mort neuronale (Robberecht
et Philips 2013).

15
Figure 4: Les mécanismes de cytotoxicité participant à la pathogénèse de la SLA
A. La délocalisation cytoplasmique de TDP43 et la formation d’agrégats de protéines
associées à la SLA causent une chaîne de mécanismes cytotoxiques menant à la mort des
motoneurones. B. Les protéines associées à la SLA causent un stress du réticulum
endoplasmique entraînant le mauvais repliement d’autres protéines et provoquent le maintien
de ces dernières en bloquant la machinerie du protéasome et de l’autophagie. C. Les protéines
associées à la SLA empêchent l’importation de protéines mitochondriales, bloquent la chaîne
respiratoire menant à une production d’espèces réactives de l’oxygène (un stress oxydatif) et
un mauvais repliement de protéines, ajoutant au fardeau des mécanismes de dégradation
protéique. D. Dans le corps cellulaire et dans l’axone, s’accumulent des neurofilaments
hyperphosphorylés. En conséquence, le cytosquelette devient désorganisé, le transport
axonal de composants cellulaires importants fait défaut, menant à la destruction des JNM. E.
Les protéines mal repliées peuvent être sécrétées par les neurones moteurs et propager leur
conformation aberrante à d’autres neurones, à la manière d’un prion.
1.1.3.1. Dérégulation du cytosquelette, du transport et de la survie axonale
La désorganisation du cytosquelette, mais plus précisément celle des neurofilaments est une
caractéristique de la SLA. Dans la SLA, les neurofilaments sont hyperphosphorylés ce qui
déclencherait leur accumulation dans le soma et les axones des motoneurones (Fig.5A)
(Manetto et al. 1988). Les neurofilaments sont nécessaires pour que l’axone et le
motoneurone (par conséquent) conserve leur intégrité structurelle. Ainsi, la dérégulation dans
la phosphorylation des neurofilaments et leur agrégation subséquente favorisent la rétraction
de l’axone et l’axonopathie typique de la SLA. Aussi, TD43 et FUS peuvent lier l’ARNm de

16
NFL, la plus courte protéine de la famille des neurofilaments et réduire son expression en
empêchant sa traduction (Bergeron et al. 1994). La présence de mutations dans les gènes des
filaments intermédiaires (NFH et PRPH) ou dans les composantes des microtubules
(TUBA4) est associée au développement de la SLA dans quelques rares cas familiaux
(Fig.5B) (Robberecht et Philips 2013, Taylor et al. 2016, Burk et Pasterkamp 2019). Les
mutations dans ces gènes promeuvent l’agrégation de leur protéine respective (Figlewicz et
al. 1994, Tomkins et al. 1998, Gros-Louis et al. 2004, Leung et al. 2004) et provoquent un
arrangement aberrant du cytosquelette (Fig.5C) (Robberecht et Philips 2013).
Les transports axonaux antérograde et rétrograde dépendants des microtubules et des
neurofilaments sont affectés dans la SLA. Ceci est mis en évidence dans des modèles murins
où ce phénomène précède largement l’apparition des symptômes dans ces modèles animaux
(Williamson et Cleveland 1999, Perlson et al. 2009, Bilsland et al. 2010, Taylor et al. 2016).
Le transport antérograde est nécessaire à l’acheminement vers la synapse des mitochondries,
des vésicules de neurotransmetteurs et d’ARNm qui seront traduits en protéines essentielles
à la fonction de cette synapse, (Taylor et al. 2016, Moller et al. 2017) Une altération dans le
transport antérograde mènerait donc à la destruction de la synapse et la perte de la JNM.
Quelques rares cas de SLA familiale sont causés par une perte de fonction de la kinésine
KIF5A impliquée dans le transport antérograde. De plus, FUS mutante dérégule l’expression
des ARNm des kinésine KIF5C, KIF1B et KIF3A (Burk et Pasterkamp 2019). Le transport
axonal rétrograde est aussi affecté dans la SLA avant l’apparition des symptômes. Ainsi, des
mutations dans le gène DCTN1, sont retrouvées dans des cas sporadiques et familiaux
affectant ce transport axonal (Taylor et al. 2016, Burk et Pasterkamp 2019). L’altération du
transport rétrograde mène à l’accumulation de certaines protéines dans les neurites, tels que
des récepteurs AMPA, NMDA et TRKB (Bilsland et al. 2010, Burk et Pasterkamp 2019).
Ces récepteurs sont normalement retirés de la synapse par endocytose, mais leur
accumulation dans la SLA mène à une plus grande excitabilité neuronale et à l’excitotoxicité
(expliquée plus en détail dans la section 1.2.2.). De façon générale, l’altération des transports
antérograde et rétrograde cause l’accumulation, dans l’axone, de sphéroïdes toxiques
contenant vésicules de neurotransmetteurs, endosomes, mitochondries et granules d’ARN
(Fig.5D) (De Vos et Hafezparast 2017, Burk et Pasterkamp 2019).

17
La santé de l’axone de la JNM dans la SLA est donc un important aspect de la maladie et
pourrait être modulée. Les protéines influençant la croissance axonale sont régulées dans la
SLA. Par exemple, l’inhibiteur de croissance axonal NOGO-A est surexprimé durant le stade
symptomatique de la maladie (Dupuis et al. 2002, Jokic et al. 2006). L’expression d’EPHA4,
un autre inhibiteur de croissance axonal, est négativement corrélée avec la survie chez
l’humain (Fig.5E) (Robberecht et Philips 2013, Pampalakis et al. 2019). La présence
d’inhibiteurs de croissance axonale empêche une JNM détruite de se reformer. L’inhibition
de la croissance axonale peut être contrée par un traitement avec VEGF (vascular endothelial
growth factor) qui aide à préserver la JNM (Storkebaum et al. 2005).
Figure 5: La dérégulation du cytosquelette menant à la perte des jonctions neuromusculaires
A. L’hyperphosphorylation et l’accumulation de neurofilaments est une des caractéristiques
principales de la SLA (Murayama et al. 1989). B. Des mutations dans les gènes du
cytosquelette promeuvent l’agrégation cytoplasmique (dans le soma et l’axone) des protéines
issues de ces gènes. Cela résulte en une désorganisation du cytosquelette, une formation de
sphéroïdes toxiques découlant des transports axonaux antérograde et rétrograde altérés et
contenant mitochondries, vésicules de neurotransmetteurs, endosomes, granules d’ARN, etc.
(C-D). E. Ces mécanismes toxiques convergent vers la perte des JNM et la rétraction de
l’axone.

18
1.1.3.2. Dérégulation de la protéostasie
La protéostasie se définie comme l’ensemble des mécanismes moléculaires et cellulaires
impliqués dans la production, le repliement ou la dégradation des protéines. Dans la SLA,
plusieurs composantes de ce système sont dérégulées. Ainsi, on retrouve un stress du
réticulum endoplasmique, une réponse de choc thermique, une dysfonction du protéasome et
une dysfonction de l’autophagie (Robberecht et Philips 2013, Taylor et al. 2016).
Le stress du réticulum endoplasmique est un phénomène cellulaire associé à la SLA depuis
un certain temps (Atkin et al. 2008) et dont le mécanisme pathogénique a été confirmé pour
la première fois dans un modèle murin exprimant la protéine SOD1 mutée (Nishitoh et al.
2008, Taylor et al. 2016). La forme mutante de SOD1 interagit avec la membrane du
réticulum endoplasmique, plus précisément avec la portion cytosolique de la protéine Derlin-
1. Cette protéine est responsable du recrutement de la machinerie de la dégradation associée
au réticulum endoplasmique (endoplasmic reticulum associated degradation, ERAD). Ainsi,
par cette interaction, SOD1 empêche Derlin-1 d’acheminer les protéines mal repliées vers la
machinerie d’ERAD et le protéasome et promeut l’accumulation de ces protéines mal
repliées dans le lumen du réticulum endoplasmique (Fig.6A) (Nishitoh et al. 2008). Les
modèles murins exprimant d’autres gènes associés à la SLA (ATXN2, FUS, TDP43,
UBQLN2, VABP) présentent aussi un stress du réticulum endoplasmique avec un mécanisme
d’activation semblable à celui trouvé dans le modèle SOD1 (Matus et al. 2013, Halloran et
al. 2019). La dérégulation de l’ERAD est aussi accompagnée par la phosphorylation de eIF2α
et l’accumulation de CHOP, PERK, et de chaperonnes (Matus et al. 2013). L’accumulation
et l’activation de chaperonnes, plus précisément les protéines de choc thermique (heat shock
proteins, HSP), est un phénomène parallèle au stress du réticulum endoplasmique (Kalmar
et Greensmith 2017). Durant un stress cellulaire et une dérégulation de la protéostasie, les
HSPs tentent de corriger le mauvais repliement des protéines mais sont surchargées par le
caractère chronique de la maladie. Par exemple, Hsp70 est recrutée pour chaperonner les
protéines mal repliées en se détachant des senseurs du stress du réticulum endoplasmique tels
que PERK ou IRE1 (Kalmar et Greensmith 2017). Les senseurs du stress du réticulum
endoplasmique participent à l’activation de la réponse aux protéines mal repliées (unfolded
protein response, UPR). L’UPR est activée par différentes voies de signalisation à la suite

19
du stress du réticulum endoplasmique. L’UPR est normalement responsable du repliement
adéquat des protéines. Si l’UPR échoue à cette tâche, elle activera la dégradation de ces
protéines mal repliées grâce au système ubiquitin-protéasome (ubiquitin-proteasome system,
UPS) et à l’autophagie (Matus et al. 2013, Taylor et al. 2016). Ces mécanismes sont altérés
par les protéines issues des gènes associés à la SLA. Comme mentionné précédemment,
l’UPS et l’autophagie peuvent être indirectement affectés par ces protéines à travers le stress
soutenu du réticulum endoplasmique. L’activation de l’UPR et la libération des senseurs du
stress du réticulum endoplasmique mènent à l’activation des voies de l’inflammation et de
l’apoptose (Nishitoh et al. 2008, Kalmar et Greensmith 2017), contribuant directement à la
mort neuronale (Fig.6A).
Deux composantes de l’UPR, l’UPS et l’autophagie, peuvent être directement affectées par
d’autres gènes associés à la SLA (C9orf72, CHMP2, OPTN, SOD1 SQSTM1, TBK1,
UBQLN2, VCP, VAPB) (Urushitani et al. 2002, Parkinson et al. 2006, Chen et al. 2010,
Johnson et al. 2010, Maruyama et al. 2010, Deng et al. 2011, Fecto et al. 2011, Taylor et al.
2016, Oakes et al. 2017, Leskelä et al. 2019). Par exemple, dans la SLA, la protéine mutante
UBQLN2 ne remplit plus son rôle dans la livraison des protéines ubiquitylées à l’UPS
(Fig.6B) (Chang et Monteiro 2015, Osaka et al. 2016, Renaud et al. 2019). Aussi, SOD1
mutante, bien qu’ubiquitylée elle aussi, pourrait encombrer le protéasome, suffisamment
pour causer sa dysfonction (Urushitani et al. 2002). Comparativement à d’autres types
neuronaux, la vulnérabilité supérieure des neurones moteurs à une inhibition du protéasome
expliquerait, en partie, leur dégénérescence préférentielle dans la SLA (Urushitani et al.
2002, Saxena et al. 2009).
OPTN2, p62 et UBQLN2, protéines issues de gènes liées à la SLA, sont aussi responsables
d’acheminer les protéines et d’autres composants cellulaires à dégrader vers
l’autophagosome, probablement en liant LC3 (Markovinovic et al. 2017, Renaud et al. 2019).
Dans la SLA, les mutations dans les gènes de OPTN et UBQLN2 mènent donc à une
altération dans l’autophagie en empêchant la maturation de l’autophagosome et la livraison
adéquate des composants cellulaires à dégrader (Fig.6C) (Markovinovic et al. 2017, Renaud
et al. 2019). En général, le stress du réticulum endoplasmique, la dysfonction de l’autophagie

20
et du protéasome exacerbent la protéinopathie et l’accumulation de protéines mal repliées,
déjà présentes dans les neurones.
Figure 6: La dérégulation de la protéostasie
A. Les protéines mutantes mal repliées interagissent avec la membrane du réticulum
endoplasmique (par la protéine Derlin-1 entre autres) et bloque la production et le contrôle
qualité des protéines. Ceci mène directement à l’activation de voies de signalisation
apoptotique, affectant directement la mort neuronale. B. Les protéines mutantes interagissent
de façon aberrante avec le protéasome ou les protéines ubiquitylées empêchant la dégradation
de ces dernières. C. Les protéines mutantes bloquent l’autophagie de protéines mal repliées
et d’autres composants cellulaires. Ainsi, la dérégulation de la protéostasie mène à une
augmentation de la quantité de protéines mal repliées dans le cytoplasme. (ERAD :
Dégradation associée au réticulum endoplasmique)
1.1.3.3. Stress oxydatif et dysfonction mitochondriale
Une des caractéristiques de la SLA est la production excessive et/ou l’échec de la clairance
des ROS causant une forte oxydation des protéines, de l’ADN et des lipides dans les tissus
des patients atteints de la SLA (Shaw et al. 1995, Shibata et al. 2001, Coppedè 2011, D'Amico
et al. 2013). Le stress oxydatif trouvé dans les tissus humains est retrouvé aussi dans les
modèles murins (Bozzo et al. 2017) ou dans les cultures cellulaires exprimant des protéines
mutées (Duan et al. 2010). Le stress oxydatif présent dans les cellules affectées par la SLA
exacerbe le mauvais repliement protéique (Bozzo et al. 2017) notamment en promouvant le
potentiel prionique de la protéine SOD1 (Grad et Cashman 2014) ou la délocalisation

21
cytoplasmique de TDP43 (Cohen et al. 2015) et de FUS (Vance et al. 2013), favorisant la
formation de granules de stress par ces deux dernières protéines (Bozzo et al. 2017). Par la
phosphorylation oxydative, les mitochondries sont les principales productrices des ROS dans
les cellules et sont susceptibles à une dysfonction causée par les protéines associées à la SLA
(Fig.7A) (Bozzo et al. 2017, Greco et al. 2019). Chez les patients atteints de la SLA, on
retrouve des mitochondries vacuolées, agglutinées et présentant une morphologie altérée
(Smith et al. 2019). Dans les modèles murins de SOD1, la détérioration mitochondriale,
détectable par un changement morphologique, apparaît relativement très tôt alors que dans
les modèles murins de TDP43, cette détérioration apparaît après l’apparition des symptômes
(Magrané et al. 2014). La TDP43 cytosolique peut entrer dans les mitochondries et mène à
la libération de l’ARN mitochondrial dans le cytoplasme et l’activation de la voie cGAS-
STING, contribuant ainsi à la neuroinflammation (Yu et al. 2020). La détérioration
mitochondriale peut s’expliquer aussi par l’interaction entre la membrane mitochondriale et
les protéines associées à la SLA (SOD1, TDP43, FUS, C9orf72, CHCHD10) et par la
séquestration cytosolique de chaperonnes mitochondriales par ces protéines (Fig.7B)
(Beddoe et Lithgow 2002, Smith et al. 2019). Ces chaperonnes ne sont pas seulement
nécessaire au bon repliement des protéines mitochondriales (ajoutant à la dérégulation de la
protéostasie, vue à la section précédente), mais sont aussi importantes pour le transport des
protéines traduites dans le cytoplasme vers les mitochondries (Beddoe et Lithgow 2002). De
plus, alors que la forme sauvage de SOD1 est recrutée dans l’espace intermembranaire
mitochondrial à l’aide du récepteur d’import mitochondriale TOM, la forme mutante de
SOD1 s’accumule de façon aberrante dans cet espace et affecte le fonctionnement de la
chaîne respiratoire (Greco et al. 2019) Aussi, les agrégats de SOD1 mutée interagissent et
bloquent TOM sur la membrane externe de la mitochondrie en empêchant l’importation de
protéines (Fig.7B) (Mattiazzi et al. 2002, Li et al. 2010, Greco et al. 2019). En plus d’être
causée par la présence excessive de ROS et la dérégulation de la protéostasie, la dysfonction
mitochondriale est accentuée par une déficience dans le cytosquelette qui, en séquestrant les
mitochondries, l’empêche de produire de l’ATP pour des régions clefs des motoneurones et
d’importer les protéines mitochondriales (Greco et al. 2019). La détérioration mitochondriale
dans la SLA, mène donc à un transport d’électrons altéré dans la chaîne respiratoire et la
production de ROS (Fig.7B). Les ROS s’attaquent à leur tour aux protéines et leur repliement,

22
exacerbant la dysfonction mitochondriale (Federico et al. 2012). Dû à leurs altérations
fonctionnelles et morphologiques, les mitochondries libèrent le cytochrome C et AIF. SOD1
mutée interagit aussi avec BCL2 et VDAC1 sur la mitochondrie. Ces mécanismes mènent
ultimement à l’activation de voies de signalisation apoptotique dans les neurones moteurs
(Fig.7C) (Pasinelli et al. 2004, Federico et al. 2012).
Le déséquilibre de l’homéostasie redox dans les neurones moteurs est d’abord causé par une
surproduction de ROS (Greco et al. 2019). Les ROS, notamment l’anion superoxyde, peuvent
réagir avec l’oxyde nitrique et former des espèces réactive de l’azote (reactive nitrogen
species, RNS) ou d’autres molécules pour former des ROS secondaires. À l’état
physiologique, des enzymes comme SOD1 ou la glutathion peroxydase (GPX) sont
responsable de la détoxification progressive des ROS. Dans la SLA, la perte de fonction de
SOD1 mutée est encore sujet à controverse, comme mentionné à la section 1.1.2.1 (Saccon
et al. 2013). L’effet de SOD1 mutée sur l’homéostasie redox vient peut-être plus d’un gain
de fonction toxique dû à sa conformation aberrante. Sur la membrane externe de la
mitochondrie, la forme mutée de SOD1 peut interagir avec BCL2 et exposer le domaine BH3,
favorisant l’interaction du canal ionique VDAC1 avec BCL2 (Pasinelli et al. 2004). SOD1
peut aussi directement interagir avec la portion cytosolique de VDAC1. L’interaction SOD1-
BCL2-VDAC1 et SOD1-VDAC1 cause une rupture dans le flot d’ATP de la mitochondrie
vers le cytoplasme par l’obstruction de VDAC1. Ceci cause un déséquilibre dans le ratio
ATP/ADP menant à une surproduction de ROS (Greco et al. 2019).
La surproduction de ROS et de RNS est accompagnée par l’épuisement de molécules
antioxydantes (Fig.7A). Le glutathion (GSH) est la molécule antioxydante par excellence
dans les cellules (Rojo et al. 2014). Il est utilisé par GPX pour réduire le peroxyde
d’hydrogène en eau et en oxygène. Le glutathion est, par la suite, réduit à son tour par
l’enzyme glutathion réductase (GR) pour être par la suite réutilisé par GPX. Lorsque
l’environnement cellulaire est particulièrement oxydant, le GSH peut être produit en plus
grande quantité par l’enzyme glutathion synthétase (GSS) (Dringen 2000, Rojo et al. 2014).
Dans la SLA, malgré la présence d’un stress oxydatif, le GSH n’est retrouvé qu’en quantité
diminuée, quand il est mesuré dans le sang (Blasco et al. 2017, Wang, Bai, et al. 2019) et le

23
cortex (Weiduschat et al. 2014) des patients atteints de la SLA, dans les modèles murins et
cultures cellulaires (Muyderman et al. 2009). Aussi, la réduction artificielle de la production
de GSH dans des modèles murins SOD1 exacerbe la progression des symptômes (Vargas et
al. 2011, Killoy et al. 2018). De plus, alors que le niveau d’expression de GR ne change pas
chez les patients atteints de la SLA, son niveau activité est diminué, augmentant l’épuisement
de la réserve de GSH (Przedborski et al. 1996, Wang, Bai, et al. 2019) Le niveau d’activité
de GR ne corrèle toutefois pas avec la progression de la maladie (Przedborski et al. 1996,
Cudkowicz et al. 2002). L’administration de GSH réduit pour le traitement de la SLA n’a
malheureusement pas d’effet sur la survie des patients (Chiò et al. 1998). Cependant, le
renforcement de l’activité réductrice des molécules antioxydantes est une voie thérapeutique
intéressante. Le médicament edaravone, récemment autorisé pour le traitement de la SLA,
est justement un collecteur de radicaux libres, compensant pour la faible activité réductrice
présente dans les cellules et tissus des patients atteints de la SLA (Rothstein 2017).
Ainsi, le stress oxydatif dans les neurones moteurs est le chaînon d’un cercle vicieux menant
au mauvais repliement de protéines, dont certaines, associées directement à la SLA. Les
protéines mal repliées encombrent la chaîne respiratoire favorisant une surproduction de
ROS et l’épuisement des molécules antioxydantes. Le déséquilibre de l’homéostasie redox
cause le stress oxydatif, le tout participant à la mort neuronale.

24
Figure 7: Le stress oxydatif et la dysfonction mitochondriale
A. Le stress oxydatif présent dans les motoneurones atteints de la SLA est causé à la fois par
la surproduction de ROS et l’épuisement des molécules antioxydantes dans le cytosol et la
mitochondrie. B. Les ROS contribuent au mauvais repliement des protéines associées à la
SLA et à l’oxydation des protéines de la cellule en général. Les protéines mal repliées
ségréguent les chaperonnes mitochondriales, contribuant à la dérégulation de la protéostasie.
Les protéines mal repliées associées à la SLA interagissent avec la membrane mitochondriale
et bloquent l’importation de protéines. Aussi, les protéines mal repliées affectent la chaîne
de transport des électrons, produisant encore plus de ROS. C. L’interaction entre SOD1,
BCL2 et VABP1 mène à l’activation des voies de signalisation apoptotiques (ROS : Espèces
réactives de l’oxygène, GSH : Glutathion)
1.1.3.4. Sécrétion des protéines agrégées
Comme mentionné précédemment, les protéines issues des gènes associées à la SLA peuvent
s’oligomériser et former des agrégats toxiques. Le mauvais repliement causant cette
agrégation est bien compris lorsque le gène en question présente une mutation susceptible
d’affecter la conformation de sa protéine. Toutefois, dans les tissus de cas sporadiques
s’accumulent aussi des protéines mal repliées, même sans l’existence d’une mutation de-
novo. Un stress cellulaire pourrait être à l’origine du mauvais repliement originel d’une unité
protéique. Ce mauvais repliement peut se transmettre, par la suite, à d’autres unités à la
manière d’un prion (Fig. 8A) (Ayers et Cashman 2018). Aussi, cette réplication prionique ne
reste pas intrinsèque à un seul motoneurone. En effet, le mauvais repliement de SOD1 (Grad

25
et Cashman 2014), ou de TPD43 (Furukawa et al. 2011, Smethurst et al. 2015) peut se
propager de neurones en neurones en partant de la région du SNC affectée en premier.
Pour propager leur conformation aberrante vers d’autres neurones, les protéines mal repliées
doivent se retrouver à l’extérieur de leur neurone d’origine. Elles peuvent y arriver quand le
neurone d’origine dégénère et le contenu cytoplasmique de celui-ci fuit dans l’espace
extracellulaire. Autrement, les protéines mal repliées peuvent aussi être activement sécrétées
dans des exosomes par les neurones moteurs (Fig.8B) (Urushitani et al. 2006, Iguchi et al.
2016). Ce mode de propagation a été confirmé pour les protéines SOD1 et TDP43. SOD1
mutée, mais pas la forme sauvage, peut interagir avec les chromogranines, composantes de
la membrane des exosomes, (Urushitani et al. 2006) et ainsi être sécrétée. La surexpression
artificielle de la chromogranine A accélère l’apparition des symptômes dans des modèles
murins pour SOD1 suggérant un rôle possible des chromogranines dans la stabilisation de
SOD1 mutée et dans la stimulation de sa sécrétion (Ezzi et al. 2010). TDP43 est aussi
retrouvée dans les exosomes de neurones atteints de la SLA, mais pas dans les exosomes des
astrocytes ou des cellules microgliales. Le traitement de cellules neuronales en culture avec
des exosomes contenant la protéine TDP43 mal repliée est suffisant pour causer la
délocalisation cytoplasmique de TDP43 dans ces cellules (Fig.8C) (Iguchi et al. 2016). La
protéine FUS, partageant avec TDP43 certains domaines structurels, pourrait être sécrétée et
se propager de la même manière que TDP43 (Ayers et Cashman 2018). Finalement, la
présence extracellulaire de protéines mal repliées affecte non-seulement les neurones
moteurs mais aussi d’autres types cellulaires du SNC, les astrocytes et la microglie par
exemple; en les activant ou en promouvant certains des mécanismes cytotoxiques énumérés
dans la section 1.1.3. (Fig.8C) (Ilieva et al. 2009, Robberecht et Philips 2013). Les autres
types cellulaires du SNC participent, eux aussi, à la pathogénèse de la SLA (Henkel et al.
2004, Furukawa et al. 2006, Ilieva et al. 2009, Appel et al. 2011, Robberecht et Philips 2013,
Zhao et al. 2015), tel qu’il sera discuté dans la prochaine section.
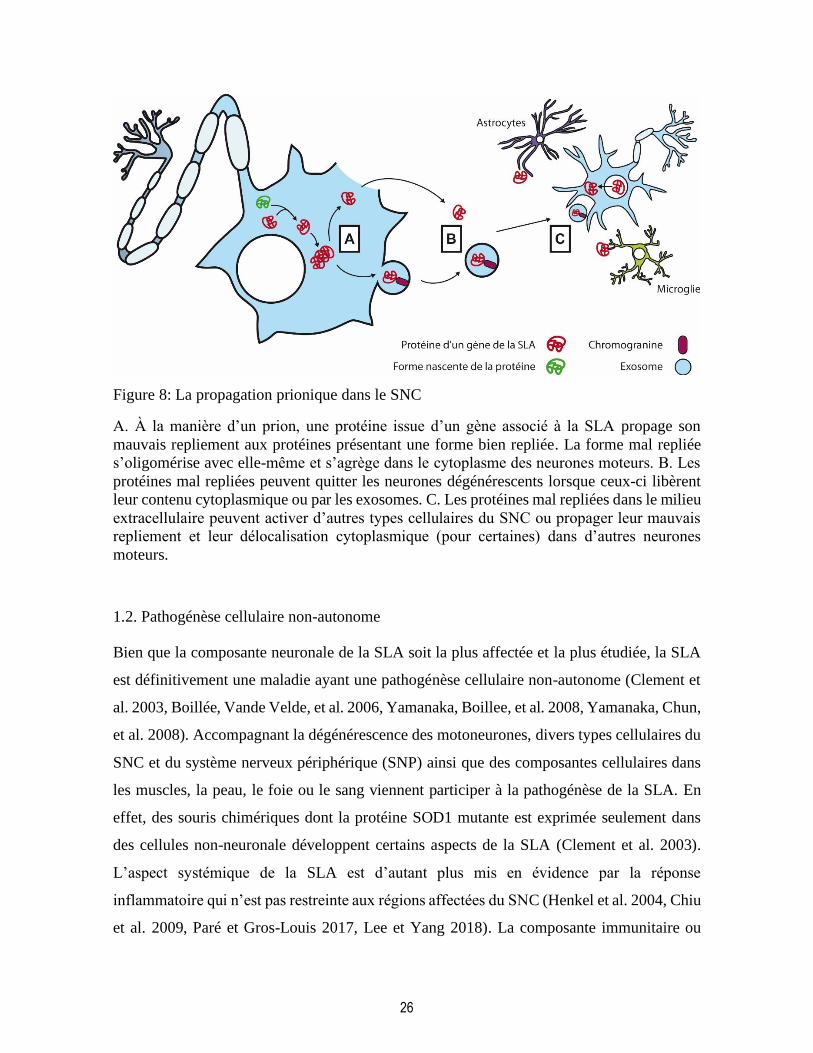
26
Figure 8: La propagation prionique dans le SNC
A. À la manière d’un prion, une protéine issue d’un gène associé à la SLA propage son
mauvais repliement aux protéines présentant une forme bien repliée. La forme mal repliée
s’oligomérise avec elle-même et s’agrège dans le cytoplasme des neurones moteurs. B. Les
protéines mal repliées peuvent quitter les neurones dégénérescents lorsque ceux-ci libèrent
leur contenu cytoplasmique ou par les exosomes. C. Les protéines mal repliées dans le milieu
extracellulaire peuvent activer d’autres types cellulaires du SNC ou propager leur mauvais
repliement et leur délocalisation cytoplasmique (pour certaines) dans d’autres neurones
moteurs.
1.2. Pathogénèse cellulaire non-autonome
Bien que la composante neuronale de la SLA soit la plus affectée et la plus étudiée, la SLA
est définitivement une maladie ayant une pathogénèse cellulaire non-autonome (Clement et
al. 2003, Boillée, Vande Velde, et al. 2006, Yamanaka, Boillee, et al. 2008, Yamanaka, Chun,
et al. 2008). Accompagnant la dégénérescence des motoneurones, divers types cellulaires du
SNC et du système nerveux périphérique (SNP) ainsi que des composantes cellulaires dans
les muscles, la peau, le foie ou le sang viennent participer à la pathogénèse de la SLA. En
effet, des souris chimériques dont la protéine SOD1 mutante est exprimée seulement dans
des cellules non-neuronale développent certains aspects de la SLA (Clement et al. 2003).
L’aspect systémique de la SLA est d’autant plus mis en évidence par la réponse
inflammatoire qui n’est pas restreinte aux régions affectées du SNC (Henkel et al. 2004, Chiu
et al. 2009, Paré et Gros-Louis 2017, Lee et Yang 2018). La composante immunitaire ou

27
inflammatoire de la SLA pourrait, en partie, retarder la neurodégénérescence et contribuer à
l’apparition tardive de la maladie, même dans les cas familiaux. À l’apparition des
symptômes de la SLA, la composante immunitaire s’emballe et exacerbe la progression de
la maladie. De plus, certains facteurs de risque liés à la composante immunitaire sont des
déterminants du pronostic de la maladie (Lopez-Lopez et al. 2014, Beers et al. 2017, Wosiski-
Kuhn et al. 2019). L’étude des différents types cellulaires des composantes immunitaires et
inflammatoire de la SLA, tels que la microglie, l’astroglie, l’oligodendroglie et les
leucocytes, est primordial pour la compréhension de la pathogénèse et de l’évolution de la
maladie.
1.2.1. Cellules microgliales
Les cellules microgliales sont les macrophages résidents du SNC et sont la composante
immunitaire de la SLA la plus étudiée. Isolées des populations myéloïdes périphériques
durant le développement embryonnaire (Ginhoux et al. 2010) les cellules microgliales ne
peuvent pas compter sur un progéniteur dans la moelle osseuse, mais possèdent un
progéniteur dans le SNC pour assurer leur renouvellement en conditions normales (Elmore
et al. 2014, Askew et al. 2017). Dans ces conditions, l’ensemble de cellules microgliales, la
microglie, présente un phénotype dit « de repos » ou « quiescent ». Ce phénotype est
caractérisé morphologiquement par un corps cellulaire relativement petit et de longs
filopodes motiles (Nimmerjahn et al. 2005). Malgré le nom de ce phénotype, la microglie a
un rôle très actif dans le maintien de l’homéostasie du SNC. Grâce à ses longs
embranchements, les cellules microgliales sont aptes à sonder le milieu extracellulaire en
quête de signaux de danger (Davalos et al. 2005). Aussi, durant le développement du SNC,
la microglie joue un rôle important dans la formation des synapses entre les nouveaux
neurones et dans l’élagage des synapses surnuméraires (Paolicelli et al. 2011). Dans un
cerveau adulte, la maturation des synapses est toujours de mise dans certaines régions du
SNC où de nouveaux neurones sont retrouvés tel que le bulbe olfactif ou l’hippocampe (Hong
et al. 2016, Reshef et al. 2017). Dans ces régions, les cellules microgliales sont donc plus
nombreuses et présentent un état activé pour compléter leurs fonctions. Une perturbation des
fonctions homéostatiques microgliales peut mener à différentes maladies développementales

28
et neurologiques comme l’épilepsie, la schizophrénie ou la maladie d’Alzheimer (Hong et al.
2016, Andoh et al. 2019, Sellgren et al. 2019). Ainsi, dans le cerveau adulte, la microglie est
plutôt hétérogène en morphologie et en concentration, indice de ses divers rôles à travers le
SNC (Lawson et al. 1990). La microglie a aussi le rôle d’assurer la survie des neurones. Elle
le fait en sécrétant divers facteurs de croissance comme le brain-derived neurotrophic factor
(BDNF), le glial-derived neurotrophic factor (GDNF) ou l’insulin-like growth factor 1 (IGF-
1) (Fig.9A) (Kettenmann et al. 2013, Cherry et al. 2014a). Ce rôle est rendu possible par le
dialogue entre les cellules microgliales et les neurones. Ces dernières atténuent l’activation
microgliale par une co-stimulation négative à la fois par la liaison de CD200 sur les neurones
et CD200R sur les cellules microgliales et par la reconnaissance de la fractalkine (CX3CL1)
sécrétée par les neurones et reconnue par son récepteur CX3CR1 sur la microglie (Fig.9A)
(Hellwig et al. 2013). Finalement, la microglie régule sa propre homéostasie et prolifération
par une signalisation autocrine impliquant IL-34 et CSF1, ligands pour CSF1R (Fig.9A)
(Easley-Neal et al. 2019).
Avenant un dommage dans le SNC soit par une infection virale ou bactérienne ou par un
destruction aigue des neurones, les cellules microgliales sont responsables de compléter la
clairance des débris et de promouvoir le retour à l’homéostasie à la suite de la
neuroinflammation. Grâce à son sensome, soit l’ensemble des récepteurs exprimés par une
cellule, la microglie peut reconnaître des motifs moléculaires associés à des pathogènes
(pathogen associated molecular patterns, PAMP) comme l’ARN et l’ADN viral ou des
composantes membranaires des bactéries. De plus, le sensome microglial permet d’identifier
des motifs moléculaires associés au danger (danger associated molecular patterns, DAMP)
qui sont généralement des débris de cellules mortes ou endommagées provenant du
cytoplasme mais maintenant retrouvés dans le milieu extracellulaire, ou encore des protéines
agrégées (Fig.9B). Les récepteurs des PAMPs et des DAMPs les reconnaissant d’une façon
plutôt non-spécifique. Par exemple, les récepteurs TLR peuvent à la fois reconnaître le
lipopolysaccharide (LPS) mais aussi les versions mutantes et agrégées des protéines SOD1
et TDP43 (Roberts et al. 2013, Zhao et al. 2015). Parmi les autres DAMPs communs, il est
possible de lister l’ATP, les produits avancés de la glycation (advanced glycation end-
product, AGE) ou encore les HSP qui peuvent être reconnus, entre autres, par les récepteurs

29
purinergiques, les récepteurs pour les AGE ou TREM2 (Fig.9B) (Kigerl et al. 2014, Juranek
et al. 2015, Painter et al. 2015). Les mêmes récepteurs sont utilisés par la microglie lors d’une
inflammation aigue et lors d’une inflammation chronique. Cependant, la réponse de la
microglie à l’activation de récepteurs diffère dans ces deux cas. Dans la neuroinflammation
aigue, les cellules microgliales changent d’aspect en rétractant leurs filopodes et en devenant
nettement amiboïde, aidant à leur fonction phagocytaire (Kettenmann et al. 2011). Le
changement de morphologie est accompagné par la production et la sécrétion de molécules
pro-inflammatoires, tels que les ROS et les RNS, le tumor necrosis factor α (TNFα) ou
l’interleukin-1β (IL-1β) (Fig.9B) (Kettenmann et al. 2011). Le phénotype pro-inflammatoire,
originellement appelé « M1 » est responsable de la clairance des débris cellulaires et de
l’arrêt de la production de nouveaux PAMPs et DAMPs. Durant la neuroinflammation aigue,
le phénotype pro-inflammatoire est normalement suivi par un changement vers un phénotype
anti-inflammatoire (ou « M2 ») responsable du retour vers l’homéostasie en supprimant la
réponse inflammatoire et en promouvant la réparation cellulaire (Cherry et al. 2014b). Ce
deuxième phénotype est accompagné par la production et la sécrétion de cytokines anti-
inflammatoires et immunosuppressives (IL-4, IL-10) de facteurs neurotrophiques comme le
BDNF ou l’IGF-1 et d’autres protéines telles qu’arginase-1, YM1, ou CD163 (Fig.9C)
(Cherry et al. 2014b). De plus, la microglie réacquiert sa co-stimulation négative avec les
neurones. Une fois l’inflammation résolue, la cellule microgliale retrouve son phénotype
quiescent.
La terminologie M1 et M2 servant à désigner respectivement les phénotypes pro-
inflammatoire et anti-inflammatoire de la microglie sont des termes plutôt archaïques
d’abord utilisés pour désigner les phénotypes acquis par des macrophages en culture à la suite
de différentes stimulations (Huang et al. 2018). Ces phénotypes sont basés sur un certain
profil de marqueurs moléculaires et supposent une distinction tranchée entre M1 et M2.
Toutefois, ceci représente difficilement la complexité phénotypique des macrophages et des
cellules microgliales, étant donné que ceux-ci expriment fréquemment les marqueurs des
deux phénotypes simultanément (Gopal et al. 2018, Huang et al. 2018, Singhal et al. 2019,
Sun, Hei, et al. 2019). De plus, les différentes conditions d’activation de la microglie lui

30
permettent d’acquérir des phénotypes d’activation qui diffèrent clairement de cette
dichotomie réductrice : M1 versus M2 ou pro-inflammatoire versus anti-inflammatoire
(Nikodemova et al. 2014, Keren-Shaul et al. 2017, Deczkowska et al. 2018). C’est d’autant
plus le cas, lors d’une activation chronique de la microglie, caractéristique des maladies
neurodégénératives (Perry et al. 2010).

31
Figure 9: L’activation microgliale dans la neuroinflammation aigue
A. La microglie maintient l’homéostasie dans le SNC en sécrétant des facteurs trophiques.
Sous des conditions normales, l’activation microgliale est atténuée par les neurones par une
costimulation négative. B. Les différents signaux de dangers sont identifiés par des récepteurs
microgliaux. Ces derniers promeuvent l’activation microgliale et permettent une clairance
des débris cellulaires. C. Lorsque que les dangers ont été éliminés, la microglie entre dans
une phase de réparation, aidée par les neurones survivants et une co-stimulation négative
retrouvée. L’inflammation est résolue et la microglie retourne à l’état de repos. Figure
adaptée de «Immunity in amyotrophic lateral sclerosis: blurred lines between excessive
inflammation and inefficient immune responses» (Béland et al. 2020).
L’activation chronique de la microglie dans la SLA diffère nettement de l’activation aigue
décrite plus haut. Dans la SLA, la microglie est connue pour passer d’un phénotype
neuroprotecteur et anti-inflammatoire durant la phase présymptomatique à un phénotype
neurotoxique durant la phase symptomatique de la maladie (Henkel et al. 2009). Durant la
phase présymptomatique de la maladie, les cellules microgliales présentent un phénotype
anti-inflammatoire, similaire à ce qui a été décrit plus haut (Fig.9C et Fig.10A). Durant cette
phase de la maladie, la microglie perçoit probablement les premiers signes de
neurodégénérescence mais est capable de maintenir un environnement neuroprotecteur
(Henkel et al. 2009, Appel et al. 2011, Liao et al. 2012). Durant le stade présymptomatique,
la microglie anti-inflammatoire produit CD163, YM1, arginase 1, des facteurs trophiques
(BDNF, GDNF, IGF-1) et des cytokines anti-inflammatoires (IL-10, TGFβ) (Fig.10A)
(Henkel et al. 2009, Appel et al. 2011). Le phénotype présymptomatique de la microglie
permet de retarder l’apparition des symptômes et de ralentir la progression de la maladie. En
effet, lors de la déplétion de ces cellules avant l’apparition des symptômes, on assiste à une
baisse de la survie dans un modèle murin de la maladie (Audet et al. 2012). Ce phénotype
microglial est soutenu par un dialogue avec les motoneurones qui maintiennent une co-
stimulation négative en sécrétant CX3CL1. Toutefois, la mort neuronale, précédant
l’apparition des symptômes, induit une baisse dans la production de CX3CL1 (Fig.10A-B)
(Zhang, Liu, et al. 2018). La perte de la co-stimulation négative entre les motoneurones et la
microglie ainsi que l’augmentation de la production de DAMPs par les motoneurones morts
ou mourant participent au changement de phénotype microglial. Par exemple, les cellules
microgliales s’activeront en détectant les formes mal repliées, mutées ou oxydées de la

32
protéine SOD1 (Henkel et al. 2004, Furukawa et al. 2006, Appel et al. 2011) et des formes
mal repliées de la protéine TDP43 (Zhao et al. 2015) grâce au co-récepteurs CD14 et TLR2/4.
Le phénotype microglial de la phase symptomatique a précédemment été décrit comme
correspondant au phénotype pro-inflammatoire classique « M1 ». En effet, à ce stade, la
microglie devient proliférative et acquiert une forme amiboïde. De plus, on retrouve une
augmentation de la sécrétion de cytokines pro-inflammatoires (IL-1β, IL-6, TNFα) et de
ROS/RNS dans le milieu extracellulaire (Fig.10B) (Henkel et al. 2009, Appel et al. 2011,
Zhao et al. 2015). La production de molécules inflammatoires est rendue possible par
l’activation de la voie de signalisation NF-κB dans la microglie à la suite de la reconnaissance
des DAMPs par les co-récepteurs CD14 et TLR2/4 (Henkel et al. 2009, Frakes et al. 2014).
De plus, l’inactivation artificielle de cette voie de signalisation spécifiquement chez la
microglie prolonge la survie dans un modèle murin. Ainsi, il est possible d’affirmer que le
phénotype microglial symptomatique est neurotoxique (Henkel et al. 2009, Liao et al. 2012).
Plusieurs études ont démontré qu’en stimulant positivement ou négativement la microglie il
est possible de modifier la pathogénèse de la SLA chez des modèles murins. Par exemple, en
stimulant la prolifération microgliale dans un modèle murin dès l’apparition des symptômes,
on assiste à une production accrue des cytokines TNFα et IL-1β ainsi qu’à une progression
plus rapide de la maladie et une survie réduite (Gowing et al. 2009). De plus, le knock-down
du gène mutant associé à la SLA dans les cellules microgliales ou le remplacement de la
microglie par des cellules WT dans un modèle murin permet, de ralentir la progression de la
maladie (Beers et al. 2006, Boillée, Yamanaka, et al. 2006, Lee, Seong, et al. 2012, Martínez-
Muriana et al. 2016). Finalement, la stimulation de la microglie symptomatique par IL-4, une
cytokine anti-inflammatoire, ou par CX3CR1 et le blocage du potentiel prolifératif
microglial, confère une certaine neuroprotection (Cardona et al. 2006, Zhao et al. 2006). Les
auteurs des précédentes études concluent que la cytotoxicité de la microglie provient de son
phénotype pro-inflammatoire.

33
En tenant compte du supposé phénotype pro-inflammatoire de la microglie durant la phase
symptomatique de l’ALS, l’utilisation de drogues anti-inflammatoires pourrait protéger les
motoneurones de la microglie. Malheureusement, les traitements anti-inflammatoires sont
des échecs lors des essais cliniques (Benatar 2007, DeLoach et al. 2015). Par exemple, les
médicaments anti-inflammatoires comme le celecoxib, la minocycline, la thalidomide ou le
plioglitazone ont échoué en phase clinique (DeLoach et al. 2015). Ainsi, bien qu’il soit clair
que la microglie soit neurotoxique au stade symptomatique, des recherches plus récentes
démontrent que le phénotype microglial est plus complexe, ou du moins, diffère
substantiellement du phénotype pro-inflammatoire. Cela pourrait expliquer, en partie, l’échec
des médicaments anti-inflammatoires lors d’essais cliniques.
L’activation microgliale durant le stade symptomatique de la SLA diffère du phénotype pro-
inflammatoire classique. Cette différence pourrait s’expliquer à la fois par la nature chronique
de l’activation ainsi que par l’expression intrinsèque de protéines mutée et/ou mal repliée,
générant un stress cellulaire (Boillée, Yamanaka, et al. 2006). Plusieurs études récentes
écartent le phénotype d’activation de la microglie symptomatique du phénotype pro-
inflammatoire classique auquel la microglie a été généralement associée. Premièrement,
Nikodemova et al. montrent que, bien que la microglie soit proliférative dans la moelle
épinière d’animaux symptomatiques, le profil pro-inflammatoire classique est altéré :
l’expression de certains marqueurs pro-inflammatoires (IL-6, TNFα) était faible alors que
d’autres marqueurs pro-inflammatoires (MCP-1, COX2, iNOS) sont surexprimés
(Nikodemova et al. 2014). Le profil microglial est complété par une forte expression de
galectin-3, de VEGF, et d’ostéopontine, associée à plusieurs fonctions microgliales comme
la prolifération cellulaire (Nikodemova et al. 2014, Rahimian, Béland, et al. 2018). De plus,
la microglie symptomatique ne semble pas pouvoir monter une réponse efficace à la suite
d’une stimulation par le LPS (Nikodemova et al. 2014). Une réponse inflammatoire
émoussée indique probablement une fatigue microgliale à la suite de leur activation
chronique. Deuxièmement, les travaux de recherche sur les « Aberrant Astrocytes » montrent
que la microglie symptomatique exprime des marqueurs cellulaires autrement réservés aux
astrocytes (GFAP, S100β, GJA1) et que son potentiel prolifératif est dépendant à CSF1R.

34
(Díaz-Amarilla et al. 2011, Trias, Díaz-Amarilla, et al. 2013, Trias et al. 2016). Au lieu de
cibler l’aspect inflammatoire de la microglie, prévenir la prolifération microgliale en
bloquant le récepteur de CSF1 réduit la progression de la maladie dans un modèle murin de
la maladie (Trias et al. 2016). Finalement, un phénotype microglial d’abord identifié dans
une sous-population de cellule microgliale dans la maladie d’Alzheimer est aussi trouvé dans
une panoplie de maladies neurodégénératives dont la sclérose en plaques (dans le modèle
d’encéphalomyélite auto-immune expérimentale) et dans la SLA et est donc nommé
« microglie associée à la maladie » (disease associated microglia, DAM) (Weissmann et al.
2016, Keren-Shaul et al. 2017, Krasemann et al. 2017, Deczkowska et al. 2018, Kang et al.
2018). Ce phénotype a été découvert grâce au profilage transcriptomique spécifique à la
microglie et semble être activé de façon dépendante au récepteur de DAMPs, TREM2
(Fig.10B) (Krasemann et al. 2017). Il est associé à la régulation négative de gènes
homéostatiques (CX3CR1, P2RY12, TMEM119) et à la régulation positive de gènes liés à
l’inflammation, à la phagocytose ou aux fonctions microgliales en général (APOE, CCL3,
CLEC7A, CST7, GPNMB, ITGAX, LGALS3, LILRB4, etc.) (Fig.10B). Malgré un rôle
bénéfique de la phagocytose dans les maladies neurodégénérative pour la clairance des
DAMPS, le rôle exact de DAM sur la progression de la SLA n’est pas encore connu.
Cependant, bien que le transcriptome de DAM soit relativement bien connu, un écart en le
transcriptome et ce qui est effectivement traduit existe dans plusieurs cellules, dont la
microglie (Boutej et al. 2017, Seyfried et al. 2017). Ainsi, une étude plus approfondie du
profil protéomique de DAM est nécessaire avant de conclure sur rôle dans la pathogénèse de
la SLA.
Ainsi, étant donné la présence du phénotype DAM dans la SLA et l’expression aberrante de
marqueurs cellulaires, il est possible d’affirmer que, bien que la microglie symptomatique
soit neurotoxique, son phénotype d’activation ne correspond pas au phénotype pro-
inflammatoire ou « M1 ». Une meilleure compréhension du phénotype microglial durant la
phase symptomatique de la SLA est donc nécessaire.

35
1.2.2. Astrocytes
Les astrocytes sont un des éléments de la glie du SNC et sont, tout comme les neurones,
d’origine neuroépithéliale (Pringle et al. 2003). Sous des conditions normales, les astrocytes
remplissent plusieurs fonctions nécessaires au développement normal du SNC et son
homéostasie. Par exemple, les astrocytes sont impliqués dans la synaptogénèse et dans la
formation de la barrière hémato-encéphalique (BHE) (Abbott et al. 2010, Molofsky et al.
2012). Ils fournissent également un support trophique et énergétique aux neurones en plus de
moduler la transmission synaptique en recapturant certains neurotransmetteurs, dont le
Figure 10: L'activation chronique de la microglie dans la SLA
Le dommage initial durant la phase présymptomatique de la maladie induit une transition de
phénotype vers un phénotype anti-inflammatoire. A. Durant cette phase, la microglie est apte à contenir l’inflammation et sécrète diverses molécules anti-inflammatoires. De plus, co-stimulation
entre les neurones et la microglie est maintenue. B. À la suite d’une stimulation chronique de la
microglie par les DAMPs, le phénotype microglial change pour un phénotype neurotoxique en même temps que l’apparition des symptômes, la co-stimulation négative est perdue et la mort neuronale est
exacerbée par la sécrétion de molécules cytotoxiques. Figure adaptée de «Immunity in amyotrophic
lateral sclerosis: blurred lines between excessive inflammation and inefficient immune responses»
(Béland et al. 2020).

36
glutamate (Pellerin et Magistretti 2012, Verkhratsky et Parpura 2016, Pajarillo et al. 2019).
Aussi, les astrocytes sont la principale source de la cytokine anti-inflammatoire TGFβ dans
le SNC. Ce faisant, ils aident la microglie à maintenir son phénotype de repos et
l’homéostasie du SNC (Diniz et al. 2019).
Dans la SLA, les astrocytes acquièrent un phénotype réactif et neurotoxique et prolifèrent
durant le stade symptomatique. L’activation astrocytaire peut être partiellement expliquée
par leur détection de l’IFNγ produit par les neurones dégénérescents puis par les astrocytes
eux même (Fig.11A) (Bowerman et al. 2015, Liu et al. 2015). De plus, les astrocytes sont,
eux aussi, sensibles à certains mécanismes de toxicité cellulaires initiés par les protéines mal
repliées dont la dérégulation de la protéostasie et la dysfonction mitochondriale, contribuant
à leur transformation vers un phénotype réactif (Bruijn et al. 1997, Cassina et al. 2008). Les
astrocytes activés perdent leurs fonctions homéostatiques (recapture du glutamate, sécrétion
de GDNF) et deviennent toxiques, spécifiquement contre les neurones moteurs et non pas
contre les autres types de neurones (Nagai et al. 2007, Yamanaka et Komine 2018). La
sensibilité des neurones moteurs à la toxicité astrocytaire viendrait d’une baisse d’expression
du complexe majeur d’histocompatibilité de classe I (CMHI) sur ces premiers. La baisse
d’expression du CMHI est détectée par les astrocytes grâce au récepteur KIR3DL2 (Killer
cell immunoglobulin-like cell receptor 3 DL 2) (Song et al. 2016).
La neurodégénérescence due au phénotype réactif des astrocytes est causée par la sécrétion
de facteurs toxiques par ces derniers (Ramírez-Jarquín et al. 2017). Parmi ceux-ci, on
retrouve IL-1β, IL-6, IL-18, ROS ou CD163 (Fig.11A) (Marchetto et al. 2008, Bowerman et
al. 2015, Johann et al. 2015). La production et la sécrétion de ces facteurs sont stimulées par
l’activation de l’inflammasome NLRP3, de TNF-like weak inducer of apoptosis (Tweak) et
de NF-κB dans les astrocytes de modèles murins et chez l’humain (Bowerman et al. 2015,
Johann et al. 2015, Ouali Alami et al. 2018, Liu et al. 2020).
La toxicité des astrocytes dans la SLA est, par elle-même, suffisante pour causer la
neurodégénération, tel que le démontrent des co-cultures d’astrocytes et de motoneurones
ainsi que des modèles murins exprimant le transgène SOD1G93A ou le transgène TDP43M377V

37
spécifiquement dans les astrocytes (Di Giorgio et al. 2007, Nagai et al. 2007, Papadeas et al.
2011, Tong et al. 2013). Aussi, des astrocytes prélevés directement sur des tissus post-mortem
de patients présentant la forme familiale ou la forme sporadique de la SLA sont toxiques pour
des motoneurones en co-culture (Haidet-Phillips et al. 2011).
En plus de la sécrétion de facteurs toxiques, les astrocytes exacerbent la dégénérescence des
neurones moteurs par leur incapacité à recapturer le glutamate. Celui-ci reste présent dans
l’espace synaptique et cause des décharges neuronales surabondantes, menant à une
excitotoxicité dans les motoneurones, celle-ci liée aux mécanismes de toxicité cellulaire
énumérés dans la section 1.2. En effet, la suractivation des récepteurs AMPA et NMDA par
le glutamate cause une entrée excessive de Ca2+ dans le neurone post-synaptique menant à la
production de ROS et à un stress du réticulum endoplasmique (encart de la figure 11) (Ilieva
et al. 2009). La recapture du glutamate est normalement assurée par le récepteur EAAT2
présent sur les astrocytes entourant les synapses (Rothstein et al. 1995, Yang et al. 2009). La
baisse d’activité et d’expression du récepteur EAAT2 retrouvée dans la SLA peut être
expliquée par la perte de synapse entre les neurones moteurs et inférieurs menant à une baisse
d’expression de HNRNPK, régulateur de EAAT2 ou par la production d’une forme tronquée
et inactive de EAAT2 à la suite de son clivage par la caspase-3 (encart de la figure 11)
(Boston-Howes et al. 2006, Ilieva et al. 2009, Yang et al. 2009). La modulation de la
recapture du glutamate est une stratégie thérapeutique importante. En effet, le médicament
riluzole, qui été pendant plus de 20 ans le seul traitement contre la SLA, bloque la
transmission glutamatergique réduisant l’excitotoxicité liée au glutamate (Doble 1996).
L’activation astrocytaire a un effet indirect sur la pathogénèse de la SLA en participant à
l’activation microgliale. En effet, bien que l’inactivation du gène muté associé à la SLA
spécifiquement dans les astrocytes, n’a pas d’effet sur l’apparition des symptômes chez un
modèle murin, la baisse dans la sécrétion de facteurs toxiques réduit l’activation microgliale
menant à une réduction dans la progression de la maladie (Yamanaka, Chun, et al. 2008).
L’activation de Tweak, surexprimée par les astrocytes réactifs, induit la production et la
sécrétion d’IL-6 chez ceux-ci en aggravant la prolifération et l’activation microgliale
(Fig.11B) (Bowerman et al. 2015). L’activation de la voie de signalisation NF-κB dans les

38
astrocytes est d’abord avantageuse car elle aide à maintenir le phénotype microglial anti-
inflammatoire associé à la phase présymptomatique de la SLA. À la fin de la phase
présymptomatique, l’activation de la voie NF-κB spécifiquement dans les astrocytes induit
la prolifération microgliale par la voie de signalisation Wnt. Toutefois, comme chez la
microglie, l’activation chronique de cette voie est finalement nuisible et accélère la
progression de la maladie (Ouali Alami et al. 2018). Alors que les astrocytes sont aptes à
moduler l’activation de la microglie, l’inverse est aussi vrai. En effet, la sécrétion de facteurs
inflammatoires, dont IL-1α, IL-1β, TNF et C1q, par la microglie contribue à la transformation
vers le phénotype réactif des astrocytes (Fig.11B) (Welser-Alves et al. 2011, Liddelow et al.
2017). Ainsi, le dialogue microglie-astrocyte semble contribuer à leur activation et leur
prolifération respective ainsi que la perte de leurs fonctions normales (Liddelow et al. 2017).
Cette escalade d’activation participe à l’apparition de l’astrogliose et de la microgliose
chroniques et excessives en plus de l’inflammation en général présente dans les tissus de
modèles animaux et des patients atteints de la SLA.

39
Figure 11: Le dialogue neurone-glie et la mort neuronale
Le dialogue entre les différents types cellulaires du SNC est un élément clef de la pathogénèse
de la SLA. A. Les astrocytes sont activés par les neurones lorsque ces derniers sécrètent IFNγ
et ont une expression réduite du CMH I. En retour, les astrocytes cessent de sécréter des
facteurs trophiques et sécrètes des molécules neurotoxiques. De plus, les astrocytes
provoquent une excitotoxicité liée au glutamate dans les neurones menant au stress oxydatif
et au stress du réticulum endoplasmique (encart). Les astrocytes promeuvent la prolifération
microgliale en sécrétant IL-6 et la microglie, à son tour, contribue au changement
phénotypique des astrocytes en sécrétant une panoplie de molécules inflammatoires. La
stimulation réciproque des astrocytes et de la microglie contribue à la mort neuronale.
1.2.3. Oligodendrocytes et cellules de Schwann
Les oligodendrocytes sont le dernier élément de la glie dans le SNC et sont responsables de
myéliniser les axones des neurones pour assurer l’acheminement rapide du potentiel d’action
vers l’arborisation terminale. La survie des oligodendrocytes est assurée, entre autres, par les
diverses fonctions de la protéine TDP43 (Wang, Ho, et al. 2018). Ainsi, la perte de fonction
de TDP43, associée à la SLA, est probablement la cause de la dégénérescence de
l’oligodendroglie et de la démyélinisation axonale, précédant la mort neuronale (Kang et al.
2013, Philips et al. 2013, Wang, Ho, et al. 2018). Cependant, la toxicité des oligodendrocytes
envers les motoneurones ne semble pas être seulement dépendante de la perte de fonction de

40
TDP43 (Wang, Ho, et al. 2018), mais plutôt d’un gain de fonction de la protéine SOD1
(Ferraiuolo et al. 2016). L’implication de la protéine SOD1 dans la cytotoxicité propre aux
oligodendrocytes a été confirmée dans des co-cultures utilisant les cellules de modèle murins
pour SOD1 mutée ou en utilisant des oligodendrocytes dérivés de cellules souche
pluripotentes induites de patients atteints de la forme familiale et de la forme sporadique de
la maladie. Toutefois, la protéine SOD1 ne semble pas être impliquée dans la cytotoxicité
propre aux oligodendrocytes chez les patients possédant une expansion hexanucléotidique
dans le gène C9orf72 (Ferraiuolo et al. 2016). Les oligodendrocytes peuvent exacerber la
mort neuronale en causant l’hyperexcitabilité neuronale, un phénomène trouvé dans le cortex
des patients atteints de la SLA et qui précède l’apparition des symptômes (Vucic et al. 2008)
ou encore par une dérégulation dans la production et la sécrétion de lactate, un substrat
métabolique utilisé par les neurones moteurs. Le transporteur de lactate MCT1, normalement
fortement exprimé par les oligodendrocytes, est dérégulé chez les patients atteints de la SLA
(Lee, Morrison, et al. 2012, Ferraiuolo et al. 2016). Dans les modèles murins exprimant la
mutation SOD1G93A, les précurseurs des oligodendrocytes sont néanmoins prolifératifs mais
ne peuvent pas atteindre leur maturation et ainsi sont inaptes à remplacer les oligodendrocytes
dégénérescents (Kang et al. 2013, Philips et al. 2013, Ferraiuolo et al. 2016).
Les cellules de Schwann sont impliquées dans la myélinisation des axones dans le SNP. Peu
d’information est disponible quant au rôle de ces cellules dans la pathogénèse de la SLA. Les
cellules de Schwann myélinisantes dégénèrent avec la progression de la maladie et
l’environnement inflammatoire du SNP empêche leur régénération et la remyélinisation,
participant au développement des troubles moteurs associés à la SLA (Deng et al. 2018). Une
sous-population de cellules de Schwann, appelée cellules de Schwann périsynaptiques (CSP)
est retrouvée à la JNM et est responsable du maintien de son intégrité structurelle et
fonctionnelle. Les CSP subissent des changements morphologiques et fonctionnels selon
l’état d’innervation de la JNM. Dans la SLA, des changements aberrants ou l’absence de
changements des CSP exacerbent les troubles moteurs (Arbour et al. 2017). L’excitabilité
neuronale est directement corrélée avec leur rapidité à dégénérer. Ainsi, lors de la perte des
unités motrices (le neurone moteur et les fibres musculaires innervées par celui-ci) rapides-
fatigable, les unités motrices lentes ré-innervent les fibres musculaires dé-innervées à l’aide

41
des PSC. Toutefois, dans la SLA, la plasticité et la capacité de ré-innervation des PSC est
plutôt réduite. De plus, les nouvelles JNM, dont la formation est encadrée par les PSC,
pourraient ne pas être complètement fonctionnelles (Arbour et al. 2017).
1.2.4. Macrophages et monocytes
Tout comme les cellules microgliales, les macrophages et les monocytes sont d’origine
myéloïde mais, en l’absence de brèche dans la BHE, ceux-ci restent absents du parenchyme
du cerveau et de la moelle épinière. Les macrophages peuvent toutefois être retrouvés dans
les méninges, l’espace périvasculaire de la BHE et dans le plexus choroïde (Prinz et Priller
2014, Prinz et al. 2017). Dû à l’important chevauchement entre les marqueurs cellulaires de
la microglie et des macrophages du SNC, il est difficile de départir les rôles uniques de ces
derniers (Goldmann et al. 2016). Bien que n’étant pas en contact direct avec les
motoneurones dégénérescents, les macrophages du SNC participent probablement à la
neuroinflammation dans la SLA, à l’instar des cellules microgliales, en sécrétant divers
facteurs toxiques. Dans le SNP (où la BHE n’est pas présente mais remplacée par la barrière
hémato-nerveuse nettement plus perméable que la BHE (Weerasuriya et Mizisin 2011)), les
macrophages peuvent infiltrer les nerfs et y sécrètent des facteur toxiques tels des molécules
du complément (Chiu et al. 2009, Sta et al. 2011). Ces facteurs promeuvent la dégénérescence
wallérienne des nerfs, associée à la SLA (Blain et al. 2011). Le niveau d’infiltration des
macrophages dans les nerfs corrèle avec la progression de la maladie (Chiu et al. 2009,
Graber et al. 2010, Lincecum et al. 2010, Kano et al. 2012).
Les monocytes sont des cellules myéloïdes retrouvées dans le sang. À la suite d’une
stimulation appropriée, les monocytes peuvent infiltrer le parenchyme de certains organes.
Dans la SLA, il existe présentement un débat à savoir si les monocytes infiltrent bel et bien
le parenchyme du SNC et participent à la neuroinflammation. Les preuves appuyant
l’hypothèse de l’infiltration se basent d’abord sur l’augmentation de la production du
chemoattractant CCL2 dans le liquide céphalo-rachidien de modèles murins et chez les
patients (Henkel et al. 2004, Butovsky et al. 2012) et une perte d’étanchéité de la BHE
(Kakaroubas et al. 2019). L’utilisation de certains marqueurs cellulaires montrerait aussi la

42
présence de monocytes dans le parenchyme du SNC (Butovsky et al. 2012, Bennett et al.
2016, Zondler et al. 2016). Toutefois, comme mentionné précédemment, plusieurs marqueurs
des cellules myéloïdes se chevauchent, d’autant plus lorsque celles-ci sont activées
(Goldmann et al. 2016, Béland et al. 2020). Dans l’hypothèse de l’infiltration monocytaire,
il n’est encore pas tout à fait clair si cette infiltration est nuisible et exacerbe la mort neuronale
(Henkel et al. 2004, Mantovani et al. 2009, Butovsky et al. 2012) ou, au contraire, soulage
l’inflammation et aide les neurones à récupérer, dans une certaine mesure (Beers et al. 2008,
Zondler et al. 2016).
Plusieurs recherches tendent plutôt vers l’hypothèse contre l’infiltration monocytaire. Par
exemple, à la suite d’une transplantation de moelle osseuse provenant de souris exprimant la
protéine fluorescente verte (GFP) dans des souris irradiées, présentant ou non une mutation
liée à la SLA, la quantité de monocytes GFP+ retrouvée dans le SNC des souris SLA ne
diffère pas de celle retrouvée dans les souris sauvages. La présence de monocyte GFP+ est
expliquée par la plus grande perméabilité de la BHE suivant l’irradiation (Solomon et al.
2006). L’utilisation de la parabiose entre une souris GFP+ et une souris SLA mène à la même
conclusion. Ici, les seules cellules positives pour GFP retrouvées dans le SNC ne sont
présentes que dans les capillaires. L’utilisation de la parabiose ne nécessite pas l’irradiation
de l’animal recevant les nouvelles cellules et est donc plus représentative des conditions
physiologiques. Ainsi la prolifération des cellules myéloïdes du SNC dépend uniquement de
la microglie et non pas d’un progéniteur externe au SNC (Ajami et al. 2007). Finalement,
l’utilisation de cytométrie de masse de cellules individuelles utilisées dans différentes
maladies neuroinflammatoires permet d’identifier une population de cellules correspondant
aux monocytes infiltrants, absente dans l’analyse portant sur la SLA (Ajami et al. 2018).
Sans tenir compte de la possible infiltration des monocytes dans le parenchyme du SNC, les
monocytes isolés des patients SLA présentent tout de même un phénotype inflammatoire et
une activation préférentiellement classique (classique : CD14++/CD16- versus non-
classique : CD14+/CD16++) (Zondler et al. 2016). Ce phénotype est accompagné par la
production ou la sécrétion de molécules pro-inflammatoire telles qu’IL-1β, IL-8, FosB,
CXCL1 et CXCL2, corrélant avec la sévérité de la maladie. Ainsi, bien que la détérioration

43
de la BHE ne pourrait pas être suffisante pour l’infiltration de cellules dans le SNC, elle
pourrait permettre une entrée plus importante de ces facteurs inflammatoires dans le SNC
participant, de cette façon, à la neuroinflammation (Zhang et al. 2005, Zhao et al. 2017). Le
gain de phénotype inflammatoire est retrouvé chez les personnes asymptomatiques possédant
une mutation liée à la SLA, indiquant que les monocytes pourraient être impliqués dans la
pathogénèse de la SLA avant l’apparition des symptômes (Zondler et al. 2016). L’implication
des monocytes dans la clairance d’agrégats protéiques extracellulaire a été démontrée dans
la maladie d’Alzheimer où ces cellules ont une vitesse de roulement réduite, une
augmentation de l’adhésion aux capillaires et aident les cellules microgliales à se débarrasser
de ces agrégats (Michaud et al. 2013, Fani Maleki et Rivest 2019). Il est possible d’imaginer
un mécanisme semblable partagé par les maladies neurodégénératives. Ainsi, dans la SLA,
les monocytes pourraient alléger le fardeau des agrégats de protéines en début de maladie.
Toutefois, ces cellules deviennent fonctionnellement altérées avec l’apparition des
symptômes. Leur capacité phagocytaire est réduite, le trafic des endosomes vers les
lysosomes est altéré et leur adhésion est diminuée (Zondler et al. 2016).
L’infiltration des monocytes dans le parenchyme du SNC affecté par la SLA restera encore
pour plusieurs années sujette à débat. Le rôle exact des monocytes dans la neuroinflammation
et la pathogénèse de la SLA demeure toujours plutôt vague. Tout comme les cellules
microgliales, les effets directs et indirects de l’activation monocytaire sur les motoneurones
pourrait changer avec la progression de la maladie; tantôt neuroprotectrice, tantôt
cytotoxique.
1.2.5. Lymphocytes
Les lymphocytes B et T sont des cellules d’origine lymphoïde et sont des éléments de
l’immunité adaptative, plus lente en cas d’infection que l’immunité innée, mais plus précise.
Bien que les lymphocytes B ne semblent pas avoir d’influence sur la progression de la
maladie dans les modèles murins (Naor et al. 2009), on retrouve une accumulation
d’anticorps de type IgG, produits par les lymphocytes B, sur les motoneurones

44
dégénérescents et les nerfs de patients atteints de la SLA (Engelhardt et Appel 1990, Chiu et
al. 2009). L’impact de ces anticorps sur la pathogénèse de la SLA n’est pas connu
Les lymphocytes T, quant à eux, ont un rôle nettement plus actif que les lymphocytes B. Les
lymphocytes T infiltrent la moelle épinière dans les modèles murins et chez les patients
(Engelhardt et al. 1993, Banerjee et al. 2008). L’infiltration par les lymphocytes T CD4 est
neuroprotectrice et est suivie dans les derniers stades de la maladie par l’infiltration des
lymphocytes T CD8, qui eux, sont plutôt neurotoxiques (Beers et al. 2008). Les lymphocytes
T CD8 sécrètent des facteurs toxiques pour les motoneurones tels que les granzymes ou
l’IFNγ (Coque et al. 2019). Inversement, l’expansion ex-vivo puis le transfert de
lymphocytes CD4 dans les souris SLA montre que ceux-ci contribuent à ralentir la
progression de la maladie. De plus, cet effet semble être causé par les lymphocytes T
régulateurs (Treg), une sous-population des lymphocytes CD4 (Banerjee et al. 2008, Beers
et al. 2008). Les Treg sont un des principaux producteurs de la cytokine homéostatique IL-
10 et pourraient contribuer à maintenir le phénotype anti-inflammatoire de la microglie plus
longtemps et ainsi réduire la neuroinflammation en général (Appel et al. 2010, Kleinewietfeld
et Hafler 2014). Chez l’humain, le nombre de Treg est inversement proportionnel avec la
vitesse de progression de la maladie. Ainsi, l’expansion ex-vivo de Treg puis leur
transplantation chez un même patient pour être une avenue thérapeutique intéressante (Beers
et al. 2017, Sheean et al. 2018).
1.2.6. Autres cellules immunitaires
Peu d’informations est à ce jour disponible sur l’effet des autres cellules immunitaires sur la
pathogénèse de la SLA, qu’elles soient d’origine lymphoïde ou myéloïde. Les cellules
dendritiques sont en nombre réduit dans le sang des patients atteints de la SLA et possèdent
un phénotype pro-inflammatoire, par la production de CCL2 et d’IL-8 (Henkel et al. 2004,
Sta et al. 2011, Rusconi et al. 2017). Toutefois, on note une augmentation de leur infiltration
dans la moelle épinière des patients SLA. Le chevauchement des marqueurs cellulaires des
cellules d’origine myéloïde rend l’étude du rôle exact des cellules dendritiques dans la
pathogénèse de la SLA difficile.

