
Intramuscular Administration of Steroids in Treatment of Rheumatoid Arthritis
Upload
independentCategory
view
1download
0
ARTHRITIS & RHEUMATISMVol. 42, No. 7, July 1999, pp 1528–1537© 1999, American College of Rheumatology
APOPTOTIC CHONDROCYTE DEATH IN RHEUMATOID ARTHRITIS
HYUN AH KIM and YEONG WOOK SONG
Objective. Recently, chondrocytes were shown toundergo apoptosis by the addition of nitric oxide and bycoupling of Fas/Fas ligand in vitro, suggesting thepossibility that chondrocytes have an inherent pro-grammed cell death pathway that operates in adultcartilage. Chondrocyte apoptosis was verified in situ inarticular cartilage samples from humans with osteoar-thritis (OA) and from an animal model of OA. Thepresent study investigates apoptotic chondrocyte deathand the expression of Bcl-2 and Fas in rheumatoidarthritis (RA) cartilage.
Methods. Cartilage samples were obtained from13 RA patients at the time of joint replacement surgeryand from 8 normal subjects at autopsy. Apoptoticchondrocytes were observed and counted in hematoxylinand eosin–stained cartilage specimens. Apoptosis wasverified by TUNEL, electron microscopy, and DNAladder assay. Bcl-2 and Fas expression were evaluatedby immunohistochemistry.
Results. Apoptotic cells were frequently observedin RA cartilage, whereas normal cartilage rarely showedapoptotic cells (3.01% versus 0.15%, respectively), afinding that was further confirmed by TUNEL staining.On electron microscopy, numerous apoptotic cells withtypical chromatin condensation were observed in RAcartilage. DNA from RA cartilage also revealed 180-basepair nucleosome ladders on electrophoresis. Bcl-2expression was significantly lower in RA cartilage thanin normal cartilage (23.3% versus 43.1%, respectively),whereas Fas expression was not statistically different.
Conclusion. Apoptotic chondrocyte death and de-creased Bcl-2 expression were verified in RA cartilage.
They might provide a novel model system for the researchof cartilage breakdown and joint destruction in RA.
Rheumatoid arthritis (RA) is a chronic inflam-matory disease that occasionally leads to serious disabil-ity resulting from joint destruction. While most studiesof the pathogenetic mechanisms of RA have focused onsynovial hyperplasia and excessive inflammatory cellinfiltration, the pathogenesis of cartilage loss has notreceived much attention. Classically, proliferative tissueknown as pannus is thought to grow centripetally from thesynovium–cartilage junction, directly invading and destroy-ing the underlying articular cartilage (1). Since the proto-type proinflammatory cytokines, interleukin-1 (IL-1) andtumor necrosis factor a (TNFa), have been observed atthese sites of cartilage erosion (2,3), these 2 potent cyto-kines may play an important role in the degradation ofcartilage by up-regulating metalloproteinase and prosta-glandin production in synoviocytes and chondrocytes.
There has been clinical evidence, however, thatjoint destruction and signs of synovitis are not alwayscorrelated. It was reported that erosions progress evenwhen measures to suppress joint inflammation are suc-cessful (4). Systemic measures of inflammation in RApatients correlate only loosely with radiographic severityof joint erosion (5). Two explanations are possible forthis observation. First, proliferated synovium might per-sist and contribute to cartilage loss in spite of ameliora-tion of inflammation. It was recently reported thatdespite extensive DNA fragmentation in rheumatoidsynovium, only few cells actually complete apoptosis (6).This finding led to the suggestion that a defect inapoptosis is responsible for the uncontrollable prolifer-ation of rheumatoid synovium. Fibroblast-like synovio-cytes transduced with an inactivator of endogenous p53were found to be less susceptible to apoptosis and moreinvasive to cartilage extract in vitro (7). Second, cartilagemight have an endogenous degradation mechanism,which is probably activated by inflammatory stimuli andcontinues to work even if the stimuli cease to exist.
Apoptosis, or programmed cell death, of chon-
Supported in part by grants from Seoul National Universityand Seoul National University Hospital.
Hyun Ah Kim, MD, Yeong Wook Song, MD: Seoul NationalUniversity College of Medicine, Seoul, Korea.
Address reprint requests to Yeong Wook Song, MD, Depart-ment of Internal Medicine, Seoul National University Hospital, 28Yongon-Dong, Chongno-Gu, Seoul 110-744, Korea.
Submitted for publication October 28, 1998; accepted inrevised form February 23, 1999.
1528

drocytes was initially reported in relation to develop-ment and growth, such as in endochondral ossification andin hypertrophic regions of growth plates (8,9). Recently,chondrocytes were shown to undergo apoptosis by additionof nitric oxide or by coupling of Fas/Fas ligand in vitro(10,11), suggesting the possibility that chondrocytes havean inherent programmed cell death pathway that operatesin adults as well as in embryos. Since apoptosis might beresponsible for cartilage breakdown in various arthritides,we tried to define apoptotic chondrocyte death in RAcartilage. We also assessed the expression of 2 relevantmolecules of apoptosis, Bcl-2 and Fas, in RA cartilage.
