
IQGAP1 regulation and roles in cancer
8
Review IQGAP1 regulation and roles in cancer Michael Johnson, Manisha Sharma, Beric R. Henderson ⁎ Westmead Institute for Cancer Research, Westmead Millennium Institute at Westmead Hospital, University of Sydney, NSW 2145, Australia abstract article info Article history: Received 14 February 2009 Accepted 26 February 2009 Available online 6 March 2006 Keywords: IQGAP1 Beta-catenin Cancer Cell adhesion Cell migration IQGAP1 is a key mediator of several distinct cellular processes, in particular cytoskeletal rearrangements. Recent studies have implicated a potential role for IQGAP1 in cancer, supported by the over-expression and distinct membrane localisation of IQGAP1 observed in a range of tumours. IQGAP1 is thought to contribute to the transformed cancer cell phenotype by regulating signalling pathways involved in cell proliferation and transformation, weakening of cell:cell adhesion contacts and stimulation of cell motility and invasion. In this review we discuss these different functional and regulatory roles of IQGAP1 and its homologues in relation to their potential impact on tumourigenesis. © 2009 Elsevier Inc. All rights reserved. Contents 1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1471 2. Altered IQGAP1 expression and localisation correlates with cancer progression . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1472 2.1. Analysis of human primary tumours . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1472 2.2. IQGAP1 expression in cancer cell lines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1473 2.3. Animal modelling of IQGAP and cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1473 3. IQGAP1 functions in various signalling pathways implicated in cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1474 4. IQGAP1 in cell:cell adhesion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1475 5. IQGAP1 at membrane ruffle structures that regulate cell migration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1476 6. IQGAP1 in tumour cell invasion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1476 7. IQGAP1 in angiogenesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1476 8. Summary and future directions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1477 Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1477 References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1477 1. Introduction IQ-domain GTPase-activating proteins (IQGAPs) are an evolutionary conserved family of multi-domain proteins that regulate distinct cellular processes including cell adhesion, cell migration, extracellular signals and cytokinesis [1–3]. IQGAP1, the first of three human IQGAP homologues discovered [4], is ubiquitously expressed, while IQGAP2 and IQGAP3 are mainly restricted to liver and the intestinal tract, brain and testis, respectively [5,6]. This review will focus on the best studied of these proteins, IQGAP1. The majority of research has concentrated on IQGAP1 cytoskeletal regulation and its role in co-ordinating cadherin- mediated cell:cell adhesion, cell polarisation and actin reorganisation to promote cell migration. IQGAP1 also modulates signal transduction pathways, such as the mitogen-activated protein kinase (MAPK) cascade and developmental Wnt pathway, that affect cell proliferation and differentiation (for comprehensive review see [1]). The name IQGAP1 is derived from two of its many interacting domains: four centrally located IQ motifs and a C-terminal RasGAP- related domain (GRD; see Fig. 1). Traditional GTPase-activating proteins (GAPs) are regulatory effectors of GTPases such as those of Cellular Signalling 21 (2009) 1471–1478 Abbreviations: APC, adenomatous polyposis coli; Arp2/3, actin related protein; EGF, epidermal growth factor; EMT, epithelial-to-mesenchymal transition; ERK, extracellular signal-regulated kinase; GBM, glioblastoma multiforme; GAP, GTPase-activating protein; HCC, hepatocellular carcinoma; IQGAP1, IQ-domain GTPase-activating protein 1; MEK, MAP/ERK kinase; MAPK, mitogen-activated protein kinase; N-WASp, neural-Wiskott– Aldrich syndrome protein; ROS, reactive oxygen species; RTK, receptor tyrosine kinase; TCF, t-cell factor; VEGFR2, vascular endothelial growth factor receptor-2. ⁎ Corresponding author. Westmead Millennium Institute, Darcy Road (PO Box 412), Westmead NSW 2145, Australia. Tel.: +612 98459057; fax: +61 2 98459102. E-mail address: [email protected] (B.R. Henderson). 0898-6568/$ – see front matter © 2009 Elsevier Inc. All rights reserved. doi:10.1016/j.cellsig.2009.02.023 Contents lists available at ScienceDirect Cellular Signalling journal homepage: www.elsevier.com/locate/cellsig
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of IQGAP1 regulation and roles in cancer

Cellular Signalling 21 (2009) 1471–1478
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r.com/ locate /ce l l s ig
Review
IQGAP1 regulation and roles in cancer
Michael Johnson, Manisha Sharma, Beric R. Henderson ⁎Westmead Institute for Cancer Research, Westmead Millennium Institute at Westmead Hospital, University of Sydney, NSW 2145, Australia
Abbreviations: APC, adenomatous polyposis coli; Arpepidermal growth factor; EMT, epithelial-to-mesenchymsignal-regulated kinase; GBM, glioblastoma multiforme;HCC, hepatocellular carcinoma; IQGAP1, IQ-domain GTPMAP/ERK kinase; MAPK, mitogen-activated protein kinAldrich syndromeprotein; ROS, reactive oxygen species; RTt-cell factor; VEGFR2, vascular endothelial growth factor re⁎ Corresponding author. Westmead Millennium Instit
Westmead NSW 2145, Australia. Tel.: +61 2 98459057;E-mail address: [email protected]
0898-6568/$ – see front matter © 2009 Elsevier Inc. Adoi:10.1016/j.cellsig.2009.02.023
a b s t r a c t
a r t i c l e i n f oArticle history:Received 14 February 2009Accepted 26 February 2009Available online 6 March 2006
Keywords:IQGAP1Beta-cateninCancerCell adhesionCell migration
IQGAP1 is a key mediator of several distinct cellular processes, in particular cytoskeletal rearrangements.Recent studies have implicated a potential role for IQGAP1 in cancer, supported by the over-expression anddistinct membrane localisation of IQGAP1 observed in a range of tumours. IQGAP1 is thought to contribute tothe transformed cancer cell phenotype by regulating signalling pathways involved in cell proliferation andtransformation, weakening of cell:cell adhesion contacts and stimulation of cell motility and invasion. In thisreview we discuss these different functional and regulatory roles of IQGAP1 and its homologues in relation totheir potential impact on tumourigenesis.
© 2009 Elsevier Inc. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14712. Altered IQGAP1 expression and localisation correlates with cancer progression . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1472
2.1. Analysis of human primary tumours . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14722.2. IQGAP1 expression in cancer cell lines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14732.3. Animal modelling of IQGAP and cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1473
3. IQGAP1 functions in various signalling pathways implicated in cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14744. IQGAP1 in cell:cell adhesion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14755. IQGAP1 at membrane ruffle structures that regulate cell migration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14766. IQGAP1 in tumour cell invasion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14767. IQGAP1 in angiogenesis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14768. Summary and future directions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1477Acknowledgements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1477References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1477
1. Introduction
IQ-domain GTPase-activating proteins (IQGAPs) are an evolutionaryconserved family ofmulti-domainproteins that regulate distinct cellularprocesses including cell adhesion, cell migration, extracellular signals
2/3, actin related protein; EGF,al transition; ERK, extracellularGAP, GTPase-activating protein;ase-activating protein 1; MEK,ase; N-WASp, neural-Wiskott–K, receptor tyrosine kinase; TCF,ceptor-2.ute, Darcy Road (PO Box 412),fax: +61 2 98459102.(B.R. Henderson).
ll rights reserved.
and cytokinesis [1–3]. IQGAP1, the first of three human IQGAPhomologues discovered [4], is ubiquitously expressed, while IQGAP2and IQGAP3 are mainly restricted to liver and the intestinal tract, brainand testis, respectively [5,6]. This reviewwill focus on the best studied ofthese proteins, IQGAP1. The majority of research has concentrated onIQGAP1 cytoskeletal regulation and its role in co-ordinating cadherin-mediated cell:cell adhesion, cell polarisation and actin reorganisation topromote cell migration. IQGAP1 also modulates signal transductionpathways, suchas themitogen-activatedprotein kinase (MAPK)cascadeand developmental Wnt pathway, that affect cell proliferation anddifferentiation (for comprehensive review see [1]).