45
Les cellules NK (Natural Killer) infiltrent quant à elles, le SNC et leur nombre augmente
avec la progression de la maladie. Les cellules NK peuvent directement causer la mort
neuronale par le récepteur KLRK1 (Killer cell lectin-like receptor K1) reconnaissant son
ligand sur les motoneurones ou indirectement en promouvant le changement du phénotype
microglial vers le phénotype neurotoxique en sécrétant l’IFNγ (Garofalo et al. 2020).
Plus de détails sont connus sur l’implication des mastocytes et des neutrophiles dans la
pathogénèse de la SLA, notamment leur rôle dans le SNP. Tout comme les macrophages, les
mastocytes et les neutrophiles sont recrutés sur les nerfs et aux JNM (Trias et al. 2018).
L’infiltration de ces cellules corrèle avec les régions du SNP affectées et la propagation de
la maladie. Les mastocytes dégranulent à la fois au niveau des fibres musculaires
endommagées et au niveau des JNM. De plus, les mastocytes aident à recruter les neurophiles
dans le SNP. Ces derniers phagocytent les JNM dégénérescentes et, ce faisant, empêchent la
réinnervation (Trias et al. 2018). La modulation de la prolifération des mastocytes par la
drogue masitinib, réduit l’infiltration des mastocytes et neutrophiles dans le SNP et ralentit
la progression de la maladie chez les patients atteints de la SLA (Mora et al. 2020).
1.3. Inflammation et pathogénèse dans la SLA
Tel qu’abordé dans les sections précédentes, la neuroinflammation est une composante
importante de la pathogénèse de la SLA. La neuroinflammation présente dans cette maladie,
peut être décrite à la fois au niveau moléculaire, où plusieurs gènes mutants liés à la SLA
influencent directement les voies de signalisation de l’inflammation. La neuroinflammation
peut aussi être décrite au niveau cellulaire et systémique, où l’étude des différents types
cellulaires immuns et leurs interactions permet de comprendre leur effet sur la progression
de la maladie.
1.3.1. Gènes associés à la SLA et leur impact sur l’inflammation
Plusieurs protéines issues de gènes présentant des mutations associées à la SLA sont des
modulateurs directs de voies de signalisation de l’inflammation. Par exemple, la protéine

46
TDP43, lorsque mutée ou mal repliée, peut interagir avec la sous-unité de NF-κB p65 dans
les neurones et la glie de modèles murins ou de patients atteints de la SLA (Swarup et al.
2011). Dans les neurones, cette interaction mène à une plus grande vulnérabilité de ceux aux
facteurs toxiques. Dans la glie, la stimulation subséquente avec du LPS ou des ROS, conduit
à une production exacerbée de molécules pro-inflammatoire et neurotoxiques (Swarup et al.
2011). Aussi, l’agrégation des formes sauvage ou mutée de la protéine UBQLN2, promeut
l’agrégation de TDP43 et l’activation de la voie de signalisation NF-κB (Picher-Martel et al.
2015). De plus, la protéine FUS mutante est aussi un activateur de cette voie de signalisation,
interagissant, elle aussi, avec p65 (Uranishi et al. 2001). L’activation de la voie NF-κB par
FUS mutée dans les astrocytes induit l’astrogliose, et la production de facteurs toxiques, dont
le TNFα, menant à la mort neuronale (Kia et al. 2018).
Dans la SLA causée par la perte de fonction du gène OPTN codant pour la protéine
optineurine, on assiste à une activation de la voie NF-κB, une microgliose et une
augmentation dans la sécrétion de cytokines pro-inflammatoires selon la mutation d’OPTN
(Slowicka et al. 2016, Liu et al. 2018, Markovinovic et al. 2018). Dans sa forme sauvage,
OPTN est un inhibiteur de NF-κB, faisant compétition à l’activateur NEMO par une
homologie de structure (Zhu et al. 2007). OPTN est aussi responsable de la production de la
cytokine immunosuppressive IFNβ. La perte de production de cette cytokine due à
l’insuffisance d’OPTN contribue à l’activation microgliale (Markovinovic et al. 2018). La
production d’IFNβ est aussi régulée par le partenaire d’OPTN, TBK1 pour lequel on compte
aussi des mutations liées à la SLA (Munitic et al. 2013, Markovinovic et al. 2018, Béland et
al. 2020). Le gène TNIP, aussi inhibiteur de NF-κB et partenaire d’OTPN, est muté dans la
SLA et perd ses fonctions normales (Benyamin et al. 2017).
L’expression de C9orf72 est généralement plus élevée dans les cellules microgliales
suggérant un rôle possible du produit protéique de ce gène pour le fonctionnement normal de
la microglie. En effet, la perte de fonction de ce gène associée au développement de la SLA
cause une augmentation dans la production de cytokines pro-inflammatoire (O'Rourke et al.
2016). Finalement, bien qu’il ne cause pas la SLA, les polymorphismes du gène CX3CR1,
exprimé par les cellules myéloïdes, mène à une progression plus rapide de la maladie chez

47
les porteurs d’un polymorphisme (Lopez-Lopez et al. 2014). CX3CR1 mutant présenterait
une affinité réduite pour son ligand, CX3CL1, exprimé par les neurones moteurs. Tel que vu
dans la section 1.2.1 (voir Fig.10A), la co-stimulation négative entre la microglie et les
neurones moteurs permet de maintenir le phénotype de repos de la microglie. Une affinité
réduite contribue donc à exacerber la microgliose et la mort neuronale.
Les protéines issues de gènes associés à la SLA peuvent aussi indirectement induire
l’inflammation. Ainsi, tel qu’expliqué aux sections 1.1.3.4. et 1.2.1., TDP43 et SOD1 mutées
peuvent être sécrétées par les neurones dégénérescents (Urushitani et al. 2006, Ezzi et al.
2010, Zhao et al. 2015). Les protéines mal repliées sont reconnues en tant que DAMP par
TLR2/4 et leur co-récepteur CD14 et activent la voie NF-κB classique (Furukawa et al. 2006,
Henkel et al. 2009, Appel et al. 2011, Zhao et al. 2015).
1.3.2. L’inflammation excessive et la réponse immunitaire inefficace
Tel que vu dans les sections précédentes, bien que les neurones moteurs soient les principales
cellules affectées par la dégénérescence, la SLA est bel et bien une maladie systémique. En
effet, le système immunitaire et d’autres organes que le SNC (foie, peau, et muscles) sont
touchés, contribuant à la pathogenèse (Henkel et al. 2004, Chiu et al. 2009, Loeffler et al.
2016, Paré et Gros-Louis 2017, Gugliandolo et al. 2018, Béland et al. 2020). Dans la SLA,
le système immunitaire présente à la fois une inflammation excessive et incontrôlée mais
affiche aussi une réponse immunitaire inefficace ou émoussée.
Alors que la phase présymptomatique des Treg, de cellules microgliales et astrocytes anti-
inflammatoires, l’inflammation excessive et la réponse immunitaire émoussée associées au
stade symptomatique de l’ALS sont caractérisés par une microgliose et une astrocytose
importantes et par la présence de lymphocytes T CD8, de neutrophiles et de mastocytes
cytotoxiques et finalement de monocytes inflammatoires (Béland et al. 2020) (Fig.12A). Le
passage vers l’hyperinflammation et la perte des fonctions normales du système immunitaire
est difficilement localisable durant la progression de la maladie, mais il est probable que ce
passage est dû à des insultes chroniques et répétées, intrinsèques ou extrinsèques au système

48
immunitaire. Durant le stade présymptomatique, les facteurs génétiques peuvent directement
affecter l’inflammation comme vu à la section 1.3.1. Plusieurs autres insultes au système
immunitaire peuvent modifier la progression de la maladie (Fig.12B). Dans la SLA, le rôle
des facteurs environnementaux est plutôt mal compris. Le vieillissement est le seul facteur
de risque confirmé pour cette maladie (Lucin et Wyss-Coray 2009). Les cellules microgliales
âgées peuvent difficilement retourner au phénotype de repos; la baisse d’expression de
CX3CR1 rend la co-stimulation négative avec les motoneurones plus ardue dans la SLA
(Wynne et al. 2010). L’inflammation systémique ou dans le SNC causée par une infection
bactérienne peut exacerber la progression de la maladie. L’injection de LPS dans une modèle
murin SOD1 précipite les symptômes de la SLA (Nguyen et al. 2004). De plus, les traumas
au SNC ou du SNP accompagnés par la neuroinflammation, semblent prédisposés pour la
SLA (Chen et al. 2007, Schram et al. 2019). La présence de toxines dans l’organisme, telles
celles produites par la consommation de tabac, semble affecter la chance de développer la
maladie (Zhan et Fang 2019); la nicotine causant un fonctionnement altéré des cellules
microgliales (Brody et al. 2017). Finalement, le microbiome, soit l’ensemble des bactéries
symbiotiques ou commensales de l’organisme, est capable d’affecter l’homéostasie du SNC.
Dans la SLA, le microbiome est susceptible de déclencher l’agrégation protéique, de
favoriser l’activation microgliale et la sécrétion de cytokines dans le SNC par la sécrétion de
toxines (Erny et al. 2015). La composition de microbiome est altérée chez les patients atteints
de la SLA et dans les modèles murins, précipitant la maladie (Rowin et al. 2017, Blacher et
al. 2019, Burberry et al. 2020).

49
Figure 12: L'activation du système immunitaire dans la SLA
La phase symptomatique de la SLA est caractérisée par une hyperinflammation et la perte de
la réponse immunitaire normale. A. Différentes populations de cellules immunitaires sont
recrutées aux différents stades de la maladie, marquant ce changement vers une inflammation
aberrante. B. De multiples facteurs et insultes au système immunitaire sont en partie ou
potentiellement responsables de l’inflammation excessive et de la réponse immunitaire
affaiblie à différente phase de la maladie. Figure adaptée de «Immunity in amyotrophic
lateral sclerosis: blurred lines between excessive inflammation and inefficient immune
responses» (Béland et al. 2020).
Alors qu’on assiste à une inflammation excessive dans la SLA, le système immunitaire
présente, en même temps mais pas paradoxalement, une réponse émoussée. La réponse
immunitaire émoussée, ou même l’immunodéficience dans la SLA prévient les cellules
immunitaires de monter une réponse adéquate pour la clairance et l’élimination des dangers
(Béland et al. 2020). Cela est mis en évidence alors que les cellules microgliales d’un modèle
murin pour l’ALS ne développent pas de phénotype d’activation fonctionnel à la suite d’une
stimulation au LPS (Nikodemova et al. 2014). Aussi, chez les modèles animaux pour la SLA

50
présentant des déficiences pour certains composants de la voie de signalisation NF-κB
(Myd88, TRIF, TBK1, OPTN, CYLD), la progression de la maladie se fait plus rapidement
à cause d’une immunodéficience et une autophagie altérée dans les cellules de l’immunité
innée (Kang et Rivest 2007, Maruyama et al. 2010, Freischmidt et al. 2015, Komine et al.
2018, Dobson-Stone et al. 2020). La dérégulation de la voie NF-κB est accompagnée par la
production aberrante de cytokines (dont IFNβ) et une phagocytose affaiblie (Kocur et al.
2015, Brenner et al. 2019). L’autophagie dysfonctionnelle et la dérégulation de la
protéostasie mènent à l’accumulation de protéines mal repliées, de ROS et de mitochondries
endommagées, ajoutant à la déstabilisation des voies de signalisation cellulaires. La
dérégulation du système immunitaire, mais surtout de la microglie, l’empêche de monter une
réponse immune aigue en réaction aux dommages initiaux des motoneurones (Béland et al.
2020). Par le caractère chronique de la neurodégénération, une activation aigue et temporaire
du système immunitaire est impossible; l’incompétence de ce système à contenir la
neuroinflammation s’en trouve amplifiée.

51
Figure 13: La dualité de l'inflammation dans la SLA: hyperinflammation et
immunodéficience
A. L’inflammation excessive, ou hyperinflammation, des différentes cellules du système
immunitaire contribue à la mort neuronale. L’activation chronique de ce système est
accompagnée par l’activation de voie de signalisation NF-κB et la production de facteurs
neurotoxiques. B. L’immunodéficience dans la SLA est caractérisée par l’impossibilité du
système immunitaire à revenir à l’homéostasie et une incapacité à retirer les signaux de
danger du milieu cellulaire exacerbant la neuroinflammation et la neurodégénérescence.
Figure adaptée de «Immunity in amyotrophic lateral sclerosis: blurred lines between
excessive inflammation and inefficient immune responses» (Béland et al. 2020).
1.3.3. Le profilage moléculaire à spécificité cellulaire pour l’étude de l’inflammation
Étant donné l’importance du système immunitaire dans la pathogénèse de la SLA, la
recherche d’un traitement contre cette maladie s’est souvent arrêtée sur celui-ci comme cible
thérapeutique (Benatar 2007, DeLoach et al. 2015). Malheureusement, tel qu’expliqué dans
les sections précédentes, la plupart des traitements potentiels ciblant le système immunitaire
ont échoués avant les essais cliniques ou dans les premières phases de ceux-ci à cause de la
complexité (ou la dualité) de la réponse immunitaire.

52
Ainsi, pour développer des traitements efficaces ciblant cet élément hautement modulable de
la pathogénèse de la SLA qu’est le système immunitaire, et plus précisément la microglie,
son effecteur le plus important dans cette maladie, il est important de développer des outils
pour étudier le phénotype microglial au niveau moléculaire. Récemment, plusieurs nouvelles
techniques ont été développées pour le profilage moléculaire général ou spécifique à un type
cellulaire. Par exemple, le phénotype microglial peut être étudié en fonction de son
sécrétome, soit l’ensemble des facteurs sécrétés dans les tissus, qui est détectable, entre
autres, avec un cytokine array (Cordeau et al. 2016). Le cytokine array permet l’étude des
cytokines sécrétées par des tissus complexes, mais ne permet pas de différencier l’origine de
la cytokine. Cette technique sera utilisée dans le deuxième chapitre de cette thèse.
Aussi, le phénotype microglial peut être étudié au niveau de ses ARNm, son transcriptome,
ou de ses protéines, son protéome. Plusieurs techniques permettre l’isolation du
transcriptome et de protéome microglial à partir d’un tissu complexe qui seront comparées
en détail dans la discussion de cette thèse. L’immunopurification d’affinité des ribosomes en
traduction (TRAP, translating ribosome affinity immunopurification) (Heiman et al. 2008,
Heiman et al. 2014) et sa technique dérivée EDTA-TRAP utilisée dans le troisième chapitre
de cette thèse (Boutej et al. 2017), permettent l’isolation du transcriptome et du protéome
microglial sans avoir recours à des lyses physiques et chimiques pour la production de
suspensions de cellules individuelles; ces lyses pouvant stresser les cellules et changer
substantiellement leur phénotype. Dans l’EDTA-TRAP, l’expression d’un transgène pour
une sous-unité ribosomique étiquetée (avec l’étiquette HA ou Flag) spécifiquement dans un
type cellulaire (sous le promoteur de CD11b pour les cellules microgliales) permet
d’immunopurifier les poly-ribosomes microgliaux, les ARNm en traduction et les peptides
en synthèse. Cette technique utilisée par Boutej et al. a déjà permis l’étude de la régulation
des ARNm et des protéines microgliales à la suite d’une stimulation avec du LPS (Boutej et
al. 2017). Il a été possible de découvrir une forte disparité entre le transcriptome et le
protéome microglial suivant cette stimulation. De plus, cette disparité s’expliquerait par
l’action du principal régulateur génique Serine-arginine rich splicing factor 3 (SRSF3) qui
empêcherait la traduction d’ARNm fixés aux ribosomes. Ce mécanisme pourrait être
possiblement retrouvé dans les cellules microgliales provenant d’un modèle murin pour la

53
SLA. En effet, les protéines mal repliées associées à la SLA ainsi que le LPS peuvent activer
la microglie et certaines voies de signalisation cellulaire par les même récepteurs (Appel et
al. 2011). Les outils pour le profilage moléculaire sont d’importance capitale pour la
recherche car ils aident à générer une quantité remarquable de nouvelles données sans un
besoin d’informations préalables. Ainsi, il est possible d’étudier le phénotype microglial en
faisant fi de des connaissances antérieures parfois biaisées. L’étude du profil moléculaire de
la microglie est donc essentielle pour comprendre son rôle dans la neuroinflammation et la
pathogénèse de la SLA.
1.4. Hypothèses et objectifs de la thèse
Étant donné l’importance du système immunitaire, mais plus précisément de la microglie,
dans la pathogénèse de la sclérose latérale amyotrophique, et étant donné que la microglie
subit une transformation importante dans le stade symptomatique de la maladie, il serait
intéressant d’étudier le profil moléculaire des cellules microgliales avant et après l’apparition
des symptômes dans un modèle murin mimant la maladie. Ainsi, comme objectif principal
de cette thèse, il serait possible d’élucider de quelle manière la transformation phénotypique
des cellules microgliales est responsable de l’exacerbation de la maladie durant le stade
symptomatique. Aussi, cela pourrait permettre d’identifier de nouvelles protéines ou de
nouvelles voies de signalisation comme potentielles cibles thérapeutiques. De plus, le
profilage moléculaire de la microglie pourrait permettre de trouver de nouveaux
biomarqueurs nécessaires pour détecter la maladie, suivre sa progression ou bien, monitorer
l’efficacité d’un traitement. Dans cette thèse, dans le but de répondre à ces questions, nous
avons utilisé plusieurs modèles murins pour étudier le lien entre l’inflammation et la
pathogénèse de la sclérose latérale amyotrophique et pour comprendre le dynamisme de la
microglie dans cette maladie.
Bien que le phénotype microglial dans le stade symptomatique de la sclérose latérale
amyotrophique soit préférentiellement étudié, le phénotype présymptomatique microglial
reste largement inconnu. Dans le Chapitre 2, nous supposons que l’étude du profil de
cytokines produites par les cellules microgliales pourrait permettre d’identifier certaines

54
cytokines responsables du maintien du phénotype présymptomatique anti-inflammatoire. De
plus, en modulant l’expression ou l’activité d’une cytokine clé, il serait possible d’affecter
l’apparition des symptômes, changer la vitesse de progression de la maladie ou de modifier
la survie chez un modèle murin.
Avec l’apparition des symptômes, les cellules microgliales acquièrent un phénotype
neurotoxique. Ainsi, dans le Chapitre 3, nous proposons que l’étude des profils moléculaires
microgliaux, c’est-à-dire du transcriptome et du protéome, pourrait aider à comprendre les
bases du changement de phénotype et cibler certaines fonctions biologiques déficientes. Ceci
pourrait être accompli par le développement d’un modèle murin pour le profilage moléculaire
spécifique à la cellule microgliales dans la SLA. De plus, en ciblant une protéine ou un groupe
de protéines, il serait possible de modifier le phénotype microglial et rétablir certaines
fonctions biologiques.
SRSF3, identifiée dans le précédant chapitre, comme principal régulateur des fonctions
immunitaires de la cellule microgliale, est une protéine liant l’ARN, essentielle dans de
nombreuses voies cellulaires, à la fois dans le noyau et dans le cytoplasme. Une meilleure
compréhension du dynamisme de cette protéine dans la cellule est nécessaire pour saisir son
implication dans le traitement de l’ARN et dans le développement de diverses maladies. Dans
le Chapitre 4, nous avons réalisé une revue de la littérature pour comprendre les mécanismes
moléculaires du traitement de l’ARN, sous-jacents aux multiples fonctions biologiques liant
SRSF3 et ses ARN cibles. Cette revue de littérature permet de saisir comment une
dérégulation dans le niveau d’expression de SRSF3 ou dans ses changements post-
traductionnels aura un impact sur l’apparition de maladies variées.

55
Chapitre 2 - IL-10 Controls Early Microglial Phenotypes and Disease Onset in ALS
Caused by Misfolded Superoxide Dismutase 1
2.1. Résumé
Bien que la microgliose soit une marque des stades avancés de la sclérose latérale
amyotrophique (SLA), le rôle des cellules microgliales dans les moments initiant ou
précipitant le début de la maladie reste plutôt inconnu. Dans cet article, nous donnons de
nouvelles preuves d’une transformation distincte et adaptative du phénotype fonctionnel
microglial dans le stade préclinique de la maladie médiée par la superoxyde dismutase 1
(SOD1) mutante. En utilisant un modèle murin pour l’étude de l’activation microgliale par
imagerie in vivo croisé avec les modèles murins SOD1G93A et SOD1G37R, nous avons
découvert que la phase présymptomatique de la maladie médiée par SOD1 est caractérisée
par le développement d’un profil anti-inflammatoire distinct et d’une réponse immunitaire
innée atténué à la suite d’un traitement au lipopolysaccharide (LPS). Ce phénotype microglial
est associé à une surexpression de la cytokine anti-inflammatoire IL-10 16 fois supérieure au
contrôle dans des conditions de base, suivie par une augmentation de 4,5 fois à la suite du
traitement au LPS. Tandis que l’infusion d’un anticorps bloquant d’IL-10R, débuté chez les
souris à un âge de 60 jours, cause une augmentation significative de marqueurs d’activation
microgliale et précipite le début de la maladie, une surexpression d’IL-10 dans les cellules
microgliale, acheminé via un vecteur viral exprimant sous le promoteur CD11b, diffère
significativement le début de la maladie et augmente la survie chez les souris SOD1G93A.
Nous proposons que le haut niveau d’IL-10 dans la microglie résidente lors du stade
présymptomatique dans l’ALS représente un mécanisme homéostatique et compensateur
agissant comme un déterminant non-neuronal du déclenchement de la maladie.

56
2.2. Abstract
While reactive microgliosis is a hallmark of advanced stages of amyotrophic lateral sclerosis
(ALS), the role of microglial cells in events initiating and/or precipitating disease onset is
largely unknown. Here we provide novel in vivo evidence of a distinct adaptive shift in
functional microglial phenotypes in preclinical stages of superoxide dismutase 1 (SOD1)-
mutant-mediated disease. Using a mouse model for live imaging of microglial activation
crossed with SOD1G93A and SOD1G37R mouse models, we discovered that the preonset phase
of SOD1-mediated disease is characterized by development of distinct anti-inflammatory
profile and attenuated innate immune/TLR2 responses to lipopolysaccharide (LPS)
challenge. This microglial phenotype was associated with a 16-fold overexpression of anti-
inflammatory cytokine IL-10 in baseline conditions followed by a 4.5-fold increase following
LPS challenge. While infusion of IL-10R blocking antibody, initiated at day 60, caused a
significant increase in markers of microglial activation and precipitated clinical onset of
disease, a targeted overexpression of IL-10 in microglial cells, delivered via viral vectors
expressed under CD11b promoter, significantly delayed disease onset and increased survival
of SOD1G93A mice. We propose that the high IL-10 levels in resident microglia in early ALS
represent a homeostatic and compensatory “adaptive immune escape” mechanism acting as
a nonneuronal determinant of clinical onset of disease.

57
2.3. Introduction
Amyotrophic lateral sclerosis (ALS) is a late-onset neurodegenerative disorder characterized
by progressive muscle weakness leading to a lethal paralysis. The majority of ALS cases are
sporadic with no known genetic components, whereas ~10% of ALS cases are familial
(FALS). approximately one-fifth of these familial cases are caused by mutations in
superoxide dismutase (SOD1; Rosen et al., 1993). In the past decades, much effort has been
focused on elucidating the mechanisms of disease induced by SOD1. However, despite the
extensive research, the exact nature of the underlying toxicity of mutant SOD1 remains
unknown. Increasing evidence suggests that misfolded SOD1 (mSOD1) species common for
all mutations may be secreted and involved in cytotoxicity (Urushitani et al., 2006; Zhao et
al., 2010; Zetterström et al., 2011; Grad et al., 2014). Furthermore, using conformation-
specific antibodies, the misfolded SOD1 species have been detected in CSF and spinal cords
of sporadic ALS patients, potentially suggesting a common SOD1-dependent pathogenic
mechanism in ALS (Bosco et al., 2010; Zetterström et al., 2011; Synofzik et al., 2012).
As an adult-onset neurodegenerative disorder, ALS may have an asymptomatic phase
spanning over several decades; however, despite intensive research efforts, the pathological
events precipitating disease onset and the mechanisms of early disease spreading remain
unclear. Transgenic mice overexpressing superoxide dismutase 1 mutants linked to familial
ALS develop progressive motor neuron disease with many pathological features resembling
the human disease (Gurney, 1994). In addition, in SOD1 mutant mice the sequence of disease
related pathological events evolve in a predictable timely manner; therefore, this disease
model system provides a powerful tool to study early disease mechanisms. While previous
evidence revealed expression of the pathogenic misfolded SOD1 species in subset of large
motor neurons as early as postnatal day 7 (Saxena et al., 2013), the role of microglial cells in
the mechanisms influencing early disease pathogenesis is largely unknown.
In the current study, using the sensitive biophotonic ALS models for in vivo imaging of
microglial activation/innate immune response developed in our laboratory, we were able to
visualize early alterations in microglial phenotypes observed in presymptomatic SOD1G93A
and SOD1G37R mice. Our results revealed that, contrary to our expectations, the preonset

58
phase of SOD1-mediated disease is characterized by the development of a distinct anti-
inflammatory profile of microglial cells, attenuated Toll-like receptor 2 (TLR2) responses to
controlled immune challenge, and a 16-fold overexpression of anti-inflammatory cytokine
IL-10. IL-10 is a key immunoregulatory/anti-inflammatory cytokine that mediates a feedback
inhibition loop and limits excessive production of proinflammatory cytokines such as TNFα,
IL-1β, and IL-6 (Howard and O’Garra, 1992; Moore et al., 2001). To investigate, whether
early induction of IL-10 in presymptomatic SOD1 mutant microglia represents an adaptive
or maladaptive microglia polarization phenotype, we initiated treatment of ALS mice with
specific IL-10 receptor blocking antibody. Here we show that treatment with specific IL-10
receptor blocking antibody, initiated in presymptomatic mice, causes a significant increase
in markers of microglial activation and precipitates the clinical onset of disease. On the other
hand, a targeted overexpression of IL-10 in lumbar spinal cord microglia delivered via
intrathecal injection of AAV2/9 vector expressed under the CD11b promoter significantly
delayed the clinical onset of disease and increased survival in SOD1G93A mice. Together, our
results suggest that early induction of IL-10 represents a homeostatic and adaptive microglial
mechanism that may act as an endogenous, nonneuronal determinant of clinical onset of
disease. Finally, we show that therapies aiming to induce the state of microglial
immunological homeostasis, such as overexpression of IL-10 in microglial cells, may have
therapeutic potential in ALS.
2.4. Materials and Methods
2.4.1. Generation of TLR2-Fluc-AcGFP;SOD1G93A and TLR2-Fluc-AcGFP;SOD1G37R
transgenic mice
The transgenic TLR2-Fluc-AcGFP reporter mice were generated as described previously
(Lalancette-Hébert et al., 2009) and maintained heterozygous in the C57BL/6 background.
Transgenic mice overexpressing the SOD1G93A mutation (B6SJL-TgN_[SOD1-G93A]_1
Gur) were purchased from The Jackson Laboratory. Transgenic mice expressing the
SOD1G37R mutation (line 29) were a gift from Drs. P. Wong and D. Price from John Hopkins
University (Baltimore, MD). The TLR2-Fluc-AcGFP reporter mice were crossed with the
SOD1 mutants to generate TLR2- Fluc-AcGFP;SOD1G93A and TLR2-Fluc-

59
AcGFP;SOD1G37R double-transgenic mice. To avoid the effect of genetic background on
disease progression and survival, all experiments were performed on age-matched
littermates. All genotypes were assessed by PCR. The TLR2-Fluc transgenic mice were
detected by the amplification of the luciferase transgene as described previously (Lalancette-
Hébert et al., 2009), the SOD1G93A transgene was detected according to the Jackson
Laboratory protocols (Keller et al., 2009, 2011), and the SOD1G37R transgene was detected
as described previously (Gowing et al., 2009). In our experiments, we used animals of either
sex randomly divided into different experimental groups.
2.4.2. Virus construction and preparation
Self-complementary adeno associated virus (scAAV) serotype 2/9 viral vector CD11b-IL10,
encoding the protein IL-10, was prepared using the inverted terminal repeat (ITR)-CMV
vector. Mouse IL-10 cDNA tagged with Myc-DDK (OriGene Technologies) was amplified
by PCR to generate BamH1 and Not1 restriction sites and then cloned into the initial vector.
For the scAAV2/9 viral vector CD11b-IL10 used to express the IL-10 protein specifically
under the control of the human CD11b promoter, the CD11b promoter was obtained by PCR
from pBluescriptSK+-CD11b-TKmt-30 as described previously (Gowing et al.,2006) and
excised to generate a Kpn1/Mlu1 fragment. It was then cloned inside the scAAV2/9-CMV-
IL10 after removing the CMV promoter with the same restriction enzymes. Cloning was
performed in SURE Competent Cells (Stratagene) to preserve the two ITR sites of the
vectors. As described previously (Patel et al., 2014), scAAV recombinant viruses were
produced in the 293T cell line cultured in DMEM (Invitrogen) supplemented with 10%
normal bovine serum, 100 U/ml penicillin G, and 100
g/ml streptomycin.
2.4.3. In vivo bioluminescence imaging
As described previously (Maysinger et al., 2007; Cordeau et al., 2008), the images were
gathered using an IVIS 200 Imaging System (PerkinElmer). Twenty-five minutes before the
imaging session, the mice received intraperitoneal injection of the luciferase substrate D-
luciferine (150 mg/kg, dissolved in 0.9% saline; PerkinElmer). Mice were anesthetized with

60
2% isoflurane in 100% oxygen at a flow rate of 2 L/min and placed in the heated, light-tight
imaging chamber. Images were collected using a highsensitivity charge-coupled device
camera with wavelengths ranging from 300 to 600 nm. Exposition time for imaging was 1
min using different fields of view and a F/1 lens aperture. The bioluminescence emission was
normalized and displayed in photons per seconds per centimeter squared per steradian (Keller
et al., 2009, 2011).
2.4.4. Lippolysaccharide treatment
Lipopolysaccharide (LPS; serotype O55:B5) from Escherichia coli was dissolved in saline
and administered by intraperitoneal injection at 5 mg/kg. Mice were maintained on heating
pads between the LPS injection and the various imaging sessions (Lalancette-Hébert et al.,
2009).
2.4.5. Surgical procedures
2.4.5.1. Stereotaxic brain injection
Mice were anesthetized with 2% isoflurane in 100% oxygen at a flow rate of 2 L/min and
placed in a stereotaxic apparatus (David Kopf Instruments). Transgenic and wild-type (WT)
mice received intracerebroventricular injection of recombinant mutant SOD1 (1 µg/µl) into
the right ventricle. A volume of 2 µl was infused over 4 min using a microinjection pump
(model A-99; Razel Scientific Instruments).
2.4.5.2. Intracerebroventricular brain infusion
Mouse IL-10 receptor (mIL-10R) blocking antibodies (CD210; BD Pharminogen) were
infused through a brain infusion cannula connected with osmotic minipumps (model 2006;
pumping rate, 0.15 µl/h; duration of treatment, 42 d; Alzet). The osmotic pumps were filled
with either 200µl of antibody (mIL-10R, 0.5 mg/ml) or saline and preconditioned overnight
in 37°C saline. SOD1G93A mice (60 d old) were anesthetized with 2% isoflurane in 100%
oxygen at a flow rate of 2 L/min and placed in a stereotaxic apparatus. The sterile brain
infusion cannula was implanted into the right ventricle and was fixed with dental cement.
The osmotic pumps were implanted under the skin. Mice received continuous 3.6µg/d of

61
antibody for 42 d. In total, 16 SOD1G93A mice received antibodies, and 13 mice were
implanted with saline filled pumps as controls. Measurements of body weight and hindlimb
reflex were used to score the clinical effect on SOD1G93A mice. The extensibility and postural
reflex of the hindlimbs when the mice were held up by their tails were scored as described
previously (Urushitani et al., 2006). Reflex score were gathered twice a week in a blind
manner by animal technicians who had no information of the treated mice, but had experience
in grading SOD1G93A mice. The survival was defined as the age when the animal could not
stand on its feet within 30 s when placed on its side.
2.4.5.3. Intrathecal injection of AAV-CD11b-IL10
At 33 d of age, WT control mice and SOD1G93A mice were injected intrathecally with 5.75 x
1010 viral particles of scAAV2/9-Cd11b-IL10 vector or scAAV-scFvD1.3 control vector by
a 25µl Hamilton syringe slipped under the dura, where the vector was slowly released into
the CSF. To confirm the position of the syringe between the L4 and L5, the tail reflex of each
mice was assessed. Mice were all anesthetized with 2% isoflurane during surgical
procedures. To confirm the correct expression of IL-10 in lumbar spinal cord following
intrathecal injections, tissues from 90-d-old WT mice and from end-point SODG93A mice
were collected for immunohistochemistry labeling Iba1 (1:500, rabbit polyclonal anti-Iba1;
Wako Chemicals) and cMyc (1:10, homemade monoclonal mouse anti-cMyc raised against
the 9E10 epitope). The sections were washed in PBS and then incubated in corresponding
fluorescent secondary antibody (Invitrogen).
2.4.5.4. Tissue collection
Animals were anesthetized by an intraperitoneal injection of ketamine/xylazine (100/10
mg/kg) and transcardially perfused with 0.9% saline, followed by ice-cold 4%
paraformaldehyde (PFA) at pH 7.4 dissolved in PBS. Tissue samples were then postfixed
overnight in 4% PFA and equilibrated in PBS/30% sucrose for 48 h. Tissue were cut in 25
µm transverse sections on a Leica frozen microtome and kept in a cryoprotective solution at
–20°C.

62
2.4.6. Immunofluorescence
As described previously (Lalancette-Hébert et al., 2007), the sections were then incubated
overnight at room temperature using primary antibodies, 1:500 rabbit polyclonal anti-Iba1
(Wako Chemicals) and 1:250 mouse anti-TLR2 (eBioscience). After wash in PBS, the
sections were incubated in a corresponding fluorescent goat secondary antiserum
(Invitrogen).
2.4.7. In situ hybridization
The expression and localization of mRNAencoding for TLR2 and TNF-α were detected on
olfactory bulb (OB) and lumbar spinal cord sections using a 35S-labeled riboprobes. Protocols
for probe synthesis and in situ hybridization were described previously by Lalancette-Hébert
et al. (2007). Slides were dried out under a vacuum overnight, postfixed in 4%
paraformaldehyde, and digested by proteinase K (10 µg/ml in 0.1 M Tris HCl, pH 8.0, and
50 mM EDTA, pH 8.0, at 37°C for 25 min), after which the sections were rinsed in water
and by a solution of 0.1 M triethanolamine (TEA, pH 8.0), acetylated in 0.25% acetic
anhydride in 0.1 M TEA, and dehydrated. The hybridization involves 107 cpm/ml/slide of
hybridization mixture and incubation at 60°C overnight in a slide warmer. Slides were rinsed
in standard saline citrate (SSC; 1X; 0.15MNaCl, 15mM trisodium citrate buffer, pH 7.0) and
digested by RNase A at 37°C (20 µg/ml), rinsed in descending concentrations of SSC,
washed in 0.1X SSC, and dehydrated through graded concentrations of ethanol. The sections
were exposed to X-ray film (Kodak) for 18–24 h and dipped in NTB2 nuclear emulsion
(diluted 1:1 with distilled water; Kodak). Slides were kept at 4°C for 2 weeks safe from light,
developed in D19 developer (Kodak) at 14°C, washed in water, and fixed in rapid fixer
(Kodak). After exposition, tissues were rinsed in distilled water for 45 min and counterstained
with thionin (0.25%). Slides were analyzed under dark-field illumination (Nguyen et al.,
2001). Counts were made of clusters of silver grain hybridization signal in olfactory bulbs
and lumbar spinal cord sections.
2.4.7. Western blots
Following LPS and mSOD1 stimulation in vivo, tissues were collected, and whole protein
lysates were extracted by sonication in urea lysis buffer (1% SDS, 6 M urea, Tris HCl 6.8).

63
Protein concentrations were determined by the Bradford method. Protein lysates were
fractioned by SDSPAGE, YM1 was detected with 1/1000 antibody dilution (Stem Cell
Technologies, anti-mouse Ym1), and actin was used as protein load control (1/30,000
dilution; Millipore).
2.4.8 Primary adult microglia cultures
WT and SOD1G93A adult mice (8–9 weeks old) were transcardially perfused with ice-cold
saline supplemented with heparine (2 U/ml; Sigma-Aldrich). Olfactory bulb, cerebral cortex,
and lumbar spinal cord, from which meninges were removed, were collected in ice-cold
hibernate medium (Hibernate A medium; BrainBits) supplemented with B-27 (1X) and 0.5
mM GlutaMax-I, L-ananyl-L-glutamine (Invitrogen). Tissues were dissociated in Earle’s
balanced salt solution (EBSS; 2 ml) containing 2 mg/ml papain and 100 U/ml DNase
(Worthington). The enzymatic reaction was quenched by adding EBSS (0.2 ml) containing
10 mg/ml ovomucoid protease inhibitor and 10 mg/ml albumin (Worthington), and 2 ml
culture medium (DMEM with GlutaMax-I) supplemented with 15% heat-inactivated fetal
bovine serum and 1% penicillin–streptomycin (Invitrogen). The cells were filtered through a
40µmcell strainer (BD Bioscience) and separated using a discontinuous Percoll (GE
Healthcare) density gradient centrifugation. Cells collected from the Percoll gradient
interphase were washed in culture medium and plated on culture-treated glass slides (Becton
Dickinson) at a 1.25x106 cells/ml density and incubated at 37°C in 95% air/5%CO2 in a
humidified culture incubator. After 24 h in cultures, cells were challenged with 1 µg/ml LPS
in culture medium for 30 min. Cells were then either homogenized in QIAzol for RNA
extraction or fixed with 4% paraformaldehyde and processed for immunocytochemical
assays.
2.4.8.1. Primary culture: IL-10 treatments
Primary cell cultures were obtained from the brains of PN1-PN4 SOD1G93A mutant pups.
Brains were collected and placed in ice-cold PBS. Following mechanical dissociation, five
or six brains were incubated in a 0.25% Trypsine-EDTA solution (Sigma) containing 250K
U/ml DNase I (Sigma). After centrifugation, the cell pellets were placed in T-75 cm2 flasks

64
(Sarstedt) for 10 d at 37°C, 5%CO2, in DMEM high-glucose media with 10% fetal bovine
serum and antibiotic solution (Sigma). At confluence, the cells were plated onto coverslips
in 24 well plates at a concentration of 10,000 cells/well in media. Cells were incubated with
G-CSF 24 h later to allow adhesion. At 4d in vitro, wells were divided into three groups:
group 1 received mutant SOD1 (0.5 µg/ml) for 48 h; group 2 received IL-10 (30 ng/ml) from
24 h before SOD1 (0.5 µg/ml) challenge and were fixed 48 h after the SOD1 treatment; group
3 received CD210 (IL-10 receptor blocking antibody, 5 µg/ml and 15 min before adding IL-
10) plus IL-10 (30 ng/ml) from 24 h before SOD1 (0.5 µg/ml) challenge and were fixed 48
h after the SOD1 stimulation. Cells were fixed in 4% paraformaldehyde, pH 7.4, for 20 min.
Following one-step wash, cells were incubated in cold methanol for 10 min, washed,
permeabilized in 0.25% Triton-X for 10 min, blocked in 3% normal goat serum for 1 h, and
then were incubated overnight at 4°C using primary antibodies (TNFα, 1:500; Ym1, 1:500;
Cd11b, 1:500).
2.4.9. Reverse transcriptase PCR
Cells from the microglia cultures were homogenized in QIAzol buffer (Qiagen), and total
RNA was extracted using the RNeasy Micro Kit (Qiagen) non-column treatment following
the manufacturer’s instructions. RNA concentrations were measured using a NanoDrop
spectrophotometer (Thermo Fisher Scientific), and quality was assessed with an Agilent 2100
Bioanalyser (Agilent Technologies). First-strand cDNA synthesis was accomplished with
Superscript III RNase H reverse transcriptase (Invitrogen) and random hexamer
oligonucleotides. Subsequent PCR amplifications were performed using oligonucleotides
targeting IL-1β, IL-6, TNF-α, IGF-1, TGF-β1, and IL-10. All reactions were normalized
using the following reference genes: ATP synthase O subunit (ATP50), hypoxanthine
guanine phosphoribosyl transferase 1 (HPRT1), and 18S ribosomal RNA (18S). Quantitative
real-time PCR measurements were performed by the qRT-PCR platform of the Centre de
Génomique de Québec.

65
2.4.10. Cytokine array
The inflammatory cytokines expression profile was analyzed with a mouse cytokine antibody
array (RayBio Mouse Inflammation Antibody Array 1; RayBiotech). Protein lysates were
obtained from WT and SOD1G93A transgenic mice 24 h after LPS intraperitoneal injection by
homogenization of olfactory bulbs and lumbar spinal cords segments in 1X cell lysis buffer
(included in the RayBiotech kit) with a protease inhibitor cocktail (Sigma, catalog #P8340).
The protein concentration was determined for each sample and diluted at 300 µg in 1X
blocking buffer. Samples for each group (four mice/group) were pooled and incubated with
the array membrane overnight at 4°C. After washing in the supplied washing buffer,
membranes were incubated with biotin-conjugated antibodies overnight. Signal detection
was performed according to RayBiotech protocols, and membranes were exposed to X-ray
film (Biomax MR1; Kodak). The film was analyzed using the Agfa Arcus II system and
ImageJ software to measure the optical density of each spot on the membrane.
2.4.11. Flow cytometry
Blood samples were collected from WT and SOD1G93A, with and without 24 h LPS
stimulation, by submandibular cheek puncture (100 µl) in EDTA-coated vials (Sarstedt) and
maintained under continuous agitation. The blood samples were incubated with Fc Block
CD16/32 antibodies (BD Pharmingen) to minimize nonspecific fluorescent labeling. Red
blood cells were lysed using Pharm Lyse buffer (BD Bioscience). Samples were then labeled
with CD45-V500, CD4-APC, CD8-PE-CF594 and CD11b-PE, or CD45-V500, CD4-APC,
and CD25-FITC (BD Bioscience). The panel labeled for CD45, CD4, and CD25 was fixed
and permeabilized using the Mouse FoxP3 Buffer set (BD Pharmingen) and cells were then
labeled with FoxP3-V450, Il-4-PECy7, and Il-10-PE (BD Bioscience). After labeling, cells
were resuspended in stain buffer (FBS; BD Pharmingen), and FACS analysis was performed
on an LSRII flow cytometer (BD Bioscience) using FACSDiva software (BD Bioscience).

66
2.5. Results
2.5.1. TLR2-luc-GFP;SOD1G93A mice, a mouse model for live analysis of early microglia
phenotypes in ALS
To investigate preonset microglia phenotypes and to evaluate the role of innate immune
responses in early disease mechanisms, we took advantage of the TLR2 reporter mouse
model generated previously in our laboratory (Lalancette-Hébert et al., 2009). In this model,
a TLR2 induction (luciferase expression detectable as bioluminescence photon emission) can
be visualized longitudinally from the brain and/or spinal cord of the living mice using
bioluminescence/biophotonic imaging and a high-sensitivity/ high-resolution CCD camera.
It is noteworthy that previous work revealed a role of TLR2 in the pathogenesis of FALS
(Liu et al., 2009; Zhao et al., 2010). The double-transgenic TLR2-luc- GFP;SOD1G93A mice
were generated by crossing heterozygous mice carrying the SOD1G93A transgene with the
heterozygous TLR2-luc-GFP mice coexpressing a dual nonpathogenic reporter transgenes
luciferase (luc) and green fluorescent protein (GFP) under transcriptional control of the
murine TLR2 gene promoter (Fig. 1A–J). The rationale for using this mouse model emerged
from the fact that TLR2 expression is very low or undetectable in resting, nonactivated
microglia, but is strongly induced in microglial cells in response to infection or brain injury
(Lalancette- Hébert et al., 2009, 2012). Moreover, previous analysis of the TLR2-luc-GFP
mouse model clearly demonstrated expression of the TLR2-driven transgenes luc and GFP
in activated microglia, revealing that this in vivo model system can be used to measure TLR2-
mediated microglial activation (Lalancette-Hébert et al., 2009, 2012).
2.5.2. Preonset ALS is associated with marked downregulation of innate immune response
and anti-inflammatory microglia profiles
To determine in vivo functional properties of microglial cells in presymptomatic disease, we
analyzed the pattern of microglial activation. As a readout, we used
biophotonic/bioluminescent TLR2 signals from the live TLR2-luc-GFP;SOD1G93A mice. In
the first set of experiments, we investigated microglial activation profile in baseline
conditions and in response to LPS challenge. LPS is an activator of innate immunity, and it
is noteworthy that LPS-related genes are indeed upregulated in ALS patients (Zhang et al.,
2011). Importantly, LPS induces orchestrated transcriptional response in macrophages and

67
microglia that can be divided into relatively limited number of distinct patterns of gene
induction and/or repression (Medzhitov and Horng, 2009). We hypothesized that analyses of
known patterns of LPS-induced gene expression may serve as a valid readout of the
functional response phenotypes of microglial cells in presymptomatic disease. In our
experiments, we focused on two major regions of interest: (1) the olfactory bulb/brain area
and (2) the spinal cord area. The rationale for analyses of microglial cells from the OB region
came from our previous studies where we discovered that the microglia from OB, even in
control conditions, express increased levels of the TLR2 signal and may react as early sensors
of brain injury (Lalancette-Hébert et al., 2009, 2010). In addition, growing evidence suggests
OB pathology in preclinical Parkinson’s and Alzheimer’s diseases, in human patients as well
as in animal models (Lerner and Bagic, 2008; Beach et al., 2009; Wesson et al., 2010).
To analyze early temporal dynamics and activation patterns of microglial cells, we performed
a series of live imaging experiments, starting at 35–40 d of age. Because disease progression
in ALS models has been associated with the development of inflammatory and neurotoxic
microglia phenotypes, we hypothesized that presymptomatic disease will be characterized by
aberrant increases in TLR2 signals. To our surprise, analysis of the TLR2 signals revealed
no visible signal from the spinal cord of the TLR2-luc-GFP;SOD1G93A mice, whereas the
bioluminescence/biophotonic imaging from the OB/brain region of interest area revealed a
significant decrease of the baseline TLR2 signal in presymptomatic ALS mice (Fig. 1A–C),
thus suggesting a less activated microglial profiles. Next, we investigated how the observed
decrease in the baseline TLR2 signal may affect functional response of resident microglial
cells; namely, we demonstrated previously that intraperitoneal injection of LPS induces
controlled and synchronized increase in resident microglial activation (visualized from live
animals as TLR2 signal induction) peaking 24 h after initial challenge (Lalancette-Hébert et
al., 2009). The TLR2 responses were evaluated before and 24–48 h after initial injection (5
mg/kg, i.p.). As expected, in control mice, the intraperitoneal injection of LPS was associated
with a robust induction of the TLR2 signal in the OB/brain region as well as in the region of
the lumbar spinal cord (Figure 1D,F). Quantitative analysis of the biophotonic signals (Figure
1H–J) revealed significantly lower TLR2 signal intensities in the SOD1G93A mutant mice
compared to their age-matched TLR2 reporter control littermates (olfactory bulb ROI,

68
baseline, WT, 2.7 × 104 ± 0.3 × 104, n = 4; SOD1G93A, 2.14 × 104 ± 0.2 × 104, n = 8; 24 h
after LPS stimulation, WT, 4.94 × 105 ± 0.6 × 105, n = 4; SOD1G93A, 2.25 × 105 ± 0.2 × 105,
n = 8; p = 0.0006; 48 h after LPS, WT, 2.26 × 105 ± 0.2 × 105, n = 5; SOD1G93A, 1.41 × 105
± 0.2 × 105, n = 4; p = 0.0269; brain ROI, baseline, WT, 8.1 × 104 ± 0.7 × 104, n = 4;
SOD1G93A, 6.8 × 104 ± 0.7 × 104, n = 8; 24 h after LPS stimulation, WT, 2.59 × 106 ± 0.24 ×
106, n = 4; SOD1G93A, 1.28 × 106 ± 0.24 × 106, n = 8; p = 0.0065; 48 h after LPS, WT, 8.36
× 105 ± 1.8 × 105, n = 4; SOD1G93A, 9.21 × 105 ± 2.9 × 105, n = 5; lumbar spinal cord ROI,
baseline, WT, 1.34 × 104 ± 0.9 × 104, n = 5; SOD1G93A, 1.42 × 104 ± 0.1 × 104, n = 7; 24 h
after LPS stimulation, WT, 2.32 × 105 ± 0.48 × 105, n = 5; SOD1G93A, 0.85 × 105 ± 0.13 ×
105, n = 5; p = 0.0181; 48 h after LPS, WT, 8.35 × 104 ± 1.5 × 104, n = 4; SOD1G93A, 7.6 ×
104 ± 2.7 × 104, n = 5). Importantly, as revealed in Figure 1H–J, the decrease in signal
intensities was observed in all region of interest, including the OB/brain area as well as spinal
cord region, suggesting marked differences in the functional phenotypes of presymptomatic
microglia carrying SOD1 mutation (Figure 1E,G,H–J).
Because the SOD1G93A mouse model carries a high copy number of the mutated gene and its
disease phenotype is characterized by a fast disease progression (onset of clinical symptoms
at ∼90 d of age with a median survival of 150 d), we next asked whether observed functional
microglia profiles are relevant to SOD1-mutant-mediated disease and/or are linked to a
specific mouse model. To answer this question, we crossed the TLR2-luc-GFP reporter
mouse with the slow disease progressing SOD1G37R mice [onset of disease at 10 months of
age; survival, ∼52 weeks (1 year)]. The same experimental protocol was repeated with
presymptomatic 4-month-old double-transgenic TLR2-luc-GFP;SOD1G37R mice. As
revealed in Figure 1K–N, the systemic injection of LPS induced a robust increase in the
TLR2 signals in control mice, whereas the signal is markedly attenuated in presymptomatic
SOD1G37R mice. The quantitative analysis of the TLR2 signal revealed significantly lower
signal intensities in the OB/brain region and the spinal cords of presymptomatic SOD1G37R
mice compared with age-matched control littermates (olfactory bulb ROI, 24 h after LPS
stimulation, WT, 1.33 × 106 ± 0.1 × 105, n = 4; SOD1G93A, 0.80 × 106 ± 0.6 × 105, n = 4; p =
0.0114; brain ROI, 24 h after LPS stimulation, WT, 5.11 × 106 ± 0.34 × 106, n = 4; SOD1G93A,
2.85 × 106 ± 0.27 × 106, n = 4; p = 0.0347; lumbar spinal cord ROI, 24 h after LPS stimulation,

69
WT, 6.69 × 105 ± 0.64 × 105, n = 4; SOD1G93A, 2.36 × 105 ± 0.05 × 105, n = 4; p = 0.0217).
Together, our data revealed significant changes in early presymptomatic functional microglia
phenotypes in two different SOD1-mutant-mediated disease models. The observed changes
strongly suggest a disease-specific downregulation of microglial activation and innate
immune responses following standard proinflammatory LPS challenge.
Importantly, in keeping with our in vivo imaging results, the immunofluorescence and in situ
hybridization analysis of the OB and the lumbar spinal cord sections revealed attenuated
microglia activation patterns in presymptomatic SOD1G93A mice. As shown in Figure 2, A–
C and F, the immunohistological analysis of the OB sections revealed more activated
“amoeboid-like” morphology of microglial cells in controls as compared to age-matched
SOD1G93A mice. Furthermore, a double-immunofluorescence analysis revealed a
colocalization of TLR2 immunostaining with the Iba1 microglial marker, confirming that in
standard LPS proinflammatory stimuli, TLR2 is indeed induced in activated microglial cells.
Next, the quantitative analysis of in situ mRNA signal for TLR2 further confirmed significant
downregulation of the TLR2 response in the olfactory bulb and spinal cord of 60-d-old
SOD1G93A mice, suggesting that in vivo imaging data paralleled immunohistochemistry and
in situ hybridization results (Fig. 2I–N,O,Q; in situ labeled TLR2 cells/tissue section in the
olfactory bulb, WT, 2.87 ± 0.63, n = 5; SOD1G93A, 2.50 ± 0.48, n = 5; WT + LPS, 130.4 ±
13.3, n = 5; SOD1G93A + LPS, 79.3 ± 11.6, n = 5; p = 0.0052; in situ labeled TLR2 cells/tissue
section in the spinal cord, WT, 0.58 ± 0.23, n = 5; SOD1G93A, 2.21 ± 0.34, n = 5; p = 0.0067;
WT + LPS, 23.94 ± 2.47, n = 5; SOD1G93A + LPS, 14.89 ± 1.66, n = 5; p = 0.0043). Because
the downstream effects and/or targets of the TLR2 signaling pathway are transcriptional
activation of nuclear factor κB (NF-κB) and synthesis of the proinflammatory cytokines such
as TNFα, to confirm that the downstream pathway is indeed affected, we also analyzed the
levels of TNFα.
As shown in Figure 2, K, L, P, and R, and in accordance with observed downregulation of
the TLR2 pathway, the quantitative analysis of in situ signal revealed significant decrease in
TNFα mRNA levels in OB and spinal cord tissue sections of the 60-d-old SOD1G93A mice
treated with LPS (in situ labeled TNFα cells/tissue section in the olfactory bulb, WT, 0.92 ±

70
0.26, n = 5; SOD1G93A, 0.31 ± 0.10, n = 5; p = 0.0221; WT + LPS, 157.6 ± 13.3, n = 5;
SOD1G93A + LPS, 64.1 ± 6.79, n = 5; p < 0.0001; in situ labeled TNFα cells/tissue section in
the spinal cord, WT, 0.58 ± 0.22, n = 5; SOD1G93A, 0.25 ± 0.09, n = 5; WT + LPS, 1.5 ± 0.29,
n = 5; SOD1G93A + LPS, 0.75 ± 0.21, n = 5; p = 0.0376). Together, our results identify early
alterations of innate immune response and microglial phenotypes in presymptomatic disease.
The downregulation of the TLR2 as well as TNFα pathways suggest that microglial cells in
presymptomatic SOD1-mutant-mediated disease may acquire anti-inflammatory polarization
phenotypes.
2.5.3. Enhanced IL-10 production by presymptomatic resident SOD1G93A microglia
To gain more insights into these intriguing findings, we next asked whether observed changes
in microglial phenotypes are caused by affected environment (i.e., stressed neurons) and/or
represent a cell-type-specific change in phenotype caused by inherited disease. To address
this issue, we established primary murine microglial cultures derived from adult (P60, the
age of observed imaging phenotype; (Figs. 1, 2) SOD1G93A mice and WT type age-matched
littermates. The primary microglial cultures were derived from OB and lumbar spinal cord
microglia since in vivo both of the regions of interest showed changes in phenotype. The cells
were treated with (1 μg/ml) LPS for 20 min. As in in vivo experiments, the microglia
activation profile was analyzed 24 h after initial stimuli. In accordance with the results
obtained in our in vivo imaging studies, in normal culture conditions, the SOD1G93A microglia
expressed lower levels of TLR2 compared to WT cells. The similar results were obtained
following LPS stimuli. As shown in Figure 3E, the quantitative analysis of the activation
profiles after LPS stimuli revealed significantly lower levels of TLR2 immunoreactivity in
the SOD1-mutant-expressing cells (WT, 17.30 ± 0.47, n = 5; SOD1G93A, 13.51 ± 0.21, n = 5;
p < 0.0001; WT + LPS, 20.59 ± 0.55, n = 5; SOD1G93A + LPS, 12.95 ± 0.21, n = 5; p <
0.0001; values expressed in arbitrary units). Hence, initial in vitro analysis confirmed
deregulation of innate immune responses observed in presymptomatic SOD1G93A-expressing
microglia in vivo. Next, we hypothesized that observed changes in microglia polarization
phenotypes are caused by and/or associated with changes in cytokine profiles. Therefore,
using the same experimental paradigm, we performed an unbiased analysis of
proinflammatory and anti-inflammatory cytokine profiles. We investigated and compared the

71
cytokine levels of primary microglial cells derived from OB and the lumbar spinal cord
region of 60-d-old WT and SOD1 mutant mice. Interestingly, we did not observe major
changes in the levels of proinflammatory cytokines following LPS challenge. The
quantitative real-time PCR revealed a modest increase in IL-1β levels in SOD1G93A-
expressing microglia after LPS stimuli (Fig. 3F; IL-1β, olfactory bulb culture, WT, 34.3 ±
6.4; SOD1G93A, 43.9 ± 5.3; WT + LPS, 1178 ± 0.0005; SOD1G93A + LPS, 1234 ± 16.6; spinal
cord culture, WT, 41.0 ± 1.1; SOD1G93A, 109.7 ± 12.5; p = 0.0319; WT + LPS, 1355 ± 9.1;
SOD1G93A + LPS, 2639 ± 0.0005; p < 0.0001), whereas no significant difference was
observed in mRNA levels of proinflammatory cytokines (IL-6 and TNF-α) in the spinal cord
microglial cells after LPS stimuli (Fig. 3 G,H; IL-6, olfactory bulb culture, WT, 5.6 ± 3.7;
SOD1G93A, 6.2 ± 1.4; WT + LPS, 115.2 ± 2.2; SOD1G93A + LPS, 102.5 ± 1.3; p = 0.0388;
spinal cord culture, WT, 4.7 ± 1.3; SOD1G93A, 13.0 ± 0.9; p = 0.0346; WT + LPS, 219.9 ±
4.3; SOD1G93A + LPS, 256.9 ± 28.9; TNFα, olfactory bulb culture, WT, 225.1 ± 10.3;
SOD1G93A, 228.3 ± 2.2; WT + LPS, 1752 ± 17.1; SOD1G93A + LPS, 1551 ± 20.2, p = 0.0170;
spinal cord culture, WT, 183.1 ± 7.8; SOD1G93A, 316.0 ± 11.3; p = 0.0105; WT + LPS, 1608
± 99.5; SOD1G93A + LPS, 1682 ± 21.9). However, the most striking changes were observed
in levels of anti-inflammatory cytokines. In addition, the changes were more pronounced in
primary microglial cells derived from spinal cords of presymptomatic SOD1G93A mice;
namely, in addition to the modest increase in the TGF-β mRNA levels, we detected a sixfold
increase in the mRNA levels of anti-inflammatory cytokine IL-10 in baseline conditions
followed by a 4.5-fold increase in IL-10 levels following LPS challenge (Fig. 3 I,J; TGF-β1,
olfactory bulb culture, WT, 57.4 ± 0.0; SOD1G93A, 58.4 ± 3.1; WT + LPS, 50.8 ± 3.8;
SOD1G93A + LPS, 50.0 ± 6.1; spinal cord culture, WT, 42.7 ± 2.9; SOD1G93A, 45.3 ± 2.1; WT
+ LPS, 34.2 ± 0.8; SOD1G93A + LPS, 60.6 ± 2.6; p = 0.0103; IL-10, olfactory bulb culture,
WT, 0.62 ± 0.29; SOD1G93A, 1.7 ± 0.26; WT + LPS, 21.5 ± 0.1; SOD1G93A + LPS, 31.5 ±
0.1; p = 0.0002; spinal cord culture, WT, 0.19 ± 0.29; SOD1G93A, 3.0 ± 0.9; p = 0.0479; WT
+ LPS, 9.1 ± 0.03; SOD1G93A + LPS, 35.8 ± 3.9; p = 0.0206). As revealed in Figure 3K, an
increase in IL-10 levels was further confirmed at the protein level (IL-10 cytokine array from
in vivo olfactory bulb homogenates, 24 h after LPS, WT + LPS, 6.8 × 10−3 ± 0.6 × 10−3, n =
6; SOD1G93A + LPS, 19.4 × 10−3 ± 0.7 × 10−3, n = 6; p = 0.0054; lumbar spinal cord
homogenates, 24 h after LPS stimulation, WT + LPS, 3.6 × 10−3 ± 0.0 × 10−3, n = 6; SOD1G93A

72
+ LPS, 8.1 × 10−3 ± 0.2 × 10−3, n = 6; p = 0.0014). IL-10 is a key homeostatic, anti-
inflammatory cytokine that plays a critical role in modulation of inflammatory processes
(Saraiva and O'Garra, 2010). Although our results revealed that in presymptomatic SOD1G93A
mice IL-10 is overexpressed and produced by resident microglia, evidence suggests that the
major source of IL-10 is T regulatory lymphocytes (Tregs) (Kleinewietfeld and Hafler,
2014).
Therefore, to determine whether observed overexpression of IL-10 is restricted to CNS and
resident microglial cells and/or Tregs are also involved in this early immunosuppression, we
performed a flow cytometry analysis of the blood of 50- to 60-d-old SOD1G93A mice and the
age-matched littermates in control conditions and 24 h after LPS challenge. We quantified
different immune cell type subpopulations and measured the mean IL-10 levels in Tregs.
Because Beers et al. (2011) and Henkel et al. (2013) demonstrated previously that in the early
symptomatic phase of disease Tregs can modulate inflammatory response via anti-
inflammatory cytokine IL-4, we also measured levels of IL-4. As shown in Figure 4A–G, in
baseline conditions, quantitative analysis of the numbers of CD4-, CD8-, and CD11b-positive
cells revealed no difference between SOD1G93A mutant mice and WT littermates. As
expected, the LPS challenge markedly increased the number of monocytes/macrophages in
the blood; however, we did not observe any difference in the numbers of CD11b-positive
cells between two experimental groups. Finally, we identified the CD25+/FoxP3+
subpopulation of cells (specific markers for Tregs; Fig. 4H,J). Quantitative analysis revealed
no significant difference in the numbers of Tregs in control condition and following LPS
challenge. As revealed in Figure 4, K and L, we did not observed statistically significant
difference in the mean cellular levels of IL-10 and IL-4 in CD25+FoxP3+ cells. Together,
our results suggest that, contrary to marked induction of IL-10 in microglial cells found in
presymptomatic SOD1-mediated disease, no changes were observed in the profiles of
peripheral immune cells and/or IL-10 levels in baseline conditions and in response to
systemic LPS challenge. Hence, in presymptomatic SOD1G93A mice, early changes in
cytokine profiles and deregulation of the innate immune responses were restricted to a
population of the resident microglial cells.

73
2.5.4. Deregulation of TLR2 is IL-10 dependent
One of the most intriguing findings in presymptomatic SOD1 mutant mice is a marked
deregulation of the TLR2 biophotonic/bioluminescence signal observed in in vivo imaging.
The above described primary culture results suggest a role of IL-10; however, the in vitro
model may not recapitulate all aspects of the presymptomatic disease. Therefore, to
determine the relevance of this finding and to gain additional insights into microglial
activation and TLR2 regulation in early ALS, we took advantage of our established in vivo
model system, a double-transgenic SOD1/TLR2 reporter mouse model. We next asked
whether above described overexpression of IL-10 may be an underlying mechanism involved
in downregulation of TLR2 response. To answer this question, and to block effects of IL-10,
we used a specific neutralizing antibody raised against IL-10R1, the functional IL-10
receptor. This approach and antibody have been successfully tested in vivo in the model of
murine West Nile virus infection (Bai et al., 2009). Mice were injected with high dose of the
mIL-10R1 antibody (200 μg, i.p.) 24 h before LPS injection. As expected, the
bioluminescence imaging of the OB/brain region of interest revealed that the systemic LPS
induced a robust induction of the TLR2 signal in 60-d-old control mice, and the response was
attenuated in presymptomatic SOD1G93A mice (Figure 5A,B). However, as shown in Figure
5, C and D, induction of the TLR2 signal was completely restored in mice pretreated with
IL-10R neutralizing antibody (WT, 19.9 × 104 ± 1.6 × 104, n = 19; SOD1G93A, 15.0 × 104 ±
0.7 × 104, n = 24; p = 0.0047; WT + LPS; 3.66 × 106 ± 0.2 × 106, n = 24; SOD1G93A + LPS,
2.79 × 106 ± 0.3 × 106, n = 20; p = 0.0273; SOD1G93A + αIL-10R + LPS, 4.62 × 106 ± 0.8 ×
106, n = 9; p = 0.0126). The same pattern of the TLR2 signal recovery was observed in the
lumbar spinal cord. As revealed in Figure 5E–H, pretreatment with IL-10R antibody
completely restored the TLR2 signal induction, following LPS challenge, in the spinal cord
region of presymptomatic SOD1G93A mice (WT, 4.45 × 104 ± 0.3 × 104, n = 19; SOD1G93A,
3.60 × 104 ± 0.2 × 104, n = 22; p = 0.0119; WT + LPS, 3.91 × 105 ± 0.3 × 105, n = 16;
SOD1G93A + LPS, 2.98 × 105 ± 0.2 × 105, n = 13; p = 0.0351; SOD1G93A + αIL-10R + LPS,
5.91 × 105 ± 0.8 × 105, n = 8; p = 0.0005). These data clearly suggest that IL-10 has a role in
modulation of early TLR2 responses in presymptomatic ALS mice.

74
The next question is, what is the pathophysiological relevance and its role in early disease
pathogenesis? IL-10 is a major anti-inflammatory cytokine involved in suppression of
production of several proinflammatory molecules. Interestingly, work by Yanagawa and
Onoé (2007) suggests that enhanced IL-10 levels and consequent downregulation of TLR
responses may be a result of cellular priming with TLR4 and TLR2 ligands. It is noteworthy
that mutated SOD1 protein early in the disease forms misfolded species that, as shown by
Urushitani et al. (2006), can be secreted from affected neurons. Once secreted, the misfolded
SOD1 species may act as danger-associated molecular patterns (DAMPs) and act as a ligand
to TLR2 and/or TLR4 receptors. Previous work by Zhao et al. (2010) nicely demonstrated
that misfolded SOD1 protein may indeed act as a TLR2 ligand and modify properties of
microglial cells in vitro. Therefore, we hypothesized that early in disease, small amounts of
the SOD1 misfolded protein species are secreted from neurons and, once extracellular, may
act as TLR2 ligands. Preexposure to small amount of DAMPs (in this case, misfolded SOD1
species) would reduce sensitivity of microglial cells and the innate immune system to the
second LPS or DAMP challenge. Thus, if our hypothesis is correct, the second exposure to
known misfolded protein would result (as in case of the LPS challenge) in enhanced IL-10
production and attenuated microglial activation/TLR response to misfolded protein in early
presymptomatic SOD1G93A mice, whereas the response would be very robust in WT age-
matched mice. As shown in Figure 5H–L, intracerebroventricular injection of recombinant
misfolded SOD1 protein induced a robust induction of the TLR2 signals in the WT mice,
whereas the signal was attenuated in the presymptomatic SOD1 mutant mice (WT, 19.9 ×
104 ± 1.6 × 104, n = 19; SOD1G93A, 15.0 × 104 ± 0.7 × 104, n = 24; p = 0.0047; WT + LPS,
5.24 × 105 ± 0.8 × 105, n = 10; SOD1G93A + LPS, 2.51 × 105 ± 0.2 × 105, n = 6; p = 0.0221).
Importantly, pretreatment with IL-10R blocking antibody restored the microglial
activation/TLR2 response in SOD1G93A mice, suggesting IL-10-mediated downregulation of
TLR2 response (SOD1G93A + αIL-10R + LPS, 4.14 × 105 ± 0.6 × 105, n = 7; p = 0.0490).
Next, to investigate whether deregulation of the TLR2 signals observed by imaging in
presymptomatic microglial cells is associated with the development of distinct polarization
phenotypes, we analyzed by immunofluorescence brain sections of the imaged animals.
Indeed, as shown in Figure 5M–T, microglial cells from the SOD1G93A mice following LPS
or mSOD1 challenge acquire less activated phenotypes reflected by more ramified

75
morphology and by a significant decrease in Iba1 immunoreactivity, as revealed by optical
density measurement. Moreover, Western blot analysis of the brain homogenates from WT
and SOD1 mutant mice exposed to mSOD1 protein revealed a twofold increase in Ym1 levels
in SOD1 mutant mice. This increase was abolished by IL-10R blocking antibody (Fig. 5U,V).
Ym1 is well established marker of microglia/macrophage alternative activation; therefore, its
selective upregulation in presymptomatic SOD1G93A mice further supports a view of early
immunosuppression in these mice.
2.5.5. Chronic infusion with IL-10R blocking antibody precipitates disease onset in
the SOD1G93A mouse model
To determine whether attenuation of TLR2 response and overexpression of IL-10 in resident
microglial cells is an adaptive and/or maladaptive cellular phenotype in presymptomatic
disease, we initiated long-term infusion with IL-10R blocking antibodies. The treatment was
initiated at 60 d of age (n = 16 per group) using an infusion cannula connected to osmotic
minipumps (Alzet model 2006; pumping rate, 0.15 μl/h; duration of treatment, 42 d). The
sterile brain infusion cannula was implanted into the right ventricle and was fixed with dental
cement and was removed at day 105. The control mice received the infusion of sterile saline.
Because treatment was initiated before the clinical disease onset, the mice were closely
monitored by a blinded investigator. As described previously (Urushitani et al., 2006; Keller
et al., 2009), the clinical onset of disease was measured and scored as a loss of extension
reflex and confirmed by a parallel loss of body weight, and body weight was measured on a
daily basis (Fig. 6C). As shown in Figure 6, A and B, treatment with IL-10R antibody
significantly shifted the clinical onset of disease by 7 d (74 vs 81 d for saline-treated animals).
Whereas body weight measurements revealed that both experimental groups followed a
classical bell-like shape of the curve (initial incremental increase of body weight followed
by a rapid decline after clinical onset of disease; Fig. 6C), analysis of the values between the
two groups revealed that treatment with IL-10R blocking antibody, consistent with the shift
in disease onset, prevented early weight gain and precipitated weight loss at the time of the
onset. The treatment ended at day 105, and animals were kept under observation for survival
analysis. As shown in Figure 6D, the delivery of IL-10R blocking antibody from days 60 to
105 did not significantly affect the survival of SOD1G93A mice. Here it is noteworthy that the

76
Alzet pumps stopped perfusion of the antibody at day 105; thus, we may not exclude the
possibility that longer treatment may have some effects on survival. We also investigated
whether treatment with IL-10R blocking antibody affects immune cell profiles at the
periphery. Surprisingly, we did not observe any changes in the immune cell profiles and/or
levels of IL-10 and IL-4 (data not shown). Because infusion of IL-10R blocking antibody
precipitated disease in SOD1G93A mice, we hypothesized that overexpression of IL-10 in
microglial cells can be used to therapeutically modulate microglial phenotypes in early ALS
and temporally extend their adaptive/neuroprotective state, which may significantly decrease
the initial burden of disease and delay neuronal dysfunction. To therapeutically target
microglia, and to overexpress IL-10 selectively in microglial cells, a 1.3 kb IL-10 cDNA was
expressed under human CD11b promoter and inserted into AAV2/9 viral vector (Fig. 6D).
As described previously (Patel et al., 2014), the construct was injected intrathecally with a
single injection of 3 × 109 viral particles in a 10 μl volume. The recipient SOD1G93A mice
were 30- to 40-d-old, and an empty viral plasmid was used as control. As shown in Figure 6,
E and F, treatment with AAV-encoded IL-10 significantly delayed the clinical onset of
disease and prevented weight loss. In addition, AAV delivery of IL-10 significantly increased
survival in SOD1G93A mice. As further shown in Figure 6, H and I, the efficiency and
selectivity of the AAV infection was confirmed by immunodetection of the myc epitope. The
double immunofluorescence of the lumbar spinal cord sections of the SOD1G93A mice that
received AAV-CD11b-cMyc-IL-10 injection, at the end of the protocol, showed almost
perfect colocalization of Iba1 microglial markers with the cMyc epitope, suggesting a
selective and targeted overexpression of IL-10 in microglial cells. Collectively, our results
provide in vivo evidence that IL-10 signaling in microglial cells may serve as an endogenous
nonneuronal determinant of disease onset and may affect disease progression.
Finally, we investigated how changes in microglial IL-10 signaling may affect microglial
polarization phenotypes. We first investigated effects of IL-10R antibody treatment. As
shown in Figure 7A–F, analysis of the different microglia activation markers revealed a
significant increase of markers of microglial activation. When compared to controls, we
observed increased immunoreactivity for Iba1 (saline, 1.05 × 104 ± 0.5 × 104, n = 5; αIL-
10R, 1.31 × 104 ± 0.7 × 104, n = 8; p = 0.0298) and CD68 (saline, 13.63 ± 1.0, n = 6; αIl-

77
10R, 16.87 ± 0.3, n = 6; p = 0.0128) in the lumbar spinal cord sections of the treated mice.
Contrary to the proinflammatory effects of IL-10R blocking antibody, enhanced IL-10 levels
have been associated with anti-inflammatory and neuroprotective microglial phenotypes. As
shown in Figure 7, G and T, addition of recombinant IL-10 into mSOD1 exposed microglia
significantly decreased expression levels of the proinflammatory cytokine TNFα and
increased expression of an alternative microglia/macrophage activation marker, Ym1.
Together, our results suggest that enhanced IL-10 levels together with downregulation of the
TLR2 response represent an adaptive resident microglia-cell-specific mechanism that
controls early disease propagation and onset. Inversely, and in keeping with this hypothesis,
blocking the IL-10 signaling with a neutralizing antibody against its functional receptor IL-
10R1 precipitated disease onset in SOD1G93A mice and shifted the microglia activation
profiles toward the proinflammatory phenotype. Importantly, and in keeping with our
hypothesis, a targeted overexpression of IL-10 in microglial cells significantly delayed the
clinical onset of disease and increased survival in SOD1G93A mice. Hence, IL-10 may serve
as an endogenous microglial mechanism involved in the early (presymptomatic) disease
mechanisms and the control of disease onset. Moreover, targeted overexpression of IL-10 in
microglial cells may have therapeutic potential in ALS.
2.6. Discussion
To date, it has been widely established that microglial cells actively contribute to disease
progression and motor neuron degeneration in ALS (Robberecht and Philips, 2013). Yet, the
role of microglial cells in early disease pathogenesis (before appearance of clinical
symptoms) and in events initiating and/or precipitating disease onset is largely unknown. The
work presented here provides important in vivo evidence of a distinct adaptive shift in
functional microglial phenotypes in preclinical stages of SOD1-mutant-mediated disease.
Using a mouse model for live imaging of microglial activation (Lalancette-Hébert et al.,
2009) crossed with SOD1G93A mice, we discovered that, contrary to our expectations, the
preonset phase of SOD1-mediated disease is characterized by development of distinct anti-
inflammatory microglia profiles and attenuated innate immune/TLR2 responses to LPS and
misfolded protein challenge. Remarkably, the same anti-inflammatory microglia profiles in
the preonset stage of disease were observed in two different models of SOD1-mutant-

78
mediated disease, the fast-progressing SOD1G93A and the slow-progressing SOD1G37R mouse
models. Here we report (1) a 16-fold overexpression of anti-inflammatory cytokine IL-10
(SOD1G93A mutant microglia derived from 60-d-old mice) in baseline conditions followed by
a 4.5-fold increase following LPS challenge, (2) that the shift in phenotypes and associated
IL-10 overexpression was restricted to resident microglia and was not observed in peripheral
immune cells, (3) that acute treatment with IL-10R blocking antibody restored the TLR2
response in vivo, (4) that intracerebroventricular long-term infusion of the IL-10R blocking
antibody initiated at day 60 precipitated disease onset by 7 d and caused significant increase
in markers of microglial activation, and (5) that selective overexpression of IL-10 in
microglial cells using the AAV gene delivery system expressed under the human CD11b
promoter significantly delayed the clinical onset of disease and increased survival of
SOD1G93A mice. Based on these exciting findings, we propose that IL-10, a master
immunoregulatory cytokine, may serve as an endogenous modifier of microglia
(neuroprotective) phenotypes in preonset ALS and act as a nonneuronal determinant of
clinical onset of the disease.
The direct role of microglial cells in SOD1-mutant-mediated disease pathogenesis was
initially described in two landmark studies (Beers et al., 2006; Boillée et al., 2006). In the
first study, using a mouse models with deletable transgenes, Boillée et al. (2006)
demonstrated that the deletion of mutant SOD1 transgene within Cd11b-positive cells
increased the life span of SOD1G37R mice by ∼100 d. Similar findings were described in a
parallel paper (Beers et al., 2006) demonstrating that wild-type microglia extended survival
in PU.1 knock-out mice bred with SOD1G93A mutants. Although there is an agreement on the
neuroprotective role of wild-type microglia, the microglial cells carrying SOD1 mutations
are markedly different relative to WT microglial cells. For example, in vitro studies revealed
that microglial cells carrying the SOD1 mutation are more neurotoxic (Xiao et al., 2007). In
addition, a previous study by Frakes et al. (2014) clearly demonstrated (in vivo and in vitro)
NF-κB-driven activation and conversion of microglia phenotypes to neurotoxic
inflammatory cells in SOD1G93A mice. Together, there is abundant evidence that after disease
onset, microglial cells develop inflammatory and/or toxic phenotypes that may accelerate
disease progression (Henkel et al., 2009). On the other hand, the contribution of microglial

79
cells in the early disease pathogenesis remains unclear. In vivo preclinical studies using anti-
inflammatory approaches clearly suggest a major shift in microglia profiles over the course
of the disease. While treatments with anti-inflammatory compounds like minocycline or NF-
κB inhibitor Withaferin A attenuated inflammation and conferred neuroprotection when
initiated early, initiation of the treatment at later stages of disease increased inflammatory
markers, suggesting a shift in cellular responses (Keller et al., 2011; Patel et al., 2014).
Interestingly, results of our previous study (Patel et al., 2015) revealed that one of the
parameters linked with a positive therapeutic outcome was indeed an increased level of IL-
10. IL-10 is a key anti-inflammatory cytokine involved in regulation of innate and adaptive
immune response (Couper et al., 2008; Saraiva and O'Garra, 2010). Induction of IL-10
represents a homeostatic mechanism that mediates a feedback inhibition loop and limits
excessive production of proinflammatory cytokines, including TNFα, IL-1β, and IL-6
(Howard and O'Garra, 1992; Moore et al., 2001). Furthermore, evidence suggests that IL-10
signaling, through its functional IL-10R1, exerts its anti-inflammatory effects in part by
selective induction/formation of the p50/p50 homodimers, known to suppress transcriptional
NF-κB activity (Driessler et al., 2004). Although in infections an overexpression of IL-10
may induce so-called immune escape and correlate with poor pathogen control (Nylén and
Sacks, 2007; Couper et al., 2008; Bai et al., 2009; Richter et al., 2013), the immune-
regulatory properties of IL-10 conferred protection in brain injuries including ischemia
(Pérez-de Puig et al., 2013), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MTPT)-
mediated striatal toxicity (Joniec-Maciejak et al., 2014), as well as in neurodegenerative
disorders such as ALS. Our results clearly demonstrate that in SOD1-mutant-mediated ALS,
experimental inhibition of IL-10 signaling markedly increases inflammatory markers in
microglial cells and precipitates disease onset in SOD1G93A mutant mice (Figs. 6,7), whereas
targeted overexpression of IL-10 in microglial cells by AAV-mediated gene delivery confers
neuroprotection; namely, a single intrathecal injection of viral vector encoding IL-10 under
transcriptional control of the CD11b promoter in adult 30- to 40-d-old SOD1G93A mice
delayed disease onset by 9 d and increased survival by 13 d. Our data are in accordance with
the previous work of Ayers et al. (2015), who demonstrated that neonatal AAV-mediated
delivery of IL-10 into the spinal axis increased the survival of SOD1G93A mice. Moreover,
clinical evidence suggests that higher levels of IL-10 protein predict longer disease duration

80
in ALS patients (Su et al., 2013). Intriguingly, the role of IL-10 signaling seems to be more
complex and/or controversial in Alzheimer's disease. Previous studies reported that the
presence of IL-10 promoter polymorphism associated with low production of IL-10 may be
considered as an additive genetic risk factor for sporadic AD (Lio et al., 2003). However,
previous work using an APP/PS1 mouse model revealed a negative role of IL-10 on Aβ
accumulation and synaptic integrity (Chakrabarty et al., 2015; Guillot-Sestier et al., 2015).
Thus, more studies are needed to clarify the role of IL-10 and adaptive immune homeostasis
in different neurodegenerative disorders.
The questions to be addressed here are, what are the underlying mechanisms involved in 16-
fold overexpression of IL-10 observed in presymptomatic microglia, and what is the
functional relevance of IL-10-mediated downregulation of TLR2 response (Figs. 3, 4)? Work
by Zhao et al. (2010) clearly demonstrated that extracellular toxicity of mSOD1 was
mediated by microglia. Indeed, the mSOD1 extracellular protein was not directly toxic to
motoneurons; it required microglial activation using TLR and CD14 pathways, thus
suggesting that misfolded SOD1 acts as a DAMP and binds to TLRs expressed on microglial
cells (Zhao et al., 2010). If misfolded SOD1 acts as a DAMP, the repetitive exposure to small
amounts of secreted misfolded SOD1 in early stages of disease would initiate TLR2- and
TLR4-mediated microglial priming and trigger enhanced production of IL-10.
Overexpression of IL-10 in TLR2–TLR4 primed cells, as reported previously in dendritic
cells (Yanagawa and Onoé, 2007; Chang et al., 2009), would then induce an attenuated innate
immune response to pathogen-associated molecular patterns (LPS) and DAMP (misfolded
mutant SOD1) challenges (Fig. 4). It has been established that misfolded SOD1 can be
secreted in extracellular space (Urushitani et al., 2006); thus, a small amount of the
extracellular protein, in early disease, can bind to microglial TLRs and prime the cells. Our
results are in accordance with this view; namely, SOD1G93A mutant mice, when compared to
age-matched WT littermates showed attenuated sensitivity to injected misfolded SOD1
species (Fig. 4). Furthermore, the presence of small amounts of misfolded protein may
explain why changes in immune cell phenotypes and relative overexpression of IL-10 were
restricted to the CNS (Figs. 5, 6E–J). It is noteworthy that misfolded SOD1 species have not

81
been detected and/or secreted at the periphery; therefore, the TLR-mediated priming effect
in presymptomatic disease is absent (Fig. 4).
In conclusion, our results provide important in vivo evidence of a distinct adaptive shift in
functional microglial phenotypes in preclinical stages of SOD1-mutant-mediated disease.
Based on our results, we propose a distinct role for IL-10 in early disease mechanisms. Here
it is noteworthy that IL-10 production becomes deregulated with aging (Ciaramella et al.,
2011). This may suggest that in ALS, the capacity of microglial cells to adequately produce
IL-10 in response to different challenges may be compromised by aging, resulting in reactive
microgliosis and neuronal stress (Figs. 6, 7), leading to a clinical stage of disease. Finally,
our results clearly demonstrate that, early in disease, IL-10 is instrumental in the maintenance
of immune homeostasis of microglial cells. Moreover, based on our results, we propose that
targeted overexpression of IL-10 in microglia may have therapeutic potential in ALS;
namely, IL-10 modulates microglial immune phenotypes and temporally extends their
adaptive/neuroprotective state, which may significantly delay early neuronal dysfunction and
decrease the initial burden of the disease.
2.7. Footnotes
This work was supported by the Muscular Dystrophy Association, Canadian Institutes of
Health Research, and the Amyotrophic Lateral Sclerosis Society of Canada. J.K. holds a
Senior Scholarship Award from Fonds de recherche du Québec en Santé. We thank Christine
Bareil, Geneviève Roussin, and Sophie Vachon for their technical help.
The authors declare no competing financial interests.

