
Hepatocyte growth factor and its receptor are required for malaria infection
7
ARTICLES 1 Instituto Gulbenkian de Ciência, Rua da Quinta Grande 6, 2780-156 Oeiras, Portugal. 2 New York University School of Medicine, Department of Medical and Molecular Parasitology, 341 E 25 th Street, New York, New York 10010, USA. 3 Institute for Cancer Research and Treatment, University of Torino School of Medicine, Strada Provinciale 142, 10060 Candiolo (Torino), Italy. 4 Departamento de Biología, Facultad de Ciencias, Universidad Autónoma de Madrid, 28049 Madrid, Spain. Correspondence should be addressed to M.M.M. ([email protected]). Published online 12 October 2003; doi:10.1038/nm947 Hepatocyte growth factor and its receptor are required for malaria infection Margarida Carrolo 1 , Silvia Giordano 3 , Laura Cabrita-Santos 1,2 , Simona Corso 3 , Ana M Vigário 1 , Susana Silva 1 , Patricia Leirião 1 , Daniel Carapau 1 , Rosario Armas-Portela 4 , Paolo M Comoglio 3 , Ana Rodriguez 2 & Maria M Mota 1 Plasmodium, the causative agent of malaria, must first infect hepatocytes to initiate a mammalian infection. Sporozoites migrate through several hepatocytes, by breaching their plasma membranes, before infection is finally established in one of them. Here we show that wounding of hepatocytes by sporozoite migration induces the secretion of hepatocyte growth factor (HGF), which renders hepatocytes susceptible to infection. Infection depends on activation of the HGF receptor, MET, by secreted HGF. The malaria parasite exploits MET not as a primary binding site, but as a mediator of signals that make the host cell susceptible to infection. HGF/MET signaling induces rearrangements of the host-cell actin cytoskeleton that are required for the early development of the parasites within hepatocytes. Our findings identify HGF and MET as potential targets for new approaches to malaria prevention. NATURE MEDICINE VOLUME 9 | NUMBER 11 | NOVEMBER 2003 1363 After Plasmodium sporozoites are injected into the mammalian host by infected mosquitoes, hepatocyte infection is the first obligatory step in the establishment of a malarial infection. Sporozoites traverse the cytosol of several hepatocytes before finally invading one by form- ing a parasitophorous vacuole 1 . During migration through cells before infection, Plasmodium sporozoites disrupt the plasma mem- branes of the traversed cells. Some of these cells die, but most sur- vive 1 . When the plasma membrane of a cell is disrupted, growth factors and other proteins stored in the cytosol are released to the extracellular environment 2,3 . Cell wounding also increases the expres- sion of certain growth factors that are secreted later, after the wound has been repaired 4 . Thus, wounded cells release factors that influence the neighboring environment. We hypothesized that hepatocytes wounded by sporozoites release one or more factors that render neighboring hepatocytes susceptible to infection. RESULTS Release of an ‘infection susceptibility–inducing factor’ (ISIF) We first searched for such an ISIF in the supernatant of cultures con- taining the mouse hepatoma cells Hepa1-6 (ref. 5) and P. yoelii sporo- zoites (referred to as mH/Py-conditioned medium). To detect ISIF activity, we incubated new hepatoma cells with mH/Py-conditioned medium for different periods of time, followed by washes and incu- bation with P. yoelii sporozoites. Infection was examined 24 h later by staining the exoerythrocytic forms (EEFs) of the parasite. Pretreatment of cells with mH/Py-conditioned medium increased the level of infection, compared with control cells incubated with fresh medium (Fig. 1a). The greatest enhancement of susceptibility to infection was observed in hepatoma cells that were pretreated for 1 h with mH/Py-conditioned medium (Fig. 1a). Medium conditioned with heat-inactivated sporozoites did not increase susceptibility to infection (Fig. 1b). Because sporozoites are obtained by dissection of infected mosquito salivary glands, we also tested conditioned media obtained from cultures containing hepatoma cells and salivary gland material from uninfected mosquitoes. Medium conditioned in this way was unable to increase the susceptibility to infection (Fig. 1b). Release of ISIF by wounded host cells To investigate the source of ISIF (sporozoites or hepatocytes) and the requirements for its release, we wounded Hepa1-6 cells using mechanical stress. Wounded cells were placed in a tissue-culture well and supernatant was collected after 1 h. We incubated Hepa1-6 cells with this supernatant before adding P. yoelii sporozoites. We found an increase in infection in these cells, similar to that observed using mH/Py-conditioned medium (Fig. 1c). This finding indicates that ISIF is not derived from sporozoites, but is released from hepatocytes as a consequence of wounding. Host-species specificity of ISIF activity To test whether the same effect on infectivity would be observed with different Plasmodium species infecting different cell lines, we gener- ated conditioned media by culturing P. berghei sporozoites with mouse Hepa1-6 cells (mH/Pb-conditioned medium) or human hepatoma HepG2 cells (hH/Pb-conditioned medium). Both © 2003 Nature Publishing Group http://www.nature.com/naturemedicine
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Hepatocyte growth factor and its receptor are required for malaria infection

A R T I C L E S
1Instituto Gulbenkian de Ciência, Rua da Quinta Grande 6, 2780-156 Oeiras, Portugal. 2New York University School of Medicine, Department of Medical andMolecular Parasitology, 341 E 25th Street, New York, New York 10010, USA. 3 Institute for Cancer Research and Treatment, University of Torino School of Medicine,Strada Provinciale 142, 10060 Candiolo (Torino), Italy. 4Departamento de Biología, Facultad de Ciencias, Universidad Autónoma de Madrid, 28049 Madrid, Spain.Correspondence should be addressed to M.M.M. ([email protected]).
Published online 12 October 2003; doi:10.1038/nm947
Hepatocyte growth factor and its receptor are required formalaria infectionMargarida Carrolo1, Silvia Giordano3, Laura Cabrita-Santos1,2, Simona Corso3, Ana M Vigário1, Susana Silva1,Patricia Leirião1, Daniel Carapau1, Rosario Armas-Portela 4, Paolo M Comoglio3, Ana Rodriguez2 & Maria M Mota1
Plasmodium, the causative agent of malaria, must first infect hepatocytes to initiate a mammalian infection. Sporozoites migratethrough several hepatocytes, by breaching their plasma membranes, before infection is finally established in one of them. Here weshow that wounding of hepatocytes by sporozoite migration induces the secretion of hepatocyte growth factor (HGF), which rendershepatocytes susceptible to infection. Infection depends on activation of the HGF receptor, MET, by secreted HGF. The malariaparasite exploits MET not as a primary binding site, but as a mediator of signals that make the host cell susceptible to infection.HGF/MET signaling induces rearrangements of the host-cell actin cytoskeleton that are required for the early development of theparasites within hepatocytes. Our findings identify HGF and MET as potential targets for new approaches to malaria prevention.
NATURE MEDICINE VOLUME 9 | NUMBER 11 | NOVEMBER 2003 1363
After Plasmodium sporozoites are injected into the mammalian hostby infected mosquitoes, hepatocyte infection is the first obligatorystep in the establishment of a malarial infection. Sporozoites traversethe cytosol of several hepatocytes before finally invading one by form-ing a parasitophorous vacuole1. During migration through cellsbefore infection, Plasmodium sporozoites disrupt the plasma mem-branes of the traversed cells. Some of these cells die, but most sur-vive1. When the plasma membrane of a cell is disrupted, growthfactors and other proteins stored in the cytosol are released to theextracellular environment2,3. Cell wounding also increases the expres-sion of certain growth factors that are secreted later, after the woundhas been repaired4. Thus, wounded cells release factors that influencethe neighboring environment. We hypothesized that hepatocyteswounded by sporozoites release one or more factors that renderneighboring hepatocytes susceptible to infection.
