
University of Groningen Modulation of lipoxygenase activity ...

Prostaglandins, Leukotrienes and Essential Fatty Acids 85 (2011) 43–52
Contents lists available at ScienceDirect
Prostaglandins, Leukotrienes andEssential Fatty Acids
0952-32
doi:10.1
$Fun
Nationan Corr
Disorde
Blvd., B
E-m1 Pr
Medical
0495, Ja
journal homepage: www.elsevier.com/locate/plefa
Inhibition of 5-lipoxygenase activity in mice during cuprizone-induceddemyelination attenuates neuroinflammation, motor dysfunction andaxonal damage$
K. Yoshikawa 1, S. Palumbo, C.D. Toscano, F. Bosetti n
Molecular Neuroscience Unit, Brain Physiology and Metabolism Section, National Institute on Aging, National Institute of Health, Bethesda, MD, USA
a r t i c l e i n f o
Article history:
Received 4 February 2011
Received in revised form
6 April 2011
Accepted 12 April 2011
Keywords:
5-Lipoxygenase
Leukotriene
Multiple sclerosis
Cuprizone
Demyelination
Neuroinflammation
Axonal damage
78/$ - see front matter Published by Elsevier
016/j.plefa.2011.04.022
ding: This work was supported by Intramur
l Institute on Aging.
esponding author. Present address: Nation
rs and Stroke, NIH, Neuroscience Center, R
ethesda, MD 20892, USA. Tel.: þ1 301 496 12
ail address: [email protected] (F. Bosetti).
esent address: Department of Pharmacology,
University, 38 Morohongo Moroyama-mach
pan.
a b s t r a c t
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system
(CNS). Increased expression of 5-lipoxygenase (5-LO), a key enzyme in the biosynthesis of leukotrienes
(LTs), has been reported in MS lesions and LT levels are elevated in the cerebrospinal fluid of MS
patients. To determine whether pharmacological inhibition of 5-LO attenuates demyelination, MK886, a
5-LO inhibitor, was given to mice fed with cuprizone. Gene and protein expression of 5-LO were
increased at the peak of cuprizone-induced demyelination. Although MK886 did not attenuate
cuprizone-induced demyelination in the corpus callosum or in the cortex, it attenuated cuprizone-
induced axonal damage and motor deficits and reduced microglial activation and IL-6 production.
These data suggest that during cuprizone-induced demyelination, the 5-LO pathway contributes to
microglial activation and neuroinflammation and to axonal damage resulting in motor dysfunction.
Thus, 5-LO inhibition may be a useful therapeutic treatment in demyelinating diseases of the CNS.
Published by Elsevier Ltd.
1. Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinat-ing disease of the central nervous system (CNS) characterized byrecurrent and progressive demyelination/remyelination cycles,resulting in development of scleroses in both the white and graymatter of the CNS [1], axonal damage, neuroinflammation andneuronal loss [2,3]. Demyelination is accompanied by depletion ofoligodendrocyte precursor cells, loss of mature oligodendrocytes,astrogliosis, and infiltration of macrophages/microglia and Tlymphocytes [4].
The cuprizone model of demyelination [5] is characterized byapoptotic death of mature oligodendrocytes [6,7], and is accom-panied by neuroinflammation [8] and motor dysfunction [9]. Fourpatterns have been described in MS lesions, with patterns Iand II showing similarities to T-cell-mediated or T-cell plus
Ltd.
al Research Program of NIH,
al Institute of Neurological
oom 2118; 6001 Executive
97.
Faculty of Medicine, Saitama
i, Iruma-gun, Saitama 350-
antibody-mediated autoimmune encephalomyelitis, respectively,and patterns III and IV lesions suggesting a primary oligodendro-cyte damage and degeneration, reminiscent of virus- or toxin-induced demyelination rather than autoimmunity [10]. Thus, thecuprizone model of demyelination is closer to patterns III and IVlesions in reproducing a primary demyelination that is indepen-dent from the immune system, and axonal damage. Mice showprogressive demyelination when they are kept on a 0.2% cupri-zone diet, with a peak in demyelination observed after 5 weeks ofcuprizone [6,11]. Cuprizone withdrawal from the diet results in aremyelination after several weeks [12].
Omega-6 fatty acids, such as linoleic acid, g-linoleic acid andarachidonic acid, have been implicated in demyelinating disease.Linoleic acid and g-linoleic acid have shown protective effects inMS and experimental autoimmune encephalomyelitis (EAE)[13–16]. On the other hand, arachidonic acid cascade is suggestedto become activated during demyelination[17,18]. Increasedexpression of 5-lipoxygenase (5-LO) in lesions [19,20] and of5-LO-derived leukotriene (LT) products in the cerebrospinal fluid[21] has been reported in patients with MS. 5-LO, a key enzyme inthe biosynthesis of LTs [22] is activated by 5-LO-activatingprotein (FLAP) and converts arachidonic acid to LTA4, which isthen converted to LTB4 by LTA4 hydrolase [23,24], or to acysteinyl-LT (such as LTC4, LTD4 or LTE4) by LTC4 synthase [25,26].
MK886 is a 5-LO inhibitor that binds to FLAP and therebyprevents 5-LO activation [27]. In vitro, MK886 inhibits LTs

K. Yoshikawa et al. / Prostaglandins, Leukotrienes and Essential Fatty Acids 85 (2011) 43–5244
biosynthesis in leukocytes [28]. In vivo, systemic administrationof MK886 has been shown to inhibit LPS-induced hypothalamicLT production [29] and cortical cysteinyl-LT production inducedby permanent occlusion of the middle cerebral artery [30].
In this study, to determine whether 5-LO activity contributesto the pathological events associated with demyelination andassociated neuroinflammation, we administered MK886 to cupri-zone-exposed mice. We demonstrated that, although MK886 didnot attenuate cuprizone-induced demyelination in the corpuscallosum and cortex, it reduced microglial activation, IL-6 produc-tion, axonal damage and motor dysfunction. These data suggestthat the 5-LO pathway is involved in microglial activation andneuroinflammation and contributes to axonal damage and motordysfunction.