82
2.8. References
Ayers JI, Fromholt S, Sinyavskaya O, Siemienski Z, Rosario AM, Li A, Crosby KW, Cruz
PE, DiNunno NM, Janus C, Ceballos-Diaz C, Borchelt DR, Golde TE, Chakrabarty P,
Levites Y. Widespread and efficient transduction of spinal cord and brain following
neonatal AAV injection and potential disease modifying effect in ALS mice. Mol Ther.
2015;23:53–62. doi: 10.1038/mt.2014.180. [PMC free article] [PubMed] [CrossRef]
[Google Scholar]
Bai F, Town T, Qian F, Wang P, Kamanaka M, Connolly TM, Gate D, Montgomery RR,
Flavell RA, Fikrig E. IL-10 signaling blockade controls murine West Nile virus
infection. PLoS Pathog. 2009;5:e1000610. doi: 10.1371/journal.ppat.1000610. [PMC
free article] [PubMed] [CrossRef] [Google Scholar]
Beach TG, White CL, 3rd, Hladik CL, Sabbagh MN, Connor DJ, Shill HA, Sue LI, Sasse J,
Bachalakuri J, Henry-Watson J, Akiyama H, Adler CH, Arizona Parkinson's Disease
C. Olfactory bulb alpha-synucleinopathy has high specificity and sensitivity for Lewy
body disorders. Acta Neuropathol. 2009;117:169–174. doi: 10.1007/s00401-008-0450-
7. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel
SH. Wild-type microglia extend survival in PU.1 knockout mice with familial
amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi:
10.1073/pnas.0607423103. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Beers DR, Henkel JS, Zhao W, Wang J, Huang A, Wen S, Liao B, Appel SH. Endogenous
regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and
correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain.
2011;134:1293–1314. doi: 10.1093/brain/awr074. [PMC free article] [PubMed]
[CrossRef] [Google Scholar]
Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G,
Cleveland DW. Onset and progression in inherited ALS determined by motor neurons
and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511.
[PubMed] [CrossRef] [Google Scholar]
Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H,
Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady
ST, Brown RH., Jr Wild-type and mutant SOD1 share an aberrant conformation and a
common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi:
10.1038/nn.2660. [PMC free article] [PubMed] [CrossRef] [Google Scholar]
Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, DiNunno N, Rosario
AM, Cruz PE, Verbeeck C, Sacino A, Nix S, Janus C, Price ND, Das P, Golde TE. IL-
10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening
cognitive behavior. Neuron. 2015;85:519–533. doi: 10.1016/j.neuron.2014.11.020.
[PMC free article] [PubMed] [CrossRef] [Google Scholar]
Chang YC, Kao WC, Wang WY, Wang WY, Yang RB, Peck K. Identification and
characterization of oligonucleotides that inhibit Toll-like receptor 2-associated immune
responses. FASEB J. 2009;23:3078–3088. doi: 10.1096/fj.09-129312. [PubMed]
[CrossRef] [Google Scholar]
Ciaramella A, Spalletta G, Bizzoni F, Salani F, Caltagirone C, Bossù P. Effect of age on
surface molecules and cytokine expression in human dendritic cells. Cell Immunol.

83
2011;269:82–89. doi: 10.1016/j.cellimm.2011.04.010. [PubMed] [CrossRef] [Google
Scholar]
Cordeau P, Jr, Lalancette-Hébert M, Weng YC, Kriz J. Live imaging of neuroinflammation
reveals sex and estrogen effects on astrocyte response to ischemic injury. Stroke.
2008;39:935–942. doi: 10.1161/STROKEAHA.107.501460. [PubMed] [CrossRef]
[Google Scholar]
Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J
Immunol. 2008;180:5771–5777. doi: 10.4049/jimmunol.180.9.5771. [PubMed]
[CrossRef] [Google Scholar]
Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular mechanisms of
interleukin-10-mediated inhibition of NF-kappaB activity: a role for p50. Clin Exp
Immunol. 2004;135:64–73. doi: 10.1111/j.1365-2249.2004.02342.x. [PMC free
article] [PubMed] [CrossRef] [Google Scholar]
Frakes AE, Ferraiuolo L, Haidet-Phillips AM, Schmelzer L, Braun L, Miranda CJ, Ladner
KJ, Bevan AK, Foust KD, Godbout JP, Popovich PG, Guttridge DC, Kaspar BK.
Microglia induce motor neuron death via the classical NF-kappaB pathway in
amyotrophic lateral sclerosis. Neuron. 2014;81:1009–1023. doi:
10.1016/j.neuron.2014.01.013. [PMC free article] [PubMed] [CrossRef] [Google
Scholar]
Gowing G, Dequen F, Soucy G, Julien JP. Absence of tumor necrosis factor-alpha does not
affect motor neuron disease caused by superoxide dismutase 1 mutations. J Neurosci.
2006;26:11397–11402. doi: 10.1523/JNEUROSCI.0602-06.2006. [PMC free article]
[PubMed] [CrossRef] [Google Scholar]
Gowing G, Lalancette-Hébert M, Audet JN, Dequen F, Julien JP. Macrophage colony
stimulating factor (M-CSF) exacerbates ALS disease in a mouse model through altered
responses of microglia expressing mutant superoxide dismutase. Exp Neurol.
2009;220:267–275. doi: 10.1016/j.expneurol.2009.08.021. [PubMed] [CrossRef]
[Google Scholar]
Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O'Neill MA, Yanai A,
Silverman JM, Zeineddine R, Corcoran L, Kumita JR, Luheshi LM, Yousefi M,
Coleman BM, Hill AF, Plotkin SS, Mackenzie IR, Cashman NR. Intercellular
propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-
dependent and -independent mechanisms. Proc Natl Acad Sci U S A. 2014;111:3620–
3625. doi: 10.1073/pnas.1312245111. [PMC free article] [PubMed] [CrossRef]
[Google Scholar]
Guillot-Sestier MV, Doty KR, Gate D, Rodriguez J, Jr, Leung BP, Rezai-Zadeh K, Town T.
Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology.
Neuron. 2015;85:534–548. doi: 10.1016/j.neuron.2014.12.068. [PMC free article]
[PubMed] [CrossRef] [Google Scholar]
Gurney ME. Transgenic-mouse model of amyotrophic lateral sclerosis. N Engl J Med.
1994;331:1721–1722. doi: 10.1056/NEJM199412223312516. [PubMed] [CrossRef]
[Google Scholar]
Henkel JS, Beers DR, Zhao W, Appel SH. Microglia in ALS: the good, the bad, and the
resting. J Neuroimmune Pharmacol. 2009;4:389–398. doi: 10.1007/s11481-009-9171-
5. [PubMed] [CrossRef] [Google Scholar]
Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, Zhao W, Moore DH,
Powell SZ, Appel SH. Regulatory T-lymphocytes mediate amyotrophic lateral

84
sclerosis progression and survival. EMBO Mol Med. 2013;5:64–79. doi:
10.1002/emmm.201201544. [PMC free article] [PubMed] [CrossRef] [Google
Scholar]
Howard M, O'Garra A. Biological properties of interleukin 10. Immunol Today.
1992;13:198–200. doi: 10.1016/0167-5699(92)90153-X. [PubMed] [CrossRef]
[Google Scholar]
Joniec-Maciejak I, Ciesielska A, Wawer A, Sznejder-Pachołek A, Schwenkgrub J, Cudna A,
Hadaczek P, Bankiewicz KS, Członkowska A, Członkowski A. The influence of
AAV2-mediated gene transfer of human IL-10 on neurodegeneration and immune
response in a murine model of Parkinson's disease. Pharmacol Rep. 2014;66:660–669.
doi: 10.1016/j.pharep.2014.03.008. [PubMed] [CrossRef] [Google Scholar]
Keller AF, Gravel M, Kriz J. Live imaging of amyotrophic lateral sclerosis pathogenesis:
disease onset is characterized by marked induction of GFAP in Schwann cells. Glia.
2009;57:1130–1142. doi: 10.1002/glia.20836. [PubMed] [CrossRef] [Google Scholar]
Keller AF, Gravel M, Kriz J. Treatment with minocycline after disease onset alters astrocyte
reactivity and increases microgliosis in SOD1 mutant mice. Exp Neurol. 2011;228:69–
79. doi: 10.1016/j.expneurol.2010.12.010. [PubMed] [CrossRef] [Google Scholar]
Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation.
Immunol Rev. 2014;259:231–244. doi: 10.1111/imr.12169. [PMC free article]
[PubMed] [CrossRef] [Google Scholar]
Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of
proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci.
2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [PMC free article]
[PubMed] [CrossRef] [Google Scholar]
Lalancette-Hébert M, Phaneuf D, Soucy G, Weng YC, Kriz J. Live imaging of Toll-like
receptor 2 response in cerebral ischaemia reveals a role of olfactory bulb microglia as
modulators of inflammation. Brain. 2009;132:940–954. [PubMed] [Google Scholar]
Lalancette-Hébert M, Moquin A, Choi AO, Kriz J, Maysinger D. Lipopolysaccharide-QD
micelles induce marked induction of TLR2 and lipid droplet accumulation in olfactory
bulb microglia. Mol Pharm. 2010;7:1183–1194. doi: 10.1021/mp1000372. [PubMed]
[CrossRef] [Google Scholar]
Lalancette-Hébert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato
S, Kriz J. Galectin-3 is required for resident microglia activation and proliferation in
response to ischemic injury. J Neurosci. 2012;32:10383–10395. doi:
10.1523/JNEUROSCI.1498-12.2012. [PMC free article] [PubMed] [CrossRef]
[Google Scholar]
Lerner A, Bagic A. Olfactory pathogenesis of idiopathic Parkinson disease revisited. Mov
Disord. 2008;23:1076–1084. doi: 10.1002/mds.22066. [PubMed] [CrossRef] [Google
Scholar]
Lio D, Licastro F, Scola L, Chiappelli M, Grimaldi LM, Crivello A, Colonna-Romano G,
Candore G, Franceschi C, Caruso C. Interleukin-10 promoter polymorphism in
sporadic Alzheimer's disease. Genes Immun. 2003;4:234–238. doi:
10.1038/sj.gene.6363964. [PubMed] [CrossRef] [Google Scholar]
Liu Y, Hao W, Dawson A, Liu S, Fassbender K. Expression of amyotrophic lateral sclerosis-
linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2. J Biol
Chem. 2009;284:3691–3699. doi: 10.1074/jbc.M804446200. [PubMed] [CrossRef]
[Google Scholar]

85
Maysinger D, Behrendt M, Lalancette-Hébert M, Kriz J. Real-time imaging of astrocyte
response to quantum dots: in vivo screening model system for biocompatibility of
nanoparticles. Nano Lett. 2007;7:2513–2520. doi: 10.1021/nl071611t. [PubMed]
[CrossRef] [Google Scholar]
Medzhitov R, Horng T. Transcriptional control of the inflammatory response. Nat Rev
Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [PubMed] [CrossRef] [Google
Scholar]
Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-
10 receptor. Annu Rev Immunol. 2001;19:683–765. doi:
10.1146/annurev.immunol.19.1.683. [PubMed] [CrossRef] [Google Scholar]
Nguyen MD, Julien JP, Rivest S. Induction of proinflammatory molecules in mice with
amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in
neurodegeneration. Ann Neurol. 2001;50:630–639. doi: 10.1002/ana.1256. [PubMed]
[CrossRef] [Google Scholar]
Nylén S, Sacks D. Interleukin-10 and the pathogenesis of human visceral leishmaniasis.
Trends Immunol. 2007;28:378–384. doi: 10.1016/j.it.2007.07.004. [PubMed]
[CrossRef] [Google Scholar]
Patel P, Kriz J, Gravel M, Soucy G, Bareil C, Gravel C, Julien JP. Adeno-associated virus-
mediated delivery of a recombinant single-chain antibody against misfolded
superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol Ther.
2014;22:498–510. doi: 10.1038/mt.2013.239. [PMC free article] [PubMed] [CrossRef]
[Google Scholar]
Patel P, Julien JP, Kriz J. Early-stage treatment with withaferin A reduces levels of misfolded
superoxide dismutase 1 and extends lifespan in a mouse model of amyotrophic lateral
sclerosis. Neurotherapeutics. 2015;12:217–233. [PMC free article] [PubMed] [Google
Scholar]
Pérez-de Puig I, Miró F, Salas-Perdomo A, Bonfill-Teixidor E, Ferrer-Ferrer M, Márquez-
Kisinousky L, Planas AM. IL-10 deficiency exacerbates the brain inflammatory
response to permanent ischemia without preventing resolution of the lesion. J Cereb
Blood Flow Metab. 2013;33:1955–1966. doi: 10.1038/jcbfm.2013.155. [PMC free
article] [PubMed] [CrossRef] [Google Scholar]
Richter K, Perriard G, Behrendt R, Schwendener RA, Sexl V, Dunn R, Kamanaka M, Flavell
RA, Roers A, Oxenius A. Macrophage and T cell produced IL-10 promotes viral
chronicity. PLoS Pathog. 2013;9:e1003735. doi: 10.1371/journal.ppat.1003735. [PMC
free article] [PubMed] [CrossRef] [Google Scholar]
Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev
Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [PubMed] [CrossRef] [Google
Scholar]
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto
J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A,
Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, et al.
Mutations in Cu/Zn superoxide dismutase gene are associated with familial
amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0.
[PubMed] [CrossRef] [Google Scholar]
Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev
Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [PubMed] [CrossRef] [Google
Scholar]

86
Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P. Neuroprotection
through excitability and mTOR required in ALS motoneurons to delay disease and
extend survival. Neuron. 2013;80:80–96. doi: 10.1016/j.neuron.2013.07.027.
[PubMed] [CrossRef] [Google Scholar]
Su XW, Simmons Z, Mitchell RM, Kong L, Stephens HE, Connor JR. Biomarker-based
predictive models for prognosis in amyotrophic lateral sclerosis. JAMA Neurol.
2013;70:1505–1511. [PubMed] [Google Scholar]
Synofzik M, Ronchi D, Keskin I, Basak AN, Wilhelm C, Gobbi C, Birve A, Biskup S, Zecca
C, Fernández-Santiago R, Kaugesaar T, Schöls L, Marklund SL, Andersen PM. Mutant
superoxide dismutase-1 indistinguishable from wild-type causes ALS. Hum Mol
Genet. 2012;21:3568–3574. doi: 10.1093/hmg/dds188. [PubMed] [CrossRef] [Google
Scholar]
Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated
secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral
sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [PubMed] [CrossRef]
[Google Scholar]
Wesson DW, Levy E, Nixon RA, Wilson DA. Olfactory dysfunction correlates with amyloid-
beta burden in an Alzheimer's disease mouse model. J Neurosci. 2010;30:505–514. doi:
10.1523/JNEUROSCI.4622-09.2010. [PMC free article] [PubMed] [CrossRef]
[Google Scholar]
Xiao Q, Zhao W, Beers DR, Yen AA, Xie W, Henkel JS, Appel SH. Mutant SOD1(G93A)
microglia are more neurotoxic relative to wild-type microglia. J Neurochem.
2007;102:2008–2019. doi: 10.1111/j.1471-4159.2007.04677.x. [PubMed] [CrossRef]
[Google Scholar]
Yanagawa Y, Onoé K. Enhanced IL-10 production by TLR4- and TLR2-primed dendritic
cells upon TLR restimulation. J Immunol. 2007;178:6173–6180. doi:
10.4049/jimmunol.178.10.6173. [PubMed] [CrossRef] [Google Scholar]
Zetterström P, Andersen PM, Brännström T, Marklund SL. Misfolded superoxide dismutase-
1 in CSF from amyotrophic lateral sclerosis patients. J Neurochem. 2011;117:91–99.
doi: 10.1111/j.1471-4159.2011.07177.x. [PubMed] [CrossRef] [Google Scholar]
Zhang R, Hadlock KG, Do H, Yu S, Honrada R, Champion S, Forshew D, Madison C, Katz
J, Miller RG, McGrath MS. Gene expression profiling in peripheral blood mononuclear
cells from patients with sporadic amyotrophic lateral sclerosis (sALS) J
Neuroimmunol. 2011;230:114–123. doi: 10.1016/j.jneuroim.2010.08.012. [PMC free
article] [PubMed] [CrossRef] [Google Scholar]
Zhao W, Beers DR, Henkel JS, Zhang W, Urushitani M, Julien JP, Appel SH. Extracellular
mutant SOD1 induces microglial-mediated motoneuron injury. Glia. 2010;58:231–
243. doi: 10.1002/glia.20919. [PMC free article] [PubMed] [CrossRef] [Google
Scholar]

87
2.9. Figures
2.9.1. Figure 1: In vivo biophotonic/bioluminescence imaging of TLR2-SOD1 mice.

88
A, B, Representative real-time imaging of the TLR2 induction in TLR2-WT controls (A) and
TLR2-SOD1G93A double-transgenic mice (B) at 20 weeks of age (color scales represent
photons/second). C, The longitudinal quantitative analysis of the total photon emissions from
olfactory bulb and brain ROIs at various time points shows a reduced TLR2 activation in
presymptomatic SOD1G93A mice (n between 10 and 20 mice per group per time point; p <
0.05 for all time points). D–G, Real-time imaging of the TLR2 response following LPS
stimulation in presymptomatic (60 d) SOD1G93A mice shows a significant reduction in TLR2
activation in the SOD1G93A mice compared to WT littermates. H–J, Photonic quantification
from the olfactory bulb ROI 24 h after LPS stimulation (WT, n = 4; SOD1G93A, n = 8; p =
0.0006) and 48 h after LPS (WT, n = 5; SOD1G93A, n = 4; p = 0.0269; H), from the brain ROI
24 h after LPS (WT, n = 4; SOD1G93A, n = 8; p = 0.0065) and 48 h after LPS (WT, n =
4; SOD1G93A, n = 5; I), and from lumbar spinal cord ROI 24 h after LPS (WT, n =
5; SOD1G93A, n = 5; p = 0.0181) and 48 h after LPS (WT, n = 4; SOD1G93A, n = 5; J). K–N,
Real-time imaging of the TLR2 response following LPS stimulation in presymptomatic (4
month) SOD1G37R mice show a similarly significant reduction of TLR2 activation. O–Q,
Photonic quantification from the olfactory bulb ROI 24 h after LPS stimulation (WT, n =
4; SOD1G37R, n = 2; p = 0.0114; O), from the brain ROI 24 h after LPS (WT, n =
4; SOD1G37R, n = 4; p = 0.0347; P), and from the lumbar spinal cord ROI 24 h after LPS
(WT; n = 4, SOD1G37R; n = 4; p = 0.0217; Q). Error bars indicate SEM
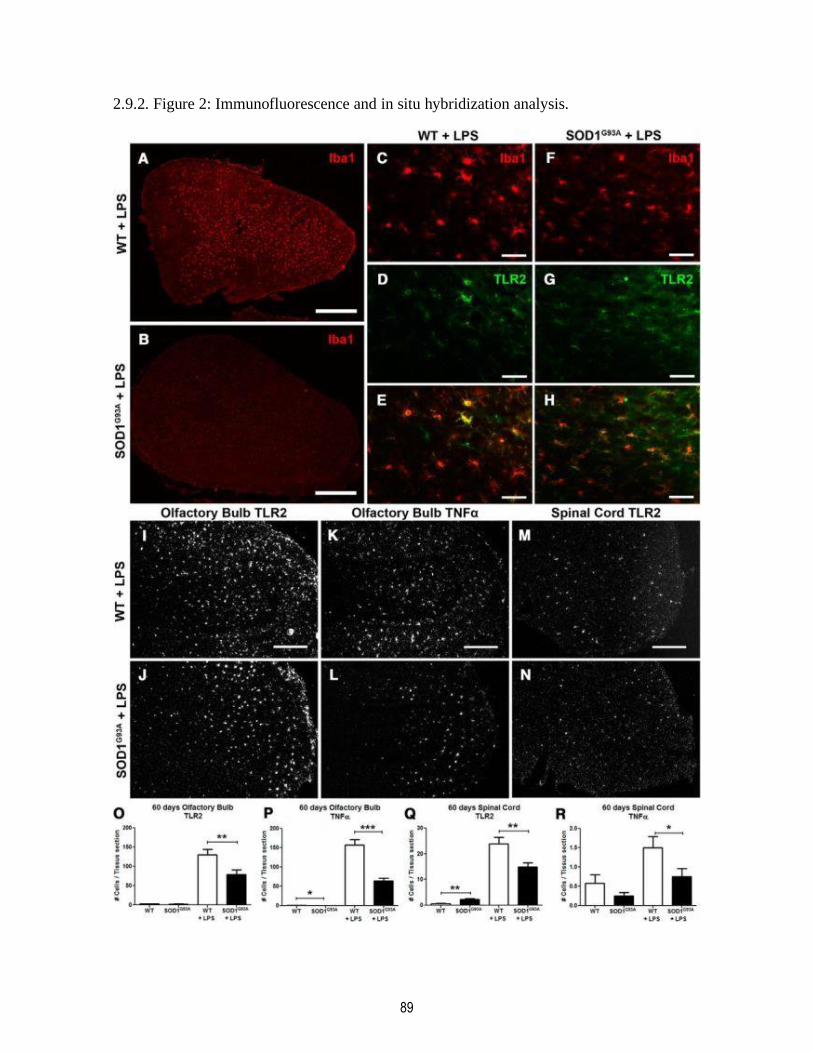
89
2.9.2. Figure 2: Immunofluorescence and in situ hybridization analysis.

90
A, B, Immunofluorescence of Iba1 staining in the olfactory bulb of WT littermates controls
(A) and presymptomatic 60-d-old SOD1G93A mice (B) 24 h after an LPS stimulation. C–H,
Higher magnification of Iba1 staining in the olfactory bulb of WT controls (C)
and SOD1G93A mice (F), TLR2 staining (D, G), and a merge showing the colocalization of
both stainings (E, H). K–N, Representative photomicrographs of the in situ hybridization of
the TLR2 expression 24 h after LPS stimulation in the olfactory bulb of WT controls (I) and
presymptomatic 60-d-old SOD1G93A mice (J), of TNFα expression in the olfactory bulb
(K, L), and TLR2 expression in the lumbar spinal cord (M, N). O–R, Quantification of in
situ labeled cells per tissue section of 60-d-old mice, from baseline and 24 h following LPS
stimulation: TLR2-labeled cells in the olfactory bulb (WT, n = 5; SOD1G93A, n = 5; WT +
LPS, n = 5; SOD1G93A + LPS, n = 5; p = 0.0052; O), TNFα-labeled cells in the olfactory bulb
(WT, n = 5; SOD1G93A, n = 5; p = 0.0221, WT + LPS, n = 5; SOD1G93A + LPS, n = 5; p <
0.0001; P), TLR2-labeled cells in the lumbar spinal cord (WT, n = 5; SOD1G93A, n = 5; p =
0.0067, WT + LPS, n = 5; SOD1G93A + LPS, n = 5; p = 0.0043; Q), and TNFα-labeled cells
in the lumbar spinal cord (WT, n = 5; SOD1G93A, n = 5; WT + LPS, n = 5; SOD1G93A +
LPS, n = 5; p = 0.0376; R). Scale bars: A, B, 500 μm; C–H, 50 μm; K–N, 250 μm. Error bars
indicate SEM.
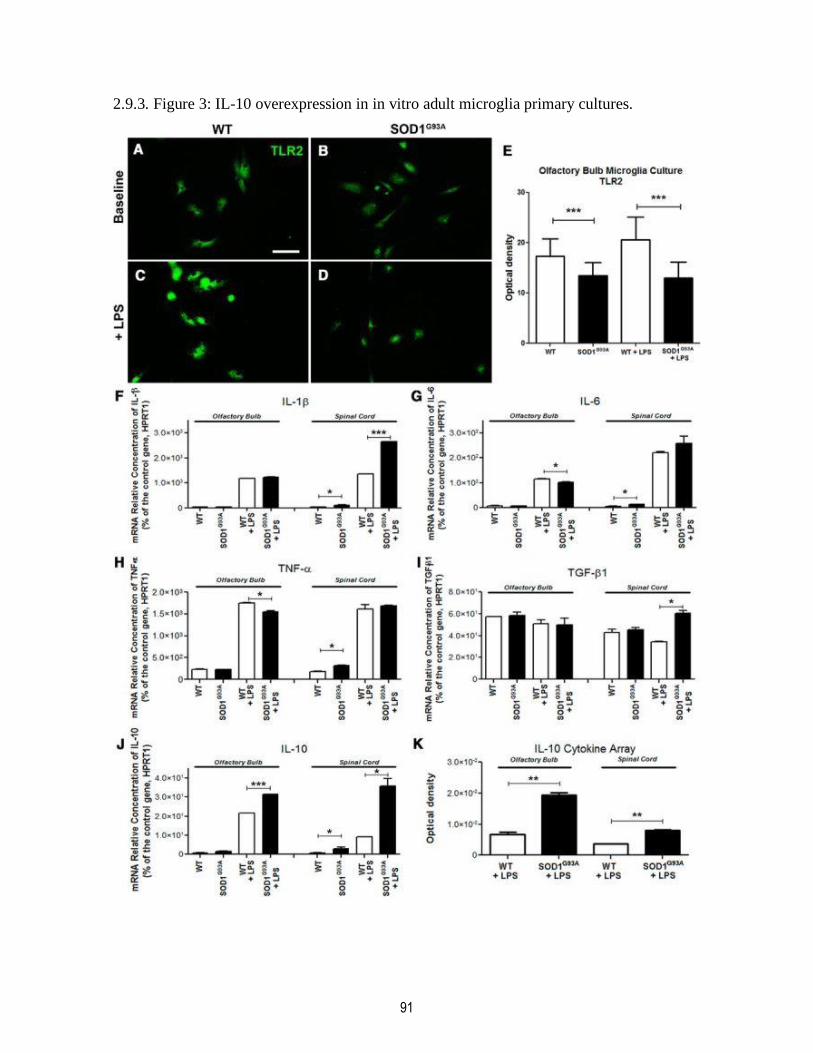
91
2.9.3. Figure 3: IL-10 overexpression in in vitro adult microglia primary cultures.

92
A–D, Baseline expression of TLR2 detected by immunofluorescent staining in adult primary
cultures derived from the olfactory bulb of 60-d-old WT controls (A) and SOD1G93A mice
(B) and TLR2 upregulation following in vitro LPS stimulation (C, D), Scale bar, 50 μm. E,
Optical density quantification of TLR2 immunofluorescent stained cultures (WT, n = 6;
SOD1G93A, n = 6; p < 0.0001; WT + LPS, n = 6; SOD1G93A + LPS, n = 6; p < 0.0001). F–J,
RT-PCR analysis of the adult microglia primary cultures from olfactory bulb and spinal cord
of WT and SOD1G93A mice, before and after LPS stimulation, looking at relative mRNA
concentrations of IL-1β (spinal cord, WT vs SOD1G93A, p = 0.0319; WT + LPS vs
SOD1G93A + LPS, p < 0.0001; F), IL-6 (olfactory bulb, WT + LPS vs SOD1G93A + LPS, p =
0.0388; spinal cord, WT vs SOD1G93A, p = 0.0346; G), TNFα (olfactory bulb, WT + LPS vs
SOD1G93A + LPS, p = 0.0170; spinal cord, WT vs SOD1G93A, p = 0.0105; H), TGF-β1 (spinal
cord, WT + LPS vs SOD1G93A + LPS, p = 0.0103; I), and IL-10 (olfactory bulb, WT + LPS
vs SOD1G93A + LPS, p = 0.0002; spinal cord, WT vs SOD1G93A, p = 0.0479; WT + LPS
vs SOD1G93A + LPS, p = 0.0206; J). All mRNA relative concentrations were normalized on
the control gene HPRT1, and n = 2 for all RT-PCR analyses. K, Quantification of the IL-10
cytokine array from in vivo olfactory bulb 24 h after LPS stimulation (WT + LPS, n = 6;
SOD1G93A + LPS, n = 6; p = 0.0054) and from in vivo lumbar spinal cord after LPS
stimulation (WT + LPS, n = 6; SOD1G93A + LPS, n = 6; p = 0.0014). Error bars indicate SEM.
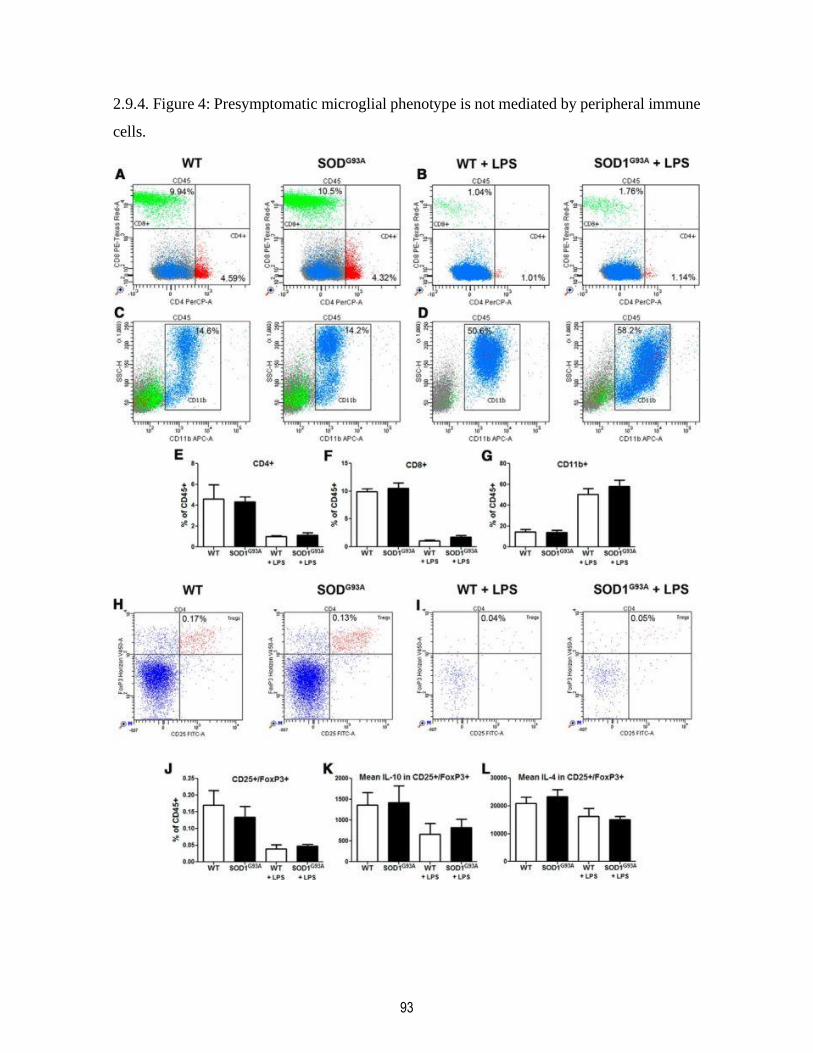
93
2.9.4. Figure 4: Presymptomatic microglial phenotype is not mediated by peripheral immune
cells.

94
Blood samples from presymptomatic (60 d) and 24 h LPS stimulated WT and SOD1G93A
mice were collected to assess the potential of immune cells in the periphery to modulate the
repressed microglial response observed in the CNS of presymptomatic SOD1G93A mice. The
blood samples were stained for CD45, CD4, CD8, and CD11b or for CD45, CD4, CD25,
FoxP3, IL-4, and IL-10 and analyzed in flow cytometry. A–D, In 60-d-old mice, the CD4+
and CD8+ lymphocyte proportions were unchanged between WT and SOD1G93A (A, B), as
were the monocyte/macrophage CD11b+ populations (C, D). CD4+, CD8+ lymphocytes and
CD11b+ monocytes/macrophages of WT and SOD1G93A mice all showed equivalent
response to LPS stimulation. E–G, Overall FACS counts suggest no deregulation in the
CD4+, CD8+, or CD11b+ population in presymptomatic 60-d-old SOD1G93A mice.
CD25+/FoxP3+ Tregs were also assessed for their production of IL-4 and IL-10. H, I, Both
at baseline (H) and after LPS stimulation (I), WT and SOD1G93A mice had similar Tregs
population ratios. J, Tregs counts suggest no deregulation between presymptomatic WT and
SOD1G93A mice at 60 d. K, L, Levels of IL-10 (K) and IL-4 (L) were estimated as mean levels
of fluorescence intensity among the Tregs cells. Again, no differences were observed
between WT and SOD1G93A presymptomatic 60 d Tregs (n = 4–6 in each groups). Error bars
indicate SEM.

95
2.9.5. Figure 5: Modulating the microglial response by blocking the IL-10 pathway.

96
A–C, E–G, Real-time imaging of TLR2 induction in the olfactory bulb and brain, 24 h after
LPS stimulation, in WT controls (A), SOD1G93A mice (B), and SOD1G93A mice treated with
αIL-10R antibodies (C) and the respective real-time imaging of the spinal cord (E–G) show
a significant LPS-induced microglial activation increase in αIL-10R-treated SOD1G93A
compared to untreated SOD1G93A mice. D, Quantitative analysis of the photonic emissions in
the head (WT, n = 19; SOD1G93A, n = 24; p = 0.0047; WT + LPS, n = 24; SOD1G93A + LPS,
n = 20; p = 0.0273; SOD1G93A + αIL-10R + LPS, n = 9; p = 0.0126). H, Quantitative analysis
of the photonic emissions in the spinal cord (WT, n = 19; SOD1G93A, n = 22; p = 0.0119; WT
+ LPS, n = 16; SOD1G93A + LPS, n = 13; p = 0.0351; SOD1G93A + αIL-10R + LPS, n = 8; p
= 0.0005). I–K, Real-time imaging of TLR2 induction in the olfactory bulb and brain, 48 h
following intracerebroventricular injection of mSOD1, in WT controls (I), SOD1G93A mice
(J), and SOD1G93A mice treated with αIL-10R antibodies (K) shows a significant mSOD1-
induced microglial activation increase in the SOD1G93A αIL-10R-treated mice. L,
Quantitative analysis of the photonic emissions in the head (WT, n = 19; SOD1G93A, n = 24;
p = 0.0047; WT + LPS, n = 10; SOD1G93A + LPS, n = 6; p = 0.0221; SOD1G93A + αIL-10R +
LPS, n = 7; p = 0.0490). M–P, Immunostaining of Iba1 after LPS stimulation in WT,
SOD1G93A, and SODG93A +αIL-10R mice (M–O) and associated optical density quantification
(P; WT + LPS, n = 5; SOD1G93A + LPS, n = 5; p = 0.0258; SOD1G93A + αIL-10R + LPS, n =
5). Q–T, Immunostaining of Iba1 after mSOD1 intracerebroventricular stimulation in WT,
SOD1G93A, and SODG93A + αIL-10R mice (Q–S) and associated optical density quantification
(T; WT + mSOD1, n = 5; SOD1G93A + mSOD1, n = 5; p = 0.0161; SOD1G93A + αIL-10R +
mSOD1, n = 5). Scale bars: 100 μm. U, V, Western blots of Ym1 in WT, SOD1G93A, and
SOD1G93A + αIL-10R brain extracts following mSOD1 intracerebroventricular stimulation
(U) and optical density quantification (V) show a significant increase of Ym1 in the
SOD1G93A + mSOD1 samples, but also a significant decrease of Ym1 levels in the SOD1G93A
+ αIL-10R + mSOD1 samples (WT + mSOD1, n = 3; SOD1G93A + mSOD1, n = 3; p = 0.0009;
SOD1G93A + αIL-10R + mSOD1, n = 3; p < 0.0126). Error bars indicate SEM.
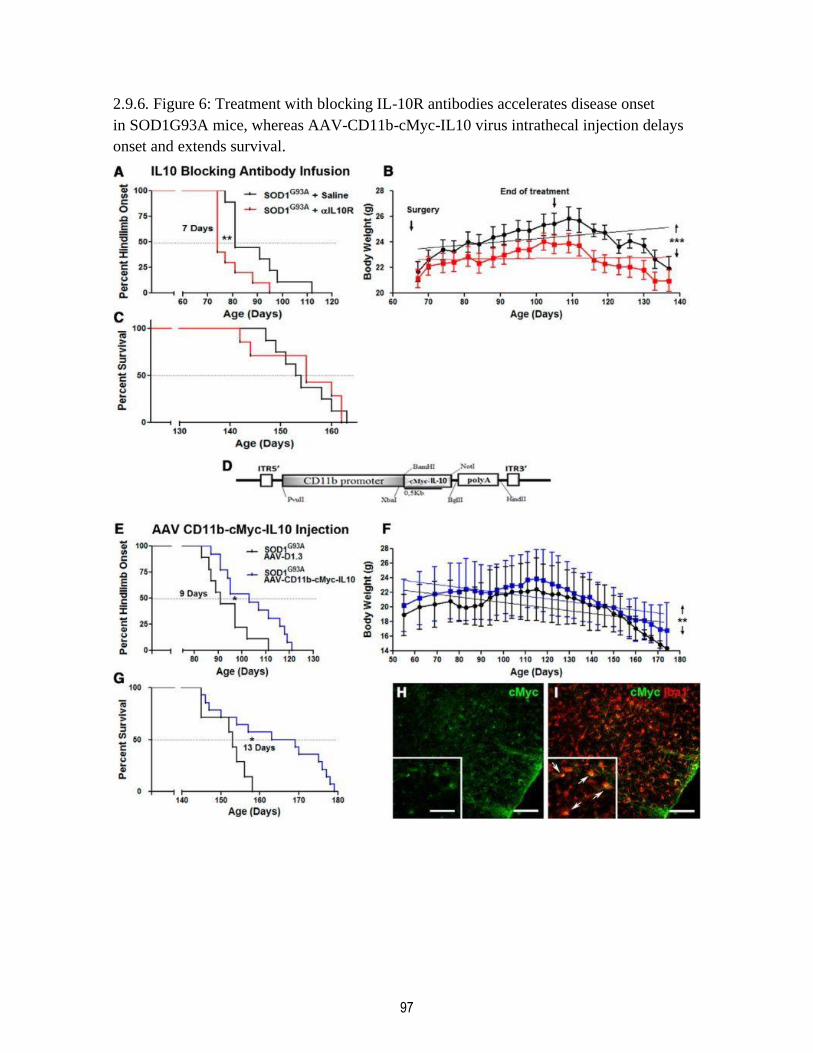
97
2.9.6. Figure 6: Treatment with blocking IL-10R antibodies accelerates disease onset
in SOD1G93A mice, whereas AAV-CD11b-cMyc-IL10 virus intrathecal injection delays
onset and extends survival.

98
Longitudinal analysis of SOD1G93A mice subjected to a 42 d intracerebroventricular infusion
of saline (black curves) or αIL-10R antibodies (red curves) started at presymptomatic 60 d
of age. A, Kaplan–Meier analysis of the extension reflex revealed a significant median onset
7 d earlier in the αIL-10R infused group (saline treated mean onset, 81 d; n = 9; αIL-10R
treated mean onset, 74 d; n = 10; p = 0.0101). B, Animals receiving the αIL-10R antibodies
failed to gain weight like the saline-treated controls and maintained an overall significant
lower weight through their lifespan (linear regression analysis shows that the elevation
between the two curves is significant; p < 0.0001). C, The presymptomatic αIL-10R
treatment did not however affect the survival rate between the two groups. D, Diagram of the
viral AAV construct expressing cMyc-IL-10 under the control of the human CD11b gene
promoter. Longitudinal analysis of SOD1G93A mice subjected to an intrathecal AAV virus
injection, with an empty plasmid control virus (black curves) or AAA-CD11b-cMyc-IL10
virus (blue curves) injected at presymptomatic 30–35 d of age. E, Kaplan–Meier analysis of
the extension reflex revealed a significant 9 d delayed median onset in the AAV-CD11b-
cMyc-IL10-infected group (control AAV mean onset, 90 d; n = 8; AAV-Cd11b-cMyc-IL10
mean onset, 99 d; n = 14; p = 0.0157). F, Animals receiving the AAV-CD11b-cMyc-IL10
virus maintained an overall significantly higher weight through their lifespan (linear
regression analysis shows that the elevation between the two curves is significant; p =
0.0039). G, The presymptomatic AAV-CD11b-cMYC-IL10 intrathecal injection also
significantly increased survival by 13 d (control AAV mean survival, 153 d; n = 7; AAV-
Cd11b-cMyc-IL10 mean survival, 166 d; n = 14; p = 0.0166). H, I, Immunostaining of cMyc
in the spinal cord of AAV-CD11b-cMyc-IL10-injected mice shows colocalization with Iba1,
confirming a microglial expression of the viral construct. Scale bars: 100 μm; insets, 50 μm.
Error bars indicate SEM.
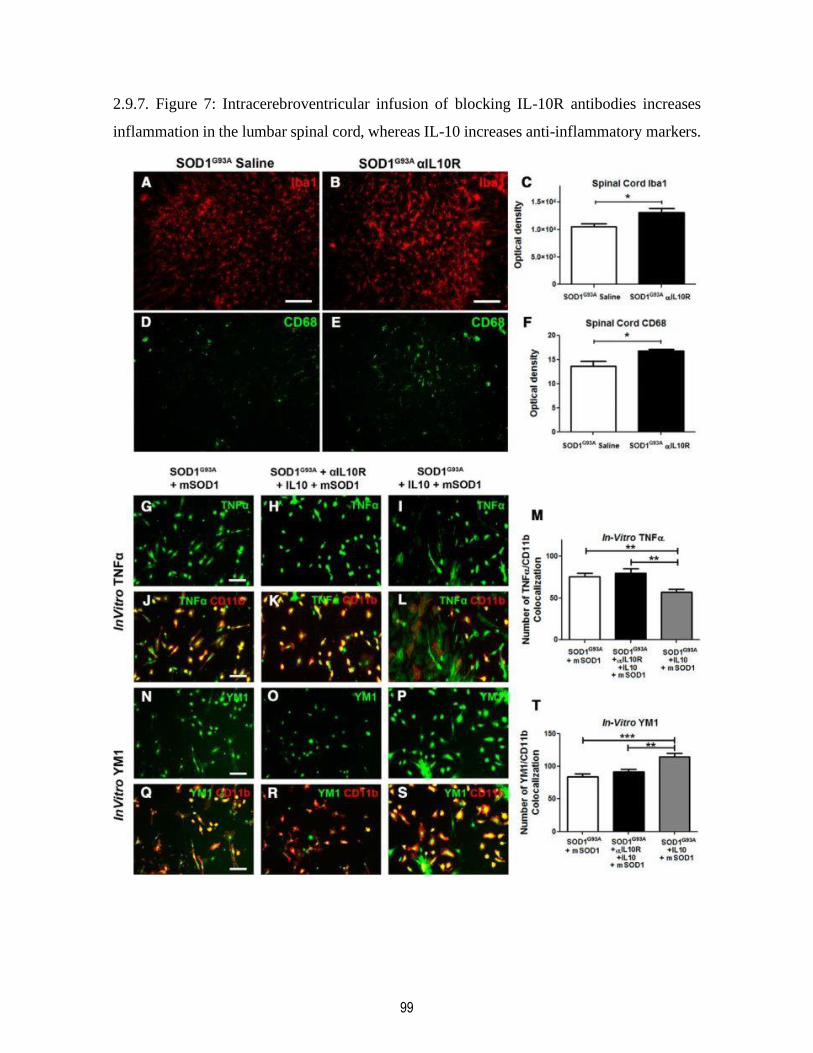
99
2.9.7. Figure 7: Intracerebroventricular infusion of blocking IL-10R antibodies increases
inflammation in the lumbar spinal cord, whereas IL-10 increases anti-inflammatory markers.

100
A–C, Immunostaining of Iba1 in the lumbar spinal cord ventral horn of mice following 42 d
of saline (A) or αIL-10R (B) intracerebroventricular infusion in SOD1G93A mice reveals,
by optical density quantification of the fluorescent signal, an increased Iba1
immunoreactivity in the αIL-10R-treated mice (C; saline, n = 5; αIL-10R, n = 8; p = 0.0298).
D–F, Immunostaining for Cd68, a marker of microglial activation (D, E), similarly shows,
by optical density quantification, an increased CD68 expression in αIL-10R-infused
SOD1G93A mice (F; saline, n = 6; αIl-10R, n = 6; p = 0.0128). G–M, TNFα immunostaining
following in vitro mSOD1 stimulation on SOD1G93A, SOD1G93A + αIL10R + IL10, and
SOD1G93A + IL10 primary cultures and relative numbers of TNFα/CD11b colocalization (M)
show a significant decrease of TNFα cells in the IL10-treated group (SOD1G93A + mSOD1,
n = 16; SOD1G93A + αIL-10R + IL10 + mSOD1, n = 16; SOD1G93A + IL10 + mSOD1, n =
16; p = 0.0041). N–T, Ym1 immunostaining following in vitro mSOD1 stimulation on
SOD1G93A, SOD1G93A + αIL-10R + IL10, and SOD1G93A + IL10 cultures and relative
numbers of YM1/CD11b colocalization (T) show a significant increase of YM1 positive cells
following IL10 treatment (SOD1G93A +mSOD1, n = 16; SOD1G93A +αIL-10R +Il10
+mSOD1, n = 16; SOD1G93A + IL10 +mSOD1, n = 16, p = 0.0002). Scale bars: 50 μm. Error
bars indicate SEM.

101
Chapitre 3 - SRSF3 acts as a ribosome-based checkpoint and regulates translation of
immune mRNAs in ALS microglia
3.1. Résumé
Nous avons récemment décrit un nouveau mécanisme cellulaire impliqué dans la traduction
de gène immunitaires et dans l’activation microgliale. Ce mécanisme se base sur la
suppression spécifique de la traduction des ARNm hautement régulés par l’interaction de
leur 3’UTR et de la protéine liant l’ARN SRSF3. Dans cette étude, nous décrivons le rôle de
SRSF3 dans la transformation pathogénique de la microglie dans la SLA. En utilisant un
modèle-système pour l’analyse de l’état traductionnel des ribosomes microgliaux à différent
stade de la maladie, nous montrons que SRSF3 se lie bel et bien aux 3’UTR des ARNm de
gène immunitaires hautement régulés et fonctionne comme un suppresseur de la traduction,
menant à une dissociation chronique des réseaux du transcriptome et du protéome de la
microglie SLA. L’analyse de section de moelle épinière de souris, rats et patients sporadiques
atteints de la SLA révèle une augmentation proportionnelle à la progression de la maladie de
la forme phosphorylée de SRSF3, restreinte au cellules Iba+. Le knockdown ciblé de SRSF3
rétablit le potentiel phagocytaire de la microglie SLA. De plus, ce traitement anti-SRSF3,
débuté durant la phase symptomatique de la maladie (140 jours), dans le modèle murin
G93ASOD allège l’arrêt traductionnel des ARNm sélectionnés, induit la synthèse de-novo
des protéines immunitaires et exerce un effet significatif sur la progression de la maladie.
Ainsi, nos recherches suggèrent de nouvelles avenues pour la modulation thérapeutique de
la réponse immunitaire dans la SLA.

102
3.2. Abstract
We recently described a novel ribosome-based regulatory mechanism/check point involved
in the control of innate immune gene translation and microglial activation. The mechanism
is based on the specific translational suppression of highly expressed mRNAs through a
3’UTR-mediated mechanism involving the RNA binding protein SRSF3. Here we describe
a role SRSF3 in the pathogenic transformation of microglia in ALS. Using a model-system
for analysis of the dynamic translational state of microglial ribosomes with mRNAs as input
and newly synthesized peptides as output, at different stages of disease, we show that SRSF3
binds to 3’UTR of highly regulated immune mRNAs and acts as a suppressor of translation,
resulting in a chronic dissociation of mRNA and protein networks in ALS microglia.
Analyses of spinal cord sections from ALS mouse and rat and human sALS reveal a disease-
associated increase in expression of effective phosho-SRSF3 restricted to Iba+ cells.
Targeted knockdown of SRSF3 restored phagocytic properties of ALS microglia, while the
treatment with anti-SRSF3 morpholinos, initiated at late symptomatic disease (140 days), in
SOD1G93A mouse model alleviated translational arrest of selected genes, induced de novo
synthesis of immune proteins and exerted a significant disease modifying effects. Together,
our findings suggest novel avenues for therapeutic modulation of immune response in ALS.

103
3.3. Introduction
Amyotrophic lateral sclerosis (ALS) is a late onset neurodegenerative disorder characterized
by a progressive muscle weakness and paralysis, leading to death 3-5 years following initial
diagnosis (Swinnen and Robberecht, 2014). To this date, only two drugs have been approved
for the treatment of ALS; the glutamatergic modulator riluzole, which improves survival by
2 to 3 months (Miller et al., 2012), and the free-radical scavenger edaravone (Hardiman and
van den Berg, 2017), that slows progression but has no significant effect on survival.
Although major clinical symptoms in ALS arise from neurodegeneration and death of motor
neurons, it is now well established that non-neuronal cells such as microglia play an
important role in disease pathogenesis. One of the prominent features of ALS is a chronic
reactive microgliosis detected in the spinal cord sections of patients and SOD1 mutant mice,
a model of inherited disease used in this study (McGeer et al., 1991, Hall et al., 1998, Gurney
et al., 1994). A current view is that over the course of disease, microglia change their
phenotypes from initially beneficial into highly toxic and aberrant cells (Trias et al., 2013,
Frakes et al., 2014, Henkel et al., 2009, Keren-Shaul et al., 2017) resistant to any conventional
immune-modulatory therapeutic interventions (Patel et al., 2015, Keller et al., 2011).
However, the underlying cause and the molecular mechanisms involved in this apparent shift
in microglia toxicity remain unclear. Recent studies employing cell-specific
transcriptomics/RNAseq, revealed a new microglial phenotype that has been identified
across many neurodegenerative diseases including Alzheimer’s disease, experimental
autoimmune encephalomyelitis, ALS as well as in aging (Holtman et al., 2015, Keren-Shaul
et al., 2017, Krasemann et al., 2017). This particular molecular signature has been
characterized by upregulation of distinct RNAs such as Clec7a, Cst7, Apoe etc. While to
date, the most of microglia profiling studies have been focused on the analysis of microglial
transcripts, the actual disease-associated microglia proteome has not been well characterized.
We recently described a novel ribosome-based check-point mechanism involved in
translation control of innate immune genes and microglia activation (Boutej et al., 2017). The
mechanism is based on a selective 3’UTR-mediated translational repression of highly
expressed, ribosome-bound and “actively translating” mRNAs orchestrated by RNA binding

104
protein SRSF3 (Boutej et al., 2017, Kim et al., 2014). SRSF3 belongs to the Serine-arginine-
rich (SR) proteins family that contains several RNA binding proteins with a functional
implication in RNA metabolism (Manley and Krainer, 2010). Like other SR proteins, SRSF3
is involved in alternative splicing events, however, recent reports emphasize its role in the
mechanisms involved in post-transcriptional regulation, such as mRNA export, surveillance,
stability and translation (Kim et al., 2014).
In the current study we investigate the role of SRSF3 in the pathogenic transformation of
microglia in ALS. Using a model-system for analysis of the dynamic translational state of
microglial ribosomes with mRNAs as input and newly synthesized peptides as output, at
different stages of disease, we show that SRSF3 binds to 3’UTR of highly regulated immune
mRNAs including Clec7a, Cst7 and/or Lilrb4a and acts as a suppressor of translation, leading
to a chronic dissociation of mRNA and protein networks in ALS microglia. It is noteworthy
that expression of active SRFS3 (phospho-SRSF3) increases with disease progression and it
is restricted to activated microglia in the lumbar spinal cord of ALS mouse and rat as well as
in human sporadic disease. Targeted knockdown of SRSF3 alleviates translational arrest of
immune mRNAs, induces de novo synthesis of selected immune proteins and restores
microglia phagocytic capacity. Furthermore, intrathecal delivery of anti-SRSF3 morpholinos
initiated at advanced stages of disease in SOD1G93A mice slows disease progression and
significantly extends survival of ALS mice. Taken together, our study opens new avenues in
therapeutic reprograming of immune response in ALS.
3.4. Results
3.4.1. Creation of in vivo Model-System for Analysis of Microglia Translational Dynamics
in ALS
To generate in vivo model-system for analysis of microglia RNA and protein molecular
signatures in ALS we took advantage of the CD11b-Flag-EGFP-mRPL10a (CD11brGFP)
mouse line, recently developed and validated in our laboratory. As previously reported
(Boutej et al. 2017), in this transgenic mouse model, a fusion protein composed of the Flag
peptide and enhanced green fluorescent protein (EGFP) attached to the N-terminus of the

105
murine large subunit ribosomal protein 10a (mRPL10a) is expressed under transcriptional
control of the myeloid cell gene promoter CD11b (Figure 1A). As a next step, the
CD11brGFP were cross-bred with SOD1G93A mice to generate a double transgenic
CD11brGFP;SOD1G93A ALS line, thus allowing a high affinity immunoprecipitation of the
ribosome-bound mRNAs and peptides from CD11b-positive spinal cord microglia at any
stage of disease. Of note, previous work revealed that in the CNS of neurodegenerative
disease models, including SOD1G93A mice, the CD11b expression is largely limited to a
population of resident microglia cells (Ajami et al. 2007, Ajami et al. 2018). As schematically
presented in Figure 1B the spinal cord tissue homogenates were immunoprecipitated using an
anti-Flag agarose affinity resin and the polyribosome complexes were used either for mRNA
extraction followed by Affymetrix Mouse Genome 430 analysis or peptide extraction
followed by a high-resolution label-free quantitative proteomic analysis. Next, double
immunofluorescence analyses of the lumbar spinal cord sections of the
CD11brGFP;SOD1G93A mice at pre-symptomatic, moderate symptomatic and advanced
symptomatic stage of disease (Figures 1C-1K) revealed the expected expression of the
CD11b driven EGFP transgene in CD11b-positive microglia. Consistent with the parameters
of our internal SOD1G93A colony, the disease onset of CD11brGFP;SOD1G93A mice was
at 96 days and the median survival was 155 days (n=20), (Figures 1L-1M respectively).
3.4.2. Chronic Dissociation of mRNA and Protein Molecular Signatures in ALS Microglia
We recently described a marked dissociation of mRNA and protein immune networks in
acutely activated microglia (Boutej et al. 2017). To assess whether chronically activated
microglia in ALS exert divergence in mRNA and protein molecular signatures, we took
advantage of the CD11brGFP;SOD1G93A mouse model and performed a high affinity
immunopurification on lumbar spinal cord tissue homogenates using an anti-Flag agarose
affinity resin to isolate polyribosomes. As previously presented in Figure 1B, the collected
polyribosomes complexes were used either for mRNA extraction followed by Affymetrix
Mouse Genome 430 analysis or for peptide extraction followed by high resolution, label-free
quantitative proteomic analysis. To understand temporal dynamics of the disease-associated
changes in microglia phenotypes, a parallel transcriptome and proteome analyses were

106
performed at the distinct time points of disease including presymptomatic, moderate
symptomatic as well as advanced stages of disease.
As shown in Figure 2A-2C, the quantitative analysis of microglia transcripts revealed a
gradual increase in the number of upregulated genes when compared to age-matched wild-
type littermates. More precisely, we found only two transcripts significantly upregulated over
the 2-fold change threshold at the presymptomatic stage (Figure 2A) compared with 1504
upregulated transcripts at the moderate symptomatic stage (Figure 2B) and 3590 upregulated
transcripts at the advanced symptomatic stage (Figure 2C, see also Table S1). In the
presymptomatic stage of ALS, Cdc42 and Rfwd3 were the only regulated transcripts at the
level of 2-fold or more when compared to controls (Figure 2A) and they are both known to
be implicated in the cell cycle (Gong et Chen 2011, Chircop 2014). At the moderate
symptomatic stage (in SOD1 mutant mice phenotypically associated with an initial hind-limb
weakness), we observed a shift towards a “more regulated transcriptome” (Figure 2B), along
with an increased expression of genes implicated in immune functions and immune
regulation. At this stage, we can observe increased expression of genes implicated in immune
response such as Ccl3 (10.32-fold change) and Cd68 (6.96-fold change) known to be found
in inflammatory microglia (Zhu et al. 2016, Chistiakov et al. 2017). Interestingly however,
we observed a marked increase of Lilrb4a (11.55-fold change), a gene implicated in
macrophage maturation as well as regulation of NF-κB and associated inflammatory
pathways (Kamphuis et al. 2016, Zoller et al. 2018) and Gpnmb upregulated 7.66-fold as
compared to control. Gpnmb has been detected in a subset of activated microglia in
Alzheimer’s disease (Hüttenrauch et al. 2018). It is thought to be a marker of alternative
macrophages polarization exerting certain neuroprotective functions (Budge et al. 2018,
Liguori et al. 2020). A similar molecular signature at mRNA level has been identified at the
advanced stage of the disease (Figure 2C), however, with a generally higher level of
regulation for the transcripts. The expression of Lilrb4a and Gpnmb was increased by 16.57
and 16.52-fold respectively. As the most upregulated transcript at the advanced stage of
disease we identified Clec7a (38.04-fold change). Clec7a is found to be implicated in
neurodegenerative diseases (Haure-Mirande et al. 2019), along with Cst7 (14.16-fold
change) and Itgax (11.91-fold change), the identified upregulated transcripts at advance

107
stages have already been associated with a particular microglial phenotype found in
neurodegenerative diseases, including Alzheimer’s disease (Keren-Shaul et al. 2017).
Surprisingly, the analysis of the sequenced peptides showed a completely different point of
view. At the presymptomatic stage, we found 34 proteins that were regulated above the 1.2-
fold change threshold (Figure 2D). At the moderate symptomatic and advanced symptomatic
stages, we found respectively 121 and 57 upregulated proteins (Figure 2E-2F, see also Table
S1). Furthermore, at the symptomatic stages of disease, the volcano plots are more spread to
the sides, suggesting that the levels of regulation are generally greater as compared to
presymptomatic disease. Among the most upregulated proteins at the presymptomatic stage
we found GOT1 (3.05-fold change): involved in macrophage polarization (Jha et al. 2015),
MVP (2.75-fold change): has been shown to have neuroprotective functions (Noorimotlagh
et al. 2017) and SOD1 (2.63-fold change), a well-known ALS causative gene (Rosen et al.
1993). We can also find the core paraspeckles components upregulated together; PSCP1
(1.66-fold change), NONO (1.55-fold change) and SFPQ (1.53-fold change) (Bond et Fox
2009) (Figure 2G). During the moderate symptomatic stage, the top upregulated protein is
LGALS3 (11.61-fold increase), a well-known marker for microglia activation and
proliferation (Lalancette-Hebert et al. 2012, Rahimian, Beland, et al. 2018) and potential
ALS-related biomarker of microglia activation (Zhou et al. 2010, Ashraf et Baeesa 2018). In
addition, we identified other proteins involved in ALS pathogenesis such as APOE, an
important causative gene of Alzheimer’s and recently linked to ALS (Krasemann et al. 2017,
Belloy et al. 2019), complements proteins C1QB and C1QC (3.54- and 5.70-fold change
respectively) (Heurich et al. 2011) (Figure 2H). At the advanced symptomatic stage of the
disease, we identified complement protein C1QA (9.06-fold change) and MTPN (11.83-fold
change). Interestingly, GFAP (5.39-fold change), well-identified astrocytic marker, belongs
to the top regulated proteins at the advanced symptomatic stage of the disease. It is consistent
with recent reports by Wilhemsson et al. (Wilhelmsson et al. 2017) revealing that following
injury and inflammation distinct subset of cells may express both microglia and astrocytes
cellular markers. Furthermore, GFAP has been also associated to diseased CD11b-positive
microglia in the late stages of ALS in a rat model of disease (Diaz-Amarilla et al. 2011, Trias,
Diaz-Amarilla, et al. 2013) (Figure 2I). Finally, we also identified upregulated proteins such
as CTSD, whose protein family is already linked to the disease associated microglial

108
phenotype (Keren-Shaul et al. 2017), NUP214 which is linked to proliferation (Sandén et al.
2013), and GSN, an actin-regulating protein with potential immune-modulatory function
important inflammation marker (Piktel et al. 2018).
To obtain a general view on how disease affects biological functions of activated microglia
in SOD1G93A, we used Cytoscape and ClueGO cluster analysis (Shannon et al. 2003,
Bindea et al. 2009, Bindea et al. 2013) and mapped biological functions from the top 50
significantly upregulated transcripts and peptides for each time points of disease. While at
presymptomatic stage (Figure 2G), the top biological microglia functions were linked to
cellular homeostasis, at the moderate symptomatic (Figure 2H) the top functions are related
to immunity and immune response. A similar mRNA molecular profile and/or top biological
functions were found at the advanced symptomatic stage (Figure 2I), revealing a strong
inclination towards immune functions and regulation. Interestingly, a parallel analysis of
microglia proteome at the same time-points of the disease revealed a marked divergence in
networks and biological functions when compared to molecular signature at mRNA level. In
contrast to highly specialized immune mRNA response, the top biological functions at
protein level were highly enriched in function related to RNA metabolism, response to stress
and cytoskeleton organisation (Figure 2J-2K-2L). Moreover, the term “Innate immune
system” is at the 9th position in the microglial proteome (Figure 2K). Together, our results
suggest that during disease progression microglial cells acquire functionally two distinct
molecular and functional signatures. While RNA networks were highly specialized and
enriched in Immune system related terms and functions, the top biological functions of
upregulated proteins were related to protein assembly, RNA and cell metabolisms and
cellular stress.
3.4.3. Highly Upregulated Transcripts in ALS Microglia Are Not Translated
As listed in Table 1 the top 10 upregulated transcripts detected at presymptomatic, moderate
and advanced stages of disease (135d and 158d) were not detected as among sequenced
proteins of the proteomic analysis, with the exception of GPNMB and EIF4A2, an eukaryotic
translation initiation factor which function is not related to immune response. In fact, none

109
of the highly up-regulated immune transcripts related to immune-modulatory and phagocytic
microglia functions were detected among the sequenced peptides. This was further confirmed
by western blot analysis. As shown in Figure 3A-3E, we validated protein levels of the most up-
regulated mRNAs such as Clec7a, Lilrb4a, Cst7, Cd68 and Ccl3 and we did not observe any
significant changes in their expression level when compared to age-matched controls.
Suggesting that translation of selected highly upregulated transcripts is actively suppressed
in microglial cells. The exception among the top regulated genes with immune function was
Gpmnb. As shown in Table 1, and as further validated by western blot, the significant
increase at mRNA level was followed by a significant increase in GPNMB protein expression
levels in the lumbar spinal cord tissue homogenates from symptomatic ALS mice, suggesting
that different regulatory mechanisms may be implicated in translational control of Gpnmb
(Figure 3F).
It is noteworthy that the observed translational arrest was restricted to a cluster of genes,
highly regulated whereas other less regulated and/or unregulated ribosome bound mRNAs
were translated, detected by quantitative mass spectrometry and validated by western blot,
revealing the expression at expected levels (see Figure S1).
3.4.5. SRSF3 is Responsible for the Translational Repression of Immune mRNAs by Binding
to their 3’UTR
We previously reported that translational repression of acutely regulated LPS genes is
orchestrated by SRSF3. We showed that the 3’UTRs of highly regulated LPS genes contain
multiple putative binding sites for SRSF3 that could be targeted for regulation (Boutej et al.,
2017). We next asked whether the same, SRSF3/3’UTR-mediated molecular mechanism is
involved in the translational repression of immune genes in chronically activated ALS
microglia. We took advantage of bioinformatics tools, the RBP map with high stringency and
conservation filter (Paz et al., 2014), and searched for potential SRSF3 binding sites within
the 3’UTR of the selected genes (see Table 1). The analyses revealed that 3’UTRs of the
selected transcripts are highly enriched in putative binding sites for SRSF3 (see Table S2).
As shown in Figure 3G, the comparison of 3’UTR sequences of confirmed arrested vs. non-

110
arrested genes revealed SRSF3 preference to target genes containing higher density of SRSF3
binding sites on their 3’UTR (p=0,0086) (Figure 3G). Interaction between SRSF3 and the
selected genes was further validated in the series of proof-of concept cross-linking
immunoprecipitation (CLIP) experiments. As shown in Figure 3H, we successfully co-
precipitated SRSF3 cross-linked to 3’UTRs of Clec7a, Cst7 and Lilrb4a (Figure 3H, see
Table S3), revealing that SRSF3 binds to 3’UTRs of highly regulated and ribosome- bound
immune transcripts. Further bioinformatics characterization of 3’UTR sequences revealed
that the genes preferentially targeted by SRSF3 have relatively short 3’UTRs. As revealed in
Figure 3I (where each dot on the graph represents the 3’UTR of a different gene), in
comparison with other RNA binding proteins (RBPs), SRSF3 possesses a large number of
binding sites on 3’UTRs, but it will preferentially bind shorter 3’UTR (Figure 3J). As
schematically presented in Figure 3J, 75% of genes whose translation is preferentially
regulated by SRSF3 possess 3’UTRs with less than 700 nucleotides and 63% less than 500
nucleotides. Interestingly, a large number of highly regulated and ribosome-bound mRNAs
arrested in translation following acute immune challenge and/or chronic ALS mediated
inflammation would fall within this category. The exception is Clec7a (3’UTR of 1540 bp),
However, our analyses revealed that Clec7a 3’UTR contains one of the highest number of
the SRSF3 binding sites when compared to other SRSF3 regulated mRNAs. Taken together
SRSF3 would preferentially bind to – and supress translation of mRNAs having 3’UTRs
under 700 nucleotides and/or with high densities of binding site for this RBP. The identified
immune transcripts have generally shorter 3’UTRs that are heavily enriched in SRSF3
binding sites, which in turn may explain why these genes could be preferentially targeted for
SRSF3-mediated translational regulation in activated microglia.
3.4.6. Upregulation of pSRSF3 in Lumbar Spinal Cord Microglia Correlates with Clinical
Onset of Disease
The activities of SRs proteins are regulated by cycles of phosphorylation and
dephosphorylation (Misteli and Spector, 1997). Indeed, our recent study suggest that
neuroinflammatory conditions are associated with a significant increase in phosphoSRSF3
(pSRSF3) levels in LPS activated microglial cells (Boutej et al., 2017). pSRSF3 is
preferentially cytoplasmic, allowing its interaction with ribosome-bound transcripts and

111
consequently controlling translation. To determine whether chronic activation of microglia
in ALS is associated with changes in SRSF3 activation patterns, we first analyzed pSRSF3
expression levels in lumbar spinal cord of the SOD1G93A mice at presymptomatic, moderate
symptomatic and advanced symptomatic stages of the disease. As revealed by western blot
analysis, the expression of pSRSF3 starts to increase after disease onset, at the moderate stage
of disease (p=0.036 vs controls and p=0.0137 vs presymptomatic stage) and is still higher
during the advanced stage (p=0.0097 vs controls and p=0.003 vs presymptomatic stage)
(Figure 4A). In order to establish the proportion of effectively phosphorylated protein, we
established the ratio of pSRSF3 vs. SRSF3 (Figure 4B). Consistent with the disease
associated increase in pSRSF3 expression levels, pSRSF3/SRSF3 ratios were significantly
changed at the moderate stage of the disease (p=0.0279 vs controls and p=0.0004 vs
presymptomatic stage) and at the advanced stage of the disease (p=0.0095 vs controls and
p=0.0004 vs presymptomatic stage). Together these data suggest an important shift in SRSF3
phosphorylation patterns between moderate and advanced stages of disease. In the
SOD1G93A mouse model this correlates with the onset of hind limb paralysis.
Next, to assess the cellular specificity of pSRSF3 expression patterns, we performed a
double-immunofluorescence analysis followed by confocal microscopy. Analysis was
performed on the lumbar spinal cord sections of the SOD1 mutant mice at different stages of
disease. As further shown in figure 4C-4J, and consistent with the results from western blot
analysis, pSRSF3 staining markedly increases in symptomatic disease, it starts to appear at
moderate stage of disease while pSRSF3 staining was robustly induced at advanced
symptomatic ALS. Importantly, a double-immunofluorescence analysis revealed that
pSRSF3 expression was mostly restricted to CD11b-positive cells with amoeboid
morphology characteristic of activated microglia. The lumbar spinal cord sections of controls
and presymptomatic mice were devoid of pSRSF3 immunoreactivity. Because in
physiological conditions, SRSF3 is predominantly nuclear protein, as expected SRSF3
immunostaining in controls and presymptomatic mice seems to be restricted to neuronal and
microglial nuclei (Figure 4C-4J).

