Characterizing Parent–Child Interactions in Families of Autistic ...

ORIGINAL ARTICLE
Involvement of the PRKCB1 gene in autistic disorder:significant genetic association and reduced neocorticalgene expressionC Lintas1,2,14, R Sacco1,2,14, K Garbett3, K Mirnics3,4, R Militerni5, C Bravaccio6, P Curatolo7, B Manzi7,
C Schneider8, R Melmed9, M Elia10, T Pascucci11,12, S Puglisi-Allegra11,12, K-L Reichelt13 and
AM Persico1,2
1Laboratory of Molecular Psychiatry and Neurogenetics, University ‘Campus Bio-Medico’, Rome, Italy; 2Laboratory ofMolecular Psychiatry and Psychiatric Genetics, Department of Experimental Neurosciences, I.R.C.C.S. ‘Fondazione SantaLucia’, Rome, Italy; 3Department of Psychiatry, Vanderbilt University, Nashville, TN, USA; 4Kennedy Center for Research onHuman Development, Vanderbilt University, Nashville, TN, USA; 5Department of Child Neuropsychiatry, II University ofNaples, Naples, Italy; 6Department of Child Neuropsychiatry, University ‘Federico II’, Naples, Italy; 7Department of ChildNeuropsychiatry, University ‘Tor Vergata’, Rome, Italy; 8Center for Autism Research and Education, Phoenix, AZ,USA; 9Southwest Autism Research and Resource Center, Phoenix, AZ, USA; 10Unit of Neurology and ClinicalNeurophysiopathology, I.R.C.C.S. ‘Oasi Maria S.S.’, Troina (EN), Italy; 11Department of Psychology, University ‘La Sapienza’,Rome, Italy; 12Laboratory of Behavioral Neurobiology, Department of Experimental Neurosciences, I.R.C.C.S. ‘FondazioneSanta Lucia’, Rome, Italy and 13Department of Pediatric Research, Rikshospitalet, University of Oslo, Oslo, Norway
Protein kinase C enzymes play an important role in signal transduction, regulation of geneexpression and control of cell division and differentiation. The fsI and bII isoenzymes resultfrom the alternative splicing of the PKCb gene (PRKCB1), previously found to be associatedwith autism. We performed a family-based association study in 229 simplex and 5 multiplexfamilies, and a postmortem study of PRKCB1 gene expression in temporocortical gray matter(BA41/42) of 11 autistic patients and controls. PRKCB1 gene haplotypes are significantlyassociated with autism (P < 0.05) and have the autistic endophenotype of enhancedoligopeptiduria (P < 0.05). Temporocortical PRKCB1 gene expression was reduced on averageby 35 and 31% for the PRKCB1-1 and PRKCB1-2 isoforms (P < 0.01 and < 0.05, respectively)according to qPCR. Protein amounts measured for the PKCbII isoform were similarlydecreased by 35% (P = 0.05). Decreased gene expression characterized patients carrying the‘normal’ PRKCB1 alleles, whereas patients homozygous for the autism-associated allelesdisplayed mRNA levels comparable to those of controls. Whole genome expression analysisunveiled a partial disruption in the coordinated expression of PKCb-driven genes, includingseveral cytokines. These results confirm the association between autism and PRKCB1 genevariants, point toward PKCb roles in altered epithelial permeability, demonstrate a significantdownregulation of brain PRKCB1 gene expression in autism and suggest that it couldrepresent a compensatory adjustment aimed at limiting an ongoing dysreactive immuneprocess. Altogether, these data underscore potential PKCb roles in autism pathogenesis andspur interest in the identification and functional characterization of PRKCB1 gene variantsconferring autism vulnerability.Molecular Psychiatry (2009) 14, 705–718; doi:10.1038/mp.2008.21; published online 4 March 2008
Keywords: autism; pervasive developmental disorders; PRKCB1; protein kinase C-b; temporalcortex; TGF-b
Introduction
Autism is a severe neuropsychiatric disorder char-acterized by impaired language, communication and
social skills, and by repetitive and stereotypicbehaviors.1 Family and twin studies support stronggenetic contributions to this disease.2 However, theheterogeneity of clinical symptoms and the complex-ity of underlying pathogenetic processes have untilnow undermined efforts to achieve reproduciblegenotype–phenotype correlations. On one hand, thephenotypic expression of autism-predisposing genesspans from minimal autistic traits to full-blownautism, identifying a broad clinical entity referred toas ‘autism-spectrum disorder’ (ASD).2,3 On the other
Received 4 August 2007; revised 23 January 2008; accepted 23January 2008; published online 4 March 2008
Correspondence: Dr AM Persico, Laboratory of MolecularPsychiatry and Neurogenetics, University ‘Campus Bio-Medico’,Via Alvaro del Portillo 21, I-00128 Rome, Italy.E-mail: [email protected] authors contributed equally to this study.
Molecular Psychiatry (2009) 14, 705–718& 2009 Nature Publishing Group All rights reserved 1359-4184/09 $32.00
www.nature.com/mp

hand, the genetic underpinnings of autism encompasssignificant interindividual heterogeneity, numerouscontributing loci, epistasis and likely gene–environ-ment interactions.2 Incomplete penetrance (that is,individuals carrying autism genes, but not fulfillingdiagnostic criteria for the ‘affected’ status) andphenocopies (individuals carrying no genetic predis-position and fulfilling diagnostic criteria for ‘autism’solely due to environmental factors) further decreasethe statistical power of genetic analyses by introdu-cing false-negative and false-positive ‘affection status’definitions. In this complex scenario, the probabilityof success can be maximized by employing in parallelboth genetic analyses and postmortem assessments ofbrain gene expression patterns. This strategy canindeed promote a better understanding of single-genecontributions to complex disorders, as recentlydemonstrated for the MET gene in autism.4,5
Several lines of evidence suggest that autismshould be viewed as a multiorgan systemic disorderwith a prenatal onset. On one hand, autism does notsolely affect the central nervous system (CNS),despite encompassing obvious neurodevelopmentalcomponents: systemic signs and symptoms includemacrosomy,6 excessive intestinal permeability andnonspecific enterocolitis,7–9 immune dysreactivity9
and renal oligopeptiduria.10 On the other hand, theneurodevelopmental mechanisms underlying theCNS abnormalities found in postmortem studies,which include reduced programmed cell death and/or increased cell proliferation, and altered neuronalmigration, differentiation and synaptogenesis, withthe exception of the latter, are all active prenatally,especially during the first trimester of pregnancy.2,11,12
Indeed, many children later diagnosed with ASDdisplay motor abnormalities13 and/or excessive bodygrowth6 already on the day of birth or in earlyneonatal life. Therefore, genes characterized by anearly onset of expression and encoding proteinsinvolved in the control of cell division, differentiationand migration can represent attractive candidates forautistic disorder even if their tissue distributionpatterns are not necessarily restricted to the CNS.
The PRKCB1 gene, located in human ch. 16p11.2,represents an interesting locus displaying expressionpatterns not restricted to the CNS and previouslyfound associated with autism.14 In general, proteinkinase C enzymes play an important role in signaltransduction, regulation of gene expression andcontrol of cell division and differentiation. The alter-native splicing of PRKCB1 generates two mRNAisoforms named PRKCB1-1 and PRKCB1-2, yieldingthe two PKCb isoenzymes bI and bII. Interestingly,either or both isoforms are expressed in the districtsmost affected in autistic disorder, namely the CNS,immune system, digestive tract and kidney. In parti-cular, brain PRKCB1 is expressed in hippocampus,striatum, suprachiasmatic nucleus and cerebellargranule cells, where PKCbI influences circadianrhythms, learning and memory, whereas PKCbII isinvolved in fear conditioning.15–19 In the immune
system, PKCb isoenzymes play a critical role in B-cellreceptor-mediated responses, T-cell migration andcytokine secretion, dendritic cell differentiation andmonocyte and macrophage functioning.20–25 In thegut, PKCbI and bII are expressed by epithelial cells,where they regulate intestinal permeability andcell proliferation, respectively.26–28 In the kidney,both PKCb isoforms are expressed by mesangial cellsand play a major role in diabetic nephropathy andalbuminuria, possibly through oxidative stress.29–31
Interestingly, many of the non-neural signs andsymptoms that often accompany autism strikinglyoverlap with the pathophysiological roles played byPKCb isoforms in districts outside of the CNS.
The chromosomal region encompassing the PRKCB1locus was initially identified using a direct identity-by-descent mapping method as one of seven regionslinked to autism, each spanning only between 0.75 and4.0 Mb.14 The same study described an associationbetween PRKCB1 gene variants and autism bothin Caucasian-Americans and in Mexican-Americans,albeit with the two ethnic groups carrying vulnerabilityalleles marked by different haplotypes.14 This resultwas later not replicated in an Irish sample (seeDiscussion).32 Still, the presence of a significant geneticassociation in two different ethnic groups, coupledwith the functional involvement of PRKCB1 in manypathophysiological processes potentially underlyingdisease-related endophenotypes, spur interest intoadditional studies of PRKCB1 in autism. In accordancewith the strategy outlined above and previouslyemployed successfully with the MET gene,4,5 this studywas designed to replicate the initial genetic findingsin an independent sample and to extend these studiesby (a) assessing PRKCB1 gene expression in postmortemtemporocortical gray matter (BA41/42) from 11 pairs ofASD patients and sex-, age-, and postmortem interval-matched controls; (b) correlating PRKCB1 gene expres-sion with PRKCB1 genotypes in these postmortem brainsamples and (c) assessing the functional consequencesof dysregulated PRKCB1 gene expression using genome-wide expression array technology. Our results confirmthe existence of a significant association betweenPRKCB1 gene variants and autism, describe for the firsttime an association with the biochemical endopheno-type defined by enhanced urinary peptide excretionrates, detect a significant reduction of PRKCB1 mRNAand protein levels in postmortem autistic brains, reveala dysregulation of gene expression patterns delineatinga possible compensatory adjustment aimed at limitingan ongoing immune dysreactive process and identify alack of downregulation in gene expression as the mostlikely functional correlate of PRKCB1 alleles conferringautism vulnerability.
