
Investigation of the Biosynthesis of Ganefromycins and ...
177
Investigation of the Biosynthesis of Ganefromycins and Rishirilides Dissertation to obtain the doctorate the Faculty of Chemistry, Pharmacy, and Earth Sciences the Albert-Ludwigs-Universität Freiburg im Breisgau submitted by Xiaohui Yan from Hubei, China 2012
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Investigation of the Biosynthesis of Ganefromycins and ...

Investigation of the Biosynthesis of
Ganefromycins and Rishirilides
Dissertation
to obtain the doctorate
the Faculty of Chemistry, Pharmacy, and Earth Sciences
the Albert-Ludwigs-Universität Freiburg im Breisgau
submitted by
Xiaohui Yan
from Hubei, China
2012


Dekan: Prof. Dr. Andreas Bechthold
Vorsitzender des Promotionsausschusses: Porf. Dr. Thorsten Koslowski
Referent: Prof. Dr. Andreas Bechthold
Korreferent: Prof. Dr. Irmgard Merfort
Datum der Promotion: 05.07.2012


Herrn Prof. Dr. Andreas Bechthold danke ich für sein
stetes Interesse und viele wertvolle Diskussionen, die den
Weg zu der vorliegenden Arbeit begleitet haben.


Publications and Posters
Publications
1) Ostash, B., Yan, X., Fedorenko, V., Bechthold, A. 2010. Chemoenzymatic and
bioenzymatic synthesis of carbohydrate containing natural products.
Top. Curr. Chem. 297, 105.
2) Yan, X., Probst, K., Linnenbrink, A. Arnold M., Paululat, T., Zeeck, A.,
Bechthold, A. 2012. Cloning and herterologous expression of three type II
PKS gene clusters from Streptomyces bottropensis. Chembiochem. 13(2),
224.
3) Bechthold, A., Yan, X. 2012. SnoaW/SnoaL2: A Different Two-Component
Monooxygenase. Chemistry & Biology. 19, 549.
Poster:
Investigation of three PKS type two clusters from Streptomyces Go C4/4,
Irseer Naturstofftage, Irsee, 23.-25.02.2011


Abstract I
Abstract
Ganefromycins and rirshirilides are polyketides produced by Streptomyces spp.: the
former are biosynthesized by the modular polyketide synthases and display a very
narrow spectrum of antibacterial activity against human pathogens, while the latter,
which have highly-oxygenated anthracene skeleton and are able to inhibit plasma
α2-macroglobulin, are formed by aromatic polyketide synthases. The chemical
structures and the bioactivities of ganefromycins and rishirilides have been
characterized for more than thirty years, but their biosynthetic pathways have not been
reported so far. In this thesis, gene clusters responsible for the biosynthesis of
ganefromycins and rishirilides were identified by gene inactivation and heterologous
expression. Based on the results from the gene inactivation experiments, the putative
biosynthetic pathways for ganefromycins and rishirilides were proposed.
The whole ganefromycins biosynthetic gene cluster was obtained by the contigs of the
three cosmids identified by H. Weiss (cosmid26, cosmid201 and cosmid200) and the
two cosmids found in this thesis (cosmid21 and cosmid2H19). The loss of
ganefromycins production in the PKS-disrupted mutant clearly proves that this gene
cluster is responsible for the biosynthesis of ganefromycins.
Rishirilide A and B are α2-macroglobulin inhibitors. They both consist of an anthracene
structure with three side chains, which is quite rare for aromatic polyketides. Rishirilide
A was once isolated from the culture broth of S. bottropensis, but attempts to reproduce
this compound failed. In this thesie, the cos4 which contains probably the whole
rishirilides biosynthetic gene cluster was introduced into the heterologous expression
host S. albus, to obtain S. albus::cos4. This new strain can produce rishirilide B in
various media steadily and high efficiently. Analysis of the genes in the rishirilide
cluster reveals the existence of a unique priming ketosynthase (RslK4), two rare
luciferase-like monooxygenases (RslO1 and RslO6) as well as four transcriptional
regulators (RslR1-R4).

II Abstract
Inactivation of rslK4 in cos4 resulted in the production of two novel rishirilide B
derivatives with shorter side chains. This result is consistent with the presumption that
RslK4 is responsible for selecting the starter unit and priming the polyketide
biosynthesis. Together with an acyltransferase (RslA), RslK4 catalyzes the
condensation between the amino acid-derived isobutyryl-CoA and malonyl-ACP, to
form the 3-oxo-isohexanoyl-ACP intermediate. Without RslK4, the minimal PKS is
able to utilize acetyl-CoA and propionyl-CoA as starter units.
Inactivation of rslO1 and rslO6 led to unexpected results. Two similar compounds with
only two side chains but the same number of carbons as rishirilides were isolated from
the rslO1-deleted mutant. Based on the structures of the derivatives as well as the
putative function of RslO1, a Favorskii-like oxidative rearrangement is proposed to be
involved in the biosynthsis of rishirilides, which also clarifies the origin of the three
side chains in rishirilides. In the rslO6-deleted mutant, rishirilide B was still produced.
Therefore this gene is proposed to participate in the conversion of rishirilide B to
rishirilide A.
Analysis of the four regulators by expression and transcriptional fusion with gusA gene
showed that RslR3 and RslR4 occupy a higher position in the regulatory casacade for
rishirilides production, while RslR1 and RslR2, two SARP regulators, are in the lower
position and might function by directly binding to the promoter regions of the structural
and resistance genes. Even more, transcription of rslR1 is only regulated by RslR4, and
transcription of rslR2 is only controlled by RslR3. This complicated regulatory network
might be able to explain the irreproducibility of rishirilide A in the native producer, S.
bottropensis.

Table of contents III
Table of contents
Abstract ..................................................................................................................................... I
1 Introduction .......................................................................................................................... 1
1.1 Natural products as a source of therapeutic agents ......................................................... 1
1.2 Biosynthesis of polyketides by polyketide synthases ..................................................... 3
1.2.1 Polyketide ................................................................................................................ 3
1.2.2 Type I PKS .............................................................................................................. 6
1.2.3 Type II PKS ............................................................................................................. 8
1.2.4. Type III PKS ......................................................................................................... 11
1.3. Post-PKS modifications ............................................................................................... 12
1.3.1 Oxygenases ............................................................................................................ 13
1.3.2 Methyltransferases ................................................................................................. 18
1.3.3 Glycosyltransferaes ................................................................................................ 19
1.4 Regulation of polyketide biosynthesis by SARP regulators ......................................... 20
1.5 The Polyketides Ganefromcyins and Rishirilides ......................................................... 21
1.6 Aims of this study ......................................................................................................... 22
2 Materials and Methods ...................................................................................................... 25
2.1 Chemicals, media components ...................................................................................... 25
2.2 Enzymes and Kits .......................................................................................................... 26
2.3 Media, Buffers and Solutions ........................................................................................ 27
2.3.1 Media for bacterial culture ..................................................................................... 27
2.3.2 Buffers ................................................................................................................... 29
2.3.3 Solutions of Antibiotics ......................................................................................... 31
2.3.4 Solutions for blue/white selection of E. coli .......................................................... 32
2.3.5 Buffers for preparation of protoplast ..................................................................... 32
2.3.6 Buffers for protein purification .............................................................................. 33
2.3.7 Staining regents for the staining of TLC plates ..................................................... 34
2.4 Bacterial strains ............................................................................................................. 34
2.4.1 E. coli strains ......................................................................................................... 34

IV Table of contents
2.4.2 Streptomyces strains ............................................................................................... 35
2.5 General cultivation of Streptomyces strains .................................................................. 35
2.5.1 Preparation of permanent culture and spore suspension ........................................ 36
2.5.2 Production of secondary metabolites ..................................................................... 36
2.6 Vectors, cosmids and plasmids ..................................................................................... 36
2.7 Methods in Molecular Biology and Biochemistry ........................................................ 38
2.7.1 Methods in isolation, concentration and analysis of DNA .................................... 38
2.7.2 Methods in DNA cloning ....................................................................................... 41
2.7.3 Inactivation of genes on the cosmid by λ RED-mediated recombination .............. 41
2.7.4 PCR amplication .................................................................................................... 43
2.7.5 Transformation of DNA into E. coli ...................................................................... 46
2.7.6 Methods for introducing DNA into Streptomyces ................................................. 48
2.8 Method of biochemistry ................................................................................................ 50
2.8.1 SDS-PAGE ............................................................................................................ 50
2.8.2 Expression and purification of RslK4 from E. coli ................................................ 50
2.8.3 Expression and purification of RslK4 from S. lividans 1326 ................................ 50
2.8.4 Transcriptional fusion analysis with the gusA gene ............................................... 51
2.9 Production, isolation and characterization of secondary metabolites ........................... 51
2.9.1 Production and extraction of Ganefromycin .......................................................... 51
2.9.2 Production of Rishirilide B and its derivatives in S. albus and S. lividans ............ 52
2.9.4 Purification of seconadry metabolites for structural measurement with NMR...... 54
2.9.5 Structure elucidation by NMR ............................................................................... 57
2.10 Softwares, databases and online tools ......................................................................... 58
3 Results.................................................................................................................................. 61
3.1 Indentification of the Ganefromycins biosynthetic gene cluster ................................... 61
3.1.1 Analysis of the DNA sequence of cosmid 21 ........................................................ 62
3.1.2 Screening and analysis of cosmid 2H19 ................................................................ 62
3.1.3 Analysis of the putative gan-cluster ...................................................................... 66
3.2 Identification of the ganefromycin biosynthetic gene cluster ....................................... 70

Table of contents V
3.2.1 Conjugation of S. lydicus ....................................................................................... 70
3.2.2 Verification of the gan-Cluster by disrupting ganAIV ........................................... 71
3.3 Production of Rishirilide B and analysis of the rishirilide biosynthetic gene cluster ... 73
3.3.1 Production of rishirilide B ..................................................................................... 74
3.3.2 Analysis of the rishirilide biosynthetic gene cluster .............................................. 78
3.4 Inactivation of genes in the rsl-cluster .......................................................................... 87
3.4.1 Inactivation of rslK4- the priming ketosynthase gene ........................................... 87
3.4.2 Inactivation of rslO1- a luciferase-like monooxygenase gene............................... 90
3.4.3 Inactivation of rslO6- a luciferase-like monooxygenase gene............................... 93
3.5 Expression of RslK4 in E. coli and S. lividans.............................................................. 94
3.6 Investigation of the rishirilide regulators ...................................................................... 95
3.6.1 Overexpression of the regulatory genes ................................................................. 95
3.6.2 Investigation of the regulatory hierarchy amongst the regulators .......................... 97
4. Discussion ......................................................................................................................... 101
4.1 Biosynthesis of ganefromycin ..................................................................................... 101
4.1.1 Sequencing and analysis of the ganefromycin biosynthetic gene cluster ............ 101
4.1.2 Biosynthesis of the ganefromycin polyketide chain ............................................ 102
4.1.3 Biosynthesis and attachment of the three deoxy sugars ....................................... 104
4.2 Heterologous production of rishirilide B in S. albus ................................................... 105
4.3 Overview of key proteins involved in rishirilide biosynthesis .................................... 107
4.3.1 RslK4 and RslA provide the starter unit .............................................................. 107
4.3.2 The two luciferase-like monooxygenases ............................................................ 109
4.3.3 Aromatization and cyclization catalyzed by the three cyclases ........................... 112
4.3.4 Regulation of rishirlide biosynthesis ................................................................... 113
4.4 Inactivation of rslK4, rslO1 and rslO6 ....................................................................... 114
4.4.1 Inactivation of the ketoacylsynthase gene rslK4 ................................................. 114
4.4.2 Inactivation of rslO1 and rslO6 ........................................................................... 115
4.5 Proposed biosynthetic pathway to rishirilide A .......................................................... 119
5. References ........................................................................................................................ 123

VI Table of contents
6 Appendix ........................................................................................................................... 141
6.1 List of abbreviations .................................................................................................... 141
6.2 Maps of plasmids ........................................................................................................ 144
6.2.1 pKCXY01 and pKCXY02 ................................................................................... 144
6.2.2 pKCganAIVEP ..................................................................................................... 145
6.2.3 pSOK804, pCDFDuet and pKC1218E ................................................................ 145
6.2.4 pGUS and its derived plasmids ............................................................................ 147
6.2.5 pUWL-H .............................................................................................................. 148
6.3 NMR and MS spectra of the compounds characterized in this thesis ......................... 149
Acknowledgements .................................................................................................................. A
Curriculum vitae ..................................................................................................................... C

Introduction 1
1 Introduction
1.1 Natural products as a source of therapeutic agents
Natural products refer to naturally occurring substances derived from living organisms,
such as plants and microorganisms. The majority of natural products are secondary
metabolites- small molecules that play significant roles in the relationships between the
hosts and their surroundings. It’s believed that the primary function of secondary
metabolites is to increase the likelihood of its producer’s procreation and survival by
repelling or attracting other organisms.[1]
Therefore, a lot of natural products are toxic
against humans, animals and microorganisms. It has been estimated that there are more
than 300000 natural products in the earth and they play many important roles in our
Scheme 1. Representative natural drugs from animals, plants and microorganisms.

2 Introduction
daily lives (Scheme 1). The use of natural compounds derived from plants to control
diseases can be traced to ancient Egypt and Mesopotamia. The WHO (World Health
Organization) estimated that perhaps 80% of the world’s population mainly depends on
traditional medicines for primary health care.[2]
Microorganisms are currently one major source for the production of bioactive natural
products. An important reason for the widely use of microorganisms to produce
natural products is that they are generally easy to cultivate in large scales with a
relatively short time and low costs, facilitating a cheap and steady supply of the
products. Of all the microorganisms, actinomycetes, a group of gram-positive,
falimentous and mainly soil-inhabit bacteria with high GC-content, produced almost
80% of the world’s antibiotics (Table 1).[3]
It was estimated that the number of
antibiotics characterized so far is less than 5% of the total, making the actinomycetes
group, especially streptomyces, an important source for drug discovery in the future.[4]
Table 1: New antibacterial drugs launched since 2000 from microorganisms
Year Name Class Lead Source
2002 Biapenem β-Lactam ( carbapenem) Thienamycin Actinomycete
2002 Ertapenem β-Lactam ( carbapenem) Thienamycin Actinomycete
2005 Doripenem β-Lactam ( carbapenem) Thienamycin Actinomycete
2009 Tebipenem pivoxil β-Lactam ( carbapenem) Thienamycin Actinomycete
2008 Ceftobiprole medocaril β-Lactam ( Cephalosporin) Cephalosporin Fungus
2010 Ceftaroline fosamil β-Lactam ( carbapenem) Cephalosporin Fungus
2001 Telithromycin Macrolide (Ketolide) Erythromycin Actinomycete
2003 Daptomycin Lipopeptide Daptomycin Actinomycete
2005 Tigecycline Tetracycline Tetracycline Actinomycete
2007 Retapamulin Pleuromutilin Pleuromutilin Fungus
2009 Telavancin Glycopeptide Vancomycin Actinomycete
Over the last two decades, advances in structures analysis techniques such as X-ray
crystallography, nuclear magnetic resonance (NMR), high-resolution mass spectro-
scopy (HRMS), circular dichrosim (CD) and new drug discovery methods such as
rational drug design (RDD), high-throughput screening (HTS), fragment-based drug
discovery (FBDD) and combinatiorial chemistry have attracted most of the attention in

Introduction 3
pharmaceutical industries.[5]
Most major pharmaceutical companies have terminated or
scaled down their NP operations. However, natural products are still an important
source of novel drugs in some therapeutic areas such as anti-infection, oncology,
immunosuppression and metabolic diseases.[6]
According to a report of Newman and
Cragg, approximately 70% of the new approved drugs between 1981 and 2006 were
derived, more or less, from natural products.[7]
1.2 Biosynthesis of polyketides by polyketide synthases
1.2.1 Polyketide
Polyketides are an extraordinarily valuable family of natural products. They are
widely distributed in bacteria, fungi and plants, with a number of more than 10000 for
characterized polyketides. Medically important polyketides include antibiotics
Scheme 2. Bioactive complex polyketides with diverse structure and function.

4 Introduction
(erythromycin A, monensin A, tetracycline), anticancer (epothilones and doxorubicin)
anti-parasitics (avermectin), anti-fungals (amphotericin B), immunesupperssants
(rapamycin), and cholesterol-lowering (lovastatin) agents (Scheme 2). It has been
estimated that the sales of the more than 40 polyketide medicines total more than
US$20 billion a year, close to the total sales of protein therapeuticals.[6]
Because of
their importance and great potential, there has been a great interest in finding new
polyketide molecules in the last 60 years.
The broad spectrum of biological activities of polyketides arises from their
considerable structural diversity. Some polyketides are highly rigidified and aromatic
compounds, for example, doxorubicin. They derive from similar polyketone chains by
alternative modes of folding, cyclization and post-PKS modifications. Others,
represented by erythromycin A, are diversified molecules with different compositions
of starter and extender units for the poly-β-ketone skeletons and different
conformations formed by intramolecular end-to-end cyclization. The post-assembly
modifications of polyketides, including oxidation, reduction, epimerization, acylation
and glycosylation, give rise to the structural and functional diversity of the resulting
compounds.[8]
Polyketides are produced by polyketide synthases (PKSs) via repetitive Claisen
condensations of extender units derived from malonyl-coenzyme A (malonyl-CoA)
with an activated carboxylic acid starter unit in a manner resembling the biosynthesis
of fatty acid.[9]
The typical starter unit and extender unit for fatty acid synthases
(FASs) is an acetyl moiety and malonyl-CoA, repectively, whereas PKSs can utilize a
variety of starter units including acetyl-, ethyl-, propionyl-, and butyryl-CoA, and also
variable extender units such as malonyl-, methylmalonyl-, or occasionally
ethylmalonyl-CoA. While the end result of FAS-catalyzed fatty acid biosynthesis is
typically a fully reduced carbon chain of a defined length, polyketides usually have
varying degrees of reduction at different carbon centers within the chain. Based on
protein architecture, PKSs can be classified into four groups: modular type I PKSs,
iterative type I PKSs, type II PKSs, and type III PKSs (Table 2 and Figure 1.1).
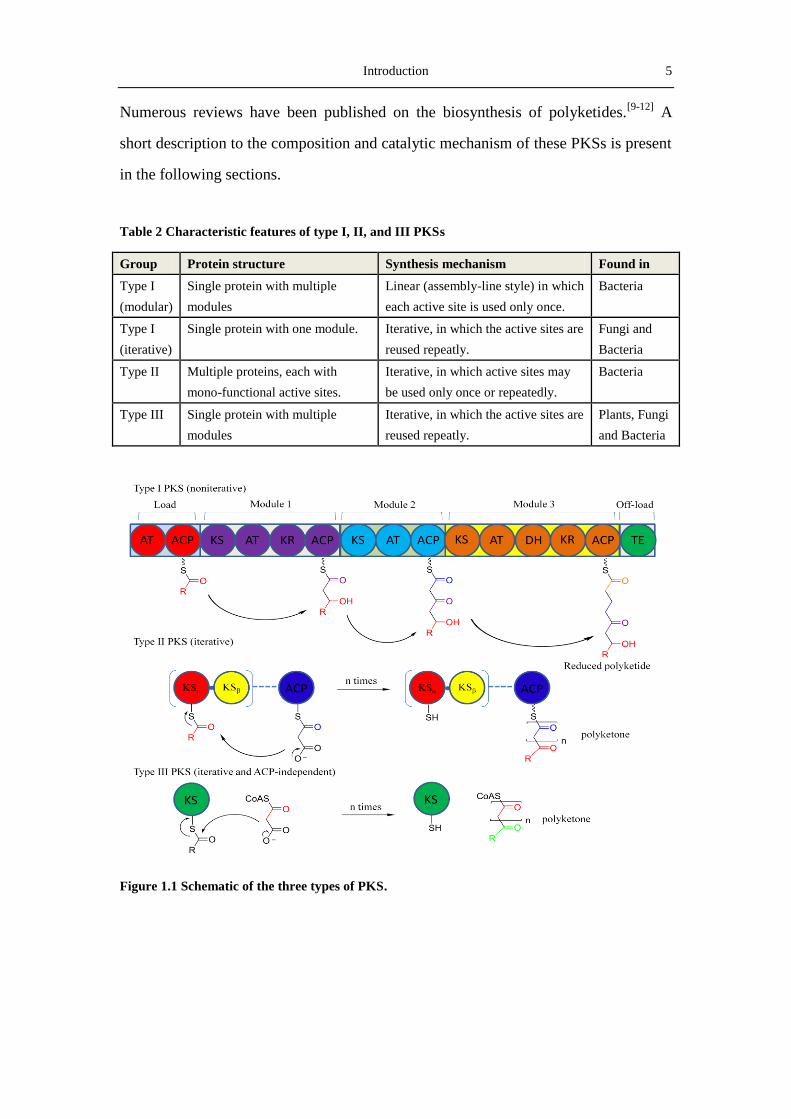
Introduction 5
Numerous reviews have been published on the biosynthesis of polyketides.[9-12]
A
short description to the composition and catalytic mechanism of these PKSs is present
in the following sections.
Table 2 Characteristic features of type I, II, and III PKSs
Group Protein structure Synthesis mechanism Found in
Type I
(modular)
Single protein with multiple
modules
Linear (assembly-line style) in which
each active site is used only once.
Bacteria
Type I
(iterative)
Single protein with one module. Iterative, in which the active sites are
reused repeatly.
Fungi and
Bacteria
Type II Multiple proteins, each with
mono-functional active sites.
Iterative, in which active sites may
be used only once or repeatedly.
Bacteria
Type III Single protein with multiple
modules
Iterative, in which the active sites are
reused repeatly.
Plants, Fungi
and Bacteria
Figure 1.1 Schematic of the three types of PKS.

6 Introduction
1.2.2 Type I PKS
Type I PKSs consist of one or more proteins that contain different active domains.
Each domain is responsible for one reaction in the polyketide chain assembly and
modification. The domains in a module are covalently fused by the linkers. Each PKS
module consist of at least three core domains: an acyltransferase (AT) domain that
captures a nucleophilic β-carboxyacyl-CoA extender unit and transfers it to the
phosphorpantetheine arm of the acyl carrier proteins (ACP) domain (Figure 1.2),
where a thioester bond is formed to fix the growing polyketide to the synthase; and a
ketosynthase (KS) domain, which is responsible for the decarboxylic condensation
between the extender unit on the ACP domain of the same module and the polyketide
intermediate bound to the ACP domain of the preceding module. Optional domains
such as ketoreductase (KR), oxidation (Ox), dehydratase (DH), methyltransferase
(MT) and enoylreductase (ER) modify the growing polyketide molecule before it is
transferred to the next module in the assembly line. In the last step for the
biosynthesis of the polyketide skeleton, the full-length polyketide chain is transferred
to a thioesterase (TE) domain, where the polyketide is released. The variation of these
chain modifying reactions, together with the variety of the starter units and extender
units lead to the structural diversity of type I polyketides.
Type I PKSs can be divided into two groups, the modular type I PKSs of bacteria and
the iterative type I PKSs of fungi. The modular PKSs typically exhibit what is called
co-linearity with their products, that is to say each module is made-up of adjacent
domains, and modules and domains are utilized in the order suggested by the gene
organization.[13]
From the sequence of modules of the PKS, one can make a
reasonably accurate prediction about the structure of the resulting product.[14]
The best
example showing how modular PKS organization is reflected in polyketide structure
is the multienzyme complex known as 6-deoxyerythronolide B synthase (DEBS),
which synthesizes the aglycon core (6-deoxyerythronolide B) of erythromycin.[15]
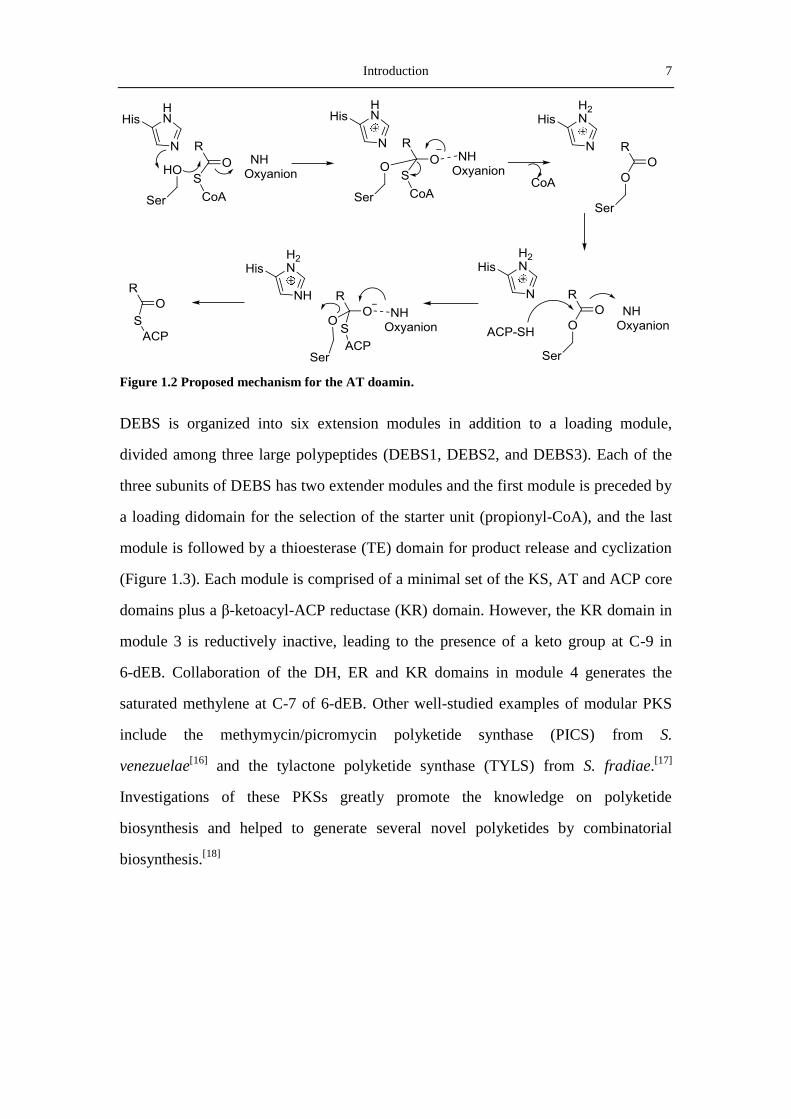
Introduction 7
Figure 1.2 Proposed mechanism for the AT doamin.
DEBS is organized into six extension modules in addition to a loading module,
divided among three large polypeptides (DEBS1, DEBS2, and DEBS3). Each of the
three subunits of DEBS has two extender modules and the first module is preceded by
a loading didomain for the selection of the starter unit (propionyl-CoA), and the last
module is followed by a thioesterase (TE) domain for product release and cyclization
(Figure 1.3). Each module is comprised of a minimal set of the KS, AT and ACP core
domains plus a β-ketoacyl-ACP reductase (KR) domain. However, the KR domain in
module 3 is reductively inactive, leading to the presence of a keto group at C-9 in
6-dEB. Collaboration of the DH, ER and KR domains in module 4 generates the
saturated methylene at C-7 of 6-dEB. Other well-studied examples of modular PKS
include the methymycin/picromycin polyketide synthase (PICS) from S.
venezuelae[16]
and the tylactone polyketide synthase (TYLS) from S. fradiae.[17]
Investigations of these PKSs greatly promote the knowledge on polyketide
biosynthesis and helped to generate several novel polyketides by combinatorial
biosynthesis.[18]

8 Introduction
Figure 1.3 Modular organization of 6-deoxyerythronolide B synthase (DEBS) and putative
intermediates.
Like DEBS, the polyketide synthases responsible for biosynthesis of the fungal
metabolites 6-MSA (by 6-MSAS)[19]
and lovastatin (synthesized by LNKS) [20]
have a
type I or covalent architecture. However, whereas DEBS contains a distinct module
for each round of chain extension, the single modules of 6-MSAS and the lovastatin
nonaketide synthase are used iteratively to assembly a polyketide product. These
fungal aromatic polyketide are known as iterative type I PKSs.
1.2.3 Type II PKS
In contrast to Type I PKSs, type II PKSs comprise of several discrete enzymes that
are monofunctional and iteratively used to generate aromatic compounds, by a mech-
anism analogous to type II bacterial and plant FASs. Type II PKSs were only found in
gram-positive actinomycetes until the recent discovery of the alkaloid aurachin PKS
from gram-negative myxobacterium.[21]
All type II PKSs contain a minimal set of

Introduction 9
iteratively used enzymes, which are generally called minimal PKS, to catalyze the
iterative decarboxylative condensation of malonyl-CoA extendr units with an acyl
starter unit (Figure 1.4). The minimal PKS consists of two ketosynthease units (KSα
and KSβ) and an ACP, which serves as an anchor for the growing polyketide chain.[10]
With a few exceptions, genes encoding these three proteins are grouped together, and
show a typical KSα/KSβ/ACP architecture. Both the KSα and KSβ subunits are highly
similar in sequence, but in the KSβ component, the active cysteine residue which is
crucial for polyketide assembly is missing. Khosla and coworkers demonstrated that
KSα and KSβ form a heterodimer in analogy to the KS homodimers (FabF) from
bacterial FAS.[22]
Furthermore, they also proved that the KSα subunit catalyzes
Claisen-type C-C bond formation from activated acyl and malonyl building blocks,
while the KSβ subunit is responsible for the loading of malonyl CoA to form acetyl
KS[23]
and for the determination of carbon chain length [therefore it is also called as
‘chain length factor’ (CLF)].[24, 25]
Beside chain assembly, the minimal PKS can
Figure 1.4 Organization of the act genes of S.coelicolar and a proposed biosynthetic pathway
for actinorhodin.

10 Introduction
partially control the regiochemistry of the first cyclization. Another opinion believes
that other than the three “minimal PKSs”, a malonyl-CoA:ACP transferase (MCAT)
is involved in the basic component of type II PKS. Since genes encoding MCATs are
not found in most type II PKS gene clusters, it was proposed that an endogenous
MCAT from the bacteria fatty acid biosynthesis pathway is recruited to participate the
polyketide biosynthesis.[26]
The crystal structure of the KSα/KSβ heterodimer showed that these two proteins form
highly complementary contacts in the interface. It was deduced that a cleft between
KSα/KSβ keeps the nascent polyketide chain extended.[27]
Observation from the
cervimycin (cer) PKS and the act PKS suggested that the length of polyketide chain is
determined by a protein cavity located at the interface of the KSα/CLF dimer by
measuring the size of the chain.[25, 28]
However, Moore and Hunter found that the
chain length is determined not only by the CLF, but also by the whole PKS complex
including the cyclase and the aromatase.[29, 30]
Most of the type II PKSs use
malonyl-CoA as primer. It is proposed that biosynthesis of aromatic polyketide starts
by decarboxylation of a malonyl unit to form an acetyl-S-KS intermediate, which is
then processed by the minimal PKS. However, plenty of type II PKSs can use other
starter units such as propionate, (iso)butyrate, malonamate and benzoate.[31]
After modifying by the minimal PKS, The highly reactive linear poly-β-carbonyl
intermediate is then subjected to a series of downstream modifications such as
ketoreduction by KR, regioselctive cyclization and aromatization by cyclase (CYC)
and aromatase (ARO) (Figure 1.5), oxidation by the oxygenase (OXY) and so on, to
yield the final aromatic compounds. In the absence of these post-PKS enzymes, the
highly labile polyketide chain undergoes spontaneous cyclization to form a series of
shunt products, as exemplified by the case of actinorhodin derivatives.[32]
The minimal PKSs can only generate very limited core polyketide structures by
varying the chain length. However, the combination of different starter units, different

Introduction 11
types and positions of cyclization, as well as different post-PKS modifications make
the type II PKSs possible to produce the numerous aromatic compounds.
Figure 1.5 The three types of CYC/AROs
1.2.4. Type III PKS
The most well-known type III PKS are the enzymes for the biosynthesis of chalcones
(CHS) and stilbenes (STS) (Figure 1.6). Members of this family are proteins of 40-70
kDa that function as homodimers and carry out iterative condensation reactions with
malonyl-CoA.[33]
It was long believed that type III PKS only exists in plant, but
recently Horinouchi and Thomashow found type III PKS from S. griseus[34]
and from
Pseudomonas fluorescens [35]
, respectively. Fujii and coworker found a type III PKS
in the fungi Aspergillus oryzae.[36]
Comparing to CHS and STS, which choose
cinnamoyl-CoA as starter unit, the type III PKS from bacteria and fungi prefer shorter
chains such as acetyl-CoA and maloyl-CoA as the primers.

12 Introduction
Figure 1.6 Biosynthesis of chalcone and stilbene by type III PKS.
1.3. Post-PKS modifications
The tailoring steps catalyzed by oxidoreductases and group transferases, such as
methyltransferases (MTs), glycosyltransferase (GTs), halogenases and acyltrans-
ferases (ATs) are responsible for adding important groups to polyketide skeletons and
are crucial for the structural diversity and biological activity of polyketides. The
biosynthetic pathway of ansamitocin P-3, a potent antitumor agent produced by
Actinosynnema pretiosum, is an excellent example to show how an amateur
polyketide (proansamitocin) is converted into the active end-product by a series of
post-PKS modifications. The biosynthetic pathway of ansamitocin, revealing by
isotopic feeding experiments and by manipulating genes of the ansamitocin (asm)
biosynthetic gene cluster, involves the assembly of an initial macrocylic polyketide,
the hypothetical proansamitocin. Proansamitocin then undergoes a six post-PKS
modification steps to introduce a chlorine, two methyl groups, a cyclic carbamate, an
ester side chain, and an epoxide function, to give ansamitocin P-3 (Figure 1.7).
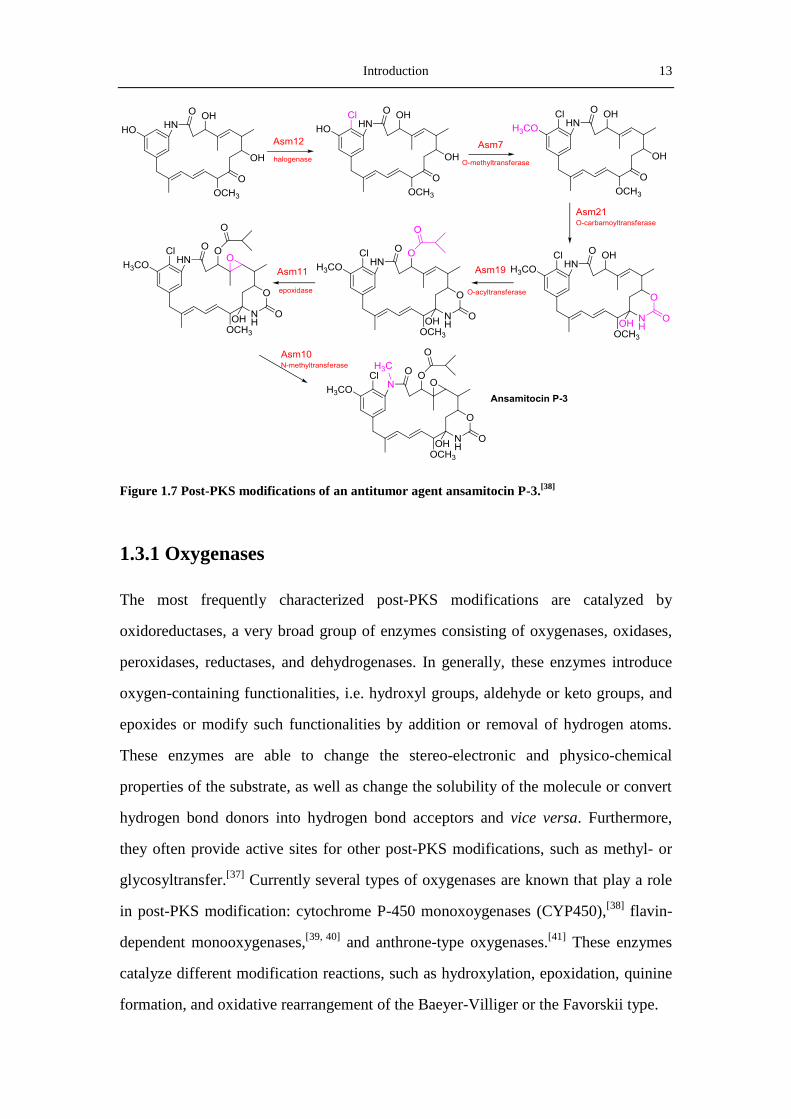
Introduction 13
Figure 1.7 Post-PKS modifications of an antitumor agent ansamitocin P-3.[38]
1.3.1 Oxygenases
The most frequently characterized post-PKS modifications are catalyzed by
oxidoreductases, a very broad group of enzymes consisting of oxygenases, oxidases,
peroxidases, reductases, and dehydrogenases. In generally, these enzymes introduce
oxygen-containing functionalities, i.e. hydroxyl groups, aldehyde or keto groups, and
epoxides or modify such functionalities by addition or removal of hydrogen atoms.
These enzymes are able to change the stereo-electronic and physico-chemical
properties of the substrate, as well as change the solubility of the molecule or convert
hydrogen bond donors into hydrogen bond acceptors and vice versa. Furthermore,
they often provide active sites for other post-PKS modifications, such as methyl- or
glycosyltransfer.[37]
Currently several types of oxygenases are known that play a role
in post-PKS modification: cytochrome P-450 monoxoygenases (CYP450),[38]
flavin-
dependent monooxygenases,[39, 40]
and anthrone-type oxygenases.[41]
These enzymes
catalyze different modification reactions, such as hydroxylation, epoxidation, quinine
formation, and oxidative rearrangement of the Baeyer-Villiger or the Favorskii type.