RESULTSRelease of an ‘infection susceptibility–inducing factor’ (ISIF)We first searched for such an ISIF in the supernatant of cultures con-taining the mouse hepatoma cells Hepa1-6 (ref. 5) and P. yoelii sporo-zoites (referred to as mH/Py-conditioned medium). To detect ISIFactivity, we incubated new hepatoma cells with mH/Py-conditionedmedium for different periods of time, followed by washes and incu-bation with P. yoelii sporozoites. Infection was examined 24 h later by staining the exoerythrocytic forms (EEFs) of the parasite.Pretreatment of cells with mH/Py-conditioned medium increased thelevel of infection, compared with control cells incubated with fresh
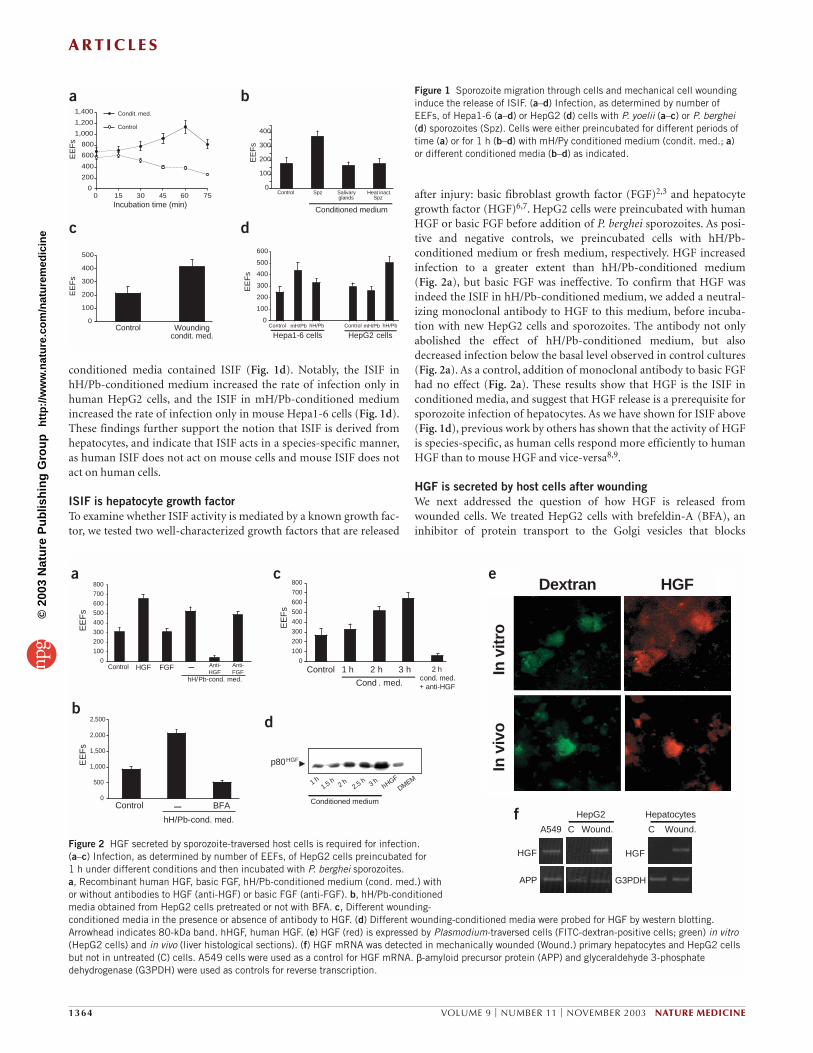
medium (Fig. 1a). The greatest enhancement of susceptibility toinfection was observed in hepatoma cells that were pretreated for 1 hwith mH/Py-conditioned medium (Fig. 1a). Medium conditionedwith heat-inactivated sporozoites did not increase susceptibility toinfection (Fig. 1b). Because sporozoites are obtained by dissection ofinfected mosquito salivary glands, we also tested conditioned mediaobtained from cultures containing hepatoma cells and salivary glandmaterial from uninfected mosquitoes. Medium conditioned in thisway was unable to increase the susceptibility to infection (Fig. 1b).
Release of ISIF by wounded host cellsTo investigate the source of ISIF (sporozoites or hepatocytes) and therequirements for its release, we wounded Hepa1-6 cells usingmechanical stress. Wounded cells were placed in a tissue-culture welland supernatant was collected after 1 h. We incubated Hepa1-6 cellswith this supernatant before adding P. yoelii sporozoites. We found anincrease in infection in these cells, similar to that observed usingmH/Py-conditioned medium (Fig. 1c). This finding indicates thatISIF is not derived from sporozoites, but is released from hepatocytesas a consequence of wounding.
Host-species specificity of ISIF activityTo test whether the same effect on infectivity would be observed withdifferent Plasmodium species infecting different cell lines, we gener-ated conditioned media by culturing P. berghei sporozoites withmouse Hepa1-6 cells (mH/Pb-conditioned medium) or humanhepatoma HepG2 cells (hH/Pb-conditioned medium). Both
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rem
edic
ine
A R T I C L E S
conditioned media contained ISIF (Fig. 1d). Notably, the ISIF inhH/Pb-conditioned medium increased the rate of infection only inhuman HepG2 cells, and the ISIF in mH/Pb-conditioned mediumincreased the rate of infection only in mouse Hepa1-6 cells (Fig. 1d).These findings further support the notion that ISIF is derived fromhepatocytes, and indicate that ISIF acts in a species-specific manner,as human ISIF does not act on mouse cells and mouse ISIF does notact on human cells.
ISIF is hepatocyte growth factorTo examine whether ISIF activity is mediated by a known growth fac-tor, we tested two well-characterized growth factors that are released
after injury: basic fibroblast growth factor (FGF)2,3 and hepatocytegrowth factor (HGF)6,7. HepG2 cells were preincubated with humanHGF or basic FGF before addition of P. berghei sporozoites. As posi-tive and negative controls, we preincubated cells with hH/Pb-conditioned medium or fresh medium, respectively. HGF increasedinfection to a greater extent than hH/Pb-conditioned medium (Fig. 2a), but basic FGF was ineffective. To confirm that HGF wasindeed the ISIF in hH/Pb-conditioned medium, we added a neutral-izing monoclonal antibody to HGF to this medium, before incuba-tion with new HepG2 cells and sporozoites. The antibody not onlyabolished the effect of hH/Pb-conditioned medium, but alsodecreased infection below the basal level observed in control cultures(Fig. 2a). As a control, addition of monoclonal antibody to basic FGFhad no effect (Fig. 2a). These results show that HGF is the ISIF in conditioned media, and suggest that HGF release is a prerequisite forsporozoite infection of hepatocytes. As we have shown for ISIF above(Fig. 1d), previous work by others has shown that the activity of HGFis species-specific, as human cells respond more efficiently to humanHGF than to mouse HGF and vice-versa8,9.
HGF is secreted by host cells after woundingWe next addressed the question of how HGF is released fromwounded cells. We treated HepG2 cells with brefeldin-A (BFA), aninhibitor of protein transport to the Golgi vesicles that blocks
1364 VOLUME 9 | NUMBER 11 | NOVEMBER 2003 NATURE MEDICINE
0
100
200
300
400
EE
Fs
Control Spz Salivary glands
Heat inact.Spz
Conditioned medium
0
100
200
300
400
500
600E
EF
s
Control mH/Pb hH/Pb
Hepa1-6 cellsControl mH/Pb hH/Pb
HepG2 cells
0
100
200
300
400
500
EE
Fs
Woundingcondit. med.
