
Increased apoptosis and increased clonogenic survival of 12V-H-ras transformed rat fibroblasts in...
13
Apoptosis 2000; 5: 355–367 C 2000 Kluwer Academic Publishers Increased apoptosis and increased clonogenic survival of 12V-H-ras transformed rat fibroblasts in response to cisplatin K. Viktorsson, T. Heiden, M. Molin, G. Akusj¨ arvi, S. Linder and M. C. Shoshan Radiumhemmet’s Research Laboratory, Cancer Center Karolinska, Karolinska Institute and Hospital, S-171 76 Stockholm, Sweden (K. Viktorsson, S. Linder, M. C. Shoshan); Department of Medical Radiobiology, Karolinska Institute, S-171 76 Stockholm, Sweden (T. Heiden); Department of Medical Biochemistry and Microbiology, BMC, Uppsala University, S-751 23 Uppsala, Sweden (M. Molin, G. Akusj¨ arvi); Department of Medical Biochemistry and Microbiology, BMC, Uppsala University, S-751 23 Uppsala, Sweden (M. Molin, G. Akusj ¨ arvi) Mutationally activated Ras is involved in tumor progres- sion and likely also in drug resistance. Using survival, viability and apoptosis assays, we have here compared the cisplatin sensitivities of FR3T3 rat fibroblasts and a 12V-H-ras transformed subline (Ras2:3). Around 24 h after cisplatin treatment Ras2:3 cells showed higher apopto- sis levels and lower viability than FR3T3. This increased sensitivity correlated with weaker cisplatin-induced ac- tivation of Jun N-terminal kinase (JNK). In contrast to apoptosis assays, colony formation assays showed that Ras2:3 were more resistant to cisplatin than were FR3T3. This was partly due to the increased cisplatin sensitivity of FR3T3 seeded at low densities, as required in colony formation assays. In addition, Ras2:3 cisplatin survivors had a higher relative proliferative capacity. Cell cycle anal- yses showed that FR3T3 cells initially responded with a dose-dependent G2 arrest, while Ras2:3 accumulated in S-phase. Experiments with an anti-apoptotic mutant of MEKK1 suggested that the apoptotic response of Ras2:3 cells is not specific to the S-phase fraction. In summary, the cisplatin response of ras-transformed fibroblasts is distinct from that of parental cells, in that they show in- creased apoptosis, a different cell cycle response and in- creased post-treatment proliferative capacity. The results illustrate the need to carefully consider methods and pro- tocols for in vitro studies on chemotherapy sensitivity. Keywords: apoptosis; chemotherapy resistance; clonogeni- city; ras. Introduction Cisplatin is widely used in anticancer therapy, and its main antiproliferative effect is believed to be caused by DNA- intrastrand crosslinks, although potential cisplatin targets Correspondence to: M. C. Shoshan, Radiumhemmet’s Research Laboratory, Cancer Center Karolinska, Karolinska Institute and Hospital, S-171 76 Stockholm, Sweden. also involve RNA and protein. 1 Cisplatin has in several re- ports been shown to induce a G2/M cell cycle arrest 1,2 but G1 arrest and S-phase delay have also been reported. 2 -5 Cisplatin has furthermore been shown to induce apopto- sis in various cell types. 1 -6 How cisplatin-induced DNA damage is translated into apoptotic or antiproliferative signaling is not entirely clear. Oncogenic mutant Ras is commonly found in human cancers and has long been known to have profound effects on cellular signalling, e.g., on regulation of proliferation, cell cycle and gene expression. The influence of consti- tutively active mutant Ras on responses to chemother- apy is therefore likely of great importance for therapeu- tic outcomes. However, reports on Ras-mediated effects on cisplatin responses vary. Inducible expression of trans- fected oncogenic Ras was found to confer cisplatin re- sistance in NIH3T3 cells; 7 similarly, expression of onco- genic Ras correlated with resistance in human tumor cell lines. 8,9 It has also been reported that Ras potentiates cisplatin sensitivity, 10 while a study of sixteen human ovarial cancer cell lines did not reveal any correlation between Ras overexpression or -mutation and cisplatin sensitivity. 11 We have here studied the cisplatin responses of 12V- ras transformed rat fibroblasts (Ras2:3) and the parental FR3T3 cells. The results show that the ras-transformed cells respond to cisplatin with a different type of cell cycle arrest and are highly sensitive to cisplatin-induced apop- tosis compared to parental cells, in which the main anti- proliferative effect is not apoptosis but cell cycle arrest. Determination of relative sensitivities by in vitro assays is of vital importance for chemotherapy research. How- ever, interpretation of results of in vitro drug sensitivity assays can be a major problem, and depends on the frac- tion of arrested cells, on the extent and rate of apoptosis, Apoptosis · Vol 5 · No 4 · 2000 355
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Increased apoptosis and increased clonogenic survival of 12V-H-ras transformed rat fibroblasts in...

Apoptosis 2000; 5: 355–367C© 2000 Kluwer Academic Publishers
Increased apoptosis and increased clonogenicsurvival of 12V-H- ras transformed rat fibroblastsin response to cisplatin
K. Viktorsson, T. Heiden, M. Molin, G. Akusj ¨ arvi, S. Linder and M. C. Shoshan
Radiumhemmet’s Research Laboratory, Cancer Center Karolinska, Karolinska Institute andHospital, S-171 76 Stockholm, Sweden (K. Viktorsson, S. Linder, M. C. Shoshan);Department of Medical Radiobiology, Karolinska Institute, S-171 76 Stockholm, Sweden(T. Heiden); Department of Medical Biochemistry and Microbiology, BMC, UppsalaUniversity, S-751 23 Uppsala, Sweden (M. Molin, G. Akusjarvi); Department of MedicalBiochemistry and Microbiology, BMC, Uppsala University, S-751 23 Uppsala, Sweden(M. Molin, G. Akusjarvi)
Mutationally activated Ras is involved in tumor progres-sion and likely also in drug resistance. Using survival,viability and apoptosis assays, we have here comparedthe cisplatin sensitivities of FR3T3 rat fibroblasts and a12V-H-ras transformed subline (Ras2:3). Around 24 h aftercisplatin treatment Ras2:3 cells showed higher apopto-sis levels and lower viability than FR3T3. This increasedsensitivity correlated with weaker cisplatin-induced ac-tivation of Jun N-terminal kinase (JNK). In contrast toapoptosis assays, colony formation assays showed thatRas2:3 were more resistant to cisplatin than were FR3T3.This was partly due to the increased cisplatin sensitivityof FR3T3 seeded at low densities, as required in colonyformation assays. In addition, Ras2:3 cisplatin survivorshad a higher relative proliferative capacity. Cell cycle anal-yses showed that FR3T3 cells initially responded with adose-dependent G2 arrest, while Ras2:3 accumulated inS-phase. Experiments with an anti-apoptotic mutant ofMEKK1 suggested that the apoptotic response of Ras2:3cells is not specific to the S-phase fraction. In summary,the cisplatin response of ras-transformed fibroblasts isdistinct from that of parental cells, in that they show in-creased apoptosis, a different cell cycle response and in-creased post-treatment proliferative capacity. The resultsillustrate the need to carefully consider methods and pro-tocols for in vitro studies on chemotherapy sensitivity.
