
Fine mapping of canine parvovirus B cell epitopes
12
Journal of General Virology (1991), 72, 2445-2456. Printed in Great Britain 2445 Fine mapping of canine parvovirus B cell epitopes Jos~ A. L6pez de Turiso, Elena Cortes, Ana Ranz, Julita Garcia, Antonio Sanz, Carmen Vela and Jos~ Ignacio Casal* INGENASA, Inmunologia y Genetica Aplicada, S.A., Hermanos Garcia Noblejas, 41-2 °, 28037 Madrid, Spain In this report we describe the topological mapping of neutralizing domains of canine parvovirus (CPV). We obtained 11 CPV-specific monoclonal antibodies (MAbs), six of which are neutralizing. The reactivities were as determined by ELISA and Western blot (immunoblot) analysis. VP2, the most abundant pro- tein of the CPV capsid, seemed to contain all the neutralization sites. Also, an almost full-length geno- mic clone of CPV was constructed in the bacterial plasmid pUC18 to enable expression of CPV proteins. All the neutralizing MAbs recognized recombinant VP2 when it was expressed as a free protein in Escherichia coli but not when expressed as a fusion protein with glutathione-S-transferase. When two large fragments containing about 85 % and 67 % of the C terminus of VP2 were expressed, no neutralization sites were detected. When fusion proteins containing the N terminus were expressed, two linear determi- nants were mapped, one between residues 1 to 10 of VP2, and the other between amino acids 11 and 23. The peptide 11 GQPAVRNERATGS 23, recognized by MAb 3C9, was synthesized chemically and checked for immunogenicity, not being able to induce neutraliz- ing activity. Although the antibody response in rabbits to all the fusion proteins was uniformly high, the anti- CPV response was very variable. Protein from pCPVExl 1, which contains a T cell epitope (peptide PKIFINLAKKKKAG) present in the VPl-specific region as well as the B cell epitopes, seemed to be the most effective in inducing virus neutralization. Introduction Canine parvovirus (CPV) is a member of the parvovirus genus which replicates autonomously, causes severe enteritis in dogs of all ages and myocarditis in pups less than 12 weeks of age. CPV, first isolated in 1978 (Appel et al., 1979; Burtonboy et al., 1979), is believed to have arisen as a natural host-range variant of feline panleuko- penia virus (FPLV). Reports on protein and DNA sequences, and serological studies show extensive anti- genic and genetic similarity between CPV, FPLV, mink enteritis virus and raccoon parvovirus (Carlson et al., 1985; Flower et al., 1980; Tratschin et al., 1982; Parrish et al., 1982, 1988; Reed et al., 1988). Despite this similarity, they each have a distinct host range in vivo, although all the viruses replicate in feline kidney cells in vitro (Appel et al., 1979; Tratschin et al., 1982). The organization of the CPV capsid is similar to that of other autonomous parvoviruses in that it contains two largely overlapping proteins, VP1 (82K) and VP2 (65K) (Paradiso et al., 1982; Surleraux & Burtonboy, 1984; Reed et al., 1988). There is a third protein, VP3 (63K), which is produced by proteolytic processing of VP2. The 22 nm parvovirus capsid comprises about 10 copies of VPI and 60 to 70 copies of VP2 (Tattersall et al., 1977; Wobble et al., 1984) which may be arranged as homo- or heterodimers (Paradiso, 1983), although the precise capsid structure is not known. For instance, it is not known whether the VP1 domains are exposed on the surface of the capsid. CPV infection is quite common and the disease is usually controlled using a live or inactivated vaccine (Pollock & Carmichael, 1982a). However, because bitches are usually vaccinated before pregnancy, mater- nally derived antibodies may block the replication of live attenuated vaccines in puppies having temporary immunity (Pollock and Carmichael, 1982b). Autono- mous parvoviruses should be a good target for the development of synthetic or subunit vaccines owing to the relative structural simplicity of the capsid proteins (there is no glycosylation, phosphorylation or acetyla- tion) and because the humoral response seems to control the virus infection adequately. Therefore, a synthetic vaccine may be useful in the control of the diseases induced by canine or feline parvoviruses. To define the B cell epitopes present on the viral capsid we prepared a collection of monoclonal antibodies (MAbs) specific for the structural proteins of CPV. Also, an almost full- length genomic clone was constructed in a bacterial plasmid to allow the isolation and expression of the 0001-0106 © 1991 SGM
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Fine mapping of canine parvovirus B cell epitopes

Journal of General Virology (1991), 72, 2445-2456. Printed in Great Britain 2445
Fine mapping of canine parvovirus B cell epitopes
Jos~ A. L6pez de Turiso, Elena Cortes, Ana Ranz, Julita Garcia, Antonio Sanz, Carmen Vela and Jos~ Ignacio Casal*
I N G E N A S A , Inmunologia y Genetica Aplicada, S.A., Hermanos Garcia Noblejas, 41-2 °, 28037 Madrid, Spain
In this report we describe the topological mapping of neutralizing domains of canine parvovirus (CPV). We obtained 11 CPV-specific monoclonal antibodies (MAbs), six of which are neutralizing. The reactivities were as determined by ELISA and Western blot (immunoblot) analysis. VP2, the most abundant pro- tein of the CPV capsid, seemed to contain all the neutralization sites. Also, an almost full-length geno- mic clone of CPV was constructed in the bacterial plasmid pUC18 to enable expression of CPV proteins. All the neutralizing MAbs recognized recombinant VP2 when it was expressed as a free protein in Escherichia coli but not when expressed as a fusion protein with glutathione-S-transferase. When two large fragments containing about 85 % and 67 % of the
C terminus of VP2 were expressed, no neutralization sites were detected. When fusion proteins containing the N terminus were expressed, two linear determi- nants were mapped, one between residues 1 to 10 of VP2, and the other between amino acids 11 and 23. The peptide 11 GQPAVRNERATGS 23, recognized by MAb 3C9, was synthesized chemically and checked for immunogenicity, not being able to induce neutraliz- ing activity. Although the antibody response in rabbits to all the fusion proteins was uniformly high, the anti- CPV response was very variable. Protein from pCPVExl 1, which contains a T cell epitope (peptide PKIFINLAKKKKAG) present in the VPl-specific region as well as the B cell epitopes, seemed to be the most effective in inducing virus neutralization.
Introduction
Canine parvovirus (CPV) is a member of the parvovirus genus which replicates autonomously, causes severe enteritis in dogs of all ages and myocarditis in pups less than 12 weeks of age. CPV, first isolated in 1978 (Appel et al., 1979; Burtonboy et al., 1979), is believed to have arisen as a natural host-range variant of feline panleuko- penia virus (FPLV). Reports on protein and DNA sequences, and serological studies show extensive anti- genic and genetic similarity between CPV, FPLV, mink enteritis virus and raccoon parvovirus (Carlson et al., 1985; Flower et al., 1980; Tratschin et al., 1982; Parrish et al., 1982, 1988; Reed et al., 1988). Despite this similarity, they each have a distinct host range in vivo, although all the viruses replicate in feline kidney cells in vitro (Appel et al., 1979; Tratschin et al., 1982).
The organization of the CPV capsid is similar to that of other autonomous parvoviruses in that it contains two largely overlapping proteins, VP1 (82K) and VP2 (65K) (Paradiso et al., 1982; Surleraux & Burtonboy, 1984; Reed et al., 1988). There is a third protein, VP3 (63K), which is produced by proteolytic processing of VP2. The 22 nm parvovirus capsid comprises about 10 copies of VPI and 60 to 70 copies of VP2 (Tattersall et al., 1977;
Wobble et al., 1984) which may be arranged as homo- or heterodimers (Paradiso, 1983), although the precise capsid structure is not known. For instance, it is not known whether the VP1 domains are exposed on the surface of the capsid.
