
Novel variants identified in methyl-CpG-binding domain genes in autistic individuals
13
ORIGINAL ARTICLE Novel variants identified in methyl-CpG-binding domain genes in autistic individuals Holly N. Cukier & Raquel Rabionet & Ioanna Konidari & Melissa Y. Rayner-Evans & Mary L. Baltos & Harry H. Wright & Ruth K. Abramson & Eden R. Martin & Michael L. Cuccaro & Margaret A. Pericak-Vance & John R. Gilbert Received: 1 May 2009 / Accepted: 26 October 2009 # Springer-Verlag 2009 Abstract Misregulation of the methyl-CpG-binding protein 2 (MECP2) gene has been found to cause a myriad of neurological disorders including autism, mental retardation, seizures, learning disabilities, and Rett syndrome. We hypothesized that mutations in other members of the methyl-CpG-binding domain (MBD) family may also cause autistic features in individuals. We evaluated 226 autistic individuals for alterations in the four genes most homolo- gous to MECP2: MBD1, MBD2, MBD3, and MBD4.A total of 46 alterations were identified in the four genes, including ten missense changes and two deletions that alter coding sequence. Several are either unique to our autistic population or cosegregate with affected individuals within a family, suggesting a possible relation of these variations to disease etiology. Variants include a R23M alteration in two affected half brothers which falls within the MBD domain of the MBD3 protein, as well as a frameshift in MBD4 that is predicted to truncate almost half of the protein. These results suggest that rare cases of autism may be influenced by mutations in members of the dynamic MBD protein family. Keywords Autism . Rett syndrome . MeCP2 Introduction Autism is a neurodevelopmental disorder with a triad of typical features: (1) impairments in social relatedness, (2) problems in communication, and (3) the presence of repetitive stereotyped behaviors or restricted interests [1]. Autism has an incidence of approximately one in every 1,000 individuals in the general population and has a skewed male to female ratio of about 4:1 [2]. Autism is part of a spectrum of disorders which includes Asperger ’ s syndrome, pervasive developmental disorder not otherwise specified, childhood disintegrative disorder, and Rett syndrome. Combined, autism spectrum disorders occur at a rate of approximately one in 150 individuals [3]. A high concordance of autism in monozygotic twins signifies a strong genetic component and heritability of the disorder [4–6]; however, linkage and association studies have been largely unable to uncover a single causative gene that accounts for an appreciable percentage of autism cases [7]. This is thought to result from one of two scenarios: (1) distinct, rare variants in an unknown number of genes whose strong effect results in a clinical phenotype or (2) common variants that have little effect by themselves, but acting in concert with modifiers or additional loci, may converge to promote the manifestation of autism. The first hypothesis is demonstrated by distinct cases of autism that have been shown to stem from single gene mutations, such Electronic supplementary material The online version of this article (doi:10.1007/s10048-009-0228-7) contains supplementary material, which is available to authorized users. H. N. Cukier : I. Konidari : M. Y. Rayner-Evans : M. L. Baltos : E. R. Martin : M. L. Cuccaro : M. A. Pericak-Vance : J. R. Gilbert (*) John P. Hussman Institute for Human Genomics, University of Miami, 1501 NW 10th Avenue, Miami, FL 33136, USA e-mail: [email protected] R. Rabionet Genes and Disease Program, Centre de Regulació Genòmica and CIBER en Epidemiología y Salud Pública, Barcelona, Spain H. H. Wright : R. K. Abramson University of South Carolina School of Medicine, Columbia, SC, USA Neurogenetics DOI 10.1007/s10048-009-0228-7
Transcript of Novel variants identified in methyl-CpG-binding domain genes in autistic individuals

ORIGINAL ARTICLE
Novel variants identified in methyl-CpG-binding domaingenes in autistic individuals
Holly N. Cukier & Raquel Rabionet & Ioanna Konidari & Melissa Y. Rayner-Evans &
Mary L. Baltos & Harry H. Wright & Ruth K. Abramson & Eden R. Martin &
Michael L. Cuccaro & Margaret A. Pericak-Vance & John R. Gilbert
Received: 1 May 2009 /Accepted: 26 October 2009# Springer-Verlag 2009
Abstract Misregulation of the methyl-CpG-binding protein2 (MECP2) gene has been found to cause a myriad ofneurological disorders including autism, mental retardation,seizures, learning disabilities, and Rett syndrome. Wehypothesized that mutations in other members of themethyl-CpG-binding domain (MBD) family may also causeautistic features in individuals. We evaluated 226 autisticindividuals for alterations in the four genes most homolo-gous to MECP2: MBD1, MBD2, MBD3, and MBD4. Atotal of 46 alterations were identified in the four genes,including ten missense changes and two deletions that altercoding sequence. Several are either unique to our autisticpopulation or cosegregate with affected individuals within afamily, suggesting a possible relation of these variations todisease etiology. Variants include a R23M alteration in twoaffected half brothers which falls within the MBD domainof the MBD3 protein, as well as a frameshift in MBD4 that
is predicted to truncate almost half of the protein. Theseresults suggest that rare cases of autism may be influencedby mutations in members of the dynamic MBD proteinfamily.
Keywords Autism . Rett syndrome .MeCP2
Introduction
Autism is a neurodevelopmental disorder with a triad oftypical features: (1) impairments in social relatedness, (2)problems in communication, and (3) the presence ofrepetitive stereotyped behaviors or restricted interests [1].Autism has an incidence of approximately one in every1,000 individuals in the general population and has askewed male to female ratio of about 4:1 [2]. Autism is partof a spectrum of disorders which includes Asperger’ssyndrome, pervasive developmental disorder not otherwisespecified, childhood disintegrative disorder, and Rettsyndrome. Combined, autism spectrum disorders occur ata rate of approximately one in 150 individuals [3]. A highconcordance of autism in monozygotic twins signifies astrong genetic component and heritability of the disorder[4–6]; however, linkage and association studies have beenlargely unable to uncover a single causative gene thataccounts for an appreciable percentage of autism cases [7].This is thought to result from one of two scenarios: (1)distinct, rare variants in an unknown number of geneswhose strong effect results in a clinical phenotype or (2)common variants that have little effect by themselves, butacting in concert with modifiers or additional loci, mayconverge to promote the manifestation of autism. The firsthypothesis is demonstrated by distinct cases of autism thathave been shown to stem from single gene mutations, such
Electronic supplementary material The online version of this article(doi:10.1007/s10048-009-0228-7) contains supplementary material,which is available to authorized users.
H. N. Cukier : I. Konidari :M. Y. Rayner-Evans :M. L. Baltos :E. R. Martin :M. L. Cuccaro :M. A. Pericak-Vance :J. R. Gilbert (*)John P. Hussman Institute for Human Genomics,University of Miami,1501 NW 10th Avenue,Miami, FL 33136, USAe-mail: [email protected]
R. RabionetGenes and Disease Program, Centre de Regulació Genòmicaand CIBER en Epidemiología y Salud Pública,Barcelona, Spain
H. H. Wright : R. K. AbramsonUniversity of South Carolina School of Medicine,Columbia, SC, USA
NeurogeneticsDOI 10.1007/s10048-009-0228-7

as those in NLGN3, NLGN4, and SHANK3 [8, 9]. Evidencesupporting the latter idea comes from studies that haveidentified a common CNTNAP2 variant in families withmale affected individuals and from genome-wide associationstudies that recognized a novel region of interest onchromosome 5 [10–13].
While there are many autism candidate genes, the autismspectrum disorder Rett syndrome results almost exclusivelyfrom alterations in a single gene, methyl-CpG-bindingprotein 2 (MECP2) [14]. Rett syndrome differs from classicautism in several ways, the most obvious being that amajority of patients are female. Due to MEPC2’s locationon the X chromosome, many mutations are heterozygousviable in females but hemizygous lethal in males. Rettpatients typically acquire the hallmark feature of repetitivehand wringing or clasping [15, 16]. Rett syndrome is alsocharacterized by developmental regression, a feature onlyseen in a subset of autistic patients [1, 17]. On the otherhand, Rett patients are similar to autistic individuals in thatthey demonstrate increased anxiety in novel situations andapathy toward people and objects around them [18].
Point mutations, deletions, and duplications in MECP2cause more than 85% of Rett syndrome cases [14, 19]. Whilethese mutations have been found throughout the gene in Rettpatients, they tend to occur in domains of functionalimportance: the methyl-CpG-binding domain (MBD) or thetranscriptional repression domain (TRD) [20]. Currentevidence demonstrates that the MeCP2 protein modulatesgene expression through chromatin remodeling, acting bothas an enhancer and a repressor of gene activity [21].
