
JNK2 and IKKβ Are Required for Activating the Innate Response to Viral Infection
11
Immunity, Vol. 11, 721–731, December, 1999, Copyright 1999 by Cell Press JNK2 and IKKb Are Required for Activating the Innate Response to Viral Infection other than viral infection, are relatively well understood (reviewed by Karin and Hunter, 1995; Verma et al., 1995), but those that control their activity as well as that of IRF Wen-Ming Chu,* Derek Ostertag, ² Zhi-Wei Li,* Lufen Chang,* Yi Chen,* Yinling Hu,* Bryan Williams, ‡ Jacques Perrault, ² proteins following viral infection have yet to be eluci- and Michael Karin* § dated (Juang et al., 1998; Wathelet et al., 1998; Yone- * Laboratory of Signal Transduction yama et al., 1998). NF-kB and AP-1 are also involved in and Gene Regulation the induction of proinflammatory and cytotoxic cyto- Department of Pharmacology kines (Barnes and Karin, 1997; Ip and Davis, 1998). University of California, San Diego NF-kB proteins are regulated through interaction with La Jolla, California 92093-0636 specific inhibitors, the IkBs, which retain them in the ² Department of Biology cytoplasm of nonstimulated cells (Verma et al., 1995; Molecular Biology Institute Baldwin 1996). In response to proinflammatory stimuli San Diego State University including viral infection, the IkBs undergo rapid phos- San Diego, California 92182 phorylation at conserved N-terminal sites that trigger ‡ Department of Cancer Biology their ubiquitin-dependent degradation and subsequent The Lerner Research Institute nuclear translocation of the liberated NF-kB dimers (Del- Cleveland, Ohio 44195 hase and Karin, 1999). The signal-responsive protein kinase that phosphorylates the IkBs and thereby causes NF-kB activation was molecularly identified (DiDonato Summary et al., 1997; Mercurio et al., 1997; Re ´ gnier et al., 1997). This protein kinase, IKK, is composed of two catalytic Viral infection or double-stranded (ds) RNA induce in- subunits, IKKa and IKKb, and one regulatory subunit, terferons (IFN) and other cytokines. Transcription fac- IKKg/NEMO (Zandi et al., 1997; 1998; Rothwarf et al., tors mediating IFN induction are known, but the signal- 1998; Yamaoka et al., 1998; Delhase and Karin, 1999). ing pathways that regulate them are less clear. We IKKg/NEMO is required for activation of NF-kB by many now describe two such pathways. The first pathway stimuli, including the proinflammatory cytokines tumor leading to NF-kB depends on the dsRNA-responsive necrosis factor a (TNFa) and IL-1, bacterial lipopolysac- protein kinase (PKR), which in turn activates IkB ki- charide (LPS), and dsRNA (Yamaoka et al., 1998). Sur- nase (IKK) through the IKKb subunit. The second viral- prisingly, however, only the IKKb catalytic subunit is and dsRNA-responsive pathway is PKR independent required for IKK and NF-kB activation by proinflamma- and involves Jun kinase (JNK) activation leading to tory cytokines and LPS (Delhase et al., 1999; Li et al., stimulation of AP-1. Both IKKb and JNK2 are essential 1999, 1999; Tanaka et al., 1999), whereas IKKa mediates for efficient induction of type I IFN and other cytokines a response to a yet-to-be identified developmental sig- in response to viral infection or dsRNA. This study nal that controls keratinocyte differentiation (Hu et al., establishes a general role for these kinases in activa- 1999; Takeda et al., 1999). Which catalytic subunit medi- tion of innate immune responses. ates the response to virus and dsRNA was heretofore unknown. Introduction Unlike NF-kB, c-Jun:ATF2 (and other AP-1) hetero- dimers are constitutively nuclear, but their transactivat- Viral infection of mammalian cells results in activation ing potential is rapidly stimulated by phosphorylation of of an innate antiviral response mediated by type I inter- their activation domains (Bine ´ truy et al., 1991; Smeal et ferons (IFNs), which impair viral gene expression and al., 1991; Gupta et al., 1995) by the Jun kinases (JNKs, replication (Sen and Lengyel, 1992; Mu ¨ ller et al., 1994), Hibi et al., 1993; De ´ rijard et al., 1994; Kyriakis et al., as well as by other cytokines, such as interleukin (IL)-6 1994; Karin, 1995). As other MAP kinases (MAPKs), the (Pulliam et al., 1995) and IL-12 (Orange and Biron, 1996), JNKs are activated via protein kinase cascades involv- which activate cell-mediated cytotoxic responses. Reg- ing two JNK kinases (JNKKs) and a much larger number ulation of IFN expression has been the subject of intense of upstream kinases (reviewed by Minden and Karin, investigation, and several transcription factors and ar- 1997). Both JNK1 and JNK2 respond to T cell activators chitectural proteins that bind IFN gene promoters were (Su et al., 1994) and are required for induction of T cell identified (Reis et al., 1992; Du et al., 1993; Matsuyama cytokines (Dong et al., 1998; Yang et al., 1998; Sabapa- et al., 1993; Thanos and Maniatis, 1995). One of these thy et al., 1999). proteins, HMGI(Y), facilitates assembly of an enhanceo- The signaling pathways that are activated by viral in- some that contains several signal-responsive transcrip- fection and lead to activation of NF-kB and c-Jun:ATF2 tion factors, including NF-kB (p50:p65), c-Jun:ATF2 (AP-1) are rather nebulous. In the case of NF-kB, a role in the heterodimers, and members of the IRF family (Thanos viral response was ascribed to the dsRNA-dependent and Maniatis, 1995; Wathelet et al., 1998). The control of kinase PKR (Kumar et al., 1994). PKR contains two RNA NF-kB and c-Jun:ATF2 activities, in response to stimuli binding regulatory domains at its N terminus and a ser- ine/threonine protein kinase domain at its C terminus (reviewed by Clemens and Elia, 1997; Williams, 1997). § To whom correspondence should be addressed (e-mail: karinoffice@ ucsd.edu). PKR expression is transcriptionally induced by IFNs,
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of JNK2 and IKKβ Are Required for Activating the Innate Response to Viral Infection

Immunity, Vol. 11, 721–731, December, 1999, Copyright 1999 by Cell Press
JNK2 and IKKb Are Required for Activatingthe Innate Response to Viral Infection
other than viral infection, are relatively well understood(reviewed by Karin and Hunter, 1995; Verma et al., 1995),but those that control their activity as well as that of IRF
Wen-Ming Chu,* Derek Ostertag,† Zhi-Wei Li,*Lufen Chang,* Yi Chen,* Yinling Hu,*Bryan Williams,‡ Jacques Perrault,†
proteins following viral infection have yet to be eluci-and Michael Karin*§
dated (Juang et al., 1998; Wathelet et al., 1998; Yone-*Laboratory of Signal Transductionyama et al., 1998). NF-kB and AP-1 are also involved inand Gene Regulationthe induction of proinflammatory and cytotoxic cyto-Department of Pharmacologykines (Barnes and Karin, 1997; Ip and Davis, 1998).University of California, San Diego
NF-kB proteins are regulated through interaction withLa Jolla, California 92093-0636specific inhibitors, the IkBs, which retain them in the†Department of Biologycytoplasm of nonstimulated cells (Verma et al., 1995;Molecular Biology InstituteBaldwin 1996). In response to proinflammatory stimuliSan Diego State Universityincluding viral infection, the IkBs undergo rapid phos-San Diego, California 92182phorylation at conserved N-terminal sites that trigger‡Department of Cancer Biologytheir ubiquitin-dependent degradation and subsequentThe Lerner Research Institutenuclear translocation of the liberated NF-kB dimers (Del-Cleveland, Ohio 44195hase and Karin, 1999). The signal-responsive proteinkinase that phosphorylates the IkBs and thereby causesNF-kB activation was molecularly identified (DiDonatoSummaryet al., 1997; Mercurio et al., 1997; Regnier et al., 1997).This protein kinase, IKK, is composed of two catalyticViral infection or double-stranded (ds) RNA induce in-subunits, IKKa and IKKb, and one regulatory subunit,terferons (IFN) and other cytokines. Transcription fac-IKKg/NEMO (Zandi et al., 1997; 1998; Rothwarf et al.,tors mediating IFN induction are known, but the signal-1998; Yamaoka et al., 1998; Delhase and Karin, 1999).ing pathways that regulate them are less clear. WeIKKg/NEMO is required for activation of NF-kB by manynow describe two such pathways. The first pathwaystimuli, including the proinflammatory cytokines tumorleading to NF-kB depends on the dsRNA-responsivenecrosis factor a (TNFa) and IL-1, bacterial lipopolysac-protein kinase (PKR), which in turn activates IkB ki-charide (LPS), and dsRNA (Yamaoka et al., 1998). Sur-nase (IKK) through the IKKb subunit. The second viral-prisingly, however, only the IKKb catalytic subunit isand dsRNA-responsive pathway is PKR independentrequired for IKK and NF-kB activation by proinflamma-and involves Jun kinase (JNK) activation leading totory cytokines and LPS (Delhase et al., 1999; Li et al.,stimulation of AP-1. Both IKKb and JNK2 are essential1999, 1999; Tanaka et al., 1999), whereas IKKa mediatesfor efficient induction of type I IFN and other cytokinesa response to a yet-to-be identified developmental sig-in response to viral infection or dsRNA. This studynal that controls keratinocyte differentiation (Hu et al.,establishes a general role for these kinases in activa-1999; Takeda et al., 1999). Which catalytic subunit medi-tion of innate immune responses.ates the response to virus and dsRNA was heretoforeunknown.