CPV infection is quite common and the disease is usually controlled using a live or inactivated vaccine (Pollock & Carmichael, 1982a). However, because bitches are usually vaccinated before pregnancy, mater- nally derived antibodies may block the replication of live attenuated vaccines in puppies having temporary immunity (Pollock and Carmichael, 1982b). Autono- mous parvoviruses should be a good target for the development of synthetic or subunit vaccines owing to the relative structural simplicity of the capsid proteins (there is no glycosylation, phosphorylation or acetyla- tion) and because the humoral response seems to control the virus infection adequately. Therefore, a synthetic vaccine may be useful in the control of the diseases induced by canine or feline parvoviruses. To define the B cell epitopes present on the viral capsid we prepared a collection of monoclonal antibodies (MAbs) specific for the structural proteins of CPV. Also, an almost full- length genomic clone was constructed in a bacterial plasmid to allow the isolation and expression of the
0001-0106 © 1991 SGM

2446 J. A. L6pez de Turiso and others
complete VP2 gene and defined fragments in order to identify neutralizing epitopes. Subgenomic fragments were generated by digestion at suitable cleavage sites. These recombinant polypeptides and synthetic peptides were tested for their immunogenicity, which showed that they are able to induce CPV-specific antibodies and a neutralizing response in vitro.
Methods
Cells and viruses. A permanent line of feline kidney cells (CRFK) (ATCC no. CCL 94) was used for virus production and preparation of replicative form (RF) DNA. CRFK cells were propagated in Dulbecco's modification of Eagle's medium (Flow Laboratories) supplemented with 10% foetal calf serum and antibiotics.
Attenuated virus strain CPV-c/780916 (Carmichael et al., 1981) was obtained from the ATCC (VR-953). FPLV (Rossete strain) was a kind gift from Dr L. Carmichael (Cornell University, Ithaca, N.Y., U.S.A.).
Virus purification. Virus particles were purified from infected monolayers of CRFK cells, according to the procedure described previously for porcine parvovirus (PPV) (Molitor et al., 1983). Briefly, CPV was recovered from infected cells by three cycles of freezing and thawing, and then precipitated with calcium chloride. Sedimented virions were resuspended in 50 mM-Tris-HC1 pH 8.7, 25 mM-EDTA and centrifuged to equilibrium in CsCI gradients; fractions were collected and assayed for haemagglutination (HA) activity with pig erythrocytes as described previously (Carmichael et aL, 1980).
MAbs and polyelonal antibodies. CPV-specific MAbs were prepared from BALB/c mice immunized with purified, full virus particles. Protocols for immunization, and the preparation of MAbs and ascitic fluids have been described previously (Sanz et aL, 1985; Vela et al., 1986). All hybridomas were cloned by limiting dilution at least three times. Hybridoma supernatants were screened by ELISA and, when necessary, an immunofluorescence assay. The class and subclass of the MAbs were determined by immunodiffusion with antisera specific for murine antibody heavy and light chains.
For immunization of mice, peptide was coupled to keyhole limpet haemocyanin (KLH) as a protein carrier with N-maleimidobenzoyl N- hydroxysuccinimide through the cysteine of the peptide. Peptide (10 to 20 ~tg) coupled to KLH or bovine serum albumin (BSA), included in liposomes or free, was used for each immunization, the first in complete Freund's adjuvant (CFA) (Difco) and two further doses in incomplete Freund's adjuvant (IFA) at 2 week intervals.
Rabbit polyclonal sera were prepared in New Zealand White female rabbits at 3 to 4 weeks of age according to standard protocols. To produce antibodies against fusion proteins the same protocol as above was followed using 150 ~tg purified protein for each immunization. Rabbit polyclonal anti-CPV serum was prepared in female New Zealand White rabbits at 3 to 4 weeks of age, according to standard protocols.
ELISA. The specificity of the MAbs was determined by indirect ELISA using purified virus as the antigen source. Briefly, polystyrene plates (Inotech Diagnostik) were coated with 0.5 ~tg virions per well overnight in 100 ~tl 0-05 M-carbonate buffer pH 9.6 at 4 °C. Plates were washed with PBS (0.15 M-NaCI in 0-1 M-sodium phosphate pH 7.4) containing 0.02% Tween 20 and incubated with the first antibody for 2 h at 37 °C, washed again and incubated with a biotin-labelled second antibody. Finally, streptavidin-peroxidase was added for 30 rain at room temperature. The reaction was developed for 10 rain in darkness using o-phenylenediamine (Sigma) as substrate and read at 450 nm on a Titertek Multiskan (Flow Laboratories).
Western blot (immunoblot) analysis. For Western blot analysis, proteins were transferred to a nitrocellulose membrane as previously described (Towbin et al., 1979; Burnette, 1981). Protein transfer was done using the PhastSystem apparatus (Pharmacia), usually at 25 mA/gel for 10 to 15 min. The nitrocellulose sheets were blocked with 3~ non-fat dried milk in 20 mM-Tris-HCl pH 7.5, 500 mM-NaC1 (TBS) for 30 min at room temperature. Strips were incubated for 1 h at room temperature, or overnight at 4 °C, with undiluted supernatant from cultured hybridoma cells or purified immunoglobulin (50 ~tg/ml), washed with TBS, 0.05 ~ Tween 20 for 20 min at room temperature and incubated with a rabbit anti-mouse serum at a 1:1000 dilution. When necessary, the antisera were pre-adsorbed with whole bacteria for 1 h at 4 °C. Strips were washed again and allowed to react with peroxidase- labelled Protein A for 1 h at room temperature. After extensive washing, strips were developed with a solution of TBS containing 0-5 mg/ml 4-chloro-l-naphthol (Sigma), 17~ (v/v) methanol and 0.015~ hydrogen peroxide in TBS until bands appeared. The reaction was stopped by rinsing the strips with distilled water.
Competitive antibody binding and neutralization activity assays. Plates were coated with purified virions as in ELISA and washed with PBS containing 0-02~ Tween 20. Twofold dilutions from 10 ~tg unlabelled purified inhibitor MAb per well were added and incubated for 1 h at 37 °C. Then, 50 ~tl was removed and replaced by 50 ~tl of peroxidase- labelled indicator antibody. The plates were incubated for 1 h at room temperature, washed and a substrate solution containing o-phenylene- diamine was added. The reaction was stopped and read as previously described for ELISA.
Neutralization activity was determined by assaying the cell monolayer protection ability of an antibody. Briefly, a known quantity of input virus [100 HA units (HAU)] was incubated with a single MAb or antiserum at different dilutions for 2 h at 37 °C. Samples were then inoculated onto indicator CRFK cell monolayers for 90 min at 37 °C. Monolayers were overlaid with 1 ml 1 ~ agarose in fresh medium. Cells were fixed with 10~ formaldehyde in PBS for 20 min 5 days post- infection (p.i.), the agarose was removed and the remaining cells were stained with 1 ~ crystal violet in 50~ ethanol for 20 min. The level of protection was evaluated by visual screening of the infected mono- layers. All incubations were done in the absence of complement.
Cloning o f the CP V genome and construction of recombinant plasmids. Restriction and other DNA-modifying enzymes were purchased from Boehringer Mannheim or New England Biolabs and were used as directed by the manufacturers. Standard methods (Maniatis et al., 1982) were used to prepare plasmid DNA.
Virus RF DNA was isolated from infected CRFK cell monolayers 24 to 48 h p.i., as previously described for PPV (Molitor et al., 1984; Ranz et al., 1989), and was used as the starting material for cloning experiments. The strategy for cloning was similar to that described previously for PPV (Casal et al., 1990)~ A collection of expression clones covering the whole or several regions of the VP2 gene were constructed in the pKK233-2 (Amann & Brosius, 1985), pGEX (Smith & Johnson, 1988) or, by preference, pET vector systems (Studier & Moffatt, 1986). One characteristic of the pET system is the use of Escherichia coli JMI09 (or other similar recA strains) for cloning and maintenance of DNA, after which BL21 is used to express the DNA when the final construct is in the right orientation. BL21 (F- ompTr~ m~) is RecA + and conveniently is deficient in the Ion and ompT proteases, although it has a somewhat lower transformation efficiency.
