
In vivo hepatocyte proliferation is inducible through a TNF and IL-6-independent pathway
Upload
independentCategory
view
2download
0
Genes regulated by hepatocyte growth factor as targetsto sensitize ovarian cancer cells to cisplatin
Martina Olivero,1 Tina Ruggiero,1 Silvia Saviozzi,3
Andrea Rasola,2 Nadia Coltella,1 Stefania Crispi,4
Ferdinando Di Cunto,5 Raffaele Calogero,3
and Maria Flavia Di Renzo1
1Laboratory of Cancer Genetics and 2Division of MolecularOncology, Institute for Cancer Research and Treatment,University of Torino School of Medicine, Candiolo (Turin), Italy;3Department of Clinical and Biological Sciences, University ofTorino, Orbassano (Turin), Italy; 4Gene Expression Core-HumanMolecular Genetics Laboratory, Institute of Genetics andBiophysics ‘‘A.B.T.,’’ Consiglio Nazionale delle Ricerche, Naples,Italy; and 5Department of Genetics, Biology, and Biochemistry,University of Torino, Turin, Italy
AbstractAdvanced ovarian cancers are initially responsive tochemotherapy with platinum drugs but develop drugresistance in most cases. We showed recently thathepatocyte growth factor (HGF) enhances death of humanovarian cancer cell lines treated with cisplatin (CDDP) andthat this effect is mediated by the p38 mitogen-activatedprotein kinase. In this work, we integrated genome-wideexpression profiling, in silico data survey, and functionalassays to identify transcripts regulated in SK-OV-3 ovariancancer cells made more responsive to CDDP by HGF.Using oligonucleotide microarrays, we found that HGFpretreatment changes the transcriptional response toCDDP. Quantitative reverse transcription-PCR not onlyvalidated all the 15 most differentially expressed genesbut also confirmed that they were primarily modulated bythe combined treatment with HGF and CDDP and reversedby suppressing p38 mitogen-activated protein kinaseactivity. Among the differentially expressed genes, wefocused functional analysis on two regulatory subunits of
the protein phosphatase 2A, which were down-modulatedby HGF plus CDDP. Decrease of each subunit by RNAinterference made ovarian cancer cells more responsive toCDDP, mimicking the effect of HGF. In conclusion, weshow that HGF and CDDP modulate transcription inovarian cancer cells and that this transcriptional responseis involved in apoptosis regulation. We also provide theproof-of-concept that the identified genes might betargeted to either increase the efficacy of chemothera-peutics or revert chemotherapy resistance. [Mol CancerTher 2006;5(5):1126–35]
IntroductionDrug resistance remains a major clinical challenge forcancer treatment. Patients suffering from advanced ovar-ian carcinoma are in most cases initially responsive to thecombined cytoreductive and chemotherapy treatment.However, they later experience disease relapse due toeventual tumor recurrence and emergence of drug-resistant tumor cells. There are several mechanisms bywhich tumor cells resist to cytotoxic agents (1, 2). One isrelated to resistance to apoptosis, a type of cell death thatis triggered in response to common chemotherapy andradiotherapy regimens. Evasion from apoptosis is also oneof the fundamental hallmarks of cancer (3). A corollary ofthis is that if the apoptotic pathway inactivated in tumordevelopment is the same as, or overlaps, that leading tocell death by drug, most cancers would be expected to beresistant to drug-induced apoptosis from their onset.Therefore, therapeutic interventions that can lower thethreshold for apoptosis of tumor cells could becomeuseful approaches to treat cancer when used either as asingle agent or in combination with other therapeuticmodalities (4).In ovarian cancer, apoptosis is impaired by the anti-
apoptotic action of several oncogenes, which includegrowth factor receptors and other kinases that transducesignals from the membrane to the nucleus (5–7). We haveshown recently (8) that the hepatocyte growth factor (HGF)sensitizes several ovarian carcinoma cell lines to cisplatin(CDDP) and Taxol, commonly used as first-line chemo-therapy in advanced ovarian cancer. HGF increases cancercell death at very low drug doses. This was a novel andsurprising finding. HGF elicits a distinctive biologicalprogram known as ‘‘invasive growth’’ by orchestratingcell survival, proliferation, and motility through activationof its receptor, encoded by the MET oncogene, and ofseveral downstream signaling pathways, such as theextracellular signal-regulated kinase 1/2 mitogen-activatedprotein kinases (MAPK) and phosphatidylinositol 3-kinase/AKT and the AKT substrate mammalian target ofrapamycin (9, 10). Nevertheless, we have shown thatactivation of these survival pathways does not hinder the
Received 1/9/06; revised 3/10/06; accepted 3/24/06.
Grant support: Italian Ministry of University and Research, RegionePiemonte, and Italian Association for Cancer Research (M.F. Di Renzo),Ministero Salute and Italian Association for Cancer Research (R. Calogero),and Regione Piemonte Ricerca Scientifica Applicata fellowship (A. Rasola).
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely toindicate this fact.
Note: The present address for T. Ruggiero is Gene Expression RegulationLaboratory, Experimental Oncology, Istituto Nazionale Ricerca sul Cancro,Genova, Italy. The present address for A. Rasolas is Department ofBiomedical Sciences, University of Padova, Padova, Italy.
Requests for reprints: Maria Flavia Di Renzo, Laboratory of CancerGenetics, Institute for Cancer Research and Treatment, University ofTorino School of Medicine, SP 142, KM 3.95, 10060, Candiolo (Torino),Italy. Phone: 39-11-993-3343; Fax: 39-11-993-3524.E-mail: [email protected]
Copyright C 2006 American Association for Cancer Research.
doi:10.1158/1535-7163.MCT-06-0013
1126
Mol Cancer Ther 2006;5(5). May 2006
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

ability of HGF to enhance drug-dependent apoptosis inovarian cancer cells. In these cells, HGF simultaneouslyactivates the p38 MAPK, which is further activated bydrugs and leads to cell death (11).As we have shown that in ovarian cancer cell lines HGF-
dependent sensitization to drugs required long-term expo-sure (8), we hypothesized that HGF effect could be coupledto transcriptional regulation of apoptosis-related genes. Toexplore this possibility, we have studied the transcriptionaltargets of HGF and CDDP in an ovarian cancer cell line. Theidentified molecules were sought as regulators of ovariancancer cell apoptotic death and can be prospectively viewedas targets for therapy. Here, we show the identification oftranscripts modulated in cells committed to CDDP-inducedapoptosis by HGF. Among them, we found subunits ofprotein phosphatase 2A (PP2A) that we targeted to enhanceovarian cancer cell response to the drug.
