
Inhibition of IGF-I–induced Erk 1 and 2 activation and mitogenesis in mesangial cells by...
10
Kidney International, Vol. 62 (2002), pp. 412–421 Inhibition of IGF-I–induced Erk 1 and 2 activation and mitogenesis in mesangial cells by bradykinin CELINE ALRIC,CHRISTIANE PECHER,ERIC CELLIER,JOOST P. SCHANSTRA,BRUNO POIRIER, JACQUES CHEVALIER,JEAN-LOUP BASCANDS, and JEAN-PIERRE GIROLAMI INSERM U388, Institut Louis Bugnard, CHU Rangueil, Toulouse, and INSERM U430, Ho ˆ pital Broussais, Paris, France Inhibition of IGF-I–induced Erk 1 and 2 activation and mito- leading to diabetic nephropathy. Insulin-like growth fac- genesis in mesangial cells by bradykinin. tor-I (IGF-I) and to a lesser extent insulin, both induce Background. The beneficial effects of therapeutic angioten- MC proliferation and collagen secretion most likely via sin-converting enzyme (ACE) inhibitor treatment against the activation of the IGF-I receptor. Insulin thus may be worsening of glomerulosclerosis during the course of diabetic involved in the early steps of the glomerulosclerotic pro- nephropathy have been widely documented. ACE inhibitors inhibit both angiotensin II formation and bradykinin (BK) cess [1–4]. The renoprotective effect of angiotensin-con- degradation, thereby reducing angiotensin II type 1 (AT1) verting enzyme (ACE) inhibitors on the progression of receptor activity and favoring B2-kinin receptor (B2 receptor) several renal diseases, including diabetic nephropathy, activation. Since the involvement of growth factors such as is supported by a large number of reports [5–12]. Several insulin-like growth factor (IGF-I) has been implicated in the early steps of diabetic nephropathy, we investigated the effect mechanisms were proposed to account for the beneficial of BK on Erk 1 and 2 activation and cell proliferation by IGF-I. effects of ACE inhibitors. ACE inhibitors can inhibit the Methods. The activation of Erk 1 and 2 in mesangial cells angiotensin II (Ang II)-induced increase in glomerular (MCs) and isolated glomeruli (IG) was investigated by immu- capillary resistance but also act on the Ang II-stimulated noprecipitation and Western blotting during activation of the secretion of matrix components by renal cells, indepen- IGF-I receptor in the presence or absence of BK and of protein kinase C (PKC), tyrosine-kinase and phosphatase selective in- dently of any hemodynamic effects. Besides the Ang II- hibitors. Mesangial cell proliferation was assessed in vitro by dependent mechanism, ACE inhibitors reduce the enzy- cell counting. matic degradation of bradykinin (BK), thereby favoring Results. In untreated MCs and IG, when added separately, the stimulation of the B2 receptor (B2 receptor). This BK and IGF-I both activated Erk 1 and 2. In contrast, in MCs is an additional potential mechanism in the protective and IG pretreated with BK, the IGF-I–induced Erk 1 and 2 ac- tivation was dose-dependently reduced. The inhibitory effect of role of ACE inhibitor that has been poorly investigated. BK on IGF-I–induced activation of Erk 1 and 2 was completely In order to document the role of Ang II in the develop- abolished by addition of a B2 antagonist, by chelation of intra- ment of diabetic glomerulosclerosis, the combined ef- cellular calcium and by tyrosine phosphatase inhibition. Addi- fects of Ang II and insulin on their signaling pathways tionally, BK reduced MC proliferation induced by IGF-I. has been investigated in mesangial cells (MCs) [13]. It Conclusions. A new inhibitory pathway of the early steps of IGF-I signaling by the B2 receptor is found both in cultured was established that Ang II and insulin are additive in MCs and in IG, which involves a calcium-dependent tyrosine stimulating the expression of transforming growth factor- phosphatase activity. Recruitment of this mechanism may ac- beta (TGF-), a major prosclerotic cytokine up-regu- count for the beneficial effects of ACE inhibitor treatment on lated by hyperglycemia. In contrast, possible cross-talk glomerulosclerosis associated with diabetic nephropathies. between BK and insulin in the kidney has never been investigated. In the past years, our group has docu- Glomerular expansion during glomerulosclerosis re- mented in detail the presence of the BK receptors in the sulting from mesangial cell (MC) hypertrophy and mes- kidney and in cultured MCs [reviewed in 14]. Initially, angial matrix accumulation is believed to be a key event we observed that B2 receptor stimulation induced prolif- eration of quiescent MCs [15], whereas our subsequent study demonstrated that BK also was able to reduce Key words: insulin-like growth factor, diabetic nephropathy, MAP ki- nase, tyrosine phosphatase, cell proliferation, glomerulosclerosis. MC proliferation induced by fetal calf serum (FCS) [16]. These opposite effects of BK have been confirmed by Received for publication August 15, 2001 other groups. The stimulating effect of BK on mitogen- and in revised form March 14, 2002 Accepted for publication March 19, 2002 activated protein (MAP) kinase has been demonstrated in MCs [17, 18], smooth muscle cells [19] and in cell lines 2002 by the International Society of Nephrology 412
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Inhibition of IGF-I–induced Erk 1 and 2 activation and mitogenesis in mesangial cells by...