Subjects and methods
Subjects recruited for the family-based associationstudyA total of 229 simplex and 5 multiplex families witha non-syndromic autistic proband were recruited for
PRKCB1 gene and autistic disorderC Lintas et al
706
Molecular Psychiatry
this study. The composition of our clinical sample,encompassing 239 autistic patients and 90 unaffectedsiblings, is summarized in Table 1. Demographic andclinical characteristics as well as diagnostic screeningprocedures used to exclude syndromic autism havebeen previously described.33 Briefly, patients fulfill-ing DSM-IV diagnostic criteria for Autistic Disorder1
were screened for non-syndromic autism usingmagnetic resonance imaging, electroencephalogram,audiometry, urinary aminoacid, and organic acidmeasurements, cytogenetic and fragile-X testing.Patients with dysmorphic features were excludedeven in the absence of detectable cytogenetic altera-tions. Patients with sporadic seizures (that is, < 1every 6 months) were included; patients with fre-quent seizures or focal neurological deficits wereexcluded. Autistic behaviors were assessed using theofficial Italian version of the Autism DiagnosticObservation Schedule34 and the Autism DiagnosticInterview-Revised (ADI-R);35 adaptive functioningwas assessed using the Vineland Adaptive BehaviorScales; I.Q. was determined using either the Griffith
Mental Developmental Scales, the Coloured RavenMatrices, the Bayley Developmental Scales or theLeiter International Performance Scale.33 All parentsgave written informed consent for themselves and fortheir children, using the consent form approved bythe I.R.B. of University Campus Bio-Medico (Rome,Italy).
GenotypingFour single nucleotide polymorphisms (SNPs) loca-ted in intron 2 of the PRKCB1 gene were genotyped,including the three SNPs previously found associatedwith autism in Caucasians,14 and one additional SNPlocated approximately 18 kb upstream (Table 2A).SNPs were genotyped using the TaqMan method(Applied Biosystems, Foster City, CA, USA), withprobes purchased from the manufacturer and usedaccording to the manufacturer’s guidelines. DNA wasPCR-amplified with denaturation at 95 1C for 10 min,40 cycles at 92 1C for 15 s, 60 1C for 1 min and 72 1Cfor 45 s, followed by elongation at 72 1C for 5 min.TaqMan assays were then read on a 7900HT Fast
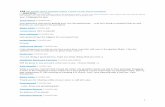
Table 1 Genetic sample composition
Site Number of individualswith autism
Number of families Number ofcomplete trios
Simplex Multiplex
II University of Naples (Naples, Italy) 51 51 — 51I.R.C.C.S. ‘Oasi Maria S.S.’ (Troina, Italy) 37 35 1 36I.R.C.C.S. ‘Ospedale Bambino-Gesu’ (Rome, Italy) 33 33 — 33University ‘Federico II’ (Naples, Italy) 28 28 — 28Southwest Autism Research Center (Phoenix, AZ) 27 21 3 24II University of Rome ‘Tor Vergata’ (Rome, Italy) 22 20 1 21U.C.B.M. (Rome, Italy) 21 21 — 21University of Milan (Milan, Italy) 15 15 — 15University of Turin (Turin, Italy) 4 4 — 4A.S.L. of Rimini (Rimini, Italy) 1 1 — 1Total sample 239 229 5 234
Table 2 Single nucleotide polymorphisms assayed in this study: (A) position on chromosome 16 relative to Build 36.2, allelicfrequencies, and size of the sample genotyped; (B) linkage disequilibrium, expressed as values of D0 (range: �1 to þ 1)/r2 (range:0–1, in italics)
rs number hCV ID Position on ch16
Allele 1 Allele 2 Allele 1frequency
Allele 2frequency
Number of familiesgenotyped (total N = 234)
(A)rs3785392 hCV11192725 23 851 984 G A 0.464992 0.535008 226rs3785387 hCV1936137 23 869 794 A G 0.438466 0.561534 222rs196002 hCV946275 23 870 738 A G 0.462079 0.537921 223rs1873423 hCV11895960 23 878 178 C T 0.158919 0.841081 228
(B)rs number rs3785392 rs3785387 rs196002 rs1873423rs3785392 — 0.905/0.736 0.712/0.495 0.349/0.027rs3785387 0.905/0.736 — 0.654/0.389 0.367/0.033rs196002 0.712/0.495 0.654/0.389 — 0.285/0.018rs1873423 0.349/0.027 0.367/0.033 0.285/0.018 —
PRKCB1 gene and autistic disorderC Lintas et al
707
Molecular Psychiatry

Real-Time PCR System (Applied Biosystems), andalleles were called using the SDS software (AppliedBiosystems).
Biochemical and morphological endophenotypesBlood samples for 5-HT levels were obtained fromall family members and centrifuged within 20 minof venipuncture at 140 g for 25 min at 4 1C; 1 mlof supernatant (that is, platelet-rich plasma) wasstored at �80 1C and assessed by high-performanceliquid chromatography, as described.36 Urinary pep-tide excretion analysis was performed by high-perfor-mance liquid chromatography on the first morningfrom urine samples of all family members, dilutedto 250 nm creatinine, as described.10 The total areaof peaks under the 215 nm absorption curve in thepeptide region following the hippuric acid peak wascalculated and expressed in mm2. Excess peptiduriawas defined on the basis of previously publishednormality ranges determined in population con-trols.10 Head circumference was measured in autisticpatients and unaffected siblings by trained physiciansusing a non-stretchable plastic measuring tapegraded in millimeters, placed over the maximumfronto-occipital head perimeter; head circumferencemeasures were transformed into percentiles usingsex- and age-specific standard tables, as described.6
Statistical analysesHardy–Weinberg equilibrium was tested using the w2
statistic, as implemented by the HAPLOVIEW soft-ware (available at http://www.broad.mit.edu/mpg/haploview/index.php)37 for the total sample andby HWE (available at http://linkage.rockefeller.edu) toanalyze separately autistic patients, mothers, fathers,and unaffected siblings. Family-based single-markerand haplotype association tests were performed usingthe FBAT statistic (S =STX, where T is the phenotypictrait and X the marker value), as implemented bythe FBAT software package (available at http://www.biostat.harvard.edu/~fbat/fbat.htm), under an addi-tive model (option �e).38 The FBAT statistic stemsfrom the transmission/disequilibrium test (TDT),where preferential allelic transmission from hetero-zygous parents to affected offspring is tested by apply-ing the (b�c)2/(bþ c) statistics and the w2 (‘McNemartest’).39 Quantitative traits were analyzed by quanti-tative TDT, as implemented by the FBAT software,38
with T as the quantitative trait of interest (instead ofa dichotomic affected/unaffected status as in theTDT), and by parametric or nonparametric ANOVA,based on genotype distributions. The HBAT commandin FBAT was also employed to estimate haplotypefrequencies and linkage disequilibrium (LD) frompedigree data.38 TDT analyses controlling for quanti-tative covariates were performed using the multi-nomial logistic regression model underpinning theretrospective likelihood method implemented by theUnphased software package, applying the ‘confoun-der’ and ‘uncertain haplotypes’ options.40 To mini-mize the impact of confounding variables, such as
drug therapy, puberal status and differential growthrates, quantitative analyses were restricted to patientsaged < 11 years and not taking selective 5-HT reup-take inhibitors for 5-HT blood levels, and to patientsaged < 16 years for head size, as in our previousstudies.6,33,36 Statistical analyses were performedmerging together our 210 Italian and 24 Caucasian-American families (Table 1) after population structureanalyses provided no evidence of major geneticdyshomogeneity in a subset of 155 Italian and 24Caucasian-American patients randomly chosen oneper family, genotyped at 90 unlinked SNPs distribu-ted genome-wide and analyzed using the Structureprogram, as described (see Supplementary Methods).41
Data are expressed as mean±s.e.m., except for headcircumference and urinary peptide excretion rates,expressed as median percentile±interquartilic range.Two-tail P-values are reported, with significance levelset at P < 0.05. No correction for repeated measureswas implemented in our family-based associationstudy, because (a) it has been undertaken to replicatepreviously published positive findings;14 (b) we docu-ment the relative genetic homogeneity of our Italianand Caucasian-American patients; (c) the four SNPsused to define a single haplotype, previously foundassociated with autism,14 are characterized by shortintermarker physical distances, significant LD andnon-independent marker segregation (see Resultssection); (d) many endophenotypic measures assessedin the same autistic patients are non-independent.Instead, statistical significance was set at P < 0.01 fortests of Hardy–Weinberg equilibrium (that is, < 0.05/5comparisons), whereas nominal P-values are reportedfor exploratory genotype–phenotype correlationsregarding clinical measures, due to the large numberof clinical variables tested.
Patient brain tissue information
Postmortem studies were performed using frozenbrain tissue samples dissected from the superiortemporal gyrus (BA 41/42) of 11 patient–control pairs,obtained through the Autism Tissue Program fromthe Maryland NICHD Brain Tissue Center and theHarvard Brain Tissue Resource Center. This neo-cortical region was chosen because it hosts well-documented structural and functional abnormalitiesin autism.42 These tissue samples largely overlapwith those employed in our recent study of the METpathway.5 Clinical and demographic information,family history and autopsy reports were obtainedfrom the Autism Tissue Program web site (www.atpportal.org) and are summarized in Table 3. Thepresence of mental retardation (MR) was defined onthe basis of a full-scale IQ < 70. ASD cases fulfilledDSM-IV diagnostic criteria,1 confirmed using theAutism Diagnostic Interview-Revised (ADI-R).35
Controls were selected to match patients on sex,age (±2 years, with the exception of ±4 years in pairnine) and postmortem interval, as much as possible(Table 3).