14 Introduction
Figure 1.8 Reactions catalyzed by the anthrone-type oxygenases TcmH and ActVA-orf6.
1.3.1.1 Anthrone-type oxygenases.
The anthrone-type monooxygenases don’t require cofactor for their catalysis. They
typically use their substrates as reducing equivalent for the reduction of an oxygen
atom (from dioxygen) to water.[41]
Hutchinson and coworkers characterized an
anthrone-type monooxygenase TcmH in the tetracenomycin biosynthetic gene cluster
from S. glaucescens. TcmH oxidizes the C-5 on naphthacenone tetracenomcin F1 to
form 5,12-naphthacenequinone tetracenomycin D3, using a radical process which
includes the generation of a superoxide anion radical.[42]
Incorporation studies with
18O and anthraquinone derivatives showed that one of the two oxygen atoms of the
quinine moiety is derived from molecular oxygen (Figure 1.8).[43]
Other well-studied
anthrone-type monooxygenases include ActVA-orf6 from the actinorhodin biosyn-
thetic pathway,[44]
AknX from the aklavinone biosynthetic pathway,[45]
and SnoaB
from the nogalamycin biosynthetic cluster.[46]
1.3.1.2 Flavin-dependent monooxygenases.
Flavin-dependent monooxyenases are attractive tailoring enzymes because of their
versatility, controllability and high enantio- and region-selectivity.[47]
Proteins of this
family incorporate an oxygen atom from molecular oxygen into the substrates with
the help of the electron rich flavin cofactor. They are widely present in a lot of
polyketides biosynthetic gene clusters[48]
. For most flavoproteins, a reactive

Introduction 15
C(4a)-hydroperoxy-flavin intermediate, which is derived from the adduction of a
molecular oxygen to the C(4a) of the flavin, is able to promote either a nucleophilic or
an electrophilic attack on the polyketide chain. As a result, one atom from molecular
oxygen is incorporated into the polyketide, while the other oxygen atom is reduced to
form water (Figure 1.9).[49]
Reactions catalyzed by the flavin-dependent monooxy-
genases include hydroxylations, epoxidations, Baeyer-Villiger and Favorskii-like
oxidative rearrangements.
Figure 1.9 Catalytic cycle of flavin-dependent monooxygenases
Baeyer-Villiger Monooxygenases [50]
The Baeyer-Villiger oxidation, a reaction oxidizing a ketone or aldehyde to an ester or
lactone, is a useful transformation in organic synthesis. The Baeyer-Villiger mono-
oxygenases (BVMO), one type of flavin-dependent monoxygenases, can catalyze the
insertion of an oxygen atom into a carbon-carbon chain of a carbonylic compound
with the help of NADPH.[50]
Rohr and coworkers studied the crystal structure of
MtmOIV, a BVMO from the mithramycin biosynthetic pathway, and provided a good
evidence for the catalytic mechanism of BVMOs.[51]
An In vitro assay in a system
contaning MtmOIV, NADPH, FAD and O2 showed that MtmOIV promotes the
cleavage of C-C bond in premithramycin B, to form an intermediate premithramycin
B-lactone, proving the Baeyer-Villiger reaction in the biosynthesis of mithramycin.
The Bayer-Villiger rearrangement is also reported to be involved in the biosynthesis

16 Introduction
of urdamycin[52]
, jadomycin,[53]
gilvocarcin (Figure 1.10),[54]
and 5-alkenyl-3,3(2H)
-furanones (E-837, E-492 and E975)[55]
, and play an important role in the biological
activity of these polyketide products.
Favorskiiase
In contrast to the widely occurrence of of Baeyer-Villiger rearrangement, the
Favorskii–type oxidative rearrangement is much rare. This reaction is named for a
reaction in which the alpha-halogenated ketones are rearranged in the presence of
base to form carboxylic acids with loss of halide. Until now only a few oxygenases
that can catalyze Favorskii rearrangement have been identified. The most famous
Favorskii-like oxgenase, EncM, which catalyzes a C-C bond rearrangement in the
polyketide chain of enterocin, has been extensively characterized.[56]
Moore and
coworkers showed that EncM catalyzes the oxidation at C-12 on the C-9 reduced
octaketide to form an 11,12,13-trione intermediate (Figure 1.10). Moreover, EncM
also catalyzes two aldol condensations between C-6&C-11 and C-7&C-14.[57]
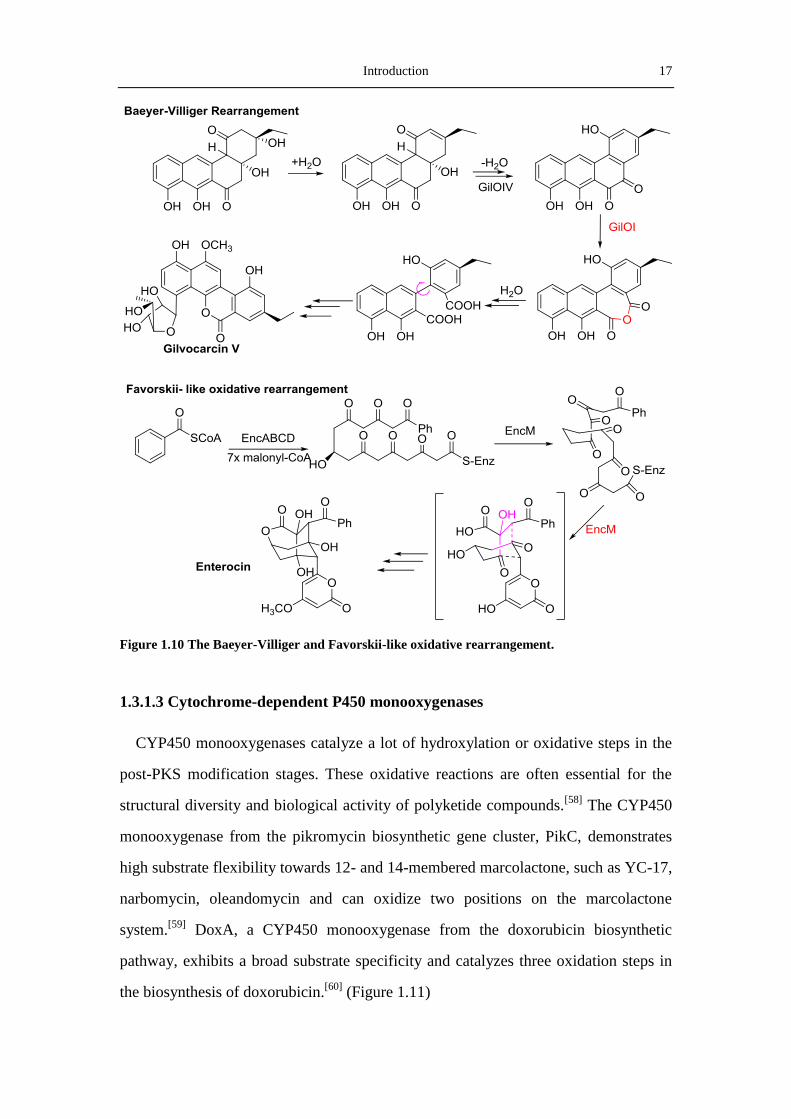
Introduction 17
Figure 1.10 The Baeyer-Villiger and Favorskii-like oxidative rearrangement.
1.3.1.3 Cytochrome-dependent P450 monooxygenases
CYP450 monooxygenases catalyze a lot of hydroxylation or oxidative steps in the
post-PKS modification stages. These oxidative reactions are often essential for the
structural diversity and biological activity of polyketide compounds.[58]
The CYP450
monooxygenase from the pikromycin biosynthetic gene cluster, PikC, demonstrates
high substrate flexibility towards 12- and 14-membered marcolactone, such as YC-17,
narbomycin, oleandomycin and can oxidize two positions on the marcolactone
system.[59]
DoxA, a CYP450 monooxygenase from the doxorubicin biosynthetic
pathway, exhibits a broad substrate specificity and catalyzes three oxidation steps in
the biosynthesis of doxorubicin.[60]
(Figure 1.11)
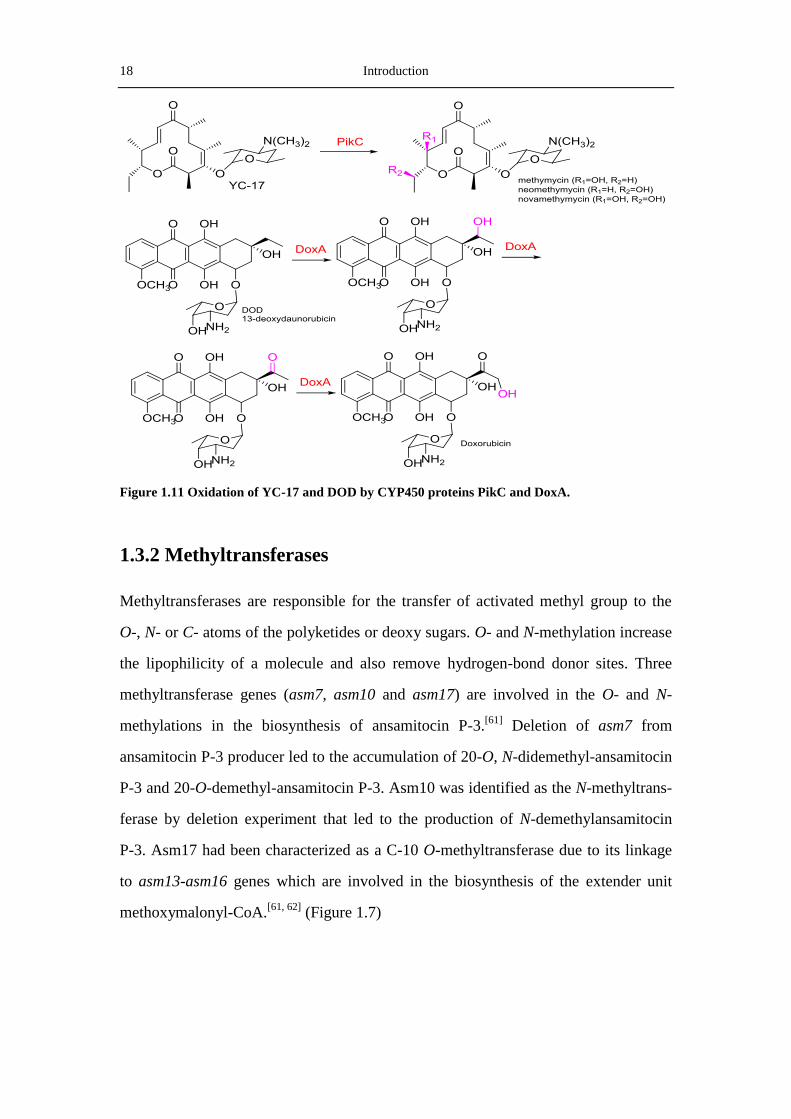
18 Introduction
Figure 1.11 Oxidation of YC-17 and DOD by CYP450 proteins PikC and DoxA.
1.3.2 Methyltransferases
Methyltransferases are responsible for the transfer of activated methyl group to the
O-, N- or C- atoms of the polyketides or deoxy sugars. O- and N-methylation increase
the lipophilicity of a molecule and also remove hydrogen-bond donor sites. Three
methyltransferase genes (asm7, asm10 and asm17) are involved in the O- and N-
methylations in the biosynthesis of ansamitocin P-3.[61]
Deletion of asm7 from
ansamitocin P-3 producer led to the accumulation of 20-O, N-didemethyl-ansamitocin
P-3 and 20-O-demethyl-ansamitocin P-3. Asm10 was identified as the N-methyltrans-
ferase by deletion experiment that led to the production of N-demethylansamitocin
P-3. Asm17 had been characterized as a C-10 O-methyltransferase due to its linkage
to asm13-asm16 genes which are involved in the biosynthesis of the extender unit
methoxymalonyl-CoA.[61, 62]
(Figure 1.7)

Introduction 19
1.3.3 Glycosyltransferaes
A large number of polyketides are glycosylated compounds, including erythromycin,
amphotericinB, avermectins and doxorubicin. These sugars contribute to the structural
biodiversity of compounds and participate in the interaction between the drug and the
cellular targets. The glycosylation reactions, in which the NDP-activated sugar donors
are attached to the acceptor molecules (aglycone), are carried out by glycosyltrans
ferases (GTs). Urdamycin A and landomycin A are two examples of glycosylated
aromatic polyketides (Figure 1.12). In most of the cases, attachment of the sugars to
the aglycone occurs through O-glycosidic linkages. But examples of C- (such as in
urdamycin) and N-glycosidic (such as in ansamitocins) linkages also exist.
Figure 1.12 Attachmentof the sugar side chains of landomycin A and urdamycin A.

20 Introduction
1.4 Regulation of polyketide biosynthesis by SARP
regulators
Streptomyces Antibiotics Regulatory Proteins (SARPs) are a novel family of
transcriptional regulators that activate the expression of specific antibiotic
biosynthetic gene clusters. Members of this family exhibit a winged helix-turn-helix
(HTH) motif near the N-termini that resembles the DNA-binding domain in the
C-terminus of the OmpR family of regulators[63]
. Some of these proteins are proved to
activate transcription by binding to the heptameric repeats within the -35 region of
their cognate promoters, and then initiate transcription by recruitment of RNA
polymerase to the appropriate sites.[64]
A well-studied SARP regulator, DnrI, has been
shown to bind to the direct-repeat sequence 5’-TCGAGC(G/C)-3’ near the -35 region
of the promoters it controls(Figure 1.13). Expression of SARP genes has been
reported to increase the production of many secondary metabolites.[65]
DnrI, the
SARP regulator from the mithramycin biosynthetic pathway can not only improve the
production of mithramycin in S. argillaceus, but also the production of actinorhodin
in S.coelicolor.[66]
Recent studies have shown that the SARP family contains not only
pathway-specific regulators but also some pleiotropic regulatory proteins, for example
AfsR from S. coelicolor.[67]
Figure 1.13 Alignment of the N-terminal amino acid sequence of DnrI with the C-terminal
DNA-binding domain of OmpR. Regions of OmpR sequence that binds to DNA (double underline)
and RNAP (dashed underline) are marked.[69]
Right: Ribbon diagram of OmpR.[70]
Introduction 21
1.5 The Polyketides Ganefromcyins and Rishirilides
Ganefromycins (ganefromycin α and ganefromycin β), a family of elfamycin
antibiotics produced by Streptomyces lydicus spp. Tanzanius Lechevalier (currently
named as S. lydicus ssp. Tanzanius NRRL18036 because of its high similarity to the
strain S. lydicus ISP 5461),[68]
have shown strong growth-promoting activity in
animals. The mechanism of the biological activity of ganefromycins is supposed to be
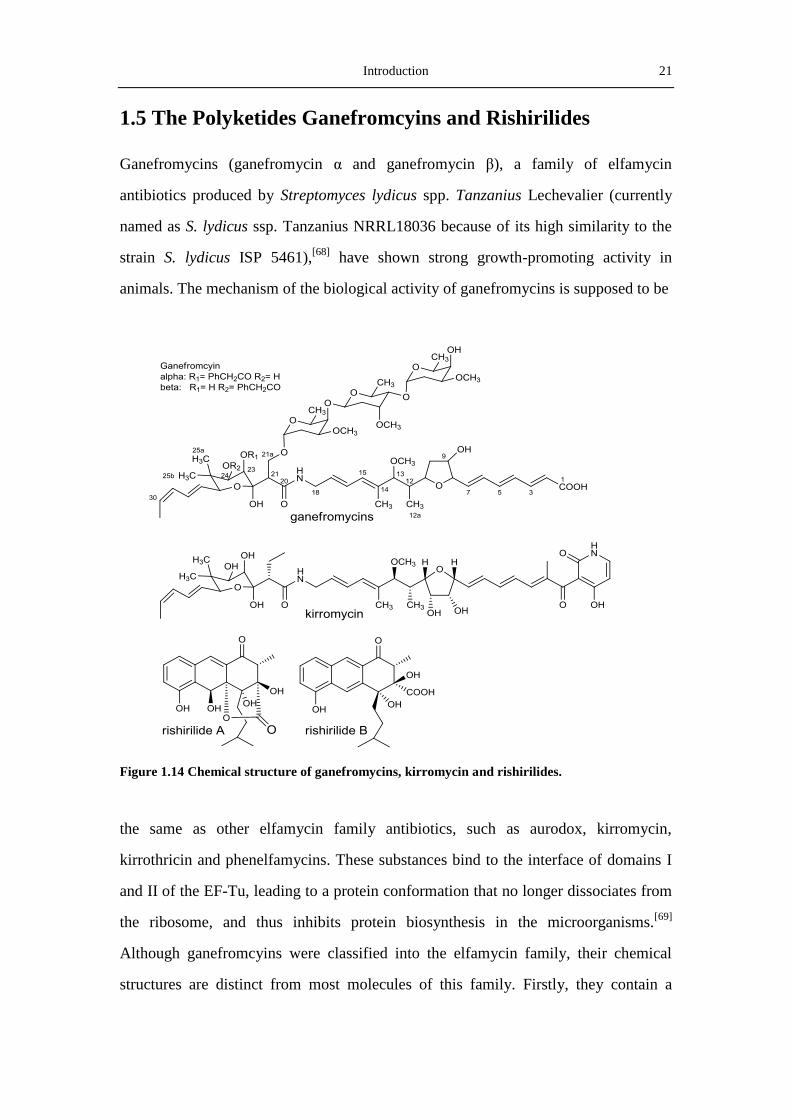
Figure 1.14 Chemical structure of ganefromycins, kirromycin and rishirilides.
the same as other elfamycin family antibiotics, such as aurodox, kirromycin,
kirrothricin and phenelfamycins. These substances bind to the interface of domains I
and II of the EF-Tu, leading to a protein conformation that no longer dissociates from
the ribosome, and thus inhibits protein biosynthesis in the microorganisms.[69]
Although ganefromcyins were classified into the elfamycin family, their chemical
structures are distinct from most molecules of this family. Firstly, they contain a

22 Introduction
trisaccharide moiety at O-21a other than O-24 for other members with deoxy sugars.
Secondly, the presence of a carboxylic acid on the truncated backbone structure is
also rare, because most of the elfamycin antibiotics contain a pyridine unit in the end
of molecules. Lastly, the phenacyl group at O-23 or O-24 is also not often (Figure
1.14). All the properties make ganefromycin an interesting target for investigating the
biosynthesis as well as the relationship between the structure and the mechanism of
action in the elfamycin antiobiotics.[70]
Rishirilides A and B, two α2-macroglobulin inhibitors, were firstly discovered in 1984
by Iwaki and coworkers from Streptomycs rishiriensis OFR-1056 (Figure 1.14).[71]
They were found to inhibit α2-macroglobulin with IC50‘s of 100 μg/ml for rishirilide
A and 35μg/ml for rishirilide B. Because α2-macroglobulin can inhibit protease via a
unique trapping mechanism, inhibitors of α2-macroglobulin are potential drugs in
treating thrombosis caused by fibrinolytic accentuation.[72]
During his work in the
discovery of the antitumor agent mensacarcin from the strain Streptomyces
bottropensis (formerly named as Streptomyces sp. Gö C4/4), Arnold Moritz also
isolated rishirilide A from the fermentation broth of S. bottropensis.[73]
However,
attempts to reproduce rishirilide A in this strain failed.
1.6 Aims of this study
Because of their therapeutic importance and structural diversity, polyketide has
always been a hot-spot both for the discovery of novel leading drugs and for the
investigation of the mechanism behind their biosynthesis. The biosynthesis of
ganefromycins by the type I PKS and rishirilides by the type II PKS are attractive, due
to their special biological activities and their unusual structures.
The aims of the ganefromycin project were:
1. Cloning and sequencing of the ganefromycin biosynthetic gene cluster.
2. Deducing of the functions of the ganefromycin biosynthetic genes.
3. Finding a method to introduce DNA into the ganefromycin producer.

Introduction 23
4. Disruption of the genes to verify the identity of the putative ganefromycin
biosynthetic gene cluster.
The aims of the rishirilides project were:
1. Production of rishirilides by heterologous expression.
2. Deletion and characterization of the rishirilides biosynthetic genes.
3. Investigation of the regulation of rishirilides biosynthetic genes.


Materials and Methods 25
2 Materials and Methods
2.1 Chemicals, media components
Table 2-1 Chemicals and media components used in this thesis
Chemical/Media Component Supplier
Aceton
Acetonitril
Acrylamide
Agar
Agarose
Ammonium persulfate (APS)
Arabinose
Bromophenol blue
Chlorofrom
Coomassie Brilliant Blue G250
Dextrin
1,4-Dithiothreitol (DTT)
Dimethyl sulfoxide (DMSO)
D-mannitol
Ethidium bromide
Ethyl acetate
Ethylene diamine tetraacetic acid (EDTA)
Glacial acetic acid
Glucose
Glycerol
Hygromycin B
Isopropanol
Isopropyl-β-thiogalactoside (IPTG)
Kanamycin sulfate
LB medium (Lennox)
Malt extract
3(N-Morpholino)-propanesulfonic acid (MOPS)
Phenol/Chloroform/Isoamylalkohol (25:24:1)
Rotiphorese Gel 30% (M/V)
Sodium dodecyl sulfate (SDS)
Roth (Karlsruhe, Germany)
26 Materials and Methods
N,N,N’,N’-Tetramethylethylenediamine (TEMED)
Tris(hydroxymethyl)aminomethane (Tris base)
Tris(hydroxymethyl)aminomethane hydrochloride
(Tris-HCl)
N-Tris-(Hydroxymethyl)-methyl-2-aminoethane
sulfonic acid (TES)
Tryptic soy broth (TSB)
Yeast extract
Roth (Karlsruhe, Germany)
Apramycin
5-Bromo-4-chlor-3-indolyl-β-D-galactopyranoside
(X-gal)
Carbenicillin
Spectionmycin
AppilChem (Darmstadt, Germany)
5-Bromo-4-chloro-3-indolyl β-D-glucuronide (X-gluc)
Phenylmethylsulfonyl fluoride (PMSF)
Phosphomycin disodium salt
Tetracycline
Thiostrepton
Sigma-Aldrich (Deisenhofen, Germany)
Coomassie Brilliant Blue R250 Serva (Heidelberg, Germany)
Casaminoacids
Malt extract
Peptone
Trypton
Becton-Difco, Heidelberg, Germany
Saccharose Suedzucker (Mannheim, Germany)
Soybean flour W. Schoenenberger GmbH (Magstadt,
Germeny)
Chloramphenicol Fluka (Ulm, Germany)
2.2 Enzymes and Kits
Table 2-2 Enyzmes and enzymatic buffers
Enzyme Supplier
Lysozyme from chicken egg white Fluka (Taufkirchen, Germany)
Oligo primers for PCR Eurofins MWG Operon (Ebersberg, Germany)
RNAse A Qiagen (Hilden)
Bovine serum albumin (BSA)
dNTP mixer
1 kb DNA ladder
Proteinase K
Restriction endonucleases
T4-DNA-Ligase
Promega (Mannheim, Germnay) or
NEB (Ipswich, US)

Materials and Methods 27
Pfu-Polymerase (5 U/μl)
Pfu-Polymerase reaction buffer (10x)
Taq-Polymerase (5 U/μl)
Taq-Polymerase reaction buffer
Lab-made
Table 2-3 Kits used for isolation and ligation of DNA
Kits Supplier
Pure Yield Plasmid Midiprep System
Wizard SV Minipreps DNA Purification System
Wizard SV Gel and PCR Clean-up System
pGEM-T Easy Vector System
Promega (Mannheim, Germnay)
Rapid DNA Ligation Kit Roche Diagnostics (Mannheim, Germnay)
2.3 Media, Buffers and Solutions
2.3.1 Media for bacterial culture
The components of the media used in the current dissertation were described as
follow. The pH of the media was adjusted by adding 1 M HCl or 1 M NaOH solution.
After well-mixed, the media were autoclaved for 20 min at 121 ℃ (15 psi). For media
used in Petri dish, 20 g/L agar was added before autoclave. If supplementary
components were required, they were commonly autoclaved seperatedly and added
into the sterile media at the time of use. Liquid media were kept at room temperature,
generally less than two weeks and Agar plates were stored at 4 ℃ for no more than
one month.
Table 2-4 Medium for cultivation of E. coli
Medium Components Note
LB-medium (Luria-Bertani
Medium)
LB medium 20 g
Restilled water 1000 mL
pH 7.3

28 Materials and Methods
Table 2-5 Media for cultivation of Streptomyces strains
Medium Components Note
CRM-medium Sucrose 103.0 g
Tryptic soy broth 20.0 g
MgCl2.6H2O 10.12 g
Yeast extract 10.0 g
Distilled water 1000 mL
CaCl2 10 mM
Glycine 0.75% (m/V)
pH 7.0, Sterile CaCl2
and Glycine
separatedly, then add to
the medium before use.
DNMP-medium Soytone 7.5 g
Baker’s yeast 5 g
MOPS 21 g
Distilled water 1000 mL
pH 6.8
HA-medium Glucose 4 g
Yeast extract 4 g
Malt extract 10 g
Tap water 1000 mL
pH 7.2
MS-medium Soybean flour 20 g
D-Mannito 20 g
Tap water 990 mL
MgCl2 10 mM
pH 7.2, autoclave
MgCl2 separately and
add at the time of use.
NL19 Soybean flour 20.0 g
D-Mannitol 20.0 g
Tap water 1000 mL
pH 7.2
PM-medium Soybean flour 10 g
Mannitol 10 g
CaCO3 5 g
Tap water 1000 mL
pH 7.3
R2YE-medium Sucrose 103.0 g
K2SO4 0.25 g
MgCl2.6H2O 10.12 g
Glucose 10.0 g
Casaminoacids 0.1 g
Trace elements solution 2.0 mL
Yeast extract 5.0 g
TES 5.73 g
Agar 20.0 g
Distill water 1000 mL
After autoclave, add
KH2PO4 (0.5%) 10 mL
CaCl2.2H2O (5M) 4 mL
L-Proline (20% (w/V)) 15 mL
NaOH (1M) 7 mL
Materials and Methods 29
R3 soft agar Sucrose 171.0 g
Glucose 10.0 g
Peptone 4.0 g
KCl 0.5 g
CaCl2.2H2O 2.2 g
MgCl2.6H2O 8.1 g
Agar 8.0 g
Distilled water 1000 mL
After autoclave, add
KH2PO4 (0.5%) 40 mL
TES (0.25 M, pH 7.2) 100 mL
SG-medium Soy peptone 10.0 g
Glucose 20.0 g
L-Valine 2.34 g
CaCO3 2.0 g
CoCl2-solution 1mg/mL 1 mL
Tap water 1000 mL
pH 7.2
Soft nutrient agar Nutrient broth 8 g
Agar 5 g
Distilled water 1000 mL
Trace elements solution ZnCl2 40 mg
FeCl3.6H2O 200 mg
CuCl2.2H2O 10 mg
MnCl2.4H2O 10mg
Na2B4O6.10H2O 10 mg
(NH4)6Mo7O24.4H2O 10 mg
Distilled water 1000 mL
TSB-Medium Tryptic Soy Broth 30 g
Tap water 1000 mL
pH 7.2
YEME medium Yeast extract 3.0 g
Peptone 5.0 g
Malt extract 3.0 g
Glucose 10.0 g
Sucrose 340.0 g
Distilled water 1000 mL
Add sterile
MgCl2.6H2O to end
concentration 5 mM
after autoclave.
2.3.2 Buffers
2.3.2.1 Buffers for isolation of plasmid from E.coli
Unless otherwise stated, the buffers were prepared with distilled water and stored at
room temperature.

30 Materials and Methods
Table 2-6 Buffers for isolation of plasmid from E.coli
Name Component Note
P1 Tris
EDTA
RNAse A
50 mM
10 mM
100 μg/mL
pH 7.8, add RNAse A just before use. Store at 4 ℃.
P2 NaOH
SDS
0.2 M
1% (m/V)
P3 KOAc 3 M pH 5.2, store at 4 ℃.
TE Tris
EDTA
10 mM
1 mM
pH 7.6
2.3.2.2 Buffers for isolation of genomic DNA from Streptomyces spp.
Table 2-7 Buffers for isolation of genomic DNA from Streptomyces strains
Puffer Component Note
SET-buffer Tris-HCl 20 mM
EDTA 25 mM
NaCl 75 mM
pH 8
Lysozyme solution Lysozyme 50 mg/mL Dissolved in SET buffer
Proteinase K solution Proteinase K 20 mg/ML Dissolved in SET buffer
SDS solution SDS 10%
NaCl solution NaCl 5 M
2.3.2.3 Buffers for DNA gel electrophoresis
Table 2-8 Buffers for DNA gel electrophoresis
Buffer Components Note
50 x TAE Tris base 2M
EDTA (0.5 M, pH 8.0) 0.05M
Glacial acetic acid 52.5 mL
Adjust the pH to 8.0 with glacial
acetic acid.
Load buffer Glycerol 30% (w/V)
Bromophenol blue 0.25% (w/V)
Store at 4 ℃
Agarose 0.7% (m/V) Agarose 7 g
TAE-buffer (1x) 1000 mL
Dissolve the agarose thorough in the
microwave oven, then store at 55 ℃.
Ethidium bromide
staining buffer
Ethidium bromide 10 μg/mL

Materials and Methods 31
2.3.2.4 Buffers and solutions for protein gel electrophoresis (SDS-PAGE) and for
Coomassie staining
Table 2-9 buffers and solutions for SDS-PAGE and Coomassie staining
Buffer/ Solution Component Note
Coomassie Brilliant Blue
G-250 solution
Coomassie Brilliant Blue G-250
Acetic acid
Methanol
Distilled water
0.25% (w/V)
10% (V/V)
45% (V/V)
45% (V/V)
Fixing buffer Acetic acid
Methanol
Distilled water
10% (V/V)
20% (V/V)
70% (V/V)
Resolving gel (10%) Distilled water
1.5 M Tris-HCl (pH 8.8)
10% (w/V) SDS
Rotiphorese® Gel 30
10% (w/V) APS
TEMED
4.1 mL
2.5 mL
0.1 mL
3.3 mL
50 μL
5 μL
Mixed all the
components except
APS and TEMED
sufficiently. Then
add APS and
TEMED just before
preparing the gel. Stacking gel (4%) Distilled water
0.5 M Tris-HCl (pH 6.8)
10% (w/V) SDS
Rotiphorese® Gel 30
10% (w/V) APS
TEMED
6.1 mL
2.5 mL
0.1 mL
1.3 mL
50 μL
10 μL
Sample buffer Distilled water
0.5 M Tris-HCl (pH 6.8)
Glycerol
10% SDS (w/V)
0.5% Bromophenol blue
3.55 mL
1.25 mL
2.5 mL
2.0 mL
0.2 mL
Add 50μL
β-mercapto- ethanol
to 950 μL sample
buffer prior to use.
10 x Running buffer Tris base
Glycine
SDS
Distilled water
30.0 g
144.0 g
10.0 g
1000 mL
Store at 4 ℃.
Stripping solution Distilled water
Acetic acid
Methanol
45% (w/V)
10% (w/V)
45% (w/V)
2.3.3 Solutions of Antibiotics
Antibiotics were dissolved in appropriate solvents at stock solution and kept at -20℃.
The aqueous solutions were sterilized by filtrated through 0.22 μm filter. The
32 Materials and Methods
solutions in ethanol and DMSO were sterile by the solvents. For antibiotic selection,
the described antibiotics were added to the media in appropriate concention unless
otherwise stated.
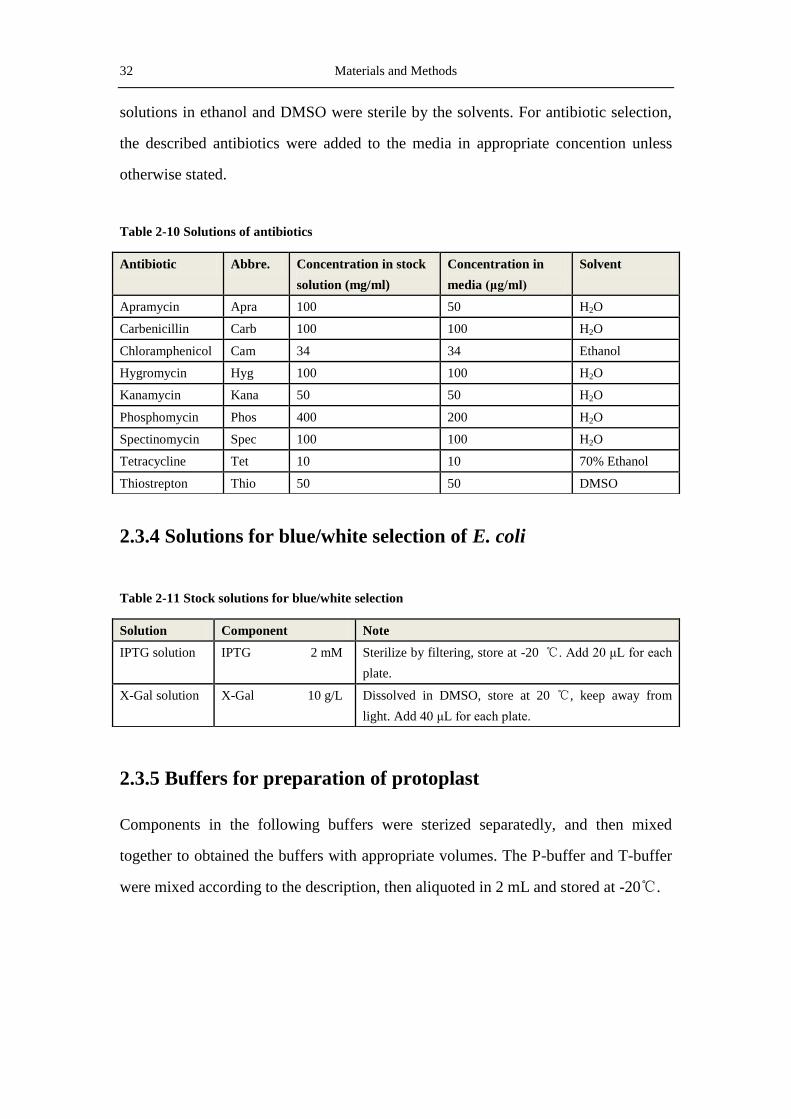
Table 2-10 Solutions of antibiotics
2.3.4 Solutions for blue/white selection of E. coli
Table 2-11 Stock solutions for blue/white selection
2.3.5 Buffers for preparation of protoplast
Components in the following buffers were sterized separatedly, and then mixed
together to obtained the buffers with appropriate volumes. The P-buffer and T-buffer
were mixed according to the description, then aliquoted in 2 mL and stored at -20℃.
Antibiotic Abbre. Concentration in stock
solution (mg/ml)
Concentration in
media (μg/ml)
Solvent
Apramycin Apra 100 50 H2O
Carbenicillin Carb 100 100 H2O
Chloramphenicol Cam 34 34 Ethanol
Hygromycin Hyg 100 100 H2O
Kanamycin Kana 50 50 H2O
Phosphomycin Phos 400 200 H2O
Spectinomycin Spec 100 100 H2O
Tetracycline Tet 10 10 70% Ethanol
Thiostrepton Thio 50 50 DMSO
Solution Component Note
IPTG solution IPTG 2 mM Sterilize by filtering, store at -20 ℃. Add 20 μL for each
plate.
X-Gal solution X-Gal 10 g/L Dissolved in DMSO, store at 20 ℃, keep away from
light. Add 40 μL for each plate.

Materials and Methods 33
Table 2-12 Buffers for preparation and transformation of Streptomyces protoplast
2.3.6 Buffers for protein purification
Table 2-13 Buffers for protein purification (Stored at 4 ℃)
Buffer Component Note
P (protoplast) buffer 12% Sucrose (w/V) 85.5 mL
MgCl2.6H2O (1 M) 1.0 mL
K2SO4 (140 mM) 1.0 mL
Trace elements solution 0.2 mL
KH2PO4 (40 mM) 1.0 mL
CaCl2.2H2O (250 mM) 1.0 mL
TES (0.25 M, pH7.2) 10.0 mL
Autoclave separately. Then
mix the components
according to the description
and aliquot the buffer in 2
mL and store at -20 ℃.
T (transformation) buffer 25% (w/V) Sucrose 1.0 mL
Trace elements solution 0.03 mL
K2SO4 (140 mM) 0.1 mL
KH2PO4 (40 mM) 0.1 mL
MgCl2.6H2O (1 M) 0.1 mL
CaCl2.2H2O (5 M) 1.0 mL
Tris-maleate (0.5 M, pH 8.0) 1.0 mL
50% (w/V) PEG1000 5.0 mL
Autoclave separately. Then
mix the components
according to the description
and aliquot the buffer in 2
mL and store at -20 ℃.
Denaturing reagent 25x TE buffer 400 μL
EDTA (0.1 M) 100 μL
Glycerol 5 mL
Ethyleneglycol 5 mL
Buffer Component
Elution buffer NaH2PO4/Na2HPO4 50 mM (pH 8.0)
NaCl 300 mM
Imidazol 250 mM
G1 buffer Tris-HCl 50 mM (pH 8.0)
DTT 5 mM
PMSF 50 μM
G2 buffer NaCl in G1 buffer 150 mM
Lysis buffer NaH2PO4/Na2HPO4 50 mM (pH 8.0)
NaCl 300 mM
Lysozyme 4 mg/mL
Imidazol 10-15 mM
Triton X-100 (1% (w/V)) 0.2%
PMSF stock solution PMSF in isopropanol 50 mM

34 Materials and Methods
2.3.7 Staining regents for the staining of TLC plates
Table 1-14 Staining regents for the staining of TLC plates
2.4 Bacterial strains
Cultivation of E. coli was always carried out in the LB medium. Single clones from
the LB plates were picked with toothpick and inoculated into test tubes containing 4
mL LB liquid medium, or into 300 mL Erlenmeyer flasks containing 100 mL LB
liquid medium. For plasmid isolation, the E. coli cells were grown overnight at 37 ℃
on a ratary shaker with the speed 180 rpm. For the λ Red-mediated recombination, E.
coli DH5α cells harboring the pBADαβγ plasmid were cultivated at 30 ℃. In order to
obtain single clone, E. coli cells were incubated on LB agar plates overnight at 37 ℃.
When a temperature sensitive plasmid (such as pSC101) is used, the E. coli cells were
incubated at 28 ℃ or 30 ℃.
2.4.1 E. coli strains
Storage buffer Tris-HCl 0.2 M (pH 7.5)
Glycerol 15%
Wash buffer NaH2PO4/Na2HPO4 50 mM (pH 8.0)
NaCl 300 mM
Imidazol 20-30 mM
Name Component
Anisaldehyde Anisaldehyde 1.0 mL
Methanol 85 mL
Acetic acid 10 mL
Concentrated sulfuric acid 5 mL
Vanillin/H2SO4 Vanillin 1.0 g
Concentrated sulfuric acid 100 mL
Ehrlichs reagent 4-dimethylaminobenzaldehyde 1.0 g
Hydrochloride acid (36%) 25 mL
Methanol 75 mL
Materials and Methods 35
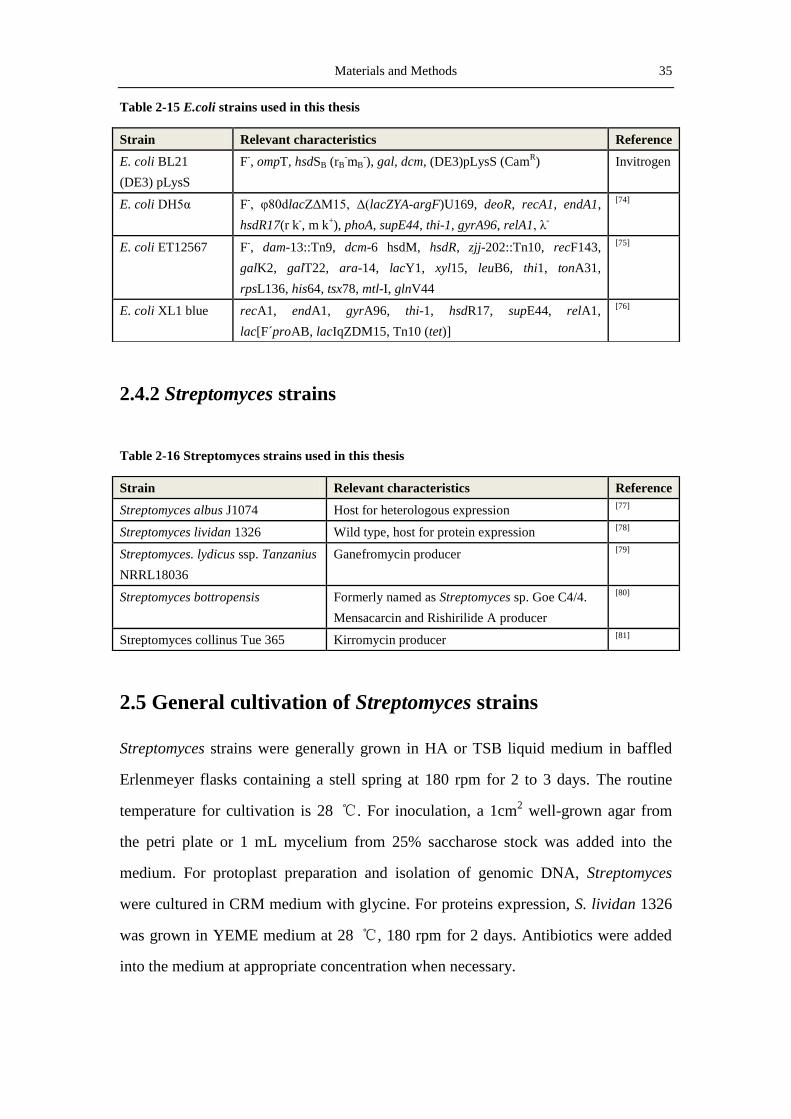
Table 2-15 E.coli strains used in this thesis
2.4.2 Streptomyces strains
Table 2-16 Streptomyces strains used in this thesis
2.5 General cultivation of Streptomyces strains
Streptomyces strains were generally grown in HA or TSB liquid medium in baffled
Erlenmeyer flasks containing a stell spring at 180 rpm for 2 to 3 days. The routine
temperature for cultivation is 28 ℃. For inoculation, a 1cm2 well-grown agar from
the petri plate or 1 mL mycelium from 25% saccharose stock was added into the
medium. For protoplast preparation and isolation of genomic DNA, Streptomyces
were cultured in CRM medium with glycine. For proteins expression, S. lividan 1326
was grown in YEME medium at 28 ℃, 180 rpm for 2 days. Antibiotics were added
into the medium at appropriate concentration when necessary.
Strain Relevant characteristics Reference
E. coli BL21
(DE3) pLysS
F-, ompT, hsdSB (rB
-mB
-), gal, dcm, (DE3)pLysS (Cam
R) Invitrogen
E. coli DH5α F-, φ80dlacZΔM15, Δ(lacZYA-argF)U169, deoR, recA1, endA1,
hsdR17(r k-, m k
+), phoA, supE44, thi-1, gyrA96, relA1, λ
-
[74]
E. coli ET12567 F-, dam-13::Tn9, dcm-6 hsdM, hsdR, zjj-202::Tn10, recF143,
galK2, galT22, ara-14, lacY1, xyl15, leuB6, thi1, tonA31,
rpsL136, his64, tsx78, mtl-I, glnV44
[75]
E. coli XL1 blue recA1, endA1, gyrA96, thi-1, hsdR17, supE44, relA1,
lac[F´proAB, lacIqZDM15, Tn10 (tet)]
[76]
Strain Relevant characteristics Reference
Streptomyces albus J1074 Host for heterologous expression [77]
Streptomyces lividan 1326 Wild type, host for protein expression [78]
Streptomyces. lydicus ssp. Tanzanius
NRRL18036
Ganefromycin producer [79]
Streptomyces bottropensis Formerly named as Streptomyces sp. Goe C4/4.
Mensacarcin and Rishirilide A producer
[80]
Streptomyces collinus Tue 365 Kirromycin producer [81]

36 Materials and Methods
2.5.1 Preparation of permanent culture and spore suspension
In order to prepare permanent culture, 10-50 mL well-grown culture in HA or TSB
medium was harvested by centrifugation (4,000 rpm, 10 min). After wash with 20 mL
of 25% sterile saccharose, the cells were resuspended in 10 mL 25% Saccharose. The
storage was carried out at -80 ℃.
For preparation of spore suspension, the Streptomyces were grown on HA agar or MS
agar plates at 28 ℃ until full sporulation. 5 mL sterile H2O was added to the top of
each plate and then the spores were scraped off and transferred from the plates to a 10
mL falcon tube. After vigorous vortex and wash by centrifugation, the spores were
separated from the mycelium by passing the suspension through sterile cotton pluged
in a disposable syringe. Spores were collected by centrifugation (4,000 rpm, 10 min, 4
℃), washed with 10 mL 25% saccharose and resuspended in 5 mL 25% saccharose.
Spores suspension was kept at -80 ℃.
2.5.2 Production of secondary metabolites
For the production of secondary metabolites, Streptomyces strains were precultured in
50 mL HA or TSB medium at 28 ℃ and 180 rpm for one or two days. 10 mL of this
preculture were inoculated into 500 mL baffled flask containing 100 mL appropriate
medium and grown at 28 ℃ and 180 rpm for 5-7 days.
2.6 Vectors, cosmids and plasmids
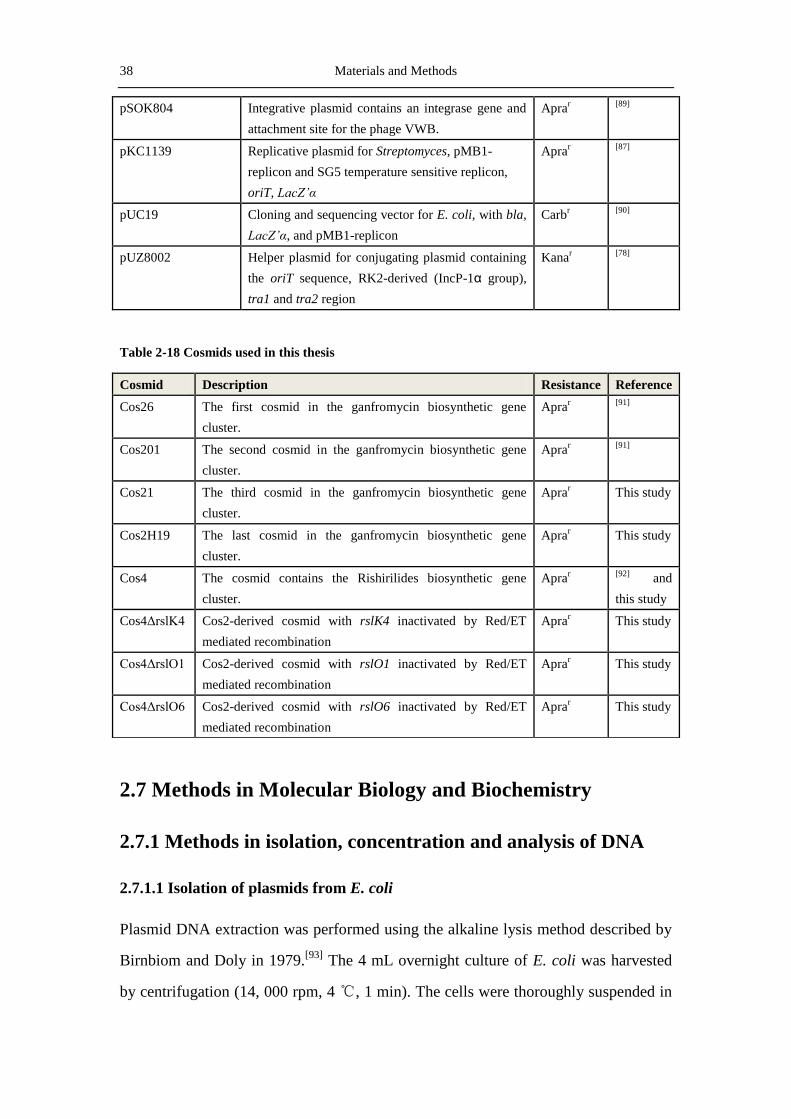
Table 2-17 Vectors and plasmids used in this thesis
Name Description Resistance Reference
pBADαβγ Vector containing code-optimized protein for the
λ-Red recombination. red-exo, red-bet, red-gam
from the λ-phage; temperature sensitive replicon
from plasmid pSC101
Tetr
[82]
pBluescript SK(-) Cloning vector, Ampr, lacZ’(α-complementation),
f1(-)-origin, ColE1-origin
Carbr Stratagene