Kidney International, Vol. 62 (2002), pp. 412–421
Inhibition of IGF-I–induced Erk 1 and 2 activation andmitogenesis in mesangial cells by bradykinin
CELINE ALRIC, CHRISTIANE PECHER, ERIC CELLIER, JOOST P. SCHANSTRA, BRUNO POIRIER,JACQUES CHEVALIER, JEAN-LOUP BASCANDS, and JEAN-PIERRE GIROLAMI
INSERM U388, Institut Louis Bugnard, CHU Rangueil, Toulouse, and INSERM U430, Hopital Broussais, Paris, France
Inhibition of IGF-I–induced Erk 1 and 2 activation and mito- leading to diabetic nephropathy. Insulin-like growth fac-genesis in mesangial cells by bradykinin. tor-I (IGF-I) and to a lesser extent insulin, both induce
Background. The beneficial effects of therapeutic angioten- MC proliferation and collagen secretion most likely viasin-converting enzyme (ACE) inhibitor treatment against theactivation of the IGF-I receptor. Insulin thus may beworsening of glomerulosclerosis during the course of diabeticinvolved in the early steps of the glomerulosclerotic pro-nephropathy have been widely documented. ACE inhibitors
inhibit both angiotensin II formation and bradykinin (BK) cess [1–4]. The renoprotective effect of angiotensin-con-degradation, thereby reducing angiotensin II type 1 (AT1) verting enzyme (ACE) inhibitors on the progression ofreceptor activity and favoring B2-kinin receptor (B2 receptor)
several renal diseases, including diabetic nephropathy,activation. Since the involvement of growth factors such asis supported by a large number of reports [5–12]. Severalinsulin-like growth factor (IGF-I) has been implicated in the
early steps of diabetic nephropathy, we investigated the effect mechanisms were proposed to account for the beneficialof BK on Erk 1 and 2 activation and cell proliferation by IGF-I. effects of ACE inhibitors. ACE inhibitors can inhibit the
Methods. The activation of Erk 1 and 2 in mesangial cells angiotensin II (Ang II)-induced increase in glomerular(MCs) and isolated glomeruli (IG) was investigated by immu-
capillary resistance but also act on the Ang II-stimulatednoprecipitation and Western blotting during activation of thesecretion of matrix components by renal cells, indepen-IGF-I receptor in the presence or absence of BK and of protein
kinase C (PKC), tyrosine-kinase and phosphatase selective in- dently of any hemodynamic effects. Besides the Ang II-hibitors. Mesangial cell proliferation was assessed in vitro by dependent mechanism, ACE inhibitors reduce the enzy-cell counting. matic degradation of bradykinin (BK), thereby favoring
Results. In untreated MCs and IG, when added separately,the stimulation of the B2 receptor (B2 receptor). ThisBK and IGF-I both activated Erk 1 and 2. In contrast, in MCsis an additional potential mechanism in the protectiveand IG pretreated with BK, the IGF-I–induced Erk 1 and 2 ac-
tivation was dose-dependently reduced. The inhibitory effect of role of ACE inhibitor that has been poorly investigated.BK on IGF-I–induced activation of Erk 1 and 2 was completely In order to document the role of Ang II in the develop-abolished by addition of a B2 antagonist, by chelation of intra- ment of diabetic glomerulosclerosis, the combined ef-cellular calcium and by tyrosine phosphatase inhibition. Addi-
fects of Ang II and insulin on their signaling pathwaystionally, BK reduced MC proliferation induced by IGF-I.has been investigated in mesangial cells (MCs) [13]. ItConclusions. A new inhibitory pathway of the early steps
of IGF-I signaling by the B2 receptor is found both in cultured was established that Ang II and insulin are additive inMCs and in IG, which involves a calcium-dependent tyrosine stimulating the expression of transforming growth factor-phosphatase activity. Recruitment of this mechanism may ac- beta (TGF-�), a major prosclerotic cytokine up-regu-count for the beneficial effects of ACE inhibitor treatment on
lated by hyperglycemia. In contrast, possible cross-talkglomerulosclerosis associated with diabetic nephropathies.between BK and insulin in the kidney has never beeninvestigated. In the past years, our group has docu-
Glomerular expansion during glomerulosclerosis re- mented in detail the presence of the BK receptors in thesulting from mesangial cell (MC) hypertrophy and mes- kidney and in cultured MCs [reviewed in 14]. Initially,angial matrix accumulation is believed to be a key event we observed that B2 receptor stimulation induced prolif-
eration of quiescent MCs [15], whereas our subsequentstudy demonstrated that BK also was able to reduceKey words: insulin-like growth factor, diabetic nephropathy, MAP ki-
nase, tyrosine phosphatase, cell proliferation, glomerulosclerosis. MC proliferation induced by fetal calf serum (FCS) [16].These opposite effects of BK have been confirmed by
Received for publication August 15, 2001other groups. The stimulating effect of BK on mitogen-and in revised form March 14, 2002
Accepted for publication March 19, 2002 activated protein (MAP) kinase has been demonstratedin MCs [17, 18], smooth muscle cells [19] and in cell lines 2002 by the International Society of Nephrology
412

Alric et al: Cross-talk between bradykinin and IGF-I 413
overexpressing the B2 receptor [20, 21]. More recently were lysed under stirring for 30 minutes in 1 mL of ice-BK-induced inhibition of smooth muscle cell prolifera- cold lysis buffer [10 mmol/L Tris/HCl (pH 7.4 containingtion was demonstrated; however, while this effect was 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraace-independent of phospholipase C (PLC) and phospholi- tic acid (EDTA), 1 mmol/L egtazic acid (EGTA), 0.2pase A2 (PLA2), the authors were not able to propose mmol/L orthovanadate, 1% NP 40 (vol/vol), 1% Tritona more precise mechanism [22, 23]. X-100 (vol/vol), 0.2 mmol/L phenylmethylsulfonyl fluo-
We hypothesized that the effect of BK on cell prolifer- ride (PMSF), 1 �g/mL leupeptin, 1 �g/mL aprotinin]. In-ation may be dependent upon growth factor receptor soluble material was removed by centrifugation at 13,000activation. IGF-I is a peptide growth factor synthesized rpm for 20 minutes. Protein concentration was deter-by many cells and tissues including MCs. An increase mined by the Bradford protein assay. Proteins of the sol-in renal IGF-I levels has been implicated during the uble extract were boiled in Laemli buffer for five minutesdevelopment of diabetic glomerulosclerosis by promot- at 100�C and stored frozen until sodium dodecyl sulfate-ing cell proliferation and collagen secretion [24–26]. In polyacrylamide gel electrophoresis (SDS-PAGE).the present article, we extend the knowledge of themechanism of BK-induced activation of MAP kinase in Isolated glomerulus preparationMCs in the absence and the presence of IGF-I. We found Freshly isolated glomeruli are the appropriate ex vivothat BK alone induced a transient activation of Erk 1 preparation to extend the in vitro observations obtainedand 2, whereas it significantly reduced IGF-I-induced with cultured MCs to a more physiological environment.Erk 1 and 2 activation, both in MCs and in IG. We Isolated glomeruli were prepared as routinely performedpropose a mechanism for this inhibitory cross-talk in- in the laboratory by graded sieving. Briefly, male Sprague-volving a calcium-dependent tyrosine phosphatase. Fi- Dawley rats (12 weeks of age) were exsanguinated andnally, we were able to demonstrate that BK reduced MC the kidneys were quickly removed. The cortex was forcedproliferation induced by IGF-I. through three consecutive steel sieves with decreasing
pore sizes (180, 125 and 75 �m) and the glomeruli werecollected on the 75 �m sieve. About 12,000 glomeruliMETHODSper kidney were obtained. Under light microscopy, moreRat MC culturesthan 90% of the glomeruli appeared to be decapsulated
Mesangial cells were obtained as outgrowths of de- and free of surrounding tubules and arterioles. The glo-capsulated collagenase-digested glomeruli obtained by meruli were resuspended in RPMI culture medium andgraded sieving as described previously [27]. Glomerular
redistributed in experimental tubes containing aboutexplants were allowed to grow to confluency at 37�C
5000 glomeruli per tube. After the appropriate incuba-in a humidified atmosphere of 5% CO2 in RPMI 1640tion time at 37�C in the presence of various concentra-medium containing 15% FCS, 100 U/mL penicillin, 100tions of BK and/or IGF-I, the incubation was stopped by
�g/mL streptomycin and d-valine substituted for l-valineadding 1 mL of ice-cold PBS containing 1 mmol/L of theto prevent fibroblast development. With this method,tyrosine phosphatase inhibitor orthovanadate. Then theMCs appear in the culture after two to three weeks.tubes were centrifuged (15,000 rpm, 4�C, 2 min) andThey were characterized as previously described by mor-the supernatant was discarded. The pellet containing thephological criteria, that is, the presence of multilayers,glomeruli was resuspended in 100 �L of lysis buffer,resistance to puromycin (10 �g/mL), sensitivity to mito-sonicated for 10 seconds and centrifuged (15,000 rpm,mycin (5 �g/mL), presence of myosin filaments revealed4�C, 15 min). Insoluble material was removed and theby specific antibodies, presence of actin filaments re-proteins of the soluble extracts were boiled in Laemlivealed by fluorescent NBD phallacidin, positive stainingbuffer for five minutes and stored frozen until SDS-for the MC marker Thy-1.1, negative staining for thePAGE. Protein concentration was determined by theendothelial cell marker factor VIII, and functional crite-Bradford protein assay.ria such as an increase in intracellular calcium induced by
Ang II [27]. Experiments were performed with primarySDS-PAGE and Western blottingcultures and passaged cells (passages 4 to 10).