The name IQGAP1 is derived from two of its many interactingdomains: four centrally located IQ motifs and a C-terminal RasGAP-related domain (GRD; see Fig. 1). Traditional GTPase-activatingproteins (GAPs) are regulatory effectors of GTPases such as those of

Fig. 1. IQGAP1 protein map with binding domains. CHD, calponin-homology domain; coiled coil, predictedα-helical structure; WW, poly-proline protein–protein domain; IQ, four IQmotifs; GRD, Ras GTPase-activating protein related domain; RGCt, RasGAP C-terminus.
1472 M. Johnson et al. / Cellular Signalling 21 (2009) 1471–1478
the Ras superfamily. Ras proteins act as signalling switches by cyclingbetween active GTP-bound and inactive GDP-bound states. GAPsnegatively regulate Ras proteins by catalysing the switch from theGTP-active to GDP-inactive state [7]. IQGAP1, however, does not act asa traditional GAP and is able to stabilise small Rho GTPases Rac1 andCdc42 in the GTP-bound state (reviewed in [3]). IQGAP1 activity itselfis controlled by the ubiquitous calcium-binding protein, calmodulin,via the IQ motifs – classical calmodulin-interacting domains found innumerous proteins. It was hypothesised that calmodulin alters IQGAP1activity by inducing a conformational changewhich influences IQGAP1-protein interactions and/or the subcellular localisation of IQGAP1 (forextensive review see [8]). IQGAP1 also possesses a calponin-homologydomain (CHD) that binds F-actin, putative coil–coil homodimerisationdomains, a tryptophan repeat motif (WW) of undefined function and aRasGAP_C-terminus (RGCt) that binds numerous proteins includingAPC, E-cadherin and β-catenin (see Fig. 1). The broad range of inter-acting partners places IQGAP1 as a key mediator of numerous cellularprocesses and signalling pathways.
There is mounting evidence to suggest a role for IQGAP1 in cancerprogression. Here, we draw on the available clinical data and discuss
Table 1Studies on IQGAP1 in cancer cells and tumour samples.
Cancer Cell line / Tumour grade Assay Expression
Lung Squamous cell carcinoma Microarray (suppressionsubtractive hybridization)
IQGAP1 uptissue
Adenocarcinoma Immunohistochemistry High in mestructures
Adenocarcinoma:well-differentiated
Immunohistochemistry Increased
Adenocarcinoma:poorly-differentiated
Low
Endometrial Well-differentiated Immunohistochemistry andWestern blot
N/APoorly-differentiated
Ovarian Adenocarcinoma Immunohistochemistry andWestern blot
Increase
HO-8910PM (invasive) cells RT-PCR and Western blot HighGastric Adenocarcinoma Microarray (comparative
genomic hybridization)IncreasedE-cadherin
Human signet ring cell gastriccancer cell lines
Northern and Western blot, Increases i
ImmunocytochemistryWell-differentiated Immunohistochemistry and
Western blotN/A
Poorly-differentiated N/AColon Adenocarcinoma Immunohistochemical Deeper thi
strong at iHCC Well-differentiated primary
tumoursImmunohistochemistry andWestern blot
Up-regulatβ-catenin
Breast MDA-MB-231 (invasive) cellline
Western blot;immunocytochemistry
~2.7-fold icells
Infiltrating ductal carcinoma Western blot Higher comGBM Primary tumour tissue Microarray Marker of
and tumouMelanoma Secondary metastases from
microinjected cells into miceMicroarray N2.5-fold i
how increased expression patterns and specific subcellular distribu-tions of IQGAP1 may help drive progression of several cancers. Wedescribe how a surplus of IQGAP1 in a neoplastic environment canalter the physiological functions of IQGAP1, in particular cell adhesionand cell migration/invasion, themorphological changes that transpireand what effects may take place at various cellular compartments. Inaddition, we evaluate how IQGAP1 may stimulate specific pathways,such as the β-catenin/Wnt system, that may impact on cell trans-formation and cancer.
2. Altered IQGAP1 expression and localisation correlates withcancer progression
2.1. Analysis of human primary tumours
The altered localisation and increased expression of IQGAP1 arefrequently observed in tumour tissue samples and various cancer celllines. IQGAP1 is located on chromosome 15q26, a hotspot for geneamplification, particularly in diffuse-type gastric cancers [9,10]. Todate, no clinically significant mutations linked to cancer have been
level characteristics Localisation pattern Reference
-regulated compared to normal N/A [74]
tastasis-linked histological Basolateral membranes andcytoplasm
[75]
Cytoplasmic [17]
Membranous
Little E-cadherin co-localisation [15]Good E-cadherin co-localisationFocally at cell:cell contact sites atinvasive front
[14]
N/A [76]IQGAP1 and Rac1 with inversedlevels
Membranous [77]
n mRNA and protein expression Membranous [12]
Membranous [18]
Cytoplasmicrds of tumours show increasing levels;nvasive front
Strong membranous, cytoplasmic [16]
ion of IQGAP1 and nuclearin IQGAP2-null mice
Enhanced cytoplasmic pattern, lossfrom membrane
[26]
ncrease compared to normal breast High at free plasma membrane,peri-nuclear
[21,69]
pared to normal breast N/A [21]amplifying neural progenitorsr cells; up-regulated
Cytoplasmic [19,20]
ncrease N/A [23]

1473M. Johnson et al. / Cellular Signalling 21 (2009) 1471–1478
reported for IQGAP1 [11]. Microarray and immunohistochemicalanalyses of tumours give some insight into the nature of IQGAP1 inneoplastic conditions. A review of the ONCOMINE cancer microarraydatabase revealed disparity between IQGAP1 and IQGAP2 expressionpatterns [12]. IQGAP1 mRNAwas consistently up-regulated in varioustumours compared to normal tissue; whereas IQGAP2 displayedfrequent down-regulation in poorly-differentiated tumours comparedto well-differentiated tumours. Comparative genomic hybridizationanalysis of gastric adenocarcinoma cell lines identified IQGAP1 as atarget gene of the 15q26 amplicon with corresponding increases inIQGAP1 mRNA and protein levels (see Table 1; [12]). On the otherhand, IQGAP2 expression is frequently lost in gastric cancer cells duein part to gene silencing through promoter methylation [13]. The lossof IQGAP2 correlated with increased invasive potential and poorprognosis. Thus, IQGAP1 and IQGAP2 are differentially regulated ingastric cancer cells.
