
Incidence and clinicobiologic characteristics of leukemic B-cell chronic lymphoproliferative...
10
doi:10.1182/blood-2003-01-0045 Prepublished online June 26, 2003; 2003 102: 2994-3002 Jesus-Maria Hernandez, Jesus F. San Miguel and Alberto Orfao Barbon, Alejandro Martin, Pilar de la Fuente, Guillermo Martin-Nuñez, Javier Fernandez-Calvo, Ana Balanzategui, Maria-Consuelo Lopez-Berges, Josep Nomdedeu, Teresa Vallespi, Marcos Maria-Luz Sanchez, Julia Almeida, David Gonzalez, Marcos Gonzalez, Maria-Antonia Garcia-Marcos, lymphoproliferative disorders with more than one B-cell clone Incidence and clinicobiologic characteristics of leukemic B-cell chronic http://bloodjournal.hematologylibrary.org/content/102/8/2994.full.html Updated information and services can be found at: (3201 articles) Clinical Trials and Observations (4217 articles) Neoplasia Articles on similar topics can be found in the following Blood collections http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requests Information about reproducing this article in parts or in its entirety may be found online at: http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprints Information about ordering reprints may be found online at: http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtml Information about subscriptions and ASH membership may be found online at: Copyright 2011 by The American Society of Hematology; all rights reserved. Washington DC 20036. by the American Society of Hematology, 2021 L St, NW, Suite 900, Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.org From
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Incidence and clinicobiologic characteristics of leukemic B-cell chronic lymphoproliferative...

doi:10.1182/blood-2003-01-0045Prepublished online June 26, 2003;2003 102: 2994-3002
Jesus-Maria Hernandez, Jesus F. San Miguel and Alberto OrfaoBarbon, Alejandro Martin, Pilar de la Fuente, Guillermo Martin-Nuñez, Javier Fernandez-Calvo,Ana Balanzategui, Maria-Consuelo Lopez-Berges, Josep Nomdedeu, Teresa Vallespi, Marcos Maria-Luz Sanchez, Julia Almeida, David Gonzalez, Marcos Gonzalez, Maria-Antonia Garcia-Marcos, lymphoproliferative disorders with more than one B-cell cloneIncidence and clinicobiologic characteristics of leukemic B-cell chronic
http://bloodjournal.hematologylibrary.org/content/102/8/2994.full.htmlUpdated information and services can be found at:
(3201 articles)Clinical Trials and Observations � (4217 articles)Neoplasia �
Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.Washington DC 20036.by the American Society of Hematology, 2021 L St, NW, Suite 900, Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly
For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.orgFrom

NEOPLASIA
Incidence and clinicobiologic characteristics of leukemic B-cell chroniclymphoproliferative disorders with more than one B-cell cloneMaria-Luz Sanchez, Julia Almeida, David Gonzalez, Marcos Gonzalez, Maria-Antonia Garcia-Marcos, Ana Balanzategui,Maria-Consuelo Lopez-Berges, Josep Nomdedeu, Teresa Vallespi, Marcos Barbon, Alejandro Martin, Pilar de la Fuente,Guillermo Martin-Nunez, Javier Fernandez-Calvo, Jesus-Maria Hernandez, Jesus F. San Miguel, and Alberto Orfao
Leukemic B-chronic lymphoproliferativedisorders (B-CLPDs) are generally be-lieved to derive from a monoclonal B cell;biclonality has only occasionally beenreported. In this study, we have exploredthe incidence of B-CLPD cases with 2 ormore B-cell clones and established boththe phenotypic differences between thecoexisting clones and the clinicobiologicfeatures of these patients. In total, 53B-CLPD cases with 2 or more B-cell cloneswere studied. Presence of 2 or more B-cell clones was suspected by immunophe-notype and confirmed by molecular/
genetic techniques in leukemic samples(n � 42) and purified B-cell subpopula-tions (n � 10). Overall, 4.8% of 477 con-secutive B-CLPDs had 2 or more B-cellclones, their incidence being especiallyhigher among hairy cell leukemia (3 of13), large cell lymphoma (2 of 10), andatypical chronic lymphocytic leukemia(CLL) (4 of 29). In most cases the 2 B-cellsubsets displayed either different surfaceimmunoglobulin (sIg) light chain (n � 37of 53) or different levels of the same sIg(n � 9 of 53), usually associated with otherphenotypic differences. Compared with
monoclonal cases, B-CLL patients with 2or more clones had lower white blood cell(WBC) and lymphocyte counts, more fre-quently displayed splenomegaly, and re-quired early treatment. Among these, thecases in which a CLL clone coexisted with anon-CLL clone were older and more oftendisplayed B symptoms, a monoclonal com-ponent, and diffuse infiltration of bone mar-row and required early treatment more fre-quently than cases with monoclonal CLL or2 CLL clones. (Blood. 2003;102:2994-3002)
© 2003 by The American Society of Hematology
Introduction
B-cell chronic lymphoproliferative disorders (B-CLPDs) are a heteroge-neous group of diseases that result from the proliferation and accumula-tion of mature-appearing aberrant B lymphocytes arrested at a givenstage of differentiation.1-5 B-CLPDs are generally believed to resultfrom the monoclonal expansion of a single transformed B lymphocyte.6
Therefore, the tumoral B lymphocytes constitute a clone in which allcells are related by possessing the original transforming mutation,possessing identical VDJ rearrangements of the immunoglobulin heavychain (IgH) gene, and showing restricted Ig light chain expression.Additionally, distinctive genetic and phenotypic features may be gainedduring the evolution of the disease. In this sense, the presence of 2 ormore morphologically different populations of neoplastic lymphocytesin the same patient, detected either simultaneously or at different timepoints, is usually interpreted as reflecting either different maturationstages or subclone formation within the original malignant tumor stemcell line.7 Although most lymphoproliferative disorders are consideredto be monoclonal, biclonality has been occasionally reported in theliterature,8-19 frequently corresponding to rare cases of B-cell non-Hodgkin lymphomas (NHLs), which more commonly develop inimmunocompromised individuals.8-11 In most instances these reportsdescribe the coexistence of 2 distinct diseases such as mantle celllymphoma (MCL) and follicular lymphoma (FL),17 FL and smalllymphocytic lymphoma (SLL),17 MCL and chronic lymphocytic leuke-
mia/SLL (CLL/SLL),17 SLL and large cell lymphoma (LCL),18 andhairy cell leukemia (HCL) and CLL/SLL.19 Therefore, these caseswould not correspond to oligoclonal expansions of B cells, which havelong been claimed to precede the monoclonal disease state.12
Despite this, to the best of our knowledge, no study has beenreported so far in which either the presence of 2 or more differentB-cell clones has been systematically analyzed in a large series ofpatients with leukemic B-CLPD or the clinical and biologiccharacteristics of patients with 2 or more B-cell clones have beencompared with those of monoclonal cases.
The aim of the present study was to establish the incidence ofcases with 2 or more B-cell clones based on a large series of 477consecutive leukemic B-CLPD patients, to establish the differentphenotypic patterns of the coexisting neoplastic B-cell clones, andto compare the clinical and biologic disease features of cases with 2or more B-cell clones versus monoclonal B-CLPD patients.
Patients, materials, and methods
Patients
A total of 507 newly diagnosed untreated patients suffering from leukemicB-CLPD were analyzed in this study. From them, 477 corresponded to
From the Servicio General de Citometria y Departamento de Medicina,Universidad de Salamanca; Centro de Investigacion del Cancer, Universidadde Salamanca; Servicio de Hematologia, Hospital Universitario de Salamanca;Servicio de Hematologia, Hospital Santa Creu i Sant Pau, Barcelona; Serviciode Hematologia, Hospital Valle del Hebron, Barcelona; Servicio deHematologia, Hospital Virgen Blanca, Leon; Servicio de Hematologia, HospitalVirgen de la Concha, Zamora; Servicio de Hematologia, Hospital GeneralYague, Burgos; Servicio de Hematologia, Hospital Virgen del Puerto,Plasencia; and Servicio de Hematologia, Hospital Clınico de Valladolid, Spain.