MeCP2 is a member of the MBD family of proteins.Along with MeCP2, there are four other closely relatedMBD proteins designated in humans: MBD1, MBD2,MBD3, and MBD4 (Fig. 1) [22]. MBD1, MBD2, MBD4,and MeCP2 each have the ability to bind DNA at methylatedCpG sites while MBD3 requires additional factors in order tobe recruited to DNA [23]. All of the MBD proteins havedemonstrated transcriptional repression activity. The MBD4protein is unique in that it has preferential binding for G–Tmismatches and acts to repair DNA damage [24]. TheMBD proteins are also capable of simultaneously bindingthe same promoter regions, implying either a functionalinterdependence or redundancy between these proteins [25,
26]. The latter can be seen where MBD2 and MBD3 act ina mutually exclusive manner to form distinct Mi-2/NuRDchromatin-remodeling complexes [27]. Therefore, it ispossible that dysfunction in one of these MBD proteinscould titrate away core components of the Mi-2/NuRDcomplex, leading to an imbalance of the protein componentsavailable to bind to the second MBD protein.
Significantly, mutations in the MECP2 gene have alsobeen identified in autistic patients without classic Rettsyndrome [28–30]. Specifically, our laboratory identifiedtwo autistic females carrying de novo MECP2 alterationsthat result in coding changes [28]. Further studies haveshown that MECP2 mutations can cause a spectrum ofclinical presentations including mild learning disabilities,mental retardation, schizophrenia, Angelman-like symptoms,and mental retardation presenting with infantile seizures [29–37]. These findings demonstrate that there is a broad range ofphenotypes caused by misregulation of the MeCP2 protein.Moreover, a recent study demonstrated an associationbetween markers within MECP2 and autism [38].
In addition to clinical data, there is accumulatingevidence from animal models demonstrating the severeeffects of MBD misregulation [39–44]. The Mecp2308/y
knock-in model generated by Shahbazian and colleaguescreates a truncated protein similar to that found in Rettpatients, and the mice exhibit anxiety, seizures, andabnormal social and nesting behavior [40, 45, 46].Furthermore, Mecp2308/y mice are defective in social,spatial, and contextual fear memory [47]. Another MBDmodel, the Mbd1−/− mouse, is also deficient in a wide rangeof social behaviors, showing increased anxiety, greatersusceptibility to depression, and a reduced prepulseinhibition of the startle response, a feature found in autisticpatients [41, 48]. Interestingly, these mice were also found tohave an increase in the RNA and protein levels of serotoninreceptor 2c [48]. Both of these models demonstrate thatmutating or completely eliminating a single MBD proteincan result in autistic features in mice.
Based on the findings of autistic patients with mutationsin MeCP2 and results from animal models, we decided toinvestigate additional members of the MBD family todetermine whether rare variations within these genes mightbe associated with autism. It is possible that the MBD
Fig. 1 The methyl-CpG-binding domain (MBD) protein family. The five MBD proteins, each containing an MBD domain, are illustrated. Alongwith this domain, each protein contains at least one other functional domain that contributes to the protein's function
Neurogenetics

domain may perform a similar function within each MBDprotein, and when the gene is misregulated, could lead tosymptoms that fall along the autism spectrum. To date, onlya single study limited to a Japanese population of 65autistic cases has evaluated the possible role of mutations inthe MBD genes in autistic patients [49]. One variation ofinterest was identified, R269C in MBD1 [49]. In this study,we evaluated MBD1, MBD2, MBD3, and MBD4 and foundmultiple alterations that cause amino acid changes andpotentially lead to autistic features.
Materials and methods
Patient ascertainment We randomly selected DNA from226 unrelated autistic individuals (195 Caucasian (CA) and31 African-American (AA) probands) and their familymembers, when available, from our dataset of autismspectrum disorder families. This cohort was comprised of180 males and 46 females.
Participants were recruited, enrolled, and sampledaccording to the Institutional Review Board protocols forthe University of Miami and the University of SouthCarolina. Individuals entered the study through well-developed referral sources (e.g., clinics) as well as throughfamily support groups, schools, and practitioners. Partic-ipants with autism were ascertained on the basis of apreviously existing diagnosis of autism and met thefollowing minimal inclusion criteria: (1) age between 3and 21 years, (2) absence of severe sensory or significantmotor impairments, and (3) absence of identified metabolic,genetic, or progressive neurological disorders. The researchdiagnosis for participants with autism was assigned usingDiagnostic and Statistical Manual of Mental Disorders IVcriteria [50], supported in most cases by the AutismDiagnostic Interview-Revised, and when available, theAutism Diagnostic Observation Schedule [51, 52]. In aminority of cases, neither measure was available, and theresearch diagnosis was made by our expert clinical panel.We also administered the Vineland Adaptive BehaviorScales (VABS) [53], a measure of developmental function-ing to assure that the participants with autism met a basicdevelopmental age of 18 months. This developmentalthreshold ensures that the diagnostic instruments are valid.Detailed family and medical histories were obtained,including information about psychiatric, developmental,and behavioral problems in family members. A completedescription of the study was provided to the subjects, andwritten informed consent was obtained. Whole blood wascollected from all participants by venipuncture.
The Caucasian control group consisted of 245 individualsthat had no known indicators of behavioral, developmental, orcognitive abnormalities (100 males and 145 females). The
human random control panel (Sigma-Aldrich) encompassedDNA from 96 Caucasian individuals collected in the UnitedKingdom (66 males and 30 females). The African-Americancontrols were a combination of the Coriell African-Americancontrol panel (NDPT031), which was comprised of 94individuals (25 males and 69 females) and 86 samplescollected at the University of Miami (49 males and 37females). Both of these datasets only contained individualsthat were negative for evidence of neurological disease.
Statistical analysis In order to determine our ability todetect variants within our autistic patient dataset, we usedthe binomial distribution to compute the probability that atleast one variant allele (with frequency p) was observed in asample of N chromosomes. We assumed sample sizes ofN=390 and N=62, which represent the number of chromo-somes in our affected CA and AA samples, respectively, andconsidered allele frequencies of 0.01 and 0.05. Theprobabilities were computed using the binomial cumulativedensity function.
Denaturing high-performance liquid chromatography GenomicDNA was isolated from whole frozen blood according tostandard protocol [54]. DNA from 226 probands wasassembled into 47 distinct pools of three to five sampleseach. Thirty-two Caucasian control samples were assembledinto five pools of three to five samples. Mutation screeningwas performed by denaturing high-performance liquidchromatography (DHPLC) using the WAVE™ DNA frag-ment analysis system (Transgenomic, Omaha). Primers weredesigned to amplify all exons for the MBD1, MBD2, MBD3,and MBD4 genes in each proband and control pool(Supplementary Table 1). The polymerase chain reaction(PCR) products were evaluated via agarose gel electropho-resis. For heteroduplex molecule formation, all PCR prod-ucts were subjected to denaturation for 3 min followed bygradual reannealing in the thermocycler for 45 min. Thetemperature for successful resolution of the heteroduplexmolecules was selected using Transgenomic software beforeproceeding to DHPLC analysis. Homozygous and heterozy-gous peak profiles were identified using Transgenomicsoftware for all proband and control pools. Pools of interestfor both homozygous and heterozygous profiles were brokendown, and each sample within the pool was sequenced.
Sequencing Individuals with possible variants identified byDHPLC were sequenced using Applied Biosystems BigDye Terminator version 3.1 on the ABI 3730 sequencer,and the primers listed in Supplementary Table 1. Wheneverpossible, family members of the proband sample were alsoexamined by sequencing. Additionally, a custom TaqManallelic discrimination assay (Applied Biosystems) wasdesigned for each novel variant. These newly identified
Neurogenetics

variants were evaluated in 150 Caucasian control individuals,and when appropriate, 180 African-American individuals.Three variants were tested in the Sigma-Aldrich Caucasianhuman random control panel instead of the 150 CA controls.For novel variants that altered the amino acid sequence andwere absent in the initial controls tested, an additional 95Caucasian controls were sequenced across the region.
