
Layered zirconium-tin phosphates
14
AppliedCatalysis, 68(1991)55-68 55 ElsevierSciencePublishers B .V ., Amsterdam Layeredzirconium-tinphosphates I .Chemicalandphysicalcharacterization G .Bagnasco DipartimentodiIngegneriaChimica,UaiversitdFedericoIIdiNapoli,Naples(Italy) P .Ciambelli DiportimentodiChimica,UniuersittFederico11diNapoli,Naples(Italy) A .Frezza,P .GalliandA .LaGinestra DipartimentodiChimica,UniversitdaLaSapienza,Rome(Italy) and M .Turco* DipartimentodiIngegneria Chimiea, Universit¢FedericoIIdiNapoli,Naples(Italy) (Received17April1990,revisedmanuscriptreceived7September1990) Abstract Crystallinezirconium-tinphosphatesofformulaZr,Sn, ._,(HPO,),,, °H,0 (0<x< ])weresynthesized andcharacterizedintermsoftheirphysicalandchemicalproperties .FromX-rayandthermalanalysis itwasfoundthatsolidsolutionsareformedforeverycomposition .Thetin contentinmixedphaseshas amarkedinfluenceonthermalbehaviour,speedingupthekineticsoftransformation tolayeredpyro- phosphates (L-Py) . Mixedphosphatespossesssurfaceareaswhicharemarkedlyhigherthanpurephos- phates-Themethodofammoniathermodesorption(TPD)wasemployedfortheacidity measurements . Thismethodhasallowedtheevaluationoftheconcentrationandstrength ofsurfaceacidsitespresent ontheexternalsurfaceof L-Py ormixedhydrogen-L-Pyphasesobtainedafteravarietyofthermal treatments,Theacidstrengthof L-Py phasesincreaseswithincreasingtincontent . Keywords : layeredphosphates,tin-zirconium,solidsolutions,surfaceacidity, INTRODUCTION Inrecentyearslayeredacidphosphatesoftetravalentmetalshavereceived increasingattentionbecauseoftheirpotentialuses,mainlyasionexchangers andcatalysts .Infacttheyareabletocatalyzeseveralreactionsofinterestin petrochemicalprocesses,suchasoxidation,isomerization,anddehydration [1-7] ; inparticulartheircatalyticpropertieshavebeeninvestigatedinthe oxydehydrogenationofethylbenzenetostyrene [8-10] . Interestingresults 0166-9834/91/$03 .50 ©1991ElsevierSciencePublishersB .V .
Transcript of Layered zirconium-tin phosphates

Applied Catalysis, 68 (1991) 55-68
55Elsevier Science Publishers B.V ., Amsterdam
Layered zirconium-tin phosphates
I. Chemical and physical characterization
G. BagnascoDipartimento di Ingegneria Chimica, Uaiversitd Federico II di Napoli, Naples (Italy)
P. Ciambelli
Diportimento di Chimica, Uniuersitt Federico 11 di Napoli, Naples (Italy)
A. Frezza, P . Galli and A. La Ginestra
Dipartimento diChimica, UniversitdaLa Sapienza, Rome (Italy)
and
M. Turco*
Dipartimento di Ingegneria Chimiea, Universit¢Federico II di Napoli, Naples (Italy)
(Received 17 April 1990, revised manuscript received 7 September 1990)
Abstract
Crystalline zirconium-tin phosphates of formula Zr,Sn, ._, (HPO,),,, °H,0 (0<x< ])were synthesizedand characterized in terms of their physical and chemical properties . From X-ray and thermal analysisit was found that solid solutions are formed for every composition . The tin content in mixed phases hasa marked influence on thermal behaviour, speeding up the kinetics of transformation to layered pyro-phosphates (L-Py) . Mixed phosphates possess surface areas which are markedly higher than pure phos-phates- The method of ammonia thermodesorption (TPD) was employed for the acidity measurements .This method has allowed the evaluation of the concentration and strength of surface acid sites presenton the external surface of L-Py or mixed hydrogen-L-Py phases obtained after a variety of thermaltreatments, The acid strength of L-Py phases increases with increasing tin content .