112
3.4.7. Disease-Associated Increase in pSRSF3 is Conserved Across Species
To assure that disease-associated expression of pSRSF3 was not restricted to our model/and
or species, we took advantage of a previously characterized ALS rat model (Trias et al.,
2019). The rat model expresses the mutated human Sod1 gene (hSod1G93A) and develops a
progressive paralysis associated with marked neuroinflammation and activation of glial cells.
We immunostained the lumbar spinal cord sections of ALS rats and observed the same
expression pattern of pSRSF3 as in a mouse model. The pSRSF3 staining was markedly
increased in the spinal cord section at the time of clinical onset of disease and in symptomatic
disease, but not in WT littermates and presymptomatic rats. The signal was restricted to
CD11b-positive cells (Figure 4L-4N).
We next asked whether increase in microglial pSRSF3 observed in different rodent models
of ALS is associated with human disease. We took the same strategy and analyzed pSRSF3
immunoreactivity in ALS post-mortem spinal cord tissue. Along with non-ALS control
tissue, we analyzed spinal cord sections from 3 sporadic ALS patients. As revealed by
immunohistochemistry analysis (Figure 4P-4R), we detected pSRSF3 positive cells in the
spinal cord of ALS patients, but not in control samples (Figure 6Q). The pSRSF3 positive
cells share the shape and size of activated microglia positive for the microglia activation
marker: galectine-3 (LGALS3) ( Zhou et al., 2010, Lalancette-Hebert et al., 2012, Ashraf and
Baeesa, 2018,) (Figure 6S), suggesting a microglia-specific expression of the phosphorylated
forms of SRSF3 in human disease. Taken together, our results revealed that the disease-
associated alterations in pSRSF3 expression patterns are conserved across the species, thus
suggesting that SRSF3 may have an important role in deregulation of immune response in
ALS.
3.4.8. Targeting Srsf3 Restores Immune Gene Translation and Microglia Phagocytic
Function
We previously showed that SRSF3 acts as a suppressor of innate immune genes translation
in LPS-activated microglia (Boutej et al., 2017). While acute suppression of inflammation
may be protective, a chronic deregulation of immune response may be detrimental and lead
to disease. It is noteworthy, that the microglial transcriptomic profile of symptomatic SOD1

113
mutant mice obtained in our study (see Figure 2, Table1) fits the DAM phenotype described
by Keren-Shaul et al. (Keren-Shaul et al., 2017) that has been hypothesized to be
neuroprotective through its phagocytic activity (Deczkowska et al., 2018). However, here we
show (Figure 2, Figure 3 and Table1) that highly regulated key functional genes such as
Clec7a, Cst7, Lilrb4a are not translated leading to a gradual loss of microglia immune
functions and their capacity to phagocyte, which in turn may accelerate a rate of
neurodegeneration in ALS. Hence, targeting SRSF3 could be of clinical interest if the
untranslated transcripts such as Clec7a, Cst7, Lilrb4a and related functions observed in the
microglial transcriptome profile were to be restored.
To assess if the rescue of microglia phagocytic function/capacity by SRSF3 knockdown is of
therapeutic interest we targeted endogenous protein using antisense morpholinos (AMOs).
As schematically presented in Figure 5A, symptomatic ALS mice received 10µmol anti-Srsf3
AMOs or scrambled sequence delivered intrathecally (IT) via 3 separate injections (Figure
5A). The therapy was delivered late in disease (at 140 d) as significant increase of microglial
pSRSF3 occurs after clinical onset of disease in SOD1G93A mice. The efficacy of SRSF3
knockdown was evaluated by ELISA. As shown in Figure 5B, approximately 30 days after
last IT injection, we observed a reduction of 45,5% of the SRSF3 expression in the spinal
cord of the morpholino treated animals when compared to the scrambled sequence treated
animals (p<0.0001) (Figure 5b). Treatment with AMOs resulted in a slower disease
progression calculated as the score progression slope (p=0.0285) (Figure 5C) and had a
significant impact on the survival of ALS mice (p=0.0029) (Figure 5D). To understand the
molecular mechanisms underlying the therapeutic effects of the SRSF3 knockdown, we
analyzed spinal cord tissue of treated vs. non-treated mice 2 weeks after IT injections. The
collected tissue was analyzed for expression of targeted genes/proteins by western blot. As
expected, the SRSF3 and pSRSF3 expression was significantly decreased in the SRSF3
AMOs treated animals (p=0.0003 and p=0.0221 respectively) (Figure 5E). The knockdown
of SRSF3 and its effective phosphorylated form alleviated translation arrest of previously
untranslated mRNAs and drove de novo synthesis of target proteins. As revealed by western
blot analysis, we show de novo synthesis and significant increase in expression levels of
CLEC7A (p=0.0293), CST7 (p=0.0099) and LILRB4a (p=0.0231) and ITGAX (p=0.0098).

114
Of note, TLR2 is not directly regulated by SRSF3, it is used here as an additional control and
functional readout of innate immune response; TLR2 (p=0.0175).
Finally, to assess the effects of SRSF3 knockdown on the phagocytic potential of disease
associated microglia, we used primary microglia cell culture system derived from the
symptomatic SOD1G93A mouse lumbar spinal cord (Díaz-Amarilla et al., 2011). We used
phagocytic assay based on the engulfment of fluorescent latex beads (Lian et al., 2016). As
shown in figure 5F, treatment with anti SRSF3 AMOs significantly increased phagocytic
function of ALS microglia as compared to scrambled treated cells or untreated cells (Figure
5F). Indeed, the mean number of fluorescent beads engulfed was higher in cells treated
AMOs than the number beads found in cells treated with a scramble morpholino (p<0.0001)
or in control cells (p<0.0001). No significant difference was found in the number of beads in
scrambled treated vs untreated cells (p=0.2972). Along with the rescue of the phagocytic
activity, treatment with AMOs prevented increase in caspase-8 immunoreactivity suggesting
its protective potential for ALS microglia (Diaz-Amarilla et al., 2011, Trias et al., 2013)
(Figure 5G).
Taken together our results suggest that targeting SRSF3 has a marked disease modifying
effect in ALS.
3.5. Discussion
To date, it has been widely established that microglial cells actively contribute to disease
progression and motor neuron degeneration in ALS (Robberecht and Philips, 2013, Beers
and Appel, 2019, Béland et al., 2020). A current view is that over the course of disease,
microglia change their phenotypes from initially beneficial toward highly toxic and aberrant
cells resistant to any therapeutic interventions (Keller et al., 2011, Patel et al., 2015). Yet, the
molecular mechanisms involved in this apparent shift in microglia toxicity remain elusive.
We recently discovered a novel check-point mechanism mediated by RNA binding protein
SRSF3 that directly controls translation of selected and highly regulated immune mRNAs in
activated microglia (Boutej et al., 2017, Kim et al., 2014). A selective translational repression

115
of highly regulated LPS-induces mRNAs in acutely activated microglia resulted in formation
of two distinct microglia molecular signatures: a highly specialized immune and pro-
inflammatory mRNA signature and a more immune-modulatory homeostatic protein
signature. As SRSF3 rather selectively arrests translation of highly regulated immune
transcripts here it is important to ask the question: what is a potential biological significance
of this ribosome-based checkpoint control mechanisms? While in acute inflammation
supressing an exacerbated and/or unwanted immune response could be protective and
prevent tissue damage, a prolonged deregulation of immunity may be detrimental to the host
and lead to a chronic disease. There is increasing evidence suggesting that a chronic
deregulation of innate immunity may represent one of the key elements in the pathobiology
of neurodegenerative disorders including ALS.
In the current study we provide important in vivo evidence of the role of SRSF3 in the
pathogenic transformation of microglia in ALS. We show that SRSF3-mediated translation
repression of immune transcripts leads to a chronic dissociation of mRNA and protein
networks in ALS microglia and resulted in two distinct molecular signatures: i) mRNA
signature highly enriched in immune functions and ii) protein signature with enrichment in
biological functions related to RNA metabolism and stress (Figures 2 and 3). As showed
previously (Boutej et al., 2017), neuroinflammatory conditions increase the SRSF3/pSRSF3
ratio and expression levels of active pSRSF3. Here we report that: i) pSRSF3 level in the
lumbar spinal cord of SOD1 mutant mice are significantly increased in symptomatic disease
ii) the localization of pSRSF3 is restricted to CD11b/Gal-3 positive activated microglia and
it is present in rodent models as well as in human sporadic ALS, iv) targeted knockdown of
SRSF3/pSRSF3 by using antisense morpholino strategy alleviates translational suppression
of immune transcripts, induces de novo synthesis of selected proteins and restores microglia
phagocytic capacity. Finally, IT delivery of AMOs slowed down disease progression and
significantly extended survival of ALS mice, suggesting that targeting SRSF3 may represent
a valid therapeutic strategy in ALS (see schematic presentation of our model in Figure 6).
SRSF3 belongs to the Serine-arginine-rich (SR) proteins family that contains several RNA
binding proteins with a functional implication in RNA metabolism (Manley and Krainer,

116
2010). Like other SR proteins, SRSF3 is involved in alternative splicing events, however,
recent reports emphasize its role in the mechanisms involved in post-transcriptional
regulation, such as mRNA export, surveillance, stability and translation (Kim et al., 2014).
SRSF3 is predominately nuclear protein where it has a role in RNA splicing and maturation
(Ajiro et al., 2016), SRSF3, upon its phosphorylation, migrates to the cytoplasm where it can
bind the 3’UTR region of ribosome-bound mRNAs. To date, disruption of its biological
functions has been mostly associated with cancer (Zhou et al., 2020). For example, increased
levels of SRSF3 have been associated with increased cellular proliferation and cancer
aggressivity/progression (Shepard and Hertel, 2009, Corbo et al., 2013, Tang et al., 2013,
Fuentes-Fayos et al., 2020).
At the present, the role of SRSF3 in acute and/or chronic neurodegeneration is not yet well
characterized. Based on our previous work and exciting findings we propose that by targeting
SRSF3 we can actually modulate and reprogram microglia proteome. As shown here (see
Figure 5), this can rescue microglia phagocytic and immune function and exerts disease
modifying effect in ALS. In our proof–of concept experiments we analyzed effects SRSF3
on translational regulation of the several key microglia transcripts including Clec7a, Cst7,
Lirlb4a etc. Increase in expression of these transcripts in microglial cells has been linked with
disease associated phenotypes in neurodegenerative disorders and aging however this cluster
of genes have not been analyzed at protein level (Deczkowska et al., 2018, Kang et al., 2018,
Keren-Shaul et al., 2017, Krasemann et al., 2017, Weissmann et al., 2016).
Taken together, our approach differs from previous attempts to target microglia or
neuroinflammation as it does not aim to reduce microglial activity, but rather to restore
protective functions from this cell type. Therefore, based on our results, we propose that
targeting SRSF3 may open new avenue in therapeutic reprogramming of immune response
in ALS and potentially other neurodegenerative disorders.

117
3.6. Acknowledgments
We thank Florence Roux-Dalvai, Proteomics Platform; Annick Ouellet, Gene Expression
Analysis Platform; and Geneviève Soucy, Yuan Cheng-Weng, and Karine Plourde for their
technical help. This work was supported by Canadian Institutes of Health Research (CIHR
116638) and the Amyotrophic Lateral Sclerosis Society of Canada.
3.7. Author Contributions
L.C.B. designed and performed the experiments, acquired and analysed the data and wrote
the manuscript. H.B. designed and performed the experiments, acquired and analysed the
data and wrote the manuscript. R.B. performed the phagocytosis assay. E.T. performed the
rat lumbar spinal immunofluorescence and produced the photographs. S.S.T. performed the
ELISA. C.J.B. and A.D. performed the bioinformatic analysis. V.P.M. and N.D. provided the
human tissues. L.B. designed the study. J.K. designed the study and wrote the manuscript.
3.8. Declaration of interests
The authors declare no competing interests.
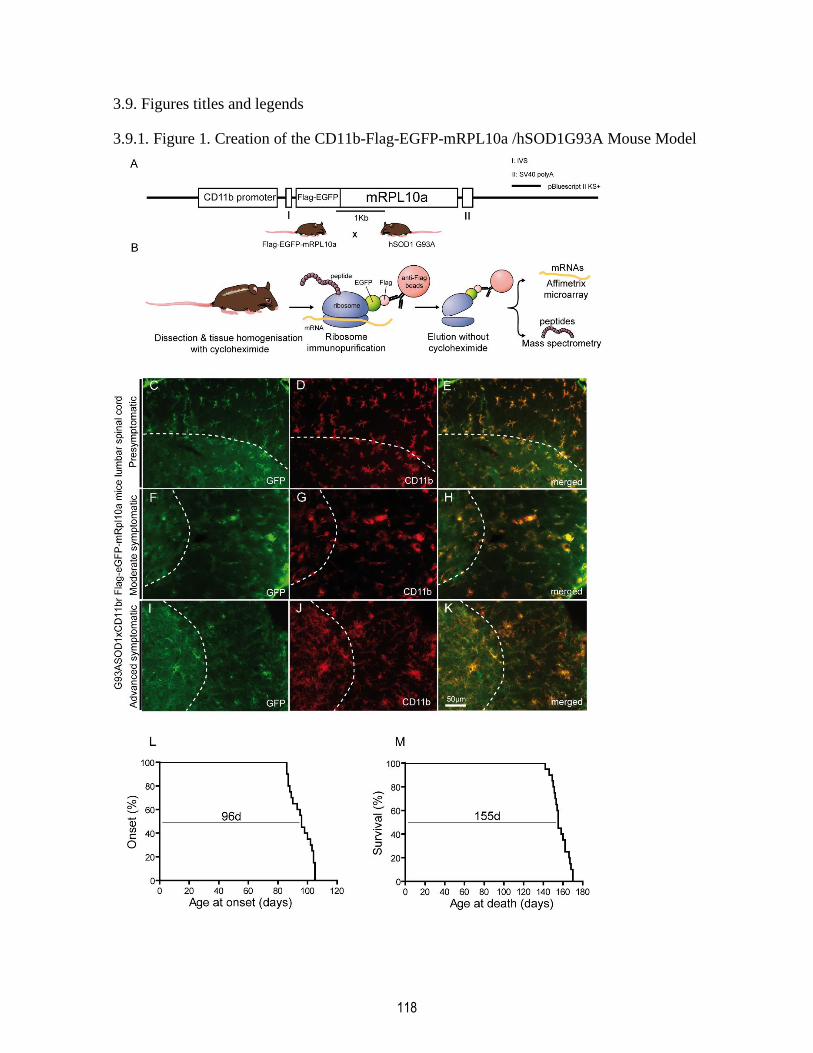
118
3.9. Figures titles and legends
3.9.1. Figure 1. Creation of the CD11b-Flag-EGFP-mRPL10a /hSOD1G93A Mouse Model

119
Diagram of the transgene microinjected in C57BL/6 mouse line. CD11b promoter drives the
expression of Flag-EGFP-mRpl10a in cells of monocytic lineage (A). Diagram of the
ribosome immunopurification protocol (B). Mice lumbar spinal cord are isolated and
processed in cycloheximide-containing lysis buffer. Ribosomes are pulled down with anti-
Flag beads. mRNAs and peptides are eluted and identified with microarray or mass
spectrometry. (C-K) Colocalization of the fusion protein Flag-EGFP-mRpl10a marked with
GFP antibody and with monocyte marker CD11b in mouse lumbar spinal cord at different
disease stages (presymptomatic C-E, early symptomatic F-H, late symptomatic I-K). G93A
SOD1 x Cd11b-Flag-EGFP-mRPL10a mouse model has a median onset of 96 days (L) and
a median survival of 155 days (M) (n=20).

120
3.9.2. Figure 2. Chronic Dissociation of mRNA and Protein Molecular Signatures in ALS
Microglia

121
Volcano plots of the mRNAs found in the transcriptomic analysis of mouse microglia from
presymptomatic stage (A), moderate symptomatic stage (B) and advanced symptomatic stage
(C). Gray dots represent non significantly regulated mRNAs (p<0.05). Black dots represent
significantly down-regulated mRNAs (fold change<0.5), blue dots, not regulated mRNAs
(0.5<fold change<2) and orange dots, significantly up-regulated mRNAs (fold change>2).
Tagged are the top regulated mRNAs for each time point. Volcano plots of the proteins found
in the proteomic analysis of mouse microglia from presymptomatic stage (D), moderate
symptomatic stage (E) and advanced symptomatic stage (F). Gray dots represent non
significantly regulated proteins (p<0.05). Black dots represent significantly down-regulated
proteins (fold change<0.83), blue dots, not regulated proteins (0.83<fold change<1,2) and
orange dots, up-regulated proteins (fold change<1,2). Tagged are the top regulated proteins
for each time point. ClueGO-generated top biological functions from the top 50 up-regulated
mRNAs at the presymptomatic stage (G), moderate symptomatic stage (H) and advanced
symptomatic stage (I). The name of the biological functions is given with the number of
mRNAs implicated in each function. ClueGO-generated top biological functions from the
top 50 up-regulated proteins at the presymptomatic stage (J), moderate symptomatic stage
(K) and advanced symptomatic stage (L).

122
3.9.3. Figure 3. Highly Upregulated Transcripts in ALS Microglia Are Translationally
Arrested by SRSF3

123
Validation of transcriptomic data through immunoblotting. The selected genes are Clecl7a
(A), Lilrb4 (B), Cst7 (C), Cd68 (D), Ccl3 (E), and Gpnmb (F). Translationally arrested genes
(n=12) have a higher SRSF3 binding site density than genes that were not translationally
arrested (n=10) (G). SRSF3 effectively binds mRNA from the immune genes Clec7a, Cst7
and Lilrb4 (H). Graph representing the number of SRSF3 binding sites on 3’UTR in function
of that 3’UTR length, where every dot represents a 3’UTR from a different gene (I). SRSF3
preferentially binds shorter 3’UTR (J). (Data is represented as mean ± SEM * p<0.05,
**p<0.01).
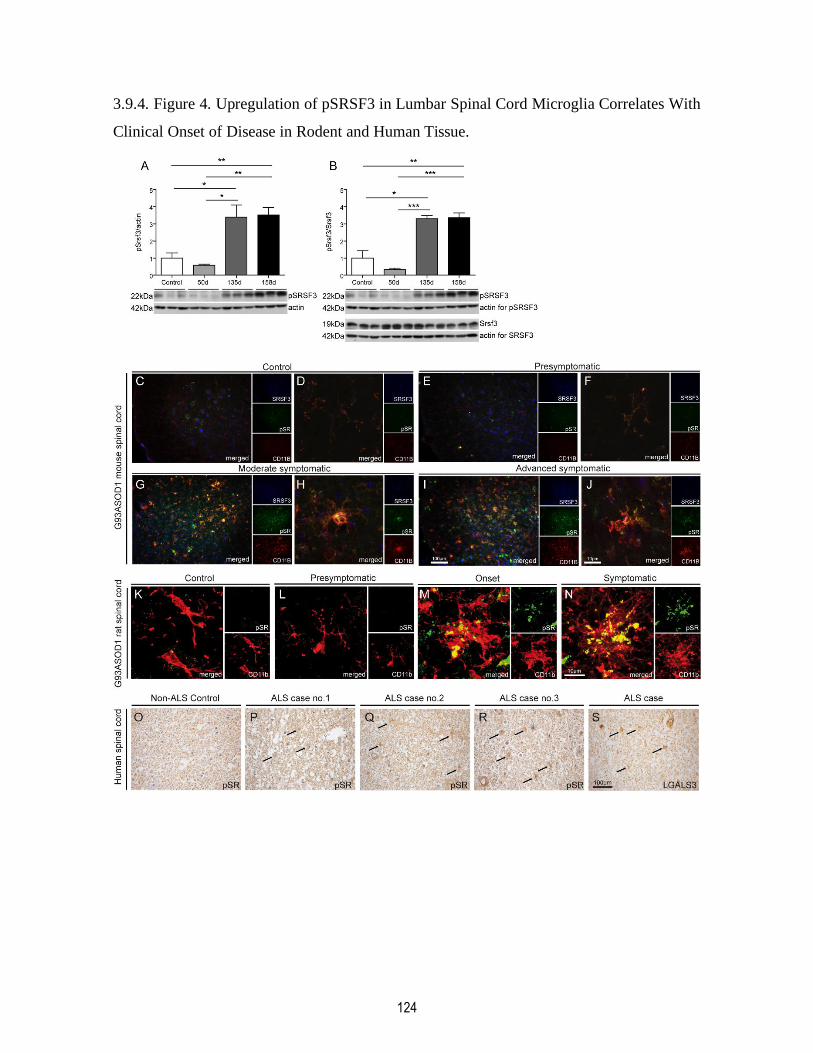
124
3.9.4. Figure 4. Upregulation of pSRSF3 in Lumbar Spinal Cord Microglia Correlates With
Clinical Onset of Disease in Rodent and Human Tissue.

125
Western blots with WT control, presymptomatic stage (SOD1 50 days old), moderate
symptomatic stage (SOD1 135 days old) and advanced symptomatic stage (158 days old),
mouse lumbar spinal cord lysates for (A) pSRSF3 or for (B) the ratio of pSRSF3/SRSF3
(Data is represented as mean ± SEM *p<0.05, **p<0.01, ***p<0.001). (C-J) Representative
microphotographs of mouse lumbar spinal cords from WT control mouse (C-D),
presymptomatic stage (E-F), moderate symptomatic stage (G-H), advanced symptomatic
stage (I-J) where SRSF3 is in blue, pSR in green and CD11b in red. Representative
microphotographs of immunofluorescence done on rat spinal cord from WT control (K),
presymptomatic stage (L), onset (M) and symptomatic stage (N) where pSR is in green and
CD11B in red. Representative microphotographs of DAB/immunochemistry against pSR
done in Non-ALS control spinal cord paraffin embedded sections (Q) and ALS patient spinal
cord paraffin embedded sections (P-R). DAB/immunochemistry against the microglial
activation marker Mac2/Galectin-3 done on ALS patients spinal cord paraffin embedded
section (S). The pSR positive cells share the morphology of activated microglial cells.
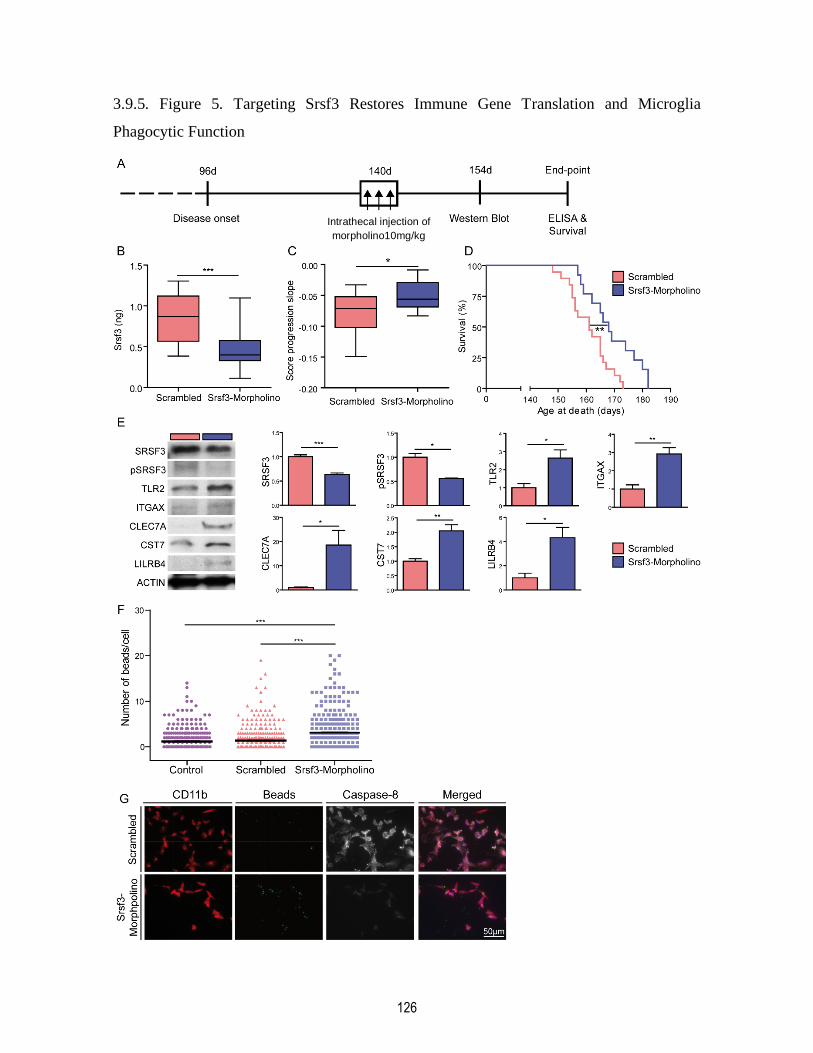
126
3.9.5. Figure 5. Targeting Srsf3 Restores Immune Gene Translation and Microglia
Phagocytic Function
Intrathecal injection of
morpholino10mg/kg

127
Srsf3-morpholino treatment protocol (A) At end-stage, SRSF3 knockdown at protein level is
measured by ELISA (n=21 for scrambled morpholino treated animals and n=23 for
morpholino-treated animals) (B). SRSF3 knockdown is accompanied by a slower score loss
(C) and an extended survival (D). Two weeks after the start of the treatment, we already
observed the knockdown of SRSF3 and reduced levels of its phosphorylated form. This is
accompanied by the de novo translation of the previously suppressed genes: TLR2, ITGAX,
CLEC7A, CST7 and LILRB4a (E) Phagocytosis assay on control, scrambled morpholino
treated or Srsf3 morpholino treated ALS adult microglial cells cultures (F). Representative
microphotographs of immunofluorescence done on scrambled- and Srsf3 morpholino treated
mice where the CD11b is in red, fluorescent latex beads in green, Caspase 8 in gray (G).
(Data is represented as mean ± SEM *p<0.05, **p<0.01, ***p<0.001)

128
3.9.6. Figure 6. Proposed Mechanisms of Microglial Rescue After Srsf3 Knockdown
(A) At the symptomatic stage of ALS, microglial cells have high levels of phosphorylated
SRSF3 in their cytoplasm. SRSF3, by binding the 3’UTR of upregulated immune transcripts
leading the ribosomal stalling and translational arrest. This leads to loss of normal microglial
phagocytic properties and immune functions, which would be found in acutely activated
microglia. Loss of those functions causes an exacerbation of the symptoms and a shorter
survival. (B) Upon Srsf3 knockdown using a Srsf3 morpholino intrathecally injected, we
were able to reduce significantly SRSF3 and its phosphorylated SRSF3 form. The effect of
pSRSF3 on translation is reduced and immune transcripts can now be synthesized to a higher
level. The phagocytic and immune functions of those genes were rescued and that lead to a
slower disease progression and an extended survival in mice.

129
3.10. Tables titles and legends
3.10.1. Table 1. Top regulated transcripts
Gene
Symbol
Gene Name Fold
Change
p-value Detected
as a
peptide
Pre
sym
pto
mati
c
Cdc42 Cell division control protein 42 homolog 2,10 0,014 x
Rfwd3 E3 ubiquitin-protein ligase RFWD3 2,05 0,009 x
Trim30d Tripartite motif-containing 30D 1,95 0,033 x
Ctnna1 Catenin beta-1 1,80 0,026 x
Pdgfra Platelet-derived growth factor receptor alpha 1,74 0,033 x
Adam10 Disintegrin and metalloproteinase domain-containing
protein 10
1,71 0,049 x
Spin1 Spindlin-1 1,70 0,018 √
Rnf125 E3 ubiquitin-protein ligase RNF125 1,67 0,033 x
Gm17655 Uncharacterized protein 1,66 0,048 x
Oas1g 2'-5' oligoadenylate synthetase 1G 1,66 0,022 x
Mo
der
ate
sy
mp
tom
ati
c
Lilrb4 Leukocyte immunoglobulin-like receptor subfamily B
member 4
11,55 0,020 x
Ccl3 C-C motif chemokine 3 10,32 0,010 x
Ms4a6c Membrane-spanning 4-domains subfamily A member
6C
8,81 0,006 x
Eif4a2 Eukaryotic initiation factor 4A-II 8,73 0,038 x
Cd48 CD48 antigen 8,01 0,034 x
Gpnmb Transmembrane glycoprotein NMB 7,66 0,021 √
Cd68 Macrosialin 6,96 0,025 x
Serpina3n Serine protease inhibitor A3N 6,61 0,023 x
Ms4a7 Membrane-spanning 4-domains, subfamily A, member
7
5,85 0,029 x
Atp6v0d2 ATPase, H+ transporting, lysosomal V0 subunit D2 5,25 0,006 x
Ad
van
ced
sym
pto
ma
tic
Clec7a C-type lectin domain family 7 member A 38,04 0,025 x
Ch25h Cholesterol 25-hydroxylase 18,26 0,002 x
Lilrb4 Leukocyte immunoglobulin-like receptor subfamily B
member 4
16,57 0,000 x
Gpnmb Transmembrane glycoprotein NMB 16,52 0,001 √
Cst7 Cystatin-F 14,16 0,010 x
Ctla2b Protein CTLA-2-beta 13,20 0,018 x
Cd68 Macrosialin 12,81 0,000 x
Eif4a2 Eukaryotic initiation factor 4A-II 12,41 0,024 x
Bcl2a1d B cell leukemia/lymphoma 2 related protein A1d 12,15 0,017 x
Itgax Integrin alpha X 11,91 0,009 x
The table presents the top 10 most upregulated genes found in the transcriptomic analysis in
the moderate symptomatic and advanced symptomatic stages of the disease with their gene
symbol, full name, fold change and associated p-value

130
3.11. STAR Methods
3.11.1 Generation and characterization of CD11b Flag-EGFP-RPL10a x G93A SOD1 mouse
As previously described (Boutej et al. 2017), CD11b promoter was amplified by PCR from
the pBluescriptSK-CD11b-TKmt-30 (Gowing et al. 2008) with the 5’ primer 5’-
GGGACGACGGGCCCCTCGAGGGGTTCAAGTGATTCTGCTGC-3’ and the 3’
primer:5’-GGGACGACGTCGACAGAACCTGGAAGGAGGTGAACC-3’. The mouse
gene coding for the 60S ribosomal protein 10a (Rpl10a) was amplified from mouse cDNA
with the 5’ primer 5'-GGGACGACGGATCCAGGTAAGTTGTTATCGTGGC-3’ and the
3’primer 5’-GGGACGACGCGGCCGCCTAATACAGACGCTGGGGCT-3’ while
generating BamH1/Not1 restriction site. Flag-EGFP was obtained from pEGFP-N3 plasmid
(Clonetech) and amplified with the 5’ primer 5’-
GGGACGACCTGCAGGAGGCAGCATGGACTACAAAGACGACGACGACAAGGTG
AGCAAGGGCGAGGA-3’ and 3' primer 3’-GGGACGA
CGGATCCCTTGTACAGCTCGTCCATGC-3’. All fragments were sequentially subcloned
into pBluescript KS+. The final vector was sequenced to verify its integrity.
From the previously detailed plasmid, a 5.2 kB DNA fragment was microinjected in C57BL/6
zygotes. The resulting CD11b-Flag-EGFP-RPL10a (CD11brGFP) transgenic mouse was
crossed with the human G93A SOD1 expressing mouse (Keller et al. 2009) that will develop
amyotrophic lateral sclerosis in a manner similar to what we find in human, i.e. motor neuron
loss, gliosis and motor deficit. The mice were kept heterozygous for both transgenes.
Presence of the Flag-EGFP-RPL10a and the hSOD1 G93A transgenes were assessed through
PCR.
Onset, disease progression, body weight loss and survival were monitored according to
Université Laval CPAUL protocol 2014-096-2. Onset was determined with the apparition of
hind limb weakness and twitching, along with the inability of the animal to correctly grasp
the cage with one hind leg. To score disease progression, we considered the animal gait, hind
limbs weakness and loss of extension. Animals were sacrificed if they reached any of the two
ALS endpoints: a loss of 30% maximum body weight or a 30s immobility.

131
3.11.2. EDTA-TRAP from Flag-EGFP-RPL10a x hSOD G93A mice.
To isolate microglial mRNAs and peptides from ALS mouse lumbar spinal cords, we used
previously described protocol (Heiman et al. 2008, Boutej et al. 2017). ALS mice of 50d,
135d, and 158d of age and their age matched controls were first anesthetized as described as
above. Lumbar spinal cords of 3 mice were rapidly dissected and pooled in dissection buffer
(1X HBSS, 2,5mM HEPES KOH pH7,4 35mM glucose, 4mM NaHCO3, 100µg/mL
cycloheximide and 1tab/10mL of EDTA free protease inhibitor cocktail). Pooled spinal cords
are then lysed with a motorized homogenizer in lysis buffer (20mM HEPES KOH pH7,4,
150nM KCl, 12mM MgCl2, 0,5mM DDT, 10U/mL RNasine, 10U/mL Superasine,
100mg/mL cycloheximide and 1tab/10mL of EDTA free protease inhibitor cocktail). Lysate
were centrifuged at 4ºC, 2000g for 10min. 1/9 volume of NP-40 10% Rnase free and 1/9
volume of DHPC (Avanti Polar Lipids) were added to the supernatant and incubated at 4ºC
for 30min on a shaker. After a centrifugation at 4ºC, 20000g for 10min, anti-Flag M2 agarose
beads (Sigma) were added to the supernatant and incubated overnight at 4ºC on a shaker.
Beads were collected by centrifugation and washed three times in high salt washing buffer
(20mM HEPES KOH pH7,4, 165mM KCl, 12mM MgCl2, 1% V/V Np-40 RNase free,
0,5mM DDT, 10µg/mL cycloheximide 1tab/10mL of EDTA free protease inhibitor
cocktail).After the last centrifugation, beads pellet can be used to isolate either mRNAs or
in-translation peptides.
3.11.3. Immunofluorescence
Correct expression of the Flag-EGFP-RPL10a fusion protein in the microglia at different
stages of the disease was checked in immunofluorescence. At 50d (presymptomatic stage),
135d (moderate symptomatic), and 158d (advanced symptomatic), mice were anesthetized
with 10mg/mL ketamine + 1mg/mL xylazine. Mice were then transcardially perfused with
60mL of PBS and 60mL of 4% phosphate buffered paraformaldehyde (PFA). Spinal cords
were dissected, post-fixed 24hrs in PFA and kept in 30% phosphate buffered sucrose. Lumbar
spinal cords were cut at 25 microns with a Leica SM200R microtome and kept in an
antifreeze solution. Sections were first blocked in PBS+0.25% Triton+10%Goat Serum GS.

132
They were then incubated overnight at room temperature with primary antibodies 1:750 rat
monoclonal anti-CD11b (Serotec) and 1:500 mouse monoclonal anti-GFP (Abcam). After
washes in PBS+0.25% Triton, the sections were incubated with their corresponding
fluorescent goat secondary antibodies.
Spinal cords from mice and rats were isolated, fixed and cut as described above. Sections
were blocked in PBS + 10% Goat Serum for 1hr and then incubated overnight in primary
antibodies: CD11b 1:500 (Serotec), 1:500 pSr (Millipore) and 1:500 Srsf3 (Abcam). Sections
were washed and incubated one 1hr in their respective secondary antibody.
3.11.4. mRNA isolation
Microglial mRNAs were isolated using the protocol and reagents from the Absolutely RNA
Nanoprep kit (Agilent). At RT, washed beads were incubated 10 min with 100µL of Lysis
Buffer + 0.7µL of β mercaptoethanol. Beads were then centrifuged at 6000rpm 15min to
collect the supernatant. Supernatent + 1V of sulfolane 80% (SigmaAldrich) was put in
centrifugation columns and spun at 12000g 1min and washed with Low Salt Buffer. RNAs
trapped in the column underwent a DNase treatment and were washed subsequently in High
Salt Buffer and Low Salt Buffer. Finally, mRNAs were eluted in 15µL Elution buffer and
kept à -80ºC
3.11.5. Affymetrix microarray
DNA microarray was performed by the Plateforme d’Expression Génique team in CHUL,
Quebec City, Canada. Before the microarray, RNA quality and quantity were measured with
a Agilent BioAnalyser (Agilent Technologies) and a ND-1000 Spectrophotometer
(NanoDrop Technologies) respectively. cDNA from the previously isolated microglial
mRNAs using cRNA hybridization cocktail (Affmetrix) following Affymetrix Gene Chip
WT cDNA Synthesis and Amplification Kit protocol. After washes, cDNA was stained and
scanned with the Affymetrix GCS 3000 7G. Resulting data was analyzed on Affymetrix
Expression Console.

133
To evaluate the regulation levels between ALS and control mice lumbar spinal cord
microglial transcripts, we used Transcriptome Analysis Console (Affymetrix). A transcript
was considered to be regulated if the absolute fold change value between ALS and control
samples was equal or higher than 2 and its p-value, lower than 0.05.
3.11.6. Peptide isolation
Microglial peptide was isolated from the anti-Flag beads from the previous section. Beads
pellet was resuspended in elution buffer (10mM HEPES KOH pH7.4, 150mM KCl, 5mM
MgCl2, 20mM EDTA and 1tab/10mL of EDTA free protease inhibitor cocktail) and
incubated at RT for 30min on a shaker. Eluate was collected after a centrifugation at RT,
7000rpm for 15min and kept at -80ºC.
3.11.7. Label-free quantification
Label free quantification by mass spectrometry and following analysis was performed by the
Plateforme de Protéomique team in CHUL. As described by Boutej et al. 2017, each sample
was injected three times (1000ng each time) in a Dionex UltiMate 3000 nanoRSLC
chromatography system (Thermo Fischer Scientific) Proteins could be quantified when found
in at least two of the three injections. Missing quantification values were replaced by the
mean of the two others replicates. If a protein was found in ALS samples but not in control
samples or vice versa, missing quantification values were replaced by the noise of
corresponding sample injection. Student test was used to evaluate if quantifications values
were significantly different between ALS samples and control samples. A protein was
considered to be regulated if the absolute fold change value between disease and control was
equal or than 1.2 and its p-value, lower than 0.05.
3.11.8. EDTA-TRAP data analysis
Volcano plots were generated by plotting all microarray data (for transcriptomic) or all label-
free quantication data (for proteomic) on the Volcano Plot application of the Origin 2018b
program (OriginLab). To generate the top 10 functions on each time point from

134
transcriptomic data and proteomic data, the 50 upregulated genes were plotted on the
application ClueGO (Bindea et al. 2009) of the Cytoscape program (Shannon et al. 2003).
using Mus Musculus ClueGO ontologies (GO Biological Process, GO Immune System
Process, GO Molecular Function, Reactome, Kyoto Encyclopedia of Genes and Genomes
(KEGG) and Wikipathways). GO term fusion was used to reduce redundancy. GO Tree
interval was set between 3 and 6. Kappa score was kept at default value. Clusters could be
generated when one gene was present (parameter: All genes or 0%). Two-sided
hypergeometric test (Enrichement/Depletion), Bonferroni adjustment and statistical
significance of less than 0.05 was used for the enrichement of terms and groups.
3.11.9. Western Blot
Mice were anesthetized as described above, perfused with cold phosphate buffered saline
(PBS) and their lumbar spinal cord were isolated. Spinal cords were lysed in Urea Lysis
Buffer (6M Urea, 1% SDS and 20mM Tris HCl pH6,8) and sonicated using 250/450 Sonifer
(Branson Ultrasonic) 10sec at power 3 and duty cycle set to 50% while kept on ice. Proteins
were quantified with Bradford colorimetric assay. 15µg of protein from 3 ALS mouse spinal
cords and 3 control littermate mouse spinal cords were resolved on SDS-PAGE and then
transferred overnight at 4ºC to an Amersham Protean 0,45µm NC nitrocellulose blotting
membrane (GE Healthcare Life Sciences). The membrane was blocked 1hr in PBS + 0,2%
Tween 20 + 5% powdered milk and incubated overnight in the recommended concentration
of primary antibody: Ccl3 1:2000 (Abcam), Cd68 1:1000 (Millipore), Dectin1 (Clec7a)
1:2500 (Abcam), Cst7 1:1000 (MyBioSource), C1qc 1:1000 (MyBiosource), Gfap 1:1000
(Sigma), Got1 1:2000 (Abcam), Gpnmb 1:500 (ThermoFischer), Hspb1 1:2000 (Cell
Signaling), Lgals3 1:3000 (Abcam), Lilrb4 1:1000 (MyBioSource), Mtpn 1:1000 (Bethyl
Laboratories), Mvp 1:1000 (Invitrogen), pSR 1:500 (Millipore), Tgm1 1:200 ( Abcam), Sod1
1:1000 (Enzo Biochem) and Srsf3 1:1000 (Abcam). After 3 washes in PBS + 0.2% Tween
20, the membrane was incubated in the corresponding peroxidase secondary antibody. The
membrane was washed again and expose to a Biomax Light Film (Sigma Aldrich) after being
treated with Pierce ECL Western Blotting Substrate (Thermo Scientific). For statistical

135
analysis, each condition was made in triplicates. Analysis was perfomed using GraphPad
Prism version 5.00.
3.11.10. Cross-linked immunoprecipitation
BV2 cells (Blasi et al. 1990) were transfected with a SRSF3-cMyc pCMV6-Entry,
mammalian vector with C-terminal Myc-DDK tag plasmid (Origene) and cultured in 15 cm
petri dishes with DMEM + 10%FBS + 0,5% sodium pyruvate + Pen/Strep. Upon confluence,
cells were treated with 1µg/mL of LPS for 4 hours to induce SRSF3 cytoplasmic localisation.
Cells were then washed in PBS and cross-linked using the UV Stratalinker 1800 (Stratagene)
at 15 000µJ. Cross-linked cells are harvested form the petri dish and the PBS supernatant is
removed. Cells are lysed in NT2 + 0.05% NP-40 lysis buffer (50mM Tris pH7.4, 150mM
NaCl, 1mM MgCl2, 0,05% NP-40, 1tab/10mL of protease inhibitor cocktail, 10U/mL
RNasine, 10U/mL Superasine, prepared in H2O DEPC) and processed first by 20 up & downs
in a 200µL tip, then by 10 up & downs in a 27G needle and finally incubated for 30min at
4°C with rotation. Lysate is centrifuged at 14000rpm for 10 min at 4°C and supernatant is
kept and quantified. 40µL/sample of Myc trap magnetic agarose beads and Binding control
magnetic agarose beads (Chromotek) are washed in PBS and blocked 1 hour in NT2 + 0,05%
NP-40 + 5% BSA. After blocking, beads are washed in NT2 + 0,05% NP-40. 1600µg of cell
lysate (volume total is completed to 500µL) is incubated O/N with the beads at 4°C with
rotation. After incubation, supernatant is removed, and the beads are washed in NT2 + 0.05%
NP-40. Beads are then incubated in 150µL of PK buffer (100mM Tris pH7.4, 50mM NaCl,
10mM EDTA) and 10µL of PK 10mg/mL for 15min at 56°C. Protease reaction is stopped at
65°C for 5 minutes. Supernatant is secured in Trizol and sent to Plateforme d’Expression
Génique team in CHUL, Quebec City, Canada for qPCR analysis of genes of interest.
3.11.11. qRT-PCR
Cells were homogenized in Qiazol buffer (Qiagen, Germantown, MD, USA) and total RNA
was extracted using the miRNeasy micro kit on-column DNase (Qiagen, Hilden, DE)
treatment following the manufacturer’s instructions. Quantity of total RNA was measured
using a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Wilmington, DE,

136
USA) and total RNA quality was assayed on an Agilent BioAnalyzer 2100 (Agilent
Technologies, Santa Clara, CA, USA). First-strand cDNA synthesis was accomplished using
0.08 to 2 ug of isolated RNA in a reaction containing 200 U of Superscript IV Rnase H-RT
(Invitrogen Life Technologies, Burlington, ON, CA), 300 ng of oligo-dT24, 50 ng of random
hexamers, 50 mM Tris-HCl pH 8.3, 75 mM KCl, 3 mM MgCl2, 500 uM deoxynucleotides
triphosphate, 5 mM dithiothreitol, and 40 U of Protector RNase inhibitor (Roche Diagnostics,
Indianapolis, IN, USA) in a final volume of 50 ul. Reaction was incubated at 25°C for 10
min, then at 50°C for 20 min, inactived at 80°C for 10 min. PCR purification kit (Qiagen,
Hilden, DE) was used to purify cDNA.Oligoprimer pairs were designed by GeneTool 2.0
software (Biotools Inc, Edmonton, AB, CA) and their specificity was verified by blast in the
GenBank database. The synthesis was performed by IDT (Integrated DNA Technology,
Coralville, IA, USA). cDNA corresponding to 5-20 ng of total RNA was used to perform
fluorescent-based Realtime PCR quantification using the LightCycler 480 (Roche
Diagnostics, Mannheim, DE). Reagent LightCycler 480 SYBRGreen I Master (Roche
Diagnostics, Indianapolis, IN, USA) was used as described by the manufacturer. The
conditions for PCR reactions were: 45 cycles, denaturation at 98°C for 10 sec, annealing at
58°C for 10 sec, elongation at 72°C for 14 sec and then 74°C for 5 sec (reading). A melting
curve was performed to assess non-specific signal. Relative quantity was calculated using
second derivative method and by applying the delta Ct method [1]. Normalization was
performed using the reference gene shown to be genes having stable expression levels from
embryonic life through adulthood in various tissues [2]: hypoxanthine guanine
phosphoribosyl transferase 1 (HPRT1) and glyceraldehyde-3-phosphate dehydrogenase
(GAPDH). Quantitative Real-Time PCR measurements were performed by the CHU de
Québec Research Center (CHUL) Gene Expression Platform, Quebec, Canada and were
compliant with MIQE guidelines.
3.11.12. 3’UTR analysis
For this analysis we used upregulated mRNAs obtained from different EDTA-TRAP
experiments. Different contexts were used to increase the n have a significant result. The 3'
UTR sequences from selected genes (4079 3’UTR sequences from 1642 genes) were

137
extracted using the TxDb.Mmusculus.UCSC.mm10.knownGene v3.10.0,
BSgenome.Mmusculus.UCSC.mm10 v1.4.0 and org.Mm.eg.db v3.11.4 packages in R
v4.0.0. Sequences were processed using the RBPmap web tool on August 8 2020
(http://rbpmap.technion.ac.il/) using the Mouse genome and all the Human / Mouse motifs.
Graphs were produced in R v4.0.0 using the ggplot2 v3.3.2. (Paz et al. 2014, Huber et al.
2015, Boutej et al. 2017)
3.11.13. Human sections immunochemistry
3 ALS patient and and 1 control spinal cords (fixed and mounted on microscope slides) were
obtained from Centre hospitalier affilié universitaire de Québec. Sections were first
deparaffinized and were treated for 15min in 0.6% H2O2. After washes in PBS + 0.25%
Triton X100, sections were blocked in PBS + 0,25% Triton X100 + 10% Goat Serum. They
were then incubated overnight at room temperature in PBS + 0.25% Triton X100 + 1% Goat
Serum + 1:500 pSR antibody. Sections were incubated in PBS + 0.25% Triton X100 + 1%
Goat Serum +1:500 biotylated mouse antibody for 1hr30 and then 1hr in avidin-biotin
complex. Lastly sections were exposed 5 min to DAB before a quick wash and being stained
with hematocyline and dehydrated in xylene.
3.11.14. Phagocytosis assay
A primary culture of microglia from 3 symptomatic mice was prepared as described by Diaz-
Amarilla (Diaz-Amarilla et al. 2011). After 5 days of culture, primary microglial cells were
either left untreated or treated with 5µM of scrambled or Srsf3 morpholino for 24hrs. Cells
then followed a phagocytosis assay (Lian et al. 2016). Briefly, conditioned cell culture
medium was replaced by a culture medium containing 0.2% v/v 1µm fluorescent latex beads
(Sigma-Aldrich) for an incubation time of 1 hour. Cultures were then rinsed and fixed in 4%
PFA. Immunofluorescence was done as described above, against CD11b (Serotec) and
Caspase-8 (Cell Signaling). Representative confocal were acquired on A1r HD confocal
microscope with the NIS Element program (Nikon). Beads in cells from 5 representative
fields of view/animal/treatment were counted and data analysed on GraphPad Prism version
5.00.

138
3.11.15. Morpholino treatment
Animals of 135 days were injected intrathecally with 10µmol of either scrambled or Srsf3
morpholino using a 25µL Hamilton syringe. The animals had three of these injections, spaced
within 10 days. Mice were either sacrificed at the second week or followed for survival using
the same criteria listed above. Score loss progression was measured as the slope value of the
score in function of time over the first 4 weeks. Survival significance was assessed using log-
rank test (Mantel-Cox). Lumbar spinal cords of the animals were dissected and either lysed
in Urea Lysis Buffer for the study of de novo synthesis of proteins following Srsf3-
morpholino treatment or lysed in RIPA Buffer for SRSF3 ELISA.
3.11.16. SRSF3 ELISA
250 ng of lumbar spinal cords lysate from scrambled or Srsf3 morpholino treated was loaded
on the mouse serine/arginine rich splicing factor 3 ELISA kit (MyBiosource). ELISA was
made following provider protocol. Plate was read on EnSpire 2300 Multilabel Reader
(PerkinElmer).

139
3.11.17. STAR Methods References
Bindea, Gabriela, Bernhard Mlecnik, Hubert Hackl, Pornpimol Charoentong, Marie Tosolini,
Amos Kirilovsky, Wolf-Herman Fridman, Franck Pagès, Zlatko Trajanoski, and
Jérôme Galon. 2009. 'ClueGO: a Cytoscape plug-in to decipher functionally grouped
gene ontology and pathway annotation networks', Bioinformatics (Oxford, England),
25: 1091-93.
Blasi, E., R. Barluzzi, V. Bocchini, R. Mazzolla, and F. Bistoni. 1990. 'Immortalization of
murine microglial cells by a v-raf/v-myc carrying retrovirus', J Neuroimmunol, 27:
229-37.
Boutej, H., R. Rahimian, S. S. Thammisetty, L. C. Beland, M. Lalancette-Hebert, and J. Kriz.
2017. 'Diverging mRNA and Protein Networks in Activated Microglia Reveal SRSF3
Suppresses Translation of Highly Upregulated Innate Immune Transcripts', Cell Rep,
21: 3220-33.
Gowing, G., T. Philips, B. Van Wijmeersch, J. N. Audet, M. Dewil, L. Van Den Bosch, A.
D. Billiau, W. Robberecht, and J. P. Julien. 2008. 'Ablation of proliferating microglia
does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by
mutant superoxide dismutase', J Neurosci, 28: 10234-44.
Heiman, M., A. Schaefer, S. Gong, J. D. Peterson, M. Day, K. E. Ramsey, M. Suarez-Farinas,
C. Schwarz, D. A. Stephan, D. J. Surmeier, P. Greengard, and N. Heintz. 2008. 'A
translational profiling approach for the molecular characterization of CNS cell types',
Cell, 135: 738-48.
Huber, Wolfgang, Vincent J. Carey, Robert Gentleman, Simon Anders, Marc Carlson,
Benilton S. Carvalho, Hector Corrada Bravo, Sean Davis, Laurent Gatto, Thomas
Girke, Raphael Gottardo, Florian Hahne, Kasper D. Hansen, Rafael A. Irizarry,
Michael Lawrence, Michael I. Love, James MacDonald, Valerie Obenchain, Andrzej
K. Oleś, Hervé Pagès, Alejandro Reyes, Paul Shannon, Gordon K. Smyth, Dan
Tenenbaum, Levi Waldron, and Martin Morgan. 2015. 'Orchestrating high-
throughput genomic analysis with Bioconductor', Nature methods, 12: 115-21.
Keller, A. F., M. Gravel, and J. Kriz. 2009. 'Live imaging of amyotrophic lateral sclerosis
pathogenesis: disease onset is characterized by marked induction of GFAP in
Schwann cells', Glia, 57: 1130-42.
Lian, H., E. Roy, and H. Zheng. 2016. 'Microglial Phagocytosis Assay', Bio Protoc, 6.
Paz, Inbal, Idit Kosti, Manuel Ares, Jr., Melissa Cline, and Yael Mandel-Gutfreund. 2014.
'RBPmap: a web server for mapping binding sites of RNA-binding proteins', Nucleic
Acids Res, 42: W361-W67.
Shannon, P., A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B.
Schwikowski, and T. Ideker. 2003. 'Cytoscape: a software environment for integrated
models of biomolecular interaction networks', Genome Res, 13: 2498-504.
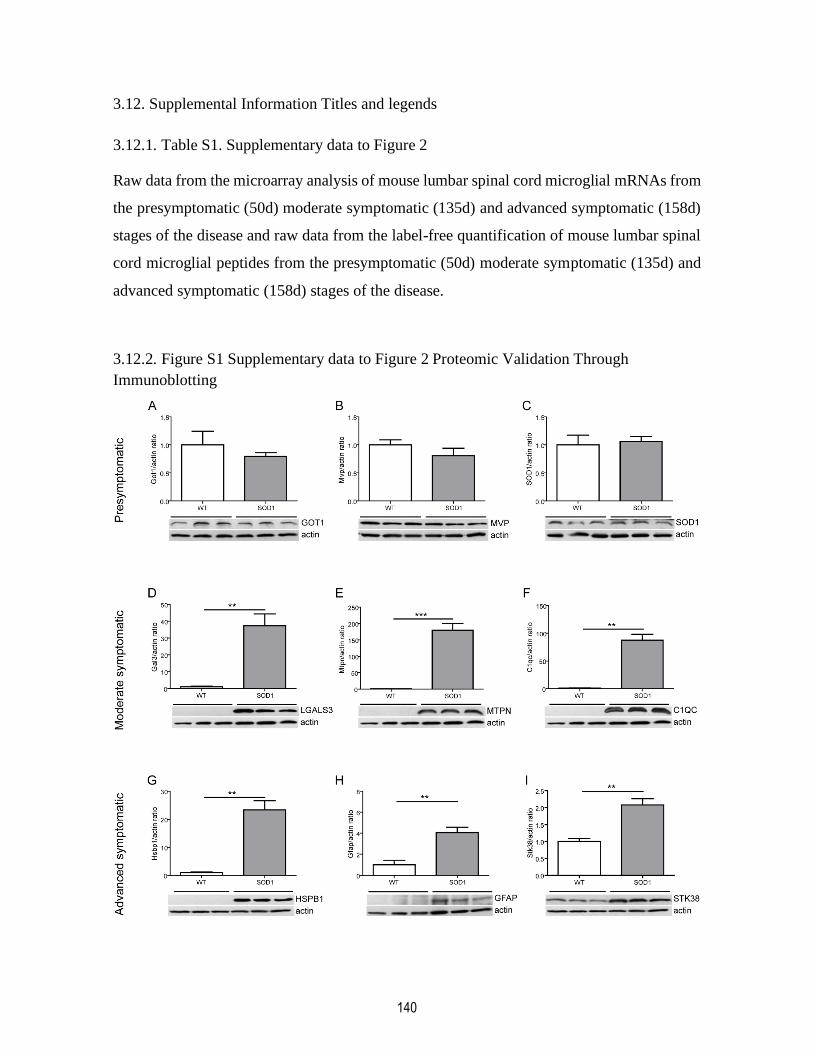
140
3.12. Supplemental Information Titles and legends
3.12.1. Table S1. Supplementary data to Figure 2
Raw data from the microarray analysis of mouse lumbar spinal cord microglial mRNAs from
the presymptomatic (50d) moderate symptomatic (135d) and advanced symptomatic (158d)
stages of the disease and raw data from the label-free quantification of mouse lumbar spinal
cord microglial peptides from the presymptomatic (50d) moderate symptomatic (135d) and
advanced symptomatic (158d) stages of the disease.
3.12.2. Figure S1 Supplementary data to Figure 2 Proteomic Validation Through
Immunoblotting

141
Validation of the presymptomatic stage proteomic data through immunoblotting for the
selected proteins: GOT1 (A), MVP (B) and SOD1 (C). There is no upregulation of any
presymptomatic genes at the protein level as seen in Western Blots. Validation of the
moderate symptomatic stage proteomic data through immunoblotting for the selected
proteins: LGALS3 (D), MTPN (E), and C1QC (F). Validation of the advanced symptomatic
stage proteomic data through immunoblotting for the selected proteins: HSPB1 (G), GFAP
(H) and STK38 (I). All selected symptomatic genes are upregulated at the protein level as
seen in Western Blots. (Data is represented as mean ± SEM *p<0,05, **p<0,01, ***p<0,001).
3.12.3. Table S2 Supplementary data to Figure 3G
Gene Symbol
Length of 3'UTR (nucleotides)
Number of SRSF3
binding sites
Binding sites density
(sites/nucleotides)
Arr
este
d g
enes
Ccl3 424 33 0,078
Ccl5 198 16 0,081
Cd68 174 25 0,144
Clec7a 1450 165 0,114
Cst7 447 51 0,114
Hmox1 632 63 0,100
Itgax 463 71 0,153
Lcn2 224 32 0,143
Lilrb4a 365 41 0,112
Saa3 128 24 0,188
Timp1 120 26 0,217
Trem2 252 56 0,222
No
n-a
rres
ted
gen
es
Apoe 151 7 0,046
C1qa 238 23 0,097
C1qb 163 21 0,129
C1qc 250 44 0,176
Crocc 458 20 0,044
Gfap 1341 41 0,031
Gpnmb 1868 137 0,073
Lgals3 487 46 0,094
Mvp 123 7 0,057
Pfkfb3 2760 151 0,055

142
SRSF3 arrested genes and non-arrested genes of which the protein expression level has been
checked by immunoblotting possess a different density of SRSF3 binding sites on their
3‘UTR. With each gene is given its 3’UTR length in nucleotide, the number of SRSF3
binding on that 3’UTR. The binding sites density is calculated with the number of SRSF3
binding sites in function with the 3’UTR length of that gene.
3.12.4. Table S3 Supplementary data to figure 3H
Raw data from the qPCR experiments from the Cross-linked immunopurification where
Clec7a, Lilr4a and Cst7 mRNA levels were measured comparatively to Gapdh level
3.12.5. Table S4 Primer list
Oligonucleotides
Primer name Source
pBluescriptSK-CD11b-TKmt-30 5’ primer 5’-GGGACGACGGGCCCCTCGAGGGGTTCAAGTGATTCTGCTGC-3’
This lab / Gowing et al. 2008
pBluescriptSK-CD11b-TKmt-30 3’primer 5’-GGGACGACGTCGACAGAACCTGGAAGGAGGTGAACC-3’
This lab / Gowing et al. 2008
Rpl10a 5’ primer 5'-GGGACGACGGATCCAGGTAAGTTGTTATCGTGGC-3’
This lab / Boutej et al. 2017
Rpl10a 3’primer 5’-GGGACGACGCGGCCGCCTAATACAGACGCTGGGGCT-3’
This lab / Boutej et al. 2017
Flag-eGFP 5’ primer 5’-GGGACGACCTGCAGGAGGCAGCATGGACTACAAAGACGACGACGACAAGGTGAGCAAGGGCGAGGA-3’
This lab / Boutej et al. 2017
Flag-eGFP 3' primer 5’-GGGACGA CGGATCCCTTGTACAGCTCGTCCATGC-3’
This lab / Boutej et al. 2017
Cst7 5’primer 5’ TTTTGTTCAAGGAGTCCCATGTCAGC 3’
This lab
Cst7 3’ primer 5’ GCAGGGTTGGTTTGGAAGTCACA 3’ This lab
Clec7a 5’primer 5’ AAAAGGCCCAGGGGATCAGAG 3’ This lab
Clec7a 3’primer 5’ CCCAGCACTGCAGCAACCACT 3’ This lab
Lilrb4 5’ primer 5’ AAGGCCAAGTTCAACATTCCAAGC 3’
This lab
Lilrb4 3’ primer 5’ CAAATACTATGGATGAGCTGCAACTAA
This lab
Primers used in this study with their name and their source

143
3.13. References
AJAMI, B., BENNETT, J. L., KRIEGER, C., TETZLAFF, W. & ROSSI, F. M. 2007. Local
self-renewal can sustain CNS microglia maintenance and function throughout adult
life. Nat Neurosci, 10, 1538-43.
AJAMI, B., SAMUSIK, N., WIEGHOFER, P., HO, P. P., CROTTI, A., BJORNSON, Z.,
PRINZ, M., FANTL, W. J., NOLAN, G. P. & STEINMAN, L. 2018. Single-cell mass
cytometry reveals distinct populations of brain myeloid cells in mouse
neuroinflammation and neurodegeneration models. Nature Neuroscience, 21, 541-
551.
AJIRO, M., JIA, R., YANG, Y., ZHU, J. & ZHENG, Z. M. 2016. A genome landscape of
SRSF3-regulated splicing events and gene expression in human osteosarcoma U2OS
cells. Nucleic Acids Res, 44, 1854-70.
ASHRAF, G. M. & BAEESA, S. S. 2018. Investigation of Gal-3 Expression Pattern in Serum
and Cerebrospinal Fluid of Patients Suffering From Neurodegenerative Disorders.
Front Neurosci, 12, 430.
BEERS, D. R. & APPEL, S. H. 2019. Immune dysregulation in amyotrophic lateral sclerosis:
mechanisms and emerging therapies. Lancet Neurol, 18, 211-220.
BÉLAND, L.-C., MARKOVINOVIC, A., JAKOVAC, H., DE MARCHI, F., BILIC, E.,
MAZZINI, L., KRIZ, J. & MUNITIC, I. 2020. Immunity in amyotrophic lateral
sclerosis: blurred lines between excessive inflammation and inefficient immune
responses. Brain Communications.
BELLOY, M. E., NAPOLIONI, V. & GREICIUS, M. D. 2019. A Quarter Century of APOE
and Alzheimer's Disease: Progress to Date and the Path Forward. Neuron, 101, 820-
838.
BINDEA, G., GALON, J. & MLECNIK, B. 2013. CluePedia Cytoscape plugin: pathway
insights using integrated experimental and in silico data. Bioinformatics, 29, 661-3.
BINDEA, G., MLECNIK, B., HACKL, H., CHAROENTONG, P., TOSOLINI, M.,
KIRILOVSKY, A., FRIDMAN, W. H., PAGÈS, F., TRAJANOSKI, Z. & GALON,
J. 2009. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene
ontology and pathway annotation networks. Bioinformatics, 25, 1091-3.
BLASI, E., BARLUZZI, R., BOCCHINI, V., MAZZOLLA, R. & BISTONI, F. 1990.
Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J
Neuroimmunol, 27, 229-37.
BOND, C. S. & FOX, A. H. 2009. Paraspeckles: nuclear bodies built on long noncoding
RNA. J Cell Biol, 186, 637-44.
BOUTEJ, H., RAHIMIAN, R., THAMMISETTY, S. S., BELAND, L. C., LALANCETTE-
HEBERT, M. & KRIZ, J. 2017. Diverging mRNA and Protein Networks in Activated
Microglia Reveal SRSF3 Suppresses Translation of Highly Upregulated Innate
Immune Transcripts. Cell Rep, 21, 3220-3233.
BUDGE, K. M., NEAL, M. L., RICHARDSON, J. R. & SAFADI, F. F. 2018. Glycoprotein
NMB: an Emerging Role in Neurodegenerative Disease. Mol Neurobiol, 55, 5167-
5176.
CHIRCOP, M. 2014. Rho GTPases as regulators of mitosis and cytokinesis in mammalian
cells. Small GTPases, 5.

144
CHISTIAKOV, D. A., KILLINGSWORTH, M. C., MYASOEDOVA, V. A., OREKHOV,
A. N. & BOBRYSHEV, Y. V. 2017. CD68/macrosialin: not just a histochemical
marker. Lab Invest, 97, 4-13.
CORBO, C., ORRÙ, S. & SALVATORE, F. 2013. SRp20: an overview of its role in human
diseases. Biochem Biophys Res Commun, 436, 1-5.
DECZKOWSKA, A., KEREN-SHAUL, H., WEINER, A., COLONNA, M., SCHWARTZ,
M. & AMIT, I. 2018. Disease-Associated Microglia: A Universal Immune Sensor of
Neurodegeneration. Cell, 173, 1073-1081.
DIAZ-AMARILLA, P., OLIVERA-BRAVO, S., TRIAS, E., CRAGNOLINI, A.,
MARTINEZ-PALMA, L., CASSINA, P., BECKMAN, J. & BARBEITO, L. 2011.
Phenotypically aberrant astrocytes that promote motoneuron damage in a model of
inherited amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A, 108, 18126-31.
FRAKES, A. E., FERRAIUOLO, L., HAIDET-PHILLIPS, A. M., SCHMELZER, L.,
BRAUN, L., MIRANDA, C. J., LADNER, K. J., BEVAN, A. K., FOUST, K. D.,
GODBOUT, J. P., POPOVICH, P. G., GUTTRIDGE, D. C. & KASPAR, B. K. 2014.
Microglia induce motor neuron death via the classical NF-κB pathway in
amyotrophic lateral sclerosis. Neuron, 81, 1009-1023.
FUENTES-FAYOS, A. C., VÁZQUEZ-BORREGO, M. C., JIMÉNEZ-VACAS, J. M.,
BEJARANO, L., PEDRAZA-ARÉVALO, S., F, L. L., BLANCO-ACEVEDO, C.,
SÁNCHEZ-SÁNCHEZ, R., REYES, O., VENTURA, S., SOLIVERA, J.,
BREUNIG, J. J., BLASCO, M. A., GAHETE, M. D., CASTAÑO, J. P. & LUQUE,
R. M. 2020. Splicing machinery dysregulation drives glioblastoma
development/aggressiveness: oncogenic role of SRSF3. Brain.
GONG, Z. & CHEN, J. 2011. E3 ligase RFWD3 participates in replication checkpoint
control. J Biol Chem, 286, 22308-13.
GOWING, G., PHILIPS, T., VAN WIJMEERSCH, B., AUDET, J. N., DEWIL, M., VAN
DEN BOSCH, L., BILLIAU, A. D., ROBBERECHT, W. & JULIEN, J. P. 2008.
Ablation of proliferating microglia does not affect motor neuron degeneration in
amyotrophic lateral sclerosis caused by mutant superoxide dismutase. J Neurosci, 28,
10234-44.
GURNEY, M. E., PU, H., CHIU, A. Y., DAL CANTO, M. C., POLCHOW, C. Y.,
ALEXANDER, D. D., CALIENDO, J., HENTATI, A., KWON, Y. W., DENG, H.
X. & ET AL. 1994. Motor neuron degeneration in mice that express a human Cu,Zn
superoxide dismutase mutation. Science, 264, 1772-5.
HALL, E. D., OOSTVEEN, J. A. & GURNEY, M. E. 1998. Relationship of microglial and
astrocytic activation to disease onset and progression in a transgenic model of familial
ALS. Glia, 23, 249-56.
HARDIMAN, O. & VAN DEN BERG, L. H. 2017. Edaravone: a new treatment for ALS on
the horizon? Lancet Neurol, 16, 490-491.
HAURE-MIRANDE, J. V., WANG, M., AUDRAIN, M., FANUTZA, T., KIM, S. H., HEJA,
S., READHEAD, B., DUDLEY, J. T., BLITZER, R. D., SCHADT, E. E., ZHANG,
B., GANDY, S. & EHRLICH, M. E. 2019. Integrative approach to sporadic
Alzheimer's disease: deficiency of TYROBP in cerebral Abeta amyloidosis mouse
normalizes clinical phenotype and complement subnetwork molecular pathology
without reducing Abeta burden. Mol Psychiatry, 24, 431-446.
HEIMAN, M., SCHAEFER, A., GONG, S., PETERSON, J. D., DAY, M., RAMSEY, K. E.,
SUAREZ-FARINAS, M., SCHWARZ, C., STEPHAN, D. A., SURMEIER, D. J.,

145
GREENGARD, P. & HEINTZ, N. 2008. A translational profiling approach for the
molecular characterization of CNS cell types. Cell, 135, 738-48.
HENKEL, J. S., BEERS, D. R., ZHAO, W. & APPEL, S. H. 2009. Microglia in ALS: the
good, the bad, and the resting. J Neuroimmune Pharmacol, 4, 389-98.
HEURICH, B., EL IDRISSI, N. B., DONEV, R. M., PETRI, S., CLAUS, P., NEAL, J.,
MORGAN, B. P. & RAMAGLIA, V. 2011. Complement upregulation and activation
on motor neurons and neuromuscular junction in the SOD1 G93A mouse model of
familial amyotrophic lateral sclerosis. J Neuroimmunol, 235, 104-9.
HOLTMAN, I. R., RAJ, D. D., MILLER, J. A., SCHAAFSMA, W., YIN, Z., BROUWER,
N., WES, P. D., MOLLER, T., ORRE, M., KAMPHUIS, W., HOL, E. M.,
BODDEKE, E. W. & EGGEN, B. J. 2015. Induction of a common microglia gene
expression signature by aging and neurodegenerative conditions: a co-expression
meta-analysis. Acta Neuropathol Commun, 3, 31.
HUBER, W., CAREY, V. J., GENTLEMAN, R., ANDERS, S., CARLSON, M.,
CARVALHO, B. S., BRAVO, H. C., DAVIS, S., GATTO, L., GIRKE, T.,
GOTTARDO, R., HAHNE, F., HANSEN, K. D., IRIZARRY, R. A., LAWRENCE,
M., LOVE, M. I., MACDONALD, J., OBENCHAIN, V., OLEŚ, A. K., PAGÈS, H.,
REYES, A., SHANNON, P., SMYTH, G. K., TENENBAUM, D., WALDRON, L.
& MORGAN, M. 2015. Orchestrating high-throughput genomic analysis with
Bioconductor. Nature methods, 12, 115-121.
HÜTTENRAUCH, M., OGOREK, I., KLAFKI, H., OTTO, M., STADELMANN, C.,
WEGGEN, S., WILTFANG, J. & WIRTHS, O. 2018. Glycoprotein NMB: a novel
Alzheimer's disease associated marker expressed in a subset of activated microglia.
Acta neuropathologica communications, 6, 108-108.
JHA, A. K., HUANG, S. C., SERGUSHICHEV, A., LAMPROPOULOU, V., IVANOVA,
Y., LOGINICHEVA, E., CHMIELEWSKI, K., STEWART, K. M., ASHALL, J.,
EVERTS, B., PEARCE, E. J., DRIGGERS, E. M. & ARTYOMOV, M. N. 2015.
Network integration of parallel metabolic and transcriptional data reveals metabolic
modules that regulate macrophage polarization. Immunity, 42, 419-30.
KAMPHUIS, W., KOOIJMAN, L., SCHETTERS, S., ORRE, M. & HOL, E. M. 2016.
Transcriptional profiling of CD11c-positive microglia accumulating around amyloid
plaques in a mouse model for Alzheimer's disease. Biochim Biophys Acta, 1862,
1847-60.
KANG, S. S., EBBERT, M. T. W., BAKER, K. E., COOK, C., WANG, X., SENS, J. P.,
KOCHER, J. P., PETRUCELLI, L. & FRYER, J. D. 2018. Microglial translational
profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J Exp
Med, 215, 2235-2245.
KELLER, A. F., GRAVEL, M. & KRIZ, J. 2009. Live imaging of amyotrophic lateral
sclerosis pathogenesis: disease onset is characterized by marked induction of GFAP
in Schwann cells. Glia, 57, 1130-42.
KELLER, A. F., GRAVEL, M. & KRIZ, J. 2011. Treatment with minocycline after disease
onset alters astrocyte reactivity and increases microgliosis in SOD1 mutant mice. Exp
Neurol, 228, 69-79.
KEREN-SHAUL, H., SPINRAD, A., WEINER, A., MATCOVITCH-NATAN, O., DVIR-
SZTERNFELD, R., ULLAND, T. K., DAVID, E., BARUCH, K., LARA-ASTAISO,
D., TOTH, B., ITZKOVITZ, S., COLONNA, M., SCHWARTZ, M. & AMIT, I.

