
Acute Encephalitis, a Poliomyelitis-like Syndrome and ... - NCBI
Upload
independentCategory
view
2download
0
HIPPOCAMPAL SYNAPTIC DYSFUNCTION IN A MURINE MODEL OFHUMAN IMMUNODEFICIENCY VIRUS TYPE 1 ENCEPHALITIS
E. R. ANDERSON,a,b J. BOYLE,a,b W. E. ZINK,a,b
Y. PERSIDSKY,a,b H. E. GENDELMANa,b,c ANDH. XIONGa,b*aCenter for Neurovirology and Neurodegenerative Disorders, Univer-sity of Nebraska Medical Center, Omaha, NE 68198-5215, USAbDepartment of Pathology and Microbiology, University of NebraskaMedical Center, Omaha, NE 68198, USAcDepartment of Internal Medicine, University of Nebraska MedicalCenter, Omaha, NE 68198, USA
Abstract—Alterations in hippocampal physiology affect cog-nition in human immunodeficiency virus type 1 (HIV-1)-asso-ciated dementia (HAD). The mechanism for how this occurs isnot well understood. To address this, we investigated howchanges in synaptic transmission and plasticity are affectedby viral infection and macrophage activation using a severecombined immunodeficiency mouse model of human HIV-1encephalitis (HIVE). HIVE was induced in mice by stereotacticinjection of HIV-1-infected human monocyte-derived macro-phages (MDM) into the striatum. Animals were sacrificed after3, 7 and 15 days. Hippocampal slices were prepared fromHIV-1, MDM- and sham-injected animals. Electrically evokedfield excitatory postsynaptic potentials were recorded in theCA1 region of the hippocampus. Neuronal physiology wasassessed by input–output and by long-term potentiation(LTP) assays. We observed that a higher stimulation intensity(mA) was required to induce a 1-mV response in the HIVEmice (0.32�0.06) compared with shams (0.17�0.01) at day 7.The stimulation intensities at day 15 were 0.44�0.07 and0.23�0.05 in the HIVE and shams, respectively. An impair-ment of synaptic function was detected through measuringsynaptic responses induced by stimuli with different intensi-ties. Paired-pulse facilitation (PPF) showed deficits in HIVEmice at days 3, 7, and 15. At day 3, PPF ratios were 1.13�0.02and 1.24�0.04 in HIVE and sham. The induction and mainte-nance of LTP was also impaired in HIVE mice. The averagemagnitude of LTP was 131.23�15.26% of basal in HIVE ascompared with sham animals of 232.63�24.18%. MDM-in-jected mice showed an intermediate response. Taken to-gether, the results show a range of neuronal synaptic trans-mission and plasticity changes in HIVE mice that may reflect
the mechanisms of cognitive dysfunction in human HAD.© 2003 IBRO. Published by Elsevier Science Ltd. All rightsreserved.
Key words: HIV-1-associated dementia, SCID mice, CA1, ex-citatory postsynaptic potential, monocyte-derived macro-phages.
The acquired immunodeficiency syndrome (AIDS) is as-sociated with significant impairments in cell-mediatedimmunity followed in parallel by opportunistic infectionsand in select patients, cognitive impairment termed hu-man immunodeficiency virus type 1 (HIV-1)-associateddementia (HAD). It is estimated that approximately 20%of the adults and 50% of the children infected with HIV-1can develop HAD (Navia et al., 1986; Epstein et al.,1988; Janssen et al., 1989, 1992). HAD is the leadingcause of dementia in people less than 60 years of age(Janssen et al., 1992; McArthur et al., 1993). Althoughthe incidence of HAD has dropped significantly as aresult of widespread use of highly active anti-retroviraltherapy (Sacktor et al., 2001), neurological dysfunctionremains prevalent. In infected individuals, as resistanceto antiviral therapy grows with viral strain mutation andthe impaired ability of drugs to penetrate the blood–brain barrier (BBB). This strongly suggests that HADremains a significant complication of advanced HIV-1disease (McArthur et al., 1999; Carpenter et al., 2000;Krebs et al., 2000). Unfortunately, there are at presentno effective treatments for preventing HIV-1 associatedcognitive impairments. Thus, a better understanding ofHAD pathogenesis should prove useful in the diagnosis,prevention and treatment of HAD and for the develop-ment of new therapeutic strategies.
HAD is characterized by progressive neuropsychiat-ric and neurological disturbances including cognitive im-pairment which begins with forgetfulness and apathy,eventually leading to florid hallucinations, coma anddeath. HIV-1 encephalitis (HIVE) is the pathological cor-relate of HAD (Wiley and Achim, 1994) and is charac-terized by viral infection of perivascular blood-derivedmacrophages, the formation of multinucleated giantcells, microglial nodules, myelin pallor, astrogliosis andneuronal dropout. A striking feature of HAD is synapticdamage (Everall et al., 1991; Masliah et al., 1992). Theinfiltration of immune competent macrophages into thebrain predicts the tempo of disease (Glass et al., 1993a).Brain mononuclear phagocyte (MP; macrophages andmicroglial) activation, as demonstrated, in part, by majorhistocompatibility complex class II expression, is the
*Correspondence to: H. Xiong, Center for Neurovirology and Neuro-degenerative Disorders, University of Nebraska Medical Center,Omaha, NE 68198-5215, USA. Tel: �1-402-559-5140; fax: �1-402-559-8922.E-mail address: [email protected] (H. Xiong).Abbreviations: ACSF, artificial cerebral spinal fluid; AD, Alzheimer’sdisease; AIDS, acquired immunodeficiency syndrome; BBB, blood–brain barrier; fEPSP, field excitatory postsynaptic potential; GFAP,glial fibrillary acid protein; HAD, HIV-1-associated dementia; HFS,high-frequency stimulation; HIV, human immunodeficiency virus;HIV-1, HIV type 1; HIVE, HIV-1 encephalitis; IO, input-output; IPI,interpulse interval; LTP, long-term potentiation; MAP-2, microtubule-associated protein 2; MDM, monocyte-derived macrophage; MP,mononuclear phagocyte; PPF, paired-pulse facilitation; PTP, post-tetanic potentiation; SCID, severe combined immunodeficiency; STP,short-term potentiation.
Neuroscience 118 (2003) 359–369
0306-4522/03$30.00�0.00 © 2003 IBRO. Published by Elsevier Science Ltd. All rights reserved.doi:10.1016/S0306-4522(02)00925-9
359

best predictor of disease severity for HAD (Glass et al.,1993a; Persidsky et al., 1999). In addition, progressiveviral infection, in both the periphery and in the nervoussystem, appear necessary, but not sufficient, to effectneurological impairments for disease (Lipton and Gen-delman, 1995; Persidsky et al., 1999).