Materials andMethodsFlow CytometryAnalysis of Apoptosis InductionExperiments were done on SK-OV-3 ovarian carcinoma
cells, which showed resistance to platinum drugs (12).Exponentially growing cells were cultured for 48 hours inthe presence of pure recombinant HGF (R&D Systems,Minneapolis, MN) at the concentration of 50 ng/mL orcontrol medium. Apoptosis was then induced by addingfresh medium, with or without HGF, supplemented with10 Amol/L CDDP. Flow cytometry recordings of severalindependent apoptotic changes were done by a single-tubeanalysis as described (8, 13).
Microarray Sample PreparationTotal RNA was extracted and purified from SK-OV-3 cell
lines at each time point using the Concert CytoplasmicRNA Purification Reagent (Invitrogen, Carlsbad, CA) assuggested by the manufacturer. RNAs were then quantifiedand inspected by Bioanalyzer (Agilent Technologies,Waldbrom, Germany) analysis. cRNAs were generatedand hybridized on 16 HGU133a Affymetrix (Santa Clara,CA) DNA chips according to the Affymetrix protocol. Thechips were scanned with a specific scanner (Affymetrix) togenerate digitized image data files.
Microarray Data AnalysisTwo of the time-course experiments, done independently
(TR5 and TR7), were analyzed. From digitized image datafiles of raw data, background-normalized image data (CELfiles) were generated. Microarray quality control andstatistical validation was done using Bioconductor (14).The presence of hybridization/construction artifacts wasevaluated with the fitPLM function (Bioconductor packageaffyPLM). The probes (PM) intensity distribution wasevaluated using hist function (Bioconductor package affy).Only two arrays, CDDP 6 hours and CDDP/HGF 6 hoursin TR7 experiment were discarded due to low quality. TR5and TR7 experiments were then analyzed separately. Probeset intensities were obtained by robust multiarray average(15), and normalization was done according to quantilesmethod (16). The HGU133a 22283 probe sets in TR5 and
TR7 were filtered separately to have an interquantile rangefor each probe set greater than 0.4 (17). This filteringyielded 3,893 probe sets in common between the twoexperiments. TR5 and TR7, represented as ratio withrespect to probe sets mean across the experiments, werecombined in a unique exprSet object. The identification ofdifferentially expressed genes associated to the HGF effectwas addressed using linear modeling approach (18). Forassessing differential expression, an empirical Bayesmethod (18) was used to moderate the SE of the estimatedlog-fold changes together with the BH correction (19) of thefalse discovery rate. Four hundred six probe sets (334Entrez Genes) were identified as differentially expressed asconsequence of HGF effect (adjusted P V 0.05). For each ofthe two experiments, fold change variation versus the timepoint 0 of the CDDP treatment was calculated followed bythe calculation of fold change variation depending on HGFpretreatment (e.g., HGF/CDDP 6 hours � CDDP 6 hours).Principal component analysis and hierarchical clustering
(ST, Euclidean distance, average clustering, and 5,000jackknife resampling steps) were done using TMEV 3.1software.6
Coexpression AnalysisThe coexpression analysis was done essentially as
described (20). Briefly, we downloaded all the humancDNA microarray experiments deposited in the StanfordMicroarray Database (4,129 experiments on May 2005) andidentified the probes corresponding to the 15 top rankeddifferentially expressed genes. For each probe, we thencalculated the Pearson correlation coefficient with eachprobe in the database normalized for the number ofcommon experiments. The normalized Pearson coefficientwas then used to rank all the probes in the database, and thetop 1% of the ranked database was selected as the list ofcoexpressed probes. We chose a 1% cutoff as we showedpreviously that on average most of the biologicallysignificant coexpression is concentrated in this interval(20). We thus obtained a total of 89 coexpression lists thatwere merged to evaluate the recurrence of probes repre-senting the 334 differentially expressed genes. The observedfrequency was then compared with that expected by chance,and an overrepresentation P was calculated (m2 test).
Quantitative ReverseTranscription-PCRcDNA was synthesized from 1 Ag RNA using MMLV-
RT(H�) enzyme (Promega, Madison, WI). Quantitativereverse transcription-PCR (RT-PCR) was done on an ABIPRISM 7900HT Sequence Detection System (PE Biosystems,Foster City, CA) in 384-well plates assembled by Biorobot8000 (Qiagen, Hilden, Germany) using a final volume of 20AL. All quantitative RT-PCR (qPCR) mixtures contained 20ng retrotranscribed RNA, 1� SYBR Green PCR Master Mix(2�, Applera, Foster City, CA), and 150 Amol/L of eachtarget-specific primer. The primer sequences are reportedin Supplementary Table S1B.7 The expression of each
6 http://www.tigr.org/software/7 Supplementary materials for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
Molecular Cancer Therapeutics 1127
Mol Cancer Ther 2006;5(5). May 2006
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

target genes was evaluated using a relative quantificationapproach (�DDCT method; ref. 21) with cyclophilin A asinternal reference. The consistency of cyclophilin A(NM_021130) was assessed in a study aimed at definingthe most stable expressed genes of seven putative referencegenes. This study consisted of qPCR of the seven genes onthe same samples (data not shown) followed by analysiswith the genNorm software (22).
Short Hairpin RNAStable ExpressionSK-OV-3 and amphotropic phoenix retroviral producer
cells were both from American Type Culture Collection(Manassas, VA). For production of retroviral stocks,phoenix cells were seeded at 4 � 106 per 10-cm dish andtransfected using CaPO4 precipitation of 15 Ag pRetroSuper-short hairpin RNA (shRNA) plasmid DNA. The followingsequences were subcloned: PPP2R1B (NM_002716) AGGA-CCCGAAGTGAATTGT and PPP2R5E (NM_006246)CCAGTTATTAGGGGGTTAA. Forty-eight hours after infec-tion, cells were selected using 1 Ag/mL puromycin; typically95% to 100% of the cells were infected and drug resistant.