PRKCB1 gene and autistic disorderC Lintas et al
708
Molecular Psychiatry
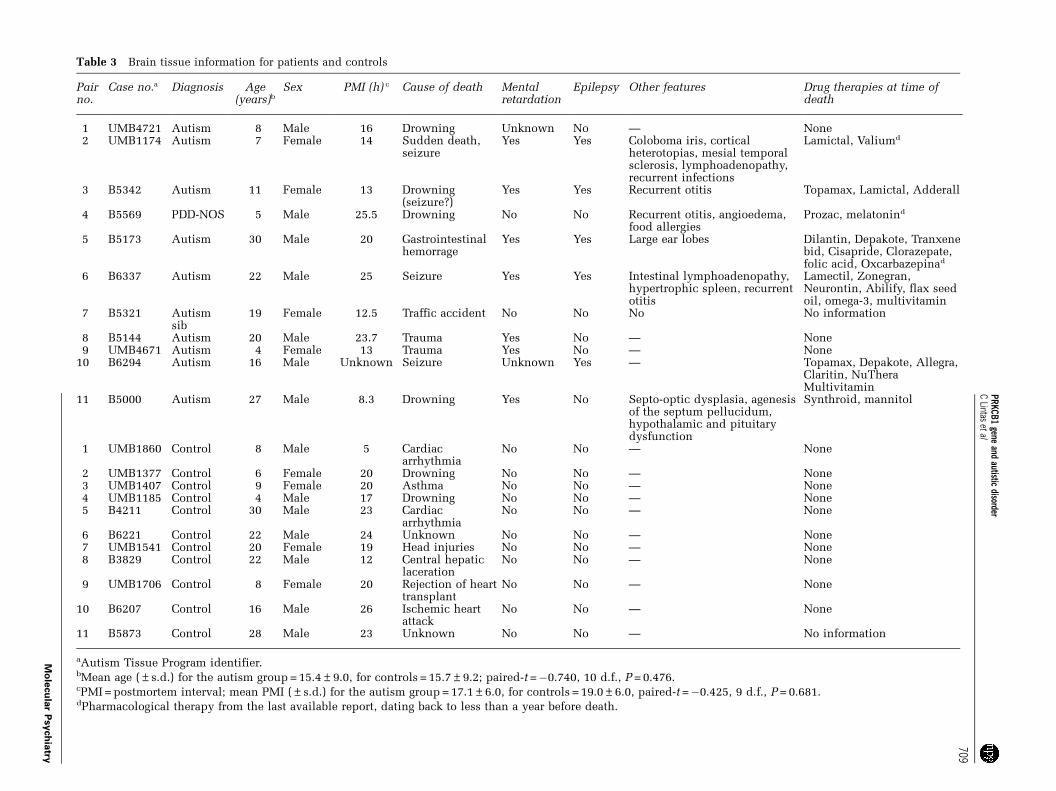
Table 3 Brain tissue information for patients and controls
Pairno.
Case no.a Diagnosis Age(years)b
Sex PMI (h) c Cause of death Mentalretardation
Epilepsy Other features Drug therapies at time ofdeath
1 UMB4721 Autism 8 Male 16 Drowning Unknown No — None2 UMB1174 Autism 7 Female 14 Sudden death,
seizureYes Yes Coloboma iris, cortical
heterotopias, mesial temporalsclerosis, lymphoadenopathy,recurrent infections
Lamictal, Valiumd
3 B5342 Autism 11 Female 13 Drowning(seizure?)
Yes Yes Recurrent otitis Topamax, Lamictal, Adderall
4 B5569 PDD-NOS 5 Male 25.5 Drowning No No Recurrent otitis, angioedema,food allergies
Prozac, melatonind
5 B5173 Autism 30 Male 20 Gastrointestinalhemorrage
Yes Yes Large ear lobes Dilantin, Depakote, Tranxenebid, Cisapride, Clorazepate,folic acid, Oxcarbazepinad
6 B6337 Autism 22 Male 25 Seizure Yes Yes Intestinal lymphoadenopathy,hypertrophic spleen, recurrentotitis
Lamectil, Zonegran,Neurontin, Abilify, flax seedoil, omega-3, multivitamin
7 B5321 Autismsib
19 Female 12.5 Traffic accident No No No No information
8 B5144 Autism 20 Male 23.7 Trauma Yes No — None9 UMB4671 Autism 4 Female 13 Trauma Yes No — None
10 B6294 Autism 16 Male Unknown Seizure Unknown Yes — Topamax, Depakote, Allegra,Claritin, NuTheraMultivitamin
11 B5000 Autism 27 Male 8.3 Drowning Yes No Septo-optic dysplasia, agenesisof the septum pellucidum,hypothalamic and pituitarydysfunction
Synthroid, mannitol
1 UMB1860 Control 8 Male 5 Cardiacarrhythmia
No No — None
2 UMB1377 Control 6 Female 20 Drowning No No — None3 UMB1407 Control 9 Female 20 Asthma No No — None4 UMB1185 Control 4 Male 17 Drowning No No — None5 B4211 Control 30 Male 23 Cardiac
arrhythmiaNo No — None
6 B6221 Control 22 Male 24 Unknown No No — None7 UMB1541 Control 20 Female 19 Head injuries No No — None8 B3829 Control 22 Male 12 Central hepatic
lacerationNo No — None
9 UMB1706 Control 8 Female 20 Rejection of hearttransplant
No No — None
10 B6207 Control 16 Male 26 Ischemic heartattack
No No — None
11 B5873 Control 28 Male 23 Unknown No No — No information
aAutism Tissue Program identifier.bMean age (±s.d.) for the autism group = 15.4±9.0, for controls = 15.7±9.2; paired-t =�0.740, 10 d.f., P = 0.476.cPMI = postmortem interval; mean PMI (±s.d.) for the autism group = 17.1±6.0, for controls = 19.0±6.0, paired-t =�0.425, 9 d.f., P = 0.681.dPharmacological therapy from the last available report, dating back to less than a year before death.
PRKCB1
geneand
autisticdisorder
CLintas
etal
709
Mo
lecu
lar
Psych
iatry

Expression analysis by real-time PCRTotal RNA was extracted from all 11 case–controlpairs (Table 3) using the TRIzol reagent (Invitrogen,Carlsbad, CA, USA) according to standard methods,and RNA quality was checked using a Bioanalyzer(Agilent, Santa Clara, CA, USA). Reverse transcrip-tion was performed using the QuantiTect ReverseTranscription kit (QIAGEN, Hilden, Germany),employing random hexamers as primers. The amountof each specific cDNA was measured using an iQ5Multicolor Real-Time PCR Apparatus according to astandard DDCt SYBR Green protocol (BioRad, Hercules,CA, USA).43 CyclophilinA cDNA was measured inparallel with both PRKCB1 isoforms and used as astandard normalizer. The following isoform-specificprimers were designed: PRKCB1-1F AGACACCTCCAACTTCGACAA; PRKCB1-1R CAACGATGGAGTTTGCATTC; PRKCB1-2F AAGCTCAACGGCTATTGTGG;PRKCB1-2R GCCATCTGCATAATCCCATC; cyclophi-linA-F GCAGACAAGGTCCCAAAG; cyclophilinA-RGAAGTCACCACCCTGACAC. Statistical significancewas calculated using the Wilcoxon test.
Western blottingWestern blotting was performed on 8 of the 11 case–control pairs (no. 1–8 in Table 3) because of limitedtissue availability. Sixty micrograms of protein extractwere diluted in 2� SDS/loading sample buffer,boiled for 5 min, loaded onto a 5% stacking/7.5%separating polyacrylamide gel, run at 100 V for 2 h,and transferred onto a nitrocellulose membrane at350 mA for 90 min. Membranes were blocked with 5%milk in 0.1% Tris-buffered saline Tween-20 (TBS-T)for 1 h, washed 3�10 min in 0.01% TBS-T, incubatedovernight at 4 1C with primary antibody againstPKCbII (sc-13149, Santa Cruz Biotechnology, SantaCruz, CA, USA) diluted 1:600 in 0.05% TBS-T with5% milk. Following 3�10 min washes in 0.01% TBS-T, membranes were incubated for 1 h at roomtemperature with horseradish peroxidase-conjugatedgoat antimouse IgG (sc-2005, Santa Cruz Biotechno-logy) diluted 1:5000 in 0.1% TBS-T and 3% milk.Following 3� 10 min washes in 0.01% TBS-T, pro-teins were visualized using the enhanced chemilu-minescent (ECL) detection method and Hyperfilm(Amersham Biosciences, Piscataway, NJ, USA). Den-sitometry was performed using the VersaDoc model4000 imaging system (BioRad).
To control for protein loading, membranes werewashed 3� 10 min in 0.01% TBS-T, and incubatedwith mouse AC15 anti-b-actin monoclonal antibody(A5441, Sigma-Aldrich, St Louis, MO, USA) diluted1:5000, and with goat antimouse IgG (AP124P,Chemicon International, Temecula, CA, USA) diluted1:5000. Data are presented as PKCbII normalized tob-actin (mean±s.d.). Statistical significance wasdetermined using the Wilcoxon test.
Microarray analysisTotal RNA isolation, reverse transcription and in vitrotranscription were performed, as previously described.44
RNA samples from six case–control pairs (no. 2, 4, 5, 7,8 and 11 in Table 3) provided RIN values > 6.9 and werethus hybridized to HG_U133plus2 human AffymetrixGeneChips, with segmentation analysis performedusing Microarray Analysis Suite 5.0 (MAS5). Normal-ization of data was performed using GC-RMA.45 Datawere converted to a linear scale by log2 transformation,with gene clustering performed using [email protected] Gene set analysis was performed using GeneSet Enrichment Analysis (GSEA) software (version 2)47
with array probe sets collapsed to gene symbol and genesets generated from Biocarta gene set list (convertedfrom probe set list with manual curation), using ASDversus control phenotypes and 1000 permutations/analysis. Statistical significance was calculated usinga paired Student’s t-test. False discovery rate thresholdin GSEA was set at q < 0.05.48 Correlation analyses wereperformed by Pearson r.
Results
PRKCB1 gene variants are associated with autismThe four SNPs, located in intron 2 of the PRKCB1locus, are in Hardy–Weinberg equilibrium both in theentire data set of 795 individuals (SupplementaryTable S1), and also when analyzing separately 239autistic patients, 234 mothers, 232 fathers, and 90unaffected siblings. The same four SNPs are also inLD, as summarized in Table 2B; their LD pattern inour sample is superimposable to the LD pattern foundby the International HapMap Project in their Cauca-sian CEPH population (that is, Utah residents withancestry from northern and western Europe; seewww.hapmap.org).