Keywords: apoptosis; chemotherapy resistance; clonogeni-city; ras.
Introduction
Cisplatin is widely used in anticancer therapy, and its mainantiproliferative effect is believed to be caused by DNA-intrastrand crosslinks, although potential cisplatin targets
Correspondence to: M. C. Shoshan, Radiumhemmet’s ResearchLaboratory, Cancer Center Karolinska, Karolinska Institute andHospital, S-171 76 Stockholm, Sweden.
also involve RNA and protein.1 Cisplatin has in several re-ports been shown to induce a G2/M cell cycle arrest1,2 butG1 arrest and S-phase delay have also been reported.2−5
Cisplatin has furthermore been shown to induce apopto-sis in various cell types.1−6 How cisplatin-induced DNAdamage is translated into apoptotic or antiproliferativesignaling is not entirely clear.
Oncogenic mutant Ras is commonly found in humancancers and has long been known to have profound effectson cellular signalling, e.g., on regulation of proliferation,cell cycle and gene expression. The influence of consti-tutively active mutant Ras on responses to chemother-apy is therefore likely of great importance for therapeu-tic outcomes. However, reports on Ras-mediated effectson cisplatin responses vary. Inducible expression of trans-fected oncogenic Ras was found to confer cisplatin re-sistance in NIH3T3 cells;7 similarly, expression of onco-genic Ras correlated with resistance in human tumor celllines.8,9 It has also been reported that Ras potentiatescisplatin sensitivity,10 while a study of sixteen humanovarial cancer cell lines did not reveal any correlationbetween Ras overexpression or -mutation and cisplatinsensitivity.11
We have here studied the cisplatin responses of 12V-ras transformed rat fibroblasts (Ras2:3) and the parentalFR3T3 cells. The results show that the ras-transformedcells respond to cisplatin with a different type of cell cyclearrest and are highly sensitive to cisplatin-induced apop-tosis compared to parental cells, in which the main anti-proliferative effect is not apoptosis but cell cycle arrest.Determination of relative sensitivities by in vitro assaysis of vital importance for chemotherapy research. How-ever, interpretation of results of in vitro drug sensitivityassays can be a major problem, and depends on the frac-tion of arrested cells, on the extent and rate of apoptosis,
Apoptosis · Vol 5 · No 4 · 2000 355

K. Viktorsson et al.
the rate of elimination of apoptotic cells, and the rate ofgrowth of surviving cells. The present study takes intoaccount two modes of sensitivity involving different timescales, and the results highlight the need to carefullyconsider the choice of experimental protocols and assaymethods.
Materials and methods
Cells and cisplatin treatment
FR3T3, a clonal immortalized rat fibroblast cell line, andfour subclones obtained by transformation of FR3T3 by12V-H-ras and selection for neomycin resistance were allmaintained at +37◦C and 7% CO2 and in Dulbecco’sModified Eagle’s Medium supplemented with 5% fetalbovine serum (both from Life Technologies), streptomycin,penicillin and L-glutamate. The cisplatin sensitivities ofthe parental FR3T3 and of the subclones were assessed (seebelow). The subclone Ras2:3 was then studied in greaterdetail. For the experiments, cells were treated with cis-platin one day after seeding; after 1 h treatment withindicated doses, they were carefully rinsed twice withphosphate-buffered saline (PBS) and postincubated for theindicated time intervals.
Assessment of apoptosis
Adherent and floating cells were always pooled for quan-tification of apoptosis by assessment of nuclear fragmen-tation. Control and cisplatin-treated cells were harvestedand suspended in a small volume of hypotonic salt solu-tion with 30 mM glycerol. Smears were made on glassslides and after drying the cells were fixed in acetone-methanol (2 : 1) for 5 min. DNA was stained with a so-lution of ethidium bromide in distilled water (5 ng/ml).After rinsing in tap water, nuclei were counted in UVmicroscopy and apoptosis assessed as the percentage offragmented nuclei in each sample. About 150 nuclei persample were counted. Apoptosis was also confirmed byterminal deoxynucleotidyl transferase-mediated nick endlabeling (TUNEL) staining (not shown), increased sub-G1populations in flow cytometry analyses, and by negativegrowth (see Results).
Clonogenic assay
Cells were seeded at 300 cells per 50 mm-dish (16 cells/cm2), treated for 1 h with cisplatin at different doses andpostincubated for 7 days. The resulting colonies wereGiemsa-stained and counted. The experiment was per-formed four times with Ras2:3.
Cell viability assay
Cells were seeded at indicated densities in 96-well plates,treated in triplicate samples with indicated cisplatin con-centrations the next day. Cell viability was assessed after48 h according to the manufacturer’s instructions withCell Titer 96 Assay kit (Promega) based on the MTT assay.The resulting absorbance is proportional to the numberof viable cells in the sample.
Cell growth
Cells were treated for 1 h with 40µM cisplatin and postin-cubated in new medium for up to 6 days. On the third day,the medium was changed. For counting cells at the indi-cated timepoints, duplicate samples of treated and controlcells for each timepoint were trypsinized in 1 ml trypsin-EDTA. Using a hemocytometer, the total number of cellsin each dish was calculated from three separate count-ings. The experiment was performed three times. On thesixth day, samples were also prepared for flow cytometryanalysis.
