
Heterogeneous Effects of IL2 on Collagen-Induced Arthritis
8
of August 16, 2016. This information is current as Collagen-Induced Arthritis Heterogeneous Effects of IL-2 on D. Finkelman and Raphael Hirsch Sherry Thornton, Gregory P. Boivin, Kwang N. Kim, Fred http://www.jimmunol.org/content/165/3/1557 doi: 10.4049/jimmunol.165.3.1557 2000; 165:1557-1563; ; J Immunol References http://www.jimmunol.org/content/165/3/1557.full#ref-list-1 , 27 of which you can access for free at: cites 55 articles This article Subscriptions http://jimmunol.org/subscriptions is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/ji/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/cgi/alerts/etoc Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved. Copyright © 2000 by The American Association of 9650 Rockville Pike, Bethesda, MD 20814-3994. The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on August 16, 2016 http://www.jimmunol.org/ Downloaded from by guest on August 16, 2016 http://www.jimmunol.org/ Downloaded from
Transcript of Heterogeneous Effects of IL2 on Collagen-Induced Arthritis

of August 16, 2016.This information is current as
Collagen-Induced ArthritisHeterogeneous Effects of IL-2 on
D. Finkelman and Raphael HirschSherry Thornton, Gregory P. Boivin, Kwang N. Kim, Fred
http://www.jimmunol.org/content/165/3/1557doi: 10.4049/jimmunol.165.3.1557
2000; 165:1557-1563; ;J Immunol
Referenceshttp://www.jimmunol.org/content/165/3/1557.full#ref-list-1
, 27 of which you can access for free at: cites 55 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2000 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on August 16, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on August 16, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

Heterogeneous Effects of IL-2 on Collagen-Induced Arthritis1
Sherry Thornton,* Gregory P. Boivin, † Kwang N. Kim,* Fred D. Finkelman, ‡§ andRaphael Hirsch2*
IL-2 is generally considered a pro-inflammatory cytokine that exacerbates Th1-mediated disease states, such as autoimmunearthritis. Consistent with this role for IL-2, recent studies from our laboratory demonstrate that IL-2 mRNA is markedly increasedduring the acute stage of collagen-induced arthritis (CIA), an animal model of rheumatoid arthritis. To further define the role ofIL-2 in CIA, the levels of IL-2 protein and its receptor and the effects of IL-2 administration were analyzed during CIA. IL-2protein and IL-2R were preferentially expressed at disease onset, compared with later stages of disease. Administration of re-combinant human IL-2 (rhIL-2) at, or just before, disease onset exacerbated disease; surprisingly, rhIL-2 given before diseaseonset inhibited CIA, associated with reduced cellular and humoral responses to type II collagen. Determination of in vivo serumlevels of Th1 and Th2 cytokines in response to rhIL-2 treatment demonstrated that IFN-g, but not IL-4, was markedly up-regulated in response to IL-2. In mice treated with anti-IFN-g Ab, both early and late IL-2 administration exacerbated CIA. Thus,IL-2 can have two opposite effects on autoimmune arthritis, a direct stimulatory effect and an indirect suppressive effect that ismediated by IFN-g. The Journal of Immunology,2000, 165: 1557–1563.
Cytokines are major mediators of inflammation in autoim-mune arthritides such as rheumatoid arthritis (RA)3 (re-viewed in Ref. 1). A myriad of cytokines are found
within the rheumatoid joint. Some appear to exacerbate disease,whereas others may down-regulate inflammation. In many cases,their precise roles are, as yet, incompletely defined. A growingbody of evidence from both animal models of arthritis and humanRA suggests that Th2-type cytokines, such as IL-4 and IL-10, canprotect against arthritis, whereas Th1-type cytokines, such as IL-2and IFN-g, can be pro-inflammatory (2–12).
Support for this concept comes from a number of observations.The RA synovium is infiltrated with CD41 T cells of the Th1phenotype (13). In murine collagen-induced arthritis (CIA), amodel of RA, administration of IFN-g can exacerbate disease (14,15), and administration of anti-IL-2R Abs can inhibit disease onset(16). Thus, it is surprising that most studies of cytokines in RAhave failed to detect IL-2 protein in RA synovial fluid (17). Recentstudies from our laboratory suggest that the failure to detect IL-2may relate to the timing of synovial sampling. Because of thedifficulties in serially sampling the synovium of patients with RA,our laboratory has used the murine collagen-induced arthritismodel to analyze mRNA cytokine patterns at various stages ofautoimmune arthritis. These studies demonstrated that IL-2 mRNAlevels are dramatically up-regulated in early CIA and that the ex-
pression of IL-2 mRNA declines during the chronic stage of dis-ease (18). Further support for a possible role for IL-2 in earlyarthritis comes from the recent demonstration that IL-2 protein issignificantly increased in peripheral blood mononuclear cells frompatients with early, but not late, RA (19).
IL-2 has been shown to regulate autoimmune processes in mice.IL-2-deficient mice develop autoimmune disorders, such as ulcer-ative colitis-like disease and generalized autoimmune hemolyticanemia (20, 21). Interestingly, these IL-2-deficient mice exhibitincreased proliferation of both T and B cells, even though IL-2 hasbeen characterized as a positive regulator of T cell proliferation.The development of autoimmune diseases in IL-2-deficient micesuggests a negative regulatory role for IL-2 in autoimmune dis-eases that may involve regulation of T and B cell proliferation.
These observations suggest that IL-2 might play a more importantrole in autoimmune arthritis than has previously been appreciated. Tofurther define the role of IL-2 in CIA, the levels of IL-2 protein andits receptor, as well as the effects of IL-2 administration, were ana-lyzed during the course of disease. These studies resulted in the un-expected observation that, although IL-2 is pro-inflammatory in es-tablished disease, it can play an anti-inflammatory role during earlystages of disease induction. Furthermore, our studies revealed thatIL-2-induced IFN-g mediates this early suppression of CIA, demon-strating the complex regulatory network of cytokine interactions in-volved in autoimmune arthritis.
Materials and MethodsAnimals
Male DBA/1 mice were purchased from The Jackson Laboratory (Bar Har-bor, ME). Mice were cared for according to American Association of Lab-oratory Animal Care guidelines. Mice were injected i.p. twice daily with30,000 IU of recombinant human IL-2 (rhIL-2; Chiron, Emoryville, CA) in0.5 ml of HBSS. All experimental procedures were approved by the An-imal Care Committee of the Children’s Hospital Research Foundation ofCincinnati.