Immunohistochemical studies show altered expression and loca-lisation patterns of IQGAP1 in tumours, particularly at the peripheralregions of tumours, otherwise known as the “invasive front” [14–18].Cells at the invasive front – notably budding or sprouting cells – ofcolorectal and ovarian tumours displayed intense IQGAP1 staining,especially at the membrane along the sites of cell:cell contact,compared to central tumour regions and normal tissue (see Fig. 3;[14,16]). In several cancer types, prognosis and survival were poor inpatients with high IQGAP1 staining intensity and correlations werealso made between altered staining pattern (cytoplasmic to mem-brane) and higher anaplastic tumour grades (see Table 1) [14,15,17–19]. When IQGAP1 staining was explicit at the membrane there wasreduced expression of adherens junction molecules and this wasthought to contribute to reduced cell adhesion, or the “loosening up” oftumour cells (discussed in Section 4; [18]).
IQGAP1 is also consistently up-regulated in glioblastoma multi-forme (GBM). One of themost malignant tumours of the brain, GBM ischaracterised by rapid growth, extensive invasiveness and angiogen-esis [19,20]. Of particular interest was the specific expression ofIQGAP1 in a sub-type of amplifying glioblastoma cells [20]. TheIQGAP1+ tumour cell phenotype was similar to that of neuralprogenitors and represented the most aggressive population withinthe tumour mass. It is not yet knownwhy the proliferating populationof the tumour mass specifically expressed IQGAP1. One proposedexplanation is that the close proximity of IQGAP1+ cells to endothelialcells is important for cellular invasion into the brain parenchyma viathe neural vasculature [20].
2.2. IQGAP1 expression in cancer cell lines
Several invasive cancer cell lines express high levels of IQGAP1protein (see Table 1). Highly metastatic MDA-MB-231 human breastcarcinoma cells express a ~3-fold increase of IQGAP1 comparedto less invasive MCF-7 breast tumour epithelial cells and the non-tumourigenic immortalised MCF-10A breast cells [21]. Likewise,increased IQGAP1 expression correlates with the invasive potentialof human ovarian cancer cell lines and silencing IQGAP1 expressionreduced the migration and invasive potential of metastatic HO-8910PMcells [22]. More direct correlations between IQGAP1 expressionand cell invasion and metastasis have been made. Clark et al. [23]studied highly metastatic mouse melanoma cells selected frompulmonary metastases originally derived from a poorly metastaticmouse melanoma cell line injected subcutaneously into mice. Ofthe 6347 genes arrayed, IQGAP1 was one of the 32 genes whoseexpression levels were consistently increased ≥2.5-fold comparedto the parent cell line, emphasising a potential role for IQGAP1 inthe malignant and invasive phenotype. Furthermore, the stable 3-fold over-expression of IQGAP1 in MCF-7 cells induced a mea-surable increase in the in vitro and in vivo invasive behaviourcompared to control cells [21,24]. This increased invasive poten-
tial was lost upon targeted silencing of IQGAP1 [24], in starkcontrast to the effect of silencing IQGAP2 in gastric cancer cells[13].
Does IQGAP1 over-expression initiate tumour formation, or is theelevated expression observed in tumours an indirect consequence oftumour progression? The potential causative role for IQGAP1 in cancerhas not yet been directly addressed. Jadeski et al. [21] showed that astably-transfected MCF-7 breast cancer cell line that over-expressedIQGAP1 had greater proliferative potential and that knock-down ofIQGAP1 reduced certain tumour cell traits, such as independentgrowth in soft agar; however the manipulation of IQGAP1 in suchtumour-derived cells will not address its role in cell transformation.Currently, no published study has yet over-expressed IQGAP1 inprimary non-transformed cells to test its role in immortalisation,or tested whether ectopic IQGAP1 can induce cell transformationin a non-tumourigenic cell line. It will be important to performthese types of investigation and determine the stage at which IQGAP1is up-regulated and furthermore how this might contribute to thetumour cell phenotype.
2.3. Animal modelling of IQGAP and cancer
Several animal model studies have demonstrated a link betweenaltered IQGAP1/2 expression and cancer. The loss of IQGAPs did notprevent animal survival as IQGAP1-, IQGAP2-knockout and IQGAP1/IQGAP2 double-knockout mice showed no embryonic developmentaldefects [25,26]. The IQGAP1-knockout mice did express an increasedfrequency of late on-set gastric hyperplasia or benign polyps [25],suggesting an important in vivo role for IQGAP1 in maintaining thegastric epithelium.While themechanism bywhich IQGAP1 loss affectsmouse gastric cancers is not yet defined, it may reflect aberrantproliferation or hindered homeostatic shedding from the villus.Currently, we are not aware of any clinical finding of silenced IQGAP1expression in human cancer.
In contrast to IQGAP1, the targeted silencing of murine IQGAP2waslinked not to gastric hyperplasia, but rather to the development ofhepatocellular carcinoma (HCC, also known as malignant hepatoma;[25,26]). HCCs in IQGAP2-null mice characteristically expressincreased levels of cytoplasmic IQGAP1 and loss of E-cadherin andβ-catenin from the hepatocyte membrane. Potential activatingmutations in the β-catenin gene were also observed in IQGAP2-nullHCC hepatocytes and this correlated with an increase in the β-catenintranscription target, cyclin D1 [26]. The development of these tumourswas proposed to be IQGAP1-dependent based on a comparison withIQGAP1/IQGAP2 double-knockout mice, which possessed smallertumours and a survival rate similar to wild-type mice. Theconcomitant loss of IQGAP2 and increase in IQGAP1 expression inHCCs of this mouse model is comparable to that observed in humangastric cancer cell lines (see Section 2.2). The levels of IQGAP1 ingastric tissue in IQGAP2-knockout mice were unfortunately notassessed. It will be interesting to determine whether loss of IQGAP2and concomitant up-regulation of IQGAP1 is a common neoplasticevent and a key switch in driving progression of specific cancers. Theanimal studies indicate that the regulation of IQGAP1 levels isimportant in maintaining normal cellular phenotype.
The various observations made from experiments with tumoursamples, cell lines and mouse studies indicate contrasting effectsbetween the expression of IQGAP1 and IQGAP2 and cancer. Up-regulation of IQGAP1 or loss of IQGAP2 expression both correlate withthe invasive and, in some cases, the expansive potential of tumourcells. It is possible that IQGAP1 may exert an oncogenic activity whileIQGAP2 could function as a putative tumour suppressor. The tumourforming or suppressing roles of both IQGAPs require further testing. Inthe following sections we discuss how IQGAP1 contributes to otheraspects of the aggressive tumour cell phenotype in which it isconsistently observed to be up-regulated.
1474 M. Johnson et al. / Cellular Signalling 21 (2009) 1471–1478
3. IQGAP1 functions in various signalling pathways implicatedin cancer
IQGAP1 assists in both ERK- and β-catenin-dependent signalling.IQGAP1 binds B-Raf, MEK and ERK to facilitate their sequentialactivation and propagation of the MAPK cascade [27,28]. Constitutiveactivation of the MAPK pathway is a common oncogenic trigger inseveral cancers, notably those caused by Ras and B-Raf activatingmutations. Whilst IQGAP1 possesses a Ras-GRD, it is not yet clearwhether IQGAP1 binds to Ras. Early studies found no detectablebinding with Ras family members H-Ras or R-Ras [29,30], but positiveinteraction with active M-Ras [31]. Furthermore, IQGAP3, not IQGAP1,was recently shown to interact with activated H-Ras [5] and found tobe an upstream regulator of ERK-dependent proliferative signalling[5]. Therefore, IQGAPs may bind to alternate Ras members and thisselectivity may determine the fate of particular Ras-mediated signals.