Keywords : layered phosphates, tin-zirconium, solid solutions, surface acidity,
INTRODUCTION
In recent years layered acid phosphates of tetravalent metals have receivedincreasing attention because of their potential uses, mainly as ion exchangersand catalysts . In fact they are able to catalyze several reactions of interest inpetrochemical processes, such as oxidation, isomerization, and dehydration[1-7] ; in particular their catalytic properties have been investigated in theoxydehydrogenation of ethylbenzene to styrene [8-10] . Interesting results
0166-9834/91/$03 .50
© 1991 Elsevier Science Publishers B .V .

56
concerning conversion and selectivity to styrene have been achieved on zirco-nium phosphate [81, and different metal pyrophosphates tI1 1 .
The Me(IV) phosphates with general formula A(HPO,) 2-nH2O (whereA= Ge, Ti, Sn or Zr) can crystallize in different forms : a and y phases are thebest known [ 12 ] . Mixed Me (IV) phosphates or arsenates have recently beenprepared, in order to modify or to improve some of the properties of the singlecompounds. The mixed systems may give rise to solid solutions over a widerange of composition, such as the case of Ti-Zr [ 13 ] and Ge-Zr phosphates[ 141, but we have also found that stable solid solutions for every compositioncan be obtained in the case of the Zr arsenophosphates [15] and for Zr-Snphosphates [16] . These materials are generally isomorphous with a-zirco-nium phosphate and exhibit similar ion exchange properties ; the catalytic be-haviour depends on their composition and on their modulated acidic properties .
In this work we have studied the physicochemical and catalytic propertiesof pure and mixed systems based on zirconium and tin phosphates in the ox-ydehydrogenation of ethylbenzene to styrene . In Part I we report on the phys-icochemical characterization of these materials with reference to their thermalbehaviour and crystal structure modifications at various temperatures . Theproperties which mainly influence the catalytic behaviour, such as surface area,pore volume, and acidity were also investigated .
In part II [ 161 we report the catalytic results obtained in oxydehydrogena-tion of ethylbenzene to styrene .
EXPERIMENTAL
Preparation of materials
Pure zirconium and tin phosphates were prepared with the refluxing methodfollowing the procedure described in previous reports [3,18] . Mixed zirco-nium-tin phosphates were obtained by coprecipitation from solutions of SnCl,and ZrOCl2 in different ratios by adding phosphoric acid (8 M) and nitric acid(4 M); the mixture was refluxed for 100 h and the white precipitate was filteredand washed with ethanol [161 . The tin content was determined by atomicadsorption spectroscopy, and phosphorus was determined colorimetrically, asdescribed in Ref. 19, Zr being calculated by difference .
Physical characterization
X-ray analysis was carried out by a Philips diffractometer using Ni-filteredCu Ka radiation .Simultaneous DTA and TG thermal analysis was performed by a Stanton
mod. 781 thermoanalyzer (Pt-Pt/Rh thermocouples) at 2-5°C/min heatingrates. SEM micrographs were obtained by Hitachi 2300 apparatus .

Surface area and pore volume were measured by nitrogen adsorption at-196° C using a Carlo Erba Sorptomatic 1800 .
Acidity measurements
The acidity measurements were performed by ammonia temperature-pro-grammed desorption (TPD), using a flow apparatus with a thermal conduc-tivity detector [20] . The ammonia adsorption was effected at room tempera-ture. The heating rate for the desorption was 10°C/min ; the water evolvedfrom the samples during the desorption was removed by a potassium hydroxidetrap located before the detector for ammonia analysis . The acidity measure-ments were carried out on the samples pretreated at 450 and 600°C, becausethe data referring to samples pretreated. at lower temperatures are greatly af-fected by ammonia intercalation [20] .