Type II collagen (CII) immunization
Bovine CII (Elastin Products, Owensville, MO) was dissolved in 0.1 Macetic acid at a concentration of 2 mg/ml and stored at270°C until use. Forimmunization, 100mg of CII was emulsified with an equal volume of CFA(2 mg/ml) and administered intradermally at the base of the tail of mice
*William S. Rowe Division of Rheumatology, Children’s Hospital Medical Center,Cincinnati, OH 45229;†Department of Pathology and Laboratory Medicine, Univer-sity of Cincinnati, Cincinnati, OH 45267;‡University of Cincinnati College of Med-icine, Cincinnati, OH 45267; and§Cincinnati Veterans Administration Medical Cen-ter, Cincinnati, OH 45220
Received for publication March 8, 2000. Accepted for publication May 17, 2000.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported in part by National Institutes of Health Grants AI34958,AR44059, and AR42632, by the Schmidlapp Foundation, and by the Children’s Hos-pital Research Foundation of Cincinnati.2 Address correspondence and reprint requests to Dr. Raphael Hirsch, Division ofRheumatology, Children’s Hospital Medical Center, Pavilion Building 2-129, 3333Burnet Avenue, Cincinnati, OH 45229. E-mail address: [email protected] Abbreviations used in this paper: RA, rheumatoid arthritis; CIA, collagen-inducedarthritis; rhIL-2, recombinant human IL-2; CII, type II collagen.
Copyright © 2000 by The American Association of Immunologists 0022-1767/00/$02.00
by guest on August 16, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

6–8 wk of age. A booster, administered as above, was given 21 days afterthe primary injection. CII for footpad injections was prepared as above andconsisted of one 100-ml injection into each footpad.
Clinical assessment of arthritis
Mouse paws were scored for arthritis, as previously described (22), usinga macroscopic scoring system ranging from 0 to 4 (0, no swelling or red-ness; 1, swelling/redness of paw or one joint; 2, two joints involved; 3,more than two joints involved; and 4, severe arthritis of the entire paw andjoints). The arthritic score for each mouse is the sum of the scores of allfour paws.
RNase protection assay
Paws were quick frozen in liquid nitrogen and stored at270°C. Frozenpaws were homogenized with a polytron Tissue Tearor (Biospec Products,Bartlesville, OK) in appropriate volumes of RNA Stat 60 (Tel-Test,Friendswood, TX). RNA was extracted from the tissue homogenates ac-cording to the manufacturer’s instructions. RNA concentrations were de-termined by spectrophotometry.
Quantitation of IL-2 mRNA was performed on 5mg of total RNA uti-lizing the Riboquant Multiprobe RNase Protection Assay System (PharM-ingen, San Diego, CA) following manufacturer’s instructions. [a-32P]UTP-labeled anti-sense RNA probes were synthesized by in vitro transcriptionof cDNA templates for IL-2 and GAPDH. DNA templates were degradedby DNase I digestion, and probes were purified by phenol/chloroform ex-traction and ethanol precipitation, with subsequent hybridization to totalRNA at 56°C overnight. Samples were treated with RNase A1T1, anddouble-stranded RNA was purified by phenol/chloroform extraction andprecipitated with ethanol and salt. Samples were resuspended in loadingdye and electrophoresed on a 5% denaturing polyacrylamide gel. The gelwas dried and subjected to PhosphorImage analysis using a Storm 860 andImageQuant software (Molecular Dynamics, Sunnyvale, CA). The mRNAlevel of IL-2 is expressed as the ratio of the PhosphorImager units of IL-2to those of GAPDH from the same RNA sample.
Protein extraction from paws
Mice were sacrificed and paws minced in 1 ml of ice-cold PBS containing2 mM PMSF and 1% Triton X-100. Tissues were placed on ice and ho-mogenized. Homogenates were centrifuged at 2000 rpm and 4°C for 5 minto pellet cellular debris, and the supernatant was frozen in aliquots at270°C until assayed. Protein concentrations were determined with bicin-choninic acid solution (Sigma, St. Louis, MO) using BSA as a standard.
IL-2 ELISA
IL-2 levels in paw homogenates were determined by ELISA as follows: a96-well flat-bottom plate was coated with anti-IL-2 mAb (JES6-1A12;PharMingen) at a concentration of 5mg/ml in PBS and incubated at 4°Covernight. Plates were washed with PBS-Tween 20 and blocked for 40 minwith PBS containing 1% BSA. After washing, 40-ml aliquots of thawedpaw homogenate were added to duplicate wells and incubated for 40 min.Plates were washed again, and biotin-labeled anti-IL-2 (JES6-5H4; Phar-Mingen) at a concentration of 5mg/ml in PBS was added and incubated for40 min. After washing, streptavidin-peroxidase (Kirkegaard & Perry Lab-oratories, Gaithersburg, MD) was added. Plates were developed with Per-oxidase Substrate System 2,29-azino-bis(3-ethylbenzthiazoline-6-sulfonicacid) (Kirkegaard & Perry Laboratories). The plates were read at 405 nmin a microtiter plate reader (Dynex, Chantilly, VA). OD readings for theduplicate wells were averaged. Units of IL-2 in experimental samples werecalculated by using a standard curve generated with dilutions of purifiedrecombinant mouse IL-2 (PharMingen).
Immunohistochemistry
Staining for IL-2Ra was performed on 8-micron frozen sections of pawsusing a Techmate 500 automatic immunostainer (Ventana, Tucson, AZ).All steps were performed at room temperature, and all rinses were with 13PBS. Sections were blocked with CAS block (Zymed, San Francisco, CA)for 8 min. A biotin-conjugated rat anti-mouse CD25 mAb was used as theprimary Ab at 2mg/ml (PharMingen). The slides were exposed to theprimary Ab for 1 h and then to streptavidin-peroxidase for 15 min. Ami-noethyl carbazole (Zymed; four incubations at 5 min each) was used as thesubstrate. Sections were counterstained with Mayer’s hematoxylin.
Anti-CII Ab titers
Titers of anti-CII Abs in the serum samples were determined by ELISA, aspreviously described (22). All samples were measured in duplicate. Per-
oxidase-labeled goat anti-mouse IgG (Kirkegaard & Perry Laboratories)was used to measure CII-specific total IgG. IgG1 and IgG2a were measuredusing biotinylated rat anti-mouse IgG1 or IgG2a (PharMingen) and thenstreptavidin-peroxidase. Plates were developed with Peroxidase SubstrateSystem 2,29-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid). The plateswere read at 405 nm, as described above. OD readings for the duplicatewells were averaged. A serum sample consisting of pooled serum fromcontrol mice was tested at various dilutions and used as a standard togenerate a curve from which relative titers of the other serum samples werecalculated.
