
Delivery and Quantification of Hydrogen Peroxide Generated ...
Upload
independentCategory
view
2download
0
FULL PAPER
DOI:10.1002/ejic.201402767
Hydrolytic Stability and Hydrogen Peroxide Activation ofZirconium-Based Oxoclusters
Francesco Faccioli,[a] Matthias Bauer,[b] Danilo Pedron,[a]
COVER PICTUREAntonio Sorarù,[a] Mauro Carraro,*[a] and Silvia Gross*[a,c]
Keywords: Cluster compounds / Zirconium / Peroxides / Oxidation / EXAFS spectroscopy
The hydrolytic stability of [Zr6(OH)4O4{O(O)CC(CH3)=CH2}12] (Zr6), and [Zr6O4(OH)4{O(O)CCH2CH=CH2}12]2·6[CH2=CHCH2C(O)OH] (Zr12) oxoclusters in different envi-ronments was thoroughly investigated by FTIR, Raman, andX-ray photoelectron spectroscopy (XPS). Specific informationabout the local structures around the Zr centers during thestability tests was achieved by in situ extended X-ray absorp-tion fine structure (EXAFS) measurements, and the exactcompositions were determined by inductively coupledplasma MS (ICP-MS) and elemental analysis. By this multi-dimensional spectroscopic approach, an overview on the
Introduction
Oxoclusters of the early transition metals are a class ofpolynuclear compounds built from a limited number ofgroup 3–5 metal atoms, typically in their highest oxidationstate, linked by oxygen bridges and coordinated by biden-tate organic ligands. Some examples also involve a secondalkaline earth metal (e.g., Ba, Mg).[1–3] They display dif-ferent nuclearities (n = 3–12), coordination numbers of themetal atoms (varying between six and nine), and connecti-vity modes (corner, edge, or face sharing). These com-pounds are globally neutral and discrete species with thegeneral formula [MyOx(OH)w{O(O)CR}z] (R = organicgroup). During the synthesis (a controlled hydrolysis andcondensation of a metal alkoxide), carboxylic acids are in-troduced to stabilize the structure of the inorganic core.[1–3]
Oxoclusters have been extensively used as building blocksfor inorganic–organic hybrid materials,[2,3] as molecular
[a] Dipartimento di Scienze Chimiche, Università degli Studi diPadova,via Marzolo 1, 35131 Padova, ItalyE-mail: [email protected]/mauro.carraro
[b] Department Chemie, Universität Paderborn,Warburger Str. 100, 33106 Paderborn, Germany
[c] Istituto per l’Energetica e le Interfasi, IENI-CNR and INSTM,UdR Padova,via Marzolo 1, 35131 Padova, ItalyE-mail: [email protected]/silvia.grossSupporting information for this article is available on theWWW under http://dx.doi.org/10.1002/ejic.201402767.
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim210
structures formed after different treatments could be gained.The stability of the oxoclusters was then investigated in thepresence of hydrogen peroxide, and the formation of peroxo–metal complexes was detected. Thus, a kinetic study wasperformed in acetonitrile to evaluate the performances of theoxoclusters as oxygen transfer catalysts. The oxidation ofmethyl p-tolyl sulfide to the corresponding sulfoxide andsulfone was chosen as a model reaction; in some cases, aninteresting selectivity towards the formation of the sulfonewas found over more than 4700 catalytic cycles.
precursors for the controlled nucleation of nanostructuredoxides,[4] and as secondary building units (SBU) for metal–organic frameworks (MOFs).[5] Among the first reportedexamples of these oxoclusters, one of the earliest to bestructurally characterized was [Ti6O4(OR)8{O(O)CMe}8],which was obtained by the reaction of Ti(OR)4 (R = alkylgroup) with acetic acid.[6] In recent years, the synthesis ofdifferent early transition and main group metal polynuclearcomplexes (with O–M–O moieties) has been extensively ex-plored, and oxoclusters based on Zr, Hf, Ti, Ti–Zr, Ag–Zr,Y, T–-Hf, Zr–Ti–Hf, Ba, Ba–Ti, Ti–Y, Nb, and Sn havebeen obtained.[1,3] Since the 1990s, Hubert-Pfalzgraf etal.[7,8] have pioneered this development by exploring origi-nal routes for the synthesis of a plethora of mono- and po-lymetallic (e.g., Pb–Zr, Pb–Ti, Cu–Y, Y, lanthanides, La–Zn, Ba–Ce, Bi–Ba, Zn–Ta, Y–Pr, Sm–Ti, and others) mixedalkoxides and oxoclusters as potential single-source precur-sors for the corresponding mixed functional oxides (e.g.,perovskites). Further examples of metal oxide clustersbased on other transition elements (Fe, Cr) that feature atriangular M3O core are also known.[9] The chemical prop-erties, the tailored synthesis and modification, and thestructural issues of these oxoclusters as well as their use asbuilding blocks for the preparation of hybrid materials havebeen thoroughly described in some reviews and researchpapers.[1–3]
Typically, these oxoclusters are characterized by the pres-ence of d0 metal ions such as TiIV, ZrIV, HfIV, or NbV. Ithas been widely reported that complexes containing high-oxidation-state early transition metals display unique cata-

www.eurjic.org FULL PAPER
lytic properties.[10] In several cases, they can activatehydrogen peroxide and organic peroxides to promote theoxidation of different substrates (olefins, sulfides and sulf-oxides, and alcohols)[11] and can also enable enantioselec-tive transformations.[12] However, although peroxo speciesinvolving zirconium atoms have been widely described[13]
and interesting reactivities were observed for Zr-substitutedpolyoxometalates,[14] there are still no reports of the use ofZr oxoclusters as oxidation catalysts in the presence ofhydrogen peroxide. Hence, this works aims to shed light onthe catalytic potential of such compounds. In addition, asone of the difficulties in using these Zr oxoclusters for cata-lytic purposes is their reactivity towards water or moisture,which leads to uncontrolled hydrolysis of the highly reactiveM–O–M bonds, their stability under operative conditionshas been assessed.
Herein, the attention has been focused on the use of twowell-known zirconium oxoclusters, [Zr6(OH)4O4{O(O)CC-(CH3)=CH2}12] (Zr6),[1g,1h] and [Zr6O4(OH)4{O(O)CCH2-CH=CH2}12]2·6CH2=CHCH2C(O)OH (Zr12),[1i] for cata-lytic H2O2 activation. The two oxoclusters have been chosenas model systems characterized by the same metal (Zr) butby different nuclearities (i.e., the number of metal atoms)and structures. Zr12 can be regarded as a dimeric form ofthe Zr6 species in which the two subunits are connected byfour carboxylate bridges (the structures are reported in theSupporting Information, Figure S1). This latter feature, to-gether with the different nuclearities and different func-tional moieties on the surface (i.e, methacrylate vs. vinyl-acetate groups), can account for the different hydrolytic be-havior shown by the two species (vide infra). Formally, ashighlighted in ref.,[1i] the Zr12 clusters can be constructedby opening two edge-bridging μ2-carboxylate ligands of Zr6
and letting two such units dimerize. Nevertheless, they arestructurally well defined and distinct polynuclear complexesthat cannot be interconverted, and their chemical behaviorhas been the topic of a thorough investigation by Schubertet al.[1i] However, the hydrolytic stability and possible useof these polynuclear complexes in catalysis has not beeninvestigated. Our present investigation encompasses the as-sessment of (1) their hydrolytic stability and (2) their ac-tivity as sulfoxidation catalysts. As mentioned above, theformer point also entails a broader interest owing to theuse of these oxoclusters as effective versatile building blocksfor a plethora of different inorganic,[4] polymer-based,[1–3]
and porous hybrid materials.[5]
The hydrolytic stability of polynuclear metal complexesis attracting growing interest because of the implicationsthat this has on their functional behavior, for example, inthe field of catalysis. For instance, the hydrolytic stability ofZr-based polynuclear systems, in this case Zr-monosubsti-tuted monomeric and dimeric polyoxometalates in aqueoussolutions at different pH values, has been the topic of arecent publication by Carbò et al.[14h]
As reactions such as polymerizations with co-monomersto obtain class II organic–inorganic hybrid materials areusually performed in organic solvents, the stability of[Zr4O2{O(O)CC(CH3)=CH2}12] (Zr4)[15] and Zr6
[1g,1h] in
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim211
benzene and tetrahydrofuran (THF), two poorly coordinat-ing solvents, has already been reported.[16] In these studies,after the complete dissolution of the two compounds, theprecipitation of white powders was observed. Both the fil-tered solutions and the dried precipitates were analyzed byextended X-ray absorption fine structure (EXAFS) spec-troscopy at the ZrK-edge and by Raman spectroscopy.[17]
Although the structures of the oxoclusters remained un-changed in solution, the X-ray absorption spectra of thepowders revealed several differences with respect to thoseof the starting compounds. It was supposed that a portionof the starting compounds degraded into dimeric or tri-meric species, which precipitated because of their lower sol-ubility. Furthermore, temperature-dependent NMR spec-troscopy studies on Zr4 and [Zr6(OH)4O4{O(O)CR}12] inCD2Cl2, as well as ab initio molecular dynamics simulationson the latter oxoclusters, allowed the postulation of theexistence of rapid intramolecular exchange phenomena ofthe bidentate ligands (“carboxylate shift”) on the surface ofthe oxocluster.[17b] The solvent appears to play a major rolein the structural modifications of oxoclusters, and its contri-bution depends on its polarity and acidic features.
In the present work, we have focused on the effect of aprotic and polar solvent such as water on the structuralmodification of Zr-based oxoclusters and, therefore, ontheir stability. To accomplish this task, Zr6 and Zr12 were(1) treated with aqueous solutions at different pH values,(2) treated with a diluted hydrogen peroxide aqueous solu-tion (30%), and (3) exposed to atmospheric air (Table 1).The resulting compounds were analyzed by attenuated totalreflectance (ATR) FTIR, Raman, and EXAFS spec-troscopy.[19–21] The possible changes in their morphologiesand structure after the treatments were investigated bySEM, and their chemical compositions were determined byinductively coupled plasma MS (ICP-MS) and CHNS ele-mental analysis. The catalytic activity of the zirconium-based oxoclusters towards H2O2 activation was finallytested in the oxidation of methyl p-tolyl sulfide to investi-gate the formation of metalloperoxides and catalyst sta-bility. In this scenario, EXAFS and FTIR spectroscopy be-fore and after catalysis allowed the assessment of the behav-ior of the compounds under turnover conditions.
Table 1. Sample labeling and experimental parameters used for theZr6 and Zr12 hydrolytic treatments.