2. Materials and methods
2.1. Animal procedures
All animal experiments were performed under a NIH approvedanimal protocol (NICHD #08-026) approved by the NIH, NICHDAnimal Care and Use Committee, in accordance with the NIHguidelines on the care and use of laboratory animals. C57BL/6male mice (Taconic Farms, Germantown, NY) were received at ourfacility at 8–10 weeks of age and were fed ad libitum a powdereddiet (Purina #5002; formulated by Research Diets, New Bruns-wick, NJ) containing 0.2% cuprizone (bis-cyclohexanone oxaldihy-drazone; Sigma, St. Louis, MO). In a preliminary experiment, micewere fed with cuprizone diet for 6 weeks to investigate time-dependent changes of 5-LO gene and protein expression duringthe demyelination process (Fig. 1A). In the following experiments,mice were fed with cuprizone diet up to 5 weeks, whichrepresents the peak of demyelination, to investigate the effectsof 5-LO inhibition on demyelination, neuroinflammation andmotor function (Fig. 1D). Mice were maintained on a 12/12 hlight/dark cycle. Mice (n¼7–8 per group) were euthanized withNembutal and forebrain containing frontal cortex and corpuscallosum was dissected on ice. Cerebellum, thalamus, hippocam-pus, striatum and olfactory bulb were excluded from the dis-sected samples. Forebrain was rapidly frozen in 2-methylbutaneat �50 1C, and stored at �80 1C until use for molecular analysis.For histology, mice (n¼5 per group) were intracardially perfusedwith 4% paraformaldehyde. Brains were postfixed overnight in 4%paraformaldehyde, subsequently cryoprotected in a 30% sucrosesolution, snap frozen and stored at �80 1C until use [31].
2.2. Treatment with MK886
MK886 (Cayman Chemical, Ann Arbor, MI) was dissolved insaline with 5% DMSO, 25% polyethylene glycol-15-hydroxystea-rate (Solutol, BASF, Ludwigshafen, Germany). MK886 was admi-nistered at a dose of 3 mg/kg by intraperitoneal (i.p.) injectiononce-daily for the last 7 days (weeks 4–5) of cuprizone exposure.Control mice on a normal cuprizone-free diet were treated inparallel with MK886 at the same dose once-daily for 7 days.
2.3. Western blotting
The cytosolic fraction was prepared from forebrain as des-cribed [32]. Forebrains (n¼6 per group) were homogenized in ahomogenizing buffer containing 20 mM Tris–HCl (pH 7.5), using aPolytrons homogenizer. The supernatant was centrifuged at100,000g for 60 min at 4 1C. The supernatant was collected andprotein concentration was measured using a Dc Protein Assay kit(Bio-Rad, Richmond, CA). Western blotting was performed as
previously described [32]. Briefly, proteins (50 mg) were loadedon Criterion gels (Bio-Rad), transferred onto a polyvinylidenedifluoride membrane (Bio-Rad) and immunoblotted with anti-bodies against 5-LO (1:500; Cayman Chemical) and b-actin(1:3000; Sigma) as loading control. An Odyssey Infrared ImagingSystem (Licor Biosciences, Lincoln, NB,) was used to detect andquantify protein levels.
2.4. Measurement of IL-6 levels
Forebrains (n¼5 per group) were homogenized in a lysisbuffer containing 25 mM Tris–HCl, pH 7.8, 150 mM NaCl, 1 mMEDTA with a complete protease inhibitor cocktail (Roche, India-napolis, IN). The homogenates were centrifuged at 14,000g for20 min, and the supernatant was immediately assayed using amouse IL-6 ELISA kit (Invitrogen, Carlsbad, CA).
2.5. Histology
Thirty mm coronal brain sections were cut on a cryostat(Bright Instrument Company, Ltd.; Huntingdon, England) andmounted on gelatin-coated glass slides. Sections were stainedwith Black Gold II (Histo-Chem, Jefferson, AR) as previouslydescribed [33]. Briefly, sections were incubated in a 0.2% BlackGold II solution for 12 min, rinsed in distilled water, fixed in 2%sodium thiosulfate, rinsed in tap water and air-dried. Sectionswere then cleared in Histo-Clear (National Diagnostics, Atlanta,GA) and coverslipped using DPX (Sigma) mounting medium.Immunohistochemistry was performed using rat anti-mouseCD11b (1:200; Serotec, Oxford, UK) as primary antibody atroom temperature overnight and visualized using VECTASTAINABC kit (Vector Laboratories, Burlingame, CA) and counter-stained them with VECTOR hematoxylin QS (Vector). Doubleimmunofluorescence was performed using anti-mouse amyloidprecursor protein (APP) (1:200; Chemicon, Temecula, CA) andanti-rabbit neurofilament 200 (NF200) (1:80, Sigma), as fol-lows. Sections were incubated with a mixture of two primaryantibodies at room temperature overnight, followed by incuba-tion at room temperature for 1 h with a mixture of the twosecondary antibodies (Alexa Fluor 594 goat anti-mouse IgG andAlexa Fluor 488 goat anti-rabbit IgG; 5 mg/ml, Invitrogen).Stained sections were imaged with a Leica TCS SP5confocalmicroscope system (Leica Microsystems, Wetzlar, Germany).All images were imported into Image J, CD11b-positive cells(corpus callosum and cortex) and APP-NF200 double positiveaxons in the corpus callosum were counted, and densities(counts/mm2) calculated.
2.6. Quantification of demyelination in the corpus callosum and
cortex
Black Gold stained sections were selected between Bregma�0.22 and �0.58 mm. Section were photographed (OlympusU-CMAD3 camera) at 10� magnification, the images wereopened with Spot Advanced 4.1 software and imported intoImage J, which was used to measure the mean optical densitywithin the middle of the corpus callosum, at the level of thefimbria, and of the cortex (primary somatosensory cortex andmotor cortex). Optical density in no tissue area was used as blank(background) and blank was subtracted using Spot Advanced4.1 software. Myelin densities for each mouse were normalizedagainst optical density values in unchallenged mice using thefollowing formula: myelin score (%)¼(density reading/unchal-lenged density average)�100.

Fig. 1. Study design and 5-LO expression levels. In a preliminary experiment, mice were fed with the cuprizone diet for 6 weeks to investigate 5-LO expression levels (A).
