
Highly parallel genomic assays
13
The elucidation of the role of human biology in health and disease requires a thorough understanding of the relationship between genomic information and the corresponding phenotype. The ability to collect this genomic information — including sequence, genotypic variation, expression levels and epigenetic status — rapidly and inexpensively has long been a bottleneck to realizing this goal. Cancer biology is one example, in which the use of genomic information to stage tumours should aid initial diagnosis and subsequent treatment 1–5 . The development and application of novel, highly paral- lel genomic assay systems have put us at the cusp of a genetic information explosion that will allow medical clinics to use individualized genetic information to make diagnostic, prognostic and therapeutic decisions. The first generation of microarray platforms for highly parallel genomic analysis was developed over 15 years ago 6,7 . They facilitated the development more than 10 years ago of intrinsically parallel assays (more commonly known as gene-expression-profiling assays) to measure mRNA abundance 8,9 . However, intrinsically parallel whole-genome approaches to genotyping, epi- genetic profiling and sequencing have only recently been developed 10,11 , and are enabling scientific studies that were previously not feasible, such as whole-genome linkage disequilibrium (LD) association studies of case–control populations 12,13 . In this Review, we focus on the development of methods and platforms that have enabled highly parallel genomic assays for genotyping, copy-number measure- ments, sequencing and detecting loss of heterzygosity (LOH), allele-specific expression and methylation. We conclude with a discussion of parallel assays for epigenomics and some of the attendant challenges. We do not discuss array-based gene-expression profiling because it is a relatively mature technology with many excellent reviews (for example, for a recent review of gene-expression and tiling arrays see REF. 14). Highly parallel genotyping assays The ability to perform these highly parallel genomic assays depends on two fundamental characteristics of the assay: a highly parallel array-based read-out and an intrinsically scalable, multiplexing sample preparation. Gene-expression profiling was the first genomic assay to be parallelized, using high-density DNA arrays for read- out and a single-tube sample preparation 9,15,16 . Designing and developing a multiplexed sample preparation for genotyping has been more challenging, mainly because the need to assay a locus at single-base resolution in the context of the entire human genome introduces specific- ity problems and because of the need to detect analytes at low concentrations. A powerful way of obtaining genomic specificity is the detection of physically coincident events on genomic targets. PCR is a good example — ampli- fication of a specific locus requires coincident anneal- ing and extension of two primers at the desired locus. Other examples include ligase chain reaction (LCR) and padlock-probe amplification, both of which use an enzy- matic approach 17–22 . Despite the potential of the other assay formats, PCR has been the workhorse of genomic sample preparation because of its ability to efficiently amplify a specific locus within the context of the entire genome. Unfortunately, the ability of PCR to multiplex in array-based applications has been limited 23 . Multiplex PCR. Multiplex PCR reactions have been plagued by primer dimer (PD) formation and unequal amplification rates that depend on amplicon length, sequence and priming efficiency 24,25 . The PD effect is exacerbated by simultaneous amplification of many loci because the number of potential primer–primer *Illumina Inc., 9885 Towne Centre Drive, San Diego, California 92121, USA. ‡ Prognosys Biosciences Inc., 505 Coast Boulevard, La Jolla, California 92037, USA. Correspondence to K.L.G. e-mail: [email protected] doi:10.1038/nrg1901 Cancer staging Classification of cancer types into groups that reflect their localization, metastasis, prognosis, recommended treatment regimen and predicted clinical outcome. Linkage disequilibrium The property of two polymorphic loci in a population such that the polymorphic states at the two loci are not independent of one another, and as a result the state of the polymorphism at one locus has a higher probability of being associated with a particular state at the second locus. This association is usually measured with a metric called r 2 that ranges between zero (no linkage) and one (complete linkage). Highly parallel genomic assays Jian-Bing Fan*, Mark S. Chee ‡ and Kevin L. Gunderson* Abstract | Recent developments in highly parallel genome-wide assays are transforming the study of human health and disease. High-resolution whole-genome association studies of complex diseases are finally being undertaken after much hypothesizing about their merit for finding disease loci. The availability of inexpensive high-density SNP-genotyping arrays has made this feasible. Cancer biology will also be transformed by high-resolution genomic and epigenomic analysis. In the future, most cancers might be staged by high-resolution molecular profiling rather than by gross cytological analysis. Here, we describe the key developments that enable highly parallel genomic assays. REVIEWS 632 | AUGUST 2006 | VOLUME 7 www.nature.com/reviews/genetics © 2006 Nature Publishing Group
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Highly parallel genomic assays

The elucidation of the role of human biology in health and disease requires a thorough understanding of the relationship between genomic information and the corresponding phenotype. The ability to collect this genomic information — including sequence, genotypic variation, expression levels and epigenetic status — rapidly and inexpensively has long been a bottleneck to realizing this goal. Cancer biology is one example, in which the use of genomic information to stage tumours should aid initial diagnosis and subsequent treatment1–5. The development and application of novel, highly paral-lel genomic assay systems have put us at the cusp of a genetic information explosion that will allow medical clinics to use individualized genetic information to make diagnostic, prognostic and therapeutic decisions.
The first generation of microarray platforms for highly parallel genomic analysis was developed over 15 years ago6,7. They facilitated the development more than 10 years ago of intrinsically parallel assays (more commonly known as gene-expression-profiling assays) to measure mRNA abundance8,9. However, intrinsically parallel whole-genome approaches to genotyping, epi-genetic profiling and sequencing have only recently been developed10,11, and are enabling scientific studies that were previously not feasible, such as whole-genome linkage disequilibrium (LD) association studies of case–control populations12,13.
In this Review, we focus on the development of methods and platforms that have enabled highly parallel genomic assays for genotyping, copy-number measure-ments, sequencing and detecting loss of heterzygosity (LOH), allele-specific expression and methylation. We conclude with a discussion of parallel assays for epigenomics and some of the attendant challenges. We do not discuss array-based gene-expression profiling because it is a relatively mature technology with many
excellent reviews (for example, for a recent review of gene-expression and tiling arrays see REF. 14).
Highly parallel genotyping assaysThe ability to perform these highly parallel genomic assays depends on two fundamental characteristics of the assay: a highly parallel array-based read-out and an intrinsically scalable, multiplexing sample preparation. Gene-expression profiling was the first genomic assay to be parallelized, using high-density DNA arrays for read-out and a single-tube sample preparation9,15,16. Designing and developing a multiplexed sample preparation for genotyping has been more challenging, mainly because the need to assay a locus at single-base resolution in the context of the entire human genome introduces specific-ity problems and because of the need to detect analytes at low concentrations. A powerful way of obtaining genomic specificity is the detection of physically coincident events on genomic targets. PCR is a good example — ampli-fication of a specific locus requires coincident anneal-ing and extension of two primers at the desired locus. Other examples include ligase chain reaction (LCR) and padlock-probe amplification, both of which use an enzy-matic approach17–22. Despite the potential of the other assay formats, PCR has been the workhorse of genomic sample preparation because of its ability to efficiently amplify a specific locus within the context of the entire genome. Unfortunately, the ability of PCR to multiplex in array-based applications has been limited23.
Multiplex PCR. Multiplex PCR reactions have been plagued by primer dimer (PD) formation and unequal amplification rates that depend on amplicon length, sequence and priming efficiency24,25. The PD effect is exacerbated by simultaneous amplification of many loci because the number of potential primer–primer
*Illumina Inc., 9885 Towne Centre Drive, San Diego, California 92121, USA.‡Prognosys Biosciences Inc., 505 Coast Boulevard, La Jolla, California 92037, USA.Correspondence to K.L.G. e-mail: [email protected]:10.1038/nrg1901
Cancer stagingClassification of cancer types into groups that reflect their localization, metastasis, prognosis, recommended treatment regimen and predicted clinical outcome.
Linkage disequilibrium The property of two polymorphic loci in a population such that the polymorphic states at the two loci are not independent of one another, and as a result the state of the polymorphism at one locus has a higher probability of being associated with a particular state at the second locus. This association is usually measured with a metric called r2 that ranges between zero (no linkage) and one (complete linkage).
Highly parallel genomic assaysJian-Bing Fan*, Mark S. Chee‡ and Kevin L. Gunderson*
Abstract | Recent developments in highly parallel genome-wide assays are transforming the study of human health and disease. High-resolution whole-genome association studies of complex diseases are finally being undertaken after much hypothesizing about their merit for finding disease loci. The availability of inexpensive high-density SNP-genotyping arrays has made this feasible. Cancer biology will also be transformed by high-resolution genomic and epigenomic analysis. In the future, most cancers might be staged by high-resolution molecular profiling rather than by gross cytological analysis. Here, we describe the key developments that enable highly parallel genomic assays.
R E V I E W S
632 | AUGUST 2006 | VOLUME 7 www.nature.com/reviews/genetics
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group
Cycle 2
Cycle 1
and
and
and
Genomic priming
Cycle 3Raise annealing temperature, prime and amplify using tag
Ligase chain reactionA cyclic amplification method for amplifying a target sequence that is similar in approach to PCR except that repeated rounds of thermally controlled denaturation, annealing and ligation of a pair of adjacent oligonucleotides are carried out.
Padlock-probe amplificationA ligation-mediated bimolecular assay for a target sequence in which the two query oligonuceotides (5′ and 3′ sequences) are derived from the two ends of a contiguous oligonucleotide. Ligation of the two ends creates a circular structure that is intertwined with the target sequence.
Primer dimer A parasitic product that is formed during PCR reactions and is caused by multiple primers interacting and extending upon themselves. Appropriate design of primer sequences can reduce this effect.
Universal PCRA multiplex PCR reaction using a single or pair of universal primer sequences to amplify a broad range of target sequences that all contain common invariant 5′ and 3′ tail sequences
interactions increases roughly with the square of the number of loci26. Several approaches have been introduced to improve PCR multiplexing beyond the traditional limit of several dozen loci per reaction.