PATIENTS AND METHODS
Patient population and tissue fixation. Fifteen carti-lage samples were obtained at the time of joint replacementsurgery from 13 RA patients who met the American College ofRheumatology (formerly, the American Rheumatism Associ-ation) criteria for a diagnosis of RA (12). The age range of thepatients was 37–72 years, with a mean age of 59.3 years. Elevenof the samples were from the tibial plateau, 3 from the distalfemur, and 1 from the proximal humerus.
Articular cartilage samples were cut just above thecalcified cartilage with a scalpel. For histology and immuno-histochemistry, 4–6 sections measuring 0.5 3 0.5 cm wereobtained and were fixed in 4% paraformaldehyde at 4°Covernight. For DNA and protein analysis, cartilage distantfrom the gross cartilage–pannus junction was snap frozen andstored at 270°C so that tissues other than cartilage could beexcluded as much as possible.
From the paraformaldehyde-fixed specimens, 2–4pieces of cartilage from each patient were embedded inparaffin. The remaining 2 pieces were stored in 30% sucrose inphosphate buffered saline overnight and, on the next day, werefrozen and cut with a cryotome. Two 6-mm sections, eachfollowed by 12 consecutive 30-mm sections, were cut. The 6-mmsections were put on glass slides and stained with SafraninO–fast green–hematoxylin. The 30-mm sections were put in a24 well plate containing cryostorage solution, consisting of 1%polyvinylpyrrolidone, 30% sucrose, and 30% ethylene glycol,and stored at 220°C until used.
Eight normal tibial plateau cartilage samples frompersons with no previous history of joint disease (mean age35.4 years, range 19–50 years) were obtained at the time ofautopsy and processed as described previously.
Detection of apoptosis. Paraffin-embedded tissue, 2mm thick, was stained with hematoxylin and eosin (H&E), andapoptotic cells were counted. An apoptotic cell was defined asa cell with a condensed, pyknotic nucleus and either shrunken,deeply eosinophilic cytoplasm or fragmentation of the nucleus/cytoplasm. In situ analysis of apoptosis was also performedusing an Apoptag kit (Oncor, Gaithersburg, MD) according tothe manufacturer’s instruction, with a minor modification.Briefly, 2 adjacent 30-mm cryopreserved cartilage sectionsfrom each specimen were pretreated with bovine testicular hyal-uronidase (0.5 mg/ml) at 37°C for 60 minutes and then with
proteinase K (20 mg/ml) for 15 minutes. Endogenous hydrogenperoxidase activity was quenched in 2% hydrogen peroxide.
After a series of rinsings, DNA was end-labeled withterminal deoxynucleotidyl transferase (TdT) and digoxigenin-11-dUTP. After incubation with anti–digoxigenin–peroxidase,color was developed with diaminobenzidine tetrahydrochlo-ride. One of the sections was counterstained with hematoxylinand the other was mounted without counterstain. Incubationwithout TdT served as the negative control.
Electron microscopy. Cartilage samples from 3 RApatients were cut in 0.1 3 0.1–cm pieces and fixed in 2.5%glutaraldehyde solution overnight. After rinsing, the sampleswere postfixed in 1% osmium tetroxide, dehydrated through agraded series of ethanol, and embedded in Epoxy resin. Semithinsections (1 mm) were stained with toluidine blue, and areasshowing the most cells with nuclear condensation were chosen forcutting into ultrathin sections. Sections were counterstained withlead citrate–uranyl acetate and examined with a transmissionelectron microscope (Hitachi 7100; Hitachi, Tokyo, Japan).
DNA electrophoresis. Cartilage samples were frozenat 270°C, milled to fine particles in liquid nitrogen using aSpex 6700 freezer mill (Spex, Edison, NJ), and incubated inlysis buffer containing 100 mM NaCl, 10 mM Tris, 10 mMEDTA, 0.5% sodium dodecyl sulfate (SDS), pH 8.0, andproteinase K (0.1 mg/ml) at 55°C for 16 hours. The supernatantwas incubated with ribonuclease A (50 mg/ml) at 37°C for 1hour. After phenol:chloroform extraction, DNA was quanti-tated by spectrophotometry.
One microgram of the DNA was incubated with 4 unitsof Klenow fragment, 10 mM Tris HCl, 7 mM MgCl2, 0.1 mMdithiothreitol, and 5 mCi of 32P-dATP for 10 minutes. Thereaction was terminated by adding 10 mM EDTA, and unla-beled radioisotopes were removed with 3 cycles of ammoniumacetate precipitation. Labeled DNA was dissolved in 10 mMTris HCl, pH 7.5, 1 mM EDTA, and approximately one-thirdof the samples were run on 1.8% agarose gel at 100V. The gelwas fixed in 10% acetic acid overnight, dried, and subsequentlyexposed to x-ray film.