Submitted January 7, 2003; accepted June 10, 2003. Prepublished online as
Blood First Edition Paper, June 26, 2003; DOI 10.1182/blood-2003-01-0045.
Reprints: Alberto Orfao, Servicio General de Citometria, Hospital Universitariode Salamanca, Paseo San Vicente, 58-182, 37007 Salamanca, Spain; e-mail:[email protected].
The publication costs of this article were defrayed in part by page chargepayment. Therefore, and solely to indicate this fact, this article is herebymarked ‘‘advertisement’’ in accordance with 18 U.S.C. section 1734.
© 2003 by The American Society of Hematology
2994 BLOOD, 15 OCTOBER 2003 � VOLUME 102, NUMBER 8
For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.orgFrom

consecutive B-CLPDs studied in our laboratory and the other 30 cases topatients suspected of having 2 or more clones by phenotypic criteria, whowere referred to our center for this specific reason. Diagnosis of B-CLPDwas established in all cases on the basis of clinical, morphologic,immunophenotypic, molecular and, whenever available, also histologiccriteria, according to the World Health Organization (WHO) classifica-tion.20 In 36% of the cases diagnosis was established in a routine blood cellanalysis. Distribution according to diagnosis was as follows: 399 caseswere CLL/SLL (for simplicity they were referred as CLL) (365 were typical[typCLL] and 34 atypical [atypCLL]); prolymphocytic leukemia (PLL), 5;hairy cell leukemia (HCL), 14; lymphoplasmacytic lymphoma (LPL), 16;splenic marginal zone lymphoma (SMZL), 16; FL, 25; MCL, 20; and LCL,10 cases; 2 B-CLPD patients could not be further classified. Mean age of thepatients was 70 � 11 years (median, 71 years; range, 19-95 years); 314 weremales and 193 were females. In all cases, samples were obtained for this studyafter informed consent according to the Ethics Committee of the UniversityHospital of Salamanca. At the moment of closing of this study, the medianfollow-up of the whole series was 28 months (range, 3-75 months): 29 months forconsecutively studied patients and 21 months for referred cases.
Cytomorphologic studies
Either peripheral blood (PB) and/or bone marrow (BM) smears from EDTA(ethylenediaminetetraacetic acid)–-anticoagulated samples, prepared imme-diately after sampling, were stained with May-Grunwald-Giemsa (MGG)and cell morphology was analyzed by optical microscopy. The sampleswere examined by 2 independent observers and lymphocytes classified intothe following categories: (1) small lymphocytes, (2) cleaved lymphocytes,(3) large nongranular lymphocytes, (4) large granular lymphocytes, (5)prolymphocytes, and (6) lymphoplasmacytoid cells. The presence orabsence of “smudged cells” was also evaluated in each sample.
Immunophenotypic analyses
PB (n � 417) and BM (n � 80) samples were collected in tubes containingK3 EDTA as anticoagulant, while ascitic fluid (n � 1) was placed in a tubecontaining heparin. Lymph node samples (n � 9) were collected in anisotonic saline buffer, cut into small pieces that were then placed in RPMImedium (BioWhittaker, Walkersville, MD), and gently dispersed into singlecell suspensions; after 2 washing steps, the cell pellet was resuspended inphosphate-buffered saline (PBS) at a concentration of around 2 � 106 cellsper 100 �L. All samples were stained using a direct immunofluorescencestain-and-then-lyse technique previously described in detail21 with thefollowing 3-color combinations of monoclonal antibodies (moAb) conju-gated with fluorescein isothiocyanate (FITC)/phycoerythrin (PE)/and thePE–cyanine 5 (PE/Cy5) fluorochrome tandem: FMC7/CD5/CD19, CD22/CD23/CD19, CD103/CD25/CD19, CD10/CD11c/CD19, and sIg�/sIg�/CD19. Directly conjugated moAb reagents were purchased from BectonDickinson Biosciences (BDB, San Jose, CA): CD5-PE, CD10-PE, CD11c-FITC, CD22-FITC, CD23-PE, CD25-PE, and anti-�–FITC/anti-�–PE; fromCaltag Laboratories (San Francisco, CA): CD19-PE/Cy5; from Immunotech(Marseille, France): FMC7-FITC; and from Immunoquality Products (Gronin-gen, The Netherlands): CD103-FITC. Data acquisition was performed usingCellQuest software on a FACSort flow cytometer (BDB). In those samples with alow percentage of B lymphocytes, a selective acquisition of CD19� gated cellswas also made as previously described in detail.21 The median percentage ofCD19� clonal B cells within the lymphocyte gate was 62% (range, 3.4%-95%).For data analysis, the Paint-A-Gate PRO software program (BDB) was used. Blymphocytes were identified according to their SSClo/int/CD19� distribution, andtheir percentage was calculated after excluding cell debris and platelets accordingto conventional procedures.21
Assessment of clonality by molecular biology
Genomic DNA preparation and Southern blot analysis. High molecularweight DNA was isolated by standard proteinase K digestion, phenol-chloroform extraction, and ethanol precipitation.22 For Southern blotanalysis, 10 �g DNA was digested to completion with the BglII andBamHI/HindIII restriction enzymes, size fractionated in 0.7% agarose gels,
and blotted onto nylon membranes as previously described.23 The blotswere hybridized with the IgHJ6 digoxigenin-labeled probe.
PCR amplification and heteroduplex analysis. For amplification ofcomplete VDJH gene rearrangements 3 different sets of family-specificprimers and 1 JH consensus primer were used in 3 different multiplexedpolymerase chain reactions (PCRs) covering the 3 framework regions(FRs). Amplification of incomplete DJH gene rearrangements was per-formed in 2 different reactions using family-specific primers for DH1 toDH6 and DH7 families, respectively, together with the consensus JHprimer. All primers had been newly designed during the BIOMED-2concerted action “PCR-based clonality studies for early diagnosis oflymphoproliferative disorders” (PL96-3936).24 All reactions were carriedout in 50 �L containing 0.1 �g DNA samples and 10 pmol of each primer.
For heteroduplex analysis, PCR products were denatured at 94°C for 10minutes and subsequently cooled at 4°C for 60 minutes to induce duplexformation.23-25 The heteroduplexes and/or homoduplexes generated wereimmediately loaded on a 10% nondenaturing polyacrylamide gel in 1 �Tris (tris(hydroxymethyl)aminomethane)–acetate–EDTA (TAE) buffer, runat room temperature (RT), and visualized by ethidium bromide staining.The presence of one or more clear bands within the expected size indicatedclonal rearrangements, while polyclonal rearrangements appeared as arough smear in the gel.23-25
Sequencing and analysis of Ig genes. PCR products were eluted fromthe polyacrylamide gels and directly sequenced in an automated ABI 377DNA sequencer using Big-Dye terminators (Applied Biosystems, FosterCity, CA). To avoid nucleotide misinterpretations due to Taq errors, all productswere sequenced at least twice from different PCR reactions using VH/DH and/orJH primers. Germ line VH, DH, and JH segments from complete VDJH generearrangements were identified by comparison with the V base26 and IGMTdatabase27 using online DNAPLOT (MRC Center for Protein Engineering,Cambridge, United Kingdom). DH and JH germ line segments from incompleteDJH gene rearrangements were identified using BLAST search in the DH-JHgerm line locus sequence (accession no. EMB/X97051).
Molecular biclonalilty/triclonalilty was defined when 3 or 4 unrelatedVDJH, DJH gene rearrangements and/or translocations involving the 14q32IGH gene locus were found. Secondary rearrangements to a pre-existentVDJH or DJH gene rearrangement were not taken into account fordefinition of biclonalilty/triclonalilty.
Cytogenetic analysis
Cytogenetic studies were carried out in BM samples from 20 of the 53multiclonal cases, after culture with TPA (12-O-tetradecanoylphorbol-13-acetate) for 72 hours, according to previously reported methods.28 Inaddition, conventional interphase fluorescence in situ hybridization (iFISH)for the translocations t(11;14), t(14;18), and t(11;18) was performed inFACS-purified subsets of clonal B cells in a subgroup of 6 of these casesusing Vysis (Downers Grove, IL) LSI IgH/CCND1 dual-color, dual-fusiontranslocation probe; LSI IgH/Bcl2 dual-color, dual-fusion translocationprobe; and LSI MALT1 dual-color Breakapart probe.