Cloning mutations All insertion/deletion variants wereverified by cloning. Exons for MBD genes were amplifiedby PCR; products were cloned into the Invitrogen pCR2.1-TOPO high copy plasmid vector and transformed in Top10Escherichia coli chemically competent cells. The standardTOPO TA cloning protocol was followed with a 30-minligation using 4µl of PCR product, 30-minute incubation onice, and 75µl of transformation plated on Luria–Bertanimedium (LB)/ampicillin/X-gal. Individual colonies wereselected and grown in LB ampicillin medium, and plasmidDNAwas extracted using the Qiagen Miniprep Spin kit. Theplasmid DNA samples were sequenced on ABI 3100 usingBigDye 3.1 Sequencing kit (Applied Biosystems) and M13forward and reverse primers (Integrated DNATechnologies).
Prediction of splice factor binding sites Sequences con-taining variations within intronic and exonic regions in thisstudy were entered into the Human Splicing Analyzerversion 2.3 to reveal potential binding sites for splicingfactors (http://www.umd.be:2300/) [55].
Results
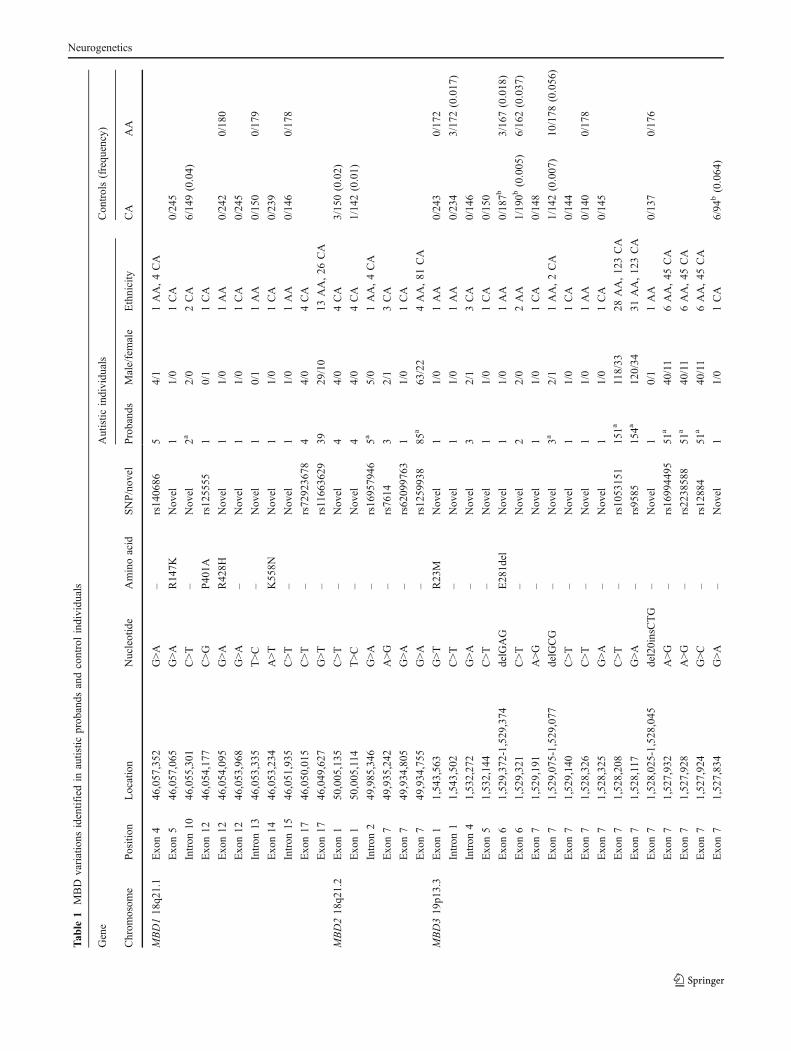
We identified 198 autistic individuals carrying geneticalterations at 46 different locations in the four MBD genesthat we studied (Table 1, Fig. 2). Twenty-five of thesechanges represent novel variations, while twenty-one aresingle nucleotide polymorphisms (SNPs) that have beenpreviously identified and incorporated into the NationalCenter for Biotechnology Information SNP database (http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp). The frequen-cies of previously recognized SNPs in both AA and CApopulations can be found in Supplementary Table 2. Of thenovel changes that were identified, 20 were singlenucleotide alterations, and five were insertion/deletions.For these 25 new variants, we tested 150 control Caucasianindividuals (300 chromosomes), and when appropriate, anadditional 180 African-American control individuals (360chromosomes) to evaluate the relative frequency of eachvariation in the general population. Fourteen of the novelsingle nucleotide changes and three of the deletions werenot found in 150 Caucasian or 180 African-Americancontrols. When a novel variant was identified that alteredthe amino acid sequence, it was tested in an additional 95
Caucasian controls (190 chromosomes). We did not detectthe MBD1 R269C variation that was previously reported ina study of Japanese autistic patients, suggesting differencesin variations due to ethnicity [49].
With our cohort of 226 autistic individuals, we calculatedthat we would have a 98.0% probability of detecting rarevariants that occurred at a 1% frequency in our 390 CAchromosomes. This probability increases to 99.9% for alleleswith a 5% frequency. We would also have a 95.8% likelihoodof detecting rare alleles occurring at a 5% frequency in our 62AA chromosomes.
MBD1
Eleven SNPs were detected in MBD1, four known andseven novel. Three of the novel SNPs as well as rs12555resulted in nonsynonymous amino acid changes (R147K,R428H, K558N, and P401A, respectively). The novelalteration c.440G>A (R147K) was identified in the Cauca-sian family no. 7801 (Fig. 2a). This alteration was found intwo of three affected brothers and an unaffected sister.Interestingly, the two affected individuals carrying thevariant appeared to be more severely affected clinicallythan the brother without the variant; while all three brotherswere found to have speech difficulties, the probanddeveloped speech later than the brother without the variant(III:5), and the third brother (III:4) remained nonverbal at45 months and had irregular breathing and gait (Table 2).The proband also demonstrated regression in language aswell as social interest and play skills, all before 5 years ofage. This alteration was inherited from the paternalgrandfather; while the father was not reported to haveautism, he did have a history of seizures. The variant wasnot identified in 245 Caucasian controls.
The second novel missense mutation, c.1283G>A(R428H), was identified in a single affected individual inthe African-American family no. 7992 and was nottransmitted maternally (Fig. 2b). This male proband wasnonverbal at the time of examination (40 months, Table 2).The third novel nonsynonymous change occurred atc.1674A>T (K558N) and was passed maternally in familyno. 7947 to a single affected son as well as an unaffectedson and daughter (Fig. 2c, Table 2). Neither of these novelSNPs were identified in control individuals. The finalnonsynonymous variant, rs125555, causes a c.1201C>Galteration and results in a P401A change. This variant wasfound in a singleton Caucasian family (no. 7978) and wascarried by two daughters—one diagnosed with autism andthe other with anxiety and speech delay (Fig. 2d). Theautistic proband presented with regression in language andsocial interest prior to 5 years of age as well as seizures andan unusual gait (Table 2). The allele is also present in boththe mother, who presented with anxiety and depression, and
Neurogenetics
Tab
le1
MBD
variations
identifiedin
autistic
prob
ands
andcontrolindividu
als
Gene
Autistic
individuals
Controls(frequency)
Chrom
osom
ePosition
Location
Nucleotide
Aminoacid
SNP/novel
Probands
Male/female
Ethnicity
CA
AA
MBD118q21.1
Exon4
46,057,352
G>A
–rs140686
54/1
1AA,4CA
Exon5
46,057,065
G>A
R147K
Novel
11/0
1CA
0/245
Intron
1046,055,301
C>T
–Novel
2a2/0
2CA
6/149(0.04)
Exon12
46,054,177
C>G
P401A
rs125555
10/1
1CA
Exon12
46,054,095
G>A
R428H
Novel
11/0
1AA
0/242
0/180
Exon12
46,053,968
G>A
–Novel
11/0
1CA
0/245
Intron
1346,053,335
T>C
–Novel
10/1
1AA
0/150
0/179
Exon14
46,053,234
A>T
K558N
Novel
11/0
1CA
0/239
Intron
1546,051,935
C>T
–Novel
11/0
1AA
0/146
0/178
Exon17
46,050,015
C>T
–rs72923678
44/0
4CA
Exon17
46,049,627
G>T
–rs11663629
3929/10
13AA,26
CA
MBD218q21.2
Exon1
50,005,135
C>T
–Novel
44/0
4CA
3/150(0.02)
Exon1
50,005,114
T>C
–Novel
44/0
4CA
1/142(0.01)
Intron
249,985,346
G>A
–rs16957946
5a5/0
1AA,4CA
Exon7
49,935,242
A>G
–rs7614
32/1
3CA
Exon7
49,934,805
G>A
–rs62099763
11/0
1CA
Exon7
49,934,755
G>A
–rs1259938
85a
63/22
4AA,81
CA
MBD319p13.3
Exon1
1,543,563
G>T
R23M
Novel
11/0
1AA
0/243
0/172
Intron
11,543,502
C>T
–Novel
11/0
1AA
0/234
3/172(0.017)
Intron
41,532,272
G>A
–Novel
32/1
3CA
0/146
Exon5
1,532,144
C>T
–Novel
11/0
1CA
0/150
Exon6
1,529,372-1,529,374
delGAG
E281del
Novel
11/0
1AA
0/187b
3/167(0.018)
Exon6
1,529,321
C>T
–Novel
22/0
2AA
1/190b
(0.005)
6/162(0.037)
Exon7
1,529,191
A>G
–Novel
11/0
1CA
0/148
Exon7
1,529,075-1,529,077
delGCG
–Novel
3a2/1
1AA,2CA
1/142(0.007)
10/178
(0.056)
Exon7
1,529,140
C>T
–Novel
11/0
1CA
0/144
Exon7
1,528,326
C>T
–Novel
11/0
1AA
0/140
0/178
Exon7
1,528,325
G>A
–Novel
11/0
1CA
0/145
Exon7
1,528,208
C>T
–rs1053151
151a
118/33
28AA,123CA
Exon7
1,528,117
G>A
–rs9585
154a
120/34
31AA,123CA
Exon7
1,528,025-1,528,045
del20insCTG
–Novel
10/1
1AA
0/137
0/176
Exon7
1,527,932
A>G
–rs16994495
51a
40/11
6AA,45
CA
Exon7
1,527,928
A>G
–rs2238588
51a
40/11
6AA,45
CA
Exon7
1,527,924
G>C
–rs12884
51a
40/11
6AA,45
CA
Exon7
1,527,834
G>A
–Novel
11/0
1CA
6/94
b(0.064)
Neurogenetics
the father, who presented with anxiety/panic and bipolardisorders.