ResultsHGF Sensitizes Ovarian Cancer Cells to CDDP-
Dependent Apoptosis via the p38MAPKIn response to CDDP, the SK-OV-3 ovarian cancer cells
enter the apoptotic program as assessed using a multi-parametric cytofluorimetric assay (8, 13), which simulta-neously measures hallmarks of early, intermediate, and latephases of apoptosis: decrease of mitochondrial membranepotential, cell shrinkage, increase in granularity, exposureof phosphatidylserine at cell surface, and plasma mem-brane permeabilization to propidium iodide. We showedpreviously that SK-OV-3 cells express the receptor for theHGF encoded by the MET oncogene, which is activatedby HGF (8, 23). As shown in Fig. 1A, a 48-hour longpretreatment with 50 ng/mL HGF caused a markedsensitization to the subsequent treatment with 10 Amol/LCDDP, with a net increase of the measured apoptoticfeatures after 48 to 72 hours. Notably, this sensitizationrequired pretreatment with HGF, as its coincubation withthe drug was ineffective (Fig. 1A). These data confirmedwhat we observed previously (8) and established the timecourse of the increased death of SK-OV-3 cells caused bythe combined treatment with HGF and CDDP.We have also observed previously that either HGF or
CDDP, alone or in combination, activate the p38 MAPK(11), which is activated by cell stress and is involved incancer cell apoptosis induced by several chemotherapeu-tics, including platinum drugs and taxanes (24, 25).We stably expressed a dominant-negative form of p38
MAPK (DN-p38) using a lentiviral vector carrying thedouble T180A/Y182F Flag-tagged p38 MAPK mutant (11).Lentiviral vectors drive the stable transgene randomintegration in cell genomic DNA, thus allowing repetitivestudies of the bulk unselected cell population. Wetransduced SK-OV-3 and assessed using Western blot andfluorescence-activated cell sorting analysis with anti-Flag
antibodies (data not shown) that f90% of cells expressedthe DN-p38. We showed previously that the DN-p38MAPK abolished the wild-type p38 MAPK activationwithout interfering with other HGF-dependent signalingpathways (11). Remarkably, we found that the inhibition ofthe p38 MAPK pathway almost abolished apoptosis elicitedby CDDP and enhanced by HGF in SK-OV-3 cells (Fig. 1B).These data indicate that p38 MAPK activation is requiredand has a central role in the induction and modulation ofapoptosis driven by CDDP and enhanced by HGF inovarian cancer cells. Thus, SK-OV-3 cells stably expressingthe DN-p38 have been used as a model to establish afunctional link between cell transcriptional response toHGF and CDDP and induction of apoptosis (see below).
HGFModulatesTranscription in Ovarian Cancer CellsCommitted to CDDP-Dependent ApoptosisAs pretreatment is required to obtain maximal sensitiza-
tion of ovarian carcinoma cells to CDDP, we assumed that
Figure 1. HGF synergizes with CDDP in activating apoptosis throughp38 MAPK in ovarian cancer cells. Apoptosis of ovarian carcinoma cells(SK-OV-3) was measured with a multiparametric fluorescence-activatedcell sorting analysis. Bottom, cells displaying mitochondrial depolarization(reduced tetramethylrhodamine methyl ester staining); right quadrant, cellsexposing phosphatidylserine on their surface (increased Annexin V-FITCstaining). Numbers, percentages of healthy cells (H, delimited in thequadrant) and dead, propidium iodide–permeable cells (D, not shown inthe diagrams). Cells undergoing apoptosis and thus outside the quadrantshould be added to the healthy cells to give 100%. A, time course (hours)of the cell apoptotic response to CDDP after 48 h of treatment withrecombinant purified HGF (50 ng/mL). Top right, response of cellssimultaneously exposed to CDDP and HGF and examined after72 h (+HGF ). Representative experiment of the seven (TR1–TR7) thatmeasured the apoptosis achievement by cells from which RNA wasextracted. B, apoptotic response to CDDP treatment (72 h) with or withoutHGF pretreatment (50 ng/mL for 48 h) of cells stably carrying andexpressing the DN-p38 MAPK transgene. Representative experiment ofthe five that measured the apoptosis achievement by cells from whichRNA was extracted. In all experiments, SK-OV-3 cells were exposed to 10Amol/L CDDP.
Targets to Sensitize Ovarian Cancer Cells to Cisplatin1128
Mol Cancer Ther 2006;5(5). May 2006
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

HGF mainly enhances the apoptotic response by modulatinggene transcription. Thus, we studied the expression profilesof ovarian carcinoma cells treated with HGF and CDDPalone or in combination. To perform genome-wide profiling,we first used microarray experiments and then validated thedifferentially expressed genes using real-time qPCR. Asshown in Fig. 1, in both cells treated with CDDP alone orwith CDDP and HGF, even the earliest apoptotic changes,such as the decrease of mitochondrial membrane potential,become manifest not earlier than after 48 hours of CDDPexposure (Fig. 1A). Therefore, time-course experiments weredone, pretreating SK-OV-3 cells with HGF, exposing HGF-treated cells to CDDP, and preparing RNA after 6, 12, and 24hours. Control cells were either exposed to control treatmentor exposed to HGF alone or CDDP alone for 6, 12, and 24hours. We analyzed the expression profiles of cells fromthree independent experiments after confirming apoptosisachievement by a sample aliquot of cells kept in culture for48 and 72 hours; a representative control experiment isshown in Fig. 1A. Each of the three time-course experiments,called TR5, TR6, and TR7, included eight experimentalpoints as follows: cells treated with control medium for 48hours (0-hour CDDP), cells treated with HGF for 48 hours(HGF/0-hour CDDP), cells pretreated with control mediumand thereafter with CDDP for 6 hours (6-hour CDDP), 12hours (12-hour CDDP), and 24 hours (24-hour CDDP), andcells pretreated with HGF and thereafter with CDDP for 6hours (HGF/6-hour CDDP), 12 hours (HGF/12-hourCDDP), and 24 hours (HGF/24-hour CDDP). mRNAs fromcells at each of the above points were analyzed. Two of thethree time-course experiments (TR5 and TR7) were analyzedusing HGU133a Affymetrix oligonucleotide microarrays.The third experiment (TR6) was added for the furthervalidation of results with qPCR (see below).Principal component analysis (26) was used to investigate