IQGAP1 can also associate with various growth factor receptorsand the hyaluronin receptor, CD44, to facilitate ERK activation (seeTable 2; [32–34]). The fate of these ERK signals is still unclear, yetsome evidence points to nuclear-related functions, either in cell-cycleregulation or activation of gene transcription. EGF promotes prefer-ential binding of IQGAP1 to MEK-1 whilst reducing MEK-2 interaction[35]. MEK-1 is thought to promote proliferation while MEK-2promotes differentiation [1]. Inhibition of this cascade blocks anIQGAP1-dependent proliferative effect in MCF-7 cells [21]. Further-more, silencing of IQGAP1 abrogates ERK-mediated phosphorylationof its nuclear target Elk-1 [34,35]. Whether IQGAP1 has a significant
Table 2Tumourigenic contribution of IQGAP1.
Tumourigenicrole
Physiologicalrole
Bindingpartners
Mechanism for tumourigenicrole
Reference
Proliferation /differentiation
Growth-factorsignalling
RTK, ?Ras,B-Raf,MEK1/2,ERK1/2.
Augments growth factor-stimulated MAPK signalling,regulating ERK-mediatedphosphorylation of Elk-1transcription factor
[27,28]
HA-signalling CD44 Stimulates ERK-mediatedactivation of Elk-1transcription factor
[34]
Wnt pathwaymediation
β-catenin Recycling β-catenin frommembrane; stimulatesβ-catenin-dependenttranscription in conditionsof stable β-catenin levels
[36,60]
Invasion Cell:celladhesion
β-catenin,E-cadherin,RTKs
Loosening of cell:cellcontacts by down-regulationof E-cadherin function viacompetition for β-cateninbinding with α-catenin, orregulating RTK-mediatedE-cadherin tyrosinephosphorylation
[37,46]
Cellmigration
F-actin,Rac1/Cdc42,N-WASp,Dia1
Orchestrates cytoskeletalrearrangement at theleading edge by cross-linkageactin-filament meshwork andstimulating N-WASp-Arp2/3-dependent actinnucleation
[59,62,78]
Cellpolarisation
APC,CLIP-170
Polarises microtubule networktoward leading edge bysequestering CLIP-170 and APCto cortical regions
[56,61]
ECMdegradation
Cdc42,Sec3/Sec8;
Exocytosis of MT1-MMP,contributing to restructuringECM at invasive front oftumours
[24,69]
Angiogenesis Endothelialcell:celladhesion
VEGFR2,VE-cadherin
Up-regulated at sites ofangiogenesis to stimulateendothelial cell migration/rearrangements
[46,57,79]
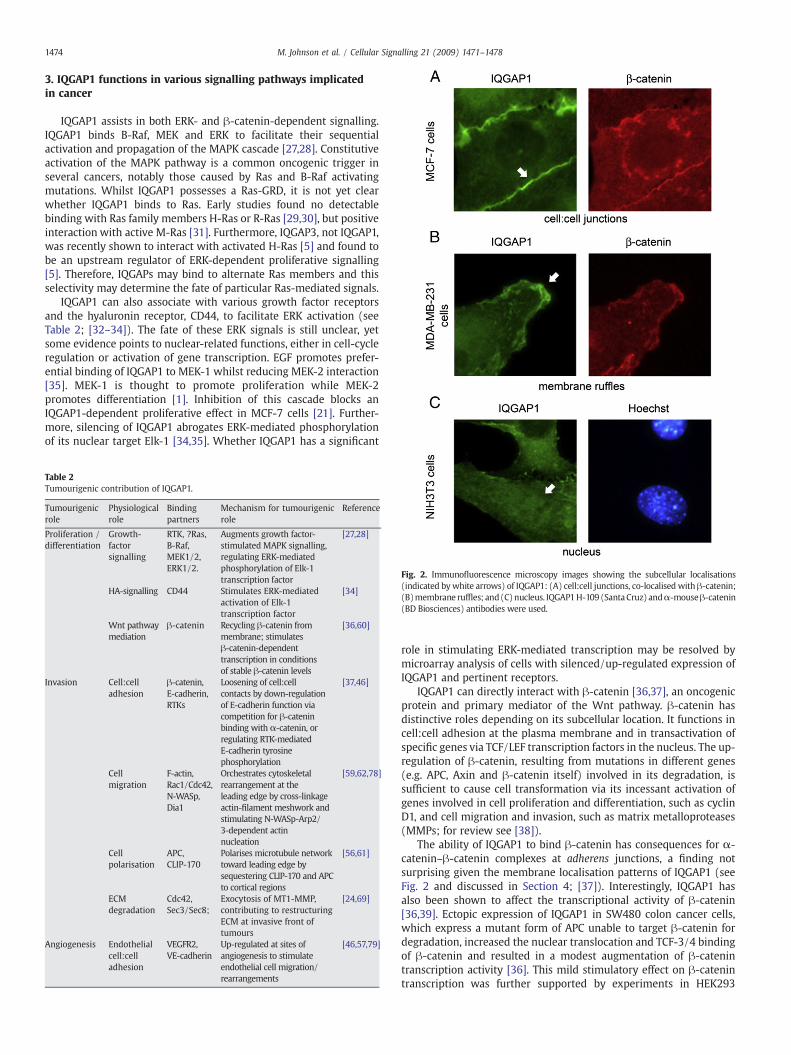
Fig. 2. Immunofluorescence microscopy images showing the subcellular localisations(indicated bywhite arrows) of IQGAP1: (A) cell:cell junctions, co-localised with β-catenin;(B)membrane ruffles; and(C)nucleus. IQGAP1H-109 (SantaCruz) andα-mouseβ-catenin(BD Biosciences) antibodies were used.
role in stimulating ERK-mediated transcription may be resolved bymicroarray analysis of cells with silenced/up-regulated expression ofIQGAP1 and pertinent receptors.
IQGAP1 can directly interact with β-catenin [36,37], an oncogenicprotein and primary mediator of the Wnt pathway. β-catenin hasdistinctive roles depending on its subcellular location. It functions incell:cell adhesion at the plasma membrane and in transactivation ofspecific genes via TCF/LEF transcription factors in the nucleus. The up-regulation of β-catenin, resulting from mutations in different genes(e.g. APC, Axin and β-catenin itself) involved in its degradation, issufficient to cause cell transformation via its incessant activation ofgenes involved in cell proliferation and differentiation, such as cyclinD1, and cell migration and invasion, such as matrix metalloproteases(MMPs; for review see [38]).
The ability of IQGAP1 to bind β-catenin has consequences for α-catenin–β-catenin complexes at adherens junctions, a finding notsurprising given the membrane localisation patterns of IQGAP1 (seeFig. 2 and discussed in Section 4; [37]). Interestingly, IQGAP1 hasalso been shown to affect the transcriptional activity of β-catenin[36,39]. Ectopic expression of IQGAP1 in SW480 colon cancer cells,which express a mutant form of APC unable to target β-catenin fordegradation, increased the nuclear translocation and TCF-3/4 bindingof β-catenin and resulted in a modest augmentation of β-catenintranscription activity [36]. This mild stimulatory effect on β-catenintranscription was further supported by experiments in HEK293

1475M. Johnson et al. / Cellular Signalling 21 (2009) 1471–1478
epithelial cells with co-expression of IQGAP1 and a β-catenin mutantresistant to APC-mediated degradation. This stimulatory effect byIQGAP1 may therefore be restricted to cells expressing stable forms ofβ-catenin and is reminiscent of HCC hepatocytes in IQGAP2-null mice.Similar effects were observed for increased expression of active Rac1[40,41]. Given that over-expression of IQGAP1 can induce expressionof active Rac1/Cdc42 [21,42], it will be interesting to test whetherIQGAP1 contributes to, or acts in concert with, a Rac1-dependentstimulation of β-catenin activity.