Control
0
200
400
600
800
1,000
1,200
1,400
EE
Fs
Incubation time (min)
Control
Condit. med.
0 15 30 60 7545
a b
c d
Figure 1 Sporozoite migration through cells and mechanical cell woundinginduce the release of ISIF. (a–d) Infection, as determined by number ofEEFs, of Hepa1-6 (a–d) or HepG2 (d) cells with P. yoelii (a–c) or P. berghei(d) sporozoites (Spz). Cells were either preincubated for different periods oftime (a) or for 1 h (b–d) with mH/Py conditioned medium (condit. med.; a)or different conditioned media (b–d) as indicated.
hH/Pb-cond. med.
0
500
1,000
1,500
2,000
2,500
EE
Fs
Control – BFA
hH/Pb-cond. med.
0
100
200
300
400
500
600
700
800
EE
Fs
Control FGFHGF Anti-HGF
Anti-FGF
–
HGF
APP
A549 C Wound.
HepG2
C Wound.
Hepatocytes
HGF
G3PDH
a
b
e
f
In v
ivo
In v
itro
Dextran HGF
0
100
200
300
400
500
600
700
800
EE
Fs
Control 1 h 2 h 3 h
Cond . med.
2 h cond. med. + anti-HGF
1 h1.5 h 2 h
3 h2.5 h
hHGF
DMEM
Conditioned medium
p80HGF
c
d
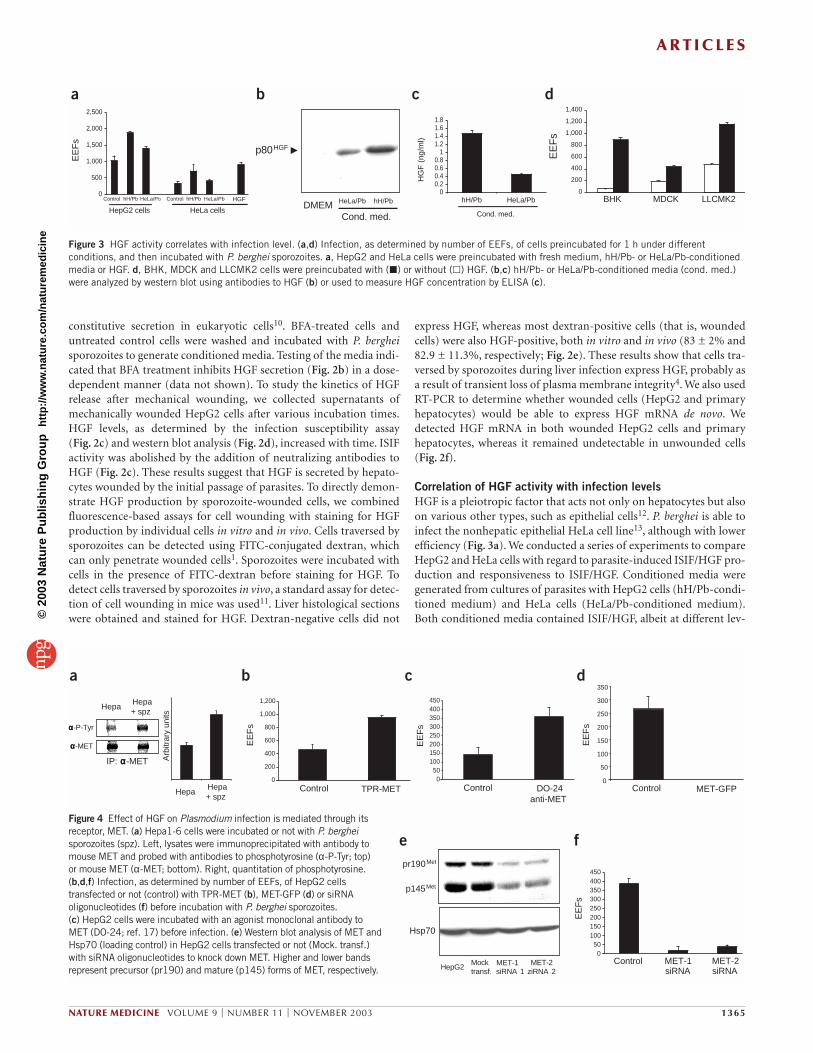
Figure 2 HGF secreted by sporozoite-traversed host cells is required for infection. (a–c) Infection, as determined by number of EEFs, of HepG2 cells preincubated for 1 h under different conditions and then incubated with P. berghei sporozoites. a, Recombinant human HGF, basic FGF, hH/Pb-conditioned medium (cond. med.) withor without antibodies to HGF (anti-HGF) or basic FGF (anti-FGF). b, hH/Pb-conditionedmedia obtained from HepG2 cells pretreated or not with BFA. c, Different wounding-conditioned media in the presence or absence of antibody to HGF. (d) Different wounding-conditioned media were probed for HGF by western blotting.Arrowhead indicates 80-kDa band. hHGF, human HGF. (e) HGF (red) is expressed by Plasmodium-traversed cells (FITC-dextran-positive cells; green) in vitro(HepG2 cells) and in vivo (liver histological sections). (f) HGF mRNA was detected in mechanically wounded (Wound.) primary hepatocytes and HepG2 cellsbut not in untreated (C) cells. A549 cells were used as a control for HGF mRNA. β-amyloid precursor protein (APP) and glyceraldehyde 3-phosphatedehydrogenase (G3PDH) were used as controls for reverse transcription.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rem
edic
ine
A R T I C L E S
constitutive secretion in eukaryotic cells10. BFA-treated cells anduntreated control cells were washed and incubated with P. bergheisporozoites to generate conditioned media. Testing of the media indi-cated that BFA treatment inhibits HGF secretion (Fig. 2b) in a dose-dependent manner (data not shown). To study the kinetics of HGFrelease after mechanical wounding, we collected supernatants ofmechanically wounded HepG2 cells after various incubation times.HGF levels, as determined by the infection susceptibility assay (Fig. 2c) and western blot analysis (Fig. 2d), increased with time. ISIFactivity was abolished by the addition of neutralizing antibodies toHGF (Fig. 2c). These results suggest that HGF is secreted by hepato-cytes wounded by the initial passage of parasites. To directly demon-strate HGF production by sporozoite-wounded cells, we combinedfluorescence-based assays for cell wounding with staining for HGFproduction by individual cells in vitro and in vivo. Cells traversed bysporozoites can be detected using FITC-conjugated dextran, whichcan only penetrate wounded cells1. Sporozoites were incubated withcells in the presence of FITC-dextran before staining for HGF. Todetect cells traversed by sporozoites in vivo, a standard assay for detec-tion of cell wounding in mice was used11. Liver histological sectionswere obtained and stained for HGF. Dextran-negative cells did not
express HGF, whereas most dextran-positive cells (that is, woundedcells) were also HGF-positive, both in vitro and in vivo (83 ± 2% and82.9 ± 11.3%, respectively; Fig. 2e). These results show that cells tra-versed by sporozoites during liver infection express HGF, probably asa result of transient loss of plasma membrane integrity4. We also usedRT-PCR to determine whether wounded cells (HepG2 and primaryhepatocytes) would be able to express HGF mRNA de novo. Wedetected HGF mRNA in both wounded HepG2 cells and primaryhepatocytes, whereas it remained undetectable in unwounded cells(Fig. 2f).
