
Amyotrophic Lateral Sclerosis Multiprotein Biomarkers in Peripheral Blood Mononuclear Cells
Upload
independentCategory
view
2download
0
Gene profiling of skeletal muscle in an amyotrophic lateral sclerosismouse model
Jose-Luis Gonzalez de Aguilar,1,2 Christa Niederhauser-Wiederkehr,3 Benoıt Halter,1,2 Marc De Tapia,1,2
Franck Di Scala,1,2 Philippe Demougin,3 Luc Dupuis,1,2 Michael Primig,3 Vincent Meininger,4
and Jean-Philippe Loeffler1,2
1Institut National de la Sante et de la Recherche Medicale, U692, Laboratoire de Signalisations Moleculaireset Neurodegenerescence, Strasbourg; 2Universite Louis Pasteur, Faculte de Medecine, Unite Mixte de Recherche enSante-692, Strasbourg, France; 3Biozentrum and Swiss Institute of Bioinformatics, Basel, Switzerland; and 4Hopital de laPitie-Salpetriere, Federation des Maladies du Systeme Nerveux, Centre Referent Maladie Rare Sclerose LateraleAmyotrophique, Paris, France
Submitted 16 January 2007; accepted in final form 12 November 2007
Gonzalez de Aguilar J-L, Niederhauser-Wiederkehr C,Halter B, De Tapia M, Di Scala F, Demougin P, Dupuis L,Primig M, Meininger V, Loeffler J-P. Gene profiling of skeletalmuscle in an amyotrophic lateral sclerosis mouse model. PhysiolGenomics 32: 207–218, 2008. First published November 13, 2007;doi:10.1152/physiolgenomics.00017.2007.—Muscle atrophy is a ma-jor hallmark of amyotrophic lateral sclerosis (ALS), the most frequentadult-onset motor neuron disease. To define the full set of alterationsin gene expression in skeletal muscle during the course of the disease,we used the G86R superoxide dismutase-1 transgenic mouse model ofALS and performed high-density oligonucleotide microarrays. Wecompared these data to those obtained by axotomy-induced denerva-tion. A major set of gene regulations in G86R muscles resembledthose of surgically denervated muscles, but many others appearedspecific to the ALS condition. The first significant transcriptionalchanges appeared in a subpopulation of mice before the onset of overtclinical symptoms and motor neuron death. These early changesaffected genes involved in detoxification (e.g., ALDH3, metallothio-nein-2, and thioredoxin-1) and regeneration (e.g., BTG1, RB1, andRUNX1) but also tissue degradation (e.g., C/EBP� and DDIT4) andcell death (e.g., ankyrin repeat domain-1, CDKN1A, GADD45�, andPEG3). Of particular interest, metallothionein-1 and -2, ATF3, ca-thepsin-Z, and galectin-3 genes appeared, among others, commonlyregulated in both skeletal muscle (our present data) and spinal motorneurons (as previously reported) of paralyzed ALS mice. The impor-tance of these findings is twofold. First, they designate the distal partof the motor unit as a primary site of disease. Second, they identifyspecific gene regulations to be explored in the search for therapeuticstrategies that could alleviate disease before motor neuron deathmanifests clinically.
atrophy; axotomy; denervation; neuromuscular disease
AMYOTROPHIC LATERAL SCLEROSIS (ALS) is a fatal adult-onsetneuromuscular disease characterized by the selective degener-ation of upper and lower motor neurons, progressive musclewasting, and paralysis. Most cases occur sporadically, butsome patients exhibit an autosomal dominant pattern of inher-itance. A small subset of these cases results from mutations inthe gene encoding Cu/Zn-superoxide dismutase (SOD1), a freeradical-scavenging enzyme protecting cells against oxidative
stress (66). When ubiquitously expressed in mice, SOD1 mu-tations trigger ALS as observed in humans (27, 65, 81).However, mutant SOD1 (mSOD1) expression exclusively inmotor neurons does not cause ALS, which suggests that thesecells may not be the only site where the mutant enzyme actsprimarily to trigger disease (44, 62). Astrocytes and microglialcells seem to play an important role in the degenerative process(5, 14). Beyond motor neuron loss, mSOD1 expression alsoinduces unexpectedly high levels of energy expenditure andmuscular metabolic rate and a reduction in adipose tissuestores. Counteracting this hypermetabolic trait with a highlyenergetic diet offers neuroprotection and extends survival (22).Altogether, these findings strongly suggest that other patholog-ical events distinct from motor neuron death do contributeto ALS.
Neuromuscular junction pathology occurs in ALS before theonset of overt clinical symptoms and motor neuron death (2,24, 25, 26). In mSOD1 muscles, increased total dismutaseactivity (41), high expression of uncoupling protein-3 (20) andother anabolic (22) and anti-oxidant enzymes (34, 50), mSOD1aggregates (73), and muscle fiber atrophy (35) also precedesignificant motor neuron loss. Interestingly, expressing a lo-cally acting IGF-I isoform in mSOD1 muscles maintains neu-romuscular junction integrity, delays motor neuron death, andextends lifespan (18). It is therefore reasonable to suggest thatALS proceeds in a distal-to-proximal dying back pattern andthat certain alterations of muscular origin could contribute toenhance axon vulnerability.
Important progress has been made to decipher the globalpattern of transcriptional modifications underlying muscle at-rophy under experimental and disease conditions. In turn,although a few studies examined large-scale gene expressionchanges in ALS spinal cord motor neurons (23, 33, 46, 60), thealterations of the muscle transcriptome in this disease havenever been investigated. We hypothesized that analysis of geneexpression in skeletal muscle may provide new insight to betterunderstand the early neuromuscular abnormalities that precedemotor neuron death in ALS. To this end, we performedhigh-density oligonucleotide microarray analysis of gene ex-pression in hindlimb skeletal muscles of SOD1(G86R) mice,one of the existing transgenic models that recapitulate many ofthe characteristics of ALS (65). To monitor denervation-de-pendent gene expression, we also determined the effects ofsciatic nerve axotomy on the muscle transcriptome. The patho-
Article published online before print. See web site for date of publication(http://physiolgenomics.physiology.org).
Address for reprint requests and other correspondence: J.-L. Gonzalez deAguilar, INSERM, U692, Universite Louis Pasteur, Faculte de Medecine, 11rue Humann, F-67085 Strasbourg, France (e-mail: [email protected]).
Physiol Genomics 32: 207–218, 2008.First published November 13, 2007; doi:10.1152/physiolgenomics.00017.2007.
1094-8341/08 $8.00 Copyright © 2008 the American Physiological Society 207

physiological implications of the observed expressionalchanges are discussed in relation to ALS neurodegeneration.
MATERIALS AND METHODS
Animals. Transgenic FVB/N males expressing the murine G86RSOD1 mutation were obtained in our animal facility and genotyped asdescribed (65). Transgenic male mice with the human G93A SOD1mutation were obtained from The Jackson Laboratory (Bar Harbor,ME). The altered transgenes contained the entire coding sequence ofeither mouse or human SOD1 and their corresponding flankingregulatory regions (27, 65). mSOD1 mRNA and protein were detectedin G86R and G93A muscles, as reported elsewhere (1, 35, 65). Micewere maintained at 23°C with a 12:12-h light-dark cycle and wereprovided water and regular rodent chow ad libitum. We had previ-ously determined the progression of disease symptoms in G86R miceaccording to a clinical rating scale going from score 4 to 0 (67). Score4 is attributed to asymptomatic G86R mice, relative to their wild-typelittermates. Score 3 corresponds to an alteration in hindlimb extensionwhen the animal is hung by the tail. Score 2 is attributed when anyslight alteration in the locomotion is observed. Score 1 represents anasymmetric or symmetric paralysis of the limbs. Score 0 correspondsto the stage at which animals are unable to roll over within 10 s afterbeing pushed on their back. For this study, hindlimb skeletal muscles,including soleus and gastrocnemius, were dissected from G86R miceat 75 days of age (score 4 or asymptomatic); at 90 days of age, when�40% of the animals present with altered hindlimb extension reflexes(score 3 or preparalyzed); and at the onset of hindlimb paralysis at105–107 days of age (score 1). Nontransgenic male littermates servedas controls. Denervated muscles were obtained from wild-type mice at90 days of age after 7 days of sciatic nerve axotomy as previouslydescribed (21). Ipsilateral and contralateral hindlimbs from axoto-mized and sham-operated animals were used. Mice were killed bydecapitation, and muscles were carefully dissected, frozen in liquidnitrogen, and stored at �80°C until use. Three to four animals werepooled per experimental condition, and each condition was done induplicate (Table 1). Animal manipulation followed present EuropeanUnion regulations and was carried out under the supervision ofauthorized investigators.