Themechanism bywhich IQGAP1 stimulates β-catenin-dependenttranscription is not obvious. It is possible that IQGAP1 binds β-cateninin the nucleus, although verification would require immunoprecipita-tion of fractionated cell extracts. The nuclear localisation of IQGAP1has not yet been characterised but is suggested by data in recentstudies [43]. In mouse oocytes and cleaving embryos, IQGAP1 wasdetected in the nucleus at purported stages of typical RNA polymeraseI-dependent transcription activity. Moreover, in mouse fibroblastsIQGAP1 accrues in the nucleus under conditions that mimic Wntsignalling (M.J. & B.R.H., unpublished data). Endogenous IQGAP1 andectopically expressed IQGAP1 also displayed some nuclear localisation(see Fig. 2; [37,44–46]; M.J. & B.R.H., unpublished data). It would also
Fig. 3. Comparison of IQGAP1 expression, subcellular distribution and function in cells of the tumadhesion complex by active Rac1/Cdc42 but cross-links and stabilises the actinmeshwork suppadhesion by disrupting adherens junctions by either linking RTK-mediated phosphorylationwith α-catenin for binding to β-catenin (curved grey arrow). In cells undergoing an EMTcomplexes or bind to N-cadherin directly. (C), migration. IQGAP1 binds plus-end proteinIQGAP1 also regulates the signalling from extracellular stimuli (short green arrow) i.e. growthnucleatingArp2/3-N-WASp complex andhelpdrive theplasmamembrane forward (black arrow(grey arrow). (D) invasion/ECM degradation. IQGAP1-exocyst interaction, promoted by active-Cpromote ECM degradation. (For interpretation of the references to colour in this figure legend,
be interesting to determine the effects of IQGAP1 over-expression onother nuclear events (e.g. gene expression by microarray) in cancercells, and to screen for potential IQGAP1–protein interactions in thenucleus using techniques such as mass spectrometry.
4. IQGAP1 in cell:cell adhesion
Epithelial-derived cancer cells undergo a transformation processtermed epithelial-to-mesenchymal transition (EMT) to gain a motileand invasive phenotype. The loss of functional adherens junctionsloosens cell:cell contacts and interferes with cellular apical-basalpolarity, contributing to the transformed phenotype [47]. IQGAP1locates to sites of cell:cell adhesion (see Fig. 2) and has emerged as aregulator of adherens junction stability (reviewed in [3]). In thecurrent model, IQGAP1 dissociates α-catenin from adherens junctionsthrough competitive binding for the same β-catenin binding site,leading to weakened cell:cell adhesion. Active Rac1/Cdc42, on theother hand, inhibits this process and causes IQGAP1 to insteadpositively regulate adherens junctions through stabilisation of localactin filaments (see Fig. 3A and B; reviewed in [3]). It is important tonote that conditions leading to over-expression of IQGAP1 result in
ourmass. (A) stable adherens junctions. IQGAP1 is sequestered from the cadherin–cateninorting the complex. (B), loosening of cell:cell contacts. IQGAP1 negatively regulates cell:cellof E-cadherin (short green arrow) or, when free of active-Rac1/Cdc42, competingand expressing N-cadherin, IQGAP1 may preferentially form N-cadherin–β-catenins APC or CLIP-170 to tether actin andmicrotubule networks for cortical polarisation cues.factors, to induce actin rearrangements at membrane ruffles/lamellipodia via the actin
). Atmembrane ruffles IQGAP1 also regulates the internalisationofWntmediatorβ-catenindc42, is necessary for invadopodia formation and exocytosis of MT1-MMP (grey arrow) tothe reader is referred to the web version of this article.)

1476 M. Johnson et al. / Cellular Signalling 21 (2009) 1471–1478
negative regulation of adherens junctions by IQGAP1 [37]. Cell scatterassays, which measure centrifugal spreading of epithelial cells, areused to model EMT processes and cell rearrangements during tissuemorphogenesis [47]. Such assays have been used to illustrate IQGAP1regulation of adherens junctions, and to show that silencing of IQGAP1reduced spreading of MDCKII cells [48]. Furthermore, decreases in α-catenin membrane localisation prior to cell:cell dissociation duringcell scattering led to reductions in α-catenin–β-catenin binding andcell adhesion [49] and increases in the IQGAP1–β-catenin interaction[49]. The concomitant reduction in α-catenin and increase in IQGAP1membrane localisation is also observed in diffuse-type gastric cancers[18]. These findings indicate an intimate role for IQGAP1 in EMTprocesses through direct binding to, and regulation of, cell:celljunctions in a manner that is determined by other binding partners,such as Rho-type GTPases.
During EMT, E-cadherin is down-regulated while the mesenchy-mal marker N-cadherin is up-regulated [50–52]. This cadherinswitch leads to more transient cell:cell adhesion and greater cellmobility. N-cadherin has been shown to bind IQGAP1 [53]. It willbe important to determine whether E- or N-cadherin binds IQGAP1more strongly, and how these associations are regulated. The E- to N-cadherin switch may prove significant for IQGAP1 function in tumourprogression; an E-cadherin–β-catenin–α-catenin complex leads tostable cell:cell junctions, whereas an N-cadherin–β-catenin–IQGAP1complex might favour cell migration and invasion (see Fig. 3B).
Adherens junction stability is also regulated by tyrosine phosphoryla-tionofβ-cateninorE-cadherin,whichprovokedisruptionof the complex[54]. This is one mechanism by which constitutively active tyrosinekinases contribute to neoplastic transformation [54,55]. In endothelialcells, IQGAP1 mediated the recruitment of vascular endothelial growthfactor receptor-2 (VEGFR2) to the VE-cadherin–catenin complex afterVEGF stimulation [46]. This mechanism was linked to VEGF-stimulatedreactive oxygen species (ROS)-dependent tyrosine phosphorylation ofVE-cadherin and disruption of the adhesion complex. This IQGAP1-mediated effect is an exciting advance in understanding themechanismof phospho-tyrosine-mediated regulation of adherens junctions(see Fig. 3B), but awaits demonstration in other cell model systems.
5. IQGAP1 at membrane ruffle structures that regulatecell migration
The over-expression and silencing of IQGAP1 promote andabrogate cell migration, respectively [24,56,57]. This results fromchanges in cytoskeletal rearrangements by IQGAP1 at the leading edgeof migrating cells. We recently examined the dynamics of IQGAP1 atmembrane ruffles, active membrane associated with cell migration,and found that ectopic IQGAP1 turned over quite slowly at thesestructures relative to β-catenin, which displayed a rapid turnover [58].This stable localisation of IQGAP1 is consistent with its role in cross-linking actin filaments with other cytoskeletal components andanchorage of β-catenin, APC and N-cadherin at membrane ruffles[59,60]. Moreover, IQGAP1 displayed a slower rate of macropinocy-tosis in NIH3T3 fibroblasts when compared with β-catenin, APC andN-cadherin [60], in line with the regulatory role of IQGAP1 in thisinternalisation process (see Fig. 3C).