Introduction Unlike NF-kB, c-Jun:ATF2 (and other AP-1) hetero-dimers are constitutively nuclear, but their transactivat-
Viral infection of mammalian cells results in activation ing potential is rapidly stimulated by phosphorylation ofof an innate antiviral response mediated by type I inter- their activation domains (Binetruy et al., 1991; Smeal etferons (IFNs), which impair viral gene expression and al., 1991; Gupta et al., 1995) by the Jun kinases (JNKs,replication (Sen and Lengyel, 1992; Muller et al., 1994), Hibi et al., 1993; Derijard et al., 1994; Kyriakis et al.,as well as by other cytokines, such as interleukin (IL)-6 1994; Karin, 1995). As other MAP kinases (MAPKs), the(Pulliam et al., 1995) and IL-12 (Orange and Biron, 1996), JNKs are activated via protein kinase cascades involv-which activate cell-mediated cytotoxic responses. Reg- ing two JNK kinases (JNKKs) and a much larger numberulation of IFN expression has been the subject of intense of upstream kinases (reviewed by Minden and Karin,investigation, and several transcription factors and ar- 1997). Both JNK1 and JNK2 respond to T cell activatorschitectural proteins that bind IFN gene promoters were (Su et al., 1994) and are required for induction of T cellidentified (Reis et al., 1992; Du et al., 1993; Matsuyama cytokines (Dong et al., 1998; Yang et al., 1998; Sabapa-et al., 1993; Thanos and Maniatis, 1995). One of these thy et al., 1999).proteins, HMGI(Y), facilitates assembly of an enhanceo- The signaling pathways that are activated by viral in-some that contains several signal-responsive transcrip- fection and lead to activation of NF-kB and c-Jun:ATF2tion factors, including NF-kB (p50:p65), c-Jun:ATF2 (AP-1) are rather nebulous. In the case of NF-kB, a role in theheterodimers, and members of the IRF family (Thanos viral response was ascribed to the dsRNA-dependentand Maniatis, 1995; Wathelet et al., 1998). The control of kinase PKR (Kumar et al., 1994). PKR contains two RNANF-kB and c-Jun:ATF2 activities, in response to stimuli binding regulatory domains at its N terminus and a ser-
ine/threonine protein kinase domain at its C terminus(reviewed by Clemens and Elia, 1997; Williams, 1997).§ To whom correspondence should be addressed (e-mail: karinoffice@
ucsd.edu). PKR expression is transcriptionally induced by IFNs,

Immunity722
while its catalytic activity is stimulated by binding in either IKKa (Hu et al., 1999) or IKKb (Li et al., 1999). Weused primary embryonic fibroblasts (MEFs) from Ikka2/2dsRNAs, which are produced during viral infection. Asand Ikkb2/2 embryos to investigate the involvement ofmost cells express basal levels of PKR, its regulationthe two catalytic subunits in the response to dsRNAmay involve a feed-forward positive autoregulatory cas-and virus. No substantial differences in IKK activationcade in which dsRNA produced during viral transcriptionin response to either dsRNA (Figure 1C) or infectionor replication activates preexisting PKR, which then acti-with vesicular stomatitis virus ([VSV]; Figure 1D) werevates transcription factors that lead to IFN induction.detected between wt or IKKa-deficient MEFs. By con-Newly synthesized IFNs activate the JAK-STAT signal-trast, loss of the IKKb subunit severely impaired IKKing cascade (Darnell et al., 1994; Ihle et al., 1994), re-activation by either dsRNA (Figure 1E) or VSV (Figuresulting in induction of more PKR and other IFN-respon-1F). As a result, very little NF-kB binding activity wassive gene products. This model is supported by analysisinduced by either dsRNA or VSV in IKKb-deficient cells.of PKR-deficient mice and cells (Yang et al., 1995; KumarAs a negative control, we measured JNK activity in theet al., 1997). These studies also suggest that inductiondifferent cells and much to our surprise found it to beof IRF-1 is dependent at least in part on NF-kB (Kumarinduced by both dsRNA and VSV (Figures 1C–1F). Theet al., 1997). Although it was suggested that PKR maykinetics of JNK activation were a bit slower from thosedirectly phosphorylate IkBs (Kumar et al., 1994), NF-kBof IKK but were not affected by the loss of either IKKais activated by TNFa in PKR-deficient cells (Yang et al.,or IKKb. A similar, but even more robust, JNK activation1995; Kumar et al., 1997), whereas IKKg/NEMO-deficientresponse was detected in HEK293 cells (data notcells do not respond to dsRNA (Yamaoka et al., 1998).shown).However, it was also observed that PKR-deficient mice
Next, we examined the dependence of IKK activationand cells exhibit residual IFN induction responses toon PKR, using two immortalized mouse fibroblast cellviral infection and dsRNA, thus suggesting the existencelines derived from either wt (Pkr1/1) or PKR knockoutof additional, PKR-independent, signaling pathwaysmice (Pkr2/2) (Yang et al., 1995; Kumar et al., 1997).(Yang et al., 1995). The signaling pathways that activateWhile incubation of Pkr1/1 cells with dsRNA resulted inc-Jun:ATF2 and other forms of AP-1 in response to viralIKK activation, there was no appreciable increase in IKKinfection are largely unknown.activity during the first 4 hr of treatment with dsRNA inWe investigated signaling pathways responsive to vi-Pkr2/2 cells (Figure 2A), but after 4 hr there was a smallral infection or dsRNA that can lead to induction ofbut reproducible increase in IKK activity in Pkr2/2 cellstype I IFNs and other cytokines. We obtained evidence(data not shown). Both cell lines, however, were respon-indicating that a major pathway that leads to NF-kBsive to TNFa, an established IKK activator. It should beactivation is indeed dependent on PKR, but in this path-noted that in mouse fibroblasts, unlike HEK293 cells, theway PKR acts upstream to IKK, targeting its IKKb sub-response of IKK to dsRNA is weaker than its response tounit rather than IKKa. We demonstrate that IKKb is es-TNFa (see also Figure 1E). As previously describedsential for induction of type I IFNs and other cytokines.(Yang et al., 1995), Pkr2/2 cells exhibited a defective NF-We identified a second signaling pathway elicited bykB activation response to dsRNA but a normal responseviral infection or dsRNA that leads to JNK activation.to TNFa (Figure 2A). Surprisingly, incubation of bothThis pathway can enhance the transcriptional activity ofPkr1/1 and Pkr2/2 cells with dsRNA resulted in efficientc-Jun:ATF2 and other AP-1 dimers, and its importance isJNK activation that was similar in magnitude to the re-illustrated by JNK2-deficient cells, which exhibit a defectsponse to TNFa, one of the most potent JNK activators.in induction of type I IFNs in response to viral infectionThe defect in IKK activation by dsRNA was indeedor dsRNA.caused by the absence of functional PKR, as coex-pression of an epitope-tagged IKKb with wt PKR in
Results Pkr2/2 cells restored robust IKK activation by dsRNA(Figure 2B).