A possible case of germline mosaicism was identified inMBD1. A novel synonymous change at c.1410G>A wasfound within exon 12 in the Caucasian family no. 7660 inaffected identical twin brothers and one unaffected sisterwhich was not identified in either of the parents (Fig. 2e,Table 2). Previous genetic testing with this family did notdemonstrate Mendelian inconsistencies, ruling out non-paternity as the cause for unusual results and suggestinginstead germline mosaicism at this locus. While thisvariation does not produce a change in amino acidsequence, the alteration is predicted to affect binding sites ofsplicing factors (Supplementary Table 2). A second novelnoncoding variant of interest was identified within intron 15in the African-American family no. 7948 (Fig. 2f). Thec.1779-27C>T change (chr18:46,051,935) is concordant withdisease in three affected brothers, two of whom are identicaltwins. While the mother does not carry the change, the fatherwas unavailable for testing. Neither variant was present inCaucasian controls nor was the c.1779-27C>T alterationpresent in African-American controls.
MBD2
We identified two novel and four previously reported SNPswithin MBD2. Five of the six changes occurred inuntranslated regions of the gene, and the remainingalteration did not result in a change of amino acid sequenceor generate a readily identified cryptic splice site.
MBD3
Variations discovered in the MBD3 gene included fiveknown SNPs, ten novel SNPs, and four insertions/deletions.Only three variants occurred in the coding region, two ofwhich altered the amino acid sequence of MBD3. Ofparticular interest is a c.68G>T (R23M) change identifiedin two affected half brothers in an African-American family(no. 7974, Fig. 2g). This variation falls within the methyl-CpG-binding domain and changes the charged arginine to anonpolar methionine. The alteration was inherited from thematernal grandmother, suggesting a possible sex-specificeffect. Both brothers were late to acquire speech and onlyone developed functional language (Table 2). The motherwas diagnosed with anxiety/panic disorder. This variationwas not identified in either the African-American orCaucasian controls.
MBD4
Alterations in our autistic population were discoveredwithin the MBD4 gene at eight previously identified SNPsT
able
1(con
tinued)
Gene
Autistic
individuals
Controls(frequency)
Chrom
osom
ePosition
Location
Nucleotide
Aminoacid
SNP/novel
Probands
Male/female
Ethnicity
CA
AA
Exon7
1,527,785-1,527,805
Deletion
–Novel
11/0
1AA
0/145
0/163
MBD43q21.3
Intron
2130,639,226
T>C
–rs140692
3829/9
11AA,27
CA
Intron
2130,638,887
C>G
–rs3138340
55/0
4AA,1CA
Exon3
130,638,360
G>A
A273T
rs10342
2719/8
2AA,25
CA
Exon3
130,638,232-130,638,235
delAAGA
E314fsX
316
Novel
10/1
1AA
0/242
0/163
Exon3
130,638,153
T>C
S342P
rs2307289
9a9/0
8AA,1CA
Exon3
130,638,141
G>A
E346K
rs140693
33/0
3CA
Exon3
130,638,104
T>C
I358T
rs2307298
5a3/2
5CA
Intron
3130,637,926
T>G
–rs3138341
55/0
4AA,1CA
Exon5
130,635,394
A>G
N467S
Novel
21/1
2CA
0/244
Exon6
130,634,779
C>T
–rs140696
3829/9
11AA,27
CA
aThese
variations
werealso
identifiedin
theDHPLCCaucasian
controlsamples
bThe
Caucasian
Sigmacontrolsamples
wereused
forthesevariations
Neurogenetics

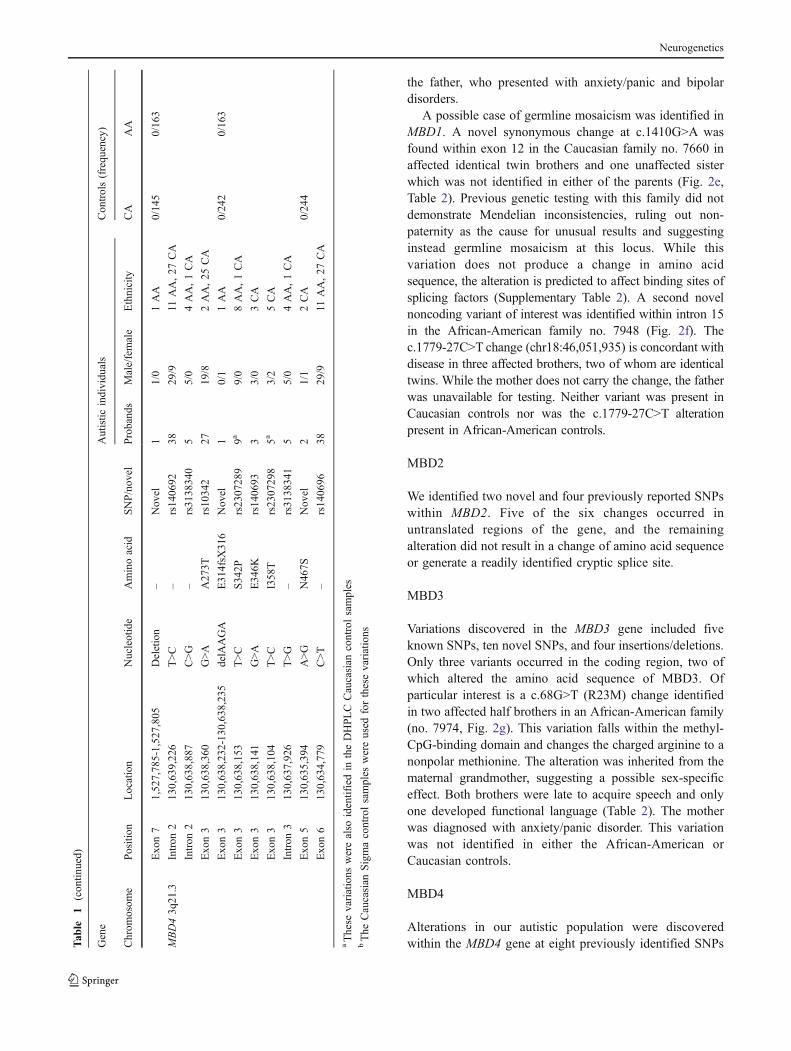
E. Family #7660 (MBD1 c.1410G>A)
G/A G/A G/A
G/G G/G
A. Family #7801 (MBD1 R147L) B. Family #7992 (MBD1 R428H)
G/A
G/G
C. Family #7947 (MBD1 K558N)
A/T
A/T
A/T A/A A/T
A/A
D. Family #7978 (MBD1 P401A)
C/GC/G bipolar
C/G C/G
I. Family #7605 (MBD4 E314fsX316)
+/del
+/del
+/+
A/G
A/G simple phobias
A/A
J. Family #7941 (MBD4 N467S) K. Family #7921 (MBD4 N467S)
A/G
A/AA/G
A/A
H. Family #7730 (MBD3 E281del)
+/+
+/+
+/+
+/del +/del
+/+
anxiety/panic disorder
depression
speech delay
autism
anxiety & depression
anxiety & speech delay
G. Family #7974 (MBD3 R23M)
G/G G/T G/T
G/T
G/Tschizophrenia
F. Family #7948 (MBD1 c.1779-27C>T)
C/C fear of heights claustrophobia
C/T C/T C/T
L. Family #7710 (MBD4 E346K) N. Family #7970 (MBD4 E346K)
G/A
G/A learning
disabilities
G/Glearning
disabilities ADHD
G/A
G/GG/A
G/A
M. Family #7785 (MBD4 E346K)
G/A
G/GG/A
G/G G/AG/G
depression & speech delay
G/G G/A
G/G
G/G G/GG/A
G/A G/GG/A G/G
G/G
G/A seizures
Fig. 2 Pedigrees of autistic families identified with mutations inMBD genes. Alterations identified in MBD1 (a–f), MBD3 (g–h), andMBD4 (i–n) are shown. Each nucleotide change is marked under the
individual in whom it occurs. In families with multiple affectedindividuals, the initially identified autistic individual (proband) issignified with an arrow
Neurogenetics

Tab
le2
Clin
ical
features
ofaffected
individu
alsidentifiedwith
variations
inMBD
genes
Variant
Fam
ilyIndividu
alAllele
Age
atexam
aAge
towalka
1st
words
aPhrase
speech
aVerbal
VABS
Regression
Midlin
emov
ements
Seizures
Irregu
lar
breathing
Unu
sual
gait
Head
circum
ference
incm
(age
a )
MBD1R14
7L78
01Maleprob
and
G/A
126
1160
96Y
–Y
NN
NY
57.0
(114
)
Brother
(III:4)
G/A
4514
––
N68
NN
NY
Y52
.5(36)
Brother
(III:5)
G/G
6212
3648
Y–
NN
NN
N50
.5(21)
MBD1P40
1A79
78Fem
aleprob
and
C/G
9213
2272