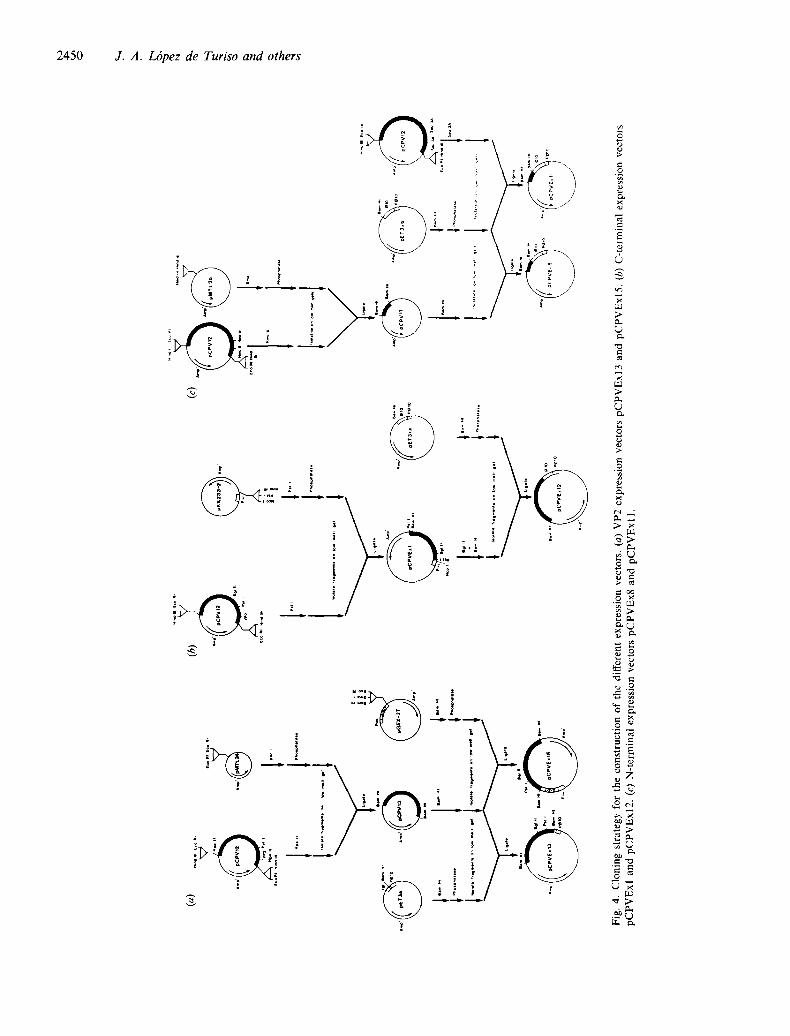
The whole VP2 gene, including 36 nucleotides of the VPI coding region, was expressed as a fusion protein with the 5' region of the gene /0-encoded protein of the E. coli phage T7 using the pET3a vector (Studier & Moffatt, 1986), in which the first 11 codons of gene 10 are inserted downstream of the insertion under the control of the ¢10 promoter and in-frame with the translation initiation codon. The VP2 gene was also cloned in pGEX-2T, to be expressed as a fusion protein
Identification o f C P V B cell epitopes 2447
with the 5' region of glutathione-S-transferase under control of the tac promoter (Fig. 4a). In all cases, the ligation mixtures were used to transform strain JM109 according to standard procedures (Hanahan, 1983).
The C-terminal region of VP2 was expressed as a free protein using pKK233-2 (Amann & Brosius, 1985), or as a fusion protein, using pET3xa (Studier & Moffatt, 1986) (Fig. 4b), yielding plasmids pCPVExl and pCPVExl2, respectively.
N-terminal portions of VP2, including the adjacent region of VP1 (Fig. 4c), were expressed as a fusion protein with the 5' region of the gene/0-encoded protein of phage T7 using pET3xb (Studier & Moffatt, 1986), in which the first 260 codons of gene 10 are inserted downstream of the insertion under control of the gplO promoter. The Sau3A Sau3A fragment was isolated from plasmid pCPV12 and inserted into the BamHl site of pET3xb as shown in Fig. 4(c). Plasmid pCPVExl 1 containing the VP2 gene fragment was characterized by restriction enzyme mapping and the insert was shown to be in the right orientation for expression in E. coli strain BL21. The HincII-Hincll fragment was also isolated from plasmid pCPV12 and ligated into the SmaI site of plasmid pMTL25 to provide flanking BamHI sites to allow it to be cloned into the BamHI site ofpET3xb (Fig. 4c). The resultant plasmid, pCPVEx8, was characterized by restriction enzyme mapping and the insert was shown to be in the right orientation for expression in BL21.
Induction of fi~sion proteins. E. coli strains JMI05, for constructs derived from pKK233-2 and pGEX, or BL21, for pET, were grown to a density of 0.6 (OD600) at 37°C. IPTG was added to a final concentration of 0.5 mM and synthesis was continued for a further 2 h, after which the cells were pelleted by centrifugation and lysed by sonication, and the inclusion bodies were recovered by centrifugation, resuspended in SDS-polyacrylamide gel buffer and analysed on a 10 to 15% polyacrylamide gel. Detection of fusion proteins was performed by staining with Coomassie blue and immunoblotting as described previously.
For immunization experiments, inclusion bodies were washed once with 50 mM-Tris HCI pH 8.0, 0.5% glycerol, 1 ~-NaC1 (buffer A) and then solubilized in 50 mM-Tris-HCl pH 8.0, 0.5 % glycerol containing 4 M-guanidinium chloride (buffer B). The solubilized protein was then
purified on a Sephacryl S-200 column (1-6 x 100 cm) (Pharmacia) and, finally, dialysed against buffer A and concentrated.
Peptide synthesis. Peptide was synthesized in bulk according to the solid phase method on an LKB apparatus using F-moc chemistry. To facilitate linking to the carrier protein, a cysteine was added to the N terminus.
R e s u l t s
Purification of CP V
Virus pa r t i c l e s w e r e c e n t r i f u g e d to e q u i l i b r i u m in CsC1
g r a d i e n t s and the H A a c t i v i t y o f the f r a c t i o n s was
ana lysed . T w o p e a k s w e r e found , o n e at 1.38 g / m l a n d
the o t h e r at 1.33 g /ml . T h e first p e a k usua l ly h a d an H A
t i t re b e t w e e n two- a n d 10-fold g r e a t e r t h a n the o ther , a n d
c o r r e s p o n d e d to full ( D N A - c o n t a i n i n g ) c a p s i d s ; t he
s e c o n d p e a k c o r r e s p o n d e d to e m p t y caps ids . W h e n the
p o l y p e p t i d e s f r o m b o t h p e a k s w e r e a n a l y s e d by S D S -
P A G E , th ree m a j o r s t ruc tu ra l p r o t e i n s o f Mr 82K, 6 5 K
a n d 6 3 K (VP1, V P 2 a n d V P 3 , r e spec t ive ly ) w e r e found .
T h e p r o p o r t i o n s o f V P 2 a n d V P 3 w e r e d i f f e r en t in e m p t y
a n d full caps ids . Ful l c aps ids a lways c o n t a i n e d m o r e
VP2 , a l t h o u g h the r e l a t i ve p r o p o r t i o n s o f these p r o t e i n s
m a y c h a n g e f r o m b a t c h to b a t c h o f pur i f i ed v i rus (resul ts
n o t shown) .
Production and general properties o f CP V-specific MAbs
T h e speci f ic i ty , i so type , t i t re , H A i n h i b i t i o n a n d
n e u t r a l i z a t i o n ac t i v i t i e s o f t he 11 C P V - s p e c i f i c M A b s
o b t a i n e d f r o m five d i f f e ren t fus ions a re s h o w n in T a b l e
1. T h e spec i f ic i t i es were d e t e r m i n e d by i m m u n o b l o t
Tab le 1. Characterization of CPV-specific MAbs
Titre
Hybridoma Isotype* Supernatant Ig (1 mg/ml)
1F11 IgM 625 10 -3 2B4 IgG 1 3125 10 -4 3A6 IgM 125 10 -3 3C10 IgG1 15625 10 -6 3C9 IgG 1 3125 10 -s 4AG6 IgG2a 625 i0 -6 4EC3 IgG 1 625 10 -4 4EA8 IgG1 3125 10 .6 4B8C6 IgG 1 15 625 10 -6 5BF7 IgG1 15 625 10 -6 5F8A9 IgG 1 76125 10 -7
Biological activity
Neutralization:[: HA
Specificity~ inhibition CPV FPLV
- § Neg[I - - VP1, VP2 Neg - - VP1, VP2 + + + VP1, VP2 Neg + - VPI, VP2 Neg + + VP1, VP2 Neg - -
ND¶ Neg - - VP1, VP2 + + +
_ _ + +
VP1, VP2 Neg - - VP1, VP2 + + +
* Determined by radial immunodiffusion. t Determined by immunoblotting.