RESULTS AND DISCUSSION
X-ray and thermal analysis
Table 1 lists the composition of the materials as found from chemical analysis .X-ray powder patterns of pure and mixed phosphates, without thermal
treatment and after 12 h heating at 450'C, 600 ° C and at 1000 ° C, are reportedin Fig. 1 .
The X-ray spectra of the hydrated phases show that the mixed phosphatesare solid solutions throughout the whole range of composition examined . Thisis indicated by the gradual shift of the do,, values, reported in Table 2, bychanging the composition of samples, which accords well with Vegard's law .The X-ray diffraction patterns of heat treated samples show deep structuralmodifications connected with the transformation to the pyrophosphate phase,as deduced by TG results. Such a process has already been described for pureZrP and SnP [18,21] . After treatment at 600°C the condensation process iscomplete for pure ZrP and SnP, giving rise to well-defined layered pyrophos-
TABLE 1
Chemical analysis of Zr-Sn phosphates
Sample Chemical composition
ZrP
Zr(HPO,)2 H,OZrSn31 Zr,,,,Sn o .a,(HPO,),-HLOZrSn1I Zros4Sna,,a (HPO a),-H,OZrSn13
Zr,,, I Sn„_os(HPO,) z . H,OSnP
Sn(HPO,), L5H,0
57

58
5 7
d(4)
f
Fig. 1 . X-ray analysis of Zr-Sn phosphates obtained upon different thermal treatanents .
phates phases (L-Py ) . This is clearly indicated by the d„o2 values of 6.1 and6.25 A (Table 2), which correspond to the interlayer distances of Zr and Snpyrophosphate, respectively . However the d(, a2 values of mixed phosphates pre-treated at 600`C are not intermediate between those of pure L-Py phases butslightly higher (Table 2), suggesting that the condensation to L-Py phases is
Z r P haarad12 hr. ar 65d°cZrSn31„ 6506 C2rSn11
++ 650°C,Zr Sn13
6500c,-SnP
11 650° C I
IZrP,1 1000 ° C iZr Sn31
10000C+ . (
IZrSn11„ 1000° C
_
J._Z rSn13„ 1000°CSnP„ 1000 ° C
7
G7 tnl
Zr P
ZrSn31
ZrSn11
I ZrSn13
`SnP IZ r P haarad12 hr., 450°C
_
IZrSn31
450° Cl ZrSn11
430° CZr Sn13
~- n 450°CSn P„ 450°C

TABLE2
d., values of Zr-Sn phosphates after different thermal treatments
59
not complete. Moreover the low intensity of the signals is indicative of a strongamorphization of the materials .
After treatment at 450°C the XRD results indicate the presence of mixedhydrogen phosphate-pyrophosphate phases together with a certainamorphization .
After treatment at 1000°C ZrP and SnP samples show the signals of cubicpyrophosphates [211 . The d values of mixed phases are intermediate betweenthose of pure compounds, showing a gradual shift on changing the composi-tion, indicating that pyrophosphate solutions have been formed .