Flow cytometry sorting of B-cell subsets
In 10 cases in which different B-cell subsets displaying aberrant phenotypeswere identified, specific fluorescence-activated cell sorting (FACS) of eachof the 2 B-cell subpopulations was performed in a FACS Vantage flowcytometer (BDB). Discrimination between the 2 cell subsets was based ontheir distinct expression for 1 or more B-cell markers, according towell-established methods. Molecular and cytogenetic clonality based onIgH gene rearrangements as well as on the presence of different chromo-some translocations was further assessed in the sorted B cells using thetechniques described elsewhere in this paper. The purity of the sortedfractions was 98% � 1.6% (range, 95%-99.5%).
Statistical methods
Either relative frequencies or mean values and their standard deviation, aswell as range, were calculated for each categorical or continuous variableunder study using the SPSS software program (SPSS 10.0, Chicago, IL). To
LEUKEMIC B-CLPDs WITH 2 OR MORE CLONES 2995BLOOD, 15 OCTOBER 2003 � VOLUME 102, NUMBER 8
For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.orgFrom

Table 1. Molecular characteristics of B-CLPD with 2 or more B-cell clones
Case no. Diagnosis
Molecular studies
Southern blot PCR
Pattern ofIgH gene
rearrangementNo. of
clones
Pattern ofIgH gene
rearrangementNo. of
clones
1 typCLL RR RG 2 RR RG 2
2 typCLL RR RR 2 RR RG 2
3 typCLL RR RG 2 RR RR 2
4 typCLL RR RG 2 RR RG 2
5 typCLL RR RR RG 3 RR RG 2
6 typCLL NA NA NA NA
7 typCLL RR RR 2 RR RR 2
8 typCLL RR 1 RR RG 2
9 typCLL RR 1 RR RR 2
10 typCLL RR 1 RR RR 2
11 typCLL RR RR 2 RR RG 2
12 typCLL RR RG 2 RR RR 2
13 typCLL RR RG 2 RR 1
14 typCLL NA NA RR RG 2
15 typCLL RR 1 RR RR 2
16 typCLL NA NA RR RR 2
17 typCLL NA NA RR RG 2
18 typCLL NA NA RR RG 2
19 typCLL NA NA RR RG 2
20 typCLL NA NA RG RG 2
21 typCLL NA NA RR RG 2
22 typCLL NA NA RR RR 2
23 typCLL NA NA NA NA
24 typCLL NA NA NA NA
25 atypCLL RR 1 RR RG 2
26 atypCLL RR RG 2 RR 1
27 atypCLL NA NA NA NA
28 atypCLL RR RR 2 RR RR 2
29 atypCLL RR RR 2 RR RR RR 3
30 atypCLL RR 1 RR RG 2
31 atypCLL NA NA RR RR 2
32 atypCLL RR 1 RR RG 2
33 atypCLL NA NA NA NA
34 HCL RR RG 2 NA NA
35 HCL NA NA NA NA
36 HCL NA NA RR RR 2
37 HCL NA NA RR RG 2
38 LPL NA NA RR RG 2
39 LPL RR 1 RR RR 2
40 LPL NA NA NA NA
41 LPL NA NA RR RG 2
42 SMZL RR 1 RR RR 2
43 SMZL NA NA RR RG 2
44 SMZL NA NA NA NA
45 FL NA NA NA NA
46 FL RR RR 2 RR RG 2
47 FL GG 0 RR RR 2
48 MCL RR RR 2 RR 1
49 MCL RR RR RR 3 RR RR 2
50 LCL RR RR 2 RR 1
51 LCL RG 1 RR RR 2
52 Unclassifiable RR RR 2 RR RG 2
53 Unclassifiable RR 1 RR RG 2
typCLL indicates typical chronic lymphocytic leukemia/small lymphocytic lymphoma; R, rearranged allele; G, germinal configuration; NA, not analyzed; atyp CLL, atypicalCLL/small lymphocytic lymphoma; HCL, hairy cell leukemia; LPL, lymphoplasmacytic lymphoma; SMZL, splenic marginal zone lymphoma; FL, follicular lymphoma; MCL,mantle cell lymphoma; and LCL, large cell lymphoma.
2996 SANCHEZ et al BLOOD, 15 OCTOBER 2003 � VOLUME 102, NUMBER 8
For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.orgFrom
explore for the potential statistical significance of differences foundbetween groups of B-CLPD patients, the Student t and �2 tests (SPSS 10.0.)were used for continuous and categorical variables, respectively. Curvesrepresenting the cases requiring treatment within each subgroup of patientswere plotted according to the method of Kaplan and Meier, and thecomparison between curves was performed using the log-rank test. P .05was considered to be associated with statistical significance.
Results
Diagnosis of B-CLPD with 2 or more B-cell clones
The presence of 2 or more B-cell clones was primarily suspectedbased on immunophenotypic studies in a total of 53 B-CLPD cases.From these patients, 23 were from the group of 477 B-CLPD casesconsecutively analyzed in our laboratory and 30 corresponded toselected patients whose samples were specifically referred to ourlaboratory for confirmation of suspected presence of 2 or moreB-cell clones. In all these 53 cases, 2 different neoplastic B-cellpopulations were observed, which showed a different pattern ofexpression for 1 or more phenotypic markers. Clonality molecularstudies devoted to confirm the existence of 2 different tumoralB-cell clones—based on the coexistence of 3 or more distinctrearrangements of the IgH gene—were carried out in a total of 44
of these 53 cases. In the remaining 9 cases, molecular studies couldnot be performed due to shortage of the sample. In all 44 casesstudied, molecular techniques confirmed the presence of at least 2different B-cell clones, based on the criteria defined above (Table1). In addition, in 10 of these cases, molecular analysis and/or iFISHstudies were performed individually on each of the 2 phenotypicallydistinct B-cell subpopulations purified by fluorescence-activated cellsorting, confirming that each of the 2 B-cell fractions had different IgHgene rearrangements and/or chromosome translocations (Figure 1). Toexclude the presence of 2 or more B-cell clones in the remaining 454cases that showed a uniform antigenic profile (continuous expression ofmarkers), we randomly selected and studied a cohort of 54 cases. Noneof them showed 3 or more IgH rearrangements. The distribution of the53 cases with 2 or more B-cell clones according to their diagnosis was asfollows: B-CLL, 33 cases (24 typical and 9 atypical); HCL,4 patients; LPL, 4 cases; SMZL, 3 cases; FL, 3 patients; MCL,2 cases; and LCL, 2 cases; the remaining 2 B-CLPD patients could notbe further subclassified.
Incidence of B-CLPD with 2 or more B-cell clones
The overall incidence of cases with 2 or more B-cell clones foundwas 4.8% (23 of the 477 consecutive B-CLPD patients analyzed).As shown in Table 2, the incidence of patients with 2 or more B-cellclones within each diagnostic group was rather variable. Accord-ingly, a significantly (P � .05) higher percentage of HCL (23.1%),LCL (20%), and atypCLL patients (13.8%) showed 2 differentneoplastic B-cell clones, whereas this was a much less frequentfinding among patients with SMZL (7.1%), MCL (5.3%), andtypCLL (3.4%). None of the PLL, FL, and LPL patients studiedconsecutively showed the presence of 2 or more B-cell clones.