Materials and Methods 37
pCDFDuet Cloning vector, template for spectinomycin
resistant gene
Specr Novagen
pET-28a(+) Vector for protein expression. N-terminal His-Tag/
Thrombin/T7 Tag, with optional C-terminal His
Tag sequence. f1 origin, pBR322 origin, lacI,
Kanar Novagen
pGEM-T Linearized vector with T-overhang, for direct
cloning of PCR Fragments with A-overhang.
Carbr Promega
pHPΩ45aac pBR322 derivative vector carrying the resistant
gene for spectinomycin and streptomycin, flanked
by transcription terminator.
Carbr
Specr
[83]
pIJ778 Redirect cassette with aadA gene and FRT-sites
into pBSK+, oriT
Carbr
Specr
[84]
pKC1132 Non-replicative vector in Streptomyces Aprar
[85]
pKCorf20EP Insert the 6.6 kb EcoRI + PstI fragment from orf20
of ganefromycin into the pKC1132 vector.
Aprar This study
pKCXY02 Vector for double cross-over screening. A codon
optimized GUS gene under the control of tipA was
inserted into pKC1132 into the BglII site.
Aprar This study
pUWL-H Streptomyces expression vector, LacZα, ermE* up
promoter, pIJ101-origin, ColE1-origin
Carbr
Hygr
[86]
pUWL-H-rslR1 Plasmid for rslR1 overexpression Carbr Hyg
r This study
pUWL-H-rslR2 Plasmid for rslR2 overexpression Carbr Hyg
r This study
pUWL-H-rslR3 Plasmid for rslR3 overexpression Carbr Hyg
r This study
pUWL-H-rslR1R2R3 Plasmid for overexpression of rslR1, rslR2 and
rslR3.
Carbr Hyg
r This study
pUWL-H-rslR4 Plasmid for rslR4 overexpression Carbr Hyg
r This study
pSET152 Integrative plasmid for Streptomyces, LacZ’α,
pMB1-replicon, Containing integration system from
from phage φC31, oriT RK2
Aprar
[87]
pSETGUS pSET152 based integrative plasmid for checking
gusA activity in Streptomyces spp.
Aprar
[88]
pGUS-PR-rslR1 pSETGUS-based plasmid with the ~500 bp
promoter region of rslR1 in front of the gusA gene
Aprar This study
pGUS-PR-rslR2 pSETGUS-based plasmid with the ~500 bp
promoter region of rslR2 in front of the gusA gene
Aprar This study
pGUS-PR-rslR3 pSETGUS-based plasmid with the ~500 bp
promoter region of rslR3 in front of the gusA gene
Aprar This study
pGUS-PR-rslR4 pSETGUS-based plasmid with the ~500 bp
promoter region of rslR4 in front of the gusA gene
Aprar This study
pKC1128E Replicative plasmid for Streptomyces,
pMB1-replicon and SCP*- replicon, oriT, LacZ’α
Aprar
[87]
38 Materials and Methods
pSOK804 Integrative plasmid contains an integrase gene and
attachment site for the phage VWB.
Aprar
[89]
pKC1139 Replicative plasmid for Streptomyces, pMB1-
replicon and SG5 temperature sensitive replicon,
oriT, LacZ’α
Aprar
[87]
pUC19 Cloning and sequencing vector for E. coli, with bla,
LacZ’α, and pMB1-replicon
Carbr
[90]
pUZ8002 Helper plasmid for conjugating plasmid containing
the oriT sequence, RK2-derived (IncP-1α group),
tra1 and tra2 region
Kanar
[78]
Table 2-18 Cosmids used in this thesis
2.7 Methods in Molecular Biology and Biochemistry
2.7.1 Methods in isolation, concentration and analysis of DNA
2.7.1.1 Isolation of plasmids from E. coli
Plasmid DNA extraction was performed using the alkaline lysis method described by
Birnbiom and Doly in 1979.[93]
The 4 mL overnight culture of E. coli was harvested
by centrifugation (14, 000 rpm, 4 ℃, 1 min). The cells were thoroughly suspended in
Cosmid Description Resistance Reference
Cos26 The first cosmid in the ganfromycin biosynthetic gene
cluster.
Aprar
[91]
Cos201 The second cosmid in the ganfromycin biosynthetic gene
cluster.
Aprar
[91]
Cos21 The third cosmid in the ganfromycin biosynthetic gene
cluster.
Aprar This study
Cos2H19 The last cosmid in the ganfromycin biosynthetic gene
cluster.
Aprar This study
Cos4 The cosmid contains the Rishirilides biosynthetic gene
cluster.
Aprar
[92] and
this study
Cos4ΔrslK4 Cos2-derived cosmid with rslK4 inactivated by Red/ET
mediated recombination
Aprar This study
Cos4ΔrslO1 Cos2-derived cosmid with rslO1 inactivated by Red/ET
mediated recombination
Aprar This study
Cos4ΔrslO6 Cos2-derived cosmid with rslO6 inactivated by Red/ET
mediated recombination
Aprar This study

Materials and Methods 39
200 μL P1 buffer by vortex. Add 200 μL P2 buffer to the suspension and mix gently
by inversion until the solution is clear. Incubation at RT for about 5 min, add 200μL
P3 buffer and incubate the solution for another 10 min. After centrifugation (14, 000
rpm, 4 ℃, 10 min), the supernatant was poured into a new Eppendorf tube. DNA was
precipitated by adding 500 μL ice-cold isopropanol and centrigation (14, 000 rpm, 4
℃, 30 min). DNA pellet was washed once with 500 μL 70% ethanol and air-dried for
10-15 min. Finally, the plasmid DNA was dissolved in 30 μL distilled water and
stored at -20 ℃.
Analytic DNA extraction with the “Wizard SV Minipreps DNA Purification System”
or preparative extraction with the “Pure Yield Plasmid Midiprep System” kits from
Promega was performed according to the manufacter’s protocol.
2.7.1.2 Isolation of genomic DNA from Streptomyces
Genomic DNA from Streptomyces was isolated as described by Pospiech et al.[78]
2
mL of 24 h culture in HA or TSB medium was harvested by centrifugation (4,000
rpm, 4 ℃, 10 min). The cells were washed with 1 mL H2O and resuspended in 500 μL
SET buffer containing 4 mg/mL lysozyme and 100 μL RNase by vortexing. The
suspension was incubated at 37 ℃ for 30 min, with occasional inversion. Then
added 50 μL 10% SDS solution and 14 μL Proteinase K solution, and incubated at 55
℃ for 1-2 hours. After adding and mixing with 200 μL 5 M NaCl solution and 500
μL chloroform, the lysate was centrifuged (14,000 rpm, 4 ℃ , 10 min). Then
transferred the supernatant to a fresh tube, added 0.6 vol isopropanol and mixed by
inversion. After about 3 min, genomic DNA was spooled onto a sealed Pasteur
pipette, rinsed with 5 mL 70% ethanol, air dried and dissolved in 1 mL TE buffer.
2.7.1.3 Agarose gel elxctrophoresis of DNA
DNA fragment separation and size determination was performed by electrophoresis
on 0.7% (w/V) agarose gel. The buffer used for electrophoresis was 1x TAE buffer.
The DNA samples and the 1 kb DNA ladder from Pormega were added into the caves
on the gel. After the running, the gel was stained with ethidium bromide and detected

40 Materials and Methods
under the UV light at 312 nm. The size of the DNA fragments could be evaluated by
comparing to the DNA ladder.
For DNA fragment preparation, the agarose band containing the aim DNA was cut out
from the gel and dissolved in the column binding solution of the “Wizard SV Gel and
PCR Clean-Up System” by heating. Further steps were performed according to the
manufacturer’s protocol.
Figure 2. 1 kb DNA ladder from Promega.
2.7.1.4 Methods in concentration and quantification of DNA
Concentration of DNA was carried out according to the method of Maniatis et al.[94]
1/10 volume of the P3 buffer for plasmid isolation was added into the DNA solution.
Then 1 volume of isoprapanol was added to precipitate the DNA. After thorough
vortex and incubation on ice for 20 min, the DNA was precipitated by centrifugation
(4 ℃, 14,000 rpm, 10 min). The supernatant was discarded and the resulting DNA
pellet was washed with 70% ethanol. Finally, dry the DNA pellet in the incubator at
60 ℃ for 10 min and dissolved in 30-100 μL sterile H2O.
2.7.1.5 DNA denaturation for ssDNA transformation in Streptomyces lydicus
The single stand DNA used for protoplast transformation was prepared by alkaline
treatment of plasmid DNA.[95]
9 μL dsDNA in H2O was mixed with 2 μL 1 M NaOH
and incubated for 10 min at 37 ℃. The mixture was cooled on ice and the reaction
was terminated by adding 2 μL 1 M HCl.

Materials and Methods 41
2.7.2 Methods in DNA cloning
2.7.2.1 DNA restriction
Restriction of DNA with endonucleases was performed according to the
manufacturer’s instructions. For a typical analytic digestion, a total volume of 20 μL
was used, while for preparative digestion, a total volume of 50 μL was used. Unless
otherwise stated, the restriction was carried out at 37 ℃.
Table 2-19 Composition for typical restriction reactions.
2.7.2.2 DNA ligation
DNA ligation was carried out using T4-DNA ligase at RT for 2-4 h or at 16 ℃
overnight. The ligation system contains 1 U T4-DNA ligase, 1x ligase buffer and
appropriate insert and vector, with a total volume of 20 μL.
2.7.3 Inactivation of genes on the cosmid by λ RED-mediated
recombination
λ RED-mediated recombination was first developed by Detsenko and Wanner to
promote the efficiency of homologous recombination in E. coli with linear DNA.[96]
Gust et al. adapted this tool in streptomycetes and had established a method for gene
inactivation in Streptomyces.[97]
Gene inactivation was first performed on a cosmid in
E. coli with a PCR product. Then the cosmid contains the inactivated gene was
introduced into the Streptomyces strain by intergeneric conjugation. In this thesis, the
Name Analytic digestion Preparative digestion
BSA (10 ×) 2.0 μL 8.0 μL
DNA 2.0 μL 20 μL
10 × restriction buffer 2.0 μL 8.0 μL
H2O 14 μL 40 μL
Enzyme 1 1.0 μL (for single enzyme, use 1.5μL) 4.0 μL
Enzyme 2 (optional) 1.0 μL 4.0 μL
Total 20 μL 80 μL

42 Materials and Methods
plasmid pBADαβγ that contains the λ RED genes redα, redβ, redγ was used to
promote gene inactivation. Details for the principles in λ RED-mediated gene
replacement please refer to the article by Gust et al.[84]
In order to obtain the PCR fragment for gene inactivation, two 64 bp primers were
designed, synthesized and purified by the HPLC method. The primers contain a 5’ 39
bp homologous arm identical to the upstream or downstream bases of the aim gene
and overlaps with the aim gene by the first three (the start codon) or the last three
bases (the stop codon). In the middle of the primers are 6 nucleotides for the
restriction enzyme (NheI in this thesis) which will be used to cut out the resistant gene
(aadA) in the PCR product. In the 3’ end bases of the primers are identical to the
beginning and the end of the resistant cassette. With these primers and the plasmid
containing the resistant cassette as template, a fragment in which the resistant gene
(aadA) is flanked by the 39 bp homologous arms could be obtained by PCR. The
plasmid pBADαβγ was introduced into DH5α by CaCl2-mediated transformation and
incubated on the LB plate containing tetracycline for 16 h at 28 ℃. Single colony was
picked from the plate, inoculated into 10 mL LB medium and cultured at 28 ℃, 180
rpm for 14-16 h. Then 1 mL of this preculture was inoculated into 100 mL of fresh
LB medium containing 20 μg/mL tetracycline and cultured at the same conditions
until the OD600 reached 0.6. After washing twice with ice-cold 10% glycerol, the
DH5α/pBADαβγ cells were used as competent cells for the transformation of cos4 by
electroporation, to obtain DH5α/ pBADαβγ/cos4.
A single colony of DH5α/pBADαβγ/cos4 was inoculated into 10 mL LB medium
containing 20 μg/mL of tetracycline and 100μg/mL of apramycin and cultured
overnight at 28 ℃, 180 rpm. Then 1 mL of this preculture was inoculated into 100 mL
of fresh LB medium containing 20 μg/mL tetracycline, 100μg/mL apramycin and 10
mM freshly prepared arabinose at cultured at the same conditions until the OD600
reached 0.6. These cells were made competent with the method for preparation of
electroporation competent cells. 10 μL of the purified PCR product was used to
transform the DH5α/pBADαβγ/cos4 competent cells. After the addition of 700 μL of

Materials and Methods 43
LB medium and incubated at 28 ℃ for 2 h, the cells were plated on LB plate with 100
μg/mL apramycin and 100 μg/mL spectinomycin and incubated at 37 ℃ overnight.
The plasmid pBADαβγ was eliminated by incubation at 37 ℃. The colonies on the LB
plate were inoculated on a fresh LB plate with the same antibiotics and cultured again
for 16-20 h, to completely remove the pBADαβγ plasmid and the false colonies. The
cells with the gene-inactivated cos4 were inoculated into LB medium containing 100
μg/mL apramycin and 100 μg/mL spectinomycin and cultivated at 37 ℃ for 16 h. The
cosmid in which the aim gene was replaced by the aadA gene was isolated with the
“Wizard SV Minipreps System” kit and the aadA gene was then excised by digestion
with NheI. The linear cosmid DNA was religated with T4-DNA ligase for 4 h and
transformed into DH5α competent cells. The plate for this transformation contained
only apramycin. The mutant cosmid (cos4Δx) could be isolated from these cells.
During these procedures, the replacement and the excision of the aadA gene in the
cosmid was controlled by colony PCR or PCR using the isolated cosmids as template.
2.7.4 PCR amplication
PCR amplification was performed on the Mastercycler Epgradient (Eppendorf, Ham-
burg, Germany) and Gene Amp®
PCR System 9700 (Applied Biosystems, Forster
City, USA). The components for PCR mixture and amplification conditions are given
in Table 2-20.
Table 2-20 Composition of a typical PCR reaction system
Component Final concentration Note
DMSO 10% Used for GC-rich templates
Polymerase reaction buffer 1 x 10 x or 5 x
dNTP-mix 0.2 mM each
Primer 1 20 pmol
Primer 2 20 pmol
Template DNA 250 ng (1 μL) For genomic DNA, use 10 μL
Polymerase 5 U (1μL) Taq or Pfu (5 U/μL)
H2O Add to total volume of 100 μL For analytic purpose use 50μL
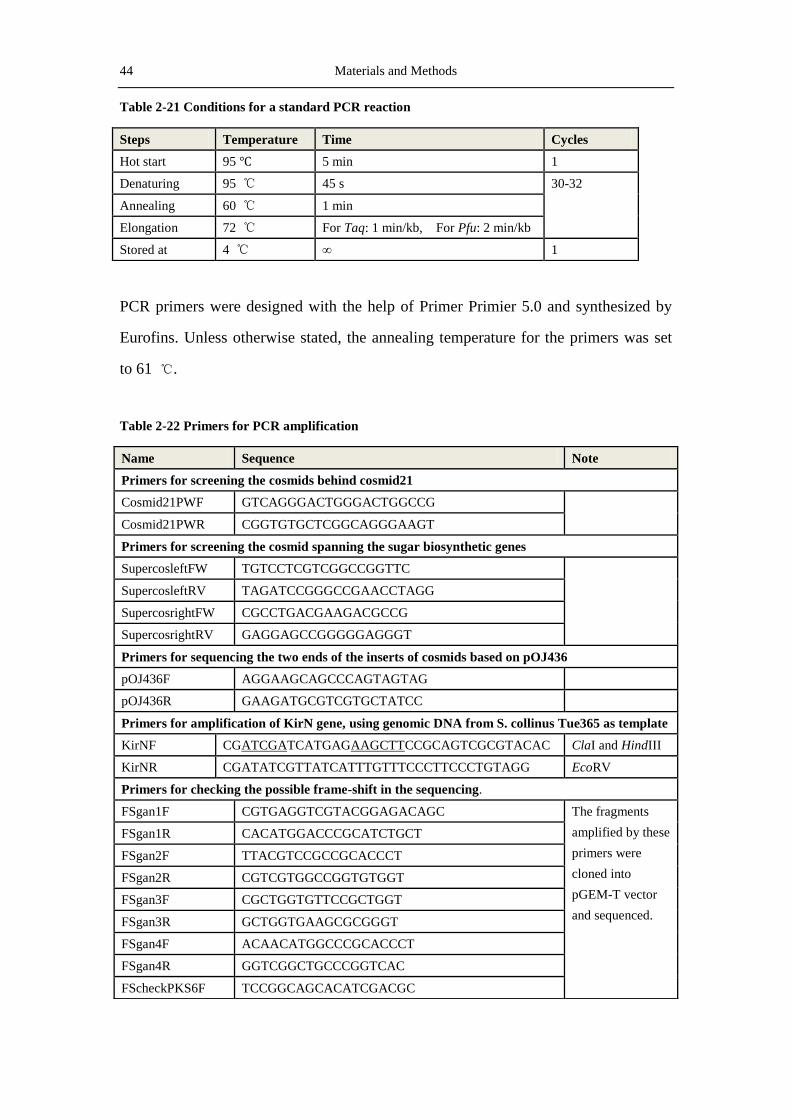
44 Materials and Methods
Table 2-21 Conditions for a standard PCR reaction
PCR primers were designed with the help of Primer Primier 5.0 and synthesized by
Eurofins. Unless otherwise stated, the annealing temperature for the primers was set
to 61 ℃.
Table 2-22 Primers for PCR amplification
Steps Temperature Time Cycles
Hot start 95 ℃ 5 min 1
Denaturing 95 ℃ 45 s 30-32
Annealing 60 ℃ 1 min
Elongation 72 ℃ For Taq: 1 min/kb, For Pfu: 2 min/kb
Stored at 4 ℃ ∞ 1
Name Sequence Note
Primers for screening the cosmids behind cosmid21
Cosmid21PWF GTCAGGGACTGGGACTGGCCG
Cosmid21PWR CGGTGTGCTCGGCAGGGAAGT
Primers for screening the cosmid spanning the sugar biosynthetic genes
SupercosleftFW TGTCCTCGTCGGCCGGTTC
SupercosleftRV TAGATCCGGGCCGAACCTAGG
SupercosrightFW CGCCTGACGAAGACGCCG
SupercosrightRV GAGGAGCCGGGGGAGGGT
Primers for sequencing the two ends of the inserts of cosmids based on pOJ436
pOJ436F AGGAAGCAGCCCAGTAGTAG
pOJ436R GAAGATGCGTCGTGCTATCC
Primers for amplification of KirN gene, using genomic DNA from S. collinus Tue365 as template
KirNF CGATCGATCATGAGAAGCTTCCGCAGTCGCGTACAC ClaI and HindIII
KirNR CGATATCGTTATCATTTGTTTCCCTTCCCTGTAGG EcoRV
Primers for checking the possible frame-shift in the sequencing.
FSgan1F CGTGAGGTCGTACGGAGACAGC The fragments
amplified by these
primers were
cloned into
pGEM-T vector
and sequenced.
FSgan1R CACATGGACCCGCATCTGCT
FSgan2F TTACGTCCGCCGCACCCT
FSgan2R CGTCGTGGCCGGTGTGGT
FSgan3F CGCTGGTGTTCCGCTGGT
FSgan3R GCTGGTGAAGCGCGGGT
FSgan4F ACAACATGGCCCGCACCCT
FSgan4R GGTCGGCTGCCCGGTCAC
FScheckPKS6F TCCGGCAGCACATCGACGC

Materials and Methods 45
FScheckPKS6R CCCTCGAAGTACGTCCGCCAGT
Primers for verifying the single cross-over of pKCorf20.
Orf20scoprobeFw CACGTCTCGCCGCGCACCC Using genomic DNA of the
mutant as template. Orf20scoprobeRv TCCTGGAGGAGCCGCCCGAG
Primers for over expression the regulators in pUWL-H. msc24: rslR1; msc30: rslR2; msc31 rslR3.
msc24overExfw CCCATCGATTCTCTTAAGGACCACGGAAGCCGCACC ClaI, AflII,
msc24overExrv GAACAGAAGCTTACGGCCGGCGCCGG HindIII
msc30overExfw ATTCCGAAGCTTCAAGCCAGCCCTGGAGG HindIII
msc30overExrv AACACTGCAGAGCTAGCGGGGGTCAGCCGGCC NheI, PstI
msc31overExfw TACGAATTCTCGCTAGCGGAGCGGACGGCCTG EcoRI, NheI
msc31overExrv GGACTAGTCACTGCTCCCGCCACCGT SpeI
rslR4amplifFw CCATCGATGGAGGCGGACATGGCGG ClaI
rslR4amplifRv GTACTAGTACGCGCTTAAGGGGTGGC SpeI
Primers for expression the priming ketosynthase (msc17: rslK4) in E. coli by pET-28a(+)
msc17ExEcoliNF CGCATTCCATATGAGGTTCGAGGACCTGTACAT NdeI
msc17ExEcoliNR ATAAGAATGCGGCCGCTTATCATCCGGATCCCGTCTCT NotI
Primers for expression the priming ketosynthase (msc17: rslK4) in S. lividans by pUWL-H
msc17-expressionFw GGGAAGCTTGGAGGCACAGTCATGCACCACCACCACC
ACCACCACCACAGGTTCGAGGACCTGTACATCGC
HindIII
msc17-expressionRv CGCGAATTCGCATTTGGCATGCTTATTATCATCCGGAT
CCCGTCTCTCC
EcoRI
Primers for inactivating rslO1 (msc21) by λ Red-mediated recombination
msc21NheIRedFw ACACCGTCCGTCCAGCCATCACATCGAGAGGACCCC
ATGGCTAGCGGAGCGTAGCGACCGAGTG
NheI
msc21NheIRedRv GCTCGGTGCGCAAGGAAACCATCGGCGGTCCGTCCC
TCAGCTAGCGGCTATTTAACGACCCTGC
NheI
Primers for inactivating rslO6 (msc32) by λ Red-mediated recombination
msc32NheIRedFW CCCCTCCTTTCGGCCCTGTCCGTGAAGGAGACCAGCG
TGGCTAGCGGAGCGTAGCGACCGAGTG
NheI
msc32NheIRedRv GTTCCGGACATTCTTCGGCAGGCCGTCCGCTCCGCTT
CAGCTAGCGGCTATTTAACGACCCTGC
NheI
Primers for inactivating rslO1 (msc17) by λ Red-mediated recombination
msc17NheIRedFw TGGTTCCCCTGACGGCCGGTGAAAGGGCATCGGGAC
ATGGCTAGCGGAGCGTAGCGACCGAGTG
NheI
msc17nheIRedRv CCCGCTCGGCGCGCATGCGCCACCGGCGCGGCGCGG
TCAGCTAGCGGCTATTTAACGACCCTGC
NheI
Primers for amplification of the promoter regions of the regulators
PromoRslR1Fw GCTCTAGAGATGGATCAGCTGCGCGGT XbaI
PromoRslR1Rv CCGGTACCTTCCGTGGTCCTTCCAGCG KpnI
PromoRslR2Fw CCTCTAGACACCCGCACTCTGGCCAT XbaI
PromoRslR2Rv CGGGTACCGTGGCCGCTGGATCTTG KpnI

46 Materials and Methods
2.7.5 Transformation of DNA into E. coli
Transformation of plasmid DNA into E. coli was performed according to a modified
method from Maniatis et al.[94]
In current thesis, two methods for DNA transformation
were used: for small plasmids (<15 kb), CaCl2-mediated heat shock transformation
was chosen; for large plasmids (>15 kb) and cosmids, the electroporation method was
used because of its much higher transformation efficiency.
2.7.5.1 Transformation of E. coli by CaCl2-mediated heat shock
Preparation of CaCl2-competent cells
100 mL LB-medium was inoculated with 1 mL of overnight culture of E. coli and
cultured at 37 ℃, 180 rpm until the OD600 (detected by the UV-spectrometer Uvikon
933) reached 0.6 (2-4 h for DH5α, BL21 DE(3) pLysS and XL-1 blue, 6 h for ET
12567 (pUZ8002)). The cells was harvested by centrifugation (3,000 rpm, 4 ℃, 10
min), resuspended in 40 mL ice-cold 0.1 M MgCl2 and centrifugated again (3,000
rpm, 4 ℃, 10 min). The cell pellet was resuspended in 20 mL ice-cold CaCl2 (0.1 M)
and incubated on ice for 30 min. After centrifugation (3,000 rpm, 4 ℃, 10 min), the
pellet was suspended in 2-5 mL buffer containing 0.1 M CaCl2 and 15% glycerol. The
competent cells could be used immediately or stored at -80 ℃ in 100 μL aliquots in
the 1.5 mL eppis.
PromoRslR3Fw GGTCTAGAGGAGGCCCGGCGGGTC XbaI
PromoRslR3Rv CCGGTACCGTGCCCAGCGCCACAG KpnI
PromoRslR4Fw CGTCTAGACGGCGAGCAGATAGCCG XbaI
PromoRslR4Rv GGGGTACCGCTCACTCCCCCTCCACTAT KpnI
Primers for complementation of the inactivated genes (the genes were expressed under the ermE
promoter of pUWL-H with the restriction enzymes ClaI and SpeI)
rslK4CFw CCATCGATGGAGGCGGGACATGAGGTTCGAGGAC ClaI
rslK4CRv CCACTAGTCATCCGGATCCCGTCTCTC SpeI
rslO1CompleFw CTATCGATGGAGGACCCCATGAAGTTCGGC ClaI
rslO1CompleRv GCACTAGTCCCTCAGTCGTTCGCTGC SpeI
rslO6CFw TCATCGATGAAGGAGACCAGCGTGAAACTG ClaI
rslO6CRv GGACTAGTCCGCTCCGCTTCACGCG SpeI

Materials and Methods 47
CaCl2-mediated transformation
5 μL plasmid DNA was added to the eppi containing 100 μL competent cells and
incubated on ice for 30 min, with occasional inversion. Then eppi was put into the
water bath at 42 ℃ for 90 seconds and cooled down on ice (about 3 min). 700 μL
LB medium was pipette into the eppi and incubated in the 37 ℃ water bath for 1 h.
The cells were harvested by centrifugation (6,000 rpm, 10 min). The supernatant was
discarded and the pellet was resuspended in the remaining medium. Finally plate the
pellet on a LB agar plate containing appropriate antibiotic. Colonies could be seen
after incubation for 12-16 h
2.7.5.2Transformation by electroporation
Preparation of electroporation competent cells
100 mL LB-medium was inoculated with 1 mL of overnight culture of E. coli and
cultured at 37 ℃, 180 rpm until the OD600 (detected by the UV-spectrometer Uvikon
933) reached 0.6 (2-4 h for DH5α, BL21 DE(3) pLysS and XL-1 blue, 6 h for ET
12567/pUZ8002). The cells was harvested by centrifugation (3,000 rpm, 4 ℃, 10
min), washed with 40 mL 10% glycerol (ice-cold). After centrifugation (3,000 rpm, 4
℃ , 10 min) and washed with 15 mL ice-cold 10% glycerol, the pellet was
resuspended in 1-2 mL cold 10% glycerol and stored in 80 μL aliquots at -80 ℃.
Transformation by electroporation
The electroporation competent cells were defrozen on ice. Then 5-10 μL plasmid
DNA or PCR fragment was added into the eppi and incubated on ice for about 15
min. Afterwards the cells and DNA were transferred into a pre-cooled electroporation
cuvette (0.1 cm gap). Electroporation was carried out with the Bio-Rad E. coli pulser
with the voltage 1.8 kv and the duration 5 ms. After the electroporation, 700 μL fresh
LB medium was added into the cuvette, mixed with the cells by pipetting, transferred
into a sterile 1.5 mL eppi and incubated at the 37 ℃ water bath for 2 h. The cells
were harvested by centrifugation (6,000 rpm, 10 min). The supernatant was discarded

48 Materials and Methods
and the pellet was resuspended in the remaining medium. Finally plate the pellet on a
LB agar plate containing appropriate antibiotic. Colonies could be seen after
incubation for 14-18 h.
2.7.6 Methods for introducing DNA into Streptomyces
2.7.6.1 DNA transfer by PEG-mediated protoplast transformation
Preparation of protoplasts
Protoplasts preparation was performed according to the modified mothod From
Maniatis et al.[94]
200 μL Streptomyces spores or mycelia from the 25% saccharose
stock was inoculated into 100 mL CRM medium and grown at 28 ℃, 180 rpm for
24-48 h. The cells was harvested by centrifugation (3,000 rpm, 4 ℃, 10 min), washed
first with 10 mL 10% saccharose and then with 10 mL P-buffer. The cell pellet was
resuspended in P-buffer with lysozyme (4 mg/mL) and incubated at 28 ℃, 30 rpm for
30-60 min with interval control the protoplast formation under microscopy. When
majority of the cells became protoplast, the reaction was stopped by adding 20 mL
ice-cold P buffer and incubation of ice for 10 min. The cell wall and intact mycelia
was removed by filtered through sterile cotton. The resulting protoplasts were
harvested by centrifugation at 4 ℃, 3,000 rpm for 10 min. The supernatant was
poured off and the pellet was resuspended in 1 mL P buffer. The protoplasts could be
used immediately or stored at -80 ℃ in 200 μL aliquots in 1.5 mL Eppi.
PEG-mediated protoplast transformation
The plasmid DNA used for protoplast transformation was first propagated in the
dam-& dcm
- E. coli strain ET12567 to avoid the methyl-specific restriction system in
Streptomyces. In the case of non-replicative plasmid (suicide plasmid), which was
introduced into Streptomyces to obtain single cross-over colony by homologous
recombination, the non-methylated DNA was further denatured by the treatment with
NaOH to form single strand DNA, because the ssDNA efficiently facilitates the
homologous recombination.

Materials and Methods 49
200 μL of protoplasts were added with 10-15 μL unmethylated DNA and 500 μL
T-buffer, mixed by gentle inversion and incubate for less than 1 min. The the mixture
was added on the top of R2YE agar plates (each plate 200-250 μL), followed by
covering with about 5 mL pre-warm R3 soft agar for each plate. After 16-20 h
incubation at 28 ℃, the plates were overlaid with 5 mL of soft nutrient agar
containing appropriate antibiotics for selection of mutants. The plates were grown in
the incubator at 28 ℃ for 5-10 days. In order to check the quality of the protoplasts,
control experiments were also necessary. Negative control was carried out by mixing
the protoplasts with sterile H2O and then plating on the R2YE plate, while positive
control was done by covering the R2YE plate with 5 mL soft nutrient agar without
any antibiotic.
2.7.6.2 DNA transfer by intergeneric conjugation
Intergeneric transfer of plasmids from methylation-deficient E. coli strain ET12567/
pUZ8002 to Streptomyces strains is getting more and more popular, because this
method is much easier to work with comparing to the PEG-mediated protoplast
transformation. The vectors contain oriT from the IncP-group plasmid RP4 are first
transformed into ET12567, and then transferred in trans from the ET12567 donor
strain to Streptomyces acceptor strain with the help of the non-transmissible pUZ8002
plasmid. In this thesis, this conjugal transfer of DNA is carried out according to a
modified method of Flett et al.[98]
10 mL culture of ET12567/pUZ8002 containing the aim plasmid was grown to an
OD600 of 0.4-0.6. The cells were pelleted by centrifugation, washed in fresh LB
medium, pelleted again and finally resuspended in 200 μL of LB. 50 mL overnight
culture of Streptomyces strain was washed three times with 1 volume, 0.1 volume and
0.01 volume of TSB medium sequentially. Then the E. coli cells and Streptomyces
cells were combined in a 1.5 mL eppi and thoroughly mixed by vortex. After
centrifugation (6,000 rpm, 1 min) and discarding the supernatant, the pellet was
resuspended with 200 μL of TSB and plated on the MS agar plate. The conjugation

50 Materials and Methods
plate was incubated for 12-16 h at 28 ℃, then overlaid with 1 mL sterile H2O
containing 8 mg phosphomycin and 2 mg of the antibiotic for plasmid maintaining.
The plate was then incubated for a further 5-7 days 28 ℃.
2.8 Method of biochemistry
2.8.1 SDS-PAGE
SDS-PAGE was carried out according to the method of Laemmli.[99]
5% and 12.5%
polyacrylamide gel were used as stacking gel and resolving gel respectively. Gel
electrophoresis was performed with the Mini-PROTEAN II Electrophoresis system
(Bio-Rad, Muechen, Germany). All the buffers for the electrophoresis were listed in
Table 2-9. Before the running, the proteins were mixed with an equal volume of
loading buffer and cooked at 100 ℃ for 5 min. After centrifugation (4 ℃, 14,000
rpm, 5 min), the protein samples were added to the slots of the polyacrylamide gel. In
order to determine the size of the proteins, protein markers were also added into the
slots. The gel electrophoresis was carriedout at 40 mA until the front blue line reached
the bottom of the gel. Then the gel was stained with Coomassie Brilliant Blue by
heating 30 s in the microwave oven. The stained gel was decolorized in the stripping
solution. The size of the proteins could be estimated by comparing to the protein
markers.
2.8.2 Expression and purification of RslK4 from E. coli
Overexpression and purification of 6×His-tagged RslK4 from E. coli BL21 were
performed as described in the user manual of Qiagen (Hilden, Germany): “A
Handbook for High Level Expression and Purification of 6×His-tagged proteins”.
2.8.3 Expression and purification of RslK4 from S. lividans 1326
Expression of RslK4 with the pUWL-H vector was conducted in S. lividans 1326, in
which this protein was believed to have good activity due to the high rishirilide B
production in this strain. The protein purification from S. lividans 1326 was carried out

Materials and Methods 51
using a modified method described by Enguita et al.[100]
S. lividans harboring the
expression plasmid pUWL-A-rslK4 was pre-cultured in TSB medium with apramycin
(100 μg/mL) at 28 ℃, 180 rpm, for 24-36 h. Then 1 mL of the preculture was
inoculated into a 500 mL flask containing 100 mL of YEME medium with Apramycin
and grown for 48 h. The cells were harvested by centrifugation (4,000 rpm, 4 ℃, 10
min) and washed twice with fresh ice-cold TSB medium. After incubating the cells
with 5 mL ice-cold lysis buffer for 30 min, the cell suspension was sonicated for 6
min at intervals of 45 s ultrasonication followed by 45 s break. The cell debris was
removed by centrifugation (25,000 rpm, 4 ℃ , 60 min) and the yield of the
recombinant protein was determined by SDS-PAGE.
2.8.4 Transcriptional fusion analysis with the gusA gene
In order to reveal the relationships among the regulators in the rishirilide biosynthetic
gene cluster, transcriptional fusions of the promoter regions of these regulators with
the gusA gene on the promoter probe vector pMAXGUS were performed. Promoters
of the regulatory genes were fused to the gusA gene using the restriction enzymes
KpnI and XbaI, to obtain the pGUS-PR-rslR1, pGUS-PR-rslR2, pGUS-PR-rslR3, and
pGUS-PR-rslR4 plasmids. Together with the plasmid for negative control
(pMAXGUS), all these five plasmids were introduced into S. albus WT, respectively.
The resulting exconjugants were inoculated and incubated on MS or HA agar plates at
28 ℃ for 24 h. Then each plate was overlaid with 1 mL of 1 mM X-gluc
(5-bromo-4-chloro-3-indolyl-β-D-glucuronide) solution and incubated for 24 h.
2.9 Production, isolation and characterization of secondary
metabolites
2.9.1 Production and extraction of Ganefromycin
For ganefromycins production, the S. lydicus ssp. Tanzanius NRRL18036 strain was
first cultivated in 50 mL of TSB medium for 24 h at 28 ℃, 180 rpm in 300 mL
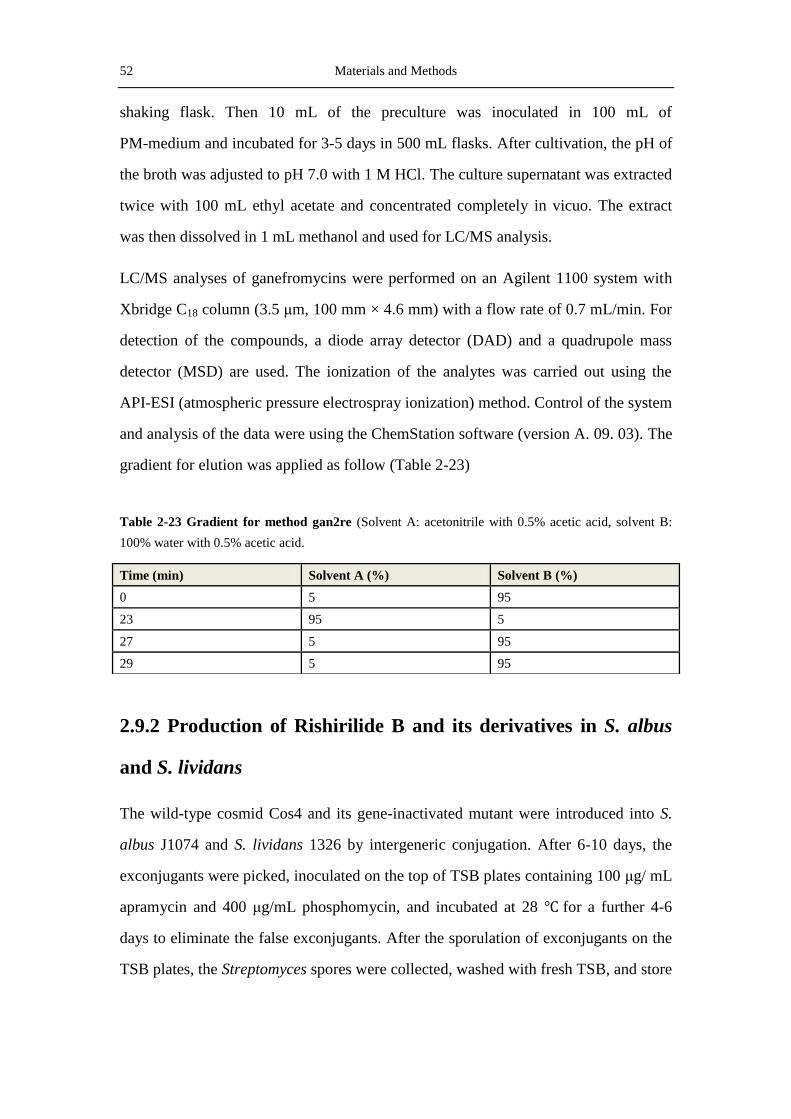
52 Materials and Methods
shaking flask. Then 10 mL of the preculture was inoculated in 100 mL of
PM-medium and incubated for 3-5 days in 500 mL flasks. After cultivation, the pH of
the broth was adjusted to pH 7.0 with 1 M HCl. The culture supernatant was extracted
twice with 100 mL ethyl acetate and concentrated completely in vicuo. The extract
was then dissolved in 1 mL methanol and used for LC/MS analysis.
LC/MS analyses of ganefromycins were performed on an Agilent 1100 system with
Xbridge C18 column (3.5 μm, 100 mm × 4.6 mm) with a flow rate of 0.7 mL/min. For
detection of the compounds, a diode array detector (DAD) and a quadrupole mass
detector (MSD) are used. The ionization of the analytes was carried out using the
API-ESI (atmospheric pressure electrospray ionization) method. Control of the system
and analysis of the data were using the ChemStation software (version A. 09. 03). The
gradient for elution was applied as follow (Table 2-23)
Table 2-23 Gradient for method gan2re (Solvent A: acetonitrile with 0.5% acetic acid, solvent B:
100% water with 0.5% acetic acid.
2.9.2 Production of Rishirilide B and its derivatives in S. albus
and S. lividans
The wild-type cosmid Cos4 and its gene-inactivated mutant were introduced into S.
albus J1074 and S. lividans 1326 by intergeneric conjugation. After 6-10 days, the
exconjugants were picked, inoculated on the top of TSB plates containing 100 μg/ mL
apramycin and 400 μg/mL phosphomycin, and incubated at 28 ℃ for a further 4-6
days to eliminate the false exconjugants. After the sporulation of exconjugants on the
TSB plates, the Streptomyces spores were collected, washed with fresh TSB, and store
Time (min) Solvent A (%) Solvent B (%)
0 5 95
23 95 5
27 5 95
29 5 95
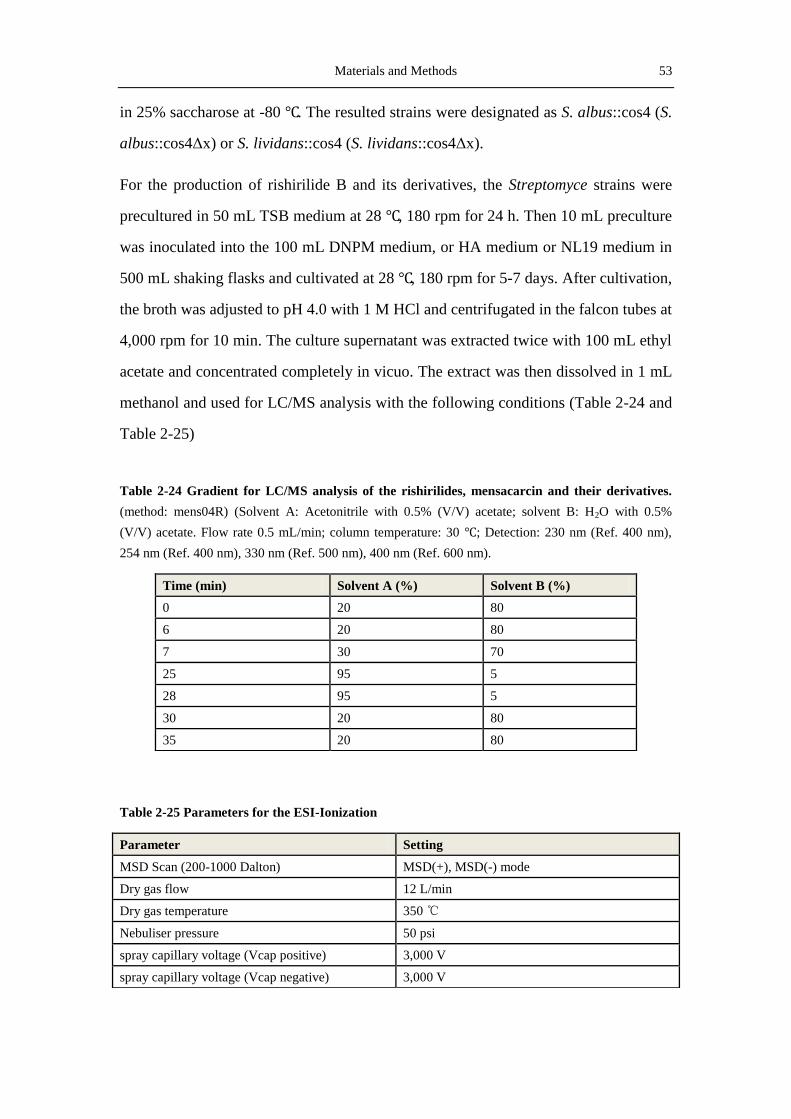
Materials and Methods 53
in 25% saccharose at -80 ℃. The resulted strains were designated as S. albus::cos4 (S.
albus::cos4Δx) or S. lividans::cos4 (S. lividans::cos4Δx).
For the production of rishirilide B and its derivatives, the Streptomyce strains were
precultured in 50 mL TSB medium at 28 ℃, 180 rpm for 24 h. Then 10 mL preculture
was inoculated into the 100 mL DNPM medium, or HA medium or NL19 medium in
500 mL shaking flasks and cultivated at 28 ℃, 180 rpm for 5-7 days. After cultivation,
the broth was adjusted to pH 4.0 with 1 M HCl and centrifugated in the falcon tubes at
4,000 rpm for 10 min. The culture supernatant was extracted twice with 100 mL ethyl
acetate and concentrated completely in vicuo. The extract was then dissolved in 1 mL
methanol and used for LC/MS analysis with the following conditions (Table 2-24 and
Table 2-25)
Table 2-24 Gradient for LC/MS analysis of the rishirilides, mensacarcin and their derivatives.
(method: mens04R) (Solvent A: Acetonitrile with 0.5% (V/V) acetate; solvent B: H2O with 0.5%
(V/V) acetate. Flow rate 0.5 mL/min; column temperature: 30 ℃; Detection: 230 nm (Ref. 400 nm),
254 nm (Ref. 400 nm), 330 nm (Ref. 500 nm), 400 nm (Ref. 600 nm).
Table 2-25 Parameters for the ESI-Ionization
Time (min) Solvent A (%) Solvent B (%)
0 20 80
6 20 80
7 30 70
25 95 5
28 95 5
30 20 80
35 20 80
Parameter Setting
MSD Scan (200-1000 Dalton) MSD(+), MSD(-) mode
Dry gas flow 12 L/min
Dry gas temperature 350 ℃
Nebuliser pressure 50 psi
spray capillary voltage (Vcap positive) 3,000 V
spray capillary voltage (Vcap negative) 3,000 V