SAPK/JNK activity
The combined activity of stress-activated protein kinases(SAPK) JNK1 and JNK2 in immunocomplexes precipi-tated from cell extracts (500 µg total protein each) wasassayed using a recombinant GST-c-Jun fusion proteinas a substrate (antibodies and substrate from Santa CruzBiotechnology Inc.). Immunocomplexes were incubatedfor 20 min in RIPA buffer containing leupeptin and apro-tinin (10 mg/ml each), PMSF, DTT (1 mM each), β-glycerophosphate, sodium vanadate (10 mM each),[γ -32P]-ATP (0.8 µCi/sample) and 1.3 µg GST-c-Jun/sample. The supernatants were analyzed by SDS-polyacrylamide gel electrophoresis, and phosphorylationof the GST-c-Jun was quantified by scintillation assay ofthe excised GST-c-Jun band.
Flow cytometry
Control and cisplatin-treated cells were harvested withcell dissociation solution (Sigma Chemical Co.), pelletedand resuspended in 4 ml of fresh buffered 4% formalde-hyde solution. After 30 min fixation at room temperature,the cells were pelleted, resuspended in dropwise added ice-cold 95% ethanol during vortexing, and stored at−20◦C.Flow cytometric analysis was done with a Partec Flow Cy-tometer PASII (Partec, Munster, Germany). Aliquots ofthe cell material that was fixed with formaldehyde andstored in ethanol were washed once in tap water. The cellswere transferred to 0.2 ml subtilisin Carlsberg solution
356 Apoptosis · Vol 5 · No 4 · 2000

12V-H-ras transformed rat fibroblasts
(0.1% Sigma protease XXIV, 0.1 M Tris, 0.007 M NaCl,pH 7.2) and incubated for 45 min at +30◦C to releasecell nuclei. These were stained by direct addition of 4,6′-diamidino-2-phenylindole dihydrochloride (DAPI) solu-tion. At least 10,000 cell nuclei were analyzed per sample.The cell cycle composition was calculated with the Mul-ticycleAV program (Phoenix Flow Systems, San Diego,Cal., USA).
Inducible expression of a dominant-negativemutant of MEKK1
The cDNA of dominant negative mutant of MAPK/ERKkinase kinase 1 (MEKK1) (37 kDa kinase fragment witha K432M point mutation; plasmid obtained from Dr. T.Maniatis) was cloned into a recombinant adenoviral vec-tor to be used in a tetracycline-inducible adenoviral genetransfer system.12 The system consists of two recombi-nant adenovirus vectors, one encoding a transactivatingprotein and another the protein of interest, in our case thedominant negative MEKK1 mutant. Briefly, the activa-tor protein rtTA is constitutively expressed but requires
Figure 1 . Cisplatin-induced fragmentation of nuclei. (A) Cells were treated with 40 µM cisplatin for 60 min and nuclear fragmentationwas assessed at the indicated timepoints thereafter. Filled bars: FR3T3; open bars: Ras2:3. (B) Cells were treated with the indicateddoses of cisplatin for 60 min. Nuclear fragmentation was assessed 24 h later. Filled bars: FR3T3; open bars: Ras2:3; hatched bars:Ras2:4; dotted bars: Ras2:5. (C) Cells were incubated with the indicated doses of cisplatin for 16 h. Filled bars: FR3T3; open bars:Ras2:3. All results are the averages from two experiments.
an inducer (doxycycline, a tetracycline derivative; DOX)to undergo a conformational change which allows it tobind to the promoter and induce expression of the geneof interest.
For the infection, 1 million cells/100 mm-dish wereseeded and after 4 h infected with 5 pfu/cell of both con-structs. DOX (0.5 µM) as then added to the medium.After 20 h cells were rinsed in PBS and either postin-cubated with DOX only, or treated with cisplatin andpostincubated for 20 h in the presence of DOX. To confirmexpression of 37 kDa mutant MEKK1, western blottingof cell extracts was performed with an antibody recog-nizing the C-terminal kinase domain of the wt MEKK1protein (Santa Cruz Biotechnology, Inc.).
Results
Increased cisplatin-induced nuclearfragmentation in ras-transformed cells
In order to study the acute apoptotic effects of cis-platin, apoptosis, seen as the fraction of cells showing
Apoptosis · Vol 5 · No 4 · 2000 357

K. Viktorsson et al.
Figure 1 . (Continued ).
358 Apoptosis · Vol 5 · No 4 · 2000
12V-H-ras transformed rat fibroblasts
fragmentation of ethidium bromide-stained nuclei, wasquantitated at 24 h after cisplatin treatment in FR3T3rat fibroblasts and in three 12V-ras transformed deriva-tive cell lines. Some basal apoptosis was seen in untreatedcells of all lines in some, but not all, experiments. Nuclearfragmentation in Ras2:3 cells had started by 17 h aftertreatment and the fraction of apoptotic cells increased forat least another 12 h, while FR3T3 cells showed very littleapoptosis (Figure 1A). TUNEL assay of similarly treatedcells also indicated higher apoptosis levels in Ras2:3 thanin FR3T3 (not shown). The levels of apoptosis in FR3T3cells and three ras-transformed subclones (Ras2:3, Ras2:4and Ras2:5) were assessed 24 hours after a 60 min ex-posure to different concentrations of cisplatin, and werethroughout the transformed cell lines found to be higherthan in FR3T3. Ras2:3 cells in the continuous presenceof cisplatin also showed more apoptosis than similarlytreated FR3T3 cells (Figure 1C).