Differential antigen expression in B-CLPD with2 or more B-cell clones
The specific phenotypic differences observed for each B-cell clonein B-CLPD cases with 2 or more B-cell clones are detailed in Table3 and summarized in Table 4. As shown in Table 3, in 24 of 53 cases(45%), B cells displayed both a different phenotype and a distinctsurface immunoglobulin (sIg) light chain in each of the 2 popula-tions, 1 being sIg�� and the other sIg��. In 13 cases (25%)although the sIg light chain isotype was also different, theexpression of all antigens studied was identical in the 2 B-cellpopulations. In another 9 cases (17%), the sIg light chain isotypewas the same in the 2 B-cell populations but could be distinguishedbased on either a different intensity of sIg expression alone (n � 1)or the intensity of sIg expression and coexisting differential
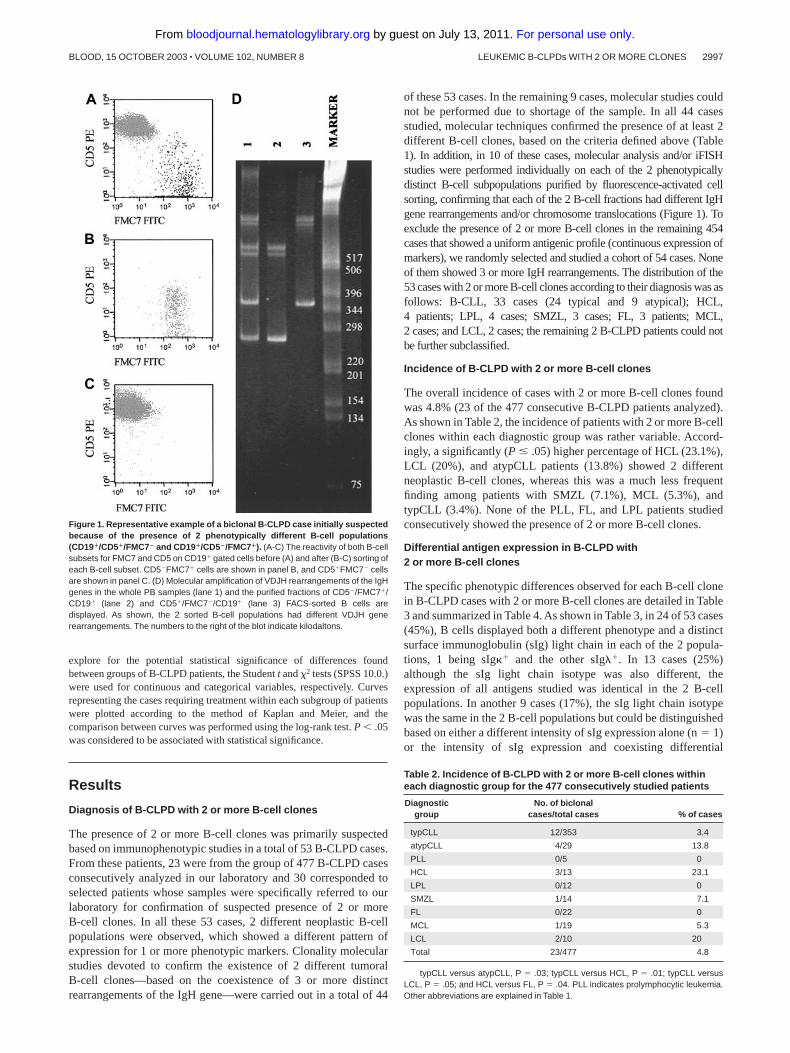
Figure 1. Representative example of a biclonal B-CLPD case initially suspectedbecause of the presence of 2 phenotypically different B-cell populations(CD19�/CD5�/FMC7� and CD19�/CD5�/FMC7�). (A-C) The reactivity of both B-cellsubsets for FMC7 and CD5 on CD19� gated cells before (A) and after (B-C) sorting ofeach B-cell subset. CD5FMC7� cells are shown in panel B, and CD5�FMC7 cellsare shown in panel C. (D) Molecular amplification of VDJH rearrangements of the IgHgenes in the whole PB samples (lane 1) and the purified fractions of CD5/FMC7�/CD19� (lane 2) and CD5�/FMC7/CD19� (lane 3) FACS-sorted B cells aredisplayed. As shown, the 2 sorted B-cell populations had different VDJH generearrangements. The numbers to the right of the blot indicate kilodaltons.
Table 2. Incidence of B-CLPD with 2 or more B-cell clones withineach diagnostic group for the 477 consecutively studied patients
Diagnosticgroup
No. of biclonalcases/total cases % of cases
typCLL 12/353 3.4
atypCLL 4/29 13.8
PLL 0/5 0
HCL 3/13 23.1
LPL 0/12 0
SMZL 1/14 7.1
FL 0/22 0
MCL 1/19 5.3
LCL 2/10 20
Total 23/477 4.8
typCLL versus atypCLL, P � .03; typCLL versus HCL, P � .01; typCLL versusLCL, P � .05; and HCL versus FL, P � .04. PLL indicates prolymphocytic leukemia.Other abbreviations are explained in Table 1.
LEUKEMIC B-CLPDs WITH 2 OR MORE CLONES 2997BLOOD, 15 OCTOBER 2003 � VOLUME 102, NUMBER 8
For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.orgFrom

Table 3. Phenotypic characteristics differentially expressed by each of the neoplastic B populations found in B-CLPDwith 2 or more B-cell clones
Caseno. Diagnosis
Morphology:no. of majorlymphocytepopulations
Phenotype of population 1 (% from the totalleukocytes; compatible diagnosis)
Phenotype of population 2 (% from the totalleukocytes; compatible diagnosis)
1 typCLL 3 CD19� FMC7 CD5� CD22 CD23�d CD11c/�het sIg
(28%; B-CLL)
CD19�� FMC7�� CD5 CD22� CD23 CD11c ��
(6.7%; LPL)
2 typCLL 2 CD19� FMC7�d CD5� CD22�d CD23� CD25�d
(39%; B-CLL)
CD19�� FMC7�� CD5 CD22� CD23 CD25
(6%; SMZL)
3 typCLL 2 FSC/SSClo CD25�d sIg (25%; B-CLL) FSC/SSCint CD25� ��d (29%; B-CLL)
4 typCLL 3 CD23� ��d (50%; B-CLL) CD23 �� (9%; MALT)
5 typCLL 1 ��d (34%; B-CLL) �� (13%; BCLL)
6 typCLL 2 FSC/SSClo FMC7�d CD5� CD22�d CD23� CD25�
CD11c�het slg (15%; B-CLL)
FSC/SSCint FMC7� CD5�� CD22� CD23 CD25
CD11c ��d (12%; MCL)
7 typCLL 2 FSC/SSClo CD19�d FMC7�d CD5� CD22�d CD23�
CD25�d CD11c ��d (24%; BCLL)
FSC/SSCint CD19� FMC7�� CD5 CD22�� CD23
CD25 CD11c�� ��d (22%; SMZL)
8 typCLL 1 FSC/SSClo CD5� CD11c ��d (9%; BCLL) FSC/SSCint CD5�� CD11c� ��d (10%; B-CLL)
9 typCLL 1 �� (25%; B-CLL) �� (8%; B-CLL)
10 typCLL 2 CD5�� ��d (63%; B-CLL) CD5� �� (2%; B-CLL)
11 typCLL 2 ��d (72%; B-CLL) �� (7%; B-CLL)
12 typCLL NA �� (2.4%; B-CLL) �� (38%; B-CLL)
13 typCLL 2 FSC/SSClo CD5�� CD25 CD11c�d (36%; B-CLL) FSC/SSCint CD5��� CD25�d CC11c� (37%; B-CLL)
14 typCLL 2 CD5 CD11c (31%; B-CLL) CD5� CD11c� (7%; B-CLL)
15 typCLL 2 �� (48%; B-CLL) �� (6%; B-CLL)
16 typCLL 3 CD19� FMC7 (18%; B-CLL) CD19�� FMC7� (44%; B-CLL)
17 typCLL NA ��d (38%; B-CLL) ��d (8%; B-CLL)
18 typCLL NA FSC/SSClo CD5�� CD25�d �� (39%; B-CLL) FSC/SSCint CD5��� CD25� ��d (16%; B-CLL)
19 typCLL NA �� (8%; B-CLL) �� (20%; B-CLL)
20 typCLL NA FMC7�� CD5 CD22� CD23 CD25 ��� (9.7%; LPL) FMC7 CD5�� CD22�d CD23�� CD25� ���
(55%; B-CLL)
21 typCLL 2 FSC/SSClo CD19� FMC7 CD5�� CD22�d CD23� CD25�
CD11c/�het �� (36%; B-CLL)
FSC/SSCint CD19�� FMC7� CD5� CD22� CD23 CD25
CD11c� ��� (8%; MCL)
22 typCLL 2 FMC7 CD5��� CD22�d CD23� �� (28%; B-CLL) FMC7��CD5�� CD22� CD23 ��� (8.5%; MCL)
23 typCLL NA FSC/SSClo CD19� CD5� CD22 CD23�d CD25 ��d
(10%; B-CLL)
FSC/SSCint CD19�� CD5�� CD22�d CD23� CD25� ��d
(45%; B-CLL)
24 typCLL NA �� (31%; B-CLL) �� (42%; B-CLL)
25 atypCLL 2 FSC/SSClo CD19�d FMC7 CD5� CD23� slg
(3%; B-CLL)
FSC/SSCint CD19� FMC7�� CD5 CD23�d �� (9%; LPL)
26 atypCLL 2 FSC/SSClo CD19� FMC7�d CD5� CD23� CD25� ��
(23.2%; B-CLL)
FSC/SSCint CD19�� FMC7�� CD5�d CD23�d CD25 ���
(62.8%; B-CLL)
27 atypCLL � 3 CD5 CD23 CD11c �� (7.4%; LPL) CD5� CD23� CD11c�� ��d (2.3%; B-CLL)
28 atypCLL NA FSC/SSClo CD19�d FMC7�d CD5�� CD22�d CD23��
CD25�� �� (10%; B-CLL)
FSC/SSCint CD19� FMC7�� CD5� CD22� CD23�
CD25� ��� (15%; MALT)
29 atypCLL 2 CD19� FMC7 CD5�d CD22�d CD23� ��d (60%; B-CLL) CD19�� FMC7� CD5 CD22� CD23 �� (7%; FL)
30 atypCLL 2 CD5�� ��d (16%; B-CLL) CD5� �� (42%; MCL)
