
A novel angiogenic pathway mediated by non-neuronal nicotinic acetylcholine receptors

1
Inhibitory effects of beta-amyloid on the nicotinic receptors which
stimulate glutamate release in rat hippocampus: the glial contribution
Alessia Salamonea,1, Elisa Murab,1, Stefania Zappettinia, Massimo Grillia, Guendalina Oliveroa,
Stefania Predab, Stefano Govonib, Mario Marchia,c,*
a Section of Pharmacology and Toxicology, Department of Pharmacy, University of Genoa, Genoa,
Italy
b Department of Drug Sciences, Centre of Excellence in Applied Biology, University of Pavia,
Pavia, Italy
c Center of Excellence for Biomedical Research, University of Genoa, Italy
* Corresponding author. Section of Pharmacology and Toxicology, Department of Pharmacy,
University of Genoa, Genoa, Italy. Tel: +39 010 3532657; Fax:+39 010 3993360
E-mail address: [email protected] (M. Marchi)
1 These authors contributed equally to this work.
This work was published by Elsevier in European journal of pharmacology 11/2013; DOI:10.1016/j.ejphar.2013.11.011

2
ABSTRACT
_______________________________________________________________________________
We investigated on the neuronal nicotinic acetylcholine receptor subtypes involved in the
cholinergic control of in vivo hippocampal glutamate (GLU), aspartate (ASP) and inhibitory γ-aminobutyric acid (GABA) overflow. We also investigated on the possible contribution of nicotinic
acetylcholine receptors subtypes present on astrocytes in the regulation of the three neurotransmitter
amino acids overflow using hippocampal gliosomes and on the effects of beta-amyloid (Aβ) 1-40
on the nicotinic control of amino acid neurotransmitter release. Nicotine was able to enhance the in
vivo overflow of the three amino acids being more potent in stimulating GLU overflow. The α7
selective agonist PHA543613 induced an overflow very similar to that of nicotine. The α4β2
selective agonist 5IA85380 was significantly less potent in inducing GLU overflow while the
overflow of ASP and GABA were almost inconsistent. Aβ1-40 inhibited the neurotransmitter
overflow stimulated by PHA543613 but not the one evoked by 5IA85380. In hippocampal
gliosomes nicotine elicited selectively GLU overflow which was also evoked by 5IA85380 and by
the α7 selective agonist choline. Nicotine- and choline-induced glutamate overflow in gliosomes
was inhibited by Aβ1-40. In conclusion nicotine administration in vivo elicits hippocampal GLU
release mostly through α7 nicotinic acetylcholine receptors likely present both on neurons and
astrocytes. Aβ inhibitory effect on the nicotinic-control of GLU release seems to depend primarily
to the inhibition of α7 nicotinic acetylcholine receptors functional responses.
Keywords: β-amyloid; aminoacid release; α7 nicotinic receptors; α4β2 nicotinic receptors;
microdialysis; isolated nerve endings

3
1. Introduction
It is well established that the cholinergic system modulates the function of glutamatergic
system in several brain areas mainly through the activation of neuronal nicotinic acetylcholine
receptor subtypes. These receptors are associated to permeability to Ca2+ thereby facilitating events
such regulation of second messenger cascades (Khiroug et al., 2003), cell survival (Mechawar et al.,
2004) and apoptosis (Berger et al., 1998) as well as the direct stimulation glutamate (GLU) release
(Toth, 1996; Fedele et al., 1998; Marchi et al., 2002). Moreover, specific intracellular mechanisms
have been also shown to be involved in the nicotinic modulation of GLU release (Lambe et al.,
2003; Dickinson et al., 2007; Zappettini et al., 2010). This modulatory effect may also be relevant
given that, in the hippocampus, glutamatergic and cholinergic systems have undoubtedly a
fundamental role in the mechanisms of learning and memory (Picciotto et al., 1995; Levin and
Simon, 1998; Giacobini, 2003; Errico et al., 2011; Parri et al., 2011) and both systems undergo age-
and pathology-associated changes such as attention and memory (Woodruff-Pak and Gould, 2002;
Mattson, 2008)..
Recent studies in vivo and in vitro have shown that beta-amyloid (Aß) was able to modulate
the function of neuronal nicotinic acetylcholine receptor subtypes (Dougherty et al., 2003; Liu and
Wu, 2006; Puzzo et al., 2008; Tong et al., 2011; Lilja et al., 2011; Ni et al., 2013). Moreover, non-
neurotoxic Aβ 1-40 concentrations were able to modulate the nicotine-evoked release of both
excitatory GLU and aspartate (ASP) and inhibitory γ-aminobutyric acid (GABA) and glycine (Mura
et al., 2010, 2012; Zappettini et al., 2012). It is important to recall that GLU is essential to memory
formation so its function might be pivotal to Alzheimer disease (AD) progression (Revett et al.,
2013).
The relevance of the activation of different neuronal nicotinic acetylcholine receptor
subtypes which stimulate in vivo the release of GLU has been so far poorly investigated.
Interestingly, nicotinic acetylcholine receptors are present in several non-neuronal cells including

4
astrocytes (Sharma and Vijayaraghavan, 2001; Lim and Kim, 2003; Patti et al., 2007) and therefore
part of the GLU which is released in vivo might result also from these cells (Santello and Volterra,
2009; Santello et al., 2011). In the present study we have first comparatively investigated, in vivo,
the stimulatory effects of two selective α4β2 and α7 neuronal nicotinic acetylcholine receptor
agonists on the overflow of endogenous GLU, GABA and ASP and the modulatory effects of the
Aβ peptide 1-40 on the function of these neuronal nicotinic acetylcholine receptor subtypes. We
have then studied the effects of selective neuronal nicotinic acetylcholine receptors agonists and Aβ
peptide on amino acid release using hippocampal gliosomes an in vitro preparation originating
from adult astrocytes (Stigliani et al., 2006; Patti et al., 2007; Milanese et al., 2010; Matos et al.,
2012a,b).
From our results we can conclude that the administration in vivo of nicotine and of two
selective α4β2 and α7 nicotinic acetylcholine receptor agonists elicits a significant release of GLU,
ASP and GABA in the rat hippocampus. The inhibitory effect of Aβ on the modulation of the three
amino acid release in vivo seems to depend primarily on the interaction with the α7 nicotinic
acetylcholine receptors. Moreover, both α7 and α4β2 nicotinic acetylcholine receptors are present
on rat hippocampal gliosomes and modulate the release of GLU but not that of ASP and GABA and
Aβ is able to inhibit only the α7 but not the α4β2 nicotinic acetylcholine receptor subtype.