the overall effects of HGF and CDDP in TR5 and TR7experiments. This technique represents the global geneexpression changes between samples as coordinates in amultidimensional space in which the axes are called the‘‘principal components.’’ In TR5 and TR7 experiments, thefirst and second components account for the greatestpart of the variability within the data set (73.5%); thus,the graphical representation of the first two components(Fig. 2) is sufficient to show the differences betweensamples. Figure 2 shows that HGF pretreatment above allstrongly influenced the overall gene expression variation,as experiments simply clustered based on this factor (abovethe dashed line in Fig. 2). Conversely, the effect of CDDPwas time dependent, although the two experiments wereonly partially synchronized.Therefore, we focused the subsequent statistical analysis
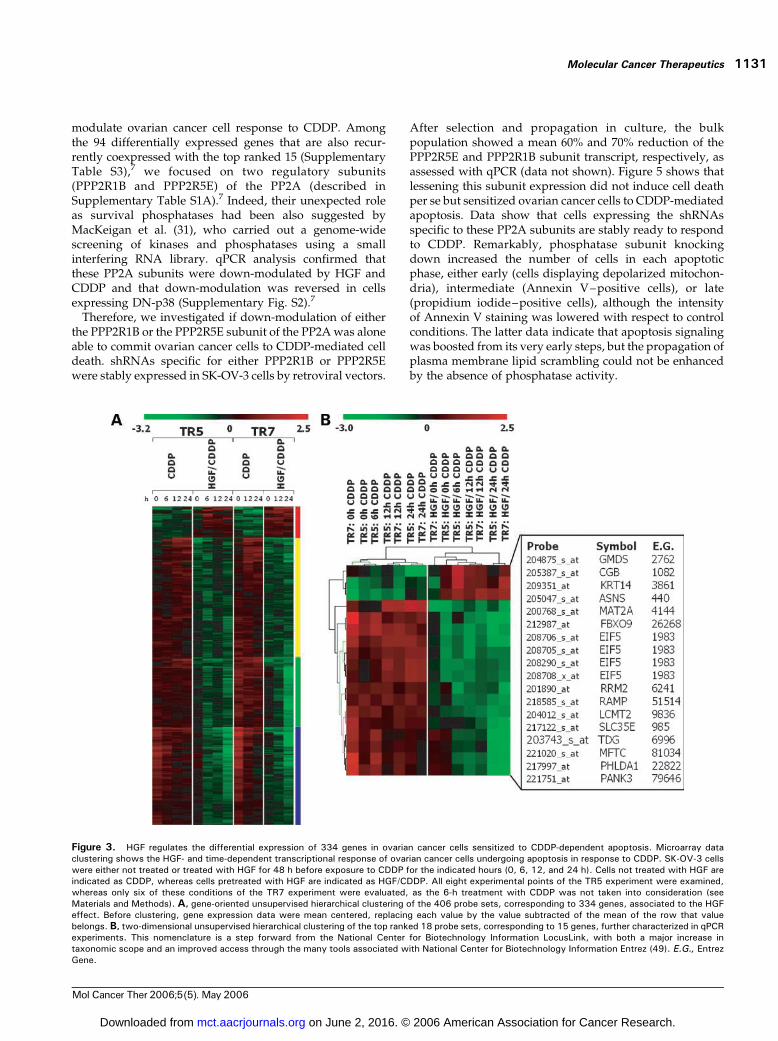
on the identification of probe sets differentially expressedin cells undergoing CDDP-dependent apoptosis as aconsequence of the HGF pretreatment. Using linearmodeling analysis (18), we identified 406 probe setsassociated to the HGF pretreatment. Unsupervised hierar-chical clustering of these 406 probe sets (Fig. 3A),corresponding to 334 genes, again showed HGF effect on
CDDP-regulated transcripts in both TR5 and TR7 experi-ments. Subclustering related to the timing of CDDP effectcan be also observed (Fig. 3A), more evidently in onecluster (blue bar in Fig. 3A).For qPCR validation, we selected the 18 probe sets, of 406,
which were characterized by the largest differential expres-sion (>2-fold change in both TR5 and TR7 experiments;Fig. 3B). The 15 genes corresponding to these probe sets arelisted in Fig. 3B and described in detail in SupplementaryTable S1A. Differential expression of all these 15 transcriptswas confirmed by qPCR not only in the TR5 and TR7experiments but also in the TR6 experiment similarly carriedout (see above) but not analyzed with microarrays. TheqPCR validation of each of the 15 genes is shown in Fig. 4and Supplementary Figs. S1 and S2.7 It is noteworthy that allthe 15 top ranked differentially expressed transcriptsshowed a quantitative regulation, which paralleled thatfound in microarrays. However, by comparing the foldchange in expression measured using microarray analysis(Fig. 3B) and that measured using qPCR (calculated by theequation �DDCT; Fig. 4; Supplementary Figs. S1 and S2),7
the differential expression came out to be two to four timeshigher using qPCR probably due to the higher sensitivity ofthe latter technique. qPCR also confirmed the consistency oftranscript regulation in all the three time-course experiments(Fig. 4; Supplementary Figs. S1 and S2).7
Furthermore, qPCR allowed a better classification of the15 top ranked differentially expressed genes. Some weremainly regulated by the combined treatment with HGF andCDDP (e.g., RAMP and RRM2 ; Fig. 4). Others were alreadymodulated by HGF alone (e.g., MAT2A and EIF5 ; Fig. 4); inthe latter case, in fact, consistent differences in expressionwere already measured after 48 hours HGF treatment (i.e.,before the addition of CDDP; 0-hour point in Fig. 4). UsingqPCR, we confirmed that 12 of 15 top ranked genes weredown-modulated by CDDP plus HGF (Figs. 3 and 4;Supplementary Figs. S1 and S2).7 Two of the 15 (CGB andGMDS ; Fig. 4; Supplementary Fig. S1)7 were down-modulated by CDDP but reversed by HGF. Only 1 of 15genes (KRT14 ; Fig. 4) was consistently and strongly up-regulated by both HGF alone and HGF plus CDDP.Supplementary Table S27 lists all the genes regulated >2-fold by HGF alone after 48 hours of treatment. This listincluded the above-mentioned MAT2A and EIF5 and theother genes (KRT14, ASNS, CGB, FBX09, SLC35E2, LCMT2 ,and PHDLA1), which we validated as regulated by HGF inqPCR (Fig. 4; Supplementary Figs. S1 and S2).7
Transcriptional Regulation by HGF and CDDP Relieson p38MAPK ActivationAs p38 MAPK regulates several transcription factors
(27–30), we asked if HGF and CDDP transcriptionmodulation requires p38 MAPK activation, which medi-ates their apoptotic effect. The bulk unselected SK-OV-3cell population, stably expressing the DN-p38, was treatedwith either CDDP alone or CDDP and HGF, and the 15top ranked transcripts were quantified using qPCR at eachtime point after treatment. The actual apoptotic responsein the time-course experiments was confirmed on a
Molecular Cancer Therapeutics 1129
Mol Cancer Ther 2006;5(5). May 2006
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

sample aliquot of cells kept in culture for 72 hours(Fig. 1B). As shown in Fig. 4 and Supplementary Figs. S1and S2,7 all the 15 transcripts were differently regulated incells where p38 MAPK was inactive. In most cases,inactivation of the p38 MAPK reverted the transcriptregulation by HGF and CDDP. These data show that theHGF-dependent transcriptional regulation of ovariancancer cells committed to CDDP-induced apoptosismostly relies on p38 MAPK activation.
Identification of Genes Coregulated with the Tran-scriptionalTargets of HGF and CDDPThe finding that transcription of 15 genes was consis-
tently modulated by HGF and CDDP and reverted by DN-p38 raised the interesting possibility that they could bepart of a wider transcriptional response functionallyrelated to p38-dependent apoptosis. Furthermore, wenoticed that the degree of differential expression of the15 genes assessed using microarray analysis was remark-ably lower than that measured with qPCR. Altogether,data suggested that a wider transcriptional response couldinclude other genes possibly underestimated by themicroarray analysis. If this was the case, the genesbelonging to the group of the 15 top ranked differentiallyexpressed genes, as well as many of the other genesmodulated to a lesser extent by the HGF pretreatment,would be expected to undergo significant coregulationunder unrelated experimental conditions.