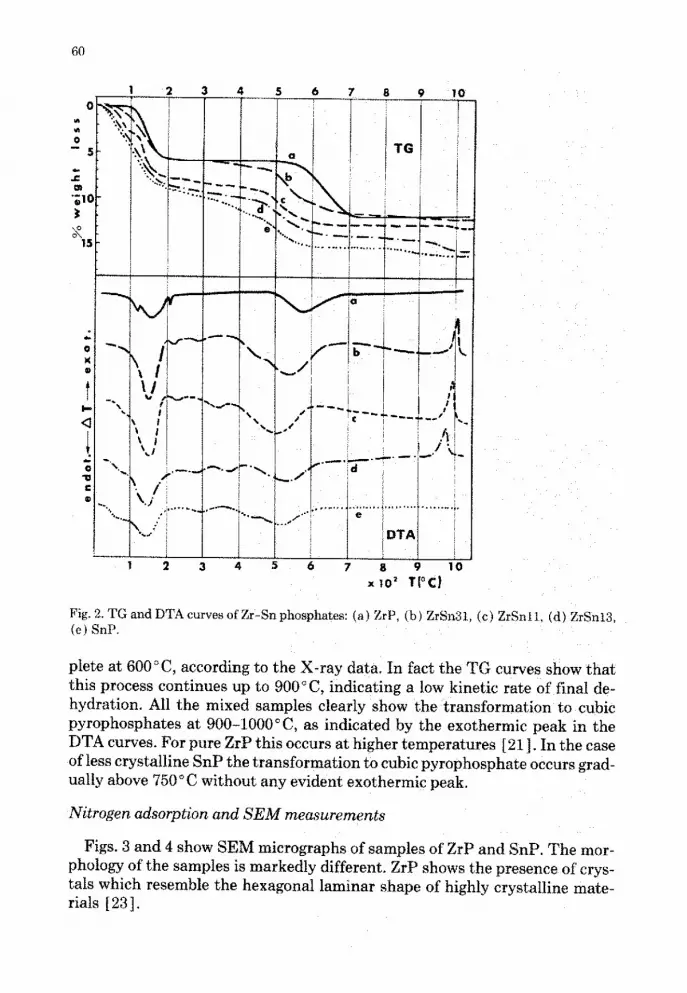
The DTA and TG curves are shown in Fig . 2. All the materials undergo adehydration process between 40 and 200'C, with weight loss corresponding toca . 1-1 .5 molecules of water. We can observe the influence of tin content onthe dehydration process, since SnP dehydrates more easily than ZrP, probablybecause of the greater interlayer distance . Moreover, by increasing the tin con-tent the gradual disappearance of the anhydrous phase transition can be ob-served; this is particularly evident in the case of pure ZrP as an endothermicpeak at around 200°C .
The condensation process to layered pyrophosphates occurs between 250and 650'C . From the TG and DTA curves we can observe that this process isquite different for ZrP and SnP . For ZrP it occurs in one step between 550 and650'C, whereas for SnP it takes place in two or three steps in a temperaturerange from 250 to 650'C. The condensation process of SnP begins at lowertemperature with respect to ZrP, probably because of the greater interlayerdistance. As the condensation proceeds the interlayer distance decreases, hin-dering the removal of water and slowing down the rate of condensation of re-sidual HPO, groups . This could explain why this process occurs in severalsteps. The behaviour of Zr-Sn mixed phosphates is intermediate in regard tothe range of temperature and the steps through which the condensation pro-cess occurs . However, the condensation to L-Py phases seems still to be incom-
Sample d.4 (A) at pretreatment temperature (°C)
25 600
ZrP 7.57 6.10ZrS1131 7.63 6.27ZrSnll 7.67 6.50ZrSnl3 7.78 6.60Sap 7.82 6.25
60
sma103
15
VC
\l
234i
f
a
N /'
I
Ia
I
TG
10
3
7
10.i02 Tr°C;
Fig. 2 . TG and DTA curves of Zr-Sn phosphates : (a) ZrP, (b) ZrSn3 c) ZrSnll (d) ZrSnll,(e) SnP .
plete at 600°C, according to the X-ray data. In fact the TG curves show thatthis process continues up to 900'C, indicating a low kinetic rate of final de-hydration. All the mixed samples clearly show the transformation to cubicpyrophosphates at 900-1000°C, as indicated by the exothermic peak in theDTA curves. For pure ZrP this occurs at higher temperatures [211 . In the caseof less crystalline SnP the transformation to cubic pyrophosphate occurs grad-ually above 750°C without any evident exothermic peak .
Nitrogen adsorption and SEM measurements
Figs. 3 and 4 show SEM micrographs of samples of ZrP and SnP . The mor-phology of the samples is markedly different . ZrP shows the presence of crys-tals which resemble the hexagonal laminar shape of highly crystalline mate-rials [23] .