PKR Acts Upstream to IKK Pkr2/2 cells also exhibited a defect in IKK and NF-We established pools of HEK293 cells, stably expressing kB activation in response to VSV infection. While VSVeither wild-type (wt) or catalytically inactive [IKKa (KM) infection of wt fibroblasts resulted in IKK and NF-kBand IKKb (KA)] epitope-tagged forms of IKKa and IKKb. activation within 3 hr, the response of Pkr2/2 fibroblastsStable transfection of either wt IKKa or wt IKKb did to VSV infection was considerably attenuated and de-not considerably potentiate IKK or NF-kB activation by layed (Figure 2C). IKK and NF-kB activities were in-either dsRNA or TNFa (Figure 1A). However, expression creased only after 7 hr post infection in Pkr2/2 cells, andof IKKa (KM) resulted in a small decrease in IKK activity, even at that point both responses were weaker thanwhereas the IKKb (KA) mutant almost completely abol- those in Pkr1/1 cells. These results were not due toished dsRNA or TNFa induced IKK activity and markedly differences in susceptibility of the two cell lines to VSVreduced NF-kB activation. Although considerable IKK infection. For instance, the kinetics of viral RNA replica-activation is observed after a 10 min incubation with tion, following infection at an moi of 10, determined bydsRNA, kinase activity peaks only after 3 hr (Figure 1B). hybridization to the VSV L gene probe, were similar inTherefore, the response to dsRNA is considerably the two cell lines (see below). In addition, VSV infectionslower than the response to TNFa, which peaks within of both Pkr1/1 and Pkr2/2 cells resulted in similar levels15 to 20 min and decays within 30 to 60 min (Zandi et of JNK activation detected at 5–7 hr post infection (Fig-al., 1997; Delhase et al., 1999). IKK activation is maximal ure 2C).at 12.5 mg/ml of dsRNA. These experiments indicate that PKR acts upstream
to IKK in the response pathway elicited by either virusWe recently generated mutant mice that are deficient

JNK and IKK Required for Interferon Induction723
Figure 1. Involvement of IKK in NF-kB Acti-vation by dsRNA and Virus
(A) Parental HEK293 cells and pools ofHEK293 cells stably transfected with eitherwt or catalytically inactive IKK [IKKa (KM) andIKKb (KA)] expression vectors were treatedwith dsRNA (50 mg/ml) for 3 hr or TNFa (20 ng/ml) for 10 min and then lysed. Lysates wereassayed for IKK kinase activity (KA) by animmunecomplex kinase assay. Recovery ofIKK was determined by immunoblotting (IB)with anti-IKKa antibody. NF-kB DNA bindingactivity was measured by an electrophoreticmobility shift assay (EMSA).(B) IKK activity was measured at differenttime points after treatment of HEK293 cellswith 50 mg/ml dsRNA (left panel) or at 3 hrafter treatment with different concentrationsof dsRNA (right panel). For comparison, cellswere treated with TNFa (20 ng/ml) for 10 min.(C) MEFs derived from wt (Ikka1/1) andIkka2/2 embryos were treated or not withdsRNA. At the indicated time points, cellswere lysed, IKK was isolated by IKKg antibod-ies to measure IkB kinase activity (KA), andJNK was isolated with anti-JNK1 antibodiesto measure c-Jun kinase activity. Recoveryof IKK and JNK was determined by immu-noblotting (IB) with anti-IKKg and anti-JNK1,respectively.(D) Ikka1/1 and Ikka2/2 MEFs were infectedwith VSV (moi 5 10), and at the indicatedtimes IKK and JNK activities were measuredas described above.(E) MEFs derived from wt (Ikkb1/1) and Ikkb2/2
embryos were examined for activation of IKK,NF-kB, and JNK by dsRNA as describedabove.(F) Ikkb1/1 and Ikkb2/2 MEFs were examinedfor IKK, NF-kB, and JNK activation in re-sponse to VSV infection (moi 5 10).
infection or dsRNA. However, residual and delayed IKK serines were replaced by alanines was no longer respon-sive to either wt or catalytically inactive PKR, either inand NF-kB activation can occur in the absence of PKR.
Efficient IKK activation by VSV or dsRNA requires the vivo (Figure 3A) or in vitro (Figure 3B). Neither form ofPKR activated JNK1, JNK2 (Figure 3C), or p38 (data notIKKb catalytic subunit. On the other hand, PKR is not
involved to any extent in viral- or dsRNA-induced JNK shown). We also found that JNK activation by dsRNAis inhibited in cells, which stably express inactive mu-activation.tants of JNKK1 or JNKK2 (data not shown). We interpretthese results to suggest that PKR catalytic activity isDifferential Response of IKK and JNK to PKR
To further examine the relationships between PKR and mostly required for its autoactivation (Clemens and Elia1997; Williams, 1997), and once sufficient amounts ofthe IKK and JNK activation cascades, we cotransfected
expression vectors encoding epitope-tagged IKKb, JNK1, PKR are present it may activate IKK by direct protein–protein interaction.or JNK2 with either wt or catalytically inactive (a K296R
mutant) PKR expression vectors. Coexpression of HA-IKKb with wt PKR resulted in efficient dose-dependentIKK activation (Figure 3A). When expressed at low levels, JNK2 and IKKb Are Required for Optimal IFN
and Cytokine InductionPKR (K296R) led to poor IKK activation, but, surprisingly,when expressed at higher levels PKR (K296R) activated To determine whether the newly discovered PKR-inde-
pendent dsRNA-responsive signaling pathway that leadsIKK. To examine this point further, we tested whetherrecombinant wt PKR and PKR (K296R) purified from to JNK activation contributes to type I IFN gene induc-
tion, we used nonimmortalized (MEF) and immortalizedEscherichia coli can directly activate recombinant IKKbpurified from Sf9 cells. Although recombinant IKKb dis- (3T3) fibroblasts derived from wt, Jnk12/2, and Jnk22/2
E12 mouse embryos. We compared the induction ofplays considerable basal activity (Zandi et al., 1998), itsincubation with either wt PKR or PKR (K296R) further IFNa1 and IFNb mRNA in these cells to their induction
in Pkr2/2 fibroblasts and IKK-deficient cells. As pre-increased its IkB kinase activity (Figure 3B). This activa-tion required phosphorylation at Ser-177 and Ser-181 viously described (Yang et al., 1995), little induction of
IFNa1 or IFNb mRNA was observed in Pkr2/2 fibroblasts(Delhase et al., 1999), because a mutant in which these
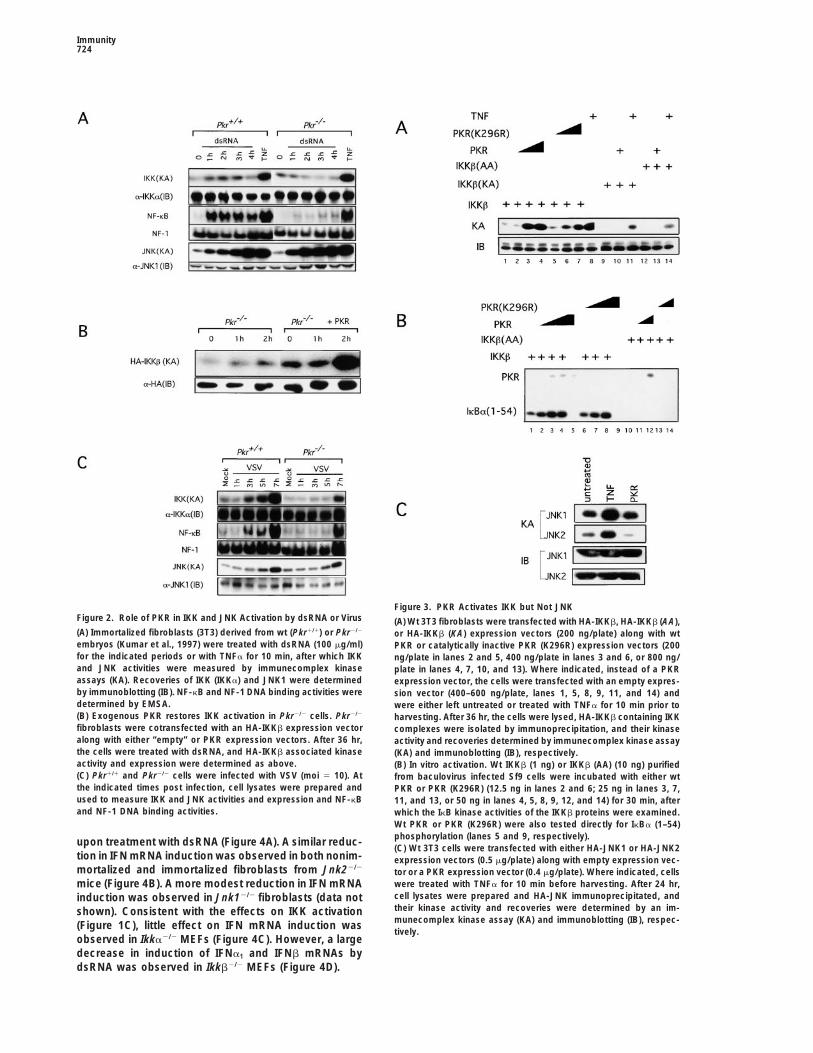
Immunity724
Figure 3. PKR Activates IKK but Not JNKFigure 2. Role of PKR in IKK and JNK Activation by dsRNA or Virus (A) Wt 3T3 fibroblasts were transfected with HA-IKKb, HA-IKKb (AA),(A) Immortalized fibroblasts (3T3) derived from wt (Pkr1/1) or Pkr2/2 or HA-IKKb (KA) expression vectors (200 ng/plate) along with wtembryos (Kumar et al., 1997) were treated with dsRNA (100 mg/ml) PKR or catalytically inactive PKR (K296R) expression vectors (200for the indicated periods or with TNFa for 10 min, after which IKK ng/plate in lanes 2 and 5, 400 ng/plate in lanes 3 and 6, or 800 ng/and JNK activities were measured by immunecomplex kinase plate in lanes 4, 7, 10, and 13). Where indicated, instead of a PKRassays (KA). Recoveries of IKK (IKKa) and JNK1 were determined expression vector, the cells were transfected with an empty expres-by immunoblotting (IB). NF-kB and NF-1 DNA binding activities were sion vector (400–600 ng/plate, lanes 1, 5, 8, 9, 11, and 14) anddetermined by EMSA. were either left untreated or treated with TNFa for 10 min prior to(B) Exogenous PKR restores IKK activation in Pkr2/2 cells. Pkr2/2 harvesting. After 36 hr, the cells were lysed, HA-IKKb containing IKKfibroblasts were cotransfected with an HA-IKKb expression vector complexes were isolated by immunoprecipitation, and their kinasealong with either “empty” or PKR expression vectors. After 36 hr, activity and recoveries determined by immunecomplex kinase assaythe cells were treated with dsRNA, and HA-IKKb associated kinase (KA) and immunoblotting (IB), respectively.activity and expression were determined as above. (B) In vitro activation. Wt IKKb (1 ng) or IKKb (AA) (10 ng) purified(C) Pkr1/1 and Pkr2/2 cells were infected with VSV (moi 5 10). At from baculovirus infected Sf9 cells were incubated with either wtthe indicated times post infection, cell lysates were prepared and PKR or PKR (K296R) (12.5 ng in lanes 2 and 6; 25 ng in lanes 3, 7,used to measure IKK and JNK activities and expression and NF-kB 11, and 13, or 50 ng in lanes 4, 5, 8, 9, 12, and 14) for 30 min, afterand NF-1 DNA binding activities. which the IkB kinase activities of the IKKb proteins were examined.