Immunohistochemistry. Each 30-mm cryopreservedsection was pretreated with chondroitinase ABC (Sigma, St.Louis, MO) at 0.0125 units/50 ml per section for 90 minutes at37°C and were quenched of endogenous hydrogen peroxidaseactivity in 2% hydrogen peroxide. After 30 minutes of blockingin 10% normal goat serum, sections were incubated withmouse anti–Bcl-2 (Neomarker, Fremont, CA) at a dilution of1:600 or with mouse anti-Fas (Transduction Laboratory, Lex-ington, KY) at a dilution of 1:1,000 for 14 hours at 4°C. Abiotin-conjugated goat anti-mouse IgG (Vector, Burlingame,CA) was used as secondary antibody.
The presence and distribution of Bcl-2 and Fas weredetermined by the avidin–biotin–peroxidase complex method(Vector), and diaminobenzidine tetrahydrochloride was usedto develop color. Each staining session included control sec-tions incubated with normal mouse nonimmune serum.
Western blots. Cartilage was milled and extracted intriple-detergent lysis buffer containing 50 mM Tris Cl, 150 mMsodium chloride, 0.1% volume/volume SDS, 1% v/v TritonX-100, 1 mM phenylmethylsulfonyl fluoride, and 1 mg/ml ofaprotinin at 4°C overnight. The protein obtained was concen-trated to 2–5 mg/ml with ice-cold ethanol and resuspended.Protein concentrations were determined with the BCA protein
CHONDROCYTE APOPTOSIS IN RA 1529

assay kit (Pierce, Rockford, IL) and bovine serum albumin asa standard. Samples (20 mg) were combined with gel loadingbuffer (50 mM Tris Cl, pH 6.8, 2% SDS, 10% glycerol, 0.1%bromphenol blue), boiled for 5 minutes, and electrophoresedon a Bio-Rad minigel (12% separating gel; Bio-Rad, Hercules,CA) in 25 mM Tris base, 0.25M glycine, and 0.1% SDS.
The proteins in the other gel were then electrophoreti-cally transferred to polyvinylidene difluoride membrane (Bio-Rad) for 1 hour at 60V in 48 mM Tris, 39 mM glycine, 0.03%SDS, and 20% methanol. The blot was blocked with Trisbuffered saline (50 mM Tris HCl, pH 7.4, 0.15M NaCl)containing 5% nonfat milk at 4°C overnight and incubated withanti–Bcl-2 (Neomarker), 1:1,000, at room temperature for 1hour. The blot was rinsed and incubated with 1:5,000peroxidase-conjugated goat anti-mouse IgG (Bio-Rad) for 1hour. Bound immunoglobulin was detected with enhancedchemiluminescence kit (Amersham, Little Chalfont, UK).Equal loading of protein was verified by Coomassie bluestaining of the gel.
Statistical analysis. Data are expressed as the mean 6SD. Bcl-2– and Fas-positive cells were quantified by a modifi-cation of a previously published method (13). In normal
sections and RA sections in which the cartilage layer waspreserved, sections were divided into 2 zones: the superficialand upper intermediate layers versus the lower intermediateand deeper zone. The former includes chondrocytes charac-terized by a cuboidal appearance following the splitline patternof the dominant fibrillar orientation and the upper 50% of thetransitional zone characterized by round or spheroidal cells;the latter includes the cells of the lower 50% of the transitionalzone and those aligned in column.
Each cartilage section was divided into 6 microscopicfields (403 magnification; Olympus, Tokyo, Japan), 3 fieldsfrom each zone. The percentage of positive chondrocytes werecounted at 4003 magnification within these defined fields andwere estimated separately for each layer of cartilage and forthe full-thickness cartilage (superficial and deep layer). If thecartilage layer was not definable in RA cartilage, 6 microscopicfields at 403 magnification were chosen, and the percentage ofpositive chondrocytes were counted by the same method.
For counting apoptotic cells in H&E-stained specimens,the fields were selected in the same manner as for immunohisto-chemistry counting. Apoptotic cells were counted at 1,0003magnification. The percentages of apoptotic cells and of chon-drocytes positive for Bcl-2 and Fas were compared between 8normal and 11 RA tibial plateau cartilage samples for statisticalanalyses by Wilcoxon’s rank sum test. Statistical significance wasestablished at the 95% confidence level (P , 0.05).
RESULTS
Morphology and apoptotic index. Grossly, RAcartilage samples were markedly thinned, and somesamples had only a scant amount of paper-thin cartilageleft on the subchondral bone. Four of the 11 tibialplateau samples had areas of mild involvement, withonly surface irregularities. In these cases, specimenswere collected separately from severely and mildly in-volved areas. Microscopic examination revealed cartilage–pannus junctions, loss of proteoglycan, and disorganiza-tion of the cartilage architecture. In contrast toosteoarthritis (OA) samples, in which deep cartilagefibrillation is one of the hallmark pathologic findings, itwas found in only 33% of the samples examined. Chon-drocyte cloning was observed only rarely. Smooth, ema-ciated cartilage covered with hyperplastic synovium was
Figure 1. Hematoxylin and eosin–stained rheumatoid arthritis carti-lage samples cut into 2-mm sections. A, Arrow shows an apoptoticchondrocyte adjacent to 2 normal chondrocytes. B, Arrows showclustering of apoptotic chondrocytes in a degenerated cartilage matrix.(Original magnification 3 1,000.)