Data analysis. Microarray data files were uploaded to the EuropeanBioinformatics Institute ArrayExpress repository at http://www.ebi.ac.uk/arrayexpress/ (accession no. E-TABM-195). CEL datafiles were computed using the statistical algorithm implemented inMicroarray Suite 5.0. Data were further analyzed using programsdeveloped in R 1.8.0 (Ref. 30 and http://www.r-project.org). TheRobust Multichip Average method as implemented in the BioCon-ductor package “affy” (Refs. 6, 31 and http://www.bioconductor.org)or GeneSpring 7.2 (Silicon Genetics, Redwood City, CA) was em-ployed for data normalization, background correction, and summari-zation. Gene expression signal calculation was based on the PerfectMatch values from each probe set as previously reported (31). Toremove transcripts with unreliable measurements (i.e., very close tobackground levels), we used the “Filter on Expression Level” tool inGeneSpring 7.2 and kept transcripts whose raw intensities were higherthan 50 in at least 2 of the 20 microarrays. To isolate highly regulatedgenes, an unsupervised selection of transcripts was performed basedon the calculation of the standard deviation (SD) of every single probeset (or transcript) across all microarrays (Fig. 1). Those probe setswith SD �0.8 were retained. This cutoff was arbitrarily fixed to obtaina handleable, small subset of genes (i.e., �100) displaying highlydifferential expression in at least one of the experimental conditions.To learn about previously unknown relationships among the experi-mental conditions we tested, the 20 samples were ordered accordingto the expression patterns of the selected transcripts using Ward’shierarchical clustering method. Although there is no definite rule as towhich type of clustering to use, we preferred the Ward’s methodbecause it is particularly adapted to large-scale studies and producesan ordering of objects, which may be informative for data display.Since Ward’s method always tries to minimize within-groups vari-ance, it tends to generate small clusters, which may be helpful fordiscovery (77). To identify typical transcriptional patterns amongthose transcripts, they were subgrouped into smaller categories (orclusters) according to their overall transcription patterns but not signalintensities using partitioning around medoids (PAM) (37). Afterseveral rounds of analysis with increasing numbers of clusters, thebest significance was obtained with six clusters, as assessed bycomparing the degree of similarity of a given pattern to those withinits own cluster and to those in all other clusters (determined by thesilhouette plots method; data not shown). To evaluate the implicationof different functional categories of genes as represented in thepreselected group of transcripts, the whole database was screened fordifferentially expressed genes between the pathological or denerva-tion condition and their corresponding controls (Fig. 1). Only geneswhose expression was changed at least twofold were kept. Thetwofold cutoff was selected because it is the most stringent onesuggested by the GeneChip manufacturer to reduce false positives(47). Functional categorization was performed using the AffymetrixGene Ontology Data Mining Tool, and additional information wasobtained from the National Center for Biotechnology Informationdatabase and the basic local alignment search tool BLAST-N. Themost significant changes in G86R muscles at the onset of paralysisand axotomy-induced denervated muscles were highlighted by apply-ing the Welch t-test using error model variances in combination withthe Benjamini and Hochberg false discovery rate for multiple testingcorrections, as implemented in GeneSpring 7.2.
Protocols for RNA extraction, cRNA target synthesis, GeneChiphybridization, real-time RT-PCR, and Western blot and assessment ofoxidative stress are given in the Supplemental Methods (supplementalmaterials are available at the online version of this article). For thereal-time RT-PCR experiments, statistical analysis was accomplishedusing ANOVA followed by Tukey’s multiple comparisons test.
RESULTS
Methodological considerations. Because RNA degradationcauses data to be less reproducible and makes it more difficult
Table 1. Experimental groups
Sample No. Genotype Operation Age, days Code No. of Mice*
1 Wild type 75 Wt75A 32 Wild type 75 Wt75B 33 G86R 75 M75A 34 G86R 75 M75B 35 Wild type 90 Wt90A 36 Wild type 90 Wt90B 37 G86R 90 M90E† 38 G86R 90 M90L† 39 Wild type 105 Wt105A 4
10 Wild type 105 Wt105B 411 G86R Onset‡ OSA 412 G86R Onset OSB 413 Wild type Ø 90 AXO-CA 414 Wild type Ø 90 AXO-CB 415 Wild type Axotomy 90 AXO-IA 416 Wild type Axotomy 90 AXO-IB 417 Wild type Ø 90 SHAM-CA 418 Wild type Ø 90 SHAM-CB 419 Wild type Sham 90 SHAM-IA 420 Wild type Sham 90 SHAM-IB 4
*No. of mice pooled per sample. †M90E and M90L represent the two90-day-old samples referred to in the text as the early and late preparalysisphases, respectively. ‡Paralysis onset was at �105 days of age. Ø, nonoperatedmuscle.
208 SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org

to detect low-abundance transcripts, all total RNA and cRNAtarget molecule preparations made from each muscle samplewere analyzed for their concentration and overall length. Sup-plemental Fig. S1 shows that reproducibility between samplesis of very high quality. Another critical aspect introducingunwanted gene expression variability is the heterogeneousorigin of the samples. Although the use of animal models ofdisease greatly minimizes this variability, the course of ALS inthe mSOD1 mice is not exempt from a certain degree ofheterogeneity. To reduce interindividual differences in diseaseprogression, we used two replicates per experimental condi-tion, each containing three to four pooled samples (Table 1).Finally, we applied an innovative approach that only uses thePerfect Match values and does not take into account theMismatch values to calculate gene expression levels. Thismethod increases the sensitivity of the measures because theMismatch oligonucleotide signal is thought not only to reflectnonspecific hybridization events but also to contain true ex-pression information that is lost when the default statisticalalgorithm for data analysis provided by the manufacturer isapplied (6, 31).
We used in this study two hindlimb muscles: the soleus,which is composed of 60% slow-twitch fibers and 40% fast-twitch fatigue-resistant fibers, and the gastrocnemius, which is�10 times bigger than the soleus and contains 55% fast-twitchfatigable fibers, 30% fast-twitch fatigue-resistant fibers, and10% slow-twitch fibers (8, 12). Fast-twitch fibers are morevulnerable and weaken at earlier stages of ALS than slow-twitch fibers (1, 17). In addition, fast muscles of mSOD1 micebecome slower and fatigue resistant, and show an age-depen-dent increase in their oxidative capacity as a consequence ofthe disease progression (69). Because the contractile featuresand phenotype of individual fibers in a muscle are constantlychanging during the course of the disease, we assumed thatthere is no simple correlation between gene expression changesin isolated fibers and the overall genetic signature characteriz-ing the atrophic process of the whole limb, and thereforeanalyzed the transcriptional alterations expressed by bothsoleus and gastrocnemius all together.
Finally, we studied here the transcriptome of ALS andacutely denervated muscles. Increased expression of acetylcho-line receptor �- and �-subunits is very noticeable after 7 daysof sciatic nerve axotomy (80), and, in our hands, such modi-fications were found to be very similar to those observed inparalyzed G86R mice (Supplemental Fig. S2), which makesthe two denervation conditions comparable.