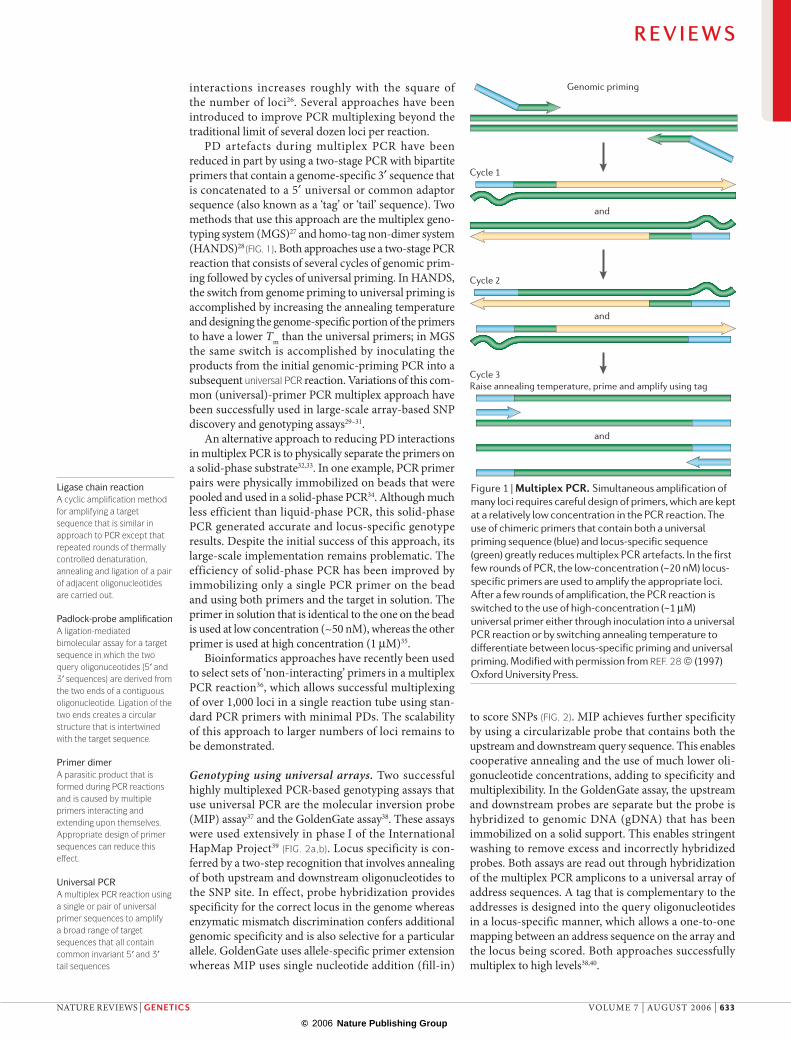
PD artefacts during multiplex PCR have been reduced in part by using a two-stage PCR with bipartite primers that contain a genome-specific 3′ sequence that is concatenated to a 5′ universal or common adaptor sequence (also known as a ‘tag’ or ‘tail’ sequence). Two methods that use this approach are the multiplex geno-typing system (MGS)27 and homo-tag non-dimer system (HANDS)28 (FIG. 1). Both approaches use a two-stage PCR reaction that consists of several cycles of genomic prim-ing followed by cycles of universal priming. In HANDS, the switch from genome priming to universal priming is accomplished by increasing the annealing temperature and designing the genome-specific portion of the primers to have a lower Tm than the universal primers; in MGS the same switch is accomplished by inoculating the products from the initial genomic-priming PCR into a subsequent universal PCR reaction. Variations of this com-mon (universal)-primer PCR multiplex approach have been successfully used in large-scale array-based SNP discovery and genotyping assays29–31.
An alternative approach to reducing PD interactions in multiplex PCR is to physically separate the primers on a solid-phase substrate32,33. In one example, PCR primer pairs were physically immobilized on beads that were pooled and used in a solid-phase PCR34. Although much less efficient than liquid-phase PCR, this solid-phase PCR generated accurate and locus-specific genotype results. Despite the initial success of this approach, its large-scale implementation remains problematic. The efficiency of solid-phase PCR has been improved by immobilizing only a single PCR primer on the bead and using both primers and the target in solution. The primer in solution that is identical to the one on the bead is used at low concentration (~50 nM), whereas the other primer is used at high concentration (1 µM)35.
Bioinformatics approaches have recently been used to select sets of ‘non-interacting’ primers in a multiplex PCR reaction36, which allows successful multiplexing of over 1,000 loci in a single reaction tube using stan-dard PCR primers with minimal PDs. The scalability of this approach to larger numbers of loci remains to be demonstrated.
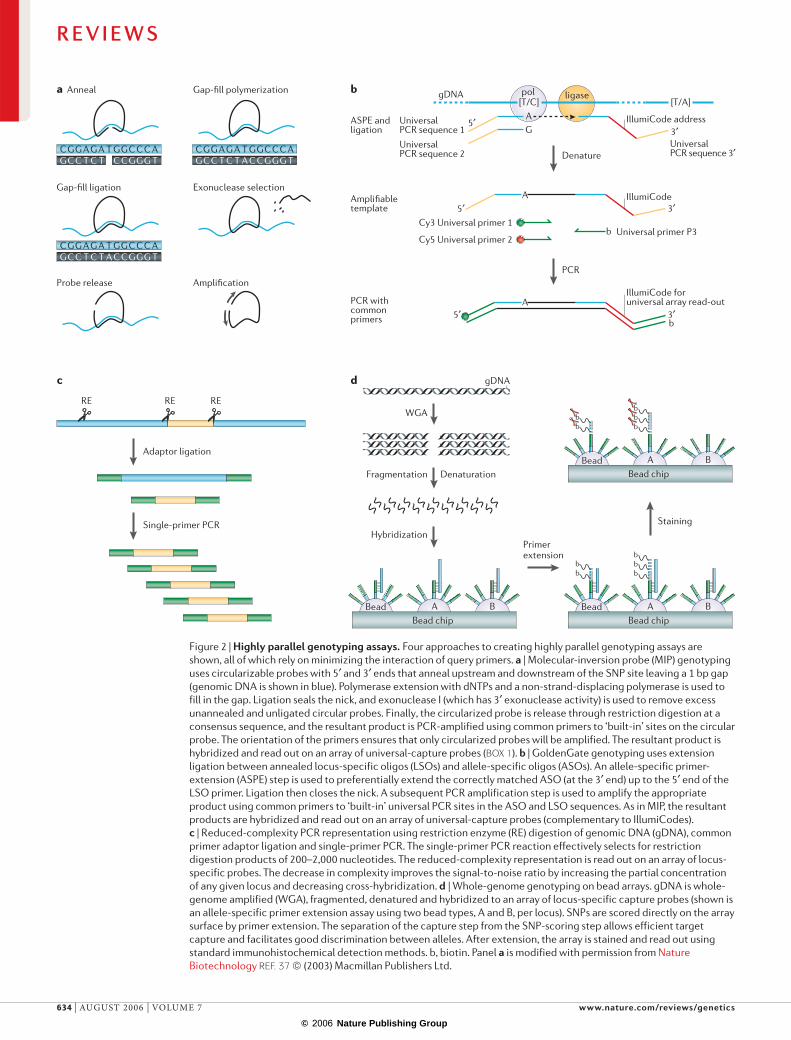
Genotyping using universal arrays. Two successful highly multiplexed PCR-based genotyping assays that use universal PCR are the molecular inversion probe (MIP) assay37 and the GoldenGate assay38. These assays were used extensively in phase I of the International HapMap Project39 (FIG. 2a,b). Locus specificity is con-ferred by a two-step recognition that involves annealing of both upstream and downstream oligonucleotides to the SNP site. In effect, probe hybridization provides specificity for the correct locus in the genome whereas enzymatic mismatch discrimination confers additional genomic specificity and is also selective for a particular allele. GoldenGate uses allele-specific primer extension whereas MIP uses single nucleotide addition (fill-in)
to score SNPs (FIG. 2). MIP achieves further specificity by using a circularizable probe that contains both the upstream and downstream query sequence. This enables cooperative annealing and the use of much lower oli-gonucleotide concentrations, adding to specificity and multiplexibility. In the GoldenGate assay, the upstream and downstream probes are separate but the probe is hybridized to genomic DNA (gDNA) that has been immobilized on a solid support. This enables stringent washing to remove excess and incorrectly hybridized probes. Both assays are read out through hybridization of the multiplex PCR amplicons to a universal array of address sequences. A tag that is complementary to the addresses is designed into the query oligonucleotides in a locus-specific manner, which allows a one-to-one mapping between an address sequence on the array and the locus being scored. Both approaches successfully multiplex to high levels38,40.
Figure 1 | Multiplex PCR. Simultaneous amplification of many loci requires careful design of primers, which are kept at a relatively low concentration in the PCR reaction. The use of chimeric primers that contain both a universal priming sequence (blue) and locus-specific sequence (green) greatly reduces multiplex PCR artefacts. In the first few rounds of PCR, the low-concentration (~20 nM) locus-specific primers are used to amplify the appropriate loci. After a few rounds of amplification, the PCR reaction is switched to the use of high-concentration (~1 µM) universal primer either through inoculation into a universal PCR reaction or by switching annealing temperature to differentiate between locus-specific priming and universal priming. Modified with permission from REF. 28 © (1997) Oxford University Press.
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 7 | AUGUST 2006 | 633
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group
Bead ABead chip
B Bead ABead chip
B
bb
bbb
Bead A B
bb
b
b
bb
Primerextension
Staining
DenaturationFragmentation
WGA
gDNA
Bead chip
CGG A G A T GGC C C AGC C T C T C CGGG T
CGG A G A T GGC C C AGC C T C T A C CGGG T
CGG A G A T GGC C C AGC C T C T A C CGGG T
Probe release
Gap-fill ligation
a Anneal
Amplification
Exonuclease selection
Gap-fill polymerization
Adaptor ligation
RE RE RE
c
gDNA
Cy3 Universal primer 1
Cy5 Universal primer 2Universal primer P3
A
A
GA
3′
5′ 3′
5′
3′5′
b
[T/A]pol ligase
[T/C]
UniversalPCR sequence 2
UniversalPCR sequence 3′
IllumiCode address
Denature
PCR
d
PCR withcommon primers
b
UniversalPCR sequence 1
ASPE andligation
IllumiCode
IllumiCode for universal array read-out
Amplifiabletemplate
HybridizationSingle-primer PCR
Figure 2 | Highly parallel genotyping assays. Four approaches to creating highly parallel genotyping assays are shown, all of which rely on minimizing the interaction of query primers. a | Molecular-inversion probe (MIP) genotyping uses circularizable probes with 5′ and 3′ ends that anneal upstream and downstream of the SNP site leaving a 1 bp gap (genomic DNA is shown in blue). Polymerase extension with dNTPs and a non-strand-displacing polymerase is used to fill in the gap. Ligation seals the nick, and exonuclease I (which has 3′ exonuclease activity) is used to remove excess unannealed and unligated circular probes. Finally, the circularized probe is release through restriction digestion at a consensus sequence, and the resultant product is PCR-amplified using common primers to ‘built-in’ sites on the circular probe. The orientation of the primers ensures that only circularized probes will be amplified. The resultant product is hybridized and read out on an array of universal-capture probes (BOX 1). b | GoldenGate genotyping uses extension ligation between annealed locus-specific oligos (LSOs) and allele-specific oligos (ASOs). An allele-specific primer-extension (ASPE) step is used to preferentially extend the correctly matched ASO (at the 3′ end) up to the 5′ end of the LSO primer. Ligation then closes the nick. A subsequent PCR amplification step is used to amplify the appropriate product using common primers to ‘built-in’ universal PCR sites in the ASO and LSO sequences. As in MIP, the resultant products are hybridized and read out on an array of universal-capture probes (complementary to IllumiCodes). c | Reduced-complexity PCR representation using restriction enzyme (RE) digestion of genomic DNA (gDNA), common primer adaptor ligation and single-primer PCR. The single-primer PCR reaction effectively selects for restriction digestion products of 200–2,000 nucleotides. The reduced-complexity representation is read out on an array of locus-specific probes. The decrease in complexity improves the signal-to-noise ratio by increasing the partial concentration of any given locus and decreasing cross-hybridization. d | Whole-genome genotyping on bead arrays. gDNA is whole-genome amplified (WGA), fragmented, denatured and hybridized to an array of locus-specific capture probes (shown is an allele-specific primer extension assay using two bead types, A and B, per locus). SNPs are scored directly on the array surface by primer extension. The separation of the capture step from the SNP-scoring step allows efficient target capture and facilitates good discrimination between alleles. After extension, the array is stained and read out using standard immunohistochemical detection methods. b, biotin. Panel a is modified with permission from Nature Biotechnology REF. 37 © (2003) Macmillan Publishers Ltd.