146
2017. A Unique Microglia Type Associated with Restricting Development of
Alzheimer's Disease. Cell, 169, 1276-1290.e17.
KIM, J., PARK, R. Y., CHEN, J. K., KIM, J., JEONG, S. & OHN, T. 2014. Splicing factor
SRSF3 represses the translation of programmed cell death 4 mRNA by associating
with the 5'-UTR region. Cell death and differentiation, 21, 481-490.
KRASEMANN, S., MADORE, C., CIALIC, R., BAUFELD, C., CALCAGNO, N., EL
FATIMY, R., BECKERS, L., O'LOUGHLIN, E., XU, Y., FANEK, Z., GRECO, D.
J., SMITH, S. T., TWEET, G., HUMULOCK, Z., ZRZAVY, T., CONDE-
SANROMAN, P., GACIAS, M., WENG, Z., CHEN, H., TJON, E., MAZAHERI, F.,
HARTMANN, K., MADI, A., ULRICH, J. D., GLATZEL, M., WORTHMANN, A.,
HEEREN, J., BUDNIK, B., LEMERE, C., IKEZU, T., HEPPNER, F. L., LITVAK,
V., HOLTZMAN, D. M., LASSMANN, H., WEINER, H. L., OCHANDO, J.,
HAASS, C. & BUTOVSKY, O. 2017. The TREM2-APOE Pathway Drives the
Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative
Diseases. Immunity, 47, 566-581.e9.
LALANCETTE-HEBERT, M., SWARUP, V., BEAULIEU, J. M., BOHACEK, I.,
ABDELHAMID, E., WENG, Y. C., SATO, S. & KRIZ, J. 2012. Galectin-3 is
required for resident microglia activation and proliferation in response to ischemic
injury. J Neurosci, 32, 10383-95.
LIAN, H., ROY, E. & ZHENG, H. 2016. Microglial Phagocytosis Assay. Bio Protoc, 6.
LIGUORI, M., DIGIFICO, E., VACCHINI, A., AVIGNI, R., COLOMBO, F. S., BORRONI,
E. M., FARINA, F. M., MILANESI, S., CASTAGNA, A., MANNARINO, L.,
CRAPAROTTA, I., MARCHINI, S., ERBA, E., PANINI, N., TAMBORINI, M.,
RIMOLDI, V., ALLAVENA, P. & BELGIOVINE, C. 2020. The soluble
glycoprotein NMB (GPNMB) produced by macrophages induces cancer stemness
and metastasis via CD44 and IL-33. Cell Mol Immunol.
MANLEY, J. L. & KRAINER, A. R. 2010. A rational nomenclature for serine/arginine-rich
protein splicing factors (SR proteins). Genes Dev, 24, 1073-4.
MCGEER, P. L., MCGEER, E. G., KAWAMATA, T., YAMADA, T. & AKIYAMA, H.
1991. Reactions of the immune system in chronic degenerative neurological diseases.
Can J Neurol Sci, 18, 376-9.
MILLER, R. G., MITCHELL, J. D. & MOORE, D. H. 2012. Riluzole for amyotrophic lateral
sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev,
Cd001447.
MISTELI, T. & SPECTOR, D. L. 1997. Protein phosphorylation and the nuclear organization
of pre-mRNA splicing. Trends Cell Biol, 7, 135-8.
NOORIMOTLAGH, Z., BABAIE, M., SAFDARIAN, M., GHADIRI, T. & RAHIMI-
MOVAGHAR, V. 2017. Mechanisms of spinal cord injury regeneration in zebrafish:
a systematic review. Iran J Basic Med Sci, 20, 1287-1296.
PATEL, P., JULIEN, J. P. & KRIZ, J. 2015. Early-stage treatment with Withaferin A reduces
levels of misfolded superoxide dismutase 1 and extends lifespan in a mouse model of
amyotrophic lateral sclerosis. Neurotherapeutics, 12, 217-33.
PAZ, I., KOSTI, I., ARES, M., JR., CLINE, M. & MANDEL-GUTFREUND, Y. 2014.
RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic
acids research, 42, W361-W367.

147
PIKTEL, E., LEVENTAL, I., DURNAŚ, B., JANMEY, P. A. & BUCKI, R. 2018. Plasma
Gelsolin: Indicator of Inflammation and Its Potential as a Diagnostic Tool and
Therapeutic Target. Int J Mol Sci, 19.
RAHIMIAN, R., BELAND, L. C. & KRIZ, J. 2018. Galectin-3: mediator of microglia
responses in injured brain. Drug Discov Today, 23, 375-381.
ROBBERECHT, W. & PHILIPS, T. 2013. The changing scene of amyotrophic lateral
sclerosis. Nat Rev Neurosci, 14, 248-64.
ROSEN, D. R., SIDDIQUE, T., PATTERSON, D., FIGLEWICZ, D. A., SAPP, P.,
HENTATI, A., DONALDSON, D., GOTO, J., O'REGAN, J. P., DENG, H. X. & ET
AL. 1993. Mutations in Cu/Zn superoxide dismutase gene are associated with familial
amyotrophic lateral sclerosis. Nature, 362, 59-62.
SANDÉN, C., AGEBERG, M., PETERSSON, J., LENNARTSSON, A. & GULLBERG, U.
2013. Forced expression of the DEK-NUP214 fusion protein promotes proliferation
dependent on upregulation of mTOR. BMC Cancer, 13, 440.
SHANNON, P., MARKIEL, A., OZIER, O., BALIGA, N. S., WANG, J. T., RAMAGE, D.,
AMIN, N., SCHWIKOWSKI, B. & IDEKER, T. 2003. Cytoscape: a software
environment for integrated models of biomolecular interaction networks. Genome
Res, 13, 2498-504.
SHEPARD, P. J. & HERTEL, K. J. 2009. The SR protein family. Genome biology, 10, 242.
SWINNEN, B. & ROBBERECHT, W. 2014. The phenotypic variability of amyotrophic
lateral sclerosis. Nat Rev Neurol, 10, 661-70.
TANG, Y., HORIKAWA, I., AJIRO, M., ROBLES, A. I., FUJITA, K., MONDAL, A. M.,
STAUFFER, J. K., ZHENG, Z. M. & HARRIS, C. C. 2013. Downregulation of
splicing factor SRSF3 induces p53beta, an alternatively spliced isoform of p53 that
promotes cellular senescence. Oncogene, 32, 2792-8.
TRIAS, E., BEILBY, P. R., KOVACS, M., IBARBURU, S., VARELA, V., BARRETO-
NUNEZ, R., BRADFORD, S. C., BECKMAN, J. S. & BARBEITO, L. 2019.
Emergence of Microglia Bearing Senescence Markers During Paralysis Progression
in a Rat Model of Inherited ALS. Front Aging Neurosci, 11, 42.
TRIAS, E., DIAZ-AMARILLA, P., OLIVERA-BRAVO, S., ISASI, E., DRECHSEL, D. A.,
LOPEZ, N., BRADFORD, C. S., IRETON, K. E., BECKMAN, J. S. & BARBEITO,
L. 2013. Phenotypic transition of microglia into astrocyte-like cells associated with
disease onset in a model of inherited ALS. Front Cell Neurosci, 7, 274.
WEISSMANN, R., HUTTENRAUCH, M., KACPROWSKI, T., BOUTER, Y., PRADIER,
L., BAYER, T. A., KUSS, A. W. & WIRTHS, O. 2016. Gene Expression Profiling
in the APP/PS1KI Mouse Model of Familial Alzheimer's Disease. J Alzheimers Dis,
50, 397-409.
WILHELMSSON, U., ANDERSSON, D., DE PABLO, Y., PEKNY, R., STÅHLBERG, A.,
MULDER, J., MITSIOS, N., HORTOBÁGYI, T., PEKNY, M. & PEKNA, M. 2017.
Injury Leads to the Appearance of Cells with Characteristics of Both Microglia and
Astrocytes in Mouse and Human Brain. Cereb Cortex, 27, 3360-3377.
ZHOU, J. Y., AFJEHI-SADAT, L., ASRESS, S., DUONG, D. M., CUDKOWICZ, M.,
GLASS, J. D. & PENG, J. 2010. Galectin-3 is a candidate biomarker for amyotrophic
lateral sclerosis: discovery by a proteomics approach. J Proteome Res, 9, 5133-41.
ZHOU, Z., GONG, Q., LIN, Z., WANG, Y., LI, M., WANG, L., DING, H. & LI, P. 2020.
Emerging Roles of SRSF3 as a Therapeutic Target for Cancer. Front Oncol, 10,
577636.

148
ZHU, X., WEI, D., CHEN, O., ZHANG, Z., XUE, J., HUANG, S., ZHU, W. & WANG, Y.
2016. Upregulation of CCL3/MIP-1alpha regulated by MAPKs and NF-kappaB
mediates microglial inflammatory response in LPS-induced brain injury. Acta
Neurobiol Exp (Wars), 76, 304-317.
ZOLLER, T., ATTAAI, A., POTRU, P. S., RUSS, T. & SPITTAU, B. 2018. Aged Mouse
Cortical Microglia Display an Activation Profile Suggesting Immunotolerogenic
Functions. Int J Mol Sci, 19.

149
Chapitre 4 – The molecular and biological functions of SRSF3 and its implication in
diseases
4.1. Résumé
Le métabolisme de l’ARN est une pièce maîtresse de la biologie moléculaire dans la cellule.
Sa régulation est le rôle plusieurs protéines liant l’ARN dont les protéines de la famille SR.
Les protéines SR sont une famille de 12 membres suivant le même modèle structurel soit un
ou deux motifs de reconnaissance de l’ARN contigus suivi d’un domaine RS d’une longueur
variable. SRSF3, la plus petite protéine de cette famille, partage plusieurs fonctions avec les
autres membres de la famille SR et est impliqué dans l’épissage alternatif, la transcription, la
maturation des miRNA, l’exportation nucléaire des ARNm et la traduction. SRSF3, par la
nature des pre-ARNm et des ARNm avec lesquels il interagit, est ainsi impliqué dans une
multitude de mécanismes cellulaires tels que le métabolisme, l’inflammation, la
gamétogénèse ou la prolifération cellulaire, pour en nommer quelques-uns. La dérégulation
des fonctions de SRSF3 peut mener à la perte de l’homéostasie cellulaire et le développement
de plusieurs maladies liées aux mécanismes cellulaires mentionnés précédemment. L’étude
du fonctionnement de SRSF3 est importante pour comprendre les mécanismes sous-jacents
aux maladies et cibler de nouvelles thérapies.

150
4.2. Abstract
RNA metabolism is a centerpiece of molecular biology in the cell. Its regulation is the role
of many RNA binding proteins; the SR protein family are amongst them. The SR proteins
are a family composed of 12 members which follow the same structural model, namely, one
or two contiguous RNA recognition motifs followed by a RS domain of variable length.
SRSF3, the smallest member of the SR proteins, shares many of its functions with the other
member of his protein family and is implicated in alternative splicing, transcription, miRNA
maturation, mRNA nuclear export and translation. SRSF3, by the nature of the pre-mRNA
and mRNA it binds, is hence implicated in the regulation of a multitude of biological
pathways such as metabolism, inflammation, gametogenesis or cellular proliferation, to name
a few. The dysregulation of SRSF3 function can lead to the loss of cellular homeostasis and
to the development of many diseases linked to the aforementioned biological pathways. The
study of SRSF3 function is important to understand disease mechanisms and aim possible
therapies.

151
4.3. Introduction
Generating a large proteomic complexity from a narrow and restricted genome is of high
importance for cell differentiation, tissue development or immunity (Baralle et Giudice
2017). The most prominent way to achieve proteomic diversity is through alternative
splicing, to which close to 95% of human intron-containing genes are subjected (Wang et al.
2008). Alternative splicing is a cellular event instigated, in part, by a family of RNA binding
and phosphorylatable proteins known as SR proteins. Transcripts isoforms from alternative
splicing may present a gain, reduction or loss of function in biological pathways and, thus, a
dysregulation is this cellular mechanism can lead to diseases such as cancer or tauopathies,
to which SR proteins have been previously linked (Liu et Gong 2008, da Silva et al. 2015).
Older studies have focused on understanding the molecular mechanisms by which SR
proteins affect RNA processing, by studying their expression, phosphorylation and protein
and RNA partners. In the last decade or so, studies instead started to delve into the implication
of the SR proteins in biological functions and in disease response. In the human genome,
twelve genes code for the different members of the SR protein family. They present a similar
structure, namely one or two RNA recognition motif (RRM) at the N-terminus and, on the
C-terminus, an arginine-serine rich sequence (RS) (Zhou et Fu 2013, Sunjoo 2017). Their
main role is centered around RNA processing and they are implicated in RNA splicing,
constitutive or alternative, RNA nuclear export and even translation (Sanford et al. 2004,
Long et Caceres 2009). The smallest member of this family, SRSF3, has largely been studied
for its role in alternative splicing and more precisely, in the alternative splicing of cell
proliferation-liked genes and its implication in cancer for its oncogenic capabilities.
In this review, we will first explore the structural characteristics of the SR proteins. The
different domains of the SR proteins allow them to engage in RNA-protein interaction and
protein-protein interaction upon specific phosphorylation. This phosphorylation of SR
proteins regulates their localization in the cell, either their translocation to and from nuclear
speckles or their transport through the nuclear membrane. SR protein phosphorylation will
also activate certain functions from this family, functions linked to RNA processing. We will

152
then delve into the molecular functions of the protein SRSF3 in the nucleus, mainly its role
in pre-mRNA maturation, that is, its role in splicing and alternative splicing, in pre-mRNA
polyadenylation and in the nucleocytoplasmic transport of those mature mRNAs. These
functions are also mostly directed through post-translation modification of SRSF3. The post-
translational modification of SRSF3 also regulates its shuttling to and from the cytoplasm
where SRSF3 is also implicated in various phases of translation with the help of other
proteins (Long et Caceres 2009). Finally, we will link the molecular functions of SRSF3 to
biological pathways and disease mechanisms where this protein plays a prominent role.
SRSF3 is implicated in cancer where it can regulate the alternative splicing of proto-
oncogenes and promotes cancer development and growth (Ajiro et al. 2016). Moreover,
SRSF3 can stimulate the translation of viral transcripts in infected cells (Bedard et al. 2007).
Furthermore, SRSF3 was recently linked to translational suppression of immune mRNAs in
myeloid cells following inflammation (Boutej et al. 2017). The importance of SRSF3 in
numerous human diseases indicates that its expression or phosphorylation could be
modulated as a therapeutic approach.
4.4. SR protein family
4.4.1. SR protein conservation
Because the presence of introns is a common characteristic of eukaryotic organisms, most
animals, plant and fungi possess a certain number of SR proteins (Shepard et Hertel 2009).
Their presence and numbers seems to correlate with the need for splicing, and more precisely,
alternative splicing (Kress et al. 2008, Shepard et Hertel 2009). For example, we find low
numbers of SR proteins in ancestral unicellular eukaryotes which generally only use
constitutive splicing (Richardson et al. 2011). Meanwhile, plants usually present a higher
number of SR proteins than animals. Moreover, the number of SR proteins could be
underestimated due to the alternative splicing of the SR transcripts themselves and the
generation of more isoforms of SR transcripts and proteins. (Palusa et al. 2007, Richardson
et al. 2011). SR proteins subfamilies, based on their structures, are not shared between plants
and animals due to divergent evolution, except for SRSF2 (Richardson et al. 2011).

153
The human genome comprises 12 genes coding for SR proteins, which have orthologous
genes in other evolutionary close species. While their number and their sequence vary in the
more ancestral metazoans, “higher” vertebrates, more precisely gnathostomes (jaw-bearing
animals), possess around the same number of SR genes with highly conserved exonic
sequence when compared to the respective orthologous human gene. As shown in Table 1,
species representative of the mammalian taxon (Pan troglodytes, Mus musculus, Sus scrofa
and Felis catus) have the highest conservation percentage of exonic sequence, while the non-
gnathostome vertebrate (Petromyzon marinus) has less orthologous SR genes and less
sequence conservation when compared to the orthologous human genes. The two
representative species from the protostome taxon, Drosophila melanogaster and
Caenorhabditis elegans, show the lowest resemblance to the human set of SR genes. Instead,
they possess many paralogous SR genes that do not necessarily fit any orthologous gene in
the human genome. Even though the SR proteins change in nature and in number in between
taxa, they are indeed found and, hence, conserved in all metazoans. The diversity in SR
proteins and their structures comes from ancient gene duplication making orthologous genes
closer to each other than paralogous genes (Cáceres et al. 1998). The high level of
conservation also justifies the use of animal models in research on SR proteins. The high
level of conservation and the plethora of SR proteins across the species highlights the
importance of this protein family in cell physiology and homeostasis.
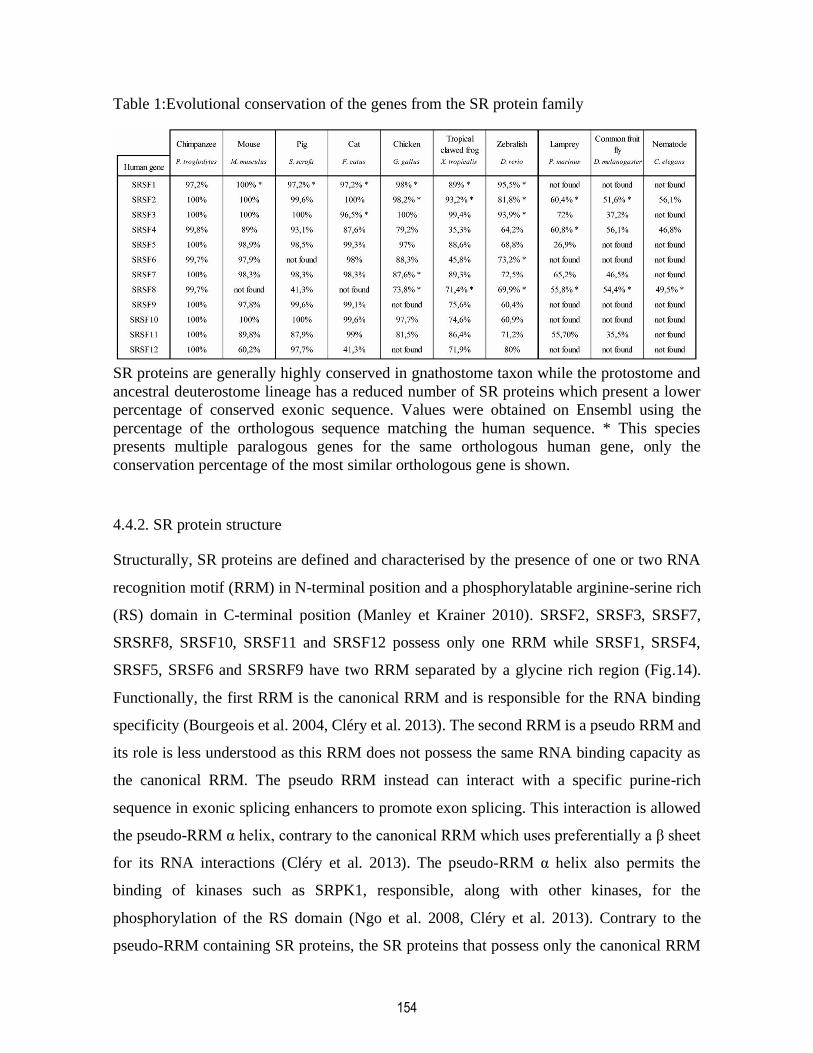
154
Table 1:Evolutional conservation of the genes from the SR protein family
SR proteins are generally highly conserved in gnathostome taxon while the protostome and
ancestral deuterostome lineage has a reduced number of SR proteins which present a lower
percentage of conserved exonic sequence. Values were obtained on Ensembl using the
percentage of the orthologous sequence matching the human sequence. * This species
presents multiple paralogous genes for the same orthologous human gene, only the
conservation percentage of the most similar orthologous gene is shown.
4.4.2. SR protein structure
Structurally, SR proteins are defined and characterised by the presence of one or two RNA
recognition motif (RRM) in N-terminal position and a phosphorylatable arginine-serine rich
(RS) domain in C-terminal position (Manley et Krainer 2010). SRSF2, SRSF3, SRSF7,
SRSRF8, SRSF10, SRSF11 and SRSF12 possess only one RRM while SRSF1, SRSF4,
SRSF5, SRSF6 and SRSRF9 have two RRM separated by a glycine rich region (Fig.14).
Functionally, the first RRM is the canonical RRM and is responsible for the RNA binding
specificity (Bourgeois et al. 2004, Cléry et al. 2013). The second RRM is a pseudo RRM and
its role is less understood as this RRM does not possess the same RNA binding capacity as
the canonical RRM. The pseudo RRM instead can interact with a specific purine-rich
sequence in exonic splicing enhancers to promote exon splicing. This interaction is allowed
the pseudo-RRM α helix, contrary to the canonical RRM which uses preferentially a β sheet
for its RNA interactions (Cléry et al. 2013). The pseudo-RRM α helix also permits the
binding of kinases such as SRPK1, responsible, along with other kinases, for the
phosphorylation of the RS domain (Ngo et al. 2008, Cléry et al. 2013). Contrary to the
pseudo-RRM containing SR proteins, the SR proteins that possess only the canonical RRM

155
cannot bind kinases the same way. SRSF3, the prototypic single RRM SR protein, will
instead bind SRPKs through its RS domain (Long et Caceres 2009).
Figure 14: SR protein family structure
The SR protein family contains 12 members which follows the same two structural rules:
they must possess one or two N-terminal RNA recognition motif and one C-terminal RS
domain containing multiple phosphorylatable serine-arginine dipeptide repeats. Potential
phosphorylation sites on serine, tyrosine and threonine residues, were identified using the
generic prediction tool of NetPhos 3.1.

156
4.4.3. SR protein phosphorylation
The phosphorylation of the RS domain dictates the subcellular and subnuclear localisation
of SR proteins (Misteli et Spector 1996) and therefore part of their cellular functions. Thus,
a combination of phosphatases and kinases are implicated in the phosphorylation of the many
serine residues found in the RS domain of the SR proteins (Fig.14), phosphatases such as
PP1 (Misteli et Spector 1996, Novoyatleva et al. 2007) and kinases from CLK and SRPK
families (Gui et al. 1994, Duncan et al. 1997). While SRPKs are found in either cytoplasm
and nucleus, CLKs are found only in the nucleus (Zhou et Fu 2013). Therefore, they will
regulate the phosphorylation state of SR proteins at different moment and the activation of
sequential functions. The phosphorylation of the SR proteins affects their protein-protein and
their RNA-protein interaction (Amrein et al. 1994, Xiao et Manley 1997). Upon their
translation, the SR proteins will be phosphorylated by SRPKs in the cytoplasm (Ding et al.
2006). SRPK, after binding the RS domain of SR proteins, will hyperphosphorylate them in
a processive manner (Long et Caceres 2009, Ghosh et Adams 2011). The processive
phosphorylation of proteins implies that most of the available serines of the protein target
will phosphorylate before the kinase dissociates from the substate (Patwardhan et Miller
2007) (Fig.14). The phosphorylated RS domain of SR proteins can now act as a nuclear
localisation signal (NLS). The nuclear import of SR proteins is only possible during active
transcription (Cáceres et al. 1998) and is mediated by the proteins transportin-SR1 and
transportin-SR2 (TRN-SR1 and TRN-SR2, respectively) which bind on SRSF3 NLS (Lai et
al. 2001) (Fig.15A). Upon their entry in the nucleus TRN-SR2 also has been reported to
direct SR protein subnuclear localisation either in the nucleoplasm or in the nuclear speckles
(Lai et al. 2001). In the nucleus, SR protein phosphorylation is adjusted by kinases (SRPKs
and CLKs) or by phosphatases (PP1) to fulfill the cellular needs in splicing factors at different
points within the splicing machinery. At this point, an intermediate level of phosphorylation
will promote the distribution of SR proteins in the nuclear speckles (Misteli et Spector 1996,
Aubol et al. 2017) and cause the recruitment of SR protein to the spliceosome (Zhou et Fu
2013). PP1 is responsible for the partial dephosphorylation of SR proteins and their
delocalisation from the nuclear speckles (Misteli et Spector 1996). It does so by binding a
specific sequence (RVxF) in the canonical RRM, RRM1, present in all SR proteins (Aubol
et al. 2017) (Fig.14). Hypophosphorylation of SR proteins is required for mature mRNA

157
bound to SR proteins to be exported into the cytoplasm where SR proteins can be recycled
and re-phosphorylated again (Zhou et Fu 2013). Moreover, the hyperphosphorylation of the
SR proteins leads to their cytoplasmic accumulation where they are implicated in other
cellular mechanisms (Misteli et Spector 1996, Cáceres et al. 1998, Lai et al. 2001).
4.5. SRSF3 controls multiple molecular functions in the nucleus and the cytoplasm.
SRSF3, also known as Srp20, is the smallest member of the SR protein family and share most
of its functions with the rest of its protein family. Although functionally redundant, the need
for multiple SR protein comes for their individual target binding specificity. SR proteins
contact their RNA targets by their RRM and without the need for their RS domain (Tacke et
Manley 1999). The RRMs sequence gives each SR protein their specificity although they
often work cooperatively (Müller-McNicoll et al. 2016). SRSF3 RRM recognizes two
specific and degenerated motifs on its RNA targets, WCWWC and CUCKUCY
(K=G/U,W=A/U, Y=C/U) (Cartegni et al. 2002, Churbanov et al. 2006). The binding of these
motifs allows SRSF3 to accomplish its many functions in the nucleus and the cytoplasm and
affects many aspects of the RNA processing, whether it be splicing, transcription, nuclear
export or translation.

158
Figure 15: SRSF3 molecular functions in the cell
A. Upon its translation, SRSF3 is phosphorylated by SRPKs in the cytoplasm and imported
into the nucleus by TRN-SR. SRSF3 phosphorylation state is adjusted in the nucleus by
SRPKs or CLKs before SRSF3 is recruited into active transcription splicing sites. After
transcription initiation, phosphorylated SRSF3 associates with Pol II to oversee splicing
events. B. SRSF3 is implicated in diverse forms of alternative splicing; alternative promoter
choice, intron retention, cassette exon inclusion or alternative polyadenylation, where it
recruits the spliceosome components. A phase of dephosphorylation by PP1 is necessary for
this step to occur. C. During transcription termination, SRSF3 recruits XRN-2 for the 3' end
formation and facilitates Pol II dissociation. D. In the nucleus, SRSF3 also participates in

159
miRNA maturation by recruiting DROSHA to pri-miRNA. E. The mRNA bound the SRSF3
during splicing and certain intronless mRNA (not represented) can be exported in the
cytoplasm after the recruitment of NXF1 on the mRNA. F. In the cytoplasm, SRSF3 can
repress translation by targeting the 5'UTR bound mRNAs to P-bodies or by stalling
polyribosomes during the translation of the 3'UTR bound mRNAs.
4.5.1. Alternative splicing
As SRSF3 name indicates, splicing, or more precisely alternative splicing, is its main
function. The SR proteins often work in collaboration with each other and have genome-wide
targets. SRSF3 alternative splicing is known to affect many genes implicated, among other
functions, in the cell cycle (Ajiro et al. 2016) in metabolism (Sen et al. 2009, Walsh et al.
2013a) and in SRSF3 splicing itself (Jumaa et al. 1997). The alternative splicing by SR
proteins alters the stability and function of the mRNA and augment the diversity of the
transcriptome and the proteome of cells and tissues. Along with the rest of the SR proteins,
SRSF3 can act as activator and repressor of alternative splicing events. SRSF3 distinguish
itself from the other SR proteins by being mainly an activator of splicing events, events in
the form of cassette exon inclusion and intron retention, the latter more usually repressed by
other SR proteins (Bradley et al. 2015).
SRSF3 is recruited to nascent RNAs during transcription and is not preassembled with RNA
polymerase II (Pol II) bound to chromatin (Sapra et al. 2009). The protein/RNA interaction
is mediated by SRSF3 RRM which will bind exonic splicing enhancers sequences on the
nascent RNA. SRSF3 will then promote the recruitment of the spliceosome components, such
as U1 snRNP, through protein-protein interaction through its RS domain, upstream and
downstream of exonic sequence, which, in turn, will reinforce SRSF3 protein/RNA
interaction (Graveley 2001, Bradley et al. 2015). It also has been reported that the C-terminal
domain (CTD) of Pol II mediates the recruitment of SRSF3 on nascent mRNA and that the
CTD/SRSF3 interaction is necessary for a correct alternative splicing activity from SRSF3
(de la Mata et Kornblihtt 2006) (Fig.15B).
As said in the previous section, the phosphorylation state of SRSF3 dictates its role in splicing
in a step-dependant manner. Indeed, the phosphorylation will change SRSF3 affinity to

160
snRNPs, to nascent RNA and to other splicing factors necessary for spliceosome complex
formation (Mermoud et al. 1994, Manley et Tacke 1996, Lin et Fu 2007). For example,
hypophosphorylated SRSF3 has high affinity to nascent mRNAs. Further phosphorylation
by SRPK and CLK will lead an hyperphosphorylated SRSF3 and nascent RNA to be
recruited to the spliceosome complex proteins, U1 snRNP for example (Zhou et Fu 2013).
Assembly in the spliceosome must be followed by SRSF3 dephosphorylation in order for the
actual splicing to occur, as both hypo- and hyperphosphorylated state of SRSF3 inhibits its
alternative splicing activity, in favor of the constitutive splicing of the nascent RNA (Prasad
et al. 1999).
During transcription initiation, SRSF3 is implicated in the promoter site selection. In D.
melanogaster, SRSF3 homologous gene, Xl6, choice of promoter (distal or proximal)
depends on Xl6 own level of expression (Bradley et al. 2015). The alternative choice of
promoter by SRSF3 will results in a different first exon and induces variability in the 5’ end
of the spliced mRNA (Faustino et Cooper 2003) (Fig.15B).
SRSF3 is also implicated in polyadenylation site selection (Bradley et al. 2015). Alternative
polyadenylation, just as the alternative splicing and the promoter site selection (and thus the
first exon selection) by SRSF3 can affect the diversity of the transcriptome and the proteome
from a limited genome (Fig.15B). The D. melanogaster SRSF3 homolog, expressed at
constitutive levels, will preferentially uses distal polyadenylation sites contrary to some other
SR proteins (Bradley et al. 2015, Müller-McNicoll et al. 2016, Shen et al. 2018, Shen et al.
2019). Upon knock down, a proximal site for polyadenylation will be chosen by SRSF3
resulting in a shorter protein product sometimes unfunctional. Moreover, SRSF3 seems to
work in tandem with a coupled Pol II to select the distal polyadenylation site (Shen et al.
2018). As in its other roles in the nucleus, SRSF3 is, in alternative polyadenylation, mainly
an adaptor protein acting as an early scaffold unit to recruit the complete cellular machinery
necessary for the current RNA processing step. SRSF3 binds a specific polyadenylation
enhancer sequence before recruiting other proteins such as CPSF6 (CF Im) or 64kDa CSTF
subunit.(Lou et al. 1998, Dettwiler et al. 2004). SRSF3 SR domain can bind CPSF6 through

161
the latter RS-like domain (Dettwiler et al. 2004) and CPSF6 will then recruits poly(A)
polymerase (PAP) to complete 3’ end polyadenylation (Fig.15B).
Alternative first exon selection, cassette-exon inclusion or skipping, intron retention and
polyadenylation site selection are different methods by which SRSF3 and SR proteins in
general can generate diversity in the proteome from a restricted genome. Moreover, the
differently spliced isoforms can be truncated or elongated by these mechanisms and this
results in gain, reduction, or loss of function. For example, SRSF3 activity can lead to the
presence of a premature termination codon and nonsense mediated decay of the spliced RNA
(da Costa et al. 2017). SRSF3 can hence change both the nature and the level of expression
of RNA transcripts.
4.5.2. Transcription
Although a lesser role for our protein of interest, transcription can be mediated by SRSF3.
SRSF3 was first reported to colocalize with RNA polymerase II in active transcription sites
in 1997 (Neugebauer et Roth 1997). Although, it was not possible to determine whether
SRSF3 actively affects transcription or that it colocalized with Pol II only because early
splicing events start during transcription. More recent studies have linked SRSF3 to the
transcription termination (Cui et al. 2008). More precisely, SRSF3 binds to the 3’ of the
nascent mRNA and the phosphorylated Pol II CTD and then could either participate in the 3’
end formation by recruiting the exonuclease XRN2 or facilitate the dissociation of Pol II
from the DNA template (Zeng et Berget 2000, Cui et al. 2008) (Fig,15C). Moreover, the
recruitment of SRSF3 to Pol II CTD could happen during transcription elongation, where
phosphorylated histone H3 tail could transfer SRSF3 to Pol II (Zhong et al. 2009). SRSF3
also has an effect on transcripts level of expression (Bradley et al. 2015). Upon knocking out
a SRSF3 homolog in a D. melanogaster cell line, a certain number of transcripts linked to
cell growth were upregulated while transcripts linked to housekeeping did not see a change
in their level of expression. The regulation of transcript levels could be linked to an increased
mRNA stability while linked to an SR protein. As said previously, the effects of SRSF3 on

162
the transcription machinery are, in the end, intertwined with its main role and will serve in
pre-mRNA splicing at the same time.
4.5.3. miRNA maturation
SRSF3 is implicated in micro RNA (miRNA) maturation, a lesser known role for this protein
in the nucleus. In the previous section, we explored how SRSF3 can directly influence the
diversity and level of expression of transcripts and proteins by driving RNA processing
mechanisms such as alternative splicing and promoter or polyadenylation site selection.
SRSF3 can also indirectly influence diversity and level of expression of transcripts and
proteins by helping in miRNA maturation, responsible for translational repression of
sequence-specific mRNAs. SRSF3 is involved in the first step of miRNA maturation (Kim
et al. 2018). On the primary miRNA (pri-miRNA), SRSF3 binds to the CNNC RNA motif
and recruits DROSHA to the basal junction of the primary miRNA (Auyeung et al. 2013,
Kim et al. 2018) which leads to the productive cleavage of this pri-miRNA (Fig.15D). SRSF3
secures DROSHA basal binding instead of the apical binding which produces unproductive
cleavage of the pri-miRNA.
As we note from the previous sections, SRSF3 is an important modulator of transcripts and
protein diversity through its roles in transcription, diverse pre-mRNA processing steps
(promoter site selection, alternative splicing or alternative polyadenylation) and miRNA
maturation. The effects of SRSF3 are usually linked to its expression level and its
phosphorylation state. This implies that outside its normal range of expression and without
its phosphorylation cycle, the dysregulation of pre-mRNA processing by SRSF3 could lead
to many defects in cellular physiology.
4.5.4. mRNA nuclear export
Depending on their phosphorylation state, SRSF3, along with SRSF1 and SRSF7 can shuttle
in between the cytoplasm and the nucleus (Cáceres et al. 1998). This allows the shuttling SR
proteins to export mRNA to the cytoplasm where it can also assist in translation. Among the
shuttling SR proteins, SRSF3 possesses the highest number of unique mRNA targets for

163
export. Moreover, while the other SR proteins can compensate for each other, SRSF3’s RRM
specificity seems unique and the other SR proteins cannot counterweigh its loss following
SRSF3 knockdown (Müller-McNicoll et al. 2016). While the nature of RRM is not essential
to mRNA nuclear export, the nature of the RS domain is. Although, the nature of the RS
domain is not sufficient for the RNA nuclear export as the binding between the RRM and
mRNA is required. This was demonstrated by replacing the RS domain of a non-shuttling
SR protein by the RS domain of a shuttling SR protein and showing that the new chimera
protein can be exported to the cytoplasm. (Cáceres et al. 1998).
After splicing, SRSF3 stays bound to spliced mRNA exonic sequences and then can contact
receptors proteins (NXF1, also known as TAP) which will facilitate export through the
nuclear pore complex. However, SRSF3 is also implicated in the export of the less common
intronless mRNAs. On the intronless mRNAs, SRSF3 recognises a 22 nucleotide sequence
and can bind it simultaneously with SRSF7 for a synergistic effect on this mRNA export
(Huang et Steitz 2001, Masuyama et al. 2004) after the recruitment of the same export
machinery used for spliced mRNAs. On spliced mRNAs, binding sites for SRSF3 are found
in all the exons, but SRSF3 will preferentially bind mRNA and promote recruitment of the
export machinery on the last exon (the 3’ UTR) (Müller-McNicoll et al. 2016) (Fig.15E).
Bound to its mRNA, SRSF3, in a dephosphorylated or hypophosphorylated state, recruits
NXF1 after splicing is concluded. The interaction happens between an arginine-rich region
in NXF1 N-terminus and a small peptide sequence in the C-terminus of SRSF3 RRM
(Hargous et al. 2006, Hautbergue et al. 2008). Interestingly, the strongest interaction between
NXF1 and any SR proteins is with SRSF3. This mRNA-SRSF3-NXF1 complex is then
exported in the cytoplasm(Huang et al. 2003, Müller-McNicoll et al. 2016).After the nuclear
export of the mRNA, SRSF3 can be reimported to the nucleus in the presence of active
transcription and following a new phosphorylation cycle or can assist in their translation
(Cáceres et al. 1998).

164
4.5.5. Translation
Although the implication of SR proteins in translation are known, few are the specific roles
of SRSF3 in that molecular event (Zhong et al. 2009). SRSF1 can promote the translation of
the mRNA it exported from the nucleus in normal condition and is found to bind polysomes
in vitro. SRSF3 was not found to initiate translation or to bind polysomes under the same
conditions (Sanford et al. 2004). Instead, SRSF3 is implicated in translational repression. In
the cytoplasm, exported mRNA are bound by their 5’UTR by SRSF3 and targeted to P-bodies
where they are silenced and degraded (Kim et al. 2014) (Fig.15E). This SRSF3 mechanism
would happen after the export of mRNA into the cytoplasm, but before its translation. SRSF3
can also repress translation during translation elongation possibly by stalling the ribosome.
Using a translating ribosome affinity purification mouse (TRAP) model to study cell-specific
molecular profiles, a recent study has found a high discrepancy between transcriptome and
proteome, as the highly upregulated mRNAs were not translated to proteins (Boutej et al.
2017). As the detection of peptides and mRNAs in EDTA-TRAP needs them to be bound to
translating or stalled ribosomes, the mechanism underlying the transcriptome/proteome
discrepancy must happen after translation initiation. SRSF3 binds the 3’UTR of those
mRNAs and, by an unknown mechanism, stalls their translation (Fig.15E). The knockdown
of SRSF3 allowed the translation of the previously silenced, yet highly regulated mRNAs.
Translational repression is the last way SRSF3 can work as a gene expression checkpoint and
can modify proteome diversity. It does so by either stopping the translation ever initiating by
helping with mRNA degradation or by stalling ongoing translation, preventing the production
of complete and functional protein products.
4.6. SRSF3 regulates many biological pathways in health and disease.
We saw previously how SRSF3, through different molecular mechanisms, can affect RNA
processing and stability in order to modify the proteome diversity. SRSF3 has a target
specificity, given by its RRM sequence, that usually don’t overlap other SR protein target
specificity (Müller-McNicoll et al. 2016). The nature of those targets during the different
steps of mRNA processing gives a clue to the biological functions SRSF3 regulates. As it is

165
sometimes the case, SRSF3 activity in these biological functions can be dysregulated (level
of expression, phosphorylation, subcellular localisation, etc.) and leads to the development
of numerous diseases.
4.6.1. Cellular proliferation and cancer
SRSF3 is known for a long time to be implicated in the cell cycle. Its upregulation is a
hallmark of most cancers and sometimes associated with a poor prognosis. SRSF3 oncogenic
properties seem to be linked to an increased in its phosphorylation and expression levels
which will affect RNA processing of genes linked to cell proliferation, cell cycle and
metabolism. (Jumaa et al. 1997, Jia et al. 2010, Song et al. 2019). SRSF3 silencing in some
cancer cell lines can lead to apoptosis (He et al. 2011, Kuranaga et al. 2018).
During the G1 phase, SRSF3 is upregulated in preparation to the S phase due to this activity
of E2F1 on SRSF3 promoter. This will affect splice site selection by SRSF3, as it is closely
related to the expression level of SRSF3, and the number of alternative splicing events. As
mentioned in a previous section, SRSF3 also regulates the alternative splicing of the exon 4
of its own pre-mRNA along with other splicing factors (SRSF1 or SLU7 for example),
affecting its biological functions and activities (Jumaa et al. 1997, Che et Fu 2020). Exon 4
is normally skipped and its inclusion through alternative splicing results in a premature stop
codon rendering SRSF3 RS domain useless (Jimenez et al. 2019, Che et Fu 2020). This, of
course, could serve as a negative feedback loop to the SRSF3 upregulation in the G1 phase.
An SRSF3 expression over physiological levels during the G1/S checkpoint leads to
development of cancer (Kurokawa et al. 2014). In cancer cells, SRSF3 regulates the
expression of genes linked the G1/S phase progression such as CCND1, CCND3, E2F1 or
E2F7 at both RNA and protein level. Contrary to other SR proteins (SRSF1 for example),
the role of SRSF3 during transcription initiation is less clear. Instead, it is possible that the
lower gene expression at RNA level comes from alternative splicing coupled to nonsense
mediated decay which will then impact protein output levels.SRSF3 is also implicated in
G2/M phase progression (Jia et al. 2010). In cancer cells, it regulates the expression at RNA
and protein levels of G2/M phase progression linked genes such as the transcriptional factor

166
FOXM1. SRSF3 higher expression favours the alternative splicing of FOXM1 into
functionally active FOXM1b and FOXM1c, instead of FOXM1a. SRSF3 activity is also
linked to the expression of other cancer/cell proliferation-related genes such as ETV1, NDE1
or RAC1 (Goncalves et al. 2009, Song et al. 2019). SRSF3 regulates the alternative splicing
of apoptosis inducers HIPK2 (Kurokawa et al. 2014) and TP53 (Tang et al. 2013) and the
alternative polyadenylation of PTEN (Shen et al. 2019) to prevent cellular senescence.
SRSF3 also regulates the alternative splicing, mRNA export and the translation of the anti-
oncogene PDCD4 (Park et Jeong 2016). In cancer cells, SRSF3 is linked to the nonsense
mediated decay of PDCD4 isoform 2 and reduces the export of both PDCD4 isoforms.
SRSF3 also blocks translation initiation of the isoform 1 by binding PDCD4 mRNA 5’UTR.
Along with the regulation of alternative splicing of cell-cycle or cell proliferation-linked
mRNAs, SRSF3 also regulates the maturation of some miRNA in cancer cells such as miR-
21 (Ajiro et al. 2016).
SRSF3 is responsible for carbohydrate metabolism disturbance that predispose to cancer (Sen
et al. 2013, Sen et al. 2015, Kuranaga et al. 2018). In cancer cells, SRSF3 alternatively splices
PKM predominantly to its PKM2 isoform (Kuranaga et al. 2018) which allows cancer cell to
preferentially use glycolysis as energy source. Moreover, a hepatocyte-specific knockout of
SRSF3 in mice induces hepatocellular carcinoma after developing chronic liver disease due
to alternatively splicing of metabolism-linked genes such as Ern1, Hmgcs1, Hnf1α or Xbp1
(Sen et al. 2015). Lastly, SRSF3 also regulates the splicing of Insr, which aberrant splicing
is responsible for abnormal cell proliferation in hepatocellular carcinoma and correlates with
the risk of tumor in patients (Sen et al. 2013, Wang, Lekbaby, et al. 2019).
Finally, dysregulation of SRSF3 activity and alternative splicing can also disrupt autophagy.
Overexpression of SRSF3 in cancer cells inhibits the cell capacity for autophagic activity
(Zhou et al. 2019). A SRSF3 overexpression reduces RELA (p65) and FOXO1 transcripts
and protein level, probably by affecting their mRNA stability. p65 and FOXO1 are
transcription factors that target, amongst others, the gene BECN1, necessary for cellular
autophagic activity.

167
SRSF3, through its implication in many steps of RNA processing, impacts cancer
development by disrupting autophagy, modifying cell metabolism and promoting cell growth
and survival. SRSF3 directly or indirectly (through its target transcripts) influences cell
proliferation pathways such as JNK, PI3-kinase/Akt or Wnt/β-catenin signaling pathways,
making SRSF3, in most cancers, an important oncogene (Goncalves et al. 2009, Kano et al.
2013).
4.6.2. Viral infection
Viruses have to ability to hack cellular machinery in order to replicate. Being a central part
of the RNA processing machinery, SRSF3 is used by many viruses to handle their genetic
material for transcription, export to the cytoplasm and translation. Viruses control the
upregulation of SRSF3 to help in these cellular processes. For example, the transcription
factor E2 from human papillomavirus (HPV) drives SRSF3 expression directly through its
promoter (Mole et al. 2009, Klymenko et al. 2016). This upregulation is necessary for the
correct splicing and transcription of HPV capsid protein L1 and regulates the early-to-late
infection phase switch (Jia et al. 2009). Moreover, human immunodeficiency virus requires
the correct expression level of SRSF3 for an accurate and balanced splicing and stability of
its pre-mRNAs (Wong et al. 2013).
SRSF3 is responsible for the export of viral transcripts into the cytoplasm. Herpes simplex
virus 1 regulatory protein ICP27 directly interacts with NXF1 for viral RNA export.
SRSF3,as another partner of NXF1, seems important to viral RNA export as it also bind
ICP27 and its knockdown reduced viral yield (Escudero-Paunetto et al. 2010). Although the
precise mechanism by which SRSF3 is involved in viral transcript export is not known,
SRSF3 could possibly bind the intronless viral transcripts through one of its binding motif
sequences as it does in non-infected cells. During Varicella-Zoster virus infection , the viral
protein IE4 interacts with SRSF3 in a RNA independent manner and helps recruit NXF1 to
the RNA/protein complex to promote viral RNA export (Ote et al. 2009).

168
As said previously, the role of SRSF3 during translation initiation is not quite clear, contrary
to other SR proteins. Nonetheless, SRSF3 has a role in the translation initiation initiated by
internal ribosome entry site (IRES) (Bedard et al. 2007, Fitzgerald et al. 2013). Although
IRES initiated translation is not common in constitutive translation initiation, its use is
widespread method during viral replication. SRSF3 RS domain interacts with the protein
PCBP2 on the IRES to help recruit translation machinery. SRSF3 knockdown lead to a
decrease in viral genes translation. Some RNA viruses such as picornaviruses actively
promotes the cytoplasmic accumulation of SRSF3 for IRES initiated mediated translation
through the expression of its 2A proteinase (Fitzgerald et al. 2013). 2A proteinase cleaves
nuclear pore proteins during infection and possibly preventing SRSF3 to enter the nucleus,
resulting in SRSF3 cytoplasmic accumulation. During papillomavirus infection, cytoplasmic
SRSF3 is targeted to cytoplasmic foci (Fitzgerald et Semler 2013). Although SRSF3 is
known to be a structural components of stress granules during cellular stress (Twyffels et al.
2011, Jayabalan et al. 2016), papillomavirus-induced cytoplasmic foci seem to be of another
nature as they sometimes lack the protein TIA-1, a common stress granule marker (Fitzgerald
et Semler 2013). The SRSF3-containing cytoplasmic foci could be used to regulate viral
translation and/or viral RNA replication.
4.6.3. Gametogenesis and development
SRSF3 has in important role in gamete maturation and embryonic development. During
development, the expansion of the proteome from a limited genome though alternative
splicing is of utmost importance for cell and tissue differentiation (Baralle et Giudice 2017).
As a master regulators of alternative splicing, SR proteins and SRSF3, in particular, are
responsible to generate the variance in transcripts necessary for the cellular and tissular
diversity.
SRSF3 is essential for embryonic development. In mice, the lack of SRSF3 in zygotes lead
to a failure to form blastocytes as the embryo dies during the morula stage (Jumaa et al.
1997).SRSF3 presence seems to be critical even before fertilisation as a maternally inherited
factor. During oocyte maturation, SRSF3 is upregulated during the prophase I (GV) and

169
meiosis II which are both preceded by active transcription state (Do et al. 2018). The loss of
SRSF3 in mouse oocytes during GV prevents zygotes to develop further than a two cells
stage, showing the necessity for SRSF3 in embryo preimplantation phase. During GV,
SRSF3 controls exon inclusion events in genes linked to GV breakdown, namely BRD8 and
PDLIM7. During early embryonic development, SRSF3 controls cellular pluripotency. It is
involved in the mRNA stability of KIF4, MYC, POU5F1, or SOX2 transcripts, to name a
few. Moreover, SRSF3 influences pluripotency independently of splicing or mRNA stability
by binding the mRNA of pluripotency transcription factor NANOG in the nucleus to facilitate
its nuclear export which affect NANOG protein output (Ratnadiwakara et al. 2018). It is
suggested that this mechanism might be shared by cancer cells to acquire cellular plasticity.
Although SRSF3 knock out is embryonically lethal, cell-type specific deletion of SRSF3
allows to study its role in the formation of different organs. SRSF3 is crucial for liver
development. Hepatocyte-specific knockdown of SRSF3 stops hepatocyte maturation,
prevents the liver acquiring the correct cellular architecture, alters lipid and glucose
metabolism and causes perinatal death in mice (Sen et al. 2013). Splicing and expression of
genes linked to liver development is changed upon SRSF3 knockdown. For example,
transcription factors such as HNF1A, HNF3B, HNF6A or TBX3, while the splicing of
HNF1A is prominently altered. This results in a changed expression of the transcription
factors targets such as the growth hormone receptor, leading to growth defects. SRSF3 is also
critical for heart development. During development, SRSF3 expression reaches a peak mid-
gestation and goes down with age. Cardiomyocyte-specific knockdown of SRSF3 in mice
lead to reduced cardiomyocyte proliferation and embryonic lethality. Inducible knockdown
of SRSF3 in adult mice induced aberrant heart contraction and death (Ortiz-Sánchez et al.
2019). The effects of SRSF3 knock out in embryos and adult mice were cause by a
dysregulation of contraction linked genes as SRSF3 controls the expression or the RNA
stability of SERCA2A, TNNC1 or TNNT2. Furthermore, SRSF3 is responsible for the
adequate splicing of the kinase mTOR in cardiomyocytes. An aberrantly spliced mTOR
promotes the de-capping of other mRNAs, resulting in SRSF3 indirectly affecting the
stability of those transcripts. Finally, SRSF3 is essential to B cell maturation. Its B cell
specific knockdown resulted in a drastic reduction of B cells in the bone marrow, spleen, and

170
thymus. The actual gene targets of SRSF3 necessary for B cells proliferation are not yet
known.
4.6.4. Metabolism
SRSF3 role in metabolism has already been mentioned since dysregulation in metabolism
can affect or be affected by cancer or development. As said previously, SRSF3 is involved
in hepatocytes maturation and liver cancer by controlling the alternative splicing of genes
and transcription factor directly or indirectly linked to metabolism (ERN1, HMGCS1,
HNF1A or XBP1) (Sen et al. 2013, Sen et al. 2015). In normal conditions, hormones and
nutrients levels control SRSF3 expression. Insulin promotes SRSF3 phosphorylation which
in turn splices G6PD to promote de novo lipogenesis (Walsh et al. 2013b). Interestingly,
while insulin affects SRSF3 activity, SRSF3 affects insulin receptor gene (INSR) processing
by alternatively splicing INSR exon 11 resulting in the IR-B isoform (Sen et al. 2009).
Contrary to the IR-A isoforms, IR-B cannot bind insulin-like growth factor, but only binds
insulin, keeping IR-B role solely to glucose homeostasis. Furthermore, SRSF3 is implicated
in energy production and glycolysis by promoting the expression of PKM2 isoform of the
PKM gene. As said, previously, it is a mechanism used by cancer cells (Kuranaga et al. 2018)
but also a way to protect cells against glucose deprivation conditions (Jiang et al. 2018).
Dysregulation of SRFS3 in the liver, namely a loss of expression of SRSF3, leads to non-
alcoholic fatty liver disease, non-alcoholic steatohepatitis and cirrhosis (Kumar et al. 2019).
Lipid excess, a hallmark of those diseases induces oxidative stress in hepatocytes that leads
to proteasomal degradation of SRSF3. As splice site selection by SRSF3 is regulated by its
expression level, some of its target genes (INSR, SLK, FN1 or MYO1B) were aberrantly
spliced affecting cell function and survival.
4.6.5. Inflammation
SRSF3 is implicated in inflammation regulation. As said previously, SRSF3 is involved in
liver disease which is accompanied with overt inflammation. In an alcoholic liver injury
model, SRSF3 splicing and expression is modulated by SLU7 (Wang, Kainrad, et al. 2018).

171
SLU7 favours the inclusion of SRSF3 exon 4, rendering SRSF3 useless. The functional
knockdown of SRSF3 lead to steatosis, fibrosis and hepatitis (Sen et al. 2015, Wang, Kainrad,
et al. 2018). Moreover, SRSF3 expression can be modulated by the growth factor TGFβ, also
implicated in fibrosis (Hallgren et al. 2012).
In monocytes, SRSF3 works as a negative regulator for interleukins (IL) secretion upon
bacterial infection (Moura-Alves et al. 2011, Zhang, Niu, et al. 2018). This regulation acts
downstream of NF-κB reporter-dependant transcription of interleukins mRNA After
infection, SRSF3 mRNA and protein levels are downregulated which increases IL-1β and
IL-8 mRNA stability. Moreover, SRSF3 downregulation also increases the mRNA stability
and activity of caspase-1 which is responsible for the cleavage and maturation of IL-1β prior
to its secretion (Moura-Alves et al. 2011). Interestingly, SRSF3 downregulation also affects
the splicing of IL-4, as it upregulates its different isoforms (Zhang, Niu, et al. 2018). Hence,
upon infection, SRSF3 downregulation affects the expression of both pro-inflammatory and
anti-inflammatory cytokines. It is yet unknown if theses mechanisms happen at the same time
or are regulated at different time points after infection. Finally, SRSF3 is involved in the
splicing of the pathogen-associated molecular pattern receptor TLR4. TLR4 is responsible
for the detection of bacterium through some membrane components such as
lipopolysaccharides (LPS) and is upstream of the Nf-κB signaling pathway. Interestingly,
SRSF3 thus controls many steps of inflammatory signaling via splicing and mRNA stability.
4.6.6. Central nervous system homeostasis and disorders
In the central nervous system (CNS), different SR proteins regulate neural cell differentiation
through the expression of distinct gene sets and a mix of splicing, mRNA stability and mRNA
export of these genes. (Ankö et al. 2010). In the adult brain, SRSF3 is responsible for the
adequate splicing of the microtubule associated protein tau (MAPT) (Yu et al. 2004). SRSF3
modulates the splicing of MAPT exon 10. The inclusion of the exon 10 of MAPT is
associated with frontotemporal dementia with parkinsonism and other tauopathies. Contrary
to its usual splicing behavior, it was found that a higher expression of SRSF3 would promote
the exclusion of exon 10, preventing the possible apparition of tauopathies. In Alzheimer’s

172
disease, SRSF3 is responsible for the upregulation of neuron specific, Alzheimer’s disease
associated protein TRKB (Wong et al. 2012). SRSF3 regulates TRKB splicing into the
TRKB-SHC variant by promoting the inclusion of its exon 19. Reciprocally, the higher rate
of exon inclusion was accompanied with SRSF3 upregulation in vitro and in Alzheimer’s
patient hippocampus.
In another context, SRSF3 seems to be also implicated in bipolar disorder where it is
upregulated in the blood of patients during the depressive and remissive phases (Watanuki et
al. 2008). The cause of SRSF3 upregulation or its gene targets in bipolar disorder are not yet
known. Moreover, the there was not significant upregulation of SRSF3 in major depressive
disorder patient, making SRSF3 expression level a possible molecular method to distinguish
the two disorders even at the first depressive episode in bipolar disorder patients. Instead, in
major depressive disorder SRSF3 seems to be implicated in the splicing of the glucocorticoid
receptor (GR), a protein associated to mood disorder. SRSF3 changes the ratio of GR-β/GR-
α, favouring the GR-β isoform.
In the spinal cord, SRSF3 modulation is associated with a better outcome of spinal muscular
atrophy (SMA). In SMA, the gene SMN1 is deficient on both alleles and normally cannot be
replaced by SMN2 as a lack of SMN2 exon 7 renders the protein mostly inactive and prone
to proteasomal degradation. An artificial overexpression of SRSF3 seems to induce a more
frequent inclusion of SM2 exon 7 which stabilised the protein product in vitro (Yuo et al.
2008). Interestingly, a previous study showed no correlation in exon 7 inclusion with SRSF3
overexpression (Young et al. 2002). Strangely enough, a more recent study proposed that
SRSF3 is instead a negative regulator of exon 7 inclusion (Wee et al. 2014). The knockdown
of SRSF3 increased the exon 7 inclusion and abundance of the SMN protein product in SMA
patient-derived cells. The discrepancy on the role of SRSF3 on the inclusion of SMN2’s 7th
exon could be explain the difference in the cellular model used in these studies or by the
expression of other probable cell-type specific SR proteins, such as SRSF9/Srp30c (Young
et al. 2002).

173
SRSF3 is a potent negative regulator of immune transcripts translation in microglial cells,
the resident macrophage of the CNS. As explained in a previous section, SRSF3 can bind the
3’UTR of translating transcripts and stalls their translation in microglial cells upon
stimulation with LPS (Boutej et al. 2017). This was put in evidence by the mouse model used
in this study. An EDTA-translating ribosome affinity purification allows the isolation and
identification of ribosome-bounds mRNAs and in-translation peptides. While highly
upregulated immune mRNAs such as Saa3, Lcn2, Ccl5, or Ccl3 could be found by
microarray, there was no trace of their protein product. The incomplete peptides from the
stalled translation are degraded through a yet unknown mechanism. Following, LPS
stimulation SRSF3 expression did not change but its phosphorylation was greater than
control tissue. Upon a knockdown of SRSF3 in mice brain, de novo translation of the previous
silenced transcripts was achieved. The stimulation of microglia by LPS is possible through
TLRs which are responsible for the detection of pathogen associated molecular patterns and
danger associated molecular patterns, in manner similar to the microglial activation in many
neurogenerative disorders such as amyotrophic lateral sclerosis (ALS), indicating a possible
role for SRSF3 in those diseases. Although, there is a nuclear segregation of SRSF3 found
in ALS cultured neurons (Kim et al. 2017), a high quantity of phosphorylated SRSF3 is
observed in microglial cytoplasm of symptomatic mice lumbar spinal cord (See Chapter 3).
As for transcripts from LPS-stimulated microglia, highly upregulated immune transcripts of
ALS microglia were not translated. The protein product of transcripts such as CLEC7A,
CST7, LILRB4, ITGAX or CCL3, implicated among others in phagocytosis, could be
rescued upon SRSF3 knockdown. Moreover, phagocytosis by microglial cells along with
mice disease progression and survival was ameliorated. The same set of microglial immune
transcripts has also been identified in microglia-specific transcriptome in aging and diseases
affecting the CNS such as Alzheimer’s disease, multiple sclerosis or Parkinson’s disease
(Keren-Shaul et al. 2017, Krasemann et al. 2017, Kang et al. 2018, Haure-Mirande et al.
2019), although SRSF3 implication on translational stalling in these conditions, bridging
inflammation and CNS health, has not yet been studied.

174
Figure 16: Gene targets of SRSF3 in biological pathways
Non-exhaustive list of SRSF3 gene targets. SRSF3, through its different molecular functions
is implicated in homeostasis and in the onset, progression and outcome of many diseases by
modulating the splicing, the mRNA stability, the mRNA export or the translation of many
genes. Gene targets whose name are in red are found in different biological pathways or are
affected by SRSF3 through different mechanisms.
4.7. Conclusion
From the previous section, it is now obvious that SRSF3 possesses a prominent role in
modulating the outcome of different diseases. SRSF3 expression or phosphorylation could
be adjusted as a therapeutic approach as many studies already demonstrated. Modulating
SRSF3 could help restoring the proper balance between spliced isoforms, increase or
decrease mRNA stability and recover the right amount of protein output of gene targets. In
fact, SRSF3 knockdown by anti-sense nucleotides of siRNAs already showed promises to
alleviate a SRSF3 dysregulation in vitro and in animal models in different diseases such as
cancer or inflammatory diseases (See Chapter 3) (Bedard et al. 2007, Wee et al. 2014, Boutej

175
et al. 2017, Kuranaga et al. 2018, Sun, Yan, et al. 2019). Moreover, some classical drugs also
have been shown to reduce SRSF3 expression. For example, Resveratrol, was found to
induce SRSF3 exon 4 inclusion and directly or indirectly SMN2 exon 7 inclusion, showing
its possible relevance in the treatment of spinal muscular atrophy (Markus et al. 2011).
Moreover, Digoxin suppresses viral replication of HIV1 by altering SRSF3 phosphorylation
(Wong et al. 2013) although the effect of the drug on the actual change of SRSF3 activity is
not yet clear (Anderson et al. 2012, Wong et al. 2013).
In conclusion, while the molecular mechanisms underlying SRSF3 role in splicing and
alternative splicing are relatively well known, there is still some work to do to understand by
which means SRSF3 affect RNA processing in the cytoplasm, before and during translation.
From the nucleus to the cytoplasm, SRSF3 ways to expand and modify the transcriptome and
proteome are intertwined with each other and the relation between each step of RNA
processing by SRSF3 must be comprehended in order to develop efficient treatment targeting
this RNA binding protein.