A central tenet of HIV-1 neuropathogenesis is theabnormal secretion of immune “neurotoxic” factors fromvirus-infected and immune-competent MPs (Desbailletset al., 1994; Conant et al., 1998; Cotter et al., 1999b;Persidsky, 1999). Following HIV-1 infection, macro-phages become primed for subsequent activation result-ing in neurotoxic secretory activities (Lipton and Gen-delman, 1995). Due to the diminished cytotoxic T lym-phocyte responses combined with the widespread glialeffector responses in the brain, a paracrine and auto-crine amplification of macrophage secretory productsensues, inciting neuronal demise (Lipton and Gendel-man, 1995; Kaul et al., 2001). Recently, the relation-ships between glial immunity and neurodegenerationhave received increased attention as both Alzheimer’s(AD) and Parkinson’s disease pathogenesis are alsoassociated with brain MP activation and inflammatoryimmune responses (Dickson et al., 1993; Saitoh et al.,1997; Giulian, 1999).
A principal question in HAD pathogenesis is, howcan relatively few infected cells lead to widespread neu-ral dysfunction? Specifically, many have questionedhow alterations in hippocampal function occur when viralinfection is predominantly in the caudate and putamen.As the hippocampus is the site for memory acquisitionand learning, such questions are relevant for HADpathogenesis (Bellinger et al., 1993; Bagetta et al.,1997; Krucker et al., 1998; Xiong et al., 1999). Theycould be addressed through studies of synaptic plasticityand function in the hippocampus of animals with dis-ease. In support of this, recent studies demonstratedimpairment in long-term potentiation (LTP) and paired-pulse facilitation (PPF) in mouse models of AD (Larson,1999). An altered spatial memory and LTP deficits werealso found in rats injected with HIV-1 gp120 (Sanchez-Alavez et al., 2000). Interestingly, such synaptic deficitswere accompanied by the appearance of amyloidplaques (Larson, 1999). Impairments in synaptic func-tion could affect memory dysfunction (Hsia et al., 1999;Larson, 1999). Despite the difference in etiology of dis-ease, both AD and HAD share brain MP activation (Stolland Jander, 1999). Thus we investigated whetherchanges in synaptic transmission and plasticity in thehippocampus could explain cognitive deficits seen in amouse model of HIVE. Our results demonstrated thatinjection of HIV-1 infected human monocyte-derivedmacrophages (MDM) into the caudate and putamen ofsevere combined immunodeficient (SCID) mice impairssynaptic function in the hippocampus and this impair-ment may underlie cognitive dysfunction seen in humanHAD.
EXPERIMENTAL PROCEDURES
Isolation and culture of monocytes
Human monocytes were recovered from peripheral blood mono-nuclear cells of HIV-1, 2 and hepatitis B seronegative donors (fromOmaha metropolitan area) after leukophoresis and purified bycountercurrent centrifugal elutriation (Gendelman et al., 1988).Monocytes were cultured as adherent monolayers (3.3�106 cells/well in six-well plastic culture plates) in Dulbecco’s Modified Eaglemedium (Sigma Chemical Co., St. Louis, MO, USA) containing10% heat-inactivated pooled human serum, 50 �g/ml gentamicin,10 �g/ml ciprofloxacin (Sigma), and 1000 U/ml highly purifiedrecombinant human macrophage colony stimulating factor (a gen-erous gift from Genetics Institute, Inc., Cambridge, MA, USA). Alltissue reagents were screened and found negative for endotoxin(�10 pg/ml; Associates of Cape Cod, Inc., Woods Hole, MA, USA)and mycoplasma contamination (Gen-probe II; Gen-probe Inc.,San Diego, CA, USA). The cell procurements are in compliancewith all required state and federal regulations and approved by theUniversity of Nebraska Medical Center Human Subjects institu-tional review board (IRB 162-93).
HIV-1 infection of MDM
Seven days after plating, MDM were infected with macrophage-tropic HIV-1ADA at a multiplicity of infection of 0.1. Culture fluidswere half exchanged every 2–3 days. Reverse transcriptase ac-tivity was determined in triplicate samples of culture fluids aspreviously described (Kalter et al., 1991). Under these conditions,peak viral replication occurred at 7 days following HIV-1 infection.The ADA strain of HIV-1 was obtained from the peripheral bloodmononuclear cells of a patient with advanced HIV-1 disease, aspreviously described (Gendelman et al., 1988).
SCID mouse model of HIVE
The preparation of HIV-1 infected MDM and subsequent injectioninto the putamen of SCID mice mirrors human HIVE (Persidsky etal., 1996). SCID mice (male C.B-17/IcrCrl-scidBR, 3–4 weeks old)were purchased from Charles River Laboratories (Wilmington,MA, USA). The caudate and putamen (subcortex) were injectedwith HIV-1ADA infected MDM, uninfected MDM, or sham-operated(sham). All animal use procedures were strictly reviewed by theInstitutional Animal Use and Care Committee (IACUC) of Univer-sity of Nebraska Medical Center (IACUC 94-126-08).
Hippocampal slices and electrophysiology
Mouse hippocampal brain slices were prepared as previouslydescribed (Xiong et al., 1996). Mice were transported in microiso-lator cages to the electrophysiology laboratory within a biosafetylevel 3 infection-containment facility, anesthetized with metofaneand decapitated, after which, brains were quickly removed fromthe cranial cavities. All efforts were made to minimize the numberof animals used and their suffering. Hippocampi ipsilateral and/orcontralateral to the injection site were separated and placed inice-cold (4 °C) oxygenated artificial cerebrospinal fluid (ACSF)prior to sectioning. Previous studies performed by our laboratoryhave shown no difference in responses yielded from slices derivedfrom the hippocampus ipsilateral and/or contralateral from theinjection site (unpublished), therefore either hippocampi wereused without preference. No hippocampi used in the experimen-tation showed any signs of needle trauma.
Transverse slices (400 �m thick) were cut from each dis-sected hippocampus using a tissue chopper. The slices were keptin a humidified/oxygenated holding chamber at room temperaturefor at least 1 h before being transferred into a recording chamber.In the recording chamber single hippocampal slices were fullysubmerged in a continuously perfused ACSF solution at a con-
E. R. Anderson et al. / Neuroscience 118 (2003) 359–369360

stant flow rate of 2 ml/min with a peristaltic pump (Rainin Instru-ment Co., Woburn, MA, USA). The ACSF contained (in mM): NaCl(124), KCl (3), CaCl2 (2), MgCl2 (2), NaH2PO3 (1), NaCO3 (26),and glucose (10). ACSF was equilibrated with 95% O2 and 5%CO2, giving a pH of 7.35–7.5. The temperature of the perfusatewas maintained at 30�1 °C with an automatic temperature con-troller (Warner Instrument Corporation, Hamden, CT, USA). Fieldexcitatory postsynaptic potentials (fEPSPs) were elicited by con-stant-current, low-frequency orthodromic stimulation (0.05 Hz) ofSchaffer-collateral–commissural axons with the use of an insu-lated bipolar tungsten electrode (except for the tip). The stimula-tion intensity was adjusted to generate approximately 40–50% ofa maximal response. The evoked fEPSPs were recorded with anAxopatch-1D amplifier (Axon Instruments, Inc., Union City, CA,USA) in the CA1 dendrite field (stratum radium). The recordingmicroelectrodes were made from borosilicated glass capillarieswith inner filaments that enabled quick backfilling. The tip diameterof the microelectrode was approximately 5.0 �m and had a resis-tance of 1–5 M� when filled with ACSF. Input–output (IO) testswere conducted by setting the stimulus intensity to evoke 40–50%of maximal fEPSPs. The pulse duration was then incremented by20 �s from 40 �s to 100 �s and the resultant fEPSPs wererecorded and analyzed. The stimulus strength needed to evoke a1.0-mV fEPSP was determined in each slice for comparisonsbetween groups at different time points.