5
2. Materials and methods
2.1. Animals and brain tissue preparation
Adult male Wistar rats (200–250 g, Harlan, Udine) were used for both in vivo experiments
and as brain tissue source for in vitro experiments. Animals were housed at constant temperature
(22 ± 1°C) and relative humidity (50%) under a regular light–dark schedule (light 7 a.m.–7 p.m.).
The in vitro experimental procedures were approved by the Ethical Committee of the Pharmacology
and Toxicology Section, Department of Pharmacy, in accordance with the European legislation
(European Communities Council Directive of 24 November 1986, 86/609/EEC) and were approved
by Italian legislation on animal experimentation (Decreto Ministeriale number 124/2003-A). The in
vivo protocol was approved by Ethical Committee of Pavia’s University (session of October 11th
2011, minutes 3/2011) according to international regulations for the care and treatment of laboratory
animals, to the Italian Act (D.L. n. 116, GU, suppl 40, 18 February, 1992) and to EEC Council
Directive (86/609, OJ L 358, 1, 12 December, 1987). All efforts were made to minimize animal
suffering and to use the minimal number of animals necessary to produce reliable results
2.2. In vivo experiments
2.2.1. Microdialysis probe implantation
Rats were anesthetized with Equithesin 3 ml/kg (pentobarbital 9.7 g, chloral hydrate 42.5 g,
MgSO4 21.3 g for 1L, 10% ethanol, 40% propylene glycol v/v) administered intraperitoneally and
placed in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, USA). The skin was
shaved, disinfected, and cut with a sterile scalpel to expose the skull. A hole was drilled to allow the

6
implantation of the probe into the brain parenchyma. The probe was implanted in the hippocampus
(CA1/CA2 regions; AP -5.8 mm, ML ± 5.0 mm from bregma and DV−8.0mm from dura) according
to the Paxinos and Watson atlas (1986), and secured to the skull with one stainless steel screw and
dental cement. All in vivo experiments were performed using microdialysis probes, made in our
laboratory according to the original method described by Di Chiara (1990) (EmophanBellco
Artificial OR-internal diameter 200 µm, cut-off 40 KDa; Bellco, Mirandola, Modena, Italy), with a
nominal active length of 5mm. Finally, the skin was sutured, and the rats were allowed to recover
from anaesthesia for at least 24 h before the neurotransmitter release study. Previous
immunohistochemical analysis has shown that the administration of Aβ1-40 through the dialysis
probe at the concentrations tested in vivo (1 µM and 10 µM) allowed the delivery of the peptide
within the hippocampus. Despite the fact that we do not know the exact amounts of Aβ reaching the
tissue, there was a visible positive correlation between the concentration administered and the signal
of Aβ immunoreactivity in the tissue. Moreover, immunohistochemical analysis shows that no
evident signs of apoptosis were observed within the area of amyloid diffusion as shown by Hoechst
staining (Mura et al., 2012).
2.2.2. Microdialysis samples collection
Microdialysis experiments were performed on conscious freely moving rats. On the day of
the experiments (24 hrs after the surgical procedure), the probe was perfused with artificial CSF
containing 145 mM NaCl, 3.0 mM KCl, 1.26 mM CaCl2, 1.0 mM MgCl2, 1.4 mM Na2HPO4,
buffered at pH 7.2–7.4 and filtered through a Millipore 0.2 µm pore membrane. In all experiments,
the microdialysis membrane was allowed to stabilize for 1 h at the flow rate of 4 µl/min, without
collecting samples. At the end of the stabilization period, three samples were collected to evaluate
baseline release of GLU, ASP and GABA and then the specific treatment started. All treatments
were administered by manually switching syringes and tubing connections to allow drugs diluted in
artificial CSF to flow through the probes. Tubing switches were performed taking care to maintain

7
constant flow rates and collection volumes. Both basal and treatment samples were collected every
20 min in 100 µl Eppendorf tubes. The flow rate of 4 µl/min was maintained using a 1000 µl
syringe (Hamilton) and a microinjection pump (CMA/100, CMA/Microdialysis AB). In vitro
recovery of the probe for GLU was 15.21 ± 0.42 (Mura et al., 2012). Each rat was used for only one
microdialysis session. At the end of each experiment animals were sacrificed by guillotine, rat
brains were removed and the position of the microdialysis probe was verified by histological
procedures, slicing the tissues by a cryostat microtome (LEICA CM 1510). Only data from rats in
which probe tracks were exactly located in the target area were used for statistical analysis.
2.3. In vitro experiments
2.3.1.Experiments of release
Rats were killed by decapitation and the hippocampus rapidly removed at 0–4°C. Purified
gliosomes were prepared essentially as described by Nakamura et al. (1993) with minor
modifications (Stigliani et al., 2006). The tissue was homogenized in 10 volumes of 0.32 M
sucrose, buffered to pH 7.4 with Tris (final concentration 0.01 M) using a glass Teflon tissue
grinder (clearance 0.24 mm). The homogenate was centrifuged at 1000 g for 5 min, to remove
nuclei and cellular debris, and the supernatant was gently stratified on a discontinuous Percoll
gradient (2, 6, 10 and 20% (v/ v) in Tris-buffered sucrose) and centrifuged at 33,500 g for 5 min.
The layer between 2 and 6% Percoll (gliosomal fraction) was collected, washed by centrifugation
and resuspended in physiological medium containing 125 mM NaCl, 3 mM KCl, 1.2 mM MgSO4,
1.2 mM CaCl2, 1 mM NaH2PO4, 22 mM NaHCO3 and 10 mM glucose, pH 7.2–7.4 (aeration with
95% O2 and 5% CO2). Gliosomes were incubated at 37°C for 15 min and at the end of the
incubation period, identical portions of the gliosomal suspension was layered on microporous filters
at the bottom of parallel superfusion chambers thermostated at 37°C (Raiteri and Raiteri, 2000;

8
Superfusion System, Ugo Basile, Comerio, Varese, Italy). Gliosomes were superfused at 1 ml/min
with standard physiological medium as previously described. The system was first equilibrated
during 36.5 min of superfusion; subsequently, four consecutive 90 s fractions of superfusate were
collected and the endogenous GLU, ASP and GABA content were measured by high performance
liquid chromatography as below described. Gliosomes were exposed to agonists for 90 s starting
from the second fraction collected (t = 38 min), with antagonists being added 8 min before agonists.
The evoked overflow was calculated by subtracting the corresponding basal release from each
fraction and was expressed as pmol/mg of synaptosomal proteins.
2.4 Endogenous amino acids determination
In both dialysates and fractions collected from gliosomes in superfusion levels of
endogenous GABA, GLU and ASP were measured by HPLC analysis following precolumn
derivatization with o-phthalaldehyde and resolution through a C18-reverse phase chromatographic
column (1064.6 mm, 3 mm; Chrompack, Middleburg, The Netherlands) coupled with fluorometric
detection (excitation wavelength 350 nm; emission wavelength 450 nm). Homoserine was used as
internal standard. Buffers and gradient program were prepared and executed as follows: solvent A,
0.1 M sodium acetate (pH 5.8)/methanol, 80:20; solvent B, 0.1 M sodium acetate (pH
5.8)/methanol, 20:80; solvent C, sodium acetate (pH 6.0)/methanol, 80:20; gradient program, 100%
C for 4 min from the initiation of the program; 90% A and 10% B in 1 min; 42% A and 58% B in
14 min; 100% B in 1 min; isocratic flow 2 min; 100% C in 3 min; flow rate 0.9 ml/min.