Monolayer protection. § Not assigned, weak reaction. II Neg, Negative. ¶ ND, Not done.

2448 J. A. L6pez de Turiso and others
1 2 3 4 5 6 7 8
~ 8 2 K ~ 6 5 K
Fig. 1. Reactivity of MAbs against native virus. Purified virus was electrophoresed on a 10 to 15% SDS-polyacrylamide gel and transferred to nitrocellulose. Lanes 1 to 6, MAbs 4B8, 5F8, 4EA8, 3A6, 3C9 and 3C 10; lane 7, no MAb; a CPV-specific mouse antiserum (lane 8) was used as a positive control.
analysis. All the MAbs reacted simultaneously with structural proteins VPI and VP2 (Fig. 1) except MAbs 1F11 and 4B8, which did not react clearly in the Western blot analysis. No VPl-specific MAbs were obtained. Most of the epitopes seem to resist SDS and 2- mercaptoethanol denaturation.
The most frequent isotype was IgG 1. The MAb titre, determined by ELISA of culture supernatant and ascitic fluid, ranged from 102 to 105 and 103 to 107 , respectively.
Table 1 shows that five MAbs (3A6, 3C9, 4B8, 4EA8 and 5F8) were able to neutralize CPV and FPLV, as determined by cell monolayer protection, with a titre ranging from 103 to 106. MAb 3C10 neutralized the virus very weakly (titre 10~). All these neutralization activity assays were done in the absence of complement.
To study the interaction between these MAbs and to construct a tentative epitope map, we did a competitive binding assay with CPV virions as the immobilized antigen. No MAb competed with the binding of the others to CPV (data not shown), although 3C9 and 3C10 showed weak competition. However, they must recog- nize different epitopes because their ability to neutralize CPV and FPLV differed (Table 1).
Since the MAbs were used at high concentration in the competition experiments, the different avidity of the MAbs should not have affected the final results. Therefore, it seems that each neutralizing MAb recog- nized a different epitope, defining at least five different neutralization sites.
x E , t x t
I.It~t* , ht i
• - O
rig.l*
An, p ' ~
E¢o m
x t i~l . I,.ller4ntl on I~, ~ ~1
RI I I I
~ Lie*t* tcQ AI
u ~ , * t,.9,~,~l Amp' i
t©o w
( c o m
Fig. 2. Schematic representation of the construction of genomic clone pCPV9 showing the insertion of an almost full-length copy of the CPV genome into bacterial plasmid pUC18.
Construction of the genomic clone
Double-stranded RF DNA from CPV strain c/780916 was the starting material for cloning experiments. The overall strategy, similar to that described previously for PPV (Casal et al., 1990), is shown in Fig. 2. To clone the 3' end, we treated the extended form of CPV RF DNA with the ssDNA-specific mung bean nuclease to blunt the DNA ends and then with EcoRI. The central region was cloned by digesting CPV RF DNA with PstI and EcoRI. The 5' end of the RF DNA was filled and repaired with the Klenow fragment of E. coli DNA polymerase I, and digested with PstI. The individual DNA fragments were isolated from 1 ~o low melting point agarose gels, ligated to appropriately digested pUC18 and transformed into E. coli strain JM109 as described previously (Hanahan, 1983). We obtained three plasmids, pCPV7, pCPV2 and pCPV5, which contained the 3' end, the central region and the 5' end of VP2, respectively. Plasmids pCPV2 and pCPV5 were

Identification of CPV B cell epitopes 2449
digested with EcoRI and PstI, and the respective fragments were isolated from agarose gels and ligated to each other. The resulting construct was cloned into EcoRI-digested pUC18, yielding pCPV6. Finally, the EcoRI insert was isolated from pCPV6 and ligated into EcoRI-digested, alkaline phosphatase-treated pCPV7 to produce pCPV9. This clone contains all the sequences of the CPV genome, except 400 bp of the 5' end, as shown by restriction mapping (data not shown), and allows single-step excision of the viral genome with Sail or BamHI.
i <_=< - = .. . . . . ," ¥ , ~pCPV12 vP2
~pCPVExI3 , ~pCPVExl
~pCPVExl2 ~ pCPVEx I 1 i i pCPVEx8
Peptide ll 23
Fig. 3. Schematic representation of the CPV genome region cloned into plasmid pCPV12. The length of the various inserts cloned in the different expression plasmids is shown. Restriction sites and VP2 initiation site are indicated.
Expression of VP2
Since all our neutralizing MAbs recognized the VP2 region only (Fig. 1), we concentrated on expressing VP2 sequences and several defined VP2 fragments as free as possible from contaminating VP1 fragments. To facili- tate this expression, a fragment from pCPV6 (Fig. 2) was recloned in pUC 18 in such a way that two polylinkers flanked the VP2 coding region, yielding plasmid pCPV12 (Fig. 3). This plasmid was the starting material for all the expression experiments. Figure 4 (a) shows the strategy followed to express VP2 in two different E. coli expression systems, pGEX2T and pET3a. VP2 spans nucleotides 2786 to 4534 of the CPV genome and can be isolated by digestion of pCPV12 with HpaII. This fragment encodes the entire VP2 polypeptide sequence plus 12 amino acids of VPI at its N terminus. Since most of the expression vectors used in this study have a unique BamHI insertion site, we cloned the HpaII fragment into the AccI sites of pMTL24. This approach produced an insertion of the VP2 fragment flanked by two BamHI sites.
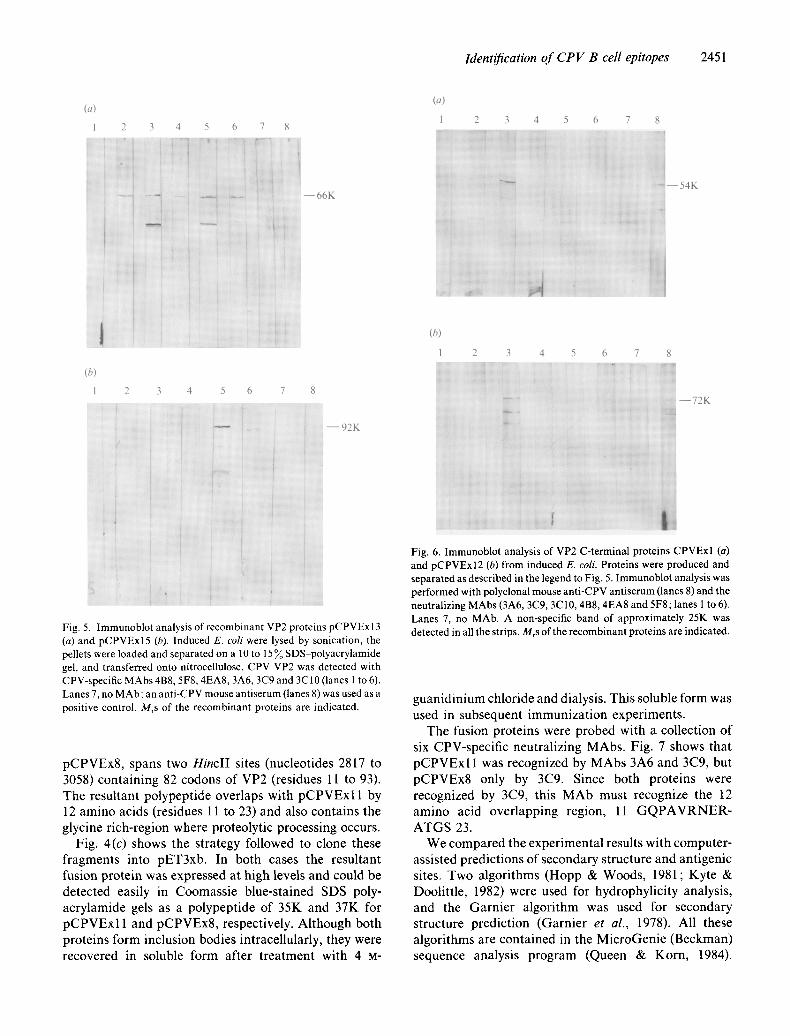
The expression of VP2 was very efficient in both systems tested. In pGEX2T, the resultant plasmid was designated pCPVExl5. VP2 was expressed as a fusion protein with glutathione-S-transferase, which facilitated its purification by affinity chromatography (Smith & Johnson, 1988). The fusion product was expressed in E. coli and could be detected easily in Coomassie blue- stained SDS-polyacrylamide gels as a polypeptide of 92K. In pET3a, the expression of VP2 was as good and as stable as that in pGEX2T, but the product from pCPVExl 3 was insoluble. In Western blot analyses, both proteins were recognized by monospecific rabbit anti- serum raised against complete CPV. To determine whether the recombinant VP2s were identical to viral VP2 antigenically, they were probed with all the neutralizing MAbs (Fig. 5). This produced an indistin- guishable pattern in the case of the protein isolated from pCPVExl3. However, most MAbs did not recognize VP2 from pCPVExl5 when it was tested as a fusion protein (Fig. 5).