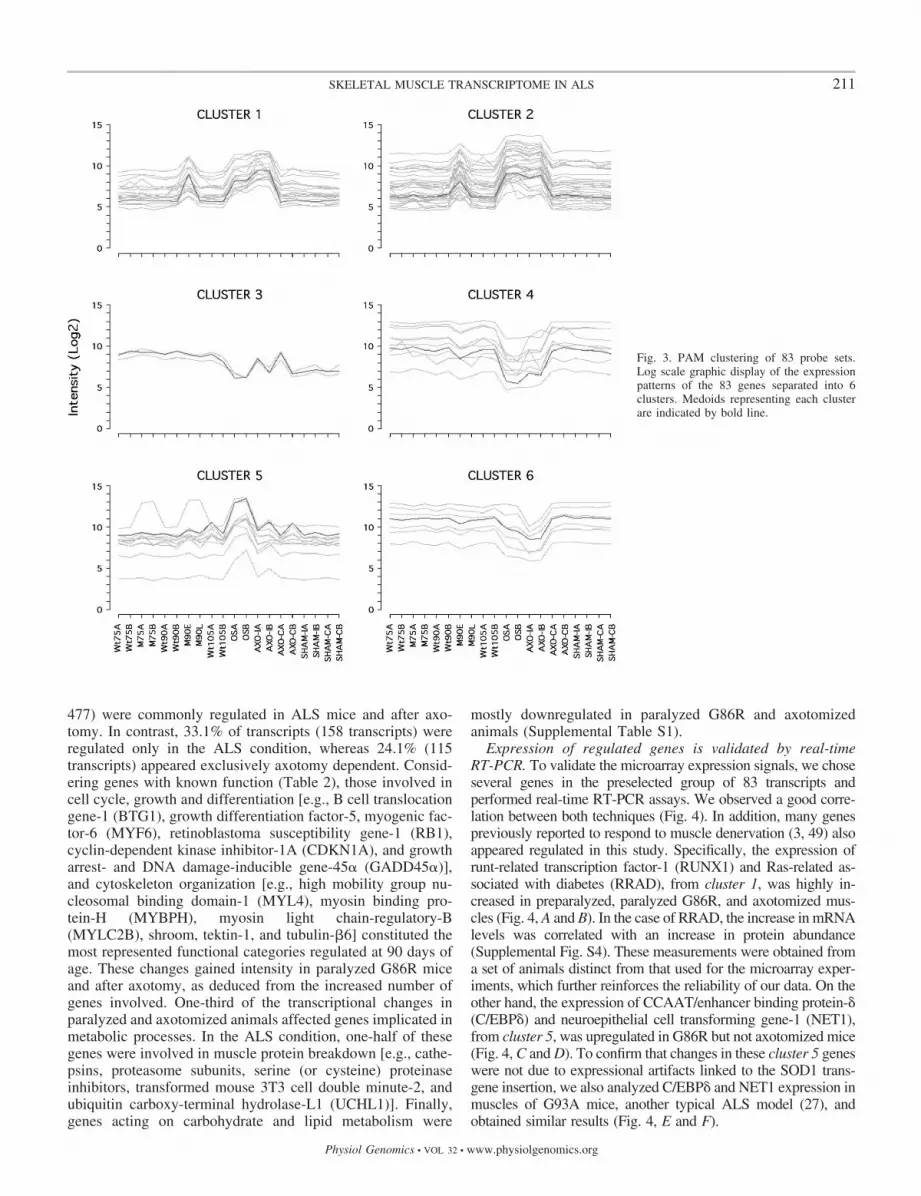
The transcriptome of ALS muscle is altered at preparalysisstages. The scatterplot matrix of the totality of the probe sets inthe MG-U74Av2 GeneChips indicated that samples fromG86R mice at the onset of paralysis and those suffering fromaxotomy resembled each other but differed importantly fromthe others (Supplemental Fig. S3). In addition, one of thesample replicates of 90-day-old G86R mice presented signifi-cant differences with respect to the other 90-day-old sample.To further assess these observations, we performed unsuper-vised hierarchical clustering of the 20 samples and 83 tran-scripts whose expression signals displayed an SD �0.8 acrossthe 20 data sets (Fig. 2). Although this approach yields a veryconservative estimate of the number of differentially expressedgenes, the two 90-day-old sample replicates appeared clearlyseparated: one sample looked more like the wild-type andcontrol samples and represented an early preparalysis phase ofthe disease, whereas the other one more closely resembled thesamples from paralyzed and axotomized mice and representeda late preparalysis phase (Fig. 2, dendrogram at top). We thenapplied the PAM algorithm to identify typical transcriptionalpatterns among the 83 preselected transcripts. Using centeredand scaled data to minimize the impact of expression levels, wedefined six typical transcriptional patterns (or medoids) thatgave rise to six different groups (or clusters) of genes (Fig. 3).Clusters 1 and 2 contained 18 and 34 transcripts, respectively,that were upregulated in samples from G86R mice during thelate preparalysis phase and at the onset of paralysis as well asin those suffering from axotomy. The three transcripts con-tained in cluster 3 presented very variable expression levels,even among the several control conditions, and were notconsidered. Clusters 4 and 6 contained nine and seven tran-scripts, respectively, that were downregulated mostly in sam-
Fig. 1. Flowchart of data mining. SD, standard devia-tion; PAM, partitioning around medoids.
209SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org

ples from paralyzed G86R and axotomized mice. Finally,cluster 5 contained 11 transcripts that appeared mostly upregu-lated in ALS but not after experimental denervation.
To gain further insight into the gene expression changes ina subset of mice at 90 days of age, we searched for differen-tially regulated genes in the entire database that presented atleast a twofold change between the pathological or denervationcondition and their corresponding controls (Supplemental
Table S1). We did not find any significant alterations insteady-state mRNA abundance in G86R mice at 75 days of ageor in the subset of 90-day-old mice at the early preparalysisphase. In contrast, 66 transcripts were regulated during the latepreparalysis phase at 90 days of age, and this list expanded,with 360 transcripts at the onset of paralysis. Sciatic nerveaxotomy induced transcriptional changes in 318 transcripts. Onthe basis of the applied cutoff, 42.8% of transcripts (204 of
Fig. 2. Hierarchical clustering of 20 samplesand 83 probe sets. A heat map of 83 tran-scripts and 2 dendograms that group genes(left) and samples (top) together are shown.Each line is a gene, and each column is asample. Expression signal intensities areshown in red and blue, indicating high andlow expression, respectively. Names of the83 genes are indicated at right.
210 SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org
477) were commonly regulated in ALS mice and after axo-tomy. In contrast, 33.1% of transcripts (158 transcripts) wereregulated only in the ALS condition, whereas 24.1% (115transcripts) appeared exclusively axotomy dependent. Consid-ering genes with known function (Table 2), those involved incell cycle, growth and differentiation [e.g., B cell translocationgene-1 (BTG1), growth differentiation factor-5, myogenic fac-tor-6 (MYF6), retinoblastoma susceptibility gene-1 (RB1),cyclin-dependent kinase inhibitor-1A (CDKN1A), and growtharrest- and DNA damage-inducible gene-45� (GADD45�)],and cytoskeleton organization [e.g., high mobility group nu-cleosomal binding domain-1 (MYL4), myosin binding pro-tein-H (MYBPH), myosin light chain-regulatory-B(MYLC2B), shroom, tektin-1, and tubulin-�6] constituted themost represented functional categories regulated at 90 days ofage. These changes gained intensity in paralyzed G86R miceand after axotomy, as deduced from the increased number ofgenes involved. One-third of the transcriptional changes inparalyzed and axotomized animals affected genes implicated inmetabolic processes. In the ALS condition, one-half of thesegenes were involved in muscle protein breakdown [e.g., cathe-psins, proteasome subunits, serine (or cysteine) proteinaseinhibitors, transformed mouse 3T3 cell double minute-2, andubiquitin carboxy-terminal hydrolase-L1 (UCHL1)]. Finally,genes acting on carbohydrate and lipid metabolism were
mostly downregulated in paralyzed G86R and axotomizedanimals (Supplemental Table S1).
Expression of regulated genes is validated by real-timeRT-PCR. To validate the microarray expression signals, we choseseveral genes in the preselected group of 83 transcripts andperformed real-time RT-PCR assays. We observed a good corre-lation between both techniques (Fig. 4). In addition, many genespreviously reported to respond to muscle denervation (3, 49) alsoappeared regulated in this study. Specifically, the expression ofrunt-related transcription factor-1 (RUNX1) and Ras-related as-sociated with diabetes (RRAD), from cluster 1, was highly in-creased in preparalyzed, paralyzed G86R, and axotomized mus-cles (Fig. 4, A and B). In the case of RRAD, the increase in mRNAlevels was correlated with an increase in protein abundance(Supplemental Fig. S4). These measurements were obtained froma set of animals distinct from that used for the microarray exper-iments, which further reinforces the reliability of our data. On theother hand, the expression of CCAAT/enhancer binding protein-�(C/EBP�) and neuroepithelial cell transforming gene-1 (NET1),from cluster 5, was upregulated in G86R but not axotomized mice(Fig. 4, C and D). To confirm that changes in these cluster 5 geneswere not due to expressional artifacts linked to the SOD1 trans-gene insertion, we also analyzed C/EBP� and NET1 expression inmuscles of G93A mice, another typical ALS model (27), andobtained similar results (Fig. 4, E and F).
Fig. 3. PAM clustering of 83 probe sets.Log scale graphic display of the expressionpatterns of the 83 genes separated into 6clusters. Medoids representing each clusterare indicated by bold line.
211SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org

Genes involved in stress mechanisms. One early transcrip-tional change in preparalyzed G86R mice was the upregulationof lysyl oxidase (LOX), an extracellular enzyme catalyzing thefirst step of the cross-linking of collagen and elastin to obtainmature insoluble fibers and assumed to generate high amountsof H2O2 as a by-product of its catalytic reaction (43). On theother hand, we observed increased expression of a series ofantioxidant genes, including heat shock 27-kDa protein-8(HSPB8), heat shock protein-4, metallothionein-I (MT1) and -II(MT2), thioredoxin-1, sestrin-1, which participates in reestab-lishing the antioxidant firewall that must be transiently disabledto allow physiological H2O2-dependent signaling (7), and al-dehyde dehydrogenase-3 (ALDH3), which is believed to de-toxify aldehydes from lipid peroxidation (64). The expressionof other antioxidant genes such as microsomal glutathioneS-transferase-3 and selenoprotein-X1 was, however, down-regulated in paralyzed G86R and axotomized mice (Supple-mental Table S1). Of note, the whole of these transcriptionalregulations was correlated in vivo with an increase in reactiveoxygen species in muscles from preparalyzed and paralyzedG86R mice and after axotomy, compared with muscles ofwild-type animals (Supplemental Fig. S5), which stronglysuggests that an important number of genes with antioxidantproperties appeared regulated in response to that oxidativeinsult.