To test this hypothesis, and to identify other genespossibly belonging to the same transcriptional module, wedecided to perform a coexpression analysis using acollection of 4,129 cDNAmicroarray experiments depositedin the Stanford Microarray Database.In particular, for each of the above 15 genes, we obtained
coexpression ranked lists containing the 1% probes of thedatabase showing the highest expression profile similarity(see Materials and Methods; see ref. 20). Consistent withour hypothesis, in these lists, the recurrence of the 15 topranked genes was much higher than expected by chance(cumulative P = 2.7 � 10�6, m2 test), thus ruling out thatthe expression regulation observed in our settings wasmerely due to cell death.We then calculated an overrepresentation P for each
gene in the obtained coexpression lists and used thisnumber as an independent filter to select those genes that,among the 334 differentially expressed genes, were aboveall recurrent (threshold P < 0.01). The selected 94 genes,listed in Supplementary Table S3,7 are the most likely to beimportant in the functional response induced by the HGFand CDDP combined treatment.
Functional Evaluation of Genes DifferentiallyExpressed in Ovarian Cancer Cells Committed to Apo-ptosisWe reasoned that genes regulated by HGF and CDDP in
cells undergoing apoptosis could be directly targeted to
Figure 2. HGF above all regulates transcription in ovarian cancer cells sensitized toCDDP-dependent apoptosis. Microarray experiments are analyzed usingprincipal component analysis that is a data reduction method to identify patterns in a data matrix and reflects the variance within the experimental data.Principal component analysis was done on overall expression data of cells either not treated or treated with HGF for 48 h before exposure to CDDP for theindicated hours (0, 6, 12, and 24 h). Cells not treated with HGF are indicated by closed symbols, whereas cells treated with HGF are indicated by opensymbols. All eight time point conditions of the TR5 experiment (diamonds ) were examined, whereas only six of these conditions of the TR7 experiment(circles ) were evaluated, as the 6-h treatment with CDDPwas not taken into consideration (see Materials and Methods). The first two principal components,PC1 and PC2, account for 58% and 15.5% of the total variance, respectively. HGF effect is visible in both experiments, allowing the separation of the plusHGF (+HGF ) with respect to the minus (-HGF ) condition, positioned above and below the dashed line, respectively. The effect of increasing length of CDDPtreatment is visible as projection on PC1. Trends are similar within the two experiments, but they are only partially synchronized.
Targets to Sensitize Ovarian Cancer Cells to Cisplatin1130
Mol Cancer Ther 2006;5(5). May 2006
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
modulate ovarian cancer cell response to CDDP. Amongthe 94 differentially expressed genes that are also recur-rently coexpressed with the top ranked 15 (SupplementaryTable S3),7 we focused on two regulatory subunits(PPP2R1B and PPP2R5E) of the PP2A (described inSupplementary Table S1A).7 Indeed, their unexpected roleas survival phosphatases had been also suggested byMacKeigan et al. (31), who carried out a genome-widescreening of kinases and phosphatases using a smallinterfering RNA library. qPCR analysis confirmed thatthese PP2A subunits were down-modulated by HGF andCDDP and that down-modulation was reversed in cellsexpressing DN-p38 (Supplementary Fig. S2).7
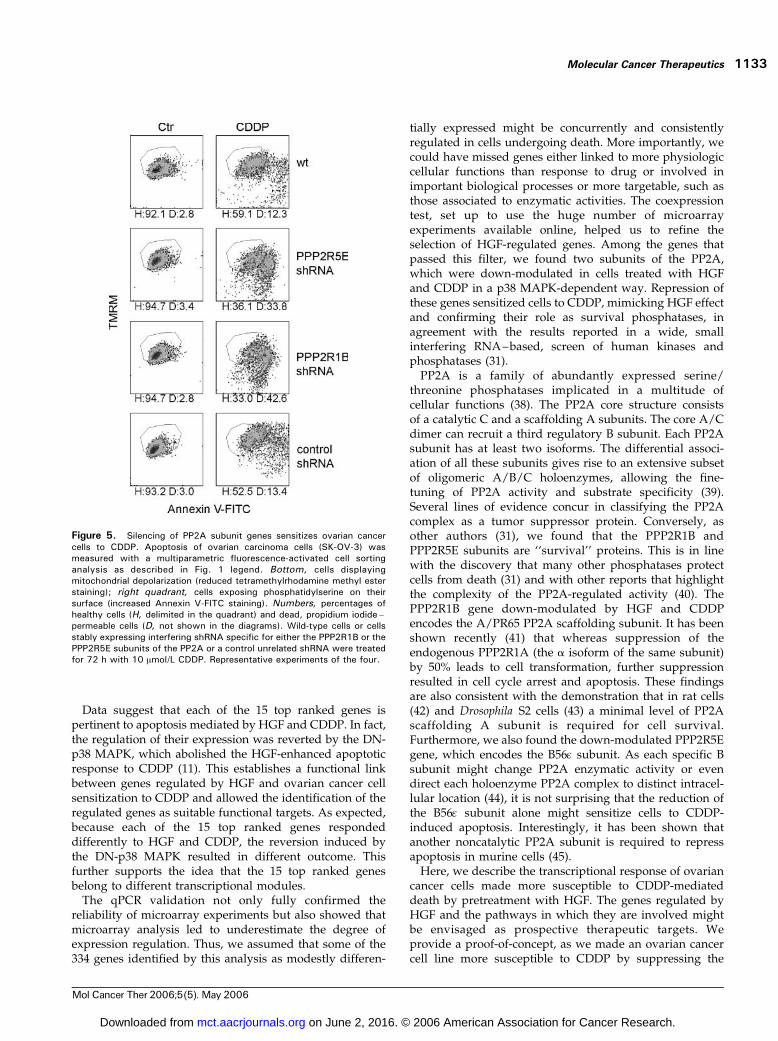
Therefore, we investigated if down-modulation of eitherthe PPP2R1B or the PPP2R5E subunit of the PP2Awas aloneable to commit ovarian cancer cells to CDDP-mediated celldeath. shRNAs specific for either PPP2R1B or PPP2R5Ewere stably expressed in SK-OV-3 cells by retroviral vectors.
After selection and propagation in culture, the bulkpopulation showed a mean 60% and 70% reduction of thePPP2R5E and PPP2R1B subunit transcript, respectively, asassessed with qPCR (data not shown). Figure 5 shows thatlessening this subunit expression did not induce cell deathper se but sensitized ovarian cancer cells to CDDP-mediatedapoptosis. Data show that cells expressing the shRNAsspecific to these PP2A subunits are stably ready to respondto CDDP. Remarkably, phosphatase subunit knockingdown increased the number of cells in each apoptoticphase, either early (cells displaying depolarized mitochon-dria), intermediate (Annexin V–positive cells), or late(propidium iodide–positive cells), although the intensityof Annexin V staining was lowered with respect to controlconditions. The latter data indicate that apoptosis signalingwas boosted from its very early steps, but the propagation ofplasma membrane lipid scrambling could not be enhancedby the absence of phosphatase activity.