PRKCB1 haplotypes display a significant associa-tion with autism, as shown in Table 4A (whole markerpermutation test, P < 0.05 after 32271 iterations).Haplotype 2-2-2-1 is transmitted from heterozygousparents to their autistic offspring significantly moreoften than expected by chance (P = 0.007), at theexpense of the complementary haplotype 1-1-1-2(Table 4A). Unaffected siblings display the oppositetrend, with an undertransmission of haplotype 2-2-2-1 reaching a borderline P = 0.053 (SupplementaryTable S2) and yielding a highly significant differencein transmission rates between autistic and non-autistic offspring (Z = 16.23, P<0.00001). Despitetheir lower statistical power, single-marker FBATanalyses also support the existence of LD betweenthe most 50 SNP, rs3785392 and the PRKCB1gene variant contributing to autism vulnerability(Table 4B). Superimposable trends in allelic transmis-sion rates are found in Italian and Caucasian-American families analyzed separately. No parent-of-origin effect is consistently present (data notshown). Preliminary analyses performed on clinicalsigns and symptoms suggest a possible influenceof PRKCB1 alleles on the ‘presence/absence of verbalor motor stereotypies at intake,’ with the ‘risk’ allelesignificantly blunting stereotypic behaviors. In fact,stereotypies were noticed at intake in 17/26 (65.4%)
PRKCB1 gene and autistic disorderC Lintas et al
710
Molecular Psychiatry

patients with the 1/1 genotype at rs3785392, in 33/67(49.3%) 1/2 individuals and in 14/47 (29.8%) 2/2individuals (w2 = 9.196, 2 d.f., nominal P = 0.01);for rs3785387, stereotypies were recorded at intakein 17/25 (68.0%) 1/1 patients, 30/64 (46.9%) 1/2individuals and 18/51 (35.3%) 2/2 individuals(w2 = 7.224, 2 d.f., nominal P = 0.03).
PRKCB1 gene variants are associated with enhancedoligopeptiduria in autism
Macrocephaly/macrosomy, hyperserotoninemia andenhanced peptiduria are among the best-characterizedmorphological and biochemical endophenotypesin autism.6,10,36 Performing a quantitative TDT, asimplemented by the FBAT program,38 we founda significant association between parent-to-autisticoffspring transmission rates of PRKCB1 gene variantsmarked by the two most 50 SNPs, rs3785392and rs3785387, and urinary peptide excretion rates(Table 5and Supplementary Table S3). ANOVAsbased on genotype distributions provide furtherevidence of semidominant effects for ‘risk’ allele 2 atSNP rs3785387, associated with enhanced peptiduria(genotype 1/1, n = 20, 218.5±251mm2; 1/2, n = 59,277.0±220mm2; 2/2, n = 43, 353.0±208mm2; Kruskal–Wallis w2 = 6.083, 2 d.f., P<0.05). In contrast, no
evidence of PRKCB1 gene variants affecting peptiduriaor any other quantitative endophenotype could befound in the unaffected siblings of autistic patients(Supplementary Table S4).
Incorporating peptiduria as a covariate into haplo-typic and single-marker analyses for affection statusessentially did not change their outcome: the wholemarker permutation test for haplotypic analysisyielded a P = 0.054, while single-marker analyses forrs3785392 and rs3785387 yielded P = 0.02 and 0.11,respectively (compare with Tables 4A and 4B). Theseresults indicate that the overtransmission of PRKCB1gene variants to autistic offspring is largely, thoughnot entirely, independent of peptiduria.
PRKCB1 gene expression is significantly decreasedin postmortem brains of autistic patients
PRKCB1 gene expression is significantly decreasedin the temporal neocortex of ASD patients comparedto their matched controls. PRKCB1 mRNA levels aredecreased by 26% according to oligonucleotide DNAmicroarray analysis (pairwise t- and rank-testsP < 0.05). Using isoform-specific primers, quantitativePCR unveils 35 and 31% mean reductions for thePRKCB1-1 and PRKCB1-2 isoforms, yielding P < 0.01and < 0.05, respectively (Figure 1). In particular,
Table 4 The PRKCB1 gene variant marked by haplotype 2-2-2-1 is associated with autism: (A) haplotype and (B) single-markerfamily-based association tests performed using FBAT, under an additive model (�e)38
4-Marker Haplotypesa Estimated frequency N. of families S E(S) Var(S) Z P-value
(A)2-2-2-2 0.388 128.0 164.218 156.899 50.805 1.027 0.3044861-1-1-2 0.258 125.3 91.847 105.174 48.876 �1.906 0.0566161-1-1-1 0.092 58.5 44.434 39.090 14.703 1.394 0.1634022-2-1-2 0.070 45.2 25.699 27.726 13.405 �0.554 0.5797961-1-2-2 0.062 43.2 21.570 25.451 11.209 �1.159 0.2463242-2-2-1 0.052 37.4 32.083 23.042 11.549 2.660 0.0078031-2-1-2 0.040 32.0 15.666 17.000 7.778 �0.478 0.6324652-1-2-2 0.017 13.0 8.980 7.490 4.740 0.684 0.493720
(B)Marker/alleles Autistic patients
N. of families S E(S) Var(S) Z P-value
rs3785392 1 146 124.000 139.500 58.250 �2.031 0.0425732 146 172.000 156.500 58.250 2.031
rs3785387 1 141 126.000 135.500 60.000 �1.162 0.2452782 141 160.000 151.000 60.000 1.162
rs196002 1 135 111.000 124.000 62.250 �1.711 0.08700712 135 163.000 149.500 62.250 1.711
rs1873423 1 90 67.000 58.500 29.250 1.572 0.1160322 90 119.000 127.000 29.250 �1.572
FBAT output variables: S, test statistic (that is, genotypic distribution in the offspring conditioned on affection status andparental genotypes); E(S), expected value for S; VAR(S), variance of S under the null hypothesis of no linkage and noassociation; N, number of informative nuclear familiesP-values < 0.05 are highlighted in bold.aWhole marker permutation test, P < 0.05 after 32271 iterations.
PRKCB1 gene and autistic disorderC Lintas et al
711
Molecular Psychiatry
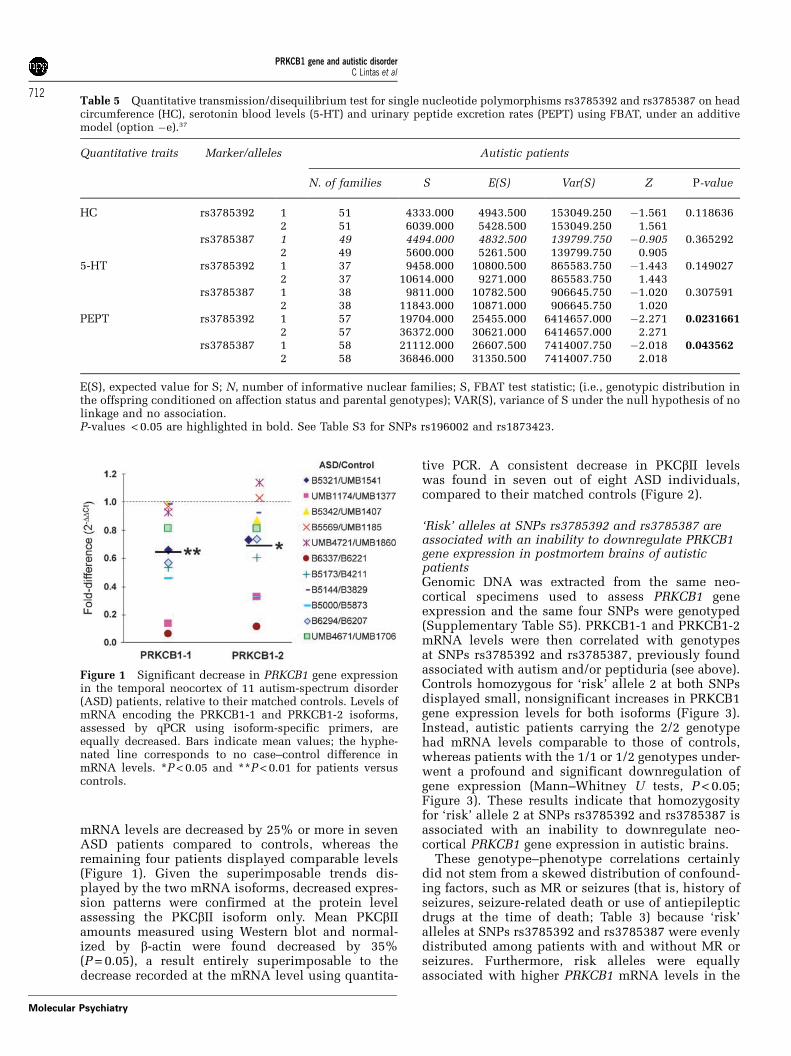
mRNA levels are decreased by 25% or more in sevenASD patients compared to controls, whereas theremaining four patients displayed comparable levels(Figure 1). Given the superimposable trends dis-played by the two mRNA isoforms, decreased expres-sion patterns were confirmed at the protein levelassessing the PKCbII isoform only. Mean PKCbIIamounts measured using Western blot and normal-ized by b-actin were found decreased by 35%(P = 0.05), a result entirely superimposable to thedecrease recorded at the mRNA level using quantita-
tive PCR. A consistent decrease in PKCbII levelswas found in seven out of eight ASD individuals,compared to their matched controls (Figure 2).