PPF curves were generated by testing the Schaffer-collateralpathway with twin pulses at a fixed pulse duration of 40 �s andvarying interpulse interval (IPI) at 50-ms increment from 50 to 400ms. The paired pulses were delivered at 20-s intervals and sixconsecutive responses were averaged at each PPF test. Thedegree of facilitation was determined as the increase in ratio of theamplitude (or initial slope) of the second response over the firstresponse in each pair. Since there was no significant differencebetween measurement in amplitude and initial slope, the datawere pooled.
The ability of high-frequency stimulation (HFS) to induce LTPin the CA1 region of the hippocampus was examined. After a30-min control, LTP was induced by a fashioned stimulus protocol(Seabrook, 1997), which consisted of a weak tetanic stimulation(10 events at 100 Hz) followed 45 min later by a burst stimulation(4�10 events at 100 Hz, delivered every 20 s). The initial slopesof the fEPSPs were measured and expressed in percentage ofbasal level. Post-tetanic potentiation (PTP), short-term potentia-tion (STP) and LTP were determined at 1, 15 and 40 min afterHFS. Results from slices with large fluctuation (�2 S.D.) wererejected.
Immunohistochemistry
Whole mouse brains were collected at necropsy at 3, 7, and 15days after injection. Tissue was fixed in 4% paraformaldehyde for48 h and embedded in paraffin. Serial coronal sections (5 �m)were cut, and dendritic arbor was detected with mouse monoclo-nal antibody against microtubule associated protein 2 (MAP-2)(Chemicon, Temecula, CA, USA). Mouse astrocytes were recog-nized with polyclonal Abs against glial fibrillary acid protein(GFAP) (Dako, Carpinteria, CA, USA; and used at 1:1000 dilu-tion). HIV-1 p24 antigen (Dako) at a 1:10 dilution (monoclonalantibodies) was used to detect viral gene products. In order todetect primary antibodies, sections were incubated with avidin–biotin complex (ABC, Vector, Burlingame, CA, USA) for 30 min atroom temperature, developed with 3,3'-diaminobenzidine (Vector)peroxidase substrate, and counterstained with Mayer’s hematox-ylin. Dako EnVision kit was used as the detection system for oneor two antigens. Sections were viewed using a Nikon Eclipse(E800) microscope and images were gathered using a MagnaFiredigital camera and software (Optronics, Goleta, CA, USA). Totalarea occupied by peroxidase positivity for MAP-2 staining was
quantified using Image-Pro Plus software (Media Cybernetics,Silver Spring, MD, USA).
Statistical tests
Data were analyzed to determine significance using two-tailedt-tests and Turkey ANOVA. The level of significance was deter-mined at P�0.05.
RESULTS
The histopathological features of human HIVE were repro-duced in SCID mice injected with HIV-1 infected humanMDM. Neuro-inflammatory responses associated with en-cephalitis first developed at 3 days and were sustained upto 21 days after stereotactic injection of infected MDM (Fig.1). Immunohistochemical tests performed with antibodiesto HIV-1 p24 and GFAP demonstrated the formation ofmultinucleated MDM expressing HIV-1 p24 antigens sur-rounded by reactive astrocytes, confirming the similaritiesbetween the xenograft model and human HIVE.
In order to explore possible alterations in neuronalfunction in areas of the brain most associated with learningand memory, thought to be involved in human HIVE, westudied electrophysiology in the CA1 region of the hip-pocampus of this SCID mouse HIVE model. Three groupsconsisted of mice injected with HIV-1-infected MDM(HIVE), uninfected MDM, and sham-operated animals. Atdays 3, 7 and 15 after injection the mice were killed andhippocampal sections dissected. Subsequently, brain sec-tions underwent a series of electrophysiological examina-tions, including IO tests and LTP studies. Due to consid-erable biohazards involved in performing electrophysiolog-ical studies in HIV-1-infected SCID mice with encephalitis,animals did not always survive the monitoring period and itwas not possible to use the same numbers of animals ateach time point.
The fEPSPs were recorded in the Schaffer-collateralpathway to CA1 synapses. When stimulated, the ability ofa neuronal population to produce an fEPSP with an am-plitude of 1mV is dependent upon the strength (currentintensity and pulse duration) of the stimulation applied. Theresultant recordings demonstrated that the HIVE hip-pocampal slices required a higher current intensity (with afixed pulse duration of 40 �s) to achieve a 1-mV responsethan did sham and MDM controls (Fig. 2). At day 3, therewere no differences between groups in stimulation inten-sity needed to generate a 1-mV fEPSP. However, thestimulation intensity for the HIVE animals at day 7 was0.32�0.06 mA (n�5) in comparison with MDM (0.21�0.01mA, n�6) and sham (0.17�0.01mA, n�10) animals. Atday 15, the stimulation intensity for the HIVE animalsincreased to 0.44�0.07mA (n�5) compared with MDM(0.28�0.04 mA, n�3) and sham (0.23�0.05mA, n�8).The differences were significant at days 7 and 15 (P�0.05by ANOVA).
To assess synaptic function, we performed twopaired-pulse IO tests. The first test employed a fixed IPIwith variable stimulus duration from 40 to 100 �s in20-�s increments. By using a constant current and vary-ing the time of exposure, this test served as a measure
E. R. Anderson et al. / Neuroscience 118 (2003) 359–369 361

of stimulation intensity. Due to the variability of re-sponses between mice, a ratio between the pairedpulses was calculated to serve as a standard. Propor-tional responses were generated between animalgroups as an indicator of neuronal function. Subsequent
20-�s increments in pulse duration demonstrated signif-icant differences between the HIVE, MDM and shamanimals at all time points (days 3, 7 and 15). Dataobtained from day 3 are shown in Table 1 and arerepresentative of the other time points.
Fig. 1. Neuropathological and immunohistochemical features of HIVE in SCID mice. SCID mice 15 days after injection of HIV-1ADA-infected MDMwere killed and brain tissue sectioned for immunohistochemical tests. A representative 5-�m section of subcortical brain tissue from the HIVE mousedouble immunostained with antibodies to HIV-1 p24 (DAB, brown) and GFAP (Fast Red, red/magneto) is shown (Dako EnVision Doublestain System,using the labeled polymer method). The HIV-1 p24 stains demonstrate viral gene expression in multinucleated giant cells. A marked astrocytosissurrounds the multinucleated giant cells. Brain sections were counterstained with Mayer’s hematoxylin. Original magnification is (�400).