Genes involved in alternative pathways of ATP production.Our previous work demonstrated a constant decrease in theproduction of muscle ATP starting in G86R mice as early as 75days of age (20). This metabolic shutdown seems to underliesome transcriptional adaptations, because the expression ofDNA damage-inducible transcript-4 (DDIT4, also known asREDD1), which is involved in the response to hypoxia andenergy depletion, was upregulated in preparalyzed mice. En-
ergy stress-induced REDD1 expression leads to suppression ofcell growth by dephosphorylation of key mTOR substrates,including eukaryotic translation initiation factor-4E bindingprotein-1 (EIF4EBP1) and ribosomal protein-S6 kinase-1 (70).We found that the expression of EIF4EBP1 and mTOR (iden-tified herein as FK506 binding protein-12 rapamycin-associ-ated protein-1) was upregulated in paralyzed G86R mice (butnot after axotomy) (Supplemental Table S1). It is thereforetempting to suggest that the early stimulation triggered by ATPloss of inhibitors of the mTOR pathway, such as REDD1,could play a significant role in ALS muscle pathology.
In paralyzed G86R and axotomized mice, gene expressioninvolved in carbohydrate and lipid metabolism was in generaldownregulated, further aggravating the metabolic stress at thisstage of the disease. The concerted action of adenylate kinase,which catalyzes the conversion of two molecules of ADP toATP and AMP, and AMP deaminase, responsible for deami-nation of AMP to IMP and ammonia, constitutes an additionalpathway to produce ATP in stressed cells (29). We detectedincreased expression of AMP deaminase-3 (AMPD3) as earlyas 90 days of age in G86R mice as well as in denervatedmuscles. In paralyzed and axotomized mice, however, adenyl-ate kinase-1 expression was decreased (Supplemental TableS1). It is also known that the purine salvage pathway stimulatesthe conversion of hypoxanthine to IMP, which can in turn bereaminated via the purine nucleotide cycle and eventuallyresult in ATP resynthesis (48). We found in G86R and axoto-mized mice increased expression of hypoxanthine phosphori-bosyltransferase-1 (HPRT1), which catalyzes the conversion ofhypoxanthine to IMP, although the expression of adenylosuc-cinate synthetase-like-1, involved in generating AMP (71),appeared downregulated (Supplemental Table S1).
Genes involved in protein degradation. Several transcriptionfactors involved in muscle proteolysis, such as C/EBP� and -�and forkhead box-O3a, were upregulated in G86R mice. Alongwith this, we also observed, particularly in paralyzed G86Rmice, increased expression of many proteasome subunits and,as early as 90 days of age, increased expression of UCHL1,involved in recycling ubiquitin from substrates that have beencommitted to degradation by the proteasome (79). Proteolysisby intralysosomal cathepsins is another type of protein degra-dation. mRNA levels of cathepsin-L and -Z were increased inmuscles of paralyzed G86R mice as well as those of cathep-sin-L and -S in surgically denervated muscles. In contrast, theexpression of skeletal muscle-specific calpain-3, involved inselective calcium-dependent proteolysis (15), appeared down-regulated (Supplemental Table S1).
Genes involved in cell cycle, growth, differentiation, and celldeath. The expression of key regulators of myogenesis, such asBTG1, appeared early to be upregulated in G86R mice. BTG1induces myoblast differentiation by stimulating the transcrip-tional activity of triiodothyronine and all-trans retinoic acidreceptors, c-JUN, and myogenic factors including myogenicdifferentiation antigen-1 (MYOD1), myogenic factor-5, andmyogenin (9). In turn, MYOD1 induces cell cycle arrest bystimulating the expression of CDKN1A and RB1, which is alsoa downstream target of CDKN1A (28, 52). Although MYOD1expression was unchanged in G86R mice (but upregulated indenervated muscle), CDKN1A and RB1 mRNAs appearedupregulated in preparalyzed animals (Supplemental Table S1).
Table 2. Classification of genes with known functiondifferentially regulated at least twofold in G86Rand axotomized mice
M90L* (51) Onset† (259) Axotomy (235)
Extracellular matrix 3.9 (2) 3.5 (9) 2.1 (5)Cell adhesion 3.9 (2) 3.9 (10) 3.8 (9)Neuromuscular junction 5.9 (3) 2.3 (6) 2.1 (5)Ion channels 2.3 (6) 2.1 (5)Cell cycle, growth, differentiation 15.7 (8) 8.9 (23) 10.6 (25)Cytoskeleton 13.7 (7) 7.3 (19) 14.0 (33)Stress response 9.8 (5) 5.0 (13) 3.4 (8)Immune response, inflammation 9.8 (5) 6.2 (16) 5.5 (13)Circadian rhythm 1.2 (3) 0.4 (1)Oxygen metabolism 2.0 (1) 1.2 (3) 0.4 (1)Metabolism (carbohydrates) 5.0 (13) 7.2 (17)Metabolism (lipids) 2.0 (1) 4.2 (11) 6.8 (16)Metabolism (proteins) 3.9 (2) 15.8 (41) 8.1 (19)Tricarboxylic acid cycle 0.4 (1) 2.1 (5)Oxidative phosphorylation 1.5 (4) 2.5 (6)Metabolism (nucleotides) 3.9 (2) 1.9 (5) 2.5 (6)Metabolism (miscellaneous) 3.9 (2) 4.6 (12) 7.2 (17)DNA processing 2.3 (6) 2.5 (6)RNA processing 3.1 (8) 2.1 (5)Transcription regulation 11.8 (6) 9.6 (25) 7.2 (17)Signal transduction 9.8 (5) 9.6 (25) 6.8 (16)
Data are expressed as percentages of the total no. of genes that appeareddifferentially regulated within a given experimental condition, with absolutenos. of genes in parentheses. *M90L represents the 90-day-old sample referredto in the text as the late preparalysis phase. †Onset indicates onset of paralysis.
212 SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org
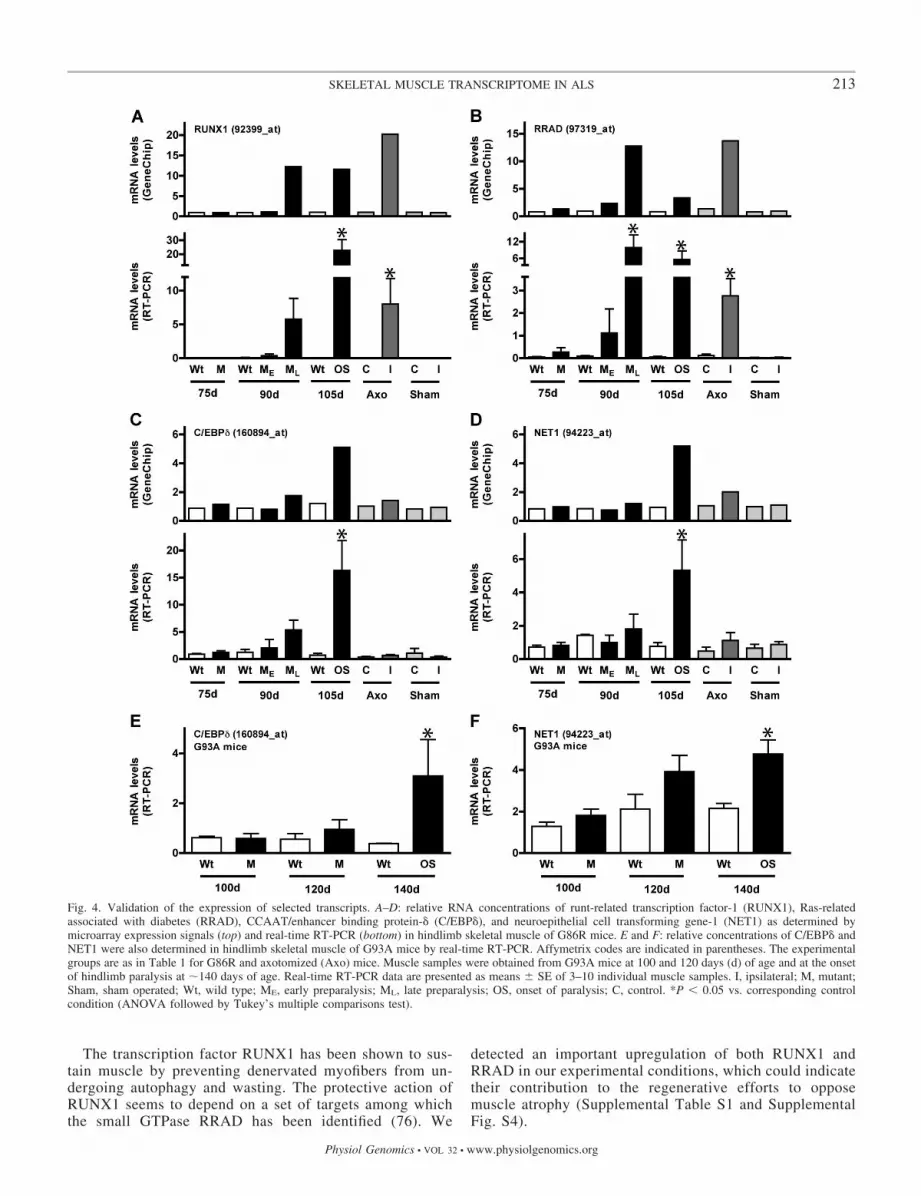
The transcription factor RUNX1 has been shown to sus-tain muscle by preventing denervated myofibers from un-dergoing autophagy and wasting. The protective action ofRUNX1 seems to depend on a set of targets among whichthe small GTPase RRAD has been identified (76). We
detected an important upregulation of both RUNX1 andRRAD in our experimental conditions, which could indicatetheir contribution to the regenerative efforts to opposemuscle atrophy (Supplemental Table S1 and SupplementalFig. S4).