Y63
YN
YN
Y–
MBD1R
428H
7992
Maleprob
and
G/A
4013
––
N–
NN
NN
Y52
.5(46)
MBD1K
558N
7947
Maleprob
and
A/T
140
1536
40Y
54N
NN
NN
53.2
(148
)
MBD1c.14
10G>A
7660
Maleprob
and
G/A
123
–36
54Y
35–
––
––
–
Identical
twin
brother
G/A
178
1148
57Y
31N
NY
NY
–
MBD1c.17
79-27C
>T
7948
Maleprob
and
C/T
192
1836
60Y
–N
NN
NY
–
MBD3R23
M79
74Maleprob
and
G/T
9812
78-
N43
NY
NN
N54
.0(91)
Halfbrother
G/T
4111
3636
Y93
NN
NN
N48
.26(18)
MBD3E28
1del
7730
Maleprob
and
+/del
209
--
--
--
--
--
-
Brother
(III:3)
+/del
176
–84
–N
–Y
NN
NN
–
MBD4E31
4fsX
316
7605
Fem
aleprob
and
+/del
142
1348
60N
–Y
NN
NY
53.0
(132
)
MBD4E34
6K77
10Maleprob
and
G/A
5617
3942
N72
NN
NN
Y55
.9(140
)
Brother
(II:2)
–60
––
––
––
––
––
56.1
(109
)
MBD4E34
6K77
85Maleprob
and
G/A
818
3636
Y–
NN
NN
N–
Brother
(II:2)
G/G
5310
36–
N–
YN
NN
Y–
Brother
(II:3)
G/G
4910
––
N–
YY
NN
Y–
MBD4E34
6K79
70Maleprob
and
G/A
3213
––
N73
YN
NN
N49
.5(31)
MBD4N46
7S79
41Fem
aleprob
and
A/G
109
1857
57N
34Y
NN
NN
56.0
(184
)
MBD4N46
7S79
21Maleprob
and
A/G
63–
––
Y48
––
––
––
End
ashthetraitwas
notevaluated
aAge
measuredin
mon
ths
Neurogenetics

and two novel variants—a deletion and a single nucleotidechange. Six of these variants result in a coding change. Ofspecial interest is the deletion, c.942_945delAAGA(E314fsX316), transmitted maternally in a single femaleproband in the African-American family no. 7605 (Fig. 2i).The affected daughter demonstrated regression in play,social skills, and communication to the extent of becomingnonverbal (Table 2). This alteration is predicted to cause aframeshift within exon 3 at amino acid 314, encoding twoaberrant amino acids and then a premature stop codon.While retaining the methyl-CpG-binding domain, thistruncation is expected to remove almost half of the protein’s580 amino acids including the DNA glycosylase domain.No Caucasian or African-American controls were identifiedwith the alteration.
Five out of the nine single nucleotide variants result inmissense changes within MBD4. The novel c.1400A>G(N467S) variant is interesting due to its location within thefunctional DNA glycosylase domain [24]. This alterationoccurred in two Caucasian singleton families, one beinginherited from mother to daughter (family no. 7941, Fig. 2j)and the other passing from father to son (family no. 7921,Fig. 2k). The novel variation changes an asparagine to aserine and was not found in 244 Caucasian controlindividuals. The remaining four missense changes occur atc.817G>A (A273T), c.1024T>C (S342P), c.1036G>A(E346K), and c.1073T>C (I358T). Each change alterseither the polarity or charge of the original residue. TheE346K alteration (rs140693) was found in three familiesand was absent in the DHPLC control DNA (Fig. 2l–n).While this variant is not concordant with disease in thefamilies, it is interesting that in family no. 7710, theunaffected sister who also inherited the E346K change wasfound to have a delayed speech acquisition. The S342P andI358T variants were each identified in controls, suggestingthat these alterations are likely not related to autism.
Discussion
While it is unlikely that every alteration identified in thisstudy is causative of autism, several variants appearespecially interesting in their relationship to this disease.Specifically, there are two alterations which may alter theamino acid sequence sufficiently to interfere with proteinfunction. The first is the R23M alteration in MBD3. Thisalteration falls within the methyl-CpG-binding domain ofthe protein and replaces the basic arginine with a nonpolarmethionine. While this amino acid is not completelyconserved in the other members of the MBD family,another basic amino acid, lysine, is found in the sameposition for the MeCP2, MBD1 and MBD2 proteins. Analteration of the charge at this position could interfere with
the function of the MBD domain. It is also interesting thatthe variation is inherited from the maternal grandmotherthrough a mother presenting with an anxiety disorder to twoaffected half brothers. This leaves open the possibility thatthere is a sex-specific effect for this variation.
The second variation of interest is E314fsX316 withinMBD4. This maternally inherited allele is predicted tocause a frameshift at amino acid 314, encode two aberrantamino acids, and then a stop codon, resulting in aprematurely truncated protein. The mutant protein wouldcontain the MBD domain, but the DNA glycosylasedomain would be absent. While it is likely that much ofthe abnormal RNA would be removed by nonsense-mediated decay [56], it is conceivable that any truncatedprotein generated with an intact MBD domain may be ableto interfere in the normal function of all of the MBDproteins. Indeed, Bader and colleagues recently demon-strated in vitro that a MBD4 protein truncated at amino acid313 could inhibit the glycosylase activity of the wild-typeMBD4 protein as well as uracil DNA glycosylase [57]. Analternative scenario is that an irregular phenotype couldsimply result from haploinsufficiency of the wild-typeprotein. This change warrants further investigation.