Gene expression of 5-LO relative to pgk-1, as determined by real-time PCR (B) and protein levels as determined by western blotting (C) in the forebrain of cuprizone-
exposed up to 6 weeks and control mice. Data are means7SEM, n¼6. Statistical analysis was performed using one-way ANOVA followed by post-hoc Newman–Keuls test
(*po0.05, ***po0.001 vs. week 0). In the following experiments, mice were fed with cuprizone diet up to 5 weeks and injected with MK886 i.p. once-daily for the last
7 days (weeks 4–5) of cuprizone exposure (D).
K. Yoshikawa et al. / Prostaglandins, Leukotrienes and Essential Fatty Acids 85 (2011) 43–52 45
2.7. RNA extraction and quantitative real-time PCR (Q-PCR)
Fresh frozen forebrains (n¼7–8 per group) were processed forRNA extraction using the Qiagen RNeasy Lipid Tissue Mini kit(Qiagen, Valencia, CA) following the manufacturer’s procedure.DNase treatment was performed during RNA purification to avoidgenomic DNA contamination and RNA purity was verified byexamining the 260/280 nm ratio using a spectrophotometer.Extracted RNA was resuspended in RNase free molecular gradewater and stored at �80 1C until usage. For Q-PCR, total RNA(5 mg) was reverse transcribed using a High Capacity cDNAArchive kit (Applied Biosystems, Foster City, CA).
Q-PCR was performed using the ABI PRISM 7000 SequenceDetection System (Applied Biosystems). Q-PCR results werenormalized to phosphoglycerate kinase 1 (PGK1; Mm_00435617_m1) expression levels, as previously reported [32].Gene expression was analyzed using the following Assays onDemand: Myelin Basic Protein (MBP; Mm01262035_m1); GlialFibrillary Acidic Protein (GFAP; Mm01253033_m1); Integrinalpha M (CD11b; Mm00434455_m1); Interleukin 6 (IL-6;Mm01210733_m1); Interleukin 1b (IL-1b; Mm99999061_m1);Tumor necrosis factor a (TNFa; Mm00443258_m1); andG protein-coupled receptor 17 (GPR17; Mm02619401_s1).Briefly, Taqman Universal PCR Master Mix, Assay-On-Demand
primers and cDNA samples were mixed in RNAse-free water andadded to an optical 96-well reaction plate (Applied Biosystems).All primers from assay-on-demands (Applied Biosystems) weredesigned by the company avoiding contaminating genomic DNAamplification by positioning one of the primers over the exon/intron boundary. Negative controls containing no cDNA and astandard curve spanning 3 orders of magnitude of dilution wererun on each plate in duplicate. Q-PCR conditions were 50 1C for2 min, 95 1C for 10 min, followed by 40 cycles of 15 s at 95 1C and1 min at 60 1C. The amount of target gene expression wascalculated by using the DDCT method [34]. Data were analyzedusing relative quantification technique. Relative changes in geneexpression were expressed as percent of expression in untreatedmice, as previously reported [31].
2.8. Rotarod test
We used an accelerating rotarod treadmill for mice (Mouserotarod, Ugo Basile, Comerio, Italy) to measure motor balance andcoordination. For training, the first day mice (n¼10 per group)were placed on the rod for 5 min at 16, then at 24 and finally at32 rpm. On the second day, mice were placed on the rod at 16 and24 rpm (for 5 min), and allowed to rest for 1 h. Then mice weretested on the rod at 32 rpm for 5 min. The number of falls from

K. Yoshikawa et al. / Prostaglandins, Leukotrienes and Essential Fatty Acids 85 (2011) 43–5246
the cylinders were counted and the time each mouse was able tostay on the rod (latency time) was recorded by a trip switch underthe floor of each rotating drum with a maximum recording timeof 300 s, and computed as the latency to fall [9].
2.9. Statistics
The number of falls from the rotarod was analyzed by anonparametric Kruscal–Wallis test. All other data were analyzedby one-way ANOVA followed by Newman–Keuls’ post-hoc test.All data were analyzed using GraphPad Prism Ver. 4.00 (GraphPadSoftware, Inc., San Diego, CA) and expressed as means7SEM.p values o0.05 were considered statistically significant.
3. Results
3.1. Gene and protein expression levels of 5-LO
Mice were exposed to cuprizone diet for up to 6 weeks andexpression of 5-LO in the forebrain during cuprizone exposurewas analyzed. mRNA and protein expression of 5-LO peakedbetween weeks 4 and 5 (Fig. 1B and C), concomitant to the peakin cuprizone-induced demyelination [6,11].
3.2. Gene expression of glial markers and CD11b
immunohistochemistry
To investigate the effects of 5-LO inhibition on neuroin-flammation associated with demyelination, we measured geneexpression of astrocytic (GFAP) and microglial (CD11b) mar-kers after 5 weeks of cuprizone. mRNA levels of both GFAP andCD11b were increased (po0.001) by cuprizone. While MK886did not significantly affect cuprizone-induced GFAP upregula-tion (Fig. 2A), it almost completely inhibited cuprizone-induced increase in CD11b mRNA expression (po0.001;Fig. 2B). We showed by immunostaining that these changesin gene expression were accompanied by changes in proteinexpression. In normal control mice, microglia immunostainingwas only sporadically seen in the corpus callosum and cortex(Fig. 2D and G). Mice exposed to cuprizone showed hyper-trophic microglia with enlarged cell bodies and thickenedprocesses (Fig. 2E and H), which were attenuated afterMK886 administration (Fig. 2F and I). Cuprizone induced anincrease in CD11b-positive cells in the corpus callosum andcortex. The number of CD11b-positive cells was inhibited byMK886 administration in the corpus callosum (po0.01) andcortex (po0.001) (Fig. 2J and K). In MK886-treated mice notexposed to cuprizone, CD11b-positive cells in the corpuscallosum and cortex were similar as in control vehicle-treatedmice (data not shown).