Table 1. Percentages of apoptotic, Bcl-2–positive, and Fas-positivecells in RA and normal chondrocytes from tibial plateau cartilage*
RA cartilage(n 5 11)
Normal cartilage(n 5 8) P
Apoptotic cells 3.01 6 1.1 0.15 6 0.25 ,0.001Bcl-2–positive cells 23.3 6 15.9 43.1 6 14.9 0.023Fas-positive cells 35.5 6 16.3 34.2 6 20.6 NS
* Values are the mean 6 SD. Statistical analysis was done usingWilcoxon’s rank sum test. RA 5 rheumatoid arthritis; NS 5 notsignificant.
1530 KIM AND SONG

found in 40% of samples, and the cartilage layer was notdiscernible in many specimens.
While normal cartilage showed rare apoptoticcells (mean 6 SD 0.15 6 0.25%, range 0–0.7), RA
cartilage frequently showed apoptotic chondrocytes withtypical intensely basophilic chromatin condensationalong the nuclear membrane and shrunken cytoplasm(3.01 6 1.1%) (Figure 1A and Table 1). There was a
Figure 2. TUNEL staining of normal and rheumatoid arthritis (RA) cartilage. A, Normal cartilage, showing only backgroundstaining. B, RA cartilage, showing numerous TUNEL-positive chondrocytes distributed in the entire cartilage layer. C, Hematoxylincounterstaining reveals ;30% TUNEL-positive chondrocytes in RA cartilage. D, High-power view of boxed area in C. E, Negativecontrol (terminal deoxynucleotidyl transferase omitted) section of RA cartilage. Results are representative of 8 normal and 15 RAcartilage samples. (Original magnification 3 100 in A, B, C, and E; 3 400 in D.)
CHONDROCYTE APOPTOSIS IN RA 1531
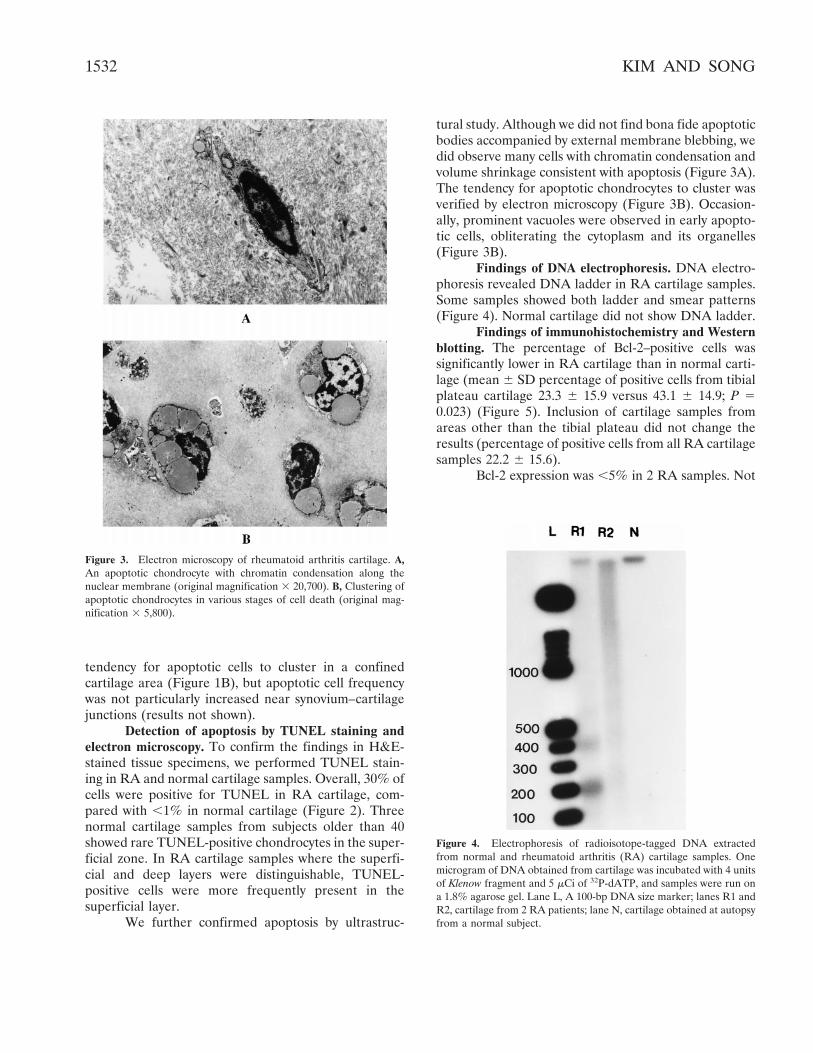
tendency for apoptotic cells to cluster in a confinedcartilage area (Figure 1B), but apoptotic cell frequencywas not particularly increased near synovium–cartilagejunctions (results not shown).