Fig. 4. Validation of the expression of selected transcripts. A–D: relative RNA concentrations of runt-related transcription factor-1 (RUNX1), Ras-relatedassociated with diabetes (RRAD), CCAAT/enhancer binding protein-� (C/EBP�), and neuroepithelial cell transforming gene-1 (NET1) as determined bymicroarray expression signals (top) and real-time RT-PCR (bottom) in hindlimb skeletal muscle of G86R mice. E and F: relative concentrations of C/EBP� andNET1 were also determined in hindlimb skeletal muscle of G93A mice by real-time RT-PCR. Affymetrix codes are indicated in parentheses. The experimentalgroups are as in Table 1 for G86R and axotomized (Axo) mice. Muscle samples were obtained from G93A mice at 100 and 120 days (d) of age and at the onsetof hindlimb paralysis at �140 days of age. Real-time RT-PCR data are presented as means � SE of 3–10 individual muscle samples. I, ipsilateral; M, mutant;Sham, sham operated; Wt, wild type; ME, early preparalysis; ML, late preparalysis; OS, onset of paralysis; C, control. *P � 0.05 vs. corresponding controlcondition (ANOVA followed by Tukey’s multiple comparisons test).
213SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org

Other transcriptional regulations seemed, however, to pro-mote cell death pathways. For instance, the increasing upregu-lation of CDKN1A and GADD45� could mediate apoptosis ofmyonuclei and hence trigger muscle atrophy (10). Similarly,cytokeratins, the largest subgroup of intermediate filamentproteins, have also been involved in apoptosis (38). The in-crease in cytokeratin-endoB expression observed in our exper-imental conditions could therefore be associated with muscleatrophy. Polyamines, including putrescine, spermidine andspermine, are small aliphatic cations that promote cell prolif-eration, growth, and differentiation but also induce apoptosiswhen they occur in abnormally elevated levels. Ornithinedecarboxylase (ODC) and S-adenosylmethionine decarboxyl-ase (AMD) are the key enzymes responsible for polyaminesynthesis (32). In our hands, ODC expression stayed un-changed, but we observed in both G86R and axotomized micea dramatic decrease in the levels of different AMD isoenzymemRNAs (Supplemental Table S1), which could lead to theaberrant toxic accumulation of putrescine. We also observed ahigh increase in the expression of ankyrin repeat domain-containing protein-1 (ANKRD1) and, to a lesser extent,ANKRD2 only in paralyzed G86R mice. These findings cor-roborate previous observations of altered protein contents ofANKRD1 in atrophic myofibers in cases of ALS (57). Theseankyrin repeat proteins are typically regulated following mus-cle stress (53). For example, ANKRD2 has been shown to bindp53 and enhance p53-induced CDKN1A upregulation (40).Interestingly, we also found increased mRNA levels of pater-nally expressed gene-3 (PEG3) (Supplemental Table S1), atranscription factor that functions downstream of p53 to regu-late Bax redistribution into the cell and promote apoptosis (16).Of note, this is consistent with other studies that showed Baximmunoreactivity as a fine granular precipitate at the sarco-lemma and within the sarcosol of atrophic myofibers in sam-ples of sporadic ALS (68).
DISCUSSION
Muscle gene regulations precede the onset of paralysis inmSOD1 mice. The goal of this study was to determine geneexpression changes occurring in skeletal muscle of G86R miceduring the course of ALS. At 75 days of age, we did not detectsignificant transcriptional changes, which may contrast withour own previous studies showing muscle gene expressionalterations at early presymptomatic stages (22). These tran-scriptional changes could not pass the several filtering stepsemployed in this study because we used the stringent twofoldcriteria to eliminate uncertain regulations and uncover the mostsignificant changes relevant to the disease. By this means, thefirst expressional modifications characterizing muscle pathol-ogy appeared in a subset of 90-day-old G86R mice, coincidentat the histological level with the onset of the reduction inmyofiber size (35) but not motor neuron loss. Indeed, noreduction in the number of lumbar motor neurons is observedin animals at 80–90 days of age, whereas a significant decreasestarts at �105 days of age, when mice display motor deficits inat least one limb (19, 54, 65). By comparing the transcriptomesfrom G86R and surgically denervated muscles, we showed thatmost transcriptional changes appearing at 90 days of age inG86R mice were common to those observed following 7 daysof sciatic nerve axotomy. These findings establish a parallel
between the rupture of neuromuscular transmission originatedby cutting the nerve in axotomized mice and the existence ofdenervation in preparalyzed G86R mice. Because axotomy-induced motor neuron death, if it occurs, takes more than aweek in the adult mouse (39), one can infer that the first signsof denervation and atrophy in G86R muscle are not caused bydegeneration of the motor neuron cell bodies but are rather theresult of axonal dysfunction. Consistent with this notion, ac-cumulation of light chain neurofilament subunits (55) andaltered expression of the fast axonal transport regulator KIF3-associated protein-3 (19) occur early in G86R motor neurons,thus providing evidence of premature axonal impairment. Sev-eral previous studies had pointed to axonal dysfunction asprimarily causing a progressive deterioration of the neuromus-cular function that would eventually lead to muscle atrophyand motor neuron death (2, 24, 25). Recent research has furtherdemonstrated that clinical symptoms in mSOD1 mice resultspecifically from damage to the distal motor axon and not fromactivation of death pathways in the cell soma (26, 67). Ourpresent findings now provide a set of genes differentiallyregulated in preparalyzed G86R mice that characterizes theinitiatory mechanisms underlying muscle pathology in ALS.The increased expression of genes implicated in detoxification(e.g., ALDH3, MT2, and thioredoxin-1), energy balance (e.g.,AMPD3, DDIT4, and HPRT1), and injury response (e.g.,ANKRD1 and HSPB8) is consistent with previous studiesshowing increased amounts of malondialdehyde and proteincarbonyls, as well as high levels of manganese SOD andcatalase activity in G93A muscles (50). We also show here thein vivo accumulation of muscle reactive oxygen species start-ing before overt paralysis, which further highlights the impor-tance of mechanisms involved in facing oxidative and energystress. The induction of genes implicated in myogenesis (e.g.,BTG1, MYF6, RB1, RUNX1, and RRAD) and cytoskeletonorganization (e.g., MYL4, MYBPH, MYLC2B, and shroom)can play an important role in stimulating the regenerativecapacity of muscle during the first steps of atrophy. In contrast,the upregulation of others underlying cell death (e.g.,CDKN1A, GADD45�, and PEG3) and cell degradation (e.g.,MAP1LC3� and UCHL1) can promote apoptotic and proteo-lytic pathways that characterize the fatal end of the atrophicprocess.
Common gene regulations occur in muscles and motorneurons of paralyzed mSOD1 mice. Although there are somedifferences among the existing mSOD1 mouse lines (4), theyall present with almost the same pathological hallmarks andclinical progression. We looked for similar gene expressionchanges operating in muscle and motor neurons at the time ofparalysis by comparing our present data with those obtainedusing laser-captured microdissected motor neurons (23, 46, 60)(Table 3). Notably, upregulation of MT1 and MT2, involved inmetal homeostasis/detoxification and free radical scavenging,appears as a general transcriptional event, occurring not only inmuscle and isolated motor neurons but also in whole spinalcord (59) and motor cortex of sporadic ALS patients (42).Reduced expression of metallothioneins by genetic meansaggravated disease in mSOD1 mice (56, 63), which stronglysuggests that their upregulation may be regarded as a keyprotective mechanism. Also of interest is the increased expres-sion of LOX and thioredoxin-1 in muscle and human ALS
214 SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org

spinal cord (51), which points to common pathways of oxida-tive damage in the whole motor unit.