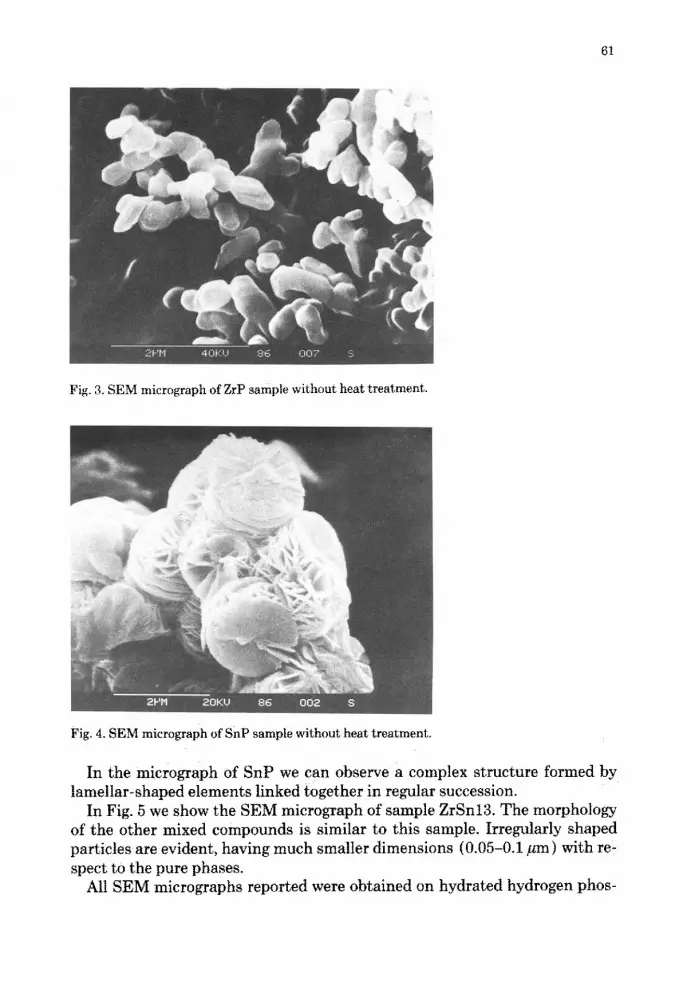
Fig . 3 . SEM micrograph of ZrP sample without heat treatment.
Fig . 4 . SEM micrograph of Sap sample without heat treatment .
In the micrograph of SnP we can observe a complex structure formed bylamellar-shaped elements linked together in regular succession .
In Fig. 5 we show the SEM micrograph of sample ZrSn13 . The morphologyof the other mixed compounds is similar to this sample . Irregularly shapedparticles are evident, having much smaller dimensions (0 .05-0.1 um) with re-spect to the pure phases .All SEM micrographs reported were obtained on hydrated hydrogen phos-
6 1

62
Fig. 5 . SEM micrograph of ZrSn13 sample without heat treatment .
TABLE 3
Surface area and pore volume of Zr-Sn phosphates after different thermal treatments
phate samples; no morphological modifications were observed after heat treat-ment up to 600'C .
The surface areas and pore volumes measured on the original samples, andafter 12 h treatment at 450 and 600°C, are reported in Table 3 . The surfacearea and pore volume values of mixed phosphates are higher than those of purephosphates . Moreover, by changing the Zr : Sn molar ratio, there is a slightincrease of surface area, reaching a maximum value for sample ZrSnl3 .
