
Protein biomarkers for amyotrophic lateral sclerosis
14
Review 10.1586/14789450.5.2.249 © 2008 Future Drugs Ltd ISSN 1478-9450 249 www.future-drugs.com Protein biomarkers for amyotrophic lateral sclerosis Expert Rev. Proteomics 5(2), 249–262 (2008) Henrik Ryberg and Robert Bowser † † Author for correspondence University of Pittsburgh School of Medicine, Department of Pathology, Center for ALS Research; and Pittsburgh Institute for Neurodegenerative Diseases, Pittsburgh, PA 15261, USA Tel.: +1 412 383 7819 Fax: +1 412 648 1916 [email protected] Amyotrophic lateral sclerosis (ALS) is a progressive motor neuron disease with largely unknown pathogenesis that typically results in death within a few years from diagnosis. There are currently no effective therapies for ALS. Clinical diagnosis usually takes several months to complete and the long delay between symptom onset and diagnosis limits the possibilities for effective intervention and clinical trials. The establishment of protein biomarkers for ALS may aid an earlier diagnosis, facilitating the search for effective therapeutic interventions and monitoring drug efficacy during clinical trials. Biomarkers could also be used to discriminate between subtypes of ALS, to measure disease progression and to detect susceptibility for developing ALS or monitor adverse effects of drug treatment. The present review will discuss the opportunities and proteomic platforms used for biomarker discovery efforts in ALS, summarizing putative ALS protein biomarkers identified in different biofluids. KEYWORDS: cerebrospinal fluid • mass spectrometry • motor neuron disease • plasma • proteomics • serum Amyotrophic lateral sclerosis Motor neuron diseases are a group of progres- sive neurodegenerative diseases primarily affecting the motor neurons in the cortex, brainstem and spinal cord. Approximately 90% of all motor neuron disease cases are amyotrophic lateral sclerosis (ALS); the differ- ential diagnosis for the various motor neuron diseases is usually based on clinical findings, and for the diagnosis of ALS, both upper and lower motor neurons must be affected [1]. The limited degeneration of specific groups of motor neurons corresponds to the differential diagnosis of primary lateral sclerosis (upper motor neuron involvement only), progressive muscular atrophy (lower motor neuron involvement only) or progressive bulbar palsy (bulbar involvement only). However, many patients start with the involvement of only one group of motor neurons, such as the upper motor neuron, lower motor neuron or bulbar involvement, and later develop symp- toms consistent with ALS [2]. It remains debatable whether the different motor neuron diseases are unique disorders or if they could be grouped as one disorder. Further discus- sion regarding the genetics of ALS and other clinical features of the disease can be found elsewhere [1,3–6]. This review will focus on protein biomark- ers for ALS, since this disease is by far the most common and investigated of the motor neuron diseases. Other types of ALS biomark- ers, including clinical measures and metabolic biomarkers, have been thoroughly discussed elsewhere and will not be the focus of this arti- cle [7,8]. Furthermore, this review will focus on protein biomarkers in blood and cerebrospinal fluid (CSF) as these biofluids are the most rel- evant in a clinical setting. Genomic biomark- ers will be discussed with respect to their relevance to proteomics-based biomarkers. The pathological characteristics of ALS include degeneration of motor neurons in the cerebrum, brain stem and spinal cord. How- ever, the degeneration seems to be selective as some motor neurons are spared, as for exam- ple those involved in oculomotor control. The notion that spared motor neurons contain higher levels of calcium-binding proteins has generated a theory that intracellular calcium homeostasis is an important determinant for motor neuron degeneration [9,10]. Other pathological features include gliosis and an accumulation of phosphorylated neuro- filament and other types of intermediate fila- ments in the perikarya and proximal axons of motor neurons. Inclusion bodies such as cystatin C (Cys C) containing Bunina bodies,
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Protein biomarkers for amyotrophic lateral sclerosis

Review
10.1586/14789450.5.2.249 © 2008 Future Drugs Ltd ISSN 1478-9450 249www.future-drugs.com
Protein biomarkers for amyotrophic lateral sclerosisExpert Rev. Proteomics 5(2), 249–262 (2008)
Henrik Ryberg and Robert Bowser†
†Author for correspondenceUniversity of Pittsburgh School of Medicine, Department of Pathology, Center for ALS Research; and Pittsburgh Institute for Neurodegenerative Diseases, Pittsburgh, PA 15261, USATel.: +1 412 383 7819Fax: +1 412 648 [email protected]
Amyotrophic lateral sclerosis (ALS) is a progressive motor neuron disease with largely unknownpathogenesis that typically results in death within a few years from diagnosis. There arecurrently no effective therapies for ALS. Clinical diagnosis usually takes several months tocomplete and the long delay between symptom onset and diagnosis limits the possibilities foreffective intervention and clinical trials. The establishment of protein biomarkers for ALS mayaid an earlier diagnosis, facilitating the search for effective therapeutic interventions andmonitoring drug efficacy during clinical trials. Biomarkers could also be used to discriminatebetween subtypes of ALS, to measure disease progression and to detect susceptibility fordeveloping ALS or monitor adverse effects of drug treatment. The present review will discussthe opportunities and proteomic platforms used for biomarker discovery efforts in ALS,summarizing putative ALS protein biomarkers identified in different biofluids.