Sample Precursor Treatment Appearance
Zr6_ac Zr6_pre aq. solution at pH 2 colorless solutionZr6_ne Zr6_pre aq. solution at pH 7 white suspensionZr6_ba Zr6_pre aq. solution at pH 13 white precipitateZr6_ox Zr6_pre 30% aq. solution of H2O2 white precipitateZr6_air Zr6_pre atmospheric air same as Zr6_preZr12_ac Zr12_pre aq. solution at pH 2 colorless solutionZr12_ne Zr12_pre aq. solution at pH 7 white suspensionZr12_ba Zr12_pre aq. solution at pH 13 white precipitateZr12_ox Zr12_pre 30% aq. solution of H2O2 pale yellow precipitateZr12_air Zr12_pre atmospheric air same as Zr12_pre

www.eurjic.org FULL PAPER
Results and Discussion
1. Hydrolytic Stability of the Zr6 and Zr12 Oxoclusters
Zr6 and Zr12 were synthesized according to literatureprocedures, and their identities were confirmed by FTIRand Raman spectroscopy. Although the Raman spec-troscopy characterization of Zr6 had already been reportedby Kickelbick et al.,[17] the Raman spectrum of Zr12 is re-ported for the first time in this work. The FTIR and Ramanspectra of Zr6 are shown in Figure 1 (a and b). As reportedpreviously,[16,17,19–21] the stretching modes of the C–Hbonds of the methyl and olefinic groups of the methacrylateligands can be observed between ν = 3099 and 2927 cm–1.The whole area shows an overlaid O–H stretching absorp-
Figure 1. (a) FTIR and (b) Raman spectra of Zr6.
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim212
tion, which is characteristic of the residual free acid mol-ecules randomly dispersed in the voids of the crystal lat-tice.[1g,1h] The stretching of the carbonyl group of these non-coordinated methacrylic acid molecules is responsible forthe strong absorption at ν = 1701 cm–1 (Figure 1, a), andthe peak at ν = 795 cm–1 in the Raman spectrum (Figure 1,b) can also be attributed to the vibrations of this com-pound.[16,17] The C=C stretching of the ligands occurs at ν= 1645 cm–1. At ν = 1574 and 1423 cm–1, the asymmetricand symmetric stretching modes of the COO– group of themethacrylate anion coordinated to the inorganic core of theoxocluster can be observed, even if they are partially over-laid with the bending frequency of the methyl groups. Theabsorption at ν = 937 cm–1 can be ascribed to the out-of-plane bending mode of the two C–H bonds of the olefinic

www.eurjic.org FULL PAPER
moiety of the methacrylate ligands.[15] The region of the Zr6
Raman spectrum below ν = 400 cm–1 (Figure 1, b) is ofhigh concern for the issues treated in the present work, asit can be considered as the “fingerprint” of oxocluster-likecompounds. The Zr–O–Zr vibration modes are found inthis region and can be related to the two broad peaks cen-tered at ν = 250 and 189 cm–1,[16,17] as found in the vi-brational spectra of crystalline zirconia[22] and amorphouszirconium oxohydroxide.[23–25] The IR shoulder at ν ≈790 cm–1 may be an additional spectral feature of the Zr–O–Zr bonds.[14a,26]
The FTIR and Raman spectra of Zr12 (Figure 2, a and b)maintain the main features observed for the Zr6 oxocluster,although the different acid used for the synthesis (vinyl-acetic acid) results in some key differences in the spectra.Firstly, the absorptions attributed to the stretching modes
Figure 2. (a) FTIR and (b) Raman spectra of Zr12.
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim213
of the methyl groups are replaced by those characteristic ofmethylene groups in the range between ν = 3080 and3022 cm–1, together with those of the olefinic C–H bonds.
The broad absorption between ν = 3000 and 3500 cm–1
can be ascribed to the O–H stretching mode of the sixcrystallization molecules of vinylacetic acid per formulaunit.[1i] The stretching of the C=O group of these moleculesis responsible for the sharp absorption at ν = 1714 cm–1,and the vibrational mode of the carbon–carbon doublebond can be found at ν = 1643 cm–1. The remaining partof the spectrum is characterized by the stretching modes ofthe COO– group and by the bending modes of the methyl-ene and olefinic moieties.
In analogy with the Zr6 oxocluster, we can assume thatthe strong scattering tail observed in the Raman spectrumbelow ν = 300 cm–1 could be related to the Zr–O–Zr vi-

www.eurjic.org FULL PAPER
brational modes. The latter bridging bonds may also beresponsible for the IR band at ν = 720 cm–1.[14a,26]
To test the hydrolytic stability of the title compounds,each zirconium oxocluster was left for a determined time in(1) water at three distinct pH values of 2, 7 and 13 (seeExperimental Section), (2) a hydrogen peroxide aqueoussolution (30% w/w), and (3) atmospheric air for compari-son (Table 1). After the treatments, the compounds werecollected and analyzed by ATR FTIR, Raman, andEXAFS spectroscopy.
The resulting spectra were then compared with those ofthe reference compounds stored under a controlled argonatmosphere (Zr6_pre and Zr12_pre).
1.1 Treatment of Zr6 and Zr12 under Acidic Conditions
The aqueous solution chemistry of ZrIV is quite com-plex.[24,25] The presence of mononuclear Zr4+ cations insolution is improbable, even at extremely low pH values. Asthe ion is relatively large and highly charged, slow equilibriabetween polymeric partially hydrolyzed species occur, andtheir nuclearity depends on the strength of the acid em-ployed.[25] In 2.8 m HCl solution, the most-stable ion ap-pears to be [Zr3(OH)6Cl3]3+, whereas the main species in1–2 m perchloric acid are thought to be [Zr3(OH)4]3+ and[Zr4(OH)8]8+.[25] Although the stable form of the hydrolyzedion is widely reported as the “zirconyl cation” [ZrO]2+,[24–28]
no crystallographic evidence of such a group has beenfound in any of its salts.[24,25] For example, hydrochloridesolutions of zirconyl oxychloride (ZrOCl2·8H2O) containthe tetrameric species [Zr4(OH)8(H2O)16]8+,[24,25] as hasbeen confirmed by X-ray diffraction and absorption mea-surements.[29–34] In our experiments, no solid could be re-trieved from the acidic solution (pH 2), and the Ramanspectra of Zr6_ac and of Zr12_ac were of little use owing tothe strong scattering of the solvent (Figure S2). Conse-quently, the most valuable information came from theEXAFS analyses of the liquids (Table 2).[34] For both oxo-
Table 2. Structural parameters obtained by fitting the experimentalEXAFS data of Zr6_ac and Zr12_ac with theoretical models.
Entry Sample Abs–Bs[a] N(Bs)[b] R(Abs–Bs) [Å][c] σ [Å2][d] Ef [eV](R)[e]
Zr6_pre Zr–O 2 2.11�0.02 0.0041 Zr6_pre Zr–O 6 2.27�0.02 0.013 5.2 (27.8)
Zr6_pre Zr–Zr 4 3.52�0.04 0.012Zr6_ac Zr–O 4.1�0.4 2.14�0.02 0.002
2 Zr6_ac Zr–O 4.1�0.4 2.31�0.02 0.002 3.9 (22.6)Zr6_ac Zr–Zr 3.0�0.6 3.54�0.04 0.012
Zr12_pre Zr–O 2 2.13�0.02 0.0043 Zr12_pre Zr–O 6 2.28�0.02 0.013 5.6 (29.9)
Zr12_pre Zr–Zr 4 3.53�0.04 0.014Zr12_ac Zr–O 4.4�0.4 2.15�0.02 0.006
4 Zr12_ac Zr–O 4.4�0.4 2.31�0.02 0.005 5.3 (23.9)Zr12_ac Zr–Zr 3.0�0.6 3.54�0.04 0.012
[a] Abs = X-ray absorbing atom, Bs = backscatterer. [b] Coordina-tion number. [c] Interatomic distance Abs–Bs. [d] Debye–Waller-like factor. [e] Ef accounts for a shift between the experimental andcalculated EXAFS function, R is the quality of fit (deviation in %).
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim214
clusters, the total Zr–O coordination number remains un-changed, but the distribution of the eight oxygen atomswithin the first two Zr coordination spheres is modified.Unlike the precursor, for which the oxygen distributionaround the Zr absorbing atom results in a 2+6 motif, a 4+4motif was observed, and the distances of the first two shellsof backscatters increased slightly (Table 2, Entry 2).
The coordination number of the Zr atoms in the thirdshell is slightly decreased and their distance remaining un-changed within error. The average number of backscatter-ing oxygen atoms (N = 4 + 4) at 2.14–2.15 and 2.31 Å andzirconium atoms (N = 3) at a distance of 3.54 Å from thecentral absorbing atom seems to be compatible with thecited tetramer [Zr4(OH)8(H2O)16]8+,[28] but the presence ofmore-complex oligomeric species likely involving the meth-acrylate ligands cannot be excluded.
1.2 Treatment of Zr6 in Atmospheric Air and under
Neutral, Basic, and Oxidative Conditions
For Zr6, the ATR FTIR and Raman spectra (Figure 3)show no appreciable degradation of the oxocluster samplesexposed to atmospheric air or to neutral aqueous solution.The Raman spectra of Zr6_pre, Zr6_air, and Zr6_ne in thefingerprint zone below ν = 500 cm–1 are definitely the same(Figure 3, a�, b�, and c�), and the absorption and scatteringbands related to the C–H and C=C bonds and the COO–
group of the methacrylate ligands in Zr6_air and Zr6_nealso remain unchanged compared to those of Zr6_pre (Fig-ure 3, b, b�, c, and c�). The broadening of the two bandsbelonging to the two different asymmetric stretching modesof the COO– group results in a band centered at ν =1549 cm–1 in the ATR FTIR spectra of Zr6_air and Zr6_ne(Figure 3, b and c); this can be explained by a slight modifi-cation of the coordination mode of the carboxylate ligandsto the central inorganic cluster core.
An effect of the treatments is the weakening of the bandsof the free acid molecules randomly dispersed into the voidsof the lattice at ν = 1701 and 795 cm–1,[1g,1h] which are pres-ent in both the ATR FTIR and Raman spectra.
The solvating power of a protic and polar solvent suchas water (or atmospheric moisture) could have promotedthe dissolution of these adsorbed molecules without in-fluencing the main oxocluster structure.