‘Risk’ alleles at SNPs rs3785392 and rs3785387 areassociated with an inability to downregulate PRKCB1gene expression in postmortem brains of autisticpatientsGenomic DNA was extracted from the same neo-cortical specimens used to assess PRKCB1 geneexpression and the same four SNPs were genotyped(Supplementary Table S5). PRKCB1-1 and PRKCB1-2mRNA levels were then correlated with genotypesat SNPs rs3785392 and rs3785387, previously foundassociated with autism and/or peptiduria (see above).Controls homozygous for ‘risk’ allele 2 at both SNPsdisplayed small, nonsignificant increases in PRKCB1gene expression levels for both isoforms (Figure 3).Instead, autistic patients carrying the 2/2 genotypehad mRNA levels comparable to those of controls,whereas patients with the 1/1 or 1/2 genotypes under-went a profound and significant downregulation ofgene expression (Mann–Whitney U tests, P < 0.05;Figure 3). These results indicate that homozygosityfor ‘risk’ allele 2 at SNPs rs3785392 and rs3785387 isassociated with an inability to downregulate neo-cortical PRKCB1 gene expression in autistic brains.
These genotype–phenotype correlations certainlydid not stem from a skewed distribution of confound-ing factors, such as MR or seizures (that is, history ofseizures, seizure-related death or use of antiepilepticdrugs at the time of death; Table 3) because ‘risk’alleles at SNPs rs3785392 and rs3785387 were evenlydistributed among patients with and without MR orseizures. Furthermore, risk alleles were equallyassociated with higher PRKCB1 mRNA levels in the
Table 5 Quantitative transmission/disequilibrium test for single nucleotide polymorphisms rs3785392 and rs3785387 on headcircumference (HC), serotonin blood levels (5-HT) and urinary peptide excretion rates (PEPT) using FBAT, under an additivemodel (option �e).37
Quantitative traits Marker/alleles Autistic patients
N. of families S E(S) Var(S) Z P-value
HC rs3785392 1 51 4333.000 4943.500 153049.250 �1.561 0.1186362 51 6039.000 5428.500 153049.250 1.561
rs3785387 1 49 4494.000 4832.500 139799.750 �0.905 0.3652922 49 5600.000 5261.500 139799.750 0.905
5-HT rs3785392 1 37 9458.000 10800.500 865583.750 �1.443 0.1490272 37 10614.000 9271.000 865583.750 1.443
rs3785387 1 38 9811.000 10782.500 906645.750 �1.020 0.3075912 38 11843.000 10871.000 906645.750 1.020
PEPT rs3785392 1 57 19704.000 25455.000 6414657.000 �2.271 0.02316612 57 36372.000 30621.000 6414657.000 2.271
rs3785387 1 58 21112.000 26607.500 7414007.750 �2.018 0.0435622 58 36846.000 31350.500 7414007.750 2.018
E(S), expected value for S; N, number of informative nuclear families; S, FBAT test statistic; (i.e., genotypic distribution inthe offspring conditioned on affection status and parental genotypes); VAR(S), variance of S under the null hypothesis of nolinkage and no association.P-values < 0.05 are highlighted in bold. See Table S3 for SNPs rs196002 and rs1873423.
Figure 1 Significant decrease in PRKCB1 gene expressionin the temporal neocortex of 11 autism-spectrum disorder(ASD) patients, relative to their matched controls. Levels ofmRNA encoding the PRKCB1-1 and PRKCB1-2 isoforms,assessed by qPCR using isoform-specific primers, areequally decreased. Bars indicate mean values; the hyphe-nated line corresponds to no case–control difference inmRNA levels. *P < 0.05 and **P < 0.01 for patients versuscontrols.
PRKCB1 gene and autistic disorderC Lintas et al
712
Molecular Psychiatry
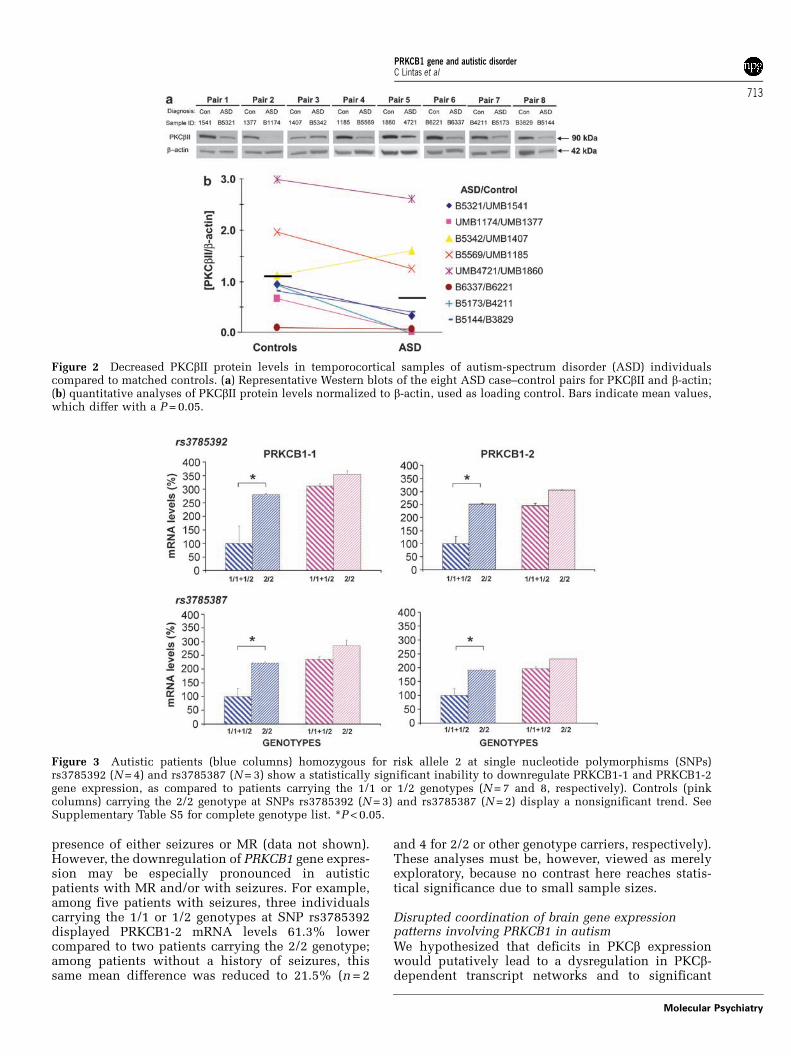
presence of either seizures or MR (data not shown).However, the downregulation of PRKCB1 gene expres-sion may be especially pronounced in autisticpatients with MR and/or with seizures. For example,among five patients with seizures, three individualscarrying the 1/1 or 1/2 genotypes at SNP rs3785392displayed PRKCB1-2 mRNA levels 61.3% lowercompared to two patients carrying the 2/2 genotype;among patients without a history of seizures, thissame mean difference was reduced to 21.5% (n = 2
and 4 for 2/2 or other genotype carriers, respectively).These analyses must be, however, viewed as merelyexploratory, because no contrast here reaches statis-tical significance due to small sample sizes.
Disrupted coordination of brain gene expressionpatterns involving PRKCB1 in autismWe hypothesized that deficits in PKCb expressionwould putatively lead to a dysregulation in PKCb-dependent transcript networks and to significant
Figure 2 Decreased PKCbII protein levels in temporocortical samples of autism-spectrum disorder (ASD) individualscompared to matched controls. (a) Representative Western blots of the eight ASD case–control pairs for PKCbII and b-actin;(b) quantitative analyses of PKCbII protein levels normalized to b-actin, used as loading control. Bars indicate mean values,which differ with a P = 0.05.
Figure 3 Autistic patients (blue columns) homozygous for risk allele 2 at single nucleotide polymorphisms (SNPs)rs3785392 (N = 4) and rs3785387 (N = 3) show a statistically significant inability to downregulate PRKCB1-1 and PRKCB1-2gene expression, as compared to patients carrying the 1/1 or 1/2 genotypes (N = 7 and 8, respectively). Controls (pinkcolumns) carrying the 2/2 genotype at SNPs rs3785392 (N = 3) and rs3785387 (N = 2) display a nonsignificant trend. SeeSupplementary Table S5 for complete genotype list. *P < 0.05.
PRKCB1 gene and autistic disorderC Lintas et al
713
Molecular Psychiatry

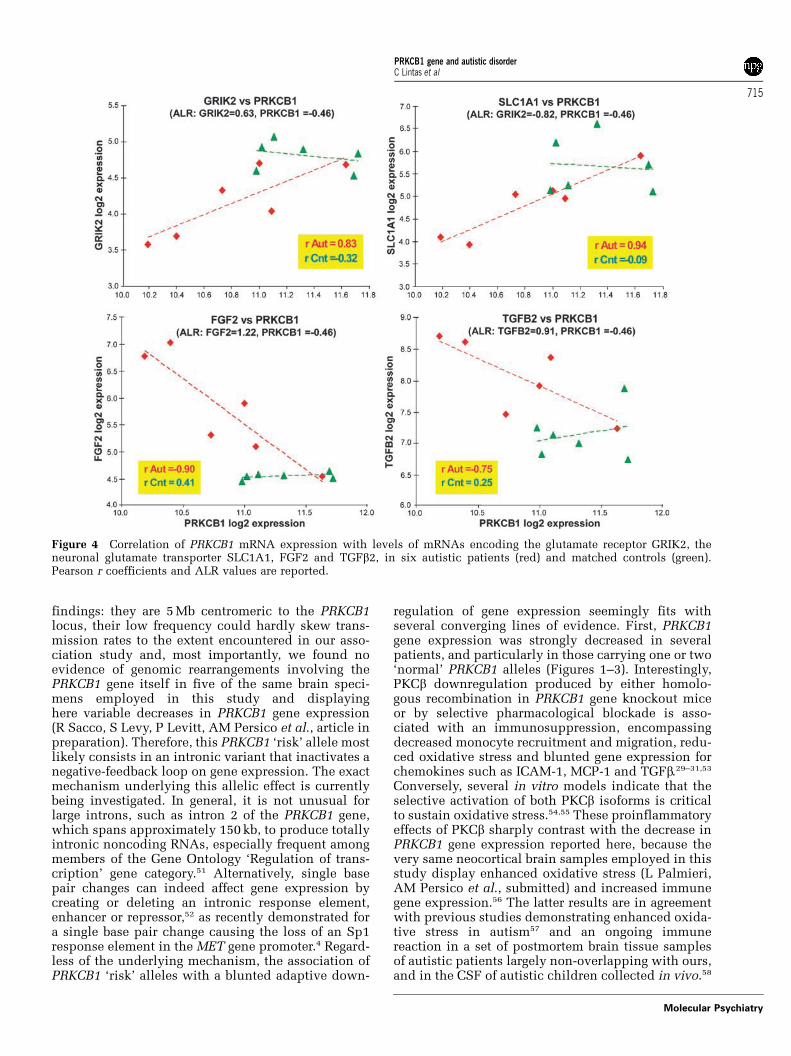
downstream alterations in neuronal function. To testthis hypothesis, we explored the temporocorticalexpression of PRKCB1-related transcript networks inautistic patients and matched control subjects usingoligonucleotide DNA microarray analysis. First, themicroarrays employed in this study encompass twoindependent probe sets for PRKCB1 (227817_at and209685_s_at), which consistently demonstratedsuperimposable expression differences between post-mortem autistic and control brains (ALR =�0.44 and�0.46, respectively). In addition, the two probe setsdisplayed high positive correlations (r = 0.98 and 0.86for autistics and controls, respectively), furthervalidating our findings.