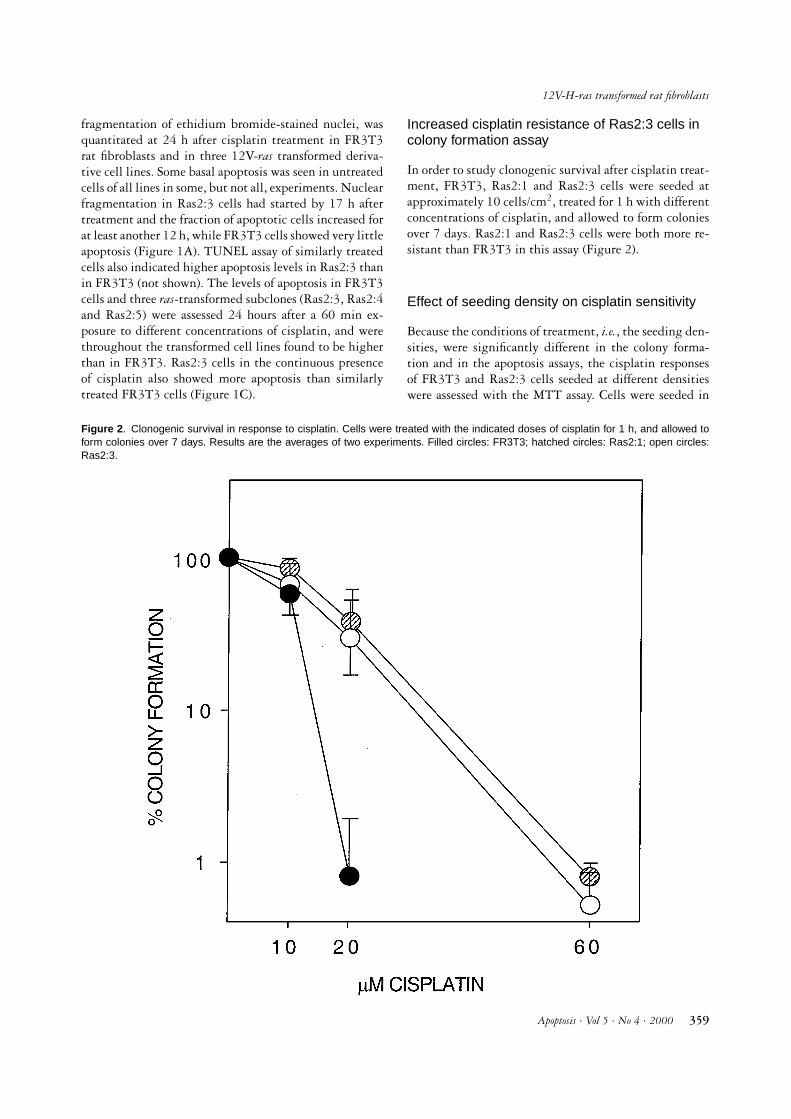
Figure 2 . Clonogenic survival in response to cisplatin. Cells were treated with the indicated doses of cisplatin for 1 h, and allowed toform colonies over 7 days. Results are the averages of two experiments. Filled circles: FR3T3; hatched circles: Ras2:1; open circles:Ras2:3.
Increased cisplatin resistance of Ras2:3 cells incolony formation assay
In order to study clonogenic survival after cisplatin treat-ment, FR3T3, Ras2:1 and Ras2:3 cells were seeded atapproximately 10 cells/cm2, treated for 1 h with differentconcentrations of cisplatin, and allowed to form coloniesover 7 days. Ras2:1 and Ras2:3 cells were both more re-sistant than FR3T3 in this assay (Figure 2).
Effect of seeding density on cisplatin sensitivity
Because the conditions of treatment, i.e., the seeding den-sities, were significantly different in the colony forma-tion and in the apoptosis assays, the cisplatin responsesof FR3T3 and Ras2:3 cells seeded at different densitieswere assessed with the MTT assay. Cells were seeded in
Apoptosis · Vol 5 · No 4 · 2000 359

K. Viktorsson et al.
Figure 3 . Seeding density-dependent viability in response to cisplatin. Cells were seeded in 96-well plates, at the indicated numbers perwell, and corresponding to a range of 1,500–12,000 cells/cm2. After treatment with 40 µM cisplatin for 60 min, cells were postincubatedfor 48 h and viability was assessed using the MTT assay. The ratios of absorbance of cisplatin-treated samples to those of untreatedcontrols were calculated.
96-well plates at densities ranging from 1,500 to 12,000cells/cm2, and treated with 40 µM cisplatin for 60 min.After 48 h, triplicate samples for each cell density were as-sayed for viability. The results (Figure 3) show that Ras2:3cells are more sensitive to cisplatin than FR3T3 at mostseeding densities. However, FR3T3 showed a density-dependent decrease in viability after cisplatin treatment.At the lowest seeding density tested, there was no appar-ent difference in viability of FR3T3 and Ras2:3 cells.
Growth rates of survivors after cisplatin treatment
In order to follow actual growth of cisplatin-treated cellsat standard densities, multiple dishes of both cell lineswere treated with 40 µM cisplatin. The total number ofcells in each sample was then counted at different time-points up to a week after treatment (Figure 4A). In ac-cordance with higher apoptosis levels during the first twodays after treatment, Ras2:3 cells show negative growthcompared to FR3T3. Between 50 h post-treatment tothe end of the experiment, surviving Ras2:3 cells resumegrowth (Figure 4A). The average population doublingtimes for Ras2:3 cells during growth phases, i.e., in the
day 3–6 interval, in three different experiments were 12 hfor controls and 20 h for cisplatin-treated cells. Growth incisplatin-treated FR3T3 was more reduced (Figure 4B);the average doubling times for FR3T3 cells being 16 and37 h, respectively. Altogether, at 6 days post-treatmentthere were more Ras2:3 survivors than FR3T3 survivors.
Flow cytometry analysis showed that by day 6, the cellcycle distribution of cisplatin-treated FR3T3 cells wasalmost identical to that in control FR3T3 (Figure 5A).To assess growth beyond this timepoint, cisplatin-treatedFR3T3 were trypsinized and reseeded on the 6th day, andtheir doubling time between days 7 and 9 was then foundto be 24 h (data not shown). Flow cytometry analysisof cisplatin-treated Ras2:3 cells on day 6 showed someincrease in G1 (Figure 5A), in accordance with slowergrowth. When these cells were trypsinized and reseeded,their doubling time between days 7 and 8 was 12 h (datanot shown).