Fig. 2. Comparison of average stimulus intensity required to evoke a 1-mV fEPSPs in the hippocampus of HIVE and control (MDM and sham) mice.The stimulus strength (mA) required to evoke 1-mV fEPSPs at each time point is illustrated. Electrophysiological recordings from the hippocampi ofHIVE mice demonstrate a requirement of much higher stimulus strength in order to elicit a 1-mV fEPSP. In comparison with uninfected groups, thedifference in stimulus strength is statistically significant at days 7 and 15 (P�0.05 and P�0.01 respectively, ANOVA). Data points representmean�S.E.M. *Denotes significant differences.
E. R. Anderson et al. / Neuroscience 118 (2003) 359–369362

The second paired-pulse IO test utilized a protocol withvariable IPIs and constant stimulus duration, and assessedthe ability of the initial pulse to facilitate the second one.The IPI was varied from 50 ms to 400 ms at 50-ms incre-ments. The facilitation ratios of the secondary over theprimary responses were measured over each of the inter-vals. The PPF tests revealed a distinct difference betweenthe HIVE group and the controls (MDM and sham). In-creases in IPI caused a proportional dissimilarity betweenexperimental groups such that statistical significance wasreached at all time intervals (Table 2). An example illus-trating a decreased facilitation ratio in HIVE animals at day7 is shown in Fig. 3.
Next, the differences between LTP induced by a weaktetanus and a burst stimulation were compared. Prior tothe delivery of tetanus stimulation, a 30-min recording ofthe responses induced by a low-frequency stimulation(0.05 Hz) was treated as the control baseline. LTP wasthen induced stepwise by an initial weak tetanus stimula-tion (10 events at 100 Hz�1) followed by a burst stimulus(10 events at 100 Hz�4 at 20-s intervals) 45 min after the
initial weak tetanus stimulation. Synaptic potentiation wasmeasured at different phases. Weak tetanus failed to in-duce LTP in all time points, but induced moderate PTP. Incontrast, burst stimulation induced LTP with a robust PTPfollowed by an STP. No significant differences were ob-served at the day 3 time point among the three groups.However, an inhibition of LTP and reduction of PTP andSTP were detected at day 7, and continued into day 15 inHIVE mice. Impairment in HIVE mice appeared to be ex-acerbated at day 7 for both weak and burst tetanus-in-duced LTP (Fig. 4). Significant differences were most ap-parent in HIVE when compared against sham groups in allphases of potentiation at day 7 (P�0.05) (Table 3). HIVEand MDM mice exhibited a diminished capacity to maintainLTP at day 7. This was transient in MDM as it was notevident at day 15 and was significantly lower than shamand MDM (P�0.05). PTP was severely impaired in theHIVE group at day 15. In contrast, at day 15 the MDMgroup recovered from any transient deficiencies expressedat earlier time points.
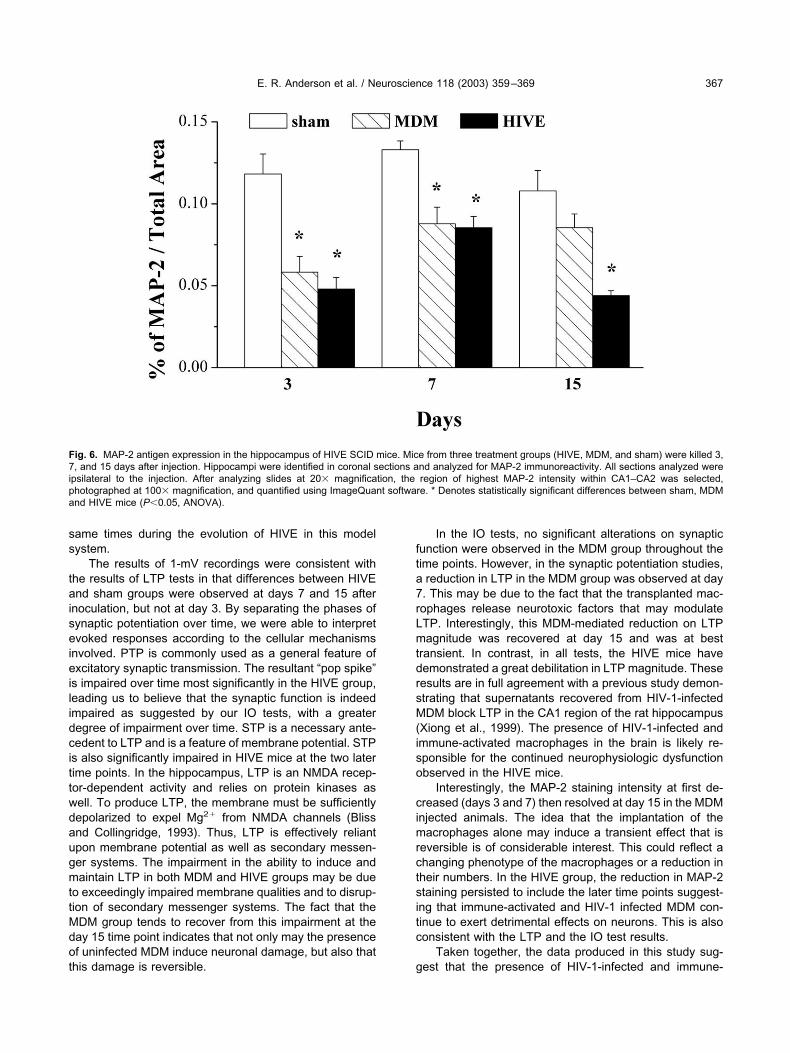
To correlate the electrophysiological changes to mor-phological features of disease, neuronal numbers wereassayed in the hippocampal region of HIVE and controlanimals from 3, 7 and 15 days. MDM or sham injectionsserved as controls. The Schaffer-collateral pathway fromCA2 to CA1 was examined for MAP-2 expression, as anindicator of neuronal morphological integrity, in the hip-pocampi ipsilateral and contralateral to the injection site.Differences between hippocampal areas stained forMAP-2 were apparent at day 3 and persisted on to day 15(Fig. 5). Significantly, reduced MAP-2 was observed in theHIVE animals at days 3 and 7. At day 15, the MDM groupexpressed MAP-2 at similar levels than that observed insham animals, whereas the HIVE group showed signifi-cantly reduced levels of MAP-2 immunoreactivity (Fig. 6).This correlated, in part, with reduced numbers of viablehuman MDM (data not shown).
DISCUSSION
The neuropathogenesis of HIV-1 infection revolves aroundinflammatory immune responses produced by virus-in-fected and/or immune-competent MP (perivascular andparenchymal brain macrophages and microglia). Diseasepersists through the transendothelial migration of mono-cytes into the brain and is mediated in part by glial che-mokines and breaches in the BBB induced by MP neuro-toxins. The increased numbers of immune-competentmacrophages in brain result in significant inflammation anddiminished cognitive function (Glass et al., 1995; Cunning-ham et al., 1997; Orenstein et al., 1997; Cotter et al.,1999a; Stoll and Jander, 1999).