R E V I E W S
634 | AUGUST 2006 | VOLUME 7 www.nature.com/reviews/genetics
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group

Whole-genome representationA representation with a sequence complexity that is similar to that of the entire genome from which it was derived.
Reduced-complexity genomic representationA representation with a sequence complexity that is a fraction of the original sample nucleic-acid complexity. In its simplest version, PCR of adaptor-ligated or restriction-enzyme-digested genomic DNA intrinsically generates a reduced-complexity representation.
DNA-array featureAn individual resolvable element of a DNA array that contains a defined sequence. This element can be created in several ways such as spotting, in situ synthesis or deposition of beads that harbour immobilized DNA sequences.
Tag SNP and tagging SNPA tag SNP is defined as a SNP that proxies for a set of SNPs in linkage disequilibrium with itself (that is, they are in the same linkage disequilibrium bin). A haplotype tagging SNP, by contrast, is based on the haplotype block concept, in which a set of tagging SNPs are used to uniquely define the variation of all SNPs that reside in the haplotype block.
In spite of the advantages of highly multiplexed SNP genotyping assays, data from over 2.4 million SNPs from phase II of the International HapMap Project were col-lected using a relatively low-multiplex long-range PCR that consisted of over 300,000 individual PCR reactions per sample41,42. About 8–9 amplicons were multiplexed per reaction, but the extended amplicon size covered, on average, about 8 SNPs for a total of ~64–72 SNPs per reaction. Amplified products were pooled in sets of ~6,250 amplicons and hybridized to a collective set of 49 SNP tiling arrays to generate the genotypes. About 92% of the genome was amplified and hybridized to the array using this approach. Although an impressive effort, this approach does not seem cost effective for large-scale genotyping projects.
Genotyping with genomic representations. Whole-genome representations are created by universal adaptor PCR of adaptor-ligated, restriction-enzyme-digested gDNA, a process that results in a reproducible fraction of the genome being amplified. The approach was originally described by Kinzler and Vogelstein in 1989 for the selec-tion of nucleic-acid sequences that are bound to regula-tory proteins43 (FIG. 2c). The fraction of the genome that is amplified depends on the restriction enzymes that are used and the PCR conditions. PCR-based, reduced-complexity genomic representations have been used for genomic pro-filing of tumour samples44–46 and cataloguing of copy-number variation in normal individuals47. Lucito et al. created both low-complexity and high-complexity rep-resentations using PCR of adaptor-ligated gDNA that had been created by digestion with restriction enzymes of 4–6-base recognition motifs. PCR amplification using universal primers provides an inherent size selection because representation inserts larger than ~1 kb are poorly amplified.
The reduced-complexity representation approach has also been successfully used in genome-wide SNP genotyping assays. Kennedy et al. and Matsuzaki et al. developed whole-genome sampling analysis (WGSA) in which gDNA is digested with XbaI, ligated to an adaptor, amplified by PCR and hybridized to DNA genotyping arrays48,49 (FIG. 2c). The effective complex-ity (the amount of sequence from the human genome) of this representation was ~60 Mb, and provided suf-ficient signal to noise when hybridized to 25-mer oligonucleotide probe arrays to call genotype accu-rately. With an additional restriction enzyme and enhanced PCR conditions, WGSA has been scaled up to genotype several hundred thousand SNPs in repre-sentations of ~300 Mb complexity on a single array50. This complexity can potentially be further increased by using more sets of restriction enzymes and/or increas-ing the average duration of PCR. A drawback of this approach is that the ability to select SNPs is limited because only a portion of the genome is represented. Moreover, that portion is to a large extent randomly selected. Nonetheless, high-density arrays of random SNPs have been used successfully to identify loci that harbour disease-predisposing genetic variants through genome-wide association scans51,52.
Whole-genome genotyping. A more global approach to genotyping can be accomplished if gDNA can be directly hybridized to an array of locus-specific capture probes and scored on the array using enzymatic allelic discrimi-nation, such as primer extension or ligation. This direct approach should allow access to most SNPs in the genome and eliminate the multiplexing bottleneck in sample preparation, making assay scalability solely dependent on array-feature density. Obtaining single-base resolution in the context of the sequence complexity and low molar concentration that is inherent in gDNA requires an assay design with high specificity and sensitivity.
In whole-genome genotyping (WGG)53,54, specificity and sensitivity were achieved in a direct hybridization assay by using a combination of design elements (FIG. 2d). Sensitivity was greatly enhanced by first amplifying the gDNA in a whole-genome amplification (WGA) array55,56 reaction to effectively increase the molar concentration of genomic loci. Specificity was achieved using the com-bination of a stringent 50-mer hybridization capture step followed by an ‘on-array’ polymerase primer-extension step. This combination of elements conferred both locus and allelic specificity on the assay. Finally, an array-based signal-amplification protocol further increased assay sensitivity. Two advantages of WGG are minimal constraints on SNP selection, which allows selection of maximally informative SNPs such as HapMap tag SNPs, and effectively unlimited multiplexing from a single sample preparation. The WGG assay has been used to develop several high-density SNP-genotyping arrays (BOX 1), including two different tag-SNP arrays that allow genotyping of over 317,000 and 550,000 tag SNPs on a single slide57.
High-resolution SNP-CGH. Array technology can also be used to characterize copy-number aberrations. Comparative genomic hybridization (CGH) to DNA arrays is a powerful approach for detecting chromosomal aberrations. In CGH, differentially labelled gDNAs (one reference and one subject sample) are co-hybridized to an array of probes. Initially, CGH used metaphase chro-mosome spreads as the chromosomal yardstick, which limited the resolution to 10–20 Mb58. More recently array CGH was developed using DNA arrays of cDNA or BAC clones, which brought the resolution down to 100 kb59–61. High-density oligonucleotide arrays, some with as many as 385,000 probes per slide, have also been used for CGH62–64. Oligonucleotide probes (~25–85 nucleotides) are much shorter than BAC probes (~100kb) and therefore their hybridization can be variable and sequence-dependent65. However, oligonucleotide arrays are easier to manufacture and the short length of oligo-nucleotides provides better spatial resolution and locus discrimination. Furthermore, any drawbacks with sen-sitivity and precision of oligonucleotide probes can be minimized by averaging the signal from several probes.
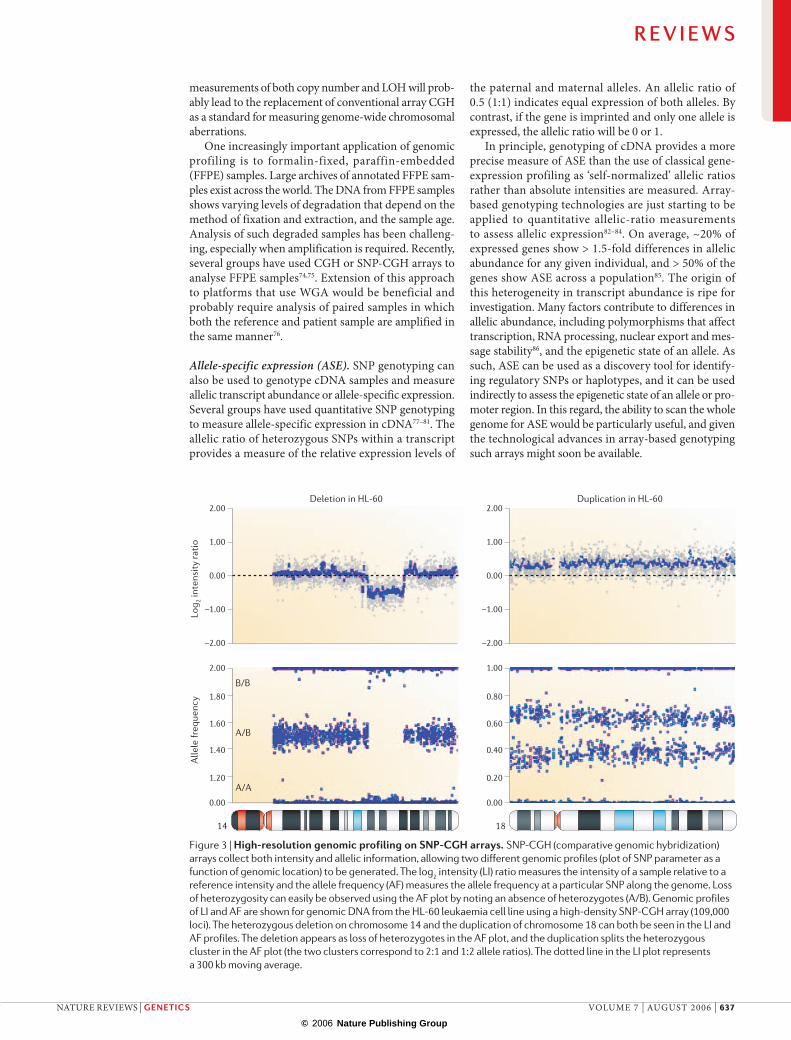
Oligonucleotide-based SNP-genotyping arrays have recently been co-opted for CGH applications to measure both physical copy-number aberrations and genetic aberrations such as LOH (FIG. 3); we refer to this application as SNP-CGH66–70. The ability of SNP-CGH
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 7 | AUGUST 2006 | 635
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group

A T G AA
CUp to70 nt
Repeat G G
GG
G
C C
a Ordered arrays
GC GCG G
Chimeric assay product
gDNA or cDNA
Universal-capture probe15–25 nt
Locus-specific probe26–70 nt
b Random arrayc dUniparental disomy
This rare genetic condition can arise constitutionally through non-disjunction during meiosis that ultimately leads to a duplication of a segment or of the entire maternal or paternal chromosome in the affected individual. A form of apparent uniparental disomy can arise in the course of normal cell division (mitosis) through mitotic recombination (a rare crossover event during mitosis).
arrays, unlike conventional CGH, to detect copy-neutral genetic anomalies such as uniparental disomy (UPD) and mitotic recombination is important in understanding the aetiology of cancer (for example, see REFS 71,72). The second advantage of SNP-CGH arrays is their ability to collect allelic information on deletions, duplications and amplifications. One recent paper that used SNP-CGH describes how most observed amplifications in lung cancer arise as a result of monoallelic amplification73. It will be informative to see whether particular haplotypes are associated with increased incidence of monoallelic amplifications, LOH or deletions.