Equal amounts of proteins (30 �g) were separated byCell extract preparation 10% SDS-PAGE in Tris/glycine buffer under 150 volts
and 30 mA current in a Bio-Rad miniature gel apparatusConfluent cells were washed three times with serum-(Mini-protean; Bio-Rad Laboratories, Richmond, CA,free culture medium and exposed to various agents inUSA). Proteins were then transferred to a nitrocelluloseserum-free culture medium at 37�C. The reactions weremembrane (Amersham SA, Orsay, France). The mem-stopped by rapid aspiration of the medium and rinsingbrane was blotted with the appropriate antibody. Proteinsthree times with ice-cold phosphate-buffered saline
(PBS) containing 1 mmol/L sodium orthovanadate. Cells were visualized using a horseradish peroxidase-conjugated

Alric et al: Cross-talk between bradykinin and IGF-I414
goat anti-rabbit immunoglobulin and an enhanced chemi- served five minutes after stimulation with 100 nmol/Lluminescence (ECL) kit (Amersham SA). BK (Fig. 1B). After 10 minutes of stimulation with BK,
Erk 1 and 2 phosphorylation returned to levels detectedMitogen-activated protein kinase activity in the absence of BK. Total Erk 1 and 2 expression,
Mitogen-activated protein kinase activity was assessed studied with an antibody against phosphorylated andby Western blotting using antiphospho-Erk antibodies non-phosphorylated forms of Erk, remained unchanged(Promega, Madison, WI, USA), which recognize the ac- during stimulation with BK. In order to determine thetive form of Erk 1 (MAP-kinase 1, molecular wt � 44 receptor subtype involved in the BK-induced stimulationkD) and Erk 2 (MAP kinase 2, molecular wt � 42 kD). of Erk 1 and 2 phosphorylation, MCs were pretreatedThe amount of total Erk also was visualized as a control with B1 and B2 antagonists, as shown in Figure 1C. Theusing an antibody that recognizes total Erk 1 and 2 pro- B1 and B2 antagonists had no effect on Erk 1 and 2teins (Santa Cruz Biotechnology, Le Perray, France) in- phosphorylation when given alone (Fig. 1C, lanes 15dependently of their level of phosphorylation. In a pre- and 16). Pretreatment of MCs with 1 �mol/L of theliminary study, we established that in our experimental B2 antagonist HOE-140 (Fig. 1C, lane 14) but not withconditions the detection of MAP kinase activity by this 1 �mol/L of the B1 antagonist des-Arg9-Leu8-BK (Fig.method was highly correlated with the incorporation of
1C, lane 17), completely abolished the BK-induced stim-radioactive phosphorus in the myelin basic protein.
ulation of Erk 1 and 2 phosphorylation (Fig. 1C, lane 14vs. lane 13).Proliferation studies
Mesangial cell proliferation was studied by counting Involvement of protein kinase C via a pertussis toxin-the number of cells (Coulter cell counter ZM; Flow Lab- dependent signaling pathway in the BK-inducedoratory, Paris, France). MCs were seeded in 12-well phosphorylation of Erk 1 and 2plates (Nunc; Polylabo, Strasbourg, France) at a density
When cells were pretreated for 24 hours with pertussisof 40,000 cells per well. After a 48-hour initial period intoxin (PTX; 100 ng / mL), the stimulating effect of 100the presence of culture medium containing 15% FCS tonmol/L BK on Erk 1 and 2 phosphorylation was inhibitedallow adhesion of the cells, they were rendered quiescent(Fig. 2, lane 4 vs. lane 2) whereas PTX alone had noby 48 hours of serum deprivation in 0.5% FCS medium.effect (Fig. 2, lane 3). In addition, the stimulating effectThen the effects of IGF-I in the presence and absenceof 100 nmol/L BK also was completely abolished by 10of BK were tested. The number of cells in triplicate
wells was determined after 48 or 72 hours as previously �mol/L U73122, a phospholipase C (PLC) inhibitor (Fig.described [15]. The results are expressed as absolute 2, lane 8 vs. lane 6). Next, we investigated the involve-values � SEM. ment of protein kinase C (PKC; Fig. 3). A five-minute
treatment with 0.3 �mol/L phorbol 12-myristate 13-ace-Statistical analysis tate (PMA), resulting in translocation of isoforms �, �
Results are expressed as mean � SEM. The t test for and ε but not � (data not shown), induced a 35-foldunpaired data was used for comparisons between two increase in Erk 1 and 2 phosphorylation (Fig. 3A, lanegroups. For multiple comparisons, results were analyzed 1 vs. lane 3). In these conditions where PKC was highlyusing a one-way analysis of variance (ANOVA) with a stimulated, no additional stimulation of Erk 1 and 2Tukey-Kramer post-hoc test. Means were considered to phosphorylation was observed in response to stimulationbe significantly different when P 0.05. All analyses with 100 nmol/L BK for five minutes (Fig. 3A, lane 2were performed with Statview 5� software (SAS Insti- vs. lane1). The stimulating effect observed after a fivetute Inc., Cary, NC, USA).
minute treatment with 100 nmol/L BK was preventedby prior incubation with 5 �mol/L PKC inhibitor GF
RESULTS 109203X (Fig. 3B, lane 7 vs. lane 5). Moreover, whenthe MCs were pre-incubated for 18 hours with 0.3 �mol/LEffect of BK on Erk 1 and 2 phosphorylation inPMA, a situation associated with complete down-regula-quiescent mesangial cellstion of PKC (data not shown), the stimulating effect ofThe dose- and time-dependent effects of BK on theBK on Erk 1 and 2 was no longer observed (Fig. 3C,phosphorylation of Erk 1 and 2 were studied by Westernlane 11 vs. lane 9). All these data indicate that Erk 1blot analysis (Fig. 1). Under our cell culture conditions,and 2 phosphorylation can be directly triggered by acti-BK induced Erk 1 and 2 phosphorylation in a dose-vation of PKC and that BK-induced Erk1 and 2 phos-dependent manner from 1 to 100 nmol/L, reaching aphorylation is mediated via PLC and PKC recruitment.maximum stimulation of sixfold (Fig. 1A). The BK-In all these experiments the total form of Erk 1 and 2induced stimulation of Erk 1 and 2 phosphorylation was
transient, and the maximum sixfold stimulation was ob- remained unchanged.