Wt PKR or PKR (K296R) were also tested directly for IkBa (1–54)phosphorylation (lanes 5 and 9, respectively).upon treatment with dsRNA (Figure 4A). A similar reduc-(C) Wt 3T3 cells were transfected with either HA-JNK1 or HA-JNK2
tion in IFN mRNA induction was observed in both nonim- expression vectors (0.5 mg/plate) along with empty expression vec-mortalized and immortalized fibroblasts from Jnk22/2
tor or a PKR expression vector (0.4 mg/plate). Where indicated, cellswere treated with TNFa for 10 min before harvesting. After 24 hr,mice (Figure 4B). A more modest reduction in IFN mRNAcell lysates were prepared and HA-JNK immunoprecipitated, andinduction was observed in Jnk12/2 fibroblasts (data nottheir kinase activity and recoveries were determined by an im-shown). Consistent with the effects on IKK activationmunecomplex kinase assay (KA) and immunoblotting (IB), respec-(Figure 1C), little effect on IFN mRNA induction wastively.
observed in Ikka2/2 MEFs (Figure 4C). However, a largedecrease in induction of IFNa1 and IFNb mRNAs bydsRNA was observed in Ikkb2/2 MEFs (Figure 4D).

JNK and IKK Required for Interferon Induction725
Figure 4. Defective IFN mRNA Induction bydsRNA in Cells Lacking PKR, JNK2, or IKKb
(A) Wt (Pkr1/1) and Pkr2/2 3T3s were left un-treated or incubated with dsRNA (100 mg/ml)for 1 hr. After washing with PBS, the cellswere incubated in complete medium for 3 hr,after which total RNA was extracted. IFNa1,IFNb, and GAPDH mRNA levels were deter-mined by Northern blot analysis.(B) Wt (Jnk21/1) and Jnk22/2 MEFs and 3T3fibroblasts were left untreated or treated withdsRNA as above. IFNa1, IFNb, and GAPDHmRNA levels were determined as describedabove.(C) Ikka1/1 and Ikka2/2 MEFs or (D) Ikkb1/1
and Ikkb2/2 MEFs were treated as above andanalyzed for induction of IFN mRNA.
We also compared induction of IFN mRNA in response or replication, because VSV genomic RNA accumulatedwith slightly faster kinetics in both Ikkb2/2 and Jnk22/2to VSV infection of the different fibroblast cultures. In-
duction of IFNa1 and IFNb mRNA was normal in Ikka2/2 cells than in wt cells. A considerable reduction in IFNgene induction in response to VSV infection also wascells (Figure 5A). However, a considerable reduction in
IFN gene induction was observed in Ikkb2/2 cells (Figure observed in Pkr2/2 cells (Figure 5D).In addition to type I IFNs, treatment of cells with5B). A partial decrease in both IFNa1 and IFNb mRNA
induction was also observed in Jnk22/2 fibroblasts (Fig- dsRNA results in induction of cytokines such as TNFa,IL-1, IL-6, and IL-12 (Gendelman et al., 1990; Verdijk eture 5C). The reduction in IFN induction in both Ikkb2/2
and Jnk22/2 cells was not due to a defect in viral infection al., 1999). We found that JNK2- and IKKb-deficient cells
Figure 5. Defective IFN mRNA Induction inResponse to VSV Infection in PKR-, IKK-, andJNK-Deficient Cells
(A) Ikka1/1 and Ikka2/2 MEFs were infectedwith VSV at moi 5 10 or mock infected. Atthe indicated times post infection (7 hr formock infected cells), total RNA was extractedand analyzed for expression of IFNa1, IFNb,and GAPDH mRNAs. VSV replication (Rep)was monitored by hybridization to a probefor the L gene.(B) Ikkb1/1 and Ikkb2/2 MEFs, (C) Jnk21/1 andJnk22/2 MEFs; and (D) Pkr1/1 and Pkr2/2 3T3swere infected and analyzed as describedabove.

Immunity726
and 7E). While both promoters were highly responsiveto VSV in wt cells, very little activation of either promoterwas observed in either Jnk22/2 or Ikkb2/2 cells, thusconfirming the results of the RNA analysis.