A third advantage of SNP-CGH arrays is their ease of manufacture and their intrinsic ability to scale to higher feature densities with improvements in array manu-facture. This increase in feature density is important for oligonucleotide arrays because, as desribed above, oligonucleotide probes generally have intrinsically higher noise, which necessitates averaging across 5–10 probes. The current SNP-CGH array densities of more than 500,000 SNPs per slide allow an effective resolu-tion of less than 50 kb. In the future, arrays with higher density will further improve this resolution. In summary, the ability of SNP-CGH arrays to make high-resolution
Box 1 | Primer-on-DNA array technologies
The two basic types of array that are used in genomic analysis are ordered arrays and random arrays. Ordered arrays are created by spotting or synthesizing known feature elements in a defined pattern on a planar surface. There are several methods for creating such ordered arrays including deposition of oligonucleotides with pins or an ink jet printer (see figure, part a, left). Alternatively, in situ oligonucleotide synthesis can be used to generate arrays of defined features by local delivery of oligonucleotide synthesis reagents or by local deprotection chemistry using photolithography or electrochemical-based deprotection (see figure, part a, right). Random arrays are created by self-assembly of bead-based feature elements. In this approach, oligonucleotides are individually immobilized on beads, pooled and assembled onto a patterned planar substrate (see figure, part b). The identities of the assembled beads are subsequently determined by a hybridization-based stepwise decoding scheme156 that uses sets of combinatorially labelled complements to the bead sequences. Both random and ordered arrays can be either universal or locus-specific probes. A universal-capture probe (see figure, part c) binds to its complementary (address) sequence that is present in the products of the genomic assay. This address sequence creates a one-to-one correspondence between a locus and a particular feature on the array. A locus-specific probe (see figure, part d) is used in direct hybridization assays such as gene-expression assays — cDNA — or whole-genome genotyping — genomic DNA (gDNA).
R E V I E W S
636 | AUGUST 2006 | VOLUME 7 www.nature.com/reviews/genetics
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group
–2.00
–1.00
0.00
1.00
2.00
–2.00
–1.00
0.00
1.00
2.00
0.00
1.20
1.60
1.40
1.80
2.00
0.00
0.20
0.60
0.40
0.80
1.00
A/A
14 18
A/B
B/B
Log 2 i
nten
sity
rati
oA
llele
freq
uenc
y
Deletion in HL-60 Duplication in HL-60
measurements of both copy number and LOH will prob-ably lead to the replacement of conventional array CGH as a standard for measuring genome-wide chromosomal aberrations.
One increasingly important application of genomic profiling is to formalin-fixed, paraffin-embedded (FFPE) samples. Large archives of annotated FFPE sam-ples exist across the world. The DNA from FFPE samples shows varying levels of degradation that depend on the method of fixation and extraction, and the sample age. Analysis of such degraded samples has been challeng-ing, especially when amplification is required. Recently, several groups have used CGH or SNP-CGH arrays to analyse FFPE samples74,75. Extension of this approach to platforms that use WGA would be beneficial and probably require analysis of paired samples in which both the reference and patient sample are amplified in the same manner76.
Allele-specific expression (ASE). SNP genotyping can also be used to genotype cDNA samples and measure allelic transcript abundance or allele-specific expression. Several groups have used quantitative SNP genotyping to measure allele-specific expression in cDNA77–81. The allelic ratio of heterozygous SNPs within a transcript provides a measure of the relative expression levels of
the paternal and maternal alleles. An allelic ratio of 0.5 (1:1) indicates equal expression of both alleles. By contrast, if the gene is imprinted and only one allele is expressed, the allelic ratio will be 0 or 1.
In principle, genotyping of cDNA provides a more precise measure of ASE than the use of classical gene-expression profiling as ‘self-normalized’ allelic ratios rather than absolute intensities are measured. Array-based genotyping technologies are just starting to be applied to quantitative allelic-ratio measurements to assess allelic expression82–84. On average, ~20% of expressed genes show > 1.5-fold differences in allelic abundance for any given individual, and > 50% of the genes show ASE across a population85. The origin of this heterogeneity in transcript abundance is ripe for investigation. Many factors contribute to differences in allelic abundance, including polymorphisms that affect transcription, RNA processing, nuclear export and mes-sage stability86, and the epigenetic state of an allele. As such, ASE can be used as a discovery tool for identify-ing regulatory SNPs or haplotypes, and it can be used indirectly to assess the epigenetic state of an allele or pro-moter region. In this regard, the ability to scan the whole genome for ASE would be particularly useful, and given the technological advances in array-based genotyping such arrays might soon be available.
Figure 3 | High-resolution genomic profiling on SNP-CGH arrays. SNP-CGH (comparative genomic hybridization) arrays collect both intensity and allelic information, allowing two different genomic profiles (plot of SNP parameter as a function of genomic location) to be generated. The log2 intensity (LI) ratio measures the intensity of a sample relative to a reference intensity and the allele frequency (AF) measures the allele frequency at a particular SNP along the genome. Loss of heterozygosity can easily be observed using the AF plot by noting an absence of heterozygotes (A/B). Genomic profiles of LI and AF are shown for genomic DNA from the HL-60 leukaemia cell line using a high-density SNP-CGH array (109,000 loci). The heterozygous deletion on chromosome 14 and the duplication of chromosome 18 can both be seen in the LI and AF profiles. The deletion appears as loss of heterozygotes in the AF plot, and the duplication splits the heterozygous cluster in the AF plot (the two clusters correspond to 2:1 and 1:2 allele ratios). The dotted line in the LI plot represents a 300 kb moving average.
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 7 | AUGUST 2006 | 637
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group

PolonyContraction of ‘polymerase colony’ that is created by growing DNA colonies from single DNA ‘seed’ molecules through the use of a PCR reaction on DNA molecules that are diffusely imbedded in a polymer matrix that contains DNA polymerase, primers and appropriate reagents.
BEAMingA process of cloning on beads in which a library of clones is grown on beads through the use of compartmentalized emulsion PCR. DNA and beads are diluted such that, on average, only a single bead and a single target molecule co-occupy a single compartment. PCR amplification grows a clonal population of molecules on the bead starting from the single target sequence.
Massively parallel signature sequencingThis enables digital transcript counting in a cDNA sample. It is accomplished by cloning a 17–20 base signature sequence tag onto micro-beads that are subsequently fixed in a single layer array in a flow cell. The sequence on the bead is then read out using a ligation-based cycle-sequencing assay.
Highly parallel sequencingThe development of high-throughput automated capil-lary sequencers enabled rapid sequencing of the human genome87. Resequencing arrays have contributed to the rapid collection of genetic information29,42,88,89. Nevertheless, the task of cataloguing human genetic variation and correlating it with susceptibility to disease is daunting and expensive. Today, a single mammalian genome can be analysed by random shotgun sequenc-ing to tenfold coverage at a cost of ~US$15M–20M. The short-term goal is to resequence the human genome at a cost 3–4 orders of magnitude less, or ~US$100,000; the ultimate goal is to reduce this cost to ~US$1,000. Moving beyond the first goal will require several tech-nical advances. Fortunately, the same basic principles of readout parallelization and sample multiplexing that proved so powerful for gene-expression and SNP genotyping analysis are also being successfully applied to large-scale sequencing.
Parallelization of the sample preparation is of para-mount importance in reducing costs. One bottleneck in sequencing is preparing the DNA samples from the bacte-rial colonies that harbour the DNA library. In the Human Genome Project, each clone was individually picked and grown-up, and the DNA was extracted or amplified out of the clone. The read-out of bases by capillary sequenc-ing is relatively slow compared with the potential of an array-based read-out (for recent reviews of array-based read-outs, miniaturized capillary sequencing, nanopore sequencing and other approaches see REFS 11,90).
One of the most promising approaches to sample preparation is the generation of polonies or clonal beads. Polonies are discrete clonal amplifications of a single DNA molecule, grown on a solid-phase surface91,92. This approach greatly improves the signal-to-noise ratio. Polonies can be generated using several techniques that include solid-phase PCR in polyacrylamide gels92, bridge PCR33,93, rolling-circle amplification94, BEAMing (beads, emulsions, amplification and magnetics)-based cloning on beads95 and massively parallel signature sequencing (MPSS) to generate clonal bead arrays96,97.
The first step in sample preparation is to create a library of amplifiable sequences from the original target DNA (for example, gDNA or cDNA). This can be done in a locus-specific, representative or random-shotgun fashion. In the targeted approach, the library can be made from pools of amplicons, and in the representative approach from gDNA or cDNA signature tags (that is, serial analysis of gene expression (SAGE) tags)98,99. In the shotgun approach, gDNA is randomly sheared by sonication, blunt-ended and ligated to a set of universal adaptors. These adaptor sequences are used to prime a bead-based emulsion PCR amplification reaction, and later serve as primers in cycle sequencing.
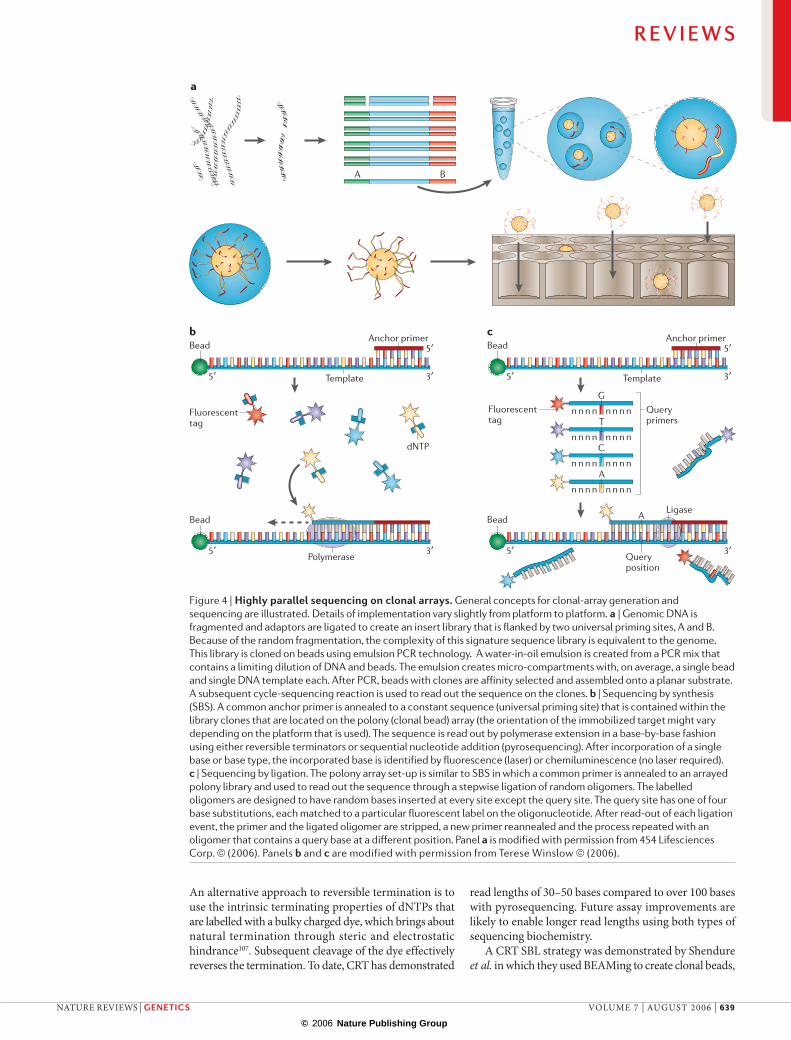
The BEAMing (FIG. 4a) approach that was developed by Dressman and Vogelstein95 shows great promise as a robust and flexible platform for polony sequencing. Single clonal polonies are grown on beads using a digi-tal emulsion PCR reaction95. To achieve this, a library is created with common primers that flank an insert; the library (single-stranded) is spiked into a PCR mix that
contains the appropriate universal primers and universal beads (immobilized with one universal primer). A ‘water in oil’ emulsion is created from this PCR mix to generate millions of aqueous micro-reactor compartments. The concentration of the library and beads is optimized to generate a maximal number of compartments that con-tain just one bead and one DNA molecule. Depending on the size of the aqueous compartments that are gener-ated during the emulsification step (from a few microns to over 100 microns), up to 3 billion individual PCR reactions per microlitre can be conducted simultane-ously in the same tube. Owing to its efficiency, BEAMing has already been implemented for highly parallel array-based cycle sequencing100,101.