KEYWORDS: cerebrospinal fluid • mass spectrometry • motor neuron disease • plasma • proteomics • serum
Amyotrophic lateral sclerosisMotor neuron diseases are a group of progres-sive neurodegenerative diseases primarilyaffecting the motor neurons in the cortex,brainstem and spinal cord. Approximately90% of all motor neuron disease cases areamyotrophic lateral sclerosis (ALS); the differ-ential diagnosis for the various motor neurondiseases is usually based on clinical findings,and for the diagnosis of ALS, both upper andlower motor neurons must be affected [1]. Thelimited degeneration of specific groups ofmotor neurons corresponds to the differentialdiagnosis of primary lateral sclerosis (uppermotor neuron involvement only), progressivemuscular atrophy (lower motor neuroninvolvement only) or progressive bulbar palsy(bulbar involvement only). However, manypatients start with the involvement of onlyone group of motor neurons, such as theupper motor neuron, lower motor neuron orbulbar involvement, and later develop symp-toms consistent with ALS [2]. It remainsdebatable whether the different motor neurondiseases are unique disorders or if they couldbe grouped as one disorder. Further discus-sion regarding the genetics of ALS and otherclinical features of the disease can be foundelsewhere [1,3–6].
This review will focus on protein biomark-ers for ALS, since this disease is by far themost common and investigated of the motorneuron diseases. Other types of ALS biomark-ers, including clinical measures and metabolicbiomarkers, have been thoroughly discussedelsewhere and will not be the focus of this arti-cle [7,8]. Furthermore, this review will focus onprotein biomarkers in blood and cerebrospinalfluid (CSF) as these biofluids are the most rel-evant in a clinical setting. Genomic biomark-ers will be discussed with respect to theirrelevance to proteomics-based biomarkers.
The pathological characteristics of ALSinclude degeneration of motor neurons in thecerebrum, brain stem and spinal cord. How-ever, the degeneration seems to be selective assome motor neurons are spared, as for exam-ple those involved in oculomotor control. Thenotion that spared motor neurons containhigher levels of calcium-binding proteins hasgenerated a theory that intracellular calciumhomeostasis is an important determinant formotor neuron degeneration [9,10]. Otherpathological features include gliosis and anaccumulation of phosphorylated neuro-filament and other types of intermediate fila-ments in the perikarya and proximal axons ofmotor neurons. Inclusion bodies such ascystatin C (Cys C) containing Bunina bodies,

250 Expert Rev. Proteomics 5(2), (2008)
Review Ryberg & Bowser
skein-like as inclusions, Lewy-like bodies and ubiquitinatedinclusions, are common in ALS spinal cord tissue [11]. RecentlyTAR-DNA binding protein 43 (TDP-43) has been identifiedas a component in inclusion bodies in frontotemporal dementiawith ubiquitin-positive, tau-negative and α-synuclein-negativeinclusions, as well as in inclusions in sporadic ALS [12]. Interest-ingly, no TDP-43-positive inclusions were found in cases withsuperoxide dismutase (SOD)1 familial ALS [13,14]. This mightbe a histopathological feature that distinguishes familial andsporadic ALS. The immunohistochemical data suggest thatTDP-43 might be useful as a biomarker for ALS. However, nodata are yet available on the levels of TDP-43 in CSF andblood, and whether TDP-43 in those biofluids could serve as aspecific biomarker for ALS.