Discussion
Role of JNK in the Antiviral ResponseViral infection or dsRNA treatment results in inductionof type I IFNs (IFNa1 and IFNb), which in turn confer anantiviral state (Sen and Lengyel, 1992). Initially, it wasassumed that the only signaling molecule activated inresponse to virus or synthetic dsRNA, a mimic of earlyproducts of viral infections, is the dsRNA-dependentprotein kinase PKR. Not only is PKR activity and expres-sion induced by dsRNA and viral infections (Clemensand Elia, 1997; Williams, 1997), but its overexpressionprotects cultured cells from vaccinia virus (Lee and Es-teban, 1993) and encephalomyocarditis virus but notfrom VSV (Meurs et al., 1992). In addition, several viruseshave evolved different strategies to inhibit PKR activityand thereby became IFN resistant (Katze, 1992; Tayloret al., 1999). The use of certain protein kinase inhibitors,which are not particularly specific to PKR, suggestedthat PKR is essential for IFN induction (Zinn et al., 1988).It was therefore surprising that PKR-deficient mice gen-Figure 6. Defective Cytokine Induction in JNK2- and IKKb-Deficienterated by gene targeting exhibited nearly normal induc-Fibroblaststion of type I IFNs in response to viral infection or dsRNAMEFs from wild-type, Jnk22/2, and Ikkb2/2 mice were treated with
either IL-1 (20 ng/ml), LPS (10 mg/ml), or dsRNA (100 mg/ml) or left (Yang et al., 1995) and normal antiviral responses (Yanguntreated. After 4 hr, total RNA was isolated and expression of IL-6, et al., 1995; Abraham et al., 1999). The results, however,IL-12, IFNb, and GAPDH determined by Northern blot hybridization. turn out to depend on the type of virus used, and PKR-
deficient mice were recently found to be highly sensitiveto VSV infection (N. Sonnenberg, personal communica-
were also defective in induction of IL-6 and IL-12 by tion). MEFs derived from PKR-deficient mice exhibit de-dsRNA, IL-1, or LPS (Figure 6). As expected, IL-1 and fective IFN gene induction, in response to either virusLPS did not induce INFb expression in these cells. or dsRNA. This defect can be rescued by “priming” with
low levels of type I IFN (Yang et al., 1995). It was thereforesuggested that the response of Pkr2/2 mice to dsRNA isJNK2 Is Required for IFNa and IFNb
Promoter Activation caused by low-level IFN production due to backgroundexposure to environmental pathogens. Nevertheless, itTo determine whether the defect in IFN mRNA induc-
tion in Jnk22/2 cells occurs at the level of IFN gene tran- was also suggested that there must be another, PKR-independent activation pathway involved in IFN genescription, we transfected established wt, Jnk12/2, and
Jnk22/2 fibroblasts with a luciferase (LUC) reporter induction (Yang et al., 1995; Abraham et al., 1999). Ourresults identify one such signaling pathway as the onedriven by the IFNb promoter (truncated at 2125; Yone-
yama et al., 1998). After normalization for transfection that leads to JNK activation. We demonstrate that JNKactivity is potently stimulated by dsRNA or infectionefficiency (which was quite similar in the different cell
lines), the IFNb promoter was approximately 4-fold less with the RNA virus VSV, in a manner that is entirelyindependent of PKR. Furthermore, JNK activity is notactive in Jnk22/2 cells, although a residual response to
dsRNA was still observed (Figure 7A). In Jnk12/2 cells, responsive to overexpression of PKR. Using MEFs andimmortalized (3T3) fibroblast lines derived from JNK2-the response was similar to that in wt fibroblasts. Similar
results were obtained using an IFNb-CAT reporter (trun- deficient mice (Sabapathy et al., 1999), we provide ge-netic evidence that JNK activation is required for optimalcated at 2110; Figure 7B). Induction of the IFNb pro-
moter by dsRNA in Jnk22/2 cells, and to a lesser extent induction of IFNa1 and IFNb transcription.The discovery of this physiological function for thebasal promoter activity, were potentiated by cotransfec-
tion of a JNK2 vector (Figure 7C). By contrast, cotrans- JNK pathway fits nicely with previous analysis of theIFNb promoter, which contains four positive regulatoryfection of a nonactivatable JNK2 mutant, JNK2 (APF)
(Kallunki et al., 1994), reduced basal IFNb promoter ac- domains (PRDs) required for its induction by virus ordsRNA (Du et al., 1993; Thanos and Maniatis, 1995).tivity and completely prevented its induction. This mu-
tant most likely acts as a dominant-negative inhibitor of PRD IV is recognized by a heterodimer of c-Jun andATF2, as well as the structural protein HMGI(Y) (Du et al.,the residual JNK activity present in Jnk22/2 cells.
We also compared the activation of the IFNa1 (trun- 1993). c-Jun and ATF2, which belong to the AP-1/ATFfamily of transcription factors, are the most extensivelycated at -140; Yoneyama et al., 1998) and IFNb promot-
ers by VSV in wt, Jnk22/2, and Ikkb2/2 cells (Figures 7D characterized JNK substrates (Karin, 1995). In both

JNK and IKK Required for Interferon Induction727
Figure 7. JNK2 Is Involved in Control of IFNb
Gene Transcription
(A) Wt, Jnk12/2, and Jnk22/2 3T3 cells weretransiently transfected with an 2125IFNb-Luc reporter (125-Luc; Yoneyama et al.,1998). After 36 hr, the cells were left untreatedor incubated with dsRNA (100 mg/ml) for 4hr. The cells were collected and lysed, andluciferase activity was determined. The re-sults shown are averages of two independentexperiments normalized for transfection effi-ciency determined by cotransfection of aSV40-LacZ reporter and measurement ofb-galactosidase activity.(B) Wt and Jnk22/2 3T3 cells were cotrans-fected with -110IFNb-CAT (Wathelet et al., 1998)and SV40-LacZ reporters. After 48 hr, the cellswere treated or not with dsRNA for 4 hr. Thecells were collected and lysed, and CAT andb-galactosidase activities were determined.Shown are the results of a typical CAT assayand the relative fold-induction of CAT activitynormalized to b-galactosidase expression.(C) Jnk22/2 3T3 cells were cotransfected with2125IFNb-Luc and SV40-LacZ reporters andeither empty, HA-JNK2, or HA-JNK2 (APF) ex-pression vectors. After 24 hr, the cells weretreated or not with dsRNA, and cell lysateswere prepared 4 hr later. Luciferase andb-galactosidase activities were determined.Shown are average luciferase activities fromtwo independent experiments normalized tob-galactosidase levels.
(D) Wt, Ikkb2/2, and Jnk22/2 cells were transfected with 2140IFNa1-Luc (La-Luc; Yoneyama et al. 1998) and SV40-LacZ reporters. After 46hr, the cells were either mock infected or infected with VSV at m.o.i 5 10. After 9 more hr, lysates were prepared and luciferase andb-galactosidase activities determined as above.(E) Similar experiment to the one above except that an 2125IFNb-Luc reporter was used.
cases, JNK-mediated phosphorylation of specific sites points. After several hours, residual NF-kB activationcan be observed in Pkr2/2 cells. This delayed responsewithin their activation domains enhances their ability to
activate transcription (Smeal et al., 1991; Hibi et al., could, in principle, be due to the priming effect dis-cussed above or to activation of a PKR-independent1993; Derijard et al., 1994; Gupta et al., 1995). Our pres-
ent results explain how viral infection results in stimula- pathway, possibly the one that leads to JNK activation.As NF-kB binds to PRDII of the IFNb promoter (Lenardotion of c-Jun:ATF2 activity.
The inability of Pkr2/2 MEFs to respond to dsRNA with et al., 1989; Visvanathan and Goodbourn, 1989), its de-fective activation can explain the defect in IFNb induc-IFN gene induction can be remediated by priming with
small amounts of IFN (Yang et al., 1995). We therefore tion in Pkr2/2 cells. However, Pkr2/2 cells also exhibit adefect in IFNa1 induction (Yang et al., 1995; Figures 4examined whether JNK activation by dsRNA or virus
may be a secondary response that depends on low-level and 5), despite the absence of NF-kB binding sites inthe proximal IFNa1 promoter (MacDonald et al., 1990).IFN induction, which may occur even in the absence
of PKR. However, JNK activation was not inhibited by The similar reduction in both IFNa1 and IFNb mRNAinduction and promoter activation in Ikkb2/2 MEFspretreatment with the protein synthesis inhibitor cyclo-
heximide and treatment of 3T3 or HEK293 cells with strongly suggests that NF-kB is, nonetheless, involvedIFNa or IFNg did not activate JNK (W.-M. C. and M. K., in IFNa1 induction. Although the IFNa1 promoter mayunpublished data). In addition, the loss of IKKb, which lack NF-kB binding sites, NF-kB may still be recruited tocauses a substantial reduction in IFN induction (Figures this promoter via an interaction with other transcription4 and 5), has no effect on JNK activation (Figure 1). factors, such as AP-1 (Stein et al., 1993).Thus, it is unlikely that JNK activation is a consequence The defect in NF-kB activation in PKR-deficient cells isof IFN induction. due to their inability to rapidly activate the IKK complex.
Previous work has demonstrated that the IKKg/NEMOregulatory subunit is required for NF-kB activation byRole of IKK and Other Signaling PathwaysdsRNA (Yamaoka et al., 1998). Here, we show that IKKIn addition to the defect in IFN gene induction, Pkr2/2
catalytic activity is stimulated by either dsRNA or viralMEFs can not mount a normal NF-kB activation re-infection and that this response is largely dependent onsponse to dsRNA (Yang et al., 1995). We show that thisPKR. As previously shown for TNFa and IL-1 (Delhasedefect can be extended to VSV as an inducer. However,et al., 1999; Li et al., 1999, 1999; Tanaka et al., 1999),with both dsRNA and VSV, the defect in NF-kB activation
is not absolute and is mostly restricted to early time the stimulation of IKK activity by dsRNA or VSV and

Immunity728
consequently activation of NF-kB are dependent on AP-1 and NF-kB transcription factors in vertebrates (Ipand Davis, 1998; Hoffmann et al., 1999). The role of NF-IKKb, but not IKKa. In cotransfection experiments, we
find that PKR efficiently stimulates phosphorylation of kB in the control of innate immune and inflammatoryresponses in mammals is well recognized (Baldwin,IKKb at the same activating phosphoacceptor sites used
by the TNFa pathway (W.-M. C. and M. K., unpublished 1996; May and Ghosh, 1998). Recent results have estab-lished the role of the JNK pathway in induction of severaldata). As high levels of catalytically inactive PKR can
result in IKK activation, both in vivo and in vitro, we important cytokines, including IL-2, IL-4, and IFNg, inresponse to T cell activation (Dong et al., 1998; Yang etsurmise that PKR activates IKK via protein–protein inter-
action that stimulates the autophosphorylation of IKKb al., 1998; Sabapathy et al., 1999). It is also well estab-lished that JNK and IKK are activated by LPS or the(Delhase and Karin, 1999), rather than by direct phos-
phorylation of IKKb. In support of this interpretation of proinflammatory cytokines. The results presented heredemonstrate that JNK and IKK are also activated inthe results shown in Figure 3, we obtained evidence
suggesting a physical interaction between PKR and IKK response to viral infection and dsRNA. Furthermore,JNK2 and IKKb are essential elements of the innate(W.-M. C. and M. K., unpublished data). Most likely,
the catalytic activity of PKR is required for its initial response to viral infection or dsRNA, required for induc-tion of type I IFNs and other cytokines, including IL-6activation that eventually results in its transcription in-
duction and elevated expression (Clemens and Elia, and IL-12. Whereas IFNs inhibit virus replication, IL-6and IL-12, which are also induced by bacterial infec-1997; Williams, 1997).