The higher surface area values of mixed phosphates can be related to thesmaller dimensions of their particles, which could be due to the lower rate ofcrystal growth during the refluxing in H 3PO4 solution, with respect to the for-mation of pure phases . We must take into account the difference between theZr and Sn ionic radii, which makes it difficult to obtain highly crystalline solid
Sample Surface area (m'/g)at pretreatment, temperature ( 'C)
Pore volume (cm'/g)at pretreatment temperature ('C)
25 450 600 25 450 600
10.0 10.0 10.0 0.03 . 0 .03 0.03ZrSn31 67.0 68 .1 70.0 0.40 0.40 0.40ZrSn11 73.0 73.8 88.4 0.35 0.36 0,38ZrSnl3 74.9 75 .1 92 .4 0.31 0.31 0.52Sup 10.0 10.5 19 .6 . 0 .06 0.05 0.10

63
solutions, as indicated by the broad reflections observed in the X-ray powderpatterns .
Moreover the above data suggest that in mixed phases the particles containsome porosity. In fact, from the dimensions observed in the SEM micrographsand taking account of the fact that the density values range from 2 .70 (ZrP )to 3.12 g/cm 3 (SnP ), for a spheroidal shape, we can estimate that the geomet-rical surface areas lie between about 20 and 40 m2/g; values which are quite alot smaller than those obtained from nitrogen adsorption . This is also con-firmed by the higher pore volumes exhibited by the mixed phases (Table 3) :,
Such a porosity should be negligible in pure phosphates ; in fact the geomet-rical areas, evaluated on the supposition of disk-shaped crystals for SnP (di-ameter=2 .00 um, thickness =0.05µm) and laminar shaped crystals for ZrP(diameter=0.5 ptm, thickness=0.10 µm), are about 10 m 2/g, in good agree-ment with the BET values .
The increase of surface areas from pure to mixed phosphates has also beenobserved by other authors for the Sr-Ti phosphate system [ 13 ] .
Both. surface area and pore volume were unchanged after treatment at 450'C,whereas in some cases it increased after treatment at 600 ° C . This effect is verymarked for pure SnP, marked for mixed phases, and negligible for ZrP (Table3) .
Such an increase cannot at present be readily explained . Many factors cancontribute to this behaviour, such as the hydrolyzability or the crystal order ofthe compounds. It is well known that SnP shows a lower degree of crystallinitywith respect to ZrP due to a less organized and regular lattice order [1.8] : thismay be the main cause of the lack of anhydrous phase transition present inZrP, TiP and GeP between 200 and 400° [23] . This leads us to think thatweaker interlayer linkings in SnP may enable a more extensive breakage ofthe linkages between the layers during the condensation process, leading tomore extended surface areas and porosity .
Acidity measurements
TPD spectra of ammonia adsorbed at 20'C on pure and mixed phosphatespretreated at 450 and 600°C for 12 h are reported in Figs . 6-9 .
First of all the reader should note the marked difference in behaviour be-tween mixed and pure phosphates . In fact the mixed phosphates show a greatercomplexity of spectra and stronger intensity of the ammonia desorption peaks(Figs. 6 and. 7) . After treatment at 450°C the TPD spectra of mixed phos-phates exhibit, besides a low temperature peak, two higher temperature signals(Fig. 7) while in the spectra of pure phosphates only one high temperaturepeak is present (Fig . 6) .
According to conclusions drawn previously [20] the low temperature peakrepresents ammonia adsorbed on the surface sites, while the higher tempera-

6 4
200
400
600
800T (°C)
Fig . 6. Ammonia TPD curves of (a) ZrP and (b) SnP samples heat treated at 450'C .
700
400
600
600T(T)
Fig . 7 . Ammonia TPD curves of (a) ZrSn31, (b) ZrSnl1, and (c) ZrSn13 samples heat treated at450'C .

ture peaks represent ammonia interacting with internal acid sites . The ad-sorption of ammonia on internal sites is greatly affected by diffusive resistance[20} . Therefore, the smaller dimensions of the particles of mixed phosphates,affording ammonia more ready access to the internal sites, could explain thelarger amount of ammonia adsorbed on these centers as well as the lower tem-perature at which desorption commences .
Owing to the presence of such diffusive phenomena the amounts of ammoniacorresponding to high temperature peaks can not give an effective measure ofinternal acidity.