Correlation of HGF activity with infection levelsHGF is a pleiotropic factor that acts not only on hepatocytes but alsoon various other types, such as epithelial cells12. P. berghei is able toinfect the nonhepatic epithelial HeLa cell line13, although with lowerefficiency (Fig. 3a). We conducted a series of experiments to compareHepG2 and HeLa cells with regard to parasite-induced ISIF/HGF pro-duction and responsiveness to ISIF/HGF. Conditioned media weregenerated from cultures of parasites with HepG2 cells (hH/Pb-condi-tioned medium) and HeLa cells (HeLa/Pb-conditioned medium).Both conditioned media contained ISIF/HGF, albeit at different lev-
NATURE MEDICINE VOLUME 9 | NUMBER 11 | NOVEMBER 2003 1365
0
500
1,000
1,500
2,000
2,500
EE
Fs
HepG2 cells HeLa cells
HGFControl ControlhH/Pb hH/PbHeLa/Pb HeLa/Pb0
200
400
600
800
1,000
1,200
1,400
3BHK MDCK LLCMK2
EE
Fs
00.20.40.60.8
11.21.41.61.8
hH/Pb
HG
F (
ng/m
l)
HeLa/Pb
Cond. med.
Cond. med.
HeLa/Pb hH/PbDMEM
p80HGF
a b c d
Figure 3 HGF activity correlates with infection level. (a,d) Infection, as determined by number of EEFs, of cells preincubated for 1 h under differentconditions, and then incubated with P. berghei sporozoites. a, HepG2 and HeLa cells were preincubated with fresh medium, hH/Pb- or HeLa/Pb-conditionedmedia or HGF. d, BHK, MDCK and LLCMK2 cells were preincubated with (�) or without (�) HGF. (b,c) hH/Pb- or HeLa/Pb-conditioned media (cond. med.)were analyzed by western blot using antibodies to HGF (b) or used to measure HGF concentration by ELISA (c).
0
200
400
600
800
1,000
1,200
EE
Fs
Control TPR-MET0
50100150200250300350400450
EE
Fs
Control DO-24anti-MET
0
50
100
150
200
250
300
350
EE
Fs
Control MET-GFP
050
100150200250300350400450
EE
Fs
Control MET-1siRNA siRNA siRNA 1
MET-2ziRNA 2
Mocktransf.
HepG2
Hepa
IP: αααα-MET
Hepa+ spz
αααα-MET
αααα-P-Tyr
0
2 0
4 0
6 0
8 0
100
120
HepaHepa+ spz
Arb
itrar
y un
its
pr190Met
p145Met
Hsp70
MET-1 MET-2
a b c d
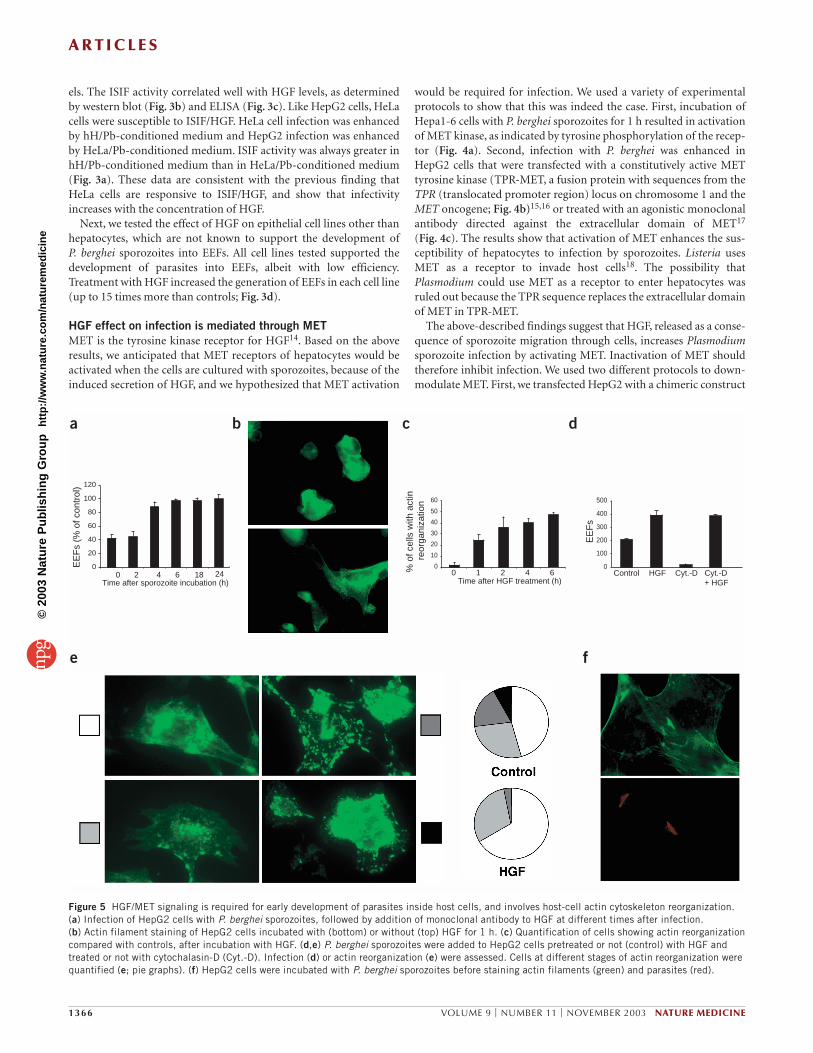
e fFigure 4 Effect of HGF on Plasmodium infection is mediated through itsreceptor, MET. (a) Hepa1-6 cells were incubated or not with P. bergheisporozoites (spz). Left, lysates were immunoprecipitated with antibody tomouse MET and probed with antibodies to phosphotyrosine (α-P-Tyr; top) or mouse MET (α-MET; bottom). Right, quantitation of phosphotyrosine.(b,d,f) Infection, as determined by number of EEFs, of HepG2 cellstransfected or not (control) with TPR-MET (b), MET-GFP (d) or siRNAoligonucleotides (f) before incubation with P. berghei sporozoites. (c) HepG2 cells were incubated with an agonist monoclonal antibody toMET (DO-24; ref. 17) before infection. (e) Western blot analysis of MET andHsp70 (loading control) in HepG2 cells transfected or not (Mock. transf.)with siRNA oligonucleotides to knock down MET. Higher and lower bandsrepresent precursor (pr190) and mature (p145) forms of MET, respectively.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rem
edic
ine
A R T I C L E S
els. The ISIF activity correlated well with HGF levels, as determinedby western blot (Fig. 3b) and ELISA (Fig. 3c). Like HepG2 cells, HeLacells were susceptible to ISIF/HGF. HeLa cell infection was enhancedby hH/Pb-conditioned medium and HepG2 infection was enhancedby HeLa/Pb-conditioned medium. ISIF activity was always greater inhH/Pb-conditioned medium than in HeLa/Pb-conditioned medium(Fig. 3a). These data are consistent with the previous finding thatHeLa cells are responsive to ISIF/HGF, and show that infectivityincreases with the concentration of HGF.
Next, we tested the effect of HGF on epithelial cell lines other thanhepatocytes, which are not known to support the development ofP. berghei sporozoites into EEFs. All cell lines tested supported thedevelopment of parasites into EEFs, albeit with low efficiency.Treatment with HGF increased the generation of EEFs in each cell line(up to 15 times more than controls; Fig. 3d).