Alric et al: Cross-talk between bradykinin and IGF-I 415
Fig. 1. Dose- (A) and time-dependent (B) effect of bradykinin (BK) and pharmacological profile (C ) of BK activation of Erk 1 and 2 phosphoryla-tion. Serum-depleted rat MCs were incubated: (A) for 5 min in the presence of increasing concentrations of BK (0 to 1000 nmol/L); (B) in thepresence of 100 nmol/L BK for different times (from 1 to 10 min); (C) in the presence of 100 nmol/L BK with either 1 �mol/L of the B2 antagonistHOE 140 (B2A) or 1 �mol/L of the B1 antagonist des-Arg9-Leu8-BK (B1A). Erk 1 and 2 phosphorylation (P-Erk) was measured by Westernblotting with an antibody against the phosphorylated forms. Analyses were performed on an equal amount of protein (30 �g) as measured withan antibody against total-Erk (phosphorylated and non-phosphorylated forms). Erk 1 (�) and Erk 2 (�) phosphorylation (P-Erk 1 and 2) wasexpressed by the fold increase of the ratio P-Erk versus total-Erk and is shown as mean � SE of the scanning densitometry of five experiments.*P 0.05; **P 0.01 when compared to the level of P-Erk in the absence of BK. ��P 0.01 when compared to maximal P-Erk level detectedin MCs stimulated with 100 nmol/L BK (lane 13).
we evaluated the effects of two different tyrosine kinaseinhibitors: PD098059, an inhibitor of MEK1 (MAP ki-nase kinase); and PPI, an inhibitor of the src proteinkinase family. Figure 4 shows that both 20 �mol/L PPI(Fig. 4, lanes 3 and 4) and 50 �mol/L PD098059 (Fig. 4,lanes 7 and 8) abolished the stimulating effect of BK,indicating that a tyrosine kinase activity also is responsi-ble for the action of the B2 receptor on MAP kinaseactivation. The total forms of Erk 1 and 2 remainedunchanged in all experiments.
Effect of BK on Erk 1 and 2 activation in MCsstimulated with IGF-I
Figure 5A demonstrates that IGF-I induced a dose-dependent increase in Erk 1 and 2 phosphorylation inFCS-free incubation medium. Moreover, when the ef-
Fig. 2. Effects of pertussis toxin (PTX) and phospholipase C inhibitor fects of IGF-I were compared with that of insulin (data(U73122) on BK-stimulated Erk 1 and 2 phosphorylation. Serum-not shown), it appeared that both insulin and IGF-Idepleted rat MCs were incubated in the presence or absence of 100
nmol/L BK for 5 min and in the presence of PTX (lanes 3 and 4) or stimulated phosphorylation of Erk 1 and 2. However,of 10 nmol/L U73122 (lanes 7 and 8). Western blotting was performed
IGF-I showed a much higher efficacy (10 nmol/L) thanas described in the legend of Fig. 1. Symbols are: (�) Erk 1; (�) Erk2. N � 5; *P 0.05, **P 0.01 when compared to the level of P-Erk in insulin (1 �mol/L) confirming previous results indicatingthe absence of BK. ��P 0.01 when compared to maximal P-Erk level that insulin probably acts through the stimulation of thedetected in MCs stimulated with 100 nmol/L BK (lanes 2 and 6).
IGF-I receptor in MCs [2, 3]. Interestingly, when MCswere pretreated with 100 nmol/L BK for five minutes
BK-induced phosphorylation of Erk 1 and 2 requires prior to stimulation with 10 �mol/L IGF-I, the maximumtyrosine kinase activity IGF-I–induced stimulation of Erk 1 and 2 phosphoryla-
tion (Fig. 5B, lane 5) was significantly and dose-depen-To determine whether a tyrosine kinase activity wasinvolved in BK-induced Erk 1 and 2 phosphorylation, dently reduced to reach a 60 � 8% (P 0.05) maximum

Alric et al: Cross-talk between bradykinin and IGF-I416
Fig. 3. Effects of protein kinase C stimulation(A) and inhibition (B and C ) on BK-stimulatedErk 1 and 2 phosphorylation. Serum-depleted ratMCs were incubated: (A) in the presence or ab-sence of 100 nmol/L BK for 5 min either in thepresence of PMA for 5 min (lanes 1 and 2); (B)in the presence of GF109203, a PKC inhibitor(lanes 6 and 7); and (C) after pretreatment withPMA for 18 hours (lanes 10 and 11). Westernblotting was performed as described in the legendof Fig. 1. Symbols are: (�) Erk 1; (�) Erk 2. N �5, *P 0.05, **P 0.01 when compared to thelevel of P-Erk in the absence of BK. ��P 0.01when compared to maximal P-Erk level detectedin MCs stimulated with 100 nmol/L BK (lanes 5and 9).
tase inhibitors. The inhibitory effect of 100 nmol/L BKon 10 �mol/L IGF-I-induced Erk 1 and 2 phosphoryla-tion (Fig. 6; lane 3 vs. lane 2) was not altered by 100�mol/L okadoic acid, a serine-threonine phosphataseinhibitor (Fig. 6, lane 5 vs. lane 3), okadoic acid alonehaving no effect on the stimulation by 10 �mol/L IGF-I(Fig. 6, lane 4 vs. lane 2). In contrast, incubation in thepresence of the tyrosine phosphatase inhibitor orthovan-adate significantly increased the levels of phosphorylatedErk 1 and 2 (Fig. 6, lane 6 vs. lane 2) and interestinglyabolished the inhibitory effect of BK on IGF-I–inducedphosphorylation of Erk 1 and 2 (Fig. 6, lanes 7 and 8 vs.lane 3). In addition, incubation in the presence of theintracellular calcium chelator BAPTA/AM, increasedthe level of P-Erk 1 and 2 after stimulation by IGF-I
Fig. 4. Effect of tyrosine kinase and src kinase inhibition on BK-stimu- (Fig. 6, lane 12 vs. lane 10), but also completely abolishedlated Erk 1 and 2 phosphorylation. Serum-depleted rat MCs were incu- the inhibition by BK of Erk 1 and 2 phosphorylationbated in the presence or absence of 100 nmol/L BK for 5 min and PPI,
(Fig. 6, lane 13 vs. 11). The specific effect of BK andan inhibitor of the src family protein (lanes 3 and 4) or PD090059, anErk 1 and 2 kinase (MEK) inhibitor (lanes 7 and 8). Western blotting BAPTA/AM on phosphorylated Erk 1 and 2 is demon-was performed as described in the legend of Fig. 1. Symbols are: (�)
strated by the unchanged levels of total Erk 1 and 2.Erk 1; (�) Erk 2. N � 5, **P 0.01 when compared to the level ofP-Erk in the absence of BK. ��P 0.01 when compared to maximalP-Erk level detected in MCs stimulated with 100 nmol/L BK (lanes 2 Effect of BK on IGF-I induced MC proliferationand 6).
The effect of BK on MC proliferation is shown inFigure 7. As expected in quiescent MCs, only a highconcentration of BK (1 �mol/L) induced a slight increase
reduction (Fig. 5B, lanes 6 to 8). This effect was pre- in MC proliferation (Fig. 7A). In MCs that were serum-vented by B2 antagonist (Fig. 5C, lanes 10 to 12 vs. lanes
depleted to remove stimuli for cell proliferation, IGF-I7 and 8), which had no effect alone on IGF-I (Fig. 5C,induced time- and dose-dependent MC proliferationlane 9). Moreover, the level of total Erk 1 and 2 remained(not shown). The effect of BK was tested in the presenceunaffected.of the peak IGF-I stimulation (10 nmol/L for 48 h). It
Involvement of a calcium-dependent can be shown that the IGF-I–induced stimulation of celltyrosine phosphatase proliferation was significantly and dose-dependently re-
duced in the presence of BK (1 nmol/L to 1 �mol/L;As the decrease in Erk 1 and 2 phosphorylation sug-Fig. 7 B). Moreover, no evidence of cell detachment orgests the activation of tyrosine phosphatase, we tested
the effect of both serine-threonine and tyrosine phospha- change in cell viability could be detected.