Differential cell cycle effects
When differences in death and survival responses werethus ascertained, we then went on to study differences in
360 Apoptosis · Vol 5 · No 4 · 2000

12V-H-ras transformed rat fibroblasts
Figure 4 . Regrowth of cisplatin-treated cells. After treatment with 40 µM cisplatin for 1 h, the absolute numbers of cells at each time pointwas calculated in duplicate samples per time point. The experiment was performed three times. Circles: untreated controls; squares:cisplatin-treated cells. Filled symbols: FR3T3; open symbols: Ras2:3. A. Initial negative growth of Ras2:3 and subsequent rapid regrowthof survivors, as compared to FR3T3. B. Filled symbols: FR3T3 cells. Confirmation of slow regrowth of cisplatin-treated FR3T3. See textfor average population doubling times.
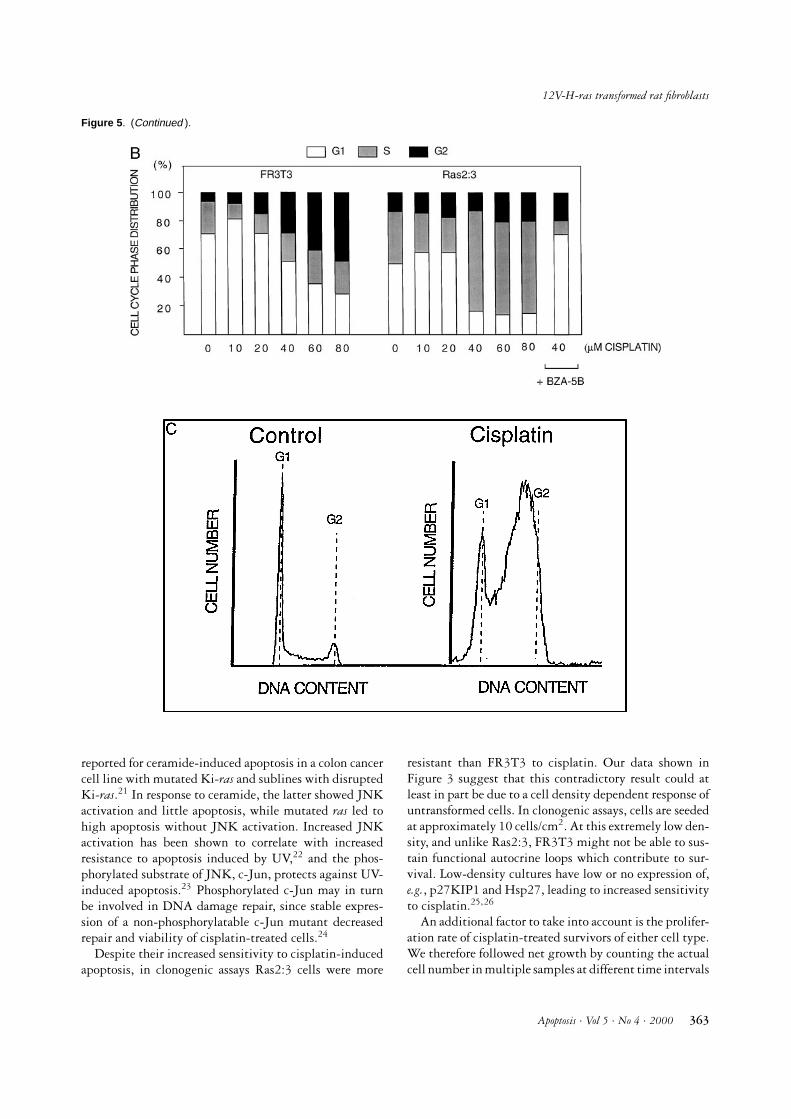
cell cycle phase distribution and cell cycle responses tocisplatin treatment. Flow cytometry analysis of untreatedFR3T3 and Ras2:3 cells showed that the latter have agreater proportion of S + G2 cells (Figure 5A). It wasalso found that at 24 h after cisplatin treatment FR3T3cells showed a dose-dependent G2 arrest, whereas Ras2:3cells showed a striking accumulation of cells in S-phase(Figure 5A–C).
To ascertain the role of Ras in this accumulation, Ras2:3cells were preincubated for 48 h with 100 µM BZA-5B, a Ras farnesylation inhibitor.13 Subsequent cisplatintreatment (40µM for 60 min) led to a G2 arrest at 24 h, asin parental FR3T3 cells, instead of S-phase accumulation(Figure 5B). In addition, cisplatin did not elicit a sub-G1 population in pretreated Ras2:3 cells (not shown),indicating that apoptosis was also abrogated.
Roles of stress-activated pathways in apoptosisand cell cycle arrest
Activation of stress-activated protein kinase (SAPK)/c-Jun N-terminal kinase (JNK) is induced by many typesof cellular stress, including cisplatin. We show here that
cisplatin-induced SAPK/JNK activation was greater inFR3T3 cells than in Ras2:3 (Figure 6), suggesting that inthis system, JNK activation has a protective effect.