HGF effect on infection is mediated through METMET is the tyrosine kinase receptor for HGF14. Based on the aboveresults, we anticipated that MET receptors of hepatocytes would beactivated when the cells are cultured with sporozoites, because of theinduced secretion of HGF, and we hypothesized that MET activation
would be required for infection. We used a variety of experimentalprotocols to show that this was indeed the case. First, incubation ofHepa1-6 cells with P. berghei sporozoites for 1 h resulted in activationof MET kinase, as indicated by tyrosine phosphorylation of the recep-tor (Fig. 4a). Second, infection with P. berghei was enhanced inHepG2 cells that were transfected with a constitutively active METtyrosine kinase (TPR-MET, a fusion protein with sequences from theTPR (translocated promoter region) locus on chromosome 1 and theMET oncogene; Fig. 4b)15,16 or treated with an agonistic monoclonalantibody directed against the extracellular domain of MET17
(Fig. 4c). The results show that activation of MET enhances the sus-ceptibility of hepatocytes to infection by sporozoites. Listeria usesMET as a receptor to invade host cells18. The possibility thatPlasmodium could use MET as a receptor to enter hepatocytes wasruled out because the TPR sequence replaces the extracellular domainof MET in TPR-MET.
The above-described findings suggest that HGF, released as a conse-quence of sporozoite migration through cells, increases Plasmodiumsporozoite infection by activating MET. Inactivation of MET shouldtherefore inhibit infection. We used two different protocols to down-modulate MET. First, we transfected HepG2 with a chimeric construct
1366 VOLUME 9 | NUMBER 11 | NOVEMBER 2003 NATURE MEDICINE
0
20
40
60
80
100
120
EE
Fs
(% o
f con
trol
)
Time after sporozoite incubation (h)0 2 4 18 6 24
0
100
200
300
400
500
Control HGF Cyt.-D Cyt.-D+ HGF
EE
Fs
0
10
20
30
40
50
60
Time after HGF treatment (h)0 1 2 4 6%
of c
ells
with
act
inre
orga
niza
tion
a b c d
e f
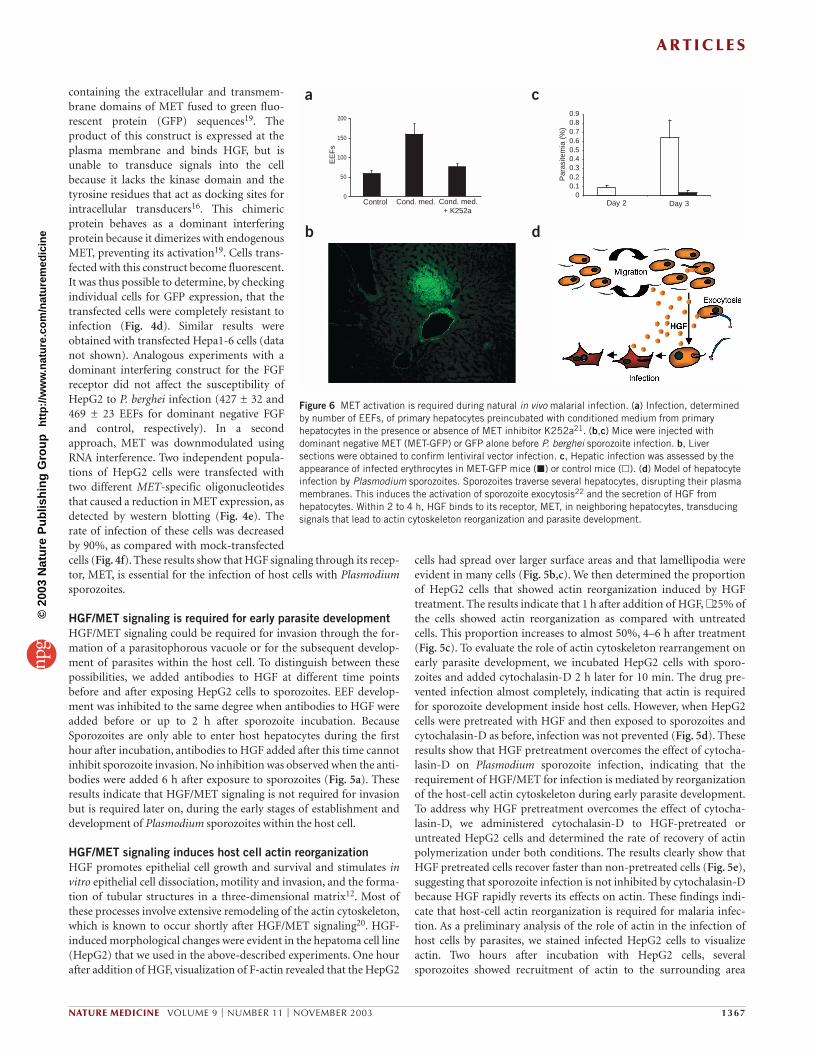
Figure 5 HGF/MET signaling is required for early development of parasites inside host cells, and involves host-cell actin cytoskeleton reorganization.(a) Infection of HepG2 cells with P. berghei sporozoites, followed by addition of monoclonal antibody to HGF at different times after infection. (b) Actin filament staining of HepG2 cells incubated with (bottom) or without (top) HGF for 1 h. (c) Quantification of cells showing actin reorganizationcompared with controls, after incubation with HGF. (d,e) P. berghei sporozoites were added to HepG2 cells pretreated or not (control) with HGF andtreated or not with cytochalasin-D (Cyt.-D). Infection (d) or actin reorganization (e) were assessed. Cells at different stages of actin reorganization werequantified (e; pie graphs). (f) HepG2 cells were incubated with P. berghei sporozoites before staining actin filaments (green) and parasites (red).
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rem
edic
ine
A R T I C L E S
containing the extracellular and transmem-brane domains of MET fused to green fluo-rescent protein (GFP) sequences19. Theproduct of this construct is expressed at theplasma membrane and binds HGF, but isunable to transduce signals into the cellbecause it lacks the kinase domain and thetyrosine residues that act as docking sites forintracellular transducers16. This chimericprotein behaves as a dominant interferingprotein because it dimerizes with endogenousMET, preventing its activation19. Cells trans-fected with this construct become fluorescent.It was thus possible to determine, by checkingindividual cells for GFP expression, that thetransfected cells were completely resistant toinfection (Fig. 4d). Similar results wereobtained with transfected Hepa1-6 cells (datanot shown). Analogous experiments with adominant interfering construct for the FGFreceptor did not affect the susceptibility ofHepG2 to P. berghei infection (427 ± 32 and469 ± 23 EEFs for dominant negative FGFand control, respectively). In a secondapproach, MET was downmodulated usingRNA interference. Two independent popula-tions of HepG2 cells were transfected withtwo different MET-specific oligonucleotidesthat caused a reduction in MET expression, asdetected by western blotting (Fig. 4e). Therate of infection of these cells was decreasedby 90%, as compared with mock-transfectedcells (Fig. 4f). These results show that HGF signaling through its recep-tor, MET, is essential for the infection of host cells with Plasmodiumsporozoites.
HGF/MET signaling is required for early parasite developmentHGF/MET signaling could be required for invasion through the for-mation of a parasitophorous vacuole or for the subsequent develop-ment of parasites within the host cell. To distinguish between thesepossibilities, we added antibodies to HGF at different time pointsbefore and after exposing HepG2 cells to sporozoites. EEF develop-ment was inhibited to the same degree when antibodies to HGF wereadded before or up to 2 h after sporozoite incubation. BecauseSporozoites are only able to enter host hepatocytes during the firsthour after incubation, antibodies to HGF added after this time cannotinhibit sporozoite invasion. No inhibition was observed when the anti-bodies were added 6 h after exposure to sporozoites (Fig. 5a). Theseresults indicate that HGF/MET signaling is not required for invasionbut is required later on, during the early stages of establishment anddevelopment of Plasmodium sporozoites within the host cell.