After treating the materials at 600'C the TPD spectra (Figs . 8 and 9) showsignals only from the external acid sites, which appear as asymmetric peakswith maxima at 135-170°C .
The concentration of surface acid sites have been evaluated from the areasof the low temperature peaks . These are reported in Table 4 for the samplestreated at 450 and 600°C. We observe that the concentration of sites in purezirconium and tin phosphates does not change with the temperature, despitethe large increase of surface area of SnP after treatment at 600°C . However,in mixed phosphates the slight variations of site concentration observed uponchanging the pretreatment temperature from 450 to 600°C are probably dueto the uncertainty in evaluating the low temperature peaks on samples treatedat 450'C, the tails of which are covered by the overlapping high temperaturepeaks. It may thus be presumed that, in the mixed phosphates, too, no signif-icant variations in site concentrations occur when varying the treatment tem-perature from 450 to 600°C. This may imply that no condensation of surface
E
209
400
600
800T (°C)
Fig. 8 . Ammonia TPI) curves of . (a) ZrP and (b) SnP samples heat treated at 600°C .
0
6 5

66
00 z00
400
600 BooT (°C)
Fig. 9 . Ammonia TPD curves of (a) ZrSn3l, (b) ZrSnll and (c) ZrSnl3 samples heat treated at600 , C .
TABLE 4
Surface acidity of Zr-Sn phosphates after treatment at 450 and 600'C
OH groups would occur in this temperature range . In fact the condensationmust be quite unlikely because of the large distance between two adjacent-POH groups [12] .
The expected concentration of acid sites which could be calculated on thebasis of the distribution of HPO 4 groups on an ideal layer of a-phosphate givesvalues ranging from 4 .2 (ZrP) to 4 .7 10 14 sites per cm' (SnP), attributing toeach hydroxyl group an area of 24.0 and 21.4 A2 , respectively [12] .
In some cases, particularly for pure tin and zirconium phosphates, we haveobtained lower values than expected . At present we have no plausible expla-nation for this .
The influence of tin content on the acid strength can. be attributed to the
Sample Tp't~=450'C 7'p,„, .,=600'C
Site cony(em2 X10-") . ( 1 C)
Site cone .(cm-2X10 -") (`C)
ZrP 3.0 13 .5 3 .0 135ZrSn31 3.7 133 4 .2 137ZrSn11 4.2 130 4 .6 147ZrSnl3 3.1 - 136 3 .7 167SnP 2.2 140 2 .2 170

higher electronegativity of tin with respect to zirconium, which leads to anincrease of ionicity of the PO-H bond .
It is worthwhile noting, from Figs. 6-9, the asymmetry of the low tempera-ture peaks, which indicates a surface heterogeneity having a wide distributionof surface acid site strengths . Thus, besides weak sites, medium and highstrength sites are also present, which should be the most effective in acid-catalyzed reactions .
We can observe that tin phosphate, which shows more tailed low tempera-ture peaks than zirconium phosphate, has a higher fraction of medium-highstrength sites (Figs . 6, 8) . In mixed phosphates the complexity of spectra, ob-tained after treatment at 450'C . makes it hard to detect the medium and highstrength surface sites (Fig . 7) . In the same samples, treated at 600°C, no sig-nificant differences in peak tailing are evident (Fig . 9) .
From the data of Table 4 we deduce that no significant difference in peakmaximum temperatures is evident in the materials treated at 450°C .
The treatment at 600°C leads to a general increase of peak maximum tem-peratures, which is more evident the higher the tin content is, A similar effectof treatment temperature on the acid strength has also been observed withcatalytic tests, employed as indirect acidity measurements on pure phosphates[3,241 . This behaviour was ascribed to a greater ionic character of the surfaceacid sites due to the formation of P-O-P bonds between the layers .
CONCLUSIONS
We have shown that the zirconium-tin phosphate system gives solid solu-tions for all the compositions we have examined . Moreover, the materials canbe synthesized with much higher surface areas than the pure phosphates .