Figure 3. HGF regulates the differential expression of 334 genes in ovarian cancer cells sensitized to CDDP-dependent apoptosis. Microarray dataclustering shows the HGF- and time-dependent transcriptional response of ovarian cancer cells undergoing apoptosis in response to CDDP. SK-OV-3 cellswere either not treated or treated with HGF for 48 h before exposure to CDDP for the indicated hours (0, 6, 12, and 24 h). Cells not treated with HGF areindicated as CDDP, whereas cells pretreated with HGF are indicated as HGF/CDDP. All eight experimental points of the TR5 experiment were examined,whereas only six of these conditions of the TR7 experiment were evaluated, as the 6-h treatment with CDDP was not taken into consideration (seeMaterials and Methods). A, gene-oriented unsupervised hierarchical clustering of the 406 probe sets, corresponding to 334 genes, associated to the HGFeffect. Before clustering, gene expression data were mean centered, replacing each value by the value subtracted of the mean of the row that valuebelongs. B, two-dimensional unsupervised hierarchical clustering of the top ranked 18 probe sets, corresponding to 15 genes, further characterized in qPCRexperiments. This nomenclature is a step forward from the National Center for Biotechnology Information LocusLink, with both a major increase intaxonomic scope and an improved access through the many tools associated with National Center for Biotechnology Information Entrez (49). E.G., EntrezGene.
Molecular Cancer Therapeutics 1131
Mol Cancer Ther 2006;5(5). May 2006
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

DiscussionIn this work, we combined different approaches to identifygenes whose expression mediate the ability of HGF tosensitize ovarian cancer cells to drug-induced apoptosis.The aim was to find genes whose modulation was per seable to predispose cells to better respond to drugs. We hereprovide proof-of-concepts that we have been able toidentify genes that might be targeted to increase theresponse of ovarian cancer cells to CDDP.We combined in vitro and in silico analyses to identify the
transcripts modulated in cells committed by HGF torespond to CDDP and relying on the p38 MAPK activationas we have shown previously that p38 MAPK mediatesHGF and CDDP combined effects (11). The associationbetween each of the most differentially expressed genesand cell death regulation is confirmed by the descriptionreported in Supplementary Table S1A.7
Genes modulated only in response to the combinedtreatment with HGF and CDDP, but not by the singletreatment, are particularly interesting as they could bebetter related toHGF-mediated sensitization toCDDP. In fact,this is a distinctive response of ovarian cancer cells as shownin several ovarian cancer cell lines (8, 11). In this group, thoseabove all down-modulated were PANK3, RAMP, RRM2,and TDG. Most of them have been already associated to cellproliferation and apoptosis and even to cancer cell resistanceto chemotherapy as described in detail in SupplementaryTable S1A (32–35).7 Among the most differentiallyexpressed genes, we identified a second group regulated byHGF alone and further modulated, in most cases, aftercell treatment with CDDP. This group includes MAT2A,ASNS, EIF5, FBX09, LCMT2, SLC35E2 , and PHDLA1 ,which have been associated to cell proliferation andsurvival, as described in Supplementary Table S1A (36, 37).7
Figure 4. p38 MAPK activitymediates gene expression regulationby HGF in ovarian cancer cells under-going CDDP-dependent apoptosis.qPCR validation of 6 of the 15 mostdifferentially expressed genes identi-fied with microarray analysis (theother 9 genes are shown in Supple-mentary Figs. S1 and S2; availableonline at http://mct.aacrjournals.org/).Expression variations of the genewere measured in wild-type (wt ) SK-OV-3 cells pretreated for 48 h eitherwith pure recombinant HGF (n) orwith control medium (5) and thencultured with 10 Amol/L CDDP forthe indicated times (0, 6, 12, and24 h). Transcripts were also mea-sured in SK-OV-3 cells stablyexpressing the DN-p38 MAPK trans-gene (DN-p38) and similarly treated.4, control medium; E, +HGF.Points, mean expression variationsmeasured at each time point usingmRNAs prepared from three experi-ments carried out independently;bars, SD. The mean fold change inexpression of the target gene intreated cells at each time point versusuntreated cells at time 0 was cal-culated using the formula: �DCT =�[(CT,target � CT,cyclophilin A)time x �(CT,target � CT,cyclophilin A)time 0].
Targets to Sensitize Ovarian Cancer Cells to Cisplatin1132
Mol Cancer Ther 2006;5(5). May 2006
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Data suggest that each of the 15 top ranked genes ispertinent to apoptosis mediated by HGF and CDDP. In fact,the regulation of their expression was reverted by the DN-p38 MAPK, which abolished the HGF-enhanced apoptoticresponse to CDDP (11). This establishes a functional linkbetween genes regulated by HGF and ovarian cancer cellsensitization to CDDP and allowed the identification of theregulated genes as suitable functional targets. As expected,because each of the 15 top ranked genes respondeddifferently to HGF and CDDP, the reversion induced bythe DN-p38 MAPK resulted in different outcome. Thisfurther supports the idea that the 15 top ranked genesbelong to different transcriptional modules.The qPCR validation not only fully confirmed the
reliability of microarray experiments but also showed thatmicroarray analysis led to underestimate the degree ofexpression regulation. Thus, we assumed that some of the334 genes identified by this analysis as modestly differen-
tially expressed might be concurrently and consistentlyregulated in cells undergoing death. More importantly, wecould have missed genes either linked to more physiologiccellular functions than response to drug or involved inimportant biological processes or more targetable, such asthose associated to enzymatic activities. The coexpressiontest, set up to use the huge number of microarrayexperiments available online, helped us to refine theselection of HGF-regulated genes. Among the genes thatpassed this filter, we found two subunits of the PP2A,which were down-modulated in cells treated with HGFand CDDP in a p38 MAPK-dependent way. Repression ofthese genes sensitized cells to CDDP, mimicking HGF effectand confirming their role as survival phosphatases, inagreement with the results reported in a wide, smallinterfering RNA–based, screen of human kinases andphosphatases (31).PP2A is a family of abundantly expressed serine/