Expression of the C terminus of VP2
Fig. 4(b) shows the subcloning of different fragments of VP2 into pKK233-2 and pET3xa. The C-terminal PstI fragment was excised from pCPVI2 and ligated in-phase into the PstI site of pKK233-2 (Fig. 4b) under the control of the Trc promoter, yielding pCPVExl . The expression product of pCPVExl was obtained as an insoluble polypeptide and attempts to recover it as a soluble peptide failed. As is the case for expression of VP2, the yield of recombinant peptide was very high. When tested for antigenicity using the whole panel of neutralizing MAbs, only 3C 10 recognized the peptide by immunoblot analysis (Fig. 6). However, the non-neutralizing MAb 2B4 yielded a positive signal of similar strength (data not shown). Since 3C 10 neutralized only weakly, this implied that none of the MAbs which neutralize more strongly reacted with this peptide, although it represents about 85~o of VP2 and is expressed as a free peptide.
We do not believe that the lack of reaction was due to the insolubility of the peptide, because when we expressed the BglII-C terminus fragment (67 ~ of VP2) in pCPVExl2 as a fusion polypeptide with the 5' region of the gene 10 product of T7, we obtained a soluble peptide that was recognized by the CPV-specific polyclonal rabbit antiserum and 3C10, but not by any other MAb (Fig. 6).
Expression and characterization of the N terminus of VP2
Previous results have shown that the C-terminal region of CPV VP2 does not seem to contain a sequential epitope. Accordingly, we focused on the expression of VP2 N-terminal fragments. Owing to the nested struc- ture of the coat proteins of CPV, we included the VP1 regions immediately adjacent to the initiation codon of VP2. We made two different constructs. One spans two Sau3A sites (nucleotides 2656 to 2850) and contains 42 codons of the VPl-specific region and the first 22 codons of VP2, and was named pCPVExl 1. The other clone,
t~
4~
(a)
1 .....
1 ......
~ o°..,
L~
....
l ,..,
l p.
,,
(c)
ig,.
~ • .
...
ii@
E,°
~,7"
1 .......
......
1
1 .....
........
........
1 ..
..........
..........
.. l
Fig.
4.
Clo
ning
str
ateg
y fo
r th
e co
nstr
ucti
on o
f th
e di
ffer
ent
expr
essi
on v
ecto
rs.
(a)
VP
2 ex
pres
sion
vec
tors
pC
PV
Exl
3 an
d pC
PV
Exl
5.
(b)
C-t
erm
inal
exp
ress
ion
vect
ors
pCP
VE
xl
and
pCP
VE
xl2.
(c
) N
-ter
min
al e
xpre
ssio
n ve
ctor
s pC
PV
Ex8
and
pC
PV
Exl
1.
Identification of CP V B cell epitopes 2451
(a)
1 2 3 4 5 6 7 8
(a)
l 2 3 4 5 6 7 8
i i ~,! . . . . . 66K i I • i
" i z i ,::,
(b)
1
: i
2 3
i ,
4 5 6 7
i . . . . . . . . . . .
- - 92K
Fig. 5. Immunoblot analysis of recombinant VP2 proteins pCPVExl3 (a) and pCPVExl5 (b). Induced E. coli were lysed by sonication, the pellets were loaded and separated on a 10 to 15 % SDS-polyacrylamide gel, and transferred onto nitrocellulose. CPV VP2 was detected with CPV-specific MAbs 4B8, 5F8, 4EA8, 3A6, 3C9 and 3C10 (lanes 1 to 6). Lanes 7, no MAb; an anti-CPV mouse antiserum (lanes 8) was used as a positive control. Mrs of the recombinant proteins are indicated.
pCPVEx8, spans two HinclI sites (nucleotides 2817 to 3058) containing 82 codons of VP2 (residues 11 to 93). The resultant polypeptide overlaps with pCPVExl 1 by 12 amino acids (residues 11 to 23) and also contains the glycine rich-region where proteolytic processing occurs.
Fig. 4(c) shows the strategy followed to clone these fragments into pET3xb. In both cases the resultant fusion protein was expressed at high levels and could be detected easily in Coomassie blue-stained SDS-poly- acrylamide gels as a polypeptide of 35K and 37K for pCPVExl 1 and pCPVEx8, respectively. Although both proteins form inclusion bodies intracellularly, they were recovered in soluble form after treatment with 4 M-
(b)
1 2 3 4 5 6 7 8
Fig. 6. Immunoblot analysis of VP2 C-terminal proteins CPVExl (a) and pCPVExI2 (b) from induced E. coli. Proteins were produced and separated as described in the legend to Fig. 5. Immunoblot analysis was performed with polyclonal mouse anti-CPV antiserum (lanes 8) and the neutralizing MAbs (3A6, 3C9, 3C10, 4B8, 4EA8 and 5F8; lanes 1 to 6). Lanes 7, no MAb. A non-specific band of approximately 25K was detected in all the strips. Mrs of the recombinant proteins are indicated.
guanidinium chloride and dialysis. This soluble form was used in subsequent immunization experiments.
The fusion proteins were probed with a collection of six CPV-specific neutralizing MAbs. Fig. 7 shows that pCPVExl 1 was recognized by MAbs 3A6 and 3C9, but pCPVEx8 only by 3C9. Since both proteins were recognized by 3C9, this MAb must recognize the 12 amino acid overlapping region, 11 GQPAVRNER- ATGS 23.
We compared the experimental results with computer- assisted predictions of secondary structure and antigenic sites. Two algorithms (Hopp & Woods, 1981; Kyte & Doolittle, 1982) were used for hydrophylicity analysis, and the Garnier algorithm was used for secondary structure prediction (Garnier et al., 1978). All these algorithms are contained in the MicroGenie (Beckman) sequence analysis program (Queen & Korn, 1984).
2452 J. A. Lbpez de Turiso and others
2 3 4 5 6 7 8
(b)
1 2 3 4 6 7
l " i
~ : ~ i [ : : ~ ~..
,:,, ] ,: .g, : : . . . . . . . .