54 Materials and Methods
2.9.3 Production and extraction of Rishirilide B and Mensacarcin from S.
bottropensis
Mensacarcin and rishirilides were produced by S. bottropensis in the HA medium and
the NL19 medium. One eighth piece of a well-covered agar from S. bottropensis was
inoculated into a 250 mL Erlenmeyer flask containing 50 mL of TSB medium and
cultured at 28 ℃, 180 rpm for 24-48 h. Then 10 mL of the preculture was added into
500 mL flasks containing 100 mL of HA or NL19 medium and cultivated for 5-7
days. After the production, the broth was adjusted to pH 4.0 with 1 M HCl and
centrifugated in the falcon tubes at 4,000 rpm for 10 min. The culture supernatant was
extracted twice with 100 mL ethyl acetate and concentrated completely in vicuo. The
extract was then dissolved in 1 mL methanol and used for LC/MS analysis with the
following conditions:
2.9.4 Purification of seconadry metabolites for structural
measurement with NMR
Before the structure determination with NMR and the element analysis with high
resulation mass spectrum (HRMS), the attractive compounds were produced in large
volume (5-10 L) and purified by the combination of solid phase extraction (SPE), thin
layer chromatography (TLC)/or silica gel column chromatography, preparative HPLC
and column chromatography with Sephadex LH-20 (Amersham Biosciences,
Freiburg, Germany). The amount and purity for the purified compounds was
controlled by LC/MS and proton NMR (when necessary) during the purification
procedures.
2.9.4.1 Prepurification of the compounds by SPE
After the production and extraction steps, the crude extract which is dissolved in
methanol was first purified with the Oasis® HLB20 35 cc (6g) extraction cartridge.
Before the purification, the cartridge was equilibrated with methanol solutions: first
with 100 mL 100% methanol, then with the same volume of 50% methanol and 20%

Materials and Methods 55
methanol respectively. After the equilibration, the cartridge was sinked in 20%
methanol overnight. Then the crude extract was dissolved in as less as possible 20%
methanol and loaded on the equilibrated cartridge. When all the crude extract was
loaded, the chromatography could be fractioned with the solvents from 100 mL of
20% methanol to 100 mL 100% methanol, and each fraction was collected in one
round bottom flask. The concentration of methanol for each fraction was increased by
10%. The resulting fractions were evaporated to dryness in the rotary evaporator and
analyzed by LC/MS with the method MENS04R. If the amount of the analyte was too
much to elute with 100 mL solvent, a large volume (200 mL or 300 mL) can also be
used for some fractions.
2.9.4.2 Purification of the compounds by TLC and silica gel column
chromatography
After SPE, the aim compounds were further purified by the Kieselgel 60 F254 TLC
plates (20 ×20 cm, 2 mm layer thick; Merck, Darmstadt, Germany) or the column
chromatography using silica gel 60 (40-63 μm, Roth). The solvent for TLC was
dichloromethane: methanol 9:1 with 0.05% acetic acid. In order to determine the
position of the band for the compounds, the TLC plates were check under the UV
light at the wavelength 254 nm and 366 nm. In the prerun with analytic TLC plates,
the plates could also be stained by the anisaldehyde staining reagents. The bands
containing the aim compounds were cut out with knife and extracted three time with
methanol. The particles from the TLC plates were removed by centrifugation (4 ℃,
14,000 rpm, 10 min). The supernatant was concentrated to dryness by the rotary
evaporator and redissolved in 1 mL methanol.
The chromatography in silica gel column was carried out using 2 → 15%
MeOH/CH2Cl2. The resulting fractions were collected in the 20 mL test tubes and
checked by analytic TLC plates. The fractions contain the aim product were
evaporated to dryness in the rotary evaporator, dissolved in 1 mL methanol and
analyzed by LC/MS using the method MENS04R.

56 Materials and Methods
2.9.4.3 Purification of the compounds by preparative HPLC
In order to obtain compounds pure enough for the NMR measurement, the partially
purified compounds were further purified by preparative HPLC. The preparative
HPLC system used in this thesis was from Waters equipped with 717 plus
autosampler, 2 Waters 515 pumps, and a Waters 2996 photodioden array detector.
The separation of the metabolites was achieved on an Aligent Zorbox C18 pre-column
(50 mm × 9.4 mm; particle size 5 μm) and a main column (150 mm × 9.4 mm;
particle size 5 μm). The software controlling the HPLC operation was “Empower
2002 Waters Corporation” The methanol solution containing the compounds were
first filtrated through a Rotilabo® syringe filter (0.45 μm, PVDF) to remove large
particles. Then 30-100 μl of the MeOH solution was injected into the HPLC system
and the aim compounds were collected two seconds after the appreance of the peak on
the screen. The collected fractions were concentrated and tested with LC/MS. The
gradient for the preparative HPLC was as Table 2-26.
Table 2-26 Gradient for the preparative HPLC. (Solvent A: Acetonitrile with 0.5% acetic acid;
Solvent B: H2O with 0.5% acetic acid).
2.9.4.4 Column chromatography using Sephadex® LH-20
Before the NMR measurement, the purified products were often further purified with
Sephadex LH-20. The Sephadex LH-20 was first suspended with methanol or aceton,
loaded onto a glass column (100 × 2.6 cm), equilibrated for 2 days in the solvent and
was then ready to use. The producted were dissolved in the same solvent as that in the
Time (min) Solvent A (%) Solvent B (%)
0 20 80
6 20 80
7 30 70
25 95 5
28 95 5
30 20 80
35 20 80
Flow rate 2 mL/min; Detection: 254 nm and 361 nm.

Materials and Methods 57
column and loaded onto the column. Elution was performed with the same solvent as
the column with a flow rate of about 1 mL/min. Each 4 mL elute was collected by
fraction collector and controlled by LC/MS. The right fractions were pooled and
concentrated in vacuum.
2.9.5 Structure elucidation by NMR
Nuclear magnetic resonance (NMR) was employed to elucidate the structures of
rishirilide B and its derivatives from the mutants. The 1H (400 MHz) and
13C-NMR
(100 MHz) spectra of these compounds were detected on the Bruker DRX-500
spectrometers (Bruker, Karlsruhe, Germany). In addition of 1D NMR, the 2D NMR
including 1H/
1H-COSY (correlation spectroscopy), HSQC (Heteronuclear Single
Quantum Coherence), HMBC (Heteronuclear Multiple Bond Correlation) and NOESY
(Nuclear Overhauser Effect Spectroscopy) were also measured to determine the
structures of the compounds. Analsysis of the NMR data and interpretation of the
structures were undergone with the help of Renato Murillo from Costa Rica. In this
thesis, the NMR data for the following 4 compounds were obtained (Table 2.27)
Table 2-27 Compounds measured by NMR in the thesis
Compound Produced by Solvent Time of measurement
Rishirilide B S. albus::cos4 CD3OD 09.2011
Rishi1a S. albus::cos4ΔrslK4 CD3OD 02.2012
Rishi2a S. albus::cos4ΔrslO1 CDCl3 03.2012
Rishi2b S. albus::cos4ΔrslO1 CDCl3 03.2012
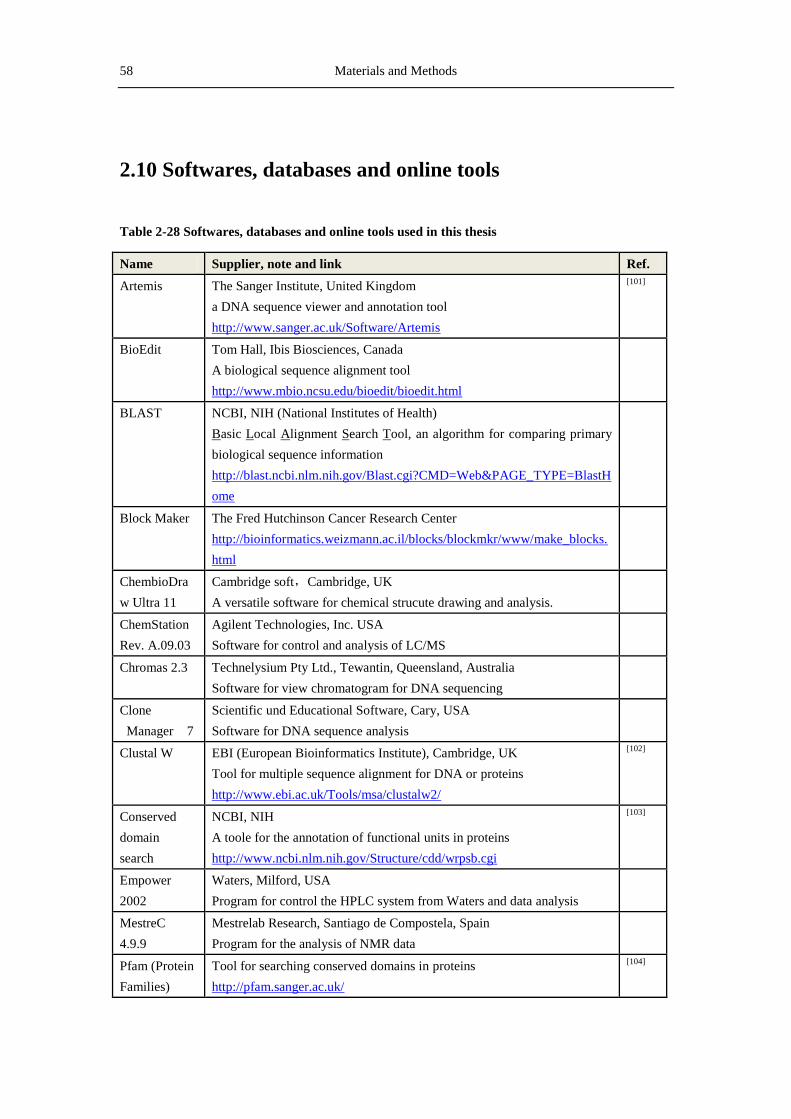
58 Materials and Methods
2.10 Softwares, databases and online tools
Table 2-28 Softwares, databases and online tools used in this thesis
Name Supplier, note and link Ref.
Artemis The Sanger Institute, United Kingdom
a DNA sequence viewer and annotation tool
http://www.sanger.ac.uk/Software/Artemis
[101]
BioEdit Tom Hall, Ibis Biosciences, Canada
A biological sequence alignment tool
http://www.mbio.ncsu.edu/bioedit/bioedit.html
BLAST
NCBI, NIH (National Institutes of Health)
Basic Local Alignment Search Tool, an algorithm for comparing primary
biological sequence information
http://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastH
ome
Block Maker The Fred Hutchinson Cancer Research Center
http://bioinformatics.weizmann.ac.il/blocks/blockmkr/www/make_blocks.
html
ChembioDra
w Ultra 11
Cambridge soft,Cambridge, UK
A versatile software for chemical strucute drawing and analysis.
ChemStation
Rev. A.09.03
Agilent Technologies, Inc. USA
Software for control and analysis of LC/MS
Chromas 2.3 Technelysium Pty Ltd., Tewantin, Queensland, Australia
Software for view chromatogram for DNA sequencing
Clone
Manager 7
Scientific und Educational Software, Cary, USA
Software for DNA sequence analysis
Clustal W EBI (European Bioinformatics Institute), Cambridge, UK
Tool for multiple sequence alignment for DNA or proteins
http://www.ebi.ac.uk/Tools/msa/clustalw2/
[102]
Conserved
domain
search
NCBI, NIH
A toole for the annotation of functional units in proteins
http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi
[103]
Empower
2002
Waters, Milford, USA
Program for control the HPLC system from Waters and data analysis
MestreC
4.9.9
Mestrelab Research, Santiago de Compostela, Spain
Program for the analysis of NMR data
Pfam (Protein
Families)
Tool for searching conserved domains in proteins
http://pfam.sanger.ac.uk/
[104]
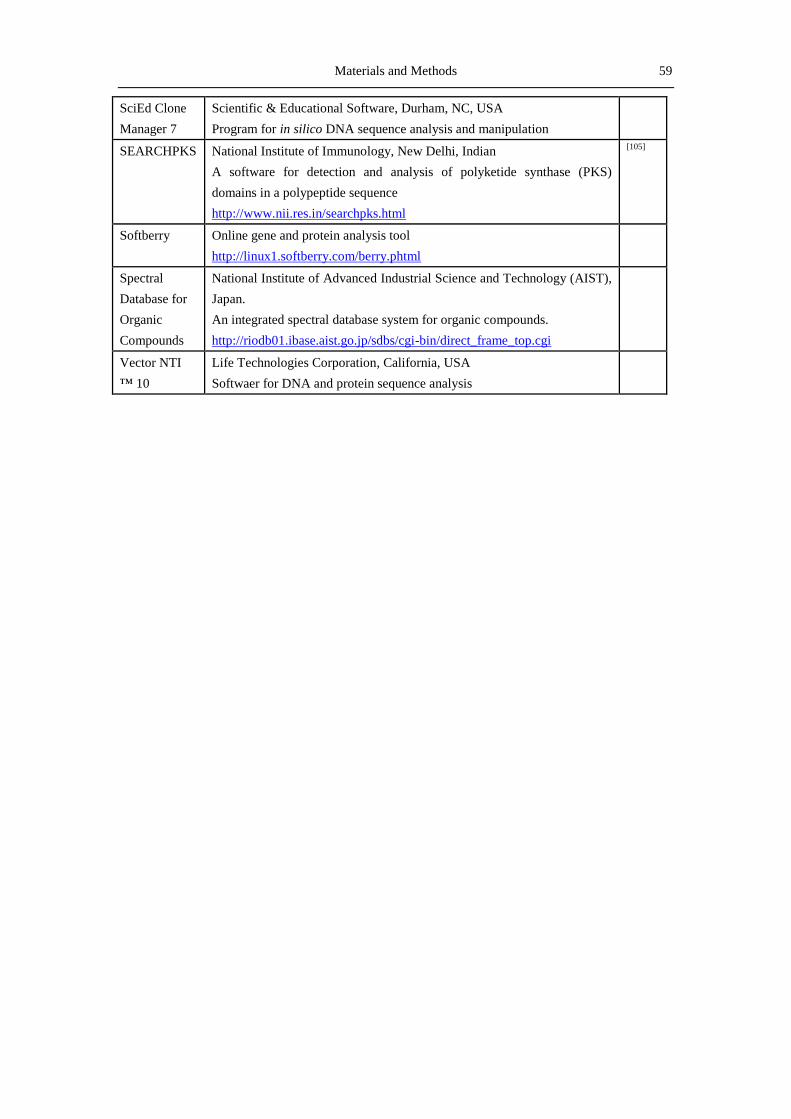
Materials and Methods 59
SciEd Clone
Manager 7
Scientific & Educational Software, Durham, NC, USA
Program for in silico DNA sequence analysis and manipulation
SEARCHPKS National Institute of Immunology, New Delhi, Indian
A software for detection and analysis of polyketide synthase (PKS)
domains in a polypeptide sequence
http://www.nii.res.in/searchpks.html
[105]
Softberry Online gene and protein analysis tool
http://linux1.softberry.com/berry.phtml
Spectral
Database for
Organic
Compounds
National Institute of Advanced Industrial Science and Technology (AIST),
Japan.
An integrated spectral database system for organic compounds.
http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi
Vector NTI
™ 10
Life Technologies Corporation, California, USA
Softwaer for DNA and protein sequence analysis

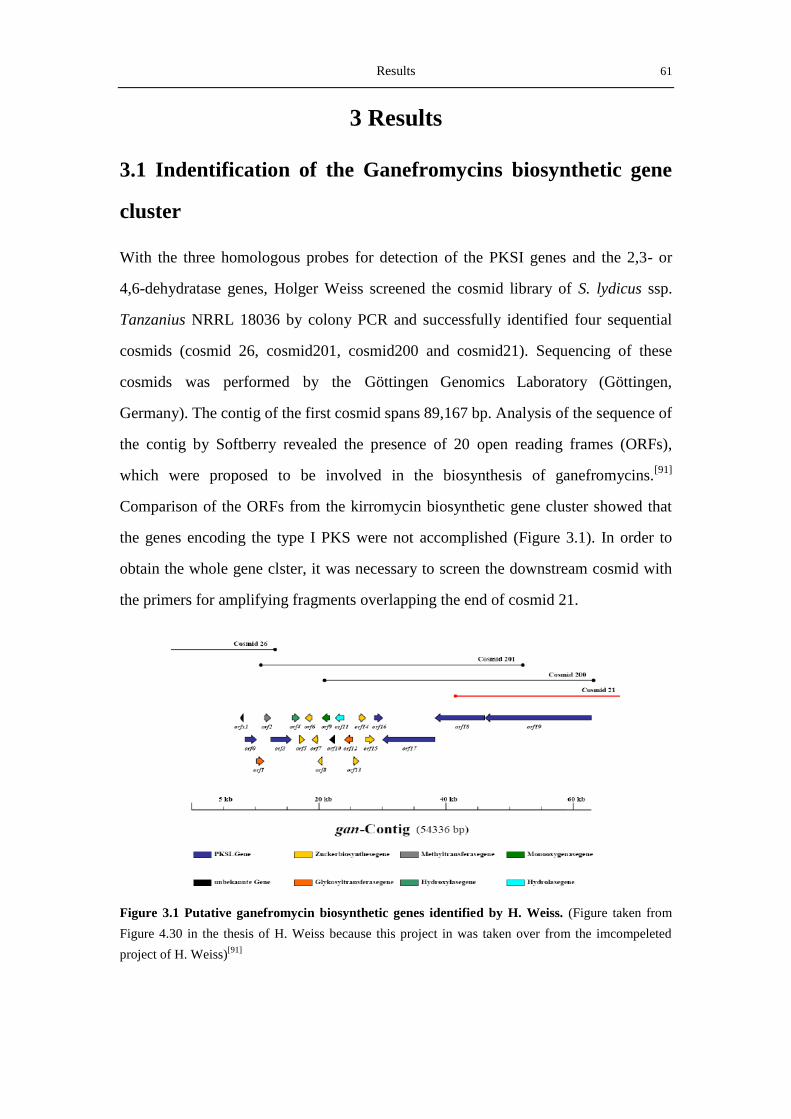
Results 61
3 Results
3.1 Indentification of the Ganefromycins biosynthetic gene
cluster
With the three homologous probes for detection of the PKSI genes and the 2,3- or
4,6-dehydratase genes, Holger Weiss screened the cosmid library of S. lydicus ssp.
Tanzanius NRRL 18036 by colony PCR and successfully identified four sequential
cosmids (cosmid 26, cosmid201, cosmid200 and cosmid21). Sequencing of these
cosmids was performed by the Göttingen Genomics Laboratory (Göttingen,
Germany). The contig of the first cosmid spans 89,167 bp. Analysis of the sequence of
the contig by Softberry revealed the presence of 20 open reading frames (ORFs),
which were proposed to be involved in the biosynthesis of ganefromycins.[91]
Comparison of the ORFs from the kirromycin biosynthetic gene cluster showed that
the genes encoding the type I PKS were not accomplished (Figure 3.1). In order to
obtain the whole gene clster, it was necessary to screen the downstream cosmid with
the primers for amplifying fragments overlapping the end of cosmid 21.
Figure 3.1 Putative ganefromycin biosynthetic genes identified by H. Weiss. (Figure taken from
Figure 4.30 in the thesis of H. Weiss because this project in was taken over from the imcompeleted
project of H. Weiss)[91]

62 Results
The screening and analysis of the remaining cosmids for ganefromycins biosynthesis
were continued by the author of this thesis from July, 2009.
3.1.1 Analysis of the DNA sequence of cosmid 21
The cosmid 21 spans 43,923 bp. Prediction of the ORFs (with the help of Dr. Tilmann
Weber, University of Tuebingen, Germany) showed that this cosmid contains three
intact genes encoding the type I PKSs in the middle and two partial genes also
encoding type I PKSs in the head and tail of the sequence, respectively. The first 2
ORFs were located in the overlap region of cosmid 200 and cosmid 21, while the
other 3 ORFs were situated in cosmid 21. The first 2 ORFs were identical, partially or
entirely, to the genes named by H. Weiss as orf18 and orf19. In order to facilitate
understanding of the names of the genes, orf18 and orf19 were named ganAV and
ganAIV in this thesis, according to their sequence in the modular PKSs. The other 3
genes were designated as ganAIII, ganAII and ganAI, respectively. The encoded
proteins by these three genes were analyzed by the SEARCHPKS software (see 2.10).
In GanAIII, 5 intact modules for type I PKSs were present, whereas in GanAII and
GanAI, the numbers of modules were 1 and 5 respectively. No exceptionally, these
proteins possess typical domain organization and several modules were located in two
adjacent genes. The identities of these proteins with the type I PKSs in the kirromycin
biosynthetic gene cluster[81]
were 55% (GanAIV with KirAIV), 56% (GanAIII with
KirAIII), and 51% (GanAII with KirAII), respectively.
3.1.2 Screening and analysis of cosmid 2H19
3.1.2.1 Screening of 2H19
The cosmids containing DNA sequence in the downstream of cosmid21 were
screened by colony PCR with the primer pairs Cosmid21PWF/Cosmid21PWR. 14
colonies showed positive PCR product for the first PCR with these primers and the
colonies from the 24 plates of the cosmid library constructed by the Combinature
Biopharm AG (Berlin, Germany) (Figure 3.2). A second round of PCR was conducted

Results 63
using the cosmid DNA isolated from these colonies to verify the reliability of the
colony PCR.
Figure 3.2 Results for colony PCR Screening of the cosmids located in the downstream of
cosmid21. The right colonies should have PCR product at the size of 1.0 kb. Ladder: 1 kb DNA ladder
from Promega.
Figure 3.3 Pattern of the 14 cosmids digested with BamHI. L: 1 kb DNA ladder from Promega. 1-14:
The 14 cosmids with which the aim 1 kb fragment was obtained for the colony PCR, showing their
overlapping with cos21.

64 Results
After colony PCR, all the possible colonies were cultivated and the cosmids inside were
isolated. Digestion of these cosmids with BamHI led to the identification of 14 cosmid
which had different restriction patterns among each other (Figure 3.3).
Among the 14 different cosmids, 6 were selected ramdonly for primer-walking
sequencing to determine the length of their overlapping regions with cosmid21.
However, localization of the ends of 3 cosmids failed due to strong noise in the
sequencing, the positions of the front end of other 3 cosmids (named as 2H19, 3B10
and 4C12, according to the location of the colonies which contain these cosmids,
respectively.). The cosmid 2H19 possesses the smallest overlapping region (14 kb) and
was selected to undergo the shotgun sequencing.
3.1.2.2 Analysis of the sequence of 2H19
The insert of cosmid 2H19 spans 39,320 kb and was predicted to include 20 ORFs by
Softberry and BLAST analysis (See 2.10). The first two putative ORFs were identical
to the last two ORFs of the cosmid 21, representing the overlap region of these two
cosmids. The next three ORFs putatively encoded a LuxR family transcriptional
regulator, a thioesterase and an elongation factor Tu. These three types of proteins often
exist in the gene clusters for the biosynthesis of type I polyketides. The LuxR family
transcriptional regulators are generally able to activate the key enzymes for polyketide
biosynthesis.[106]
Thioesterase (TE) domains or proteins catalyze the release of
polyketide chain from the PKSs.[107]
The elongation factor Tu in the putative
gan-cluster is proposed to confer resistance to the produced antibiotic, because
ganefromycins action by interfering the binding of elongation factor Tu and GTP.[108]
The other genes were responsible for encoding the 30S or 50S ribosomal proteins,
DNA-directed RNA polymerase subunits and ion-transporting ATPases. As the
ribosomal proteins and polymerase proteins are important for the growth of the cells
and belong the the primary metabolic pathway, the gene encodes the elongation factor
Tu was most possible the last one in the ganefromycin biosynthetic gene cluster.

Results 65
Table 3-1 Proteins involved in ganefromycin biosynthesis
Protein Aa Putative function Blast Score
/E Value
Highest Similarity to Ref.
ORF(-1) 188 hypothetical protein 157/4e-45 hypothetical protein
(Streptomyces hygroscopicus
ATCC 53653)
ganAT1 575 Acyltransferase 309/2e-95 KirCII (Streptomyces collinus) [81]
ganGT1 409 Glycoslytransferase 310/2e-98 AveBI (Streptomyces
avermitilis MA-4680)
[109]
ganM 317 SAM-dependent
methyltransferase
318/3e-104 KirM (Streptomyces collinus) [81]
ganAT2 1091 Acyltransferase 1482/0.0 KirC1 (Streptomyces cattleya
NRRL 8057)
[110]
ganO1 402 Putative cytochrome
P450 hydroxylase
492/9e-170 EryF (Streptomyces cattleya
NRRL 8057)
[110]
GanS1 265 dTDP-6-deoxy-L-
hexose
3-O-methyltransferase
409/2e-141 CalS11 (Micromonospora
echinospora)
[111]
GanS2 328 dTDP-4-keto-6-deoxy-
hexose 2,3- reductase
459/2e-159 VinE (Streptomyces halstedii) [112]
GanS3 321 Putative NDP-hexose
4-ketoreductase
216/3e-64 PokS6 (Streptomyces
diastatochromogenes)
[113]
GanS4 197 Putative NDP-hexose
3,5-epimerase
218/1e-68 PokS7 (Streptomyces
diastatochromogenes)
[113]
GanO2 394 cytochrome P450
monooxygenase
298/7e-94 PtmO5 (Streptomyces
platensis)
[114]
GanH 286 hypothetical protein 387/3e-132 KirHVI (Streptomyces
collinus)
[81]
GanI 473 Indoleacetamide
hydrolase
635/0.0 Bam (Streptomyces cattleya
NRRL 8057)
[110]
GanGT2 424 Glycosyltransferase 411/3e-137 DesVII (Streptomyces
venezuelae)
[115]
GanS5 290 NDP-glucose-synthase 398/2e-136 AclY (Streptomyces galilaeus) [116]
GanS6 335 NDP-glucose
4,6-dehydratase
428/6e-147 RfbB (Streptomyces olivaceus) [117]
GanS7 477 NDP-hexose
2,3-dehydratase
449/2e-150 SnogH (Streptomyces
nogalater)
[118]
GanAT3 427 Putative acyltransferase 272/1e-83 PldC (Streptomyces platensis) [119]
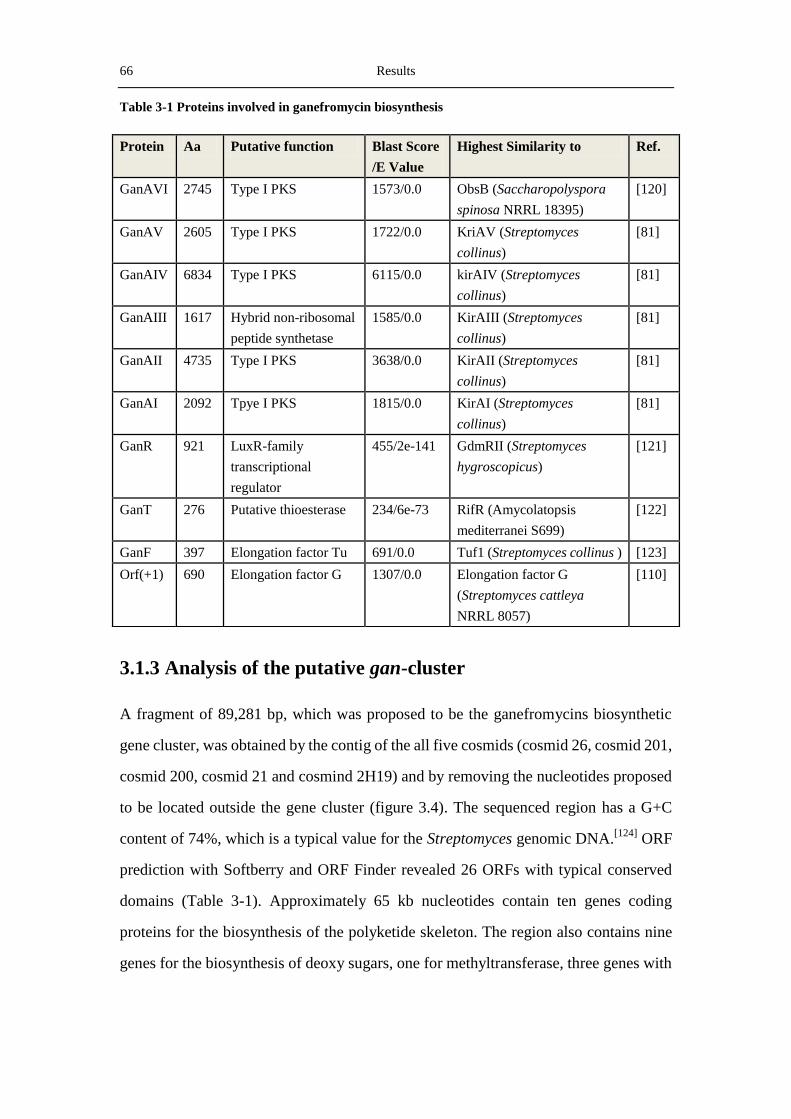
66 Results
Table 3-1 Proteins involved in ganefromycin biosynthesis
3.1.3 Analysis of the putative gan-cluster
A fragment of 89,281 bp, which was proposed to be the ganefromycins biosynthetic
gene cluster, was obtained by the contig of the all five cosmids (cosmid 26, cosmid 201,
cosmid 200, cosmid 21 and cosmind 2H19) and by removing the nucleotides proposed
to be located outside the gene cluster (figure 3.4). The sequenced region has a G+C
content of 74%, which is a typical value for the Streptomyces genomic DNA.[124]
ORF
prediction with Softberry and ORF Finder revealed 26 ORFs with typical conserved
domains (Table 3-1). Approximately 65 kb nucleotides contain ten genes coding
proteins for the biosynthesis of the polyketide skeleton. The region also contains nine
genes for the biosynthesis of deoxy sugars, one for methyltransferase, three genes with
Protein Aa Putative function Blast Score
/E Value
Highest Similarity to Ref.
GanAVI 2745 Type I PKS 1573/0.0 ObsB (Saccharopolyspora
spinosa NRRL 18395)
[120]
GanAV 2605 Type I PKS 1722/0.0 KriAV (Streptomyces
collinus)
[81]
GanAIV 6834 Type I PKS 6115/0.0 kirAIV (Streptomyces
collinus)
[81]
GanAIII 1617 Hybrid non-ribosomal
peptide synthetase
1585/0.0 KirAIII (Streptomyces
collinus)
[81]
GanAII 4735 Type I PKS 3638/0.0 KirAII (Streptomyces
collinus)
[81]
GanAI 2092 Tpye I PKS 1815/0.0 KirAI (Streptomyces
collinus)
[81]
GanR 921 LuxR-family
transcriptional
regulator
455/2e-141 GdmRII (Streptomyces
hygroscopicus)
[121]
GanT 276 Putative thioesterase 234/6e-73 RifR (Amycolatopsis
mediterranei S699)
[122]
GanF 397 Elongation factor Tu 691/0.0 Tuf1 (Streptomyces collinus ) [123]
Orf(+1) 690 Elongation factor G 1307/0.0 Elongation factor G
(Streptomyces cattleya
NRRL 8057)
[110]
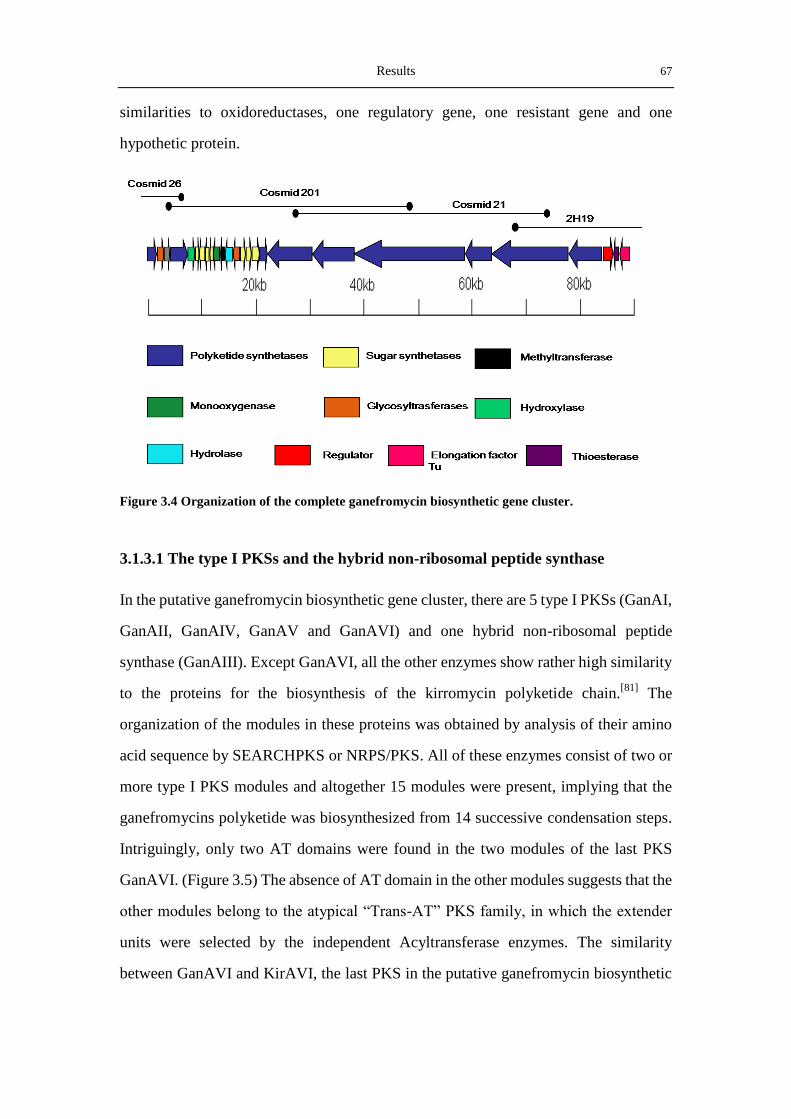
Results 67
similarities to oxidoreductases, one regulatory gene, one resistant gene and one
hypothetic protein.
Figure 3.4 Organization of the complete ganefromycin biosynthetic gene cluster.
3.1.3.1 The type I PKSs and the hybrid non-ribosomal peptide synthase
In the putative ganefromycin biosynthetic gene cluster, there are 5 type I PKSs (GanAI,
GanAII, GanAIV, GanAV and GanAVI) and one hybrid non-ribosomal peptide
synthase (GanAIII). Except GanAVI, all the other enzymes show rather high similarity
to the proteins for the biosynthesis of the kirromycin polyketide chain.[81]
The
organization of the modules in these proteins was obtained by analysis of their amino
acid sequence by SEARCHPKS or NRPS/PKS. All of these enzymes consist of two or
more type I PKS modules and altogether 15 modules were present, implying that the
ganefromycins polyketide was biosynthesized from 14 successive condensation steps.
Intriguingly, only two AT domains were found in the two modules of the last PKS
GanAVI. (Figure 3.5) The absence of AT domain in the other modules suggests that the
other modules belong to the atypical “Trans-AT” PKS family, in which the extender
units were selected by the independent Acyltransferase enzymes. The similarity
between GanAVI and KirAVI, the last PKS in the putative ganefromycin biosynthetic

68 Results
gene cluster and the the kirromycin biosynthetic gene cluster respectively, was little
lower (42%). The three new type I PKSs found in current thesis were proposed to be
Figure 3.5 The predicted type I PKS modules in the six genes (GanAI-GanAVI). The modules
span two adjacent genes were marked with a black circle. X in the 4th module (M4) means the function
of the domain is not clear. In ganAVI, there is a incomplete module downstreams module 15, but its
function in the biosynthesis of ganefromycin could not be proposed.
responsible for the initiation of the polyketide biosynthesis. The hybrid non-ribosomal
peptide synthase (GanAIII) was responsible for the condensation of the polyketide
chain with glycine. This condensation yielded the amide bond and the polyketide chain
was further extended by the action of GanAIV-VI.
3.1.3.2 The LuxR-family transcriptional regulator
GanR, the only regulatory gene in the gan-cluster, encodes a protein of 921 amino
acids. In BLASTP analysis, GanR shows high similarity to the LuxR family
transcriptional regulator GdmRII in Saccharopolyspora erythraes NRRL 2338,
implying the identity of GanR as a member of the novel large ATP- binding regulators
of the LuxR family (LAL). The LAL family, represented by the regulator of maltose
regulon in E. coli MalT,[125]
contain an N-terminal ATP/GTP-binding domain,[126]
and
a C-terminal helix-turn-helix motif. Several members of the LAL family have been

Results 69
reported to be present in Streptomyces antibiotic gene cluster, such as PikD from the
pilromycin biosynthetic gene cluster,[127]
RapH from the rapamycin pathway[128]
and
PimR from the pimaricin biosynthetic pathway.[129]
3.1.3.3 The Thioesterase GanT
GanT consists of 276 aa and is most similar to RifR, a characterized type II
thioesterase.[130]
Type I TEs are covalently attached to the terminal module of the PKSs
and usually unload the final polyketide from the polyketide assembly line complex,
while type II TEs are independent proteins and they can remove intermediates from any
module. Structural and biochemical studies of RifR showed that this protein were able
to hydrolyze both carboxylated and decarbolylated acyl thioesters. Especially, RifR
preferred aberrant decarboxylated acyl thioester over natural building blocks of
rifamycin, consistent with the function of type II TEs as a “housekeeper” for ensuring
the extension of the polyketide chain in the right manner.
3.1.3.4 GanF: an elongation factor Tu
The kirromycin and elfamycin family antibiotics were reported to inhibit bacteria
protein synthesis by binding to elongation factor Tu (EF-Tu).[131]
In some of the
kirromycin-type antibiotics producers, such as Streptomyces cinnamomeus (produces
kirrothricin), Streptomyces lactamdurans (produces efrotomycin) and Streptovertici-
llium mobaraense (produces pulvomycin), the resistace to this family of antibiotics was
attributed to drug-insensitive EF-Tu. Mutagensis analysis of the EF-Tu in S.
cinnamoneus showed that the Thr378
residue in this protein was an important
determinant of kirromycin resistance.[132]
Alignment analysis of the sequence of GanF
with other kirromycin-resistant EF-Tu showed that the Thr amino acid was also found
in GanF, imply that this protein was responsible for the resistance to the product (Figure
3.6).