threonine phosphatases implicated in a multitude ofcellular functions (38). The PP2A core structure consistsof a catalytic C and a scaffolding A subunits. The core A/Cdimer can recruit a third regulatory B subunit. Each PP2Asubunit has at least two isoforms. The differential associ-ation of all these subunits gives rise to an extensive subsetof oligomeric A/B/C holoenzymes, allowing the fine-tuning of PP2A activity and substrate specificity (39).Several lines of evidence concur in classifying the PP2Acomplex as a tumor suppressor protein. Conversely, asother authors (31), we found that the PPP2R1B andPPP2R5E subunits are ‘‘survival’’ proteins. This is in linewith the discovery that many other phosphatases protectcells from death (31) and with other reports that highlightthe complexity of the PP2A-regulated activity (40). ThePPP2R1B gene down-modulated by HGF and CDDPencodes the A/PR65 PP2A scaffolding subunit. It has beenshown recently (41) that whereas suppression of theendogenous PPP2R1A (the a isoform of the same subunit)by 50% leads to cell transformation, further suppressionresulted in cell cycle arrest and apoptosis. These findingsare also consistent with the demonstration that in rat cells(42) and Drosophila S2 cells (43) a minimal level of PP2Ascaffolding A subunit is required for cell survival.Furthermore, we also found the down-modulated PPP2R5Egene, which encodes the B56q subunit. As each specific Bsubunit might change PP2A enzymatic activity or evendirect each holoenzyme PP2A complex to distinct intracel-lular location (44), it is not surprising that the reduction ofthe B56q subunit alone might sensitize cells to CDDP-induced apoptosis. Interestingly, it has been shown thatanother noncatalytic PP2A subunit is required to repressapoptosis in murine cells (45).Here, we describe the transcriptional response of ovarian
cancer cells made more susceptible to CDDP-mediateddeath by pretreatment with HGF. The genes regulated byHGF and the pathways in which they are involved mightbe envisaged as prospective therapeutic targets. Weprovide a proof-of-concept, as we made an ovarian cancercell line more susceptible to CDDP by suppressing the
Figure 5. Silencing of PP2A subunit genes sensitizes ovarian cancercells to CDDP. Apoptosis of ovarian carcinoma cells (SK-OV-3) wasmeasured with a multiparametric fluorescence-activated cell sortinganalysis as described in Fig. 1 legend. Bottom, cells displayingmitochondrial depolarization (reduced tetramethylrhodamine methyl esterstaining); right quadrant, cells exposing phosphatidylserine on theirsurface (increased Annexin V-FITC staining). Numbers, percentages ofhealthy cells (H, delimited in the quadrant) and dead, propidium iodide–permeable cells (D, not shown in the diagrams). Wild-type cells or cellsstably expressing interfering shRNA specific for either the PPP2R1B or thePPP2R5E subunits of the PP2A or a control unrelated shRNA were treatedfor 72 h with 10 Amol/L CDDP. Representative experiments of the four.
Molecular Cancer Therapeutics 1133
Mol Cancer Ther 2006;5(5). May 2006
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

PP2A subunits, which we found down-modulated by HGFand CDDP in that cell line. Therefore, this work proposes avaluable protocol to identify potential targets that shouldbe validated in several additional settings and possiblytailored to ovarian cancer subsets. We found that most ofthe genes that we considered most likely to be important inthe apoptotic response induced by the HGF and CDDPcombined treatment are not reported in the signatures thathave been proposed to classify either chemoresistant orchemosensitive ovarian cancers (12, 35, 46–48). This wasnot surprising, as the steady-state expression level of theidentified genes is not expected to mark ovarian cancercells sensitivity to CDDP. Nevertheless, HGF-regulatedgenes are of potential value to modify response to CDDP,the most active chemotherapeutic in ovarian cancertreatment. The potential use of low-dose chemotherapy inchemotherapy-sensitive cancers, like ovarian carcinomas, isimportant, as lower dosage is easily attainable and lesslikely to cause toxicity in patients. In addition, in ovariancancer, chemotherapy resistance is the major cause ofsecondary failure of conventional chemotherapy. There-fore, the discovery of novel pathways and moleculesinvolved in the apoptotic response to drugs is valuable,provided that they are targetable either to kill cells or torevert chemotherapy resistance.
Acknowledgments
We thank Vincenzo De Sio, Raffaella Albano, and Solange Tienga fortechnical help, Elena Grassi for the enthusiastic help in data mining, andDr. Enzo Medico for providing the PPP2R1B and PPP2R5E specific shRNAviruses.
References
1. Agarwal R, Kaye SB. Ovarian cancer: strategies for overcomingresistance to chemotherapy. Nat Rev Cancer 2003;3:502–16.
2. Brown JM, Attardi LD. The role of apoptosis in cancer development andtreatment response. Nat Rev Cancer 2005;5:231–7.
3. Hanahan D,Weinberg RA. The hallmarks of cancer. Cell 2000;100:57–70.
4. Green DR, Kroemer G. Pharmacological manipulation of cell death:clinical applications in sight? J Clin Invest 2005;115:2610–7.
5. Shayesteh L, Lu Y, Kuo WL, et al. PIK3CA is implicated as an oncogenein ovarian cancer. Nat Genet 1999;21:99–102.
6. Yuan ZQ, Sun M, Feldman RI, et al. Frequent activation of AKT2 andinduction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Aktpathway in human ovarian cancer. Oncogene 2000;19:2324–30.
7. Cuello M, Ettenberg SA, Clark AS, et al. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast andovarian cancer cell lines that overexpress erbB-2. Cancer Res 2001;61:4892–900.
8. Rasola A, Anguissola S, Ferrero N, et al. Hepatocyte growth factorsensitizes human ovarian carcinoma cell lines to paclitaxel and cisplatin.Cancer Res 2004;64:1744–50.
9. Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met,metastasis, motility and more. Nat Rev Mol Cell Biol 2003;4:915–25.
10. Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors:cell signalling for invasive growth. Nat Rev Cancer 2002;2:289–300.
11. Coltella N, Rasola A, Nano E, et al. p38 MAPK turns hepatocytegrowth factor to a death signal that commits ovarian cancer cells tochemotherapy-induced apoptosis. Int J Cancer 2006;118:2981–90.
12. Roberts D, Schick, J, Conway S, et al. Identification of genesassociated with platinum drug sensitivity and resistance in human ovariancancer cells. Br J Cancer 2005;92:1149–58.
13. Rasola A, Gramaglia D, Boccaccio C, Comoglio PM. Apoptosisenhancement by the HIV-1 Nef protein. J Immunol 2001;166:81–8.
14. Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: opensoftware development for computational biology and bioinformatics.Genome Biol 2004;5:R80–1.
15. Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, andsummaries of high density oligonucleotide array probe level data.Biostatistics 2003;4:249–64.
16. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison ofnormalization methods for high density oligonucleotide array data basedon variance and bias. Bioinformatics 2003;19:185–93.
17. von Heydebreck A, Huber W, Gentleman R. Differential expressionwith the Bioconductor Project. Bioconductor Project Working Papers2004; Working Paper 7. Available from:http://www.bepress.com/ bioconductor/paper7.
18. Wettenhall JM, Smyth GK. limmaGUI: a graphical user interface forlinear modeling of microarray data. Bioinformatics 2004;20:3705–6.
19. Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressedgenes using false discovery rate controlling procedures. Bioinformatics2003;19:368–75.