At membrane ruffles and lamellipodia IQGAP1 acts as a scaffold totether actin and microtubule networks to plus-end proteins APC [56]and CLIP-170 [61], stimulates cytoskeletal regulators N-WASp [32,62]and Dia1 [63] and regulates involution of adherens junction compo-nents β-catenin and N-cadherin [60,64]. IQGAP1 also promotesformation of invasive structures with exocytic proteins (discussed inSection 7). Interestingly, all of these protein partners bind the RGCtdomain of IQGAP1 (the binding site of N-WASp is still under debate;see Fig. 1). The role for, and regulation of, IQGAP1 in cell migrationwasrecently reviewed [59]. Distinct signalling events are speculated toalter IQGAP1 conformation and may consequently determine which
interacting partner binds to the RGCt domain [59,62,65]. It is not yetknown whether up-regulated IQGAP1 levels alter the preference forany one of these RGCt binding partners. This may prove important inunderstanding how IQGAP1 behaves in tumourigenic conditions,particularly at plasma membrane sites.
The direct interaction of IQGAP1 with tumour suppressor APC –
mutated in N95% of colon cancers – provides an important link betweenIQGAP1, cell migration and tumour progression. A study undertaken bythe Gumbiner lab characterised the two (major) soluble APC proteincomplexes in colon tumour epithelial cells. The 20S complex signifi-cantly comprised members of the β-catenin destruction complex, aswell as some IQGAP1,whereas theheavier 60S complexcontainedminorfractions ofα-tubulin, γ-tubulin and IQGAP1 that specifically associatedwith APC [66]. Thus, a significant pool of APC associates with IQGAP1.APC and IQGAP1 localise interdependently tomembrane ruffles and thecomplex formed is required for directed cell migration in Verofibroblasts [56]. This occurs through the proposed tethering ofmicrotubule and actin networks (see Fig. 3C; [56]). The majority ofcancer mutations truncate the C-terminus of APC, producing a mutantAPC that still binds to IQGAP1 via the Arm domain (see [67]). It is notknown whether these cancer-causing mutants have altered bindingaffinity for IQGAP1 – perhaps competing with other RGCt bindingpartners – or whether their increased mobility within the cell mightinfluence the subcellular distribution and function of IQGAP1. It ispossible that these N-terminal APC mutants, which lose directmicrotubule-binding capability, may hinder IQGAP1-APC complex-mediated tethering of actin and microtubule networks, and therebyimpede directed cell migration.
6. IQGAP1 in tumour cell invasion
The IQGAP1-induced invasive potential of tumour cells may result notonly from advanced motile potential but also degradation of extracellularmatrix (ECM). ECM degradation is essential for invading tumour cells toundergo metastasis [68]. IQGAP1 has been implicated in the targeting ofmembrane type-1 matrix metalloproteinase (MT1-MMP) to smallmembrane protrusions termed invadopodia (summarised in Table 2 andFig. 3D; [69]). Invadapodia are small fillapodial-like structures of plasmamembrane involved in exocytosis and ECMdegradation [70]. IQGAP1wasfound to bind to, and act upstream of, exocyst complex components Sec3and Sec8 to target MT1-MMP to invadopodia. This process was regulatedby active Cdc42 and RhoA GTPase. The precise role for IQGAP1 at theseinvasive structures is yet to be completely defined. IQGAP1 may tethermicrotubule plus-ends to the actin meshwork via APC to allow exocystvesicledelivery, or stimulateArp2/3-N-WASp-dependentactinnucleationpromoting invadopodial protrusion and exocytic structures.
An IQGAP1–CD44 interaction may also be important for theinvasive potential of tumours. CD44 is a hyaluronon receptor involvedin cell:matrix signalling events. It is critical for oncogenic signalling,particularly in stimulating cell migration and invasion [71], and CD44over-expression in various cancers is related to metastatic potential[72]. IQGAP1 has been shown to interact directly with CD44 andmediate not only nuclear-destined ERK-2 signalling, but also relay HA-stimulated cytoskeletal rearrangements to drive cell migration (seeTable 2; [34]). Therefore, synergistic up-regulation of IQGAP1 andCD44 may assist in the lethality of the malignant phenotype of thesecancers.
7. IQGAP1 in angiogenesis
Neovascularisation is an essential process for the growth andmaintenance of tumours. When microinjected into immunocompro-mised mice, MCF-7 cells stably over-expressing IQGAP1 developedgreater neovascularised tumours relative to control [21]. In a separatestudy IQGAP1 expression was significantly up-regulated in a mousemodel of angiogenesis (see Table 2; [46]). Furthermore, knock-down

1477M. Johnson et al. / Cellular Signalling 21 (2009) 1471–1478
of IQGAP1 suppressed VEGF-induced angiogenesis in an in vivosystem [73]. The role for IQGAP1 in angiogenesis, however, stemsfrom its specific expression in endothelial cells and binding withVEGFR2 [57]. This interaction is crucial for endothelial cell rearrange-ments and migration [46,57]. IQGAP1 was also shown to directlyinteract with proto-oncogene c-Src to assist in VEGFR2-stimulatedproliferation via B-Raf signalling [73]. Although expression of IQGAP1in cancer cells per se has not been directly linked to an angiogenicresponse within the tumour mass, these data implicate IQGAP1 inseveral angiogenic responses of endothelial cells and as a potentialanti-angiogenic therapeutic target.
8. Summary and future directions
The many cellular processes that IQGAP1 orchestrates are highlyrelevant to cancer. IQGAP1 protein is not mutated but does display anunusually high expression in several cancer types, correlating withpoor outcome. Its main function is cytoskeletal rearrangement,regulating cell adhesion/migration and the internalisation andexocytosis of various molecules. The mechanism(s) by whichIQGAP1 contributes to cancer progression appears to involve acomplex interplay of cytoskeletal regulation and cell transformationpathways. IQGAP1 action has been characterised at themembrane andcytoplasm, but may also occur in the nucleus. The majority ofimmunohistochemical studies detect IQGAP1 at the membrane incancer cells, where it reduces cell:cell adhesion or promotes actinrearrangements at membrane ruffles to aid cell migration, or bindsvarious membrane-bound receptors to assist in the signalling ofextracellular stimuli for cell growth or invasion. In regard to IQGAP1subcellular distribution, the evidence for nuclear IQGAP1 requiresmore definitive analysis in primary tumours and tumour cell lines byconfocal microscopy or biochemical fractionation. Some of the majorunanswered questions concern how IQGAP1 contributes to theaggressive phenotype in which it is consistently up-regulated. IsIQGAP1 oncogenic?What triggers its up-regulation and is this an earlyor late event? Moreover, what key binding partner(s) augment thetumourigenic role of IQGAP1?