A very different picture was obtained after treatment inbasic conditions. Indeed, the ATR FTIR and Raman spec-tra of Zr6_ba are completely different from those of Zr6_pre(Figure 3, d and d�). In this case, the degree of hydrolysis ismarked, as can be seen by the broad absorptions centeredat ν = 3300 and 1600 cm–1 in the ATR FTIR spectrum (Fig-ure 3, d). According to a previous report, the former can beassigned to the O–H asymmetric and symmetric stretchingmodes of lattice water molecules, and the latter can be as-cribed to the corresponding H–O–H bending mode.[35] TheO–H bending mode of the hydroxy groups bonded to theZr atoms can be clearly seen at ν = 1060 cm–1 in the Ramanspectrum of Zr6_ba (Figure 3, d�), in agreement with theliterature value.[35] The two broad peaks at ν = 414 and523 cm–1 are probably ascribed to the Zr–O vibrations and
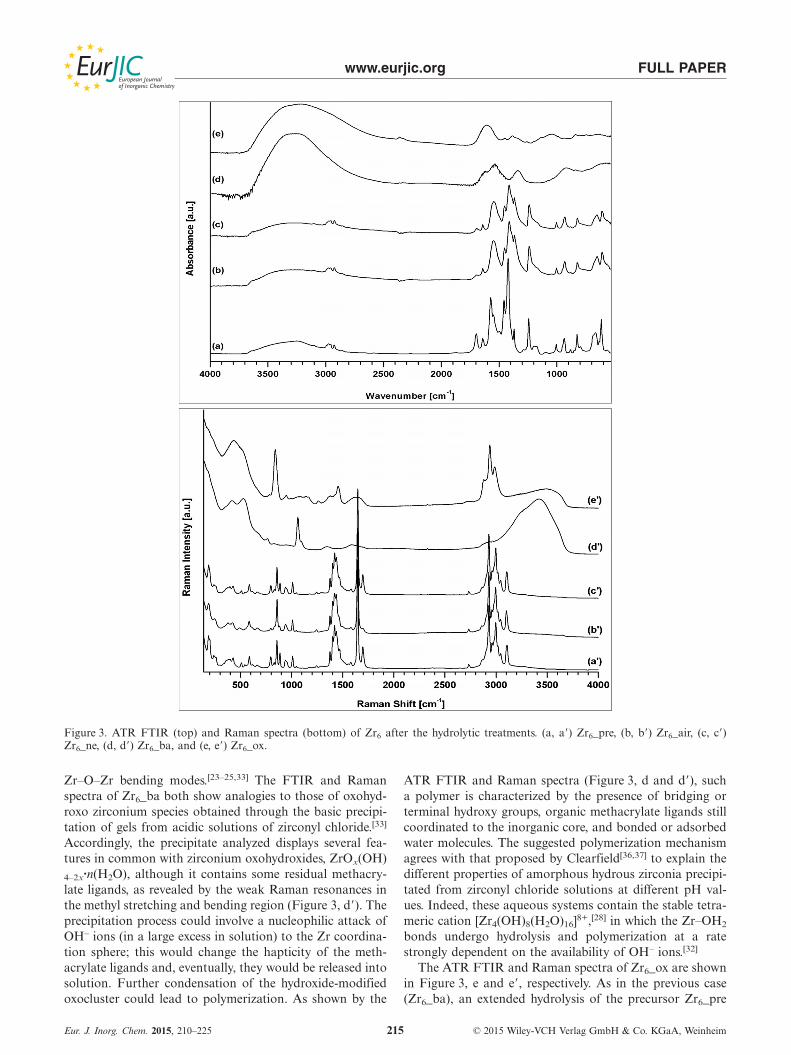
www.eurjic.org FULL PAPER
Figure 3. ATR FTIR (top) and Raman spectra (bottom) of Zr6 after the hydrolytic treatments. (a, a�) Zr6_pre, (b, b�) Zr6_air, (c, c�)Zr6_ne, (d, d�) Zr6_ba, and (e, e�) Zr6_ox.
Zr–O–Zr bending modes.[23–25,33] The FTIR and Ramanspectra of Zr6_ba both show analogies to those of oxohyd-roxo zirconium species obtained through the basic precipi-tation of gels from acidic solutions of zirconyl chloride.[33]
Accordingly, the precipitate analyzed displays several fea-tures in common with zirconium oxohydroxides, ZrOx(OH)4–2x·n(H2O), although it contains some residual methacry-late ligands, as revealed by the weak Raman resonances inthe methyl stretching and bending region (Figure 3, d�). Theprecipitation process could involve a nucleophilic attack ofOH– ions (in a large excess in solution) to the Zr coordina-tion sphere; this would change the hapticity of the meth-acrylate ligands and, eventually, they would be released intosolution. Further condensation of the hydroxide-modifiedoxocluster could lead to polymerization. As shown by the
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim215
ATR FTIR and Raman spectra (Figure 3, d and d�), sucha polymer is characterized by the presence of bridging orterminal hydroxy groups, organic methacrylate ligands stillcoordinated to the inorganic core, and bonded or adsorbedwater molecules. The suggested polymerization mechanismagrees with that proposed by Clearfield[36,37] to explain thedifferent properties of amorphous hydrous zirconia precipi-tated from zirconyl chloride solutions at different pH val-ues. Indeed, these aqueous systems contain the stable tetra-meric cation [Zr4(OH)8(H2O)16]8+,[28] in which the Zr–OH2
bonds undergo hydrolysis and polymerization at a ratestrongly dependent on the availability of OH– ions.[32]
The ATR FTIR and Raman spectra of Zr6_ox are shownin Figure 3, e and e�, respectively. As in the previous case(Zr6_ba), an extended hydrolysis of the precursor Zr6_pre

www.eurjic.org FULL PAPER
(Figure 3, a and a�) is evident, as shown by the strong andbroad absorption bands centered at ν = 3300 and 1600 cm–1
in the ATR FTIR spectrum (Figure 3, e).[35] The presenceof water molecules in the precipitate is in agreement withthe peroxidic hydrolysis of ZrIV salts with an excess of H2O2
reported by Connor et al.[13b] More detailed informationcan be gained from the Raman spectrum, in which the vi-brations of the residual C–H groups of the methacrylic acidcan be observed between ν = 2879 and 2986 cm–1 and at ν≈ 1400 cm–1. Their presence in the precipitate can be ex-plained on the basis of the hydrolysis mechanism of theoxocluster proposed before, owing to the acidity of H2O2
solutions (the pH of a 30 % aqueous solution of H2O2 is aslow as 2). A quite intense band can be observed at ν =839 cm–1 in the Raman spectrum of Zr6_ox, followed by abroader one at ν = 436 cm–1 with a shoulder at 517 cm–1
(Figure 3, e�). According to previous work on bulk zirconiatreated with hydrogen peroxide solution, the former is as-signed to peroxo ν(O–O) vibrations, and the second is at-tributed to the superimposed Zr(O2) asymmetric and sym-metric stretching modes,[35,38] although an overlap with thescattering bands related to the Zr–O–Zr vibrational modeshas to be considered. Even if the attribution of the hapticityof the peroxo ligand in metal complexes (bidentate, bridg-ing rather than terminal) through spectroscopic methods isquite difficult,[13b,13c] a comparison with the Raman andFTIR characterization of previously reported Zr complexessuch as K2[Zr(O2)(ox)2]·2H2O (ox = oxalate), K2[Zr(O2)2-(cit)]·2H2O (cit = citrate),[13j] and (NH4)3Zr(O2)F5
[13h] (allbearing η2-peroxo ligands) strengthen the hypothesis thatthe real coordination mode of the O–O ligand to zirconiumis bidentate, especially in light of the position of the O–Ostretching band at ν = 839 cm–1 in the Raman spectrum ofZr6_ox (see Table 3).
Table 3. Comparison of the vibrational frequencies of the O–O andZr(O2) bonds in Zr6_ox, Zr12_ox, and some previously reportedcompounds.
ν(O–O) [cm–1] ν[Zr(O2)] [cm–1] Reference
Zr6_ox 839 517, 436 this workZr12_ox 844 517, 446 this workK2[Zr(O2)(ox)2]·2H2O 848 565 [13j]
K2[Zr(O2)2(cit)]·H2O 853 550 [13j]
Zr(O2)(H2EDTA) 840 650, 600 [13k,35]
(NH4)2[ZrO(O2)F2] 850 640, 585 [13l,35]
(NH4)3[Zr(O2)F5] 837 550, 471 [13h,35]
H2O2 877 – [13c]
As this value is significantly different from that of freehydrogen peroxide (the last entry in Table 3), the presenceof hydrogen peroxide molecules of crystallization or ad-sorbed in the sample can be excluded. The existence of co-ordinated peroxo groups in the isolated compound has beenunequivocally verified by a qualitative iodometric assay[27]
on Zr6_ox previously washed with deionized water.The results of the EXAFS analysis (Table 4) confirm the
findings of the vibrational spectroscopy characterizations.
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim216
Table 4. EXAFS data of Zr6 samples after hydrolytic treatment (seeTable 2 for acronyms used).
Entry Sample Abs–Bs N(Bs) R(Abs–Bs) σ Ef [eV][Å] [Å2] (R)
1 Zr6_pre Zr–O 2 2.11�0.02 0.004Zr6_pre Zr–O 6 2.27�0.02 0.013 5.2 (27.8)Zr6_pre Zr–Zr 4 3.52�0.04 0.012
2 Zr6_air Zr–O 2. 5�0.3 2.11�0.02 0.004Zr6_air Zr–O 6.5�0.7 2.27�0.02 0.013 4.8 (26.1)Zr6_air Zr–Zr 3.5�0.7 3.53�0.04 0.012
3 Zr6_ne Zr–O 2.6�0.3 2.12�0.02 0.004Zr6_ne Zr–O 6.5�0.7 2.27�0.02 0.013 4.9 (25.8)Zr6_ne Zr–Zr 3.2�0.6 3.54�0.04 0.012
4 Zr6_ba Zr–O 2.8�0.3 2.10�0.02 0.004Zr6_ba Zr–O 5.2�0.5 2.23�0.02 0.013 5.9 (30.6)Zr6_ba Zr–Zr 0.8�0.1 3.45�0.03 0.012
5 Zr6_ox Zr–O 8.7�0.9 2.18�0.02 0.016 7.8 (22.9)Zr6_ox Zr–Zr 0.9�0.1 3.36�0.03 0.016
The treatments in neutral water and in atmospheric air(Zr6_ne and Zr6_air) yield identical structural parametersto those of Zr6_pre within error.
On the contrary, basic conditions induce significantchanges; the most striking changes are for the higher Zr–Oand Zr–Zr shells; the Zr–Zr coordination number in par-ticular suggests that the initial Zr core is reduced to a di-meric motif,[17] and the distance parameter changes from3.52 to 3.45 Å.
For Zr6_ox, a Zr–O distance of 2.18 Å is also characteris-tic of Zr–peroxo groups.[13p,14] However, as only one averageZr–O shell could be fitted, no further information aboutpossible peroxo groups can be extracted from the EXAFSmeasurements.
Additionally, only one Zr–Zr shell is found at 3.36 Å.This picture corroborates the presence of peroxo ligands co-ordinated to the zirconium atoms in Zr6_ox, as alreadyindicated by the analysis of its Raman spectrum.
Finally, a significant structural alteration is found inZr6_ox, as only one Zr–O shell with 8.7� 0.9 atoms can befound. The corresponding distance of 2.18 Å lies betweenthe original Zr–O distances of the Zr6_pre sample.