Next, in a data-driven approach, we identified 1009gene transcripts that were highly coregulated withPKCb in the brain of control subjects (r > 0.9). Inaddition, we identified 131 transcripts that (a)displayed correlation differences of DrAUT�CONX0.8with PRKCB1 between patients and controls (rAUT�rCON) and (b) reported a significant expressiondifference between the autistics and control post-mortem temporal cortices (|ALR| > 0.585 and P < 0.05in pairwise t-test). Among these, 71 genes showed apositive correlation with PRKCB1 levels in ASD casesand an opposite or unchanged trend in controls,including the kainate 2 ionotropic glutamate receptor(GRIK2) and the neuronal/epithelial high affinityglutamate transporter (SLC1A1). Even more interest-ingly, 60 mRNAs that were positively correlatedwith PRKCB1 levels in controls showed a loss of thepositive correlation with PRKCB1 levels in ASD cases.Among others, these included genes encodingmultiple neurodevelopmentally relevant molecules(for example, FGF2 and FGF-R1) and cytokines (forexample, TGFb2) (Figure 4).
Finally, we explored a knowledge-based correlationbetween mRNA levels of PRKCB1 and geneswhose expression has been shown to be under director indirect PKCb control.23,24,26,28–31 These resultsrevealed that coordinated expression patterns arelost for several cytokines and enzymes involved inoxidative stress, maintained to a large extent forothers, and entirely preserved for extracellular matrixproteins (Table 6). In addition to the loss of corre-lation with PKCb transcript levels, many of thesegenes also displayed significant expression differ-ences between autistic and control brains, includingTGFb2, MCP1, collagen IV and fibronectin 1 (Table 6).
Discussion
This study (a) replicates in an independent samplethe positive association between PRKCB1 genevariants and autism previously reported by Philippiet al.;14 (b) extends family-based genetic findingsby demonstrating a significant association also witha biochemical endophenotype characterized by en-hanced oligopeptiduria; (c) provides novel evidenceshowing significantly reduced PRKCB1 mRNA andprotein levels, as well as disrupted PKCb-related gene
expression patterns in postmortem neocortical tissuesamples of autistic patients compared to age-, sex-,and postmortem interval-matched controls and (d)identifies a blunted downregulation of PRKCB1 geneexpression in postmortem brains as the functionalcorrelate of PRKCB1 ‘risk’ alleles associated withautism. These functional abnormalities in PRKCB1gene expression nicely parallel genetic evidence ofPRKCB1 gene contributions to autism pathogenesisand lend further support to PKCb involvement in thepathogenetic processes underlying autistic disorder.
Our family-based linkage/association study essen-tially confirms the existence of a significant asso-ciation between autism and PRKCB1 gene variantsmarked in Caucasians by polymorphisms located inintron 2. The C-G-T haplotype found associated withautism in Caucasians by Philippi et al.14 is identical toour risk haplotype for rs3785387 and rs196002,whereas it differs at SNP rs1873423. Considering theprogressive decrease of LD going from 50 to 30 in thisregion (Table 2B), and the much stronger associationof 50 markers both with autism and with oligopepti-duria in this study (Table 4 and Supplementary TableS3), it seems likely that haplotypes associated withautism in our sample and in Caucasian-Americans14
are essentially marking the same PRKCB1 gene vari-ant. Moreover, a comparison of LD patterns presentin our sample and in the 171 Irish families assessedby Yang et al.32 fosters a more cautious interpretationof their allegedly negative association findings.SNPs rs3785387, rs196002 and rs1873423 displayhere D0 values ranging from 0.285 to 0.654 (Table 2B),whereas D0 values range only from 0.045 to 0.195 inthe Irish sample.32 These population genetic differen-ces can alone explain why polymorphisms employedin this and in previous studies14,32 are apt to mark theautism liability conferring PRKCB1 gene variant inthe Italian and Caucasian-American populations, butare uninformative in the Irish population.
Our postmortem study has conclusively demons-trated a significant decrease in PRKCB1 gene expres-sion (Figures 1 and 2). Potential confounding factorsare inevitably intermingled with autism in our post-mortem sample, as they are at the phenotypic level.Approximately 75% of autistic patients indeed haveMR and 25% a positive history of seizures.2 In ourpostmortem sample, MR and seizures could modulatePRKCB1 allelic effects on gene expression, but do notappear to produce a spurious association. Instead,this significant downregulation in gene expressionis interestingly associated with ‘normal’ PRKCB1alleles, not with alleles conferring autism liability.A thorough mutational search for non-conservative,coding mutations and for splice junction variantspreviously provided negative results.14 Recently, a593 kb region located in human ch. 16p11.2 has beenshown to undergo a recurrent microdeletion or areciprocal microduplication in approximately 1% ofautistic patients.49,50 These genomic rearrangements,typically de novo but in some cases also inherited, areunlikely to explain our expression and association
PRKCB1 gene and autistic disorderC Lintas et al
714
Molecular Psychiatry
findings: they are 5 Mb centromeric to the PRKCB1locus, their low frequency could hardly skew trans-mission rates to the extent encountered in our asso-ciation study and, most importantly, we found noevidence of genomic rearrangements involving thePRKCB1 gene itself in five of the same brain speci-mens employed in this study and displayinghere variable decreases in PRKCB1 gene expression(R Sacco, S Levy, P Levitt, AM Persico et al., article inpreparation). Therefore, this PRKCB1 ‘risk’ allele mostlikely consists in an intronic variant that inactivates anegative-feedback loop on gene expression. The exactmechanism underlying this allelic effect is currentlybeing investigated. In general, it is not unusual forlarge introns, such as intron 2 of the PRKCB1 gene,which spans approximately 150 kb, to produce totallyintronic noncoding RNAs, especially frequent amongmembers of the Gene Ontology ‘Regulation of trans-cription’ gene category.51 Alternatively, single basepair changes can indeed affect gene expression bycreating or deleting an intronic response element,enhancer or repressor,52 as recently demonstrated fora single base pair change causing the loss of an Sp1response element in the MET gene promoter.4 Regard-less of the underlying mechanism, the association ofPRKCB1 ‘risk’ alleles with a blunted adaptive down-
regulation of gene expression seemingly fits withseveral converging lines of evidence. First, PRKCB1gene expression was strongly decreased in severalpatients, and particularly in those carrying one or two‘normal’ PRKCB1 alleles (Figures 1–3). Interestingly,PKCb downregulation produced by either homolo-gous recombination in PRKCB1 gene knockout miceor by selective pharmacological blockade is asso-ciated with an immunosuppression, encompassingdecreased monocyte recruitment and migration, redu-ced oxidative stress and blunted gene expression forchemokines such as ICAM-1, MCP-1 and TGFb.29–31,53
Conversely, several in vitro models indicate that theselective activation of both PKCb isoforms is criticalto sustain oxidative stress.54,55 These proinflammatoryeffects of PKCb sharply contrast with the decrease inPRKCB1 gene expression reported here, because thevery same neocortical brain samples employed in thisstudy display enhanced oxidative stress (L Palmieri,AM Persico et al., submitted) and increased immunegene expression.56 The latter results are in agreementwith previous studies demonstrating enhanced oxida-tive stress in autism57 and an ongoing immunereaction in a set of postmortem brain tissue samplesof autistic patients largely non-overlapping with ours,and in the CSF of autistic children collected in vivo.58
Figure 4 Correlation of PRKCB1 mRNA expression with levels of mRNAs encoding the glutamate receptor GRIK2, theneuronal glutamate transporter SLC1A1, FGF2 and TGFb2, in six autistic patients (red) and matched controls (green).Pearson r coefficients and ALR values are reported.