A potentially overall unifying theme for motor neuron dis-eases, and other adult onset neurodegenerative diseases, is pro-tein misfolding and aggregation that contributes to the diseasepathogenesis. Such protein abnormalities might be interesting asbiomarkers for ALS and other neurodegenerative diseases, inwhich increased protein misfolding and aggregation could beimportant. Altered post-translational modifications may triggeror contribute to protein misfolding and aggregation [15]. Recentstudies identified modified or misfolded SOD1 protein withinaggregates contained in motor neurons of both sporadic andfamilial ALS [16,17]. Protein aggregates are a significant histo-pathologic feature in ALS and other neurodegenerative diseases,but their relationship to disease onset or progression oftenremains unclear. Thus, different modified protein variants couldserve as biomarkers for neurodegenerative diseases.
Opportunity for biomarkers in ALS
Biomarkers are generally defined as any objective measure thatcan be used as an indicator of a biological process or effect oftreatment [7]. Furthermore, for a biomarker to be useful fordisease diagnosis or treatment evaluation, it should reflect animportant feature of the disease [18]. Biomarkers can be used as
diagnostic biomarkers if they have highlevels of sensitivity and selectivity. Forexample, frequent criteria for a diagnosticbiomarker are that the biomarker shouldfind 90% or more of the disease cases (inthis case, ALS) and at the same time rec-ognize 90% or more of the healthy casesand disease control cases (disease controlfor ALS could be other neurological dis-eases) as not related to that specific diag-nosis (in this case as non-ALS) [19]. Otherpossible uses for biomarkers are to pre-dict disease susceptibility, identify effica-cious treatments, monitor disease pro-gression or evaluate toxicity of proposedtherapies (FIGURE 1).
Given the lack of diagnostic tests forALS and limited understanding of the disease pathogenesis,many opportunities exist for protein biomarkers of ALS.Furthermore, the fast development in the proteomics area dur-ing the last decade facilitates the discovery of new proteinbiomarkers. The mean diagnostic delay from symptom onsetto a diagnosis of ALS has been reported to be between 8 and15 months [20,21]. Thus, considering the average disease dura-tion of approximately 36 months, the disease has significantlyprogressed prior to diagnosis, limiting the potential for effec-tive therapeutic intervention. Early in the disease process, ALSmay resemble numerous other neurological conditions. Amore rapid diagnosis would increase the likelihood for a suc-cessful intervention of the disease and reduce stress associatedwith patients waiting for a final clinical diagnosis. Thus, thereare obvious and important roles for biomarkers in the treat-ment of ALS, as well in the research for new pharmacologicalinterventions [7,22]. Protein biomarkers in biofluids, such asplasma and CSF, might also be useful as surrogate end pointmarkers. For a biomarker to be regarded as a surrogate endpoint, it must be a substitute for a clinically meaningful endpoint [23]. Surrogate end point markers could be used in early-phase clinical trials to make more rapid ‘go–no–go’ decisionsearly in the drug development process. A surrogate end pointbiomarker could speed drug development, aid the interpreta-tion of clinical trial results and potentially increase the overallnumber of clinical trials for ALS.
Biomarkers also provide insight into the pathogenic mecha-nisms of diseases. Biofluids, such as CSF and blood, have theadvantage over tissue samples in that they can be sampled dur-ing all stages of disease (TABLE 1). Tissue-based biomarker analysisfor ALS is limited to muscle biopsy material within living sub-jects or postmortem tissue samples that may best represent theend-stage of disease [24]. Different imaging techniques, such asPET and MRI, are promising and rapidly developing [25]. How-ever, to date, they cannot be used for detecting most of the bio-logic changes in the CNS related to ALS and rely on measuringthe loss of motor neurons or corticospinal tracts.
Figure 1. Time axis showing key clinical events and their relation to different types of biomarkers. Disease initiating events likely include environmental factors, age and genetics. Susceptibility biomarkers could be genetic mutations or polymorphisms. Toxicology markers could be used for monitoring adverse effects of treatments. Diagnostic biomarkers could also be used to discriminate between different subtypes of disease.