9
2.5. Statistical analysis
2.5.1. In vivo experiments
Values were expressed either as amount of GLU measured in the dialysate (pmol/80 µL) or
as area under the curve (AUC), evaluating the cumulative release over time. AUC was used as a
measure of treatment exposure and was calculated, for each animal, using GraphPad Prism (version
4.03 GraphPad Software, San Diego, CA, USA), defining as baseline of the area the basal value
(average concentration of three consecutive samples immediately preceding the drug dose).
D’Agostino-Pearson Omnibus Test (GraphPad Prism, version 4.03, GraphPad Software, San Diego,
CA, USA) and Grubb’s Test (GraphPadQuickCalcs, online calculator for scientists at
http://www.graphpad.com/quickcalcs/, GraphPad Software, San Diego, CA, USA) were used as
preliminary tests in order to evaluate whether data were sampled from a Gaussian distribution and
to detect outliers respectively. All outliers were excluded from the analysis. Data were then
analyzed by analysis of variance (one- or two-way ANOVA) followed, when significant, by an
appropriate post hoc comparison test. Data were considered significant for P < 0.05. The reported
data are expressed as means ± S.E.M. The number of animals used for each experiment is reported
in the legend to figures.
2.5.2. In vitro experiments
Multiple comparisons were performed with one-way ANOVA followed by an appropriate
post hoc test (Dunnett’s and Bonferroni). Data were considered significant for P < 0.05, at least.
2.6. Preparation of beta-amyloid solutions
We used only Aβ 1–40 peptide in our experiments, for two main reasons. First,
physiologically the 40-aminoacid long peptide is the most abundant form (Pearson and Peers,

10
2006). Second, Aβ1–42 has been reported to aggregate faster than Aβ1–40 and thus it is considered
as the most neurotoxic species. With the aim of exploring new effects of Aβ other than the
neurotoxic ones, we chose to avoid this potentially confounding element. In the case of both in vivo
and in vitro experiments, synthetic human Aβ1–40 was dissolved in aCSF at a concentration of 100
µM (stock solution). Then, this solution was filtered through a Millipore 0.2 mm pore membrane
and stocked in small aliquots at -80° C. Working solutions were freshly prepared by diluting an
aliquot of Aβ1–40 stock solution at the final concentrations (10 µM, 1 µM, or 100 nM Aβ1–40 for
in vivo experiments, 100 nM, 10 nM, 1 nM, or 100 pM for in vitro analysis), just before the
administration.
It is well known that the most likely explanation for the many discrepancies observed
between reports using Aβ preparations may be attributable to differences in the aggregation state of
the Aβ used. As far as the characteristics of the Aβ peptides delivered through the dialysis probe we
have previously shown, by western blot procedure, that we administered at least predominantly, Aβ
monomers although we cannot completely exclude that small amounts of Aβ oligomers are also
present and may participate to produce the observed effects (Mura et al., 2012). In regard to the in
vitro Aβ preparations, since we did not observe aggregation at the concentrations and the timing (up
to 40 min) analyzed in vivo, we also do not expect to observe aggregation at the lower
concentrations and shorter times used in vitro in light of the fact that aggregation is a concentration
and time-dependent process.
2.7. Chemicals
Beta-amyloid (1-40; 40-1), Percoll®, choline, dimethyl sulfoxide, nicotine hydrogen tartrate
salt (Sigma-Aldrich, St Louis, MO, USA); 5IA85380, PHA543613, (Tocris Bioscience, Bristol,
UK); all salts used for the preparation of aCSF (NaCl, KCl, CaCl2, MgCl2, Na2HPO4) and for

11
Equithesin (MgSO4) were purchased at Merck KGaA, Darmstadt, Germany; chloral hydrate,
ethanol 96% and propylene glycol were used for the preparation of Equithesin and were obtained at
VWR BDH Prolabo, Belgium.