70 Results
Figure 3.6 Alignment of selected Streptomyces EF-Tu sequences. The important residues which are
important for the resistance to kirromycin-family antibiotics were marked below the sequence and
labelled red.
3.2 Identification of the ganefromycin biosynthetic gene
cluster
3.2.1 Conjugation of S. lydicus
After the identification of the genes for ganefromycin biosynthesis, genetic operation
on the type I PKS genes as well as the post-PKS modification genes was conducted.
Due to the special polyketide structure and the presence of the phenyl acetic acid group
and the three sugars, investigation of the ganefromycin biosynthetic enzymes would be
of great importance in revealing the mechanism for type I polyketide biosynthesis and
for the post-PKS modification reactions. During his work on ganefromycin, Holger
Weiss constructed several plasmids for inactivating genes in the gan-cluster, however,
no conjugation or protoplast transformation was successful.
In this thesis, protoplast transformation and conjugation experiments were carried out
first with two integrative plasmids pSET152 (φ31-based) and pSOK804 (VWB-based),

Results 71
as well as three replicative plasmids (pKC1139, pKC1218, and pUWL201) with
different replicons. Although pSET152 was able to integrate into the chromosome of
most Streptomyces, no exconjugants were obtained by conjugation of S. lydicus with
pSET152. The plasmid pSOK804 was able to integrate into the genome of S. lydicus
efficiently. Ostash et al. had shown that a putative tRANArg
(AGG) gene served as the
integration site for VWB-based plasmid.[133]
The different ability of pSET152 and
pSOK804 in integration into the chromosome of S. lydicus implies the presence of the
VWB-integration site and the absence of the attB site in this strain. In the case of
transformation by replicative plasmids, only the SCP*2-based pKC1218 was able to
replicate in S. lydicus. (Figure 3.7)
Figure 3.7 Plates for conjugation of S. lydicus with pKC1218 and pSOK804.
3.2.2 Verification of the gan-Cluster by disrupting ganAIV
Although domain prediction of the type I PKSs in the putative gan-cluster fitted
perfectly with the PKSs from the kirromycin biosynthetic pathway, and the presence of
the ganefromycin-insensitive EF-Tu in this cluster strongly suggest that the current
cluster is responsible for the biosynthesis of ganefromycins, direct proofs from gene
inactivation are still required to verify the hypothesis. In order to carry out the gene
disruption experiments, the two glycosyltransferase genes (ganGT1 and ganGT2) and
one PKS gene, ganAIV, were selected.
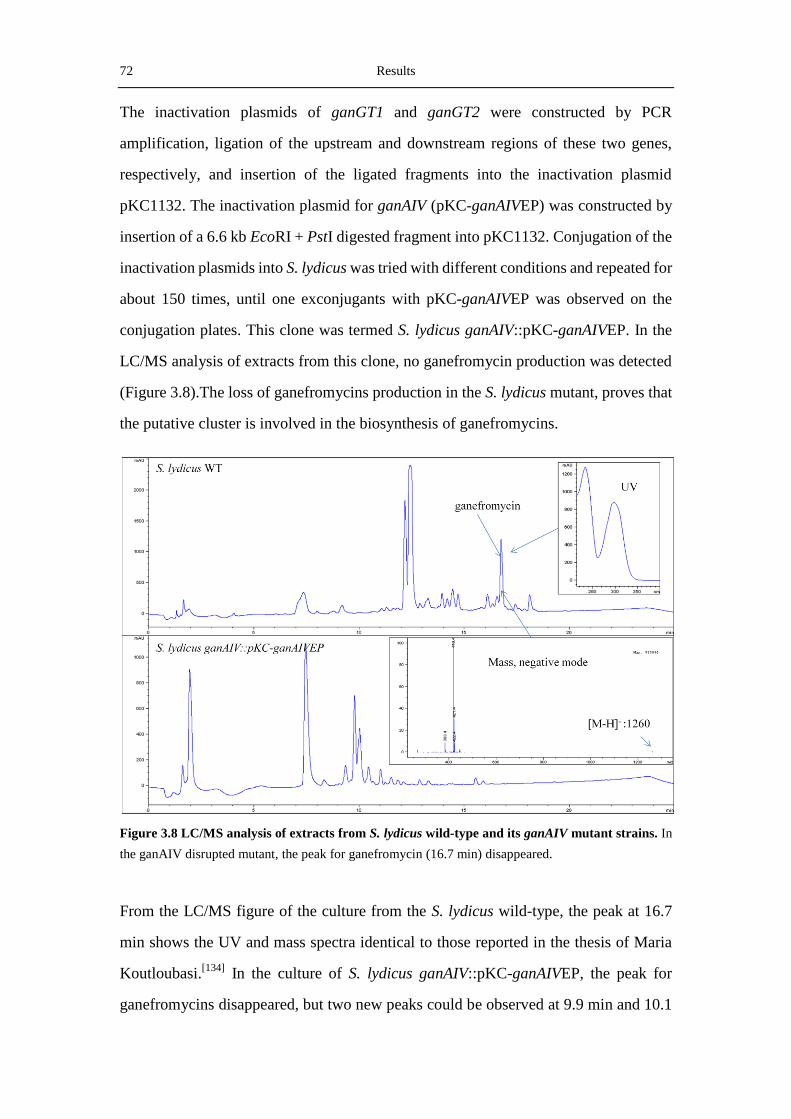
72 Results
The inactivation plasmids of ganGT1 and ganGT2 were constructed by PCR
amplification, ligation of the upstream and downstream regions of these two genes,
respectively, and insertion of the ligated fragments into the inactivation plasmid
pKC1132. The inactivation plasmid for ganAIV (pKC-ganAIVEP) was constructed by
insertion of a 6.6 kb EcoRI + PstI digested fragment into pKC1132. Conjugation of the
inactivation plasmids into S. lydicus was tried with different conditions and repeated for
about 150 times, until one exconjugants with pKC-ganAIVEP was observed on the
conjugation plates. This clone was termed S. lydicus ganAIV::pKC-ganAIVEP. In the
LC/MS analysis of extracts from this clone, no ganefromycin production was detected
(Figure 3.8).The loss of ganefromycins production in the S. lydicus mutant, proves that
the putative cluster is involved in the biosynthesis of ganefromycins.
Figure 3.8 LC/MS analysis of extracts from S. lydicus wild-type and its ganAIV mutant strains. In
the ganAIV disrupted mutant, the peak for ganefromycin (16.7 min) disappeared.
From the LC/MS figure of the culture from the S. lydicus wild-type, the peak at 16.7
min shows the UV and mass spectra identical to those reported in the thesis of Maria
Koutloubasi.[134]
In the culture of S. lydicus ganAIV::pKC-ganAIVEP, the peak for
ganefromycins disappeared, but two new peaks could be observed at 9.9 min and 10.1
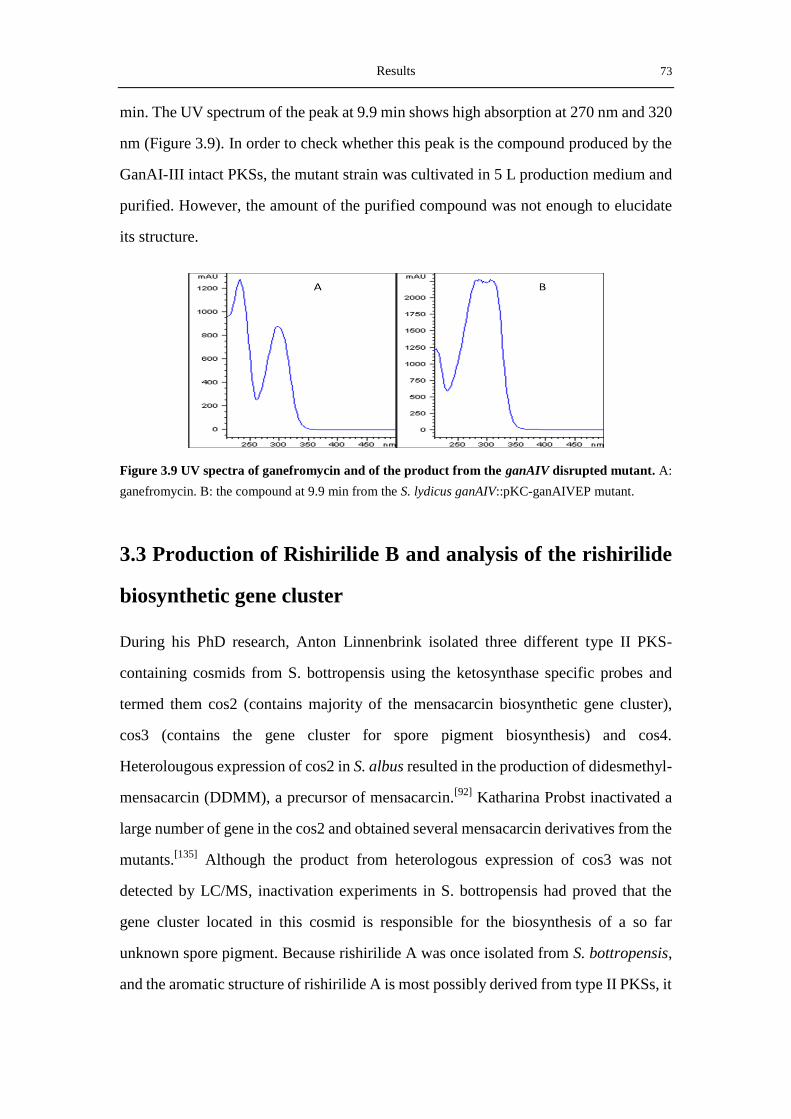
Results 73
min. The UV spectrum of the peak at 9.9 min shows high absorption at 270 nm and 320
nm (Figure 3.9). In order to check whether this peak is the compound produced by the
GanAI-III intact PKSs, the mutant strain was cultivated in 5 L production medium and
purified. However, the amount of the purified compound was not enough to elucidate
its structure.
Figure 3.9 UV spectra of ganefromycin and of the product from the ganAIV disrupted mutant. A:
ganefromycin. B: the compound at 9.9 min from the S. lydicus ganAIV::pKC-ganAIVEP mutant.
3.3 Production of Rishirilide B and analysis of the rishirilide
biosynthetic gene cluster
During his PhD research, Anton Linnenbrink isolated three different type II PKS-
containing cosmids from S. bottropensis using the ketosynthase specific probes and
termed them cos2 (contains majority of the mensacarcin biosynthetic gene cluster),
cos3 (contains the gene cluster for spore pigment biosynthesis) and cos4.
Heterolougous expression of cos2 in S. albus resulted in the production of didesmethyl-
mensacarcin (DDMM), a precursor of mensacarcin.[92]
Katharina Probst inactivated a
large number of gene in the cos2 and obtained several mensacarcin derivatives from the
mutants.[135]
Although the product from heterologous expression of cos3 was not
detected by LC/MS, inactivation experiments in S. bottropensis had proved that the
gene cluster located in this cosmid is responsible for the biosynthesis of a so far
unknown spore pigment. Because rishirilide A was once isolated from S. bottropensis,
and the aromatic structure of rishirilide A is most possibly derived from type II PKSs, it

74 Results
was proposed that the putative gene cluster (A. Linnenbrink termed it msc-cluster) in
cos4 is responsible for the synthesis of rishirilide A. However, no product from the
msc-cluster had been identified.
3.3.1 Production of rishirilide B
3.3.1.1 Heterlogous production of rishirilide B in S. albus and S. lividans
Cos4 was introduced into S.albus J1074 and S. lividans 1326 by intergeneric
conjugation with ET12567/pUZ8002, respectively (see method 2.7.6.2). For each
conjugation, one exconjugant that was arpamycin resistant was picked and tested for
the integration of the cosmid into their chromosomes by colony PCR. The PCR results,
which showed the correct fragment size of the specifc genes from cos4, proved the
integration of cos4 into the genome of these two strains.
In order to check the production of rishirilides by heterologous expression, the two
strains S. albus::cos4 and S. lividans::cos4 were cultivated in HA-, SG- and NL19-
media respectively (see 2.9.2). LC/MS analysis of the extracts showed the presence of a
peak with the characteristic UV spectra of rirhirilide B (218 nm, 264 nm, 305 nm and
370 nm) and the mass of rishirilide B in negative mode ([M-H]-= 371.2) at 22.1 min
(Figure 3.10).
In order to prove that this new compound is rishirilide B, S. albus::cos4 was cultured
in 5 L HA-medium (See 2.9.2). Then the main compound was sequentially purified by
SPE, preparative HPLC and column chromatography by Sephadex LH20. Finally 3.2
mg of compound was obtained and measured by NMR. The 1H and
13C NMR data of
this compound is in good accordance to the data reported for synthetic rashirilide B,[136]
unequivocally proving the identity of this compound to be rishirilide B (Table 3-2 and
Figure 3.11). Because rishirilide A is not reproducible in S. bottropensis, cultivation of
S. albus::cos4 and S. lividans::cos4 in the HA-medium were repeated independently for
three times. Peak of rishirilide B could be detected all the time at very close peak area,
implying the repeatability and stability of rishirilide B production in the two

Results 75
heterlogous expression hosts. However, no peak for rishirilide A was found in these
cultures.
Figure 3.10 LC/MS analysis of the cos4-integrated strains in HA medium. (UV detection at λ =
254 nm); The mass of the main peak was shown in negative mode. The UV-absorption characteristics
of S. albus and S. lividans wild-type in HA medium was almost identical due to an unknown reason.

76 Results
Table 3-2 The 1H and
13C NMR data of rishirilide B isolated from S. albus::cos4.
Figure 3.11 Structure of rishirilide B.
3.3.1.2 Production of rishirilide B in S. bottropensis
S. bottrpensis from the saccharose stock solution was cultured in the HA-medium and
the NL111-medium in attempt to verify its identity. Interestingly, LC/MS analysis of
the culture revealed the production of mensacarcin in the HA-medium and the
production of rishirilide B in the NL111 medium (Figure 3.12). Although only
rishirilide A was isolated from the culture broth of S. bottropensis in the dissertation
of M. Arnold,[73]
the isolation of rishirilide B in the broth of S. bottropensis in the
NL111 medium was not a surprise, because the latter was proposed to be a precursor
of rishirilide A. The reason for the production of rishirilide B instead of A could be
attributed to the oxygen concentration in the broth, or to the difference in the
extraction processes for the fermentation broth.
Interestingly, in the raw extract of S. bottropensis from the NL111 medium, one peak
(17.2 min) with the same mass as bottromycin A2, a potent antibiotic against
Pos. C
[ppm]
H (J Hz) Pos. C
[ppm]
H (J Hz) Pos. C
[ppm]
H (J Hz)
1 197.3 6 119.6 6.92 d (7.4) 11 48.0 1.63-1.49 m
2 35.1 2.95 (q,6.6) 7 125.6 7.28 (dd, 8.1, 7.4) 12 31.2 2.30-2.15 m
3 77.0 8 119.8 7.45 d (8.1) 13 29.0 1.33-1.21 m
4 83.5 9 126.4 8.27 s 14 22.8 0.76 d (6.6)
4a 140.6 9a 126.1 15 22.5 0.65 d (6.6)
5 153.2 10 109.8 8.25 s 16 174.2
5-OH 10.25 10a 130.2 17 10.2 1.17 d (6.6)

Results 77
methicillin-resistant Streptococcus aureus (MRSA) and vancomycin-resistant
Enterococci (VRE) strains, was also detected[137, 138]
(Figure 3.13). But the UV
spectrum of this peak was not clear due to the low yield in the NL111 medium. This
result is consistent with the discovery of bottromycins in S. bottropensis.
Figure 3.12 The LC/MS profiles of S. bottropensis in NL111-medium and HA-medium.
Figure 3.13 Production of bottromycin by S. bottropensis in NL111-medium.

78 Results
3.3.2 Analysis of the rishirilide biosynthetic gene cluster
Analysis of the sequence in cos4 by Softberry and BLASTP resulted in the
identification of 42 ORFs. The first 11 genes enconde peptide with high identities
(>77%) to proteins for the biosynthesis of exoplysaccharide in Streptomyces sp.
139.[139]
Therefore, they are supposed to be involved in the biosynthesis of
exoploysaccharide in S. bottropensis. The last 3 ORFs in cos4 encode two peptides
similar to putative transcriptional anti-termination regulator from S. ambofaciens
ATCC 23877[140]
and one hypothetic protein. The rishirilide biosynthetic gene cluster
(rsl-cluster) is located in the middle of cos4 with 28 ORFs, including 20 structural
genes, 4 regulatory genes (rslR1-R4) and 4 resistance-related genes (rslT1-T4).
Five of the 20 structural genes were predicted to be responsible for the biosynthesis of
the polyketide chain, including genes encoding the the “minimal PKS” (rslK1-k3),
and genes encoding proteins for the selection of the unusual starter unit (rslK4 and
rslA). Besides these genes, there are three cyclase/aromatase-encoding genes
(rslC1-C3), ten genes encoding oxidoreductases, as well as a putative phosphotrans-
ferase-encoding gene (rslP) and a putative amidohydrolase-encoding gene (rslH).Two
of the four regulatory genes (rslR1 and rlsR2) encode proteins of the SARP-family,
while the other two genes encode a LAL-family regulator (RslR3) and a MarR family
transcriptional regulator (RslR4), respectively. Proteins encoded by rslT1, rslT2, and
rslT3 were proposed to form a three-component ABC-transporter. The RslT4 protein
shows homology to drug resistance transporter. These two transporters were supposed
to be responsible for the formation of self-resistance to rishirilide A and B. The
organization of the rsl-cluster and the putative proteins are shown in Figure 3.14 and
Table 3-3.
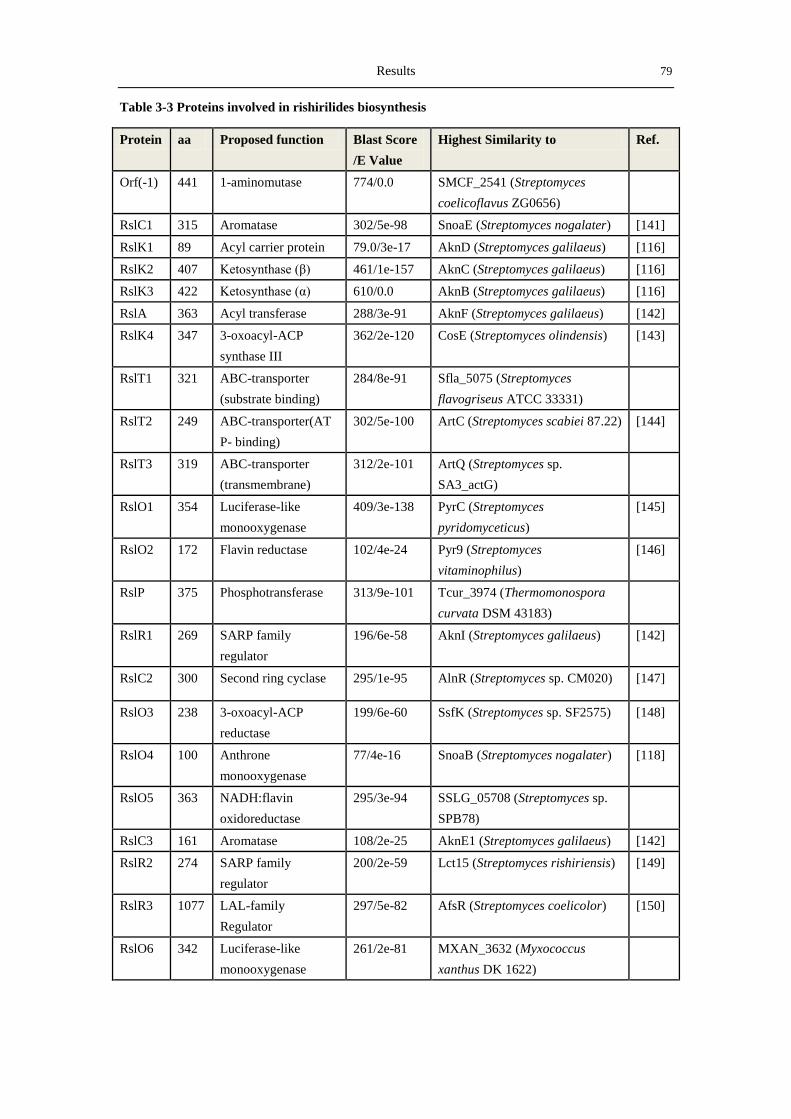
Results 79
Table 3-3 Proteins involved in rishirilides biosynthesis
Protein aa Proposed function Blast Score
/E Value
Highest Similarity to Ref.
Orf(-1) 441 1-aminomutase 774/0.0 SMCF_2541 (Streptomyces
coelicoflavus ZG0656)
RslC1 315 Aromatase 302/5e-98 SnoaE (Streptomyces nogalater) [141]
RslK1 89 Acyl carrier protein 79.0/3e-17 AknD (Streptomyces galilaeus) [116]
RslK2 407 Ketosynthase (β) 461/1e-157 AknC (Streptomyces galilaeus) [116]
RslK3 422 Ketosynthase (α) 610/0.0 AknB (Streptomyces galilaeus) [116]
RslA 363 Acyl transferase 288/3e-91 AknF (Streptomyces galilaeus) [142]
RslK4 347 3-oxoacyl-ACP
synthase III
362/2e-120 CosE (Streptomyces olindensis) [143]
RslT1 321 ABC-transporter
(substrate binding)
284/8e-91 Sfla_5075 (Streptomyces
flavogriseus ATCC 33331)
RslT2 249 ABC-transporter(AT
P- binding)
302/5e-100 ArtC (Streptomyces scabiei 87.22) [144]
RslT3 319 ABC-transporter
(transmembrane)
312/2e-101 ArtQ (Streptomyces sp.
SA3_actG)
RslO1 354 Luciferase-like
monooxygenase
409/3e-138 PyrC (Streptomyces
pyridomyceticus)
[145]
RslO2 172 Flavin reductase 102/4e-24 Pyr9 (Streptomyces
vitaminophilus)
[146]
RslP 375 Phosphotransferase 313/9e-101 Tcur_3974 (Thermomonospora
curvata DSM 43183)
RslR1 269 SARP family
regulator
196/6e-58 AknI (Streptomyces galilaeus) [142]
RslC2 300 Second ring cyclase 295/1e-95 AlnR (Streptomyces sp. CM020) [147]
RslO3 238 3-oxoacyl-ACP
reductase
199/6e-60 SsfK (Streptomyces sp. SF2575) [148]
RslO4 100 Anthrone
monooxygenase
77/4e-16 SnoaB (Streptomyces nogalater) [118]
RslO5 363 NADH:flavin
oxidoreductase
295/3e-94 SSLG_05708 (Streptomyces sp.
SPB78)
RslC3 161 Aromatase 108/2e-25 AknE1 (Streptomyces galilaeus) [142]
RslR2 274 SARP family
regulator
200/2e-59 Lct15 (Streptomyces rishiriensis) [149]
RslR3 1077 LAL-family
Regulator
297/5e-82 AfsR (Streptomyces coelicolor) [150]
RslO6 342 Luciferase-like
monooxygenase
261/2e-81 MXAN_3632 (Myxococcus
xanthus DK 1622)
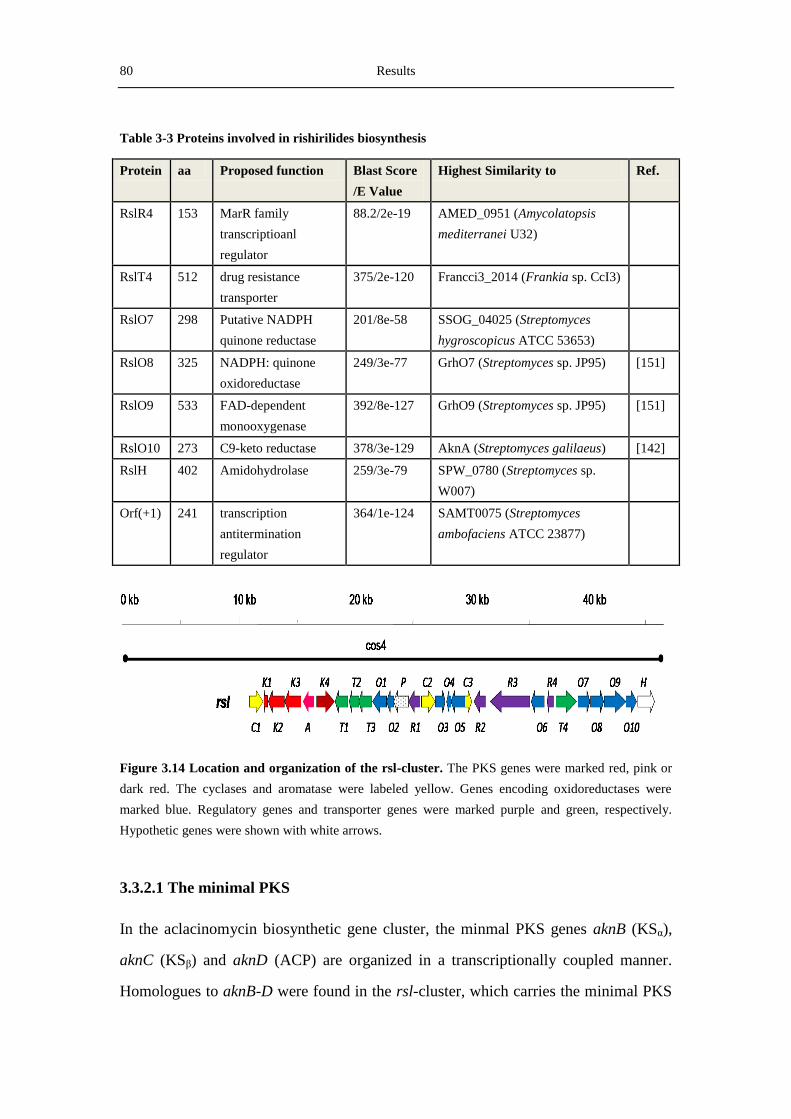
80 Results
Figure 3.14 Location and organization of the rsl-cluster. The PKS genes were marked red, pink or
dark red. The cyclases and aromatase were labeled yellow. Genes encoding oxidoreductases were
marked blue. Regulatory genes and transporter genes were marked purple and green, respectively.
Hypothetic genes were shown with white arrows.
3.3.2.1 The minimal PKS
In the aclacinomycin biosynthetic gene cluster, the minmal PKS genes aknB (KSα),
aknC (KSβ) and aknD (ACP) are organized in a transcriptionally coupled manner.
Homologues to aknB-D were found in the rsl-cluster, which carries the minimal PKS
Table 3-3 Proteins involved in rishirilides biosynthesis
Protein aa Proposed function Blast Score
/E Value
Highest Similarity to Ref.
RslR4 153 MarR family
transcriptioanl
regulator
88.2/2e-19 AMED_0951 (Amycolatopsis
mediterranei U32)
RslT4 512 drug resistance
transporter
375/2e-120 Francci3_2014 (Frankia sp. CcI3)
RslO7 298 Putative NADPH
quinone reductase
201/8e-58 SSOG_04025 (Streptomyces
hygroscopicus ATCC 53653)
RslO8 325 NADPH: quinone
oxidoreductase
249/3e-77 GrhO7 (Streptomyces sp. JP95) [151]
RslO9 533 FAD-dependent
monooxygenase
392/8e-127 GrhO9 (Streptomyces sp. JP95) [151]
RslO10 273 C9-keto reductase 378/3e-129 AknA (Streptomyces galilaeus) [142]
RslH 402 Amidohydrolase 259/3e-79 SPW_0780 (Streptomyces sp.
W007)
Orf(+1) 241 transcription
antitermination
regulator
364/1e-124 SAMT0075 (Streptomyces
ambofaciens ATCC 23877)

Results 81
genes in the following order: KSα (rslK3), KSβ (rslK2) and ACP (rslK1) (Figure
3.13). The gene rslK3 encodes a peptide of 422 amino acids with highly conserved
domains for KS and AT. rslK2 encodes a 407-amino acid protein with about 42%
similarity to rslK3, but it lacks the active centers for KS and AT like other KSβ
proteins. rslK1 encodes a 89 amino acid peptide. Sequence analysis by BLASTP
showed that RslK1is an ACP. In rslK2, the highly conserved glutamine residue,
which is important for the polyketide initation is present. Interestingly, rslK3 overlaps
with rslK2 by 3 bp and rslK2 overlaps with rslK1 by 4 bp.
3.3.2.2 Enzymes for the selection and biosynthesis of the starter unit
Like the gene clusters for the biosynthesis of R1128, daunorubicin and aclacinomy-
cins, the rsl-cluster contains two genes encoding an acyltransferase (RslA) and a
starter unit-specific β-ketoacyl:ACP synthase III (RslK4), respectively. RslA consists
of 363 amino acids and show high similarities (63%) to AknF from the aclacinomycin
biosynthetic pathway[142]
and DpsD from the daunorubicin pathway[152]
. RslK4
contains 347 amino acids and shows 55% identity to CosE from the cosmomycin
pathway[143]
and DpsC from the daunorubicin pathway[152]
.Alignment results of RslK4
with other KASIII enzymes showed that RslK4 contains a Cys-His-Asp catalytic
triads (Figure 3.15). rslO3 encodes a peptide similar to a 3-oxoacyl-ACP reductase,
ssfK. SsfK is an enzyme catalyzing the β-ketoreduction of 2-methyl-acetoacetyl-CoA
to form 3-hydroxyl-2-methyl-butyryl-CoA.[148]
Therefore, RslO3 is proposed to
participate in the biosynthesis of the starter unit, converting a 3-oxoacyl-ACP
reductase substrate to its hydroxylation product, which is recognized by the minimal
PKS to start the extension reactions with malonyl-CoA.
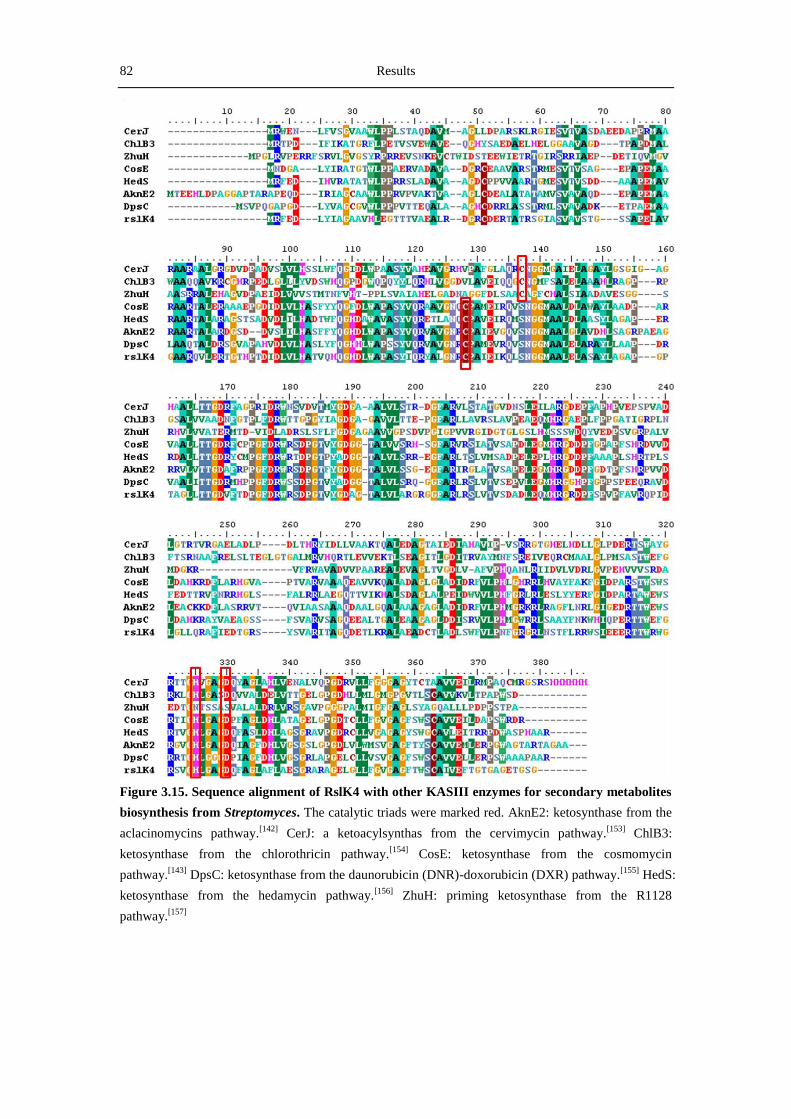
82 Results
Figure 3.15. Sequence alignment of RslK4 with other KASIII enzymes for secondary metabolites
biosynthesis from Streptomyces. The catalytic triads were marked red. AknE2: ketosynthase from the
aclacinomycins pathway.[142]
CerJ: a ketoacylsynthas from the cervimycin pathway.[153]
ChlB3:
ketosynthase from the chlorothricin pathway.[154]
CosE: ketosynthase from the cosmomycin
pathway.[143]
DpsC: ketosynthase from the daunorubicin (DNR)-doxorubicin (DXR) pathway.[155]
HedS:
ketosynthase from the hedamycin pathway.[156]
ZhuH: priming ketosynthase from the R1128
pathway.[157]

Results 83
3.3.2.3 The three aromatases and cyclases
In the rsl-cluster, there are three aromatase/cyclase encoding genes: rslC1, rslC2 and
rslC3. RslC1 encodes 315 amono acids protein which shows high similarity to SnoaE
from the nogalamycin cluster. SnoaE was shown by Metsä-Ketelä et al. to be
responsible for the formation of the first ring of nogalamycin. RslC1 is also similar to
ChaF and RmdK, aromatases from the type II polyketide biosynthetic pathways.
rslC2 encodes a enzyme with 300 amino acids. BLASTP analysis shows that RslC2 is
homologous to cyclases responsible for the second ring closure in polyketide
biosynthesis, such as AlnR from the alnumycin pathway, SsfY2 from the SF2575
pathway and ActIV from the actinorhodin pathway. Therefore, RslC2 is proposed to
be involved in the formation of the second ring in rishirlide biosynthesis. rslC3
encodes a 161 amino acid protein, which shows high similarity to several cyclases,
such as AknE1 from S. galilaeus, CmmX from S. griseus, OxyI from S. rimosus and
SnoO from S. nogalater. (See figure 3.16)
Figure 3.16 Phylogenetic tree analysis of the three cyclases/Aromatases with other cyclases from
aromatic polyketide biosynthesis gene clusters. RslC1, RslC2 and RslC3: the three cyclases from the
rsl-cluster. RdmK: aromatase from the rhodomycin cluster.[142]
SnoaE: An aromatase responsible for
the aromization of the first ring in nogalamycin biosynthesis.[158]
ChaF: a putative aromatase from the
chartreusin cluster.[159]
CmmX: a putative cyclase proposed to be involved in the forth ring closure.[160]
SnoO: a putative cyclase from the nogalamycin cluster. OxyI: a putative cyclase from the
oxytetracycline biosynthetic pathway.[161]
AlnR: cyclase from the alnumycin gene cluster[147]
SsfY2:
second ring cyclase from the SF2575 biosynthetic pathway.[148]
ActIV: cyclase from the actinorhodin
biosynthetic pathway.[162]

84 Results
3.3.2.4 The tailoring genes
In the rsl-cluster, there are ten oxidoreductase-encoding genes. rslO1 encodes a
354-amino acid peptide, which is highly similar (58% identity) to luciferase-like
monooxygenase PyrC from S. pyridomyceticus. The luciferase-like monooxygenases
utilize FMN or F420 as cofactor to catalyze the oxidation of their substrates. The
structures of several luciferase-like monooxygenases have been elucidated. All these
enzymes consist of an (αβ)8 TIM barrel, which is proposed to form the flavin-binding
cave, however, non structure of these enzymes in complex with their coenzyme, FMN
or F420 was obtained. Modeling of RslO1 based on a F420-dependent secondary
alcohol dehydrogenase (Adf) from Methanoculleus thermophilicus by SWISS-
MODEL showed that RslO1 also consists of a TIM barrel (Figure 3.17).
Figure 3.17 Comparison of RslO1 modelling (A) with crystal structure of Adf from Methano-
culleus thermophilicus (B)[163]
. Modelling of RslO1 was perfomed based on the crystal structure of a
monooxygenase (Pdb ID: 2I7G) with SWISS-MODEL. Adf is a F420-dependent secondary alcohol
dehydrogenase.[164]
Ribbon diagram of the Adf dimer shows the TIM barrel architecture of the
monomer (B). In the modeling of RslO1, the TIM barrel can also be seen (A).
RslO2 is a putative flavin reductase. The close localization of rslO1 and rslO2 might
imply that RslO1/RslO2 is a two-component flavin-dependent monooxygenase, and
that RslO1 possibly utilizes FMN as cofactor. The protein encoded by rslO4 is highly
similar (45% identity, 56% similarity) to SnoaB, an anthrone monooxygenase from
the nogalamycin pathway. SnoaB catalyzes the conversion of 12-deoxy-nogalonic
acid to nogalonic acid in the biosynthesis of aromatic polyketide nogalamycin in S.

Results 85
nogalater (Figure 3.18).[165]
Beside SnoaB, RslO4 also has homologues in
biosynthetic pathways of other aromatic polyketides, such as AknX in aclacinomycin
biosynthesis,[116]
DnrG/DauG in daunomycin biosynthesis[166]
and MsnO6 in
mensacarcin biosynthesis.[167]
Therefore, RslO4 is proposed to be responsible for the
formation of the anthracene quinone intermediate in the biosynthesis of rishirilides.
RslO5 is a putative NADH:flavin oxidoreductase, an enzyme that catalyzes the
reduction of FMN by NADH to give NAD and FMNH2. Proteins of the NADH:flavin
oxidoreductase family usually act as auxillary enzymes for the supplying reduced
FMN to another enzyme in oxidative chemistry.
rslO6 encodes a luciferase-like monooxygenase, which is homologous to MsnO8
from the mensacarcin biosynthetic cluster.[135]
BLASTP analysis showed that both
RslO6 and MsnO8 contain a COG2141 domain. This domain is characteristic for
coenzyme F420-dependent N5,N10-methylene tetrahydromethanopterin (methylene-
H4MPT) reductase and related flavin-dependent oxidoreductase. The methylene
H4MPT reductase forms a homodimer and catalyzes the oxidative reaction using F420
cofactor.[168]
Analysis of RslO7 by BLASTP didn’t provide clear hint for its function. RslO8 is a
putative NADPH:quinone reductase, resembling several well-studied enzymes such as
ActVI-ORF2 from actinorhodin pathway[169]
and GrhO7 from the Griseorhodin A
biosynthetic pathway.[151]
ActVI-ORF2 recognizes (S)-DNPA (4- hydro-9-hydroxy-
1-methyl-10-oxo-3-H-naphtho-[2,3-c]-pyran-3-(S)-acetic acid) as a substrate for
stereospecific reduction at C-15 (Figure 3.18). Disrupton of ActVI-ORF2 leads to no
actinorhodin production.
RslO9 is a putative FAD-dependent monooxygenase, with 40% identity to GrhO9 in
the biosynthesis of Griseorhodin A[151]
and 39% identity to XanO5 from the
Xantholipin biosynthetic pathway.[170]
XanO5 catalyzes the C4 hydroxylation of the
carbon backbone in the biosynthesis of Xantholipin (Figure 3.18).

86 Results
rslO10 encodes a C-9 keto reductase, like AknA from the aclacinomycin pathway.
AknA is proposed to be responsible for reducing the keto group at C2 of the
polyketide to a hydroxyl group that is removed during closing and aromatizing of the
first ring by ARO. Further mutational analysis will provide further knowledge about
their functions in the biosynthesis of rishirilides. In the rsl-cluster, there is one
hypothetic protein (rslH) which shows low similarity to other known proteins.
Figure 3.18 Functions of the homologues of RslO4, RslO8 and RslO9 in the biosynthesis of
aromatic polyketides.
3.3.2.5 Genes involved in transportation of rishirlides
There are 4 genes involved in the export of rishirilides in the rsl-cluster. The first
three genes (rslT1-T3) encode peptides belong to the ABC-transporter family. The
sequence similarity, conserved domains and hydrophobicity profiles strongly imply
that the rslT1T2T3 gene products are integrated into a typical three-component ABC
transporter system, which consists of the substrate-binding subunit RslT1, the
ATP-binding subunit RslT2 and the membrane fusion protein RslT3. Beside these
three proteins, a putative resistance gene, rslT4, is found in the rsl-cluster. The
deduced protein of rslT4 is homologous to members of the EmrB/QacA family drug

Results 87
resistance transporter. These proteins may be involved in rishirilides export and
self-protection against the products.
3.3.2.6 Genes involved in regulation
Intriguingly, four regulators are present in the rsl-cluster, including two SARP-family
regulators (RslR1 and RslR2), one LAL-family global regulators and one MarR
(multiantibiotic resistance) family transcriptional regulator (RslR4). BLASTP analysis
showed that RslR1 and RslR2 were similar to AknI from the aclacinomycin
pathway[151]
and Lct15 from the lactonamycin pathway[149]
, respectively. Both of
AknI and Lct15 are activator proteins, therefore, RslR1 and RslR2 are also proposed
to be activator for rishirilides biosynthesis. rslR3 encodes a 1077-amino acid peptide
with high similarity (28%) to AfsR from S. coelicolor.[171]
AfsR is a putative
pleitropic regulator protein that participates in the activation of ActII-ORF4 and of
RedD, two pathway-specific regulatory proteins required for the production of the
antibiotics undecylprodigiosin (Red) and actinorhodin (Act), respectively. Therefore,
it is proposed that RslR3 is a pleitropic regulator. RslR4 shows homology to MarR
family transcriptional regulators. In E. coli, the MarR protein represses expression of
the multiple antibiotic resistance operon.[172]
The MarR family consists largely of
negative regulators but also includes a number of positive regulators. No TTA codon
was found in all of these four regulators.
3.4 Inactivation of genes in the rsl-cluster
After the identification of the rsl-cluster by heterologous expression of cos4 in S.
albus, mutational analysis of several key genes in the rishirilide biosynthetic pathway
was carried out in S. albus::cos4 using the Red/ET recombination (See Materials and
Methods 2.7.3).[84]
3.4.1 Inactivation of rslK4- the priming ketosynthase gene
Using the Red/ET recombination, rslK4 in cos4 was inactivated to generate a mutant
cosmid cos4ΔrslK4. Then cos4ΔrslK4 was introduced into S. albus WT by
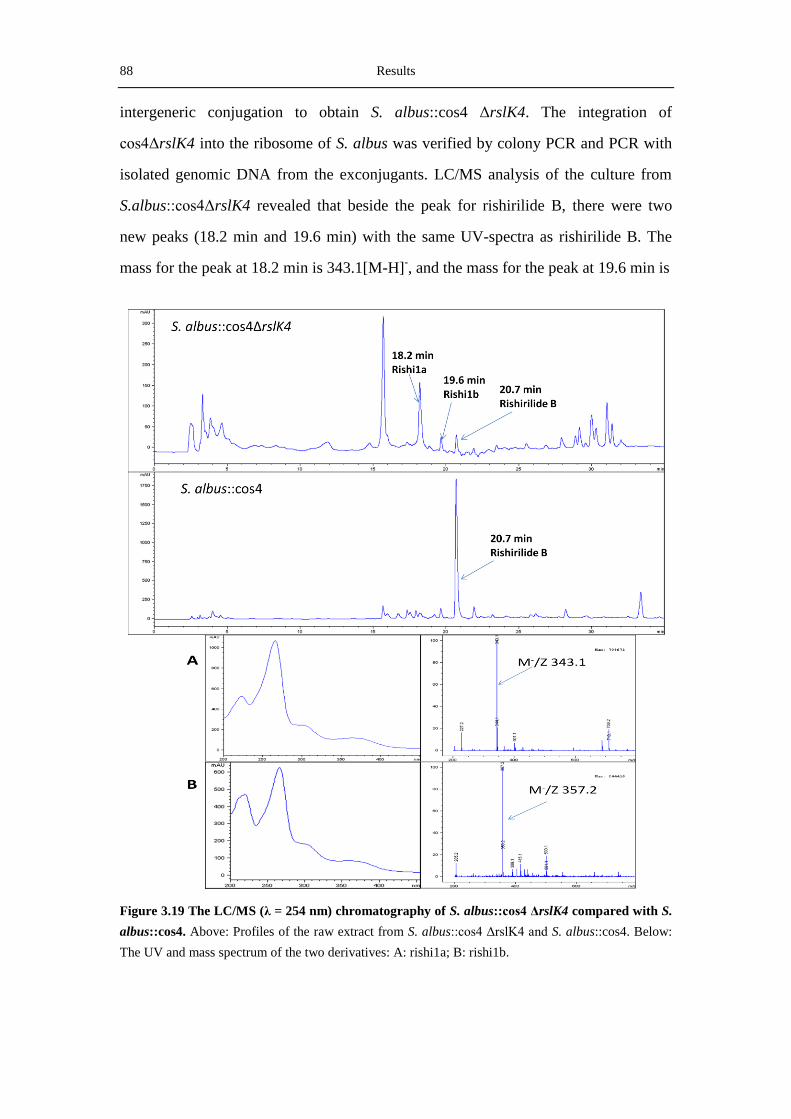
88 Results
intergeneric conjugation to obtain S. albus::cos4 ΔrslK4. The integration of
cos4ΔrslK4 into the ribosome of S. albus was verified by colony PCR and PCR with
isolated genomic DNA from the exconjugants. LC/MS analysis of the culture from
S.albus::cos4ΔrslK4 revealed that beside the peak for rishirilide B, there were two
new peaks (18.2 min and 19.6 min) with the same UV-spectra as rishirilide B. The
mass for the peak at 18.2 min is 343.1[M-H]-, and the mass for the peak at 19.6 min is
Figure 3.19 The LC/MS (λ = 254 nm) chromatography of S. albus::cos4 ΔrslK4 compared with S.
albus::cos4. Above: Profiles of the raw extract from S. albus::cos4 ΔrslK4 and S. albus::cos4. Below:
The UV and mass spectrum of the two derivatives: A: rishi1a; B: rishi1b.