Diagnostic biomarkers Toxicology biomarkers
Healthy
Biomarkers for susceptibility Surrogate markers for disease progression
Disease progression End stage
Treatment startsSymptom onset
DiagnosisDisease initiating event

Protein biomarkers for amyotrophic lateral sclerosis Review
www.future-drugs.com 251
Sample types for protein biomarker discovery efforts
There are several important issues to address when choosingthe starting material for a biomarker discovery study (TABLE 2).The most important is perhaps the availability of samples ofsufficient quality and high relevance for the disease studied.Tissues primarily affected by the disease are assumed to containthe most disease-specific protein alternation. Relevant tissuesample types for ALS are spinal cord, brain stem, motor cortexand muscle. Other nonaffected areas of the CNS could be usedas controls to demonstrate specificity. However, the availabilityof CNS tissue samples is low and typically represents autopsyderived materials at the end stage of disease. The post-morteminterval also affects the quality of the samples and, therefore,integrity of potential protein biomarkers. The use of CNS tis-sue is generally limited to an area believed to be primarilyaffected by the disease and excluding potential biomarkersoriginating from other CNS regions or tissues.
Biofluids such as blood and CSF have the advantage of beingmore readily available and can be sampled under more control-led conditions. Thus, for blood and CSF it is easier and morecost effective to collect the usually large number of samplesneeded for biomarker discovery efforts. Biomarkers found in
such sample types are also more applicable in a clinical setting.One disadvantage is that a potential protein biomarkerexpressed and released from a specific cell type will be dilutedinto the relatively large volumes of CSF or blood, which couldconceal changes in protein levels. On the other hand, the dilu-tion effect also makes it possible to detect processes taking placein many areas of the CNS or several organs in one single sam-ple. Of the two biofluids, CSF may best reflect events takingplace in the CNS and contains secreted proteins and proteinsreleased into the extracellular fluid by cell types such as acti-vated glia or degenerating neurons. In contrast to the CSF, theblood compartment is physically separated from the CNS bythe blood–brain barrier. However, since CSF circulation emp-ties into the dural venous sinuses and ultimately into the bloodcirculation, there is the potential to detect CNS-relatedbiomarkers in the blood. Another advantage of blood-basedbiomarker studies is the potential to discover muscle-derivedbiomarkers released into the bloodstream. One of the mainchallenges with using either blood or CSF for protein bio-marker discovery studies is the huge dynamic range of proteinswithin these fluids. Thus, many of the interesting disease-spe-cific proteins exist at several magnitudes lower levels as com-pared with the most abundant proteins (TABLE 2). Methods to
Table 1. Summary of different sample types relevant for biomarker discovery in amyotrophic lateral sclerosis.
Type of sample Advantages Disadvantages
Plasma – Readily available– Well-studied proteome– Provides information about systemic events– Short time from sampling to processing
– Complex matrix– Huge dynamic range*– Dilution of tissue-specific protein– Processing might cause hemolysis
Serum – Readily available– Well-studied proteome– Provides information about systemic events– Enzyme cascade sometimes needed
– Complex matrix– Huge dynamic range*– Dilution of tissue-specific protein – Enzymatic cascade might cause artifacts
Cerebrospinal fluid – Only sample type from the CNS that can be routinely sampled from living subjects– Well-studied proteome
– Not as accessible as blood– Huge dynamic range*– Might be contaminated with blood– Limited amount can be sampled
Urine – Noninvasively sampled– Very easily sampled
– High salt content– Lower protein content– Might have little relevance for ALS– Little known about this proteome
Saliva – Noninvasively sampled– Very easily sampled
– Might have little relevance for ALS– Little known about the this proteome
Brain tissue (spinal cord), post-mortem
– Gives cell type and area-specific information– Gives information from the primary affected tissue
– Represents only the end stage– Might take a long time from death to processing– Expensive
Other tissue (e.g., muscle), post-mortem
– Gives cell type and area-specific information – Represents only the end stage– Might take a long time from death to processing– Expensive
Biopsies – Shorter processing time compared with post-mortem – Usually no biopsies from CNS tissue
*Dynamic range represents the difference in concentration between the most abundant and least abundant proteins. For more information on dynamic range in cerebrospinal fluid see TABLE 2.ALS: Amyotrophic lateral sclerosis.

252 Expert Rev. Proteomics 5(2), (2008)
Review Ryberg & Bowser
deplete abundant proteins or enrich proteins of special interestfor the biomarker discovery study often have to be employedduring these biomarker discovery efforts [26].
The blood–brain barrier is a selective physical barrier and itsrestriction of passive diffusion varies greatly between differentmolecules and some proteins are actively transported across thebarrier [27]. The result is a complex interchange of proteinsbetween the blood, CNS and CSF compartments, potentiallyconfounding interpretation of the origin for biomarker candi-dates. Therefore, levels of the putative biomarker proteinshould be measured in both biofluids, and tissue stainingshould be performed to facilitate interpretation of the results.