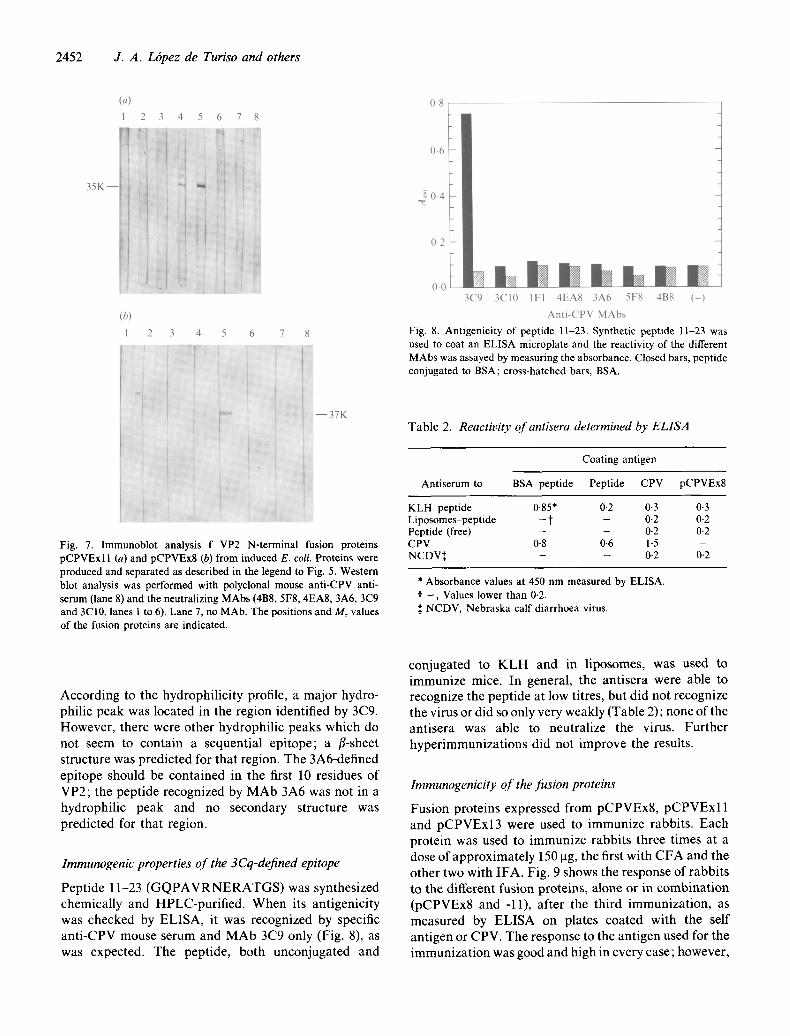
Fig. 7. Immunoblot analysis f VP2 N-terminal fusion proteins pCPVExl 1 (a) and pCPVEx8 (b) from induced E. coll. Proteins were produced and separated as described in the legend to Fig. 5. Western blot analysis was performed with polyclonal mouse anti-CPV anti- serum (lane 8) and the neutralizing MAbs (4B8, 5F8, 4EA8, 3A6, 3C9 and 3C10, lanes 1 to 6). Lane 7, no MAb. The positions and Mr values of the fusion proteins are indicated.
(a) 1
0.8
0.6
80 .4
0.2
0-0 t 3C9
I, Ihm b m Im 3C10 1F1 4EA8 3A6 5F8 4B8 ( - )
Anti-CPV MAbs
Fig. 8. Antigenicity of peptide 11-23. Synthetic peptide 11-23 was used to coat an ELISA microplate and the reactivity of the different MAbs was assayed by measur ing the absorbance. Closed bars, peptide conjugated to BSA; cross-hatched bars, BSA.
Table 2. Reactivity of antisera determined by ELISA
Coating antigen
Ant iserum to BSA peptide Peptide CPV pCPVEx8
K L H peptide 0.85* 0-2 0.3 0.3 Liposomes-pept ide - t - 0.2 0-2 Peptide (free) - - 0.2 0-2 CPV 0.8 0.6 1-5 - NCDV:~ - - 0.2 0.2
* Absorbance values at 450 n m measured by ELISA. t - , Values lower than 0-2. :~ NCDV, Nebraska calf diarrhoea virus.
According to the hydrophilicity profile, a major hydro- philic peak was located in the region identified by 3C9. However, there were other hydrophilic peaks which do not seem to contain a sequential epitope; a fl-sheet structure was predicted for that region. The 3A6-defined epitope should be contained in the first 10 residues of VP2; the peptide recognized by MAb 3A6 was not in a hydrophilic peak and no secondary structure was predicted for that region.
Immunogenic properties of the 3Cq-defined epitope
Peptide 11-23 ( G Q P A V R N E R A T G S ) was synthesized chemically and HPLC-purified. When its antigenicity was checked by ELISA, it was recognized by specific anti-CPV mouse serum and MAb 3C9 only (Fig. 8), as was expected. The peptide, both unconjugated and
conjugated to K L H and in liposomes, was used to immunize mice. In general, the antisera were able to recognize the peptide at low titres, but did not recognize the virus or did so only very weakly (Table 2); none of the antisera was able to neutralize the virus. Further hyperimmunizat ions did not improve the results.
Immunogenicity of the fusion proteins
Fusion proteins expressed from pCPVEx8, pCPVExl 1 and pCPVExl3 were used to immunize rabbits. Each protein was used to immunize rabbits three times at a dose of approximately 150 ~tg, the first with CFA and the other two with IFA. Fig. 9 shows the response of rabbits to the different fusion proteins, alone or in combinat ion (pCPVEx8 and -11), after the third immunization, as measured by ELISA on plates coated with the self antigen or CPV. The response to the antigen used for the immunization was good and high in every case; however,
Identification of CPV B cell epitopes 2453
2.5 Discussion
2.0
1.5
1-0
0,5
0.0 Ex8 Exl l E x S + l l Exl3 CPV (-)
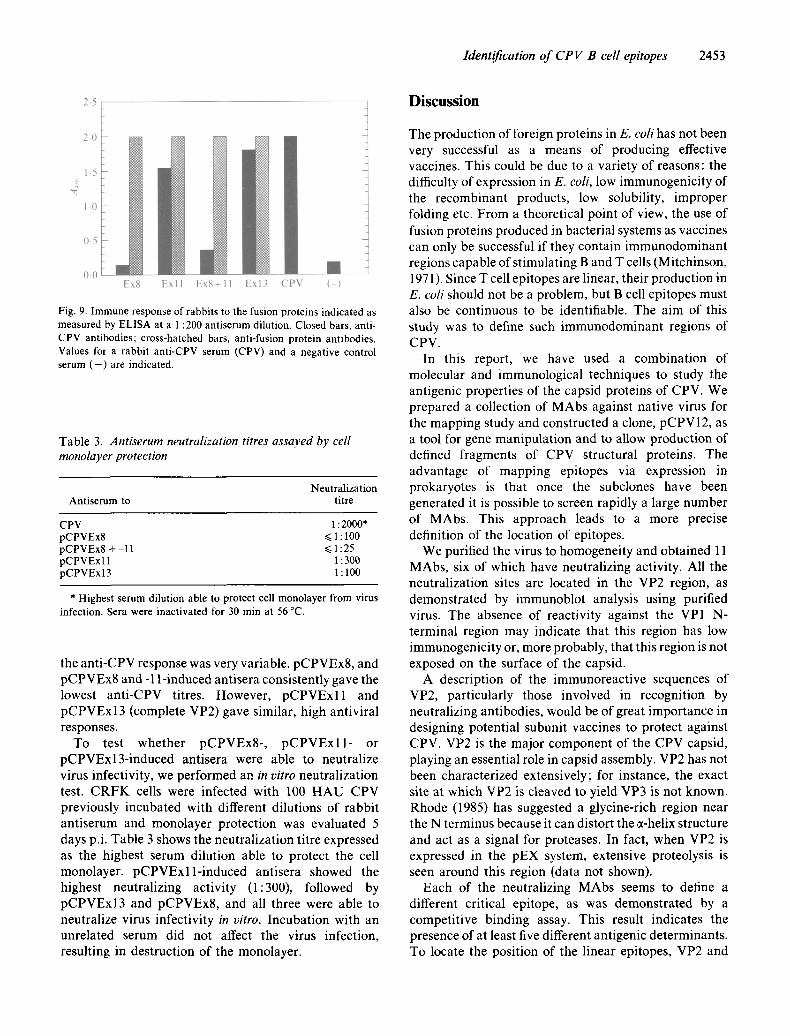
Fig. 9. Immune response of rabbits to the fusion proteins indicated as measured by ELISA at a 1:200 antiserum dilution. Closed bars, anti- CPV antibodies; cross-hatched bars, anti-fusion protein antibodies. Values for a rabbit anti-CPV serum (CPV) and a negative control serum ( - ) are indicated.
Table 3. Antiserum neutralization titres assayed by cell monolayer protection
Neutralization Ant iserum to titre
CPV 1 : 2000* pCPVEx8 ~< 1 : 100 pCPVEx8 + 11 ~< 1:25 pCPVExl 1 1:300 pCPVExI3 1 : 100
* Highest serum dilution able to protect cell monolayer from virus infection. Sera were inactivated for 30 min at 56 °C.
the anti-CPV response was very variable, pCPVEx8, and pCPVEx8 and -1 l-induced antisera consistently gave the lowest anti-CPV titres. However, pCPVExll and pCPVExl 3 (complete VP2) gave similar, high antiviral responses.