HGF/MET signaling induces host cell actin reorganizationHGF promotes epithelial cell growth and survival and stimulates invitro epithelial cell dissociation, motility and invasion, and the forma-tion of tubular structures in a three-dimensional matrix12. Most ofthese processes involve extensive remodeling of the actin cytoskeleton,which is known to occur shortly after HGF/MET signaling20. HGF-induced morphological changes were evident in the hepatoma cell line(HepG2) that we used in the above-described experiments. One hourafter addition of HGF, visualization of F-actin revealed that the HepG2
cells had spread over larger surface areas and that lamellipodia wereevident in many cells (Fig. 5b,c). We then determined the proportionof HepG2 cells that showed actin reorganization induced by HGFtreatment. The results indicate that 1 h after addition of HGF, ∼ 25% ofthe cells showed actin reorganization as compared with untreatedcells. This proportion increases to almost 50%, 4–6 h after treatment(Fig. 5c). To evaluate the role of actin cytoskeleton rearrangement onearly parasite development, we incubated HepG2 cells with sporo-zoites and added cytochalasin-D 2 h later for 10 min. The drug pre-vented infection almost completely, indicating that actin is requiredfor sporozoite development inside host cells. However, when HepG2cells were pretreated with HGF and then exposed to sporozoites andcytochalasin-D as before, infection was not prevented (Fig. 5d). Theseresults show that HGF pretreatment overcomes the effect of cytocha-lasin-D on Plasmodium sporozoite infection, indicating that therequirement of HGF/MET for infection is mediated by reorganizationof the host-cell actin cytoskeleton during early parasite development.To address why HGF pretreatment overcomes the effect of cytocha-lasin-D, we administered cytochalasin-D to HGF-pretreated oruntreated HepG2 cells and determined the rate of recovery of actinpolymerization under both conditions. The results clearly show thatHGF pretreated cells recover faster than non-pretreated cells (Fig. 5e),suggesting that sporozoite infection is not inhibited by cytochalasin-Dbecause HGF rapidly reverts its effects on actin. These findings indi-cate that host-cell actin reorganization is required for malaria infec-tion. As a preliminary analysis of the role of actin in the infection ofhost cells by parasites, we stained infected HepG2 cells to visualizeactin. Two hours after incubation with HepG2 cells, several sporozoites showed recruitment of actin to the surrounding area
NATURE MEDICINE VOLUME 9 | NUMBER 11 | NOVEMBER 2003 1367
0
50
100
150
200
Control Cond. med. Cond. med.+ K252a
EE
Fs
00.10.20.30.40.50.60.70.80.9
Par
asite
mia
(%
)
Day 2 Day 3
a
b
c
d
Figure 6 MET activation is required during natural in vivo malarial infection. (a) Infection, determinedby number of EEFs, of primary hepatocytes preincubated with conditioned medium from primaryhepatocytes in the presence or absence of MET inhibitor K252a21. (b,c) Mice were injected withdominant negative MET (MET-GFP) or GFP alone before P. berghei sporozoite infection. b, Liversections were obtained to confirm lentiviral vector infection. c, Hepatic infection was assessed by theappearance of infected erythrocytes in MET-GFP mice (�) or control mice (�). (d) Model of hepatocyteinfection by Plasmodium sporozoites. Sporozoites traverse several hepatocytes, disrupting their plasmamembranes. This induces the activation of sporozoite exocytosis22 and the secretion of HGF fromhepatocytes. Within 2 to 4 h, HGF binds to its receptor, MET, in neighboring hepatocytes, transducingsignals that lead to actin cytoskeleton reorganization and parasite development.
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rem
edic
ine

A R T I C L E S
(Fig. 5f). This recruitment was observed consistently in all experi-ments (4.2 ± 1.1 sporozoites with actin recruitment; n = 6) and wasnever detected when cells were treated with cytochalasin-D 2 h afterinfection (0 ± 0 sporozoites with actin recruitment; n = 6). Together,these findings show that HGF-induced reorganization of the actincytoskeleton in host cells is required for sporozoite infection.
Inactivation of MET inhibits Plasmodium liver infectionTo investigate the in vivo relevance of our findings, we conductedexperiments using freshly prepared primary hepatocytes from mice.Medium conditioned by incubating sporozoites with these primaryhepatocytes had ISIF activity similar to that in media that were con-ditioned by the hepatocyte cell lines (Fig. 6a). Because blocking mon-oclonal antibodies are not available for mouse HGF, we used atyrosine kinase inhibitor (K525a) that is known to inhibit MET21.This inhibitor abolished ISIF activity (Fig. 6a). If activation of theHGF/MET system is indeed required for productive infection, itwould be possible to interfere with Plasmodium infection in mice byblocking MET activation. To prove this assumption, we injected micewith a lentivirus expressing a MET-GFP fusion protein that repre-sents a dominant negative form of MET19. Control mice were injectedwith a lentivirus expressing GFP alone (Fig. 6b). The lentivirus infec-tion efficacy was determined by analyzing GFP expression in hepato-cytes. Approximately 10% of the total liver volume was infected, andinfection was confined to areas near major vessels. Two days afterlentivirus infection, mice were infected with P. berghei sporozoites. Toassess the hepatic development of the parasites, we examined theappearance of infected erythrocytes. Two days after the administra-tion of sporozoites, blood-stage infection was detectable in controlmice but not in mice expressing the dominant negative form of MET.At three days, infection was still 20 times higher in mice infected withthe control lentivirus (Fig. 6c). These findings indicate that lentivirusinfection prevented MET signaling and parasite development in asubstantial number of hepatocytes. We thus conclude that MET sig-naling is required for the hepatic development of P. berghei in mice.
DISCUSSIONOur previous work has shown that migration through hepatocytesinduces the exocytosis of sporozoite apical organelles, a prerequisitefor infection involving formation of a vacuole22. Here we show that thehost responds to the traveling parasites by secreting HGF, which ren-ders hepatocytes susceptible to infection (Fig. 6d). HGF is known toact as an autocrine cytokine in various cancer cells and precancerouscell types23,24, but it is not normally expressed by hepatocytes or bymost other MET-expressing epithelial cells. Previous in situ hybridiza-tion studies revealed that HGF-producing cells in the liver are non-parenchymal, whereas the HGF receptor is selectively expressed byhepatocytes25. Here we showed that HepG2 cells and hepatocytesfreshly isolated by mouse liver perfusion produce HGF de novo whenthey are wounded mechanically or by sporozoite migration. In cir-rhotic patients, HGF expression is also detected in hepatocytes26.Because cirrhosis leads to liver injury, HGF expression may be inducedby the same mechanisms that are activated during Plasmodium infec-tion. In addition, the present data clearly show that signaling by activeHGF is required for infection of hepatocytes by sporozoites.
Infection requires the entry of sporozoites into hepatocytesthrough the formation of a parasitophorous vacuole. This process ispoorly understood. Its initial steps involve heparan sulfate proteogly-cans27,28 and CD81 (ref. 29), at least for some Plasmodium species.Our findings suggest that infection requires reorganization of theactin cytoskeleton of the host cell, a process initiated by MET signals.
However, because antibodies to HGF interfered with the infection ofhepatocytes even when added 2 h after exposure to sporozoites, weassume that MET signals are required for the proper function of theparasitophorous vacuole and/or early establishment of infection,rather than for its initial formation.
The requirement of MET signaling for malaria infection hasimportant clinical implications. It may explain why severe malariacases are more frequent in hepatitis B virus carriers, who have ele-vated HGF levels30, than in matched controls31. It also suggests newstrategies for the prevention of malarial infections. The recentlydescribed beneficial effect of vitamin A in malaria infection32 couldbe attributed at least in part to the inhibition of HGF production byretinoic acid33, a metabolite of vitamin A. Although this remains to beshown, it is conceivable that malaria prophylaxis might be achievedwith other agents that inhibit HGF synthesis or activation, or agentsthat interfere with HGF-induced MET signals.