3.3. Proinflammatory cytokine levels
Cuprizone exposure for 5 weeks increased the mRNA expres-sion of the proinflammatory cytokines IL-1b, TNF-a and IL-6(po0.001; Fig. 3). Although the expression level of IL-1b andTNF-a remained unchanged after MK886 treatment, MK886inhibited cuprizone-induced increase in IL-6 mRNA and proteinexpression (po0.001; Fig. 3C and D). Treatment with MK886alone did not change IL-6 protein level (0.09570.019 pg/mg oftissue) compared to control mice.
3.4. Myelin content
To determine whether the reduction in the neuroinflammatoryresponse to cuprizone by MK886 was accompanied by reduced
demyelination, we quantified myelin content in the corpuscallosum and cortex with Black Gold staining and measured themRNA expression of myelin basic protein (MBP). After 5 weeks ofcuprizone exposure, we found significant demyelination of thecorpus callosum and cortex (Fig. 4A–H), accompanied by adecrease in MBP mRNA expression (po0.001; Fig. 4I). However,treatment with MK886 did not significantly change cuprizone-induced demyelination. MK886-treated control mice not exposedto cuprizone showed the same level of myelin in the corpuscallosum and cortex as control vehicle-treated mice (data notshown).
3.5. Axonal damage and motor performance
To determine whether a reduction in neuroinflammation byMK886 attenuated cuprizone-induced axonal damage, we per-formed double immunofluorescence of APP as a marker of axonaldamage [35,36] and the axonal marker NF 200 to detect damagedaxon (Fig. 5A). Because APP is also expressed in astrocytes [37],we counted APP and NF 200 double positive axons in the corpuscallosum. MK886 treatment significantly reduced the number ofAPP positive axons after 5 weeks of cuprizone (po0.05) (Fig. 5B).Furthermore, GPR17, a newly reported CysLT receptor, has beenshown to act as a sensor for neuronal damage [38,39]. Cuprizoneexposure induced GPR17 mRNA expression and MK886 treatmentattenuated its level (po0.05) (Fig. 5C). Next, we assessed loco-motor coordination and balance on a rotarod apparatus. Com-pared to normal controls, mice exposed to cuprizone recorded asignificant decline in locomotion time (po0.05), which wasrescued by MK886 treatment (Fig. 5D). MK886 treatment alsorescued the cuprizone-induced increase in the number of fallsfrom the rod (Fig. 5E). Overall, MK886 treatment significantlyattenuated cuprizone-induced impairment of motor performance.
4. Discussion and conclusions
Using the 5-LO inhibitor MK886 we tested whether 5-LO playsa causative role in the cuprizone-induced demyelination.Although MK886 did not attenuate cuprizone-induced demyeli-nation, it attenuated the increase in IL-6 production and micro-glial activation. These data suggest that the 5-LO pathway isinvolved in microglial activation and neuroinflammation inde-pendently of the demyelination process. We also demonstratedthat inhibition of neuroinflammation by MK886 attenuated axo-nal damage and motor dysfunction during demyelination, sug-gesting that 5-LO inhibition may be a useful therapeutictreatment in MS.
IL-6 is known to be involved in neuroinflammatory responsesand produced at high level in active MS lesions [40,41]. We foundthat MK886 administration inhibited cuprizone-induced IL-6production and microglial activation. Supporting our result LTB4
has been reported to induce IL-6 transcription, mRNA expressionand protein production [42,43]. In addition, studies in culturedmicroglia [44], IL-6-deficient mice [45] and IL-6 transgenic mice[46] revealed that IL-6 is an important factor in microglialactivation. Furthermore, 5-LO pathway may play a key role inmicroglial activation directly. Supporting this concept, culturedmicroglia express cysteinyl-LT receptors (CysLT1 and CysLT2) [47]and inhibition of the 5-LO pathway in vivo prevented microglialactivation induced by LPS administration or by spreading depres-sion [48,49]. LTB4 has been shown to upregulate CD11b expres-sion in monocytes [50]. Thus, microglia may also activate CD11bexpression via LTB4. Moreover, suppression of IL-6 production andmicroglial activation probably result in attenuation of axonaldamage and motor dysfunction. Blockade of IL-6 receptor

Fig. 2. Relative gene expression of glial markers and CD11b immunostaining. Gene expression of GFAP (A) and CD11b (B) relative to pgk-1, as determined by real-time PCR
in the forebrain after 5 weeks of cuprizone exposure. Data are means7SEM, n¼7–8. Statistical analysis was performed using one-way ANOVA followed by post-hoc
Newman–Keuls test (***po0.001 vs. normal controls: ###po0.001 vs. CPZþvehicle). Schematic diagram of the mouse brain in coronal section. The red line marks the area
of corpus callosum and cortex analyzed for CD11b-positive cells after 5 weeks of cuprizone (C). Immunostaining with anti-CD11b (D–I) showed microglial activation.
Coronal brain sections of corpus callosum (D: normal control, E: CPZ, F: CPZþMK886) and cortex (G: normal control, H: CPZ, I: CPZþMK886). Scale bars¼50 mm. Number
of microglia in the corpus callosum (J) and cortex (K). Data are means7SEM, n¼5, respectively. Statistical analysis was performed using one-way ANOVA followed by
post-hoc Newman–Keuls test (**po0.01, ***po0.001 vs. normal controls: ###po0.001, ##po0.01 vs. CPZþvehicle).
K. Yoshikawa et al. / Prostaglandins, Leukotrienes and Essential Fatty Acids 85 (2011) 43–52 47

Fig. 3. Proinflammatory cytokine levels. Gene expression of IL-1b (A), TNF-a (B) and IL-6 (C) relative to pgk-1, as determined by real-time PCR in the forebrain after
5 weeks of cuprizone. Data are means7SEM, n¼7–8. Statistical analysis was performed using one-way ANOVA followed by post-hoc Newman–Keuls test (**po0.01,
***po0.01 vs. normal controls: ###po0.001 vs. CPZþvehicle). IL-6 protein levels (D) in the forebrain. Data are means7SEM, n¼5. Statistical analysis was performed using
one-way ANOVA followed by post-hoc Newman–Keuls test (*po0.05 vs. normal controls: ##po0.01 vs. CPZþvehicle).