Array-based cycle sequencing involves repeated rounds of sequencing on the array surface. After each round, data are collected by imaging. Several formats of cycle sequencing have been described, including sequencing by synthesis (SBS), sequencing by liga-tion (SBL) and sequencing by hybridization (FIG. 4b,c). SBS consists of repeated rounds of polymerase-based nucleotide insertion and fluorescent/chemiluminescent read-out. SBS has two formats: single nucleotide addition (SNA), which uses cycles of dNTP incorporation and imaging, and cyclic reversible termination (CRT), which uses cycles of incorporation of terminators, imaging and deblocking102,103.
Versions of SNA that use fluorescent dNTPs have been developed by several groups. The use of fluores-cent dNTPs requires the removal or destruction of the fluorescence signal after each incorporation cycle. In their original paper on polony amplification and fluo-rescent in situ sequencing, Mitra et al. used a disulphide linker between the fluorophore and the dNTP to remove fluorescence92. In a similar approach, but using single molecules, Braslavsky et al. used basewise addition of BODIPY (boron dipyrromethene difluoride)-labelled dNTPs and destroyed the fluorescence by photobleach-ing before addition of the next base104. One of the main challenges with fluorescent SNA is the poor read-out of homopolymeric runs owing to fluorescent quenching.
An alternative SNA approach that uses lumines-cence rather than fluorescence is pyrosequencing105, which has been successfully implemented in a highly parallel bead-array-based sequencer that can sequence over 20 million bases within 2–4 hours101. The read length (> 100 bases), accuracy and throughput of this instrument are impressive, although a couple of techni-cal issues remain to be solved before the approach can be scaled to human genome sequencing. First, the current size of the clonal beads (~35 µm) limits the array density and second, long homopolymeric runs of bases remain a challenge to read out accurately.
The use of fluorescently labelled reversible ter-minators in CRT enables, in theory, read-out of homopolymeric runs. The main challenge with CRT is the development of fluorescently labelled reversible terminators that can be cleaved and deblocked under mild conditions and incorporated efficiently by a poly-merase103. Solexa is currently developing a commercial sequencer using this approach on polony-based arrays106.
R E V I E W S
638 | AUGUST 2006 | VOLUME 7 www.nature.com/reviews/genetics
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group
Anchor primer
Template
Bead
Query position
A
n n n n
G
n n n n
n n n n
T
n n n n
n n n n
C
n n n n
n n n n
A
n n n n
Ligase
Queryprimers
Fluorescenttag
Fluorescenttag
dNTP
Anchor primer
Template
Bead
Polymerase
a
A B
5′ 3′
5′ 3′
5′
5′ 3′
5′ 3′
5′
bBead
cBead
An alternative approach to reversible termination is to use the intrinsic terminating properties of dNTPs that are labelled with a bulky charged dye, which brings about natural termination through steric and electrostatic hindrance107. Subsequent cleavage of the dye effectively reverses the termination. To date, CRT has demonstrated
read lengths of 30–50 bases compared to over 100 bases with pyrosequencing. Future assay improvements are likely to enable longer read lengths using both types of sequencing biochemistry.
A CRT SBL strategy was demonstrated by Shendure et al. in which they used BEAMing to create clonal beads,
Figure 4 | Highly parallel sequencing on clonal arrays. General concepts for clonal-array generation and sequencing are illustrated. Details of implementation vary slightly from platform to platform. a | Genomic DNA is fragmented and adaptors are ligated to create an insert library that is flanked by two universal priming sites, A and B. Because of the random fragmentation, the complexity of this signature sequence library is equivalent to the genome. This library is cloned on beads using emulsion PCR technology. A water-in-oil emulsion is created from a PCR mix that contains a limiting dilution of DNA and beads. The emulsion creates micro-compartments with, on average, a single bead and single DNA template each. After PCR, beads with clones are affinity selected and assembled onto a planar substrate. A subsequent cycle-sequencing reaction is used to read out the sequence on the clones. b | Sequencing by synthesis (SBS). A common anchor primer is annealed to a constant sequence (universal priming site) that is contained within the library clones that are located on the polony (clonal bead) array (the orientation of the immobilized target might vary depending on the platform that is used). The sequence is read out by polymerase extension in a base-by-base fashion using either reversible terminators or sequential nucleotide addition (pyrosequencing). After incorporation of a single base or base type, the incorporated base is identified by fluorescence (laser) or chemiluminescence (no laser required). c | Sequencing by ligation. The polony array set-up is similar to SBS in which a common primer is annealed to an arrayed polony library and used to read out the sequence through a stepwise ligation of random oligomers. The labelled oligomers are designed to have random bases inserted at every site except the query site. The query site has one of four base substitutions, each matched to a particular fluorescent label on the oligonucleotide. After read-out of each ligation event, the primer and the ligated oligomer are stripped, a new primer reannealed and the process repeated with an oligomer that contains a query base at a different position. Panel a is modified with permission from 454 Lifesciences Corp. © (2006). Panels b and c are modified with permission from Terese Winslow © (2006).
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 7 | AUGUST 2006 | 639
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group

mRNA
GGGAC NNNNNNNNNN ˆCCCTG NNNNNNNNNNNNNN ˆ- 5′
CAGCAG NNNNNNNNNNNNNNNNNNNNNNN ˆGTCGTC NNNNNNNNNNNNNNNNNNNNNNNNN ˆ- 5′
SAGETag Gene name
Gene A
Gene B
Gene C
Gene D
No.
1
4
6
1
1
4
6
1
Gene-expression profile Tag annotation usingDNA database
Concentrate tags andcarry out sequencing
BsmFI (14 bp for SAGE)
EcoP15I (26 bp for SuperSAGE)
Genomic DNA Transcription Tags
Cell Extract tag sequences from mRNAs with a tagging enzyme
Count the number of tags
Type IIS restriction enzymeRestriction enzymes that primarily exist as monomers and require only Mg2+ as a cofactor. Recognition sites are nonpalindromic, nearly always contiguous and without ambiguities; at least one strand is cleaved outside the recognition sequence.
followed by repeated rounds of primer annealing, liga-tion of labelled oligomers, imaging and stripping to read out the sequence100 (FIG. 4c). Ligation can sequence from both anchor (universal) sequences and does not require terminator and polymerase engineering. Currently, the major disadvantage of the described approach is the limited read length (< 20 bases), although alternative schemes might extend this read length108.
Digital sequence analysisHighly parallel array technology has at least one main weakness — limited sensitivity to detect a rare sequence variant in the presence of a large quantity of wild-type variant. This ‘needle in a haystack’ problem applies to the characterization of rare variants and low-abundance messages. Examples include the detection of a rare allele in a background of wild-type alleles, of chromosomal aberrations in tumour samples that are contaminated with high levels of normal tissue and of low-level mosaicism in tumours. This poor sensitivity is due to the ‘analogue’ mode of signal collection from a popula-tion of molecules. Detection becomes more sensitive if instead of measuring signal from an aggregate of molecules, the molecules are counted one-by-one in a digital fashion109,110.
This concept was first applied to gene expression. Velculescu et al. developed SAGE for carrying out digital analysis of cDNA signature sequences that serve as proxies for transcripts98. In SAGE, short, representa-tive signature sequence tags are generated by digesting cDNA samples with a 4-base-motif restriction enzyme, ligating adaptors that contain a ‘built-in’ type IIS restriction site and digesting the sample with the corresponding type IIS restriction enzyme (FIG. 5). These signature sequences are then concatenated and counted using serial analysis by standard Sanger sequencing. Recently, the exclusivity of these signature sequences
has been improved by using SuperSage tags111,112. Digital analysis of signature sequences has also been devel-oped for karyotype analysis and characterization of chromosomal aberrations99,113.
With the recent development of highly parallel array-based sequencing technologies11,100,101,114, char-acterization of these signature sequences is becoming much less expensive and is a harbinger of future genomic analysis in which sequencing-based digital analysis of arrayed clones or possibly single molecules will have a central role in characterization of gene expression, mutation detection, genomic profiling and detection of rare variants for diagnostic applications115,116. BEAMing technology has already been used to detect rare mutant APC alleles in the plasma of patients with colon cancer116. This approach has a direct clinical application in detect-ing variants that are derived from tumour cells by monitoring appropriate bodily fluids.
Optical mappingIn the technologies that are described above, the parallel read-out platform is created by various methods that produce an array of DNA fragments. These fragments represent probes or the target sequence that is to be analysed but do not preserve the long-range order of the genome from which they were originally derived. In a way, the ultimate array or read-out platform is the genome itself, because it contains this long-range information, which includes rearrangements such as inversions and translocations. Optical mapping is a relatively high-resolution method that is based on the concept of using gDNA itself as an array platform for intrinsically parallel read-out117–119. In this approach, gDNA is deposited on a surface in a way that results in the DNA becoming teth-ered in an elongated form120. The DNA is then digested with a restriction enzyme. This results in a slight relax-ation, which produces a gap at the site of digestion. Stained
Figure 5 | Digital analysis of gene expression using serial analysis of gene expression (SAGE). Signature sequences are extracted from transcripts by using two restriction-enzyme digestion steps, the first using anchoring enzymes NlaIII or Sau3A1 and the second using a type IIS restriction enzyme, the consensus sequence of which is incorporated in the ligated adaptor (shown in pink). The type IIS restriction enzyme extracts a 14–26 bp signature sequence adjacent to the most 3′ anchor enzyme motif. Modified with permission from REF. 112 © (2005) Blackwell Publishing Ltd.
R E V I E W S
640 | AUGUST 2006 | VOLUME 7 www.nature.com/reviews/genetics
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group

ChIP-on-chipChromatin immunoprecipitation (ChIP) combined with microarray detection. Usually, cells are treated with a crosslinking reagent (for example, formaldehyde), which is used to covalently link protein complexes in situ to DNA. The crosslinked chromatin is then isolated and fragmented. An immunoprecipitation step is used to enrich the protein of interest together with crosslinked DNA fragments. To identify and quantify these DNA fragments, the crosslinks are reversed and the DNA fragments are usually labelled with a fluorescent dye and hybridized to microarrays with probes that correspond to genomic regions of interest.