Another viral- and dsRNA-responsive transcription tions, elicit cytotoxic responses needed for eliminationof intracellular pathogens. Thus, JNK and IKK serve asfactor involved in type I IFN gene induction is IRF3 or
IRF7 (Juang et al., 1998; Schafer et al., 1998; Wathelet universal activators of innate immune responses to bac-teria and viruses.et al., 1998; Yoneyama et al., 1998). These closely related
members of the IRF family bind to PRDI and PRDIII of Our results may also explain how dsRNA can inducecostimulation of T cells (Hoffmann et al., 1999) andthe IFNb promoter and to related elements in the IFNa1
promoter. In response to viral infection, IRF3 is phos- upregulate expression of numerous cytokines (Gendel-man et al., 1990; Verdijk et al., 1999). Thus, JNK andphorylated at a cluster of serines near its C terminus,
and this modification induces its nuclear entry and inter- IKK, being responsive to LPS and dsRNA, provide animportant link between innate and adaptive immunity. Inaction with the coactivator CBP/p300 (Juang et al., 1998;
Yoneyama et al., 1998). The protein kinase responsible this context, it is appealing to draw an analogy betweendsRNA and LPS. While the first serves as a genericfor IRF3 phosphorylation has not been identified, but
examination of the identified C-terminal phosphoaccep- signal for viral infection, the second serves as a genericsignal indicating infection by Gram-negative bacteria.tor sites suggests that they are not a target for PKR,
JNK, or IKK. Indeed, we have not been able to detect LPS and other byproducts of bacterial infections arerecognized by the Toll family of pattern recognition re-efficient phosphorylation of the C-terminal portion of
IRF3 by JNK or IKK (W.-M. C. and M. K., unpublished ceptors (Medzhitov and Janeway, 1997; Hoffmann etal., 1999). It is possible that dsRNA, whose chemicaldata).
Induction of type I IFN in most cell types is a specific properties resemble those of LPS (being a negativelycharged polysaccharide), may also activate membersresponse to viral infection that does not occur following
cell stimulation with TNFa (Thanos and Maniatis, 1995), of this rapidly growing family.IL-1, or LPS (Figure 6). However, TNFa, IL-1, and LPS
Experimental Proceduresare potent activators of the IKK and JNK pathways,which lead to stimulation of NF-kB and AP-1 activities.
PlasmidsTherefore, other transcription factors, most likely IRF3Expression vectors encoding HA-tagged wt and mutant IKKa and
and/or IRF7, that interact with IFN gene promoters, must IKKb, HA-tagged JNK1 and JNK2, wt PKR, and catalytically inactivebe responsible for this response specificity. By contrast, PKR (296R) were previously described (Kallunki et al., 1994; Kumar
et al., 1997; Zandi et al., 1997). Reporter plasmids containing IFNbactivation of AP-1 and NF-kB may be sufficient for induc-and IFNa1 promoter were kindly provided by Drs. T. Maniatis (-110tion of other cytokines, such as IL-6 and IL-12, whoseIFN-CAT; Thanos and Maniatis, 1995) and T. Fujita (-125-Luc-; -140promoters contain binding sites for these factorsLa-Luc; Yoneyama et al., 1998). IFNa1 and IFNb cDNA plasmids(Sanceau et al., 1995).were kindly provided by Dr. Y.-L. Yang.
Cell Culture, Transfection, dsRNA Treatment,The Role of JNK and IKK in Innateand Viral InfectionImmune ResponsesCells were cultured in DMEM supplemented with 10% FBS andInfection with bacterial, fungal, and viral pathogens re-antibiotics. Cells were transfected using Lipofectamine (Gibco) orsults in activation of innate immune responses, whichSuperfect (Qiagen), according to manufacturers’ recommendations.
provide the first line of defense against infection and Cells were washed twice with PBS before incubating with dsRNAalert the adaptive immune system to the presence of in serum-free medium containing dextran sulfate (5 mg/ml). VSV
strain T1026R1 (obtained from Dr. J. Lucas-Lenard, U. of Connecti-pathogens (Medzhitov and Janeway, 1997). Innate im-cut, Storrs) was used in all experiments, as it is a good IFN inducermunity has appeared early in evolution, preceding the(Boulares et al., 1996). High-titer virus stocks were prepared in BHKappearance of adaptive immunity, which is unique tocells from a single plaque isolate. Mouse cell monolayers grown tovertebrates (Hoffmann et al., 1999). Analysis of innate80%–90% confluency on 6 cm dishes were infected at an moi of
immune responses to bacteria and fungi in Drosophila 10 (based on pfu titer determined on BHK cells). Virus was absorbedhave implicated the involvement of signaling pathways at room temperature for 30 min in 0.5 ml of medium. Incubation at
378C was initiated concurrent with addition of 3.5 ml of mediumthat are highly similar to those that lead to activation of

JNK and IKK Required for Interferon Induction729
(time 0). In some experiments, resulting virus titers were determined Boulares, A.H., Ferran, M.C., and Lucaslenard, J. (1996). NF-kappa-Bactivation is delayed in mouse L929 cells infected with interferonat about 24 hr post infection (p.i.) by plaque assay on BHK cells.suppressing, but not inducing, vesicular stomatitis strains. Virology218, 71–80.Immunoprecipitation, Kinase, Electrophoretic Mobility,Clemens, M.J., and Elia, A. (1997). The double-stranded RNA-depen-and Reporter Assaysdent protein kinase PKR: structure and function. J. Interferon Cyto-Lysates were prepared and incubated with antibody at 48C over-kine Res. 17, 503–524.night. Immunoprecipitates were washed and used either for kinase
assays or for immunoblotting. Immunecomplex kinase assays, im- Darnell, J.E., Kerr, I.M., and Stark, G.R. (1994). Jak-STAT pathwaysmunoblot analysis, and electrophoretic mobility shift assay (EMSA) and transcriptional activation in response to IFNs and other extracel-were carried out as described (Zandi et al., 1997; Rothwarf et al., lular signaling proteins. Science 264, 1415–1421.1998). Wt and catalytically inactive PKR were expressed and purified Delhase, M., and Karin, M. (1999). The IkB kinase: a master regulatorfrom E. coli, while IKKb was expressed and purified from Sf9 cells of NF-kB, innate immunity and epidermal differentiation. Cold Spring(Zandi et al., 1998). The proteins were coincubated in kinase buffer Harb. Symp. Quant. Biol. 64, in press.in the presence of ATP and then the IkB kinase activity of IKKb was
Delhase, M., Hayakawa, M., Chen, Y., and Karin, M. (1999). Positiveassayed by standard procedures. CAT and Luciferase assays wereand negative regulation of IkB kinase activity through IKKb subunit
performed as described (Wathelet et al., 1998).phosphorylation. Science 284, 309–313.