The upregulation of cathepsins, intralysosomal proteolyticenzymes important for normal protein turnover and involved ina number of pathological conditions (72), appears as a generaltranscriptional event in ALS. In particular, increased expres-sion of cathepsin-Z (also named cathepsin-X) was detected in
muscles and isolated motor neurons (Table 3). Consistently,increases in cathepsin-Z protein content and enzymatic activitywere also observed in mSOD1 mouse spinal cord (78), al-though its exact role in the pathological process is presentlyunknown. Interestingly, several serpins, serine (or cysteine)proteinase inhibitors, were commonly upregulated in muscles(our present data), isolated spinal motor neurons (23, 60), and
Table 3. Genes with common or opposed transcriptional regulation between muscle and spinal cord of paralyzedmSOD1 mice
Name Symbol Ref. Nos. and Remarks
Genes upregulated in muscle and spinal cord
Lectin, galactose binding, soluble 1 LGALS1 muscleLectin, galactose binding, soluble 3 LGALS3 23, 60Cyclin-dependent kinase inhibitor 1A, P21 CDKN1A 60GADD45 protein gene GADD45� 46, 60Myocyte enhancer factor 2A MEF2A muscleMyocyte enhancer factor 2C MEF2C 60 (spinal cord)Troponin T2, cardiac TNNT2 muscleTroponin T3, skeletal, fast TNNT3 60 (spinal cord)Tubulin, beta 6 TUB�6 46, 60Heat shock 27 kDa protein 8 HSPB8 60Metallothionein-I MT1 23, 60Metallothionein-II MT2 60Chemokine (C-X-C motif) ligand 13 CXCL13 muscleChemokine (C-X-C motif) ligand 14 CXCL14 23P lysozyme structural LZP-S 23, 46, 60S100 calcium binding protein A8, calgranulin A S100A8 60 (down in 90-day-old muscle)Cathepsin Z CTSZ 23, 60Serine (or cysteine) proteinase inhibitor, clade A, member 3n SERPINA3N 23Serine (or cysteine) proteinase inhibitor, clade B, member 1a SERPINB1A 60 (spinal cord)Serine (or cysteine) proteinase inhibitor, clade B, member 6a SERPINB6A muscleTransformed mouse 3T3 cell double minute 2 MDM2 23Ribonucleotide reductase M2 RRM2 60Activating transcription factor 3 ATF3 46, 60CCAAT/enhancer binding protein, delta C/EBP� 60RIO kinase 3, yeast RIOK3 60CD68 antigen CD68 60
Genes downregulated in muscle and spinal cord
Potassium voltage-gated channel, shaker-related, beta member 1 KCNA�1 musclePotassium voltage-gated channel, shaker-related, member 7 KCNA7 musclePotassium voltage-gated channel, shaker-related, member 6 KCNA6 60 (spinal cord)CD24a antigen CD24A 23Immunoglobulin heavy chain 6, heavy chain of IgM IGH-6 23, 46, 60Cytochrome c, somatic CYCS 23Transcription elongation factor A (SII), 1 TCEA1 23 (spinal cord)Transcription elongation factor A (SII), 3 TCEA3 muscle3-Monooxygenase/tryptophan 5-monooxygenase activation protein- YWHAG muscle3-Monooxygenase/tryptophan 5-monooxygenase activation protein-ε, YWHAE,Q 23 (spinal cord)Integral membrane protein 2A ITM2A 23, 46, 60
Genes differentially regulated in muscle and spinal cord
Fibromodulin FMOD 60, up (spinal cord), down (muscle)Procollagen, type I, alpha 2 COL1�2 60, up (spinal cord), down (muscle)Actin, alpha, cardiac ACTC1 60, up (spinal cord), down (muscle)Kinesin family member 1B KIF1B 23, up (spinal cord), down (muscle)Kinesin family member 3A KIF3A 60, up (spinal cord)S100 calcium binding protein A9, calgranulin B S100A9 60, up (spinal cord), down (muscle)Fatty acid binding protein 3, muscle and heart FABP3 down (muscle)Fatty acid binding protein 7, brain FABP7 60, up (spinal cord)TAF9 RNA polymerase II, TATA box binding protein-associated factor TAF9 23, down (spinal cord), up (muscle)ERBB receptor feedback inhibitor 1 ERRFI1 23, down (spinal cord), up (muscle)Aquaporin 4 AQP4 60, up (spinal cord), down (muscle)Leucine rich repeat protein 1, neuronal LRRN1 60, down (spinal cord), up (muscle)
Genes were selected when expression was changed at least 2-fold compared with corresponding controls. No remark implies that the regulation was detectedin muscle and spinal cord (in some cases, the regulation of related genes was only observed in muscle or spinal cord, as indicated). mSOD1, mutantCu/Zn-superoxide dismutase-1.
215SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org

motor cortex of sporadic ALS patients (42). These transcrip-tional regulations can be relevant to the disease because certainserpins can inhibit cathepsin activity and have been involved inpreventing cell death in response to oxidative stress (45).Moreover, several serpins were found in the neurofilamentouslesions characteristic of ALS motor neurons (13) and weresuggested to participate locally in neuromuscular junctionremodeling (74).
Another noticeable finding is the upregulation of the stress-inducible gene-activating transcription factor-3 (ATF3) inmuscles and motor neurons (Table 3). Interestingly, high levelsof ATF3 mRNA and protein were found to precede death ofALS motor neurons (75). Although it is not clear whetherATF3 upregulation can promote or prevent cell death in ALS,transgenic mice expressing ATF3 in cardiomyocytes showedconduction and contractile abnormalities (58). If expression ofATF3 correlates with cellular injury, deciphering the moleculartargets and pathways dependent on this immediate early genecould help with the identification of potential therapeutic in-terventions.
Also of interest is the increased expression of lectin galac-toside binding soluble-3 (or galectin-3) in muscles and motorneurons, together with that of galectin-1 in muscles (Table 3).Galectin-1 was found to accumulate in the neurofilamentouslesions of the spinal cord of sporadic and familial ALS subjects(36), and its administration as an oxidized molecule to mSOD1mice ameliorated impairment of motor function and extendedsurvival (11). In contrast to these findings, the reduced form ofgalectin-1 has been shown to participate in the degeneration ofpresynaptic nerve terminals after axonal lesion in vivo (61).The aberrant muscle expression of such lectins in the ALScondition could therefore contribute to the degenerative pro-cess.
Muscle atrophy is the major cause of motor disability andsuffering in ALS patients. To determine efficient therapeuticstrategies that could alleviate muscle pathology, it is impera-tive to identify the specific molecular effectors that altermuscle function during the course of the disease. The set oftranscriptional regulations we describe herein, appearing earlyat preparalysis stages and, in some cases, in common withthose occurring in spinal cord motor neurons, can channelizeour efforts to palliate neuromuscular dysfunction before theirrevocable loss of motor neuron cell bodies.
ACKNOWLEDGMENTS
We thank A. Picchinenna and M. J. Ruivo for excellent technical assistance.We also thank Dr. C. R. Kahn (Joslin Diabetes Center, Boston, MA) forproviding anti-RRAD antiserum.
GRANTS
The Institut National de la Sante et de la Recherche Medicale-U692laboratory is supported by Alsace Biovalley, Association Francaise contre lesMyopathies, Association pour la Recherche sur la Sclerose Laterale Amyotro-phique, and Fondation pour la Recherche sur le Cerveau. B. Halter is supportedby Region Alsace and Association pour la Recherche et le Developpement deMoyens de Lutte contre les Maladies Neurodegeneratives.
REFERENCES
1. Atkin JD, Scott RL, West JM, Lopes E, Quah AK, Cheema SS.Properties of slow- and fast-twitch muscle fibres in a mouse model ofamyotrophic lateral sclerosis. Neuromuscul Disord 15: 377–388, 2005.
2. Azzouz M, Leclerc N, Gurney M, Warter JM, Poindron P, Borg J.Progressive motor neuron impairment in an animal model of familialamyotrophic lateral sclerosis. Muscle Nerve 20: 45–51, 1997.
3. Batt J, Bain J, Goncalves J, Michalski B, Plant P, Fahnestock M,Woodgett J. Differential gene expression profiling of short and long termdenervated muscle. FASEB J 20: 115–117, 2006.
4. Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, BrannstromT, Rehnmark A, Marklund SL. Overloading of stable and exclusion ofunstable human superoxide dismutase-1 variants in mitochondria of mu-rine amyotrophic lateral sclerosis models. J Neurosci 26: 4147–4154,2006.
5. Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA,Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inher-ited ALS determined by motor neurons and microglia. Science 312:1389–1392, 2006.
6. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison ofnormalization methods for high density oligonucleotide array data basedon variance and bias. Bioinformatics 19: 185–193, 2003.
7. Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM.Regeneration of peroxiredoxins by p53-regulated sestrins, homologs ofbacterial AhpD. Science 304: 596–600, 2004.
8. Burkholder TJ, Fingado B, Baron S, Lieber RL. Relationship betweenmuscle fiber types and sizes and muscle architectural properties in themouse hindlimb. J Morphol 221: 177–190, 1994.
9. Busson M, Carazo A, Seyer P, Grandemange S, Casas F, PessemesseL, Rouault JP, Wrutniak-Cabello C, Cabello G. Coactivation of nuclearreceptors and myogenic factors induces the major BTG1 influence onmuscle differentiation. Oncogene 24: 1698–1710, 2005.