DNA molecules are detected by fluorescence imaging, and the fragments are identified and their lengths deter-mined by computational algorithms. The data are used to construct restriction maps of gDNA. The method is intrinsically parallel — the entire genome array can be generated from a single sample preparation. Because it is based on single-molecule analysis, random noise (due, for example, to some sites not being cut) must be overcome by over sampling. The method is intrinsically scalable, and is now in routine use to analyse the struc-ture of microbial genomes, although promising results have also been obtained with mammalian genomes121. A limitation of the current method is that only restriction sites are directly accessible for analysis. There is clearly potential to carry out other types of analysis on the array, including highly parallel sequencing119. Although there are several technical hurdles to be overcome, such a method would have the advantage of simultaneously providing sequence information and the relative position and orientation of sequences in the genome.
Finding functional elements in the genomeAs well as collecting genome-wide sequencing and genotyping information, there remains a requirement for accurate transcript annotation, mapping and character-ization of functional elements and genome structure to provide a better understanding of gene regulation during development, differentiation and disease. In 2003, The US National Institutes of Health (NIH) launched The ENCODE (Encyclopedia of DNA Elements) Project122, which aims to identify all functional elements in the human genome. An international consortium was formed to develop and apply high-throughput tech-nologies for detecting all sequence elements that confer biological function. The results of this pilot phase (which was focused on ~1% of the genome) will guide future efforts to analyse the entire human genome. Significant efforts have been made towards this goal, including genome-wide mapping of DNase hypersensitive sites using MPSS123 and transcriptome characterization and genome annotation using a gene-identification sig-nature analysis124. Also, high-density oligonucleotide arrays have been made to experimentally annotate the human genome. For example, whole-chromosome or whole-genome tiling arrays have been used to validate and refine computational gene predictions, define full-length transcripts and alternatively spliced exons125, and to discover novel transcripts126,127.
Genomic mapping of protein location: ChIP-on-chip. Genome-wide location and functional analysis of DNA-binding proteins has been widely used to study transcriptional regulatory networks128,129. ChIP-on-chip is proving useful for collecting genome-wide information on binding of proteins to DNA regulatory regions. This approach has been used to build a high-resolution map of active promoters in the human genome130 and other mammalian genomes131, using either a whole-genome til-ing array or arrays that target specific genomic regions. It has also been used to map the binding sites in the genome for a range of transcription factors132–134.
Post-translational histone modifications135 lead to changes in chromatin structure and activation or repres-sion of transcription136. ChIP-on-chip analysis has been used to construct a genome-wide map of nucleosome acetylation and methylation in yeast137 and in genome-wide comparative analysis of histone modifications in human and mouse138.
Mapping epigenetic regulatory networksRecent advances in epigenetic research139,140 have dem-onstrated the power of high-throughput technologies for analyses of DNA cytosine methylation patterns and histone modification marks. The Human Epigenome Project aims to identify all the chemical changes and relationships among chromatin constituents that provide function to the DNA code. The project will allow a better understanding of normal development, ageing, abnor-mal gene control in cancer, diabetes, psychiatric diseases and other diseases, as well as the role of the environment in human health141,142.
Methylation profiling of CpG dinucleotides in DNA. DNA methylation has a crucial role in the regulation of gene expression143,144. Microarray-based DNA-methylation profiling technologies have been developed to access the methylation status for many genes or the entire genome. These methods can be categorized into three main classes on the basis of how the methylation status is interrogated: discrimination of bisulphite-induced C to T transi-tion145–148; cleavage of gDNA by methylation-sensitive restriction enzymes149–151; and immunoprecipitation with methyl-binding protein or antibodies against methylated cytosines152. Although methylation-sensitive enzymes do not interrogate every CpG site, about a third of all CpGs in the genome can be assayed using a combination of enzymes150; therefore, this provides a powerful approach for genome-wide methylation profiling when coupled with a high-density array read-out. The immunopre-cipitation method overcomes the sequence-dependent limitation of all restriction-digestion-based approaches. However, it cannot provide methylation information at single-base resolution for any targeted sequence. For the bisulphite-based approach, the challenges lie in dealing effectively with the reduced genome complexity and increased sequence redundancy that ensues after bisul-phite conversion of the gDNA. Target-specific probe selection and hybridization specificity remain the main technical hurdles. Efforts have been made to improve the assay specificity by incorporation of an enzymatic dis-crimination step, such as oligonucleotide ligation148 and allele-specific extension147, therefore allowing multiplexed profiling of CpG methylation status in several hundred genes. Although significant challenges must be overcome, more comprehensive, high-resolution genome-wide methods for epigenomic analysis are feasible.
Integrated genomic profiling of cancersIn all forms of cancer, genomic changes — often specific to a particular type or stage of cancer — cause disrup-tions within pathways that result in uncontrolled cell growth153. In 2005, NIH launched an initiative to obtain a
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 7 | AUGUST 2006 | 641
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group

comprehensive description of the genetic basis of human cancer using genome-scale analysis technologies. The Cancer Genome Atlas Pilot Project will assess the fea-sibility of a high-resolution, genome-wide effort to systematically identify and characterize sites of genomic alteration that are involved in all main types of human cancer. Towards this end, one near-term application of highly parallel genomic assays will be to characterize the molecular basis of cancer. In the longer term, integrated analysis of the cancer genome will probably include large-scale genome sequencing; cataloguing somatic mutations; high-resolution SNP genotyping and genomic loss and amplification analysis; epigenetic studies, such as DNA methylation and chromatin modification analysis; inte-grative analysis of the cancer transcriptome154,155, such as profiling of mRNA, microRNA and other non-coding RNA expression and monitoring of alternative splicing; and gene-regulation studies, such as location analysis. We predict an even broader application of whole-genome technologies if they can be used to analyse scarce and degraded samples such as those from FFPE tissues. Tapping into this resource for retrospective studies will be important for correlating molecular profiles with response to treatment and with clinical outcomes.
ConclusionsThe combination of highly multiplexed assays with a highly parallel read-out represents a relatively new but already successful paradigm in high-throughput analy-sis. The power of this approach is based on its ability to scale according to the principle that underlies Moore’s Law: costs are reduced with increasing miniaturization of the read-out platform, whereas sample costs for highly multiplexed assays remain relatively constant. This means that with the right combination of platform and assay the cost per unit of information can be decreased by orders of magnitude whereas throughput is increased by orders of magnitude over traditional assays. This is exemplified by SNP genotyping: 5 years ago, the price per genotype was about $0.30 whereas today it is a hun-dredfold less at about $0.002–$0.003. Concomitantly, the number of SNPs per assay has increased from hundreds to hundreds of thousands.
High-resolution, relatively comprehensive disease studies that might require about a billion genotypes or more would have been too expensive several years ago. Today, such a study would cost only a few million dollars, and is clearly within reach. Moreover, the qual-ity and completeness of the data sets would also be far higher, and genotyping can be carried out in a few weeks
instead of many years. Therefore, the ability to redesign and reformat SNP assays for high-density arrays is enabling a revolution in human genetics. Specifically, it is enabling large-scale whole-genome association studies that will have unprecedented power to detect common genetic variants with a significant role in human disease and pharmacogenomic responses.
The fundamental concepts that underlie highly par-allel assays are broadly applicable, requiring only the ability to resolve a complex mixture of sequences. This has been carried out successfully by sequence-specific hybridization to arrays and emulsion PCR on bead sub-strates; other approaches based on physical separation are also possible. These general concepts have already been applied to several assays, including gene-expression profiling, SNP genotyping, genomic copy-number analysis, and genome mapping of DNA binding sites and methylation status. Perhaps most significantly, systems that are designed around parallel assays are assisting a renaissance in the field of DNA sequencing, and will enable powerful new comprehensive sequencing studies that are too expensive today.
One of the more unexpected lessons from the development of highly parallel assays is that increased complexity does not necessarily result in decreased data quality. This observation has flown in the face of conven-tional wisdom. There used to be a strong consensus that data quality of array-based approaches would inevitably deteriorate with increasing sample complexity, owing to nonspecific hybridization and signal-to-noise ratio issues. Indeed, early genomic assays, such as gene-expression profiling, which relied only on sequence-specific probe hybridization to confer specificity, did face some of these problems. The next generation of assays have made use of enzymatic discrimination in addition to hybridization to increase specificity and to enable assay designs that extract more information. As a result, they have managed to circumvent some of these problems. Parallel assays also reap the benefit of the simplicity of single-tube sample preparation. Strikingly, the most complex and scalable SNP genotyping assay today, in which essentially the entire human genome is hybridized to an array, is also among the most robust, reproducible and accurate.
The development of highly parallel genomic assays is still a relatively young field, and more exciting advances can be expected in the future. One of the great promises of this approach is that the ability to carry out comprehensive genomic analyses easily, inexpensively, accurately and rap-idly with high sensitivity should also help to create a new generation of routine diagnostic and prognostic tools.
1. Golub, T. R. et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286, 531–537 (1999).This paper demonstrates the utility and power of whole-genome gene-expression profiling for cancer classification.
2. van ‘t Veer, L. J. et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536 (2002).
3. Ramaswamy, S., Ross, K. N., Lander, E. S. & Golub, T. R. A molecular signature of metastasis in primary solid tumors. Nature Genet. 33, 49–54 (2003).
4. Kildal, W. et al. Evaluation of genomic changes in a large series of malignant ovarian germ cell tumors — relation to clinicopathologic variables. Cancer Genet. Cytogenet. 155, 25–32 (2004).
5. Sanchez-Carbayo, M., Socci, N. D., Lozano, J., Saint, F. & Cordon-Cardo, C. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J. Clin. Oncol. 24, 778–789 (2006).
6. Southern, E. M., Maskos, U. & Elder, J. K. Analyzing and comparing nucleic acid sequences by hybridization to arrays of oligonucleotides: evaluation using experimental models. Genomics 13, 1008–1017 (1992).
7. Pease, A. C. et al. Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proc. Natl Acad. Sci. USA 91, 5022–5026 (1994).
8. Schena, M., Shalon, D., Davis, R. W. & Brown, P. O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270, 467–470 (1995).
9. Lockhart, D. J. et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nature Biotechnol. 14, 1675–1680 (1996).The landmark paper that described high-density oligonucleotide arrays for gene-expression profiling.
R E V I E W S
642 | AUGUST 2006 | VOLUME 7 www.nature.com/reviews/genetics
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group

10. Syvanen, A. C. Toward genome-wide SNP genotyping. Nature Genet. 37, S5–S10 (2005).This article provides an excellent background on genotyping technologies.
11. Shendure, J., Mitra, R. D., Varma, C. & Church, G. M. Advanced sequencing technologies: methods and goals. Nature Rev. Genet. 5, 335–344 (2004).This article provides an excellent background on novel sequencing technologies.
12. Zondervan, K. T. & Cardon, L. R. The complex interplay among factors that influence allelic association. Nature Rev. Genet. 5, 89–100 (2004).
13. Hirschhorn, J. N. & Daly, M. J. Genome-wide association studies for common diseases and complex traits. Nature Rev. Genet. 6, 95–108 (2005).
14. Mockler, T. C. et al. Applications of DNA tiling arrays for whole-genome analysis. Genomics 85, 1–15 (2005).
15. Van Gelder, R. N. et al. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proc. Natl Acad. Sci. USA 87, 1663–1667 (1990).
16. Phillips, J. & Eberwine, J. H. Antisense RNA amplification: a linear amplification method for analyzing the mRNA population from single living cells. Methods 10, 283–288 (1996).