In past studies, we reproduced several features ofHIVE in our SCID mouse model of human disease (Per-sidsky et al., 1996; Persidsky and Gendelman, 1997).Indeed, this animal model recapitulates human HIVE fol-lowing the injection of immune-activated virus-infectedMDM into the basal ganglia, an area severely affected inHIVE (Glass et al., 1993b; Wiley and Achim, 1994). The
Table 1. Synaptic responses generated by stimuli with variable pulsedurations
Potentiation ratio
Pulse duration(�s)
40 60 80 100
Sham 2.05�0.13 1.95�0.11 1.90�0.11 1.81�0.12MDM 2.31�0.12 2.09�0.10 2.00�0.10 1.91�0.09HIVE 1.61�0.10 1.48�0.08 1.41�0.08 1.34�0.08
Columns depict the ratio of the secondary response over the primaryresponse in a paired-pulse stimulation paradigm with varying stimula-tion duration (40, 60, 80, 100 �s). All stimulation increments at eachtime point were found to be statistically significant (P�0.05, analysis ofvariance) in the HIVE as compared to the control (MDM and sham)groups. The responses were significantly lowered in HIVE animals atdays 3, 7, and 15 at each pulse duration. The day 3 time point wasillustrated and is representative of what was observed at days 7 and15. At all time points, the measurements were statistically significantamong the experimental and control groups (HIVE n�14, MDM n�31,sham n�15; P�0.05). MDM, monocyte-derived macrophage; HIVE,human immunodeficiency virus type 1 encephalitis.
Table 2. Synpatic responses evoked by paired pulses with variableIPIs
Potentiation ratio
IPI (ms) 50 100 200 400Sham 1.99�0.13 1.80�0.10 1.53�0.07 1.24�0.04MDM 2.01�0.09 1.77�0.07 1.54�0.07 1.33�0.04HIVE 1.50�0.10 1.42�0.06 1.29�0.05 1.13�0.02
Data illustrate the facilitation ratios of primary and secondary re-sponses recorded with interpulse intervals (IPIs) of 50 ms, 100 ms,200 ms, and 400 ms which were recorded at day 3. Significant differ-ence between HIVE and control groups was found at all IPIs (P�0.05)(these differences remained consistent at each of the time points: days3, 7, and 15; data not shown). Significantly lower ratios were observedin HIVE mice compared with controls (HIVE n�14, MDM n�34, shamn�14; P�0.05, analysis of variance). MDM, monocyte-derived mac-rophage; HIVE, human immunodeficiency virus type 1 encephalitis.
E. R. Anderson et al. / Neuroscience 118 (2003) 359–369 363

region of study, the CA1 area of the hippocampus, islocated approximately 250 �m from the site of injection. Inthe HIVE mouse, microglial activation spreads as far as200–400 �m and astrocytosis as far as 1500 �m from theimplanted human cells (Persidsky et al., 1996). The evi-dence of hippocampal dysfunction supports the contentionthat small numbers of HIV-1-infected MP, strategically po-sitioned within the brain, can cause widespread neuronaldamage. The electrophysiological examination of hip-pocampal physiology shown in this report supports thisnotion and extends our previously published results thatinfected cells can amplify, through paracrine and autocrinemechanisms, pro-inflammatory processes and are capableof producing significant neural damage in the infectedhuman host.
In the presence of ongoing viral infection, glial inflam-matory factors and their resultant synaptic deficits areenhanced. Viral particles, or HIV-1 proteins (gp120, gp41,tat, rev and nef) secreted from infected cells can instigateneuronal death (Kaul and Lipton, 1999; Zheng et al.,1999). In our SCID mouse model of HIVE, alterations inneuronal physiology occur as a consequence of viral rep-lication and inflammatory responses. Impairment in hip-pocampal function was shown by IO function tests, anddeficits in LTP, which may underlie the cognitive dysfunc-tion seen in a patient with AIDS. The importance of virusesin altering neuronal physiology is highlighted by animalbehavior studies demonstrating that HIV-1 gp120 alonecan cause altered cognition in rodents (Hill et al., 1993;Brenneman et al., 1994; Sanchez-Alavez et al., 2000), and
Fig. 3. Impairments in PPF in the CA1 region of HIVE SCID mice. (A) Traces of fEPSPs at varying IPIs recorded at day 7 were taken as arepresentative sample. The IPIs in milliseconds were (i) 50, (ii) 100, (iii) 200, and (iv) 400, respectively. (B) Superimposed traces with different IPIs.Note a decrease in second responses as IPI increases. (C) Line graph plots the magnitude of PPF versus IPIs. The magnitude of PPF is expressedas PPF ratio, which is calculated by dividing the second EPSP of the pair by the first EPSP of the pair. Significant difference between HIVE and controlgroups was found at all IPIs (P�0.05) and these differences remained consistent at the other two time points: days 7 and 15 (data not shown).*Denotes significant differences.
E. R. Anderson et al. / Neuroscience 118 (2003) 359–369364

has been found previously in SCID HIVE mice (Avgero-poulos et al., 1998). Most lentiviral infections in their nat-
ural hosts including Visna-maedi, caprine arthritis enceph-alitis virus, feline immunodeficiency virus and simian im-
Fig. 4. Comparison between LTP induced by a weak and burst tetanus in the CA1 region of hippocampal slices taken from SCID mice inoculated withHIV-1-infected MDM and control manipulations at day 3 (panel A), day 7 (panel B) and days 14–16 (panel C). Arrows in each panel indicate the timewhen the weak stimulus (arrow on the left) and burst stimulus (arrow on the right) were delivered. Note that a significant reduction in LTP magnitudein HIVE mice at days 7 and 15, but not at day 3. No slices used for this study showed needle trauma. Panels D and E illustrate averaged potentiationmagnitudes measured in PTP, STP, and LTP induced by burst tetanus at days 7 and 15, respectively. A significant reduction in PTP, STP and LTPwas observed in SCID mice inoculated with HIV-1-infected MDM. No difference between groups was observed at day 3. Data points representmean�S.E.M. * Denotes significant differences.
E. R. Anderson et al. / Neuroscience 118 (2003) 359–369 365

munodeficiency virus, induce neuropathologic andneurocognitive abnormalities (Desrosiers et al., 1991; Zinket al., 1997; Nathanson, 1998; Steigerwald et al., 1999;
Burudi and Fox, 2001). Our study demonstrated that HIV-1infection of macrophages in brain elicits synaptic dysfunc-tion.