The thermal treatment leading to condensation to pyrophosphates does notdecrease the surface area for ZrP and produces an increase of surface areaswhich is greater as the tin content increases .
Every sample, after thermal treatment, presents su face acid sites with awide range of acidic strength . This strength seems to ncrease by increasingthe temperature of pretreatment .
The thermal treatment of the materials must therefore be carefully con-trolled since it may affect the catalytic properties of the phases formed aftertreatment .
REFERENCES
1 J.B. Moffat, Catal . Rev : Sci . Eng ., 18 (1978) 199 .2 A. Clearfield and D .S . Thakur, J . CataL, 65 (1980) 185 .3 A. La Ginestra, P . Patrono, M .L . Berardelli, P . Galli, C. Ferragina and M.A. Massucci, J .
Catal., 103 (1987) 345 .4 K. Segawa, Y . Nakajima, S . Nakata, S. Asaoka and H. Takahashi, J . Catal., 101 (1986) 81 .
67

68
56789 Y. Murakami, K. Iwayama, H. Uchioda, T. Hattori and T. Tagaka, J . Catal ., 71 (1981) 2K-
10 Y. Murakami, K . lwayama, H. Uchioda, T . Hattori and T . Tagaka, Appl . Catal ., 2 (1982) 67 .11 G.E . Vrieland, J . Catal ., 111 (1988) 1 .12 A. Clearfield, in A. Clearfield (Ed .) Inorganic Ion Exchange Materials, CRC Press, Boca
Raton, FL, 1982 .13 T.N. Frianeza and A . Clearfield, J . Coral ., 85 (1984) 398 .14 P. Galli, A. La Ginestra, M .L. Berardelli, M .A. Massucci and P. Patrono . Thermochimica
Acta, 92 (1985) 615 .15 M.L. Berardelli, P . Galli, A . La Ginestra, M .A. Massucci and K.G. Varshney, J . Chem. Soc.
Dalton Trans ., 1737 (1985) .16 P. Galli, A . La Ginestra, P . Patrono, M .A. Massucci, C. Ferragina, P . Ciambelli and G. Bag-
nasco, Italian Pat. 21587 A/86 (1986) .17 G. Bagnasco, P . Ciambelli, M . Turco, A . La Ginestra, and P. Patrono, Appl . Catal ., 68 (1991)
69 .18 A. La Ginestra, P . Patrono, M .A. Massucci, P . Galli, C . Ferragina and C. Mancini . in MA-
Phillips and M . Ternan (Eds .), Proceedings of the 9th Int . Congr . on Catalysis, Calgary(Canada.) 1988, Vol. 1, p . 499 .
19 D.N. Bernhart and A .R. Wreath, Anal . Chem ., 27 (1955) 440 .20 M. Turco, P . Ciambelli, G . Bagnasco, A. La Ginestra, P . Galli and C. Ferragina, J . Catal .. 117
(1988) 355 .21 A. La Ginestra, C . Ferragina, M .A. Massucci, P . Patrono, R. Di Rocco and A.A.G. Tomlinson,
Gazz, Chim . Ital., 113 (1983) 357.22 GR. Levi and G . Peyronel, Z . Kristallogr., 92 (1935) 190 .23 A. Clearfield and D . Smith, Inorg_ Chem., 8 (1969) 431 .24 G. harms, G . Buses, V . Lorenzelli, A . La Ginestra, P. Galli and M.A. Massucci. J . Chem . Soc .
Dalton Trans ., 881 . (1988) .
M. Iwamoto, Y. Nomura and S . Kagawa, J. Cat l., 69 (1981) 234 .G.F. Crum and S.J . Paton, U .S . Pat. 4 291 184 (1981) .G.F. Crum and S.J . Paton, U .S . Pat . 4 291 183 (1981) .G. Emig and H . Hofmann, J . Catal ., 84 (1983) 15 .




