31 atypCLL 2 �� (56%; B-CLL) ��d (8%; B-CLL)
32 atypCLL 3 FSC/SSClo CD19�d FMC7 CD5� CD22�d CD103
CD11c slg (3.8%; B-CLL)
FSC/SSCint CD19� FMC7�� CD5 CD22� CD103�
CD11c� �� (25.4%; HCL)
33 atypCLL NA �� (4%; MALT) �� (20%; MALT)
34 HCL 2 FSC/SSClo CD103 CD25 CD11c�� �� (3.7%; SMZL) FSC/SSCint CD103� CD25� CD11c��� �� (4.5%; HCL)
35 HCL NA FSC/SSClo CD19�d FMC7 CD5� CD22�d CD23�
CD103 CD25�d CD11c/�het ��d (0.8%; B-CLL)
FSC/SSCint CD19� FMC7�� CD5 CD22� CD23
CD103� CD25 CD11c�� �� (1.1%; HCL)
36 HCL 1 CD19� FMC7/�het CD11c�d �� (30.5%; MALT) CD19�� FMC7��� CD11c�� ��d (6.3%; HCLv)
37 HCL 2 CD103� (22%; SMZL) CD103�� (5%; HCL)
38 LPL NA CD19� FMC7� CD5� CD22�d CD23� CD25�� CD11c
(6.5%; B-CLL)
CD19�� FMC7�� CD5 CD22� CD23 CD25
CD11c/�het (38%; LPL)
39 LPL NA ��d (21%; B-CLL) ��� (0.6%; LPL)
40 LPL 3 FSC/SSClo FMC7 CD5� ��� (1.5%; MCL) FSC/SSCint FMC7� CD5 ��� (4.7%; LCL)
41 LPL 2 �� (46%; LPL) �� (6%; LPL)
42 SMZL 2 FSC/SSClo CD19�d FMC7�d CD5� CD22�d CD23� CD25�
slg (0.6%; B-CLL)
FSC/SSCint CD19� FMC7�� CD5 CD22� CD23 CD25
��d (3.7%; SMZL)
43 SMZL 2 CD5� (60%; MALT) CD5 (6%; SMZL)
44 SMZL 2 ��� (29%; SMZL) ��� (25%; SMZL)
45 FL NA CD5� CD22�d CD23 ��� (57%; MCL) CD5 CD22� CD23/�het �� (9%; FL)
46 FL 2 FSC/SSClo CD5 CD23 CD103 CD25 CD11c ��
(6%; FL)
FSC/SSCint CD5� CD23� CD103� CD25� CD11c��� ��
(47%; LCL)
47 FL NA �� (4.5%; FL) �� (2.2%; FL)
2998 SANCHEZ et al BLOOD, 15 OCTOBER 2003 � VOLUME 102, NUMBER 8
For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.orgFrom

expression of other phenotypic markers (n � 8). In the remaining 7patients (13%), the pattern of expression of sIg light chain wasidentical in the 2 B-cell populations that could be specificallyidentified based on the pattern of expression of the other markersand/or their light scatter properties. Representative examples ofthese phenotypic differences are illustrated in Figure 2.
Overall, within these 53 B-CLPD patients, sIg was the indi-vidual antigen most frequently (86%) found to be differentiallyexpressed in the 2 B-cell populations. CD5 and FMC7 expressionas well as forward light scatter/sideward light scatter (FSC/SSC)properties were different between the 2 B-cell clones in around halfof the patients (53%, 41%, and 41%, respectively). In approxi-mately one third of these patients the 2 B-cell clones showeddifferent reactivity for CD23, CD22, CD19, CD25, and CD11c(39%, 35%, 33%, 33%, and 31% of the patients, respectively). In alower proportion of patients both clones could be distinguished onthe basis of their expression for CD103 (10%) and CD10 (2%)(Table 4). In Table 4 differences in reactivity for individual antigensin the 2 B-cell clones are displayed specifically for each B-CLPDdiagnostic group.
In 33 of the 37 cases (89%) in which MGG-stained smears wereindependently evaluated by 2 different observers, the presence of 2
or more morphologically different populations of lymphocytes wasfound. In the other 4 cases (11%), a single population of lympho-cytes was observed by morphology (Table 3). In 3 of these latter 4patients, both tumoral clones were phenotypically compatible withtypCLL distinguishable by a different pattern of reactivity forCD11c (negative versus positive), CD5 (dim versus strong), andsIg light chain (�� versus ��) in 1 case (case 8) and a distinct sIglight chain isotype in the other 2 patients (cases 5 and 9). In thefourth individual (case 36), corresponding to an HCL, the 2 B-cellclones showed different patterns of expression for several markers,including CD19, FMC7, CD11c, and sIg� (Table 1).
Clinical and biologic characteristics of patients with 2 or moreB-cell clones versus monoclonal B-CLPD cases
To explore whether B-CLPD patients with 2 or more B-cell clonesdisplay a different clinical behavior as compared with monoclonalcases, both groups of individuals were compared within eachspecific diagnostic subgroup. However, due to the relatively lownumber of cases with 2 or more B-cell clones found in mostsubgroups, comparison was restricted to B-CLL patients. B-CLPDcases specifically referred to our center for confirmation of the
Table 4. B-CLPD with 2 or more B-cell clones: phenotypic differences in individual markers between each neoplastic B-cell subpopulationwithin each diagnostic disease group
Differingphenotypic
marker
Diagnostic group
typCLL,n � 24
atypCLL,n � 9
HCL,n � 4
LPL,n � 4
SMZL,n � 3
FL,n � 3
MCL,n � 2
LCL,n � 2
Unclassifiable,n � 2
Total, %;n � 53
sIg 20� 9� 3� 3� 2§ 3� 2� 2� 2� 87�
CD5* 13§ 8� 1¶ 2§ 2§ 2§ 1§ 0 2� 58§
FMC7 8¶ 6§ 2§ 2§ 1¶ 0 2� 1§ 1§ 44¶
CD23† 9¶ 7§ 2§ 2§ 1¶ 2§ 1§ 0 2� 49¶
CD22 8¶ 5§ 1¶ 1¶ 1¶ 1¶ 1§ 2� 1§ 39¶
CD19 6¶ 5§ 2§ 1¶ 1¶ 0 1§ 1§ 0 32¶
CD25 9¶ 2 2§ 1¶ 1¶ 1¶ 0 1§ 1§ 34¶
CD11c 7¶ 2 3� 1¶ 0 1¶ 0 2� 0 30¶
CD103‡ 0 1 3� 0 0 1¶ 0 0 0 9
CD10 0 0 0 0 0 0 0 1§ 0 2
FSC/SSC 8¶ 4¶ 2§ 1¶ 0 1¶ 2� 2� 1§ 40¶
The 2 cases of unclassified B-CLPD were excluded from this table.Abbreviations are explained in Table 1.*atypCLL versus HCL (P � .05) and LCL (P � .05).†typCLL versus atypCLL (P � .04).‡HCL versus typCLL (P � .01) and atypCLL (P � .05).§Phenotypic difference between each B-cell clone present in 50% to 74% of cases.�Phenotypic difference between each B-cell clone present in 25% to 49% of cases.¶Phenotypic difference between each B-cell clone present in 75% to 100% of cases.