12
3. Results
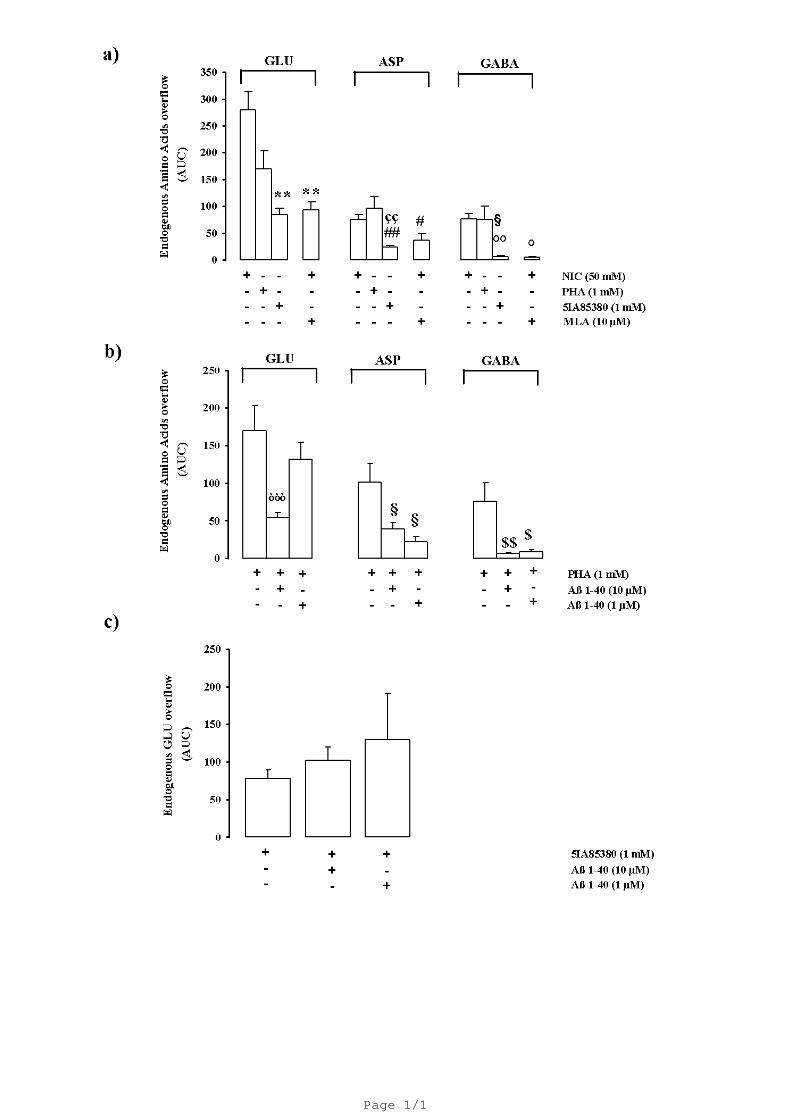
Figure 1a shows the stimulatory effect of the in vivo administration of nicotine and two
nicotinic acetylcholine receptor agonists, PHA543613 and 5-IA85380. In our experimental
conditions, 40 minutes-long administration of 50 mM nicotine was able to greatly enhance the
overflow of all the three amino acids being more potent in stimulating GLU overflow. Figure 1a
shows that the α7 agonist PHA543613 induced a significant release of amino acid. Conversely, the
amount of GLU elicited by the α4β2 nicotinic acetylcholine receptor agonist 5IA85380 was
significantly lower comparable to that elicited by nicotine (-70 %) while the overflow of ASP and
GABA was almost inconsistent. The nicotine evoked aminoacid release was strongly inhibited in
presence of the α7 antagonist MLA (10 µM) (-66 % GLU; -51 % ASP; -94% GABA). The
possibility that Aβ may differentially inhibit the two nicotinic acetylcholine receptors subtypes
which control amino acid release has been investigated. In order to verify this point we have studied
the effects of Aβ1-40 on the release elicited by the two different agonists PHA543613 and
5IA85380. Figure 1b shows that the PHA-evoked overflow of endogenous GLU, ASP and GABA
was significantly inhibited by Aβ 1-40 used at 10 µM concentration. Aβ 1-40 at 1 µM concentration
was still able to decrease ASP and GABA overflow but was ineffective on GLU overflow. Aβ1-40
(10 µM) did not modify the basal release of endogenous GLU, ASP and GABA (data not shown).
Interestingly the 5IA85380 (10 nM)-evoked in vivo overflow of GLU from rat hippocampus was
not affected by Aβ1-40 at all concentration used (Fig. 1c). Based on the in vivo data we then
analyzed the effects of Aβ1-40 on the administration of nicotine, choline (Ch) and 5IA85380,
known to act selectively on the α7 and α4β2 nicotinic acetylcholine receptor subtypes respectively
(Mukhin et al., 2000; Uteshev et al., 2003; Dickinson et al., 2007; Zappettini et al., 2010), on the
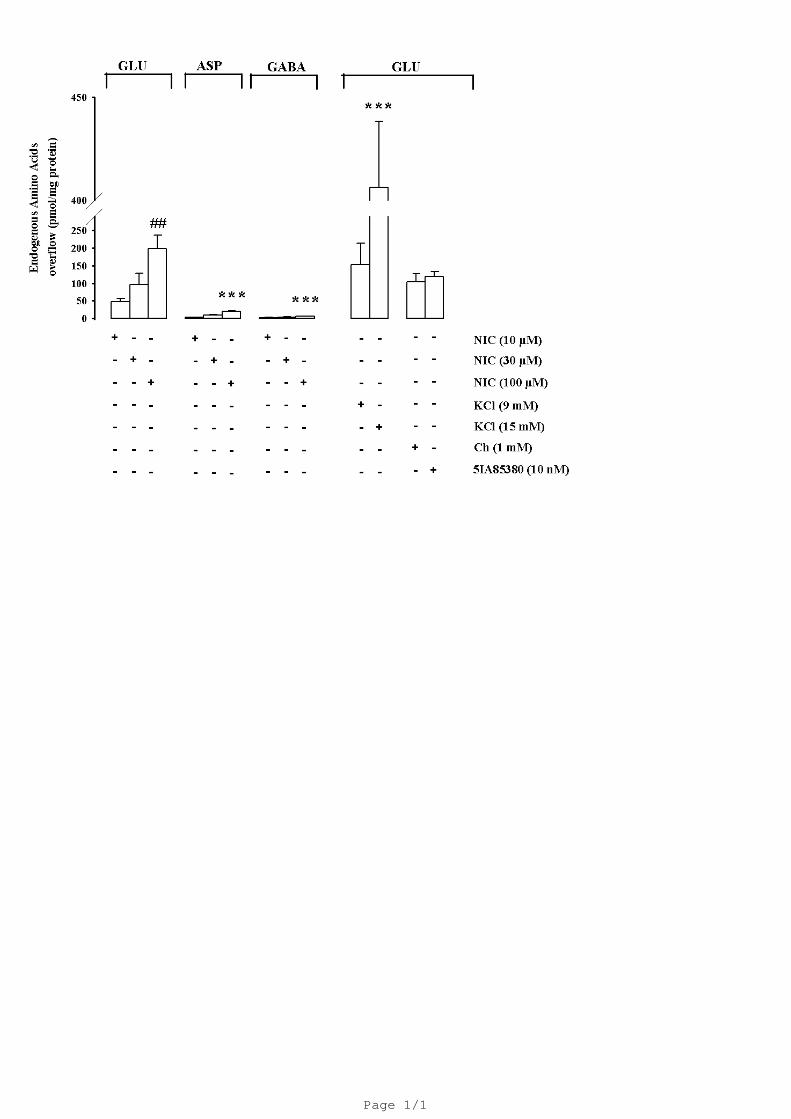
overflow of GLU, ASP and GABA from rat hippocampal gliosomes in vitro. Figure 2 shows that
nicotine, in a concentration dependent manner elicited a significant overflow of endogenous GLU
but not of ASP and GABA. This overflow was comparable to that produced by 9 mM KCl. A

13
higher KCl concentration (15 mM) produced a significant increase of GLU release (406 ± 32). Also
Ch (1 mM) and 5-IA85380 hydrochloride (5IA85380; 10 nM) evoked a significant overflow of
GLU (104 ± 23; 118 ± 15) from hippocampal gliosomes demonstrating the involvement of two
nicotinic acetylcholine receptor subtypes. The Ch (1 mM)-and 5IA85380 (10 nM)-evoked GLU
overflow was significantly Ca2+-dependent (data not shown).
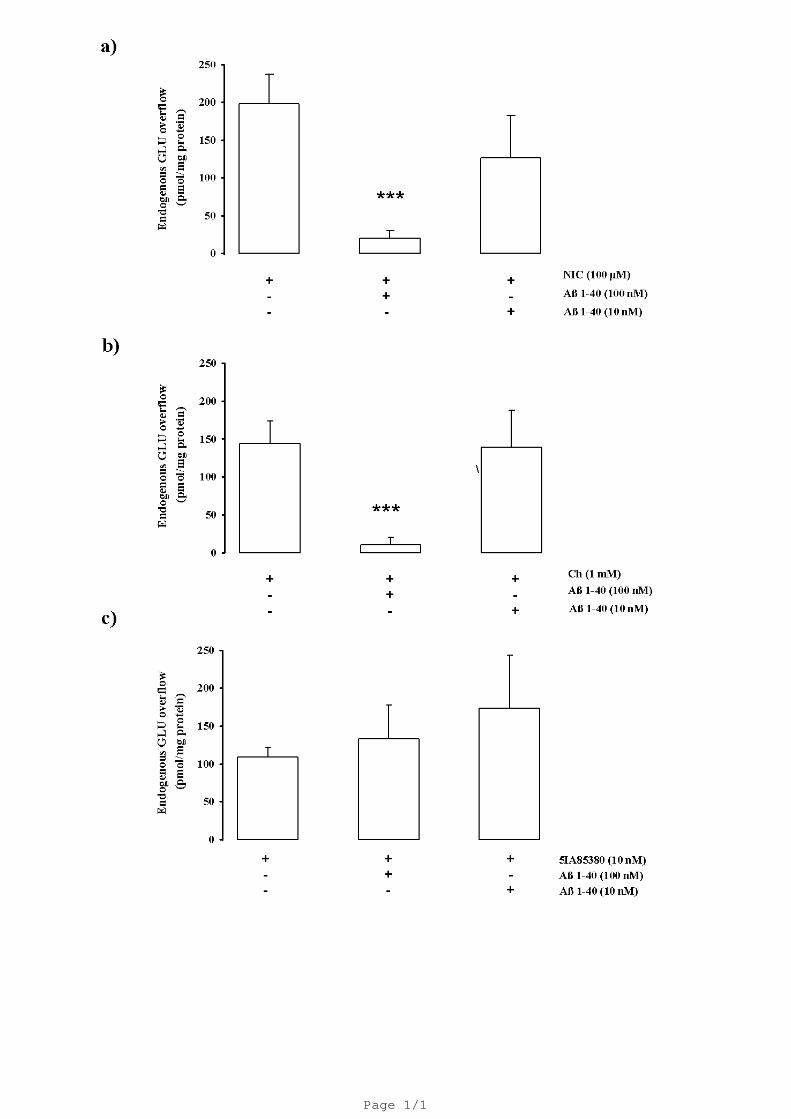
Both nicotine- and Ch-evoked GLU overflow were significantly inhibited by Aβ1-40 at 100
nM but not at 10 nM concentrations (-58%; -80 %; Fig. 3a, b). Interestingly, the 5IA85380 (10
nM)-evoked GLU overflow was unaffected in presence of Aβ1-40 (100 nM and 10 nM). Aβ1-40
(100 nM) did not modify the basal release of endogenous GLU (data not shown).