K. Yoshikawa et al. / Prostaglandins, Leukotrienes and Essential Fatty Acids 85 (2011) 43–5248
suppressed axonal damage and ameliorated functional recoveryafter spinal cord injury [51,52]. Activated microglia are alsoinvolved in axonal damage [53] by the mechanism of strippingof synaptic protein [54] and/or microglia-derived glutamateexcitotoxicity [55,56].
We also demonstrated that MK866 reduced cuprizone-induced expression of GPR17. GPR17, a newly reported Cystei-nyl-LT receptor [57], was expressed in microglia/macrophages inthe lesioned area of focal ischemia and spinal cord injury [38,39].It is possible that reduced neuroinflammation would reduceneuronal damage-associated signaling. Thus, 5-LO inhibition byMK886 leads to suppression of IL-6 production, microglial activa-tion, axonal damage and motor dysfunction by a multi-stepmechanism in the demyelination process.
While it is unclear at this point why MK886 selectivelydecreases cuprizone-induced IL-6, but not IL-1b and TNF-a, wespeculate that a possible mechanism involves selective targetingof cytokine producing cells. In a neuroinflammatory situation,microglia produce proinflammatory cytokines [58–60] and astro-cytes also produce TNFa and IL-1b [61–63]. Thus, in our model,MK886 might inhibit IL-6 production by microglia, but not TNFaand IL-1b by astrocytes.
As previously described [64], we found that cuprizone inducedalso marked cortical demyelination (Fig. 5). There was no regionaldifference in the contribution of the 5-LO pathway to demyelina-tion, since MK886 did not affect demyelination levels in eithercorpus callosum or cortex. A regional accumulation of microgliain the corpus callosum and in the cortical cell layer has beendescribed [65]. Our results indicate that activated microglia ispresent mainly in the corpus callosum and, to a lesser extent, inthe cortex. MK886 treatment attenuated microglial activation andaccumulation in both brain areas.
We found that inhibition of the 5-LO pathway did not affectcuprizone-induced demyelination. Similarly, in the EAE model,
a 5-LO inhibitor did not significantly reduce clinical score of EAE,even though the onset of EAE was delayed [66]. Furthermore,5-LO deficient mice developed more severe EAE than wild-typemice [67], although no difference in pathological parameters wasdetected between deficient mice and control EAE mice with thesame clinical score. Thus, both in the EAE and in the cuprizonemodels of demyelination, the 5-LO pathway does not appear todirectly affect myelin levels although its inhibition amelioratesthe clinical symptoms of the disease.
Our data demonstrate that the 5-LO pathway plays a key rolein microglial activation and neuroinflammation independently ofthe demyelination process. We also demonstrated that 5-LOinhibition attenuated axonal damage and motor dysfunctionduring cuprizone-induced demyelination. These results suggestthat pharmacological inhibition of 5-LO is a valuable anti-inflam-matory treatment for brain inflammation and could providetherapeutic amelioration of MS symptoms.
Competing interests
The authors declare that they have no competing interests.
Acknowledgements
This work was supported by Intramural Research Program ofNIH, National Institute on Aging. We thank Drs. Sang-Ho Choi andSaba Aid for helpful discussion and technical advice. We thankDr. Anthony Donsante for kindly providing the rotarod apparatus.We thank Drs. Mukoyama and Onitsuka for kindly providing theconfocal microscope and technical advice.

Fig. 4. Black Gold staining of myelin and MBP gene expression. Representative photomicrographs of coronal brain sections at the level of the fimbria demonstrate a
progressive demyelination of the corpus callosum (A–C) and cortex (D–F) after 5 weeks of cuprizone treatment. Black gold staining of normal control (A and D),
CPZþvehicle (B and E) and CPZþMK886 (C and F). Myelin densities of the corpus callosum (G) and cortex (H) were compared with normal controls and expressed as a
percentage of control values using the Image J analysis program (***po0.001 vs. normal controls). Gene expression of MBP (I) relative to pgk-1, as determined by real-time
PCR in the forebrain. Data are means7SEM, n¼7–8. Statistical analysis was performed using one-way ANOVA followed by post-hoc Newman–Keuls test (***po0.001 vs.
normal controls). Scale bar for (A–C)¼500 mm. Scale bar for (D–F)¼1 mm.