1.3 Treatment of Zr12 in Atmospheric Air and under
Neutral, Basic, and Oxidative Conditions
The ATR FTIR and Raman spectra of the Zr12 samplestreated in the same hydrolytic environments as Zr6 are dis-played in Figure 4. Some analogies in the behavior of thetwo oxoclusters can be pointed out: Zr12 is also quite resist-ant towards hydrolysis from the moisture in atmosphericair. In the Raman spectrum of Zr12_air, no substantialmodifications can be observed with respect to that ofZr12_pre (Figure 4, b�).
As for Zr6_air, the broadening of the two bands of thetwo different asymmetric stretching modes of the COO–
group results in the appearance of one broad band centeredat ν = 1548 cm–1 in the ATR FTIR spectrum of Zr12_air(Figure 4, b); this can be explained by a slight modification

www.eurjic.org FULL PAPER
Figure 4. ATR FTIR (top) and Raman spectra (bottom) of Zr12 after the hydrolytic treatments. (a, a�) Zr12_pre, (b, b�) Zr12_air, (c, c�)Zr12_ne, (d, d�) Zr12_ba, and (e, e�) Zr12_ox.
of the coordination mode of the vinylacetate ligands to eachof the two inorganic subunits from which Zr12 is built up.[1i]
As was already observed for Zr6, the treatment with aneutral aqueous solution seems to cause only some slightmodifications to the pristine oxocluster structure, as is evi-dent from the fingerprint region of the Zr12_ne spectrum inFigure 4 (c�). The feature common to the spectra ofZr12_air and Zr12_ne is the decreased intensity of the bandat ν = 1714 cm–1 (Figure 4, b� and c�) for crystallizationmolecules of vinylacetic acid, which could be replaced dur-ing the transition of the compound from a crystalline to anamorphous form.
In analogy with results for Zr6_ba, the treatment of Zr12
with a basic solution radically changes the structure of the
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim217
oxocluster (Figure 4, d and d�). The signs of an advancedhydrolysis are evidenced by the broad absorptions centeredat ν = 3300 and 1600 cm–1 in the ATR FTIR spectrum ofZr12_ba (Figure 4, d). However, the presence of weak peaksat ν ≈ 1400 and 2900 cm–1 in the Raman spectrum (Fig-ure 4d�) suggests that traces of vinylacetate ligands are stillpresent in the precipitate.
Moreover, the striking analogies between both the ATRFTIR and Raman spectra of Zr12_ba and Zr6_ba (Figure 5)makes us confident that the precipitate obtained in the twocases is the same, at least as far as the inorganic moiety isconcerned. The Raman spectrum of Zr12_ba presents thesame bands at ν = 1060, 414, and 523 cm–1 that charac-terized Zr6_ba (Figure 5, a� and b�).

www.eurjic.org FULL PAPER
Figure 5. ATR FTIR (top) and Raman spectra (bottom) of Zr6_ba (a and a�) and Zr12_ba (b and b�). The Raman spectra are normalizedwith respect to the peak at ν = 1060 cm–1.
As already discussed, these bands can be assigned to O–H bending,[35] Zr–O vibrations, and Zr–O–Zr bendingmodes, respectively;[23,33] therefore, it can be supposed thatthe precipitate in this case is also built up of a zirconiumoxohydroxide species of the general formula ZrOx(OH)4–2x·n(H2O) with vinylacetate residues.
The ATR FTIR and Raman spectra of Zr12_ox areshown in Figure 4, e and e�, respectively. Their featuresmostly resemble those of Zr6_ox, as can be seen from thecomparison in Figure 6. On the basis of the same consider-ations made for Zr6_ox, the peak in the Raman spectrumof Zr12_ox at ν = 844 cm–1 (Figure 4, e�) can be assigned tothe O–O stretching mode of a bidentate peroxide ligand
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim218
(Table 3), and the band at ν = 446 and the shoulder at517 cm–1 can be ascribed to superimposed Zr(O2) asym-metric and symmetric stretching modes,[35,38] which areprobably overlapped with the scattering bands of the Zr–O–Zr vibrational modes.[23]
As for Zr6_ox, the presence of peroxo groups in the com-pound has been further confirmed by a qualitative iodo-metric assay.[27] Nonetheless, as was pointed out for Zr12_baand in analogy with the Zr6_ox case, the presence of anorganic moiety in Zr12_ox is evident. The broad peaks cen-tered at ν = 1427 and 2937 cm–1 resemble those of the C–H bending and stretching modes of the vinylacetate ligandsin Zr12_pre (Figure 4, a�). Hence, a partial hydrolysis of the

www.eurjic.org FULL PAPER
Figure 6. ATR FTIR (top) and Raman spectra (bottom) of Zr6_ox (a and a�) and Zr12_ox (b and b�). The Raman spectra are normalizedwith respect to the peaks at ν = 839 and 844 cm–1.
oxocluster can also be inferred in this case, together withthe formation of peroxo species on the surface of the oxidenetwork.
The conclusions drawn from the ATR FTIR and Ramanspectroscopy characterizations seem to be confirmed by theEXAFS data (Table 5).
It may be pointed out that the treatments with atmo-spheric air and water at pH 7 (Zr12_air and Zr12_ne) do nothave any major impact on the formed structures, as was thecase for Zr6_air and Zr6_ne, because the number of back-scatterers and the distance remain unchanged. However, asEXAFS probes only the local environment of the absorbingatoms and does not provide information on the distancesbetween the zirconium atoms located in two different sub-
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim219
units of Zr12,[1i] structural rearrangements involving the de-composition of the Zr12 cluster into separate Zr6 units can-not be excluded.[39,40] Nonetheless, it can be inferred fromthe vibrational spectra of Zr12_air (Figure 4, b and b�) thatthe structure of Zr12 is not modified upon exposure to at-mospheric moisture, although the same cannot be assumedfrom the fingerprint region in the Raman spectrum ofZr12_ne (Figure 4, c�). The same trend observed throughoutthe series of the Zr6 treatments is repeated for Zr12_ba andZr12_ox. The changes to their structures are highlightedmainly by the increment of the oxygen atoms in the Zr–Oshell and the decrement of the Zr atoms in the Zr–Zr neigh-bors, as well as their distance parameters. Once more, thestructural parameters (coordination numbers and shell ra-
www.eurjic.org FULL PAPER
Table 5. EXAFS data of Zr12 samples after hydrolytic treatments(see Table 2 for acronyms used).
Entry Sample Abs–Bs N(Bs) R(Abs–Bs) σ Ef [eV][Å] [Å2] (R)
1 Zr12_pre Zr–O 2 2.13�0.02 0.004Zr12_pre Zr–O 6 2.28�0.02 0.013 5.6 (29.9)Zr12_pre Zr–Zr 4 3.53�0.04 0.014
2 Zr12_air Zr–O 2.0�0.2 2.13�0.02 0.004Zr12_air Zr–O 6.0�0.6 2.27�0.02 0.013 5.4 (29.9)Zr12_air Zr–Zr 4.0�0.8 3.54�0.04 0.014
3 Zr12_ne Zr–O 2.4�0.3 2.13�0.02 0.004Zr12_ne Zr–O 6.7�0.7 2.28�0.02 0.013 5.6 (27.3)Zr12_ne Zr–Zr 3.8�0.7 3.54�0.04 0.012
4 Zr12_ba Zr–O 3.1�0.3 2.11�0.02 0.004Zr12_ba Zr–O 5.8�0.6 2.25�0.02 0.013 6.2 (29.0)Zr12_ba Zr–Zr 1.0�0.2 3.46�0.03 0.014
5 Zr12_ox Zr–O 8.7�0.9 2.20�0.02 0.018 8.3 (24.2)Zr12_ox Zr–Zr 0.4�0.1 3.36�0.03 0.012
dius) of Zr12_ox could be compatible with the coordinationof peroxo ligands to zirconium atoms in the final com-pound, also in view of the results of the Raman spec-troscopy characterization.
2. Catalytic Tests
Several mechanistic studies of oxygen transfer reactionswith mononuclear d0 peroxometal complexes of the generalformula [LnM(O2)m] (M = TiIV, VV, MoVI, WVI) have beenreported.[11a,12a] Accordingly, we have addressed the oxid-ation of aromatic sulfides by H2O2 in the presence of a cata-lytic amount of oxocluster. The oxidation of sulfur-contain-ing compounds is an important synthetic process[41] for theproduction of sulfoxides and sulfones and is also widely ex-plored as a method for the oxidative desulfurization offuels.[42] Under such conditions, methyl p-tolyl sulfide, cho-sen as a model substrate, is oxidized to its correspondingsulfoxide and then to the sulfone by consecutive bimolecu-lar, mono-oxygenation steps (Scheme 1).[43]
The reactions were performed in acetonitrile at differenttemperatures (T = 30 and 50 °C) with Zr6 and Zr12 as cata-lysts and an excess of H2O2 (2.5 equiv.) to promote theoxidation to sulfone. With k1 and k2 as the second-orderkinetic constants of the first (from sulfide to sulfoxide, k1)and second (from sulfoxide to sulfone, k2) oxidation steps,the reactions have been compared in terms of sulfide con-version, turnover number (TON, calculated as mol of oxid-ized substrate per mol of catalyst), turnover frequency
Scheme 1. Oxidation of methyl p-tolyl sulfide to methyl p-tolyl sulfoxide and methyl p-tolyl sulfone by H2O2.
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim220
(TOF, calculated as mol of oxidized substrate per mol ofcatalyst per time unit), and selectivity ratio S = k1/k2. Theresults obtained with both oxoclusters after 3 h (when thesubstrate is completely consumed in most cases) are col-lected in Table 6.
Table 6. Oxidation of methyl p-tolyl sulfide with metal oxoclus-ters.[a]
Entry Oxocluster Catalyst Conversion TON[c] TOF[d] SO/ S[mol-%] [%][b,c] [min–1] SO2
[c] (k1/k2)
1[e] Zr12 0.08 90 1850 27 37:63 1.42[e] Zr6 0.08 90 1225 18 91:9 163[f] Zr12 0.08 �99 2350 81 12:88 1.14[f] Zr6 0.08 96 1700 47 57:43 8.75[f] Zr12 0.16 �99 1225 67 4:96 0.66[f] Zr6 0.16 97 850 25 61:39 2.27[f] Zr12 0.24 �99 817 74 4:96 0.68[f] Zr6 0.24 99 708 45 28:72 2.09[f] Zr12 0.32 �99 613 81 3:97 0.610[f] Zr6 0.32 99 513 31 35:65 1.8
[a] Reaction conditions: the specified amount of oxocluster wasadded to acetonitrile (1.2 mL) containing methyl p-tolyl sulfide(1 mmol) and H2O2 (2.5 mmol, added as 30% aqueous solution).[b] Methyl p-tolyl sulfide conversion. A blank reaction at 50 °Cwithout oxoclusters led to sulfide consumption in 24 h with no sulf-one formation. [c] Values obtained at t = 3 h. [d] Number of cata-lytic cycles per minute, measured at ca. 20% substrate conversion.[e] T = 30 °C. [f] T = 50 °C.