To test whether pCPVEx8-, pCPVExll- or pCPVExl3-induced antisera were able to neutralize virus infectivity, we performed an in vitro neutralization test. CRFK cells were infected with 100 HAU CPV previously incubated with different dilutions of rabbit antiserum and monolayer protection was evaluated 5 days p.i. Table 3 shows the neutralization titre expressed as the highest serum dilution able to protect the cell monolayer, pCPVExll-induced antisera showed the highest neutralizing activity (1:300), followed by pCPVExl3 and pCPVEx8, and all three were able to neutralize virus infectivity in vitro. Incubation with an unrelated serum did not affect the virus infection, resulting in destruction of the monolayer.
The production of foreign proteins in E. coli has not been very successful as a means of producing effective vaccines. This could be due to a variety of reasons: the difficulty of expression in E. coli, low immunogenicity of the recombinant products, low solubility, improper folding etc. From a theoretical point of view, the use of fusion proteins produced in bacterial systems as vaccines can only be successful if they contain immunodominant regions capable of stimulating B and T cells (Mitchinson, 1971). Since T cell epitopes are linear, their production in E. coli should not be a problem, but B cell epitopes must also be continuous to be identifiable. The aim of this study was to define such immunodominant regions of CPV.
In this report, we have used a combination of molecular and immunological techniques to study the antigenic properties of the capsid proteins of CPV. We prepared a collection of MAbs against native virus for the mapping study and constructed a clone, pCPV 12, as a tool for gene manipulation and to allow production of defined fragments of CPV structural proteins. The advantage of mapping epitopes via expression in prokaryotes is that once the subclones have been generated it is possible to screen rapidly a large number of MAbs. This approach leads to a more precise definition of the location of epitopes.
We purified the virus to homogeneity and obtained 11 MAbs, six of which have neutralizing activity. All the neutralization sites are located in the VP2 region, as demonstrated by immunoblot analysis using purified virus. The absence of reactivity against the VP1 N- terminal region may indicate that this region has low immunogenicity or, more probably, that this region is not exposed on the surface of the capsid.
A description of the immunoreactive sequences of VP2, particularly those involved in recognition by neutralizing antibodies, would be of great importance in designing potential subunit vaccines to protect against CPV. VP2 is the major component of the CPV capsid, playing an essential role in capsid assembly. VP2 has not been characterized extensively; for instance, the exact site at which VP2 is cleaved to yield VP3 is not known. Rhode (1985) has suggested a glycine-rich region near the N terminus because it can distort the a-helix structure and act as a signal for proteases. In fact, when VP2 is expressed in the pEX system, extensive proteolysis is seen around this region (data not shown).
Each of the neutralizing MAbs seems to define a different critical epitope, as was demonstrated by a competitive binding assay. This result indicates the presence of at least five different antigenic determinants. To locate the position of the linear epitopes, VP2 and

2454 J. A. L6pez de Turiso and others
portions covering the C and N termini have been expressed as fusion or free proteins in three different E. coli expression systems. All the neutralizing MAbs except 4B8 recognize the E. coli-derived VP2, but they fail to recognize two large fragments containing 85 ~ and 67 ~ of the C terminus of VP2 respectively. When fusion proteins containing the N-terminal region of VP2 are analysed by immunoblotting, there were two neutraliz- ing MAbs, 3C9 and 3A6, which recognize two different epitopes on the fusion protein expressed by pCPVExl 1. MAb 3C9 also reacted with fusion protein pCPVEx8, localizing the 3C9-defined epitope to residues 11 to 23 of VP2. Since MAb 3A6 did not recognize the protein from pCPVEx8 it should recognize residues 1 to 10 of VP2. Therefore, we can conclude that the N terminus is the most immunogenic region of VP2.
There could be two main reasons to explain the absence of reactivity of the neutralizing MAbs with most of VP2. First, there are no linear epitopes in the large C- terminal region, or second, the epitopes could be modified when expressed as an isolated fragment, inducing a dramatic change in the antigenicity of the protein. We support the first hypothesis, in .contrast to Parrish et al. (1988) who described an important type- common neutralizing epitope in the 64 to 73 map unit region around the BgllI restriction site. The fact that only two of the 11 MAbs obtained recognized the C-terminal region, probably indicates that it does not contain linear neutralizing epitopes. A previous study (Smith & Hailing, 1984) reached similar conclusions in that several portions of the C-terminal region failed to elicit neutralizing antibodies when expressed as/~-galactosi- dase fusion proteins in E. coli. To minimize the second possibility, we have used two different expression systems, pKK and pET3x, which yield insoluble and soluble products, respectively. In both cases we obtained an equally poor response against the C-terminal polypep- tides. Furthermore, solubility can not be the problem because VP2 expressed by pCPVExI3 is insoluble and is recognized by the same MAbs that recognize viral VP2. We conclude that most of the antigenic determinants in CPV are probably discontinuous, with exception of two epitopes at the N terminus of VP2.
From these studies we can also conclude that only two of the six neutralizing MAbs, 3C9 and 3A6, recognize sequential or linear epitopes as they are expressed in E. coli fusion proteins. The other three neutralizing MAbs probably recognize conformational or discontinuous epitopes because they recognize the whole molecule and not fragments of it. MAb 4B8 does not recognize either the viral or the E. coli-derived VP2 by immunoblot assay. These results confirm that most of the critical epitopes in CPV seem to be non-linear.
To define the structural characteristics of the two
epitopes, we made a computer prediction of the secondary structure and the hydrophilicity profile of VP2 (data not shown). The 3C9-defined epitope is contained in the peptide GQPAVRNERATGS (residues 11 to 23 of VP2). A fl-sheet conformation and a major peak of hydrophilicity are predicted for this region. None of the other predicted major hydrophilic peaks inside VP2 seem to be relevant antigenically.
To evaluate its immunogenicity, the peptide 11-23 was synthesized chemically with a cysteine coupled to the N terminus to facilitate binding to a carrier protein. The peptide was conjugated to KLH and used to immunize mice. Although the antisera raised recognize the peptide, they recognize virus very poorly and do not show any neutralizing activity, indicating that this B cell epitope by itself is insufficient to induce an adequate response.
Since they contain immunodominant regions, the fusion proteins were also checked for their immunogeni- city and ability to protect against virus infectivity in vitro. Three different expression products were used to raise polyclonal antibodies in rabbits. Protein expression in the pET system is high and protein is in a stable form, probably due to the use of a protease-deficient E. coli strain. Despite the fact that T7 gene 10 fusion products are generally insoluble, all our recombinant polypep- tides, except pCPVExl3, were recovered as soluble products after treatment with 4 M-guanidinium chloride and dialysis. In general terms, all the fusion proteins elicit antisera with good titres against themselves and a variable anti-virus titre, showing a weak correlation between anti-virus titre (ELISA) and monolayer protec- tion capability in vitro, pCPVEx8 induces sera with low neutralization titre. This result again confirms that the B cell epitope present in peptide 11-23 is insufficient by itself to elicit an adequate humoral response. Recombin- ant protein pCPVExl 1 seems to be the most effective for raising antisera with neutralizing activity, even more so than the complete VP2 contained in pCPVExI3. This is probably due to the insolubility of pCPVExl3, which makes it a weaker immunogen. We are trying to produce soluble VP2 in order to make a better comparison. The suppressive effect of pCPVEx8 on pCPVExll is not fully understood. A possible explanation for the high immunogenicity of pCPVExl 1 has been given recently. While these experiment were in progress, the location of several T cell epitopes was described (Rimmelzwaan et al., 1990a, b), some of which are located in the VP1 region contained in pCPVExll , such as peptide 119 PKIFINLAKKKKAG 132 (residues of VP1). The presence of these T cell epitopes in combination with the B cell epitopes could confer high immunogenicity to the molecule.
In summary, this work demonstrates that fusion

Identification of CPV B cell epitopes 2455
proteins containing immunogenic CPV sequences ex- pressed in a bacterial system can induce neutralizing activity. The next step will be immunization trials with dogs to check the effective potency of these fusion proteins as potential vaccines. Since all the members of the Kilham rat parvovirus group, which includes CPV, share significant sequence similarity, and a similar genome organization and replication strategy (Ranz et al., 1989), it may be possible to design subunit vaccines for other economically important parvoviruses such as PPV using the same strategy.
The authors are grateful to Dr J. M. Garcia Anton of the CID (Barcelona) for peptide synthesis. We thank Maria del Mar Pitarch for typing of the manuscript. This work was supported by funds from the Spanish CAICYT and ERCROS S.A.