20. Pellegrino M, Provero P, Silengo L, Di Cunto F. CLOE: identification ofputative functional relationships among genes by comparison of expres-sion profiles between two species. BMC Bioinformatics 2004;5:179.
21. Livak KJ, Schmittgen TD. Analysis of relative gene expression datausing real-time quantitative PCR and the 2(�DDC(T)) method. Methods2001;25:402–8.
22. Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalizationof real-time quantitative RT-PCR data by geometric averaging of multipleinternal control genes. Genome Biol 2002;3:RESEARCH0034.
23. Maggiora P, Lorenzato A, Fracchioli S, et al. The RON and METoncogenes are co-expressed in human ovarian carcinomas and cooperatein activating invasiveness. Exp Cell Res 2003;288:382–9.
24. Losa JH, Parada Cobo C, Viniegra JG, Sanchez-Arevalo Lobo VJ,Ramon y Cajal S, Sanchez-Prieto R. Role of the p38 MAPK pathway incisplatin-based therapy. Oncogene 2003;22:3998–4006.
25. Olson JM, Hallahan AR. p38 MAP kinase: a convergence point incancer therapy. Trends Mol Med 2004;10:125–9.
26. Raychaudhuri S, Stuart JM, Altman RB. Principal componentsanalysis to summarize microarray experiments: application to sporulationtime series. In: Altman RB, et al, editors. Pacific Symposium onBiocomputing 2000. World Sci. Pub. Co.; 2000, p. 655–6.
27. Wang XZ, Ron D. Stress-induced phosphorylation and activation ofthe transcription factor CHOP (GADD153) by p38 MAP kinase. Science1996;272:1347–9.
28. Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ. FGF andstress regulate CREB and ATF-1 via a pathway involving p38 MAP kinaseand MAPKAP kinase-2. EMBO J 1996;15:4629–42.
29. Morton S, Davis RJ, Cohen P. Signalling pathways involved inmultisite phosphorylation of the transcription factor ATF-2. FEBS Lett2004;572:177–83.
30. Poizat C, Puri PL, Bai Y, Kedes L. Phosphorylation-dependentdegradation of p300 by doxorubicin-activated p38 mitogen-activatedprotein kinase in cardiac cells. Mol Cell Biol 2005;25:2673–87.
31. MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen ofhuman kinases and phosphatases identifies new regulators of apoptosisand chemoresistance. Nat Cell Biol 2005;7:591–600.
32. Zhou B, Mo X, Liu X, Qiu W, Yen Y. Human ribonucleotide reductaseM2 subunit gene amplification and transcriptional regulation in ahomogeneous staining chromosome region responsible for the mechanismof drug resistance. Cytogenet Cell Genet 2001;95:34–42.
33. Zhou BS, Tsai P, Ker R, et al. Overexpression of transfected humanribonucleotide reductase M2 subunit in human cancer cells enhances theirinvasive potential. Clin Exp Metastasis 1998;16:43–9.
34. Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. RNAinterference targeting the M2 subunit of ribonucleotide reductaseenhances pancreatic adenocarcinoma chemosensitivity to gemcitabine.Oncogene 2004;23:1539–48.
35. Macleod K, Mullen P, Sewell J, et al. Altered ErbB receptor signalingand gene expression in cisplatin-resistant ovarian cancer. Cancer Res2005;65:6789–800.
36. Martinez-Chantar ML, Corrales FJ, Martinez-Cruz LA, et al. Spontaneous
Targets to Sensitize Ovarian Cancer Cells to Cisplatin1134
Mol Cancer Ther 2006;5(5). May 2006
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

oxidative stress and liver tumors in mice lacking methionine adenosyl-transferase 1A. FASEB J 2002;16:1292–4.
37. Paneda C, Gorospe I, Herrera B, Nakamura T, Fabregat I, Varela-NietoI. Liver cell proliferation requires methionine adenosyltransferase 2AmRNA up-regulation. Hepatology 2002;35:1381–91.
38. Janssens V, Goris J, Van Hoof C. PP2A: the expected tumorsuppressor. Curr Opin Genet Dev 2005;15:34–41.
39. Cohen P. The structure and regulation of protein phosphatases. AnnuRev Biochem 1989;58:453–508.
40. Van Hoof C, Goris J. Phosphatases in apoptosis: to be or not to be,PP2A is in the heart of the question. Biochim Biophys Acta 2003;1640:97–104.
41. Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. Cancer-associated PP2A Aa subunits induce functional haploinsufficiency andtumorigenicity. Cancer Res 2005;65:8183–92.
42. Strack S, Cribbs JT, Gomez L. Critical role for protein phosphatase2A heterotrimers in mammalian cell survival. J Biol Chem 2004;279:47732–9.
43. Li X, Scuderi A, Letsou A, Virshup DM. B56-associated protein
phosphatase 2A is required for survival and protects from apoptosis inDrosophila melanogaster . Mol Cell Biol 2002;22:3674–84.
44. Dagda RK, Zaucha JA, Wadzinski BE, Strack S. A developmentallyregulated, neuron-specific splice variant of the variable subunit Bh targetsprotein phosphatase 2A to mitochondria and modulates apoptosis. J BiolChem 2003;278:24976–85.
45. Kong M, Fox CJ, Mu J, et al. The PP2A-associated protein a4 is anessential inhibitor of apoptosis. Science 2004;306:695–8.
46. Lu KH, Patterson AP, Wang L, et al. Selection of potential markers forepithelial ovarian cancer with gene expression arrays and recursivedescent partition analysis. Clin Cancer Res 2004;10:3291–300.
47. Jazaeri AA, Awtrey CS, Chandramouli GV, et al. Gene expressionprofiles associated with response to chemotherapy in epithelial ovariancancers. Clin Cancer Res 2005;11:6300–10.
48. Spentzos D, Levine DA, Kolia S, et al. Unique gene expression profilebased on pathologic response in epithelial ovarian cancer. J Clin Oncol2005;23:7911–8.
49. Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res 2005;33:D54–8.
Molecular Cancer Therapeutics 1135
Mol Cancer Ther 2006;5(5). May 2006
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from

2006;5:1126-1135. Mol Cancer Ther Martina Olivero, Tina Ruggiero, Silvia Saviozzi, et al. sensitize ovarian cancer cells to cisplatinGenes regulated by hepatocyte growth factor as targets to
Updated version
http://mct.aacrjournals.org/content/5/5/1126
Access the most recent version of this article at:
Cited articles
http://mct.aacrjournals.org/content/5/5/1126.full.html#ref-list-1
This article cites 47 articles, 20 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/5/5/1126.full.html#related-urls
This article has been cited by 6 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
on June 2, 2016. © 2006 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Copyright © 2022 FDOKUMEN




