At 30 °C (Table 6, Entries 1 and 2), the two oxoclustersdisplay a different reactivity, and Zr12 is more efficient thanZr6. The statistical factor, that is, the increased number ofmetal centers in Zr12, is not responsible for this higher ac-tivity, as the turnover frequency per metal ion (TOF/Zr) is2.3 and 3 for Zr12 and Zr6, respectively (see also Table 6,Entries 3 and 6 with the same total concentration of Zrions). Thus, the hydrolysis of the dimeric structure to twohexanuclear clusters with analogous reactivity can be ex-cluded.
The different reactivity of the two oxoclusters is alsohighlighted by the different product distribution and selec-tivity ratio S, which is tenfold lower for Zr12. Indeed, al-though k1 for the dimeric oxocluster is double the corre-sponding value for Zr6, k2 is more than twenty times bigger(see Supporting Information, Table S1).
As demonstrated by the higher reaction yield and theincreased sulfone production, the reactions are faster at50 °C (Table 6, Entries 3–4 and Table S1), and a TON of2350 was obtained after 3 h with Zr12. The kinetic behaviorof the reactions with the two catalysts under such condi-
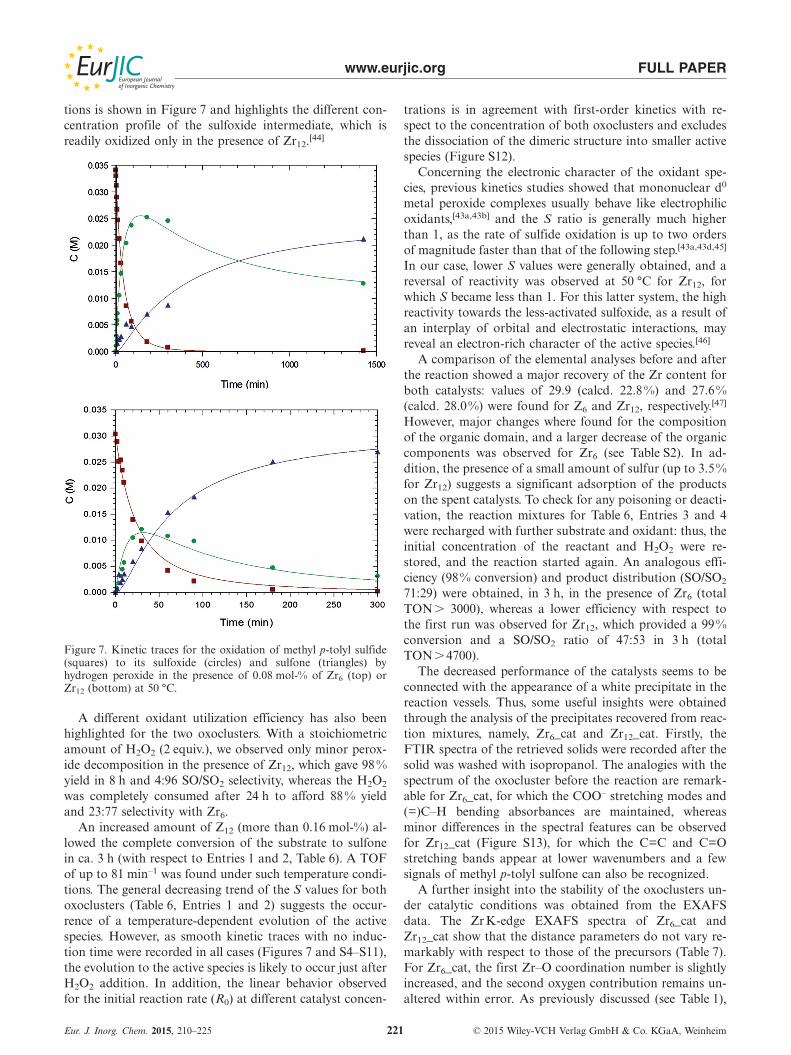
www.eurjic.org FULL PAPER
tions is shown in Figure 7 and highlights the different con-centration profile of the sulfoxide intermediate, which isreadily oxidized only in the presence of Zr12.[44]
Figure 7. Kinetic traces for the oxidation of methyl p-tolyl sulfide(squares) to its sulfoxide (circles) and sulfone (triangles) byhydrogen peroxide in the presence of 0.08 mol-% of Zr6 (top) orZr12 (bottom) at 50 °C.
A different oxidant utilization efficiency has also beenhighlighted for the two oxoclusters. With a stoichiometricamount of H2O2 (2 equiv.), we observed only minor perox-ide decomposition in the presence of Zr12, which gave 98 %yield in 8 h and 4:96 SO/SO2 selectivity, whereas the H2O2
was completely consumed after 24 h to afford 88% yieldand 23:77 selectivity with Zr6.
An increased amount of Z12 (more than 0.16 mol-%) al-lowed the complete conversion of the substrate to sulfonein ca. 3 h (with respect to Entries 1 and 2, Table 6). A TOFof up to 81 min–1 was found under such temperature condi-tions. The general decreasing trend of the S values for bothoxoclusters (Table 6, Entries 1 and 2) suggests the occur-rence of a temperature-dependent evolution of the activespecies. However, as smooth kinetic traces with no induc-tion time were recorded in all cases (Figures 7 and S4–S11),the evolution to the active species is likely to occur just afterH2O2 addition. In addition, the linear behavior observedfor the initial reaction rate (R0) at different catalyst concen-
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim221
trations is in agreement with first-order kinetics with re-spect to the concentration of both oxoclusters and excludesthe dissociation of the dimeric structure into smaller activespecies (Figure S12).
Concerning the electronic character of the oxidant spe-cies, previous kinetics studies showed that mononuclear d0
metal peroxide complexes usually behave like electrophilicoxidants,[43a,43b] and the S ratio is generally much higherthan 1, as the rate of sulfide oxidation is up to two ordersof magnitude faster than that of the following step.[43a,43d,45]
In our case, lower S values were generally obtained, and areversal of reactivity was observed at 50 °C for Zr12, forwhich S became less than 1. For this latter system, the highreactivity towards the less-activated sulfoxide, as a result ofan interplay of orbital and electrostatic interactions, mayreveal an electron-rich character of the active species.[46]
A comparison of the elemental analyses before and afterthe reaction showed a major recovery of the Zr content forboth catalysts: values of 29.9 (calcd. 22.8 %) and 27.6%(calcd. 28.0%) were found for Z6 and Zr12, respectively.[47]
However, major changes where found for the compositionof the organic domain, and a larger decrease of the organiccomponents was observed for Zr6 (see Table S2). In ad-dition, the presence of a small amount of sulfur (up to 3.5%for Zr12) suggests a significant adsorption of the productson the spent catalysts. To check for any poisoning or deacti-vation, the reaction mixtures for Table 6, Entries 3 and 4were recharged with further substrate and oxidant: thus, theinitial concentration of the reactant and H2O2 were re-stored, and the reaction started again. An analogous effi-ciency (98% conversion) and product distribution (SO/SO2
71:29) were obtained, in 3 h, in the presence of Zr6 (totalTON� 3000), whereas a lower efficiency with respect tothe first run was observed for Zr12, which provided a 99%conversion and a SO/SO2 ratio of 47:53 in 3 h (totalTON�4700).
The decreased performance of the catalysts seems to beconnected with the appearance of a white precipitate in thereaction vessels. Thus, some useful insights were obtainedthrough the analysis of the precipitates recovered from reac-tion mixtures, namely, Zr6_cat and Zr12_cat. Firstly, theFTIR spectra of the retrieved solids were recorded after thesolid was washed with isopropanol. The analogies with thespectrum of the oxocluster before the reaction are remark-able for Zr6_cat, for which the COO– stretching modes and(=)C–H bending absorbances are maintained, whereasminor differences in the spectral features can be observedfor Zr12_cat (Figure S13), for which the C=C and C=Ostretching bands appear at lower wavenumbers and a fewsignals of methyl p-tolyl sulfone can also be recognized.
A further insight into the stability of the oxoclusters un-der catalytic conditions was obtained from the EXAFSdata. The Zr K-edge EXAFS spectra of Zr6_cat andZr12_cat show that the distance parameters do not vary re-markably with respect to those of the precursors (Table 7).For Zr6_cat, the first Zr–O coordination number is slightlyincreased, and the second oxygen contribution remains un-altered within error. As previously discussed (see Table 1),

www.eurjic.org FULL PAPER
spectral changes are likely to result from modifications ofthe coordination modes or displacement of the carboxylateligands.
Table 7. EXAFS data of Zr6 and Zr12 samples after the catalyticoxidation of methyl p-tolyl sulfide (see Table 2 for acronyms used).
Sample Abs–Bs N(Bs) R(Abs–Bs) σ Ef [eV][Å] [Å2] (R)
Zr6_pre Zr–O 2 2.11�0.02 0.004Zr–O 6 2.27�0.02 0.013 5.2 (27.8)Zr–Zr 4 3.52�0.04 0.012
Zr6_cat Zr–O 2.9�0.3 2.13�0.02 0.004Zr–O 6.3�0.6 2.27�0.02 0.013 4.9 (25.8)Zr–Zr 2.4�0.5 3.55�0.04 0.012
Zr12_pre Zr–O 2 2.13�0.02 0.004Zr–O 6 2.28�0.02 0.013 5.6 (29.9)Zr–Zr 4 3.53�0.04 0.014
Zr12_cat Zr–O 2.1�0.2 2.13�0.02 0.004Zr–O 4.5�0.5 2.27�0.02 0.013 4.5 (26.4)Zr–Zr 2.0�0.4 3.55�0.04 0.014
However, the reduction of the Zr–Zr coordinationnumber to almost 50% of the precursor value indicates adistortion in the Zr–Zr shell.
Compared to the previous sample, the structural changesin Zr12_cat are more significant and affect both the secondZr–O shell and the Zr–Zr shell, for which a reduction ofthe coordination numbers is found.
This trend resembles those observed for Zr6_ba, Zr12_ba,Zr6_ox, and Zr12_ox, for which the modification of the par-ent structure of the oxoclusters has been explained with anextended hydrolysis of the precursor compounds. However,the same cannot be said about Zr6_cat and Zr12_cat, astheir infrared spectra still present many features of the pre-cursors and the oxidation of methyl p-tolyl sulfide was per-formed in an organic solvent with a low amount of water.Nonetheless, the precipitation of the analyzed samples fromthe reaction mixture should be explained by the rearrange-ment of the parent oxoclusters into oligomeric species witha lower solubility and, for Zr12, a lower catalytic perform-ance than the precursor.
To further assess the changes in the structures of theoxoclusters under oxidative conditions, the samples beforeand after the catalytic process were analyzed by SEM toprovide information on their morphologies. The collectedmicrographs are shown in Figure S14. From a comparisonof the images, it can be appreciated that the morphology ofZr6 is not dramatically affected by the catalysis process, butthe morphology of Zr12 is slightly modified after catalysis;the facets of the material are less sharp and more roundedas the catalysis process could result in a slight “etching” ofthe material. These data confirm the enhanced stability ofZr6 with respect to the dimeric Zr12.