The SAPK/JNK pathway is regulated upstream byMEKK1. Early in apoptosis, cisplatin induces cleavageof MEKK1, resulting in loss of SAPK/JNK regulationand in the formation of a pro-apoptotic kinase fragment,14
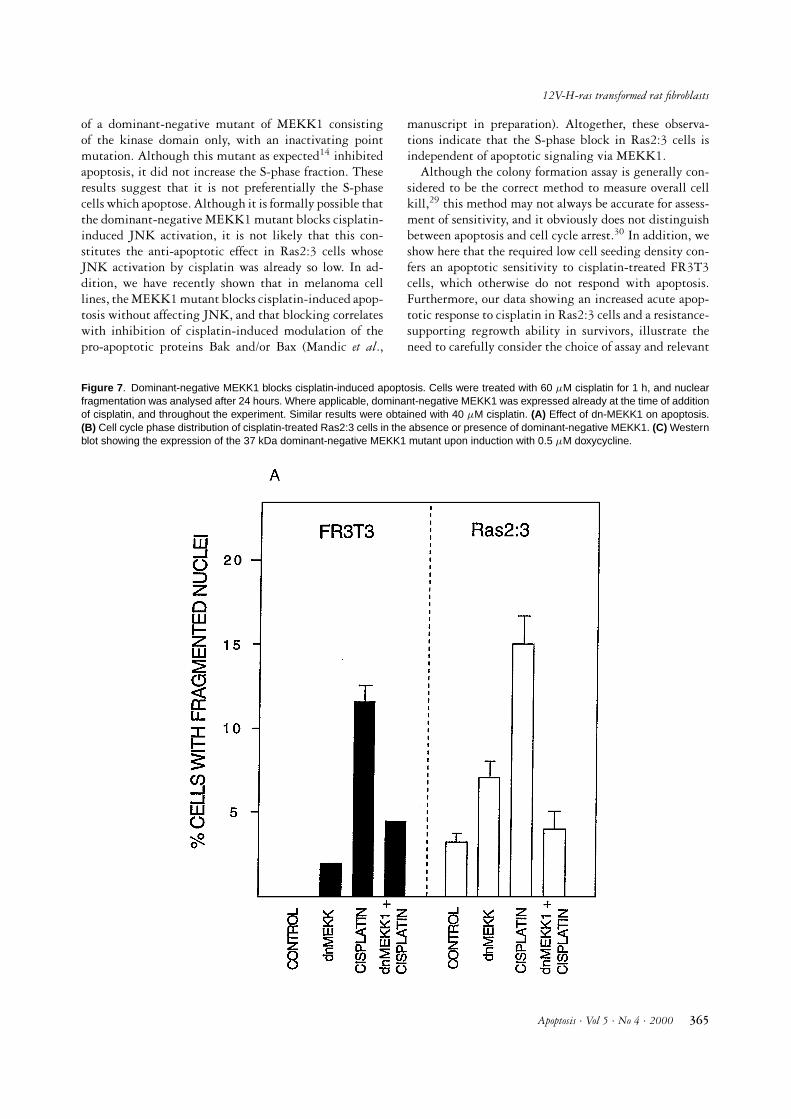
the kinase-inactive form of which has been shown to blockcisplatin-induced apoptosis in HEK293 cells.14 We there-fore used such a dominant-negative mutant of MEKK1to examine if it would affect also cell cycle/S-phase ar-rest. Twenty hours prior to cisplatin treatment, cells ofboth cell lines were infected at a multiplicity of 5 pfu/cellwith a recombinant adenovirus expressing a doxycycline-inducible dominant-negative form of MEKK1. Twentyhours after cisplatin treatment at approximately equi-toxic doses, both cell types had developed apoptosis, whilecisplatin-treated cells expressing the MEKK1 mutant asexpected showed reduced or only background apoptosis(Figure 7A). By contrast, the mutant did not affect theS-phase block (Figure 7B). Figure 7C shows doxycycline-induced expression of 37 kDa mutant MEKK1 protein inboth cell lines.
Apoptosis · Vol 5 · No 4 · 2000 361

K. Viktorsson et al.
Discussion
Oncogenic, constitutively active Ras is found in about30% of all human cancers.15 Because it may regulate anumber of pathways involved in mitogenic, stress andapoptotic signaling, respectively,16,17 active Ras can beexpected to affect tumor cell responses to anticancer ther-apy. In the case of cisplatin, pairwise examination of non-transformed and ras-transformed fibroblast cell lines hasrevealed either increased resistance in transformants,7,18
or increased sensitivity10 or no difference between thetwo.8,19
We have here compared the separate effects of cis-platin on apoptosis, growth and cell cycle of immortalizedrat fibroblasts (FR3T3) and a ras-transformed derivative(Ras2:3). We found that at 24 h after cisplatin treatment,Ras2:3 fibroblast cells exhibited significantly higher nu-clear fragmentation and lower viability than did parentalFR3T3 cells. This increase in apoptosis was seen also withTUNEL and caspase-3 activity assays (data not shown).
Figure 5 . Cell cycle phase distributions. (A) Distribution of cell cycle phases, as assessed by flow cytometry analysis of samples atindicated timepoints after 60 min treatment with 40 µM cisplatin. n = 6 for 0 and 24 h; n = 2 for 144 h. (B) Distribution of cell cyclephases 24 h after treatment with the indicated concentrations of cisplatin for 60 min. At the far right, pretreatment of Ras2:3 cells withRas farnesylation inhibitor BZA-5B is shown to abrogate the S-phase block induced by 40 µM cisplatin. Results are averages from atleast two separate experiments. (C) DNA profiles of untreated (control) and cisplatin-treated Ras2:3 cells obtained by flow cytometryanalysis. Positions of G1 and G2 peaks are indicated in both figures.
It is interesting to speculate whether the altered res-ponses of Ras2:3 cells are related to the finding that ac-tivated Ras binds to MEKK1 upstream of SAPK/JNK20
and whether this association might alter the availabilityof MEKK1 for activation or for MEKK1 substrates. It iswell established that the MEKK1-SAPK pathway is acti-vated by various types of cellular stress, including manyapoptosis-inducing agents. Both MEKK1 and JNK havepotential pro- or anti-proliferative functions; MEKK1 hasindeed been described as a molecular switch betweensurvival and death,14 in that the activated full-lengthMEKK1 regulates pro-survival SAPK/JNK activities,whereas during early apoptosis it can be proteolyticallycleaved to yield a pro-apoptotic constitutively active ki-nase fragment which is likely involved in apoptosis with-out affecting SAPK/JNK.14 We therefore comparedcisplatin-induced activation of SAPK/JNK in FR3T3 andRas2:3 cells, and found that activation was reduced inRas2:3 cells, suggesting that in this system SAPK/JNKactivation has a protective role. Similar results have been
362 Apoptosis · Vol 5 · No 4 · 2000
12V-H-ras transformed rat fibroblasts
Figure 5 . (Continued ).