Results 89
Table 3-4 The 1H and
13C NMR data of rishi1a.
Position 1H m d
13C m HMBC correlation with
1 200.0 s
2 3.05 q 6.8 49.3* d C-1, C-3, C-14, C-15
3 84.5** s
4 78.9 s
4a 140.8
5 154.6 s
6 6.89 d 8 111.2 d
7 7.28 dd 8, 8 127.6 d C-5, C-8a,
8 7.43 d 8 121.5 d C-10a
8a 131.3
9 8.39 s 127.9 d C-1, C-8, C-10a
9a 131.4
10 8.43 s 121.1 d C-4, C-5, C-8a, C-9a
10a 128.2
11 1.68 ddd 12.4, 4.4, 4.4 41.0 t
11´ 2.30 ddd 14.4, 4.4, 4.4 C-4
12 1.05 m 17.2 t
12´ 1.55 m
13 0.80 t 7.2 14.7 q C-11, C-12
14 178.0**
15 1.30 d 6.8 10.5 q C-1, C-2, C-3
* overlapped with CH3OH signal **data obtained indirectly from HMBC
Figure 3.20 The structures of rishirilide B, rishi1a and rishi1b. Rishi1a lacks the two methyl
groups in the isobutyryl side chain of rhisirlide B, while rishi1b has one more carbon than rishi1a and
lacks one of the two methyl groups in the side chain of rishirilide B.

90 Results
357.2 [M-H]-. These two compounds were termed rishi1a (the peak at 18.2 min) and
rishi1b (the peak at 19.6 min), respectively. After cultivation in 10 L DNPM-medium
and the following purification, 3.4 mg purified rishi1a was obtained and its structure
was determined by NMR (Figure 3.20). The amount of purified rishi1b was less than
0.6 mg and elucidation of its structure by NMR was not possible. Because of their
similarity in the UV and mass spectrum, the structure of rishi1b was also proposed
(Figure 3.20).
3.4.2 Inactivation of rslO1- a luciferase-like monooxygenase gene
BLASTP analysis showed that rslO1 and rslO2 encode a Flavin-dependent monooxy-
genase and a flavin reductase, repectively. Because of their close location in the rsl-
cluster and complementary properties in flavin conversion, these two proteins were
proposed to form a two-component monooxygenase, like SnoaL2/SnoaW in the
biosynthesis of nogalamycin,[173]
AlnT/AlnH from the alnumycin pathway[174]
and
ActVA-ORF5/ActVB from the actinorhodin pathway.[175]
These two-component
monooxygenases are responsible for the introduction of an oxygen atom into the
aromatic polyketides.
In this thesis, rslO1 was deleted in cos4 to yield cos4ΔrslO1. Then the rslO1 deleted
cosmid was introduced into S. albus to obtain S. albus:: cos4ΔrslO1. After
verification by colony PCR, S. albus::cos4ΔrslO1 was cultivated in DNPM medium
and its raw extract was analyzed by LC/MS (Figure 3.21). In the LC/MS
chromatography of the raw extract from S. albus::cos4ΔrslO1, the peaks at 26.9 min
and at 31.6 min were intriguing. The two peaks were absent in the raw extract of S.
albus::cos4 and showed similar mass spectra to rishirilide B. According to the time of
purification, these two compounds were termed rishi2a (31.6 min) and rishi2b (26.9
min), respectively.
In order to elucidate the structures of these two compounds, S. albus::cos4ΔrslO1 was
cultured in 6 L DNPM-medium and the raw extracts was purified by the combination

Results 91
Figure 3.21 The LC/MS (λ = 254 nm) chromatography of S. albus::cos4ΔrslO1 compared with S.
albus::cos4. Above: Raw extract from S. albus::cos4 ΔrslO1 and S. albus::cos4. A: The UV and mass
spectrum of rishi2b. B: The UV and mass spectrum of rishi2a. The mass spectra were measured by ESI
in the negative mode.
of solide phase extract (SPE), silica gel column chromatography, preparative TLC and
column chromatography with Sephadex LH20. Finally, 4 mg of rishiri2a and 3.4 mg
of rishi2b were obtained and analyzed by NMR and MS. Their structures were
elucidated based on the NMR data (Table 3-5 and Table 3-6, respectively).
Interestingly, the structures of rishi2a and rishi2b differ significantly from that of
rishirilide B. In rishiri2a and rishi2b, there are only two side chains attached to the
aromatic ring, while in rishirilide B, three groups are connected to the aromatic ring.
The total number of carbon atoms in the two derivatives is the same as that in

92 Results
rishirlide. Therefore, a carbon-carbon rearrangement reaction was proposed to occur
during the biosynthesis of rishirides.
Table 3-5 The 1H and
13C NMR data of Rishi2a
Position 1H ppm J
13C HMBC correlations
1 2,38 s 20,3 with C-2, C-3, C-15
2 145,5
3 7,69 s 122,3 with C-13, C-5, C-4
4 137,4
5 181,5
6 133,1
7 7,85 dd 11.4, 1.8 120,3 with C-11, C-5, C-9
8 7,73 dd 11.4, 11.4 137,7 with C-6, C-10
9 7,32 dd 11.4, 1.8 124,9 with C-7, C-11
10 162,7
11 115,9
12 192,7
13 114,2
14 159,4
15 136,7
16 205,8
17 2,88 m 42,5 with C-16, C-18, C-19
18 1,63 m 32,2 with C-17, C-19, C-20/21
19 1,64 m 27,8
20 0,94 d 7,2 22,5 with C-18, C-19
21 0,94 d 7,2 22,5 with C-18, C-19
OH-C-10 12,02 s withC-9, C-10, C-11
OH-C-14 with C-13, C-14, C-15
Figure 3.22 The structures of rishi2a and rishi2b.

Results 93
Table 3-6 The 1H and
13C NMR data of rishi2b
3.4.3 Inactivation of rslO6- a luciferase-like monooxygenase gene
In this thesis, rslO6 in cos4 was deleted by Red/ET recombination to generate the
mutant cosmid cos4ΔrslO6 and then this cosmid was introduced into S. albus to
obtain S. albus::cos4ΔrslO6. The raw extract of S. albus::cos4ΔrslO6 in the DNPM
medium was analyzed by LC/MS. Unexpectedly, the peak for rishirilide B could also
be seen in the culture from S. albus::cos4ΔrslO6. Actually, the yield of rishirilide B
production in the rslO6 mutant was almost the same as that in S. albus::cos4 (Figure
3.23).
Position 1H ppm J
13C HMBC correlations
1 3.75 s 63.0 with C-2, C-3, C-15
2 141.3
3 7,77 s 122,5 with C-5, C-13, C-15
4 137,8
5 181,0
6 133,4
7 7,85 dd 12.6, 1.2 120,5 with C-5, C-6, C-9
8 7,71 dd 12.6, 12.6 137,8 with C-6, C-10
9 7,33 dd 12.6, 1.8 125.1 with C-7, C-10, C-11
10 162,8
11 115,8
12 192,8
13 115,5
14 160,3
15 135.8
16 206.5*
17 3,00 m 42,4
18 1,62 m 32,4
19 1,64 m 27,8
20 0,93 d 7,2 22,5 with C-18, C-19
21 0,93 d 7,2 22,5 with C-18, C-19
OH-C-10 11,90 s with C-9, C-10, C-11
OH-C-14 12,55 s with C-13, C-14, C-15
* data obtained indirectly from HMBC.
94 Results
Figure 3.23 LC/MS profiles of the raw extracts from S. albus::cos4ΔrslO6 and S. albus::cos4.
λ=254 nm.
3.5 Expression of RslK4 in E. coli and S. lividans
To reveal the mechanism for the selection of the starter unit in rishirilide biosynthesis,
expression and crystallization of the priming ketosynthase, RslK4, was carried out.
For RslK4 expression in E. coli, the 1.3 kb PCR product of rslK4 was inserted into the
expression vector pET-28a(+), with an N-terminal 6×-His tag. Then the plasmid was
transformed into BL21/pLysS. SDS-PAGE results showed that in the lane for
insoluble proteins there was a strong band with the size of 37 kD, but in the lane for
supernatant there was only a very weak band with the size of about 37 kD. Western
blotting and mass spectrum detection of the 37 kD bands in both the soluble and
insoluble fractions showed that the protein in these band were not RslK4. Because
RslK4 was predicted to be insoluble in E. coli,[176]
the rslO4 gene was also cloned into
a high copy number Streptomyces expression vector pUWL-A-ermE* and expressed
in S. lividans 1326, however, no production of the recombinant protein was detected.

Results 95
3.6 Investigation of the rishirilide regulators
Plenty of SARP-encoding genes have been found by sequence analysis of secondary
metabolites biosynthesis gene clusters, and in the majority of cases, these clusters
contain only one SARP-encoding gene. Some clusters, such as the aclacinomycin
cluster,[142]
the griseorhodin biosynthesis cluster[151]
and the pristinamycin gene
cluster and,[177]
harbor more than one SARP-encoding gene. In S. fradiae, the tylosin
biosynthesis gene cluster contains five regulatory genes (tylP, tylQ, tylR, tylS and
tylT).[178]
Among them, two are SARP-encoding genes (tylS and tylR). It was shown
that TylS was essential for tylosin production but TylT was not. In the rishirilide
biosynthesis gene cluster, the relationship among the four regulatory proteins is
intriguing.
3.6.1 Overexpression of the regulatory genes
In order to check the influence of the regulators on the production of rishirilide B, the
four regulatory genes (rslR1-R4) were amplified by PCR, cloned into pUWL-H vector
and overexpressed in S. albus::cos4. The resulting strains were cultivated in 100 mL
HA-medium for 5 days and extracted twice with equal volume of ethyl acetate. Then
the raw extracts were dried and dissolved in 1 mL methanol. Finally 15 μL of each
solution was analyzed by LC/MS (Figure 3.24).
It can be seen from the chromatography that there was no obvious difference in the
production of rishirilide B in the stains that overexpress rslR1 or rslR4, but the yields
of rishirlide B were much higher in the strains that overpress rslR2 or rslR3 than the
strains with rslR1 or rslR4 overexpression. The amount of rishirilide B in the strain
that coexpresses rslR1R2R3 was similar to the strains that ovexpress rslR2 or rslR3.
Interestingly, three new peaks can also be seen in the strains in which rslR2 or rslR3
was overexpressed. All these peaks have similar UV spectrum as rishirilide B. The
peak at 18.3 min ([M-H]- = 387.3) shows the same mass, but different UV spectrum,
as rishirlide A. The peak at 19.7 min and 21.8 min show the M/Z value of 358.2 and

96 Results
386.3, respectively, which are 14 Dalton smaller or bigger than rishirilide B. Because
of their similarities in UV and mass spectrum, these peaks were proposed to be
rishirlide B related compounds.
Figure 3.24 LC/MS profiles of the raw extracts from S. albus::cos4 with the overexpressed
regulators and the UV and mass spectra of the new peaks in figure B and C. λ=254 nm. A:
overpression of rslR1; B: overpression of rslR2; C: overpression of rslR3; D: overpression of rslR1,
rslR2 and rslR3 in one plasmid; E: overpression of rslR4. F: the UV spectra of the peaks at 18.3 min,
19.6 min, 20.7 min (rishirilide B) and 21.8 min in figure B. G: The m/z value of the peak at 18.3 min;
H: The m/z value of the peak at 19.7 min; I: The m/z value of the peak at 20.7 min (rishirilide B); J:
The m/z value of the peak at 21.8 min; All the mass data were measured by ESI in the genative mode.

Results 97
3.6.2 Investigation of the regulatory hierarchy amongst the
regulators
Myronovskyi et al. reported a sensitive reporter system for actinomycetes that is
based on gusA, which encodes the β-glucuronidase enzyme.[88]
Several vectors for
transcriptional and translational fusion were constructed to investigate the regulatory
cascade of the phenalinolactone biosynthetic gene cluster in his paper. In order to
study the relationship amongst the regulators in the rsl-cluster, plasmid pGUS, a
promoter-probe vector based on the integrative plasmid pSET152, was used to
perform the transcriptional fusion analysis of the promoter regions of the regulators
and the gusA gene (Figure 3.25).
Figure 3.25 Schematic for the regulatoion of the promoter region (PR) of gene B by regulator A
using transcriptional fusion analysis. A fragment of about 500 bp unstream the gene B, which
propably contains the promoter of the aim gene (gene B), was amplified by PCR and cloned into the
XbaI + KpnI site of pGUS, to obtain plasmid pGUS-PR-B. On the other hand, the putative regulator
gene (gene A) was amplified and cloned into a replicative plasmid pUWL-H, to generated plasmid
pUWL-H-A. Then these two plasmids were introduced into S. albus wild-type by intergeneric
conjugation. The exconjugants were cultivated on the HA plates for 24 h, then overlaid with 1 mL 1
mM X-Gluc and incubated at 28 ℃ overnight. If regulator A indeed regulates the promoter of gene B,
the gusA gene downstream the promoter region of gene B will be trancripted and the resulting
β-glucuronidase activity will convert the colorless X-Gluc to blue. If A does not regulate the promoter
of gene B, the gusA will not be transcripted, therefore, no blue color can be seen on the HA plate
overlaid with X-Gluc.

98 Results
In order to study the relationship among rslR1-R4, the regulatory genes themselves
and their respective promoter regions were cloned into pUWL-H and into pGUS, to
generate expression plasmids pUWL-H-rslR1, pUWL-H-rslR2, pUWL-H-rslR3,
pUWL-H-rslR4, and transcriptional fusion plasmids pGUS-PR-rslR1, pGUS-PR-
rslR2, pGUS-PR-rslR3, pGUS-PR-rslR4, respectively. Then the pGUS-derived
transcriptional fusion plasmids were firstly introduced into S. albus wild-type by
conjugation. After verification of the integration of these plasmids by colony PCR, the
pGUS-integrated strains were conjugated a second time with the pUWL-H-based
expression plasmids. For negative control, pUWL-H was introduced into S. albus WT
or into S. albus contains gusA-harboring plasmids. Finally 25 different strains were
obtained and each strain contains both one integrative plasmid and one replicative
plasmid. Then the β-glucuronidase activities in these strains were indicated by the
color of the strains after overlaid with X-Gluc (Table 3-7 and Figure 3.26). In attempt to
check the reason behind the very often irreproducibility of rishirlides in S. bottropensis,
the five gusA gene harboring plasmids were also introduced into the native producer of
rishirilides, to study the level of transcription of the regulatory genes in the rsl-cluster.
Table 3-7. The GUS activities of the S. albus strains contain both the pGUS-based transcription
fusion plasmids and the pUWL-H-based plasmids for the expression of the regulatory genes.
GUS activity pUWL-H-rslR1 pUWL-H-rslR2 pUWL-H-rslR3 pUWL-H-rslR4 pUWL-H
PR-rslR1 - - - + -
PR-rslR2 - - + - -
PR-rslR3 + + + + +
PR-rslR4 + + + + +
pGUS - - - - -
It can be seen from the below figure that the promoters for rslR3 and rslR4 were
always active, whether the regulatory genes were expressed or not. In the cases of
risR1 and rslR2, the results are different. The promoter for rslR1 is active only when
rslR4 is expressed, while the promoter for rslR2 is active only when rslR3 is
expressed. These results implied that rslR3 and rslR4 were probably in the higher

Results 99
Figure 3.26 Colonies of S. albus and S. bottropensis strains overlaid with X-Gluc solution. Blue
halos are 5,5’-dibromo-4,4’-dichloro-indigo, formed by the β-glucuronidase activity. Gus means the
pGUS empty vector, PR-1 means strains contain the plasmid harboring the promoter region (PR) of
rslR1. The plate labelled with pUWL-H-rslR1 means all the five S. albus strains on the plate contain
the plasmid pUWL-H- rslR1.
hierarchy of the regulatory cascade, while rslR1 and rslR2, the two SARP regulators,
were in the lower level for the regulation. However, so far it is not clear whether rslR3
and rslR4 are pleiotropic regulators or pathway-specific regulatory proteins. Further
studies with reverse trancription PCR of specific genes and mutanenesis analysis of
these regulatory genes might be possible to elucidate the relationship amongst these
four regulators.


Discussion 101
4. Discussion
4.1 Biosynthesis of ganefromycin
4.1.1 Sequencing and analysis of the ganefromycin biosynthetic
gene cluster
The complete sequence of the putative ganefromycin biosynthetic gene cluster was
obtained by the contig of five sequential cosmids screened from the cosmid library of
S. lydicus ssp. tanzanius. Analysis of the DNA sequence by Softberry and BLASTP
revealed that the putative gan-cluster spans 89,281 bp and consists of 26 ORFs. The
putative functions of proteins encoded by these ORFs were assigned based on their
conserved domains and similarities to other characterized proteins. Disruption of
ganAIV with pKC-GanAIVEP by homologous recombination led to the abolishment
of ganefromycin production, uniequivocally proving that the putative gan-cluster is
responsible for the biosynthesis of ganefromycins.
Interestingly, ganefromycins and kirromycin are similar not only in their chemical
structures, but also in the organization of the modules in their type I PKSs. Based on
the arrangement of the type I PKS modules, a putative pathway for the biosynthesis of
ganefromycin polyketide can be proposed. Alignment analysis of the elongation
factor Tu in the gan-cluster (GanF) with other elfamycin family antibiotic-resistance
determinants showed that GanF contains all the key residues conferring the resistance
to elfamycin family antibiotics, therefore, it was proposed to be responsible for the
self-resistance of S. lydicus to ganefromycins.
The origin of the phenyl acetic acid group attached at C-23 or C-24 of ganefromycins
is also an attractive topic. No possible enzymes for the biosynthesis of phenylacetate
were found in the gan-cluster; therefore it is possibly derived from other pathways.
Because there are three acyltransferases (GanAT1, GanAT2 and GanAT3) in the
gan-cluster and only two ATs in the kirromycin biosynthetic gene cluster, GanAT3,

102 Discussion
which shows no obvious similarity to the two ATs from the kirromycin pathway, was
proposed to be responsible for the selection and transfer of phenylacetate-CoA to
C-23 or C-24 of ganefromycin polyketide skeleton. In order to test this hypothesis,
ganAT3 was cloned and heterologously expressed in S. collinus Tue365, the
kirromycin producer; however, no difference was observed from the raw extract of S.
collinus wild type and S. collinus×ganAT3.
4.1.2 Biosynthesis of the ganefromycin polyketide chain
Analysis of the six type I PKSs SEARCHPKS in the gan-cluster revealed the
existence of 15 continuous modules in these enzymes. By comparing these modules
with the modules in the type I PKSs from kirromycin pathway, a putative pathway for
the biosynthesis of the ganefromycin polyketide chain could be proposed (Figure 4.1).
Although this prediction fits quite well to ganefromycin polyketide chain in the numer
of carbons and carbon-carbon double bonds, some details such as the stereochemistry
of the hydroxyl groups, the selection of the still unknown starter unit and extender
units are still not clear.
After the condensation reactions catalyzed by the six type I PKSs and NRPS/PKS
proteins, the nascent polyketide chain was methylated at C-13 by the O-methyl
transferase GanM, and oxidized by two cytochrome-P450 oxidoreductases (GanO1
and GanO2). GanO1 and GanO2 possess 49% and 41% identities to KirOII and KirOI
from kirromycin pathway, respectively. Based on their putative functions, they were
proposed to be responsible for the formation of the tetrahydrofuran ring[81]
and the
sugar-like C-22/C-26 ring structure. Further experiments by deletion or replacement
of the modules or proteins for the polyketide e biosynthesis might be able to elucidate
the mechanism behind the formation of this polyketide structure

Discussion 103
Figure 4.1 Hypothetical biosynthetic pathway for the ganefromycin polyketide. A:adenylation
domain; ACP: acyl carrier protein; AT: acyltransferase domain; C: condensation domain; DH:
dehydratase domain; ER: enoyl reductase domain; KR: keto reductase domain; KS: keto synthase
doamin; MT: methyl transferase domain; PCP: peptidyl carrier protein; X: domain with unknown
function. The function of the last module in GanAI is not known. Except the modules in GanAI, no AT
domains were found in the the other modules, and the functions of their AT domains were
complemented by the two independent ATs (GanAT1 and GanAT2).

104 Discussion
4.1.3 Biosynthesis and attachment of the three deoxy sugars
There are 7 deoxy sugar biosynthetic genes and two glycosyltransferases in the
gan-cluster, which are responsible for the biosynthesis and the attachment of the two
L-oleandroses (sugar 1 and 3) and one L-cymarose (sugar 2) to the polyketide chain
of ganefromycins. L-oleandrose and L-cymarose are both 2,6-dideoxy-3-O-methyl-
hexoses, differing only in the position of 3-O-methyl. L-oleandrose is a common
deoxy sugar in natural products, such as oleandomyin, avermectin,[179]
while
L-cymarose is not frequently found in bioactive compounds. To our knowledge,
L-cymarose was only found in heliquinomycin, an antibiotic of the rubromycins
family (Figure 4.2).[180]
In the characterization of the sugars in oleandomycin
produced by S. antibioticus, Salas et al. presented the putative pathway from glucose
-1-phosphate to L-oleandrose.[181]
Hutchinson and coworkers also proposed a pathway
for the biosynthesis of L-oleandrose in avermectin.[179]
The difference between the
Hutchinson pathway and the Salas pathway is the step of 4-ketoreduction. GanS3, the
putative 4-ketoreductase in the gan-cluster, showed higher similarity to the
4-ketoreductase (avrE) in the avermection biosynthetic gene cluster. Therefore, the
4-ketoreduction in the biosynthesis of L-oleandrose in ganefromycins was proposed to
Figure 4.2 Structures of Avermectin A1a, Heliquinomycin andOleandomycin.

Discussion 105
occur in the late stage of the sugar biosynthesis, like the biosynthesis of TDP-L-
oleandrose in the avermectin pathway.[179]
Based on the Hutchinson pathway, the
putative pathways for the biosynthesis of L-oleandrose and L-cymarose were
proposed (Figure 4.3). After their biosynthesis, L-oleandrose and L-cymarose were
attached to the ganefromycin polyketide skeleton by the two glycosyltransferases
GanGT1 and GT2. Because there are two L-oleandroses, it could be proposed that one
of these two GTs catalyzes the attachment of the two glycosylation iteratively. Futher
mutagenesis or in vitro assay of these two GTs might be able to elucidate which GT
transfers L-oleandrose to the polyketide chain and catalyzes the iterative glycosylation
reaction.
Figure 4.3 Porposed pathways for the biosynthesis of L-oleandrose and L-cymarose by S. lydicus.
4.2 Heterologous production of rishirilide B in S. albus
Rishirilide A and B were firstly discovery by Fukuyama and coworkers in 1984 from
the culture broth of Streptomyces rishiriensis OFR-1056.[71]
In his PhD thesis, Arnold
Moritz reported the discovery of three CD-active secondary metabolites from the
fermentation broth of Streptomyces. bottropensis (formely termed S. sp. Gö C4/4) in
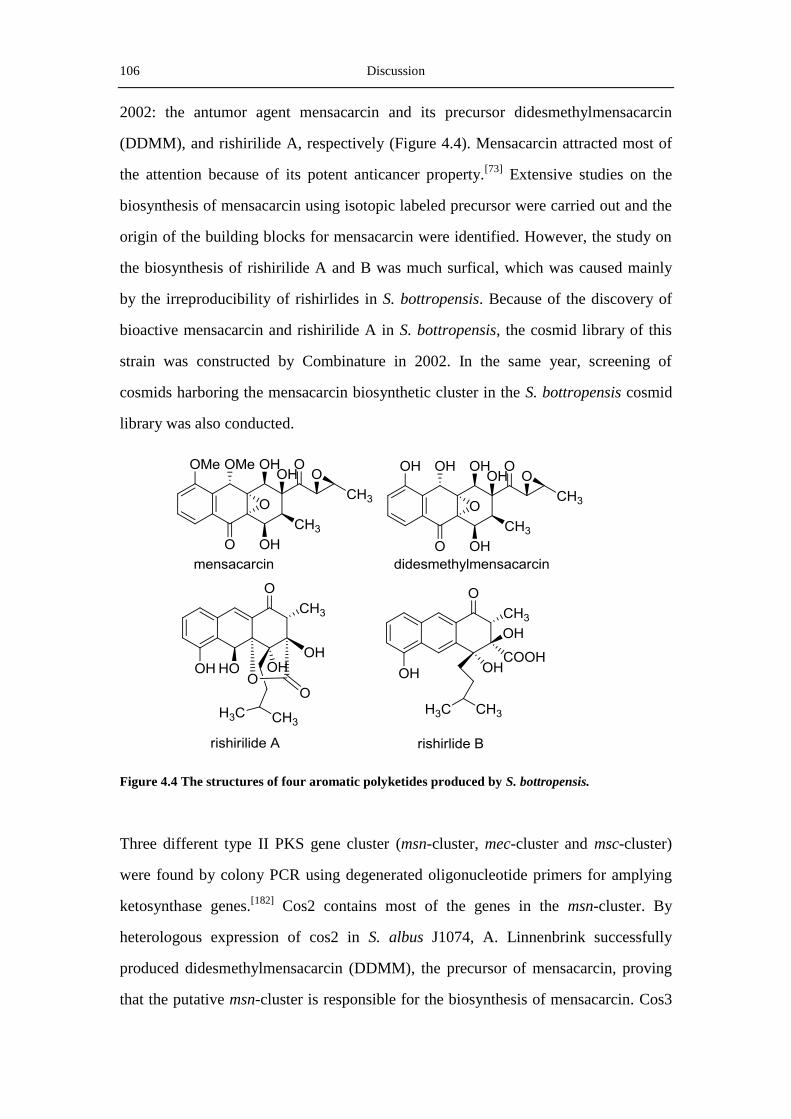
106 Discussion
2002: the antumor agent mensacarcin and its precursor didesmethylmensacarcin
(DDMM), and rishirilide A, respectively (Figure 4.4). Mensacarcin attracted most of
the attention because of its potent anticancer property.[73]
Extensive studies on the
biosynthesis of mensacarcin using isotopic labeled precursor were carried out and the
origin of the building blocks for mensacarcin were identified. However, the study on
the biosynthesis of rishirilide A and B was much surfical, which was caused mainly
by the irreproducibility of rishirlides in S. bottropensis. Because of the discovery of
bioactive mensacarcin and rishirilide A in S. bottropensis, the cosmid library of this
strain was constructed by Combinature in 2002. In the same year, screening of
cosmids harboring the mensacarcin biosynthetic cluster in the S. bottropensis cosmid
library was also conducted.
Figure 4.4 The structures of four aromatic polyketides produced by S. bottropensis.
Three different type II PKS gene cluster (msn-cluster, mec-cluster and msc-cluster)
were found by colony PCR using degenerated oligonucleotide primers for amplying
ketosynthase genes.[182]
Cos2 contains most of the genes in the msn-cluster. By
heterologous expression of cos2 in S. albus J1074, A. Linnenbrink successfully
produced didesmethylmensacarcin (DDMM), the precursor of mensacarcin, proving
that the putative msn-cluster is responsible for the biosynthesis of mensacarcin. Cos3

Discussion 107
consists of a putative spore pigment biosynthetic gene cluster (the mec-cluster), which
shows extremely high similarity (>94%) to spore pigment biosynthetic gene cluster
from Streptomyces scabies 87.22.[144]
However, no possible product of the mec-cluster
was observed from the culture broth of S. albus::cos3. A. Linnenbrink also tried to
identify the product of the msc-cluster which is located in cos4. But no product was
obtained.
In this thesis, cos4 was introduced into S. albus J1074 by intergeneric conjugation.
LC/MS analysis revealed the existence of a product with the same UV and mass
spectra as rishirilide B in the raw extract of S. albus::cos4. Measurement of the
purified product by NMR verified that this product is rishirilide B. In the broth of S.
albus::cos4, another product with the same mass as rishirlide A (m/z 388.3) was also
detected, however, the UV spectrum of the product was different from that of
rishirilide A and identical to rishirilide B. Therefore, it was proposed that this product
is an intermediate compound between rishirilide B and A. Furthermore, it was found
that the yield of rishirilide B produced by S. albus::cos4 in the DNPM-medium was
much more than that in the HA-medium. This might be attributed to the richer
nutrients and stronger buffering ability in the DNPM-medium.
4.3 Overview of key proteins involved in rishirilide
biosynthesis
4.3.1 RslK4 and RslA provide the starter unit
During the BLASTP analysis of the proteins encoded by the genes in the rsl-cluster, a
KASIII type priming ketosynthase RslK4 was discovered. RslK4 shows similarities to
a number of KS III type ketosynthases, such as AknE2 from the aclacinomycins
pathway,[142]
DpsC from the daunorubicin-doxorubicin pathway,[155]
and ZhuH from
the R1128 pathway.[157, 183]
These priming ketosynthases play crucial roles in
providing and selecting the unusual starter units, which are subsequentially
transferred to the minimal PKSs and elongated to form complete polyketide chains.

108 Discussion
Using a cell-free experiment, Mäntsälä and coworker exhibited that DpsC and DpsD
played primary roles in the selection of the starter unit and the initiation of
daunorubicin polyketide biosynthesis.[142]
In the biosynthesis of aclacinomycins, the
gene products of AknE2 and AknF also cooperated to provide the propionyl-CoA
starter unit.[184]
Because of their high similarites to AknE2 and AknF, RslK4 and RslA
are supposed to select the isobutyryl starter unit and then catalyze a condensation with
malonyl-CoA to form the 3-oxoacyl-ACP intermediate.
One of the R1128 compounds, R1128C, possesses the same isobutyryl side chain as
rishirlides (Figure 4.5). Therefore it was supposed that ZhuH and RslK4 select their
starter units in a similar mechanism. Pan et al. characterized the crystal structure of
ZhuH in 2002.[157]
It was observed that in ZhuH a primer unit acetyl-CoA was bound
in a 20 Å-long channel, which placed the acetyl starter unit against the
Cys121-His257-Asn288 catalytic traids. In the channel, the acetyl primer unit is
covalently attached to the CYS121 residue. ZhuH was proposed to be flexible to
alternative CoA-derived primer units, due to the existence of the multiple R1128
polyketide congeners. However, sequence alignment of RslK4 with ZhuH showed
that RslK4 probably does not contain the Cys-His-Asn catalytic triads. A closer
inspection of the results of the amino acid sequence alignment revealed that RslK4
features a Cys-His-Asp motif, like the catalytic triads in the primging ketosynthase
CerJ [153]
(Table 4.1).
The different substrate flexibility of ZhuH and RslK4 might to attribute to the
difference in the catalytic triads. In ZhuH, the Cys-His-Asn catalytic triads located in
the interface of the acyl group binding site and the CoA binding channel, which
determines the type of the priming acyl groups. It is proposed that in RslK4, the
interface between the CoA binding channel and the acyl group site is occupied by the
Cys-His-Asp catalytic triads, leading to the rigider acetyl substrate specificity in this
protein. Elucidation of the crystal structure of RslK4, therefore, would enable the
interpretation of the difference in the substrate specificity between RslK4 and ZhuH.

Discussion 109
Figure 4.5 Structures of aromatic polyketides with unusual starter units.
Table 4.1 Active site comparision of KS III enzymes. The catalytic residues of DpsC, ChlB3 and
RslK4 were tentatively assigned.
4.3.2 The two luciferase-like monooxygenases
In the biosynthesis of rishilide A and B, two luciferase-like monooxygenases (RslO1
and RslO6) are involved. Pair sequence alignment showed that RslO1 and RslO6 have
65% and 51% identies to MsnO2 and MsnO8, two luciferase-like monooxygenases
Protein Important residues
FabH Cys121 His274 Asn288 Ser279
CHS Cys117 His247 Asn326 Ser331
DpsC Cys109 His266 His297 Asp302
ChlB3 Cys113 Val261 His296 Asp301
CerJ Cys116 Val263 His295 Asp300
RslK4 Cys104 Val269 His293 Asp298

110 Discussion
from the mensacarcin pathway, respectively. Luciferase-like monooxygenases utilize
FMN or F420 as coenzyme for the oxidation. F420 is a deazaflavin analog of FMN,
which is distributed sparsely among prokaryotes (Figure 4.6).
Figure 4.6 Structures of FMN and F420.
Alignment of the protein sequences of RslO1 and RslO6 with other luciferase-like
monooxygeanses by Clustal-W (see 2.10) showed that the key Histidine and Arginine
residues which is important for the attachment of the coenzyme were found in both
enzymes (Figure 4.7).[92]
Sequence analysis suggests that RslO1 probably uses FMN
as coenzyme, while RslO6 utilizes F420 as coenzyme, because it belongs to the
COG2141 motif-containing family, members of which were reported to catalyze the
oxidative reactions with the help of F420. The presence of several luciferase-like
monooxygenases in one type II PKS gene cluster is quite rare in streptomycetes. Until
now only a few luciferase-like monooxygenases were reported from the secondary
metabolites biosynthetic pathways, such as MitH from the mitomycin C biosynthetic
pathway,[185]
LmbY from the lincomycin A pathway,[186]
and SnaA/SnaB (or VirN)
from the virginiamycin (pristinamycin IIA) pathway.[187]
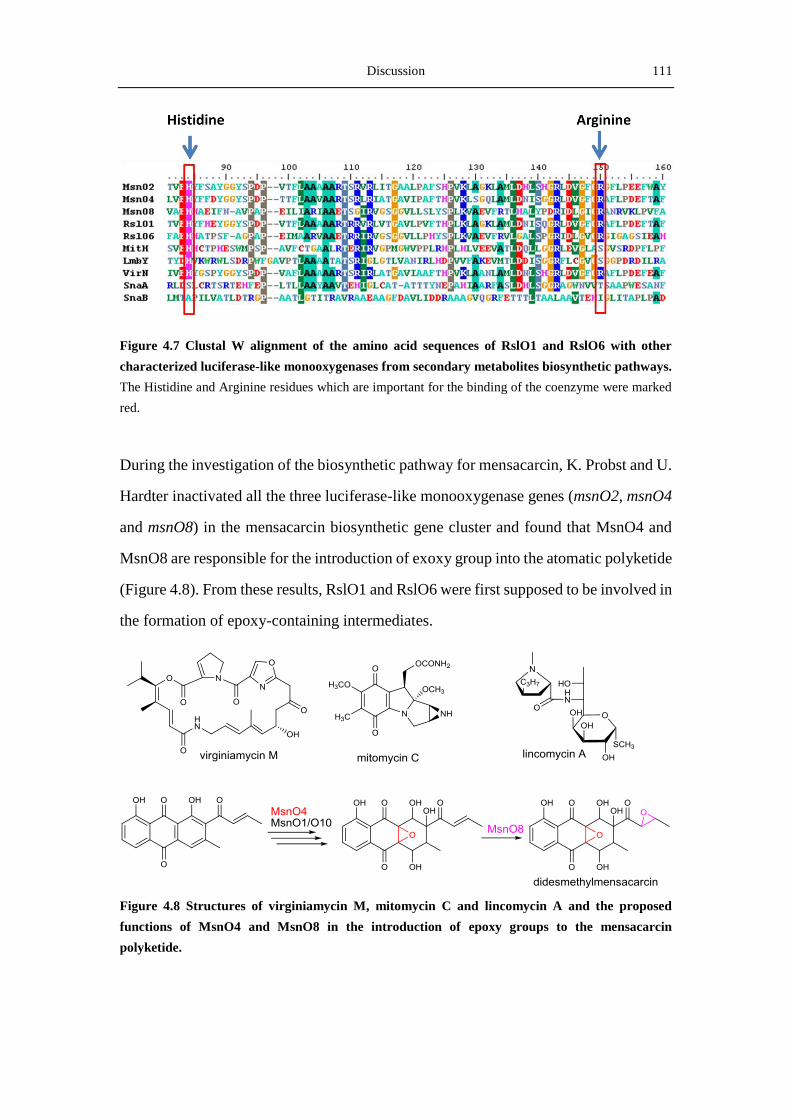
Discussion 111
Figure 4.7 Clustal W alignment of the amino acid sequences of RslO1 and RslO6 with other
characterized luciferase-like monooxygenases from secondary metabolites biosynthetic pathways.
The Histidine and Arginine residues which are important for the binding of the coenzyme were marked
red.
During the investigation of the biosynthetic pathway for mensacarcin, K. Probst and U.
Hardter inactivated all the three luciferase-like monooxygenase genes (msnO2, msnO4
and msnO8) in the mensacarcin biosynthetic gene cluster and found that MsnO4 and
MsnO8 are responsible for the introduction of exoxy group into the atomatic polyketide
(Figure 4.8). From these results, RslO1 and RslO6 were first supposed to be involved in
the formation of epoxy-containing intermediates.
Figure 4.8 Structures of virginiamycin M, mitomycin C and lincomycin A and the proposed
functions of MsnO4 and MsnO8 in the introduction of epoxy groups to the mensacarcin
polyketide.

112 Discussion
4.3.3 Aromatization and cyclization catalyzed by the three
cyclases
After the polyketie chain extension reactions, the three Aromatase/cyclases (RslC1,
RslC2 and RslC3) catalyze the aromatization and cyclization of the nascent
polyketide chain. The homologues of RslO1 (e.g. SnoaE), RslO2 (e.g. ActIV) and
RslO3 (e.g. SnoO) were shown to be responsible for the formation of the first ring, the
Figure 4.9 Hypothetical cyclization functions of the homologues of RslO1, RslO2 and RslO3.
second ring and the fourth ring in aromatic polyketides, respectively (Figure 4.9).
Therefore, based on the functions of their homologues, RslO1, RslO2 and RslO3 were
proposed to catalyze the cyclization of the first, the second and the third ring,
respectively. In order to form the first ring in nogalamycin, the polyketide chain is
first reduced by the ketoreductase SnoaD, to form the C-9 hydroxyl group and to form
the C7-C12 ring closure. Then SnoaE catalyzes the aromatization of the C-9
hydroxyl-containing intermediate to generate the first aromatic ring. In the rsl-cluster,
rslO10 encodes a protein similar to SnoaD, implying the occurrence of C-9
hydroxylation in the biosynthesis of rishirlides. From this fact, it is proposed that
RslO10 reduces the keto- group at C9 to form the hydroxyl group and the cyclized
intermediate. Then RslC1 aromatizes the intermediate to obtain the first aromatic ring
of rishirilides.

Discussion 113
4.3.4 Regulation of rishirlide biosynthesis
Four regulators are involved in the biosynthesis of rishirilides. From the results of
transcriptional analysis of the interaction between the regulatory proteins and the
promoter regions of these regulatory genes in S. albus, transcription of the two SARP-
encoding genes (rslR1 and rslR2) were shown to be inactive, while transcription of
the LAL-family regulatory gene (rslR3) and the Mar family regulatory gene (rslR4)
were active. When rslR3 is expressed, transcription of rslR2 is activated, and rslR1
transcription is activated when rslR4 is expressed. Expression of rslR1 has no effect
on rslR2 trancription, and vice versa. It is not clear whether RslR3 or RslR4 has effect
on the transcription of rslR4 or rslR3, due to the original transcription of these two
genes in S. albus. From the above results, it is proposed that RslR3 and RslR4 occupy
higher hierarchies in the regulation cascade for rishirilides biosynthesis, while RslR1
and RslR2 are in the lower position. Like most other SARP-proteins, RslR1 and
RslR2 were proposed to function by binding to the promoters of other structural and
modification genes.
What’s more, the complicated regulation network in the rsl-cluster might account for
the irreproducibility of rishirilides in S. bottropensis. It was supposed that in S.
bottropensis there are up-level regulators which might inhibit the transcription of
rslR3 and/or rslR4, leading to the inactivation of the promoters of rlsR1 and rslR2,
and the abolishment of rishirilides production in S. bottropensis. When the rsl-cluster
was expressed in S. albus, rslR3 and rslR4 can well transcripted without the up-level
inhibitors, resulting in the steady production of rishirilides B in S. albus. Based on
these facts, a hypothetical regulatory cascade for the biosynthesis of rishirilides can be
proposed (Figure 4.10).