17. Barany, F. The ligase chain reaction in a PCR world. PCR Methods Appl. 1, 5–16 (1991).
18. Nilsson, M. et al. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science 265, 2085–2088 (1994).
19. Landegren, U., Samiotaki, M., Nilsson, M., Malmgren, H. & Kwiatkowski, M. Detecting genes with ligases. Methods 9, 84–90 (1996).
20. Baner, J., Nilsson, M., Mendel-Hartvig, M. & Landegren, U. Signal amplification of padlock probes by rolling circle replication. Nucleic Acids Res. 26, 5073–5078 (1998).
21. Qi, X., Bakht, S., Devos, K. M., Gale, M. D. & Osbourn, A. L-RCA (ligation-rolling circle amplification): a general method for genotyping of single nucleotide polymorphisms (SNPs). Nucleic Acids Res. 29, E116 (2001).
22. Faruqi, A. F. et al. High-throughput genotyping of single nucleotide polymorphisms with rolling circle amplification. BMC Genomics 2, 4 (2001).
23. Edwards, M. C. & Gibbs, R. A. Multiplex PCR: advantages, development, and applications. PCR Methods Appl. 3, S65–S75 (1994).
24. Rychlik, W. Selection of primers for polymerase chain reaction. Mol. Biotechnol. 3, 129–134 (1995).
25. Schoske, R., Vallone, P. M., Ruitberg, C. M. & Butler, J. M. Multiplex PCR design strategy used for the simultaneous amplification of 10 Y chromosome short tandem repeat (STR) loci. Anal. Bioanal. Chem. 375, 333–343 (2003).
26. Vallone, P. M. & Butler, J. M. AutoDimer: a screening tool for primer-dimer and hairpin structures. Biotechniques 37, 226–31 (2004).
27. Lin, Z., Cui, X. & Li, H. Multiplex genotype determination at a large number of gene loci. Proc. Natl Acad. Sci. USA 93, 2582–2587 (1996).
28. Brownie, J. et al. The elimination of primer–dimer accumulation in PCR. Nucleic Acids Res. 25, 3235–3241 (1997).
29. Chee, M. et al. Accessing genetic information with high-density DNA arrays. Science 274, 610–614 (1996).
30. Wang, D. G. et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 280, 1077–1082 (1998).References 29 and 30 represent the first application of high-density oligonucleotide arrays for large-scale genomic resequencing, SNP identification and genotyping.
31. Fan, J. B. et al. Parallel genotyping of human SNPs using generic high-density oligonucleotide tag arrays. Genome Res. 10, 853–860 (2000).
32. Adams, C. P. & Kron, S. J. Method for performing amplification of nucleic acid with two primers bound to a single solid support. US Patent 5,641,658 (1997).
33. Adessi, C. et al. Solid phase DNA amplification: characterisation of primer attachment and amplification mechanisms. Nucleic Acids Res. 28, E87 (2000).
34. Shapero, M. H., Leuther, K. K., Nguyen, A., Scott, M. & Jones, K. W. SNP genotyping by multiplexed solid-phase amplification and fluorescent minisequencing. Genome Res. 11, 1926–1934 (2001).
35. Andreadis, J. D. & Chrisey, L. A. Use of immobilized PCR primers to generate covalently immobilized DNAs for in vitro transcription/translation reactions. Nucleic Acids Res. 28, e5 (2000).
36. Wang, H. Y. et al. A genotyping system capable of simultaneously analyzing >1000 single nucleotide polymorphisms in a haploid genome. Genome Res. 15, 276–283 (2005).
37. Hardenbol, P. et al. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nature Biotechnol. 21, 673–678 (2003).
38. Fan, J. B. et al. Highly parallel SNP genotyping. Cold Spring Harb. Symp. Quant. Biol. 68, 69–78 (2003).References 37 and 38 describe two highly multiplexed SNP genotyping assays.
39. Altshuler, D. et al. A haplotype map of the human genome. Nature 437, 1299–1320 (2005).The seminal paper that described the results of the International HapMap Project.
40. Hardenbol, P. et al. Highly multiplexed molecular inversion probe genotyping: over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res. 15, 269–275 (2005).
41. Hinds, D. A. et al. Whole-genome patterns of common DNA variation in three human populations. Science 307, 1072–1079 (2005).
42. Patil, N. et al. Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21. Science 294, 1719–1723 (2001).
43. Kinzler, K. W. & Vogelstein, B. Whole genome PCR: application to the identification of sequences bound by gene regulatory proteins. Nucleic Acids Res. 17, 3645–3653 (1989).
44. Lisitsyn, N. & Wigler, M. Cloning the differences between two complex genomes. Science 259, 946–951 (1993).
45. Lucito, R. et al. Genetic analysis using genomic representations. Proc. Natl Acad. Sci. USA 95, 4487–4492 (1998).
46. Lucito, R. et al. Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation. Genome Res. 13, 2291–2305 (2003).
47. Sebat, J. et al. Large-scale copy number polymorphism in the human genome. Science 305, 525–528 (2004).
48. Kennedy, G. C. et al. Large-scale genotyping of complex DNA. Nature Biotechnol. 21, 1233–1237 (2003).
49. Matsuzaki, H. et al. Parallel genotyping of over 10,000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res. 14, 414–425 (2004).References 48 and 49 describe the original development of whole genome sampling analysis (WGSA), which allows high-density genome-wide SNP analysis.
50. Matsuzaki, H. et al. Genotyping over 100,000 SNPs on a pair of oligonucleotide arrays. Nature Methods 1, 109–111 (2004).
51. Herbert, A. et al. A common genetic variant is associated with adult and childhood obesity. Science 312, 279–283 (2006).
52. Klein, R. J. et al. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 (2005).
53. Gunderson, K. L., Steemers, F. J., Lee, G., Mendoza, L. G. & Chee, M. S. A genome-wide scalable SNP genotyping assay using microarray technology. Nature Genet. 37, 549–554 (2005).This is the first paper that demonstrated the ability to conduct whole-genome genotyping on whole-genome amplified samples of full genome complexity.
54. Steemers, F. J. et al. Whole-genome genotyping with the single-base extension assay. Nature Methods 3, 31–33 (2006).
55. Langmore, J. P. Rubicon Genomics, Inc. Pharmacogenomics 3, 557–560 (2002).
56. Dean, F. B. et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl Acad. Sci. USA 99, 5261–5266 (2002).
57. Gunderson, K. L. et al. Whole-genome genotyping of haplotype tag single nucleotide polymorphisms. Pharmacogenomics 7, 641–648 (2006).
58. Kallioniemi, A. et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 258, 818–821 (1992).
59. Pinkel, D. et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nature Genet. 20, 207–211 (1998).The first demonstration of array technology applied to high-resolution comparative genomic hybridization (CGH) applications.
60. Pollack, J. R. et al. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nature Genet. 23, 41–46 (1999).
61. Ishkanian, A. S. et al. A tiling resolution DNA microarray with complete coverage of the human genome. Nature Genet. 36, 299–303 (2004).
62. Barrett, M. T. et al. Comparative genomic hybridization using oligonucleotide microarrays and total genomic DNA. Proc. Natl Acad. Sci. USA 101, 17765–17770 (2004).
63. Carvalho, B., Ouwerkerk, E., Meijer, G. A. & Ylstra, B. High resolution microarray comparative genomic hybridisation analysis using spotted oligonucleotides. J. Clin. Pathol. 57, 644–646 (2004).
64. Selzer, R. R. et al. Analysis of chromosome breakpoints in neuroblastoma at sub-kilobase resolution using fine-tiling oligonucleotide array CGH. Genes Chromosomes Cancer 44, 305–319 (2005).
65. Ylstra, B., van den Ijssel, P., Carvalho, B., Brakenhoff, R. H. & Meijer, G. A. BAC to the future! or oligonucleotides: a perspective for micro array comparative genomic hybridization (array CGH). Nucleic Acids Res. 34, 445–450 (2006).
66. Bignell, G. R. et al. High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res. 14, 287–295 (2004).
67. Huang, J. et al. Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum. Genomics 1, 287–299 (2004).
68. Rauch, A. et al. Molecular karyotyping using an SNP array for genomewide genotyping. J. Med. Genet. 41, 916–922 (2004).
69. Zhao, X. et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 64, 3060–3071 (2004).
70. Zhao, X. et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 65, 5561–5570 (2005).
71. Bruce, S. et al. Global analysis of uniparental disomy using high density genotyping arrays. J. Med. Genet. 42, 847–851 (2005).
72. Teh, M. T. et al. Genomewide single nucleotide polymorphism microarray mapping in basal cell carcinomas unveils uniparental disomy as a key somatic event. Cancer Res. 65, 8597–8603 (2005).
73. LaFramboise, T. et al. Allele-specific amplification in cancer revealed by SNP array analysis. PLoS Comput. Biol. 1, e65 (2005).This paper highlights the effects of high-level allele-specific amplification in cancer.
74. Lips, E. H. et al. Reliable high-throughput genotyping and loss-of-heterozygosity detection in formalin-fixed, paraffin-embedded tumors using single nucleotide polymorphism arrays. Cancer Res. 65, 10188–10191 (2005).
75. Little, S. E. et al. Array CGH using whole genome amplification of fresh-frozen and formalin-fixed, paraffin-embedded tumor DNA. Genomics 87, 298–306 (2006).
76. Bredel, M. et al. Amplification of whole tumor genomes and gene-by-gene mapping of genomic aberrations from limited sources of fresh-frozen and paraffin-embedded DNA. J. Mol. Diagn. 7, 171–182 (2005).
77. Yan, H., Yuan, W., Velculescu, V. E., Vogelstein, B. & Kinzler, K. W. Allelic variation in human gene expression. Science 297, 1143 (2002).The first systematic cataloguing of allele-specific expression in human genes.
78. Bray, N. J., Buckland, P. R., Owen, M. J. & O’Donovan, M. C. Cis-acting variation in the expression of a high proportion of genes in human brain. Hum. Genet. 113, 149–153 (2003).
79. Knight, J. C., Keating, B. J., Rockett, K. A. & Kwiatkowski, D. P. In vivo characterization of regulatory polymorphisms by allele-specific quantification of RNA polymerase loading. Nature Genet. 33, 469–475 (2003).
80. Pastinen, T. & Hudson, T. J. Cis-acting regulatory variation in the human genome. Science 306, 647–650 (2004).
81. Ronald, J. et al. Simultaneous genotyping, gene-expression measurement, and detection of allele-specific expression with oligonucleotide arrays. Genome Res. 15, 284–291 (2005).
82. Lo, H. S. et al. Allelic variation in gene expression is common in the human genome. Genome Res. 13, 1855–1862 (2003).
R E V I E W S
NATURE REVIEWS | GENETICS VOLUME 7 | AUGUST 2006 | 643
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group

83. Liljedahl, U., Fredriksson, M., Dahlgren, A. & Syvanen, A. C. Detecting imbalanced expression of SNP alleles by minisequencing on microarrays. BMC Biotechnol. 4, 24 (2004).
84. Wang, Y. et al. Allele quantification using molecular inversion probes (MIP). Nucleic Acids Res. 33, e183 (2005).
85. Pant, P. V. et al. Analysis of allelic differential expression in human white blood cells. Genome Res. 16, 331–339 (2006).