reported for ceramide-induced apoptosis in a colon cancercell line with mutated Ki-ras and sublines with disruptedKi-ras.21 In response to ceramide, the latter showed JNKactivation and little apoptosis, while mutated ras led tohigh apoptosis without JNK activation. Increased JNKactivation has been shown to correlate with increasedresistance to apoptosis induced by UV,22 and the phos-phorylated substrate of JNK, c-Jun, protects against UV-induced apoptosis.23 Phosphorylated c-Jun may in turnbe involved in DNA damage repair, since stable expres-sion of a non-phosphorylatable c-Jun mutant decreasedrepair and viability of cisplatin-treated cells.24
Despite their increased sensitivity to cisplatin-inducedapoptosis, in clonogenic assays Ras2:3 cells were more
resistant than FR3T3 to cisplatin. Our data shown inFigure 3 suggest that this contradictory result could atleast in part be due to a cell density dependent response ofuntransformed cells. In clonogenic assays, cells are seededat approximately 10 cells/cm2. At this extremely low den-sity, and unlike Ras2:3, FR3T3 might not be able to sus-tain functional autocrine loops which contribute to sur-vival. Low-density cultures have low or no expression of,e.g., p27KIP1 and Hsp27, leading to increased sensitivityto cisplatin.25,26
An additional factor to take into account is the prolifer-ation rate of cisplatin-treated survivors of either cell type.We therefore followed net growth by counting the actualcell number in multiple samples at different time intervals
Apoptosis · Vol 5 · No 4 · 2000 363

K. Viktorsson et al.
Figure 6 . JNK activation by cisplatin. The joint activity of JNK1 and JNK2 immunoprecipitated from cell extracts (500 µg total proteineach) was assessed as incorporation of [γ -32P]-ATP into a GST-c-Jun fusion protein, as described in Materials and Methods. JNKactivity is shown as fold activity relative to the activity in FR3T3 control extracts. In addition to controls, samples were taken after 60 minpost-addition of cisplatin, after 60 min cisplatin treatment followed by 90 min or 20 h postincubation in conditioned medium.
after cisplatin treatment. These experiments confirmedthat during the first 2 days after treatment, cisplatininduced negative growth in Ras2:3 cells, in accordancewith significant apoptosis for at least 29 h, whereaftersurvivors resumed growth, albeit with an increase in dou-bling time compared to controls. By contrast, cisplatin-treated FR3T3 cells did not show any negative growth,in accordance with very little apoptosis. After the secondday there was some net growth, but the doubling timewas twice that of control FR3T3. By day 6, there was stillsome G2 accumulation, which cannot alone explain theslow growth rate during the preceding days. However,these samples also showed a sub-G1 fraction, which sug-gests that in addition to G2 arrest there was also somelate-developing cell death (apoptosis or necrosis) as a con-sequence of an inability to divide. Cells damaged but not
killed by cisplatin have been shown to remain arrested andmetabolizing for several days before dying.27,28 In Ras2:3survivors, some G1 accumulation seen on day 6 likely ex-plains the reduction in growth rate during the precedingdays. Beyond day 6, survivors of both cell lines showednormal or near-normal doubling times, in accordance witha near normal cell cycle distribution on day 6.
In addition to increased apoptosis, another major dif-ference in the acute cisplatin responses of Ras2:3 cells andFR3T3 was the induction of a massive S-phase block inRas2:3, while in FR3T3 cells the S-phase fraction was un-affected at all doses tested. The S-phase fraction in Ras2:3remained unaltered at 30 h after cisplatin treatment (notshown), despite significant apoptosis by that time, indi-cating that apoptosis is not restricted to S-phase cells.We then examined the effect on cell cycle distribution
364 Apoptosis · Vol 5 · No 4 · 2000
12V-H-ras transformed rat fibroblasts
of a dominant-negative mutant of MEKK1 consistingof the kinase domain only, with an inactivating pointmutation. Although this mutant as expected14 inhibitedapoptosis, it did not increase the S-phase fraction. Theseresults suggest that it is not preferentially the S-phasecells which apoptose. Although it is formally possible thatthe dominant-negative MEKK1 mutant blocks cisplatin-induced JNK activation, it is not likely that this con-stitutes the anti-apoptotic effect in Ras2:3 cells whoseJNK activation by cisplatin was already so low. In ad-dition, we have recently shown that in melanoma celllines, the MEKK1 mutant blocks cisplatin-induced apop-tosis without affecting JNK, and that blocking correlateswith inhibition of cisplatin-induced modulation of thepro-apoptotic proteins Bak and/or Bax (Mandic et al.,
Figure 7 . Dominant-negative MEKK1 blocks cisplatin-induced apoptosis. Cells were treated with 60 µM cisplatin for 1 h, and nuclearfragmentation was analysed after 24 hours. Where applicable, dominant-negative MEKK1 was expressed already at the time of additionof cisplatin, and throughout the experiment. Similar results were obtained with 40 µM cisplatin. (A) Effect of dn-MEKK1 on apoptosis.(B) Cell cycle phase distribution of cisplatin-treated Ras2:3 cells in the absence or presence of dominant-negative MEKK1. (C) Westernblot showing the expression of the 37 kDa dominant-negative MEKK1 mutant upon induction with 0.5 µM doxycycline.
manuscript in preparation). Altogether, these observa-tions indicate that the S-phase block in Ras2:3 cells isindependent of apoptotic signaling via MEKK1.
Although the colony formation assay is generally con-sidered to be the correct method to measure overall cellkill,29 this method may not always be accurate for assess-ment of sensitivity, and it obviously does not distinguishbetween apoptosis and cell cycle arrest.30 In addition, weshow here that the required low cell seeding density con-fers an apoptotic sensitivity to cisplatin-treated FR3T3cells, which otherwise do not respond with apoptosis.Furthermore, our data showing an increased acute apop-totic response to cisplatin in Ras2:3 cells and a resistance-supporting regrowth ability in survivors, illustrate theneed to carefully consider the choice of assay and relevant
Apoptosis · Vol 5 · No 4 · 2000 365

K. Viktorsson et al.
Figure 7 . (Continued ).
366 Apoptosis · Vol 5 · No 4 · 2000

12V-H-ras transformed rat fibroblasts
timing in studies on drug efficacy as well as on cell-typedependent responses to drugs.
Acknowledgments
This work was supported by grants from the SwedishCancer Foundation (Cancerfonden) and the Gustaf VJubilee Foundation. We also thank Dr. T. Maniatis forproviding the mutant MEKK1 cDNA.
References
1. Perez RP. Cellular and molecular determinants of cisplatin re-sistance. Eur J Cancer 1998; 34: 1535–1542.
2. Zamble DB, Jacks T, Lippard SJ. p53-dependent and -inde-pendent responses to cisplatin in mouse testicular teratocarci-noma cells. Proc Natl Acad Sci USA 1998; 95: 6163–6168.