PRKCB1 gene and autistic disorderC Lintas et al
715
Molecular Psychiatry

Many genes expressing chemokines are either directlyor indirectly upregulated by PKCb-induced phosphor-ylation:29–31,53 the decrease in PRKCB1 gene expres-sion documented in our study, and the increasedexpression of immune genes like TGFb2 (Figure 4)clearly argue against PRKCB1 driving this ongoingimmune activation in patient samples. On the con-trary, a compensatory decrease in PRKCB1 geneexpression occurring in ASD patients to buffer aninappropriate activation of the immune system see-mingly represents the most plausible scenario. Inthe latter case, any genetic variant reducing thesensitivity of PRKCB1 gene expression to this nega-tive-feedback loop would result in an even morepersistent immune activation, sustaining oxidativestress, and fostering developmental damage duringearly pregnancy. It will thus be extremely importantto identify the exact sequence variant conferringautism vulnerability and to determine its functionalcorrelates in vitro. Only then it will be possible to tiethe current results into the broader picture of autismpathogenesis, also because, despite its intendedadaptive value, the decrease in PRKCB1 gene expres-sion recorded in most autistic brains could still play anegative role, for example by blocking the physio-logical relocation of both FMRP and Fmr1 mRNAin dendrites following activation of metabotropicglutamate receptors.59
The significant association recorded here betweenPRKCB1 gene variants and enhanced urinary peptide
excretion rates increases confidence in the reliabilityof our results and spurs further interest in the under-lying pathophysiology. Functional PRKCB1 promotervariants have been shown to be associated withdiabetic nephropathy and albuminuria,60,61 mediatedby increased renal oxidative stress, production offibrotic cytokines such as TGFb, and consequentlyincreased glomerular permeability.29–31 Also of inter-est is the potential gene–gene interaction with APOEe2 alleles, which contribute to the developmentand progression of diabetic nephropathy62,63 and arepreferentially transmitted to autistic children in oursample.64 A potential parallel between ‘leaky kidney’and ‘leaky gut’ syndromes is suggested by an in vitrostudy demonstrating that PKCb and calcium are bothnecessary for oxidative stress to yield abnormalpermeability in monolayers of human intestinalCaco-2 cells.26 This effect, linking calcium,65 oxida-tive stress,57 and PKCb to the stability of epithelialcytoskeleton in the gut, could conceivably explain theabnormally elevated intestinal permeability presentin many autistic patients and the subsequent activa-tion of the immune system due to the absorptionof the first foreign dietary peptides recognized by thegut immune system as ‘non-self’, namely caseinand gluten.7 Our current phenotypic data set doesnot allow us to directly test this hypothesis. None-theless, our sample encompasses 106 families withmixed genotypes at SNP rs3785392, 7 families whosemembers all carry the ‘normal’ 1/1 genotype, and 10families entirely composed of 2/2 ‘risk genotype’carriers. A positive history of ‘food allergies’ inunaffected family members of the autistic probandwas self-reported in 11/106 (11.6%) ‘mixed genotype’families, 1/7 (14.3%) ‘normal genotype’ families and4/10 (40.0%) ‘risk genotype’ families, respectively(w2 = 7.097, 2 d.f., P < 0.05). This analysis is merelysuggestive, but spurs interest into specific hypothesis-driven studies aimed at determining whether there isa pathophysiological overlap between the PKCb-dependent processes responsible for a ‘leaky kidney’and the mechanisms contributing to the ‘leaky gut’syndrome in autism.
In summary, this study confirms the existence ofa genetic association between PRKCB1 gene variantsand autism, demonstrates a significant associationbetween PRKCB1 gene variants and the quantitativeendophenotype of oligopeptiduria, shows a promi-nent reduction in PRKCB1 gene expression in thetemporal neocortex of autistic patients compared tomatched controls, demonstrates that alleles confer-ring autism vulnerability are associated with a lackof downregulation in PRKCB1 gene expression,and provides evidence of a partially uncoordinatedexpression of PRKCB1 and several PKCb-dependentgenes involved in neurodevelopment and immunefunctions. These expression changes can currently bebest interpreted as a compensatory adjustment aimedat hampering a dysregulated immune response affect-ing the CNS of autistic patients. The negative-feed-back loop mediating this compensatory adjustment is
Table 6 Pearson correlation coefficients for PRKCB1 andgenes whose expression is either directly or indirectlymodulated by PKCb,23,24,26,28–31 assessed in autistic patients(rAUT) and in matched controls (rCON); differences in mRNAlevels between cases and controls, expressed as difference inaverage log2 ratios [ALR (aut–con)], and relative paired andrank P-values; coordinated expression patterns are (A) lost forseveral cytokines and enzymes involved in oxidative stress,(B) maintained to some extent for others and (C) largelypreserved for extracellular matrix molecules
rAUT rCON ALR(aut-con)
Prpval RANKpval
ACTGF 0.51 �0.10 0.006 0.982 0.948NADPHoxidase
0.81 �0.39 �0.005 0.979 0.879
TGFb2 �0.75 0.25 0.908 0.049 0.045VEGF �0.19 0.63 0.768 0.119 0.114
BICAM1 0.26 0.50 0.040 0.608 0.732MCP1 �0.29 �0.42 3.213 0.047 0.026TGFb1 �0.37 �0.52 0.658 0.193 0.247Endothelin-1 �0.29 �0.22 0.818 0.052 0.067Cox2 �0.01 �0.40 1.161 0.193 0.124
CCollagen IV �0.58 �0.51 1.426 0.040 0.036Collagen VI �0.87 �0.89 0.650 0.071 0.097Fibronectin 1 �0.26 0.14 0.468 0.039 0.055
PRKCB1 gene and autistic disorderC Lintas et al
716
Molecular Psychiatry

seemingly inactivated in PRKCB1 alleles conferringautism vulnerability. The identification of thisPRKCB1 DNA sequence variant is being activelypursued, to achieve a thorough understanding of thefunctional implications of PKCb in autism.
Acknowledgments
We gratefully acknowledge Laura Gaita and RosannaD’Oronzio for technical assistance, Francis Rousseauand Anne Philippi (IntegraGen SA, Evry, France) forthe genotyping and the population stratificationanalysis, Carlo Lenti and Roberto Rigardetto forcontributing to patient recruitment, all the patientsand families who generously contributed to thesestudies, and the Autism Tissue Program, HarvardBrain Tissue Resource Center, and Maryland NICHDBrain Tissue Center for providing the brain tissuesamples. This work was supported by the ItalianMinistry for University, Scientific Researchand Technology (Programmi di Ricerca di InteresseNazionale, prot. no.2006058195), the NationalAlliance for Autism Research (Princeton, NJ), theCure Autism Now Foundation (Los Angeles, CA) andthe Fondation Jerome Lejeune (Paris, France) to AMP,and by VUKC Startup Fund, R01 MH079299, and K02MH070786 to KM.
References
1 American Psychiatric Association. Diagnostic and StatisticalManual of Mental Disorders, 4th edn. American PsychiatricAssociation: Washington, DC, 1994.
2 Persico AM, Bourgeron T. Searching for ways out of the autismmaze: genetic, epigenetic and environmental clues. TrendsNeurosci 2006; 29: 349–358.
3 Piven J, Palmer P, Jacobi D, Childress D, Arndt S. Broader autismphenotype: evidence from a family history study of multiple-incidence autism families. Am J Psychiatry 1997; 154: 185–190.
4 Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C,Trillo S et al. A genetic variant that disrupts MET transcriptionis associated with autism. Proc Natl Acad Sci USA 2006; 103:16834–16839.
5 Campbell DB, D’Oronzio R, Garbett K, Ebert PJ, Mirnics K, Levitt Pet al. Disruption of cerebral cortex METsignaling in autismspectrum disorder. Ann Neurol 2007; 62: 243–250.
6 Sacco R, Militerni R, Frolli A, Bravaccio C, Gritti A, Elia M et al.Clinical, morphological, and biochemical correlates of headcircumference in autism. Biol Psychiatry 2007; 62: 1038–1047.
7 D’Eufemia P, Celli M, Finocchiaro R, Pacifico L, Viozzi L,Zaccagnini M et al. Abnormal intestinal permeability in childrenwith autism. Acta Paediatr 1996; 85: 1076–1079.
8 Wakefield AJ, Ashwood P, Limb K, Anthony A. The significance ofileo-colonic lymphoid nodular hyperplasia in children withautistic spectrum disorder. Eur J Gastroenterol Hepatol 2005; 17:827–836.
9 Jyonouchi H, Geng L, Ruby A, Zimmerman-Bier B. Dysregulatedinnate immune responses in young children with autism spectrumdisorders: their relationship to gastrointestinal symptoms anddietary intervention. Neuropsychobiology 2005; 51: 77–85.
10 Reichelt WH, Knivsberg AM, Nodland M, Stensrud M, Reichelt KL.Urinary peptide levels and patterns in autistic children from sevencountries, and the effect of dietary intervention after 4 years. DevBrain Dysfunct 1997; 10: 44–55.
11 Bauman ML, Kemper TL. Neuroanatomic observations of the brainin autism: a review and future directions. Int J Dev Neurosci 2005;23: 183–187.
12 Miller MT, Stromland K, Ventura L, Johansson M, Bandim JM,Gillberg C. Autism associated with conditions characterized bydevelopmental errors in early embryogenesis: a mini review. Int JDev Neurosci 2005; 23: 201–219.
13 Teitelbaum O, Benton T, Shah PK, Prince A, Kelly JL, TeitelbaumP. Eshkol-Wachman movement notation in diagnosis: the earlydetection of Asperger’s syndrome. Proc Natl Acad Sci USA 2004;101: 11909–11914.
14 Philippi A, Roschmann E, Tores F, Lindenbaum P, Benajou A,Germain-Leclerc L et al. Haplotypes in the gene encoding proteinkinase c-beta (PRKCB1) on chromosome 16 are associated withautism. Mol Psychiatry 2005; 10: 950–960.
15 Bult A, Kobylk ME, Van der Zee EA. Differential expression ofprotein kinase C betaI (PKCbetaI) but not PKCalpha and PKCbetaII inthe suprachiasmatic nucleus of selected house mouse lines, and therelationship to arginine-vasopressin. Brain Res 2001; 914: 123–133.
16 Ashique AM, Kharazia V, Yaka R, Phamluong K, Peterson AS,Ron D. Localization of the scaffolding protein RACK1 in thedeveloping and adult mouse brain. Brain Res 2006; 1069: 31–38.
17 Wu J, Song TB, Li YJ, He KS, Ge L, Wang LR. Prenatal restraintstress impairs learning and memory and hippocampal PKCbeta1expression and translocation in offspring rats. Brain Res 2007;1141: 205–213.
18 Colombo PJ, Wetsel WC, Gallagher M. Spatial memory is related tohippocampal subcellular concentrations of calcium-dependentprotein kinase C isoforms in young and aged rats. Proc Natl AcadSci USA 1997; 94: 14195–14199.
19 Birikh KR, Sklan EH, Shoham S, Soreq H. Interaction of ‘read-through’ acetylcholinesterase with RACK1 and PKCbII correlateswith intensified fear-induced conflict behavior. Proc Natl Acad SciUSA 2003; 100: 283–288.
20 Kang SW, Wahl MI, Chu J, Kitaura J, Kawakami Y, Kato RM et al.PKCb modulates antigen receptor signaling via regulation of Btkmembrane localization. EMBO J 2001; 20: 5692–5702.