Conclusions
The hydrolytic stability and the catalytic properties ofZr6 and Zr12 oxoclusters were thoroughly investigated by amultiple-technique approach involving FTIR, Raman, and
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim222
EXAFS spectroscopy investigations. The oxoclusters arestable in air and neutral aqueous solution, whereas theyevolve to lower nuclearity complexes or to polymeric struc-tures under acidic and alkaline conditions, respectively. Asindicated by the Raman and EXAFS spectroscopy results,the zirconium oxoclusters can coordinate hydrogen perox-ide. The applicability of the resulting peroxo complexes asoxidation catalysts was demonstrated by their ability to pro-mote oxygen transfer to a model aromatic sulfide in aceto-nitrile solution. Indeed, methyl p-tolyl sulfide was smoothlyoxidized in 100–500 min to yield a mixture of the corre-sponding sulfoxide and sulfone. Under multi-turnover con-ditions, the two oxoclusters exhibit different stabilities andelectronic characters. Zr6 is generally less reactive (58 %yield of oxygenated products in 3 h with 0.08 mol-% cata-lyst, whereas Zr12 afforded 90% yield) with decreased ac-tivity towards sulfoxide oxidation and lower H2O2 utiliza-tion efficiency. However, the monomeric oxocluster seemsto be more stable under operative catalysis conditions, asthe recovered catalyst maintains the spectral features of thestarting material, and its reactivity can be restored upon theaddition of further quantities of reagent. Despite the re-duced hydrolytic stability of Zr12, this dimeric oxoclusterdisplays a higher reactivity (TOF� 80 min–1), coupled witha higher selectivity toward the oxidation of the sulfoxide(up to 97% sulfone in 3 h).
The overall increase of stability in aqueous acetonitrilesuggests that the activity and stability of the oxoclusterscould be further improved by using water-free peroxo-basedoxidants or biphasic reaction mixtures. The possibility toembed the oxoclusters within hydrophobic matrixes willalso be explored. Owing to this interesting reactivity, appli-cations in the field of oxidative desulfurization are underinvestigation in our laboratories.
Experimental Section[Zr6(OH)4O4{O(O)CC(CH3)=CH2}12] (Zr6): Zr6 was synthesizedaccording to standard literature procedures.[1g,1h] FTIR (KBr): ν =3253 (m br), 3099 [w, ν(C–H)], 3018 (w), 2991 (sh), 2975 [m,νas(CH3)], 2960 [m, νas(CH3)], 2927 [m νs(CH3)], 2858 (w), 1701[m, ν(O=COH)], 1645 [m, ν(C=C)], 1574 [s, νas(COO–)], 1549 [s,νas(COO–)], 1498 (w), 1458 [s, νs(COO–)], 1423 [s, δ(CH3)], 1400(sh), 1371 [m, δ(CH3)], 1288 (w), 1246 [s, δ(CH3)], 1201 (m), 1174(m), 1095 (w), 1007 [m δ(CH3)], 937 [m, δoop(C=CH2)], 883 (w),850 (w), 825 [m, δ(CH3)], 796 (w), 665 (m br), 636 (m), 617 [s,δ(OCO)], 565 (w), 503 (m), 467 (m), 424 (m) cm–1. Raman: ν =3267 [w, ν(O–H)], 3104 [m, ν(C–H)], 3041(w), 3016 (m), 2995 [s,νas(CH3)], 2978 (sh), 2958 (m), 2925 [s, νs(CH3)], 2874 (sh), 2728(w), 1695 [m, ν(O=COH)], 1645 [s, ν(C=C)], 1579 (w), 1494 (sh),1460 (sh), 1434 (s), 1420 (s), 1399 (s), 1373 (m), 1300 (w), 1242 (w),1116 (w), 1044 (w), 1007 (m), 938 (m), 884 (m), 853 (s), 826 (w),795 (m), 659 (w), 616 (w), 585 (m), 542 (w), 508 (w), 426 (w), 395(w), 380 (w), 361 (w), 250 (w, Zr–O–Zr), 236 (w), 189 (m, Zr–O–Zr) cm–1.
[Zr6O4(OH)4{O(O)CCH2CH=CH2}12]2·6CH2=CHCH2C(O)OH(Zr12):[1i] In a previously dried Schlenk tube, vinylacetic acid(3.71 mL, 40.94 mmol) was added dropwise under an Ar atmo-sphere to a 70% (w/w) solution of Zr(nPrO)4 (2.53 g, 5.41 mmol)

www.eurjic.org FULL PAPER
in propanol to achieve a 1:7.4 zirconium/acid molar ratio. Aftermixing at room temperature, the solution was allowed to stand atroom temperature for 3 d under an inert atmosphere; this resultedin the formation of cubic colorless crystals, which were decantedand dried under low vacuum for 2 h (yield: 96%). FTIR (KBr): ν= 3398 [m, ν(O–H)], 3255 [m br, ν(O–H)], 3080 [m, ν(C–H)], 3022[w, ν(C–H)], 2983 [w, νas(CH2)], 2964 [w, νs(CH2)], 2908 (sh), 1714[s, ν(O=COH)], 1643 [m, ν(C=C)], 1605 [s, νas(COO–)], 1554 [s,νas(COO–)], 1489 (w), 1439 [s, νs(COO–)], 1419 [s, δ(CH2)], 1396 [s,δ(CH2)], 1313 (w), 1296 (w), 1265 [s, δ(CH2)], 1200 (m), 1099 [brm, δ(CH2)], 1016 (w), 995 (m), 914 [s, δoop(C=CH2)], 812 (m br),729 (m), 648 (s), 507 (m), 469 (m), 418 (m) cm–1. Raman: ν = 3395[w, ν(O–H)], 3271 [w, ν(O–H)], 3084 [m, ν(C–H)], 3021 [s,νas(CH2)], 2984 [s, νas(CH2)], 2912 [s br, νs(CH2)], 1716 [w,ν(O=COH)], 1642 [s, ν(C=C)], 1602 (w), 1584 (w), 1558 (w), 1512(w), 1456 (w), 1420 (m), 1399 (m), 1296 (s), 1273 (w), 1291 (w),1125 (w), 996 (w), 975 (m), 936 (sh), 913 (m), 890 (m), 765 (w br),728 (w), 704 (w), 626 (w), 589 (w), 517 (m), 474 (w), 457 (w), 414(w), 398 (w), 376 (w), 351 (w), 322 (w), 298 (w), 242 (m), 226 (m),203 (m), 169 (m) cm–1.
Stability Tests: All aqueous solutions were prepared with MilliQwater at pH 7. The acidic solutions (pH 2) were prepared from a37% (w/w) solution of HCl, and the basic solutions (pH 13) wereprepared with solid NaOH. Hydrolysis tests in an oxidative envi-ronment were performed with a 30% (w/w) aqueous solution ofH2O2. The samples were prepared by grinding Zr6 or Zr12 crystals.The cluster compounds (100 mg of each) were added to the respec-tive solutions (5 mL). All of the treatments lasted 24 d, after whichthe precipitates were collected by filtration through a G4 Goochfilter, carefully washed with deionized water, and finally dried. Thehydrolysis effect of the atmospheric moisture was tested by expos-ing the ground oxoclusters (100 mg) to atmospheric air for 24 d.
Catalytic Tests: The oxidation reactions were run at a fixed tem-perature (30 and 50 °C) in a closed vial. In a typical oxidationreaction, Zr6 or Zr12 (molar percent with respect to sulfide 0.08–0.32%) was added to acetonitrile (1.2 mL). Then, methyl p-tolylsulfide (1 mmol) and H2O2 (2.5 mmol, from a 30% aqueous solu-tion) were added under magnetic stirring. Samples (50 μL) werewithdrawn at fixed time intervals, diluted in a 5 mm solution(500 μL) of n-undecane (standard) in acetonitrile, and treated withtriphenylphosphine [as a solid or supported on poly(styrene/divin-ylbenzene) (PS/DVB) resin] as an oxidant quencher. The occur-rence of unproductive decomposition of the oxidant was evaluatedin a reaction with 2 equiv. of H2O2 (T = 50 °C). A blank reactionwas performed in the absence of Zr oxoclusters at 50 °C, and itshowed a quantitative substrate conversion after 24 h with negligi-ble sulfone production.
In the “recharge” reaction, methyl p-tolyl sulfide (1 mmol) wasadded, followed by H2O2 (2 mmol).
GC analyses were performed with a Shimadzu GC2010 instrumentequipped with an ionization flame detector and a Equity-5(15 m � 0.1 mm) capillary column of poly(5% diphenyl/95% di-methylsiloxane) with 0.1 μm film thickness. Conditions: Tinj =270 °C, Tdet = 280 °C, carrier gas: He, Tinitial = 90 °C for 1 min,rate = 90 °C/min; Tfinal = 260 °C for 5 min. Retention times: n-undecane: 1.9 min, methyl p-tolyl sulfide: 2.28 min, methyl p-tolylsulfoxide: 2.83 min, methyl p-tolyl sulfone: 2.96 min. Calibrationwas performed by integration of the chromatographic peak areas(A) of the standard (S) and of the analytes (X). The concentrations(M) of methyl p-tolyl sulfide and its oxidation products were calcu-lated by using the following response factors: F(undecane) = 1,
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim223
F(methyl p-tolyl sulfide) = 0.58, F(methyl p-tolyl sulfoxide) = 0.48,F(methyl p-tolyl sulfone) = 0.57; F = (AS/MS)/(AX/MX).
The second-order kinetics constants were determined by fitting theexperimental values with the Scientist Micromath® software for thekinetics scheme (Scheme S1). A minor contribution of H2O2 de-composition (k� 4�10–5 m–1 s–1) was also considered.
The stabilities of the oxoclusters were assessed by comparison ofthe FTIR and EXAFS spectra before and after the reaction; therecovered precipitate was washed with isopropanol. To obtainenough oxoclusters to analyze after the oxidation process, the reac-tions were performed with the Zr6 and Zr12 molar ratios increasedto 1% with respect to sulfide.