Derijard, B., Hibi, M., Wu, I.-H., Barrett, T., Su, B., Deng, T., Karin,RNA Analysis
M., and Davis, R.J. (1994). JNK1: a protein kinase stimulated by UVFor Northern blot analysis, 10 mg of total RNA per lane was fraction-
light and Ha-Ras that binds and phosphorylates the c-Jun activationated on a 1% denaturing agarose gel (Yang et al., 1995). Northern
domain. Cell 76, 1025–1037.blots were hybridized with random-primed 32P-labeled probes for
DiDonato, J.A., Hayakawa, M., Rothwarf, D.M., Zandi, E., and Karin,glyceraldehyde-3-phosphate dehydrogenase (GAPDH), IFNa1, orM. (1997). A cytokine-responsive IkB kinase that activates the tran-IFNb (Yang et al., 1995). Radioactivity was quantified using a phos-scription factor NF-kB. Nature 388, 548–554.phoimager (Molecular Dynamics), and all values were normalizedDong, C., Yang, D.D., Wysk, M., Whitmarsh, A.J., Davis, R.J., andrelative to the GAPDH value in the same lane.Flavell, R.A. (1998). Defective T cell differentiation in the absenceTo analyze viral RNA expression, medium was removed from in-of Jnk1. Science 282, 2092–2095.fected cells, and monolayers were washed twice with cold PBS
before lysis in 0.2 ml of 0.05 M Tris-Cl (pH 7.6), 0.2 M NaCl, 3 mM Du, W., Thanos, D., and Maniatis, T. (1993). Mechanisms of transcrip-EDTA, 0.5% NP-40, and 10% glycerol. Nuclei were removed by tional synergism between distinct virus-inducible enhancer ele-centrifugation at 2,000 3 g for 5 min at 48C. Half of each extract ments. Cell 74, 887–898.was immediately processed for analysis of total cytoplasmic RNA Gendelman, H.E., Friedman, R.M., Joe, S., Baca, L.M., Turpin, J.A.,(viral and cellular), while the other half was first treated with micro- Dveksler, G., Meltzer, M.S., and Dieffenbach, C. (1990). A selectivecoccal nuclease (Sigma, 1.5 units, 15–30 min at 378C in the presence defect of interferon alpha production in human immunodeficiencyof 10 mM CaCl2) before RNA extraction. The latter protocol is used virus-infected monocytes. J. Exp. Med. 172, 1433–1442.to assay for VSV genomic replication, which yields encapsidated Gupta, S., Campbell, D., Derijard, B., and Davis, R.J. (1995). Tran-products resistant to nuclease digestion. RNA purification by pro- scription factor ATF2: regulation by the JNK signal transductionteinase K and phenol-chloroform as well as Northern blot analysis pathway. Science 267, 389–393.of viral RNA were carried out as described (Spadafora et al., 1996).
Hibi, M., Lin, A., Smeal, T., Minden, A., and Karin, M. (1993). Identifi-Viral genome replication was probed with 32P-labeled T7 transcriptcation of an oncoprotein-responsive and UV-responsive protein ki-containing nucleotides 286 to 2559 of L gene (plus strand).nase that binds and potentiates the c-Jun activation domain. GenesDev. 7, 2135–2148.
AcknowledgmentsHoffmann, J.A., Kafatos, F.C., Janeway, C.A., and Ezekowiz, R.A.B.(1999). Phylogenetic perspectives in innate immunity. Science 284,We thank Z. Wu for his technical assistance and T. Maniatis, T.1313–1318.Fujita, P. Pitha, P. Howley, and Y.-L. Yang for the IFN promoterHu, Y., Baud, V., Delhase, M., Zhang, P., Deerinck, T., Ellisman, M.,reporters, IRF3 expression vectors, and IFN cDNAs. We also ac-Johnson, R., and Karin, M. (1999). Abnormal morphogenesis butknowledge excellent manuscript assistance by B. Thompson. Sup-intact IKK activation in mice lacking the IKKa subunit of the IkBported by grants from the National Institutes of Health to M. K.kinase. Science 284, 316–320.(AI43477 and ES06376), J. P. (AI21572), and B. W. (AI34039). L. C.,Ihle, J.N., Witthuhn, B.A., Quelle, F.W., Yamamoto, K., Thierfelder,Y. C., and Y. H. were supported by postdoctoral fellowships fromW.E., Kreider, B., and Silvennoinen, O. (1994). Signaling by the cyto-the Giannini Foundation, the Tobacco Related Disease Researchkine superfamily: JAKs and STATs. Trends Biochem. Sci. 19,Program, and the Arthritis Foundation, respectively. M. K. is the222–227.Frank and Else Schilling-American Cancer Society Research Pro-
fessor. Ip, Y.T., and Davis, R.J. (1998). Signal transduction by the c-JunNH-2-terminal kinase (JNK)—from inflammation to development.Curr. Opin. Cell Biol. 10, 205–219.Received September 21, 1999; revised November 3, 1999.
Juang, Y.-T., Lowther, W., Kellum, M., Au, W.-C., Lin, R., Hiscott,J., and Pitha, P.M. (1998). Primary activation of interferon A andReferencesinterferon B gene transcription by interferon regulatory factor 3.Proc. Natl. Acad. Sci. USA 95, 9837–9842.Abraham, N., Stojdl, D.F., Duncan, P.I., Methot, N., Ishii, T., Dube,
M., Vanderhyden, B.C., Atkins, H.L., Gray, D.A., McBurney, M.W., Kallunki, T., Su, B., Tsigelny, I., Sluss, H.K., Derijard, B., Moore,et al. (1999). Characterization of transgenic mice with targeted dis- G., Davis, R., and Karin, M. (1994). JNK2 contains a specificity-ruption of the catalytic domain of the double-stranded RNA-depen- determining region responsible for efficient c-Jun binding and phos-dent protein kinase, PKR. J. Biol. Chem. 274, 5953–5962. phorylation. Genes Dev. 8, 2996–3007.Baldwin, A.S. (1996). The NF-kB and IkB proteins: new discoveries Karin, M. (1995). The regulation of AP-1 activity by mitogen-activatedand insights. Annu. Rev. Immunol. 14, 649–681. protein kinases. J. Biol. Chem. 270, 16483–16486.
Barnes, P.J., and Karin, M. (1997). Nuclear factor-kB—a pivotal tran- Karin, M., and Hunter, T. (1995). Transcriptional control by proteinscription factor in chronic inflammatory diseases. New Engl. J. Med. phosphorylation: signal transmission from cell surface to the nu-336, 1066–1071. cleus. Curr. Biol. 5, 747–757.
Binetruy, B., Smeal, T., and Karin, M. (1991). Ha-Ras augments c-Jun Katze, M.G. (1992). The war against the interferon-induced dsRNA-activity and stimulates phosphorylation of its activation domain. activated protein kinase: can viruses win? J. Interferon Res. 12,
241–248.Nature 351, 122–127.

Immunity730
Kumar, A., Haque, J., Lacoste, J., Hiscott, J., and Williams, B.R. Sabapathy, K., Hu, Y., Kallunki, T., Schreiber, M., David, J.-P., Jo-chum, W., Wagner, E.F., and Karin, M. (1999). JNK2 is required for(1994). Double-stranded RNA-dependent protein kinase activates
transcription factor NF-kB by phosphorylating IkB. Proc. Natl. Acad. efficient T-cell activation and apoptosis but not for normal lympho-cyte development. Curr. Biol. 9, 116–125.Sci. USA 91, 6288–6292.
Kumar, A., Yang, Y.L., Flati, V., Der, S., Kadereit, S., Deb, A., Haque, Sanceau, J., Kaisho, T., Hirano, T., and Wietzerbin, J. (1995). Trig-gering of the human interleukin-6 gene by interferon-g and tumorJ., Reis, L., Weissmann, C., and Williams, B.R. (1997). Deficient
cytokine signaling in mouse embryo fibroblasts with a targeted dele- necrosis factor-a in monocytic cells involves cooperation betweeninterferon regulatory factor-1, NFkB, and Sp1 transcription factors.tion in the PKR gene: role of IRF-1 and NF-kB. EMBO J. 16, 406–416.J. Biol. Chem. 270, 27920–27931.Kyriakis, J.M., Banerjee, P., Nikolakaki, E., Dai, T., Rubie, E.A., Ah-
mad, M.F., Avruch, J., and Woodgett, J.R. (1994). The stress-acti- Schafer, S.L., Lin, R., Moore, P.A., Hiscott, J., and Pitha, P.M. (1998).Regulation of type I interferon gene expression by interferon regula-vated protein kinase subfamily of c-Jun kinases. Nature 369,
156–160. tory factor-3. J. Biol. Chem. 273, 2714–2720.
Sen, G.C., and Lengyel, P. (1992). The interferon system. A bird’sLee, S.B., and Esteban, M. (1993). The interferon-induced double-stranded RNA-activated human p68 protein kinase inhibits the repli- eye view of its biochemistry. J. Biol. Chem. 267, 5017–5020.cation of vaccinia virus. Virology 193, 1037–1041. Smeal, T., Binetruy, B., Mercola, D., Birrer, M., and Karin, M. (1991).
Phosphorylation of cJun on serines 63 and 73 is required for onco-Lenardo, M.J., Fan, C.M., Maniatis, T., and Baltimore, D. (1989).The involvement of NF-kappa B in beta-interferon gene regulation genic and transcriptional cooperation with Ha-Ras. Nature 354,
494–496.reveals its role as widely inducible mediator of signal transduction.Cell 57, 287–294. Spadafora, D., Canter, D.M., Jackson, R.L., and Perrault, J. (1996).