Table 3. Phenotypic characteristics differentially expressed by each of the neoplastic B-populations found in B-CLPDwith 2 or more B-cell clones (continued)
Caseno. Diagnosis
Morphology:no. of majorlymphocytepopulations
Phenotype of population 1 (% from the totalleukocytes; compatible diagnosis)
Phenotype of population 2 (% from the totalleukocytes; compatible diagnosis)
48 MCL 2 FSC/SSClo CD19� FMC7� CD22� CD23� ��
(12%; MALT)
FSC/SSCint CD19�d FMC7�� CD22�d CD23 ��
(66%; MCL)
49 MCL NA FSC/SSClo FMC7�hom CD5�d ��d (11%; MCL) FSC/SSCint FMC7�het CD5� �� (22%; MCL)
50 LCL NA FSC/SSClo CD22�d CD10 CD11c ��d (23%; B-CLL) FSC/SSCint CD22� CD10�d CD11c� slg (12%; LCL)
51 LCL 2 FSC/SSClo CD19�d FMC7 CD22 CD25 CD11c ��d
(60%; LPL)
FSC/SSCint CD19� FMC7�het CD22�d CD25�d CD11c�d
�� (14%; LCL)
52 Unclassifiable 2 FSC/SSClo FMC7�d CD5� CD22�d CD23� CD25� ��
(42%; B-CLL)
FSC/SSCint FMC7�� CD5 CD22� CD23 CD25 ��
(36%; SMZL)
53 Unclassifiable 2 CD5 CD23 �� (18%; LPL) CD5�� CD23� ��d (3.5%; B-CLL)
� indicates antigen expression at normal levels; —, absence of expression; het, heterogeneous antigen expression; ��, strong antigen expression; d, dim antigenexpression; SSClo, low scatter; SSCint, intermediate scatter; ���, very strong antigen expression; HCLv, HCL variant; and hom, homogeneous antigen expression. Otherabbreviations are explained in Table 1.
LEUKEMIC B-CLPDs WITH 2 OR MORE CLONES 2999BLOOD, 15 OCTOBER 2003 � VOLUME 102, NUMBER 8
For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.orgFrom

coexistence of 2 or more B-cell clones (n � 30) showed similarclinical and biologic characteristics to those identified within thegroup of cases consecutively analyzed. Therefore, the 2 cohorts(n � 53) were pooled for comparison with monoclonal cases(n � 454). As seen in Tables 5 and 6, at diagnosis, CLL patientswith 2 or more B-cell clones showed a significantly greaterincidence of splenomegaly (P � .02) together with significantlylower white blood cell (WBC) (P � .05) and PB absolute lympho-cyte counts (P � .01) as compared with monoclonal CLL cases. Inaddition, CLL cases with 2 or more B-cell clones showed a slightlyhigher incidence of deaths as compared with monoclonal patients,although no statistically significant differences were found betweenboth groups of patients. Once cases with 2 B-cell populationsimmunophenotypically compatible with CLL (CLL�CLL) andthose who showed one B-cell clone with CLL phenotype and the secondone compatible with another B-CLPD (CLL�non-CLL) were sepa-rately considered, it was observed that CLL�non-CLL cases had asignificantly higher mean age (P � .05), more frequently presentedconstitutional symptoms (P � .03), had a serum monoclonal compo-nent (P � .002), and showed a lower incidence of diffuse pattern of BMinvolvement (P � .05) as compared with both monoclonal CLL andCLL�CLL cases. No statistically significant differences were foundbetween these groups of patients regarding the other clinical andbiologic variables studied at diagnosis.
Similar overall survival rates were found in patients with 2 or moreB-cell clones as compared with monoclonal CLL cases (data notshown); despite this, statistically significant differences (P � .01) were
found between both groups of patients once the percentage of casesrequiring treatment during follow-up was considered, this being higheramong CLL patients with 2 or more B-cell clones (Figure 3A).
Interestingly, when patients with 2 or more B-cell clones weredivided into the CLL�CLL and CLL�non-CLL subgroups, theproportion of cases that required treatment within the first 18months after diagnosis was significantly higher (P � .03) amongCLL�non-CLL individuals as compared with both CLL�CLL andmonoclonal CLL patients (Figure 3B).
Discussion
B-CLPDs are a group of diseases characterized by the proliferationof mature-appearing B cells, which are generally believed to resultfrom the monoclonal expansion of a single transformed precursorcell.1 In fact, in B-CLPD oligoclonality has been usually related tothe development of new B-cell subclones resulting from theoccurrence of additional genetic abnormalities on the neoplasticcells along the evolution of the disease29-30; reports on B-CLPDcases with 2 or more distinct unrelated B-cell clones are scanty inthe literature and restricted to between 1 and 3 patients atmaximum.12,15,17-19,31,32 In most of these cases, biclonality wassuspected based on flow cytometry immunophenotypic studies,which revealed the presence of 2 phenotypically distinct B-cellpopulations,15,17 frequently displaying a different sIg light chainisotype12,17,18,31,32; molecular analysis confirmed the presence of 2
Figure 2. Illustrative 3-dimensional dot plots of samples from patients with several different leukemic B-CLPDs in which 2 distinct neoplastic B-cell clones weredetected using immunophenotypic techniques. The neoplastic B-cell clones are named as A (one clone) and B (the second clone); other leukocytes present in the sampleare shown as gray events.
Table 5. Clinical characteristics of monoclonal CLL cases versus CLL with 2 or more B-cell clones
2 or more B-cell clones
Monoclonal CLL,n � 366 P
CLL � CLL,n � 18
CLL � non-CLL,n � 15
Age, y 66 � 11 77 � 8* 71 � 10 .05
Sex, % NS
Female 22 40 38
Male 78 60 62
ECOG greater than 2, % 7 7 2 NS
Binet clinical stage, % NS
A 69 67 81
B or C 31 33 19
B symptoms, % 9 50* 14 .03
Adenopathies, % 56 33 36 NS
Hepatomegaly, % 12 7 13 NS
Splenomegaly, % 37 27 16* .02
Monoclonal component, % 0 15* 2 .002
Diffuse pattern of BM infiltration, % 44* 0 19 .05
% of cases requiring treatment 47 50 30 NS
% of cases requiring treatment 18 mo after diagnosis 40 50 26 .03
% of cases with associated neoplasias 0 0 4 NS
% deaths due to PD 6 7 4 NS
Results expressed as percentage of cases. Values for age presented as mean � SD. PD indicates progressive disease. NS indicates that no statistically significantdifferences were observed between groups (P � .05). ECOG indicates the performance status scaled from 0 to 5 by the Eastern Cooperative Oncology Group.
*Statistically significantly different from the other 2 groups of patients.