Cell transformation assays are required to answer whether IQGAP1has oncogenic or transformation-inducing capacity (see Section 2.2).However this may be unlikely given that IQGAP1 over-expression isusually restricted to poorly-differentiated tumours and cells at theinvasive front of tumours. Further insight into the mechanism ofIQGAP1 up-regulation is necessary. IQGAP1 over-expression mayresult from earlier genetic aberrations that trigger the transformedphenotype. The amplification of IQGAP1 is regularly observed ingastric cancer and silencing of IQGAP2 in mice can cause IQGAP1 over-expression. This may reflect a feed-back effect, whereby loweringexpression of one IQGAP member elevates another. This may howeverbe restricted to organs where IQGAP2 or IQGAP3 are specificallyexpressed. Furthermore, the range of cancers in which IQGAP1 is up-regulated reflects its ubiquitous expression in the body. Its role in eachcancer type may depend on the genetic mutation status or expressionof its various binding partners. Mass spectrometry-aided peptideanalyses will help to define the repertoire of IQGAP1 binding partnersin different subcellular fractions and in cells undergoing differentstages of transformation or EMT processes. As IQGAP1 sits at thecrossroads of various cellular processes, understanding the mechan-isms behind the role(s) of IQGAP1 in promoting or sustaining thetransformed cell phenotype may lead to the development of viabletherapeutic targets for advanced tumours.
Acknowledgements
Our research into the area of IQGAP1, Wnt signalling and cancerhas been supported by the National Health and Medical ResearchCouncil of Australia and the Australian Research Council.
References
[1] M.D. Brown, D.B. Sacks, Trends Cell Biol. 16 (2006) 242.[2] L.M. Machesky, Curr. Biol. 8 (1998) R202.[3] J. Noritake, T. Watanabe, K. Sato, S. Wang, K. Kaibuchi, J. Cell Sci. 118 (2005) 2085.[4] L. Weissbach, J. Settleman, M.F. Kalady, A.J. Snijders, A.E. Murthy, Y.X. Yan,
A. Bernards, J. Biol. Chem. 269 (1994) 20517.[5] H. Nojima, M. Adachi, T. Matsui, K. Okawa, S. Tsukita, S. Tsukita, Nat. Cell Biol. 10
(2008) 971.[6] S. Wang, T. Watanabe, J. Noritake, M. Fukata, T. Yoshimura, N. Itoh, T. Harada,
M. Nakagawa, Y. Matsuura, N. Arimura, K. Kaibuchi, J. Cell Sci. 120 (2007) 567.[7] M. Fukata, M. Nakagawa, K. Kaibuchi, Curr. Opin. Cell Biol. 15 (2003) 590.[8] M.W. Briggs, D.B. Sacks, FEBS Lett. 542 (2003) 7.[9] Y. Fukuda, N. Kurihara, I. Imoto, K. Yasui, M. Yoshida, K. Yanagihara, J. Park,
Y. Nakamura, J. Inazawa, Genes Chromosomes Cancer 29 (2000) 315.[10] N. Sugimoto, I. Imoto, Y. Fukuda, N. Kurihara, S. Kuroda, A. Tanigami, K. Kaibuchi,
R. Kamiyama, J. Inazawa, J. Hum. Genet. 45 (2001) 21.[11] L.E. Morris, G.S. Bloom, H.F. Frierson, S.M. Powell, Genes Chromosomes. Genomes
42 (2005) 280.[12] http://www.oncomine.org, Oncomine (Research) – Cancer Profiling Database.[13] S.H. Jin, Y. Akiyama, H. Fukamachi, K. Yanagihara, T. Akashi, Y. Yuasa, Int. J. Cancer
122 (2008) 1040.[14] P. Dong, K. Nabeshima, N. Nishimura, T. Kawakami, T. Hachisuga, T. Kawarabayashi,
H. Iwasaki, Cancer Lett. 243 (2006) 120.[15] S. Miyamoto, H. Baba, S. Kuroda, K. Kaibuchi, T. Fukuda, Y. Maehara, T. Saito,
Br. J. Cancer 83 (2000) 1168.[16] K. Nabeshima, Y. Shimao, T. Inoue, M. Koono, Cancer Lett. 176 (2002) 101.[17] H. Nakamura, K. Fujita, H. Nakagawa, F. Kishi, A. Takeuchi, I. Aute, H. Kato, Oncol.
Rep. (2005) 427.[18] H. Takemoto, Y. Doki, H. Shiozaki, H. Imamura, T. Utsunomiya, H. Miyata, M. Yano,
M. Inoue, Y. Fujiwara, M. Monden, Int. J. Cancer 91 (2001) 783.[19] K. McDonald, M. O'Sullivan, J. Parkinson, J. Shaw, C. Payne, J. Brewer, L. Young,
D. Reader, H. Wheeler, R. Cook, M. Biggs, N. Little, C. Teo, G. Stone, B. Robinson,J. Neuropathol. Exp. Neurol. 66 (2007) 405.
[20] L. Balenci, I.D. Clarke, P.B. Dirks, N. Assard, F. Ducray, A. Jouvet, M.-F. Belin,J. Honnorat, J. Baudier, Cancer Res. 66 (2006) 9074.
[21] L. Jadeski, J.M. Mataraza, H.-W. Jeong, Z. Li, D.B. Sacks, J. Biol. Chem. 283 (2008) 1008.[22] P.X. Dong, N. Jia, Z.J. Xu, Y.T. Liu, D.J. Li, Y.J. Feng, J. Exp. Clin. Cancer Res. 27 (2008)
77.[23] E.A. Clark, T.R. Golub, E.S. Lander, R.O. Hynes, Nature 406 (2000) 532.[24] J.M. Mataraza, M.W. Briggs, Z. Li, A. Entwistle, A.J. Ridley, D.B. Sacks, J. Biol. Chem.
278 (2003) 41237.[25] S. Li, Q.Wang, A. Chakladar, R.T. Bronson, A. Bernards, Mol. Cell Biol. 20 (2000) 697.[26] V.A. Schmidt, C.S. Chiariello, E. Capilla, F. Miller, W.F. Bahou, Mol. Cell Biol. 28
(2008) 1489.[27] J. Ren, Z. Li, D.B. Sacks, PNAS 104 (2007) 10465.[28] M. Roy, Z. Li, D.B. Sacks, Mol. Cell Biol. 25 (2005) 7940.[29] M.J. Hart, M.G. Callow, B. Souza, P. Polakis, EMBO J. 15 (1996) 2997.[30] S.J. McCallum, W.J. Wu, R.A. Cerione, J. Biol. Chem. 271 (1996) 21732.[31] J. Vasilescu, X. Guo, J. Kast, Proteomics 4 (2004) 3845.[32] L.B. Benseñor, H.-M. Kan, N. Wang, H. Wallrabe, L.A. Davidson, Y. Cai, D.A.
Schafer, G.S. Bloom, J. Cell Sci. 120 (2007) 658.[33] B. Blagoev, I. Kratchmarova, S.E. Ong, M. Nielsen, L.J. Foster, M. Mann, Nat.
Biotechnol. 21 (2003) 315.[34] L.Y.W. Bourguignon, E. Gilad, K. Rothman, K. Peyrollier, J. Biol. Chem. 280 (2005)
11961.[35] M. Roy, D.B. Sacks, J. Biol. Chem. 279 (2004) 17329.[36] M.W. Briggs, Z. Li, D.B. Sacks, J. Biol. Chem. 277 (2002) 7453.[37] S. Kuroda, M. Fukata, M. Nakagawa, K. Fujii, T. Nakamura, T. Ookubo, I. Izawa, T.
Nagase, N. Nomura, H. Tani, I. Shoji, Y. Matsuura, S. Yonehara, K. Kaibuchi, Science281 (1998) 832.