10. Caiozzo VJ, Wu YZ, Baker MJ, Crumley R. Effects of denervation oncell cycle control in laryngeal muscle. Arch Otolaryngol Head Neck Surg130: 1056–1068, 2004.
11. Chang-Hong R, Wada M, Koyama S, Kimura H, Arawaka S,Kawanami T, Kurita K, Kadoya T, Aoki M, Itoyama Y, Kato T.Neuroprotective effect of oxidized galectin-1 in a transgenic mouse modelof amyotrophic lateral sclerosis. Exp Neurol 194: 203–211, 2005.
12. Chin ER, Grange RW, Viau F, Simard AR, Humphries C, Shelton J,Bassel-Duby R, Williams RS, Michel RN. Alterations in slow-twitchmuscle phenotype in transgenic mice overexpressing the Ca2� bufferingprotein parvalbumin. J Physiol 547: 649–663, 2003.
13. Chou SM, Taniguchi A, Wang HS, Festoff BW. Serpin�serine pro-tease-like complexes within neurofilament conglomerates of motoneuronsin amyotrophic lateral sclerosis. J Neurol Sci 160: S73–S79, 1998.
14. Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, RuleM, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH Jr,Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cellsextend survival of SOD1 mutant motor neurons in ALS mice. Science 302:113–117, 2003.
15. Costelli P, Reffo P, Penna F, Autelli R, Bonelli G, Baccino FM.Ca(2�)-dependent proteolysis in muscle wasting. Int J Biochem Cell Biol37: 2134–2146, 2005.
16. Deng Y, Wu X. Peg3/Pw1 promotes p53-mediated apoptosis by inducingBax translocation from cytosol to mitochondria. Proc Natl Acad Sci USA97: 12050–12055, 2000.
17. Derave W, Van Den Bosch L, Lemmens G, Eijnde BO, Robberecht W,Hespel P. Skeletal muscle properties in a transgenic mouse model foramyotrophic lateral sclerosis: effects of creatine treatment. Neurobiol Dis13: 264–272, 2003.
18. Dobrowolny G, Giacinti C, Pelosi L, Nicoletti C, Winn N, Barberi L,Molinaro M, Rosenthal N, Musaro A. Muscle expression of a local Igf-1isoform protects motor neurons in an ALS mouse model. J Cell Biol 168:193–199, 2005.
19. Dupuis L, de Tapia M, Rene F, Lutz-Bucher B, Gordon JW, MerckenL, Pradier L, Loeffler JP. Differential screening of mutated SOD1transgenic mice reveals early up-regulation of a fast axonal transportcomponent in spinal cord motor neurons. Neurobiol Dis 7: 274–285, 2000.
20. Dupuis L, di Scala F, Rene F, de Tapia M, Oudart H, Pradat PF,Meininger V, Loeffler JP. Up-regulation of mitochondrial uncouplingprotein 3 reveals an early muscular metabolic defect in amyotrophic lateralsclerosis. FASEB J 17: 2091–2093, 2003.
21. Dupuis L, Gonzalez de Aguilar JL, di Scala F, Rene F, de Tapia M,Pradat PF, Lacomblez L, Seihlan D, Prinjha R, Walsh FS, MeiningerV, Loeffler JP. Nogo provides a molecular marker for diagnosis ofamyotrophic lateral sclerosis. Neurobiol Dis 10: 358–365, 2002.
22. Dupuis L, Oudart H, Rene F, Gonzalez de Aguilar JL, Loeffler JP.Evidence for defective energy homeostasis in amyotrophic lateral sclerosis:benefit of a high-energy diet in a transgenic mouse model. Proc Natl AcadSci USA 101: 11159–11164, 2004.
216 SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org

23. Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ.Microarray analysis of the cellular pathways involved in the adaptation toand progression of motor neuron injury in the SOD1 G93A mouse modelof familial ALS. J Neurosci 27: 9201–9219, 2007.
24. Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosisis a distal axonopathy: evidence in mice and man. Exp Neurol 185:232–240, 2004.
25. Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early andselective loss of neuromuscular synapse subtypes with low sproutingcompetence in motoneuron diseases. J Neurosci 20: 2534–2542, 2000.
26. Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM,Milligan CE, Oppenheim RW. Complete dissociation of motor neurondeath from motor dysfunction by Bax deletion in a mouse model of ALS.J Neurosci 26: 8774–8786, 2006.
27. Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, AlexanderDD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neurondegeneration in mice that express a human Cu,Zn superoxide dismutasemutation. Science 264: 1772–1775, 1994.
28. Halevy O, Novitch BG, Spicer DB, Skapek SX, Rhee J, Hannon GJ,Beach D, Lassar AB. Correlation of terminal cell cycle arrest of skeletalmuscle with induction of p21 by MyoD. Science 267: 1018–1021, 1995.
29. Hellsten-Westing Y, Norman B, Balsom PD, Sjodin B. Decreasedresting levels of adenine nucleotides in human skeletal muscle afterhigh-intensity training. J Appl Physiol 74: 2523–2528, 1993.
30. Ihaka R, Gentleman R. R: a language for data analysis and graphics.J Comp Graph Stat 5: 299–314, 1996.
31. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ,Scherf U, Speed T. Exploration, normalization, and summaries of highdensity oligonucleotide array probe level data. Biostatistics 4: 249–264,2003.
32. Janne J, Alhonen L, Pietila M, Keinanen TA. Genetic approaches to thecellular functions of polyamines in mammals. Eur J Biochem 271: 877–894, 2004.
33. Jiang YM, Yamamoto M, Kobayashi Y, Yoshihara T, Liang Y, TeraoS, Takeuchi H, Ishigaki S, Katsuno M, Adachi H, Niwa J, Tanaka F,Doyu M, Yoshida M, Hashizume Y, Sobue G. Gene expression profileof spinal motor neurons in sporadic amyotrophic lateral sclerosis. AnnNeurol 57: 236–251, 2005.
34. Jokic N, Di Scala F, Dupuis L, Rene F, Muller A, Gonzalez De AguilarJL, Loeffler JP. Early activation of antioxidant mechanisms in muscle ofmutant Cu/Zn-superoxide dismutase-linked amyotrophic lateral sclerosismice. Ann NY Acad Sci 1010: 552–556, 2003.
35. Jokic N, Gonzalez De Aguilar JL, Dimou L, Lin S, Fergani A, RueggMA, Schwab ME, Dupuis L, Loeffler JP. The neurite outgrowth inhib-itor Nogo-A promotes denervation in an amyotrophic lateral sclerosismodel. EMBO Rep 7: 1162–1167, 2006.
36. Kato T, Kurita K, Seino T, Kadoya T, Horie H, Wada M, KawanamiT, Daimon M, Hirano A. Galectin-1 is a component of neurofilamentouslesions in sporadic and familial amyotrophic lateral sclerosis. BiochemBiophys Res Commun 282: 166–172, 2001.
37. Kaufman L, Rousseeuw PJ. Finding Groups in Data, an Introduction toCluster Analysis. Brussels, Belgium: John Wiley and Sons, 1990.
38. Kirfel J, Magin TM, Reichelt J. Keratins: a structural scaffold withemerging functions. Cell Mol Life Sci 60: 56–71, 2003.
39. Kiryu-Seo S, Hirayama T, Kato R, Kiyama H. Noxa is a criticalmediator of p53-dependent motor neuron death after nerve injury in adultmouse. J Neurosci 25: 1442–1447, 2005.
40. Kojic S, Medeot E, Guccione E, Krmac H, Zara I, Martinelli V, ValleG, Faulkner G. The Ankrd2 protein, a link between the sarcomere and thenucleus in skeletal muscle. J Mol Biol 339: 313–325, 2004.
41. Leclerc N, Ribera F, Zoll J, Warter JM, Poindron P, Lampert E, BorgJ. Selective changes in mitochondria respiratory properties in oxidative orglycolytic muscle fibers isolated from G93AhumanSOD1 transgenic mice.Neuromuscul Disord 11: 722–727, 2001.
42. Lederer CW, Torrisi A, Pantelidou M, Santama N, Cavallaro S.Pathways and genes differentially expressed in the motor cortex of patientswith sporadic amyotrophic lateral sclerosis. BMC Genomics 8: 26, 2007.
43. Li PA, He Q, Cao T, Yong G, Szauter KM, Fong KS, Karlsson J, KeepMF, Csiszar K. Up-regulation and altered distribution of lysyl oxidase inthe central nervous system of mutant SOD1 transgenic mouse model ofamyotrophic lateral sclerosis. Brain Res Mol Brain Res 120: 115–122,2004.
44. Lino MM, Schneider C, Caroni P. Accumulation of SOD1 mutants inpostnatal motoneurons does not cause motoneuron pathology or motoneu-ron disease. J Neurosci 22: 4825–4832, 2002.
45. Liu N, Wang Y, Ashton-Rickardt PG. Serine protease inhibitor 2Ainhibits caspase-independent cell death. FEBS Lett 569: 49–53, 2004.