The impairment of synaptic function after HIV-1-infec-tion in the brain has been demonstrated by differencesamong HIVE, MDM and sham animals in a burst-stimula-tion-induced LTP study. In this report, a two-stage step-wise examination of the phases of potentiation was imple-mented in order to determine any differences which maybe attributed to frequency-dependent modulations of syn-aptic activity. In addition to this, we also utilized IO tests toexamine neuronal function in order to detect any alter-ations induced by HIV-1 infection in the brain. These func-tional IO tests included measures of synaptic strength inresponse to different stimulation intensities (variable stim-ulation pulse duration), PPF, and the stimulation strengthrequired to evoke a 1-mV EPSP. Both the stimulation-response recording and the PPF tests showed impairmentof synaptic function after HIV-1 infection of MDM in thebrain as early as 3 days after injection and continued untilday 15. In contrast, the 1-mV IO and the LTP tests failed toproduce any noticeable differences between the HIVE andthe control groups (MDM and sham) at the day 3 timepoint. The possible cause for these apparent differences isnot clear. The results could reflect neuronal physiology atdifferent HIVE stages or may be due to the fact that not allof the physiological processes manifest deficits at the
Table 3. The phases of synaptic potentiation at different time points
Day 7 Basal level (%)
PTP STP LTPSham 261.87�24.18 245.56�24.01 232.63�24.18MDM 231.38�25.67 182.34�10.68 166.0�8.42HIVE 177.75�15.34 143.98�14.71 131.23�15.26
Day 15 Basal level (%)
PTP STP LTPSham 278.85�38.62 256.46�44.60 254.73�55.43MDM 323.04�66.75 262.89�59.13 229.40�51.90HIVE 194.91�13.26 164.67�11.93 169.24�15.47
High-frequency stimulation (HFS) (10 events at 100 Hz�4) inducedsynaptic potentiation in HIVE and control (MDM, sham) animals at day7 (upper) and day 15 (lower). PTP, STP, and LTP were determined at1, 15, and 40 min after HFS. Data are expressed as mean�S.E.M. ofthe initial slope measured in percent of basal activity. The numbers ofanimals examined at each of the time points are shown in the paren-theses and include: day 7 HIVE (6), MDM (7), sham (9), day 15 HIVE(8), MDM (8), and sham (7). PTP, post-tetanic potentiation; STP,short-term potentiation; LTP, long-term potentiation; MDM, monocyte-derived macrophage; HIVE, human immunodeficiency virus type 1encephalitis.
Fig. 5. MAP-2 expression within the hippocampal CA1–CA2 region. Mice from three treatment groups (HIVE, MDM, and sham) were killed 3, 7, and15 days after injection. Panels A–F present sections of SCID mouse brains immunostained with antibodies to MAP-2. Hippocampi were identified incoronal sections and analyzed for MAP-2 immunoreactivity. All sections shown were ipsilateral to the injection. After analyzing slides at 20�magnification, the region of highest MAP-2 intensity within CA1–CA2 was selected, photographed at 100� magnification. At day 3, MAP-2 stainingwas decreased in MDM (B) at day 3 and HIVE (C) as compared with sham (A). Similar differences were seen at day 15, when sham (D) was comparedwith MDM (E) and HIVE (F). Primary antibodies (are detected by Vectastain Elite Kit using DAB as a substrate, and tissue sections werecounterstained with Mayer’s hematoxylin. Original magnification for panels A–F�1000.
E. R. Anderson et al. / Neuroscience 118 (2003) 359–369366
same times during the evolution of HIVE in this modelsystem.
The results of 1-mV recordings were consistent withthe results of LTP tests in that differences between HIVEand sham groups were observed at days 7 and 15 afterinoculation, but not at day 3. By separating the phases ofsynaptic potentiation over time, we were able to interpretevoked responses according to the cellular mechanismsinvolved. PTP is commonly used as a general feature ofexcitatory synaptic transmission. The resultant “pop spike”is impaired over time most significantly in the HIVE group,leading us to believe that the synaptic function is indeedimpaired as suggested by our IO tests, with a greaterdegree of impairment over time. STP is a necessary ante-cedent to LTP and is a feature of membrane potential. STPis also significantly impaired in HIVE mice at the two latertime points. In the hippocampus, LTP is an NMDA recep-tor-dependent activity and relies on protein kinases aswell. To produce LTP, the membrane must be sufficientlydepolarized to expel Mg2� from NMDA channels (Blissand Collingridge, 1993). Thus, LTP is effectively reliantupon membrane potential as well as secondary messen-ger systems. The impairment in the ability to induce andmaintain LTP in both MDM and HIVE groups may be dueto exceedingly impaired membrane qualities and to disrup-tion of secondary messenger systems. The fact that theMDM group tends to recover from this impairment at theday 15 time point indicates that not only may the presenceof uninfected MDM induce neuronal damage, but also thatthis damage is reversible.
In the IO tests, no significant alterations on synapticfunction were observed in the MDM group throughout thetime points. However, in the synaptic potentiation studies,a reduction in LTP in the MDM group was observed at day7. This may be due to the fact that the transplanted mac-rophages release neurotoxic factors that may modulateLTP. Interestingly, this MDM-mediated reduction on LTPmagnitude was recovered at day 15 and was at besttransient. In contrast, in all tests, the HIVE mice havedemonstrated a great debilitation in LTP magnitude. Theseresults are in full agreement with a previous study demon-strating that supernatants recovered from HIV-1-infectedMDM block LTP in the CA1 region of the rat hippocampus(Xiong et al., 1999). The presence of HIV-1-infected andimmune-activated macrophages in the brain is likely re-sponsible for the continued neurophysiologic dysfunctionobserved in the HIVE mice.
Interestingly, the MAP-2 staining intensity at first de-creased (days 3 and 7) then resolved at day 15 in the MDMinjected animals. The idea that the implantation of themacrophages alone may induce a transient effect that isreversible is of considerable interest. This could reflect achanging phenotype of the macrophages or a reduction intheir numbers. In the HIVE group, the reduction in MAP-2staining persisted to include the later time points suggest-ing that immune-activated and HIV-1 infected MDM con-tinue to exert detrimental effects on neurons. This is alsoconsistent with the LTP and the IO test results.
Taken together, the data produced in this study sug-gest that the presence of HIV-1-infected and immune-
Fig. 6. MAP-2 antigen expression in the hippocampus of HIVE SCID mice. Mice from three treatment groups (HIVE, MDM, and sham) were killed 3,7, and 15 days after injection. Hippocampi were identified in coronal sections and analyzed for MAP-2 immunoreactivity. All sections analyzed wereipsilateral to the injection. After analyzing slides at 20� magnification, the region of highest MAP-2 intensity within CA1–CA2 was selected,photographed at 100� magnification, and quantified using ImageQuant software. * Denotes statistically significant differences between sham, MDMand HIVE mice (P�0.05, ANOVA).
E. R. Anderson et al. / Neuroscience 118 (2003) 359–369 367

activated MDM, implanted into the brain, results in impair-ments on synaptic transmission and plasticity. How thisoccurs requires further investigation, although the mecha-nisms likely involve active viral replication, immune com-petence, and release of neurotoxic substances, which col-lectively affect neuronal and synaptic function. One poten-tial mechanism underlying the impairments on synapticfunction may be due to HIV-1, or viral-related neurotoxicity-induced cell death. Several studies have shown that HIV-1proteins and related substances can cause neuronaldeath. However, our preliminary studies using terminaldeoxynucleotidyl transferase-mediated nick end labeling(TUNEL) staining on both HIV and sham mice have re-vealed that the quantity of cell death between groups wassimilar (unpublished observations). While it is possible thatthis may be the reason in part, we are led to believe thatthe HIV-1-induced synaptic function deficiencies may notnecessarily be related to cell death. If it is true that thesedecrements in synaptic responsiveness are due primarilyto a perpetuated effect by some diffusible neurotoxin, thenthe deficit may be ameliorated upon removal of the offend-ing substances or their source.