METHODSCells and parasites. Cell lines were maintained in DMEM with 10% FCS.Plasmodium sporozoites were obtained by dissection of infected Anophelesstephensi mosquito salivary glands. Primary hepatocytes were prepared asdescribed34. Briefly, hepatocytes were isolated by collagenase H perfusion ofmouse liver lobules and purified over a 60% Percoll gradient. Cells (inWilliam’s medium with 10% FCS) were seeded in Lab-Tek chamber slides andincubated at 37 °C with 5% CO2 for 24 h.
Preparation of conditioned media. Conditioned medium results from theincubation of Plasmodium sporozoites (105) with cell monolayers (2 × 105) at37 °C for 1 h (except for Fig. 2c,d, where different incubation periods wereused). We verified the absence of infectious sporozoites by adding conditionedmedia to hepatocytes and determining infection.
Sporozoite infection. Sporozoites (5 × 104) were added to cell monolayers (2 × 105) for 24 h, before fixation and staining with monoclonal antibody toEEFs (2E 6)35. Infection was quantified by counting the number of EEFs. Allinfections were done in triplicate and repeated three times.
Migration through hepatocytes in vitro and in vivo and HGF staining. For invitro staining, sporozoites were added to monolayers of cells for 1 h in the pres-ence of 1 mg/ml of FITC-conjugated, lysine-fixable dextran (Mr 10,000;Molecular Probes). Cells were washed, fixed with 4% paraformaldehyde andstained with monoclonal antibody to HGF (Sigma) followed by rhodamine-conjugated secondary antibody. For in vivo staining, BALB/c mice (6 weeks)were injected intravenously with 8 mg of FITC-dextran in PBS and 2 × 105
P. yoelii sporozoites. After 45 min, mice were anesthetized and their livers wereperfused with 20 ml of PBS and fixed with 20 ml of 8% paraformaldehyde.Livers were dissected, frozen and sectioned before quantification of dextran-positive cells in histological sections. HGF staining was done as above.
HGF evaluation. HGF production was evaluated by AN’ALYZA immunoassay(TECHNE). HGF mRNA was evaluated by RT-PCR using RNA extracted fromwounded or control cells by using the RNA Wiz kit (Ambion), followed byreverse transcription. For HepG2 and A549 cells we used the sense oligonu-cleotide 5′-CGAAATCCTCGAGGGGAAGAAGGG-3′, the antisense oligonu-cleotide 5′-AATTGCACAGTACTCCCAGCGGT-3′, an annealing temperatureof 60 °C, an elongation time of 1 min and an expected size of 324 base pairs. Asa control for reverse transcription, we amplified the β-amyloid precursor pro-tein using the sense oligonucleotide 5′-CACAGAGAGAACCACCGCA-3′, theantisense oligonucleotide 5′-ACATCCGCCGTAAAAGAATG-3′ and anexpected size of 180 base pairs. For mouse primary hepatocytes we used thesense oligonucleotide 5′-CCAGCCAGAAACAAAGACTTGAAAGACTAT-3′,the antisense oligonucleotide 5′-ACATCCACGACCAGGAACAATGACAC-CAAG-3′, an annealing temperature of 62 °C, an elongation time of 1 min andan expected size of 500 base pairs. As a control for reverse transcription, weamplified glyceraldehyde 3-phosphate dehydrogenase using the sense oligonucleotide 5′-ACCACAGTCCATGCCATCAC-3′, the antisense oligonu-
1368 VOLUME 9 | NUMBER 11 | NOVEMBER 2003 NATURE MEDICINE
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rem
edic
ine

A R T I C L E S
cleotide 5′-TCCACCACCCT GTT GCTGTA-3′ and an expected size of 500base pairs.
MET immunoprecipitation and detection of phosphorylation status. Hepa1-6cells (8 × 105) were incubated or not with 4 × 105 P. berghei sporozoites and thenlysed for 20 min at 4 °C in 200 µl of a buffer containing 50 mM HEPES (pH7.4), 5 mM EDTA, 2 mM EGTA, 150 mM NaCl, 10% glycerol and 1% Triton X-100 in the presence of protease and phosphatase inhibitors. Extracts were clari-fied at 15,000g for 15 min and incubated with protein G-Sepharose andantibody to mouse MET (1 µg/ml; Santa Cruz Biotechnology) for 4 h at 4 °Cwith gentle rotation. Samples were then centrifuged, resuspended in 50 µl of2× Laemmli buffer and boiled for 5 min. Extracts were electrophoresed on SDS-PAGE and transferred onto nitrocellulose membranes. Blots were probed withantibodies to phosphotyrosine (Calbiochem) and MET, which were detected bythe enhanced chemiluminescence system (Amersham).
Cell transfection and lentivirus production. To produce TPR-MET-contain-ing retroviruses, we transfected Phoenix cells with the pLXSN plasmid. Vectorscollected from 24-h supernatants were used to infect HepG2 cells overnight.Cells were selected with G418 and the resistant cells were pooled and exam-ined by western blotting. The MET-GFP construct was cloned into thep156RRLsin-PPT-hCMV-MCS-pre lentiviral vector as previously described19.In vitro infection was done by incubating the cells overnight in the presence ofvirus-containing supernatant. Efficiency of infection was evaluated using GFPfluorescence. For the in vivo studies, viral particles were purified by ultracen-trifugation at 25,000g. for 140 min and resuspended in sterile, endotoxin-freePBS. Viral p24 antigen concentration was determined by HIV-1 p24 CoreProfile ELISA (NEN Life Science Products). Purified p24 (15 µg) was injectedinto the tail veil of each mouse. Control mice were injected with correspon-ding amounts of viral particles containing GFP alone. All experiments weredone according to specific rules approved by the Direcção Geral de Veterinárialicensing committee.
MET modulation. MET expression was downmodulated by siRNA using the MET-1 (5′-ACUCUAGAUGCUCAGACUUTT-3′) and MET-2 (5′-GUCAUAGGAAGAGGGCAUUTT-3′) oligonucleotides. Transfections (200nM of each oligo) were done with the Oligofectamine reagent (Invitrogen).After transfection, cells were kept in culture for 72 h and then extracted withboiling Laemmli buffer. Equal amounts of proteins (evaluated by BCA; Pierce)were loaded in each lane. Blots were probed with antibodies to human MET(C12; Santa Cruz Biotechnology).
Actin staining. Cells were fixed with 4% paraformaldehyde for 10 min andpermeabilized in 0.1% Triton X-100 in PBS for 2 min. Actin filaments werevisualized by incubating cells for 20 min with 0.1 µg/ml TRITC-labeled phal-loidin diluted in 0.05% Tween (in PBS).
ACKNOWLEDGMENTSWe thank W. Haas, A. Jacinto, L. Parreira and M.C. Soares for critically reviewingthe manuscript; T. Maciag for the FGF dominant negative construct; V.E. doRosário, C. Casimiro and C. Alves for providing P. berghei-infected mosquitoes;and L. Naldini, A. Follenzi and M. Mazzone for kindly providing lentiviral vectorsand experimental support. Funding was provided by Fundação para a Ciência eTecnologia (Projects POCTI/38563/MGI/2001 and POCTI/MGI/44517/2002) andthe National Institutes of Health (grants AI 49432 and AI 53698). M.C., L.C.-S.,A.M.V., S.S., P.L., D.C. and M.M.M. are supported by Fundação para a Ciência eTecnologia.
COMPETING INTERESTS STATEMENTThe authors declare that they have no competing financial interests.
Received 28 July; accepted 22 September 2003Published online at http://www.nature.com/naturemedicine/
1. Mota, M.M. et al. Migration of Plasmodium sporozoites through cells before infec-tion. Science 291, 141–144 (2001).
2. McNeil, P.L. et al. Growth factors are released by mechanically wounded endothelialcells. J. Cell Biol. 109, 811–822 (1989).