T cell IFN-g production
Draining lymph nodes were removed from mice and pooled to preparesingle cell suspensions. Cells were plated in triplicate at a concentrationof 5 3 105 cells/well in the presence of 300mg/ml heat-denatured CII(10 min, 56°C) or 50ml of 2C11 containing culture supernatant (anti-CD3). Supernatants were harvested at 48 h and analyzed by ELISA asdescribed (22).
In vivo cytokine production
Serum levels of IL-4 and IFN-g were measured using the Cincinnati Cy-tokine Capture Assay (23). Mice were treated twice daily with rhIL-2(30,000 U) for 2 days and once on the third day. Two hours after the lastIL-2 administration, 10mg of biotin-labeled anti-IL-4 (BVD4-1D11) or 50mg of anti-IFN-g (R4-6A2) was administered i.v. via the lateral tail vein.Mice were bled 3 h later, and serum was collected and analyzed for IL-4and IFN-g levels by ELISA as described (23).
Statistical analysis
Intergroup differences were assessed for statistical significance by theMann-WhitneyU test for qualitative data. The generalized Wilcoxon (Ge-han) test was used to compare the two “survival curves.” To test frequencydata for statistical significance,x2 was used. To compare baseline charac-teristics of the two groups, we used the independent two-tailed for para-metric datat test. Comparisons of more than two means were done usingthe one-way ANOVA. Tukey’s honest significant difference multiple com-parison test was used for pair-wise comparisons if significance was foundby ANOVA. The p values less than 0.05 were considered statistically sig-nificant, and allp values shown are unadjusted for multiple comparison.All calculations were performed by means of the software packageSPSS-PC (version 8.0 for Windows, Chicago, IL).
ResultsIL-2 and IL-2R are preferentially expressed during the earlystages of CIA
After immunization of DBA/1 mice with CII on days 0 and 21,arthritis is first observed between days 26 and 35. We have pre-viously demonstrated that IL-2 mRNA levels are markedly in-creased in early CIA (18). The increase in IL-2 mRNA expressionlevels is first observed on day 28 and correlates with disease se-verity (Fig. 1A). To determine whether the presence of mRNA forIL-2 correlated with protein levels, paw homogenates from ar-thritic mice were analyzed for IL-2 at various times during diseaseevolution (Fig. 1B). IL-2 protein was detected during both acutedisease (day 35) and chronic disease (day 49).
Up-regulation of thea subunit of the IL-2R can be caused byIL-2 gene activation and the presence of IL-2 protein (24, 25). Todetermine whether IL-2Ra is expressed in synovium of arthriticmice, joints from mice with acute (day 35) and chronic (day 49)CIA were analyzed by immunohistochemistry (Fig. 2). High levelsof IL-2Ra were detected on day 35 in paws with severe arthritis(score of 4; Fig. 2A). This IL-2Ra expression required the pres-ence of active synovitis because nonarthritic, day-35 paws (scoreof 0; Fig. 2B) and pre-arthritic, day-21 paws (data not shown)expressed little IL-2Ra. In contrast to early CIA, little IL-2Ra wasobserved in chronic disease (day 49), regardless of the severity ofarthritis (Fig. 2,C andD). These observations suggest a functionalrole for IL-2 during the early stages of established CIA.
1558 ROLE OF IL-2 IN CIA
by guest on August 16, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

IL-2 exacerbates established CIA
To further define the role of IL-2 in established CIA, rhIL-2(30,000 U i.p. twice daily) was administered for 7 days to CII-immunized mice starting one day after thefirst clinical manifesta-tion of CIA (average day of onset, day 28). Mice were scored forarthritis daily. IL-2 significantly exacerbated established disease (Fig.3), demonstrating that IL-2 is pro-inflammatory in established CIA.
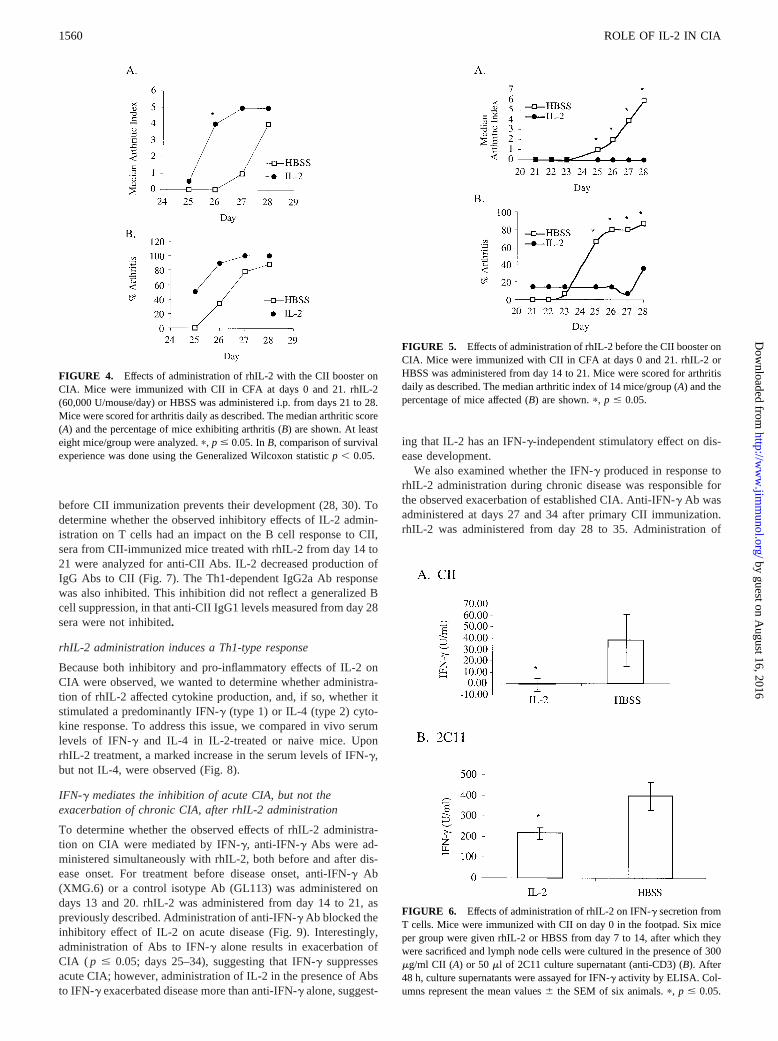
To determine whether IL-2 had an effect when administeredimmediately before clinical disease onset, rhIL-2 was administeredfor 7 days, beginning at the time of the CII booster (day 21).Treatment with rhIL-2 from day 21 to 28 significantly hastened theonset of arthritis and increased the severity of disease (Fig. 4).