86. Pastinen, T. et al. Mapping common regulatory variants to human haplotypes. Hum. Mol. Genet. 14, 3963–3971 (2005).An excellent review article that describes the origin, measurements and effects of cis-acting regulatory polymorphisms on transcription.
87. Drossman, H., Luckey, J. A., Kostichka, A. J., D’Cunha, J. & Smith, L. M. High-speed separations of DNA sequencing reactions by capillary electrophoresis. Anal. Chem. 62, 900–903 (1990).
88. Winzeler, E. A. et al. Direct allelic variation scanning of the yeast genome. Science 281, 1194–1197 (1998).
89. Albert, T. J. et al. Light-directed 5′–>3′ synthesis of complex oligonucleotide microarrays. Nucleic Acids Res. 31, e35 (2003).
90. Metzker, M. L. Emerging technologies in DNA sequencing. Genome Res. 15, 1767–1776 (2005).
91. Mitra, R. D. & Church, G. M. In situ localized amplification and contact replication of many individual DNA molecules. Nucleic Acids Res. 27, e34 (1999).
92. Mitra, R. D., Shendure, J., Olejnik, J., Edyta Krzymanska, O. & Church, G. M. Fluorescent in situ sequencing on polymerase colonies. Anal. Biochem. 320, 55–65 (2003).References 91 and 92 describe the original polony approach and associated fluorescent in situ sequencing.
93. Pemov, A., Modi, H., Chandler, D. P. & Bavykin, S. DNA analysis with multiplex microarray-enhanced PCR. Nucleic Acids Res. 33, e11 (2005).
94. Lizardi, P. M. et al. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nature Genet. 19, 225–232 (1998).
95. Dressman, D., Yan, H., Traverso, G., Kinzler, K. W. & Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl Acad. Sci. USA 100, 8817–8822 (2003).This paper describes the use of emulsion PCR for cloning on beads (BEAMing).
96. Reinartz, J. et al. Massively parallel signature sequencing (MPSS) as a tool for in-depth quantitative gene expression profiling in all organisms. Brief. Funct. Genomic. Proteomic. 1, 95–104 (2002).
97. Brenner, S. et al. Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nature Biotechnol. 18, 630–634 (2000).
98. Velculescu, V. E., Zhang, L., Vogelstein, B. & Kinzler, K. W. Serial analysis of gene expression. Science 270, 484–487 (1995).The original paper that described serial analysis of gene expression (SAGE).
99. Wang, T. L. et al. Digital karyotyping. Proc. Natl Acad. Sci. USA 99, 16156–16161 (2002).
100. Shendure, J. et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732 (2005).
101. Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005).References 100 and 101 were the first two papers to describe the sequencing of bacterial genomes using highly parallel array-based sequencing assays.
102. Tsien, R. Y., Ross, P., Fahnestock, M. & Johnston, A. J. DNA sequencing. PCT Patent WO 91/06678 (1991).
103. Metzker, M. L. et al. Termination of DNA synthesis by novel 3′-modified-deoxyribonucleoside 5′-triphosphates. Nucleic Acids Res. 22, 4259–4267 (1994).
104. Braslavsky, I., Hebert, B., Kartalov, E. & Quake, S. R. Sequence information can be obtained from single DNA molecules. Proc. Natl Acad. Sci. USA 100, 3960–3964 (2003).
105. Ronaghi, M., Uhlen, M. & Nyren, P. A sequencing method based on real-time pyrophosphate. Science 281, 363–365 (1998).
106. Bennett, S. T., Barnes, C., Cox, A., Davies, L. & Brown, C. Toward the $1000 human genome. Pharmacogenomics 6, 373–382 (2005).
107. Tebbutt, S. J. et al. Deoxynucleotides can replace dideoxynucleotides in minisequencing by arrayed primer extension. Biotechniques 40, 331–338 (2006).
108. Macevicz, S. C. DNA sequencing by parallel oligonucleotide extensions. US Patent 6,306,597 (2001).
109. Vogelstein, B. & Kinzler, K. W. Digital PCR. Proc. Natl Acad. Sci. USA 96, 9236–9241 (1999).
110. Pohl, G. & Shih I.-M. Principle and applications of digital PCR. Expert Rev. Mol. Diagn. 4, 41–47 (2004).
111. Matsumura, H. et al. Gene expression analysis of plant host–pathogen interactions by SuperSAGE. Proc. Natl Acad. Sci. USA 100, 15718–15723 (2003).
112. Matsumura, H. et al. SuperSAGE. Cell. Microbiol. 7, 11–18 (2005).
113. Tengs, T. et al. Genomic representations using concatenates of Type IIB restriction endonuclease digestion fragments. Nucleic Acids Res. 32, e121 (2004).
114. Lu, C. et al. Elucidation of the small RNA component of the transcriptome. Science 309, 1567–1569 (2005).
115. Li, M., Diehl, F., Dressman, D., Vogelstein, B. & Kinzler, K. W. BEAMing up for detection and quantification of rare sequence variants. Nature Methods 3, 95–97 (2006).
116. Diehl, F. et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl Acad. Sci. USA 102, 16368–16373 (2005).
117. Jing, J. et al. Automated high resolution optical mapping using arrayed, fluid-fixed DNA molecules. Proc. Natl Acad. Sci. USA 95, 8046–8051 (1998).
118. Aston, C., Mishra, B. & Schwartz, D. C. Optical mapping and its potential for large-scale sequencing projects. Trends Biotechnol. 17, 297–302 (1999).
119. Ramanathan, A., Pape, L. & Schwartz, D. C. High-density polymerase-mediated incorporation of fluorochrome-labeled nucleotides. Anal. Biochem. 337, 1–11 (2005).
120. Dimalanta, E. T. et al. A microfluidic system for large DNA molecule arrays. Anal. Chem. 76, 5293–5301 (2004).
121. Zody, M. C. et al. DNA sequence of human chromosome 17 and analysis of rearrangement in the human lineage. Nature 440, 1045–1049 (2006).
122. The ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306, 636–640 (2004).
123. Crawford, G. E. et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res. 16, 123–131 (2005).
124. Ng, P. et al. Gene identification signature (GIS) analysis for transcriptome characterization and genome annotation. Nature Methods 2, 105–111 (2005).
125. Shoemaker, D. D. et al. Experimental annotation of the human genome using microarray technology. Nature 409, 922–927 (2001).
126. Cheng, J. et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308, 1149–1154 (2005).
127. Kapranov, P. et al. Large-scale transcriptional activity in chromosomes 21 and 22. Science 296, 916–919 (2002).
128. Kim, T. H. et al. Direct isolation and identification of promoters in the human genome. Genome Res. 15, 830–839 (2005).
129. Ren, B. et al. Genome-wide location and function of DNA binding proteins. Science 290, 2306–2309 (2000).The first study that used a ChIP-on-chip approach for genome-wide mapping and functional analysis of DNA-binding proteins.
130. Kim, T. H. et al. A high-resolution map of active promoters in the human genome. Nature 436, 876–880 (2005).
131. Carninci, P. et al. The transcriptional landscape of the mammalian genome. Science 309, 1559–1563 (2005).
132. Carroll, J. S. et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 122, 33–43 (2005).
133. Cheng, A. S. et al. Combinatorial analysis of transcription factor partners reveals recruitment of c-MYC to estrogen receptor-α responsive promoters. Mol. Cell 21, 393–404 (2006).
134. Li, Z. et al. A global transcriptional regulatory role for c- Myc in Burkitt’s lymphoma cells. Proc. Natl Acad. Sci. USA 100, 8164–8169 (2003).
135. Plass, C. Cancer epigenomics. Hum. Mol. Genet. 11, 2479–2488 (2002).
136. van Steensel, B. Mapping of genetic and epigenetic regulatory networks using microarrays. Nature Genet. 37, S18–S24 (2005).
137. Pokholok, D. K. et al. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122, 517–527 (2005).
138. Bernstein, B. E. et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120, 169–181 (2005).
139. van Steensel, B. & Henikoff, S. Epigenomic profiling using microarrays. Biotechniques 35, 346–357 (2003).
140. Rakyan, V. K. et al. DNA methylation profiling of the human major histocompatibility complex: a pilot study for the human epigenome project. PLoS Biol. 2, e405 (2004).This work represents the first large-scale bisulphite sequencing effort for a large genomic region.
141. Jones, P. A. & Martienssen, R. A blueprint for a Human Epigenome Project: The AACR Human Epigenome Workshop. Cancer Res 65, 11241–11246 (2005).
142. Murrell, A., Rakyan, V. K. & Beck, S. From genome to epigenome. Hum. Mol. Genet. 14, R3–R10 (2005).
143. Bestor, T. H. Gene silencing. Methylation meets acetylation. Nature 393, 311–312 (1998).
144. Herman, J. G. & Baylin, S. B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 349, 2042–2054 (2003).
145. Gitan, R. S., Shi, H., Chen, C. M., Yan, P. S. & Huang, T. H. Methylation-specific oligonucleotide microarray: a new potential for high-throughput methylation analysis. Genome Res. 12, 158–164 (2002).
146. Adorjan, P. et al. Tumour class prediction and discovery by microarray-based DNA methylation analysis. Nucleic Acids Res. 30, e21 (2002).
147. Bibikova, M. et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 16, 383–393 (2006).
148. Cheng, Y. W., Shawber, C., Notterman, D., Paty, P. & Barany, F. Multiplexed profiling of candidate genes for CpG island methylation status using a flexible PCR/LDR/Universal Array assay. Genome Res. 16, 282–289 (2006).
149. Lippman, Z., Gendrel, A. V., Colot, V. & Martienssen, R. Profiling DNA methylation patterns using genomic tiling microarrays. Nature Methods 2, 219–224 (2005).
150. Schumacher, A. et al. Microarray-based DNA methylation profiling: technology and applications. Nucleic Acids Res. 34, 528–542 (2006).
151. Heisler, L. E. et al. CpG Island microarray probe sequences derived from a physical library are representative of CpG Islands annotated on the human genome. Nucleic Acids Res. 33, 2952–2961 (2005).
152. Weber, M. et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature Genet. 37, 853–862 (2005).
153. Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
154. Rhodes, D. R. & Chinnaiyan, A. M. Integrative analysis of the cancer transcriptome. Nature Genet. 37, S31–S37 (2005).
155. Rhodes, D. R. et al. Mining for regulatory programs in the cancer transcriptome. Nature Genet. 37, 579–583 (2005).
156. Gunderson, K. L. et al. Decoding randomly ordered DNA arrays. Genome Res. 14, 870–877 (2004).
AcknowledgementsIllumina, Infinium and GoldenGate are registered trademarks or trademarks of Illumina.
Competing interests statementThe authors declare competing financial interests.
FURTHER INFORMATIONCancer Genome Atlas Pilot Project: http://www.cancergenome.hih.gov ENCODE international consortium: http://www.genome.gov/12513391Human Epigenome Project: http://www.epigenome.orgAccess to this links box is available online.
R E V I E W S
644 | AUGUST 2006 | VOLUME 7 www.nature.com/reviews/genetics
© 2006 Nature Publishing Group
© 2006 Nature Publishing Group




