Detection of apoptosis by TUNEL staining andelectron microscopy. To confirm the findings in H&E-stained tissue specimens, we performed TUNEL stain-ing in RA and normal cartilage samples. Overall, 30% ofcells were positive for TUNEL in RA cartilage, com-pared with ,1% in normal cartilage (Figure 2). Threenormal cartilage samples from subjects older than 40showed rare TUNEL-positive chondrocytes in the super-ficial zone. In RA cartilage samples where the superfi-cial and deep layers were distinguishable, TUNEL-positive cells were more frequently present in thesuperficial layer.
We further confirmed apoptosis by ultrastruc-
tural study. Although we did not find bona fide apoptoticbodies accompanied by external membrane blebbing, wedid observe many cells with chromatin condensation andvolume shrinkage consistent with apoptosis (Figure 3A).The tendency for apoptotic chondrocytes to cluster wasverified by electron microscopy (Figure 3B). Occasion-ally, prominent vacuoles were observed in early apopto-tic cells, obliterating the cytoplasm and its organelles(Figure 3B).
Findings of DNA electrophoresis. DNA electro-phoresis revealed DNA ladder in RA cartilage samples.Some samples showed both ladder and smear patterns(Figure 4). Normal cartilage did not show DNA ladder.
Findings of immunohistochemistry and Westernblotting. The percentage of Bcl-2–positive cells wassignificantly lower in RA cartilage than in normal carti-lage (mean 6 SD percentage of positive cells from tibialplateau cartilage 23.3 6 15.9 versus 43.1 6 14.9; P 50.023) (Figure 5). Inclusion of cartilage samples fromareas other than the tibial plateau did not change theresults (percentage of positive cells from all RA cartilagesamples 22.2 6 15.6).
Bcl-2 expression was ,5% in 2 RA samples. Not
Figure 3. Electron microscopy of rheumatoid arthritis cartilage. A,An apoptotic chondrocyte with chromatin condensation along thenuclear membrane (original magnification 3 20,700). B, Clustering ofapoptotic chondrocytes in various stages of cell death (original mag-nification 3 5,800).
Figure 4. Electrophoresis of radioisotope-tagged DNA extractedfrom normal and rheumatoid arthritis (RA) cartilage samples. Onemicrogram of DNA obtained from cartilage was incubated with 4 unitsof Klenow fragment and 5 mCi of 32P-dATP, and samples were run ona 1.8% agarose gel. Lane L, A 100-bp DNA size marker; lanes R1 andR2, cartilage from 2 RA patients; lane N, cartilage obtained at autopsyfrom a normal subject.
1532 KIM AND SONG

only was the percentage of positive cells lower, but thestaining intensity was very weak in RA cartilage. Bcl-2expression tended to decrease in severely involved car-tilage areas compared with mildly involved areas in the
same patient, but statistical data were not calculatedbecause of the small sample number. In normal carti-lage, Bcl-2–positive cells were more frequent in thesuperficial layer (Figure 5). This result was also con-
Figure 5. Immunohistochemical localization of Bcl-2–positive cells in normal and rheu-matoid arthritis (RA) cartilage. Frozen sections of cartilage were incubated with Bcl-2monoclonal antibody and then stained by the immunoperoxidase method, followed byhematoxylin counterstaining. A, Normal cartilage, showing Bcl-2–positive cells predomi-nantly in the superficial layer. B, Normal cartilage, showing a Bcl-2–positive cell withcytoplasmic staining. C, Normal cartilage incubated with nonimmune sera, showing noimmunohistochemical staining. D, RA cartilage, with only rare Bcl-2–positive cells. Resultsare representative of 8 normal and 15 RA cartilage samples. (Original magnification 3 200in A; 3 100 in C and D; 3 1,000 in B.)
CHONDROCYTE APOPTOSIS IN RA 1533

firmed by Western blotting, which revealed a muchfainter band in RA cartilage samples than in normalsamples (Figure 6).
There was a high degree of variability in Fasexpression from specimen to specimen, but we failed todemonstrate any significant difference between RA andnormal cartilage samples (percentage of positive cells35.5 6 16.3 versus 34.2 6 20.6) (Figure 7). In contrastwith Bcl-2, RA cartilage frequently expressed Fas inmid-zone and deep-zone chondrocytes as the superficiallayer was lost.
DISCUSSION
Although RA patients experience intractablepain and discomfort resulting from chronic inflamma-tion in the synovium, it is the cartilage loss and subse-quent joint failure that leads to eventual disability. Themechanism leading to cartilage erosion in RA has beenconsidered to be a secondary phenomenon, caused byinflammatory mediators and proteolytic enzymes se-creted by chronic infiltration of cells originating from thesynovium. Most of the histologic research on RA carti-lage has concentrated on the cartilage–pannus junctionbecause this is the site where cartilage erosion begins.Very few studies have focused on chondrocytes in RAcartilage, however.