Constitutive phosphorylation of the vesicular stomatitis virus P pro-Li, Q., Van Antwerp, D., Mercurio, F., Lee, K.-F., and Verma, I.M.(1999). Severe liver degeneration in mice lacking the IkB kinase 2 tein modulates polymerase complex formation but is not essential
for transcription or replication. J. Virol. 70, 4538–4548.gene. Science 284, 321–325.
Li, Z.-W., Chu, W., Hu, Y., Delhase, M., Deerinck, T., Ellisman, M., Stein, B., Baldwin, A.S., Jr., Ballard, D.W., Green, W.C., Angel, P.,and Herrlich, P. (1993). Cross-coupling of the NF-kB p65 and Fos/Johnson, R., and Karin, M. (1999). The IKKb subunit of IkB kinase
(IKK) is essential for NF-kB activation and prevention of apoptosis. Jun transcription factors produces potentiated biological function.EMBO J. 12, 3879–3891.J. Exp. Med. 189, 1839–1845.
MacDonald, N.J., Kuhl, D., Maguire, D., Naf, D., Gallant, P., Gos- Su, B., Jacinto, E., Hibi, M., Kallunki, T., Karin, M., and Ben-Neriah,Y. (1994). JNK is involved in signal integration during costimulationwamy, A., Hug, H., Bueler, H., Chaturvedi, M., de la Fuente, J., et
al. (1990). Different pathways mediate virus inducibility of the human of T lymphocytes. Cell 77, 727–736.IFN-a 1 and IFN-b genes. Cell 60, 767–779. Takeda, K., Takeuchi, O., Tsujimura, T., Itami, S., Adachi, O., Kawai,
T., Sanjo, H., Yoshikawa, K., Terada, N., and Akira, S. (1999). LimbMatsuyama, T., Kimura, T., Kitagawa, M., Pfeffer, K., Kawakami, T.,Watanabe, N., Kundig, T.M., Amakawa, R., Kishihara, K., Wakeham, and skin abnormalities in mice lacking IKKa. Science 284, 313–316.A., et al. (1993). Targeted disruption of IRF-1 or IRF-2 results in Tanaka, M., Fuentes, M.E., Yamaguchi, K., Durnin, M.H., Dalrymple,abnormal type-I IFN gene induction and aberrant lymphocyte devel- S.A., Hardy, K.L., and Goeddel, D.V. (1999). Embryonic lethality,opment. Cell 75, 83–97. liver degeneration, and impaired NF-kB activation in IKK-b-deficient
mice. Immunity 10, 421–429.May, M.J., and Ghosh, S. (1998). Signal transduction through NF-kB. Immunol. Today 19, 80–88. Taylor, D.R., Shi, S.T., Romano, P.R., Barber, G.N., and Lai, M.M.C.
(1999). Inhibition of the interferon-inducible protein kinase PKR byMedzhitov, R., and Janeway, C.A. (1997). Innate immunity: the vir-tues of a nonclonal system of recognition. Cell 91, 295–298. HCV E2 protein. Science 285, 107–110.
Thanos, D., and Maniatis, T. (1995). Virus induction of human IFNbMercurio, F., Zhu, H., Murray, B.W., Shevchenko, A., Bennett, B.L.,Li, J., Young, D.B., Barbosa, M., Mann, M., Manning, A., and Rao, gene expression requires the assembly of an enhanceosome. Cell
83, 1091–1100.A. (1997). IKK-1 and IKK-2: cytokine-activated IkB kinases essentialfor NF-kB activation. Science 278, 860–866. Verdijk, R.M., Mutis, T., Esendam, B., Kamp, J., Melief, C.J., Brand,
A., and Goulmy, E. (1999). Polyriboinosinic polyribocytidylic acidMeurs, E.F., Watanabe, Y., Kadereit, S., Barber, G.N., Katze, M.G.,Chong, K., Williams, B.R., and Hovanesian, A.G. (1992). Constitutive (poly(I:C)) induces stable maturation of functionally active human
dendritic cells. J. Immunol. 163, 57–61.expression of human double-stranded RNA-activated p68 kinase inmurine cells mediates phosphorylation of eukaryotic initiation factor Verma, I.M., Stevenson, J.K., Schwarz, E.M., Van Antwerp, D., and2 and partial resistance to encephalomyocarditis virus growth. J. Miyamoto, S. (1995). Rel/NF-kB/IkB family: intimate tales of associa-Virol. 66, 5804–5814. tion and dissociation. Genes Dev. 9, 2723–2735.Minden, A., and Karin, M. (1997). Regulation and function of the JNK Visvanathan, K.V., and Goodbourn, S. (1989). Double-stranded RNAsubgroup of MAP kinases. Biochem. Biophys. Acta. 1333, F85–F104. activates binding of NF-kappa B to an inducible element in the
human beta-interferon promoter. EMBO J. 8, 1129–1138.Muller, U., Steinhoff, U., Reis, L.F.L., Hemmi, S., Pavlovic, J., Zinker-nagel, R.M., and Aguet, M. (1994). Functional role of type I and type Wathelet, M.G., Lin, C.H., Parekh, B.S., Ronco, L.V., Howley, P.M.,II interferons in antiviral defense. Science 264, 1918–1921. and Maniatis, T. (1998). Virus infection induces the assembly of
coordinately activated transcription factors on the IFN-b enhancerOrange, J.S., and Biron, C.A. (1996). Characterization of early IL-12,IFN-ab, and TNF effects on antiviral state and NK cell responses in vivo. Mol. Cell 1, 507–518.during murine cytomegalovirus infection. J. Immunol. 156, 4746– Williams, B.R.G. (1997). Role of the double-stranded RNA-activated4756. protein kinase (PKR) in cell regulation. Biochem. Soc. Trans. 25,
509–513.Pulliam, L., Moore, D., and West, D.C. (1995). Human cytomegalovi-rus induces IL-6 and TNFa macrophages and microglial cells: possi- Yamaoka, S., Courtois, G., Bessia, C., Whiteside, S.T., Weil, R.,ble role in neurotoxicity. J. Neurovirol. 1, 219–227. Agou, F., Kirk, H.E., Kay, R.J., and Israel, A. (1998). Complementation
cloning of NEMO, a component of the IkB kinase complex essentialRegnier, C.H., Song, H.Y., Gao, X., Goeddel, D.V., Cao, Z., and Rothe,M. (1997). Identification and characterization of an IkB kinase. Cell for NF-kB activation. Cell 93, 1231–1240.90, 373–383. Yang, Y., Reis, L.F.L., Pavlovic, J., Aguzzi, A., Schafer, R., Kumar,
A., Williams, B.R.G., Aguet, M., and Weissmann, C. (1995). DeficientReis, L.F., Harada, H., Wolchok, J.D., Taniguchi, T., and Vilcek, J.(1992). Critical role of a common transcription factor, IRF-1, in the signaling in mice devoid of double-stranded RNA-dependent protein
kinase. EMBO J. 14, 6095–6106.regulation of IFN-b and IFN-inducible genes. EMBO J. 11, 185–193.
Rothwarf, D.M., Zandi, E., Natoli, G., and Karin, M. (1998). IKKg is Yang, D.D., Conze, D., Whitmarsh, A.J., Barrett, T., Davis, R.J., Rin-con, M., and Flavell, R.A. (1998). Differentiation of CD4(1) T cells toan essential regulatory subunit of the IkB kinase complex. Nature
395, 297–300. Th1 cells requires MAP kinase JNK2. Immunity 9, 575–585.

JNK and IKK Required for Interferon Induction731
Yoneyama, M., Suhara, W., Fukuhara, Y., Fukuda, M., Nishida, E.,and Fujita, T. (1998). Direct triggering of the type I interferon systemby virus infection: activation of a transcription factor complex con-taining IRF-3 and CBP/p300. EMBO J. 17, 1087–1095.
Zandi, E., Rothwarf, D.M., Delhase, M., Hayakawa, M., and Karin,M. (1997). The IkB kinase complex (IKK) contains two kinase sub-units, IKKa and IKKb, necessary for IkB phosphorylation and NF-kB activation. Cell 91, 243–252.
Zandi, E., Chen, Y., and Karin, M. (1998). Direct phosphorylation ofIkB by IKKa and IKKb: discrimination between free and NF-kB-bound substrate. Science 281, 1360–1363.
Zinn, K., Keller, A., Whittemore, L.A., and Maniatis, T. (1988). 2-Amino-purine selectively inhibits the induction of b-interferon, c-fos, andc-myc gene expression. Science 240, 210–213.




