114 Discussion
Figure 4.10 Hypothetical transcriptional regulation network of the rsl-cluster in S. albus and S.
bottropensis.
Nevertheless, it is worth to mention that in a tentative cultivation of S. bottropensis in
the NL111 medium, production of rishirilide B was achieved. So far it is not clear the
reason behind this reproduction. Further comparison of the genome sequence of S.
albus J1074 and S. bottropensis, as well as transcriptional analysis of the genes in the
rsl-cluster might be able to explain the difference in rishirilides production between S.
albus and S. bottropensis.
4.4 Inactivation of rslK4, rslO1 and rslO6
4.4.1 Inactivation of the ketoacylsynthase gene rslK4
The ketoacylsynthases (KASIII) are crucial for providing and selecting the unusual
starter units in the biosynthesis of aromtatic polyketides. In the rsl-cluster, rslK4
encodes a KASIII similar to a series of FabH like proteins, such as DpsC, AknE2 and
BenQ from daunorubicin, aclacinomycin and benastatin biosynthetic pathways,
respectively. Benastatins A and B are inhibitors of glutathione S-transferase produced
by Streptomyces sp. MI384-DF12.[188]
Xu et al. showed that heterologous expression
of the benQ-deleted benastatin biosynthetic gene cluster in S. albus resulted in the
production of novel penta- and hexacyclic benastatin derivatives (Figure 4. 11).[189, 190]

Discussion 115
In this thesis, inactivation of rslK4 in cos4 led to the production of two novel
rishirilide B derivatives rishi1a and rishi1b. This result clearly revealed that RslK4 is
responsible for the providing and selecting of the isobutyryl side chain for rishirilides.
In the absence of RslK4, the minimal PKS recruits alternative branched and straight-
chain acyl starter units from the fatty acid biosynthesis pool, yielding rishirilide B
derivatives with three- and four-carbon side chains. The lacking of these side chains
in the presence of RslK4 indicates that the KASIII functions as a gatekeeper. When
RslK4 is expressed, it preferably selects the isobutyryl group and catalyzes the
condensation between the isobutyryl-CoA and malonyl-ACP, to form branched-chain
3-oxoisohexanoyl-ACP, which is then reduced by a 3-oxoacyl-ACP reductase
(RslO3) and other dehydratase to form isohexanoyl-CoA. Then the isohexanoyl-CoA
is accepted by the elongation enzymes.
Figure 4.11 Strucutres of benastatins A and B and the novel penta- and hexacyclic benastatin
deravatives (E-I) resulted from the BenQ-deleted mutant.[190]
4.4.2 Inactivation of rslO1 and rslO6
In order to test whether RslO1 and RslO6 are responsible for introducing epoxy
groups into the aromatic polyketide, their encoding genes were inactivated with the
help of Red/ET and the cosmid containing the resulting mutants were introduced into

116 Discussion
S. albus. Interestingly, in the rslO1 null mutant, two products with only two side
chains were isolated. Trom the point of polyketide biosynthesis, the origin of two side
chains is much easier to deduce from the PKS machinery, while the biogenesis of
three side chains on the aromatic polyketides is often difficult to predict. The
discovery of the two-side-chain derivatives from the rslO1 mutant unequivocally
deciphered the origin of the three side chains on the rishirilides polyketides, however,
a new question about how the two side chains are converted to three parts is
following. In the biosynthesis of some polyketides, such as mithramycin,[191]
methyltransferases are able to introduce a methyl group to the polyketide backbone,
however, no methyltransferase is found in the rsl-cluster.
After comparison of the difference between rishirilide B and rishiri2a, the isopentyl
side chain in rishirilide B was proposed to be formed via a Favorskii-like oxidative
rearrangement, resembling the reaction catalyzed by EncM in the biosynthesis of
enterocin.[192]
Until now only a few Favorskii-like rearrangements have been
proposed in secondary metabolic pathways based on feeding experiments. Other
examples include the aspyrone polyketide biosynthesis pathway in Aspergillus
melleus[193]
and the dinoflagellate polyether okadaic acid.[194]
EncM is a FAD-
dependent oxygenase which catalyzes the Favorskii rearrangement with the assistance
of EncK, an O-methyltransferase. Sequence alignment of RslO1 and EncM showed
that these two proteins are similar to some extent (Figure 4.12).
According to the mechanism of Favorskii rearrangement and the reaction catalyzed by
EncM, RslO1 was proposed to first introduce an oxygen atom to C-16 of the first-ring
formed rishirilide polyketide to generate a highly active intermediate, in which the
C-18 carbon atom is attached to C-15. Then a Favorskii-like rearrangement is
occurred in this intermediate and the C-18 isopentyl group was transferred to C-15, in
the mean time a carboxyl group and a hydroxyl group were formed at C-17 and C-16,
respectively, because of a Michael addition reaction between the intermediate and
water (Figure 4.13). However, it should be noticed that in a common Favorskii
rearrangement, a tri-membered ring is involved, but in proposed rishirilide

Discussion 117
rearrangement, the C-C breach happens via a four-membered ring. An in vitro assay
containing RslO1, the FMN coenzyme and the proposed substrate might be able to
make clear how the rearrangement is carried out in the biosynthesis of rishirilides.
Figure 4.12 Sequence alignment of RslO1 with EncM. The Histidine residue that was proposed to be
important for FAD binding is marked red.

118 Discussion
Figure 4.13. The proposed Favorskii-like oxidative rearrangement in the biosynthesis of
enterocin (A) and rishirilides (B).

Discussion 119
Unexpectedly, in the rslO6 deletion mutant, rishirlide B was still produced. No
obverious difference was found in the yield of rishirilide B produced by S. albus::cos4
and S. albus::cos4ΔrslO6. This result suggests that RslO6, a hypothetical
F420-utilizing luciferase-like monooxygenase, might be involved in the conversion of
rishirilide B to rishirilide A. Comparison of the composition of these two compounds
shows that rishirilide A harbors one more oxygen atom, implying the function of
RslO6 in introducing an oxygen atom. Because MsnO8, the protein with the highest
similarity to RslO6, introduces an epoxy group to the aromatic polyketide of
mensacarcin,[135]
therefore, RslO6 was proposed to introduce an oxygen atom to
rishirilide B to form an epoxy group. Actually, in the raw xtract of S. albus::cos4, a
peak with the same UV spectrum as rishirilide B and with the mass of 388.3 can be
seen. But the attempt to purify this compound was failed, maybe because of the
instability of the product. Then this putative epoxy group is proposed to be attacked
by the C-16 carboxyl group spontaneously or assisted by the putative hydrolase RslH,
to form a lactone structure (Figure 4.14). The currently ongoing experiments of
overexpressing rslO6 and rslH in S .albus::cos4 might be able to verify this
hypothesis.
Figure 4.14 Hypothetical biosynthetic pathway from rishirilide B to rishirilide A.
4.5 Proposed biosynthetic pathway to rishirilide A
Although rishirlide A and B are not able to be steadily produced by S. bottropensis,
their native producer, heterlogous expression of the rsl-cluster in S. albus led to
constant and high efficient production of rishirilide B. When the three regulators

120 Discussion
RslR1R2R3 were overexpressed in S. albus::cos4, the production of rishirilide B was
increased threefold. Twenty-eight genes involved in rishirilides biosynthesis were
identified. Their putative functions were assigned based on homology with other
genes from aromatic polyketide biosynthesis gene clusters. Among them, three
intriguing genes were inactivated using Red/ET recombination and four novel
rishirilide B derivatives were obtained and structurally characterized. The regulatory
cascade among the four transcriptional activators were investigated using gusA gene
as a reporter. Based on the results from protein sequence comparison, gene
inactivations and transcription fusion analysis, a biosynthetic pathway for rishirilides
can be proposed.
The polyketide backbone of rishirilides is presumably primed by isobutyryl-CoA,
which is probably derived from the branched-chain amind acid L-valine.[195]
Together
with an acyltransferase (RslA), a 3-oxoacyl-ACP reductase (RslO3), as well as an
ACP, a dehydratase and an enoylreductase from the bacterial fatty acid biosynthesis
pathway, RslK4 is predicted to generate an ACP-bound intermediate (isohexonyl-
ACP). This intermediate is then transferred to the KSα/KSβ heterodimer (encoded by
RslK1 and RslK2). Together with an ACP (encoded by RslK3) and a malonyl-CoA:
ACP malonyltransferase (MAT) from the bacterial fatty acid synthase, the PKSs
catalyzed eight extension reactions to generate the full-length polyketide chain. This
reactive polyketide chain is firstly reduced by a C-9 reductase (RslO10) to produce
C-9 hydroxylated intermediate, which undergoes a C7-C12 ring closure and then
aromatized by an aromatase (RslC1) to form the first aromatic ring. Then a Favorskii-
like rearrangement, which is catalyzed by a luciferase-like monooxygenase RslO1 and
a FMN reductase (RslO2), is occurred to yield a polyketide molecule with three side
chains. The second and the third cyclizations leading to the formation of the
anthracene core of rishirilides are presumably catalyzed by RslC2 and RslC3,
respectively. Then the aromatic polyketide is tailored by the various oxidoreductases,
such as RslO4, RslO7, RslO8 and RslO9, to form rishirilide B (Figure 4.15).

Discussion 121
The biosynthetic pathway from rishirilide B to rishirilide A is presumably happened
in two steps. Firstly, RslO6, another luciferase-like monooxygenase, putatively
catalyzes the incorporatin of an oxygen atom to rishirilide B to yield an epoxidized
intermediate. Secondly, the intermediate undergoes a lactonization spontaneously or
assisted by RslH, to generate rishirilide A. The ongoing experiments in feeding
isotopically labeled acetate and in activation of the tailoring genes in the rsl-cluster
would be of great help to decipher the biosynthesis pathway of these two α2-
macroglobulin inhibitors.
Figure 4.15 Proposed biosynthetic pathway for rishirilide A and B.


References 123
5. References
[1] R. Verpoorte, in Comprehensive Natural Products II: Chemistry and Biology, Vol. 3
(Eds.: L. Mander, H. W. Liu), Elsevier Science Ltd., Amsterdam, 1999.
[2] N. R Farnsworth, O. Akerele, A. S. Bingel, D. D. Soejarto, Z. Guo. Medicinal plants
in therapy. Bull. WHO. 1985, 63, 965-981.
[3] A. Raja, P. Prabakarana. Actinomycetes and Drug- A review. Am. J. Drug. Dis. Dev.
2011, 1, 75-84.
[4] S. B. Singh, F. Pelaez, F. Petersen, R. Amstutz, in Prog. Drug Res., Vol. 1 (Ed.: E.
Jucker), Birkhäuser Basel, Basel, 2008, pp. 141-174.
[5] C. W. Murray, D. C. Rees. The rise of fragment-based drug discovery. Nat. Chem.
2009, 1, 187-192.
[6] M. S. Butler. Natural products to drugs: natural product-derived compounds in
clinical trials. Nat. Prod. Rep. 2008, 25, 475-516.
[7] D. J. Newman, G. M. Cragg. Natural Products as Sources of New Drugs over the Last
25 Years. J. Nat. Prod. 2007, 70, 461-477.
[8] D. H. Kwan, F. Schulz. The Stereochemistry of Complex Polyketide Biosynthesis by
Modular Polyketide Synthases. Molecules 2011, 16, 6092-6115.
[9] B. J. Rawlings. Biosynthesis of polyketides. Nat. Prod. Rep. 1997, 14, 523-556.
[10] B. Shen. Biosynthesis of Aromatic Polyketides. Top. Curr. Chem. 2000, 209, 1-51.
[11] C. Hertweck, A. Luzhetskyy, Y. Rebets, A. Bechthold. Type II polyketide synthases:
gaining a deeper insight into enzymatic teamwork. Nat. Prod. Rep. 2007, 24,
162-190.
[12] J. Staunton, K. J. Weissman. Polyketide biosynthesis: a millennium review. Nat. Prod.
Rep. 2001, 18, 380-416.
[13] D. E. Cane, C. T. Walsh, C. Khosla. Harnessing the Biosynthetic Code:
Combinations, Permutations, and Mutations. Science 1998, 282, 63-68.
[14] G. Yadav, R. S. Gokhale, D. Mohanty. Towards Prediction of Metabolic Products of
Polyketide Synthases: An In Silico Analysis. PLoS Comput. Biol. 2009, 5, e1000351.
[15] J. Cortes, S. F. Haydock, G. A. Roberts, D. J. Bevitt, P. F. Leadlay. An unusually
large multifunctional polypeptide in the erythromycin-producing polyketide synthase
of Saccharopolyspora erythraea. Nature 1990, 348, 176-178.
[16] Y. Xue, L. Zhao, H.-w. Liu, D. H. Sherman. A gene cluster for macrolide antibiotic
biosynthesis in Streptomyces venezuelae: Architecture of metabolic diversity. Proc.
Natl. Acad. Sci. U. S. A. 1998, 95, 12111-12116.

124 References
[17] A. R. Gandecha, S. L. Large, E. Cundliffe. Analysis of four tylosin biosynthetic genes
from the tylLM region of the Streptomyces fradiae genome. Gene 1997, 184,
197-203.
[18] E. Rodriguez, H. G. Menzella, H. Gramajo, in Complex Enzymes in Microbial
Natural Product Biosynthesis, Part B: Polyketides, Aminocoumarins and
Carbohydrates, Vol. 459, 2009, pp. 339-365.
[19] S. Wattanachaisaereekul, A. E. Lantz, M. L. Nielsen, J. Nielsen. Production of the
polyketide 6-MSA in yeast engineered for increased malonyl-CoA supply. Metab.
Eng. 2008, 10, 246-254.
[20] J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas, C. R. Hutchinson.
Modulation of Polyketide Synthase Activity by Accessory Proteins During Lovastatin
Biosynthesis. Science 1999, 284, 1368-1372.
[21] A. Sandmann, J. S. Dickschat, H. Jenke-Kodama, B. Kunze, E. Dittmann, R. Müller.
A Type II Polyketide Synthase from the Gram-Negative Bacterium Stigmatella
aurantiaca is Involved in Aurachin Alkaloid Biosynthesis. Angew. Chem. Int. Edit.
2007, 46, 2712-2716.
[22] A. C. Price, C. O. Rock, S. W. White. The 1.3-Angstrom-Resolution Crystal
Structure of β-Ketoacyl-Acyl Carrier Protein Synthase II from Streptococcus
pneumoniae. J. Bacteriol. 2003, 185, 4136-4143.
[23] C. Bisang, P. F. Long, J. Cortes, J. Westcott, J. Crosby, A.-L. Matharu, R. J. Cox, T. J.
Simpson, J. Staunton, P. F. Leadlay. A chain initiation factor common to both
modular and aromatic polyketide synthases. Nature 1999, 401, 502-505.
[24] R. McDaniel, S. Ebert-Khosla, D. A. Hopwood, C. Khosla. Rational design of
aromatic polyketide natural products by recombinant assembly of enzymatic subunits.
Nature 1995, 375, 549-554.
[25] K. K. Burson, C. Khosla. Dissecting the Chain Length Specificity in Bacterial
Aromatic Polyketide Synthases using Chimeric Genes. Tetrahedron 2000, 56,
9401-9408.
[26] A.-L. Matharu, R. J. Cox, J. Crosby, K. J. Byrom, T. J. Simpson. MCAT is not
required for in vitro polyketide synthesis in a minimal actinorhodin polyketide
synthase from Streptomyces coelicolor. Chem. Biol. 1998, 5, 699-711.
[27] A. T. Keatinge-Clay, D. A. Maltby, K. F. Medzihradszky, C. Khosla, R. M. Stroud.
An antibiotic factory caught in action. Nat. Struct. Mol. Biol. 2004, 11, 888-893.
[28] K. Herold, F. A. Gollmick, I. Groth, M. Roth, K.-D. Menzel, U. Möllmann, U. Gräfe,
C. Hertweck. Cervimycin A–D: A Polyketide Glycoside Complex from a Cave
Bacterium Can Defeat Vancomycin Resistance. Chem. -Eur. J. 2005, 11, 5523-5530.

References 125
[29] Y. Shen, P. Yoon, T.-W. Yu, H. G. Floss, D. Hopwood, B. S. Moore. Ectopic
expression of the minimal whiE polyketide synthase generates a library of aromatic
polyketides of diverse sizes and shapes. Proc. Natl. Acad. Sci. U. S. A. 1999, 96,
3622-3627.
[30] H. Petkovic, A. Thamchaipenet, L.-H. Zhou, D. Hranueli, P. Raspor, P. G. Waterman,
I. S. Hunter. Disruption of an Aromatase/Cyclase from the Oxytetracycline Gene
Cluster of Streptomyces rimosus Results in Production of Novel Polyketides with
Shorter Chain Lengths. J. Biol. Chem. 1999, 274, 32829-32834.
[31] B. S. Moore, C. Hertweck. Biosynthesis and attachment of novel bacterial polyketide
synthase starter units. Nat. Prod. Rep. 2002, 19, 70-99.
[32] H. Fu, D. A. Hopwood, C. Khosla. Engineered biosynthesis of novel polyketides:
evidence for temporal, but not regiospecific, control of cyclization of an aromatic
polyketide precursor. Chem. Biol. 1994, 1, 205-210.
[33] B. S. Moore, J. N. Hopke. Discovery of a New Bacterial Polyketide Biosynthetic
Pathway. ChemBioChem 2001, 2, 35-38.
[34] N. Funa, Y. Ohnishi, I. Fujii, M. Shibuya, Y. Ebizuka, S. Horinouchi. A new pathway
for polyketide synthesis in microorganisms. Nature 1999, 400, 897-899.
[35] M. G. Bangera, L. S. Thomashow. Identification and Characterization of a Gene
Cluster for Synthesis of the Polyketide Antibiotic 2,4-Diacetylphloroglucinol from
Pseudomonas fluorescens Q2-87. J. Bacteriol. 1999, 181, 3155-3163.
[36] Y. Seshime, P. R. Juvvadi, K. Kitamoto, Y. Ebizuka, T. Nonaka, I. Fujii. Aspergillus
oryzae type III polyketide synthase CsyA is involved in the biosynthesis of
3,5-dihydroxybenzoic acid. Bioorg. Med. Chem. Lett. 2010, 20, 4785-4788.
[37] U. Rix, C. Fischer, L. L. Remsing, J. Rohr. Modification of post-PKS tailoring steps
through combinatorial biosynthesis. Nat. Prod. Rep. 2002, 19, 542-580.
[38] C. Olano, C. Mendez, J. A. Salas. Post-PKS tailoring steps in natural
product-producing actinomycetes from the perspective of combinatorial biosynthesis.
Nat. Prod. Rep. 2010, 27, 571-616.
[39] C. T. Walsh, Y. C. J. Chen. Enzymic Baeyer–Villiger Oxidations by
Flavin-Dependent Monooxygenases. Angew. Chem. Int. Edit. 1988, 27, 333-343.
[40] W. J. H. van Berkel, N. M. Kamerbeek, M. W. Fraaije. Flavoprotein
monooxygenases, a diverse class of oxidative biocatalysts. J. Biotech. 2006, 124,
670-689.
[41] S. Fetzner, R. Steiner. Cofactor-independent oxidases and oxygenases. Appl.
Microbiol. Biotech. 2010, 86, 791-804.

126 References
[42] B. Shen, C. R. Hutchinson. Tetracenomycin F1 monooxygenase: Oxidation of a
naphthacenone to a naphthacenequinone in the biosynthesis of tetracenomycin C in
Streptomyces glaucescens. Biochemistry 1993, 32, 6656-6663.
[43] S. Fetzner. Oxygenases without requirement for cofactors or metal ions. Appl.
Microbiol. Biotech. 2002, 60, 243-257.
[44] J. L. Caballero, E. Martinez, F. Malpartida, D. A. Hopwood. Organisation and
functions of the actV A region of the actinorhodin biosynthetic gene cluster of
Streptomyces coelicolor. Mol. Gen. Genet. 1991, 230, 401-412.
[45] K. Räty, T. Kunnari, J. Hakala, P. Mäntsälä, K. Ylihonko. A gene cluster from
Streptomyces galilaeus involved in glycosylation of aclarubicin Mol. Gen. Genet.
2000, 264, 164-172.
[46] K. Ylihonko, J. Tuikkanen, S. Jussila, L. Cong, P. Mäntsälä. A gene cluster involved
in nogalamycin biosynthesis from Streptomyces nogalater: sequence analysis and
complementation of early-block mutations in the anthracycline pathway. Mol. Gen.
Genet. 1996, 251, 113-120.
[47] A. H. Westphal, A. Matorin, M. A. Hink, J. W. Borst, W. J. H. van Berkel, A. J. W. G.
Visser. Real-time Enzyme Dynamics Illustrated with Fluorescence Spectroscopy of
p-Hydroxybenzoate Hydroxylase. J. Biol. Chem. 2006, 281, 11074-11081.
[48] Y.-H. Chen, C.-C. Wang, L. Greenwell, U. Rix, D. Hoffmeister, L. C. Vining, J. Rohr,
K.-Q. Yang. Functional Analyses of Oxygenases in Jadomycin Biosynthesis and
Identification of JadH as a Bifunctional Oxygenase/Dehydrase. J. Biol. Chem. 2005,
280, 22508-22514.
[49] B. Entsch, W. J. van Berkel. Structure and mechanism of para-hydroxybenzoate
hydroxylase. FASEB J. 1995, 9, 476-483.
[50] D. E. Torres Pazmino, H. M. Dudek, M. W. Fraaije. Baeyer-Villiger monooxy-
genases: recent advances and future challenges. Cur. Opion. Chem. Biol. 2010, 14,
138-144.
[51] M. P. Beam, M. A. Bosserman, N. Noinaj, M. Wehenkel, J. Rohr. Crystal Structure
of Baeyer-Villiger Monooxygenase MtmOIV, the Key Enzyme of the Mithramycin
Biosynthetic Pathway. Biochemistry 2009, 48, 4476-4487.
[52] U. Rix, L. L. Remsing, D. Hoffmeister, A. Bechthold, J. Rohr. Urdamycin L: A
Novel Metabolic Shunt Product that Provides Evidence for the Role of the urdM
Gene in the Urdamycin A Biosynthetic Pathway of Streptomyces fradiae TÜ 2717.
ChemBioChem 2003, 4, 109-111.
[53] U. Rix, C. Wang, Y. Chen, F. M. Lipata, L. L. Remsing Rix, L. M. Greenwell, L. C.
Vining, K. Yang, J. Rohr. The Oxidative Ring Cleavage in Jadomycin Biosynthesis:
A Multistep Oxygenation Cascade in a Biosynthetic Black Box. ChemBioChem 2005,
6, 838-845.

References 127
[54] T. Liu, C. Fischer, C. Beninga, J. Rohr. Oxidative Rearrangement Processes in the
Biosynthesis of Gilvocarcin V J. Am. Chem. Soc. 2004, 126, 12262-12263.
[55] A. H. Banskota, J. B. McAlpine, D. Sorensen, M. Aouidate, M. Piraee, A.-M. Alarco,
S. Omura, K. Shiomi, C. M. Farnet, E. Zazopoulos. Isolation and Identification of
Three New 5-Alkenyl-3,3(2H)-furanones from Two Streptomyces species using a
Genomic Screening Approach. J. Antibiot. 2006, 59, 168-176.
[56] L. Xiang, J. A. Kalaitzis, B. S. Moore. EncM, a versatile enterocin biosynthetic
enzyme involved in Favorskii oxidative rearrangement, aldol condensation, and
heterocycle-forming reactions. Proc. Natl. Acad. Sci. U. S. A. 2004, 101,
15609-15614.
[57] J. Piel, K. Hoang, B. S. Moore. Natural Metabolic Diversity Encoded by the
Enterocin Biosynthesis Gene Cluster. J. Am. Chem. Soc. 2000, 122, 5415-5416.
[58] L. M. Podust, H. Bach, Y. C. Kim, D. C. Lamb, M. Arase, D. H. Sherman, S. L.
Kelly, M. R. Waterman. Comparison of the 1.85 Å structure of CYP154A1 from
Streptomyces coelicolor A3(2) with the closely related CYP154C1 and CYPs from
antibiotic biosynthetic pathways Protein Sci. 2004, 13, 255-268.
[59] Y. J. Yoon, B. J. Beck, B. S. Kim, H.-Y. Kang, K. A. Reynolds, D. H. Sherman.
Generation of Multiple Bioactive Macrolides by Hybrid Modular Polyketide
Synthases in Streptomyces venezuelae. Chem. Biol. 2002, 9, 203-214.
[60] R. J. Walczak, M. L. Dickens, N. D. Priestley, W. R. Strohl. Purification, Properties,
and Characterization of Recombinant Streptomyces sp. Strain C5 DoxA, a
Cytochrome P-450 Catalyzing Multiple Steps in Doxorubicin Biosynthesis. J.
Bacteriol. 1999, 181, 298-304.
[61] P. Spiteller, L. Bai, G. Shang, B. J. Carroll, T.-W. Yu, H. G. Floss. The
Post-Polyketide Synthase Modification Steps in the Biosynthesis of the Antitumor
Agent Ansamitocin by Actinosynnema pretiosum. J. Am. Chem. Soc. 2003, 125,
14236-14237.
[62] T.-W. Yu, L. Bai, D. Clade, D. Hoffmann, S. Toelzer, K. Q. Trinh, J. Xu, S. J. Moss,
E. Leistner, H. G. Floss. The biosynthetic gene cluster of the maytansinoid antitumor
agent ansamitocin from Actinosynnema pretiosum. Proc. Natl. Acad. Sci. U. S. A.
2002, 99, 7968-7973.
[63] A. Wietzorrek, M. Bibb. A novel family of proteins that regulates antibiotic
production in streptomycetes appears to contain an OmpR-like DNA-binding fold.
Mol. Microbiol. 1997, 25, 1181-1184.
[64] R. P. Garg, R. J. Parry. Regulation of valanimycin biosynthesis in Streptomyces
viridifaciens: characterization of VlmI as a Streptomyces antibiotic regulatory protein
(SARP). Microbiology 2010, 156, 472-483.

128 References
[65] C. Olano, Felipe Lombó, Carmen Méndez, J. A. Salas. Improving production of
bioactive secondary metabolites in actinomycetes by metabolic engineering. Metab.
Eng. 2008, 10, 281-292.
[66] G. Blanco, E. Fernández, M. J. Fernández, A. F. Braña, U. Weissbach, E. Künzel, J.
Rohr, C. Méndez, J. A. Salas. Characterization of two glycosyltransferases involved
in early glycosylation steps during biosynthesis of the antitumor polyketide
mithramycin by Streptomyces argillaceus. Mol. Gen. Genet. 2000, 262, 991-1000.
[67] A. Tanaka, Y. Takano, Y. Ohnishi, S. Horinouchi. AfsR Recruits RNA Polymerase to
the afsS Promoter: A Model for Transcriptional Activation by SARPs. J. Mol. Biol.
2007, 369, 322-333.
[68] W. M. Maiese, M. P. Lechevalier, H. A. Lechevalier, J. Korshalla, J. Goodman, M. J.
Wildey, N. Kuck, S. D. Conner, M. Greenstein. LL-E19020α and β, animal growth
promoting antibiotics: Taxonomy, Fermentation and Biological activity. J. Antibiot.
1989, 42, 1489-1493.
[69] L. Vogeley, G. J. Palm, J. R. Mesters, R. Hilgenfeld, Vol. 276, 2001, pp.
17149-17155.
[70] G. T. Carter, D. W. Phillipson, R. R. West, D. B. Borders. Chemistry and Structure of
Ganefromycin. J. Org. Chem. 1993, 58, no-6588-6595.
[71] H. Iwaki, Y. Nakayama, M. Takahashi, S. Uetsuki, M. Kido, Y. Fukuyama.
Structures of Rishirilides A and B, alpha2-macroglobulin inhibitors produced by
Streptomyces rishiriensis OFR-1056. J. Antibiot. 1984, 37, 1091-1093.
[72] L. H. Mejorado, T. R. R. Pettus. Total Synthesis of (+)-Rishirilide B:Development
and Application of General Processes for Enantioselective Oxidative Dearomatization
of Resorcinol Derivatives. J. Am. Chem. Soc. 2006, 128, 15625-15631.
[73] M. Arnold,CD-aktive Metabolite aus Streptomyceten sowie Untersuchung der
Biosyntheseleistungen des Mensacarcin-Bildners Streptomyces sp. Gö C4/4.
Dissertation, University of Göttingen (Göttingen), 2002.
[74] D. Hanahan. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol.
1983, 166, 557-580.
[75] D. J. MacNeil, K. M. Gewain, C. L. Ruby, G. Dezeny, P. H. Gibbons, T. MacNeil.
Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis
utilizing a novel integration vector. Gene 1992, 111, 61-68.
[76] E. A. Raleigh, J. Benner, F. Bloom, H. D. Braymer, E. DeCruz, K. Dharmalingam, J.
Heitman, M. Noyer Weidner, A. Piekarowicz, P. L. Kretz. Nomenclature relating to
restriction of modified DNA in Escherichia coli. J. Bacteriol. 1991, 173, 2707-2709.
[77] K. F. Chater, L. C. Wilde. Streptomyces albus G Mutants Defective in the SaZGI
Restriction-midification system. J. Gen. Microbiol. 1980, 116, 323-334.

References 129
[78] D. A. Hopwood, M. J. Bibb, K. F. Chater, T. Kieser, C. J. Bruton, H. M. Kieser, D. J.
Lydiate, C. P. Smith, J. M. Ward, H. and Schrempf. Genetic manipulation of
streptomyces: A laboratory manual., The John Innes Foundation., Norwich, 1985.
[79] Maiese WM, Lechevalier MP, Lechevalier HA, Korshalla J, Goodman J, Wildey MJ,
Kuck N, Conner SD, G. M. LL-E19020 alpha and beta, animal growth promoting
antibiotics: taxonomy, fermentation and biological activity. J. Antibiot. 1989, 42,
1489-1493.
[80] M. Arnold,CD-aktive Metabolite aus Streptomyceten sowie Untersuchung der
Biosyntheseleistungen des Mensacarcin-Bildners Streptomyces sp. Gö C4/4.
Dissertation, Georg-August-Universität zu Göttingen (Göttingen), 2002.
[81] T. Weber, K. J. Laiple, E. K. Pross, A. Textor, S. Grond, K. Welzel, S. Pelzer, A.
Vente, W. Wohlleben. Molecular Analysis of the Kirromycin Biosynthetic Gene
Cluster Revealed ?Alanine as Precursor of the Pyridone Moiety. Chem. Biol. 2008, 15,
175-188.
[82] Y. M. Zhang, J. P. P. Muyrers, J. Rientjes, F. Stewart. Phage annealing proteins
promote oligonucleotide-directed mutagenesis in Escherichia coli and mouse ES cells.
BMC Mol. Biol. 2003, 4, 1-14.
[83] R. Fellay, J. Frey, H. Krisch. Interposon mutagenesis of soil and water bacteria: a
family of DNA fragments designated for in vivo insertional mutagenesis of
Gram-negative bacteria. Gene 1987, 52, 147-154.
[84] B. Gust, G. L. Challis, K. Fowler, T. Kieser, K. F. Chater. PCR-targeted Streptomyces
gene replacement identifies a protein domain needed for biosynthesis of the
sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U. S. A. 2003, 100,
1541-1546.
[85] M. Bierman, R. Logan, K. O'Brien, E. T. Seno, R. N. Rao, B. E. Schoner. Plasmid
cloning vectors for the conjugal transfer of DNA from Escherichia coli to
Streptomyces spp.. Gene 1992, 116, 43-49.
[86] M. Doumith, P. Weingarten, U. F. Wehmeier, K. Salah-Bey, B. Benhamou, C.
Capdevila, J.-M. Michel, W. Piepersberg, M.-C. Raynal. Analysis of genes involved
in 6-deoxyhexose biosynthesis and transfer in Saccharopolyspora erythraea. Mol.
Gen. Genet. 2000, 264, 477-485.
[87] M. Bierman, R. Logan, K. Obrien, E. T. Seno, R. N. Rao, B. E. Schoner. Plasmid
cloning vectors for the conjugal transfer of DNA from Escherichia coli to
Streptomyces spp. Gene 1992, 116, 43-49.
[88] M. Myronovskyi, E. Welle, V. Fedorenko, A. Luzhetskyy. Beta-Glucuronidase as a
Sensitive and Versatile Reporter in Actinomycetes. Appl. Environ. Microbiol. 2011,
77, 5370-5383.

130 References
[89] O. N. Sekurova, T. Brautaset, H. Sletta, S. E. F. Borgos, M. Jakobsen, T. E. Ellingsen,
A. R. Strøm, S. Valla, S. B. Zotchev. In Vivo Analysis of the Regulatory Genes in the
Nystatin Biosynthetic Gene Cluster of Streptomyces noursei ATCC 11455 Reveals
Their Differential Control Over Antibiotic. Biosynthesis J. Bacteriol. 2004, 186,
1345-1354.
[90] C. Yanischperron, J. Vieira, J. Messing. Improved M13 phage cloning vectors and
host strains - nucleotidesequences of the M13 MP18 and PUC19 Vectors. Gene 1985,
33, 103-119.
[91] H. Weiß, Klonierung neuer Glykosyltransferasen aus Aktinomyceten sowie
Charakterisierung zweier Polyketidbiosynthesegencluster aus Streptomyces
aureofaciens Tü117 und Streptomyces lydicus ssp. tanzanius. Dissertation,
Universität Freiburg (Freiburg), 2009.
[92] A. Linnenbrink,Gewinnung glykosylierter Naturstoffe durch Biotransformation von
Saccharothrix espanaensis sowie Charakterisierung dreier Polyketidbiosynthese
gencluster aus dem Mensacarcinproduzent Streptomyces spp. Gö C4/4. Dissertation,
Universität Freiburg (Freiburg), 2009.
[93] H. C. Birnbiom, J. Doly. A rapid alkaline extraction procedure for screening
recombinant plasmid DNA. Nucl. Acids Res. 1979, 7, 1513-1523.
[94] T. Maniatis, E. Fritsch, J. Sambrook. Molecular cloning. A laboratory manual., Cold
Spring Habor, New York, 1989.
[95] S. H. Oh, K. F. Chater. Denaturation of circular or linear DNA facilitates targeted
integrative transformation of Streptomyces coelicolor A3(2): possible relevance to
other organisms. J. Bacteriol. 1997, 179, 122-127.
[96] K. A. Datsenko, B. L. Wanner. One-step inactivation of chromosomal genes in
Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 2000, 97,
6640-6645.
[97] T. P. Korman, J. A. Hill, T. N. Vu, S.-C. Tsai. Structural Analysis of Actinorhodin
Polyketide Ketoreductase:Cofactor Binding and Substrate Specificity. Biochemistry
2004, 43, 14529-14538.
[98] F. Flett, V. Mersinias, C. P. Smith. High efficiency intergeneric conjugal transfer of
plasmid DNA from Escherichia coli to methyl DNA-restricting streptomycetes FEMS
Microbio. Lett. 1997, 155, 223-229.
[99] U. K. Laemmli. Cleavage of Structural Proteins during the Assembly of the Head of
Bacteriophage T4 Nature 1970, 227, 680-685.
[100] F. J. Enguita, J. L. Fuente, J. F. Martín, P. Liras. An inducible expression system of
histidine-tagged proteins in Streptomyces lividans for one-step purification by Ni2+
affinity chromatography FEMS Microbiol. Lett. 1996, 137, 135-140.

References 131
[101] K. Rutherford, J. Parkhill, J. Crook, T. Horsnell, P. Rice, M. A. Rajandream, B.
Barrell. Artemis: sequence visualization and annotation Bioinformatics (Oxford,
England) 2000, 16, 944-945.
[102] J. D. Thompson, D. G. Higgins, T. J. Gibson. CLUSTAL W: improving the
sensitivity of progressive multiple sequence alignment through sequence weighting,
position-specific gap penalties and weight matrix choice Nucl. Acids Res. 1994, 22,
4673-4680.
[103] A. Marchler-Bauer, S. Lu, C. Zheng, S. H. Bryant. CDD: a Conserved Domain
Database for the functional annotation of proteins Nucl. Acids Res. 2011, 39,
D225-D229.
[104] M. Punta, P. C. Coggill, A. Bateman, R. D. Finn. The Pfam protein families database.
Nucl. Acids Res. 2012, 40, D290-D301.
[105] G. Yadav, R. S. Gokhale, D. Mohanty. SEARCHPKS: a program for detection and
analysis of polyketide synthase domains Nucl. Acids Res. 2003, 31, 3654-3658.
[106] W. He, J. Lei, Y. Liu, Y. Wang. The LuxR family members GdmRI and GdmRII are
positive regulators of geldanamycin biosynthesis Streptomyces hygroscopicus17997
Arch. Microbiol. 2008, 189, 501-510.
[107] D. L. Akey, J. D. Kittendorf, J. W. Giraldes, R. A. Fecik, D. H. Sherman, J. L. Smith.
Structural basis for macrolactonization by the pikromycin thioesterase Nat. Chem.
Biol. 2006, 2, 537-542.
[108] L. N. Olsthoorn-Tieleman, R.-J. T. S. Palstra, G. P. van Wezel, M. J. Bibb, C. W. A.
Pleij. Elongation Factor Tu3 (EF-Tu3) from the Kirromycin Producer Streptomyces
ramocissimus Is Resistant to Three Classes of EF-Tu-Specific Inhibitors. J. Bacteriol.
2007, 189, 3581-3590.
[109] S. Omura, H. Ikeda, J. Ishikawa, A. Hanamoto, C. Takahashi, M. Shinose, Y.
Takahashi, H. Horikawa, H. Nakazawa, T. Osonoe, H. Kikuchi, T. Shiba, Y. Sakaki,
M. Hattori. Genome sequence of an industrial microorganism Streptomyces
avermitilis: Deducing the ability of producing secondary metabolites. Proc. Natl.
Acad. Sci. U. S. A. 2001, 98, 12215-12220.
[110] V. Barbe, M. Bouzon, S. Mangenot, B. Badet, J. Poulain, B. Segurens, D. Vallenet, P.
Marlière, J. Weissenbach. Complete Genome Sequence of Streptomyces cattleya
NRRL 8057, a Producer of Antibiotics and Fluorometabolites. J. Bacteriol. 2011, 193,
5055-5056.
[111] J. Ahlert, E. Shepard, N. Lomovskaya, E. Zazopoulos, A. Staffa, B. O. Bachmann, K.
Huang, L. Fonstein, A. Czisny, R. E. Whitwam, C. M. Farnet, J. S. Thorson. The
Calicheamicin Gene Cluster and Its Iterative Type I Enediyne PKS Science 2002, 297,
1173-1176.

132 References
[112] Y. Ogasawara, K. Katayama, A. Minami, M. Otsuka, T. Eguchi, K. Kakinuma.
Cloning, Sequencing, and Functional Analysis of the Biosynthetic Gene Cluster of
Macrolactam Antibiotic Vicenistatin in Streptomyces halstedii. Chem. Biol. 2004, 11,
79-86.
[113] M. Daum, Peintner,I., Linnenbrink,A., Frerich,A., Weber,M.,Paululat,T. and
Bechthold,A. Organisation of the biosynthetic gene cluster and tailoring enzymes in
the biosynthesis of the tetracyclic quinone glycoside antibiotic polyketomycin.
ChemBioChem 2009, 10, 1073-1083.
[114] M. J. Smanski, Z. Yu, J. Casper, S. Lin, R. M. Peterson, Y. Chen, E.
Wendt-Pienkowski, S. R. Rajski, B. Shen. Dedicated ent-kaurene and ent-atiserene
synthases for platensimycin and platencin biosynthesis. Proc. Natl. Acad. Sci. U. S. A.
2011, 108, 13498-13503.
[115] Y. Xue, L. Zhao, H. Liu, D. H. Sherman. A gene cluster for macrolide antibiotic
biosynthesis in Streptomyces venezuelae: Architecture of metabolic diversity. Proc.
Natl. Acad. Sci. U. S. A. 1998, 95, 12111-12116.
[116] J.-y. Chung, I. Fujii, S. Harada, U. Sankawa, Y. Ebizuka. Expression, Purification,
and Characterization of AknX Anthrone Oxygenase, Which Is Involved in
Aklavinone Biosynthesis in Streptomyces galilaeus. J. Bacteriol. 2002, 184,
6115-6122.
[117] A. Ramos, F. Lombó, A. F. Braña, J. R. Rohr, C. Méndez, J. A. Salas. Biosynthesis of
elloramycin in Streptomyces olivaceus requires glycosylation by enzymes encoded
outside the aglycon cluster. Microbiology 2008, 154, 781-788.
[118] S. Torkkell, K. Ylihonko, J. Hakala, M. Skurnik, P. Mäntsälä. Characterization of
Streptomyces nogalater genes encoding enzymes involved in glycosylation steps in
nogalamycin biosynthesis. Mol. Gen. Genet. 1997, 256, 203-209.
[119] K. Machida, A. Arisawa, S. Takeda, T. Tsuchida, Y. Aritoku, M. Yoshida, H. Ikeda.
Organization of the Biosynthetic Gene Cluster for the Polyketide Antitumor
Macrolide, Pladienolide, in Streptomyces platensisMer-11107. Biosci. Biotech.
Biochem. 2008, 72, 1-7.
[120] Zirkle R., Black T.A., Gorlach J., Ligon J.M., M. I. Analysis of a 108-kb region of
the Saccharopolyspora spinosa genome covering the obscurin polyketide synthase
locus. DNA Seq. 2004, 15, 123-134.
[121] A. Rascher, Z. Hu, N. Viswanathan, A. Schirmer, R. Reid, W. C. Nierman, M. Lewis,
C. R. Hutchinson. Cloning and characterization of a gene cluster for geldanamycin
production in Streptomyces hygroscopicus NRRL 3602 FEMS Microbiol. Lett. 2003,
218, 223-230.