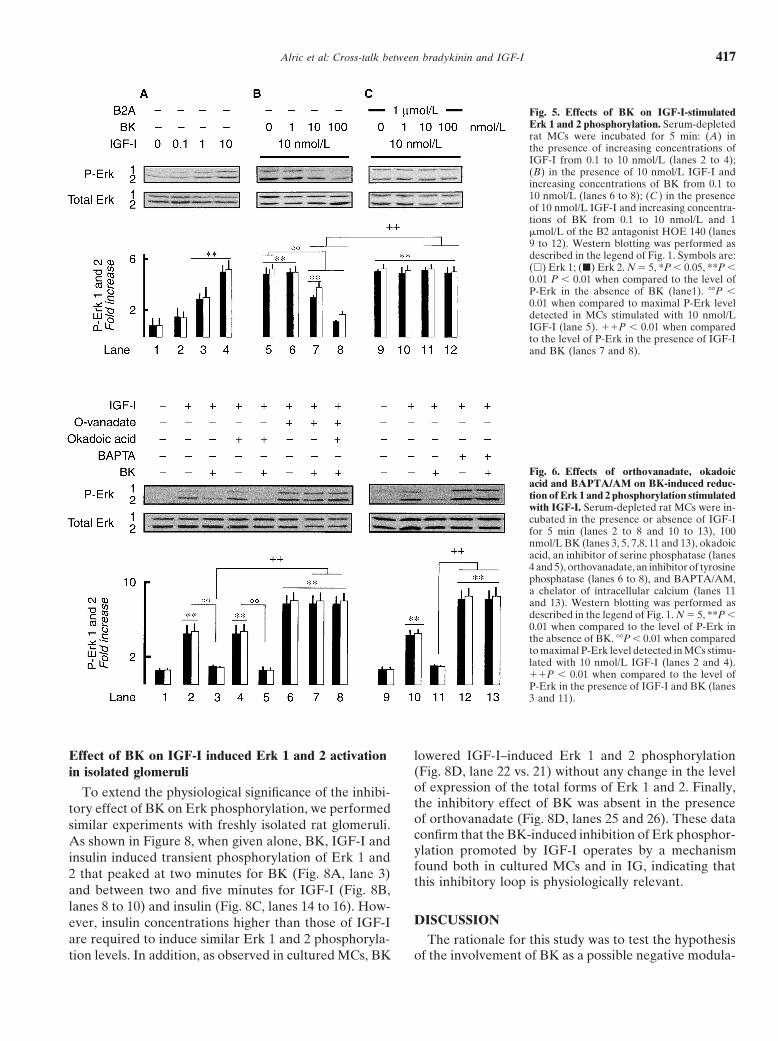
Alric et al: Cross-talk between bradykinin and IGF-I 417
Fig. 5. Effects of BK on IGF-I-stimulatedErk 1 and 2 phosphorylation. Serum-depletedrat MCs were incubated for 5 min: (A) inthe presence of increasing concentrations ofIGF-I from 0.1 to 10 nmol/L (lanes 2 to 4);(B) in the presence of 10 nmol/L IGF-I andincreasing concentrations of BK from 0.1 to10 nmol/L (lanes 6 to 8); (C ) in the presenceof 10 nmol/L IGF-I and increasing concentra-tions of BK from 0.1 to 10 nmol/L and 1�mol/L of the B2 antagonist HOE 140 (lanes9 to 12). Western blotting was performed asdescribed in the legend of Fig. 1. Symbols are:(�) Erk 1; (�) Erk 2. N � 5, *P 0.05, **P 0.01 P 0.01 when compared to the level ofP-Erk in the absence of BK (lane1). ��P 0.01 when compared to maximal P-Erk leveldetected in MCs stimulated with 10 nmol/LIGF-I (lane 5). P 0.01 when comparedto the level of P-Erk in the presence of IGF-Iand BK (lanes 7 and 8).
Fig. 6. Effects of orthovanadate, okadoicacid and BAPTA/AM on BK-induced reduc-tion of Erk 1 and 2 phosphorylation stimulatedwith IGF-I. Serum-depleted rat MCs were in-cubated in the presence or absence of IGF-Ifor 5 min (lanes 2 to 8 and 10 to 13), 100nmol/L BK (lanes 3, 5, 7,8, 11 and 13), okadoicacid, an inhibitor of serine phosphatase (lanes4 and 5), orthovanadate, an inhibitor of tyrosinephosphatase (lanes 6 to 8), and BAPTA/AM,a chelator of intracellular calcium (lanes 11and 13). Western blotting was performed asdescribed in the legend of Fig. 1. N � 5, **P 0.01 when compared to the level of P-Erk inthe absence of BK. ��P 0.01 when comparedto maximal P-Erk level detected in MCs stimu-lated with 10 nmol/L IGF-I (lanes 2 and 4).P 0.01 when compared to the level ofP-Erk in the presence of IGF-I and BK (lanes3 and 11).
Effect of BK on IGF-I induced Erk 1 and 2 activation lowered IGF-I–induced Erk 1 and 2 phosphorylation(Fig. 8D, lane 22 vs. 21) without any change in the levelin isolated glomeruliof expression of the total forms of Erk 1 and 2. Finally,To extend the physiological significance of the inhibi-the inhibitory effect of BK was absent in the presencetory effect of BK on Erk phosphorylation, we performedof orthovanadate (Fig. 8D, lanes 25 and 26). These datasimilar experiments with freshly isolated rat glomeruli.confirm that the BK-induced inhibition of Erk phosphor-As shown in Figure 8, when given alone, BK, IGF-I andylation promoted by IGF-I operates by a mechanisminsulin induced transient phosphorylation of Erk 1 andfound both in cultured MCs and in IG, indicating that2 that peaked at two minutes for BK (Fig. 8A, lane 3)this inhibitory loop is physiologically relevant.
and between two and five minutes for IGF-I (Fig. 8B,lanes 8 to 10) and insulin (Fig. 8C, lanes 14 to 16). How-
DISCUSSIONever, insulin concentrations higher than those of IGF-Iare required to induce similar Erk 1 and 2 phosphoryla- The rationale for this study was to test the hypothesis
of the involvement of BK as a possible negative modula-tion levels. In addition, as observed in cultured MCs, BK

Alric et al: Cross-talk between bradykinin and IGF-I418
initial observation, the same group revised the mecha-nism of GH-induced glomerulosclerosis and suggestedthe involvement of both GH and IGF-I during low-pro-tein-induced reduction of glomerulosclerosis [32]. Thesame authors also reported that transgenic mice for GHexhibited elevated plasmatic IGF-I concentrations asso-ciated with glomerulosclerosis in spite of normal glyce-mia [33]. Moreover, several studies have pointed outthat IGF-I acts as an initiation factor by inducing MCproliferation [3, 34] and collagen secretion [4, 29, 35],two major contributors to the development of diabeticglomerulosclerosis, whereas similar direct effects of GHon MCs have never been reported. Finally, inhibition ofearly renal growth in diabetic and uninephrectomizedrats is prevented by the IGF-I antagonist [36]. All thesearguments support rather than detract from a contribu-tion of IGF-I in diabetic nephropathies. Since MC hyper-plasia is a feature common to several human glomerulardiseases, the inhibitory effect of BK on the signalingcascade of IGF-I appears to be a potential protectiveFig. 7. Effects BK on MC proliferation. MCs were plated at a density
of 40,000 cells per well and were stimulated with increasing concentra- endogenous mechanism. This hypothesis is consistenttions of BK for 48 hours in two different conditions. (A) MCs cultured with the renoprotective effects of ACE inhibitors ob-in RPMI medium containing 0.5% FCS. (B) MCs cultured in RPMI
served in experimental models and clinical trials as com-medium containing 10 nmol/L IGF-I. Cell proliferation is shown aspercent of control values obtained in the absence of BK and is expressed pared with other antihypertensive drugs [5–12].as mean � SE of five experiments. *P 0.01, **P 0.01 when compared Moreover, beside the effects on MC proliferation andto proliferation in the absence of BK.