References
AMANN, E. & BROSIUS, J. (1985). 'ATG vectors' for regulated high-level expression of cloned genes in Escherichia coll. Gene 40, 183-190.
APPLE, M., SCOTT, F. W. & CARMICHAEL, L. R. (1979). Isolation and immunization studies of a canine parED-like virus from dogs with haemorrhagic enteritis. Veterinary Record 105, 156-179.
BURNETTE, W. N. (198 l) 'Western-blotting': electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated Protein A. Analytical Biochemistry 112, 195-203.
BURTONBOY, G., COIGNOUL, F., DELFERRIERE, N. & PASTORET, P. P. (1979). Canine haemorrhagic enteritis: detection of viral particles by electron microscopy. Archives of Virology 61, 1-11.
CARLSON, J., RUSHLOW, K., MAXWELL, I., MAXWELL, F., WINSTON, S. & HAHN, W. (1985). Cloning and sequence of DNA encoding structural proteins of the autonomous parvovirus feline panleuko- penia virus. Journal of Virology 55, 574-582.
CARMICHAEL, L. E., JOUBERT, J. C. & POLLOCK, R. (1980). Hemagglu- tination by canine parvovirus: serologic studies and diagnostic applications. American Journal of Veterinary Research 4, 784-791.
CARMICHAEL, L. E., JOUBERT, J. C. & POLLOCK, R. V. H. (1981). A modified live canine parvovirus strain with novel plaque characteris- tics. I. Viral attenuation and dog response. Cornell Veterinarian 71, 408-427.
CASAL, J. ][., DiAZ, E., RANZ, A. I. & MANCEIOS, J. J. (1990). Construction of an infectious genomic clone of porcine parvovirus: effect of the 5' end on DNA replication. Virology 177, 764-767.
FLOWER, R. L. P., WILCOX, G. E. & ROBINSON, W. F. (1980), Antigenic differences between canine parvovirus and feline panleukopenia virus. Veterinary Records 107, 254-256.
GARNIER, J., OSGUTHORPE, D. J. & ROBSON, B. (1978). Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. JournalofMolecular Biology 120, 97-120.
HANAHAN, D. (1983). Studies on transformation ofEscherichia coli with plasmids. Journal of Molecular Biology 166, 557-580.
HOPP, T. P. & WOODS, K. R. (1981). Prediction of protein antigenic determinants from amino acid sequences. Proceedings of the National Academy of Sciences, U.S.A. 78, 3824-3828.
KYTE, J. & DOOLITrLE, R. F. (1982). A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology 157, 105-132.
MANIATIS, T., FRITSCH, E. F. & SAMBROOK, J. (1982). Molecular Cloning." A Laboratory Manual. New York: Cold Spring Harbor Laboratory.
MITCHINSON, N. A. (1971). The carrier effect in the secondary response to hapten-protein conjugates. It Cellular cooperation. European Journal of lmmunology 1, 18.
MOLITOR, T. W., JOO, H. S. & COLLETT, M. S. (1983). Porcine parvovirus: virus purification and structural and antigenic proper- ties of virion polypeptides. Journal of Virology 45, 842-854.
MOEITOR, T. W., Joo, H. S. & COLLETT, M. S. (1984). Porcine parvovirus DNA : characterization of the genomic and the replica- Live form DNA of two virus isolates. Virology 137, 241-254.
PARADISO, P. R. (1983). Analysis of the protein-protein interactions in the parvovirus H-1 capsid. Journal of Virology 46, 94-102.
PARADISO, P. R., RHODE, S. L., I I I & SINGER, I. I. (1982). Canine parvovirus: a biochemical and ultrastructural characterization. Journal of General Virology 62, l 13-125.
PARRISn, C. R., CARMICHAEL, L. E. & ANTCZAK, D. F. (1982). Antigenic relationships between canine parvovirus type 2, feline panleukopenia virus and mink enteritis virus using a conventional antiserum and monoclonal antibodies. Archives of Virology 72, 267- 278.
PARRISH, C. R., AQUADRO, C. F. & CARMICHAEL, L. E. (1988). Canine host range and a specific epitope map along with variant sequences in the capsid protein gene of canine parvovirus and related feline, mink and raccoon parvoviruses. Virology 166, 293-276.
POLLOCK, R. V. H. & CARMICHAEL, L. E. (1982a). Dog response to inactivated canine parvovirus and feline panleukopenia virus vaccines. Cornell Veterinarian 72, 16-35.
POLLOCK, R. V. H. & CARMICHAEL, L. E. (1982b). Maternally derived immunity to canine parvovirus infection: transfer, decline and interference with vaccination. Journal of the American Veterinary Medical Association 180, 37-42.
Q'OE~N, C. & KORN, L. J. (1984). A comprehensive sequence analysis program for the IBM PC. Nucleic Acids Research 12, 581-599.
RANZ, A. I., MANCLUS, J. J., DIAZ-AROCA, E. & CASAL, J. I. (1989). Porcine parvovirus: DNA sequence and genome organization. Journal of General Virology 70, 2541-2553.
REED, A. P., JONES, E. V. & MILLER, T. J. (1988). Nucleotide sequence and genome organization of canine parvovirus. Journal of Virology 62, 266-276.
RHODE, S. L. 0985). Nucleotide sequence of the coat protein gene of canine parvovirus. Journal of Virology 54, 630-633.
RIMMELZWAAN, G. F., VAN DER HEIJDEN, R. W. J., TIJHAAR, E., POLLEN, M. C. M., CARLSON, J., OSTERHAUS, A. D. M. E. & UYTDEHAAG, F. G. C. M. (1990a). Establishment and characteriza- tion of canine parvovirus-specific murine CD4 + T cell clones and their use for the delineation of T cell epitopes. Journal of General Virology 71, 1095-1102.
RIMMELZWAAN, G. F., POLLEN, M. C. M., MELOEN, R. H., CARLSON, J., UYTDEHAAG, F. G. C. M. & OSTERHAIJS, A. D. M. E. (1990b). Delineation of canine parvovirus T cell epitopes with peripheral blood mononuclear cells and T cell clones from immunized dogs. Journal of General Virology 71, 2321-2329.
SANZ, A., GARCIA-BERRENO, B., NOGAL, M. L., VII~d'ELA, E. & ENJUANES, L. (1985). Monoclonal antibodies specific for African swine fever virus proteins. Journal of Virology 54, 199-206.
SMITH, D. B. & JOHNSON, K. S. (1988). Single-step purification of polypeptides expressed in E. coti as fusions with glutathione-S- transferase. Gene 67, 31-40.
SMITH, D. B. & HALLING, S. M. (1984). Expression of canine parvovirus-fl-galactosidase fusion proteins in E. coll. Gene 29, 263- 269.
STUDIER, F. W. & MOEEAT, B. A. (1986). Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. Journal of Molecular Biology 189, 113-130.
SURLERAUX, M. & BURTONBOY, G. (1984). Structural polypeptides of a canine parvovirus: study by immunoadsorption and sequential analysis of infected cells. Archives of Virology 82, 233-240.
TATTERSALE, P., SHATKIN, A. J. & WARD, D. C. (1977). Sequence homology between the structural polypeptides of minute virus of mice. Journal of Molecular Biology 111,775-794.
TOWBIN, H., STAEHELIN, T. & GORDON, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proceedings of the National Academy of Sciences, U.S.A. 76, 4350-4354.
TRATSCHIN, J.-D., MCMASTER, G. K., KRONAUER, G. & SIEGE, G. (1982). Canine parvovirus: relationship to wild-type and vaccine

2456 J. A. L6pez de Turiso and others
strains of feline panleukopenia virus and mink enteritis virus. Journal of General Virology 61, 33-41.
VELA, C., CAMBRA, M., CORTF.S, E., MORENO, P., MIGUET, J. G., PI~REZ DE SAN ROM~.N, C. & SANZ, A. (1986). Production and characteriza- tion of monoclonal antibodies for citrus tristeza virus and their use in diagnosis. Journal of General Virology 67, 91-96.
WOBBLE, C. R., MITRA, S. • RAMAKRISHNAN, V. (1984). Structure of the capsid of Kilham rat virus from small-angle neutral scattering. Biochemistry 23, 6565~569.
(Received 2 January 1991; Accepted 11 June 1991)




