There are additional examples of amino acid changesdetected in the MBD proteins that could result in functionalchanges. There are three nonsynonymous changes, MBD1R147K, MBD1 K558N, and MBD4 E346K, which eitherremove or create lysine residues. Lysine is an amino acidsubject to dramatic posttranslational modifications includingmethylation, acetylation, sumoylation, ubiquitination, andneddylation [58]. The MBD proteins have demonstrated thatthey are susceptible to such modifications [59–62]. Alteringthis key amino acid could dramatically influence the functionof a protein, affect protein regulation, or interfere withprotein–protein interactions. Another example of a variationthat may affect posttranslational modification is S342P inMBD4. The disruption of a serine could remove a potentialsite of phosphorylation or glycosylation. Likewise for thecreation of a serine site, as occurs in the MBD4 N467Svariation within the glycosylase domain. Other variantswhich alter the polarity and charge of the residues, as seenin missense mutations in MBD1 and MBD4, could interferewith a binding site on the protein or cause misfolding.
It is also possible that some of the detected MBDvariations could result in RNA processing errors bydestroying splice sites, creating cryptic splice sites, or evenchanging the ratios of alternative isoforms by eliminating orgenerating binding sites for splicing factors (SupplementaryTable 3). One example of this is the MBD1 intronic variantc.1585-12T>C, just 12 bp upstream of exon 14. Thealteration causes a change in a highly conserved nucleotide,potentially breaking a consensus site for the SRp55 splicingfactor and creating new binding sites.
Neurogenetics

Surprisingly, there appears to be a higher occurrence ofvariations with potential effects on amino acid functionwithin the African-American autistic patients as comparedto Caucasian patients. African-Americans comprise only 31of the 226 (13.7%) families participating in this study, yetthese families represent a disproportionate number of thosewith nonsynonymous alterations (24%). This could be due,in part, to the higher genetic diversity found in Africanpopulations [63, 64]. Furthermore, missense changes thatwere most abundant varied between the ethnicities, with theMBD4 A273T (rs10342) change being the most commonfor Caucasian families and the MBD4 S342P (rs2307289)alteration being predominantly found in African-Americanfamilies. These results are consistent with the HapMap datafor these known SNPs in the AA and CA populations. Inaddition, four of the five deletions identified in this studyoccurred only in African-American families, three of whichwere not identified in any control individuals. The remainingMBD3 GCG deletion was identified in both AA and CAaffected and controls while the MBD3 GAG deletion wasfound exclusively in AA affected and controls, suggestingthat both deletions are most likely not associated withautism. Nonetheless, these racial differences in variantfrequencies add to previous findings in which African-Americans with autism showed subtle differences in pheno-type and association to SNPs in GABAergic genes [65, 66].
In the families with nonsynonymous mutations, psychi-atric disorders were reported (on family history interview)in first degree relatives who carry a variation. This isconsistent with previous reports of an increased prevalenceof neuropsychiatric disorders including schizophrenia,depression, and anxiety in the parents of autistic children[67–70]. Inheritance of MBD variants from parents diag-nosed with psychiatric disorders was found in familiesnumbers 7978, 7974, 7941, and 7785 (Fig. 2d, g, j, m).Moreover, two families have additional offspring who carrythe same variant found in their affected sibling and haspsychiatric problems without a diagnosis of autism. Thesister in family no. 7978 was diagnosed with anxiety andspeech delay, and a brother in family no. 7710 alsopresented with delayed speech (Fig. 2d, l). This lendssupport to the idea that the variants identified here couldcontribute to autism and other psychological defects.Indeed, some variants were identified in both affected andunaffected individuals within a family and multiplexfamilies were not always concordant for the variant beingidentified. In some instances, as outlined above, the variantsappear in people negative for autism, but demonstratingclinical characteristic found in autism. Such is the case forfamily no. 7974 where a mother with anxiety/panic disorderpasses along the MBD3 R23M variant to two autistic halfbrothers. The variation in phenotypic presentation may alsobe related to incomplete penetrance of genetic factors. This
can be demonstrated by the fact that there is an increase inthe concordance of autism in monozygotic twins ascompared to dizygotic twins, but there is not 100%concordance in MZ twins [4–6]. Therefore, althoughgenetics plays a strong role in autism etiology, it is notthe sole component influencing presentation of disease.Lack of perfect concordance should not be used as ameasure to rule out causal mutations within complexdiseases. Other factors such as environmental or epigeneticevents may also contribute to the intricacy of autism.
It is intriguing that we observed clinical similarities inselect patients to the Rett syndrome phenotype. While, bydefinition, Rett syndrome and autism overlap clinically,there are features common to Rett syndrome which are lessfrequent in the general autistic population. For example,regression is found in most classical manifestations of Rett,but only in a minority of autistic individuals [1, 17]. Yet,we identified eight cases with a regression in language, insome cases accompanied by losses in social responsivenessor play skills. Furthermore, select individuals were reportedto have irregular breathing, unusual gait, and seizures, alltraits are commonly found in Rett patients. Also of notewere two individuals identified with unusual midlinemovements, reminiscent of the classic Rett feature wheregirls clasp their hands together at the center of their body[15, 16]. Perhaps, these patients are presenting clinically ina manner more similar to Rett individuals due to thealterations identified in this study within the MBD genes. Itis also interesting that the MBD1 R147K alterationidentified in family no. 7801 was found in the two moreseverely affected of three autistic brothers as well as anunaffected sister. This could suggest that this alterationresults in a sex-specific effect that may contribute to theautism phenotype without being the sole causative factor.
While there is mounting evidence from human andmouse studies implicating MBD genes in autism, fewpatients have been identified with alterations in MBD1,MBD2, MBD3, and MBD4 [28, 29, 39, 45, 48, 71]. Thisseeming inconsistency may be clarified by evaluating themurine phenotypes. While the Mbd1−/− mouse exhibitsdeficiencies in a wide range of social behaviors, it is able tolive an almost normal life span [48]. In contrast, Mecp2−/−
mice have a severe phenotype that is usually lethal by10 weeks of age [39, 71]. Observing the relatively mildphenotype of the Mbd1 null mice in relation to the Mecp2null mice suggests that there may be an ascertainment biasin identifying patients with recognized MECP2 mutations.MBD1 mutations may lead to such a mild phenotype thatthey rarely present as clinical abnormalities [48].
In summary, we identified genetic alterations at 46unique loci in the MBD genes. Twenty-one were previouslyrecognized SNPs, while 25 represent novel variations (fiveinsertion/deletions and 20 SNPs). Although the majority of
Neurogenetics

alterations were synonymous or noncoding variants, weidentified ten nonsynonymous changes in MBD1, MBD3,and MBD4, the deletion of a single amino acid in MBD3,and a frameshift mutation in MBD4 that is predicted totruncate almost half of the protein. These results suggestthat rare variants in these genes may contribute to autismsusceptibility. Further studies utilizing larger cohorts will berequired to determine the potential contribution of thesegenes to the autism clinical spectrum, but evidence to dateindicates that the entire MBD family may play a role inautism etiology.
Acknowledgements We are grateful to the patients with autism andtheir families for their participation, without whom, this work wouldnot be possible. A subset of the participants was ascertainedwhile several of the authors (RR, IK, MYR, ERM, MLC, MAPV,and JRG) were at Duke University. We also acknowledge fellow labmembers for their critical review of this manuscript. This research wassupported by grants from the National Institutes of Health (NS26630,NS36768, and MH080647), Autism Speaks, and by a gift from theHussman Foundation. RR is supported by the Ramon y Cajalprogram.