3. Muthukrishnan, L. et al. Basic fibroblast growth factor is efficiently released from acytosolic storage site through plasma membrane disruptions of endothelial cells. J.Cell. Physiol. 148, 1–16 (1991).
4. McNeil, P.L. & Steinhardt, R.A. Loss, restoration, and maintenance of plasma mem-brane integrity. J. Cell Biol. 137, 1–4 (1997).
5. Mota, M.M. & Rodriguez, A. Plasmodium yoelii: efficient in vitro invasion and com-plete development of sporozoites in mouse hepatic cell lines. Exp. Parasitol. 96,257–259 (2000).
6. Kinoshita, T., Hirao, S., Matsumoto, K. & Nakamura, T. Possible endocrine controlby hepatocyte growth factor of liver regeneration after partial hepatectomy.Biochem. Biophys. Res. Commun. 177, 330–335 (1991).
7. Zarnegar, R., DeFrances, M.C., Kost, D.P., Lindroos, P. & Michalopoulos, G.K.Expression of hepatocyte growth factor mRNA in regenerating rat liver after partialhepatectomy. Biochem. Biophys. Res. Commun. 177, 559–565 (1999).
8. Rong, S. et al. Tumorigenicity of the met proto-oncogene and the gene for hepato-cyte growth factor. Mol. Cell. Biol. 12, 5152–5158 (1992).
9. Bhargava, M. et al. Scatter factor and hepatocyte growth factor: activities, proper-ties, and mechanism. Cell Growth Differ. 3, 11–20 (1992).
10. Misumi, Y., Miki, K., Takatsuki, A., Tamura, G. & Ikehara, Y. Novel blockade bybrefeldin A of intracellular transport of secretory proteins in cultured rat hepato-cytes. J. Biol. Chem. 261, 11398–11403 (1986).
11. McNeil, P.L., Clarke, M.F.S. & Miyake, K. Cell motility, cell wound assays. in CurrentProtocols in Cell Biology, Supplement 2 (eds. Bonifacino, J.S., Dasso, M.,Lippincott-Schwartz, J., Harford, J.B. & Yamada, K.M.) 12.4.1–12.4.15 (JohnWiley & Sons, New York, 1999).
12. Trusolino, L. & Comoglio, P.M. Scatter-factor and semaphorin receptors: cell signal-ing for invasive growth. Nat. Rev. Cancer 2, 289–300 (2002).
13. Calvo-Calle, J.M., Moreno, A., Eling, W.M. & Nardin, E.H. In vitro development ofinfectious liver stages of P. yoelii and P. berghei malaria in human cell lines. Exp.Parasitol. 79, 362–373 (1994).
14. Naldini, L. et al. Hepatocyte growth factor (HGF) stimulates the tyrosine kinaseactivity of the receptor encoded by the proto-oncogene c-MET. Oncogene 6,501–504 (1991).
15. Park, M. et al. Mechanism of met oncogene activation. Cell 45, 895–904 (1986).16. Ponzetto, C. et al. A multifunctional docking site mediates signaling and transfor-
mation by the hepatocyte growth factor/scatter factor receptor family. Cell 77,261–271 (1994).
17. Prat, M., Crepaldi, T., Pennacchietti, S., Bussolino, F. & Comoglio, P.M. Agonisticmonoclonal antibodies against the Met receptor dissect the biological responses toHGF. J. Cell Sci. 111, 237–247 (1998).
18. Shen, Y., Naujokas, M., Park, M. & Ireton, K. InIB-dependent internalization ofListeria is mediated by the Met receptor tyrosine kinase. Cell 103, 501–510(2000).
19. Giordano, S. et al. The semaphorin 4D receptor controls invasive growth by couplingwith MET. Nat. Cell Biol. 4, 720–724 (2002).
20. Royal, I., Lamarche-Vane, N., Lamorte, L., Kaibuchi, K. & Park, M. Activation ofcdc42, rac, PAK, and rho-kinase in response to hepatocyte growth factor dofferen-tially regulates epithelial cell colony spreading and dissociation. Mol. Biol. Cell 11,1709–1725 (2000).
21. Morotti, A., Mila, S., Accornero, P., Tagliabue, E. & Ponzetto, C. K252a inhibits theoncogenic properties of Met, the HGF receptor. Oncogene 21, 4885–4893 (2002).
22. Mota, M.M., Hafalla, J.C.R. & Rodriguez, A. Migration through host cells activatesPlasmodium sporozoites for infection. Nat. Med. 8, 1318–1322 (2002).
23. Jeffers, M., Rong, S., Anver, M. & Vande Woude, G.F. Autocrine hepatocyte growthfactor/scatter factor-Met signaling induces transformation and the invasive/metas-tastic phenotype in C127 cells. Oncogene 13, 853–856 (1996).
24. Rong, S., Segal, S., Anver, M., Resau, J.H. & Vande Woude, G.F. Invasiveness andmetastasis of NIH 3T3 cells induced by Met-hepatocyte growth factor/scatter factorautocrine stimulation. Proc. Natl Acad. Sci. USA 91, 4731–4735 (1994).
25. Matsumoto, K. & Nakamura, T. Hepatocyte growth factor: molecular structure andimplications for a central role in liver regeneration. J. Gastroenterol. Hepatol. 6,509–519 (1991).
26. Ljubimova, J.Y., Petrovic, L.M., Wilson, S.E., Geller, S.A. & Demetriou, A.A.Expression of HGF, its receptor c-met, c-myc, and albumin in cirrhotic and neoplas-tic human liver tissue. J. Histochem. Cytochem. 45, 79–87 (1997).
27. Pinzon-Ortiz, C., Friedman, J., Esko, J. & Sinnis, P. The binding of the circumsporo-zoite protein to cell surface heparan sulfate proteoglycans is required forPlasmodium sporozoite attachment to target cells. J. Biol. Chem. 276,26784–26791 (2001).
28. Pradel, G., Garapaty, S. & Frevert, U. Proteoglycans mediate malaria sporozoite tar-geting to the liver. Mol. Microbiol. 45, 637–651 (2002).
29. Silvie, O. et al. Hepatocyte CD81 is required for Plasmodium falciparum andPlasmodium yoelii sporozoite infectivity. Nat. Med. 9, 93–96 (2003).
30. Naoumov, N.V. & Eddleston, A.L. Host immune response and variations in the virusgenome: pathogenesis of liver damage caused by hepatitis B virus. Gut 35,1013–1017 (1994).
31. Thursz, M.R. et al. Association of hepatitis B surface antigen carriage with severemalaria in Gambian children. Nat. Med. 1, 374–375 (1995).
32. Serghides, L. & Kain, K.C. Mechanism of protection induced by vitamin A in falci-parum malaria. Lancet 359, 1404–1406 (2002).
33. Gohda, E. Function and regulation of production of hepatocyte growth factor (HGF).Nippon Yakurigaku Zasshi 119, 287–294 (2002).
34. Mazier, D. et al. Complete development of hepatic stages of Plasmodium falciparumin vitro. Science 227, 440–442 (1985).
35. Tsuji, M., Mattei, D., Nussenzweig, R.S., Eichinger, D. & Zavala, F. Demonstration ofheat-shock protein 70 in the sporozoite stage of malaria parasites. Parasitol. Res.80, 16–21 (1994).
NATURE MEDICINE VOLUME 9 | NUMBER 11 | NOVEMBER 2003 1369
©20
03 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://w
ww
.nat
ure
.co
m/n
atu
rem
edic
ine




