14
4. Discussion
As previously mentioned there are several in vitro studies that demonstrate the existence in
hippocampus of α7 and α4β2 nicotinic acetylcholine receptor or both which modulate the release of
GLU (Bancila et al., 2009; Zappettini et al., 2011; Lozada et al., 2012). Huang et al. (2010) in an
iontophoretic study showed that some selective α7 nicotinic acetylcholine receptor agonists mimic
the effect of nicotine suggesting that the α7 nicotinic acetylcholine receptor is the major nicotinic
acetylcholine receptor subtype involved in evoking the excitation of CA3 neurons in rat
hippocampus. Using in vivo microdialysis technique it has been also reported that both GLU and
GABA are released upon activation α7 nicotinic acetylcholine receptors in rat striatum (Campos et
al., 2010). To the best of our knowledge, this is the first study that demonstrates the ability of α7
and of non-α7 nicotinic acetylcholine receptor agonists to directly modulate GLU, ASP and GABA
release in rat hippocampus in vivo. Our data showed that both α7 and α4β2 nicotinic acetylcholine
receptors are able to stimulate, in vivo, the release of GLU, ASP and GABA but the α7 nicotinic
acetylcholine receptors have a quantitatively predominant role. The nicotine-evoked aminoacid
release was indeed quantitatively relevant compared to that evoked by a selective α7 nicotinic
acetylcholine receptor agonist PHA543613 and strongly reduced in presence of MLA an inhibitor
of α7 nicotinic acetylcholine receptor. Moreover, the GLU and ASP release evoked in presence of
5IA85380, used at a concentration that normally produce the maximal effect (Livingstone et al.,
2009), was very modest (or inconsistent as in the case of GABA release). Of course, the hypothesis
that the administration of higher doses might also activate nicotinic acetylcholine receptor subtypes
different from those reported should not be excluded.
Quite interestingly the finding that the activation of α7 nicotinic acetylcholine receptor is
predominant in the in vivo release of GLU may explain also that the inhibitory effect of Aβ, in vivo,
seems to be exclusively due to the inhibition of the α7 nicotinic acetylcholine receptor. Indeed, as
mentioned before the stimulatory effect of 5IA85380, although modest, was not altered in presence

15
of Aβ while the PHA543613-evoked GLU release was strongly and specifically reduced. This result
is also in agreement with the ineffectiveness of Aβ in inhibiting the 5IA85380-evoked GLU release
from gliosomes. It has to be also mentioned that Aβ was also unable to inhibit the in vivo release of
GLY elicited by α4β2 nicotinic acetylcholine receptors agonist while it exerted an inhibition of both
nicotine- and PHA543613-stimulated GLY release (Zappettini et al., 2012).
It is now well known that astrocytes can cross talk with neighboring neurons and are
involved in the active control of synaptic transmission and brain activity (Araque et al., 1999;
Haydon, 2001; Volterra and Meldolesi, 2005). They can exhibit an increase in Ca2+ response,
(Enkvist et al., 1989; Cornell-Bell et al., 1990; Pasti et al., 1995; Harris-White et al., 1998; Parri et
al., 2011) which produce as consequence the release of gliotransmitters, such as GLU. This event
can be observed also following stimulation by several compounds including nicotine (Dani et al.,
1992; Porter and McCarthy, 1996; Parpura and Haydon, 2000; Pasti et al., 2001; Bezzi et al., 2004;
Chen et al., 2005). Indeed, astrocytes in culture express nicotinic acetylcholine receptors,
particularly those containing α7 and α4β2 subunits (Sharma and Vijayaraghavan, 2001; Xiu et al.,
2005; Shen and Yakel, 2012) and it has been demonstrated that the stimulation of these receptors
modulates GLU uptake and GLU synthase activity in astrocytes (Lim and Kim, 2003).
In a previous paper Stigliani and colleagues (2006) characterized in detail a particular
particle called gliosomes which are an in vitro vital preparation originating from mature astrocytes
present in different brain areas. Gliosomes are able to respond to exogenous stimuli and present
some differences from their neuronal counterpart (synaptosomes). Furthermore, it is well known
that GLU is also released from astrocytes and the presence of nicotinic receptors stimulating GLU
release from cortical gliosomes in mice have been already demonstrated (Patti et al., 2007). We here
report that the cholinergic modulation of GLU release on rat hippocampal gliosomes was modulated
by both α7 and α4β2 nicotinic acetylcholine receptors. Interestingly, both receptors elicited
selectively the release of GLU but not that of ASP or GABA. We do not know their physiological
importance in the intact astrocytes since information in the literature regarding to the presence of