K. Yoshikawa et al. / Prostaglandins, Leukotrienes and Essential Fatty Acids 85 (2011) 43–52 49
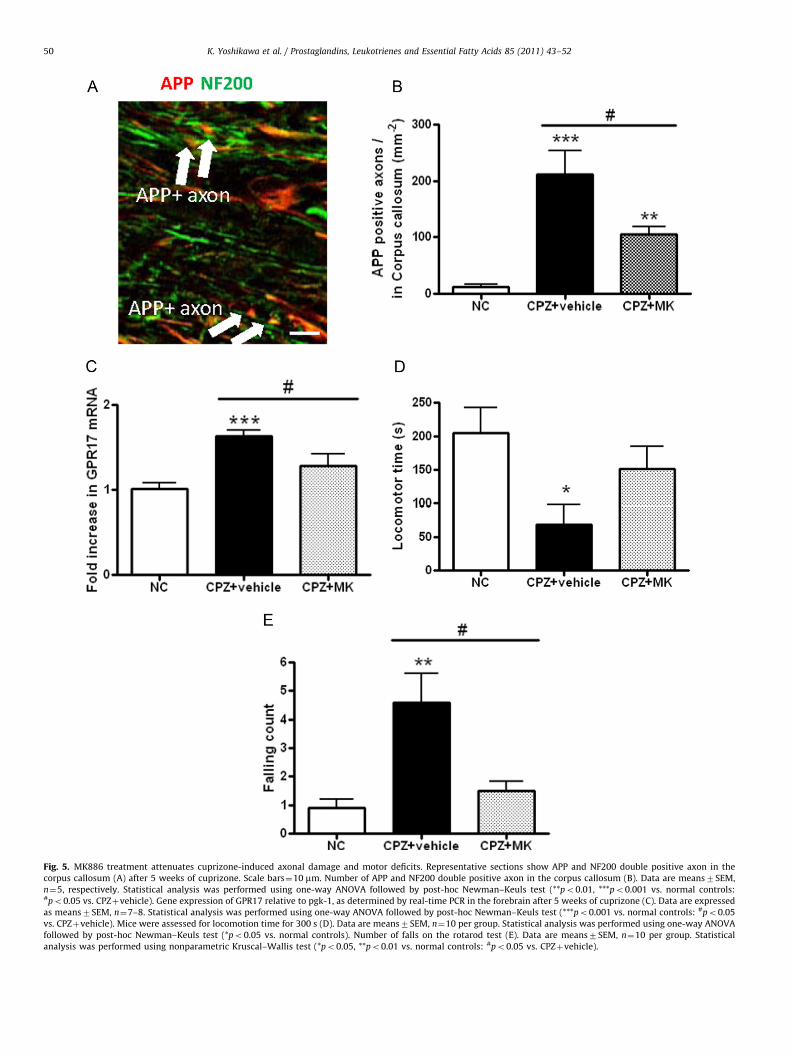
Fig. 5. MK886 treatment attenuates cuprizone-induced axonal damage and motor deficits. Representative sections show APP and NF200 double positive axon in the
corpus callosum (A) after 5 weeks of cuprizone. Scale bars¼10 mm. Number of APP and NF200 double positive axon in the corpus callosum (B). Data are means7SEM,
n¼5, respectively. Statistical analysis was performed using one-way ANOVA followed by post-hoc Newman–Keuls test (**po0.01, ***po0.001 vs. normal controls:#po0.05 vs. CPZþvehicle). Gene expression of GPR17 relative to pgk-1, as determined by real-time PCR in the forebrain after 5 weeks of cuprizone (C). Data are expressed
as means7SEM, n¼7–8. Statistical analysis was performed using one-way ANOVA followed by post-hoc Newman–Keuls test (***po0.001 vs. normal controls: #po0.05
vs. CPZþvehicle). Mice were assessed for locomotion time for 300 s (D). Data are means7SEM, n¼10 per group. Statistical analysis was performed using one-way ANOVA
followed by post-hoc Newman–Keuls test (*po0.05 vs. normal controls). Number of falls on the rotarod test (E). Data are means7SEM, n¼10 per group. Statistical
analysis was performed using nonparametric Kruscal–Wallis test (*po0.05, **po0.01 vs. normal controls: #po0.05 vs. CPZþvehicle).
K. Yoshikawa et al. / Prostaglandins, Leukotrienes and Essential Fatty Acids 85 (2011) 43–5250

K. Yoshikawa et al. / Prostaglandins, Leukotrienes and Essential Fatty Acids 85 (2011) 43–52 51
References
[1] D. Kidd, F. Barkhof, R. McConnell, P.R. Algra, I.V. Allen, T. Revesz, Corticallesions in multiple sclerosis, Brain 122 (Pt 1) (1999) 17–26.
[2] C. Lucchinetti, W. Bruck, J. Noseworthy, Multiple sclerosis: recent develop-ments in neuropathology, pathogenesis, magnetic resonance imaging studiesand treatment, Curr. Opin. Neurol. 14 (2001) 259–269.
[3] J.H. Noseworthy, C. Lucchinetti, M. Rodriguez, B.G. Weinshenker, Multiplesclerosis, N. Engl. J. Med. 343 (2000) 938–952.
[4] M.H. Barnett, J.W. Prineas, Relapsing and remitting multiple sclerosis:pathology of the newly forming lesion, Ann. Neurol. 55 (2004) 458–468.
[5] W.F. Blakemore, Demyelination of the superior cerebellar peduncle in themouse induced by cuprizone, J. Neurol. Sci. 20 (1973) 63–72.
[6] G.K. Matsushima, P. Morell, The neurotoxicant, cuprizone, as a model to studydemyelination and remyelination in the central nervous system, Brain Pathol.11 (2001) 107–116.
[7] O. Torkildsen, L.A. Brunborg, K.M. Myhr, L. Bo, The cuprizone model fordemyelination, Acta Neurol. Scand. Suppl. 188 (2008) 72–76.
[8] L. Liu, A. Belkadi, L. Darnall, et al., CXCR2-positive neutrophils are essential forcuprizone-induced demyelination: relevance to multiple sclerosis, Nat. Neu-rosci. 13 (2010) 319–326.
[9] N. Franco-Pons, M. Torrente, M.T. Colomina, E. Vilella, Behavioral deficits inthe cuprizone-induced murine model of demyelination/remyelination, Tox-icol. Lett. 169 (2007) 205–213.
[10] C. Lucchinetti, W. Bruck, J. Parisi, B. Scheithauer, M. Rodriguez, H. Lassmann,Heterogeneity of multiple sclerosis lesions: implications for the pathogenesisof demyelination, Ann. Neurol. 47 (2000) 707–717.
[11] M.M. Hiremath, Y. Saito, G.W. Knapp, J.P. Ting, K. Suzuki, G.K. Matsushima,Microglial/macrophage accumulation during cuprizone-induced demyelina-tion in C57BL/6 mice, J. Neuroimmunol. 92 (1998) 38–49.
[12] R.C. Armstrong, T.Q. Le, N.C. Flint, A.C. Vana, Y.X. Zhou, Endogenous cellrepair of chronic demyelination, J. Neuropathol. Exp. Neurol. 65 (2006)245–256.
[13] L.S. Harbige, M.K. Sharief, Polyunsaturated fatty acids in the pathogenesis andtreatment of multiple sclerosis, Br. J. Nutr. 98 (Suppl. 1) (2007) S46–553.
[14] R.H. Dworkin, D. Bates, J.H. Millar, D.W. Paty, Linoleic acid and multiplesclerosis: a reanalysis of three double-blind trials, Neurology 34 (1984)1441–1445.
[15] C.J. Meade, J. Mertin, J. Sheena, R. Hunt, Reduction by linoleic acid of theseverity of experimental allergic encephalomyelitis in the guinea pig, J.Neurol. Sci. 35 (1978) 291–308.
[16] D. Hughes, A.B. Keith, J. Mertin, E.A. Caspary, Linoleic acid therapy in severeexperimental allergic encephalomyelitis in the guinea-pig: suppression bycontinuous treatment, Clin. Exp. Immunol. 40 (1980) 523–531.