In this study, we verified chondrocyte apoptosisin RA cartilage, both morphologically and biochemi-cally. What is the implication of apoptotic chondrocytedeath in RA cartilage? Despite the widespread consen-sus that the pathway of cartilage degradation involvesenzymes such as collagenase and stromelysin and cyto-
kines that are produced by inflammatory cells andsynovial cells, some clinical observations contradict theprimary role of synovial inflammation in RA erosion.Recently, correlation between ongoing synovitis anderosive progression in individual joints of the rheuma-
Figure 6. Western blot analysis of Bcl-2 expression. Cartilage wasextracted in triple-detergent lysis buffer containing 50 mM Tris Cl, 150mM sodium chloride, 0.1% v/v sodium dodecyl sulfate, 1% v/v TritonX-100, 1 mM phenylmethylsulfonyl fluoride, and 1 mg/ml of aprotinin,and 20 mg of each sample was electrophoresed on 12% polyacrylamidegels, transferred to polyvinylidene difluoride membranes, and blottedwith anti–Bcl-2. Lane C, Control protein from tonsil; lanes R1–R3,protein from 3 rheumatoid arthritis patients’ tibial plateaus; lanes N1and N2, protein from 2 normal tibial plateaus. Molecular size markers(in kd) are shown at the left.
Figure 7. Immunohistochemical localization of Fas-positive cells innormal and rheumatoid arthritis (RA) cartilage. Frozen sections ofnormal and RA cartilage samples were incubated with anti-Fasmonoclonal antibody and stained by the immunoperoxidase method,followed by hematoxylin counterstaining. A, Normal cartilage, showingFas-positive cells predominantly in the superficial layer. B, RA carti-lage, showing Fas-positive cells mainly in the deep-zone chondrocytes.Results are representative of 8 normal and 15 RA cartilage samples.(Original magnification 3 200.)
1534 KIM AND SONG

toid hand was reported to be weak (mean correlationr 5 0.248) (14). This implies that cartilage erosions andsynovitis might not represent the same pathologic pro-cess and that there might be a possibility that chondro-cytes have an inherent and independent degradationpathway that operates by itself in diseased cartilage. Inaccordance with this assumption, chondrocytes are re-ported to behave in vitro in a way that promotes articularcartilage loss; they have receptors for cytokines such asIL-1, TNFa, and TNFb, and respond to these ligandswith increased production of prostaglandin E2 and met-alloproteinases (15).
Aside from the pathway of enzymatic or freeradical–mediated degradation of extracellular matrix,there is some evidence that chondrocyte death may beimportant in the pathogenesis of various forms of arthri-tis. Blanco et al (16) were the first to report in situapoptotic chondrocyte death in human OA cartilage byTUNEL staining and electron microscopy. We have alsoobserved increased apoptosis in severely involved areascompared with that in mildly involved areas of OAcartilage obtained from the same patient, which indi-cates the role of accelerated apoptosis in the progressionof OA lesions (Kim et al: unpublished observations). Inthe present study, the frequency of apoptotic chondro-cytes in RA cartilage was more than twice that ofseverely involved OA cartilage (percentage of apoptoticcells 3.01 versus 1.42).
There was about a 10-fold difference in thefrequency of apoptotic cells by light microscopy andTUNEL staining. Because we strictly defined apoptoticcells as those that have both chromatin condensationand cytoplasmic change and/or nuclear/cytoplasmic frag-mentation, it is possible that we counted only those cellsthat are in the final stage of apoptosis. On the otherhand, TUNEL staining detects cells that contain DNAfragmented nuclei, resulting in the detection of a widerrange of apoptotic cells (17). Because only 4 of thesamples had areas of relatively well-preserved cartilagestructure, we could not perform statistical analyses, butapoptotic chondrocytes tended to appear more fre-quently in severely involved areas than in mildly involvedareas (percentage of apoptotic cells 4.4 versus 2.6). Itseems plausible, therefore, that the rate of apoptoticchondrocyte death increases as rheumatoid cartilagelesions progress. While our RA patients were consider-ably older than our normal control subjects, it is notlikely that this increase in apoptosis results merely fromthe effect of degenerative changes that accompany ag-ing. The apoptotic rate of 30% as measured by theTUNEL method is far higher than the reported rate in
OA cartilage (16). Since apoptosis is a swift form of celldeath that spans only a few hours, this high rate wouldbe incompatible with a slowly progressing degenerativeprocess.
Due to the fleeting appearance of apoptotic cellsmorphologically and the possible risk of false-positiveresults from the TUNEL method, which occasionallystains necrotic cells (18), we further confirmed apoptosisby ultrastructural study and DNA ladder assay. Both ofthese latter studies reconfirmed that apoptosis doesoccur in RA cartilage, but we did not find bona fideapoptotic bodies with external membrane blebbing.Considering the dynamics of apoptotic cell death andthe limitations of examination by electron microscopy,numerous sections are required to catch the moment ofapoptotic cell death.