IL-2 inhibits induction of CIA
The above studies demonstrate that IL-2 is pro-inflammatory whenadministered during established CIA or just before development ofclinical disease. These findings are not unexpected, given that theonset of CIA is dependent on T cell and B cell responses to CII(26–30), which are well-established by the time of the boosteradministration on day 21 (31). Because IL-2 can enhance T cellresponses to Ag (32), we hypothesized that a similar exacerbationof CIA would be observed if IL-2 was administered between thefirst and second CII immunization. Surprisingly, when rhIL-2 wasadministered from day 14 to 21, a significant decrease in both theincidence and severity of arthritis was observed (Fig. 5).
IL-2 inhibits T and B cell responses to CII
To determine the mechanism by which early administration ofIL-2 inhibited CIA, T and B cell responses to CII were measuredin mice treated with IL-2. Mice were immunized with CII on day0 in the footpad. IL-2 was administered from day 7 to 14, at whichtime mice were sacrificed, and draining popliteal and inguinallymph nodes were harvested. Lymph node cells were stimulated invitro with CII or anti-CD3 (2C11). Administration of IL-2 signif-icantly suppressed secretion of IFN-g in response to CII and 2C11in lymph node cell cultures (Fig. 6).
Anti-CII Abs are a prominent characteristic of CIA. This Abproduction is dependent on T cell help, in that T cell depletion
FIGURE 1. IL-2 mRNA and protein expression during CIA.A, Micewere sacrificed on days indicated and IL-2 mRNA levels were examined byRNase protection assay. Symbols represent the mean6 the SEM of threeor more paw RNAs (n5 normal RNA from unimmunized mouse paws).B, Mice were sacrificed at days 35 and 49 of arthritis, and 40ml of pawextract was analyzed for IL-2 activity by ELISA. Columns represent themean values6 the SEM of four paw extracts.
FIGURE 2. IL-2Ra expression at days35 and 49 of CIA. Mice were sacrificed andpaws were frozen. Sections were stained forIL-2Ra with anti-CD25 Ab. A, Day 35,score5 0; B, day 35, score5 4; C, day 49,score5 0; D, day 49, score5 4.
FIGURE 3. Effects of administration of rhIL-2 on established CIA.HBSS (n5 15) or rhIL-2 (n5 14) was administered to CII-treated mice1 day after onset of disease. Mice were scored as described.p, p # 0.01.
1559The Journal of Immunology
by guest on August 16, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
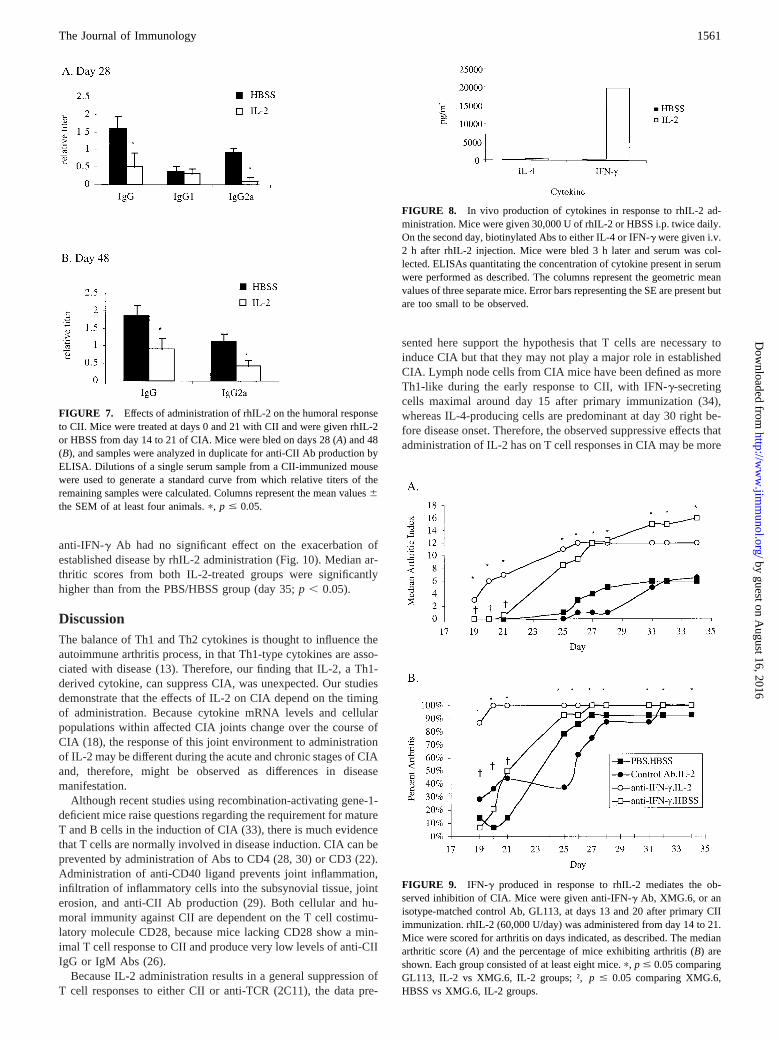
before CII immunization prevents their development (28, 30). Todetermine whether the observed inhibitory effects of IL-2 admin-istration on T cells had an impact on the B cell response to CII,sera from CII-immunized mice treated with rhIL-2 from day 14 to21 were analyzed for anti-CII Abs. IL-2 decreased production ofIgG Abs to CII (Fig. 7). The Th1-dependent IgG2a Ab responsewas also inhibited. This inhibition did not reflect a generalized Bcell suppression, in that anti-CII IgG1 levels measured from day 28sera were not inhibited.
rhIL-2 administration induces a Th1-type response
Because both inhibitory and pro-inflammatory effects of IL-2 onCIA were observed, we wanted to determine whether administra-tion of rhIL-2 affected cytokine production, and, if so, whether itstimulated a predominantly IFN-g (type 1) or IL-4 (type 2) cyto-kine response. To address this issue, we compared in vivo serumlevels of IFN-g and IL-4 in IL-2-treated or naive mice. UponrhIL-2 treatment, a marked increase in the serum levels of IFN-g,but not IL-4, were observed (Fig. 8).