3000 SANCHEZ et al BLOOD, 15 OCTOBER 2003 � VOLUME 102, NUMBER 8
For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.orgFrom

unrelated malignant clones, indicating that these cases corre-sponded to true biclonal B-CLPD.12,15-17,31,32 Nevertheless, untilnow the incidence of biclonality among B-CLPD has not beenestablished. The first goal of the present study was to determine thisincidence; for that purpose, we have analyzed at diagnosis a largeseries of 477 consecutive patients suffering from leukemic B-CLPD. Our results show that the presence of cases with 2 or moreB-cell clones is a relatively frequent finding (4.8%) in B-CLPDpatients. The presence of 2 or more B-cell clones was suspected byimmunophenotyping and confirmed by molecular analysis of bothunfractioned samples and purified B-cell populations. Accordingly,all tested samples with 2 phenotypically different B-cell subsetshad 3 or more Ig gene rearrangements, and these were different ineach of the B-cell populations identified; in contrast, none of therandomly selected cases with uniform phenotype had more than 2IgH gene rearrangements. Altogether, these results suggest theutility of the immunophenotypic criteria to detect the presence of 2or more B-cell clones. In line with previous observations,18,31,32 inmost of these patients (66%) the presence of 2 or more B-cellclones was suspected based on the expression of a different sIglight chain isotype by each of the different neoplastic B-cell clones.Interestingly, in some of these cases an sIg��/sIg�� B-cell ratiowithin the normal range was observed. Despite this, in these lattercases, a detailed analysis of the immunophenotype of both thesIg�� and sIg�� B cells for the remaining surface markers testedrevealed that both B-cell subsets were immunophenotypicallyaberrant.21 These results support the notion that the analysis of the
sIg��/sIg�� ratio by itself may not be a useful marker to detect clonalityin these patients; in contrast, its combination with the identification ofaberrant marker expression would be a useful sensitive and specific toolfor the diagnosis of leukemic B-CLPD with 2 or more B-cell clones. Thecombined use of both sIg light chain together with the detection ofphenotypic aberrations is also crucial for the identification of B-CLPDwith 2 or more B-cell clones in which the neoplastic B-cell subsetsdisplay the same sIg light chain isotype. Accordingly, in 16 cases thatshowed 2 different neoplastic B-cell subpopulations with an identicalsIg light chain isotype by phenotype, the existence of 2 populations thatexpress the same sIg light chain isotype but at different intensities wasfrequently observed in the context of distinct aberrant phenotypes foreach clone.
Previous case reports on biclonal CLPD have suggested theexistence of large phenotypic differences between the 2 expandedcell clones.15,17,19 In the present study, a detailed analysis of thephenotypic differences between the coexisting neoplastic cellclones showed that they are relatively variable from one diagnosticsubgroup to another, and cases existed in which the phenotypicdifferences between clones were restricted to a single marker,together with patients displaying more complex phenotypic pat-terns. Virtually all antigens analyzed were found to be differentiallyexpressed in the 2 coexisting B-cell clones. However, the incidenceat which each marker displays a different reactivity in thecoexisting B-cell populations varies among the different diagnosticgroups. sIg was the individual marker most frequently found to bedifferentially expressed in the 2 B-cell clones in all diagnostic
Table 6. Biologic characteristics of monoclonal CLL cases versus CLL with 2 or more B-cell clones
2 or more B-cell clones
Monoclonal CLL, n � 366 PCLL � CLL, n � 18 CLL � non-CLL, n � 15
Hemoglobin level, g/dL (range) 13.2 � 3 (5.6-17) 12.2 � 3 (6.3-15) 13.8 � 2.4 (2.1-17.4) NS
Platelet count, � 109/L (range) 153.8 � 81.5 (24-371) 178.4 � 94.2 (30-363) 184.7 � 78.7 (6-688) NS
WBC count, � 109/L (range) 24.4 � 13 (5.7-62) 26.1 � 23.4 (3.2-84.9) 41.6 � 57.4* (2.3-450) � .05
Lymphocyte count, � 109/L (range) 17.7 � 12 (2.9-53) 17.9 � 17.6 (0.9-63) 32.8 � 50* (0.9-418.5) .01
�2-microglobulin level, mg/dL (range) 2.6 � 1.5 (1.4-5.9) 3.1 � 2.6 (1.3-9.8) 2.6 � 1.9 (0.5-16) NS
LDH level, U/L (range) 343 � 121 (149-665) 1357.3 � 2163.5 (175-5806) 418.6 � 418 (127-7060) NS
CRP level, mg/L (range) 1.1 � 1 (0.2-3.2) 1 � 1.3 (0.2-2.5) 1.7 � 2.5 (0.1-15.5) NS
IgG level, mg/dL (range) 999 � 201.6 (649-1320) 1017.6 � 575 (415-1919) 1091.1 � 353.2 (109-2900) NS
IgM level, mg/dL (range) 149.4 � 151 (28.5-488) 299.1 � 265.7 (43.6-807) 100.4 � 79.1 (4.6-582) NS
IgA level, mg/dL (range) 146.5 � 101.7 (36-414) 230.1 � 74.3 (109-315) 204.2 � 114.5 (6.6-846) NS
Results expressed as mean � standard deviation. NS indicates no statistically significant differences were observed between groups (P � .05); CRP, c-reactive protein;and LDH, lactic dehydrogenase.
*Statistically significantly different from the other 2 groups of patients.
Figure 3. Cases requiring treatment. Curves represent-ing the proportion of cases requiring treatment withinmonoclonal CLL versus CLL patients with 2 or moreB-cell clones during both the overall follow-up (A) and thefirst 18 months after diagnosis (B). In panel B, CLL � CLLand CLL � non-CLL cases are separately represented.The proportion of cases requiring treatment during thefirst 18 months after diagnosis (� the 95% confidenceinterval) is in parentheses for each patient group.
LEUKEMIC B-CLPDs WITH 2 OR MORE CLONES 3001BLOOD, 15 OCTOBER 2003 � VOLUME 102, NUMBER 8
For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.orgFrom

categories, this including all atypCLL, FL, MCL, and LCL cases with2 or more B-cell clones. Markers other than sIg showed differentialexpression in the 2 B-cell clones coexisting in CLPD cases with 2 ormore B-cell clones in a variable proportion of patients, depending on thediagnostic subgroup and the individual marker considered. Notably, inall diagnostic groups the 2 B-cell clones detected by phenotype alsodisplayed different FSC/SSC values in a variable proportion of cases,ranging from one fourth of LPL to all MCL and LCL patients analyzed.Interestingly, the presence of both subclones appears to be rather stablebecause it could be confirmed in sequential studies performed in asubgroup of 8 cases between 3 and 60 months after initial diagnosis(data not shown).
The final goal of our study was to explore whether or not thepresence of 2 or more B-cell clones confers a distinct and uniqueclinical and biologic behavior as compared with monoclonal cases.Due to the relatively low number of cases with 2 or more B-cellclones included in most diagnostic groups, only CLL patients wereanalyzed in this part of the study. Overall, similar survival rateswere found once CLL patients with 2 or more B-cell clones werecompared with monoclonal cases (data not shown), probably due tothe still relatively short follow-up. Despite this, slightly differentclinical and biologic features were detected between monoclonaland CLL patients with 2 or more B-cell clones, especially oncecases in which a CLL clone coexisted with a non-CLL clone were
considered. Accordingly, a significantly higher incidence of spleno-megaly and a higher percentage of cases that required earlytreatment were found among B-CLPD cases with 2 or more B-cellCLL clones as compared with monoclonal CLL patients. In asimilar way, CLL�non-CLL cases more frequently showed B symp-toms and a serum monoclonal component than both CLL�CLL andmonoclonal CLL patients. Of particular interest was the finding thatCLL cases with 2 or more B-cell clones, particularly those in which 1 ofthe 2 coexisting clones corresponded to a B-CLPD other than CLL,required treatment within the first 18 months significantly more fre-quently than monoclonal CLL or CLL�CLL cases. This finding wouldsupport the hypothesis that, within CLL patients, the presence of 2 ormore B-cell clones (one CLL and the second non-CLL) could confer aworse prognosis. It might well be that, in these cases, clinical progres-sion is related mainly to the second B-CLPD (different from CLL) ratherthan to B-CLL itself.