matrix secretion, acute elevation of IGF-I by exogenousadministration reduces renal resistance and raises theglomerular filtration rate at least in normal rats [37],whereas the hemodynamic effects of chronic endogenoustor of IGF-I signaling pathways and IGF-I–stimulated
cell proliferation. Erk 1 and 2 activity is stimulated during elevation of IGF-I concentration remain controversial.It also has been suggested that IGF-I–induced hyperfil-the early phases of hyperglycemia in diabetic rats [27]
and in MCs exposed to elevated glucose concentrations tration can be corrected by a B2-kinin antagonist [38].However, that study was conducted for only 60 minutes[28]. The contribution of IGF-I–induced Erk 1 and 2
activation and cell proliferation is proposed as an essen- with exogenous infusion of high doses of IGF-I reaching50 nmol/L. It is clear that in this acute situation IGF-Itial initial event in the development of diabetic nephrop-
athy. IGF-I is a growth factor locally released by MCs predominantly induced a vasodilatory effect. Blockadeof the B2 receptor can reduce this effect whereas thein the glomerulus during the development of diabetic
nephropathy and acts in an autocrine and paracrine fash- effect of chronic B2 blockade on the renal hemodynamicsof diabetic rats has never been reported. The variationsion [29]. Therefore, we tested the effect of BK on IGF-I–
induced Erk 1 and 2 activation. of the activity and expression of the kallikrein-kinin sys-tem during diabetes mellitus expression have been inves-Although a BK-induced inhibition of fibroblast divi-
sion stimulated by a mixture of IGF-I and EGF has been tigated and show a biphasic pattern. During an acuteinitial phase, glomerular hyperfiltration is prevented byreported [30], our study documents a possible mecha-
nism of an inhibitory effect of BK on IGF-I–induced Erk administration of a B2 antagonist. On the other hand,renal kallikrein expression is suppressed in insulin-1 and 2 activation and cell proliferation. Interestingly, the
inhibitory effect of BK is not only observed in vitro in untreated streptozotocin (STZ)-diabetic rats and thisabnormality is reversed by insulin [39].cultured MCs, but also is detectable in a physiological
renal structure such as the isolated glomerulus. However, In order to associate the inhibition of MAP kinaseactivation with a cellular response, we investigated the ef-the importance of IGF-I in the development of glomeru-
losclerosis of diabetic nephropathy could still appear fect of BK on IGF-I–induced MC proliferation. BK in-hibits IGF-I–induced MC proliferation and this is consis-controversial. The main argument against a noxious ac-
tion of IGF-I in diabetes mellitus is the initial observation tent with the inhibition of Erk phosphorylation. Whereasthe proliferative action of BK on quiescent cell systemsshowing that transgenic mice overexpressing growth hor-
mone (GH) develop glomerulosclerosis whereas trans- has been extensively investigated, few studies report aninhibitory effect of BK in smooth muscle cells, fibroblastsgenic mice overexpressing IGF-I do not [31]. Since that
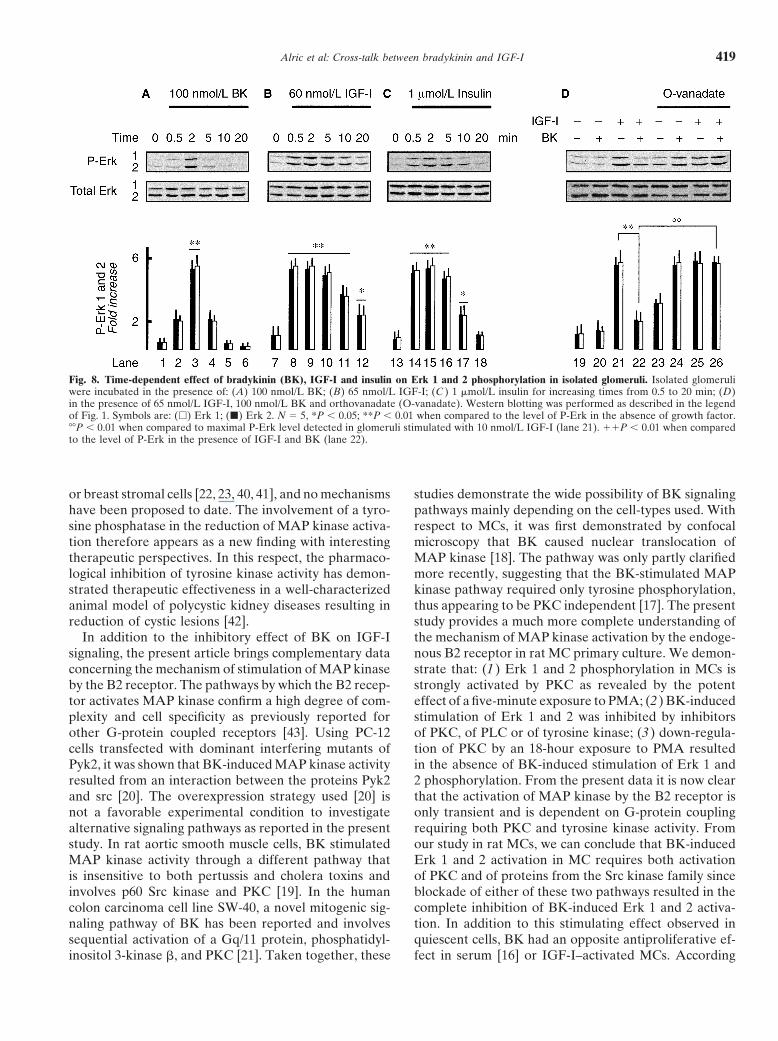
Alric et al: Cross-talk between bradykinin and IGF-I 419
Fig. 8. Time-dependent effect of bradykinin (BK), IGF-I and insulin on Erk 1 and 2 phosphorylation in isolated glomeruli. Isolated glomeruliwere incubated in the presence of: (A) 100 nmol/L BK; (B) 65 nmol/L IGF-I; (C ) 1 �mol/L insulin for increasing times from 0.5 to 20 min; (D)in the presence of 65 nmol/L IGF-I, 100 nmol/L BK and orthovanadate (O-vanadate). Western blotting was performed as described in the legendof Fig. 1. Symbols are: (�) Erk 1; (�) Erk 2. N � 5, *P 0.05; **P 0.01 when compared to the level of P-Erk in the absence of growth factor.��P 0.01 when compared to maximal P-Erk level detected in glomeruli stimulated with 10 nmol/L IGF-I (lane 21). P 0.01 when comparedto the level of P-Erk in the presence of IGF-I and BK (lane 22).