[17] A. Kalyvas, C. Baskakis, V. Magrioti, et al., Differing roles for members of thephospholipase A2 superfamily in experimental autoimmune encephalomye-litis, Brain 132 (2009) 1221–1235.
[18] S. Marusic, M.W. Leach, J.W. Pelker, et al., Cytosolic phospholipase A2 alpha-deficient mice are resistant to experimental autoimmune encephalomyelitis,J. Exp. Med. 202 (2005) 841–851.
[19] L.W. Whitney, S.K. Ludwin, H.F. McFarland, W.E. Biddison, Microarrayanalysis of gene expression in multiple sclerosis and EAE identifies 5-lipox-ygenase as a component of inflammatory lesions, J. Neuroimmunol. 121(2001) 40–48.
[20] A.T. Arthur, P.J. Armati, C. Bye, et al., Genes implicated in multiple sclerosispathogenesis from consilience of genotyping and expression profiles inrelapse and remission, BMC Med. Genet. 9 (2008) 17.
[21] I. Neu, J. Mallinger, A. Wildfeuer, L. Mehlber, Leukotrienes in the cerebrosp-inal fluid of multiple sclerosis patients, Acta Neurol. Scand. 86 (1992)586–587.
[22] B. Samuelsson, S.E. Dahlen, J.A. Lindgren, C.A. Rouzer, C.N. Serhan, Leuko-trienes and lipoxins: structures, biosynthesis, and biological effects, Science237 (1987) 1171–1176.
[23] M. Minami, S. Ohno, H. Kawasaki, et al., Molecular cloning of a cDNA codingfor human leukotriene A4 hydrolase. Complete primary structure of anenzyme involved in eicosanoid synthesis, J. Biol. Chem. 262 (1987)13873–13876.
[24] J.Z. Haeggstrom, A. Wetterholm, J.F. Medina, B. Samuelsson, Leukotriene A4hydrolase: structural and functional properties of the active center, J. LipidMediat. 6 (1993) 1–13.
[25] D.J. Welsch, D.P. Creely, S.D. Hauser, K.J. Mathis, G.G. Krivi, P.C. Isakson,Molecular cloning and expression of human leukotriene-C4 synthase, Proc.Natl. Acad. Sci. USA 91 (1994) 9745–9749.
[26] B.K. Lam, J.F. Penrose, G.J. Freeman, K.F. Austen, Expression cloning of a cDNAfor human leukotriene C4 synthase, an integral membrane protein conjugat-ing reduced glutathione to leukotriene A4, Proc. Natl. Acad. Sci. USA 91(1994) 7663–7667.
[27] P.J. Vickers, 5-Lipoxygenase-activating protein (FLAP), J. Lipid Mediat. CellSignal. 12 (1995) 185–194.
[28] R.A. Dixon, R.E. Diehl, E. Opas, et al., Requirement of a 5-lipoxygenase-activating protein for leukotriene synthesis, Nature 343 (1990) 282–284.
[29] L. Paul, V. Fraifeld, J. Kaplanski, Evidence supporting involvement of leuko-trienes in LPS-induced hypothermia in mice, Am. J. Physiol. 276 (1999)R52–R58.
[30] P. Ciceri, M. Rabuffetti, A. Monopoli, S. Nicosia, Production of leukotrienes in amodel of focal cerebral ischaemia in the rat, Br. J. Pharmacol. 133 (2001)1323–1329.
[31] S.H. Choi, R. Langenbach, F. Bosetti, Genetic deletion or pharmacologicalinhibition of cyclooxygenase-1 attenuate lipopolysaccharide-induced inflam-matory response and brain injury, FASEB J. 22 (2008) 1491–1501.
[32] C.D. Toscano, V.V. Prabhu, R. Langenbach, K.G. Becker, F. Bosetti, Differentialgene expression patterns in cyclooxygenase-1 and cyclooxygenase-2 defi-cient mouse brain, Genome Biol. 8 (2007) R14.
[33] L. Schmued, W. Slikker Jr., Black-gold: a simple, high-resolution histochem-ical label for normal and pathological myelin in brain tissue sections, BrainRes. 837 (1999) 289–297.
[34] K.J. Livak, T.D. Schmittgen, Analysis of relative gene expression data usingreal-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods 25(2001) 402–408.
[35] S.M. Gentleman, M.J. Nash, C.J. Sweeting, D.I. Graham, G.W. Roberts, Beta-amyloid precursor protein (beta APP) as a marker for axonal injury after headinjury, Neurosci. Lett. 160 (1993) 139–144.
[36] M. Lindner, J. Fokuhl, F. Linsmeier, C. Trebst, M. Stangel, Chronic toxicdemyelination in the central nervous system leads to axonal damage despiteremyelination, Neurosci. Lett. 453 (2009) 120–125.
[37] T. Clarner, J.P. Buschmann, C. Beyer, M. Kipp, Glial amyloid precursor proteinexpression is restricted to astrocytes in an experimental toxic model ofmultiple sclerosis, J. Mol. Neurosci. 43 (2010) 268–274.
[38] D. Lecca, M.L. Trincavelli, P. Gelosa, et al., The recently identified P2Y-likereceptor GPR17 is a sensor of brain damage and a new target for brain repair,PLoS One 3 (2008) e3579.
[39] S. Ceruti, G. Villa, T. Genovese, et al., The P2Y-like receptor GPR17 as a sensorof damage and a new potential target in spinal cord injury, Brain 132 (2009)2206–2218.
[40] B. Cannella, C.S. Raine, The adhesion molecule and cytokine profile ofmultiple sclerosis lesions, Ann. Neurol. 37 (1995) 424–435.
[41] M.N. Woodroofe, M.L. Cuzner, Cytokine mRNA expression in inflammatorymultiple sclerosis lesions: detection by non-radioactive in situ hybridization,Cytokine 5 (1993) 538–583.
[42] M. Rola-Pleszczynski, J. Stankova, Leukotriene B4 enhances interleukin-6(IL-6) production and IL-6 messenger RNA accumulation in human mono-cytes in vitro: transcriptional and posttranscriptional mechanisms, Blood 80(1992) 1004–1011.