References
1. Johnson CP, Myers SM (2007) American academy of pediatricscouncil on children with disabilities: identification and evaluationof children with autism spectrum disorders. Pediatrics 120(5):1183–1215
2. Fombonne E (2002) Epidemiological trends in rates of autism.Mol Psychiatry 7(Suppl 2):S4–S6
3. Autism and Developmental Disabilities Monitoring NetworkSurveillance Year 2002 Principal Investigators, Centers forDisease Control and Prevention: prevalence of autism spectrumdisorders—autism and developmental disabilities monitoringnetwork, 14 sites, United States, 2002. MMWR Surveill Summ(2007) 56(1):12–28
4. Ritvo ER, Freeman BJ, Mason-Brothers A, Mo A, Ritvo AM(1985) Concordance for the syndrome of autism in 40 pairs ofafflicted twins. Am J Psychiatry 142:74–77
5. Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E,Yuzda E, Rutter M (1995) Autism as a strongly genetic disorder:evidence from a British twin study. Psychol Med 25:63–77
6. Greenberg DA, Hodge SE, Sowinski J, Nicoll D (2001) Excess oftwins among affected sibling pairs with autism: implications forthe etiology of autism. Am J Hum Genet 69:1062–1067
7. The Autism Genome Project Consortium (2007) Mapping autismrisk loci using genetic linkage and chromosomal rearrangements.Nat Genet 39(3):319–328
8. Jamain S, Quach H, Betancur C, Rastam M, Colineaux C,Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C,Bourgeron T (2003) Mutations of the X-linked genes encodingneuroligins NLGN3 and NLGN4 are associated with autism. NatGenet 34:27–29
9. Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P,Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsater H,Sponheim E, Goubran-Botros H, Delorme R, Chabane N,Mouren-Simeoni MC, de Mas P, Bieth E, Roge B, Heron D,Burglen L, Gillberg C, Leboyer M, Bourgeron T (2007) Mutationsin the gene encoding the synaptic scaffolding protein SHANK3
are associated with autism spectrum disorders. Nat Genet 39(1):25–27
10. Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, IkedaM, Rea A, Guy M, Lin S, Cook EH, Chakravarti A (2008) Acommon genetic variant in the neurexin superfamily memberCNTNAP2 increases familial risk of autism. Am J Hum Genet 82(1):160–164
11. Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV,Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, NelsonSF, Cantor RM, Geschwind DH (2008) Linkage, association, andgene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet 82(1):150–159
12. Ma DQ, Salyakina D, Jaworski JM, Konidari I, Whitehead PL,Andersen AN, Hoffman JD, Slifer SH, Hedges DJ, Cukier HN,Griswold AJ, McCauley JL, Beecham GW, Wright HH, AbramsonRK, Martin ER, Hussman JP, Gilbert JR, Cuccaro ML, Haines JL,Pericak-Vance MA (2009) A genome-wide association study ofautism reveals a common novel risk locus at 5p14.1. Ann HumGenet73(3):263–273
13. Wang K, Haitao Z, Ma D, Bucan M, Glessner JT, Abrahams BS,Salyakina D, Imielinski M, Bradfield JP, Sleiman PMA, Kim CE,Chiavacci R, Lajonchere C, Munson J, Estes A, Korvatska O,Piven J, Sonnenblick LI, Alvarez Retuerto AI, Herman EI, DongH, Hutman T, Sigman M, Ozonoff S, Klin A, Owley T, SweeneyJA, Brune CW, Cantor RM, Bernier R, Gilbert JR, Cuccaro ML,Wassink TH, McMahon WM, Coon H, Miller J, Nurnberger JI,State MW, Haines JL, Sutcliffe JS, Cook E, Minshew N,Buxbaum JD, Dawson G, Grant SFA, Geschwind DH, Pericak-Vance MA, Schellenberg GD, Hakonarson H (2009) Commongenetic variants on 5p14.1 associate with autism spectrum disorder.Nature 459(7246):528–533
14. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U,Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. NatGenet 23:185–188
15. Hagberg B, Aicardi J, Dias K, Ramos O (1983) A progressivesyndrome of autism, dementia, ataxia, and loss of purposeful handuse in girls: Rett’s syndrome: report of 35 cases. Ann Neurol 14(4):471–479
16. Hagberg B (1985) Rett’s syndrome: prevalence and impact onprogressive severe mental retardation in girls. Acta Paediatr Scand74(3):405–408
17. Stefanatos GA (2008) Regression in autistic spectrum disorders.Neuropsychol Rev 18(4):305–319
18. Shahbazian MD, Zoghbi HY (2002) Rett syndrome and MeCP2:linking epigenetics and neuronal function. Am J Hum Genet 71(6):1259–1272
19. Moretti P, Zoghbi HY (2006) MeCP2 dysfunction in Rettsyndrome and related disorders. Curr Opin Genet Dev 16(3):276–281
20. Buschdorf JP, Stratling WH (2004) AWW domain binding regionin methyl-CpG-binding protein MeCP2: impact on Rett syndrome.J Mol Med 82(2):135–143
21. Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, ZoghbiHY (2008) MeCP2, a key contributor to neurological disease,activates and represses transcription. Science 320(5880):1224–1229
22. Roloff TC, Ropers HH, Nuber UA (2003) Comparative study ofmethyl-CpG-binding domain proteins. BMC Genomics 4(1):1
23. Jorgensen HF, Bird A (2002) MeCP2 and other methyl-CpGbinding proteins. Ment Retard Dev Disabil Res Rev 8(2):87–93
24. Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A (1999) Thethymine glycosylase MBD4 can bind to the product of deaminationat methylated CpG sites. Nature 401(6750):301–304
25. Ballestar E, Ropero S, Alaminos M, Armstrong J, Setien F, AgreloR, Fraga MF, Herranz M, Avila S, Pineda M, Monros E, Esteller
Neurogenetics

M (2005) The impact of MECP2 mutations in the expressionpatterns of Rett syndrome patients. Hum Genet 116(1–2):91–104
26. Matarazzo MR, De Bonis ML, Strazzullo M, Cerase A, FerraroM, Vastarelli P, Ballestar E, Esteller M, Kudo S, D’Esposito M(2007) Multiple binding of methyl-CpG and polycomb proteins inlong-term gene silencing events. J Cell Physiol 210(3):711–719
27. Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA,Cohen A, Lasonder E, Stunnenberg HG (2006) MBD2/NuRD andMBD3/NuRD, two distinct complexes with different biochemicaland functional properties. Mol Cell Biol 26(3):843–851
28. Carney RM, Wolpert CM, Ravan SA, Shahbazian M, Ashley-Koch A, Cuccaro ML, Vance JM, Pericak-Vance MA (2003)Identification of MeCP2 mutations in a series of females withautistic disorder. Pediatr Neurol 28:205–211
29. Shibayama A, Cook EH Jr, Feng J, Glanzmann C, Yan J,Craddock N, Jones IR, Goldman D, Heston LL, Sommer SS(2004) MECP2 structural and 3′-UTR variants in schizophrenia,autism and other psychiatric diseases: a possible association withautism. Am J Med Genet B Neuropsychiatr Genet 128(1):50–53
30. Coutinho AM, Oliveira G, Katz C, Feng J, Yan J, Yang C,Marques C, Ataide A, Miguel TS, Borges L, Almeida J, CorreiaC, Currais A, Bento C, Mota-Vieira L, Temudo T, Santos M,Maciel P, Sommer SS, Vicente AM (2007) MECP2 codingsequence and 3′UTR variation in 172 unrelated autistic patients.Am J Med Genet B Neuropsychiatr Genet 144(4):475–483
31. Wan M, Lee SS, Zhang X, Houwink-Manville I, Song HR, AmirRE, Budden S, Naidu S, Pereira JL, Lo IF, Zoghbi HY, SchanenNC, Francke U (1999) Rett syndrome and beyond: recurrentspontaneous and familiar MECP2 mutations at CpG hotspots. AmJ Hum Genet 65:1520–1529
32. Watson P, Black G, Ramsden S, Barrow M, Super M, Kerr B,Clayton-Smith J (2001) Angelman syndrome phenotype associatedwith mutations in MECP2, a gene encoding a methyl CpG bindingprotein. J Med Genet 38:224–228
33. Klauck SM, Lindsay S, Beyer KS, Splitt M, Burn J, Poustka A(2002) A mutation hot spot for nonspecific X-linked mentalretardation in the MECP2 gene causes the PPM-X syndrome. AmJ Hum Genet 70(4):1034–1037
34. Milani D, Pantaleoni C, D’Arrigo S, Selicorni A, Riva D (2005)Another patient with MECP2 mutation without classic Rettsyndrome phenotype. Pediatr Neurol 32(5):355–357
35. Kankirawatana P, Leonard H, Ellaway C, Scurlock J, Mansour A,Makris CM, Dure LS 4th, Friez M, Lane J, Kiraly-Borri C, FabianV, Davis M, Jackson J, Christodoulou J, Kaufmann WE, RavineD, Percy AK (2006) Early progressive encephalopathy in boysand MECP2 mutations. Neurology 67(1):164–166