In summary, in the present study we show that B-CLPD cases with 2or more B-cell clones are a relatively frequent finding, the phenotypicdifferences between both B-cell clones involving either the patterns ofsIg light chain expression alone or complex combinations of 2 or moremarkers. From the clinical and biologic point of view, slight differencesare observed between monoclonal and B-CLL patients with 2 or moreB-cell clones, especially if the latter correspond to cases in which anon-CLL B-cell clone coexists with the CLL clone.
References
1. Orfao A, Almeida J, Sanchez ML, San Miguel JF.Immunophenotypic diagnosis of leukemic B-cellchronic lymphoproliferative disorders other thanCLL. In: Guy B. Faguet, ed. Chronic LymphocyticLeukemia: Molecular Genetics, Biology, Diagno-sis and Management. Totowa, NJ: HumanaPress; 2003:173-190.
2. Matutes E, Worner I, Sainati L, de Oliveira MP, Ca-tovsky D. Advances in the lymphoproliferative disor-ders: review of our experience in the study of over1000 cases. Biol Clin Hematol. 1989;11:53-62.
3. Matutes E, Owusu-Ankomah K, Morilla R, et al.The immunological profile of B-cell disorders: aproposal of a scoring system for the diagnosis ofCLL. Leukemia. 1994;8:1640-1645.
4. Catovsky D. Chronic lymphoproliferative disor-ders. Curr Opin Oncol. 1995;7:3-11.
5. Montserrat E. Chronic lymphoproliferative disor-ders. Curr Opin Oncol. 1997;9:34-41.
6. Fialkow PJ. Clonal and stem cell origin of blood cellneoplasms. In: Lobue J, Gordon AS, Silber R, Mug-gia FM, eds. Contemporary Hematology/Oncology.Plenum Publishing; New York, NY; 1980:1-46.
7. Woda BA, Knowles DM. Nodular lymphocyticlymphoma eventuating into diffuse histiocytic lym-phoma: immunoperoxidase demonstration ofmonoclonality. Cancer. 1979;43:303-307.
8. Cleary ML, Sklar J. Lymphoproliferative disordersin cardiac transplant recipients are multiclonallymphomas. Lancet. 1984;2:489-493.
9. Cleary ML, Warnke R, Sklar J. Monoclonality oflymphoproliferative lesions in cardiac transplantrecipients: clonal analysis based on immuno-globulin-gene rearrangements. N Engl J Med.1984;310:477-482.
10. Schmitt-Graff A, Hummel M, Anagnostopoulos I,Stoltenburg G, Stein H. Primary brain lymphomain acquired immunodeficiency syndrome: immu-nophenotype and molecular pathologic character-ization in stereotactic biopsy, autopsy and cere-brospinal fluid cytology [in German]. Pathologe.1995;16:75-80.
11. Armes JE, Angus P, Southey MC, et al. Lympho-proliferative disease of donor origin arising in pa-tients after orthotopic liver transplantation. Can-cer. 1994;74:2436-2441.
12. Sklar J, Cleary ML, Thielemans K, Gralow J,Warnke R, Levy R. Biclonal B-cell lymphoma.N Engl J Med. 1984;311:20-27.
13. Siegelman MH, Cleary ML, Warnke R, Sklar J.Frequent biclonality and Ig gene alterationsamong B-cell lymphomas that show multiple his-tologic forms. J Exp Med. 1985;161:850-863.
14. Hairman C, Vaeemans E, Carels D, TheunissenJ, Van-Camp B, Theilemans K. Isotype switchand idiotype variation in hairy cell leukemia. Leu-kemia. 1990;4:856-862.
15. Gonzalez-Campos J, Rios-Herranz E, De Blas-Orlando JM, Martin-Noya A, Parody-Ruiz-BerdejoR, Rodriguez-Fernandez JM. Chronic lympho-cytic leukemia with two cellular populations: abiphenotypic or biclonal disease. Ann Hematol.1997;74:243-246.
16. Wong KF, So CC. Biclonal B-cell chronic lympho-cytic leukemia with inv(14)(q11q32). CancerGenet Cytogenet. 1997;94:135-137.
17. Fend F, Quintanilla-Martınez L, Kumar S, et al. Com-posite low grade B-cell lymphomas with two immuno-phenotypically distinct cell populations are true bi-clonal lymphomas.Am J Pathol. 1999;154:1857-1866.
18. Duque RE, Everett ET, Iturraspe J. Biclonal compos-ite lymphoma: a multiparameter flow cytometric anal-ysis. Arch Pathol Lab Med. 1990;114:176-179.
19. Gine E, Bosch F, Villamor N, Rozman M, et al.Simultaneous diagnosis of hairy cell leukemiaand chronic lymphocytic leukemia/small lympho-cytic lymphoma: a frequent association? Leuke-mia. 2002;16:1454-1459.
20. Harris N, Jaffe E, Diebold J, et al. World HealthOrganization classification of neoplastic diseasesof the hematopoietic and lymphoid tissues: reportof the Clinical Advisory Comittee meeting—AirlieHouse, Virginia, November 1997. J Clin Oncol.1999;17:3835-3849.
21. Sanchez ML, Almeida J, Vidriales MB, et al. Inci-dence of phenotypic aberrations in a series of467 patients with B-chronic lymphoproliferativedisorders: basis for the design of specific 4-colorstainings to be used for minimal residual diseaseinvestigation. Leukemia. 2002;16:1460-1469.
22. Gonzalez M, Gonzalez D, Lopez-Perez R, et al.Heteroduplex analysis of VDJ amplified seg-ments from rearranged IgH genes for clonalityassessments in B-cell non-Hodgkin’s lymphoma:a comparison between different strategies.Haematologica. 1999;84:779-784.
23. Beishuizen A, Verhoeven MA, Mol EJ, Breit TM,Wolvers Tettero IL, van Dongen JJ. Detection of im-munoglobulin heavy-chain gene rearrangements bySouthern blot analysis: recommendations for optimalresults. Leukemia. 1993;7:2045-2053.
24. van Dongen JM, Langerak AW, San Miguel JF, et al.PCR-based clonality studies for early diagnosis oflymphoproliferative disorders: report of the BIOMED-2concerted action [abstract]. Blood. 2001;98:129a.
25. Langerak AW, Szczepanski T, van der Burg M,Wolvers-Tettero IL, van Dongen JM. Heterodu-plex PCR analysis of rearranged T cell receptorgenes for clonality assessment in suspect T cellproliferations. Leukemia. 1997;11:2192-2199.
26. Cook GP, Tomlinson IM. The human immunoglobulinVH repertoire. Immunol Today. 1995;16:237-242.
27. Lefranc MP. IMGT, the international ImMuno-GeneT-ics database. Nucleic Acids Res. 2001;29:207-209.
28. Hernandez JM, Garcia JL, Gutierrez NC, et al.Novel genomic imbalances in B-cell splenic mar-ginal zone lymphomas revealed by comparativegenomic hybridization and cytogenetics. Am JPathol. 2001;158:1843-1850.
29. Clearly ML, Galili N, Trela M, Levy R, Sklar J.Single cell origin of bigenotypic and biphenotypicB cell proliferations in human follicular lympho-mas. J Exp Med. 1988;167:582-597.
30. Raffeld M, Neckers L, Longo DL, Cossman J. Spon-taneous alteration of idiotype in a monoclonal B-celllymphoma: escape from detection by anti-idiotype.N Engl J Med. 1985;312:1653-1658.
31. Finch CN, Nichols M, Shrimtom A, Liu D, Hutchi-son RE. Primary nodal marginal zone B-cell lym-phoma arising from more than one clonal neo-plastic population. Arch Pathol Lab Med. 2000;124:1816-1819.
32. Hsi ED, Hoeltge G, Tubbs RR. Biclonal chroniclymphocytic leukemia. Am J Clin Pathol. 2000;113:798-804.
3002 SANCHEZ et al BLOOD, 15 OCTOBER 2003 � VOLUME 102, NUMBER 8
For personal use only. by guest on July 13, 2011. bloodjournal.hematologylibrary.orgFrom




