[43] J. Stankova, M. Rola-Pleszczynski, Interleukin 6 production by mononuclearphagocytes can be stimulated by leukotrienes, Arch. Immunol. Ther. Exp.(Warsz) 40 (1992) 17–21.
[44] W.J. Streit, S.D. Hurley, T.S. McGraw, S.L. Semple-Rowland, Comparativeevaluation of cytokine profiles and reactive gliosis supports a critical rolefor interleukin-6 in neuron-glia signaling during regeneration, J. Neurosci.Res. 61 (2000) 10–20.
[45] M.A. Klein, J.C. Moller, L.L. Jones, H. Bluethmann, G.W. Kreutzberg, G. Raivich,Impaired neuroglial activation in interleukin-6 deficient mice, Glia 19 (1997)227–233.
[46] E. Fattori, D. Lazzaro, P. Musiani, A. Modesti, T. Alonzi, G. Ciliberto, IL-6expression in neurons of transgenic mice causes reactive astrocytosis andincrease in ramified microglial cells but no neuronal damage, Eur. J. Neurosci.7 (1995) 2441–2449.
[47] P. Ballerini, P. Di Iorio, R. Ciccarelli, et al., P2Y1 and cysteinyl leukotrienereceptors mediate purine and cysteinyl leukotriene co-release in primarycultures of rat microglia, Int. J. Immunopathol. Pharmacol. 18 (2005)255–268.
[48] A.O. Caggiano, R.P. Kraig, Eicosanoids and nitric oxide influence induction ofreactive gliosis from spreading depression in microglia but not astrocytes,J. Comp. Neurol. 369 (1996) 93–108.
[49] L.B. Willard, B. Hauss-Wegrzyniak, W. Danysz, G.L. Wenk, The cytotoxicity ofchronic neuroinflammation upon basal forebrain cholinergic neurons of ratscan be attenuated by glutamatergic antagonism or cyclooxygenase-2 inhibi-tion, Exp. Brain Res. 134 (2000) 58–65.
[50] K. Vaddi, R.C. Newton, Regulation of monocyte integrin expression by beta-family chemokines, J. Immunol. 153 (1994) 4721–4732.
[51] M. Mukaino, M. Nakamura, O. Yamada, et al., Anti-IL-6-receptor antibodypromotes repair of spinal cord injury by inducing microglia-dominantinflammation, Exp. Neurol. 224 (2010) 403–414.
[52] S. Okada, M. Nakamura, Y. Mikami, et al., Blockade of interleukin-6 receptorsuppresses reactive astrogliosis and ameliorates functional recovery inexperimental spinal cord injury, J. Neurosci. Res. 76 (2004) 265–276.
[53] O.W. Howell, J.L. Rundle, A. Garg, M. Komada, P.J. Brophy, R. Reynolds,Activated microglia mediate axoglial disruption that contributes to axonalinjury in multiple sclerosis, J. Neuropathol. Exp. Neurol. 69 (2010)1017–1033.
[54] S. Rasmussen, Y. Wang, P. Kivisakk, et al., Persistent activation of microglia isassociated with neuronal dysfunction of callosal projecting pathways andmultiple sclerosis-like lesions in relapsing—remitting experimental autoim-mune encephalomyelitis, Brain 130 (2007) 2816–2829.
[55] D. Pitt, P. Werner, C.S. Raine, Glutamate excitotoxicity in a model of multiplesclerosis, Nat. Med. 6 (2000) 67–70.
[56] Y. Fu, W. Sun, Y. Shi, R. Shi, J.X. Cheng, Glutamate excitotoxicity inflictsparanodal myelin splitting and retraction, PLoS One 4 (2009) e6705.

K. Yoshikawa et al. / Prostaglandins, Leukotrienes and Essential Fatty Acids 85 (2011) 43–5252
[57] P. Ciana, M. Fumagalli, M.L. Trincavelli, et al., The orphan receptor GPR17identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor,EMBO J. 25 (2006) 4615–4627.
[58] R.B. Banati, J. Gehrmann, P. Schubert, G.W. Kreutzberg, Cytotoxicity ofmicroglia, Glia 7 (1993) 111–118.
[59] D. Giulian, M. Corpuz, Microglial secretion products and their impact on thenervous system, Adv. Neurol. 59 (1993) 315–320.
[60] P.L. McGeer, E.G. McGeer, The inflammatory response system of brain:implications for therapy of Alzheimer and other neurodegenerative diseases,Brain Res. Rev. 21 (1995) 195–218.
[61] L.T. Lau, A.C. Yu, Astrocytes produce and release interleukin-1, interleukin-6,tumor necrosis factor alpha and interferon-gamma following traumatic andmetabolic injury, J. Neurotrauma 18 (2001) 351–359.
[62] E. Corsini, A. Dufour, E. Ciusani, et al., Human brain endothelial cells andastrocytes produce IL-1 beta but not IL-10, Scand. J. Immunol. 44 (1996)506–511.
[63] M. Sawada, N. Kondo, A. Suzumura, T. Marunouchi, Production of tumornecrosis factor-alpha by microglia and astrocytes in culture, Brain Res. 491(1989) 394–397.
[64] T. Skripuletz, M. Lindner, A. Kotsiari, et al., Cortical demyelination isprominent in the murine cuprizone model and is strain-dependent, Am.J. Pathol. 172 (2008) 1053–1061.
[65] V. Gudi, D. Moharregh-Khiabani, T. Skripuletz, et al., Regional differencesbetween grey and white matter in cuprizone induced demyelination, BrainRes. 1283 (2009) 127–138.
[66] S. Marusic, P. Thakker, J.W. Pelker, et al., Blockade of cytosolic phospholipaseA2 alpha prevents experimental autoimmune encephalomyelitis anddiminishes development of Th1 and Th17 responses, J. Neuroimmunol. 204(2008) 29–37.
[67] M.R. Emerson, S.M. LeVine, Experimental allergic encephalomyelitis isexacerbated in mice deficient for 12/15-lipoxygenase or 5-lipoxygenase,Brain Res. 1021 (2004) 140–145.
Copyright © 2022 FDOKUMEN




