IFN-g mediates the inhibition of acute CIA, but not theexacerbation of chronic CIA, after rhIL-2 administration
To determine whether the observed effects of rhIL-2 administra-tion on CIA were mediated by IFN-g, anti-IFN-g Abs were ad-ministered simultaneously with rhIL-2, both before and after dis-ease onset. For treatment before disease onset, anti-IFN-g Ab(XMG.6) or a control isotype Ab (GL113) was administered ondays 13 and 20. rhIL-2 was administered from day 14 to 21, aspreviously described. Administration of anti-IFN-g Ab blocked theinhibitory effect of IL-2 on acute disease (Fig. 9). Interestingly,administration of Abs to IFN-g alone results in exacerbation ofCIA ( p # 0.05; days 25–34), suggesting that IFN-g suppressesacute CIA; however, administration of IL-2 in the presence of Absto IFN-g exacerbated disease more than anti-IFN-g alone, suggest-
ing that IL-2 has an IFN-g-independent stimulatory effect on dis-ease development.
We also examined whether the IFN-g produced in response torhIL-2 administration during chronic disease was responsible forthe observed exacerbation of established CIA. Anti-IFN-g Ab wasadministered at days 27 and 34 after primary CII immunization.rhIL-2 was administered from day 28 to 35. Administration of
FIGURE 4. Effects of administration of rhIL-2 with the CII booster onCIA. Mice were immunized with CII in CFA at days 0 and 21. rhIL-2(60,000 U/mouse/day) or HBSS was administered i.p. from days 21 to 28.Mice were scored for arthritis daily as described. The median arthritic score(A) and the percentage of mice exhibiting arthritis (B) are shown. At leasteight mice/group were analyzed.p, p # 0.05. InB, comparison of survivalexperience was done using the Generalized Wilcoxon statisticp , 0.05.
FIGURE 5. Effects of administration of rhIL-2 before the CII booster onCIA. Mice were immunized with CII in CFA at days 0 and 21. rhIL-2 orHBSS was administered from day 14 to 21. Mice were scored for arthritisdaily as described. The median arthritic index of 14 mice/group (A) and thepercentage of mice affected (B) are shown.p, p # 0.05.
FIGURE 6. Effects of administration of rhIL-2 on IFN-g secretion fromT cells. Mice were immunized with CII on day 0 in the footpad. Six miceper group were given rhIL-2 or HBSS from day 7 to 14, after which theywere sacrificed and lymph node cells were cultured in the presence of 300mg/ml CII (A) or 50ml of 2C11 culture supernatant (anti-CD3) (B). After48 h, culture supernatants were assayed for IFN-g activity by ELISA. Col-umns represent the mean values6 the SEM of six animals.p, p # 0.05.
1560 ROLE OF IL-2 IN CIA
by guest on August 16, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
anti-IFN-g Ab had no significant effect on the exacerbation ofestablished disease by rhIL-2 administration (Fig. 10). Median ar-thritic scores from both IL-2-treated groups were significantlyhigher than from the PBS/HBSS group (day 35;p , 0.05).
DiscussionThe balance of Th1 and Th2 cytokines is thought to influence theautoimmune arthritis process, in that Th1-type cytokines are asso-ciated with disease (13). Therefore, our finding that IL-2, a Th1-derived cytokine, can suppress CIA, was unexpected. Our studiesdemonstrate that the effects of IL-2 on CIA depend on the timingof administration. Because cytokine mRNA levels and cellularpopulations within affected CIA joints change over the course ofCIA (18), the response of this joint environment to administrationof IL-2 may be different during the acute and chronic stages of CIAand, therefore, might be observed as differences in diseasemanifestation.
Although recent studies using recombination-activating gene-1-deficient mice raise questions regarding the requirement for matureT and B cells in the induction of CIA (33), there is much evidencethat T cells are normally involved in disease induction. CIA can beprevented by administration of Abs to CD4 (28, 30) or CD3 (22).Administration of anti-CD40 ligand prevents joint inflammation,infiltration of inflammatory cells into the subsynovial tissue, jointerosion, and anti-CII Ab production (29). Both cellular and hu-moral immunity against CII are dependent on the T cell costimu-latory molecule CD28, because mice lacking CD28 show a min-imal T cell response to CII and produce very low levels of anti-CIIIgG or IgM Abs (26).
Because IL-2 administration results in a general suppression ofT cell responses to either CII or anti-TCR (2C11), the data pre-
sented here support the hypothesis that T cells are necessary toinduce CIA but that they may not play a major role in establishedCIA. Lymph node cells from CIA mice have been defined as moreTh1-like during the early response to CII, with IFN-g-secretingcells maximal around day 15 after primary immunization (34),whereas IL-4-producing cells are predominant at day 30 right be-fore disease onset. Therefore, the observed suppressive effects thatadministration of IL-2 has on T cell responses in CIA may be more
FIGURE 7. Effects of administration of rhIL-2 on the humoral responseto CII. Mice were treated at days 0 and 21 with CII and were given rhIL-2or HBSS from day 14 to 21 of CIA. Mice were bled on days 28 (A) and 48(B), and samples were analyzed in duplicate for anti-CII Ab production byELISA. Dilutions of a single serum sample from a CII-immunized mousewere used to generate a standard curve from which relative titers of theremaining samples were calculated. Columns represent the mean values6the SEM of at least four animals.p, p # 0.05.
FIGURE 8. In vivo production of cytokines in response to rhIL-2 ad-ministration. Mice were given 30,000 U of rhIL-2 or HBSS i.p. twice daily.On the second day, biotinylated Abs to either IL-4 or IFN-g were given i.v.2 h after rhIL-2 injection. Mice were bled 3 h later and serum was col-lected. ELISAs quantitating the concentration of cytokine present in serumwere performed as described. The columns represent the geometric meanvalues of three separate mice. Error bars representing the SE are present butare too small to be observed.
FIGURE 9. IFN-g produced in response to rhIL-2 mediates the ob-served inhibition of CIA. Mice were given anti-IFN-g Ab, XMG.6, or anisotype-matched control Ab, GL113, at days 13 and 20 after primary CIIimmunization. rhIL-2 (60,000 U/day) was administered from day 14 to 21.Mice were scored for arthritis on days indicated, as described. The medianarthritic score (A) and the percentage of mice exhibiting arthritis (B) areshown. Each group consisted of at least eight mice.p, p # 0.05 comparingGL113, IL-2 vs XMG.6, IL-2 groups; †,p # 0.05 comparing XMG.6,HBSS vs XMG.6, IL-2 groups.
1561The Journal of Immunology
by guest on August 16, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

critical early in disease when Th1-type cells are dominant. Thefailure of IL-2 to suppress established disease suggests that theseT cell responses are not critical in established CIA. Once the T cellresponse has been established or after the second CII injection,IL-2 may stimulate the production of IL-2R-bearing inflammatorycells in the synovium, such as macrophages that may increase theinflammatory response to CII and result in the observed exacer-bation of disease.