To our knowledge, there has been no report ofthe expression of relevant molecules of apoptosis in RAcartilage. Our study demonstrated that Bcl-2 expressionwas markedly decreased in RA cartilage samples, whileFas did not show a significant difference between RAand normal cartilage samples. Considering the fact thatBcl-2 promotes cell survival by forming heterodimerswith Bax, which otherwise accelerates apoptosis (19,20),this decrease in Bcl-2 expression was expected. On theother hand, Erlacher et al (21) demonstrated by reversetranscriptase–polymerase chain reaction that Bcl-2 mes-senger RNA tended to be up-regulated in OA cartilagecompared with normal cartilage.
We also observed that Bcl-2 expression was sig-nificantly higher in lesional areas than in nonlesionalareas in OA cartilage (Kim et al: unpublished observa-tions). The explanation for this contradictory findingcould be that OA chondrocytes still preserve the abilityto compensate for ongoing chondrocyte death by up-regulating Bcl-2, while RA chondrocytes do not. Thisleads to the assumption that chondrocyte apoptosis playsa role of remodeling in OA cartilage, in contrast with arole of deterioration in RA cartilage. Because Bcl-2expression was prominent in the superficial layer ofnormal cartilage, decreased levels of Bcl-2 expression inRA cartilage may merely result from the loss of thislayer, without any qualitative change in chondrocytes.OA chondrocytes seem to redistribute Bcl-2 (Kim et al:unpublished observations) and Fas expression in themidzone and deep zone as the superficial area is de-stroyed (11). Although RA cartilage does redistributeFas expression as the superficial layer is lost, as reflectedby the nonsignificant difference in the level of Fasexpression compared with normal cartilage, the samedoes not hold true for Bcl-2. Whether the failure to
CHONDROCYTE APOPTOSIS IN RA 1535

express Bcl-2 in the deep zone of RA cartilage is due tothe rather swift process of joint destruction or to thepathologic process specific to RA is a matter for specu-lation.
Our cross-sectional study does not give answersabout whether apoptotic chondrocyte death is a result orthe cause of cartilage damage in RA. If chondrocytedeath occurs, matrix synthesis would cease, and thisobviously will lead to cartilage degradation. On the otherhand, it is generally accepted that except for circulatingblood cells, most cells require attachment to one anotheror to extracellular matrix for proper growth, function,and survival; otherwise, they would die by apoptosis, andthis dependence of cell growth and survival on substrateattachment is known as anchorage dependence (22).Since loss of proteoglycan and perturbation of collagenfibrils is one of the main pathologic features of cartilagedegradation, this provides an ample environment fordisturbance of chondrocyte anchorage to extracellularmatrix.
There is also the possibility that chondrocytedeath and matrix loss form a vicious cycle, the progres-sion of one having a detrimental effect on the other. Itwould be important to see the relationship betweenchondrocyte apoptosis and matrix loss prospectively topursue this issue. In addition, in vitro studies have shownthat chondrocytes can survive for weeks at high celldensity in the absence of other cell types, serum, orexogenous proteins, but they die by apoptosis at low celldensity, indicating the need for an autocrine signal fromother chondrocytes for survival (23). Since cell deathwould abolish the presumed autocrine signal to adjacentcells, our finding that chondrocyte apoptosis tends tooccur in clusters is consistent with this observation.
A couple of difficulties can be raised with regardto our study. First, although we tried to stick to theblinded protocol for assessing apoptotic cells andimmune-positive cells, the obvious morphologic changein RA cartilage defied this attempt. However, thisproblem was partly compensated for by DNA ladderanalysis and Western blotting, both of which revealedobjective findings consistent with the quantitative re-sults. Second, while we excluded gross pannus–cartilagejunction for DNA and protein preparation, a smallamount of synovium could have been processed simul-taneously, since it was very common to see a layer ofhyperplastic synovium covering the surface of RA carti-lage. For Western blotting, however, this would haveaffected the result in a way that would increase the Bcl-2signal rather than if only RA chondrocyte protein wasprocessed, because both synovial lining cells and infil-
trating T cells are reported to be an ample source ofBcl-2 protein (24,25).
In conclusion, the present study demonstrateschondrocyte apoptosis in RA cartilage by H&E staining,TUNEL staining, electron microscopy, and DNA ladderassay. This was accompanied by a down-regulation of theapoptosis-protecting molecule, Bcl-2. Considering thefact that apoptosis is a highly regulated process in abiologic system, it would be important to delineate theevents that culminate in apoptotic chondrocyte death inRA cartilage.
ACKNOWLEDGMENTS
The authors thank Drs. Sang-Cheol Seong, Dae KyungBae, and Hyung In Yang for providing patient specimens.
REFERENCES
1. Bromley M, Woolley DE. Histopathology of the rheumatoidlesion: identification of cell types at sites of cartilage erosion.Arthritis Rheum 1984;27:857–63.
2. Chu CQ, Field M, Feldmann M, Maini RN. Localization of tumornecrosis factor a in synovial tissues and at the cartilage-pannusjunction in patients with rheumatoid arthritis. Arthritis Rheum1991;34:1125–32.