EXAFS Measurements: The EXAFS and X-ray absorption nearedge structure (XANES) measurements were performed atbeamline C1 at HASYLAB (Hamburg). A Si(311) double-crystalmonochromator was used for measurements at the ZrK-edge(17.998 keV). The second monochromator crystal was tilted for op-timal harmonic rejection. The spectra were recorded in trans-mission mode by using ionization chambers and in fluorescencemode by using a passivated implanted planar silicon (PIPS) detec-tor. For the Zr6_cat and Zr12_cat samples (see main text), only thefluorescence data were useful owing to the low Zr concentration.Energy calibration was performed with Zr metal foil; however, adrift in the spectra could occur as it was not possible to measurethe Zr foil in parallel to the sample measurements. Solid sampleswere pressed into self-supporting wafers with cellulose as a binder.The solutions were measured in a sealed sample cell that can befilled under an inert atmosphere.[19,20]
Data evaluation started with the removal of background absorp-tion from the experimental absorption spectrum by subtracting aVictoreen-type polynomial. As there were several turning points inthe absorption edge, the threshold energy E0 was determined bytaking the energy at half of the edge jump.[20] To determine thesmooth part of the spectrum, corrected for pre-edge absorption, apiecewise polynomial was used. It was adjusted in such a way thatthe low-R components of the resulting Fourier transform wereminimal (Figure S3). After division of the background-subtractedspectrum by its smooth part, the photon energy was converted tophotoelectron wavenumbers k. The resulting χ(k) function wasweighted with k3 and a Fourier transform was applied with aHanning window function. Data analysis was performed in k-spaceon unfiltered data. The adjustment of the common theoreticalEXAFS expression
χ(k) = �i
Nj
kri2 S0
2(k)Fj(k)e–2k2σj2e–2rj/λsin[2krj + δj(k)]
was made according to the curved-wave formalism of theEXCURV98 program with XALPHA phase and amplitude func-tions.[21] The mean free path of the scattered electrons was calcu-lated from the imaginary part of the potential (VPI, set to–4.00 eV). An inner potential correction Ef was introduced whenthe experimental data was fitted with theoretical models that ac-count for an overall phase shift between the experimental and cal-culated spectra.
Raman Measurements: The Raman spectra were collected with ahomemade micro-Raman system with a single 320 mm focal lengthimaging spectrograph (Triax-320 Jobin Yvon) equipped with aholographic 1800 g/mm grating and a liquid-nitrogen-cooled CCDdetector. The excitation source was a Spectra Physics Ar+ ion laser(Stabilite 2017) operating at 514.5 nm, and an appropriate long-pass edge filter (Semrock Filters) was used to reduce the stray-light

www.eurjic.org FULL PAPER
level. An optical microscope (Olympus BX 40), equipped with threeobjectives (20�/0.35, 50�/0.75, and 100�/0.90) was optically cou-pled to the spectrograph and used to collect the Raman spectra inbackscattering micro configuration. In the present work, the Ra-man spectra were recorded between 100 and 4000 cm–1 with aninstrumental resolution of ca. 2 cm–1 by using the long workingdistance 20�/0.35 objective lens. The irradiation power on the sam-ple was typically 40–80 mW, and no damage to the solid sampleswas observed.
SEM Measurements: Scanning electron microscopy measurementswere performed with a field-emission SEM (Zeiss SUPRA 40VP)equipped with an energy-dispersive X-ray spectroscopy (EDXS)system (Oxford INCA).
FTIR Measurements: The infrared spectra were collected with aThermo Quest Nicolet 5700 instrument. The oxocluster or spentcatalyst (a few milligrams) was dispersed into KBr pellets.
Elemental Analyses: Inductively coupled plasma mass spectrometry(ICP-MS) was performed with an Agilent Technologies 7700x ICP-MS System to establish the Zr content of the oxoclusters. The sam-ples were mineralized upon heating at 220 °C by using a mixtureof H2SO4, HNO3, and HF in ratio of 4:8:2. CHNS analyses wereperformed with a Fison EA1108 CHNS analyzer.
Supporting Information (see footnote on the first page of this arti-cle): Structures of Zr6 and Zr12; FTIR, Raman, and EXAFS spec-tra; kinetic traces; SEM micrographs.
Acknowledgments
The Italian Foreign Affairs Ministry (Ministero degli Affari Esteri,MAE, Italy) and the National Research Council (CNR, Italy) aregratefully acknowledged by S. G. for the financial support of thiswork in the framework of the “Progetto Grande Rilevanza Italia–Slovenia 2013”. The University of Padova and the Italian Consor-tium INSTM are acknowledged for providing money and equip-ment. The authors would like to thank the Deutsches ElektronenSynchrothron (DESY) and the European Union Calipso Pro-gramme for financial support in activities at Hasylab, DESY. Theresearch leading to these results has received funding from theEuropean Union Seventh Framework Program (FP7/2007-2013)under grant agreement number 226716. M. B. acknowledges finan-cial support by the Carl-Zeiss Foundation. A. S and M. C. acknow-ledge the financial support from the Italian Fondazione Cariparo(Progetto Dottorati di Ricerca, PARO118894/11).
[1] a) U. Schubert, Macromol. Symp. 2008, 267, 1–8 and referencescited therein; b) U. Schubert, Acc. Chem. Res. 2007, 40, 730–737 and references cited therein; c) U. Schubert, in: Macromol-ecules Containing Metal and Metal-like Elements, vol. 7 (Eds.:A. Abd-El Aziz, C. Carraher, C. Pittman, M. Zeldin), Wiley,New York, 2006, p. 55; d) U. Schubert, J. Mater. Chem. 2005,15, 3701–3715, and references cited therein; e) U. Schubert, J.Sol-Gel Sci. Technol. 2004, 31, 19–24; f) U. Schubert, Chem.Mater. 2001, 13, 3487–3494; g) G. Kickelbick, U. Schubert,Chem. Ber./Recueil 1997, 130, 473–477; h) G. Kickelbick, P.Wiede, U. Schubert, Inorg. Chim. Acta 1999, 284, 1–7; i) M.Puchberger, F. R. Kogler, M. Jupa, S. Gross, H. Fric, G. Kick-elbick, U. Schubert, Eur. J. Inorg. Chem. 2006, 3283–3293.
[2] a) U. Schubert, Chem. Soc. Rev. 2011, 40, 575–582, and refer-ences cited therein; b) M. Carraro, S. Gross, Materials 2014, 7,3956–3989, and references cited therein.
[3] S. Gross, J. Mater. Chem. 2011, 21, 15853–15861, and refer-ences cited therein.
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim224
[4] M. A. Sliem, D. A. Schmidt, A. Bétard, S. B. Kalidindi, S.Gross, M. Havenith-Newen, A. Devi, R. A. Fischer, Chem.Mater. 2012, 24, 4274–4282.
[5] a) V. Guillerm, S. Gross, C. Serre, T. Devic, M. Bauer, G. Férey,Chem. Commun. 2010, 46, 767–769; b) V. Guillerm, F. Ragon,M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vi-mont, G. Clet, Q. Yang, G. Maurin, G. Férey, A. Vittadini, S.Gross, C.Serre, Angew. Chem. Int. Ed. 2012, 51, 9267–9271;Angew. Chem. 2012, 124, 9401–9405.
[6] a) I. Gautier-Luneau, A. Mosset, J. Z. Galy, Z. Kristallogr.1987, 180, 83–95; b) S. Doeuff, Y. Dromzee, F. Taulelle, C.Sanchez, Inorg. Chem. 1989, 28, 4439–4445.
[7] a) L. G. Hubert-Pfalzgraf, G. Liliane, J. Mater. Chem. 2004,14, 3113–3123; b) L. G. Hubert-Pfalzgraf, Coord. Chem. Rev.1998, 178–180, 967–997; c) L. G. Hubert-Pfalzgraf, New J.Chem. 1995, 19, 727–750.
[8] a) A. Brethon, L. G. Hubert-Pfalzgraf, J. Sol-Gel Sci. Technol.2006, 39, 159–167; b) A. Brethon, L. G. Hubert-Pfalzgraf, J. C.Daran, Dalton Trans. 2006, 250–257; c) d) L. G. Hubert-Pfalz-graf, S. Daniele, C. R. Chim. 2004, 7, 521–527; e) S. Daniele,L. G. Hubert-Pfalzgraf, P. B. Hitchcock, M. F. Lappert, Inorg.Chem. Commun. 2000, 3, 218–220; f) H. Guillon, L. G. Hubert-Pfalzgraf, J. Vaissermann, Eur. J. Inorg. Chem. 2000, 1243–1252; g) S. Daniele, L. G. Hubert-Pfalzgraf, J. Vaissermann,Polyhedron 1998, 17, 4249–4256; h) L. G. Hubert-Pfalzgraf, C.Sirio, C. Bois, Polyhedron 1998, 17, 821–830; i) S. Parola, R.Papiernik, L. G. Hubert-Pfalzgraf, C. Bois, J. Chem. Soc., Dal-ton Trans. 1998, 5, 737–739; j) S. Boulmaaz, L. G. Hubert-Pfalzgraf, S. Halut, J. C. Daran, J. Chem. Soc., Chem. Com-mun. 1994, 601–602; k) L. G. Hubert-Pfalzgraf, S. Daniele, A.Bennaceur, J. C. Daran, J. Vaissermann, Polyhedron 1997, 16,1223–1234; l) S. Daniele, L. G. Hubert-Pfalzgraf, J. C. Daran,Polyhedron 1996, 15, 1063–70; m) S. Daniele, L. G. Hubert-Pfalzgraf, J. C. Daran, S. Halut, Polyhedron 1994, 13, 927–32.
[9] a) G. Losada, M. A. Mendiola, M. T. Sevilla, Inorg. Chim.Acta 1997, 255, 125–131; b) K. L. Zhang, Y. J. Shi, X. Z. You,K. B. Yu, J. Mol. Struct. 2005, 743, 73–77; c) D. L. Long, P.Kögerler, L. J. Farrugia, L. Cronin, Chem. Asian J. 2006, 1,352–357.
[10] a) R. H. Holm, Chem. Rev. 1987, 87, 1401–1449; b) M. H.Dickman, M. T. Pope, Chem. Rev. 1994, 94, 569–584; c) I. A.Weinstock, Chem. Rev. 1998, 98, 113–170.
[11] a) V. Conte, F. Di Furia, G. Licini, Appl. Catal. A 1997, 157,335–361; b) B. S. Lane, K. Burgess, Chem. Rev. 2003, 103,2457–2473; c) R. Noyori, M. Aokib, K. Sato, Chem. Commun.2003, 1977–1986; d) N. V. Maksimchuk, M. S. Melgunov, J.Mrowiec-Białon, A. B. Jarzebski, O. A. Kholdeeva, J. Catal.2005, 235, 175–183; e) N. S. Antonova, J. J. Carbó, U. Kortz,O. A. Kholdeeva, J. M. Poblet, J. Am. Chem. Soc. 2010, 132,7488–7497; f) I. W. C. E. Arends, V. Conte, G. Licini, in: Innov-ative Catalysis in Organic Synthesis: Oxidation, Hydrogenation,and C–X Bond Forming Reactions (Ed.: P. G. Andersson),Wiley-VCH, Weinheim, Germany, 2012, p. 77–102.
[12] a) M. Bonchio, G. Licini, S. Mantovani, G. Modena, W. A.Nugent, J. Org. Chem. 1999, 64, 1326–1330; b) B. Saito, T.Katsuki, Tetrahedron Lett. 2001, 42, 3873–3876; c) K. P. Bryl-iakov, E. P. Talsi, J. Mol. Catal. A 2007, 264, 280–287; d) H.Srour, P. Le Maux, S. Chevance, G. Simonneaux, Coord. Chem.Rev. 2013, 257, 3030–3050.