21 Long A, Kelleher D, Lynch S, Volkov Y. Cutting edge: proteinkinase Cb expression is critical for export of Il-2 from T cells.J Immunol 2001; 167: 636–640.
22 Volkov Y, Long A, McGrath S, Ni Eidhin D, Kelleher D. Crucialimportance of PKCbI in LFA-1-mediated locomotion of activatedT cells. Nat Immunol 2001; 2: 508–514.
23 Cejas PJ, Carlson LM, Zhang J, Padmanabhan S, Kolonias D,Lindner I et al. Protein kinase C bII plays an essential role indendritic cell differentiation and autoregulates its own expression.J Biol Chem 2005; 280: 28412–28423.
24 Siow YL, Au-Yeung KK, Woo CW, Karmin O. Homocysteinestimulates phosphorylation of NADPH oxidase p47phox andp67phox subunits in monocytes via protein kinase Cb activation.Biochem J 2006; 398: 73–82.
25 Ma HT, Lin WW, Zhao B, Wu WT, Huang W, Li Y et al. Proteinkinase C b and d isoenzymes mediate cholesterol accumulationin PMA-activated macrophages. Biochem Biophys Res Commun2006; 349: 214–220.
26 Banan A, Fields JZ, Zhang Y, Keshavarzian A. Key role of PKC andCa2þ in EGF protection of microtubules and intestinal barrieragainst oxidants. Am J Physiol Gastrointest Liver Physiol 2001;280: G828–G843.
27 Liu Y, Su W, Thompson EA, Leitges M, Murray NR, Fields AP.Protein kinase CbII regulates its own expression in rat intestinalepithelial cells and the colonic epithelium in vivo. J Biol Chem2004; 279: 45556–45563.
28 Yu W, Murray NR, Weems C, Chen L, Guo H, Ethridge R et al.Role of cyclooxygenase 2 in protein kinase C bII-mediated coloncarcinogenesis. J Biol Chem 2003; 278: 11167–11174.
29 Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL.Characterization of protein kinase C beta isoform activation on thegene expression of transforming growth factor-beta, extracellularmatrix components, and prostanoids in the glomeruli of diabeticrats. J Clin Invest 1997; 100: 115–126.
30 Wu Y, Wu G, Qi X, Lin H, Qian H, Shen J et al. Protein kinase C binhibitor LY333531 attenuates intercellular adhesion molecule-1and monocyte chemotactic protein-1 expression in the kidney indiabetic rats. J Pharmacol Sci 2006; 101: 335–343.
31 Ohshiro Y, Ma RC, Yasuda Y, Hiraoka-Yamamoto J, Clermont AC,Isshiki K et al. Reduction of diabetes-induced oxidative stress,
PRKCB1 gene and autistic disorderC Lintas et al
717
Molecular Psychiatry

fibrotic cytokine expression, and renal dysfunction in proteinkinase Cbeta-null mice. Diabetes 2006; 55: 3112–3120.
32 Yang MS, Cochrane L, Conroy J, Hawi Z, Fitzgerald M, Gallagher Let al. Protein kinase C-b1 gene variants are not associated withautism in the Irish population. Psychiatr Genet 2007; 17: 39–41.
33 Conciatori M, Stodgell CJ, Hyman SL, O’Bara M, Militerni R,Bravaccio C et al. Morphogenetic effect of the HOXA1 A218Gpolymorphism on head circumference in patients with autism.Biol Psychiatry 2004; 55: 413–419.
34 Lord C, Rutter M, DiLavore PC, Risi S. ADOS, Autism DiagnosticObservation Schedule. Western Psychological Services: Los Angeles,2002 (Italian version ed. by Tancredi R, Saccani M, Persico AM,Parrini B, Igliozzi R and Faggioli R. Organizzazioni Speciali:Florence, 2005).
35 Rutter M, Le Couter A, Lord C. ADI-R, Autism DiagnosticInterview—Revised. Western Psychological Services: Los Angeles,2003 (Italian version ed. by Faggioli R, Saccani M, Persico AM,Tancredi R, Parrini B and Igliozzi R. Organizzazioni Speciali:Florence, 2005).
36 Persico AM, Pascucci T, Puglisi-Allegra S, Militerni R, BravaccioC, Schneider C et al. Serotonin transporter promoter variants donot explain the hyperserotoninemia in autistic children. MolPsychiatry 2002; 7: 795–800.
37 Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis andvisualization of LD and haplotype maps. Bioinformatics 2005; 21:263–265.
38 Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM.Family-based tests for associating haplotypes with generalphenotype data: application to asthma genetics. Genet Epidemiol2004; 26: 61–69.
39 Spielman RS, McGinnis RE, Ewens WJ. Transmission test forlinkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 1993; 52:506–516.
40 Dudbridge F. Pedigree disequilibrium tests for multilocus haplo-types. Genet Epidemiol 2003; 25: 115–121.
41 Pritchard JK, Stephens M, Donnelly P. Inference of populationstructure using multilocus genotype data. Genetics 2000; 155:945–959.
42 Zilbovicius M, Meresse I, Chabane N, Brunelle F, Samson Y,Boddaert N. Autism, the superior temporal sulcus and socialperception. Trends Neurosci 2006; 29: 359–366.
43 Bustin SA. Absolute quantification of mRNA using real-timereverse transcription polymerase chain reaction assays. J MolEndocrinol 2000; 25: 169–193.
44 Arion D, Sabatini M, Unger T, Pastor J, Alonso-Nanclares L,Ballesteros-Yanez I et al. Correlation of transcriptome profile withelectrical activity in temporal lobe epilepsy. Neurobiol Dis 2006;22: 374–387.
45 Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ,Scherf U et al. Exploration, normalization, and summaries of highdensity oligonucleotide array probe level data. Biostatistics 2003;4: 249–264.
46 Lepre J, Rice JJ, Tu Y, Stolovitzky G. Genes@Work: an efficientalgorithm for pattern discovery and multivariate feature selectionin gene expression data. Bioinformatics 2004; 20: 1033–1044.
47 Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL,Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles.Proc Natl Acad Sci USA 2005; 102: 15545–15550.
48 Storey JD, Tibshirani R. Statistical significance for genomewidestudies. Proc Natl Acad Sci USA 2003; 100: 9440–9445.
49 Kumar RA, Karamohamed S, Sudi J, Conrad DF, Brune C, Badner JAet al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet2007; 17: 628–638.
50 Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal Ret al. Association between microdeletion and microduplication at16p11.2 and autism. N Engl J Med 2008; 358: 667–675.
51 Nakaya HI, Amaral PP, Louro R, Lopes A, Fachel AA, Moreira YBet al. Genome mapping and expression analyses of human intronicnoncoding RNAs reveal tissue-specific patterns and enrichmentin genes related to regulation of transcription. Genome Biol 2007;8: R43.
52 Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M et al.Whole-genome cartography of estrogen receptor alpha bindingsites. PLoS Genet 2007; 3: e87.
53 Leitges M, Schmedt C, Guinamard R, Davoust J, Schaal S, Stabel Set al. Immunodeficiency in protein kinase cb-deficient mice.Science 1996; 273: 788–791.
54 Dimitri P, Domenicotti C, Nitti M, Vitali A, Borghi R, Cottalasso Det al. Oxidative stress induces increase in intracellular amyloidbeta-protein production and selective activation of bI and bIIPKCs in NT2 cells. Biochem Biophys Res Commun 2000; 268:642–646.
55 Domenicotti C, Paola D, Vitali A, Nitti M, d’Abramo C, CottalassoD et al. Glutathione depletion induces apoptosis of rat hepatocytesthrough activation of protein kinase C novel isoforms anddependent increase in AP-1 nuclear binding. Free Radic BiolMed 2000; 29: 1280–1290.
56 Garbett KA, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics Ket al. Immune transcriptome alterations in the temporal cortex ofsubjects with autism. Neurobiol Dis 2008 (in press).
57 Chauhan A, Chauhan V. Oxidative stress in autism. Pathophysio-logy 2006; 13: 171–181.
58 Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA.Neuroglial activation and neuroinflammation in the brain ofpatients with autism. Ann Neurol 2005; 57: 67–81.
59 Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ.Metabotropic glutamate receptor activation regulates fragile xmental retardation protein and FMR1 mRNA localizationdifferentially in dendrites and at synapses. J Neurosci 2004; 24:2648–2655.
60 Araki S, Ng DP, Krolewski B, Wyrwicz L, Rogus JJ, Canani L et al.Identification of a common risk haplotype for diabetic nephro-pathy at the protein kinase C-b1 (PRKCB1) gene locus. J Am SocNephrol 2003; 14: 2015–2024.
61 Araki S, Haneda M, Sugimoto T, Isono M, Isshiki K, Kashiwagi Aet al. Polymorphisms of the protein kinase C-b gene (PRKCB1)accelerate kidney disease in type 2 diabetes without overtproteinuria. Diabetes Care 2006; 29: 864–868.
62 Araki S, Koya D, Makiishi T, Sugimoto T, Isono M, Kikkawa R et al.APOE polymorphism and the progression of diabetic nephropathyin Japanese subjects with type 2 diabetes: results of a prospectiveobservational follow-up study. Diabetes Care 2003; 26: 2416–2420.
63 Araki S, Moczulski DK, Hanna L, Scott LJ, Warram JH, KrolewskiAS. APOE polymorphisms and the development of diabeticnephropathy in type 1 diabetes: results of case-control andfamily-based studies. Diabetes 2000; 49: 2190–2195.
64 Persico AM, D’Agruma L, Zelante L, Militerni R, Bravaccio C,Schneider C et al. Enhanced APOE2 transmission rates in familieswith autistic probands. Psychiatr Genet 2004; 14: 73–82.
65 Krey JF, Dolmetsch RE. Molecular mechanisms of autism: apossible role for Ca2þ signaling. Curr Opin Neurobiol 2007; 17:112–119.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
PRKCB1 gene and autistic disorderC Lintas et al
718
Molecular Psychiatry
Copyright © 2022 FDOKUMEN