3. Chu CQ, Field M, Allard S, Abney E, Feldmann M, Maini RN.Detection of cytokines at the cartilage-pannus junction in patientswith rheumatoid arthritis: implications for the role of cytokines incartilage destruction and repair. Br J Rheumatol 1992;32:653–61.
4. Scott DL, Spector TD, Pullar T, McConkey B. What should wehope to achieve when treating rheumatoid arthritis? Ann RheumDis 1989;48:256–61.
5. Plant MJ, Saklatvala J, Borg AA, Jones PW, Dawes PT. Measure-ment and prediction of radiological progression in early rheuma-toid arthritis. J Rheumatol 1994;21:1808–13.
6. Firestein GS, Nguyen K, Aupperle KR, Teo M, Boyle DL, ZvaiflerNJ. Apoptosis in rheumatoid arthritis. Am J Pathol 1996;149:2143–51.
7. Aupperle KR, Boyle DL, Hendrix M, Seftor EA, Zvaifler NJ,Barbosa M, et al. Regulation of synoviocyte proliferation, apopto-sis, and invasion by the p53 tumor suppressor gene. Am J Pathol1998;152:1091–8.
8. Gibson GJ, Kohler WJ, Schaffler MB. Chondrocyte apoptosis inendochondral ossification of chick sterna. Dev Dyn 1995;203:468–76.
9. Hatori M, Klatte KJ, Teixeira CC, Shapiro IM. End labelingstudies of fragmented DNA in the avian growth plate: evidence ofapoptosis in terminally differentiated chondrocytes. J Bone MinerRes 1995;10:1960–8.
10. Blanco FJ, Ochs RL, Schwarz H, Lotz M. Chondrocyte apoptosisinduced by nitric oxide. Am J Pathol 1995;146:75–85.
11. Hashimoto S, Setareh M, Ochs RL, Lotz M. Fas/Fas ligandexpression and induction of apoptosis in chondrocytes. ArthritisRheum 1997;40:1749–55.
12. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF,Cooper NS, et al. The American Rheumatism Association 1987revised criteria for the classification of rheumatoid arthritis.Arthritis Rheum 1988;31:315–24.
13. Pelletier J-P, Mineau F, Raynauld J-P, Woessner JF Jr, Gunja-Smith Z, Martel-Pelletier J. Intraarticular injections with methyl-prednisolone acetate reduce osteoarthritic lesions in parallel with
1536 KIM AND SONG

chondrocyte stromelysin synthesis in experimental osteoarthritis.Arthritis Rheum 1994;37:414–23.
14. Kirwan JR. The relationship between synovitis and erosions inrheumatoid arthritis. Br J Rheumatol 1997;36:225–8.
15. Woolley DE, Tetlow LC. Observations on the microenvironmentalnature of cartilage degradation in rheumatoid arthritis. AnnRheum Dis 1997;56:151–61.
16. Blanco FJ, Guitian R, Vazquez-Martul E, de Toro FJ, Galdo F.Osteoarthritis chondrocytes die by apoptosis: a possible pathwayfor osteoarthritis pathology. Arthritis Rheum 1998;41:284–9.
17. Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of pro-grammed cell death in situ via specific labeling of nuclear DNAfragmentation. J Cell Biol 1992;119:493–501.
18. Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K,Bursch W, Schulte-Hermann R. In situ detection of fragmentedDNA fails to discriminate among apoptosis, necrosis, and autolyticcell death: a cautionary note. Hepatology 1995;21:1465–8.
19. Korsmeyer SJ. Bcl-2 initiates a new category of oncogenes:regulators of cell death. Blood 1992;80:879–86.
20. Knudson CM, Tung KS, Tourtellotte WG, Brown GA, KorsmeyerSJ. Bax-deficient mice with lymphoid hyperplasia and male germcell death. Science 1995;270:96–9.
21. Erlacher L, Maier R, Ullich R, Kiener H, Aringer M, Menschik M,et al. Differential expression of the protooncogene bcl-2 in normaland osteoarthritic human articular cartilage. J Rheumatol 1995;22:926–31.
22. Ruoslahti E. Stretching is good for a cell. Science 1997;276:1345–6.
23. Ishizaki Y, Burne JF, Raff MC. Autocrine signals enable chon-drocytes to survive in culture. J Cell Biol 1994;126:1069–77.
24. Sugiyama M, Tsukazaki T, Yonekura A, Matsuzaki S, YamashitaS, Iwasaki K. Localization of apoptosis and expression of apoptosisrelated protein in the synovium of patients with rheumatoidarthritis. Ann Rheum Dis 1996;55:442–9.
25. Matsumoto S, Muller-Ladner U, Gay RE, Nishoika K, Gay S.Ultrastructural demonstration of apoptosis, Fas, and Bcl-2 expressionof rheumatoid synovial fibroblasts. J Rheumatol 1996;23:1345–52.
CHONDROCYTE APOPTOSIS IN RA 1537
Copyright © 2022 FDOKUMEN




