16
nicotinic acetylcholine receptors on these cells are different depending the species considered, the
areas and the age of animals (Xiu et al., 2005; Yu et al., 2005). However, from our findings it is
possible to hypothesize that GLU release evoked, in vivo, by nicotine or nicotinic agonists, as
shown in Fig. 1, might originate also from nicotinic stimulation of astrocytes. Conversely, the
overflow of ASP and GABA seem to be exclusively from neuronal origin.
It is significant to note that Aβ selectively inhibits, in gliosomes, the GLU overflow evoked
by α7 nicotinic acetylcholine receptor subtypes but is inactive in inhibiting that evoked by α4β2
nicotinic acetylcholine receptor. This finding is very relevant because Aβ at the same concentration
used in this paper, was able to inhibit the 5IA85380-evoked GLU overflow from isolated nerve
endings (Mura et al., 2012). Therefore there is the possibility that the 5IA85380-sensitive nicotinic
acetylcholine receptors present on gliosomes are different from those present on synaptosomes.
Although the exact nature of the Aβ interaction with nicotinic acetylcholine receptor subtypes is so
far not well understood it is quite interestingly to note that the α7 nicotinic acetylcholine receptor
subtypes modulating the overflow of GLU from gliosomes (Fig. 2) and from synaptosomes (Mura
et al., 2012) were functionally inhibited in vitro apparently to a similar extent by Aβ concentrations
in the nanomolar range.
5. Conclusions
From our results we can therefore conclude that a) both α7 that α4β2 nicotinic acetylcholine
receptors are present on rat hippocampal gliosomes and they modulate the release of GLU but not
that of ASP and GABA; b) Aβ is able to selectively inhibit only the α7 but not the α4β2 nicotinic
acetylcholine receptor subtype present on gliosomes c) the administration of nicotine in vivo is able
to elicit GLU release mostly through the activation of α7 nicotinic acetylcholine receptors present
both on neurons and astrocytes; d) the nicotinic evoked release of ASP and GABA has exclusively a
neuronal origin. We can therefore foresee that in an integrated system, where cellular networks and
their functional relationships are completely preserved and several direct and indirect processes are
simultaneously taking place in neurons and astrocytes, the effect of Aβ on the cholinergic

17
modulation of GLU function may mostly depend on the interaction of Aβ with the α7 nicotinic
acetylcholine receptors. This finding should be relevant considering that α7 nicotinic acetylcholine
receptors are strongly increased in astrocytes of patients with AD (Nagele et al., 2003) and an
increase in the proportion of astrocytes expressing α7 immunoreactivity was observed in AD
patients compared with age-matched controls (Teaktong et al., 2003).
Acknowledgements
This work was supported by from the Italian Ministero Università Ricerca to Prof. Mario Marchi
(Prot. N° 2009R7WCZS_003), by Compagnia di San Paolo, by University of Genoa ‘Progetto
Ricerca Ateneo’ and by project AROMA (ALCOTRA 2007-2013) by the European Community
Project PYRGI, project n° B51H10000000006 and by the project 7FP-KBBE BAMMBO and to
Prof. Govoni (MIUR 2009). This work is supported by a grant of the Alzheimer’s Association
(NIRG-11-205183) to Elisa Mura. We wish to thank Maura Agate for editorial assistance and Dr.
Silvia E. Smith, Ph.D. (University of Idaho, IBEST, School of Life Sciences) for reviewing the
manuscript.

18
References
Araque, A., Parpura, V., Sanzgiri, R.P., Haydon, P.G., 1999. Tripartite synapses: glia, the
unacknowledged partner. Trends Neurosci. 22, 208-215.
Bancila, V., Cordeiro, J.M., Bloc, A., Dunant, Y., 2009. Nicotine-induced and depolarization-
induced glutamate release from hippocampus mossy fibre synaptosomes: two distinct
mechanisms. J. Neurochem. 110, 570-580.
Berger, F., Gage, F.H., Vijayaraghavan, S., 1998. Nicotinic receptor-induced apoptotic cell death of
hippocampal progenitor cells. J. Neurosci. 18, 6871–6881.
Bezzi, P., Gundersen, V., Galbete, J.L., Seifert, G., Steinhauser, C., Pilati, E., Volterra, A., 2004.
Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of
glutamate. Nat. Neurosci. 7, 613-620.
Campos, F., Alfonso, M., Dura´n R., 2010. In vivo modulation of a7 nicotinic receptors on striatal
glutamate release induced by anatoxin-A. Neurochem. Int. 56, 850-855.
Chen, X., Wang, L., Zhou, Y., Zheng, L.H., Zhou, Z., 2005. "Kiss-and-run" glutamate secretion in
cultured and freshly isolated rat hippocampal astrocytes. J. Neurosci. 25, 9236-9243.
Cornell-Bell, A.H., Thomas, P.G., Smith, S.J., 1990. The excitatory neurotransmitter glutamate
causes filopodia formation in cultured hippocampal astrocytes. Glia 3, 322-334.
Dani, J.W., Chernjavsky, A., Smith, S.J., 1992. Neuronal activity triggers calcium waves in
hippocampal astrocyte networks. Neuron 8, 429-440.
Di Chiara, G., 1990. In-vivo brain dialysis of neurotransmitters. Trends Pharmacol. Sci. 11, 116-
121.
Dickinson, J.A., Hanrott, K.E., Mok, M.H., Kew, J.N., Wonnacott, S., 2007. Differential coupling
of alpha7 and non-alpha7 nicotinic acetylcholine receptors to calcium-induced calcium
release and voltage-operated calcium channels in PC12 cells. J. Neurochem. 100, 1089-
1096.

19
Dougherty, J.J., Wu, J., Nichols, R.A., 2003. Beta-amyloid regulation of presynaptic nicotinic
receptors in rat hippocampus and neocortex. J. Neurosci. 23, 6740-6747.
Enkvist, M.O., Holopainen, I., Akerman, K.E., 1989. Glutamate receptor-linked changes in
membrane potential and intracellular Ca2+ in primary rat astrocytes. Glia 2, 397-402.
Errico, F., Nisticò, R., Napoletano, F., Mazzola, C., Astone, D., Pisapia, T., Giustizieri, M.,
D'Aniello, A., Mercuri, N.B., Usiello, A., 2011. Increased D-aspartate brain content rescues
hippocampal age-related synaptic plasticity deterioration of mice. Neurobiol Aging 32,
2229-2243.
Fedele, E., Varnier, G., Ansaldo, M.A., Raiteri, M., 1998. Nicotine administration stimulates the in
vivo N-methyl-D-aspartate receptor/nitric oxide/cyclic GMP pathway in rat hippocampus
through glutamate release. Br. J. Pharmacol. 125, 1042-1048.
Giacobini, E., 2003. Cholinergic function and Alzheimer's disease. Int. J. Geriatr. Psychiatry
18(Suppl 1), S1-5.
Harris-White, M.E., Zanotti, S.A., Frautschy, S.A., Charles, A.C., 1998. Spiral intercellular calcium
waves in hippocampal slice cultures. J. Neurophysiol. 79, 1045-1052.
Haydon, P.G., 2001. GLIA: listening and talking to the synapse. Nat. Rev. Neurosci. 2, 185-193.
Huang, L.T., Sherwood, J.L., Sun, Y.J., Lodge, D., Wang, Y., 2010. Activation of presynaptic a7
nicotinic receptors evokes an excitatory response in hippocampal CA3 neurones in
anaesthetized rats: an in vivo iontophoretic study. Br. J. Pharmacol. 159, 554-565.
Khiroug, L, Giniatullin, R., Klein, R.C., Fayuk, D., Yakel, J.L., 2003. Functional mapping and Ca2+
regulation of nicotinic acetylcholine receptor channels in rat hippocampal CA1 neurons. J.
Neurosci. 23, 9024 –9031.
Lambe, E.K., Picciotto, M.R., Aghajanian, G.K., 2003. Nicotine induces glutamate release from
thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology 28, 216–225.
Levin, E., Simon, B., 1998. Nicotinic acetylcholine involvement in cognitive function in animals.
Psychopharmacology (Berl.)138, 217-230.