3. Ormerod MG, Orr RM, Peacock JH. The role of apoptosis incell killing by cisplatin: A flow cytometric study. Br J Cancer1994; 69: 93–100.
4. Wilkins DE, Ng CE, Raaphorst GP. Cell cycle perturbationsin cisplatin-sensitive and resistant human ovarian carcinomacells following treatment with cisplatin and low dose rate irra-diation. Cancer Chemother Pharmacol 1997; 40: 159–166.
5. Vaisman A, Varchenko M, Said I, Chaney SG. Cell cycle changesassociated with formation of Pt-DNA adducts in human ovar-ian carcinoma cells with different cisplatin sensitivity. Cytometry1997; 2: 54–64.
6. Sanchez-Perez I, Murguia JR, Perona R. Cisplatin inducesa persistent activation of JNK that is related to cell death.Oncogene 1998; 16: 533–540.
7. Isonishi S, Hom DK, Thiebaut FB, et al. Expression of the c-Ha-ras oncogene in mouse NIH3T3 cells induces resistance tocisplatin. Cancer Res 1991; 51: 5903–5909.
8. Warenius HM, Seabra LA, Maw P. Sensitivity to cis-diamminedichloroplatinum in human cancer cells is relatedto expression of cyclin D1 but not c-raf-1 protein. Int J Cancer1996; 67: 224–231.
9. Jansen B, Schlagbauer-Wadl H, Eichler HG, et al. Activated N-Ras contributes to the chemoresistance of human melanoma insevere combined immunodeficiency (SCID) mice by blockingapoptosis. Cancer Res 1997; 57: 362–365.
10. Basu A, Cline JS. Oncogenic transformation alters cisplatin-induced apoptosis in rat embryo fibroblasts. Int J Cancer 1995;63: 597–603.
11. Holford J, Rogers P, Kelland LR. ras mutation and platinumresistance in human ovarian carcinomas in vitro. Int J Cancer1998; 77: 94–100.
12. Molin M, Shoshan MC, Ohman-Forslund K, Linder S,Akusjarvi G. Two novel adenovirus vector systems permittingregulated protein expression in gene transfer experiments. JVirol 1998; 72: 8358–8361.
13. James GL, Goldstein JL, Brown MS, et al. Benzodiazepine pep-tidomimetics: Potent inhibitors of ras farnesylation in animalcells. Science 1993; 260: 1937–1942.
14. Widmann C, Gerwins P, Lassignal Johnson N, Jarpe MB,Johnson GL. MEK kinase 1, a substrate for DEVD-directedcaspases, is involved in genotoxin-induced apoptosis. Mol CellBiol 1998; 18: 2416–2429.
15. Bos JL. ras oncogenes in human cancer—a review. Cancer Res1989; 49: 4682–4689.
16. Joneson T, Bar-Sagi D. Ras effectors and their role in mitoge-nesis and oncogenesis. J Mol Medicine 1997; 75: 587–593.
17. Chen CY, Liou J, Forman LW, Faller DV. Differential regulationof discrete apoptotic pathways by Ras. J Biol Chem 1998; 273:16700–16709.
18. Sklar MD. Increased resistance to cis-diamminedichloroplati-num(II) in NIH3T3 cells transformed by ras oncogenes. CancerRes 1988; 48: 793–797.
19. Perez RP, Hamaguchi K, Tracey PA, Handel LM, HamiltonLM, Godwin AK. Transformation of rat ovarian epithelial andRat-1 fibroblast cell lines by RAST24 does not influence cis-platin sensitivity. Cancer Res 1993; 53: 3771–3775.
20. Russell M, Lange-Carter CA, Johnson GL. Direct interactionbetween Ras and the kinase domain of mitogen-activated pro-tein kinase kinase kinase (MEKK1), J Biol Chem 1995; 270:11757–11760.
21. Ohmori M, Shirasawa S, Furuse M, Okumura K, Sasazuki T. Ac-tivated Ki-ras enhances sensitivity of ceramide-induced apop-tosis without c-Jun NH2-terminal kinase/stress-activated pro-tein kinase or extracellular signal-regulated kinase activationin human colon cancer cells. Cancer Res 1997; 57: 4714–4717.
22. Butterfield L, Storey B, Maas L, Heasley LE. c-Jun NH2-terminal kinase regulation of the apoptotic response of smallcell lung cancer cells to ultraviolet radiation. J Biol Chem 1997;272: 10110–10116.
23. Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycleprogression and apoptosis by distinct mechanisms. EMBO J1999; 18: 188–197.
24. Potapova O, Haghighi A, Bost F, et al. The Jun kinase/stress-activated protein kinase pathway functions to regulate DNArepair and inhibition of the pathway sensitizes tumor cells tocisplatin. J Biol Chem 1997; 272: 14041–14044.
25. Garrido C, Ottavi P, Fromentin A, et al. HSP27 as a mediatorof confluence-dependent resistance to cell death induced byanticancer drugs. Cancer Res 1997; 57: 2661–2667.
26. Dimanche-Boitrel MT, Micheau O, Hammann A, Eymin B,Chauffert B, Solary E. Contribution of the cyclin-dependentkinase inhibitor p27KIP1 to the confluence-dependent resis-tance of HT29 human colon carcinoma cells. Int J Cancer 1998;77: 796–802.
27. Sorenson CM, Barry MA, Eastman A. Analysis of events asso-ciated with cell cycle arrest at G2 phase and cell death inducedby cisplatin. J Natl Cancer Inst 1990; 82: 749–755.
28. Eastman A. Activation of programmed cell death by anticanceragents: Cisplatin as a model system. Cancer Cells 1990; 2: 275–280.
29. Brown JM, Wouters BG. Does apoptosis contribute to tu-mor cell sensitivity to anticancer agents? In: Hickman JA,Dive C, eds. Apoptosis and Cancer Chemotherapy. Totowa, NJ,USA: Humana Press, 1999: 1–20.
30. Waldman T, Zhang Y, Dillehay L, et al. Cell-cycle arrest versuscell death in cancer therapy. Nature Medicine 1997; 3: 1034–1036.
Apoptosis · Vol 5 · No 4 · 2000 367




