or breast stromal cells [22, 23, 40, 41], and no mechanisms studies demonstrate the wide possibility of BK signalingpathways mainly depending on the cell-types used. Withhave been proposed to date. The involvement of a tyro-
sine phosphatase in the reduction of MAP kinase activa- respect to MCs, it was first demonstrated by confocalmicroscopy that BK caused nuclear translocation oftion therefore appears as a new finding with interesting
therapeutic perspectives. In this respect, the pharmaco- MAP kinase [18]. The pathway was only partly clarifiedmore recently, suggesting that the BK-stimulated MAPlogical inhibition of tyrosine kinase activity has demon-
strated therapeutic effectiveness in a well-characterized kinase pathway required only tyrosine phosphorylation,thus appearing to be PKC independent [17]. The presentanimal model of polycystic kidney diseases resulting in
reduction of cystic lesions [42]. study provides a much more complete understanding ofthe mechanism of MAP kinase activation by the endoge-In addition to the inhibitory effect of BK on IGF-I
signaling, the present article brings complementary data nous B2 receptor in rat MC primary culture. We demon-strate that: (1) Erk 1 and 2 phosphorylation in MCs isconcerning the mechanism of stimulation of MAP kinase
by the B2 receptor. The pathways by which the B2 recep- strongly activated by PKC as revealed by the potenteffect of a five-minute exposure to PMA; (2) BK-inducedtor activates MAP kinase confirm a high degree of com-
plexity and cell specificity as previously reported for stimulation of Erk 1 and 2 was inhibited by inhibitorsof PKC, of PLC or of tyrosine kinase; (3) down-regula-other G-protein coupled receptors [43]. Using PC-12
cells transfected with dominant interfering mutants of tion of PKC by an 18-hour exposure to PMA resultedin the absence of BK-induced stimulation of Erk 1 andPyk2, it was shown that BK-induced MAP kinase activity
resulted from an interaction between the proteins Pyk2 2 phosphorylation. From the present data it is now clearthat the activation of MAP kinase by the B2 receptor isand src [20]. The overexpression strategy used [20] is
not a favorable experimental condition to investigate only transient and is dependent on G-protein couplingrequiring both PKC and tyrosine kinase activity. Fromalternative signaling pathways as reported in the present
study. In rat aortic smooth muscle cells, BK stimulated our study in rat MCs, we can conclude that BK-inducedErk 1 and 2 activation in MC requires both activationMAP kinase activity through a different pathway that
is insensitive to both pertussis and cholera toxins and of PKC and of proteins from the Src kinase family sinceblockade of either of these two pathways resulted in theinvolves p60 Src kinase and PKC [19]. In the human
colon carcinoma cell line SW-40, a novel mitogenic sig- complete inhibition of BK-induced Erk 1 and 2 activa-tion. In addition to this stimulating effect observed innaling pathway of BK has been reported and involves
sequential activation of a Gq/11 protein, phosphatidyl- quiescent cells, BK had an opposite antiproliferative ef-fect in serum [16] or IGF-I–activated MCs. Accordinginositol 3-kinase �, and PKC [21]. Taken together, these

Alric et al: Cross-talk between bradykinin and IGF-I420
10. Lewis EJ: Angiotensin-converting enzyme inhibition in type I dia-to the state of the cellular activation we suggest thatbetic nephropathy. Contrib Nephrol 118:206–213, 1996
cross-talk between the B2 receptor and IGF-I signaling 11. Marre M, Bouhanick B, Berrut G, et al: Renal changes on hyper-glycemia and angiotensin-converting enzyme in type 1 diabetes.reduces IGF-I–stimulated MAP kinase levels through aHypertens 33:775–780, 1999mechanism involving a calcium-dependent tyrosine
12. Hoelscher D, Bakris G: Antihypertensive therapy and progres-phosphatase. Besides the inhibitory effect of BK on the sion of diabetic renal disease. J Cardiovasc Pharmacol 23(Suppl):
S34–S38, 1994signaling pathway activated by IGF-I, an additive inhibi-13. Anderson PW, Zhang XY, Tian J, et al: Insulin and angiotensintory effect of BK on IGF-I mRNA in dermal fibroblasts
II are additive in stimulating TGF-beta 1 and matrix mRNAs incould participate in the attenuation of IGF-I effects [44]. mesangial cells. Kidney Int 50:745–753, 1996
14. Schanstra JP, Alric C, Marin-Castano EM, et al: Renal bradyki-The molecular mechanism of activation by BK is cur-nin receptors: Localisation, transduction pathways and molecularrently under investigation. However, the dual effect ofbasis for a possible pathological role. (Review) Int J Mol Med 3:
BK (that is, activation or inhibition of Erk phosphoryla- 185–191, 199915. Bascands JL, Pecher C, Rouaud S, et al: Evidence for existencetion) is consistent with our recent report showing that
of two distinct bradykinin receptors on rat mesangial cells. Am JBK can both activate and inhibit tyrosine kinase activityPhysiol 264:F548–F556, 1993
by two independent pathways [45]. The activation of 16. Alric C, Pecher C, Bascands JL, Girolami JP: Effect of bradyki-nin on tyrosine kinase and phosphatase activities and cell prolifera-tyrosine kinase is abolished by PTX whereas the inhibi-tion in mesangial cells. Immunopharmacology 45:57–62, 1999tion is PTX resistant. It could have important implica-
17. El-Dahr SS, Dipp S, Yosipiv IV, Baricos WH: Bradykinin stimu-tions for treatment of diseases associated with hyperprol- lates c-fos expression, AP-1-DNA binding activity and prolifera-
tion of rat glomerular mesangial cells. Kidney Int 50:1850–1855,iferation with ACE inhibitors. Consistent with our1996results, a recent study demonstrates that ACE inhibition
18. Jaffa AA, Miller BS, Rosenzweig SA, et al: Bradykinin inducesattenuates IGF-I–induced cardiac fibroblast prolifera- tubulin phosphorylation and nuclear translocation of MAP kinase
in mesangial cells. Am J Physiol 273:F916–F924, 1997tion [46]. Whether this BK-dependent mechanism is re-19. Velarde V, Ullian ME, Morinelli TA, et al: Mechanisms ofcruited during chronic treatment with ACE inhibitor
MAPK activation by bradykinin in vascular smooth muscle cells.remains to be investigated. Am J Physiol 277:C253–C261, 1999
20. Dikic I, Tokiwa G, Lev S, et al: A role for Pyk2 and Src in linkingG-protein-coupled receptors with MAP kinase activation. NatureACKNOWLEDGMENTS383:547–550, 1996
21. Graneß A, Adomeit A, Heinze, R, et al: A novel mitogenic signal-Celine Alric was supported by a grant from MNSR and Eric Cellier isa post-doctoral fellow supported by a grant from the Medical Research ing pathway of bradykinin in the human colon carcinoma cell
lane SW-480 involves sequential activation of a Gq/11 protein,Council of Canada. This work received funding from INSERM andfrom Midi-Pyrenees grant n� 99001254. phosphatidylinositol 3-kinase beta, and protein kinase C epsilon.