The ability of IL-2 to suppress immune cell functions has alsobeen observed in other settings. For example, IL-2-deficient micehave increased T and B cell proliferation and develop autoimmunedisorders such as ulcerative colitis-like disease and autoimmunehemolytic anemia (20, 21, 35), which can be prevented by treat-ment with rhIL-2 before postnatal day 10 (36). IL-2 has also beenshown to suppress the immune response to corneal implants byprolonging allograft survival in mice (37).
A number of recent studies suggest potential mechanisms for theobserved inhibitory effects of IL-2 on CIA. For example, in vitroculture with IL-2 leads to apoptosis of activated T cells (38, 39).Other studies have implicated IFN-g, which enhances Fas and Fasligand expression, in lymphocyte apoptosis (40–42). Because theinhibitory effect of IL-2 on CIA development is IFN-g-dependentand IL-2 treatment induces a large increase in IFN-g production, itseems likely that IL-2 indirectly inhibits CIA by inducing produc-tion of IFN-g, which enhances apoptosis of lymphoid cells thatwould otherwise contribute to CIA development. Although IL-2might also suppress CIA by activating a subpopulation of CD41
IL-2R1 T cells that can induce peripheral T cell tolerance to au-toantigens (43, 44), this mechanism is not known to be IFN-gdependent. Consequently, it is less likely to be mediating suppres-sion of CIA in our system. Additional experiments to investigatethe mechanisms of IL-2 suppression of T and B cell responses arein progress.
Analysis of the role of IFN-g in animal models of arthritis havesuggested both protective and exacerbating effects of IFN-g ondisease (14, 15, 45–48). Interestingly, administration of IFN-g hasbeen shown to protect or exacerbate disease in rat adjuvant arthri-tis, depending on the time of administration (49). These results aresimilar to our observations with rhIL-2 administration. However,even though the inhibition of onset of disease was mediated byIFN-g in response to IL-2 administration, IL-2 itself mediated apro-inflammatory effect on CIA, as evidenced by the increase inarthritic scores in mice treated with both IL-2 and anti-IFN-g Abs.
Additionally, the observed exacerbation of disease by IL-2 admin-istration was not mediated by IFN-g. Other studies have demon-strated that treatment of mice with IL-2 can induce IFN-g produc-tion and subsequent in vitro killer cell activity (50, 51). Studies arecurrently underway to further characterize this in vivo IFN-g re-sponse to rhIL-2 treatment.
The observation that IL-2 mRNA levels paralleled the up-reg-ulation of IL-2Ra expression is consistent with the dependence ofIL-2Ra expression on the presence of IL-2 protein (24, 25). Theobserved increase in IL-2 mRNA contrasts with results from pre-vious studies that were unable to detect IL-2 protein in RA syno-vial fluid or CIA joints (17, 52). However, failure to detect IL-2 inthe RA synovium may reflect the tendency of investigators to bi-opsy later in the disease course. Recently, IL-2 mRNA has beendetected in RA synovial cells (53), and IL-2 protein has been foundto be significantly increased in PBMCs from RA patients withearly onset, but not late onset, synovitis (19).
Results from other recent studies also support a possible role forIL-2 in the autoimmune disease process. Weak linkage of certaincytokine genes, including IL-2, to RA (54) suggests that IL-2 mayplay a role in the disease process but that it is probably one ofseveral factors involved in disease manifestation. Additionally,mouse models of diabetes and experimental autoimmune enceph-alomyelitis have linked a region of mouse chromosome 3, includ-ing the IL-2 gene locus, to the manifestation of these autoimmunediseases in mice (55).
In summary, the results presented here demonstrate a role forIL-2 during the early stages of CIA. IL-2 can inhibit diseasethrough effects that are mediated by IFN-g, and IL-2 can exacer-bate disease, possibly by direct effects on IL-2R-bearing cells. IL-2may be integral to many autoimmune processes. Further investi-gation of the mechanisms of action of IL-2 in autoimmunity andthe IFN-g dependence of these mechanisms may lead to a betterunderstanding of the pathogenesis of autoimmune arthritis andother autoimmune disorders.
AcknowledgmentsWe thank Dr. Ed Giannini and Monica DeLay for helpful discussions andcritical review of the manuscript. We would like to acknowledge Chironfor supplying the recombinant human IL-2 used in these studies.
References1. Feldmann, M., F. M. Brennan, and R. N. Maini. 1996. Role of cytokines in
rheumatoid arthritis.Annu. Rev. Immunol. 14:397.2. Bessis, N., M. C. Boissier, P. Ferrara, T. Blankenstein, D. Fradelizi, and
C. Fournier. 1996. Attenuation of collagen-induced arthritis in mice by treatmentwith vector cells engineered to secrete interleukin-13.J. Immunol. 26:2399.
3. Hesse, M., S. Bayrak, and A. Mitchison. 1996. Protective major histocompati-bility complex genes and the role of interleukin-4 in collagen-induced arthritis.Eur. J. Immunol. 26:3234.
4. Horsfall, A. C., D. M. Butler, L. Marinova, P. J. Warden, R. O. Williams,R. N. Maini, and M. Feldmann. 1997. Suppression of collagen-induced arthritisby continuous administration of IL-4.J. Immunol. 159:5687.
5. Joosten, L. A. B., E. Lubberts, P. Durez, M. M. A. Helsen, M. J. M. Jacobs,M. Goldman, and W. B. van den Berg. 1997. Role of interleukin-4 and interleu-kin-10 in murine collagen-induced arthritis.Arthritis Rheum. 40:249.
6. Tanaka, Y., T. Otsuka, T. Hotokebuchi, H. Miyahara, H. Nakashima, S. Kuga,Y. Nemoto, H. Niiro, and Y. Niho. 1996. Effect of IL-10 on collagen-inducedarthritis in mice.Inflamm. Res. 45:283.
7. Walmsley, M., P. D. Katsikis, E. Abney, S. Parry, R. O. Williams, R. N. Maini,and M. Feldmann. 1996. Interleukin-10 inhibition of the progression of estab-lished collagen-induced arthritis.Arthritis Rheum. 39:495.
8. Allen, J. B., H. L. Wong, G. L. Costa, M. J. Bienkowski, and S. M. Wahl. 1993.Suppression of monocyte function and differential regulation of IL-1 and IL-1raby IL-4 contribute to resolution of experimental arthritis.J. Immunol. 151:4344.