[38] P. Polakis, Curr. Opin. Genet. Dev. 17 (2007) 45.[39] Y. Wang, A. Wang, F. Wang, M. Wang, M. Zhu, Y. Ma, R. Wu, Exp Mol Pathol, 85
(2008) 122.[40] S. Esufali, B. Bapat, Oncogene 23 (2004) 8260.[41] X. Wu, X. Tu, K.S. Joeng, M.J. Hilton, D.A. Williams, F. Long, Cell 133 (2008) 340.[42] J.M. Mataraza, M.W. Briggs, Z. Li, R. Frank, D.B. Sacks, Biochem. Biophys. Res.
Commun. 305 (2003) 315.[43] A. Bielak-Zmijewska, A. Kolano, K. Szczepanska, M. Maleszewski, E. Borsuk, Dev.
Biol. 322 (2008) 21.[44] S.C. Mateer, L.E. Morris, D.A. Cromer, L.B. Benseñor, G. Bloom, Cell Motil.
Cytoskeleton 58 (2004) 231.[45] S.C. Mateer, A.E. McDaniel, V. Nicolas, G.M. Habermacher, M.-J.S. Lin, D.A. Cromer,
M.E. King, G.S. Bloom, J. Biol. Chem. 277 (2002) 12324.[46] M. Yamaoka-Tojo, T. Tojo, H.W. Kim, L. Hilenski, N.A. Patrushev, L. Zhang, T. Fukai,
M. Ushio-Fukai, Arterioscler. Thromb. Vasc. Biol. 26 (2006) 1991.[47] M. Guarino, Int. J. Biochem. Cell Biol. 39 (2007) 2153.[48] J. Noritake, M. Fukata, K. Sato, M. Nakagawa, T. Watanabe, N. Izumi, S. Wang,
Y. Fukata, K. Kaibuchi, Mol. Biol. Cell 15 (2004) 1065.[49] M. Fukata, M. Nakagawa, N. Itoh, A. Kawajiri, M. Yamaga, S. Kuroda, K. Kaibuchi,
Mol. Cell Biol. 21 (2001) 2165.[50] H. Acloque, J.P. Thiery, M.A. Nieto, EMBO J. 9 (2008) 322.[51] M. Guarino, B. Rubino, G. Ballabio, Pathology 39 (2007) 305.[52] R.B. Hazan, R. Qiao, R. Keren, I. Badano, K. Suyama, Ann. N.Y. Acad. Sci.1014 (2004) 155.[53] C. Schrick, A. Fischer, D.P. Srivastava, N.C. Tronson, P. Penzes, J. Radulovic, Neuron
55 (2007) 786.

1478 M. Johnson et al. / Cellular Signalling 21 (2009) 1471–1478
[54] J. Lilien, J. Balsamo, Curr. Opin. Cell Biol. 17 (2005) 459.[55] W.J. Nelson, Biochem. Soc. Trans. 036 (2008) 149.[56] T. Watanabe, S. Wang, J. Noritake, K. Sato, M. Fukata, M. Takefuji, M. Nakagawa,
N. Izumi, T. Akiyama, K. Kaibuchi, Dev. Cell 7 (2004) 871.[57] M. Yamaoka-Tojo, M. Ushio-Fukai, L. Hilenski, S.I. Dikalov, Y.E. Chen, T. Tojo,
T. Fukai, M. Fujimoto, N.A. Patrushev, N. Wang, C.D. Kontos, G.S. Bloom, R.W. Alexander,Circ. Res. 95 (2004) 276.
[58] M. Johnson, M. Sharma, C. Jamieson, J.M. Henderson, M.T.S. Mok, L. Bendall,B.R. Henderson, Cell. Signal. 21 (2009) 339.
[59] D.T. Brandt, R. Grosse, EMBO Rep. 8 (2007) 1019.[60] M. Sharma, B.R. Henderson, J. Biol. Chem. 282 (2007) 8545.[61] M. Fukata, T. Watanabe, J. Noritake, M. Nakagawa, M. Yamaga, S. Kuroda, Y. Matsuura,
A. Iwamatsu, F. Perez, K. Kaibuchi, Cell 109 (2002) 873.[62] C. Le Clainche, D. Schlaepfer, A. Ferrari, M. Klingauf, K. Grohmanova, A. Veligodskiy,
D. Didry, D. Le, C. Egile, M.-F. Carlier, R. Kroschewski, J. Biol. Chem. 282 (2007) 426.[63] D.T. Brandt, S. Marion, G. Griffiths, T. Watanabe, K. Kaibuchi, R. Grosse, J. Cell. Biol.
178 (2007) 193.[64] S. Etienne-Manneville, A. Hall, Nature 421 (2003) 753.[65] K. Grohmanova, D. Schlaepfer, D. Hess, P. Gutierrez, M. Beck, R. Kroschewski, J. Biol.
Chem. 279 (2004) 48495.[66] S. Mahadevaiyer, C. Xu, B.M. Gumbiner, Biochim. Biophys. Acta 1773 (2007) 120.[67] M. Brocardo, B.R. Henderson, Trends Cell Biol. 18 (2008) 587.
[68] H. Sato, T. Takino, H. Miyamori, Cancer Sci. 96 (2005) 212.[69] M. Sakurai-Yageta, C. Recchi, G. Le Dez, J.-B. Sibarita, L. Daviet, J. Camonis,
C. D'Souza-Schorey, P. Chavrier, J. Cell. Biol. 181 (2008) 985.[70] S.S. Stylli, A.H. Kaye, P. Lock, J. Clin. Neurosci. 15 (2008) 725.[71] R. Marhaba, M. Zöller, J. Mol. Histol. 35 (2004) 211.[72] S. Jothy, Clin. Exp. Metastasis 20 (2003) 195.[73] R.D. Meyer, D.B. Sacks, N. Rahimi, PLoS ONE 3 (2008) e3848.[74] W. Sun, K. Zhang, X. Zhang, W. Lei, T. Xiao, J. Ma, S. Guo, S. Shao, H. Zhang, Y. Liu,
J. Yuan, Z. Hu, Y. Ma, X. Feng, S. Hu, J. Zhou, S. Cheng, Y. Gao, Cancer Lett. 212(2004) 83.
[75] T.Miyoshi, T. Shirakusa, Y. Ishikawa, A. Iwasaki, T. Shiraishi, Y.Makimoto, H. Iwasaki,K. Nabeshima, Pathol. Int. 55 (2005) 419.
[76] P.X. Dong, N. Jia, Z.J. Xu, Y.T. Liu, D.J. Li, Y.J. Feng, J. Exp. Clin. Cancer Res. 27 (2008)77.
[77] A. Walch, S. Seidl, C. Hermannstadter, S. Rauser, J. Deplazes, R. Langer, C.H. vonWeyhern, M. Sarbia, R. Busch, M. Feith, S. Gillen, H. Hofler, B. Luber, Mod. Path. 21(2008) 544.
[78] J.M. Swart-Mataraza, Z. Li, D.B. Sacks, J. Biol. Chem. 277 (2002) 24753.[79] P. Yue, D. Wong, W.Y. Ha, M.C. Fung, N.K. Mak, H.W. Yeung, H.W. Leung, K. Chan,
L. Liu, T.P.D. Fan, R. Wong, Angiogenesis 8 (2005) 205.




