176
Discussion générale
5.1. Résumé des chapitres précédents
La SLA est une maladie à la pathogénèse cellulaire non-autonome. Ainsi, plusieurs cellules,
autres que les neurones moteurs, participent au développement des symptômes et à la
progression de la maladie. Les cellules du système immunitaire sont recrutées
différentiellement selon le stade de la maladie et peuvent influencer le cours de la maladie.
Le principal composant du système immunitaire dans le SNC, la microglie, présente un
changement drastique de phénotype entre la phase présymptomatique et la phase
symptomatique de la SLA. Elle possède tout d’abord un phénotype anti-inflammatoire et
neuroprotecteur avant de changer, à l’apparition des symptômes, pour un phénotype
d’activation aberrant et neurotoxique (Henkel et al. 2009, Béland et al. 2020). La complexité
du changement phénotypique et fonctionnel de la microglie demande l’utilisation de
technique de profilage moléculaire pour déterminer la nature de ce changement. Le profilage
moléculaire permet d’étudier différents ensembles des molécules biologiques, tels que le
transcriptome, le protéome ou le sécrétome, en faisant abstraction d’informations antérieures
pouvant amener un biais de recherche. Dans cette thèse, nous démontrons l’importance de
l’utilisation de différents outils de profilage moléculaire pour examiner les phénotypes des
cellules microgliales et expliquer leurs changements à travers les stades de la SLA.
Dans le second chapitre, nous identifions la cytokine IL-10, en étudiant le sécrétome comme
responsable du maintien du phénotype anti-inflammatoire microgliale associé au stade
présymptomatique de la maladie (Gravel et al. 2016). La surexpression de cette cytokine a
été identifiée au niveau de son ARNm et de sa protéine. L’origine de cette cytokine durant le
stade présymptomatique de la maladie est microgliale. L’action de la cytokine IL-10 peut
être modulée et affecte la progression de la maladie, confirmant son rôle bénéfique dans le
maintien du phénotype anti-inflammatoire de la microglie.
Dans le 3e chapitre, nous étudions le transcriptome et le protéome microgliale à travers
différents stades de la SLA dans un modèle murin en nous assurant du caractère spécifique à
un type cellulaire du modèle utilisé. La spécificité à un type cellulaire (plus précisément la

177
spécificité à la cellule microgliale) est atteinte par l’utilisation de la technique EDTA-TRAP,
qui permet de récupérer à partir d’un même échantillon les ARNm collés aux ribosomes en
traduction et les peptides nouvellement synthétisés.(Boutej et al. 2017). L’isolation des
ribosomes microgliaux depuis un tissu complexe (la moelle épinière lombaire), puis
l’indentification subséquente des transcriptome et protéome microgliaux permet de
comprendre et d’expliquer la nature du changement phénotypique de ce type cellulaire dans
la maladie. En utilisant la méthode EDTA-TRAP, nous découvrons une disparité importante
entre le transcriptome et le protéome de la microglie et que la protéine SRSF3 pourrait être à
l’origine de cette disparité. Dans les stades symptomatiques de la maladie SRSF3 est
hautement phosphorylé, s’accumule dans le cytoplasme des cellules microgliales et est lié à
des ARNm issus de gènes immunitaires ayant des fonctions phagocytaires. Le knockdown de
SRSF3 mène à la synthèse des ARNm immunitaires précédemment réprimés, au
rétablissement de la phagocytose de la microglie et un ralentissement de la progression de la
maladie dans un modèle murin. Le potentiel thérapeutique de la régulation de SRSF3 pour
traiter de la SLA mériterait ainsi d’être approfondi.
Dans le but de mieux comprendre l’importance de SRSF3 dans la physiologie cellulaire, nous
explorons, dans le 4e chapitre, son rôle dans les divers mécanismes du métabolisme de l’ARN
et dans la régulation d’expression des gènes. Ont aussi été étudié sa phosphorylation, sa
localisation cellulaire, son niveau d’expression et l’importance de ces paramètres dans
l’homéostasie cellulaire et le développement de maladies. L’implication de SRSF3 dans tant
de phénomènes physiologiques et la complexité de sa fonction démontre l’ampleur du défi
pour son utilisation comme cible thérapeutique dans la SLA où son expression doit être
balancée.
Nous estimons que les travaux de recherches présentés dans cette thèse répondent aux
hypothèses de travail proposées dans la section 1.4. En effet, il a été possible d’enrichir les
connaissances scientifiques sur le rapport entre la microglie et la neuroinflammation dans la
SLA. Nous avons pu identifier IL-10 comme la cytokine responsable du maintien du
phénotype anti-inflammatoire microglial, présent durant la phase présymptomatique de SLA.
Aussi, nous avons pu élucider, du moins en partie, la nature du phénotype aberrant et

178
neurotoxique de la microglie durant la phase symptomatique. La neurotoxicité de la microglie
pourrait, entre autres, provenir de son incapacité à correctement phagocyter, démontré entre
autres par l’absence de traduction d’ARNm liés à la fonction phagocytaire. En modifiant
l’expression de SRSF3, un régulateur génique, il a été possible de récupérer cette fonction
cellulaire altérée. La modification indirecte de l’expression d’une multitude de gène de la
même voie de signalisation permet, chez un modèle murin, de réduire la vitesse de la
progression de la maladie et de prolonger la survie.
5.2. Profilage du sécrétome de la microglie présymptomatique
L’étude du sécrétome correspond à l’identification des facteurs sécrétés par un ou plusieurs
types cellulaires. L’étude du sécrétome est importante pour comprendre l’interaction entre
les cellules dans un tissus et la signalisation autocrine, paracrine ou endocrine. La nature des
facteurs sécrétés aura un impact sur la différentiation et l’activation des cellules avoisinantes
ou le recrutement de cellules en périphérie (Willis et al. 2020). Le sécrétome peut aussi être
étudié de façon connexe avec le sensome, soit l’ensemble des récepteurs cellulaires
permettant la détection des facteurs sécrétés (Hickman et El Khoury 2019). Dans la SLA, le
sécrétome et le sensome ont fait l’objet de plusieurs études surtout durant le stade
symptomatique de la maladie et parfois sans spécificité cellulaire. (Henkel et al. 2009, Appel
et al. 2011).
Dans le Chapitre 2, pour la première fois, le profil du sécrétome microglial, et plus
précisément le profil des cytokines de la microglie durant le stade présymptomatique de la
SLA est étudié. Le profil de cytokines est d’abord étudié au niveau de l’ARN. Le niveau de
régulation d’un groupe présélectionné de cytokines importantes a été vérifié par qRT-PCR
dans des cellules microgliales adultes en culture et validée par cytokine array, permettant la
détection simultanée de 40 cytokines. Ainsi, le niveau de régulation des cytokines est
confirmé au niveau des protéines. La spécificité cellulaire de l’expression de la protéine IL-
10 a finalement été validée par cytométrie en flux. Comme mentionné précédemment, les
expériences subséquentes de cette étude ont montré l’importance de la cytokine IL-10 dans
le maintien d’un environnement immunosuppressif et anti-inflammatoire dans la moelle

179
épinière des souris présymptomatiques. L’induction de la cytokine IL-10 par vecteur viral
durant le stade présymptomatique de la SLA a été l’objet de deux autres études qui viennent
valider nos propres résultats (Ayers et al. 2015, Strickland et al. 2020). Pour compléter
l’image du profil de cytokine de la microglie au cours de la maladie, une autre étude a fait le
rapport du profil de cytokines de la microglie durant la phase symptomatique de la maladie
(Nikodemova et al. 2014). En effet, cette étude montre que le patron de cytokines de la
microglie symptomatique ne correspond pas à un phénotype pro-inflammatoire ou à un
phénotype anti-inflammatoire chez la microglie symptomatique. En effet, les cytokines les
plus représentatives de ces deux phénotypes (pro-inflammatoire : TNFα, IL-6 et anti-
inflammatoire : IL-10, IL-4) voient leur expression baisser avec la progression de la maladie.
Nikodemova et al. proposent ainsi l’existence d’un phénotype microglial unique, spécifique
aux maladies des neurones moteurs, issu de l’activation chronique de la microglie, où la
production des facteurs pro-inflammatoires et anti-inflammatoires est aberrante. Ce profil
aberrant de la microglie a bien été étudié au niveau du chapitre 3.Outre la microglie, l’autre
source d’IL-10 dans le CNS sont les cellules Treg (Kleinewietfeld et Hafler 2014). Or, la
population de ces cellules est réduite, tout comme la microglie anti-inflammatoire dans le
stade symptomatique de la maladie (Beers et al. 2017) et leur déplétion corrèle avec la
sévérité des symptômes. L’action bénéfique des Treg est possiblement liée à sa production
d’IL-10.
Il est intéressant de voir qu’IL-10 est une cytokine importante dans plusieurs autres maladies
neurodégénératives. Par exemple, elle est sécrétée par les lymphocytes B régulateurs et réduit
l’inflammation retrouvée dans la sclérose en plaques. Un traitement anti-CD20, permet de
réduire l’ensemble des lymphocytes B, généralement neurotoxiques, et de guérir la sclérose
en plaques. Toutefois, ce traitement déplète aussi les lymphocytes B régulateurs et empêche
leur production d’IL-10 et son effet bénéfique dans les tissus affectés par la sclérose en
plaques (Wang et al. 2020). De plus, dans la maladie d’Alzheimer, une plus grande
production d’IL-10 est associée à une moins grande perte cognitive et meilleure neurogénèse
(Kiyota et al. 2012, Porro et al. 2020) Dans la maladie de Parkinson, le rôle d’IL-10 est plutôt
mitigé (Porro et al. 2020). Alors que certaines études montrent que la production d’IL-10
serait un facteur de risque pour cette maladie (Rentzos et al. 2009, Li et al. 2012), d’autres

180
études ne voient pas d’effet de cette cytokine dans la maladie de Parkinson (Menza et al.
2010, Pascale et al. 2011, Chu et al. 2012). Le rôle d’IL-10 dans ces maladies reste à être
mieux exploré. Il serait intéressant de comprendre le dynamisme de la production de cette
cytokine aux différentes phases de ces maladies et de la corréler à l’inflammation et à
l’activité microgliale. En conclusion, IL-10 est cytokine centrale dans le maintien d’un
phénotype microglial immunosuppressif et homéostatique durant la neuroinflammation.
5.3. Profilage moléculaire de la microglie dans la neuroinflammation
Le profilage moléculaire spécifique à un type cellulaire est l’outil central de cette thèse. Dans
le Chapitre 3, afin d’étudier le transcriptome et le protéome de la microglie, nous avons utilisé
l’EDTA-TRAP. Il s’agit de la technique Translating Ribosome Affinity Purification modifée
afin de permettre, depuis le même échantillon l’immunopurification des ARNm liés aux
ribosomes et des peptides qui y sont synthétisés. Pour ce faire, nous avons généré une souris
exprimant une protéine de fusion contenant la sous-unité ribosomique (RPL10a), la protéine
eGFP et l’étiquette Flag spécifiquement dans les cellules microgliales grâce au promoteur
CD11B. La présence de la cycloheximide, des inhibiteurs de RNAses et de magnésium dans
les différents tampons utilisés permet de garder les polysomes dans une état stable. Le
transcriptome microglial a été identifié par analyse de l’ARN par microarray, alors que le
protéome microglial a été identifié suite à un séquençage des peptides par spectrométrie de
masse. Les résultats générés par ces deux analyses sont ensuite comparés à plusieurs banques
de données pour y déceler l’implication de certaines fonctions biologiques des profils
microgliaux.
5.3.1. L’étude du profil transcriptomique de la microglie symptomatique
Selon notre analyse transcriptomique, la microglie symptomatique acquiert un phénotype qui
diffère du phénotype d’activation classique. Alors que l’ARN des facteurs pro-
inflammatoires courants (IL-1β ou TNFα par exemple) sont, pour la plupart, non-régulés,
l’ARN de plusieurs autres gènes sont régulés de plus en plus fortement avec la progression
de la maladie. Par exemple, nous retrouvons une surrégulation de l’ARNm Clec7a, Cst7,
Lilrb4, Gpnmb, Ccl3 Itgax ou encore Cd68. Après l’analyse des fonctions biologiques de ces

181
gènes, il est possible de souligner leur rôle dans l’inflammation et dans l’immunité. En effet,
en utilisant plusieurs banques de données disponible (Kyoto Encyclopedia for Genes and
Genomes, Wikipathways, Reactome ou Gene Ontology), on remarque la présence de ces
gènes regroupés dans des fonctions comme l’activation de la réponse immunitaire ou
inflammatoire, l’immunité médiée par les leucocytes ou encore, la régulation positive de la
phagocytose. Ainsi, selon son profil transcriptomique, la microglie symptomatique présente
un clair phénotype d’activation et elle serait capable de phagocyter les divers débris cellulaire
et DAMPs retrouvés dans les tissus SLA. Ce profil microglial décrit dans le 3e chapitre de
cette thèse a été retrouvé dans d’autres maladies par diverses équipes de recherche et
correspond au phénotype DAM (voir la prochaine section). Les quelques études qui se sont
intéressées à ce phénotype l’ont étudié au niveau de son profil transcriptomique et lui
proposent un effet bénéfique sur la progression des maladies neurodégénératives, dû à son
potentiel phagocytaire. De plus, on retrouve une forte ressemblance des marqueurs du
phénotype DAM entre les études, malgré la disparité des méthodes utilisées pour son
profilage.
5.3.2. Le phénotype DAM dans les maladies neurodégénératives
Récemment, plusieurs autres études ont aussi fait rapport d’un phénotype microglial unique
associé aux maladies neurodégénératives. Ce phénotype a été nommé microglie associée à la
maladie (disease associated microglia : DAM) ou phénotype microglial neurodégénératif
(microglial neurodegenerative phenotype : MGnD) (Chiu et al. 2013, Noristani et al. 2015,
Weissmann et al. 2016, Cooper-Knock et al. 2017, Keren-Shaul et al. 2017, Krasemann et al.
2017, Deczkowska et al. 2018, Kang et al. 2018). Comme mentionné dans l’introduction, ce
phénotype est étudié principalement au niveau de transcriptome microglial et est caractérisé
par la perte d’expression d’ARNm liés à l’homéostasie et la surexpression d’ARNm liés à
l’immunité ou l’inflammation. On retrouve une ressemblance notable des gènes régulés
caractérisant le phénotype DAM entre les différentes études tel que montré dans le Tableau
2 qui dresse une liste non-exhaustive des ARNm surexprimés et produit avec les données
supplémentaires disponibles des études énumérées. De plus, nos propres résultats du
transcriptome microglial symptomatique (voir le Chapitre 3) résument fidèlement le

182
phénotype DAM selon les gènes les plus régulés au niveau de l’ARN. Ainsi, avec les efforts
réunis de plusieurs équipes de recherche, il a été possible de décrire un nouveau phénotype
microglial selon sa signature transcriptomique. Malgré ces efforts, le phénotype DAM reste
à l’état descriptif et peu d’informations existent quant à sa fonction réelle, à son impact sur
la pathogénèse des différentes maladies neurodégénératives dans lequel il est impliqué ou
encore, à la manière dont il est induit. Certaines études montrent que APOE serait
responsable du développement de ce phénotype, modifiant en aval l’expression des autres
gènes de DAM (Krasemann et al. 2017, Kang et al. 2018), alors que qu’une autre étude laisse
ce rôle à TREM2 (Keren-Shaul et al. 2017, Krasemann et al. 2017). APOE et TREM2
modifieraient le programme transcriptionnel et post-transcriptionnel des cellules
microgliales, supprimant leurs fonctions tolérogènes et homéostatiques et activant leurs
fonctions liées au phénotype DAM (Krasemann et al. 2017). Les fonctions spécifiques des
gènes surrégulés du phénotype DAM suggéreraient un rôle dans la phagocytose et la
clairance des débris cellulaires (Deczkowska et al. 2018). Ainsi, la présence du phénotype
DAM dans l’ensemble des cellules microgliales ou seulement dans une sous-population
pourrait être bénéfique en retirant les DAMPs de l’environnement cellulaire (Deczkowska et
al. 2018)
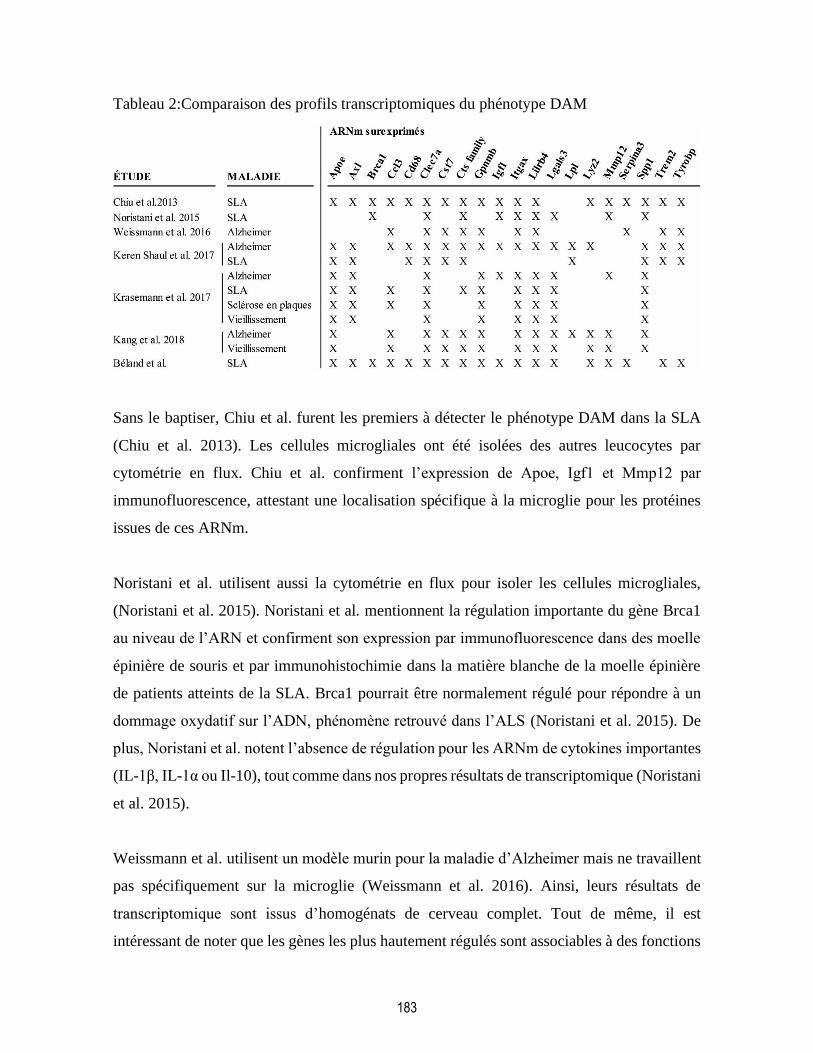
183
Tableau 2:Comparaison des profils transcriptomiques du phénotype DAM
Sans le baptiser, Chiu et al. furent les premiers à détecter le phénotype DAM dans la SLA
(Chiu et al. 2013). Les cellules microgliales ont été isolées des autres leucocytes par
cytométrie en flux. Chiu et al. confirment l’expression de Apoe, Igf1 et Mmp12 par
immunofluorescence, attestant une localisation spécifique à la microglie pour les protéines
issues de ces ARNm.
Noristani et al. utilisent aussi la cytométrie en flux pour isoler les cellules microgliales,
(Noristani et al. 2015). Noristani et al. mentionnent la régulation importante du gène Brca1
au niveau de l’ARN et confirment son expression par immunofluorescence dans des moelle
épinière de souris et par immunohistochimie dans la matière blanche de la moelle épinière
de patients atteints de la SLA. Brca1 pourrait être normalement régulé pour répondre à un
dommage oxydatif sur l’ADN, phénomène retrouvé dans l’ALS (Noristani et al. 2015). De
plus, Noristani et al. notent l’absence de régulation pour les ARNm de cytokines importantes
(IL-1β, IL-1α ou Il-10), tout comme dans nos propres résultats de transcriptomique (Noristani
et al. 2015).
Weissmann et al. utilisent un modèle murin pour la maladie d’Alzheimer mais ne travaillent
pas spécifiquement sur la microglie (Weissmann et al. 2016). Ainsi, leurs résultats de
transcriptomique sont issus d’homogénats de cerveau complet. Tout de même, il est
intéressant de noter que les gènes les plus hautement régulés sont associables à des fonctions

184
microgliales et à l’inflammation. De plus, ces gènes correspondent relativement bien au
phénotype DAM, décrit de façon spécifique par d’autres études chez la microglie uniquement
(Tableau 2) (Chiu et al. 2013, Noristani et al. 2015, Keren-Shaul et al. 2017, Krasemann et
al. 2017, Kang et al. 2018). Il est possible de suggérer que la microglie est le type cellulaire
le plus réactif du SNC lors de la neurodégénérescence. Cela pourrait expliquer la détection
facile du phénotype DAM dans un tissus complexe malgré la contribution d’autres types
cellulaires au pool d’ARNm. Dans l’étude originale, Weissmann et al. ne confirment pas
l’expression d’aucun ARNm régulé au niveau de leur protéine (Weissmann et al. 2016).
Toutefois, Gpnmb, 20e ARNm le plus surrégulé dans Weissmann et al. 2016, est la cible de
l’étude subséquente de cette équipe (Hüttenrauch et al. 2018). L’expression spécifique de
GPNMB dans la microglie est confirmée par immunofluorescence dans des tissus murins et
par immunohistochimie dans de cerveau de patients atteints de l’Alzheimer. Cependant,
aucune étude de cette équipe a tenté de vérifier l’expression des protéines issues des autres
ARNm les plus régulés sur leur liste.
Keren-Shaul et al. étudient les profils transcriptomiques de différentes sous-population de
cellules par cytométrie en flux et par MARS-Seq (Massively parrallel single-cell RNA
sequencing) (Jaitin et al. 2014, Keren-Shaul et al. 2017). Dans la maladie d’Alzheimer, Keren
Shaul et al. identifie le phénotype DAM dans une petite sous-population microgliale et un
phénotype intermédiaire entre le phénotype de repos et le phénotype DAM. La proportion de
la sous-population présentant le phénotype DAM augmente avec la progression de la
maladie. De plus, l’expression d’une certaine partie des gènes serait régulée par TREM2.
Cependant, APOE, un autre régulateur du phénotype DAM identifié ultérieurement, n’est pas
régulé par TREM2 (Keren-Shaul et al. 2017, Kang et al. 2018). Keren-Shaul et al. confirment
l’expression de CSF1, TIMP2, ITGAX, et de LPL dans les cellules microgliales par
immunofluorescence. Selon les fonctions partagées par les ARNm surrégulés, le phénotype
DAM, posséderait une fonction phagocytaire qui permettrait la clairance des débris
cellulaires (Deczkowska et al. 2018). Cependant, la microglie activée présente dans
l’Alzheimer est reconnue depuis un certain temps comme possédant une fonction
phagocytaire déficiente (Mandrekar-Colucci et Landreth 2010). Par MARS-Seq, Keren-
Shaul et al. montrent aussi la présence d’une sous-population microgliale possédant le

185
phénotype DAM (3% de la population totale) présente dans la moelle épinière d’un modèle
murin de la SLA. MARS-Seq est ainsi bon un outil pour disséquer le dynamisme et
l’hétérogénéité de la microglie au niveau de son transcriptome dans les maladies
neurodégénératives. Toutefois, aucune technique similaire ne permet l’identification du
protéome de cellules individuelles.
Krasemann et al. isolent, par cytométrie en flux, les cellules microgliales dans différents
types de neuroinflammation (maladie d’Alzheimer, SLA, sclérose en plaques et
vieillissement) et identifient le phénotype DAM (nommé MGnD) dans une sous-population
microgliale. (Krasemann et al. 2017). Des ARNm surrégulés, Krasemann et al. valident
l’expression de CLEC7A et de TREM2 au niveau de leur protéine. Aussi, Krasemann et al.
montrent, contrairement à Keren-Shaul et al., que la régulation génique du phénotype DAM
par APOE est subséquente et dépendante à TREM2 (Keren-Shaul et al. 2017, Krasemann et
al. 2017). De plus, la modification de l’activité de l’un ou l’autre régulateur génique entraîne
la perte du phénotype DAM.
Kang et al. utilisent, comme nous, la méthode TRAP pour l’identification du transcriptome
microglial, qu’ils étudient dans la maladie d’Alzheimer et le vieillissement (Kang et al.
2018). Kang et al. notent que la méthode TRAP engendrerait moins de variabilité inter-
échantillon et permet un meilleur enrichissement des ARNm microgliaux comparativement
aux différentes stratégies de production de suspension de cellules individuelles De plus, Kang
et al. réaffirment l’importance de APOE dans l’activation du phénotype DAM.
Malheureusement, Kang el al. ne valident l’expression d’aucun ARNm surexprimé dans leur
analyse.
Les différentes études énumérées plus haut se sont basées sur l’analyse de l’ARN et donc de
la transcriptomique comme premier outil d’étude du profil moléculaire de la microglie.
Toutefois, dans l’ensemble de ces études, une petite fraction des découvertes faites au niveau
de l’ARN est validée au niveau des protéines. Ainsi, le phénotype DAM pourrait, dans la
majorité de ces études, ne pas représenter effectivement le vrai phénotype microglial. De
plus, tel que mentionné plus haut, la fonction phagocytaire du phénotype DAM, donné par la

186
nature de ses ARNm les plus régulés, est en contradiction avec le paradigme actuel présentant
un épuisement microglial et une incapacité à détruire les débris cellulaires dans la maladie
d’Alzheimer (Mandrekar-Colucci et Landreth 2010, Deczkowska et al. 2018). Grâce à notre
étude comparative transcriptomique versus protéomique, nous montrons pour la première
fois que le phénotype DAM, par notre analyse transcriptomique du profil microglial au stade
symptomatique de la SLA, n’est pas le vrai phénotype de la microglie puisque tous ces gènes
fortement régulés ne figurent pas dans notre analyse protéomique. Mieux encore, nos
validations par immunobuvardage montrent justement que les protéines codées par ces
ARNm du phénotype DAM ne sont pas produites. Outre quelques gènes (GPNMB, C1QA,
C1QB, C1QC, LGALS3, par exemple), les gènes régulés au niveau de l’ARN ne sont pas
traduits ou régulés au niveau des protéines. De plus, nous trouvons que la fonction
phagocytaire du phénotype DAM n’est pas présente dans les cellules microgliales provenant
d’un modèle murin pour la SLA. Nous pensons donc que l’étude de protéome de la microglie
serait plus représentative du phénotype réel de cette cellule
5.3.3. Le dynamisme du profil protéomique de la microglie dans la pathogénèse de la SLA
Le profil transcriptomique microglial dans le stade symptomatique de la SLA a été le point
d’étude principal du Chapitre 3. En effet, il semble que rétablir la traduction des ARNm
immuns trouvés par le profilage transcriptomique aide à ralentir la progression de la maladie,
justifiant son étude approfondie. Cependant bien que le profil transcriptomique de la
microglie fût le seul à être étudié et discuté jusqu’ici, des informations importantes sur la
pathogénèse de la SLA peuvent être découvertes par l’étude du profil protéomique de la
microglie. Par exemple, l’étude des protéines les plus régulées permet l’identification de
nouveaux biomarqueurs pour le phénotype microglial dans la SLA. Ainsi, nous retrouvons
les protéines du complément (C1QA, C1QB et C1QC) comme hautement régulées dans la
microglie dans les stades symptomatiques (modéré et avancés). Nous voyons aussi une
régulation de la protéine galectine-3 (LGALS3) dans le stade symptomatique modéré. Cette
protéine est importante pour l’activation de la microglie, sa migration et sa prolifération
(Rahimian, Béland, et al. 2018). De plus, galectine-3 a été, à plusieurs reprises, liée à la SLA
et suggérée comme biomarqueur pour cette maladie (Zhou et al. 2010, Yan et al. 2016, Ashraf

187
et Baeesa 2018); nos recherches confirmant l’origine microgliale de la surexpression de
galectine-3. Nous montrons aussi que APOE, bien connue pour son rôle dans la maladie
d’Alzheimer, semble aussi surexprimée dans la SLA (Wolfe et al. 2018). APOE, aussi
régulée au niveau de son ARNm, pourrait donc être un biomarqueur intéressant pour mesurer
l’activation microgliale dans la SLA. MTPN quant à elle est impliquée dans la dimérisation
des sous-unités de NF-κB et a donc un rôle dans la régulation de l’inflammation
(Knuefermann et al. 2002). Finalement, nous retrouvons une régulation de la protéine GFAP,
normalement associée à l’astrogliose. Cette protéine a pourtant déjà été liée au
développement d’un phénotype microglial activé et aberrant dans le stade symptomatique de
la SLA (Díaz-Amarilla et al. 2011, Trias, Díaz-Amarilla, et al. 2013). Aussi, par l’étude du
protéome microglial dans son ensemble, nous trouvons un phénotype activé, mis en évidence
par les fonctions biologiques des protéines les plus régulées : Defense response, Complement
and coagulation cascades, Response to oxydative stress, Innate immune response,
Cytoskeleton organization, etc. Ce phénotype, bien que comportant certains termes
semblables au phénotype DAM, diffère dans sa fonction par l’absence de termes liés à la
phagocytose.
Bien qu’il soit possible d’étudier les protéines surexprimées une à une, il est aussi intéressant
d’étudier leur implication dans la régulation de voies de signalisation cellulaire. Par exemple,
l’activation de la voie de signalisation de l’inflammation NF-κB, spécifiquement dans la
microglie a été identifié comme chef d’orchestre de la neurodégénération dans la SLA
(Frakes et al. 2014). Ainsi il serait intéressant d’étudier le dynamisme des protéines
impliquées dans cette voie de signalisation dans les stades de la maladie entourant
l’apparition des symptômes. Dans le Tableau 3, sont énumérés les protéines impliquées dans
la voie de signalisation NF-κB (TNF-alpha NF-κB signaling pathway de Wikipathways,
correspondant à la voie d’activation classique de NF-κB retrouvée dans la SLA (Frakes et al.
2014)) et retrouvées dans l’analyse du protéome microglial.
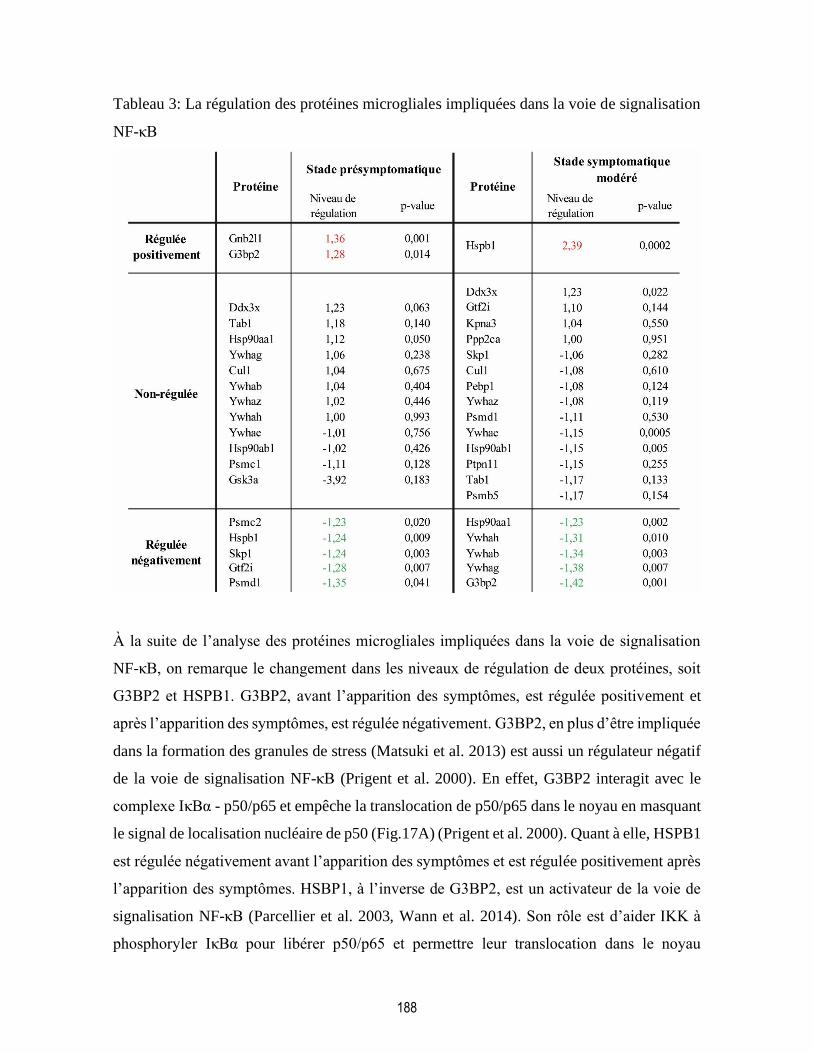
188
Tableau 3: La régulation des protéines microgliales impliquées dans la voie de signalisation
NF-κB
À la suite de l’analyse des protéines microgliales impliquées dans la voie de signalisation
NF-κB, on remarque le changement dans les niveaux de régulation de deux protéines, soit
G3BP2 et HSPB1. G3BP2, avant l’apparition des symptômes, est régulée positivement et
après l’apparition des symptômes, est régulée négativement. G3BP2, en plus d’être impliquée
dans la formation des granules de stress (Matsuki et al. 2013) est aussi un régulateur négatif
de la voie de signalisation NF-κB (Prigent et al. 2000). En effet, G3BP2 interagit avec le
complexe IκBα - p50/p65 et empêche la translocation de p50/p65 dans le noyau en masquant
le signal de localisation nucléaire de p50 (Fig.17A) (Prigent et al. 2000). Quant à elle, HSPB1
est régulée négativement avant l’apparition des symptômes et est régulée positivement après
l’apparition des symptômes. HSBP1, à l’inverse de G3BP2, est un activateur de la voie de
signalisation NF-κB (Parcellier et al. 2003, Wann et al. 2014). Son rôle est d’aider IKK à
phosphoryler IκBα pour libérer p50/p65 et permettre leur translocation dans le noyau
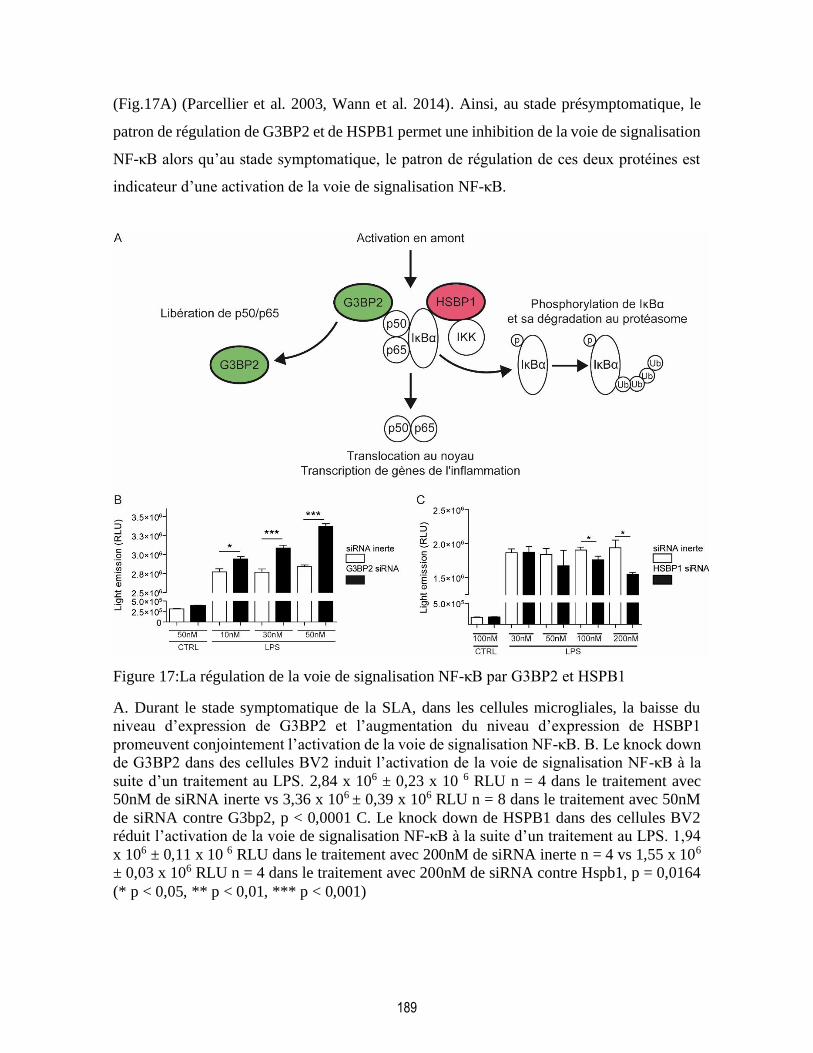
189
(Fig.17A) (Parcellier et al. 2003, Wann et al. 2014). Ainsi, au stade présymptomatique, le
patron de régulation de G3BP2 et de HSPB1 permet une inhibition de la voie de signalisation
NF-κB alors qu’au stade symptomatique, le patron de régulation de ces deux protéines est
indicateur d’une activation de la voie de signalisation NF-κB.
Figure 17:La régulation de la voie de signalisation NF-κB par G3BP2 et HSPB1
A. Durant le stade symptomatique de la SLA, dans les cellules microgliales, la baisse du
niveau d’expression de G3BP2 et l’augmentation du niveau d’expression de HSBP1
promeuvent conjointement l’activation de la voie de signalisation NF-κB. B. Le knock down
de G3BP2 dans des cellules BV2 induit l’activation de la voie de signalisation NF-κB à la
suite d’un traitement au LPS. 2,84 x 106 ± 0,23 x 10 6 RLU n = 4 dans le traitement avec
50nM de siRNA inerte vs 3,36 x 106 ± 0,39 x 106 RLU n = 8 dans le traitement avec 50nM
de siRNA contre G3bp2, p < 0,0001 C. Le knock down de HSPB1 dans des cellules BV2
réduit l’activation de la voie de signalisation NF-κB à la suite d’un traitement au LPS. 1,94
x 106 ± 0,11 x 10 6 RLU dans le traitement avec 200nM de siRNA inerte n = 4 vs 1,55 x 106
± 0,03 x 106 RLU n = 4 dans le traitement avec 200nM de siRNA contre Hspb1, p = 0,0164
(* p < 0,05, ** p < 0,01, *** p < 0,001)

190
Il a été possible de confirmer la spécificité de l’action de ces deux protéines dans des cellules
microgliale in vitro. Les cellules BV2 (cellules microgliales murines immortalisées)
transfectées de manière stable avec le gène de la luciférase exprimé sous le contrôle d’un
reporteur pour p65 émettent une quantité mesurable de photons lorsque la voie de
signalisation NF-κB est activée dans celles-ci. Lorsque l’expression de G3BP2 est diminuée
dans les cellules BV2 activée par du LPS, on assiste à une augmentation de l’activation de la
voie de signalisation NF-κB (Fig.17B). Aussi, lorsque l’expression de HSPB1 est diminuée
dans les cellules BV2 activée par du LPS, on assiste à une diminution de l’activation de la
voie de signalisation NF-κB (Fig.17C). Ainsi, nous confirmons que la régulation de la voie
de signalisation NF-κB par G3BP2 et HSPB1 est possible dans les cellules microgliales.
L’étude de voies de signalisation microgliales par la régulation des protéines de ces voies
amène de nouvelles informations sur les mécanismes cellulaires impliqués dans la
neurodégénérescence. Il serait intéressant de modifier l’expression de ces deux protéines
spécifiquement dans les cellules microgliales d’un modèle murin pour la SLA pour
comprendre l’effet exact de G3BP2 et HSPB1 sur la pathogénèse de la maladie. Il est
intéressant de noter que la majorité des protéines de la voie de signalisation TNF-alpha NF-
κB signaling pathway ne sont pas identifiées par spectrométrie de masse ou bien ne sont pas
régulées à l’un ou l’autre des stades de la maladie. De plus, les niveaux de régulation de ces
protéines régulées restent relativement faibles. Ainsi, il est possible que cette voie de
signalisation soit régulée uniquement par G3BP2 et HSPB1, ou bien que l’implication de la
voie de signalisation NF-κB dans la pathogénèse de la SLA ne soit pas aussi importante que
l’on pourrait croire. Ainsi, il est possible que les protéines les plus régulées dans nos analyses
(les protéines du complément ou galectine-3 par exemple) soient les vraies effectrices du
phénotype microglial symptomatique et soient impliquées dans d’autres voies de
signalisation. La précision de notre analyse dépend toujours de l’exactitude des banques de
données préexistantes.
La production de résultats sur le profilage moléculaire spécifique à un type cellulaire génère
une quantité appréciable de nouvelles informations qui ne peuvent être couvertes durant la
durée d’un seul doctorat, Ainsi, et bien que l’ensemble de cette thèse ait été consacré à
l’études des ARNm et des protéines surexprimés, il existe une grande quantité d’informations

191
disponible dans l’étude des ARNm et des protéines sous-exprimées par rapport à la condition
contrôle. Par exemple, il est possible de noter l’absence complète de la protéine GSS
(glutathion synthétase) dans les cellules microgliales de la moelle épinière lombaire de souris
au stade symptomatique de la SLA. Ainsi, tel que vu dans l’introduction de cette thèse, les
patients atteint de la SLA ont une diminution importante du produit de la GSS, soit le
glutathion (Weiduschat et al. 2014, Blasco et al. 2017, Wang, Bai, et al. 2019). Cette baisse
de la production de glutathion dans le SNC des patients atteints de la SLA est possiblement
d’origine microgliale. La microglie participerait donc fortement au stress oxydatif présent
dans les tissus SLA. De plus, le phénotype microglial peut être nettement modifier par l’état
redox de son environnement (Rojo et al. 2014). L’activation de la microglie par un
environnement oxydatif pourrait expliquer, entre autres, l’exacerbation de la
neuroinflammation et la neurodégénérescence dans la SLA par ce type cellulaire.
L’étude du profil protéomique microglial permet donc d’identifier de nouveaux
biomarqueurs, importants pour la détection de la maladie ou pour surveiller l’effet d’un
traitement pour la SLA sur les cellules microgliales. Aussi, l’étude du profil protéomique de
la microglie permet une étude approfondie de voie de signalisation cellulaire. Ces voies
peuvent, à la fois, être présélectionnées en fonction de connaissance antérieure sur la
pathogénèse de la SLA (comme ce fut le cas pour la voie de signalisation NF-κB) ou être
identifiée en soumettant les protéines du profil moléculaire microglial à un programme
spécialisé et des banques de données. Finalement, alors que les phénotypes cellulaires sont
souvent définis selon des protéines surexprimées, l’étude des protéines sous-exprimées peut
mettre à jour certains mécanismes cellulaires déficients, contribuant au changement du
phénotype microglial dans la SLA.
En conclusion, l’analyse du profil protéomique donne une meilleure représentation du réel
phénotype microglial que l’analyse du profil transcriptomique. Tel qu’expliqué dans le 3e
chapitre, nous notons une forte disparité entre les ces deux profils, quant à la nature des
ARNm ou des protéines qui y sont les plus régulés. Notamment, cette disparité est mise en
évidence par l’absence de la traduction d’ARNm fortement régulés au stades symptomatique
et provenant de gènes associés à l’immunité. Ainsi, au lieu du phénotype phagocytaire et

192
neuroprotecteur annoncé par le profil transcriptomique, la microglie symptomatique possède
plutôt un phénotype aberrant, activé et neurotoxique. La répression de la traduction des
ARNm immunitaires semble être due à l’action de la protéine liant l’ARN SRSF3.
5.4. La protéine SRSF3 dans la pathogénèse de la SLA
SRSF3 est un des 12 membres de la famille des protéines SR contrôlant divers aspects du
traitement de l’ARN. SRSF3, partageant plusieurs rôles avec les protéines SR, est impliquée
dans la stabilité générale des ARNm, la transcription, l’épissage alternatif (incluant la
sélection alternative du premier exon et du site de polyadénylation), la maturation des micro-
ARN (miRNA), l’exportation des ARNm ou encore la traduction (Cui et al. 2008, Kim et al.
2014, Bradley et al. 2015, Ajiro et al. 2016, Müller-McNicoll et al. 2016, Boutej et al. 2017,
Kim et al. 2018). L’altération de son rôle clef dans l’un ou l’autre de ces processus cellulaires
lie SRSF3 a de multiples maladies : les cancers, les maladies métaboliques du foie, les
infections virales, les troubles du développement, ou encore les maladies du SNC (He et al.
2011, Klymenko et al. 2016, Boutej et al. 2017, Kumar et al. 2019).
Dans la microglie, suivant une activation aigüe (Boutej et al. 2017) ou chronique (voir le
Chapitre 3), SRSF3 a été identifiée pour la première fois comme responsable de la disparité
de l’expression de gènes immuns au niveau de leur ARNm et leur protéine. À la suite d’une
injection intra-péritonéale de LPS, les ARNm les plus hautement régulés ne sont pas traduits
en protéines. pSRSF3 est retrouvé en abondance dans la microglie activée par le LPS et
pourrait se lier au 3’UTR de ces ARNm. Ces ARNm sont déjà en traduction, mais SRSF3
empêcherait la progression du ribosome sur l’ARNm, arrêtant leur traduction (Boutej et al.
2017). L’utilisation d’un siRNA contre SRSF3 permet la traduction de-novo des ARNm
précédemment réprimés et la modulation de l’inflammation. Dans la SLA, on retrouve un
phénomène semblable : SRSF3 est hautement phosphorylée dans les tissus de modèles
animaux et de patients atteints de cette maladie, et s’accumule avec la progression de la
maladie. Ceci a été vérifié par immunobuvardage en utilisant un homogénat de moelle
épinière lombaire de souris SLA, par immunofluorescence sur des coupe de moelle épinière
lombaire de souris SLA et par immunohistochimie sur des coupe de moelle épinière de
patients atteints de la SLA (voir Fig.4P-T de la section 3.8.5.). De plus, nous démontrons que

193
les ARNm réprimés par SRSF3 dans la microglie symptomatique peuvent être traduit de
nouveau lorsque l’expression de SRSF3 est diminuée à la suite d’un traitement avec un
morpholino contre l’ARNm de celui-ci. Chez la souris SLA, l’utilisation du morpholino
contre SRSF3, ralentit la progression de la maladie et augmente son espérance de vie en
rétablissant les fonctions perdues des ARNm réprimés.
5.4.1. La phosphorylation de SRSF3 dans la SLA
En conditions normales, SRSF3 est retrouvé dans le noyau pour y accomplir ses diverses
tâches liées au traitement de l’ARN. En effet, SRSF3 colocalise parfaitement avec les noyaux
de l’ensemble des cellules de la moelle épinière, tel que montré par immunofluorescence
(voir Fig.4D-K de la section 3.8.5.) Toutefois, dans la SLA, pSRSF3 s’accumule
spécifiquement dans le cytoplasme des cellules microgliales. Tel que mentionné dans le
Chapitre 4, SRSF3 peut être phosphorylée par les kinases SRPK et CLK (Gui et al. 1994,
Duncan et al. 1997). SRPK1 et SRPK2 sont responsables de la phosphorylation de SRSF3
dans le cytoplasme après la traduction de celui-ci pour l’acheminer vers le noyau. Les SRPK
vont aussi ajuster la phosphorylation de SRSF3 lorsque qu’il se retrouve de nouveau dans le
cytoplasme après y avoir exporter des ARNm (Zhou et Fu 2013). Dans le corps, les SRPK
sont préférentiellement exprimées dans différents organes, par exemple, SRPK1 est d’abord
exprimée dans le foie tandis que SRPK2 l’est dans le SNC (Wang et al. 1999). De son côté,
CLK1 est seulement retrouvée dans le noyau des cellules et ajuste la phosphorylation de
SRSF3 afin de contrôler étroitement à quelle étape du traitement de l’ARN SRSF3 est rendu
(Zhou et Fu 2013). Ainsi il serait possible d’hypothétiser un dérèglement dans l’expression
des kinases régissant la phosphorylation de SRSF3. Cependant, bien que SRPK1 semblerait
être surexprimée dans les tissus SLA comparativement aux tissus contrôles, les bandes
retrouvées par immunobuvardage sont les produits de clivage de SRPK1 (Kamachi et al.
2002) (Fig.18A). SRPK1 est spécifiquement clivée et inactivée par caspase-8 (Kamachi et
al. 2002), cette dernière étant fortement exprimée dans les cellules microgliales activées
(Kavanagh et al. 2015, Zhang, Jiang, et al. 2018). De plus, aucun changement d’expression
n’est retrouvé chez SRPK2 ou chez CLK1 par immunobuvardage en utilisant des homogénats
de moelle épinière lombaire de souris SLA (Fig.18B-C).
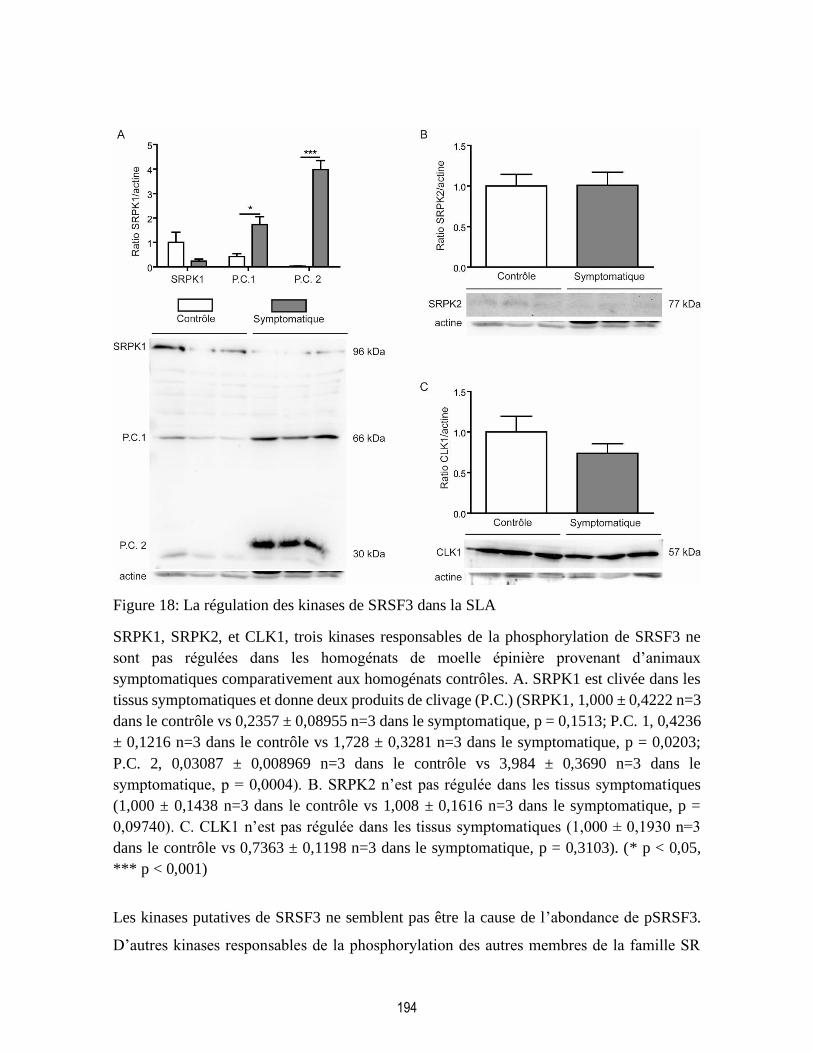
194
Figure 18: La régulation des kinases de SRSF3 dans la SLA
SRPK1, SRPK2, et CLK1, trois kinases responsables de la phosphorylation de SRSF3 ne
sont pas régulées dans les homogénats de moelle épinière provenant d’animaux
symptomatiques comparativement aux homogénats contrôles. A. SRPK1 est clivée dans les
tissus symptomatiques et donne deux produits de clivage (P.C.) (SRPK1, 1,000 ± 0,4222 n=3
dans le contrôle vs 0,2357 ± 0,08955 n=3 dans le symptomatique, p = 0,1513; P.C. 1, 0,4236
± 0,1216 n=3 dans le contrôle vs 1,728 ± 0,3281 n=3 dans le symptomatique, p = 0,0203;
P.C. 2, 0,03087 ± 0,008969 n=3 dans le contrôle vs 3,984 ± 0,3690 n=3 dans le
symptomatique, p = 0,0004). B. SRPK2 n’est pas régulée dans les tissus symptomatiques
(1,000 ± 0,1438 n=3 dans le contrôle vs 1,008 ± 0,1616 n=3 dans le symptomatique, p =
0,09740). C. CLK1 n’est pas régulée dans les tissus symptomatiques (1,000 ± 0,1930 n=3
dans le contrôle vs 0,7363 ± 0,1198 n=3 dans le symptomatique, p = 0,3103). (* p < 0,05,
*** p < 0,001)
Les kinases putatives de SRSF3 ne semblent pas être la cause de l’abondance de pSRSF3.
D’autres kinases responsables de la phosphorylation des autres membres de la famille SR

195
pourraient être en cause (CDC2P34, TOPO1) (Shepard et Hertel 2009). Bien que la
phosphorylation de SRSF3 ait été décrite comme très restreinte à certaines kinases (Gui et al.
1994, Duncan et al. 1997), d’autres kinases inhabituelles pourraient être impliquées dans la
phosphorylation de SRSF3 dans les conditions atypiques de la SLA. Aussi, la balance entre
la forme non-phosphorylée et la forme phosphorylée de SRSF3 dépend, en conditions
normales, de l’activité conjointe de ses kinases et de sa phosphatase, PP1 (Misteli et Spector
1996, Novoyatleva et al. 2007). Toutefois, la déphosphorylation de SRSF3 par les
phosphatases se fait uniquement dans le noyau pour moduler l’activité de SRSF3 dans les
étapes du traitement de l’ARN (Misteli et Spector 1996). Une baisse d’activité de la
phosphatase de SRSF3 ne pourrait donc pas expliquer le niveau de pSRSF3 retrouvé dans le
cytoplasme de la microglie symptomatique. Ainsi l’augmentation de la phosphorylation ou
la baisse de la déphosphorylation de SRSF3 ne sont potentiellement pas responsables de
l’accumulation de pSRSF3 dans le cytoplasme microglial.
5.4.2. La distribution cytoplasmique de SRSF3 dans la microglie symptomatique
En plus l’action de kinases, l’accumulation de SRSF3 dans le cytoplasme des cellules
microgliales symptomatiques pourrait aussi être due à sa rétention aberrante dans ce
compartiment. En effet, SRSF3 pourraient être incapable d’entrer dans le noyau par les pores
nucléaires. Dans le noyau, en plus de réguler l’expression d’une panoplie de gènes SRSF3
peut s’autoréguler. Tel qu’expliqué dans le Chapitre 4, lors de l’épissage de l’ARN de
SRSF3, l’exon 4 est normalement exclus. Toutefois, lorsque SRSF3 est présent en plus
grande quantité, l’exon 4 est inclus causant la formation prématurée d’un codon stop avant
le domaine RS de SRSF3 (Jumaa et al. 1997, Che et Fu 2020). Dans la SLA, SRSF3 est
possiblement exclus du noyau des cellules microgliales et ne peut plus s’autoréguler. Ainsi,
les ribosomes continuent à traduire les ARNm de SRSF3 exportés dans le cytoplasme. SRSF3
est ensuite constamment phosphorylé par ses kinases, tentant de l’acheminer vers le noyau.
Pour vérifier cet énoncé, il serait intéressant de comparer le niveau de pSRSF3 dans le noyau
et dans le cytoplasme des cellules microgliales. La composante nucléaire de SRSF3 étant
possiblement réduite dans les tissus SLA comparativement à des tissus contrôles.

196
Les mécanismes régissant une potentielle rétention aberrante de SRSF3 dans le cytoplasme
microglial ne sont présentement pas connu mais pourrait hypothétiquement s’expliquer de
plusieurs façons. Premièrement, les pores nucléaires sont possiblement défectueux. Plusieurs
gènes causant la SLA sont responsables d’un dysfonctionnement des pores nucléaires,
notamment TDP43, C9orf72 et SOD1. L’altération des pores nucléaires et de la membrane
nucléaire est un phénomène aussi retrouvé chez les patients sporadiques (Fallini et al. 2020)
Le dysfonctionnement des pores nucléaires dans la SLA a été uniquement étudié dans les
neurones moteurs. Toutefois, tel que mentionné dans la section 1.1.3, les cellules microgliales
partagent avec les motoneurones plusieurs mécanismes de toxicité cellulaire pouvant
potentiellement causer cette dysfonction. L’agrégation des protéines issues de ces gènes peut
bloquer physiquement les pores nucléaires ou réguler différentiellement la présence de
certaines nucléoporines dans ces pores (Nagara et al. 2013, Aizawa et al. 2019, Fallini et al.
2020). La disruption des pores nucléaires dans la SLA et spécifiquement dans la microglie
n’a malheureusement jamais été étudiée. Aussi, nous n’avons pas détecté de changement
dans la régulation des nucléoporines dans notre analyse du profil protéomique microglial.
Deuxièmement, les protéines impliquées dans le transport de SRSF3 ne fonctionneraient pas
correctement. Tel que vu dans le Chapitre 4, l’importation de SRSF3 dans le noyau est
dépendant de la liaison de son domaine RS (devenu un signal de localisation nucléaire à la
suite de sa phosphorylation) avec les transportines TRN-SR1 et TRN-SR2 (Lai et al. 2001).
Il est possible que leur expression soit altérée d’une certaine façon. Toutefois, nous n’avons
pas détecté de changement dans la régulation des transportines ou des importines dans notre
analyse du profil protéomique de la microglie. Cependant, durant le stress cellulaire causé
par la SLA, les transportines (et par le fait même, certaines nucléoporines), sont recrutées,
avec leur cargo, dans les granules de stress (Zhang, Daigle, et al. 2018). SRSF3 pourrait,
hypothétiquement, être séquestré aussi dans les granules de stress, lié au transportines. Des
transportines séquestrées dans les granules de stress pourraient aussi ne pas être en mesure
de lier SRSF3 est laisser cette dernière seule dans le cytoplasme.
Troisièmement, et faisant suite au précédant point, SRSF3 s’associerait anormalement avec
des composants du cytoplasme. En effet, il est déjà démontré que SRSF3 peut être recruté

197
dans les P-bodies pour y acheminer des ARNm à dégrader (Kim et al. 2014). Ce mécanisme
cellulaire implique l’interaction de SRSF3 avec le 5’UTR des ARNm. Dans la SLA, une telle
interaction n’est pas encore connue. Aussi, nous pensons que l’interaction entre SRSF3 et les
ARNm, dans la microglie symptomatique, viendrait plutôt de son interaction avec le 3’UTR
des ARNm et que ces derniers ne sont pas dégradés, car ils sont détectés sur les ribosomes.
SRSF3 pourrait aussi possiblement être recruté avec d’autres composantes de la machinerie
de la traduction dans les granules de stress, compartiment où certains ARNm peuvent
entreposés suivant le blocage du ribosome et un stress cellulaire (Buchan et Parker 2009). La
fraction de SRSF3 possiblement séquestrée dans les granules de stress n’est pas la même que
celle étudiée dans le chapitre 3, plutôt retrouvée attachée au ribosome en traduction active.
Toutefois, la séquestration de SRSF3 dans les granules de stress ou autres organites
cellulaires, l’empêcherait de réguler l’intégrité de son propre ARNm dans le noyau et
d’ajuster son niveau d’expression.
Ces trois hypothèses concernant les mécanismes de rétention de SRSF3 dans le cytoplasme
mériteraient d’être éclaircies en étudiant l’association de SRSF3 et sa forme phosphorylée
avec les différentes composantes cellulaires énumérées plus haut.
5.4.3. Les avenues thérapeutiques ciblant SRSF3
Boutej et al. utilisent un petit ARN interférent (small interfering RNA : siRNA) contre
l’ARNm de SRSF3 et peuvent rétablir l’expression au niveau des protéines des gènes
immunitaires précédemment réprimés à la suite d’une injection de LPS (Boutej et al. 2017).
Dans le Chapitre 3, nous rétablissons aussi l’expression de gènes liés à l’immunité et la
phagocytose dans les cellules microgliales à l’aide d’un morpholino contre l’ARNm de
SRSF3 (voir la Fig.5E de la section 3.8.6). Les morpholinos, comme les autres
oligonucléotides interférents, peuvent s’hybrider spécifiquement à un ARNm cible au niveau
de sa région 5’UTR et causer sa dégradation et ainsi diminuer la traduction de sa protéine.
Ils diffèrent des autres oligonucléotides interférents par leur structure chimique. En effet, au
lieu de posséder un squelette composé de ribose-phosphate, le squelette des morpholinos est
composé de morpholine-phosphorodiamidate, rendant ceux-ci moins toxiques que les siRNA

198
(Summerton 2007). De plus les morpholinos sont plus solubles dans l’eau que les autres
oligonucléotides interférents (donc administré à plus forte dose) et sont moins susceptible à
la dégradation, allongeant leur effet transitoire (Summerton 2007, Cappella et al. 2019).
Finalement, les morpholinos sont généralement plus spécifiques que les siRNA (Summerton
2007, Cappella et al. 2019). Dans un modèle murin de la SLA, l’utilisation d’un morpholino
contre SRSF3 permet de rétablir la fonction phagocytaire de la microglie et ainsi, de ralentir
la progression de la maladie. Pour contrer l’effet transitoire d’un morpholino ou d’un siRNA,
l’utilisation d’un shRNA aurait aussi pu être considérée. Celui-ci nécessite toutefois d’être
acheminé dans les cellules par vecteur viral ou bactérien (Wang et al. 2011, Cappella et al.
2019). Les shRNA peuvent être acheminés par des AAV ou des lentivirus, utiles pour une
expression à long-terme du gène transduit, mais difficilement contrôlable par la suite
(Cappella et al. 2019). L’utilisation d’un morpholino, plus modulable, est donc toujours
l’avenue la plus intéressante.
Le traitement de la SLA en ciblant l’expression de SRSF3 par son ARNm concorde avec
l’intérêt grandissant porté sur les thérapies géniques pour traiter les maladies
neurodégénératives ou la SLA en particulier (O'Connor et Boulis 2015, Schoch et Miller
2017, Cappella et al. 2019). Déjà, plusieurs tentatives se font pour cibler, par thérapie
génique, l’expression des gènes directement liés à la SLA. Par exemple, des oligonucléotides
antisens contre C9orf2 et deux formes familiales de SOD1 sont présentement en essai
clinique pour le traitement de la SLA (Smith et al. 2006, Miller et al. 2013, Jiang et al. 2016,
Cappella et al. 2019). Dans des modèles murins, les oligonucléotides interférent (siRNA,
morpholinos et shRNA) sont aussi testés pour modifier l’expression de certains gènes causant
la SLA, mais aucun traitement par oligonucléotide interférent n’est présentement en phase
clinique (Ding et al. 2003, Cappella et al. 2019). In vitro et in vivo, le système CRISPR-Cas9
a aussi été utilisé pour diminuer l’expression de SOD1 ou de C9orf72. (Pribadi et al. 2016,
Gaj et al. 2017, Cappella et al. 2019). Le système CRISPR-Cas9 n’est pas non plus rendu en
phase clinique. Les présentes thérapies géniques, qu’elles soient rendues ou non en phase
clinique, ciblent des gènes mutés causant la SLA. Les thérapies géniques présentement
testées pour guérir la SLA ne pourrait être utilisée que pour le traitement des formes
familiales de la maladie. Bien que la protéine TDP43 mutée ne soit retrouvée dans tous les

199
cas de SLA (Neumann et al. 2006), elle est toutefois majoritairement régulée dans les tissus
des patients atteints de la SLA. La modulation de son expression pourrait donc être ciblée
par une thérapie génique.
De la même façon, nous proposons que l’utilisation d’une thérapie génique ciblant SRSF3
puisse potentiellement être une avenue intéressante pour le traitement de la SLA. En effet,
l’augmentation de la phosphorylation de SRSF3 semble être retrouvée dans les cellules
microgliales de patients atteints de la SLA sans précédents familiaux (voir Fig.4P-T de la
section 3.8.5.). L’utilisation d’une thérapie génique ciblant SRSF3 serait donc intéressante
pour le traitement des cas sporadique de la maladie. Étant donné les différents rôles de SRSF3
dans le fonctionnement normal de la cellule, une thérapie génique contre SRSF3 pose certains
défis. Il est important de tenir compte de ses autres rôles dans la cellule, de l’effet d’un
knockdown sur d’autres types cellulaires et d’identifier la dose appropriée pour obtenir le
knockdown désiré. Néanmoins, dans le modèle murin pour la SLA, utilisé dans le Chapitre
3, la diminution de SRSF3 par thérapie génique semble prometteuse pour ralentir la
progression de la maladie. Ce faisant, il serait intéressant de poursuivre les recherches afin
d’amener cette technologie chez l’humain.

200
Conclusion
La pathogénèse de la SLA est fortement influencée par le changement de phénotype des
cellules microgliales. Les cellules microgliales présentent un phénotype anti-inflammatoire
et neuroprotecteur avant l’apparition des symptômes, mais développent un phénotype
aberrant et neurotoxique après l’apparition des symptômes (Henkel et al. 2009). Cependant,
la nature elle-même de ces phénotypes et les mécanismes régissant la transition de l’un vers
l’autre sont mal connus. Dans cette thèse nous avons utilisé divers outils de profilage
moléculaire afin d’élucider comment la microglie passe d’un phénotype neuroprotecteur à
un phénotype neurotoxique. Dans les Chapitres 2, 3 et 4 nous avons réussis à compléter les
objectifs énoncés dans l’introduction.
Nous avons d’abord concentré nos recherches sur l’étude du profil de cytokine des cellules
microgliales présymptomatiques. Il a été possible de découvrir une régulation positive de la
cytokine IL-10 par la microglie dans ce stade de la maladie. De plus nous avons réussi, en
modifiant la réponse à IL-10 ou la production d’IL-10 à modifier la progression de la SLA
dans un modèle murin. Premièrement, par le blocage d’IL-10R, il a été possible d’accélérer
l’apparition des symptômes. Deuxièmement, par l’augmentation de la production d’IL-10 à
l’aide d’un vecteur viral, il a été possible de retarder l’apparition des symptômes et de
prolonger l’espérance de vie de ce modèle murin. Ainsi, nous montrons que le maintien du
phénotype microglial présymptomatique par IL-10 est bénéfique car il semble retarder la
progression de la SLA.
Par la suite, nous souhaitions comprendre les mécanismes impliqués dans le changement de
phénotype et caractériser les phénotypes des cellules microgliales à travers la maladie par
l’étude de leur transcriptome et de leur protéome. Pour se faire nous avons développé un
modèle murin pour le profilage moléculaire de la microglie dans la SLA, exprimant une
protéine ribosomale étiquetée spécifiquement dans les cellules microgliales. Ce modèle nous
a permis d’isoler les ARNm et les protéines en traduction dans la microglie depuis un tissu

201
complexe. Il a été possible de découvrir une divergence importante entre le transcriptome et
le protéome microglial des stades symptomatiques. Cette divergence était d’autant plus
apparente pour les gènes immunitaires, ceux-ci, très régulés au niveau de leur ARNm mais
absents de l’analyse protéomique. Cette divergence serait causée par un arrêt traduction due
à l’interaction de la protéine SRSF3 et du 3’UTR de ces ARNm. Le knock down de SRSF3
in vitro et dans un modèle murin pour l’ALS permet de rétablir l’expression des ARNm
immunitaires au niveau de leur protéine, de rétablir le potentiel phagocytaire des cellules
microglial et de ralentir la progression de la maladie. Le traitement de la SLA par la
diminution d’expression de SRSF3 pourrait être une avenue thérapeutique intéressante mais
une connaissance plus approfondie de cette protéine et des différents mécanismes cellulaires
dans lesquelles elle est impliquée est nécessaire.
Le travail accompli durant cette thèse peut mener à l’élaboration de plusieurs projets futurs.
Par exemple, dans le Chapitre 2, le maintien du phénotype anti-inflammatoire et
neuroprotecteur de la microglie présymptomatique a été atteint par l’injection intrathécale
d’un vecteur viral exprimant la cytokine IL-10 durant le stade présymptomatique. Une telle
technique a peu d’intérêt thérapeutique car il est impossible de débuter un traitement dans le
stade présymptomatique de la maladie pour les cas sporadiques. Ainsi, il serait intéressant de
voir l’effet d’une injection intrathécale de ce même vecteur viral au stade symptomatique sur
le phénotype microgliale et la progression de la SLA dans un modèle murin. Il pourrait être
intéressant de voir l’effet d’une injection intrathécale ou intrapéritonéale du vecteur viral sur
les cellules immunitaires périphériques, les Treg par exemple. Aussi, dans les Chapitres 3 et
4 nous explorons le potentiel thérapeutique d’une diminution d’expression de la protéine
SRSF3 dans les cellules microgliales. Il serait important de vérifier l’effet de la modulation
de SRSF3 sur d’autres types cellulaires, notamment les cellules du système immunitaire
comme les monocytes ou les macrophages. De plus, il sera nécessaire de tester la viabilité de
différentes concentrations de morpholino contre SRSF3 lors de son administration chez la
souris, avant de pouvoir penser à amener cette thérapie chez l’humain. Finalement, grâce à
la grande quantité d’information générée par l’EDTA-TRAP, il sera possible de clarifier la
nature des phénotypes microgliaux et les mécanismes sous-jacents au changement

202
phénotypique. L’étude de la régulation des protéines microgliales et les voies de signalisation
cellulaires dans lesquelles ces protéines sont impliquées permettra d’approfondir nos
connaissances sur l’implication de la microglie dans la pathogénèse de la SLA et de
développer de nouvelles cibles thérapeutiques.

203
Bibliographie
Abbott, N. J., A. A. Patabendige, D. E. Dolman, S. R. Yusof, and D. J. Begley. 2010.
'Structure and function of the blood-brain barrier', Neurobiol Dis, 37: 13-25.
Aizawa, Hitoshi, Takenari Yamashita, Haruhisa Kato, Takashi Kimura, and Shin Kwak.
2019. 'Impaired Nucleoporins Are Present in Sporadic Amyotrophic Lateral Sclerosis
Motor Neurons that Exhibit Mislocalization of the 43-kDa TAR DNA-Binding
Protein', J Clin Neurol, 15: 62-67.
Ajami, B., J. L. Bennett, C. Krieger, W. Tetzlaff, and F. M. Rossi. 2007. 'Local self-renewal
can sustain CNS microglia maintenance and function throughout adult life', Nat
Neurosci, 10: 1538-43.
Ajami, Bahareh, Nikolay Samusik, Peter Wieghofer, Peggy P. Ho, Andrea Crotti, Zach
Bjornson, Marco Prinz, Wendy J. Fantl, Garry P. Nolan, and Lawrence Steinman.
2018. 'Single-cell mass cytometry reveals distinct populations of brain myeloid cells
in mouse neuroinflammation and neurodegeneration models', Nature Neuroscience,
21: 541-51.
Ajiro, M., R. Jia, Y. Yang, J. Zhu, and Z. M. Zheng. 2016. 'A genome landscape of SRSF3-
regulated splicing events and gene expression in human osteosarcoma U2OS cells',
Nucleic Acids Res, 44: 1854-70.
Amrein, H., M. L. Hedley, and T. Maniatis. 1994. 'The role of specific protein-RNA and
protein-protein interactions in positive and negative control of pre-mRNA splicing by
Transformer 2', Cell, 76: 735-46.
Andersen, P. M., L. Forsgren, M. Binzer, P. Nilsson, V. Ala-Hurula, M. L. Keränen, L.
Bergmark, A. Saarinen, T. Haltia, I. Tarvainen, E. Kinnunen, B. Udd, and S. L.
Marklund. 1996. 'Autosomal recessive adult-onset amyotrophic lateral sclerosis
associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation.
A clinical and genealogical study of 36 patients', Brain, 119 ( Pt 4): 1153-72.
Anderson, Erik S., Chia-Ho Lin, Xinshu Xiao, Peter Stoilov, Christopher B. Burge, and
Douglas L. Black. 2012. 'The cardiotonic steroid digitoxin regulates alternative
splicing through depletion of the splicing factors SRSF3 and TRA2B', RNA (New
York, N.Y.), 18: 1041-49.
Andoh, Megumi, Yuji Ikegaya, and Ryuta Koyama. 2019. 'Synaptic Pruning by Microglia in
Epilepsy', Journal of clinical medicine, 8: 2170.
Ankö, M. L., L. Morales, I. Henry, A. Beyer, and K. M. Neugebauer. 2010. 'Global analysis
reveals SRp20- and SRp75-specific mRNPs in cycling and neural cells', Nat Struct
Mol Biol, 17: 962-70.
Appel, S. H., D. R. Beers, and J. S. Henkel. 2010. 'T cell-microglial dialogue in Parkinson's
disease and amyotrophic lateral sclerosis: are we listening?', Trends Immunol, 31: 7-
17.
Appel, S. H., W. Zhao, D. R. Beers, and J. S. Henkel. 2011. 'The microglial-motoneuron
dialogue in ALS', Acta Myol, 30: 4-8.
Arai, T., M. Hasegawa, H. Akiyama, K. Ikeda, T. Nonaka, H. Mori, D. Mann, K. Tsuchiya,
M. Yoshida, Y. Hashizume, and T. Oda. 2006. 'TDP-43 is a component of ubiquitin-
positive tau-negative inclusions in frontotemporal lobar degeneration and
amyotrophic lateral sclerosis', Biochem Biophys Res Commun, 351: 602-11.

204
Arbour, D., C. Vande Velde, and R. Robitaille. 2017. 'New perspectives on amyotrophic
lateral sclerosis: the role of glial cells at the neuromuscular junction', J Physiol, 595:
647-61.
Arthur, Karissa C., Andrea Calvo, T. Ryan Price, Joshua T. Geiger, Adriano Chiò, and Bryan
J. Traynor. 2016. 'Projected increase in amyotrophic lateral sclerosis from 2015 to
2040', Nature Communications, 7: 12408-08.
Ashraf, G. M., and S. S. Baeesa. 2018. 'Investigation of Gal-3 Expression Pattern in Serum
and Cerebrospinal Fluid of Patients Suffering From Neurodegenerative Disorders',
Front Neurosci, 12: 430.
Askew, K., K. Li, A. Olmos-Alonso, F. Garcia-Moreno, Y. Liang, P. Richardson, T. Tipton,
M. A. Chapman, K. Riecken, S. Beccari, A. Sierra, Z. Molnár, M. S. Cragg, O.
Garaschuk, V. H. Perry, and D. Gomez-Nicola. 2017. 'Coupled Proliferation and
Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain', Cell Rep,
18: 391-405.
Atkin, J. D., M. A. Farg, A. K. Walker, C. McLean, D. Tomas, and M. K. Horne. 2008.
'Endoplasmic reticulum stress and induction of the unfolded protein response in
human sporadic amyotrophic lateral sclerosis', Neurobiol Dis, 30: 400-7.
Aubol, B. E., K. L. Hailey, L. Fattet, P. A. Jennings, and J. A. Adams. 2017. 'Redirecting SR
Protein Nuclear Trafficking through an Allosteric Platform', J Mol Biol, 429: 2178-
91.
Audet, J. N., G. Gowing, R. Paradis, G. Soucy, and J. P. Julien. 2012. 'Ablation of
proliferating cells in the CNS exacerbates motor neuron disease caused by mutant
superoxide dismutase', PLOS ONE, 7: e34932.
Auyeung, Vincent C., Igor Ulitsky, Sean E. McGeary, and David P. Bartel. 2013. 'Beyond
secondary structure: primary-sequence determinants license pri-miRNA hairpins for
processing', Cell, 152: 844-58.
Ayers, J. I., and N. R. Cashman. 2018. 'Prion-like mechanisms in amyotrophic lateral
sclerosis', Handb Clin Neurol, 153: 337-54.
Ayers, J. I., S. Fromholt, O. Sinyavskaya, Z. Siemienski, A. M. Rosario, A. Li, K. W. Crosby,
P. E. Cruz, N. M. DiNunno, C. Janus, C. Ceballos-Diaz, D. R. Borchelt, T. E. Golde,
P. Chakrabarty, and Y. Levites. 2015. 'Widespread and efficient transduction of spinal
cord and brain following neonatal AAV injection and potential disease modifying
effect in ALS mice', Mol Ther, 23: 53-62.
Banerjee, Rebecca, R. Lee Mosley, Ashley D. Reynolds, Alok Dhar, Vernice Jackson-Lewis,
Paul H. Gordon, Serge Przedborski, and Howard E. Gendelman. 2008. 'Adaptive
Immune Neuroprotection in G93A-SOD1 Amyotrophic Lateral Sclerosis Mice',
PLOS ONE, 3: e2740.
Baralle, Francisco E., and Jimena Giudice. 2017. 'Alternative splicing as a regulator of
development and tissue identity', Nature Reviews Molecular Cell Biology, 18: 437-
51.
Barmada, S. J., and S. Finkbeiner. 2010. 'Pathogenic TARDBP mutations in amyotrophic
lateral sclerosis and frontotemporal dementia: disease-associated pathways', Rev
Neurosci, 21: 251-72.
Bedard, K. M., S. Daijogo, and B. L. Semler. 2007. 'A nucleo-cytoplasmic SR protein
functions in viral IRES-mediated translation initiation', Embo j, 26: 459-67.
Beddoe, T., and T. Lithgow. 2002. 'Delivery of nascent polypeptides to the mitochondrial
surface', Biochim Biophys Acta, 1592: 35-9.

205
Beers, D. R., J. S. Henkel, Q. Xiao, W. Zhao, J. Wang, A. A. Yen, L. Siklos, S. R. McKercher,
and S. H. Appel. 2006. 'Wild-type microglia extend survival in PU.1 knockout mice
with familial amyotrophic lateral sclerosis', Proc Natl Acad Sci U S A, 103: 16021-6.
Beers, D. R., W. Zhao, J. Wang, X. Zhang, S. Wen, D. Neal, J. R. Thonhoff, A. S. Alsuliman,
E. J. Shpall, K. Rezvani, and S. H. Appel. 2017. 'ALS patients' regulatory T
lymphocytes are dysfunctional, and correlate with disease progression rate and
severity', JCI Insight, 2: e89530.
Beers, David R., Jenny S. Henkel, Weihua Zhao, Jinghong Wang, and Stanley H. Appel.
2008. 'CD4+ T cells support glial neuroprotection, slow disease progression, and
modify glial morphology in an animal model of inherited ALS', Proc Natl Acad Sci
U S A, 105: 15558-63.
Béland, Louis-Charles, Andrea Markovinovic, Hrvoje Jakovac, Fabiola de Marchi, Ervina
Bilic, Letizia Mazzini, Jasna Kriz, and Ivana Munitic. 2020. 'Immunity in
amyotrophic lateral sclerosis: blurred lines between excessive inflammation and
inefficient immune responses', Brain Communications.
Belloy, M. E., V. Napolioni, and M. D. Greicius. 2019. 'A Quarter Century of APOE and
Alzheimer's Disease: Progress to Date and the Path Forward', Neuron, 101: 820-38.
Belzil, V. V., P. O. Bauer, M. Prudencio, T. F. Gendron, C. T. Stetler, I. K. Yan, L. Pregent,
L. Daughrity, M. C. Baker, R. Rademakers, K. Boylan, T. C. Patel, D. W. Dickson,
and L. Petrucelli. 2013. 'Reduced C9orf72 gene expression in c9FTD/ALS is caused
by histone trimethylation, an epigenetic event detectable in blood', Acta Neuropathol,
126: 895-905.
Benatar, M. 2007. 'Lost in translation: treatment trials in the SOD1 mouse and in human
ALS', Neurobiol Dis, 26: 1-13.
Bennett, Mariko L., F. Chris Bennett, Shane A. Liddelow, Bahareh Ajami, Jennifer L.
Zamanian, Nathaniel B. Fernhoff, Sara B. Mulinyawe, Christopher J. Bohlen,
Aykezar Adil, Andrew Tucker, Irving L. Weissman, Edward F. Chang, Gordon Li,
Gerald A. Grant, Melanie G. Hayden Gephart, and Ben A. Barres. 2016. 'New tools
for studying microglia in the mouse and human CNS', Proceedings of the National
Academy of Sciences, 113: E1738.
Benussi, L., G. Rossi, M. Glionna, E. Tonoli, E. Piccoli, S. Fostinelli, A. Paterlini, R. Flocco,
D. Albani, R. Pantieri, C. Cereda, G. Forloni, F. Tagliavini, G. Binetti, and R.
Ghidoni. 2014. 'C9ORF72 hexanucleotide repeat number in frontotemporal lobar
degeneration: a genotype-phenotype correlation study', J Alzheimers Dis, 38: 799-
808.
Benyamin, Beben, Ji He, Qiongyi Zhao, Jacob Gratten, Fleur Garton, Paul J. Leo, Zhijun
Liu, Marie Mangelsdorf, Ammar Al-Chalabi, Lisa Anderson, Timothy J. Butler, Lu
Chen, Xiang-Ding Chen, Katie Cremin, Hong-Weng Deng, Matthew Devine, Janette
Edson, Jennifer A. Fifita, Sarah Furlong, Ying-Ying Han, Jessica Harris, Anjali K.
Henders, Rosalind L. Jeffree, Zi-Bing Jin, Zhongshan Li, Ting Li, Mengmeng Li,
Yong Lin, Xiaolu Liu, Mhairi Marshall, Emily P. McCann, Bryan J. Mowry, Shyuan
T. Ngo, Roger Pamphlett, Shu Ran, David C. Reutens, Dominic B. Rowe, Perminder
Sachdev, Sonia Shah, Sharon Song, Li-Jun Tan, Lu Tang, Leonard H. van den Berg,
Wouter van Rheenen, Jan H. Veldink, Robyn H. Wallace, Lawrie Wheeler, Kelly L.
Williams, Jinyu Wu, Xin Wu, Jian Yang, Weihua Yue, Zong-Hong Zhang, Dai
Zhang, Peter G. Noakes, Ian P. Blair, Robert D. Henderson, Pamela A. McCombe,
Peter M. Visscher, Huji Xu, Perry F. Bartlett, Matthew A. Brown, Naomi R. Wray,

206
and Dongsheng Fan. 2017. 'Cross-ethnic meta-analysis identifies association of the
GPX3-TNIP1 locus with amyotrophic lateral sclerosis', Nature Communications, 8:
611.
Bergeron, C., K. Beric-Maskarel, S. Muntasser, L. Weyer, M. J. Somerville, and M. E. Percy.
1994. 'Neurofilament light and polyadenylated mRNA levels are decreased in
amyotrophic lateral sclerosis motor neurons', J Neuropathol Exp Neurol, 53: 221-30.
Bilsland, L. G., E. Sahai, G. Kelly, M. Golding, L. Greensmith, and G. Schiavo. 2010.
'Deficits in axonal transport precede ALS symptoms in vivo', Proc Natl Acad Sci U
S A, 107: 20523-8.
Bindea, G., J. Galon, and B. Mlecnik. 2013. 'CluePedia Cytoscape plugin: pathway insights
using integrated experimental and in silico data', Bioinformatics, 29: 661-3.
Bindea, G., B. Mlecnik, H. Hackl, P. Charoentong, M. Tosolini, A. Kirilovsky, W. H.
Fridman, F. Pagès, Z. Trajanoski, and J. Galon. 2009. 'ClueGO: a Cytoscape plug-in
to decipher functionally grouped gene ontology and pathway annotation networks',
Bioinformatics, 25: 1091-3.
Blacher, Eran, Stavros Bashiardes, Hagit Shapiro, Daphna Rothschild, Uria Mor, Mally Dori-
Bachash, Christian Kleimeyer, Claudia Moresi, Yotam Harnik, Maya Zur, Michal
Zabari, Rotem Ben-Zeev Brik, Denise Kviatcovsky, Niv Zmora, Yotam Cohen,
Noam Bar, Izhak Levi, Nira Amar, Tevie Mehlman, Alexander Brandis, Inbal Biton,
Yael Kuperman, Michael Tsoory, Leenor Alfahel, Alon Harmelin, Michal Schwartz,
Adrian Israelson, Liisa Arike, Malin E. V. Johansson, Gunnar C. Hansson, Marc
Gotkine, Eran Segal, and Eran Elinav. 2019. 'Potential roles of gut microbiome and
metabolites in modulating ALS in mice', Nature, 572: 474-80.
Blain, C. R. V., S. Brunton, V. C. Williams, A. Leemans, M. R. Turner, P. M. Andersen, M.
Catani, B. R. Stanton, J. Ganesalingham, D. K. Jones, S. C. R. Williams, P. N. Leigh,
and A. Simmons. 2011. 'Differential corticospinal tract degeneration in homozygous
'D90A' SOD-1 ALS and sporadic ALS', Journal of neurology, neurosurgery, and
psychiatry, 82: 843-49.
Blasco, H., G. Garcon, F. Patin, C. Veyrat-Durebex, J. Boyer, D. Devos, P. Vourc'h, C. R.
Andres, and P. Corcia. 2017. 'Panel of Oxidative Stress and Inflammatory Biomarkers
in ALS: A Pilot Study', Can J Neurol Sci, 44: 90-95.
Blasi, E., R. Barluzzi, V. Bocchini, R. Mazzolla, and F. Bistoni. 1990. 'Immortalization of
murine microglial cells by a v-raf/v-myc carrying retrovirus', J Neuroimmunol, 27:
229-37.
Boillée, S., C. Vande Velde, and D. W. Cleveland. 2006. 'ALS: a disease of motor neurons
and their nonneuronal neighbors', Neuron, 52: 39-59.
Boillée, S., K. Yamanaka, C. S. Lobsiger, N. G. Copeland, N. A. Jenkins, G. Kassiotis, G.
Kollias, and D. W. Cleveland. 2006. 'Onset and progression in inherited ALS
determined by motor neurons and microglia', Science, 312: 1389-92.
Bond, C. S., and A. H. Fox. 2009. 'Paraspeckles: nuclear bodies built on long noncoding
RNA', J Cell Biol, 186: 637-44.
Bosco, Daryl A., Gerardo Morfini, N. Murat Karabacak, Yuyu Song, Francois Gros-Louis,
Piera Pasinelli, Holly Goolsby, Benjamin A. Fontaine, Nathan Lemay, Diane
McKenna-Yasek, Matthew P. Frosch, Jeffrey N. Agar, Jean-Pierre Julien, Scott T.
Brady, and Robert H. Brown, Jr. 2010. 'Wild-type and mutant SOD1 share an aberrant
conformation and a common pathogenic pathway in ALS', Nature Neuroscience, 13:
1396-403.