It is possible that viral or infected macrophage productsimpede the actions of expressed proteins that are neces-sary antecedents to or enhancers of LTP. These secretedneurotoxins may interfere with these proteins and/or theirexpression resulting in impaired synaptic responsiveness.Again, if virus and its inflammatory products are cleared,this damage may be reversed. Future studies involvingproteomic profiling may uncover diminished antecedentproteins following viral infection.
This neuronal and synaptic dysfunction caused by im-plantation of HIV-1-infected MDM into brain may underliethe pathogenesis of HIVE and HAD as seen in AIDSpatients. However, it is important to note that the revers-ibility of impairment seen in the control MDM group sug-gests that it may be possible to positively affect HIV-1mediated neuronal perturbations if virus and its ensuinginflammation are cleared. In other words, the damageinduced by HIV-1-immune activated brain macrophagesmay be reversible if interrupted in early stages of disease.
Acknowledgements—The authors extend a special thanks to MsRobin Taylor for excellent graphic and administrative support, Dr.Larisa Poluektova for her help with immunohistochemical staining,and Ms Laura McCabe for proofreading the manuscript. This workwas supported in part by NIH grants RO1 NS41862-01 to H.X.,P20 RR15635-01 to H.X. and H.E.G., and R01 NSA136127-01 toH.E.G.
REFERENCES
Avgeropoulos N, Kelley B, Middaugh L, Arrigo S, Persidsky Y, Gen-delman HE, Tyor WR (1998) SCID mice with HIV encephalitisdevelop behavioral abnormalities. J Acquir Immune Defic SyndrHum Retrovirol 18:13–20.
Bagetta G, Corasaniti MT, Costa N, Berliocchi L, Finazzi-Agro A,Nistico G (1997) The human immunodeficiency virus type 1 (HIV-1)glycoprotein gp120 reduces the expression of neuronal nitric oxidesynthase in the hippocampus but not in the cerebral cortex andmedial septal nucleus of rat. Neurosci Lett 224:75–78.
Bellinger FP, Madamba S, Siggins GR (1993) Interleukin 1 beta inhib-
its synaptic strength and long-term potentiation in the rat CA1hippocampus. Brain Res 628:227–234.
Bliss TVP, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361:31–39.
Brenneman DE, McCune SK, Mervis RF, Hill JM (1994) gp120 as anetiologic agent for NeuroAIDS: neurotoxicity and model systems.Adv Neuroimmunol 4:157–165.
Burudi EM, Fox HS (2001) Simian immunodeficiency virus model ofHIV-induced central nervous system dysfunction. Adv Virus Res56:435–468.
Carpenter CC, Cooper DA, Fischl MA, Gatell JM, Gazzard BG, Ham-mer SM, Hirsch MS, Jacobsen DM, Katzenstein DA, Montaner JS,Richman DD, Saag MS, Schechter M, Schooley RT, ThompsonMA, Vella S, Yeni PG, Volberding PA (2000) Antiretroviral therapyin adults: updated recommendations of the International AIDS So-ciety-USA Panel. JAMA 283:381–390.
Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, PowerC, Gallo RC, Major EO (1998) Induction of monocyte chemoattrac-tant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation inAIDS dementia. Proc Natl Acad Sci USA 95:3117–3121.
Cotter R, Zheng J, Gendelman HE (1999) The role of mononuclearphagocytes in neurodegenerative disorders: lessons from multiplesclerosis, Alzheimer’s disease and HIV-1 dementia. In: Advances inneurodegenerative disorders, Vol 2 (Marwah J, Teitelbaum H, eds),pp 203–241. Scottsdale, AZ: Prominent Press.
Cotter RL, Burke WJ, Thomas VS, Potter JF, Zheng J, Gendelman HE(1999b) Insights into the neurodegenerative process of Alzheimer’sdisease: a role for mononuclear phagocyte-associated inflamma-tion and neurotoxicity. J Leukoc Biol 65:416–427.
Cunningham AL, Naif H, Saksena N, Lynch G, Chang J, Li S, JozwiakR, Alali M, Wang B, Fear W, Sloane A, Pemberton L, Brew B (1997)HIV infection of macrophages and pathogenesis of AIDS dementiacomplex: interaction of the host cell and viral genotype. J LeukocBiol 62:117–125.
Desbaillets I, Tada M, de Tribolet N, Diserens AC, Hamou MF, VanMeir EG (1994) Human astrocytomas and glioblastomas expressmonocyte chemoattractant protein-1 (MCP-1) in vivo and in vitro.Int J Cancer 58:240–247.
Desrosiers RC, Hansen-Moosa A, Mori K, Bouvier DP, King NW,Daniel MD, Ringler DJ (1991) Macrophage-tropic variants of SIVare associated with specific AIDS-related lesions but are not es-sential for the development of AIDS. Am J Pathol 139:29–35.
Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C (1993) Micro-glia and cytokines in neurological disease, with special reference toAIDS and Alzheimer’s disease. Glia 7:75–83.
Epstein LG, Sharer LR, Goudsmit J (1988) Neurological and neuro-pathological features of human immunodeficiency virus infection inchildren. Ann Neurol 23:S19–23.
Everall IP, Luthert PJ, Lantos PL (1991) Neuronal loss in the frontalcortex in HIV infection. Lancet 337:1119–1121.
Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, PhippsT, Wahl LA, Lane HC, Fauci AS, Burke DS (1988) Efficient isolationand propagation of human immunodeficiency virus on recombinantcolony-stimulating factor 1-treated monocytes. J Exp Med 167:1428–1441.
Giulian D (1999) Microglia and the immune pathology of Alzheimerdisease. Am J Hum Genet 65:13–18.
Glass JD, Fedor H, Wesselingh SL, McArthur JC (1995) Immunocy-tochemical quantitation of human immunodeficiency virus in thebrain: correlations with dementia. Ann Neurol 38:755–762.
Glass JD, Wesselingh SL, Selnes OA, McArthur JC (1993a) Clinicalneuropathologic correlation in HIV-associated dementia. Neurology43:2230–2237.
Hill JM, Mervis RF, Avidor R, Moody TW, Brenneman DE (1993) HIVenvelope protein-induced neuronal damage and retardation of be-havioral development in rat neonates. Brain Res 603:222–233.
Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholo-denko D, Malenka RC, Nicoll RA, Mucke L (1999) Plaque-indepen-
E. R. Anderson et al. / Neuroscience 118 (2003) 359–369368

dent disruption of neural circuits in Alzheimer’s disease mousemodels. Proc Natl Acad Sci USA 96:3228–3233.