9. Chomarat, P., J. Banchereau, and P. Miossec. 1995. Differential effects of inter-leukins 10 and 4 on the production of interleukin-6 by blood and synoviummonocytes in rheumatoid arthritis.Arthritis Rheum. 38:1046.
10. Isomaki, P., R. Lukkainen, R. Saario, P. Toivanen, and J. Punnonen. 1996. In-terleukin-10 functions as an antiinflammatory cytokine in rheumatiod synovium.Arthritis Rheum. 39:386.
FIGURE 10. IFN-g produced in response to rhIL-2 treatment does notmediate the observed exacerbation of CIA. Mice were given anti-IFN-gAb, XMG.6, or an isotype-matched control Ab, GL113, at days 27 and 34after primary CII immunization. rhIL-2 (60,000 U/day) was administeredfrom day 27 to 35. Mice were scored for arthritis on days indicated, asdescribed. Each group consisted of at least eight mice.
1562 ROLE OF IL-2 IN CIA
by guest on August 16, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

11. Katsikas, P. D., C.-Q. Chu, F. M. Brennan, R. N. Maini, and M. Feldmann. 1994.Immunoregulatory role of interleukin 10 in rheumatoid arthritis.J. Exp. Med.179:1517.
12. van Roon, J. A., J. L. van Roy, F. H. Gmelig-Meyling, F. P. Lafeber, andJ. W. Bijlsma. 1996. Prevention and reversal of cartilage degradation in rheu-matoid arthritis by interleukin-10 and interleukin-4.Arthritis Rheum. 39:829.
13. Dolhain, R. J., A. N. van der Heiden, N. T. ter Haar, F. C. Breedveld, andA. M. Miltenburg. 1996. Shift toward T lymphocytes with a T helper 1 cytokine-secretion profile in the joints of patients with rheumatoid arthritis.ArthritisRheum. 39:1961.
14. Cooper, S. M., S. Sriram, and G. E. Ranges. 1988. Suppression of murine col-lagen-induced arthritis with monoclonal anti-Ia antibodies and augmentation withIFN-g. J. Immunol. 141:1958.
15. Mauritz, N. J., R. Holmdahl, R. Jonsson, P. H. Van der Meide, A. Scheynius, andL. Klareskog. 1988. Treatment withg-interferon triggers the onset of collagenarthritis in mice.Arthritis Rheum. 31:1297.
16. Banerjee, S., B. Wei, K. Hillman, H. S. Luthra, and C. S. David. 1988. Immu-nosuppression of collagen-induced arthritis in mice with an anti-IL-2 receptorantibody.J. Immunol. 141:1150.
17. Firestein, G. S., W. D. Xu, K. Townsend, D. Broide, J. Alvaro-Gracia,A. Glasebrook, and N. J. Zvaifler. 1988. Cytokines in chronic inflammatory ar-thritis. J. Exp. Med. 168:1573.
18. Thornton, S., G. P. Boivin, Y. Ma, and R. Hirsch. 1999. Association of the courseof collagen-induced arthritis with distinct patterns of cytokine and chemokinemessenger RNA expression.Arthritis Rheum. 42:1109.
19. Kanik, K. S., E. Hagiwara, C. H. Yarboro, H. R. Schumacher, R. L. Wilder, andD. M. Klinman. 1998. Distinct patterns of cytokine secretion characterize newonset synovitis versus chronic rheumatoid arthritis.J. Rheumatol. 25:16.
20. Sadlack, B., H. Merz, H. Schorle, A. Schimpl, A. C. Feller, and I. Horak. 1993.Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene.Cell75:253.
21. Horak, I. 1995. Immunodeficiency in IL-2-knockout mice.Clin. Immunol. Im-munopathol. 76:S172.
22. Hughes, C., J. A. Wolos, E. H. Giannini, and R. Hirsch. 1994. Induction of Thelper cell hyporesponsiveness in an experimental model of autoimmunity byusing nonmitogenic anti-CD3 monoclonal antibody.J. Immunol. 153:3319.
23. Finkelman, F. D., and S. C. Morris. 1999. Development of an assay to measurein vivo cytokine production in the mouse.Int. Immunol. 11:1811.
24. Chastagner, P., J. L. Moreau, Y. Jacques, T. Tanaka, M. Miyasaka, M. Kondo,K. Sugamura, and J. Theze. 1996. Lack of intermediate-affinity interleukin-2receptor in mice leads to dependence on interleukin-2 receptora, b andg chainexpression for T cell growth.Eur. J. Immunol. 26:201.
25. Froussard, P., D. L. Jankovic, P. Chastagner, and J. Theze. 1991. Induction ofmouse p55 interleukin-2 receptor gene expression by IL-2 and IL-4 and charac-terization of its transcription initiation sites.Mol. Immunol. 28:87.
26. Tada, Y., K. Nagasawa, A. Ho, F. Morito, O. Ushiyama, N. Suzuki, H. Ohta, andT. W. Mak. 1999. CD28-deficient mice are highly resistant to collagen-inducedarthritis.J. Immunol. 162:203.
27. Wooley, P. H., H. S. Luthra, M. M. Griffiths, J. M. Stuart, A. Huse, andC. S. David. 1985. Type II collagen-induced arthritis in mice. IV. Variations inimmunogenetic regulation provide evidence for multiple arthritogenic epitopeson the collagen molecule.J. Immunol. 135:2443.
28. Goldschmidt, T. J., M. Andersson, V. Malmstrom, and R. Holmdahl. 1992. Ac-tivated type II collagen reactive T cells are not eliminated by in vivo anti-CD4treatment: implications for therapeutic approaches on autoimmune arthritis.Im-munobiology 184:359.
29. Durie, F. H., R. A. Fava, T. M. Foy, A. Aruffo, J. A. Ledbetter, and R. J. Noelle.1993. Prevention of collagen-induced arthritis with an antibody to gp39, theligand for CD40.Science 261:1328.
30. Chu, E. B., M. V. Hobbs, C. B. Wilson, C. G. Romball, P. S. Linsley, andW. O. Weigle. 1996. Intervention of CD41 cell subset shifts and autoimmunityin the BXSB mouse by murine CT4LA4Ig.J. Immunol. 156:1262.
31. Stuart, J. M., A. S. Townes, and A. H. Kang. 1982. Nature and specificity of theimmune response to collagen in type II collagen-induced arthritis in mice.J. Clin.Invest. 69:673.
32. Theze, J., P. M. Alzari, and J. Bertoglio. 1996. Interleukin 2 and its receptors:recent advances and new immunological functions.Immunol. Today 17:481.