20
Lilja, A.M, Porras, O., Storelli, E., Nordberg, A., Marutle, A., 2011. Functional interactions of
fibrillar and oligomeric amyloid-β with alpha7 nicotinic receptors in Alzheimer's disease. J.
Alzheimers Dis. 23, 335-47.
Lim, D.K., Kim, H.S., 2003. Opposite modulation of glutamate uptake by nicotine in cultured
astrocytes with/without cAMP treatment. Eur. J. Pharmacol. 476, 179-184.
Livingstone, P.D., Srinavasan, J., Kew, J.N., Dawson, L.A., Gotti, C., Moretti, M., Shoaib, M.,
Wonnacott, S., 2009. Alpha7 and non alpha 7 nicotine acetylcholine receptors modulate
dopamine release in vitro and in vivo in the rat prefrontal cortex. Eur. J. Neurosci. 29, 539-
550.
Liu, Q., Wu, J., 2006. Neuronal nicotinic acetylcholine receptors serve as sensitive targets that
mediate beta-amyloid neurotoxicity. Acta Pharmacol. Sin. 27, 1277-1286.
Lozada, A.F., Wang, X., Gounko, N.V., Massey, K.A., Duan, J., Liu, Z., Berg, D.K., 2012.
Induction of dendritic spines by β2-containing nicotinic receptors. J. Neurosci. 32, 8391-
8400.
Marchi, M., Risso, F., Viola, C., Cavazzani, P., Raiteri, M., 2002. Direct evidence that release-
stimulating alpha7* nicotinic cholinergic receptors are localized on human and rat brain
glutamatergic axon terminals. J. Neurochem. 80, 1071-1078.
Matos, M., Augusto, E., Machado, N.J., dos Santos-Rodrigues, A., Cunha, R.A., Agostinho, P.,
2012a. Astrocytic adenosine A2A receptors control the amyloid-β peptide-induced decrease
of glutamate uptake. J. Alzheimers Dis. 31, 555-567.
Matos, M., Augusto, E., Santos-Rodrigues, A.D., Schwarzschild, M.A., Chen, J.F., Cunha, R.A.,
Agostinho, P., 2012b. Adenosine A2A receptors modulate glutamate uptake in cultured
astrocytes and gliosomes. Glia 60, 702-716.
Mattson, M.P., 2008. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann. N.
Y. Acad. Sci. 1144, 97-112.

21
Mechawar, N., Saghatelyan, A., Grailhe, R., Scoriels, L., Gheusi, G., Gabellec, M.M., Lledo, P.M.,
Changeux, J.P., 2004. Nicotinic receptors regulate the survival of newborn neurons in the
adult olfactory bulb. Proc. Natl. Acad. Sci. USA 101, 9822-9826.
Milanese, M., Zappettini, S., Jacchetti, E., Bonifacino, T., Cervetto, C., Usai, C., Bonanno, G.,
2010. In vitro activation of GAT1 transporters expressed in spinal cord gliosomes stimulates
glutamate release that is abnormally elevated in the SOD1/G93A(+) mouse model of
amyotrophic lateral sclerosis. J. Neurochem. 113, 489-501.
Mukhin, A.G., Gündisch, D., Horti, A.G., Koren, A.O., Tamagnan, G., Kimes, A.S., Chambers, J.,
Vaupel, D.B., King, S.L., Picciotto, M.R., Innis, R.B., London, E.D., 2000. 5-Iodo-A-85380,
an alpha4beta2 subtype-selective ligand for nicotinic acetylcholine receptors. Mol.
Pharmacol. 57, 642-649.
Mura, E., Lanni, C., Preda, S., Pistoia, F., Sarà, M., Racchi, M., Schettini, G., Marchi, M., Govoni,
S., 2010. Beta-amyloid: a disease target or a synaptic regulator affecting age-related
neurotransmitter changes? Curr. Pharm. Des. 16, 672-683.
Mura, E., Zappettini, S., Preda, S., Biundo, F., Lanni, C., Grilli, M., Cavallero, A., Olivero, G.,
Salamone, A., Govoni, S., Marchi, M., 2012. Dual effect of beta-amyloid on α7 and α4β2
nicotinic receptors controlling the release of glutamate, aspartate and GABA in rat
hippocampus. PLoS One 7, e29661.
Nagele, R.G., D'Andrea, M.R., Lee, H., Venkataraman, V., Wang, H.Y., 2003. Astrocytes
accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease
brains. Brain Res. 971, 197-209.
Nakamura, Y., Iga, K., Shibata, T., Shudo, M., Kataoka, K., 1993. Glial plasmalemmal vesicles: a
subcellular fraction from rat hippocampal homogenate distinct from synaptosomes. Glia 9,
48-56.
Ni, R., Marutle, A., Nordberg, A., 2013. Modulation of α7 nicotinic acetylcholine receptor and
fibrillar amyloid-β interactions in Alzheimer's disease brain. J. Alzheimers Dis. 33, 841-51.

22
Parpura, V., Haydon, P.G., 2000. Physiological astrocytic calcium levels stimulate glutamate
release to modulate adjacent neurons. Proc. Natl. Acad. Sci. USA 97, 8629-8634.
Parri, H.R., Hernandez, C.M., Dineley, K.T., 2011. Research update: Alpha7 nicotinic acetylcholine
receptor mechanisms in Alzheimer's disease. Biochem. Pharmacol. 82, 931-942.
Pasti, L., Pozzan, T., Carmignoto, G., 1995. Long-lasting changes of calcium oscillations in
astrocytes. J. Biol. Chem. 270, 15203-15210.
Pasti, L., Zonta, M., Pozzan, T., Vicini, S., Carmignoto, G., 2001. Cytosolic calcium oscillations in
astrocytes may regulate exocytotic release of glutamate. J. Neurosci. 21, 477-484.
Patti, L., Raiteri, L., Grilli, M., Zappettini, S., Bonanno, G., Marchi, M., 2007. Evidence that alpha7
nicotinic receptor modulates glutamate release from mouse neocortical gliosomes.
Neurochem. Int. 51, 1-7.
Paxinos, G., Watson, C., 1986. The Rat Brain in Stereotaxic coordinates. 2nd Edition., New York,
Academic Press.
Pearson, H.A., Peers, C., 2006. Physiological roles for amyloid β peptides. J. Physiol. 575, 5-10.
Picciotto, M., Zoli, M., Léna, C., Bessis, A., Lallemand, Y., Le Novère, N., Vincent, P., Pich, E.,
Brûlet, P., Changeux, J., 1995. Abnormal avoidance learning in mice lacking functional
high-affinity nicotine receptor in the brain. Nature 374, 65-67.
Porter, J.T., McCarthy, K.D., 1996. Hippocampal astrocytes in situ respond to glutamate released
from synaptic terminals. J. Neurosci. 16, 5073-5081.
Puzzo, D., Privitera, L., Leznik, E., Fà, M., Staniszewski, A., Palmeri, A., Arancio, O., 2008.
Picomolar amyloid-beta positively modulates synaptic plasticity and memory in
hippocampus. J. Neurosci. 28, 14537-14545.
Raiteri, L., Raiteri, M., 2000. Synaptosomes still viable after 25 years of superfusion. Neurochem.
Res. 25, 1265-1274.