36. Harvey CG, Menon SD, Stachowiak B, Noor A, Proctor A,Mensah AK, Mnatzakanian GN, Alfred SE, Guo R, Scherer SW,Kennedy JL, Roberts W, Srivastava AK, Minassian BA, Vincent JB(2007) Sequence variants within exon 1 of MECP2 occur in femaleswith mental retardation. Am J Med Genet B Neuropsychiatr Genet144(3):355–360
37. Lugtenberg D, Kleefstra T, Oudakker AR, Nillesen WM, YntemaHG, Tzschach A, Raynaud M, Rating D, Journel H, Chelly J,Goizet C, Lacombe D, Pedespan JM, Echenne B, Tariverdian G,O’Rourke D, King MD, Green A, van Kogelenberg M, Van EschH, Gecz J, Hamel BC, van Bokhoven H, de Brouwer AP (2008)Structural variation in Xq28: MECP2 duplications in 1% ofpatients with unexplained XLMR and in 2% of male patients withsevere encephalopathy. Eur J Hum Genet
38. Loat C, Curran S, Lewis C, Abrahams B, Duvall J, Geschwind D,Bolton P, Craig I (2008) Methyl-CpG-binding protein 2 polymor-phisms and vulnerability to autism. Genes Brain Behav 7(7):754–760
39. Guy J, Hendrich B, Holmes M, Martin JE, Bird A (2001) Amouse Mecp2-null mutation causes neurological symptoms thatmimic Rett syndrome. Nat Genetic 27:322–326
40. Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B,Noebels J, Armstrong D, Paylor R, Zoghbi H (2002) Mice withtruncated MeCP2 recapitulate many Rett syndrome features anddisplay hyperacetylation of histone H3. Neuron 35(2):243–254
41. Zhao X, Ueba T, Christie BR, Barkho B, McConnell MJ,Nakashima K, Lein ES, Eadie BD, Willhoite AR, Muotri AR,Summers RG, Chun J, Lee KF, Gage FH (2003) Mice lackingmethyl-CpG binding protein 1 have deficits in adult neurogenesisand hippocampal function. Proc Natl Acad Sci USA 100(11):6777–6782
42. Collins AL, Levenson JM, Vilaythong AP, Richman R, ArmstrongDL, Noebels JL, David SJ, Zoghbi HY (2004) Mild over-expression of MeCP2 causes a progressive neurological disorderin mice. Hum Mol Genet 13(21):2679–2689
43. Cukier HN, Perez AM, Collins AL, Zhou Z, Zoghbi HY, Botas J(2008) Genetic modifiers of MeCP2 function in Drosophila. PLoSGenet 4(9):e1000179
44. Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, MorettiP, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY(2008) Deletion of Mecp2 in Sim1-expressing neurons reveals acritical role for MeCP2 in feeding behavior, aggression, and theresponse to stress. Neuron 59(6):947–958
45. Moretti P, Bouwknecht JA, Teague R, Paylor R, Zoghbi HY(2005) Abnormalities of social interactions and home-cagebehavior in a mouse model of Rett syndrome. Hum Mol Genet14(2):205–220
46. McGill BE, Bundle SF, Yaylaoglu MB, Carson JP, Thaller C,Zoghbi HY (2006) Enhanced anxiety and stress-induced cortico-sterone release are associated with increased Crh expression in amouse model of Rett syndrome. Proc Natl Acad Sci USA 103(48):18267–18272
47. Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R,Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY(2006) Learning and memory and synaptic plasticity are impairedin a mouse model of Rett syndrome. J Neurosci 26(1):319–327
48. Allan AM, Liang X, Luo Y, Pak C, Li X, Szulwach KE, Chen D,Jin P, Zhao X (2008) The loss of methyl-CpG binding protein 1leads to autism-like behavioral deficits. Hum Mol Genet 17(13):2047–2057
49. Li H, Yamagata T, Mori M, Yasuhara A, Momoi MY (2005)Mutation analysis of methyl-CpG binding protein family genes inautistic patients. Brain Dev 27(5):321–325
50. American Psychiatric Association (1994) Diagnostic and statisticalmanual of mental disorders (DSM-IV). American Psychiatric Press,Inc, Washington
51. Lord C, Rutter M, DiLavore P, Risi S (1999) Autism DiagnosticObservation Schedule-WPS (WPS edition)
52. Rutter M, LeCouteur A, Lord C (2003) Autism DiagnosticInterview, Revised (ADI-R)
53. Sparrow SS, Balla D, Cicchetti D (1984) Vineland adaptivebehavior scales, interview edition. AGS Publishing, Circle Pines
54. Vance JM (1998) The collection of biological samples for DNAanalysis. In: Haines JL, Pericak-Vance MA (eds) Approaches togene mapping in complex human diseases. Wiley-Liss, New York,pp 201–211
55. Hubbard TJ, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y,Clarke L, Coates G, Cunningham F, Cutts T, Down T, Dyer SC,Fitzgerald S, Fernandez-Banet J, Graf S, Haider S, Hammond M,Herrero J, Holland R, Howe K, Howe K, Johnson N, Kahari A,Keefe D, Kokocinski F, Kulesha E, Lawson D, Longden I,Melsopp C, Megy K, Meidl P, Ouverdin B, Parker A, Prlic A,Rice S, Rios D, Schuster M, Sealy I, Severin J, Slater G, SmedleyD, Spudich G, Trevanion S, Vilella A, Vogel J, White S, Wood M,Cox T, Curwen V, Durbin R, Fernandez-Suarez XM, Flicek P,Kasprzyk A, Proctor G, Searle S, Smith J, Ureta-Vidal A, BirneyE (2007) Ensembl 2007. Nucleic Acids Res 35:D610–D617
Neurogenetics

56. Maquat LE (2004) Nonsense-mediated mRNA decay: splicing,translation and mRNP dynamics. Nat Rev Mol Cell Biol 5(2):89–99
57. Bader SA, Walker M, Harrison DJ (2007) A human cancer-associated truncation of MBD4 causes dominant negative impair-ment of DNA repair in colon cancer cells. Br J Cancer 96(4):660–666
58. Yang XJ, Seto E (2008) Lysine acetylation: codified crosstalk withother posttranslational modifications. Mol Cell 31(4):449–461
59. Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC,Jaenisch R, Greenberg ME (2003) Derepression of BDNFtranscription involves calcium-dependent phosphorylation ofMeCP2. Science 302(5646):885–889
60. Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G,Sun YE (2003) DNA methylation-related chromatin remodeling inactivity-dependent BDNF gene regulation. Science 302(5646):890–893
61. Lyst MJ, Nan X, Stancheva I (2006) Regulation of MBD1-mediated transcriptional repression by SUMO and PIAS proteins.EMBO J 25(22):5317–5328
62. Miyake K, Nagai K (2007) Phosphorylation of methyl-CpGbinding protein 2 (MeCP2) regulates the intracellular localizationduring neuronal cell differentiation. Neurochem Int 50(1):264–270
63. Salisbury BA, Pungliya M, Choi JY, Jiang R, Sun XJ, StephensJC (2003) SNP and haplotype variation in the human genome.Mutat Res 526:53–61
64. Campbell MC, Tishkoff SA (2008) African genetic diversity:implications for human demographic history, modern human
origins, and complex disease mapping. Ann Rev Genomics HumGenet 9:403–433
65. Collins AL, Ma D,Whitehead PL,Martin ER,Wright HH, AbramsonRK, Hussman JP, Haines JL, Cuccaro ML, Gilbert JR, Pericak-VanceMA (2006) Investigation of autism and GABA receptor subunitgenes in multiple ethnic groups. Neurogenetics 7:167–174
66. Cuccaro ML, Brinkley J, Abramson RK, Hall A, Wright HH,Hussman JP, Gilbert JR, Pericak-Vance MA (2007) Autism inAfrican American families: clinical-phenotypic findings. Am JMed Genet B Neuropsychiatr Genet 144B(8):1022–1026
67. Larsson HJ, Eaton WW, Madsen KM, Vestergaard M, Olesen AV,Agerbo E, Schendel D, Thorsen P, Mortensen PB (2005) Riskfactors for autism: perinatal factors, parental psychiatric history,and socioeconomic status. Am J Epidemiol 161(10):916–925,discussion 926-8
68. Daniels JL, Forssen U, Hultman CM, Cnattingius S, Savitz DA,Feychting M, Sparen P (2008) Parental psychiatric disordersassociated with autism spectrum disorders in the offspring.Pediatrics 121(5):e1357–e1362
69. Mazefsky CA, Williams DL, Minshew NJ (2008) Variability inadaptive behavior in autism: evidence for the importance of familyhistory. J Abnorm Child Psychol 36(4):591–599
70. Wallace AE, Anderson GM, Dubrow R (2008) Obstetric andparental psychiatric variables as potential predictors of autismseverity. J Autism Dev Disord 38(8):1542–1554
71. Chen RZ, Akbarian S, Tudor M, Jaenisch R (2001) Deficiency ofmethyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 27(3):327–331
Neurogenetics




