References 133
[122] T.-W. Yu, R. Müller, M. Müller, X. Zhang, G. Draeger, C.-G. Kim, E. Leistner, H. G.
Floss. Mutational Analysis and Reconstituted Expression of the Biosynthetic Genes
Involved in the Formation of 3-Amino-5-hydroxybenzoic Acid, the Starter Unit of
Rifamycin Biosynthesis in Amycolatopsis mediterranei S699. J. Biol. Chem. 2001,
276, 12546-12555.
[123] K. Mikulik, E. Zhulanova. Sequencing of the tuf1 Gene and the Phosphorylation
Pattern of EF-Tu1 during Development and Differentiation in Streptomyces collinus
Producing Kirromycin. Biochem. Biophys. Res. 1995, 213, 454-461.
[124] F. Wright, M. J. Bibb. Codon usage in the G+C-rich Streptomyces genome Gene
1992, 113, 55-65.
[125] W. Boos, H. Shuman. Maltose/maltodextrin system of Escherichia coli: transport,
metabolism, and regulation. Microbiol. Mol. Biol. Rev. 1998, 62, 204-229.
[126] J. E. Walker, M. Saraste, M. J. Runswick, N. J. Gay. Distantly related sequences in
the alpha- and beta-subunits of ATP synthase, myosin, kinases and other
ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1,
945-951.
[127] D. J. Wilson, Y. Xue, K. E. Reynolds, D. H. Sherman. Characterization and analysis
of the PikD regulatory factor in the pikromycin biosynthetic pathway of Streptomyces
venezuelae. J. Bacteriol. 2001, 183, 3468-3475.
[128] I. Molnár, J. F. Aparicio, S. F. Haydock, L. E. Khaw, T. Schwecke, A. König, J.
Staunton, P. F. Leadlay. Organisation of the biosynthetic gene cluster for rapamycin
in Streptomyces hygroscopicus: analysis
of genes Xanking the polyketide synthase. Gene 1996, 169, 1-7.
[129] N. Anton, M. V. Mendes, J. F. Martin, J. F. Aparicio. IdentiWcation of PimR as a
positive regulator of pimaricin biosynthesis in Streptomyces natalensis. J. Bacteriol.
2004, 186, 2567-2575.
[130] H. B. Claxton, D. L. Akey, M. K. Silver, S. J. Admiraal, J. L. Smith. Structure and
Functional Analysis of RifR, the Type II Thioesterase from the Rifamycin
Biosynthetic Pathway J. Biol. Chem. 2009, 284, 5021-5029.
[131] C. Glockner, H. Wolf. Mechanism of natural resistance to kirromycin-type antibiotics
in actinomycetes FEMS Microbio. Lett. 1984, 25, 121-124.
[132] C. Cappellano, F. Monti, M. Sosio, S.Donadio, E. Sarubbi. Natural kirromycin
resistance of elongation factor Tu from the kirrothricin producer Streptomyces
cinnamoneus. Microbiology 1997, 143, 617-624.
[133] B. Ostash, R. Makitrinskyy, S. Walker, V. Fedorenko. Identification and
characterization of Streptomyces ghanaensis ATCC14672 integration sites for three
actinophage-based plasmids. Plasmid 2009, 61, 171-175.

134 References
[134] M. Koutloubasi, Stammcharakterisierung von Streptomyces lydicus ssp. Tanzanius
NRRL18036 und Screening nach dem Ganefromycinbiosynthese-gencluster.
Dissertation, Universität Tübingen (Tübingen), 2004.
[135] K. Probst.,Untersuchungen zur Biosynthese von Didesmethylmensacarcin.
Dissertation, Universität Freiburg (Freiburg), 2011.
[136] K. Yamamoto, M. F. Hentemann, J. G. Allen, S. J. Danishefsky. On the Total
Synthesis and Determination of the Absolute Configuration of Rishirilide B:
Exploitation of Subtle Effects to Control the Sense of Cycloaddition of
o-Quinodimethides. Chem. Eur. J. 2003, 9, 3242-3252.
[137] H. Gouda, Y. Kobayashi, T. Yamada, T. Ideguchi, A. Sugawara, T. Hirose, S. mura,
T. Sunazuka, S. Hirono. Three-Dimensional Solution Structure of Bottromycin A2: A
Potent Antibiotic Active against Methicillin-Resistant Staphylococcus aureus and
Vancomycin-Resistant Enterococci. Chem. Pharm. Bull. 2012, 60, 169-171.
[138] J. M. Waisvisz, M. G. van der Hoeven, J. van Peppen, W. C. M. Zwennis.
Bottromycin. I. A New Sulfur-containing Antibiotic J. Am. Chem. Soc. 1957, 79,
4520-4521.
[139] X.-Q. Qi, Q.-L. Sun, L.-P. Bai, J.-J. Shan, Y. Zhang, R. Zhang, Y. Li. Identification
of α-D-glucose-1-phosphate cytidylyltransferase involved in Ebosin biosynthesis of
Streptomyces sp. 139. Appl. Microbiol. Biotech. 2009, 83, 361-368.
[140] F. Choulet, B. Aigle, A. Gallois, S. Mangenot, C. Gerbaud, C. Truong, F.-X. Francou,
C. Fourrier, M. Guérineau, B. Decaris, V. Barbe, J.-L. Pernodet, P. Leblond.
Evolution of the Terminal Regions of the Streptomyces Linear Chromosome. Mol.
Biol. Evol. 2006, 23, 2361-2369.
[141] M. Metsä-Ketelä, K. Palmu, T. Kunnari, K. Ylihonko, P. Mäntsälä. Engineering
Anthracycline Biosynthesis toward Angucyclines. Antimicrob. Agents Chemother.
2003, 47, 1291-1296.
[142] K. Räty, J. Kantola, A. Hautala, J. Hakala, K. Ylihonko, P. Mäntsälä. Cloning and
characterization of Streptomyces galilaeus aclacinomycins polyketide synthase (PKS)
cluster. Gene 2002, 293, 115-122.
[143] L. Garrido, F. Lombó, I. Baig, M. Nur-e-Alam, R. Furlan, C. Borda, A. Braña, C.
Méndez, J. Salas, J. Rohr, G. Padilla. Insights in the glycosylation steps during
biosynthesis of the antitumor anthracycline cosmomycin: characterization of two
glycosyltransferase genes. Appl. Microbiol. Biotech. 2006, 73, 122-131.
[144] D. R. D. Bignell, R. F. Seipke, J. C. Huguet-Tapia, A. H. Chambers, R. J. Parry, R.
Loria. Streptomyces scabies 87-22 Contains a Coronafacic Acid-Like Biosynthetic
Cluster That Contributes to Plant-microbe Interactions. Mol. Plant Microbe In. 2010,
23, 161-175.

References 135
[145] T. Huang, Y. Wang, J. Yin, Y. Du, M. Tao, J. Xu, W. Chen, S. Lin, Z. Deng.
Identification and Characterization of the Pyridomycin Biosynthetic Gene Cluster of
Streptomyces pyridomyceticus NRRL B-2517. J. Biol. Chem. 2011, 286,
20648-20657.
[146] X. Zhang, R. J. Parry. Cloning and Characterization of the Pyrrolomycin Biosynthetic
Gene Clusters from Actinosporangium vitaminophilum ATCC 31673 and
Streptomyces sp. Strain UC 11065. Antimicrob. Agents Chemother. 2007, 51,
946-957.
[147] T. Oja, K. Palmu, H. Lehmussola, O. Leppäranta, K. Hännikäinen, J. Niemi, P.
Mäntsälä, M. Metsä-Ketelä. Characterization of the Alnumycin Gene Cluster Reveals
Unusual Gene Products for Pyran Ring Formation and Dioxan Biosynthesis. CHem.
Biol. 2008, 15, 1046-1057.
[148] L. B. Pickens, W. Kim, P. Wang, H. Zhou, K. Watanabe, S. Gomi, Y. Tang.
Biochemical Analysis of the Biosynthetic Pathway of an Anticancer Tetracycline
SF2575. J. Am. Chem. Soc. 2009, 131, 17677-17689.
[149] X. Zhang, L. B. Alemany, H.-P. Fiedler, M. Goodfellow, R. J. Parry. Biosynthetic
Investigations of Lactonamycin and Lactonamycin Z: Cloning of the Biosynthetic
Gene Clusters and Discovery of an Unusual Starter Unit. Antimicrob. Agents
Chemother. 2008, 52, 574-585.
[150] S. Horinouchi, M. Kito, M. Nishiyama, K. Furuya, S. K. Hong, K. Miyake, T. and
Beppu. Primary structure of AfsR, a global regulatory protein for secondary
metabolite formation in Streptomyces coelicolor A3(2). Gene 1990, 95, 49-56.
[151] A. Li, J. Piel. A Gene Cluster from a Marine Streptomyces Encoding the Biosynthesis
of the Aromatic Spiroketal Polyketide Griseorhodin A. Chem. Biol. 2002, 9,
1017-1026.
[152] V. B. Rajgarhia, N. D. Priestley, W. R. Strohl. The Product of dpsC Confers Starter
Unit Fidelity upon the Daunorubicin Polyketide Synthase of Streptomyces sp. Strain
C5. Metab. Eng. 2001, 3, 49-63.
[153] T. Bretschneider, G. Zocher, M. Unger, K. Scherlach, T. Stehle, C. Hertweck. A
ketosynthase homolog uses malonyl units to form esters in cervimycin biosynthesis.
Nat. Chem. Biol. 2012, 8, 154-161.
[154] X.-Y. Jia, Z.-H. Tian, L. Shao, X.-D. Qu, Q.-F. Zhao, J. Tang, G.-L. Tang, W.
Liu. .Genetic Characterization of the Chlorothricin Gene Cluster as a Model for
Spirotetronate Antibiotic Biosynthesis Chem. Biol. 2006, 13, 575-585.
[155] A. Grimm, K. Madduri, A. Ali, C. R. Hutchinson. Characterization of the
Streptomyces peucetius ATCC 29050 genes encoding doxorubicin polyketide
synthase. Gene 1994, 151, 1-10.

136 References
[156] T. Bililign, C.-G. Hyun, J. S. Williams, A. M. Czisny, J. S. Thorson. The Hedamycin
Locus Implicates a Novel Aromatic PKS Priming Mechanism. Chem. Biol. 2004, 11,
959-969.
[157] H. Pan, S.-c. Tsai, E. S. Meadows, L. J. W. Miercke, A. T. Keatinge-Clay, J.
O'Connell, C. Khosla, R. M. Stroud. Crystal Structure of the Priming β-Ketosynthase
from the R1128 Polyketide Biosynthetic Pathway. Structure 2002, 10, 1559-1568.
[158] M. Metsä-Ketelä, J. Niemi, P. Mäntsälä, G. Schneider, K. Krohn, Vol. 282, Springer
Berlin / Heidelberg, 2008, pp. 101-140.
[159] Z. Xu, K. Jakobi, K. Welzel, C. Hertweck. Biosynthesis of the Antitumor Agent
Chartreusin Involves the Oxidative Rearrangement of an Anthracyclic Polyketide.
Chem. Biol. 2005, 12, 579-588.
[160] N. Menéndez, M. Nur-e-Alam, A. F. Braña, J. Rohr, J. A. Salas, C. Méndez.
Biosynthesis of the Antitumor Chromomycin A3 in Streptomyces griseus: Analysis of
the Gene Cluster and Rational Design of Novel Chromomycin Analogs. Chem. Biol.
2004, 11, 21-32.
[161] W. Zhang, K. Watanabe, C. C. C. Wang, Y. Tang. Investigation of early tailoring
reactions in the oxytetracycline biosynthetic pathway. J. Biol. Chem. 2007, 282,
25717-25725.
[162] R. McDaniel, S. Ebert-Khosla, D. A. Hopwood, C. Khosla. Engineered Biosynthesis
of Novel Polyketides: actVII and actIV Genes Encode Aromatase and Cyclase
Enzymes, Respectively. J. Am. Chem. Soc. 1994, 116, 10855-10859.
[163] S. W. Aufhammer, E. Warkentin, H. Berk, S. Shima, R. K. Thauer, U. Ermler.
Coenzyme Binding in F420-Dependent Secondary Alcohol Dehydrogenase, a
Member of the Bacterial Luciferase Family. Structure 2004, 12, 361-370.
[164] F. Widdel, R. S. Wolfe. Expression of secondary alcohol dehydrogenase in
methanogenic bacteria and purification of the F420-specific enzyme from
Methanogenium thermophilum strain TCI. Arch. Microbiol. 1989, 152, 322-328.
[165] T. Grocholski, H. Koskiniemi, Y. Lindqvist, P. Ma ntsa la , J. Niemi, G. Schneider.
Crystal Structure of the Cofactor-Independent Monooxygenase SnoaB from
Streptomyces nogalater: Implications for the Reaction Mechanism. Biochemistry
2010, 49, 934-944.
[166] V. B. Rajgarhia, W. R. Strohl. Minimal Streptomyces sp. strain C5 daunorubicin
polyketide biosynthesis genes required for aklanonic acid biosynthesis. J. Bacteriol.
1997, 179, 2690-2696.
[167] X. Yan, K. Probst, A. Linnenbrink, M. Arnold, T. Paululat, A. Zeeck, A. Bechthold.
Cloning and Heterologous Expression of Three Type II PKS Gene Clusters from
Streptomyces bottropensis. ChemBioChem 2012, 13, 224-230.

References 137
[168] K. Ma, R. K. Thauer. Purification and properties of N5, N10-methylenetetrahydro
methanopterin reductase from Methanobacterium thermoautotrophicum (strain
Marburg). Eur. J. Biochem. 1990, 191, 187-193.
[169] S. Okamoto, T. Taguchi, K. Ochi, K. Ichinose. Biosynthesis of Actinorhodin and
Related Antibiotics: Discovery of Alternative Routes for Quinone Formation
Encoded in the act Gene Cluster. Chem. Biol. 2009, 16, 226-236.
[170] W. Zhang, L. Wang, L. Kong, T. Wang, Y. Chu, Z. Deng, D. You. Unveiling the
Post-PKS Redox Tailoring Steps in Biosynthesis of the Type II Polyketide Antitumor
Antibiotic Xantholipin. Chem. Biol. 2012, 19, 422-432.
[171] B. Floriano, M. Bibb. afsR is a pleiotropic but conditionally required regulatory gene
for antibiotic production in Streptomyces coelicolor A3(2). Mol. Microbiol. 1996, 21,
385-396.
[172] A. S. Seoane, a. S. B. Levy. Characterization of MarR, the repressor of the multiple
antibiotic resistance (mar) operon in Escherichia coli. J. Bacteriol. 1995, 177,
3414-3419.
[173] S. T. Torkkell, T. K. Kunnari, K. P. Palmu, P. M. Mäntsälä, J. H. Hakala, K. Y.
Ylihonko. The entire nogalamycin biosynthetic gene cluster of Streptomyces
nogalater: characterization of a 20-kb DNA region and generation of hybrid
structures. Mol. Genet. Genomics 2001, 266, 276-288.
[174] T. Oja, K. D. Klika, L. Appassamy, J. Sinkkonen, P. Mäntsälä, J. Niemi, M.
Metsä-Ketelä. Biosynthetic pathway toward carbohydrate-like moieties of
alnumycins contains unusual steps for C-C bond formation and cleavage. Proc. Natl.
Acad. Sci. U. S. A. 2012, 109, 6024-6029.
[175] S. Okamoto, T. Taguchi, K. Ochi, and K. Ichinose. Biosynthesis of actinorhodin and
related antibiotics: discovery of alternative routes for quinone formation encoded in
the act gene cluster. Chem. Biol. 2009, 16, 226-236.
[176] D. L. Wilkinson, R. G. Harrison. Predicting the Solubility of Recombinant Proteins in
Escherichia coli. Nat. Biotech. 1991, 9, 443-448.
[177] M. Folcher, H. Gaillard, L. T. Nguyen, K. T. Nguyen, P. Lacroix, N. Bamas-Jacques,
M. Rinkel, C. J. Thompson. Pleiotropic Functions of a Streptomyces pristinaespiralis
Autoregulator Receptor in Development, Antibiotic Biosynthesis, and Expression of a
Superoxide Dismutase. J. Biol. Chem. 2001, 276, 44297-44306.
[178] N. Bate, A. R. Butler, A. R. Gandecha, E. Cundliffe. Multiple regulatory genes in the
tylosin biosynthetic cluster of Streptomyces fradiae. Chem. Biol. 1999, 6, 617-624.
[179] S.-E. Wohlert, N. Lomovskaya, K. Kulowski, L. Fonstein, J. L. Occi, K. M. Gewain,
D. J. MacNeil, C. R. Hutchinson. Insights about the biosynthesis of the avermectin
deoxysugar L-oleandrose through heterologous expression of Streptomyces
avermitilis deoxysugar genes in Streptomyces lividans. Chem. Biol. 2001, 8, 681-700.

138 References
[180] M. Brasholz, S. Sörgel, C. Azap, H.-U. Reißig. Rubromycins: Structurally Intriguing,
Biologically Valuable, Synthetically Challenging Antitumour Antibiotics. Eur. J. Org.
Chem. 2007, 2007, 3801-3814.
[181] I. Aguirrezabalaga, C. Olano, N. Allende, L. Rodriguez, A. F. Braña, C. Méndez, J. A.
Salas. Identification and Expression of Genes Involved in Biosynthesis of
l-Oleandrose and Its Intermediatel-Olivose in the Oleandomycin Producer
Streptomyces antibioticus. Antimicrob. Agents Chemother. 2000, 44, 1266-1275.
[182] M. Metsä-Ketelä, V. Salo, L. Halo, A. Hautala, J. Hakala, P. Mäntsälä, K. Ylihonko.
An efficient approach for screening minimal PKS genes from Streptomyces. FEMS
Microbio. Lett. 1999, 180, 1-6.
[183] T. Marti, Z. Hu, N. L. Pohl, A. N. Shah, C. Khosla. Cloning, Nucleotide Sequence,
and Heterologous Expression of the Biosynthetic Gene Cluster for R1128, a
Non-steroidal Estrogen Receptor Antagonist. J. Biol. Chem. 2000, 275, 33443-33448.
[184] K. Räty, J. Kantola, A. Hautala, J. Hakala, K. Ylihonko, P. Mäntsäläa. Cloning and
characterization of Streptomyces galilaeus aclacinomycins polyketide synthase (PKS)
cluster Gene 2002, 293, 115-122.
[185] Y. Mao, M. Varoglu, D. H. Sherman. Molecular characterization and analysis of the
biosynthetic gene cluster for the antitumor antibiotic mitomycin C from Streptomyces
lavendulae NRRL 2564. Chem. Biol. 1999, 6, 251-263.
[186] M. Koběrská, J. Kopecký, J. Olšovská, M. Jelínková, D. Ulanova, P. Man, M. Flieger,
J. Janata. Sequence analysis and heterologous expression of the lincomycin
biosynthetic cluster of the type strain Streptomyces lincolnensis ATCC 25466. Folia.
Microbiologica. 2008, 53, 395-401.
[187] V. Blanc, D. Lagneaux, P. Didier, P. Gil, P. Lacroix, J. Crouzet. Cloning and analysis
of structural genes from Streptomyces pristinaespiralis encoding enzymes involved in
the conversion of pristinamycin IIB to pristinamycin IIA (PIIA): PIIA synthase and
NADH:riboflavin 5'-phosphate oxidoreductase. J. Bacteriol. 1995, 177, 5206-5214.
[188] Takaaki Aoyagi, Takayuki Aoyama, Fukiko Kojima, Naoko Matsuda, Masato
Maruyama, Masa Hamada, T .Takeuchi. Benastatins A and B, new inhibitors of
glutathione s-transferase, produced by Streptomyces sp. MI384-DF12. I. taxonomy,
production, isolation, physico-chemical properties and biological activities. J.
Antibiot. 1992, 9, 1385-1390.
[189] Z. Xu, A. Schenk, C. Hertweck. Molecular Analysis of the Benastatin Biosynthetic
Pathway and Genetic Engineering of Altered Fatty Acid-Polyketide Hybrids. J. Am.
Chem. Soc. 2007, 129, 6022-6030.
[190] Z. Xu, M. Metsä-Ketelä, C. Hertweck. Ketosynthase III as a gateway to engineering
the biosynthesis of antitumoral benastatin derivatives. J. Biotech. 2009, 140, 107-113.

References 139
[191] M. J. F. Lozano, L. L. Remsing, L. M. Quirós, A. F. Braña, E. Fernández, C. Sánchez,
C. Méndez, J. Rohr, J. A. Salas. Characterization of Two Polyketide
Methyltransferases Involved in the Biosynthesis of the Antitumor Drug Mithramycin
by Streptomyces argillaceus. J. Biol. Chem. 2000, 275, 3065-3074.
[192] Q. Cheng, L. Xiang, M. Izumikawa, D. Meluzzi, B. S. Moore. Enzymatic total
synthesis of enterocin polyketides. Nat. Chem. Biol. 2007, 3, 557-558.
[193] T. J. Simpson, J. S. E. Holker. The biosynthesis of a pyrone metabolite of Aspergillus
melleus an application of long-range 13
c-13
c coupling constants. Tetrahedron Lett.
1975, 16, 4693-4696.
[194] J. L. C. Wright, T. Hu, J. L. McLachlan, J. Needham, J. A. Walter. Biosynthesis of
DTX-4: Confirmation of a Polyketide Pathway, Proof of a Baeyer-Villiger Oxidation
Step, and Evidence for an Unusual Carbon Deletion Process. J. Am. Chem. Soc. 1996,
118, 8757-8758.
[195] B. S. Moore, C. Hertweck. Biosynthesis and attachment of novel bacterial polyketide
synthase starter unit. Nat. Prod. Rep. 2001, 19, 70-99.


Appendix 141
6 Appendix
6.1 List of abbreviations
°C degree celsius
1H,
1H-COSY
1H,
1H-correlated spectroscopy
2D two dimensional
6×His hexahistidines
aa amino acids
aac(3)IV 3’-N-acetyltransferase
ACP acyl carrier protein
Amp ampicillin
APS ammonium persulphate
AT acyltransferase
ATP adenosine triphosphate
BLAST basic logical alignment search tool
bp base pair
BSA bovine serum albumin
CDCl3: deuterated chloroform
CoA coenzyme A
CYP450 cytochrome P450
Da Dalton
DAD diode array detector
DH dehydrogenase
DMSO dimethyl sulfoxide
DNA deoxyribonucleic acid
dNTP deoxyribonucleoside 5´-triphosphates
dsDNA double-stranded deoxyribonucleic acid

142 Appendix
DTT 1,4-dithiothreitol
E. coli Escherichia coli
EDTA ethylenediamine tetraacetic acid
ER enoylreductase
ESI electrospray ionization
eV electron volt
g gram
h hour
HAc acetic acid
HCl hydrochloric acid
HMBC heteronuclear nultiple-bond correlation
HPLC high performance liquid chromatography
HSQC heteronuclear single-quantum coherence
Hz hertz
IPTG isopropyl-β-thiogalactoside
J coupling constant
k kilo
KAc potassium acetate
kb kilobase
kDa kilodalton
KR ketoreductase
KS ketosynthase
L liter
lacZ β-galactosidase gene
LC-MS liquid chromatography-mass spectrometry
M molar
m milli-

Appendix 143
μ micro-
min minute
m/z mass-to-charge ratio
MW molecular weight
MS mass spectroscopy
n nano
NaAc sodium acetate
NaOH sodium hydroxide
Ni-NTA nickel-nitrilotriacetic acid
NMR nuclear magnetic resonance
ORF open reading frame
oriT origin of transfer
PCR polymerase chain reaction
PCP peptidyl carrier protein
PEG polyethylene glycol
PKS polyketide synthase
PMSF phenylmethylsulfonyl fluoride
RNA ribonuclear acid
RNase ribonuclease
RP reverse phase
rpm rotation per minute
RT room temperature
s second
S. Streptomyces
SDS sodium dodecyl sulphate
SDS-PAGE sodium dodecyl sulphate-polyacrylamide gel electrophoresis
ssDNA single-stranded deoxyribonucleic acid

144 Appendix
TEMED N,N,N´,N´-tetramethylethylenediamine
TES N-Tris-(hydroxymethyl)-methyl-2-aminoethanesulfonic acid
Tris 2-amino-2-(hydroxymethyl)-1,3-propanediol
Tris-maleate Tris-(hydroxymethyl)-aminomethane-maleate
UV ultraviolet
WT wild-type
X-gal 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside
X-Gluc 5-bromo-4-chloro-3-indolyl-β-D-glucuronic acid
cyclohexylammonium salt
6.2 Maps of plasmids
6.2.1 pKCXY01 and pKCXY02
These two plasmids were constructed to facilitate selection of double cross-over in
Streptomyces. The toxic gene CodA and the gusA gene were inserted into pKC1132,
respectively.
pKCXY02
5407 bp
tipA
gusA
aac(3)IV
lacZa
ori(pUC)
oriT
BamHI (7)
Cla I (2300)
Eco RI (31)
Eco RV (27)
Pst I (5400)
Xba I (1)
pKCXY01
4551 bp
ori(pUC18)
aac(3)IV
oriT
CodA(sm)
promoter
Afl II (1449)
Eco RI (1465)
HindIII (1421)
Nsi I (1459)
Spe I (1439)
Bgl II (91)
Bgl II (216)

Appendix 145
6.2.2 pKCganAIVEP
6.2.3 pSOK804, pCDFDuet and pKC1218E
pKCganAIVEP
10146 bpori(pUC18)
aac(3)IV
oriT
fragment of ganAIV
Eco RI (6603)
Pst I (1)
pSOK804
5493 bp
integrase
transcription terminator
attP site
aac3(IV)
oriT
RBS
ColE1ori

146 Appendix
pKC1218E
6121 bp
ermE
oriT aac(3)IV
rep-pUC
SCP2 minimal replicon
pCDFDuet
3781 bp
SmR
lacI
-10
-35CDF
BamHI (107)
Eco RI (113)
HindIII (144)
Nco I (70)
Pst I (136)
Xba I (2367)
Kpn I (353)
Eco RV (320)

Appendix 147
6.2.4 pGUS and its derived plasmids
pGUS-PR-rlsR1
10249 bp
GUS
aac(3)IV
aadA
int
ori (pUC18)
OriT
Xba I (1)
Kpn I (532)
pGUS-PR-rslR2
10225 bp
GUS
aac(3)IV
aadA
int
PR-rslR2
ori (pUC18)
OriT
Xba I (1)
Kpn I (508)
pGUS-PR-rslR3
10185 bp
GUS
aac(3)IV
aadA
int
ori (pUC18)
OriT
Xba I (1)
Kpn I (468)
pGUS
10228 bp
GUS
aac(3)IV
aadA
int
PR-rslR4
ori (pUC18)
OriT
Xba I (1)
Kpn I (511)
pGUS
9737 bp
GUS
aac(3)IV
aadA
int
ori (pUC18)
OriT

148 Appendix
6.2.5 pUWL-H
For the overpression of rlsR1, rslR2, rslR3, rslR1R2R3, and rslR4, the respective PCR
fragments were digested with ClaI+SpeI, and then inserted into pUWL-H.
pUWL-H
7791 bp
rep
ori
hph
'fd ter
ori ColE1
bla
ermE up
oriT
Cla I (7088)
Spe I (7045)

Appendix 149
6.3 NMR and MS spectra of the compounds characterized in
this thesis
Figure 6.1 The 1H and
13C-NMR spectra of the product from S. lydicus ganAIV::pKC-ganAIVEP.
Up: 1H -NMR spectrum. Down:
13C-NMR spectrum.

150 Appendix
Figure 6.2 The 1H and
13C-NMR spectra of rishirilide B isolated from the culture of S. albus. Up:
1H -NMR spectrum. Down:
13C-NMR spectrum.
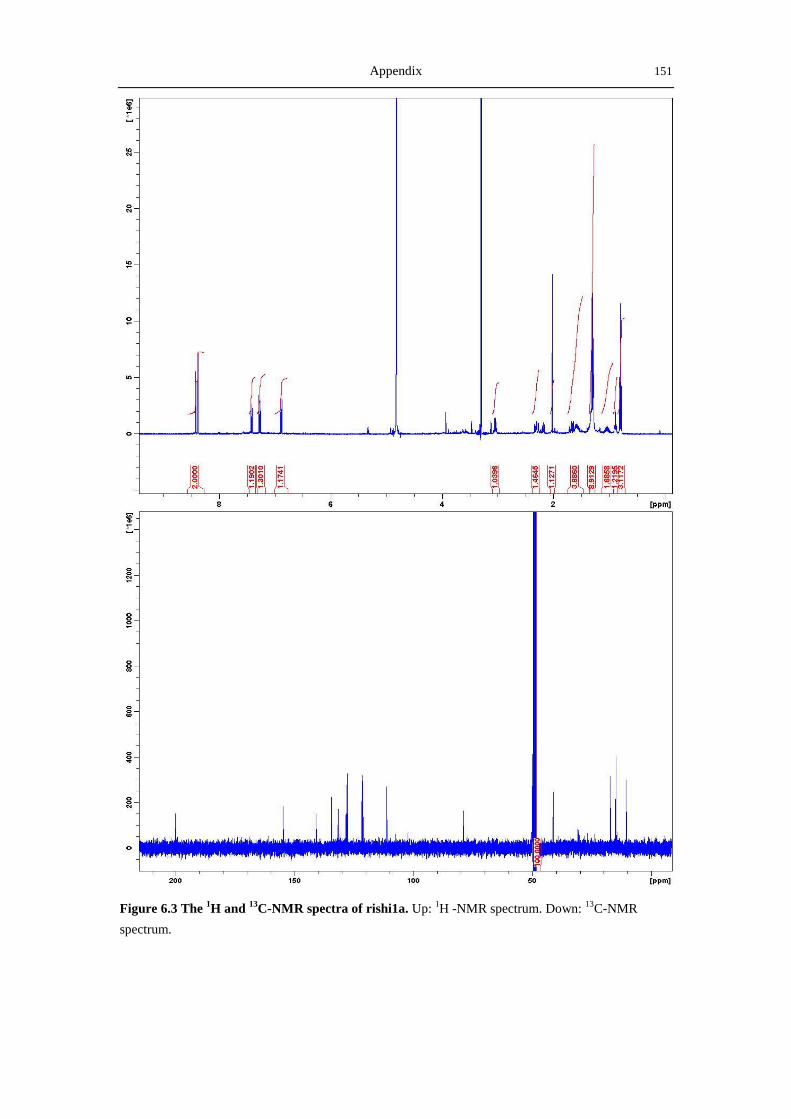
Appendix 151
Figure 6.3 The 1H and
13C-NMR spectra of rishi1a. Up:
1H -NMR spectrum. Down:
13C-NMR
spectrum.
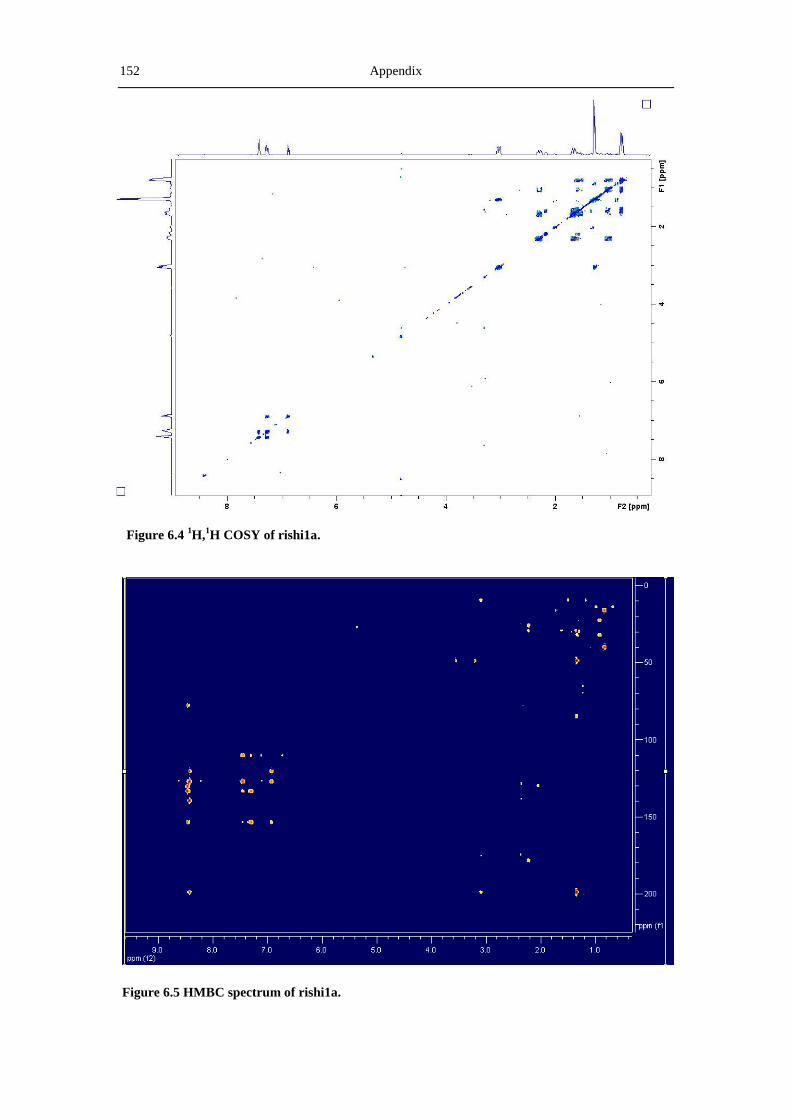
152 Appendix
Figure 6.4 1H,
1H COSY of rishi1a.
Figure 6.5 HMBC spectrum of rishi1a.

Appendix 153
Figure6.6 Mass spectrum of rishi1a.
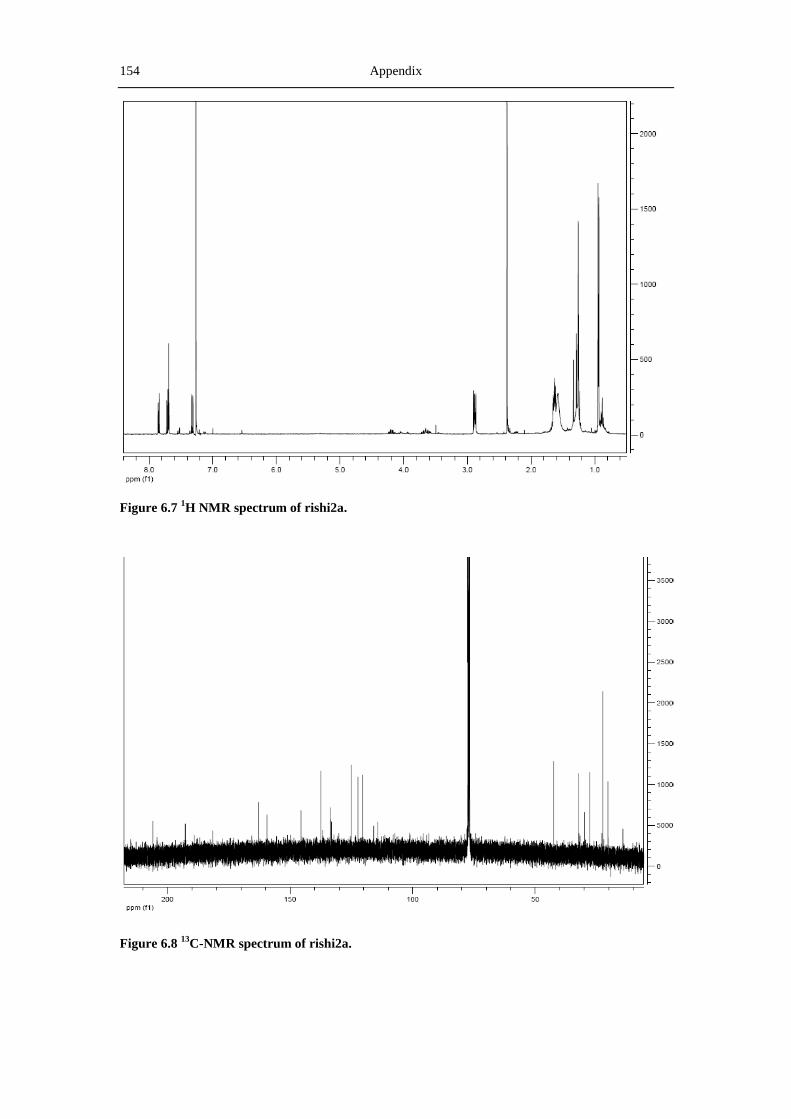
154 Appendix
Figure 6.7 1H NMR spectrum of rishi2a.
Figure 6.8 13
C-NMR spectrum of rishi2a.

Appendix 155
Figure 6.9 1H,
1H COSY of rishi2a.
Figure 6.10 HMBC of rishi2a.

156 Appendix
Figure 6.11 Mass spectrum of rishi2a by the CI method.

Appendix 157
Figure 6.12 1H NMR spectrum of rishi2b.
Figure 6.13 1H,
1H COSY of rishi2b.
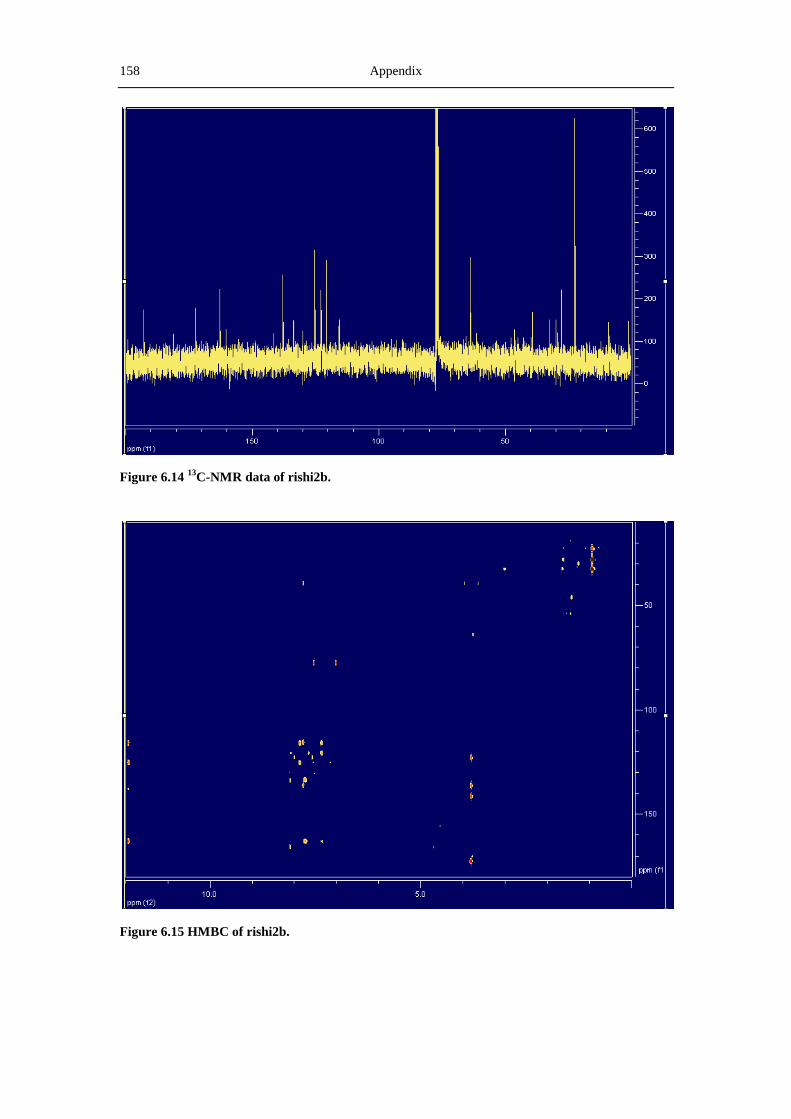
158 Appendix
Figure 6.14 13
C-NMR data of rishi2b.
Figure 6.15 HMBC of rishi2b.

Appendix 159
Figure 6.16. Mass spectrum of rishi2b by the ESI method.


Acknowledgement A
Acknowledgements
This study is performed in the Institute of Pharmaceutical Sciences, Pharmaceutical
Biology and Biotechnology, Faculty of Chemistry, Pharmacy, and Earth Sciences,
University of Freiburg from Septermber 2008 to June 2012.
I want to give my greatest thanks to my supervisor, Prof. Dr. Andreas Bechthold.
Thank him for providing the opportunity to study in this nice group and to live in such
a nice Freiburg city. Thank him for his unlimited support and encouragement, without
which the achievements in this thesis could not be obtained. More importantly, his
guidance and thinking recharged my enthusiasm in scientific research.
I am grateful to Dr. Gabriele Weitnauer and Dr. Andriy Luzhetskyy for their generous
help on the operations of and discussion about my experiments. I learned a lot from
their help.
I express my greatest gratitude to Dr. Tilmann Weber on the analysis of gene
sequence in the ganefromycin project; to Prof. Dr. Alex Zeeck on the precious
discussion about the production of rishirilides, the techniques in the feeding
experiments as well as the help on the manuscript for Chembiochem; to Dr. Thomas
Paululat for the great help on the interpretation of the NMR data for rishirilide B.
I am especially grateful to Prof. Dr. Willi Bannwarth and Mr. Stephan Mundinger for
their generous help on the purification of rishirilide B derivatives. I want to express
my greatest thanks to Prof. Dr. Renato Murillo from the Escuela de Quimica and
Ciprona, Universidad de Costa Rica, Costa Rica for his nice help on interpreting the
structures of the derivatives of rishirilide B.
I would like to thank all my colleges in the lab. The help from Anton, Arne, Holger,
Irene, Johannes, Julia, Katharina, Lutz, Sarah, Simone, Suzan, Tanja, Theresa, Tina,
and Uwe is very important and makes me feel not depressed in the difficult time. I

B Acknowledgement
also offer my warmest gratitude to the three very nice technicians in the lab:
Elizabeth, Marcus, and Miss Weber.
Last but not the least, I want to thank my dear wife, Ying. With her company, nothing
is impossible for me. Thank her for giving birth to my beloved angel, Shuman, before
I finish my study in Freiburg.
.

Curriculum vitae C
Curriculum vitae
Name Xiaohui Yan
Date of Birth 06.02.1979
Place of Birth Hubei Province, P. R. China
1986-1992 Elementary school in Macheng City, Hubei Province, P. R. China
1992-1995 Secondary school in Macheng City, Hubei Province, P. R. China
1995-1998 Huanggang middle school in Huanggang City, Hubei Province, P. R.
China
1998-2002 Department of Chemical Engineering and Process, Tianjin University.
Tianjin City, P. R. China
07.2002 Bachelor of Engineering
2002-2004 Zhenhai Refining & Chemical Company, Sinopec.
Ningbo City, Zhejiang Province, P. R. China
2005-2008 Deparment of Biological Sciences and Technology, Tsinghua University.
Beijing City, P. R. China
Supervisor: Prof. Dr. Guoqiang Chen
07.2008 Master of Science
2008-2012 PhD student at the University of Freiburg.
The faculty of Chemistry, Pharmacy, and Earth Sciences
Institute of Pharmaceutical Sciences, Biology and Biotechnology
Supervisor: Prof. Dr. Andreas Bechthold




