Janssen RS, Nwanyanwu OC, Selik RM, Stehr-Green JK (1992) Ep-idemiology of HIV encephalopathy in the United States. Neurology42:1472–1476.
Janssen RS, Saykin AJ, Cannon L, Campbell J, Pinsky PF, HessolNA, O’Malley PM, Lifson AR, Doll LS, Rutherford GW, Kaplan JE(1989) Neurological and neuropsychological manifestations ofHIV-1 infection: association with AIDS-related complex but notasymptomatic HIV-1 infection. Ann Neurol 26:592–600.
Kalter DC, Nakamura JA, Turpin JA, Baca LM, Hoover DL, Dieffen-bach C, Ralph P, Gendelman HE, Meltzer MS (1991) EnhancedHIV replication in macrophage colony-stimulating factor-treatedmonocytes. J Immunol 146:298–306.
Kaul M, Garden GA, Lipton SA (2001) Pathways to neuronal injury andapoptosis in HIV-associated dementia. Nature 410:988–994.
Kaul M, Lipton SA (1999) Chemokines and activated macrophages inHIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci USA96:8212–8216.
Krebs FC, Ross H, McAllister J, Wigdahl B (2000) HIV-1-associatedcentral nervous system dysfunction. Adv Pharmacol 49:315–385.
Krucker T, Toggas S, Mucke L, Siggins S (1998) Transgenic mice withcerebral expression of human immunodeficiency virus type-1 coatprotein gp120 show divergent changes in short- and long-termpotentiation in CA1 hippocampus. Neuroscience 83:691–700.
Larson J, Lynch G, Games D, Seubert P (1999) Alterations in synapticansmission and n-term potentiation in hippocampal slices fromyoung and aged PDAPP mice. Brain Res 840:23–35.
Lipton SA, Gendelman HE (1995) Dementia associated with the ac-quired immunodeficiency syndrome. N Engl J Med 16:934–940.
Masliah E, Achim CL, Ge N, DeTeresa R, Terry RD, Wiley CA (1992)Spectrum of human immunodeficiency virus-associated neocorticaldamage. Ann Neurol 32:321–329.
McArthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT,Graham NM, McArthur JH, Selnes OA, Jacobson LP, Visscher BR,Concha M, Saah A (1993) Dementia in AIDS patients: incidenceand risk factors: Multicenter AIDS Cohort Study. Neurology 43:2245–2252.
McArthur JC, Sacktor N, Selnes O (1999) Human immunodeficiencyvirus-associated dementia. Semin Neurol 19:129–150.
Nathanson N (1998) Towards an AIDS vaccine: the role of primatemodels. Int J STD AIDS 9:3–7.
Navia BA, Jordan BD, Price RW (1986) The AIDS dementia complex:I. Clinical features. Ann Neurol 19:517–524.
Orenstein JM, Fox C, Wahl SM (1997) Macrophage as a source of HIVduring opportunistic infections. Science 276:1857–1861.
Persidsky Y (1999) Model systems for studies of leukocyte migrationacross the blood-brain barrier. J Neurovirol 5:579–590.
Persidsky Y, Gendelman HE (1997) Development of laboratory and
animal model systems for HIV-1 encephalitis and its associateddementia. J Leukoc Biol 62:100–106.
Persidsky Y, Ghorpade A, Rasmussen J, Limoges J, Liu XJ, Stins M,Fiala M, Way D, Kim KS, Witte MH, Weinand M, Carhart L, Gen-delman HE (1999) Microglial and astrocyte chemokines regulatemonocyte migration through the blood-brain barrier in human im-munodeficiency virus-1 encephalitis. Am J Pathol 155:1599–1611.
Persidsky Y, Limoges J, McComb R, Bock P, Baldwin T, Tyor W, PatilA, Nottet HS, Epstein L, Gelbard H, Flanagan E, Reinhard J,Pirruccello SJ, Gendelman HE (1996) Human immunodeficiencyvirus encephalitis in SCID mice. Am J Pathol 149:1027–1053.
Sacktor N, Lyles RH, Skolasky R, Kleeberger C, Selnes OA, Miller EN,Becker JT, Cohen B, McArthur JC (2001) HIV-associated neuro-logic disease incidence changes: Multicenter AIDS Cohort Study,1990-1998. Neurology 56:257–260.
Saitoh T, Kang D, Mallory M, DeTeresa R, Masliah E (1997) Glial cellsin Alzheimer’s disease: preferential effect of APOE risk on scat-tered microglia. Gerontology 43:109–118.
Sanchez-Alavez M, Criado J, Gomez-Chavarin M, Jimenez-AnguianoA, Navarro L, Diaz-Ruiz O, Galicia O, Sanchez-Narvaez F, Murillo-Rodriguez E, Henriksen SJ, Elder JH, Prospero-Garcia O (2000)HIV- and FIV-derived gp120 alter spatial memory, LTP, and sleepin rats. Neurobiol Dis 7:384–394.
Seabrook GR, Easter A, Dawson GR, Bowery BJ (1997) Modulation oflong-term potentiation in CA1 region of mouse hippocampal brainslices by GABA A receptor benzodiazepine site ligands. Neuro-pharmacology 36:823–830.
Steigerwald ES, Sarter M, March P, Podell M (1999) Effects of felineimmunodeficiency virus on cognition and behavioral function incats. J Acquir Immune Defic Syndr Hum Retrovirol 20:411–419.
Stoll G, Jander S (1999) The role of microglia and macrophages in thepathophysiology of the CNS. Prog Neurobiol 58:233–247.
Wiley CA, Achim C (1994) Human immunodeficiency virus encepha-litis is the pathological correlate of dementia in acquired immuno-deficiency syndrome. Ann Neurol 36:673–676.
Xiong H, Baskys A, Wojtowicz JM (1996) Brain-derived peptides in-hibits synaptic transmission via GABAB receptors in CA1 area ofrats. Brain Res 737:188–194.
Xiong H, Zeng YC, Zheng J, Thylin M, Gendelman HE (1999) SolubleHIV-1 infected macrophage secretory products mediate blockadeof long-term potentiation: a mechanism for cognitive dysfunction inHIV-1-associated dementia. J NeuroVirol 5:519–528.
Zheng J, Ghorpade A, Niemann D, Cotter RL, Thylin MR, Epstein L,Swartz JM, Shepard RB, Liu X, Nukuna A, Gendelman HE (1999)Lymphotropic virions affect chemokine receptor-mediated neuralsignaling and apoptosis: implications for human immunodeficiencyvirus type 1-associated dementia. J Virol 73:8256–8267.
Zink MC, Amedee AM, Mankowski JL, Craig L, Didier P, Carter DL,Munoz A, Murphey-Corb M, Clements JE (1997) Pathogenesis ofSIV encephalitis. Am J Pathol 151:793–803.
(Accepted 8 November 2002)
E. R. Anderson et al. / Neuroscience 118 (2003) 359–369 369
Copyright © 2022 FDOKUMEN




