23
Revett, T.J., Baker, G.B., Jhamandas, J., Kar, S., 2013. Glutamate system, ß amyloid peptides and
tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J.
Psychiatry Neurosci. 38, 6-23.
Santello, M., Volterra, A., 2009. Synaptic modulation by astrocytes via Ca2+-dependent glutamate
release. Neuroscience 158, 253-259.
Santello, M., Bezzi, P., Volterra, A., 2011. TNFα controls glutamatergic gliotransmission in the
hippocampal dentate gyrus. Neuron 69, 988-1001.
Sharma, G., Vijayaraghavan, S., 2001. Nicotinic cholinergic signaling in hippocampal astrocytes
involves calcium-induced calcium release from intracellular stores. Proc. Natl. Acad. Sci.
USA 98, 4148 - 4153.
Shen, J.X., Yakel, J.L., 2012. Functional α7 nicotinic ACh receptors on astrocytes in rat
hippocampal CA1 slices. J. Mol. Neurosci. 48, 14-21.
Stigliani, S., Zappettini, S., Raiteri, L., Passalacqua, M., Melloni, E., Venturi, C., Tacchetti, C.,
Diaspro, A., Usai, C., Bonanno, G., 2006. Glia re-sealed particles freshly prepared from
adult rat brain are competent for exocytotic release of glutamate. J. Neurochem. 96, 656-
668.
Teaktong, T., Graham, A., Court, J., Perry, R., Jaros, E., Johnson, M., Hall, R., Perry, E., 2003
Alzheimer's disease is associated with a selective increase in alpha7 nicotinic acetylcholine
receptor immunoreactivity in astrocytes. Glia. 41, 207-211.
Tong, M., Arora, K., White, M.M., Nichols, R.A., 2011. Role of key aromatic residues in the
ligand-binding domain of α7 nicotinic receptors in the agonist action of β-amyloid. J. Biol.
Chem. 286, 34373-34381.
Toth, E., 1996. Effect of nicotine on the level of extracellular amino acids in the hippocampus of
rat. Neurochem. Res. 21, 903-907.
Uteshev, V., Meyer, E., Papke, R., 2003, Regulation of neuronal function by choline and 4OHGTS-
21 through alpha 7 nicotinic receptors. J. Neurophysiol. 89, 1797-1806.

24
Volterra, A., Meldolesi, J., 2005. Astrocytes, from brain glue to communication elements: the
revolution continues. Nat. Rev. Neurosci. 6, 626-640.
Woodruff-Pak, D.S., Gould, T.J., 2002. Neuronal nicotinic acetylcholine receptors: involvement in
Alzheimer’s disease and schizophrenia. Behav. Cogn. Neurosci. Rev. 1, 5-20.
Xiu, J., Nordberg, A., Zhang, J.T., Guan, Z.Z., 2005. Expression of nicotinic receptors on primary
cultures of rat astrocytes and up-regulation of the alpha7, alpha4 and beta2 subunits in
response to nanomolar concentrations of the beta-amyloid peptide(1-42). Neurochem. Int.
47, 281-290.
Yu, W.F., Guan, Z.Z., Bogdanovic, N., Nordberg, A., 2005. High selective expression of alpha7
nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer's disease
and patients carrying Swedish APP 670/671 mutation: a possible association with neuritic
plaques. Exp. Neurol. 192, 215-225.
Zappettini, S., Grilli, M., Salamone, A., Fedele, E., Marchi, M., 2010. Pre-synaptic nicotinic
receptors evoke endogenous glutamate and aspartate release from hippocampal
synaptosomes by way of distinct coupling mechanisms. Br. J. Pharmacol. 161, 1161-1171.
Zappettini, S., Mura, E., Grilli, M., Preda, S., Salamone, A., Olivero, G., Govoni, S., Marchi, M.,
2011. Different presynaptic nicotinic receptor subtypes modulate in vivo and in vitro the
release of glycine in the rat hippocampus. Neurochem. Int. 59, 729-738.
Zappettini, S., Grilli, M., Olivero, G., Mura, E., Preda, S., Govoni, S., Salamone, A., Marchi, M.,
2012. Beta Amyloid differently modulate nicotinic and muscarinic receptor subtypes which
stimulate in vitro and in vivo the release of glycine in the rat hippocampus. Front.
Pharmacol. 3, 146.

25
Legends to the Figures
Fig. 1. Panel a) shows the effects of the in vivo administration of nicotine (NIC), PHA543613
(PHA) and 5IA85380 on the endogenous overflows (AUC) of glutamate (GLU), aspartate (ASP)
and GABA from rat hippocampus. Data are mean ± S.E.M. of 4–12 individual rats for each
experimental group. ** P < 0.01 versus nicotine-evoked GLU overflow; # P < 0.05; ## P < 0.01
versus nicotine-evoked ASP overflow; ςς P < 0.01 versus PHA543613-evoked ASP overflow, ° P <
0.05; °° P < 0.01 versus nicotine-evoked GABA overflow, § P < 0.05 versus PHA543613-evoked
GABA overflow (One-way ANOVA followed by Bonferroni post hoc test). Panel b) shows the in
vivo effects of Aβ 1-40 on the PHA543613 (PHA)-evoked endogenous GLU, ASP and GABA
overflows from rat hippocampus. Data are mean ± S.E.M. of 4–12 individual rats for each
experimental group. òòò P < 0.001 versus PHA543613-evoked GLU overflow, § P < 0.05 versus
PHA543613-evoked ASP overflow; $ P < 0.05; $$P < 0.01 versus PHA543613-evoked GABA
overflow (One-way ANOVA followed by Dunnett's Multiple Comparison Test). Panel c) shows the
in vivo effects of Aβ 1-40 on the 5IA85380-evoked endogenous GLU overflow from rat
hippocampus. Data are mean ± S.E.M. of 4–12 individual rats for each experimental group.
Fig. 2. Effect of different concentrations of nicotine (NIC) on endogenous overflow of glutamate
(GLU), aspartate (ASP) and GABA and effect of KCl, 5IA85380 and Choline (Ch) on the
endogenous overflow of GLU from rat hippocampal gliosomes. Data are mean ± S.E.M. of 4
experiments run in triplicate. ## P < 0.01 versus NIC 10µM, *** P < 0.001 versus NIC 100µM
evoked endogenous GLU overflow (one-way ANOVA followed by Newman Keuls post hoc test).

26
Fig. 3. The figure shows the concentration dependent effect of Aβ 1–40 on the nicotine (NIC)-,
choline (Ch)- and 5IA85380- induced overflow of GLU, in rat hippocampal gliosomes (Panels a, b
and c, respectively). *** P < 0.001 vs. NIC and Ch (One-way ANOVA followed by Dunnett's
Multiple Comparison Test). Data are expressed as mean ± S.E.M. of 4 experiments run in triplicate.
Page 1/1
Page 1/1
Page 1/1
Copyright © 2022 FDOKUMEN




