46. Lobsiger CS, Boillee S, Cleveland DW. Toxicity from different SOD1mutants dysregulates the complement system and the neuronal regenera-tive response in ALS motor neurons. Proc Natl Acad Sci USA 104:7319–7326, 2007.
47. Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS,Mittmann M, Wang C, Kobayashi M, Horton H, Brown EL. Expres-sion monitoring by hybridization to high-density oligonucleotide arrays.Nat Biotechnol 14: 1675–1680, 1996.
48. Lowenstein JM. Ammonia production in muscle and other tissues: thepurine nucleotide cycle. Physiol Rev 52: 382–414, 1972.
49. Magnusson C, Svensson A, Christerson U, Tagerud S. Denervation-induced alterations in gene expression in mouse skeletal muscle. EurJ Neurosci 21: 577–580, 2005.
50. Mahoney DJ, Kaczor JJ, Bourgeois J, Yasuda N, Tarnopolsky MA.Oxidative stress and antioxidant enzyme upregulation in SOD1–G93Amouse skeletal muscle. Muscle Nerve 33: 809–816, 2006.
51. Malaspina A, Kaushik N, de Belleroche J. Differential expression of 14genes in amyotrophic lateral sclerosis spinal cord detected using griddedcDNA arrays. J Neurochem 77: 132–145, 2001.
52. Martelli F, Cenciarelli C, Santarelli G, Polikar B, Felsani A, CarusoM. MyoD induces retinoblastoma gene expression during myogenic dif-ferentiation. Oncogene 9: 3579–3590, 1994.
53. Miller MK, Bang ML, Witt CC, Labeit D, Trombitas C, Watanabe K,Granzier H, McElhinny AS, Gregorio CC, Labeit S. The muscleankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family oftitin filament-based stress response molecules. J Mol Biol 333: 951–964,2003.
54. Morrison BM, Janssen WG, Gordon JW, Morrison JH. Time course ofneuropathology in the spinal cord of G86R superoxide dismutase trans-genic mice. J Comp Neurol 391: 64–77, 1998.
55. Morrison BM, Shu IW, Wilcox AL, Gordon JW, Morrison JH. Earlyand selective pathology of light chain neurofilament in the spinal cord andsciatic nerve of G86R mutant superoxide dismutase transgenic mice. ExpNeurol 165: 207–220, 2000.
56. Nagano S, Satoh M, Sumi H, Fujimura H, Tohyama C, Yanagihara T,Sakoda S. Reduction of metallothioneins promotes the disease expressionof familial amyotrophic lateral sclerosis mice in a dose-dependent manner.Eur J Neurosci 13: 1363–1370, 2001.
57. Nakamura K, Nakada C, Takeuchi K, Osaki M, Shomori K, Kato S,Ohama E, Sato K, Fukayama M, Mori S, Ito H, Moriyama M. Alteredexpression of cardiac ankyrin repeat protein and its homologue, ankyrinrepeat protein with PEST and proline-rich region, in atrophic muscles inamyotrophic lateral sclerosis. Pathobiology 70: 197–203, 2002–2003.
58. Okamoto Y, Chaves A, Chen J, Kelley R, Jones K, Weed HG,Gardner KL, Gangi L, Yamaguchi M, Klomkleaw W, Nakayama T,Hamlin RL, Carnes C, Altschuld R, Bauer J, Hai T. Transgenic micewith cardiac-specific expression of activating transcription factor 3, astress-inducible gene, have conduction abnormalities and contractile dys-function. Am J Pathol 159: 639–650, 2001.
59. Olsen MK, Roberds SL, Ellerbrock BR, Fleck TJ, McKinley DK,Gurney ME. Disease mechanisms revealed by transcription profiling inSOD1–G93A transgenic mouse spinal cord. Ann Neurol 50: 730–740,2001.
60. Perrin FE, Boisset G, Docquier M, Schaad O, Descombes P, Kato AC.No widespread induction of cell death genes occurs in pure motoneuronsin an amyotrophic lateral sclerosis mouse model. Hum Mol Genet 14:3309–3320, 2005.
61. Plachta N, Annaheim C, Bissiere S, Lin S, Ruegg M, Hoving S, MullerD, Poirier F, Bibel M, Barde YA. Identification of a lectin causing thedegeneration of neuronal processes using engineered embryonic stemcells. Nat Neurosci 10: 712–719, 2007.
62. Pramatarova A, Laganiere J, Roussel J, Brisebois K, Rouleau GA.Neuron-specific expression of mutant superoxide dismutase 1 in trans-genic mice does not lead to motor impairment. J Neurosci 21: 3369–3374,2001.
63. Puttaparthi K, Gitomer WL, Krishnan U, Son M, Rajendran B,Elliott JL. Disease progression in a transgenic model of familial amyo-trophic lateral sclerosis is dependent on both neuronal and non-neuronalzinc binding proteins. J Neurosci 22: 8790–8796, 2002.
217SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org

64. Reisdorph R, Lindahl R. Hypoxia exerts cell-type-specific effects onexpression of the class 3 aldehyde dehydrogenase gene. Biochem BiophysRes Commun 249: 709–712, 1998.
65. Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Trans-genic mice expressing an altered murine superoxide dismutase geneprovide an animal model of amyotrophic lateral sclerosis. Proc Natl AcadSci USA 92: 689–693, 1995.
66. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A,Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Znsuperoxide dismutase gene are associated with familial amyotrophiclateral sclerosis. Nature 362: 59–62, 1993.
67. Rouaux C, Panteleeva I, Rene F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier AL, Loeffler JP. Sodiumvalproate exerts neuroprotective effects in vivo through CREB-bindingprotein-dependent mechanisms but does not improve survival in an amyo-trophic lateral sclerosis mouse model. J Neurosci 27: 5535–5545, 2007.
68. Schoser BG, Wehling S, Blottner D. Cell death and apoptosis-relatedproteins in muscle biopsies of sporadic amyotrophic lateral sclerosis andpolyneuropathy. Muscle Nerve 24: 1083–1089, 2001.
69. Sharp PS, Dick JR, Greensmith L. The effect of peripheral nerve injuryon disease progression in the SOD1(G93A) mouse model of amyotrophiclateral sclerosis. Neuroscience 130: 897–910, 2005.
70. Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR andcell growth in response to energy stress by REDD1. Mol Cell Biol 25:5834–5845, 2005.
71. Sun H, Li N, Wang X, Chen T, Shi L, Zhang L, Wang J, Wan T, CaoX. Molecular cloning and characterization of a novel muscle adenylosuc-cinate synthetase, AdSSL1, from human bone marrow stromal cells. MolCell Biochem 269: 85–94, 2005.
72. Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts andopportunities. EMBO J 20: 4629–4633, 2001.
73. Turner BJ, Lopes EC, Cheema SS. Neuromuscular accumulation ofmutant superoxide dismutase 1 aggregates in a transgenic mouse model offamilial amyotrophic lateral sclerosis. Neurosci Lett 350: 132–136, 2003.
74. Verdiere-Sahuque M, Akaaboune M, Lachkar S, Festoff BW, Jan-drot-Perrus M, Garcia L, Barlovatz-Meimon G, Hantai D. Myoblastfusion promotes the appearance of active protease nexin I on humanmuscle cell surfaces. Exp Cell Res 222: 70–76, 1996.
75. Vlug AS, Teuling E, Haasdijk ED, French P, Hoogenraad CC,Jaarsma D. ATF3 expression precedes death of spinal motoneurons inamyotrophic lateral sclerosis-SOD1 transgenic mice and correlates withc-Jun phosphorylation, CHOP expression, somato-dendritic ubiquitinationand Golgi fragmentation. Eur J Neurosci 22: 1881–1894, 2005.
76. Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR,Burden SJ. Runx1 prevents wasting, myofibrillar disorganization, andautophagy of skeletal muscle. Genes Dev 19: 1715–1722, 2005.
77. Ward JH. Hierarchical grouping to optimize an objective function. J AmStat Assoc 58: 236–244, 1963.
78. Wendt W, Zhu XR, Lubbert H, Stichel CC. Differential expression ofcathepsin X in aging and pathological central nervous system of mice. ExpNeurol 204: 525–540, 2007.
79. Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM,Pohl J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science 246: 670–673, 1989.
80. Witzemann V, Brenner HR, Sakmann B. Neural factors regulate AChRsubunit mRNAs at rat neuromuscular synapses. J Cell Biol 114: 125–141,1991.
81. Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, JenkinsNA, Sisodia SS, Cleveland DW, Price DL. An adverse property of afamilial ALS-linked SOD1 mutation causes motor neuron disease charac-terized by vacuolar degeneration of mitochondria. Neuron 14: 1105–1116,1995.
218 SKELETAL MUSCLE TRANSCRIPTOME IN ALS
Physiol Genomics • VOL 32 • www.physiolgenomics.org
Copyright © 2022 FDOKUMEN




