207
Boston-Howes, W., S. L. Gibb, E. O. Williams, P. Pasinelli, R. H. Brown, Jr., and D. Trotti.
2006. 'Caspase-3 cleaves and inactivates the glutamate transporter EAAT2', J Biol
Chem, 281: 14076-84.
Bourgeois, C. F., F. Lejeune, and J. Stevenin. 2004. 'Broad specificity of SR (serine/arginine)
proteins in the regulation of alternative splicing of pre-messenger RNA', Prog Nucleic
Acid Res Mol Biol, 78: 37-88.
Boutej, H., R. Rahimian, S. S. Thammisetty, L. C. Beland, M. Lalancette-Hebert, and J. Kriz.
2017. 'Diverging mRNA and Protein Networks in Activated Microglia Reveal SRSF3
Suppresses Translation of Highly Upregulated Innate Immune Transcripts', Cell Rep,
21: 3220-33.
Bowerman, M., C. Salsac, E. Coque, É Eiselt, R. G. Deschaumes, A. Brodovitch, L. C.
Burkly, F. Scamps, and C. Raoul. 2015. 'Tweak regulates astrogliosis, microgliosis
and skeletal muscle atrophy in a mouse model of amyotrophic lateral sclerosis', Hum
Mol Genet, 24: 3440-56.
Bozzo, F., A. Mirra, and M. T. Carrì. 2017. 'Oxidative stress and mitochondrial damage in
the pathogenesis of ALS: New perspectives', Neurosci Lett, 636: 3-8.
Bradley, Todd, Malcolm E. Cook, and Marco Blanchette. 2015. 'SR proteins control a
complex network of RNA-processing events', RNA (New York, N.Y.), 21: 75-92.
Braun, A. T., C. Caballero-Eraso, and N. Lechtzin. 2018. 'Amyotrophic Lateral Sclerosis and
the Respiratory System', Clin Chest Med, 39: 391-400.
Brenner, D., K. Sieverding, C. Bruno, P. Lüningschrör, E. Buck, S. Mungwa, L. Fischer, S.
J. Brockmann, J. Ulmer, C. Bliederhäuser, C. E. Philibert, T. Satoh, S. Akira, S.
Boillée, B. Mayer, M. Sendtner, A. C. Ludolph, K. M. Danzer, C. S. Lobsiger, A.
Freischmidt, and J. H. Weishaupt. 2019. 'Heterozygous Tbk1 loss has opposing
effects in early and late stages of ALS in mice', J Exp Med, 216: 267-78.
Brody, A. L., R. Hubert, R. Enoki, L. Y. Garcia, M. S. Mamoun, K. Okita, E. D. London, E.
L. Nurmi, L. C. Seaman, and M. A. Mandelkern. 2017. 'Effect of Cigarette Smoking
on a Marker for Neuroinflammation: A [(11)C]DAA1106 Positron Emission
Tomography Study', Neuropsychopharmacology, 42: 1630-39.
Broom, H. R., J. A. Rumfeldt, and E. M. Meiering. 2014. 'Many roads lead to Rome? Multiple
modes of Cu,Zn superoxide dismutase destabilization, misfolding and aggregation in
amyotrophic lateral sclerosis', Essays Biochem, 56: 149-65.
Broom, Helen R., Jessica A. O. Rumfeldt, Kenrick A. Vassall, and Elizabeth M. Meiering.
2015. 'Destabilization of the dimer interface is a common consequence of diverse
ALS-associated mutations in metal free SOD1', Protein science : a publication of the
Protein Society, 24: 2081-89.
Bruijn, L. I., M. W. Becher, M. K. Lee, K. L. Anderson, N. A. Jenkins, N. G. Copeland, S.
S. Sisodia, J. D. Rothstein, D. R. Borchelt, D. L. Price, and D. W. Cleveland. 1997.
'ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes
rapidly progressive disease with SOD1-containing inclusions', Neuron, 18: 327-38.
Buchan, J. Ross, and Roy Parker. 2009. 'Eukaryotic stress granules: the ins and outs of
translation', Mol Cell, 36: 932-41.
Budge, K. M., M. L. Neal, J. R. Richardson, and F. F. Safadi. 2018. 'Glycoprotein NMB: an
Emerging Role in Neurodegenerative Disease', Mol Neurobiol, 55: 5167-76.
Burberry, Aaron, Michael F. Wells, Francesco Limone, Alexander Couto, Kevin S. Smith,
James Keaney, Gaëlle Gillet, Nick van Gastel, Jin-Yuan Wang, Olli Pietilainen,
Menglu Qian, Pierce Eggan, Christopher Cantrell, Joanie Mok, Irena Kadiu, David

208
T. Scadden, and Kevin Eggan. 2020. 'C9orf72 suppresses systemic and neural
inflammation induced by gut bacteria', Nature, 582: 89-94.
Burk, Katja, and R. Jeroen Pasterkamp. 2019. 'Disrupted neuronal trafficking in amyotrophic
lateral sclerosis', Acta Neuropathologica, 137: 859-77.
Butovsky, O., S. Siddiqui, G. Gabriely, A. J. Lanser, B. Dake, G. Murugaiyan, C. E. Doykan,
P. M. Wu, R. R. Gali, L. K. Iyer, R. Lawson, J. Berry, A. M. Krichevsky, M. E.
Cudkowicz, and H. L. Weiner. 2012. 'Modulating inflammatory monocytes with a
unique microRNA gene signature ameliorates murine ALS', J Clin Invest, 122: 3063-
87.
Cáceres, J. F., G. R. Screaton, and A. R. Krainer. 1998. 'A specific subset of SR proteins
shuttles continuously between the nucleus and the cytoplasm', Genes & development,
12: 55-66.
Cappella, M., C. Ciotti, M. Cohen-Tannoudji, and M. G. Biferi. 2019. 'Gene Therapy for
ALS-A Perspective', Int J Mol Sci, 20.
Cappello, Valentina, and Maura Francolini. 2017. 'Neuromuscular Junction Dismantling in
Amyotrophic Lateral Sclerosis', Int J Mol Sci, 18: 2092.
Cardona, Astrid E., Erik P. Pioro, Margaret E. Sasse, Volodymyr Kostenko, Sandra M.
Cardona, Ineke M. Dijkstra, DeRen Huang, Grahame Kidd, Stephen Dombrowski,
RanJan Dutta, Jar-Chi Lee, Donald N. Cook, Steffen Jung, Sergio A. Lira, Dan R.
Littman, and Richard M. Ransohoff. 2006. 'Control of microglial neurotoxicity by the
fractalkine receptor', Nature Neuroscience, 9: 917-24.
Cartegni, L., S. L. Chew, and A. R. Krainer. 2002. 'Listening to silence and understanding
nonsense: exonic mutations that affect splicing', Nat Rev Genet, 3: 285-98.
Cassina, Patricia, Adriana Cassina, Mariana Pehar, Raquel Castellanos, Mandi Gandelman,
Andrés de León, Kristine M. Robinson, Ronald P. Mason, Joseph S. Beckman, Luis
Barbeito, and Rafael Radi. 2008. 'Mitochondrial Dysfunction in SOD1G93A-Bearing
Astrocytes Promotes Motor Neuron Degeneration: Prevention by Mitochondrial-
Targeted Antioxidants', The Journal of Neuroscience, 28: 4115.
Chang, L., and M. J. Monteiro. 2015. 'Defective Proteasome Delivery of Polyubiquitinated
Proteins by Ubiquilin-2 Proteins Containing ALS Mutations', PLOS ONE, 10:
e0130162.
Che, Yingying, and Lin Fu. 2020. 'Aberrant expression and regulatory network of splicing
factor-SRSF3 in tumors', Journal of Cancer, 11: 3502-11.
Chen-Plotkin, A. S., V. M. Lee, and J. Q. Trojanowski. 2010. 'TAR DNA-binding protein 43
in neurodegenerative disease', Nat Rev Neurol, 6: 211-20.
Chen, C., X. Ding, N. Akram, S. Xue, and S. Z. Luo. 2019. 'Fused in Sarcoma: Properties,
Self-Assembly and Correlation with Neurodegenerative Diseases', Molecules, 24.
Chen, H. J., G. Anagnostou, A. Chai, J. Withers, A. Morris, J. Adhikaree, G. Pennetta, and
J. S. de Belleroche. 2010. 'Characterization of the properties of a novel mutation in
VAPB in familial amyotrophic lateral sclerosis', J Biol Chem, 285: 40266-81.
Chen, H., M. Richard, D. P. Sandler, D. M. Umbach, and F. Kamel. 2007. 'Head injury and
amyotrophic lateral sclerosis', Am J Epidemiol, 166: 810-6.
Cherry, Jonathan D., John A. Olschowka, and M. Kerry O’Banion. 2014a. 'Are “Resting”
Microglia More “M2”?', Frontiers in Immunology, 5.
———. 2014b. 'Neuroinflammation and M2 microglia: the good, the bad, and the inflamed',
Journal of Neuroinflammation, 11: 98.

209
Chew, J., T. F. Gendron, M. Prudencio, H. Sasaguri, Y. J. Zhang, M. Castanedes-Casey, C.
W. Lee, K. Jansen-West, A. Kurti, M. E. Murray, K. F. Bieniek, P. O. Bauer, E. C.
Whitelaw, L. Rousseau, J. N. Stankowski, C. Stetler, L. M. Daughrity, E. A.
Perkerson, P. Desaro, A. Johnston, K. Overstreet, D. Edbauer, R. Rademakers, K. B.
Boylan, D. W. Dickson, J. D. Fryer, and L. Petrucelli. 2015. 'Neurodegeneration.
C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and
behavioral deficits', Science, 348: 1151-4.
Chiò, A., A. Cucatto, A. A. Terreni, and D. Schiffer. 1998. 'Reduced glutathione in
amyotrophic lateral sclerosis: an open, crossover, randomized trial', Ital J Neurol Sci,
19: 363-6.
Chiò, A., G. Logroscino, B. J. Traynor, J. Collins, J. C. Simeone, L. A. Goldstein, and L. A.
White. 2013. 'Global epidemiology of amyotrophic lateral sclerosis: a systematic
review of the published literature', Neuroepidemiology, 41: 118-30.
Chircop, M. 2014. 'Rho GTPases as regulators of mitosis and cytokinesis in mammalian
cells', Small GTPases, 5.
Chistiakov, D. A., M. C. Killingsworth, V. A. Myasoedova, A. N. Orekhov, and Y. V.
Bobryshev. 2017. 'CD68/macrosialin: not just a histochemical marker', Lab Invest,
97: 4-13.
Chiu, I. M., E. T. Morimoto, H. Goodarzi, J. T. Liao, S. O'Keeffe, H. P. Phatnani, M. Muratet,
M. C. Carroll, S. Levy, S. Tavazoie, R. M. Myers, and T. Maniatis. 2013. 'A
neurodegeneration-specific gene-expression signature of acutely isolated microglia
from an amyotrophic lateral sclerosis mouse model', Cell Rep, 4: 385-401.
Chiu, I. M., H. Phatnani, M. Kuligowski, J. C. Tapia, M. A. Carrasco, M. Zhang, T. Maniatis,
and M. C. Carroll. 2009. 'Activation of innate and humoral immunity in the peripheral
nervous system of ALS transgenic mice', Proc Natl Acad Sci U S A, 106: 20960-5.
Chu, K., X. Zhou, and B. Y. Luo. 2012. 'Cytokine gene polymorphisms and Parkinson's
disease: a meta-analysis', Can J Neurol Sci, 39: 58-64.
Churbanov, Alexander, Igor B. Rogozin, Jitender S. Deogun, and Hesham Ali. 2006. 'Method
of predicting Splice Sites based on signal interactions', Biology Direct, 1: 10.
Clement, A. M., M. D. Nguyen, E. A. Roberts, M. L. Garcia, S. Boillée, M. Rule, A. P.
McMahon, W. Doucette, D. Siwek, R. J. Ferrante, R. H. Brown, Jr., J. P. Julien, L. S.
Goldstein, and D. W. Cleveland. 2003. 'Wild-type nonneuronal cells extend survival
of SOD1 mutant motor neurons in ALS mice', Science, 302: 113-7.
Cléry, Antoine, Rahul Sinha, Olga Anczuków, Anna Corrionero, Ahmed Moursy, Gerrit M.
Daubner, Juan Valcárcel, Adrian R. Krainer, and Frédéric H. T. Allain. 2013. 'Isolated
pseudo-RNA-recognition motifs of SR proteins can regulate splicing using a
noncanonical mode of RNA recognition', Proc Natl Acad Sci U S A, 110: E2802-E11.
Cohen, Todd J., Andrew W. Hwang, Clark R. Restrepo, Chao-Xing Yuan, John Q.
Trojanowski, and Virginia M. Y. Lee. 2015. 'An acetylation switch controls TDP-43
function and aggregation propensity', Nature Communications, 6: 5845.
Cooper-Knock, J., C. Green, G. Altschuler, W. Wei, J. J. Bury, P. R. Heath, M. Wyles, C.
Gelsthorpe, J. R. Highley, A. Lorente-Pons, T. Beck, K. Doyle, K. Otero, B. Traynor,
J. Kirby, P. J. Shaw, and W. Hide. 2017. 'A data-driven approach links microglia to
pathology and prognosis in amyotrophic lateral sclerosis', Acta Neuropathol
Commun, 5: 23.
Coppedè, Fabio. 2011. 'An overview of DNA repair in amyotrophic lateral sclerosis',
TheScientificWorldJournal, 11: 1679-91.

210
Coque, Emmanuelle, Céline Salsac, Gabriel Espinosa-Carrasco, Béla Varga, Nicolas
Degauque, Marion Cadoux, Roxane Crabé, Anaïs Virenque, Claire Soulard, Julie K.
Fierle, Alexandre Brodovitch, Margot Libralato, Attila G. Végh, Stéphanie Venteo,
Frédérique Scamps, José Boucraut, David Laplaud, Javier Hernandez, Csilla Gergely,
Thierry Vincent, and Cédric Raoul. 2019. 'Cytotoxic CD8+ T lymphocytes
expressing ALS-causing SOD1 mutant selectively trigger death of spinal
motoneurons', Proceedings of the National Academy of Sciences, 116: 2312.
Cordeau, P., Jr., M. Lalancette-Hébert, Y. C. Weng, and J. Kriz. 2016. 'Estrogen receptors
alpha mediates postischemic inflammation in chronically estrogen-deprived mice',
Neurobiol Aging, 40: 50-60.
Cudkowicz, M. E., D. McKenna-Yasek, P. E. Sapp, W. Chin, B. Geller, D. L. Hayden, D. A.
Schoenfeld, B. A. Hosler, H. R. Horvitz, and R. H. Brown. 1997. 'Epidemiology of
mutations in superoxide dismutase in amyotrophic lateral sclerosis', Ann Neurol, 41:
210-21.
Cudkowicz, M. E., K. A. Pastusza, P. C. Sapp, R. K. Mathews, J. Leahy, P. Pasinelli, J. W.
Francis, D. Jiang, J. K. Andersen, and R. H. Brown, Jr. 2002. 'Survival in transgenic
ALS mice does not vary with CNS glutathione peroxidase activity', Neurology, 59:
729-34.
Cui, Mingxue, Mary Ann Allen, Alison Larsen, Margaret Macmorris, Min Han, and Tom
Blumenthal. 2008. 'Genes involved in pre-mRNA 3'-end formation and transcription
termination revealed by a lin-15 operon Muv suppressor screen', Proc Natl Acad Sci
U S A, 105: 16665-70.
D'Amico, E., P. Factor-Litvak, R. M. Santella, and H. Mitsumoto. 2013. 'Clinical perspective
on oxidative stress in sporadic amyotrophic lateral sclerosis', Free Radic Biol Med,
65: 509-27.
da Costa, P. J., J. Menezes, and L. Romao. 2017. 'The role of alternative splicing coupled to
nonsense-mediated mRNA decay in human disease', Int J Biochem Cell Biol, 91: 168-
75.
da Silva, Maria Roméria, Gabriela Alves Moreira, Ronni Anderson Gonçalves da Silva,
Éverton de Almeida Alves Barbosa, Raoni Pais Siqueira, Róbson Ricardo Teixera,
Márcia Rogéria Almeida, Abelardo Silva Júnior, Juliana Lopes Rangel Fietto, and
Gustavo Costa Bressan. 2015. 'Splicing Regulators and Their Roles in Cancer
Biology and Therapy', BioMed research international, 2015: 150514-14.
Davalos, D., J. Grutzendler, G. Yang, J. V. Kim, Y. Zuo, S. Jung, D. R. Littman, M. L.
Dustin, and W. B. Gan. 2005. 'ATP mediates rapid microglial response to local brain
injury in vivo', Nat Neurosci, 8: 752-8.
de la Mata, M., and A. R. Kornblihtt. 2006. 'RNA polymerase II C-terminal domain mediates
regulation of alternative splicing by SRp20', Nat Struct Mol Biol, 13: 973-80.
De Vos, K. J., and M. Hafezparast. 2017. 'Neurobiology of axonal transport defects in motor
neuron diseases: Opportunities for translational research?', Neurobiol Dis, 105: 283-
99.
Deczkowska, A., H. Keren-Shaul, A. Weiner, M. Colonna, M. Schwartz, and I. Amit. 2018.
'Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration',
Cell, 173: 1073-81.
DeLoach, A., M. Cozart, A. Kiaei, and M. Kiaei. 2015. 'A retrospective review of the
progress in amyotrophic lateral sclerosis drug discovery over the last decade and a
look at the latest strategies', Expert Opin Drug Discov, 10: 1099-118.

211
Deng, B., W. Lv, W. Duan, Y. Liu, Z. Li, Y. Ma, G. Zhang, X. Song, C. Cui, X. Qi, Y. Li,
and C. Li. 2018. 'Progressive Degeneration and Inhibition of Peripheral Nerve
Regeneration in the SOD1-G93A Mouse Model of Amyotrophic Lateral Sclerosis',
Cell Physiol Biochem, 46: 2358-72.
Deng, H. X., W. Chen, S. T. Hong, K. M. Boycott, G. H. Gorrie, N. Siddique, Y. Yang, F.
Fecto, Y. Shi, H. Zhai, H. Jiang, M. Hirano, E. Rampersaud, G. H. Jansen, S.
Donkervoort, E. H. Bigio, B. R. Brooks, K. Ajroud, R. L. Sufit, J. L. Haines, E.
Mugnaini, M. A. Pericak-Vance, and T. Siddique. 2011. 'Mutations in UBQLN2
cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia', Nature,
477: 211-5.
Deng, Han-Xiang, Hong Zhai, Eileen H. Bigio, Jianhua Yan, Faisal Fecto, Kaouther Ajroud,
Manjari Mishra, Senda Ajroud-Driss, Scott Heller, Robert Sufit, Nailah Siddique,
Enrico Mugnaini, and Teepu Siddique. 2010. 'FUS-immunoreactive inclusions are a
common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis',
Annals of Neurology, 67: 739-48.
Dettwiler, S., C. Aringhieri, S. Cardinale, W. Keller, and S. M. Barabino. 2004. 'Distinct
sequence motifs within the 68-kDa subunit of cleavage factor Im mediate RNA
binding, protein-protein interactions, and subcellular localization', J Biol Chem, 279:
35788-97.
Dewey, C. M., B. Cenik, C. F. Sephton, D. R. Dries, P. Mayer, 3rd, S. K. Good, B. A.
Johnson, J. Herz, and G. Yu. 2011. 'TDP-43 is directed to stress granules by sorbitol,
a novel physiological osmotic and oxidative stressor', Mol Cell Biol, 31: 1098-108.
Di Giorgio, F. P., M. A. Carrasco, M. C. Siao, T. Maniatis, and K. Eggan. 2007. 'Non-cell
autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS
model', Nat Neurosci, 10: 608-14.
Diaz-Amarilla, P., S. Olivera-Bravo, E. Trias, A. Cragnolini, L. Martinez-Palma, P. Cassina,
J. Beckman, and L. Barbeito. 2011. 'Phenotypically aberrant astrocytes that promote
motoneuron damage in a model of inherited amyotrophic lateral sclerosis', Proc Natl
Acad Sci U S A, 108: 18126-31.
Díaz-Amarilla, P., S. Olivera-Bravo, E. Trias, A. Cragnolini, L. Martínez-Palma, P. Cassina,
J. Beckman, and L. Barbeito. 2011. 'Phenotypically aberrant astrocytes that promote
motoneuron damage in a model of inherited amyotrophic lateral sclerosis', Proc Natl
Acad Sci U S A, 108: 18126-31.
Ding, H., D. S. Schwarz, A. Keene, B. Affar el, L. Fenton, X. Xia, Y. Shi, P. D. Zamore, and
Z. Xu. 2003. 'Selective silencing by RNAi of a dominant allele that causes
amyotrophic lateral sclerosis', Aging Cell, 2: 209-17.
Ding, J. H., X. Y. Zhong, J. C. Hagopian, M. M. Cruz, G. Ghosh, J. Feramisco, J. A. Adams,
and X. D. Fu. 2006. 'Regulated cellular partitioning of SR protein-specific kinases in
mammalian cells', Mol Biol Cell, 17: 876-85.
Diniz, L. P., I. Matias, M. Siqueira, J. Stipursky, and F. C. A. Gomes. 2019. 'Astrocytes and
the TGF-β1 Pathway in the Healthy and Diseased Brain: a Double-Edged Sword',
Mol Neurobiol, 56: 4653-79.
Do, Dang Vinh, Bernhard Strauss, Engin Cukuroglu, Iain Macaulay, Keng Boon Wee, Tim
Xiaoming Hu, Ruiz De Los Mozos Igor, Caroline Lee, Andrew Harrison, Richard
Butler, Sabine Dietmann, Ule Jernej, John Marioni, Christopher W. J. Smith,
Jonathan Göke, and M. Azim Surani. 2018. 'SRSF3 maintains transcriptome integrity

212
in oocytes by regulation of alternative splicing and transposable elements', Cell
discovery, 4: 33-33.
Doble, A. 1996. 'The pharmacology and mechanism of action of riluzole', Neurology, 47:
233S.
Dobson-Stone, C., M. Hallupp, H. Shahheydari, A. M. G. Ragagnin, Z. Chatterton, F. Carew-
Jones, C. E. Shepherd, H. Stefen, E. Paric, T. Fath, E. M. Thompson, P. Blumbergs,
C. L. Short, C. D. Field, P. K. Panegyres, J. Hecker, G. Nicholson, A. D. Shaw, J. M.
Fullerton, A. A. Luty, P. R. Schofield, W. S. Brooks, N. Rajan, M. F. Bennett, M.
Bahlo, J. E. Landers, O. Piguet, J. R. Hodges, G. M. Halliday, S. D. Topp, B. N.
Smith, C. E. Shaw, E. McCann, J. A. Fifita, K. L. Williams, J. D. Atkin, I. P. Blair,
and J. B. Kwok. 2020. 'CYLD is a causative gene for frontotemporal dementia -
amyotrophic lateral sclerosis', Brain, 143: 783-99.
Dringen, R. 2000. 'Metabolism and functions of glutathione in brain', Prog Neurobiol, 62:
649-71.
Duan, W., X. Li, J. Shi, Y. Guo, Z. Li, and C. Li. 2010. 'Mutant TAR DNA-binding protein-
43 induces oxidative injury in motor neuron-like cell', Neuroscience, 169: 1621-29.
Duncan, P. I., D. F. Stojdl, R. M. Marius, and J. C. Bell. 1997. 'In vivo regulation of
alternative pre-mRNA splicing by the Clk1 protein kinase', Molecular and Cellular
Biology, 17: 5996.
Dupuis, L., J. L. Gonzalez de Aguilar, F. di Scala, F. Rene, M. de Tapia, P. F. Pradat, L.
Lacomblez, D. Seihlan, R. Prinjha, F. S. Walsh, V. Meininger, and J. P. Loeffler.
2002. 'Nogo provides a molecular marker for diagnosis of amyotrophic lateral
sclerosis', Neurobiol Dis, 10: 358-65.
DuVal, M. G., V. K. Hinge, N. Snyder, R. Kanyo, J. Bratvold, E. Pokrishevsky, N. R.
Cashman, N. Blinov, A. Kovalenko, and W. T. Allison. 2019. 'Tryptophan 32
mediates SOD1 toxicity in a in vivo motor neuron model of ALS and is a promising
target for small molecule therapeutics', Neurobiol Dis, 124: 297-310.
Easley-Neal, Courtney, Oded Foreman, Neeraj Sharma, Ali A. Zarrin, and Robby M Weimer.
2019. 'CSF1R Ligands IL-34 and CSF1 Are Differentially Required for Microglia
Development and Maintenance in White and Gray Matter Brain Regions', Frontiers
in Immunology, 10.
Elmore, M. R., A. R. Najafi, M. A. Koike, N. N. Dagher, E. E. Spangenberg, R. A. Rice, M.
Kitazawa, B. Matusow, H. Nguyen, B. L. West, and K. N. Green. 2014. 'Colony-
stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking
a microglia progenitor cell in the adult brain', Neuron, 82: 380-97.
Engelhardt, J. I., and S. H. Appel. 1990. 'IgG reactivity in the spinal cord and motor cortex
in amyotrophic lateral sclerosis', Arch Neurol, 47: 1210-6.
Engelhardt, J. I., J. Tajti, and S. H. Appel. 1993. 'Lymphocytic infiltrates in the spinal cord
in amyotrophic lateral sclerosis', Arch Neurol, 50: 30-6.
Erny, D., A. L. Hrabě de Angelis, D. Jaitin, P. Wieghofer, O. Staszewski, E. David, H. Keren-
Shaul, T. Mahlakoiv, K. Jakobshagen, T. Buch, V. Schwierzeck, O. Utermöhlen, E.
Chun, W. S. Garrett, K. D. McCoy, A. Diefenbach, P. Staeheli, B. Stecher, I. Amit,
and M. Prinz. 2015. 'Host microbiota constantly control maturation and function of
microglia in the CNS', Nat Neurosci, 18: 965-77.
Escudero-Paunetto, Laurimar, Ling Li, Felicia P. Hernandez, and Rozanne M. Sandri-
Goldin. 2010. 'SR proteins SRp20 and 9G8 contribute to efficient export of herpes
simplex virus 1 mRNAs', Virology, 401: 155-64.

213
Ezzi, S. A., R. Larivière, M. Urushitani, and J. P. Julien. 2010. 'Neuronal over-expression of
chromogranin A accelerates disease onset in a mouse model of ALS', J Neurochem,
115: 1102-11.
Ezzi, S. A., M. Urushitani, and J. P. Julien. 2007. 'Wild-type superoxide dismutase acquires
binding and toxic properties of ALS-linked mutant forms through oxidation', J
Neurochem, 102: 170-8.
Fallini, C., B. Khalil, C. L. Smith, and W. Rossoll. 2020. 'Traffic jam at the nuclear pore: All
roads lead to nucleocytoplasmic transport defects in ALS/FTD', Neurobiol Dis, 140:
104835.
Fani Maleki, A., and S. Rivest. 2019. 'Innate Immune Cells: Monocytes, Monocyte-Derived
Macrophages and Microglia as Therapeutic Targets for Alzheimer's Disease and
Multiple Sclerosis', Front Cell Neurosci, 13: 355.
Faustino, N. A., and T. A. Cooper. 2003. 'Pre-mRNA splicing and human disease', Genes &
development, 17: 419-37.
Fecto, F., J. Yan, S. P. Vemula, E. Liu, Y. Yang, W. Chen, J. G. Zheng, Y. Shi, N. Siddique,
H. Arrat, S. Donkervoort, S. Ajroud-Driss, R. L. Sufit, S. L. Heller, H. X. Deng, and
T. Siddique. 2011. 'SQSTM1 mutations in familial and sporadic amyotrophic lateral
sclerosis', Arch Neurol, 68: 1440-6.
Federico, A., E. Cardaioli, P. Da Pozzo, P. Formichi, G. N. Gallus, and E. Radi. 2012.
'Mitochondria, oxidative stress and neurodegeneration', J Neurol Sci, 322: 254-62.
Feneberg, E., E. Gray, O. Ansorge, K. Talbot, and M. R. Turner. 2018. 'Towards a TDP-43-
Based Biomarker for ALS and FTLD', Mol Neurobiol, 55: 7789-801.
Ferraiuolo, L., K. Meyer, T. W. Sherwood, J. Vick, S. Likhite, A. Frakes, C. J. Miranda, L.
Braun, P. R. Heath, R. Pineda, C. E. Beattie, P. J. Shaw, C. C. Askwith, D. McTigue,
and B. K. Kaspar. 2016. 'Oligodendrocytes contribute to motor neuron death in ALS
via SOD1-dependent mechanism', Proc Natl Acad Sci U S A, 113: E6496-e505.
Figlewicz, D. A., A. Krizus, M. G. Martinoli, V. Meininger, M. Dib, G. A. Rouleau, and J.
P. Julien. 1994. 'Variants of the heavy neurofilament subunit are associated with the
development of amyotrophic lateral sclerosis', Hum Mol Genet, 3: 1757-61.
Fischer, L. R., D. G. Culver, P. Tennant, A. A. Davis, M. Wang, A. Castellano-Sanchez, J.
Khan, M. A. Polak, and J. D. Glass. 2004. 'Amyotrophic lateral sclerosis is a distal
axonopathy: evidence in mice and man', Exp Neurol, 185: 232-40.
Fitzgerald, Kerry D., Amanda J. Chase, Andrea L. Cathcart, Genevieve P. Tran, and Bert L.
Semler. 2013. 'Viral proteinase requirements for the nucleocytoplasmic relocalization
of cellular splicing factor SRp20 during picornavirus infections', Journal of Virology,
87: 2390-400.
Fitzgerald, Kerry D., and Bert L. Semler. 2013. 'Poliovirus infection induces the co-
localization of cellular protein SRp20 with TIA-1, a cytoplasmic stress granule
protein', Virus research, 176: 223-31.
Frakes, A. E., L. Ferraiuolo, A. M. Haidet-Phillips, L. Schmelzer, L. Braun, C. J. Miranda,
K. J. Ladner, A. K. Bevan, K. D. Foust, J. P. Godbout, P. G. Popovich, D. C.
Guttridge, and B. K. Kaspar. 2014. 'Microglia induce motor neuron death via the
classical NF-κB pathway in amyotrophic lateral sclerosis', Neuron, 81: 1009-23.
Freischmidt, A., T. Wieland, B. Richter, W. Ruf, V. Schaeffer, K. Müller, N. Marroquin, F.
Nordin, A. Hübers, P. Weydt, S. Pinto, R. Press, S. Millecamps, N. Molko, E.
Bernard, C. Desnuelle, M. H. Soriani, J. Dorst, E. Graf, U. Nordström, M. S. Feiler,
S. Putz, T. M. Boeckers, T. Meyer, A. S. Winkler, J. Winkelman, M. de Carvalho, D.

214
R. Thal, M. Otto, T. Brännström, A. E. Volk, P. Kursula, K. M. Danzer, P. Lichtner,
I. Dikic, T. Meitinger, A. C. Ludolph, T. M. Strom, P. M. Andersen, and J. H.
Weishaupt. 2015. 'Haploinsufficiency of TBK1 causes familial ALS and fronto-
temporal dementia', Nat Neurosci, 18: 631-6.
Fujioka, Y., S. Ishigaki, A. Masuda, Y. Iguchi, T. Udagawa, H. Watanabe, M. Katsuno, K.
Ohno, and G. Sobue. 2013. 'FUS-regulated region- and cell-type-specific
transcriptome is associated with cell selectivity in ALS/FTLD', Sci Rep, 3: 2388.
Furukawa, Y., R. Fu, H. X. Deng, T. Siddique, and T. V. O'Halloran. 2006. 'Disulfide cross-
linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide
dismutase aggregates in spinal cords of model mice', Proc Natl Acad Sci U S A, 103:
7148-53.
Furukawa, Y., K. Kaneko, S. Watanabe, K. Yamanaka, and N. Nukina. 2011. 'A seeding
reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation
response element (TAR) DNA-binding protein-43 inclusions', J Biol Chem, 286:
18664-72.
Fushimi, Kazuo, Charles Long, Neha Jayaram, Xiaoping Chen, Liming Li, and Jane Y. Wu.
2011. 'Expression of human FUS/TLS in yeast leads to protein aggregation and
cytotoxicity, recapitulating key features of FUS proteinopathy', Protein & cell, 2:
141-49.
Gaj, Thomas, David S. Ojala, Freja K. Ekman, Leah C. Byrne, Prajit Limsirichai, and David
V. Schaffer. 2017. 'In vivo genome editing improves motor function and extends
survival in a mouse model of ALS', Science advances, 3: eaar3952-eaar52.
Garofalo, Stefano, Germana Cocozza, Alessandra Porzia, Maurizio Inghilleri, Marcello
Raspa, Ferdinando Scavizzi, Eleonora Aronica, Giovanni Bernardini, Ling Peng,
Richard M. Ransohoff, Angela Santoni, and Cristina Limatola. 2020. 'Natural killer
cells modulate motor neuron-immune cell cross talk in models of Amyotrophic
Lateral Sclerosis', Nature Communications, 11: 1773.
Gendron, Tania F., and Leonard Petrucelli. 2018. 'Disease Mechanisms of C9ORF72 Repeat
Expansions', Cold Spring Harbor perspectives in medicine, 8: a024224.
Geser, F., N. J. Brandmeir, L. K. Kwong, M. Martinez-Lage, L. Elman, L. McCluskey, S. X.
Xie, V. M. Lee, and J. Q. Trojanowski. 2008. 'Evidence of multisystem disorder in
whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis', Arch
Neurol, 65: 636-41.
Ghosh, G., and J. A. Adams. 2011. 'Phosphorylation mechanism and structure of serine-
arginine protein kinases', Febs j, 278: 587-97.
Ginhoux, F., M. Greter, M. Leboeuf, S. Nandi, P. See, S. Gokhan, M. F. Mehler, S. J.
Conway, L. G. Ng, E. R. Stanley, I. M. Samokhvalov, and M. Merad. 2010. 'Fate
mapping analysis reveals that adult microglia derive from primitive macrophages',
Science, 330: 841-5.
Goetz, C. G. 2000. 'Amyotrophic lateral sclerosis: early contributions of Jean-Martin
Charcot', Muscle Nerve, 23: 336-43.
Goldmann, T., P. Wieghofer, M. J. Jordão, F. Prutek, N. Hagemeyer, K. Frenzel, L. Amann,
O. Staszewski, K. Kierdorf, M. Krueger, G. Locatelli, H. Hochgerner, R. Zeiser, S.
Epelman, F. Geissmann, J. Priller, F. M. Rossi, I. Bechmann, M. Kerschensteiner, S.
Linnarsson, S. Jung, and M. Prinz. 2016. 'Origin, fate and dynamics of macrophages
at central nervous system interfaces', Nat Immunol, 17: 797-805.

215
Goncalves, V., P. Matos, and P. Jordan. 2009. 'Antagonistic SR proteins regulate alternative
splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways',
Hum Mol Genet, 18: 3696-707.
Gong, Z., and J. Chen. 2011. 'E3 ligase RFWD3 participates in replication checkpoint
control', J Biol Chem, 286: 22308-13.
Gopal, R., B. Lee, K. J. McHugh, H. E. Rich, K. Ramanan, S. Mandalapu, M. E. Clay, P. J.
Seger, R. I. Enelow, M. L. Manni, K. M. Robinson, J. Rangel-Moreno, and J. F.
Alcorn. 2018. 'STAT2 Signaling Regulates Macrophage Phenotype During Influenza
and Bacterial Super-Infection', Front Immunol, 9: 2151.
Gowing, G., M. Lalancette-Hébert, J. N. Audet, F. Dequen, and J. P. Julien. 2009.
'Macrophage colony stimulating factor (M-CSF) exacerbates ALS disease in a mouse
model through altered responses of microglia expressing mutant superoxide
dismutase', Exp Neurol, 220: 267-75.
Gowing, G., T. Philips, B. Van Wijmeersch, J. N. Audet, M. Dewil, L. Van Den Bosch, A.
D. Billiau, W. Robberecht, and J. P. Julien. 2008. 'Ablation of proliferating microglia
does not affect motor neuron degeneration in amyotrophic lateral sclerosis caused by
mutant superoxide dismutase', J Neurosci, 28: 10234-44.
Graber, D. J., W. F. Hickey, and B. T. Harris. 2010. 'Progressive changes in microglia and
macrophages in spinal cord and peripheral nerve in the transgenic rat model of
amyotrophic lateral sclerosis', J Neuroinflammation, 7: 8.
Grad, L. I., and N. R. Cashman. 2014. 'Prion-like activity of Cu/Zn superoxide dismutase:
implications for amyotrophic lateral sclerosis', Prion, 8: 33-41.
Gravel, M., L. C. Beland, G. Soucy, E. Abdelhamid, R. Rahimian, C. Gravel, and J. Kriz.
2016. 'IL-10 Controls Early Microglial Phenotypes and Disease Onset in ALS Caused
by Misfolded Superoxide Dismutase 1', J Neurosci, 36: 1031-48.
Graveley, B. R. 2001. 'Alternative splicing: increasing diversity in the proteomic world',
Trends Genet, 17: 100-7.
Greco, V., P. Longone, A. Spalloni, L. Pieroni, and A. Urbani. 2019. 'Crosstalk Between
Oxidative Stress and Mitochondrial Damage: Focus on Amyotrophic Lateral
Sclerosis', Adv Exp Med Biol, 1158: 71-82.
Gregory, Jenna M., Delphine Fagegaltier, Hemali Phatnani, and Matthew B. Harms. 2020.
'Genetics of Amyotrophic Lateral Sclerosis', Current Genetic Medicine Reports, 8:
121-31.
Gros-Louis, F., R. Larivière, G. Gowing, S. Laurent, W. Camu, J. P. Bouchard, V. Meininger,
G. A. Rouleau, and J. P. Julien. 2004. 'A frameshift deletion in peripherin gene
associated with amyotrophic lateral sclerosis', J Biol Chem, 279: 45951-6.
Gugliandolo, A., S. Giacoppo, P. Bramanti, and E. Mazzon. 2018. 'NLRP3 Inflammasome
Activation in a Transgenic Amyotrophic Lateral Sclerosis Model', Inflammation, 41:
93-103.
Gui, J. F., H. Tronchère, S. D. Chandler, and X. D. Fu. 1994. 'Purification and
characterization of a kinase specific for the serine- and arginine-rich pre-mRNA
splicing factors', Proc Natl Acad Sci U S A, 91: 10824-28.
Gurney, M. E., H. Pu, A. Y. Chiu, M. C. Dal Canto, C. Y. Polchow, D. D. Alexander, J.
Caliendo, A. Hentati, Y. W. Kwon, H. X. Deng, and et al. 1994. 'Motor neuron
degeneration in mice that express a human Cu,Zn superoxide dismutase mutation',
Science, 264: 1772-5.

216
Haidet-Phillips, Amanda M., Mark E. Hester, Carlos J. Miranda, Kathrin Meyer, Lyndsey
Braun, Ashley Frakes, SungWon Song, Shibi Likhite, Matthew J. Murtha, Kevin D.
Foust, Meghan Rao, Amy Eagle, Anja Kammesheidt, Ashley Christensen, Jerry R.
Mendell, Arthur H. M. Burghes, and Brian K. Kaspar. 2011. 'Astrocytes from familial
and sporadic ALS patients are toxic to motor neurons', Nature Biotechnology, 29:
824-28.
Hallgren, Oskar, Johan Malmström, Lars Malmström, Annika Andersson-Sjöland, Marie
Wildt, Ellen Tufvesson, Peter Juhasz, György Marko-Varga, and Gunilla
Westergren-Thorsson. 2012. 'Splicosomal and serine and arginine-rich splicing
factors as targets for TGF-β', Fibrogenesis & Tissue Repair, 5: 6.
Halloran, M., A. M. G. Ragagnin, M. Vidal, S. Parakh, S. Yang, B. Heng, N. Grima, H.
Shahheydari, K. Y. Soo, I. Blair, G. J. Guillemin, V. Sundaramoorthy, and J. D.
Atkin. 2019. 'Amyotrophic lateral sclerosis-linked UBQLN2 mutants inhibit
endoplasmic reticulum to Golgi transport, leading to Golgi fragmentation and ER
stress', Cell Mol Life Sci.
Hardiman, Orla, Ammar Al-Chalabi, Adriano Chio, Emma M. Corr, Giancarlo Logroscino,
Wim Robberecht, Pamela J. Shaw, Zachary Simmons, and Leonard H. van den Berg.
2017. 'Amyotrophic lateral sclerosis', Nature Reviews Disease Primers, 3: 17071.
Hargous, Yann, Guillaume M. Hautbergue, Aura M. Tintaru, Lenka Skrisovska, Alexander
P. Golovanov, James Stevenin, Lu-Yun Lian, Stuart A. Wilson, and Frédéric H. T.
Allain. 2006. 'Molecular basis of RNA recognition and TAP binding by the SR
proteins SRp20 and 9G8', Embo j, 25: 5126-37.
Haure-Mirande, J. V., M. Wang, M. Audrain, T. Fanutza, S. H. Kim, S. Heja, B. Readhead,
J. T. Dudley, R. D. Blitzer, E. E. Schadt, B. Zhang, S. Gandy, and M. E. Ehrlich.
2019. 'Integrative approach to sporadic Alzheimer's disease: deficiency of TYROBP
in cerebral Abeta amyloidosis mouse normalizes clinical phenotype and complement
subnetwork molecular pathology without reducing Abeta burden', Mol Psychiatry,
24: 431-46.
Hautbergue, Guillaume M., Ming-Lung Hung, Alexander P. Golovanov, Lu-Yun Lian, and
Stuart A. Wilson. 2008. 'Mutually exclusive interactions drive handover of mRNA
from export adaptors to TAP', Proc Natl Acad Sci U S A, 105: 5154-59.
Hazenberg, A., H. A. M. Kerstjens, S. C. L. Prins, K. M. Vermeulen, and P. J. Wijkstra. 2016.
'Is chronic ventilatory support really effective in patients with amyotrophic lateral
sclerosis?', Journal of Neurology, 263: 2456-61.
He, X., A. D. Arslan, M. D. Pool, T. T. Ho, K. M. Darcy, J. S. Coon, and W. T. Beck. 2011.
'Knockdown of splicing factor SRp20 causes apoptosis in ovarian cancer cells and its
expression is associated with malignancy of epithelial ovarian cancer', Oncogene, 30:
356-65.
Heiman, M., R. Kulicke, R. J. Fenster, P. Greengard, and N. Heintz. 2014. 'Cell type-specific
mRNA purification by translating ribosome affinity purification (TRAP)', Nat
Protoc, 9: 1282-91.
Heiman, M., A. Schaefer, S. Gong, J. D. Peterson, M. Day, K. E. Ramsey, M. Suárez-Fariñas,
C. Schwarz, D. A. Stephan, D. J. Surmeier, P. Greengard, and N. Heintz. 2008. 'A
translational profiling approach for the molecular characterization of CNS cell types',
Cell, 135: 738-48.
Hellwig, Sabine, Annette Heinrich, and Knut Biber. 2013. 'The brain's best friend: microglial
neurotoxicity revisited', Front Cell Neurosci, 7: 71-71.

217
Henkel, J. S., D. R. Beers, W. Zhao, and S. H. Appel. 2009. 'Microglia in ALS: the good, the
bad, and the resting', J Neuroimmune Pharmacol, 4: 389-98.
Henkel, J. S., J. I. Engelhardt, L. Siklós, E. P. Simpson, S. H. Kim, T. Pan, J. C. Goodman,
T. Siddique, D. R. Beers, and S. H. Appel. 2004. 'Presence of dendritic cells, MCP-
1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord
tissue', Ann Neurol, 55: 221-35.
Heurich, B., N. B. El Idrissi, R. M. Donev, S. Petri, P. Claus, J. Neal, B. P. Morgan, and V.
Ramaglia. 2011. 'Complement upregulation and activation on motor neurons and
neuromuscular junction in the SOD1 G93A mouse model of familial amyotrophic
lateral sclerosis', J Neuroimmunol, 235: 104-9.
Hickman, S. E., and J. El Khoury. 2019. 'Analysis of the Microglial Sensome', Methods Mol
Biol, 2034: 305-23.
Hong, Soyon, Lasse Dissing-Olesen, and Beth Stevens. 2016. 'New insights on the role of
microglia in synaptic pruning in health and disease', Current Opinion in
Neurobiology, 36: 128-34.
Huang, X., Y. Li, M. Fu, and H. B. Xin. 2018. 'Polarizing Macrophages In Vitro', Methods
Mol Biol, 1784: 119-26.
Huang, Y., R. Gattoni, J. Stevenin, and J. A. Steitz. 2003. 'SR splicing factors serve as adapter
proteins for TAP-dependent mRNA export', Mol Cell, 11: 837-43.
Huang, Y., and J. A. Steitz. 2001. 'Splicing factors SRp20 and 9G8 promote the
nucleocytoplasmic export of mRNA', Mol Cell, 7: 899-905.
Huber, Wolfgang, Vincent J. Carey, Robert Gentleman, Simon Anders, Marc Carlson,
Benilton S. Carvalho, Hector Corrada Bravo, Sean Davis, Laurent Gatto, Thomas
Girke, Raphael Gottardo, Florian Hahne, Kasper D. Hansen, Rafael A. Irizarry,
Michael Lawrence, Michael I. Love, James MacDonald, Valerie Obenchain, Andrzej
K. Oleś, Hervé Pagès, Alejandro Reyes, Paul Shannon, Gordon K. Smyth, Dan
Tenenbaum, Levi Waldron, and Martin Morgan. 2015. 'Orchestrating high-
throughput genomic analysis with Bioconductor', Nature methods, 12: 115-21.
Hukema, R. K., F. W. Riemslagh, S. Melhem, H. C. van der Linde, L. A. Severijnen, D.
Edbauer, A. Maas, N. Charlet-Berguerand, R. Willemsen, and J. C. van Swieten.
2014. 'A new inducible transgenic mouse model for C9orf72-associated GGGGCC
repeat expansion supports a gain-of-function mechanism in C9orf72-associated ALS
and FTD', Acta Neuropathol Commun, 2: 166.
Hüttenrauch, Melanie, Isabella Ogorek, Hans Klafki, Markus Otto, Christine Stadelmann,
Sascha Weggen, Jens Wiltfang, and Oliver Wirths. 2018. 'Glycoprotein NMB: a
novel Alzheimer's disease associated marker expressed in a subset of activated
microglia', Acta neuropathologica communications, 6: 108-08.
Iguchi, Y., L. Eid, M. Parent, G. Soucy, C. Bareil, Y. Riku, K. Kawai, S. Takagi, M. Yoshida,
M. Katsuno, G. Sobue, and J. P. Julien. 2016. 'Exosome secretion is a key pathway
for clearance of pathological TDP-43', Brain, 139: 3187-201.
Ilieva, H., M. Polymenidou, and D. W. Cleveland. 2009. 'Non-cell autonomous toxicity in
neurodegenerative disorders: ALS and beyond', J Cell Biol, 187: 761-72.
Ishigaki, Shinsuke, and Gen Sobue. 2018. 'Importance of Functional Loss of FUS in
FTLD/ALS', Frontiers in molecular biosciences, 5: 44-44.
Jaitin, Diego Adhemar, Ephraim Kenigsberg, Hadas Keren-Shaul, Naama Elefant, Franziska
Paul, Irina Zaretsky, Alexander Mildner, Nadav Cohen, Steffen Jung, Amos Tanay,

218
and Ido Amit. 2014. 'Massively Parallel Single-Cell RNA-Seq for Marker-Free
Decomposition of Tissues into Cell Types', Science, 343: 776.
Jayabalan, Aravinth Kumar, Anthony Sanchez, Ra Young Park, Sang Pil Yoon, Gum-Yong
Kang, Je-Hyun Baek, Paul Anderson, Younghoon Kee, and Takbum Ohn. 2016.
'NEDDylation promotes stress granule assembly', Nature Communications, 7: 12125-
25.
Jha, A. K., S. C. Huang, A. Sergushichev, V. Lampropoulou, Y. Ivanova, E. Loginicheva, K.
Chmielewski, K. M. Stewart, J. Ashall, B. Everts, E. J. Pearce, E. M. Driggers, and
M. N. Artyomov. 2015. 'Network integration of parallel metabolic and transcriptional
data reveals metabolic modules that regulate macrophage polarization', Immunity, 42:
419-30.
Jia, Rong, Cuiling Li, J. Philip McCoy, Chu-Xia Deng, and Zhi-Ming Zheng. 2010. 'SRp20
is a proto-oncogene critical for cell proliferation and tumor induction and
maintenance', International Journal of Biological Sciences, 6: 806-26.
Jia, Rong, Xuefeng Liu, Mingfang Tao, Michael Kruhlak, Ming Guo, Craig Meyers, Carl C.
Baker, and Zhi-Ming Zheng. 2009. 'Control of the Papillomavirus Early-to-Late
Switch by Differentially Expressed SRp20', Journal of Virology, 83: 167.
Jiang, H., S. Zhang, T. Song, X. Guan, R. Zhang, and X. Chen. 2018. 'Trichostatin a Protects
Dendritic Cells Against Oxygen-Glucose Deprivation via the
SRSF3/PKM2/Glycolytic Pathway', Front Pharmacol, 9: 612.
Jiang, J., Q. Zhu, T. F. Gendron, S. Saberi, M. McAlonis-Downes, A. Seelman, J. E. Stauffer,
P. Jafar-Nejad, K. Drenner, D. Schulte, S. Chun, S. Sun, S. C. Ling, B. Myers, J.
Engelhardt, M. Katz, M. Baughn, O. Platoshyn, M. Marsala, A. Watt, C. J. Heyser,
M. C. Ard, L. De Muynck, L. M. Daughrity, D. A. Swing, L. Tessarollo, C. J. Jung,
A. Delpoux, D. T. Utzschneider, S. M. Hedrick, P. J. de Jong, D. Edbauer, P. Van
Damme, L. Petrucelli, C. E. Shaw, C. F. Bennett, S. Da Cruz, J. Ravits, F. Rigo, D.
W. Cleveland, and C. Lagier-Tourenne. 2016. 'Gain of Toxicity from ALS/FTD-
Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides
Targeting GGGGCC-Containing RNAs', Neuron, 90: 535-50.
Jimenez, M., R. Urtasun, M. Elizalde, M. Azkona, M. U. Latasa, I. Uriarte, M. Arechederra,
D. Alignani, M. Barcena-Varela, G. Alvarez-Sola, L. Colyn, E. Santamaria, B.
Sangro, C. Rodriguez-Ortigosa, M. G. Fernandez-Barrena, M. A. Avila, and C.
Berasain. 2019. 'Splicing events in the control of genome integrity: role of SLU7 and
truncated SRSF3 proteins', Nucleic Acids Res, 47: 3450-66.
Johann, S., M. Heitzer, M. Kanagaratnam, A. Goswami, T. Rizo, J. Weis, D. Troost, and C.
Beyer. 2015. 'NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse
model of ALS and in human sporadic ALS patients', Glia, 63: 2260-73.
Johnson, J. O., J. Mandrioli, M. Benatar, Y. Abramzon, V. M. Van Deerlin, J. Q.
Trojanowski, J. R. Gibbs, M. Brunetti, S. Gronka, J. Wuu, J. Ding, L. McCluskey, M.
Martinez-Lage, D. Falcone, D. G. Hernandez, S. Arepalli, S. Chong, J. C. Schymick,
J. Rothstein, F. Landi, Y. D. Wang, A. Calvo, G. Mora, M. Sabatelli, M. R. Monsurrò,
S. Battistini, F. Salvi, R. Spataro, P. Sola, G. Borghero, G. Galassi, S. W. Scholz, J.
P. Taylor, G. Restagno, A. Chiò, and B. J. Traynor. 2010. 'Exome sequencing reveals
VCP mutations as a cause of familial ALS', Neuron, 68: 857-64.
Johnson, V. E., W. Stewart, J. Q. Trojanowski, and D. H. Smith. 2011. 'Acute and chronically
increased immunoreactivity to phosphorylation-independent but not pathological

219
TDP-43 after a single traumatic brain injury in humans', Acta Neuropathol, 122: 715-
26.
Jokic, N., J. L. Gonzalez de Aguilar, L. Dimou, S. Lin, A. Fergani, M. A. Ruegg, M. E.
Schwab, L. Dupuis, and J. P. Loeffler. 2006. 'The neurite outgrowth inhibitor Nogo-
A promotes denervation in an amyotrophic lateral sclerosis model', EMBO Rep, 7:
1162-7.
Jonsson, P. A., K. S. Graffmo, T. Brännström, P. Nilsson, P. M. Andersen, and S. L.
Marklund. 2006. 'Motor neuron disease in mice expressing the wild type-like D90A
mutant superoxide dismutase-1', J Neuropathol Exp Neurol, 65: 1126-36.
Jumaa, H., J. L. Guenet, and P. J. Nielsen. 1997. 'Regulated expression and RNA processing
of transcripts from the Srp20 splicing factor gene during the cell cycle', Mol Cell Biol,
17: 3116-24.
Juranek, J. K., G. K. Daffu, J. Wojtkiewicz, D. Lacomis, J. Kofler, and A. M. Schmidt. 2015.
'Receptor for Advanced Glycation End Products and its Inflammatory Ligands are
Upregulated in Amyotrophic Lateral Sclerosis', Front Cell Neurosci, 9: 485.
Kakaroubas, N., S. Brennan, M. Keon, and N. K. Saksena. 2019. 'Pathomechanisms of
Blood-Brain Barrier Disruption in ALS', Neurosci J, 2019: 2537698.
Kalmar, B., and L. Greensmith. 2017. 'Cellular Chaperones As Therapeutic Targets in ALS
to Restore Protein Homeostasis and Improve Cellular Function', Frontiers in
molecular neuroscience, 10: 251.
Kamachi, M., T. M. Le, S. J. Kim, M. E. Geiger, P. Anderson, and P. J. Utz. 2002. 'Human
autoimmune sera as molecular probes for the identification of an autoantigen kinase
signaling pathway', J Exp Med, 196: 1213-25.
Kamphuis, W., L. Kooijman, S. Schetters, M. Orre, and E. M. Hol. 2016. 'Transcriptional
profiling of CD11c-positive microglia accumulating around amyloid plaques in a
mouse model for Alzheimer's disease', Biochim Biophys Acta, 1862: 1847-60.
Kang, J., and S. Rivest. 2007. 'MyD88-deficient bone marrow cells accelerate onset and
reduce survival in a mouse model of amyotrophic lateral sclerosis', J Cell Biol, 179:
1219-30.
Kang, S. H., Y. Li, M. Fukaya, I. Lorenzini, D. W. Cleveland, L. W. Ostrow, J. D. Rothstein,
and D. E. Bergles. 2013. 'Degeneration and impaired regeneration of gray matter
oligodendrocytes in amyotrophic lateral sclerosis', Nat Neurosci, 16: 571-9.
Kang, S. S., M. T. W. Ebbert, K. E. Baker, C. Cook, X. Wang, J. P. Sens, J. P. Kocher, L.
Petrucelli, and J. D. Fryer. 2018. 'Microglial translational profiling reveals a
convergent APOE pathway from aging, amyloid, and tau', J Exp Med, 215: 2235-45.
Kano, Osamu, David R. Beers, Jenny S. Henkel, and Stanley H. Appel. 2012. 'Peripheral
nerve inflammation in ALS mice: cause or consequence', Neurology, 78: 833.
Kano, S., K. Nishida, C. Nishiyama, Y. Akaike, K. Kajita, K. Kurokawa, K. Masuda, Y.
Kuwano, T. Tanahashi, and K. Rokutan. 2013. 'Truncated serine/arginine-rich
splicing factor 3 accelerates cell growth through up-regulating c-Jun expression', J
Med Invest, 60: 228-35.
Kavanagh, Edel, Miguel Angel Burguillos, Alejandro Carrillo-Jimenez, María José Oliva-
Martin, Martiniano Santiago, Johanna Rodhe, Bertrand Joseph, and Jose Luis Venero.
2015. 'Deletion of caspase-8 in mouse myeloid cells blocks microglia pro-
inflammatory activation and confers protection in MPTP neurodegeneration model',
Aging, 7: 673-89.

220
Keller, A. F., M. Gravel, and J. Kriz. 2009. 'Live imaging of amyotrophic lateral sclerosis
pathogenesis: disease onset is characterized by marked induction of GFAP in
Schwann cells', Glia, 57: 1130-42.
Keren-Shaul, H., A. Spinrad, A. Weiner, O. Matcovitch-Natan, R. Dvir-Szternfeld, T. K.
Ulland, E. David, K. Baruch, D. Lara-Astaiso, B. Toth, S. Itzkovitz, M. Colonna, M.
Schwartz, and I. Amit. 2017. 'A Unique Microglia Type Associated with Restricting
Development of Alzheimer's Disease', Cell, 169: 1276-90.e17.
Kerman, A., H. N. Liu, S. Croul, J. Bilbao, E. Rogaeva, L. Zinman, J. Robertson, and A.
Chakrabartty. 2010. 'Amyotrophic lateral sclerosis is a non-amyloid disease in which
extensive misfolding of SOD1 is unique to the familial form', Acta Neuropathol, 119:
335-44.
Kettenmann, H., U. K. Hanisch, M. Noda, and A. Verkhratsky. 2011. 'Physiology of
microglia', Physiol Rev, 91: 461-553.
Kettenmann, H., F. Kirchhoff, and A. Verkhratsky. 2013. 'Microglia: new roles for the
synaptic stripper', Neuron, 77: 10-8.
Khalfallah, Y., R. Kuta, C. Grasmuck, A. Prat, H. D. Durham, and C. Vande Velde. 2018.
'TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant
cell types', Sci Rep, 8: 7551.
Kia, Azadeh, Kevin McAvoy, Karthik Krishnamurthy, Davide Trotti, and Piera Pasinelli.
2018. 'Astrocytes expressing ALS-linked mutant FUS induce motor neuron death
through release of tumor necrosis factor-alpha', Glia, 66: 1016-33.
Kigerl, Kristina A., Juan Pablo de Rivero Vaccari, W. Dalton Dietrich, Phillip G. Popovich,
and Robert W. Keane. 2014. 'Pattern recognition receptors and central nervous system
repair', Exp Neurol, 258: 5-16.
Killoy, K. M., B. A. Harlan, M. Pehar, K. L. Helke, J. A. Johnson, and M. R. Vargas. 2018.
'Decreased glutathione levels cause overt motor neuron degeneration in hSOD1(WT)
over-expressing mice', Exp Neurol, 302: 129-35.
Kim, J., R. Y. Park, J. K. Chen, J. Kim, S. Jeong, and T. Ohn. 2014. 'Splicing factor SRSF3
represses the translation of programmed cell death 4 mRNA by associating with the
5'-UTR region', Cell Death Differ, 21: 481-90.
Kim, Jee-Eun, Yoon Ho Hong, Jin Young Kim, Gye Sun Jeon, Jung Hee Jung, Byung-Nam
Yoon, Sung-Yeon Son, Kwang-Woo Lee, Jong-Il Kim, and Jung-Joon Sung. 2017.
'Altered nucleocytoplasmic proteome and transcriptome distributions in an in vitro
model of amyotrophic lateral sclerosis', PLOS ONE, 12: e0176462-e62.
Kim, Kijun, Trung Duc Nguyen, Shaohua Li, and Tuan Anh Nguyen. 2018. 'SRSF3 recruits
DROSHA to the basal junction of primary microRNAs', RNA (New York, N.Y.), 24:
892-98.
Kiyota, T., K. L. Ingraham, R. J. Swan, M. T. Jacobsen, S. J. Andrews, and T. Ikezu. 2012.
'AAV serotype 2/1-mediated gene delivery of anti-inflammatory interleukin-10
enhances neurogenesis and cognitive function in APP+PS1 mice', Gene Ther, 19:
724-33.
Kleinewietfeld, Markus, and David A. Hafler. 2014. 'Regulatory T cells in autoimmune
neuroinflammation', Immunological reviews, 259: 231-44.
Klymenko, T., H. Hernandez-Lopez, A. I. MacDonald, J. M. Bodily, and S. V. Graham. 2016.
'Human Papillomavirus E2 Regulates SRSF3 (SRp20) To Promote Capsid Protein
Expression in Infected Differentiated Keratinocytes', J Virol, 90: 5047-58.

221
Knuefermann, P., P. Chen, A. Misra, S. P. Shi, M. Abdellatif, and N. Sivasubramanian. 2002.
'Myotrophin/V-1, a protein up-regulated in the failing human heart and in postnatal
cerebellum, converts NFkappa B p50-p65 heterodimers to p50-p50 and p65-p65
homodimers', J Biol Chem, 277: 23888-97.
Kocur, Magdalena, Reiner Schneider, Ann-Kathrin Pulm, Jens Bauer, Sonja Kropp, Michael
Gliem, Jens Ingwersen, Norbert Goebels, Judith Alferink, Timour Prozorovski,
Orhan Aktas, and Stefanie Scheu. 2015. 'IFNβ secreted by microglia mediates
clearance of myelin debris in CNS autoimmunity', Acta neuropathologica
communications, 3: 20-20.
Komine, Okiru, Hirofumi Yamashita, Noriko Fujimori-Tonou, Masato Koike, Shijie Jin,
Yasuhiro Moriwaki, Fumito Endo, Seiji Watanabe, Satoshi Uematsu, Shizuo Akira,
Yasuo Uchiyama, Ryosuke Takahashi, Hidemi Misawa, and Koji Yamanaka. 2018.
'Innate immune adaptor TRIF deficiency accelerates disease progression of ALS mice
with accumulation of aberrantly activated astrocytes', Cell Death & Differentiation,
25: 2130-46.
Krasemann, S., C. Madore, R. Cialic, C. Baufeld, N. Calcagno, R. El Fatimy, L. Beckers, E.
O'Loughlin, Y. Xu, Z. Fanek, D. J. Greco, S. T. Smith, G. Tweet, Z. Humulock, T.
Zrzavy, P. Conde-Sanroman, M. Gacias, Z. Weng, H. Chen, E. Tjon, F. Mazaheri, K.
Hartmann, A. Madi, J. D. Ulrich, M. Glatzel, A. Worthmann, J. Heeren, B. Budnik,
C. Lemere, T. Ikezu, F. L. Heppner, V. Litvak, D. M. Holtzman, H. Lassmann, H. L.
Weiner, J. Ochando, C. Haass, and O. Butovsky. 2017. 'The TREM2-APOE Pathway
Drives the Transcriptional Phenotype of Dysfunctional Microglia in
Neurodegenerative Diseases', Immunity, 47: 566-81.e9.
Kress, T. L., N. J. Krogan, and C. Guthrie. 2008. 'A single SR-like protein, Npl3, promotes
pre-mRNA splicing in budding yeast', Mol Cell, 32: 727-34.
Kryndushkin, Dmitry, Reed B. Wickner, and Frank Shewmaker. 2011. 'FUS/TLS forms
cytoplasmic aggregates, inhibits cell growth and interacts with TDP-43 in a yeast
model of amyotrophic lateral sclerosis', Protein & cell, 2: 223-36.
Kumar, D., M. Das, C. Sauceda, L. G. Ellies, K. Kuo, P. Parwal, M. Kaur, L. Jih, G. K.
Bandyopadhyay, D. Burton, R. Loomba, O. Osborn, and N. J. Webster. 2019.
'Degradation of splicing factor SRSF3 contributes to progressive liver disease', J Clin
Invest, 130: 4477-91.
Kumar, David R., Florence Aslinia, Steven H. Yale, and Joseph J. Mazza. 2011. 'Jean-Martin
Charcot: the father of neurology', Clinical medicine & research, 9: 46-49.
Kuranaga, Yuki, Nobuhiko Sugito, Haruka Shinohara, Takuya Tsujino, Kohei Taniguchi,
Kazumasa Komura, Yuko Ito, Tomoyoshi Soga, and Yukihiro Akao. 2018. 'SRSF3,
a Splicer of the PKM Gene, Regulates Cell Growth and Maintenance of Cancer-
Specific Energy Metabolism in Colon Cancer Cells', Int J Mol Sci, 19: 3012.
Kurokawa, K., Y. Akaike, K. Masuda, Y. Kuwano, K. Nishida, N. Yamagishi, K. Kajita, T.
Tanahashi, and K. Rokutan. 2014. 'Downregulation of serine/arginine-rich splicing
factor 3 induces G1 cell cycle arrest and apoptosis in colon cancer cells', Oncogene,
33: 1407-17.
Lagier-Tourenne, C., M. Polymenidou, K. R. Hutt, A. Q. Vu, M. Baughn, S. C. Huelga, K.
M. Clutario, S. C. Ling, T. Y. Liang, C. Mazur, E. Wancewicz, A. S. Kim, A. Watt,
S. Freier, G. G. Hicks, J. P. Donohue, L. Shiue, C. F. Bennett, J. Ravits, D. W.
Cleveland, and G. W. Yeo. 2012. 'Divergent roles of ALS-linked proteins FUS/TLS
and TDP-43 intersect in processing long pre-mRNAs', Nat Neurosci, 15: 1488-97.

222
Lai, M. C., R. I. Lin, and W. Y. Tarn. 2001. 'Transportin-SR2 mediates nuclear import of
phosphorylated SR proteins', Proc Natl Acad Sci U S A, 98: 10154-59.
Lalancette-Hebert, M., A. Sharma, A. K. Lyashchenko, and N. A. Shneider. 2016. 'Gamma
motor neurons survive and exacerbate alpha motor neuron degeneration in ALS', Proc
Natl Acad Sci U S A, 113: E8316-e25.
Lalancette-Hebert, M., V. Swarup, J. M. Beaulieu, I. Bohacek, E. Abdelhamid, Y. C. Weng,
S. Sato, and J. Kriz. 2012. 'Galectin-3 is required for resident microglia activation and
proliferation in response to ischemic injury', J Neurosci, 32: 10383-95.
Lawson, L. J., V. H. Perry, P. Dri, and S. Gordon. 1990. 'Heterogeneity in the distribution
and morphology of microglia in the normal adult mouse brain', Neuroscience, 39:
151-70.
Lee, J. C., J. Seong, S. H. Kim, S. J. Lee, Y. J. Cho, J. An, D. H. Nam, K. M. Joo, and C. I.
Cha. 2012. 'Replacement of microglial cells using Clodronate liposome and bone
marrow transplantation in the central nervous system of SOD1(G93A) transgenic
mice as an in vivo model of amyotrophic lateral sclerosis', Biochem Biophys Res
Commun, 418: 359-65.
Lee, S. H., and E. J. Yang. 2018. 'Relationship between Liver Pathology and Disease
Progression in a Murine Model of Amyotrophic Lateral Sclerosis', Neurodegener Dis,
18: 200-07.
Lee, Y., B. M. Morrison, Y. Li, S. Lengacher, M. H. Farah, P. N. Hoffman, Y. Liu, A.
Tsingalia, L. Jin, P. W. Zhang, L. Pellerin, P. J. Magistretti, and J. D. Rothstein. 2012.
'Oligodendroglia metabolically support axons and contribute to neurodegeneration',
Nature, 487: 443-8.
Leskelä, S., N. Huber, H. Rostalski, T. Natunen, A. M. Remes, M. Takalo, M. Hiltunen, and
A. Haapasalo. 2019. 'C9orf72 Proteins Regulate Autophagy and Undergo
Autophagosomal or Proteasomal Degradation in a Cell Type-Dependent Manner',
Cells, 8.
Leung, C. L., C. Z. He, P. Kaufmann, S. S. Chin, A. Naini, R. K. Liem, H. Mitsumoto, and
A. P. Hays. 2004. 'A pathogenic peripherin gene mutation in a patient with
amyotrophic lateral sclerosis', Brain Pathol, 14: 290-6.
Li, D., Q. He, R. Li, X. Xu, B. Chen, and A. Xie. 2012. 'Interleukin-10 promoter
polymorphisms in Chinese patients with Parkinson's disease', Neurosci Lett, 513:
183-6.
Li, Quan, Christine Vande Velde, Adrian Israelson, Jing Xie, Aaron O. Bailey, Meng-Qui
Dong, Seung-Joo Chun, Tamal Roy, Leah Winer, John R. Yates, Roderick A.
Capaldi, Don W. Cleveland, and Timothy M. Miller. 2010. 'ALS-linked mutant
superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and
decreases protein import', Proc Natl Acad Sci U S A, 107: 21146-51.
Lian, H., E. Roy, and H. Zheng. 2016. 'Microglial Phagocytosis Assay', Bio Protoc, 6.
Liao, B., W. Zhao, D. R. Beers, J. S. Henkel, and S. H. Appel. 2012. 'Transformation from a
neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS', Exp
Neurol, 237: 147-52.
Liddelow, Shane A., Kevin A. Guttenplan, Laura E. Clarke, Frederick C. Bennett,
Christopher J. Bohlen, Lucas Schirmer, Mariko L. Bennett, Alexandra E. Münch,
Won-Suk Chung, Todd C. Peterson, Daniel K. Wilton, Arnaud Frouin, Brooke A.
Napier, Nikhil Panicker, Manoj Kumar, Marion S. Buckwalter, David H. Rowitch,
Valina L. Dawson, Ted M. Dawson, Beth Stevens, and Ben A. Barres. 2017.