33. Plows, D., G. Kontogeorgos, and G. Kollias. 1999. Mice lacking mature T and Blymphocytes develop arthritic lesions after immunization with type II collagen.J. Immunol. 162:1018.
34. Doncarli, A., L. M. Stasiuk, C. Fournier, and O. Abehsira-Amar. 1997. Conver-sion in vivo from an early dominant Th0/Th1 response to a Th2 phenotype duringthe development of collagen-induced arthritis.Eur. J. Immunol. 27:1451.
35. Contractor, N. V., H. Bassiri, T. Reya, A. Y. Park, D. C. Baumgart, M. A. Wasik,S. G. Emerson, and S. R. Carding. 1998. Lymphoid hyperplasia, autoimmunity,and compromised intestinal intraepithelial lymphocyte development in colitis-free gnotobiotic IL-2-deficient mice.J. Immunol. 160:385.
36. Klebb, G., I. B. Autenrieth, H. Haber, E. Gillert, B. Sadlack, K. A. Smith, andI. Horak. 1996. Interleukin-2 is indispensable for development of immunologicalself-tolerance.Clin. Immunol. Immunopathol. 81:282.
37. Zhang, E., T. Poul, S. Bulfone-Paus, J. Wachtlin, U. Kunzendorf, andF. Hoffmann. 1998. Prolongation of corneal allograft survival by an interleukin-2immunoglobulin fusion protein in mice.Graefe’s Arch. Clin. Exp. Ophthalmol.236:486.
38. Zheng, L., C. L. Trageser, D. M. Willerford, and M. J. Lenardo. 1998. T cellgrowth cytokines cause the superinduction of molecules mediating antigen-in-duced T lymphocyte death.J. Immunol. 160:763.
39. Critchfield, J. M., M. K. Racke, J. C. Zuniga-Pflucker, B. Cannella, C. S. Raine,J. Goverman, and M. J. Lenardo. 1994. T cell deletion in high antigen dosetherapy of autoimmune encephalomyelitis.Science 263:1139.
40. Oyaizu, N., T. W. McCloskey, S. Than, R. Hu, V. S. Kalyanaraman, andS. Pahwa. 1994. Cross-linking of CD4 molecules upregulates Fas antigen ex-pression in lymphocytes by inducing interferon-g and tumor necrosis factor-asecretion.Blood 84:2622.
41. Shustov, A., P. Nguyen, F. Finkelman, K. B. Elkon, and C. S. Via. 1998. Dif-ferential expression of Fas and Fas ligand in acute and chronic graft-versus-hostdisease: up-regulation of Fas and Fas ligand requires CD81 T cell activation andIFN-g production.J. Immunol. 161:2848.
42. Maciejewski, J., C. Selleri, S. Anderson, and N. S. Young. 1995. Fas antigenexpression on CD341 human marrow cells is induced by interferong and tumornecrosis factora and potentiates cytokine-mediated hematopoietic suppression invitro. Blood 85:3183.
43. Thornton, A. M., and E. M. Shevach. 1998. CD41CD251 immunoregulatory Tcells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2production.J. Exp. Med. 188:287.
44. Sakaguchi, S., N. Sakaguchi, M. Asano, M. Itoh, and M. Toda. 1995. Immuno-logic self-tolerance maintained by activated T cells expressing IL-2 receptora-chains (CD25): breakdown of a single mechanism of self-tolerance causesvarious autoimmune diseases.J. Immunol. 155:1151.
45. Boissier, M. C., G. Chiocchia, N. Bessis, J. Hajnal, G. Garotta, F. Nicoletti, andC. Fournier. 1995. Biphasic effect of interferon-g in murine collagen-inducedarthritis.Eur. J. Immunol. 25:1184.
46. Manoury-Schwartz, B., G. Chiocchia, N. Bessis, O. Abehsira-Amar, F. Batteux,S. Muller, S. Huang, M. C. Boissier, and C. Fournier. 1997. High susceptibilityto collagen-induced arthritis in mice lacking IFN-g receptors.J. Immunol. 158:5501.
47. Kageyama, Y., Y. Koide, A. Yoshida, M. Uchijima, T. Arai, S. Miyamoto,T. Ozeki, M. Hiyoshi, K. Kushida, and T. Inoue. 1998. Reduced susceptibility tocollagen-induced arthritis in mice deficient in IFN-g receptor.J. Immunol. 161:1542.
48. Vermeire, K., H. Heremans, M. Vandeputte, S. Huang, A. Billiau, andP. Matthys. 1997. Accelerated collagen-induced arthritis in IFN-g receptor-defi-cient mice.J. Immunol. 158:5507.
49. Jacob, C. O., J. Holoshitz, P. Van der Meide, S. Strober, and H. O. McDevitt.1989. Heterogeneous effects of IFN-g in adjuvant arthritis.J. Immunol. 142:1500.
50. Weigent, D. A., G. J. Stanton, and H. M. Johnson. 1983. Interleukin 2 enhancesnatural killer cell activity through induction ofg interferon.Infect. Immun. 41:992.
51. Sayers, T., H. Rossiter, J. Chung, A. Hren, and D. Armerding. 1985. In vivoactivation of murine natural killer cell functions by human recombinant DNAinterleukin 2.Immunobiology 169:303.
52. Mussener, A., M. J. Litton, E. Lindroos, and L. Klareskog. 1997. Cytokine pro-duction in synovial tissue of mice with collagen-induced arthritis (CIA).Clin.Exp. Immunol. 107:485.
53. Buchan, G., K. Barrett, T. Fujita, T. Taniguchi, R. Maini, and M. Feldmann.1988. Detection of activated T cell products in the rheumatoid joint using cDNAprobes to interleukin 2 (IL-2) IL-2 receptor and IFN-g. Clin. Exp. Immunol.71:295.
54. John, S., A. Myerscough, A. Marlow, A. Hajeer, A. Silman, W. Ollier, andJ. Worthington. 1998. Linkage of cytokine genes to rheumatoid arthritis: evi-dence of genetic heterogeneity.Ann. Rheum. Dis. 57:361.
55. Encinas, J. A., L. S. Wicker, L. B. Peterson, A. Mukasa, C. Teuscher, R. Sobel,H. L. Weiner, C. E. Seidman, J. G. Seidman, and V. K. Kuchroo. 1999. QTLinfluencing autoimmune diabetes and encephalomyelitis map to a 0.15-cM regioncontaining Il2.Nat. Genet. 21:158.
1563The Journal of Immunology
by guest on August 16, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from




