J Biol Chem 48:32016–32022, 199822. Dixon BS, Dennis MJ: Regulation of mitogenesis by kinins inReprint requests to Dr. Jean-Pierre Girolami, INSERM U388, Institut
Louis Bugnard, CHU Rangueil, 31403 Toulouse Cedex, France. arterial smooth muscle cells. Am J Physiol 273:C7–C20, 199723. Dixon BS, Dennis MJ: Interaction of kinins and captopril in regu-E-mail: [email protected]
lating arterial smooth muscle cell proliferation. Kidney Int 52(Suppl61):S14–S17, 1997REFERENCES 24. Oemar BS, Law NM, Rosenzweig SA: Insulin-like growth factor-1induces tyrosyl phosphorylation of nuclear proteins. J Biol Chem1. Abrass CK, Raugi GJ, Gabourel LS, Lovett DH: Insulin and266:24241–24244, 1991insulin-like growth factor I binding to cultured rat glomerular mes-
25. Schreiber BD, Hughes ML, Groggel GC: Insulin-like growthangial cells. Endocrinology 123:2432–2439, 1988factor-1 stimulates production of MC matrix components. Clin2. Arnqvist HJ, Ballermann BJ, King GL: Receptors for and effectsNephrol 43:368–374, 1995of insulin and IGF-I in rat glomerular mesangial cells. Am J Physiol
26. Shankland SJ, Scholey JW: Expression of growth-related proto-254:C411–C416, 1988oncogenes during diabetic renal hypertrophy. Kidney Int 47:782–3. Conti FG, Striker LJ, Lesniak MA, et al: Studies on binding788, 1995and mitogenic effect of insulin and insulin-like growth factor I in
27. Emond C, Bascands JL, Pecher C, et al: Characterization of a B2-glomerular mesangial cells. Endocrinology 122:2788–2795, 1988bradykinin receptor in rat renal mesangial cells. Eur J Pharmacol4. Feld SM, Hirschberg R, Artishevsky A, et al: Insulin-like growth190:381–392, 1990factor I induces mesangial proliferation and increases mRNA and
28. Haneda M, Araki S, Togawa M, et al: Mitogen-activated proteinsecretion of collagen. Kidney Int 48:45–51, 1995kinase cascade is activated in glomeruli of diabetic rats and glomer-5. Heudes D, Michel O, Chevalier J, et al: Effect of chronic ANGular mesangial cells cultured under high glucose conditions. Diabe-I-converting enzyme inhibition on aging processes. I. Kidney struc-tes 46:847–853, 1997ture and function. Am J Physiol 266:R1038–R1051, 1994
29. Flyvbjerg A, Orskow H: Role of growth hormone, insulin-like6. Omata K, Kanazawa M, Sato T, et al: Therapeutic advantages ofgrowth factors (IGDs) and IGF-binding proteins in the renal com-angiotensin converting enzyme inhibitors in chronic renal disease.plications of diabetes. Kidney Int 52(Suppl 60):S12–S19, 1997Kidney Int 49(Suppl 55):S57–S62, 1996
30. Newman EL, Hydahl L, Larson O, et al: Bradykinin blocks the7. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD: The effect ofaction of EGF, but not PDGF, on fibroblast division. FEBS J 251:angiotensin-converting-enzyme inhibition on diabetic nephropa-225–229, 1989thy. The Collaborative Study Group. N Engl J Med 329:1456–1462,
31. Doi T, Striker LJ, Quaife C, et al: Progressive glomerulosclerosis1993develops in transgenic mice chronically expressing growth hor-8. Brunner HR: ACE inhibitors in renal disease. Kidney Int 42:463–mone and growth hormone releasing factor but not in those ex-479, 1992pressing insulin-like growth factor-I. Am J Pathol 131: 398–403,9. Ruiz Ortega M, Gonzalez S, Serun D, et al: ACE inhibition1988reduces proteinuria, glomerular lesions and extracellular matrix
32. Doi SQ, Rasaiah S, Tack I, et al Low-protein diet suppressesproduction in a normotensive rat model of immune complex ne-phritis. Kidney Int 48:1778–1791, 1995 insulin-like growth factor-1 and decelerates the progression of

Alric et al: Cross-talk between bradykinin and IGF-I 421
growth hormone-induced glomeruli. Am J Nephrol 21:331–339, 40. McAllister BS, Leeb-Lundberg F, Olson MS: Bradykinin inhibi-tion of EGF- and PDGF-induced DNA synthesis in human fibro-2001blasts. Am J Physiol 265:C477–C484, 199333. Doi SQ, Hattori M, Agodoa LY, et al: Glomerular lesions in
41. Patel KV, Schrey MP: Inhibition of DNA synthesis and growthnonobese diabetic mouse: Before and after the onset of hyperglyce-in human breast stromal cells by bradykinin: Evidence for indepen-mia. Lab Invest 63:204–212, 1990dent roles of B1 and B2 receptors in the respective control of cell34. Doi T, Striker LJ, Elliot SJ: Insulin-like growth factor-1 is a pro-growth and phospholipid hydrolysis. Cancer Res 52:334–340, 1992gression factor for human mesangial cells. Am J Pathol 134:395–
42. Sweeney WE, Chen Y, Nakanishi K, et al: Treatment of polycystic400, 1989kidney disease with a novel tyrosine kinase inhibitor. Kidney Int35. Lupia E, Elliot SJ, Lenz O, et al: IGF-I: Decreases collagen57:33–40, 1997degradation in diabetic NOD mesangial cells: Implication for dia-
43. Luttrell LM, Daaka Y, Lefkowitz RJ: Regulation of tyrosinebetic nephropathy. Diabetes 48:1638–1644, 1999kinase by G protein-coupled receptor. Cur Opin Cell Biol 11:177–36. Haylor JL, Hickling H, El Eter E, et al: JB3, an IGF-I receptor 183, 1999antagonist, inhibits early renal growth in diabetic and uninephrec- 44. Lowe WL Jr, Yorek M, Teasdale RB: Ligands that activate pro-
tomized rats. J Am Soc Nephrol 11: 2027–2035, 2000 tein kinase-C differ in their ability to regulate basic fibroblast37. Hirschberg R, Kopple JD, Blantz R, et al: Effect of recombinant growth factor and insulin-like growth factor-I messenger ribo-
human insulin-like growth factor I on glomerular dynamics in the nucleic acid levels. Endocrinology 132:1593–1602, 1992rat. J Clin Invest 87:1200–1206, 1991 45. Alric C, Pecher C, Schanstra JP, et al: Bradykinin-induced inhibi-
38. Jaffa AA, LeRoith D, Roberts CT, et al: Insulin-like growth tion of cell proliferation and tyrosine kinase activity in rat mesan-factor I produces renal hyperfiltration by a kinin-mediated mecha- gial cells. Int J Mol Med 5:85–93, 2000nism. Am J Physiol 266:F102–F107, 1994 46. Van Eickels M, Vetter H: Grobe C: Angiotensin-converting en-
39. Jaffa AA, Vio C, Velarde V, et al: Induction of renal kallikrein zyme (ACE) inhibition attenuates insulin-like growth factor-Iand renin gene expression by insulin and IGF-I in the diabetic (IGF-I) induced cardiac fibroblast proliferation. Br J Pharmacol
131:1592–1596, 2000rats. Diabetes 46:2049–2056, 1997




