[13] a) R. Schwartz, H. Z. Giese, Z. Anorg. Allg. Chem. 1928, 176,209–232; b) J. A. Connor, E. A. V. Ebsworth, Adv. Inorg. Chem.Radiochem. 1964, 6, 279–381; c) W. P. Griffith, T. D. Wickins,J. Chem. Soc. A 1968, 176, 397–400; d) G. D. Gupta, G. V. Jere,Indian J. Chem. 1968, 6, 54–59; e) G. V. Jere, G. D. Gupta, J.Inorg. Nucl. Chem. 1970, 32, 537–542; f) G. D. Gupta, G. V.Jere, Indian J. Chem. 1972, 10, 102; g) G. V. Jere, M. T. San-thamma, Inorg. Chim. Acta 1977, 24, 57–61; h) R. Schmidt, G.Pausewang, W. Massa, Z. Anorg. Allg. Chem. 1986, 535, 135–142; i) M. T. H. Tarafder, M. A. L. Miah, Inorg. Chem. 1986,25, 2265–2268; j) A. C. Dengel, W. P. Griffith, Polyhedron 1989,

www.eurjic.org FULL PAPER
8, 1371–1377; k) M. T. H. Tarafder, A. A. M. A. Islam, Polyhe-dron 1989, 8, 109–112; l) C. R. Bhattacharjee, M. Bhattach-arjee, M. K. Chaudhuri, M. Choudhury, Polyhedron 1990, 9,1653–1657; m) D. D. Agarwal, R. Jain, R. P. Bhatnagar, S. Sriv-astava, Polyhedron 1990, 9, 1405–1409; n) M. T. H. Tarafder,A. R. Khan, Polyhedron 1991, 10, 819–822; o) M. T. H. Tar-afder, A. R. Khan, Polyhedron 1991, 10, 973–976; p) T. Okachi,N. Murai, M. Onaka, Org. Lett. 2003, 5, 85–87; q) G. Wang,H. H. Y. Sung, I. D. Williams, W. Leung, Inorg. Chem. 2012,51, 3640–3647.
[14] a) O. A. Kholdeeva, G. M. Maksimov, R. I. Maksimovskaya,M. P. Vanina, T. A. Trubitsina, D. Yu. Naumov, B. A. Kolesov,N. S. Antonova, J. J. Carbó, J. M. Poblet, Inorg. Chem. 2006,45, 7224–7234; b) O. A. Kholdeeva, R. I. Maksimovskaya, J.Mol. Catal. A 2007, 262, 7–24; c) S. S. Mal, N. H. Nsouli, M.Carraro, A. Sartorel, G. Scorrano, H. Oelrich, L. Walder, M.Bonchio, U. Kortz, Inorg. Chem. 2010, 49, 7–9; d) C. Jahier,S. S. Mal, U. Kortz, S. Nlate, Eur. J. Inorg. Chem. 2010, 1559–1566; e) G. Al-Kadamany, S. S. Mal, B. Milev, B. G. Donoeva,R. I. Maksimovskaya, O. A. Kholdeeva, U. Kortz, Chem. Eur.J. 2010, 16, 11797–11800; f) M. Carraro, N. H. Nsouli, H. Oel-rich, A. Sartorel, A. Sorarù, S. S. Mal, G. Scorrano, L. Walder,U. Kortz, M. Bonchio, Chem. Eur. J. 2011, 17, 8371–8378; g)D. Li, H. Han, Y. Wang, X. Wang, Y. Li, E. Wang, Eur. J.Inorg. Chem. 2013, 1926–1934; h) P. Jiménez-Lozano, J. J.Carbó, A. Chaumont, J. M. Poblet, A. Rodríguez-Fortea, G.Wipff, Inorg. Chem. 2014, 53, 778–786.
[15] G. Trimmel, S. Gross, G. Kickelbick, U. Schubert, Appl. Or-ganomet. Chem. 2001, 15, 401–406.
[16] M. P. Feth, PhD Thesis, University of Stuttgart, Germany,2003.
[17] a) G. Kickelbick, M. P. Feth, H. Bertagnolli, M. Puchberger,D. Holzinger, S. Gross, J. Chem. Soc., Dalton Trans. 2002,3892–3898; b) P. Walther, F. Puchberger, F. R. Kogler, K.Schwarz, U. Schubert, Phys. Chem. Chem. Phys. 2009, 11,3640–3647.
[18] F. R. Kogler, M. Jupa, M. Puchberger, U. Schubert, J. Mater.Chem. 2004, 14, 3133–3138.
[19] a) M. Bauer, G. Heusel, S. Mangold, H. Bertagnolli, J. Syn-chrotron Radiat. 2010, 17, 273–279; b) M. Bauer, C. Gastl,Phys. Chem. Chem. Phys. 2010, 12, 5575–5584.
[20] a) M. Newville, P. Livins, Y. Yacoby, J. J. Rehr, E. A. Stern,Phys. Rev. B 1993, 47, 14126–14131; b) T. S. Ertel, H. Bertag-nolli, S. Hückmann, U. Kolb, D. Peter, Appl. Spectrosc. 1992,46, 690–698.
[21] S. J. Gurman, N. Binsted, I. Ross, J. Phys. C 1984, 17, 143–152.
[22] C. Carlone, Phys. Rev. B 1992, 45, 2079–2082.[23] V. G. Keramidas, W. B. White, J. Am. Ceram. Soc. 1974, 57,
22–24.[24] a) A. F. Holleman, E. Wiberg, Lehrbuch der Anorganische
Chemie, 101st ed., De Gruyter, Berlin, 1995, p. 1411; b) F. A.Cotton, G. Wilkinson, Advanced Inorganic Chemistry, 3rd ed.,Interscience, New York, 1972, p. 930.
[25] N. N. Greenwood, A. Earnshaw, Chemistry of the Elements,Butterworth-Heinemann Ltd., Oxford, UK, 1984.
[26] R. Villanneau, H. Carabineiro, X. Carrier, R. Thouvenot, P.Herson, F. Lemos, F. Ramôa Ribeiro, M. Che, J. Phys. Chem.B 2004, 108, 12465–12471.
Eur. J. Inorg. Chem. 2015, 210–225 © 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim225
[27] A. I. Vogel, Vogel’s Textbook Of Macro and SemiMicro Quali-tative Inorganic Analysis, 5th ed. (revised by G. Svehla), Long-man, London, 1979.
[28] A. Clearfield, P. A. Vaughan, Acta Crystallogr. 1956, 9, 555–558.
[29] A. Clearfield, Inorg. Chem. 1964, 3, 146–148.[30] T. C. W. Mak, Can. J. Chem. 1968, 46, 3491–3497.[31] M. Aberg, Acta Chem. Scand., Ser. B 1977, 31, 171–181.[32] G. T. Mamott, P. Barnes, S. E. Tarling, S. L. Jones, C. J. Nor-
man, J. Mater. Sci. 1991, 26, 4054–4061.[33] G. Y. Guo, Y. L. Chen, W. J. Ying, Mater. Chem. Phys. 2004,
84, 308–314.[34] a) M. Bauer, C. Gastl, C. Köppl, G. Kickelbick, H. Bertagnolli,
Monatsh. Chem. 2006, 137, 567–581; b) M. Bauer, S. Müller,G. Kickelbick, H. Bertagnolli, New J. Chem. 2007, 31, 1950–1959.
[35] K. Nakamoto, Infrared and Raman Spectra of Inorganic & Co-ordination Compound, 6th ed., Wiley Interscience, New York,2009.
[36] A. Clearfield, Rev. Pure Appl. Chem. 1964, 14, 91–108.[37] A. Clearfield, J. Mater. Res. 1990, 5, 161–162.[38] M. Kantcheva, C. J. Koz, Mater. Sci. 2007, 42, 6074–6086.[39] M. Bauer, H. Bertagnolli, J. Phys. Chem. B 2007, 111, 13756–
13764.[40] F. Faccini, H. Fric, U. Schubert, E. Wendel, O. Tsetsgee, K.
Müller, H. Bertagnolli, A. Venzo, S. Gross, J. Mater. Chem.2007, 17, 3297–3307.
[41] a) R. A. Sheldon, I. W. C. E. Arends, U. Hanefeld, GreenChemistry and Catalysis, Wiley-VCH, Weinheim, Germany,2007; b) K. Kaczorowska, Z. Kolarska, K. Mitka, P. Kowalski,Tetrahedron 2005, 61, 8315–8327; c) K. Sato, M. Hyodo, M.Aoki, X.-Q. Zheng, R. Noyori, Tetrahedron 2001, 57, 2469–2476.
[42] a) K. Yazu, Y. Yamamoto, T. Furuya, K. Miki, K. Ukegawa,Energy Fuels 2001, 15, 1535–1536; b) J. M. Campos-Martin,M. C. Capel-Sanchez, J. L. G. Fierro, Green Chem. 2004, 6,557–562; c) M. Carraro, G. Fiorani, L. Mognon, F. Caneva,M. Gardan, C. Maccato, M. Bonchio, Chem. Eur. J. 2012, 18,13195–13202.
[43] a) O. Bortolini, S. Campestrini, V. Conte, F. Di Furia, G. Mod-ena, J. Org. Chem. 1988, 53, 5721–5724; b) W. Adam, W. Haas,B. B. Lohray, J. Am. Chem. Soc. 1991, 113, 6202–6208; c) F. B.Ballistreri, M. Bonchio, V. Conte, M. A. De Conciliis, F.Di Furia, G. A. Tomaselli, R. M. Toscano, J. Org. Chem. 1995,60, 4475–4480; d) M. Bonchio, S. Campestrini, V. Conte, F.Di Furia, S. Moro, Tetrahedron 1995, 51, 12363–12372.
[44] Interestingly, Zr6 displays a selectivity similar to that reportedfor Zr6- and Hf6-containing polyoxotungstates, which wereused to catalyze the oxidation of methyl phenyl sulfide withH2O2 (see ref.[14e]).
[45] M. Carraro, L. Sandei, A. Sartorel, G. Scorrano, M. Bonchio,Org. Lett. 2006, 8, 3671–3674.
[46] G. Licini, C. Zonta, Angew. Chem. Int. Ed. 2013, 52, 2911–2914; Angew. Chem. 2013, 125, 2983.
[47] The experimental data agree with the following formulas forthe starting materials: [Zr6(OH)4O4{O(O)CC(CH3)CH2}12]·8[CH2C(CH3)C(O)OH]8 and [{Zr6O4(OH)4{O(O)CCH2-CHCH2}12}2]·6[CH2CHCH2C(O)OH]6.
Received: August 8, 2014Published Online: November 6, 2014
Copyright © 2022 FDOKUMEN




