223
'Neurotoxic reactive astrocytes are induced by activated microglia', Nature, 541: 481-
87.
Liguori, M., E. Digifico, A. Vacchini, R. Avigni, F. S. Colombo, E. M. Borroni, F. M. Farina,
S. Milanesi, A. Castagna, L. Mannarino, I. Craparotta, S. Marchini, E. Erba, N.
Panini, M. Tamborini, V. Rimoldi, P. Allavena, and C. Belgiovine. 2020. 'The soluble
glycoprotein NMB (GPNMB) produced by macrophages induces cancer stemness
and metastasis via CD44 and IL-33', Cell Mol Immunol.
Lin, S., and X. D. Fu. 2007. 'SR proteins and related factors in alternative splicing', Adv Exp
Med Biol, 623: 107-22.
Lincecum, J. M., F. G. Vieira, M. Z. Wang, K. Thompson, G. S. De Zutter, J. Kidd, A.
Moreno, R. Sanchez, I. J. Carrion, B. A. Levine, B. M. Al-Nakhala, S. M. Sullivan,
A. Gill, and S. Perrin. 2010. 'From transcriptome analysis to therapeutic anti-CD40L
treatment in the SOD1 model of amyotrophic lateral sclerosis', Nat Genet, 42: 392-9.
Liu, Fei, and Cheng-Xin Gong. 2008. 'Tau exon 10 alternative splicing and tauopathies',
Molecular neurodegeneration, 3: 8-8.
Liu, H. N., T. Sanelli, P. Horne, E. P. Pioro, M. J. Strong, E. Rogaeva, J. Bilbao, L. Zinman,
and J. Robertson. 2009. 'Lack of evidence of monomer/misfolded superoxide
dismutase-1 in sporadic amyotrophic lateral sclerosis', Ann Neurol, 66: 75-80.
Liu, Juan, Lina Gao, and Dawei Zang. 2015. 'Elevated Levels of IFN-γ in CSF and Serum of
Patients with Amyotrophic Lateral Sclerosis', PLOS ONE, 10: e0136937-e37.
Liu, Y., A. Pattamatta, T. Zu, T. Reid, O. Bardhi, D. R. Borchelt, A. T. Yachnis, and L. P.
Ranum. 2016. 'C9orf72 BAC Mouse Model with Motor Deficits and
Neurodegenerative Features of ALS/FTD', Neuron, 90: 521-34.
Liu, Zhengzhao, Hongming Li, Chungu Hong, Menglu Chen, Tao Yue, Chunyuan Chen,
Zhenxing Wang, Qing You, Chuanyin Li, Qinjie Weng, Hui Xie, and Ronggui Hu.
2018. 'ALS-Associated E478G Mutation in Human OPTN (Optineurin) Promotes
Inflammation and Induces Neuronal Cell Death', Frontiers in Immunology, 9: 2647-
47.
Liu, Zhouyang, Xi Cheng, Shanshan Zhong, Xiuchun Zhang, Chang Liu, Fangxi Liu, and
Chuansheng Zhao. 2020. 'Peripheral and Central Nervous System Immune Response
Crosstalk in Amyotrophic Lateral Sclerosis', Front Neurosci, 14.
Loeffler, J. P., G. Picchiarelli, L. Dupuis, and J. L. Gonzalez De Aguilar. 2016. 'The Role of
Skeletal Muscle in Amyotrophic Lateral Sclerosis', Brain Pathol, 26: 227-36.
Long, J. C., and J. F. Caceres. 2009. 'The SR protein family of splicing factors: master
regulators of gene expression', Biochem J, 417: 15-27.
López-Erauskin, J., T. Tadokoro, M. W. Baughn, B. Myers, M. McAlonis-Downes, C.
Chillon-Marinas, J. N. Asiaban, J. Artates, A. T. Bui, A. P. Vetto, S. K. Lee, A. V.
Le, Y. Sun, M. Jambeau, J. Boubaker, D. Swing, J. Qiu, G. G. Hicks, Z. Ouyang, X.
D. Fu, L. Tessarollo, S. C. Ling, P. A. Parone, C. E. Shaw, M. Marsala, C. Lagier-
Tourenne, D. W. Cleveland, and S. Da Cruz. 2018. 'ALS/FTD-Linked Mutation in
FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear
Loss-of-Function of FUS', Neuron, 100: 816-30.e7.
Lopez-Lopez, Alan, Josep Gamez, Emilio Syriani, Miguel Morales, Maria Salvado, Manuel
J. Rodríguez, Nicole Mahy, and Jose M. Vidal-Taboada. 2014. 'CX3CR1 Is a
Modifying Gene of Survival and Progression in Amyotrophic Lateral Sclerosis',
PLOS ONE, 9: e96528.

224
Lou, H., K. M. Neugebauer, R. F. Gagel, and S. M. Berget. 1998. 'Regulation of alternative
polyadenylation by U1 snRNPs and SRp20', Molecular and Cellular Biology, 18:
4977-85.
Lucin, K. M., and T. Wyss-Coray. 2009. 'Immune activation in brain aging and
neurodegeneration: too much or too little?', Neuron, 64: 110-22.
Magrané, J., C. Cortez, W. B. Gan, and G. Manfredi. 2014. 'Abnormal mitochondrial
transport and morphology are common pathological denominators in SOD1 and
TDP43 ALS mouse models', Hum Mol Genet, 23: 1413-24.
Mandrekar-Colucci, Shweta, and Gary E. Landreth. 2010. 'Microglia and inflammation in
Alzheimer's disease', CNS & neurological disorders drug targets, 9: 156-67.
Manetto, V., N. H. Sternberger, G. Perry, L. A. Sternberger, and P. Gambetti. 1988.
'Phosphorylation of neurofilaments is altered in amyotrophic lateral sclerosis', J
Neuropathol Exp Neurol, 47: 642-53.
Manley, J. L., and R. Tacke. 1996. 'SR proteins and splicing control', Genes & development,
10: 1569-79.
Manley, James L., and Adrian R. Krainer. 2010. 'A rational nomenclature for serine/arginine-
rich protein splicing factors (SR proteins)', Genes & development, 24: 1073-74.
Mantovani, S., S. Garbelli, A. Pasini, D. Alimonti, C. Perotti, M. Melazzini, C. Bendotti, and
G. Mora. 2009. 'Immune system alterations in sporadic amyotrophic lateral sclerosis
patients suggest an ongoing neuroinflammatory process', J Neuroimmunol, 210: 73-
9.
Marchetto, Maria C. N., Alysson R. Muotri, Yangling Mu, Alan M. Smith, Gabriela G. Cezar,
and Fred H. Gage. 2008. 'Non-Cell-Autonomous Effect of Human SOD1G37R
Astrocytes on Motor Neurons Derived from Human Embryonic Stem Cells', Cell
Stem Cell, 3: 649-57.
Markovinovic, A., R. Cimbro, T. Ljutic, J. Kriz, B. Rogelj, and I. Munitic. 2017. 'Optineurin
in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of
different neuroprotective mechanisms', Prog Neurobiol, 154: 1-20.
Markovinovic, A., T. Ljutic, L. C. Béland, and I. Munitic. 2018. 'Optineurin Insufficiency
Disbalances Proinflammatory and Anti-inflammatory Factors by Reducing
Microglial IFN-β Responses', Neuroscience, 388: 139-51.
Markus, M. A., F. Z. Marques, and B. J. Morris. 2011. 'Resveratrol, by modulating RNA
processing factor levels, can influence the alternative splicing of pre-mRNAs', PLOS
ONE, 6: e28926.
Martínez-Muriana, A., R. Mancuso, I. Francos-Quijorna, A. Olmos-Alonso, R. Osta, V. H.
Perry, X. Navarro, D. Gomez-Nicola, and R. López-Vales. 2016. 'CSF1R blockade
slows the progression of amyotrophic lateral sclerosis by reducing microgliosis and
invasion of macrophages into peripheral nerves', Sci Rep, 6: 25663.
Maruyama, H., H. Morino, H. Ito, Y. Izumi, H. Kato, Y. Watanabe, Y. Kinoshita, M.
Kamada, H. Nodera, H. Suzuki, O. Komure, S. Matsuura, K. Kobatake, N. Morimoto,
K. Abe, N. Suzuki, M. Aoki, A. Kawata, T. Hirai, T. Kato, K. Ogasawara, A. Hirano,
T. Takumi, H. Kusaka, K. Hagiwara, R. Kaji, and H. Kawakami. 2010. 'Mutations of
optineurin in amyotrophic lateral sclerosis', Nature, 465: 223-6.
Masuyama, K., I. Taniguchi, N. Kataoka, and M. Ohno. 2004. 'SR proteins preferentially
associate with mRNAs in the nucleus and facilitate their export to the cytoplasm',
Genes Cells, 9: 959-65.

225
Matsuki, H., M. Takahashi, M. Higuchi, G. N. Makokha, M. Oie, and M. Fujii. 2013. 'Both
G3BP1 and G3BP2 contribute to stress granule formation', Genes Cells, 18: 135-46.
Mattiazzi, M., M. D'Aurelio, C. D. Gajewski, K. Martushova, M. Kiaei, M. F. Beal, and G.
Manfredi. 2002. 'Mutated human SOD1 causes dysfunction of oxidative
phosphorylation in mitochondria of transgenic mice', J Biol Chem, 277: 29626-33.
Matus, Soledad, Vicente Valenzuela, Danilo B. Medinas, and Claudio Hetz. 2013. 'ER
Dysfunction and Protein Folding Stress in ALS', International journal of cell biology,
2013: 674751-51.
McAlary, Luke, Steven S. Plotkin, Justin J. Yerbury, and Neil R. Cashman. 2019. 'Prion-
Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral
Sclerosis', Frontiers in molecular neuroscience, 12: 262-62.
McGeer, P. L., S. Itagaki, and E. G. McGeer. 1988. 'Expression of the histocompatibility
glycoprotein HLA-DR in neurological disease', Acta Neuropathologica, 76: 550-57.
Menza, M., R. D. Dobkin, H. Marin, M. H. Mark, M. Gara, K. Bienfait, A. Dicke, and A.
Kusnekov. 2010. 'The role of inflammatory cytokines in cognition and other non-
motor symptoms of Parkinson's disease', Psychosomatics, 51: 474-9.
Mermoud, J. E., P. T. Cohen, and A. I. Lamond. 1994. 'Regulation of mammalian
spliceosome assembly by a protein phosphorylation mechanism', Embo j, 13: 5679-
88.
Michaud, J. P., M. A. Bellavance, P. Préfontaine, and S. Rivest. 2013. 'Real-time in vivo
imaging reveals the ability of monocytes to clear vascular amyloid beta', Cell Rep, 5:
646-53.
Miller, R. G., J. D. Mitchell, and D. H. Moore. 2012. 'Riluzole for amyotrophic lateral
sclerosis (ALS)/motor neuron disease (MND)', Cochrane Database Syst Rev, 2012:
Cd001447.
Miller, T. M., A. Pestronk, W. David, J. Rothstein, E. Simpson, S. H. Appel, P. L. Andres,
K. Mahoney, P. Allred, K. Alexander, L. W. Ostrow, D. Schoenfeld, E. A. Macklin,
D. A. Norris, G. Manousakis, M. Crisp, R. Smith, C. F. Bennett, K. M. Bishop, and
M. E. Cudkowicz. 2013. 'An antisense oligonucleotide against SOD1 delivered
intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1,
randomised, first-in-man study', Lancet Neurol, 12: 435-42.
Misteli, T., and D. L. Spector. 1996. 'Serine/threonine phosphatase 1 modulates the
subnuclear distribution of pre-mRNA splicing factors', Molecular biology of the cell,
7: 1559-72.
Mitchell, J. C., R. Constable, E. So, C. Vance, E. Scotter, L. Glover, T. Hortobagyi, E. S.
Arnold, S. C. Ling, M. McAlonis, S. Da Cruz, M. Polymenidou, L. Tessarolo, D. W.
Cleveland, and C. E. Shaw. 2015. 'Wild type human TDP-43 potentiates ALS-linked
mutant TDP-43 driven progressive motor and cortical neuron degeneration with
pathological features of ALS', Acta Neuropathol Commun, 3: 36.
Mitchell, J. C., P. McGoldrick, C. Vance, T. Hortobagyi, J. Sreedharan, B. Rogelj, E. L.
Tudor, B. N. Smith, C. Klasen, C. C. Miller, J. D. Cooper, L. Greensmith, and C. E.
Shaw. 2013. 'Overexpression of human wild-type FUS causes progressive motor
neuron degeneration in an age- and dose-dependent fashion', Acta Neuropathol, 125:
273-88.
Mole, Sarah, Melanie McFarlane, Thanaporn Chuen-Im, Steven G. Milligan, David Millan,
and Sheila V. Graham. 2009. 'RNA splicing factors regulated by HPV16 during
cervical tumour progression', The Journal of pathology, 219: 383-91.

226
Moller, A., C. S. Bauer, R. N. Cohen, C. P. Webster, and K. J. De Vos. 2017. 'Amyotrophic
lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of
mitochondria by reducing Miro1 levels', Hum Mol Genet, 26: 4668-79.
Molofsky, Anna V., Robert Krencik, Erik M. Ullian, Hui-hsin Tsai, Benjamin Deneen,
William D. Richardson, Ben A. Barres, and David H. Rowitch. 2012. 'Astrocytes and
disease: a neurodevelopmental perspective', Genes & development, 26: 891-907.
Mora, J. S., A. Genge, A. Chio, C. J. Estol, D. Chaverri, M. Hernández, S. Marín, J. Mascias,
G. E. Rodriguez, M. Povedano, A. Paipa, R. Dominguez, J. Gamez, M. Salvado, C.
Lunetta, C. Ballario, N. Riva, J. Mandrioli, A. Moussy, J. P. Kinet, C. Auclair, P.
Dubreuil, V. Arnold, C. D. Mansfield, and O. Hermine. 2020. 'Masitinib as an add-
on therapy to riluzole in patients with amyotrophic lateral sclerosis: a randomized
clinical trial', Amyotroph Lateral Scler Frontotemporal Degener, 21: 5-14.
Moura-Alves, Pedro, Ana Neves-Costa, Helena Raquel, Teresa Raquel Pacheco, Bruno
D'Almeida, Raquel Rodrigues, Iris Cadima-Couto, Ângelo Chora, Mariana Oliveira,
Margarida Gama-Carvalho, Nir Hacohen, and Luis F. Moita. 2011. 'An shRNA-based
screen of splicing regulators identifies SFRS3 as a negative regulator of IL-1β
secretion', PLOS ONE, 6: e19829-e29.
Müller-McNicoll, Michaela, Valentina Botti, Antonio M. de Jesus Domingues, Holger
Brandl, Oliver D. Schwich, Michaela C. Steiner, Tomaz Curk, Ina Poser, Kathi
Zarnack, and Karla M. Neugebauer. 2016. 'SR proteins are NXF1 adaptors that link
alternative RNA processing to mRNA export', Genes & development, 30: 553-66.
Munitic, I., M. L. Giardino Torchia, N. P. Meena, G. Zhu, C. C. Li, and J. D. Ashwell. 2013.
'Optineurin insufficiency impairs IRF3 but not NF-κB activation in immune cells', J
Immunol, 191: 6231-40.
Murakami, T., S. Qamar, J. Q. Lin, G. S. Schierle, E. Rees, A. Miyashita, A. R. Costa, R. B.
Dodd, F. T. Chan, C. H. Michel, D. Kronenberg-Versteeg, Y. Li, S. P. Yang, Y.
Wakutani, W. Meadows, R. R. Ferry, L. Dong, G. G. Tartaglia, G. Favrin, W. L. Lin,
D. W. Dickson, M. Zhen, D. Ron, G. Schmitt-Ulms, P. E. Fraser, N. A. Shneider, C.
Holt, M. Vendruscolo, C. F. Kaminski, and P. St George-Hyslop. 2015. 'ALS/FTD
Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible
Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function', Neuron, 88:
678-90.
Murayama, S., Y. Ookawa, H. Mori, I. Nakano, Y. Ihara, S. Kuzuhara, and M. Tomonaga.
1989. 'Immunocytochemical and ultrastructural study of Lewy body-like hyaline
inclusions in familial amyotrophic lateral sclerosis', Acta Neuropathol, 78: 143-52.
Muyderman, H., P. G. Hutson, D. Matusica, M. L. Rogers, and R. A. Rush. 2009. 'The Human
G93A-Superoxide Dismutase-1 Mutation, Mitochondrial Glutathione and Apoptotic
Cell Death', Neurochemical Research, 34: 1847-56.
Nagai, Makiko, Diane B. Re, Tetsuya Nagata, Alcmène Chalazonitis, Thomas M. Jessell,
Hynek Wichterle, and Serge Przedborski. 2007. 'Astrocytes expressing ALS-linked
mutated SOD1 release factors selectively toxic to motor neurons', Nature
Neuroscience, 10: 615-22.
Nagara, Yuko, Takahisa Tateishi, Ryo Yamasaki, Shintaro Hayashi, Mami Kawamura,
Hitoshi Kikuchi, Kyoko Motomura Iinuma, Masahito Tanaka, Toru Iwaki, Takuya
Matsushita, Yasumasa Ohyagi, and Jun-ichi Kira. 2013. 'Impaired Cytoplasmic–
Nuclear Transport of Hypoxia-Inducible Factor-1α in Amyotrophic Lateral
Sclerosis', Brain Pathology, 23: 534-46.

227
Naor, S., Z. Keren, T. Bronshtein, E. Goren, M. Machluf, and D. Melamed. 2009.
'Development of ALS-like disease in SOD-1 mice deficient of B lymphocytes', J
Neurol, 256: 1228-35.
Neugebauer, K. M., and M. B. Roth. 1997. 'Distribution of pre-mRNA splicing factors at
sites of RNA polymerase II transcription', Genes & development, 11: 1148-59.
Neumann, M., D. M. Sampathu, L. K. Kwong, A. C. Truax, M. C. Micsenyi, T. T. Chou, J.
Bruce, T. Schuck, M. Grossman, C. M. Clark, L. F. McCluskey, B. L. Miller, E.
Masliah, I. R. Mackenzie, H. Feldman, W. Feiden, H. A. Kretzschmar, J. Q.
Trojanowski, and V. M. Lee. 2006. 'Ubiquitinated TDP-43 in frontotemporal lobar
degeneration and amyotrophic lateral sclerosis', Science, 314: 130-3.
Ngo, Jacky Chi Ki, Kayla Giang, Sutapa Chakrabarti, Chen-Ting Ma, Nhat Huynh, Jonathan
C. Hagopian, Pieter C. Dorrestein, Xiang-Dong Fu, Joseph A. Adams, and
Gourisankar Ghosh. 2008. 'A sliding docking interaction is essential for sequential
and processive phosphorylation of an SR protein by SRPK1', Mol Cell, 29: 563-76.
Nguyen, M. D., T. D'Aigle, G. Gowing, J. P. Julien, and S. Rivest. 2004. 'Exacerbation of
motor neuron disease by chronic stimulation of innate immunity in a mouse model of
amyotrophic lateral sclerosis', J Neurosci, 24: 1340-9.
Nikodemova, M., A. L. Small, S. M. Smith, G. S. Mitchell, and J. J. Watters. 2014. 'Spinal
but not cortical microglia acquire an atypical phenotype with high VEGF, galectin-3
and osteopontin, and blunted inflammatory responses in ALS rats', Neurobiol Dis,
69: 43-53.
Nimmerjahn, A., F. Kirchhoff, and F. Helmchen. 2005. 'Resting microglial cells are highly
dynamic surveillants of brain parenchyma in vivo', Science, 308: 1314-8.
Nishitoh, Hideki, Hisae Kadowaki, Atsushi Nagai, Takeshi Maruyama, Takanori Yokota,
Hisashi Fukutomi, Takuya Noguchi, Atsushi Matsuzawa, Kohsuke Takeda, and
Hidenori Ichijo. 2008. 'ALS-linked mutant SOD1 induces ER stress- and ASK1-
dependent motor neuron death by targeting Derlin-1', Genes & development, 22:
1451-64.
Noorimotlagh, Z., M. Babaie, M. Safdarian, T. Ghadiri, and V. Rahimi-Movaghar. 2017.
'Mechanisms of spinal cord injury regeneration in zebrafish: a systematic review',
Iran J Basic Med Sci, 20: 1287-96.
Nordin, A., C. Akimoto, A. Wuolikainen, H. Alstermark, P. Jonsson, A. Birve, S. L.
Marklund, K. S. Graffmo, K. Forsberg, T. Brännström, and P. M. Andersen. 2015.
'Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal
and non-neuronal tissues in 18 patients with ALS or FTD', Hum Mol Genet, 24: 3133-
42.
Noristani, Harun Najib, Jean Charles Sabourin, Yannick Nicolas Gerber, Marisa Teigell,
Andreas Sommacal, Maria dM Vivanco, Markus Weber, and Florence Evelyne
Perrin. 2015. 'Brca1 is expressed in human microglia and is dysregulated in human
and animal model of ALS', Molecular neurodegeneration, 10: 34-34.
Novoyatleva, Tatyana, Bettina Heinrich, Yesheng Tang, Natalya Benderska, Matthew E. R.
Butchbach, Christian L. Lorson, Monique A. Lorson, Claudia Ben-Dov, Pascale
Fehlbaum, Laurent Bracco, Arthur H. M. Burghes, Mathieu Bollen, and Stefan
Stamm. 2007. 'Protein phosphatase 1 binds to the RNA recognition motif of several
splicing factors and regulates alternative pre-mRNA processing', Human Molecular
Genetics, 17: 52-70.

228
O'Connor, D. M., and N. M. Boulis. 2015. 'Gene therapy for neurodegenerative diseases',
Trends Mol Med, 21: 504-12.
O'Rourke, J. G., L. Bogdanik, A. Yáñez, D. Lall, A. J. Wolf, A. K. Muhammad, R. Ho, S.
Carmona, J. P. Vit, J. Zarrow, K. J. Kim, S. Bell, M. B. Harms, T. M. Miller, C. A.
Dangler, D. M. Underhill, H. S. Goodridge, C. M. Lutz, and R. H. Baloh. 2016.
'C9orf72 is required for proper macrophage and microglial function in mice', Science,
351: 1324-9.
Oakes, J. A., M. C. Davies, and M. O. Collins. 2017. 'TBK1: a new player in ALS linking
autophagy and neuroinflammation', Mol Brain, 10: 5.
Okamoto, Y., M. Ihara, M. Urushitani, H. Yamashita, T. Kondo, A. Tanigaki, M. Oono, J.
Kawamata, A. Ikemoto, Y. Kawamoto, R. Takahashi, and H. Ito. 2011. 'An autopsy
case of <em>SOD1</em>-related ALS with TDP-43 positive inclusions',
Neurology, 77: 1993.
Ortiz-Sánchez, Paula, María Villalba-Orero, M. López-Olañeta Marina, Javier Larrasa-
Alonso, Fátima Sánchez-Cabo, Carlos Martí-Gómez, Emilio Camafeita, M. Gómez-
Salinero Jesús, Laura Ramos-Hernández, J. Nielsen Peter, Jesús Vázquez, Michaela
Müller-McNicoll, Pablo García-Pavía, and Enrique Lara-Pezzi. 2019. 'Loss of SRSF3
in Cardiomyocytes Leads to Decapping of Contraction-Related mRNAs and Severe
Systolic Dysfunction', Circulation Research, 125: 170-83.
Osaka, M., D. Ito, and N. Suzuki. 2016. 'Disturbance of proteasomal and autophagic protein
degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin
2', Biochem Biophys Res Commun, 472: 324-31.
Ote, I., M. Lebrun, P. Vandevenne, S. Bontems, C. Medina-Palazon, E. Manet, J. Piette, and
C. Sadzot-Delvaux. 2009. 'Varicella-zoster virus IE4 protein interacts with SR
proteins and exports mRNAs through the TAP/NXF1 pathway', PLOS ONE, 4:
e7882.
Ouali Alami, N., C. Schurr, F. Olde Heuvel, L. Tang, Q. Li, A. Tasdogan, A. Kimbara, M.
Nettekoven, G. Ottaviani, C. Raposo, S. Röver, M. Rogers-Evans, B. Rothenhäusler,
C. Ullmer, J. Fingerle, U. Grether, I. Knuesel, T. M. Boeckers, A. Ludolph, T. Wirth,
F. Roselli, and B. Baumann. 2018. 'NF-κB activation in astrocytes drives a stage-
specific beneficial neuroimmunological response in ALS', Embo j, 37.
Painter, Meghan M., Yuka Atagi, Chia-Chen Liu, Rosa Rademakers, Huaxi Xu, John D.
Fryer, and Guojun Bu. 2015. 'TREM2 in CNS homeostasis and neurodegenerative
disease', Molecular neurodegeneration, 10: 43-43.
Pajarillo, E., A. Rizor, J. Lee, M. Aschner, and E. Lee. 2019. 'The role of astrocytic glutamate
transporters GLT-1 and GLAST in neurological disorders: Potential targets for
neurotherapeutics', Neuropharmacology, 161: 107559.
Palusa, S. G., G. S. Ali, and A. S. Reddy. 2007. 'Alternative splicing of pre-mRNAs of
Arabidopsis serine/arginine-rich proteins: regulation by hormones and stresses', Plant
J, 49: 1091-107.
Pampalakis, G., K. Mitropoulos, G. Xiromerisiou, E. Dardiotis, G. Deretzi, M. Anagnostouli,
T. Katsila, M. Rentzos, and G. P. Patrinos. 2019. 'New molecular diagnostic trends
and biomarkers for amyotrophic lateral sclerosis', Hum Mutat, 40: 361-73.
Paolicelli, R. C., G. Bolasco, F. Pagani, L. Maggi, M. Scianni, P. Panzanelli, M. Giustetto,
T. A. Ferreira, E. Guiducci, L. Dumas, D. Ragozzino, and C. T. Gross. 2011.
'Synaptic pruning by microglia is necessary for normal brain development', Science,
333: 1456-8.

229
Papadeas, S. T., S. E. Kraig, C. O'Banion, A. C. Lepore, and N. J. Maragakis. 2011.
'Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-
type motor neuron degeneration in vivo', Proc Natl Acad Sci U S A, 108: 17803-8.
Parcellier, A., E. Schmitt, S. Gurbuxani, D. Seigneurin-Berny, A. Pance, A. Chantôme, S.
Plenchette, S. Khochbin, E. Solary, and C. Garrido. 2003. 'HSP27 is a ubiquitin-
binding protein involved in I-kappaBalpha proteasomal degradation', Mol Cell Biol,
23: 5790-802.
Paré, Bastien, and François Gros-Louis. 2017. 'Potential skin involvement in ALS: revisiting
Charcot’s observation – a review of skin abnormalities in ALS', Reviews in the
Neurosciences, 28: 551-72.
Paré, Bastien, Manuela Lehmann, Marie Beaudin, Ulrika Nordström, Stephan Saikali, Jean-
Pierre Julien, Jonathan D. Gilthorpe, Stefan L. Marklund, Neil R. Cashman, Peter M.
Andersen, Karin Forsberg, Nicolas Dupré, Peter Gould, Thomas Brännström, and
François Gros-Louis. 2018. 'Misfolded SOD1 pathology in sporadic Amyotrophic
Lateral Sclerosis', Scientific reports, 8: 14223-23.
Park, Seung Kuk, and Sunjoo Jeong. 2016. 'SRSF3 represses the expression of PDCD4
protein by coordinated regulation of alternative splicing, export and translation',
Biochemical and Biophysical Research Communications, 470: 431-38.
Parkinson, N., P. G. Ince, M. O. Smith, R. Highley, G. Skibinski, P. M. Andersen, K. E.
Morrison, H. S. Pall, O. Hardiman, J. Collinge, P. J. Shaw, and E. M. Fisher. 2006.
'ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein
2B)', Neurology, 67: 1074-7.
Pascale, E., E. Passarelli, C. Purcaro, A. R. Vestri, A. Fakeri, R. Guglielmi, F. Passarelli, and
G. Meco. 2011. 'Lack of association between IL-1β, TNF-α, and IL-10 gene
polymorphisms and sporadic Parkinson's disease in an Italian cohort', Acta Neurol
Scand, 124: 176-81.
Pasinelli, P., M. E. Belford, N. Lennon, B. J. Bacskai, B. T. Hyman, D. Trotti, and R. H.
Brown, Jr. 2004. 'Amyotrophic lateral sclerosis-associated SOD1 mutant proteins
bind and aggregate with Bcl-2 in spinal cord mitochondria', Neuron, 43: 19-30.
Patwardhan, Parag, and W. Todd Miller. 2007. 'Processive phosphorylation: mechanism and
biological importance', Cell Signal, 19: 2218-26.
Paz, Inbal, Idit Kosti, Manuel Ares, Jr., Melissa Cline, and Yael Mandel-Gutfreund. 2014.
'RBPmap: a web server for mapping binding sites of RNA-binding proteins', Nucleic
Acids Res, 42: W361-W67.
Pellerin, Luc, and Pierre J. Magistretti. 2012. 'Sweet sixteen for ANLS', Journal of cerebral
blood flow and metabolism : official journal of the International Society of Cerebral
Blood Flow and Metabolism, 32: 1152-66.
Perlson, E., G. B. Jeong, J. L. Ross, R. Dixit, K. E. Wallace, R. G. Kalb, and E. L. Holzbaur.
2009. 'A switch in retrograde signaling from survival to stress in rapid-onset
neurodegeneration', J Neurosci, 29: 9903-17.
Perry, V. H., J. A. Nicoll, and C. Holmes. 2010. 'Microglia in neurodegenerative disease',
Nat Rev Neurol, 6: 193-201.
Philips, T., A. Bento-Abreu, A. Nonneman, W. Haeck, K. Staats, V. Geelen, N. Hersmus, B.
Küsters, L. Van Den Bosch, P. Van Damme, W. D. Richardson, and W. Robberecht.
2013. 'Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral
sclerosis', Brain, 136: 471-82.

230
Picher-Martel, V., K. Dutta, D. Phaneuf, G. Sobue, and J. P. Julien. 2015. 'Ubiquilin-2 drives
NF-κB activity and cytosolic TDP-43 aggregation in neuronal cells', Mol Brain, 8:
71.
Picher-Martel, V., P. N. Valdmanis, P. V. Gould, J. P. Julien, and N. Dupré. 2016. 'From
animal models to human disease: a genetic approach for personalized medicine in
ALS', Acta Neuropathol Commun, 4: 70.
Piktel, E., I. Levental, B. Durnaś, P. A. Janmey, and R. Bucki. 2018. 'Plasma Gelsolin:
Indicator of Inflammation and Its Potential as a Diagnostic Tool and Therapeutic
Target', Int J Mol Sci, 19.
Polymenidou, M., C. Lagier-Tourenne, K. R. Hutt, S. C. Huelga, J. Moran, T. Y. Liang, S.
C. Ling, E. Sun, E. Wancewicz, C. Mazur, H. Kordasiewicz, Y. Sedaghat, J. P.
Donohue, L. Shiue, C. F. Bennett, G. W. Yeo, and D. W. Cleveland. 2011. 'Long pre-
mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss
of TDP-43', Nat Neurosci, 14: 459-68.
Porro, C., A. Cianciulli, and M. A. Panaro. 2020. 'The Regulatory Role of IL-10 in
Neurodegenerative Diseases', Biomolecules, 10.
Prasad, J., K. Colwill, T. Pawson, and J. L. Manley. 1999. 'The protein kinase Clk/Sty directly
modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing',
Molecular and Cellular Biology, 19: 6991-7000.
Pribadi, Mochtar, Zhongan Yang, Tanya S. Kim, Elliot W. Swartz, Alden Y. Huang, Jason
A. Chen, Deepika Dokuru, Jaeyun Baek, Fuying Gao, Andrea T. Fua, Kevin Wojta,
Qing Wang, Anna Karydas, Jamie Fong, Ed Lezcano, Stephanie Ng, Farid F. Chehab,
Harry V. Vinters, Bruce L. Miller, and Giovanni Coppola. 2016. 'CRISPR-Cas9
targeted deletion of the <em>C9orf72</em> repeat expansion mutation
corrects cellular phenotypes in patient-derived iPS cells', bioRxiv: 051193.
Prigent, M., I. Barlat, H. Langen, and C. Dargemont. 2000. 'IkappaBalpha and IkappaBalpha
/NF-kappa B complexes are retained in the cytoplasm through interaction with a
novel partner, RasGAP SH3-binding protein 2', J Biol Chem, 275: 36441-9.
Pringle, N. P., W. P. Yu, M. Howell, J. S. Colvin, D. M. Ornitz, and W. D. Richardson. 2003.
'Fgfr3 expression by astrocytes and their precursors: evidence that astrocytes and
oligodendrocytes originate in distinct neuroepithelial domains', Development, 130:
93-102.
Prinz, M., and J. Priller. 2014. 'Microglia and brain macrophages in the molecular age: from
origin to neuropsychiatric disease', Nat Rev Neurosci, 15: 300-12.
Prinz, Marco, Daniel Erny, and Nora Hagemeyer. 2017. 'Ontogeny and homeostasis of CNS
myeloid cells', Nature Immunology, 18: 385-92.
Przedborski, S., D. M. Donaldson, P. L. Murphy, O. Hirsch, D. Lange, A. B. Naini, D.
McKenna-Yasek, and R. H. Brown, Jr. 1996. 'Blood superoxide dismutase, catalase
and glutathione peroxidase activities in familial and sporadic amyotrophic lateral
sclerosis', Neurodegeneration, 5: 57-64.
Ragagnin, Audrey M. G., Sina Shadfar, Marta Vidal, Md Shafi Jamali, and Julie D. Atkin.
2019. 'Motor Neuron Susceptibility in ALS/FTD', Front Neurosci, 13: 532-32.
Rahimian, R., L. C. Beland, and J. Kriz. 2018. 'Galectin-3: mediator of microglia responses
in injured brain', Drug Discov Today, 23: 375-81.
Rahimian, R., L. C. Béland, and J. Kriz. 2018. 'Galectin-3: mediator of microglia responses
in injured brain', Drug Discov Today, 23: 375-81.

231
Ramírez-Jarquín, Uri N., Fabiola Rojas, Brigitte van Zundert, and Ricardo Tapia. 2017.
'Chronic infusion of SOD1G93A astrocyte-secreted factors induces spinal
motoneuron degeneration and neuromuscular dysfunction in healthy rats', Journal of
Cellular Physiology, 232: 2610-15.
Ratnadiwakara, Madara, Stuart K. Archer, Craig I. Dent, Igor Ruiz De Los Mozos, Traude
H. Beilharz, Anja S. Knaupp, Christian M. Nefzger, Jose M. Polo, and Minna-Liisa
Anko. 2018. 'SRSF3 promotes pluripotency through Nanog mRNA export and
coordination of the pluripotency gene expression program', eLife, 7: e37419.
Renaud, L., V. Picher-Martel, P. Codron, and J. P. Julien. 2019. 'Key role of UBQLN2 in
pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia', Acta
Neuropathol Commun, 7: 103.
Renton, A. E., A. Chiò, and B. J. Traynor. 2014. 'State of play in amyotrophic lateral sclerosis
genetics', Nat Neurosci, 17: 17-23.
Rentzos, M., C. Nikolaou, E. Andreadou, G. P. Paraskevas, A. Rombos, M. Zoga, A.
Tsoutsou, F. Boufidou, E. Kapaki, and D. Vassilopoulos. 2009. 'Circulating
interleukin-10 and interleukin-12 in Parkinson's disease', Acta Neurol Scand, 119:
332-7.
Reshef, R., E. Kudryavitskaya, H. Shani-Narkiss, B. Isaacson, N. Rimmerman, A. Mizrahi,
and R. Yirmiya. 2017. 'The role of microglia and their CX3CR1 signaling in adult
neurogenesis in the olfactory bulb', eLife, 6.
Richardson, Dale N., Mark F. Rogers, Adam Labadorf, Asa Ben-Hur, Hui Guo, Andrew H.
Paterson, and Anireddy S. N. Reddy. 2011. 'Comparative Analysis of
Serine/Arginine-Rich Proteins across 27 Eukaryotes: Insights into Sub-Family
Classification and Extent of Alternative Splicing', PLOS ONE, 6: e24542.
Robberecht, W., and T. Philips. 2013. 'The changing scene of amyotrophic lateral sclerosis',
Nat Rev Neurosci, 14: 248-64.
Roberts, K., R. Zeineddine, L. Corcoran, W. Li, I. L. Campbell, and J. J. Yerbury. 2013.
'Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a
cytotoxic phenotype', Glia, 61: 409-19.
Rojo, A. I., G. McBean, M. Cindric, J. Egea, M. G. López, P. Rada, N. Zarkovic, and A.
Cuadrado. 2014. 'Redox control of microglial function: molecular mechanisms and
functional significance', Antioxid Redox Signal, 21: 1766-801.
Rosen, D. R., T. Siddique, D. Patterson, D. A. Figlewicz, P. Sapp, A. Hentati, D. Donaldson,
J. Goto, J. P. O'Regan, H. X. Deng, and et al. 1993. 'Mutations in Cu/Zn superoxide
dismutase gene are associated with familial amyotrophic lateral sclerosis', Nature,
362: 59-62.
Rothstein, J. D. 2017. 'Edaravone: A new drug approved for ALS', Cell, 171: 725.
Rothstein, J. D., M. Van Kammen, A. I. Levey, L. J. Martin, and R. W. Kuncl. 1995.
'Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis',
Ann Neurol, 38: 73-84.
Rowin, J., Y. Xia, B. Jung, and J. Sun. 2017. 'Gut inflammation and dysbiosis in human
motor neuron disease', Physiol Rep, 5.
Rusconi, M., F. Gerardi, W. Santus, A. Lizio, V. A. Sansone, C. Lunetta, I. Zanoni, and F.
Granucci. 2017. 'Inflammatory role of dendritic cells in Amyotrophic Lateral
Sclerosis revealed by an analysis of patients' peripheral blood', Sci Rep, 7: 7853.
Saccon, R. A., R. K. Bunton-Stasyshyn, E. M. Fisher, and P. Fratta. 2013. 'Is SOD1 loss of
function involved in amyotrophic lateral sclerosis?', Brain, 136: 2342-58.

232
Sandén, C., M. Ageberg, J. Petersson, A. Lennartsson, and U. Gullberg. 2013. 'Forced
expression of the DEK-NUP214 fusion protein promotes proliferation dependent on
upregulation of mTOR', BMC Cancer, 13: 440.
Sanford, Jeremy R., Nicola K. Gray, Karsten Beckmann, and Javier F. Cáceres. 2004. 'A
novel role for shuttling SR proteins in mRNA translation', Genes & development, 18:
755-68.
Sapra, A. K., M. L. Anko, I. Grishina, M. Lorenz, M. Pabis, I. Poser, J. Rollins, E. M.
Weiland, and K. M. Neugebauer. 2009. 'SR protein family members display diverse
activities in the formation of nascent and mature mRNPs in vivo', Mol Cell, 34: 179-
90.
Sawada, H. 2017. 'Clinical efficacy of edaravone for the treatment of amyotrophic lateral
sclerosis', Expert Opin Pharmacother, 18: 735-38.
Saxena, S., E. Cabuy, and P. Caroni. 2009. 'A role for motoneuron subtype-selective ER
stress in disease manifestations of FALS mice', Nat Neurosci, 12: 627-36.
Schoch, K. M., and T. M. Miller. 2017. 'Antisense Oligonucleotides: Translation from Mouse
Models to Human Neurodegenerative Diseases', Neuron, 94: 1056-70.
Schram, Sarah, Donald Chuang, Greg Schmidt, Hristo Piponov, Cory Helder, James Kerns,
Mark Gonzalez, Fei Song, and Jeffrey A. Loeb. 2019. 'Mutant SOD1 prevents normal
functional recovery through enhanced glial activation and loss of motor neuron
innervation after peripheral nerve injury', Neurobiology of Disease, 124: 469-78.
Sellgren, Carl M., Jessica Gracias, Bradley Watmuff, Jonathan D. Biag, Jessica M. Thanos,
Paul B. Whittredge, Ting Fu, Kathleen Worringer, Hannah E. Brown, Jennifer Wang,
Ajamete Kaykas, Rakesh Karmacharya, Carleton P. Goold, Steven D. Sheridan, and
Roy H. Perlis. 2019. 'Increased synapse elimination by microglia in schizophrenia
patient-derived models of synaptic pruning', Nature Neuroscience, 22: 374-85.
Sen, S., H. Jumaa, and N. J. Webster. 2013. 'Splicing factor SRSF3 is crucial for hepatocyte
differentiation and metabolic function', Nat Commun, 4: 1336.
Sen, S., M. Langiewicz, H. Jumaa, and N. J. Webster. 2015. 'Deletion of serine/arginine-rich
splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice',
Hepatology, 61: 171-83.
Sen, S., I. Talukdar, and N. J. Webster. 2009. 'SRp20 and CUG-BP1 modulate insulin
receptor exon 11 alternative splicing', Mol Cell Biol, 29: 871-80.
Sephton, C. F., A. A. Tang, A. Kulkarni, J. West, M. Brooks, J. J. Stubblefield, Y. Liu, M.
Q. Zhang, C. B. Green, K. M. Huber, E. J. Huang, J. Herz, and G. Yu. 2014. 'Activity-
dependent FUS dysregulation disrupts synaptic homeostasis', Proc Natl Acad Sci U
S A, 111: E4769-78.
Sévigny, M., I. Bourdeau Julien, J. P. Venkatasubramani, J. B. Hui, P. A. Dutchak, and C. F.
Sephton. 2020. 'FUS contributes to mTOR-dependent inhibition of translation', J Biol
Chem, 295: 18459-73.
Seyfried, N. T., E. B. Dammer, V. Swarup, D. Nandakumar, D. M. Duong, L. Yin, Q. Deng,
T. Nguyen, C. M. Hales, T. Wingo, J. Glass, M. Gearing, M. Thambisetty, J. C.
Troncoso, D. H. Geschwind, J. J. Lah, and A. I. Levey. 2017. 'A Multi-network
Approach Identifies Protein-Specific Co-expression in Asymptomatic and
Symptomatic Alzheimer's Disease', Cell Syst, 4: 60-72.e4.
Shan, Xiaoyang, David Vocadlo, and Charles Krieger. 2009. 'Mislocalization of TDP-43 in
the G93A mutant SOD1 transgenic mouse model of ALS', Neuroscience Letters, 458:
70-74.

233
Shannon, P., A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, N. Amin, B.
Schwikowski, and T. Ideker. 2003. 'Cytoscape: a software environment for integrated
models of biomolecular interaction networks', Genome Res, 13: 2498-504.
Sharma, Aarti, Alexander K. Lyashchenko, Lei Lu, Sara Ebrahimi Nasrabady, Margot
Elmaleh, Monica Mendelsohn, Adriana Nemes, Juan Carlos Tapia, George Z. Mentis,
and Neil A. Shneider. 2016. 'ALS-associated mutant FUS induces selective motor
neuron degeneration through toxic gain of function', Nature Communications, 7:
10465-65.
Shaw, P. J., P. G. Ince, G. Falkous, and D. Mantle. 1995. 'Oxidative damage to protein in
sporadic motor neuron disease spinal cord', Ann Neurol, 38: 691-5.
Sheean, Rebecca K., Fiona C. McKay, Erika Cretney, Christopher R. Bye, Nirma D. Perera,
Doris Tomas, Richard A. Weston, Karlene J. Scheller, Elvan Djouma, Parvathi
Menon, Stephen D. Schibeci, Najwa Marmash, Justin J. Yerbury, Stephen L. Nutt,
David R. Booth, Graeme J. Stewart, Mathew C. Kiernan, Steve Vucic, and Bradley
J. Turner. 2018. 'Association of Regulatory T-Cell Expansion With Progression of
Amyotrophic Lateral Sclerosis: A Study of Humans and a Transgenic Mouse Model',
JAMA Neurology, 75: 681-89.
Shen, Ting, Huan Li, Yifan Song, Jun Yao, Miao Han, Ming Yu, Gang Wei, and Ting Ni.
2018. 'Antisense transcription regulates the expression of sense gene via alternative
polyadenylation', Protein & cell, 9: 540-52.
Shen, Ting, Huan Li, Yifang Song, Li Li, Jinzhong Lin, Gang Wei, and Ting Ni. 2019.
'Alternative polyadenylation dependent function of splicing factor SRSF3 contributes
to cellular senescence', Aging, 11: 1356-88.
Shepard, Peter J., and Klemens J. Hertel. 2009. 'The SR protein family', Genome biology, 10:
242.
Shibata, N., R. Nagai, K. Uchida, S. Horiuchi, S. Yamada, A. Hirano, M. Kawaguchi, T.
Yamamoto, S. Sasaki, and M. Kobayashi. 2001. 'Morphological evidence for lipid
peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic
lateral sclerosis patients', Brain research, 917: 97-104.
Singhal, S., J. Stadanlick, M. J. Annunziata, A. S. Rao, P. S. Bhojnagarwala, S. O'Brien, E.
K. Moon, E. Cantu, G. Danet-Desnoyers, H. J. Ra, L. Litzky, T. Akimova, U. H.
Beier, W. W. Hancock, S. M. Albelda, and E. B. Eruslanov. 2019. 'Human tumor-
associated monocytes/macrophages and their regulation of T cell responses in early-
stage lung cancer', Sci Transl Med, 11.
Slowicka, K., L. Vereecke, C. Mc Guire, M. Sze, J. Maelfait, A. Kolpe, X. Saelens, R.
Beyaert, and G. van Loo. 2016. 'Optineurin deficiency in mice is associated with
increased sensitivity to Salmonella but does not affect proinflammatory NF-κB
signaling', Eur J Immunol, 46: 971-80.
Smethurst, P., K. C. Sidle, and J. Hardy. 2015. 'Review: Prion-like mechanisms of transactive
response DNA binding protein of 43 kDa (TDP-43) in amyotrophic lateral sclerosis
(ALS)', Neuropathol Appl Neurobiol, 41: 578-97.
Smith, Emma F., Pamela J. Shaw, and Kurt J. De Vos. 2019. 'The role of mitochondria in
amyotrophic lateral sclerosis', Neuroscience Letters, 710: 132933.
Smith, R. A., T. M. Miller, K. Yamanaka, B. P. Monia, T. P. Condon, G. Hung, C. S.
Lobsiger, C. M. Ward, M. McAlonis-Downes, H. Wei, E. V. Wancewicz, C. F.
Bennett, and D. W. Cleveland. 2006. 'Antisense oligonucleotide therapy for
neurodegenerative disease', J Clin Invest, 116: 2290-6.

234
Solomon, J. N., C. A. Lewis, B. Ajami, S. Y. Corbel, F. M. Rossi, and C. Krieger. 2006.
'Origin and distribution of bone marrow-derived cells in the central nervous system
in a mouse model of amyotrophic lateral sclerosis', Glia, 53: 744-53.
Song, S., C. J. Miranda, L. Braun, K. Meyer, A. E. Frakes, L. Ferraiuolo, S. Likhite, A. K.
Bevan, K. D. Foust, M. J. McConnell, C. M. Walker, and B. K. Kaspar. 2016. 'Major
histocompatibility complex class I molecules protect motor neurons from astrocyte-
induced toxicity in amyotrophic lateral sclerosis', Nat Med, 22: 397-403.
Song, X., X. Wan, T. Huang, C. Zeng, N. Sastry, B. Wu, C. D. James, C. Horbinski, I.
Nakano, W. Zhang, B. Hu, and S. Y. Cheng. 2019. 'SRSF3-Regulated RNA
Alternative Splicing Promotes Glioblastoma Tumorigenicity by Affecting Multiple
Cellular Processes', Cancer Res, 79: 5288-301.
Spiller, K. J., C. R. Restrepo, T. Khan, M. A. Dominique, T. C. Fang, R. G. Canter, C. J.
Roberts, K. R. Miller, R. M. Ransohoff, J. Q. Trojanowski, and V. M. Lee. 2018.
'Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a
mouse model of TDP-43 proteinopathy', Nat Neurosci, 21: 329-40.
Sta, M., R. M. Sylva-Steenland, M. Casula, J. M. de Jong, D. Troost, E. Aronica, and F. Baas.
2011. 'Innate and adaptive immunity in amyotrophic lateral sclerosis: evidence of
complement activation', Neurobiol Dis, 42: 211-20.
Stephenson, Jodie, and Sandra Amor. 2017. 'Modelling amyotrophic lateral sclerosis in
mice', Drug Discovery Today: Disease Models, 25-26: 35-44.
Storkebaum, E., D. Lambrechts, M. Dewerchin, M. P. Moreno-Murciano, S. Appelmans, H.
Oh, P. Van Damme, B. Rutten, W. Y. Man, M. De Mol, S. Wyns, D. Manka, K.
Vermeulen, L. Van Den Bosch, N. Mertens, C. Schmitz, W. Robberecht, E. M.
Conway, D. Collen, L. Moons, and P. Carmeliet. 2005. 'Treatment of motoneuron
degeneration by intracerebroventricular delivery of VEGF in a rat model of ALS', Nat
Neurosci, 8: 85-92.
Strickland, M. R., K. R. Ibanez, M. Yaroshenko, C. C. Diaz, D. R. Borchelt, and P.
Chakrabarty. 2020. 'IL-10 based immunomodulation initiated at birth extends
lifespan in a familial mouse model of amyotrophic lateral sclerosis', Sci Rep, 10:
20862.
Summerton, J. E. 2007. 'Morpholino, siRNA, and S-DNA compared: impact of structure and
mechanism of action on off-target effects and sequence specificity', Curr Top Med
Chem, 7: 651-60.
Sun, Y., M. Hei, Z. Fang, Z. Tang, B. Wang, and N. Hu. 2019. 'High-Mobility Group Box 1
Contributes to Cerebral Cortex Injury in a Neonatal Hypoxic-Ischemic Rat Model by
Regulating the Phenotypic Polarization of Microglia', Front Cell Neurosci, 13: 506.
Sun, Y., L. Yan, J. Guo, J. Shao, and R. Jia. 2019. 'Downregulation of SRSF3 by antisense
oligonucleotides sensitizes oral squamous cell carcinoma and breast cancer cells to
paclitaxel treatment', Cancer Chemother Pharmacol, 84: 1133-43.
Sunjoo, Jeong. 2017. 'SR Proteins: Binders, Regulators, and Connectors of RNA', Molecules
and Cells, 40: 1-9.
Swarup, V., J. N. Audet, D. Phaneuf, J. Kriz, and J. P. Julien. 2012. 'Abnormal regenerative
responses and impaired axonal outgrowth after nerve crush in TDP-43 transgenic
mouse models of amyotrophic lateral sclerosis', J Neurosci, 32: 18186-95.
Swarup, V., D. Phaneuf, N. Dupré, S. Petri, M. Strong, J. Kriz, and J. P. Julien. 2011.
'Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-
mediated pathogenic pathways', J Exp Med, 208: 2429-47.

235
Tacke, Roland, and James L. Manley. 1999. 'Determinants of SR protein specificity', Curr
Opin Cell Biol, 11: 358-62.
Tafuri, Francesco, Dario Ronchi, Francesca Magri, Giacomo P. Comi, and Stefania Corti.
2015. 'SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral
sclerosis pathogenesis', Front Cell Neurosci, 9: 336-36.
Talbott, E. O., A. M. Malek, and D. Lacomis. 2016. 'The epidemiology of amyotrophic lateral
sclerosis', Handb Clin Neurol, 138: 225-38.
Tan, C. F., H. Eguchi, A. Tagawa, O. Onodera, T. Iwasaki, A. Tsujino, M. Nishizawa, A.
Kakita, and H. Takahashi. 2007. 'TDP-43 immunoreactivity in neuronal inclusions in
familial amyotrophic lateral sclerosis with or without SOD1 gene mutation', Acta
Neuropathol, 113: 535-42.
Tandan, Rup, and Walter G. Bradley. 1985. 'Amyotrophic lateral sclerosis: Part 1. Clinical
features, pathology, and ethical issues in management', Annals of Neurology, 18: 271-
80.
Tang, Y., I. Horikawa, M. Ajiro, A. I. Robles, K. Fujita, A. M. Mondal, J. K. Stauffer, Z. M.
Zheng, and C. C. Harris. 2013. 'Downregulation of splicing factor SRSF3 induces
p53beta, an alternatively spliced isoform of p53 that promotes cellular senescence',
Oncogene, 32: 2792-8.
Taylor, J. Paul, Robert H. Brown, and Don W. Cleveland. 2016. 'Decoding ALS: from genes
to mechanism', Nature, 539: 197-206.
Tomkins, J., P. Usher, J. Y. Slade, P. G. Ince, A. Curtis, K. Bushby, and P. J. Shaw. 1998.
'Novel insertion in the KSP region of the neurofilament heavy gene in amyotrophic
lateral sclerosis (ALS)', Neuroreport, 9: 3967-70.
Tong, J., C. Huang, F. Bi, Q. Wu, B. Huang, X. Liu, F. Li, H. Zhou, and X. G. Xia. 2013.
'Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous
motor neuron death in rats', Embo j, 32: 1917-26.
Trias, E., P. Diaz-Amarilla, S. Olivera-Bravo, E. Isasi, D. A. Drechsel, N. Lopez, C. S.
Bradford, K. E. Ireton, J. S. Beckman, and L. Barbeito. 2013. 'Phenotypic transition
of microglia into astrocyte-like cells associated with disease onset in a model of
inherited ALS', Front Cell Neurosci, 7: 274.
Trias, E., P. Díaz-Amarilla, S. Olivera-Bravo, E. Isasi, D. A. Drechsel, N. Lopez, C. S.
Bradford, K. E. Ireton, J. S. Beckman, and L. Barbeito. 2013. 'Phenotypic transition
of microglia into astrocyte-like cells associated with disease onset in a model of
inherited ALS', Front Cell Neurosci, 7: 274.
Trias, E., S. Ibarburu, R. Barreto-Núñez, J. Babdor, T. T. Maciel, M. Guillo, L. Gros, P.
Dubreuil, P. Díaz-Amarilla, P. Cassina, L. Martínez-Palma, I. C. Moura, J. S.
Beckman, O. Hermine, and L. Barbeito. 2016. 'Post-paralysis tyrosine kinase
inhibition with masitinib abrogates neuroinflammation and slows disease progression
in inherited amyotrophic lateral sclerosis', J Neuroinflammation, 13: 177.
Trias, E., P. H. King, Y. Si, Y. Kwon, V. Varela, S. Ibarburu, M. Kovacs, I. C. Moura, J. S.
Beckman, O. Hermine, and L. Barbeito. 2018. 'Mast cells and neutrophils mediate
peripheral motor pathway degeneration in ALS', JCI Insight, 3.
Twyffels, Laure, Cyril Gueydan, and Véronique Kruys. 2011. 'Shuttling SR proteins: more
than splicing factors', The FEBS Journal, 278: 3246-55.
Uranishi, H., T. Tetsuka, M. Yamashita, K. Asamitsu, M. Shimizu, M. Itoh, and T. Okamoto.
2001. 'Involvement of the pro-oncoprotein TLS (translocated in liposarcoma) in

236
nuclear factor-kappa B p65-mediated transcription as a coactivator', J Biol Chem,
276: 13395-401.
Urushitani, M., J. Kurisu, K. Tsukita, and R. Takahashi. 2002. 'Proteasomal inhibition by
misfolded mutant superoxide dismutase 1 induces selective motor neuron death in
familial amyotrophic lateral sclerosis', J Neurochem, 83: 1030-42.
Urushitani, M., A. Sik, T. Sakurai, N. Nukina, R. Takahashi, and J. P. Julien. 2006.
'Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to
amyotrophic lateral sclerosis', Nat Neurosci, 9: 108-18.
Vance, C., E. L. Scotter, A. L. Nishimura, C. Troakes, J. C. Mitchell, C. Kathe, H. Urwin, C.
Manser, C. C. Miller, T. Hortobágyi, M. Dragunow, B. Rogelj, and C. E. Shaw. 2013.
'ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within
cytoplasmic stress granules', Hum Mol Genet, 22: 2676-88.
Vargas, M. R., D. A. Johnson, and J. A. Johnson. 2011. 'Decreased glutathione accelerates
neurological deficit and mitochondrial pathology in familial ALS-linked
hSOD1(G93A) mice model', Neurobiol Dis, 43: 543-51.
Verkhratsky, Alexei, and Vladimir Parpura. 2016. 'Astrogliopathology in neurological,
neurodevelopmental and psychiatric disorders', Neurobiology of Disease, 85: 254-61.
Vucic, S., G. A. Nicholson, and M. C. Kiernan. 2008. 'Cortical hyperexcitability may precede
the onset of familial amyotrophic lateral sclerosis', Brain, 131: 1540-50.
Walsh, C. M., A. L. Suchanek, T. J. Cyphert, A. B. Kohan, W. Szeszel-Fedorowicz, and L.
M. Salati. 2013a. 'Serine arginine splicing factor 3 is involved in enhanced splicing
of glucose-6-phosphate dehydrogenase RNA in response to nutrients and hormones
in liver', J Biol Chem, 288: 2816-28.
Walsh, Callee M., Amanda L. Suchanek, Travis J. Cyphert, Alison B. Kohan, Wioletta
Szeszel-Fedorowicz, and Lisa M. Salati. 2013b. 'Serine arginine splicing factor 3 is
involved in enhanced splicing of glucose-6-phosphate dehydrogenase RNA in
response to nutrients and hormones in liver', J Biol Chem, 288: 2816-28.
Wang, A., O. Rojas, D. Lee, and J. L. Gommerman. 2020. 'Regulation of neuroinflammation
by B cells and plasma cells', Immunological reviews.
Wang, E. T., R. Sandberg, S. Luo, I. Khrebtukova, L. Zhang, C. Mayr, S. F. Kingsmore, G.
P. Schroth, and C. B. Burge. 2008. 'Alternative isoform regulation in human tissue
transcriptomes', Nature, 456: 470-6.
Wang, H., B. Lekbaby, N. Fares, J. Augustin, T. Attout, A. Schnuriger, A. M. Cassard, G.
Panasyuk, G. Perlemuter, I. Bieche, S. Vacher, J. Selves, J. M. Peron, B. Bancel, P.
Merle, D. Kremsdorf, J. Hall, I. Chemin, and P. Soussan. 2019. 'Alteration of splicing
factors' expression during liver disease progression: impact on hepatocellular
carcinoma outcome', Hepatol Int, 13: 454-67.
Wang, H. Y., K. C. Arden, J. R. Bermingham, Jr., C. S. Viars, W. Lin, A. D. Boyer, and X.
D. Fu. 1999. 'Localization of serine kinases, SRPK1 (SFRSK1) and SRPK2
(SFRSK2), specific for the SR family of splicing factors in mouse and human
chromosomes', Genomics, 57: 310-5.
Wang, J., W. Y. Ho, K. Lim, J. Feng, G. Tucker-Kellogg, K. A. Nave, and S. C. Ling. 2018.
'Cell-autonomous requirement of TDP-43, an ALS/FTD signature protein, for
oligodendrocyte survival and myelination', Proc Natl Acad Sci U S A, 115: E10941-
e50.

237
Wang, J., N. Kainrad, H. Shen, Z. Zhou, P. Rote, Y. Zhang, L. E. Nagy, J. Wu, and M. You.
2018. 'Hepatic Knockdown of Splicing Regulator Slu7 Ameliorates Inflammation
and Attenuates Liver Injury in Ethanol-Fed Mice', Am J Pathol, 188: 1807-19.
Wang, Z., Z. Bai, X. Qin, and Y. Cheng. 2019. 'Aberrations in Oxidative Stress Markers in
Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis', Oxid Med
Cell Longev, 2019: 1712323.
Wang, Z., D. D. Rao, N. Senzer, and J. Nemunaitis. 2011. 'RNA interference and cancer
therapy', Pharm Res, 28: 2983-95.
Wann, A. K. T., J. P. Chapple, and M. M. Knight. 2014. 'The primary cilium influences
interleukin-1β-induced NFκB signalling by regulating IKK activity', Cell Signal, 26:
1735-42.
Watanuki, T., H. Funato, S. Uchida, T. Matsubara, A. Kobayashi, Y. Wakabayashi, K.
Otsuki, A. Nishida, and Y. Watanabe. 2008. 'Increased expression of splicing factor
SRp20 mRNA in bipolar disorder patients', J Affect Disord, 110: 62-9.
Wee, C. D., M. A. Havens, F. M. Jodelka, and M. L. Hastings. 2014. 'Targeting SR proteins
improves SMN expression in spinal muscular atrophy cells', PLOS ONE, 9: e115205.
Weerasuriya, A., and A. P. Mizisin. 2011. 'The blood-nerve barrier: structure and functional
significance', Methods Mol Biol, 686: 149-73.
Wegorzewska, I., and R. H. Baloh. 2011. 'TDP-43-based animal models of
neurodegeneration: new insights into ALS pathology and pathophysiology',
Neurodegener Dis, 8: 262-74.
Weiduschat, N., X. Mao, J. Hupf, N. Armstrong, G. Kang, D. J. Lange, H. Mitsumoto, and
D. C. Shungu. 2014. 'Motor cortex glutathione deficit in ALS measured in vivo with
the J-editing technique', Neurosci Lett, 570: 102-7.
Weissmann, Robert, Melanie Hüttenrauch, Tim Kacprowski, Yvonne Bouter, Laurent
Pradier, Thomas A. Bayer, Andreas W. Kuss, and Oliver Wirths. 2016. 'Gene
Expression Profiling in the APP/PS1KI Mouse Model of Familial Alzheimer’s
Disease', Journal of Alzheimer's Disease, 50: 397-409.
Welser-Alves, Jennifer V., Stephen J. Crocker, and Richard Milner. 2011. 'A dual role for
microglia in promoting tissue inhibitor of metalloproteinase (TIMP) expression in
glial cells in response to neuroinflammatory stimuli', Journal of Neuroinflammation,
8: 61-61.
Weskamp, Kaitlin, and Sami J. Barmada. 2018. 'TDP43 and RNA instability in amyotrophic
lateral sclerosis', Brain research, 1693: 67-74.
Wilhelmsson, U., D. Andersson, Y. de Pablo, R. Pekny, A. Ståhlberg, J. Mulder, N. Mitsios,
T. Hortobágyi, M. Pekny, and M. Pekna. 2017. 'Injury Leads to the Appearance of
Cells with Characteristics of Both Microglia and Astrocytes in Mouse and Human
Brain', Cereb Cortex, 27: 3360-77.
Williamson, T. L., and D. W. Cleveland. 1999. 'Slowing of axonal transport is a very early
event in the toxicity of ALS-linked SOD1 mutants to motor neurons', Nat Neurosci,
2: 50-6.
Willis, C. M., A. M. Nicaise, L. Peruzzotti-Jametti, and S. Pluchino. 2020. 'The neural stem
cell secretome and its role in brain repair', Brain research, 1729: 146615.
Wolfe, C. M., N. F. Fitz, K. N. Nam, I. Lefterov, and R. Koldamova. 2018. 'The Role of
APOE and TREM2 in Alzheimer's Disease-Current Understanding and Perspectives',
Int J Mol Sci, 20.

238
Wong, J., B. Garner, G. M. Halliday, and J. B. Kwok. 2012. 'Srp20 regulates TrkB pre-
mRNA splicing to generate TrkB-Shc transcripts with implications for Alzheimer's
disease', J Neurochem, 123: 159-71.
Wong, P. C., C. A. Pardo, D. R. Borchelt, M. K. Lee, N. G. Copeland, N. A. Jenkins, S. S.
Sisodia, D. W. Cleveland, and D. L. Price. 1995. 'An adverse property of a familial
ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar
degeneration of mitochondria', Neuron, 14: 1105-16.
Wong, Raymond W., Ahalya Balachandran, Mario A. Ostrowski, and Alan Cochrane. 2013.
'Digoxin suppresses HIV-1 replication by altering viral RNA processing', PLoS
pathogens, 9: e1003241-e41.
Wosiski-Kuhn, Marlena, Mac Robinson, Jane Strupe, Phonepasong Arounleut, Matthew
Martin, James Caress, Michael Cartwright, Robert Bowser, Merit Cudkowicz, Carl
Langefeld, Gregory A. Hawkins, and Carol Milligan. 2019. 'IL6
receptor<sup>358</sup>Ala variant and trans-signaling are disease
modifiers in amyotrophic lateral sclerosis', Neurology - Neuroimmunology
Neuroinflammation, 6: e631.
Wynne, Angela M., Christopher J. Henry, Yan Huang, Anthony Cleland, and Jonathan P.
Godbout. 2010. 'Protracted downregulation of CX3CR1 on microglia of aged mice
after lipopolysaccharide challenge', Brain, Behavior, and Immunity, 24: 1190-201.
Xiao, S. H., and J. L. Manley. 1997. 'Phosphorylation of the ASF/SF2 RS domain affects
both protein-protein and protein-RNA interactions and is necessary for splicing',
Genes & development, 11: 334-44.
Yamanaka, K., S. Boillee, E. A. Roberts, M. L. Garcia, M. McAlonis-Downes, O. R. Mikse,
D. W. Cleveland, and L. S. Goldstein. 2008. 'Mutant SOD1 in cell types other than
motor neurons and oligodendrocytes accelerates onset of disease in ALS mice', Proc
Natl Acad Sci U S A, 105: 7594-9.
Yamanaka, K., S. J. Chun, S. Boillee, N. Fujimori-Tonou, H. Yamashita, D. H. Gutmann, R.
Takahashi, H. Misawa, and D. W. Cleveland. 2008. 'Astrocytes as determinants of
disease progression in inherited amyotrophic lateral sclerosis', Nat Neurosci, 11: 251-
3.
Yamanaka, K., and O. Komine. 2018. 'The multi-dimensional roles of astrocytes in ALS',
Neurosci Res, 126: 31-38.
Yan, J., Y. Xu, L. Zhang, H. Zhao, L. Jin, W. G. Liu, L. H. Weng, Z. H. Li, and L. Chen.
2016. 'Increased Expressions of Plasma Galectin-3 in Patients with Amyotrophic
Lateral Sclerosis', Chin Med J (Engl), 129: 2797-803.
Yang, Yongjie, Oguz Gozen, Andrew Watkins, Ileana Lorenzini, Angelo Lepore, Yuanzheng
Gao, Svetlana Vidensky, Jean Brennan, David Poulsen, Jeong Won Park, Noo Li
Jeon, Michael B. Robinson, and Jeffrey D. Rothstein. 2009. 'Presynaptic regulation
of astroglial excitatory neurotransmitter transporter GLT1', Neuron, 61: 880-94.
Young, Philip J., Christine J. DiDonato, Diane Hu, Rashmi Kothary, Elliot J. Androphy, and
Christian L. Lorson. 2002. 'SRp30c-dependent stimulation of survival motor neuron
(SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2β1', Human
Molecular Genetics, 11: 577-87.
Yu, C. H., S. Davidson, C. R. Harapas, J. B. Hilton, M. J. Mlodzianoski, P.
Laohamonthonkul, C. Louis, R. R. J. Low, J. Moecking, D. De Nardo, K. R. Balka,
D. J. Calleja, F. Moghaddas, E. Ni, C. A. McLean, A. L. Samson, S. Tyebji, C. J.
Tonkin, C. R. Bye, B. J. Turner, G. Pepin, M. P. Gantier, K. L. Rogers, K. McArthur,

239
P. J. Crouch, and S. L. Masters. 2020. 'TDP-43 Triggers Mitochondrial DNA Release
via mPTP to Activate cGAS/STING in ALS', Cell, 183: 636-49.e18.
Yu, Q., J. Guo, and J. Zhou. 2004. 'A minimal length between tau exon 10 and 11 is required
for correct splicing of exon 10', J Neurochem, 90: 164-72.
Yuo, Chung-Yee, Hui-Hua Lin, Ya-Sian Chang, Wen-Kuang Yang, and Jan-Gowth Chang.
2008. '5-(N-ethyl-N-isopropyl)-amiloride enhances SMN2 exon 7 inclusion and
protein expression in spinal muscular atrophy cells', Annals of Neurology, 63: 26-34.
Zeng, C., and S. M. Berget. 2000. 'Participation of the C-terminal domain of RNA
polymerase II in exon definition during pre-mRNA splicing', Molecular and Cellular
Biology, 20: 8290-301.
Zhan, Y., and F. Fang. 2019. 'Smoking and amyotrophic lateral sclerosis: A mendelian
randomization study', Ann Neurol, 85: 482-84.
Zhang, C. J., M. Jiang, H. Zhou, W. Liu, C. Wang, Z. Kang, B. Han, Q. Zhang, X. Chen, J.
Xiao, A. Fisher, W. J. Kaiser, M. A. Murayama, Y. Iwakura, J. Gao, J. Carman, A.
Dongre, G. Dubyak, D. W. Abbott, F. D. Shi, R. M. Ransohoff, and X. Li. 2018.
'TLR-stimulated IRAKM activates caspase-8 inflammasome in microglia and
promotes neuroinflammation', J Clin Invest, 128: 5399-412.
Zhang, J., Y. Liu, X. Liu, S. Li, C. Cheng, S. Chen, and W. Le. 2018. 'Dynamic changes of
CX3CL1/CX3CR1 axis during microglial activation and motor neuron loss in the
spinal cord of ALS mouse model', Transl Neurodegener, 7: 35.
Zhang, K., C. J. Donnelly, A. R. Haeusler, J. C. Grima, J. B. Machamer, P. Steinwald, E. L.
Daley, S. J. Miller, K. M. Cunningham, S. Vidensky, S. Gupta, M. A. Thomas, I.
Hong, S. L. Chiu, R. L. Huganir, L. W. Ostrow, M. J. Matunis, J. Wang, R. Sattler,
T. E. Lloyd, and J. D. Rothstein. 2015. 'The C9orf72 repeat expansion disrupts
nucleocytoplasmic transport', Nature, 525: 56-61.
Zhang, K., J. C. Grima, J. D. Rothstein, and T. E. Lloyd. 2016. 'Nucleocytoplasmic transport
in C9orf72-mediated ALS/FTD', Nucleus, 7: 132-7.
Zhang, Ke, J. Gavin Daigle, Kathleen M. Cunningham, Alyssa N. Coyne, Kai Ruan, Jonathan
C. Grima, Kelly E. Bowen, Harsh Wadhwa, Peiguo Yang, Frank Rigo, J. Paul Taylor,
Aaron D. Gitler, Jeffrey D. Rothstein, and Thomas E. Lloyd. 2018. 'Stress Granule
Assembly Disrupts Nucleocytoplasmic Transport', Cell, 173: 958-71.e17.
Zhang, R., R. Gascon, R. G. Miller, D. F. Gelinas, J. Mass, K. Hadlock, X. Jin, J. Reis, A.
Narvaez, and M. S. McGrath. 2005. 'Evidence for systemic immune system
alterations in sporadic amyotrophic lateral sclerosis (sALS)', J Neuroimmunol, 159:
215-24.
Zhang, W., C. Niu, R. Y. Fu, and Z. Y. Peng. 2018. 'Mycobacterium tuberculosis H37Rv
infection regulates alternative splicing in Macrophages', Bioengineered, 9: 203-08.
Zhao, W., D. R. Beers, S. Bell, J. Wang, S. Wen, R. H. Baloh, and S. H. Appel. 2015. 'TDP-
43 activates microglia through NF-κB and NLRP3 inflammasome', Exp Neurol, 273:
24-35.
Zhao, W., W. Xie, Q. Xiao, D. R. Beers, and S. H. Appel. 2006. 'Protective effects of an anti-
inflammatory cytokine, interleukin-4, on motoneuron toxicity induced by activated
microglia', J Neurochem, 99: 1176-87.
Zhao, Weihua, David R. Beers, Kristopher G. Hooten, Douglas H. Sieglaff, Aijun Zhang,
Shanker Kalyana-Sundaram, Christopher M. Traini, Wendy S. Halsey, Ashley M.
Hughes, Ganesh M. Sathe, George P. Livi, Guo-Huang Fan, and Stanley H. Appel.

240
2017. 'Characterization of Gene Expression Phenotype in Amyotrophic Lateral
Sclerosis Monocytes', JAMA Neurology, 74: 677-85.
Zhong, Xiang-Yang, Pingping Wang, Joonhee Han, Michael G. Rosenfeld, and Xiang-Dong
Fu. 2009. 'SR proteins in vertical integration of gene expression from transcription to
RNA processing to translation', Mol Cell, 35: 1-10.
Zhou, J. Y., L. Afjehi-Sadat, S. Asress, D. M. Duong, M. Cudkowicz, J. D. Glass, and J.
Peng. 2010. 'Galectin-3 is a candidate biomarker for amyotrophic lateral sclerosis:
discovery by a proteomics approach', J Proteome Res, 9: 5133-41.
Zhou, L., J. Guo, and R. Jia. 2019. 'Oncogene SRSF3 suppresses autophagy via inhibiting
BECN1 expression', Biochem Biophys Res Commun, 509: 966-72.
Zhou, Yueqin, Songyan Liu, Guodong Liu, Arzu Öztürk, and Geoffrey G. Hicks. 2013. 'ALS-
Associated FUS Mutations Result in Compromised FUS Alternative Splicing and
Autoregulation', PLOS Genetics, 9: e1003895.
Zhou, Z., and X. D. Fu. 2013. 'Regulation of splicing by SR proteins and SR protein-specific
kinases', Chromosoma, 122: 191-207.
Zhu, G., C. J. Wu, Y. Zhao, and J. D. Ashwell. 2007. 'Optineurin negatively regulates
TNFalpha- induced NF-kappaB activation by competing with NEMO for
ubiquitinated RIP', Curr Biol, 17: 1438-43.
Zhu, X., D. Wei, O. Chen, Z. Zhang, J. Xue, S. Huang, W. Zhu, and Y. Wang. 2016.
'Upregulation of CCL3/MIP-1alpha regulated by MAPKs and NF-kappaB mediates
microglial inflammatory response in LPS-induced brain injury', Acta Neurobiol Exp
(Wars), 76: 304-17.
Zoller, T., A. Attaai, P. S. Potru, T. Russ, and B. Spittau. 2018. 'Aged Mouse Cortical
Microglia Display an Activation Profile Suggesting Immunotolerogenic Functions',
Int J Mol Sci, 19.
Zondler, L., K. Müller, S. Khalaji, C. Bliederhäuser, W. P. Ruf, V. Grozdanov, M. Thiemann,
K. Fundel-Clemes, A. Freischmidt, K. Holzmann, B. Strobel, P. Weydt, A. Witting,
D. R. Thal, A. M. Helferich, B. Hengerer, K. E. Gottschalk, O. Hill, M. Kluge, A. C.
Ludolph, K. M. Danzer, and J. H. Weishaupt. 2016. 'Peripheral monocytes are
functionally altered and invade the CNS in ALS patients', Acta Neuropathol, 132:
391-411.




