Alternative biofluids for protein biomarker discovery in ALSare urine and saliva. The advantage of using these fluids is thattheir collection is minimally invasive. On the other hand,there is no obvious connection between neurodegenerationand the proteome in these fluids, but it cannot be excludedthat for ALS, there are relevant processes that could be moni-tored using these sample types. Urine is perhaps a more suita-ble source for metabolomic biomarker discoveries, but doescontain significant amounts of proteins and peptides andcould be used for protein biomarker studies [28].
The sampling procedures and storage conditions of biofluidsare important factors in protein biomarker discovery efforts thatoften are overlooked. These factors could be especially importantfor larger multicenter studies. Using nonstandardized and sub-optimal conditions for sampling and storage could lead to errone-ous identification of biomarkers. The time between sampling andsample processing at different temperatures has been investigatedboth for serum and CSF [29,30]. From these studies, it is suggestedthat protein artifacts are created if the serum or CSF samples havebeen at room temperature for longer than 1–2 h or at 4°C longerthan 6–8 h. Another important factor is the tubes in which sam-ples are stored. It is recommended that different factors, such asthe influence of tube coating to minimize protein binding, areproperly investigated before starting a protein discovery effort.
Experimental platforms suitable for protein biomarker discovery in ALS
There are a number of technological platforms for the simul-taneous analysis of multiple proteins that can be used forbiomarker discovery efforts in ALS. Such approaches are typi-cally denoted as proteomics or peptidomics. A proteome canbe defined as all the different proteins and their variantsexpressed in, for example, a certain cell type, tissue or biofluidat a certain time point. A peptidome is the correspondingconcept for the low-molecular-weight proteome. There aremany different separation and detection techniques that canbe used in biomarker discovery efforts. However, in this sec-tion we will review a few of the typical proteomic methodolo-gies used in biomarker discovery efforts for neurodegenerationand ALS.
Most proteomic platforms are based on different protein sep-aration techniques combined with mass spectrometry (MS) forprotein detection and identification. Biomarker discovery isdependent on quantification of proteins and peptides. SinceMS-based techniques do not usually allow direct quantifica-tion, this issue must be solved by either relative quantificationduring the protein separation steps prior to MS, isotope labe-ling of peptides or by using special software that, via normaliza-tion, alignment and statistical analyses of mass peak spectra,can calculate relative levels for peptide and protein peaks.
Proteomic applications for MS usually require powerful sepa-ration of the different proteins and peptides prior to MS analy-sis. Mass spectrometers using electrospray ionization coupled toliquid chromatography (LC) can separate a very large number ofpeptides, but such systems usually also need separation of pro-teins prior the enzymatic digestion. One of the most powerfulseparation techniques is 2D gel electrophoresis [31]. The advan-tage with this technique is that the quantification of the differ-ent proteins can be performed through fluorescence labeling ofproteins combined with imaging techniques and softwareprocessing. Samples from control and disease groups can be ana-lyzed on the same gel, increasing the ability to find differentiallyexpressed proteins between two subject groups. The main dis-advantages are that 2D gels are labor intensive and relatively fewindividual samples can be assayed using this technique.
The desire to move away from gel-based platforms has gener-ated a number of alternative methods suitable for biomarkerdiscovery. One such system is SELDI-TOF-MS. This technol-ogy is based on the selective absorption of proteins to differ-ently charged or chemically modified chip surfaces, combinedwith a highly automated collection of mass spectra and softwarefor processing the spectra. The advantages are that a largenumber of samples can be processed at the same time and thatthe system is relatively easy to use. The disadvantage is thatidentifying the differentially expressed protein peaks found withthe platform can be quite labor intensive and often requires theuse of additional mass spectrometers. The mass spectrometerused by SELDI-TOF-MS also has limited resolution.
Table 2. Reported concentrations of representative proteins in human cerebrospinal fluid.
Protein Concentration (mg/l) Ref.
Total protein 320 [101]
Albumin 177 [101]
IgG 53 [102]
Transferrin 18.8 [103]
Apolipoprotein E 12.4 [101]
IgM 0.82 [104]
Typical biomarker <0.01 [73,83,85]
The large dynamic range in protein concentrations makes it difficult to see the changes for the most interesting pathology-specific proteins. The typical biomarker might be present at levels 104–107-times lower than the most abundant proteins.

Protein biomarkers for amyotrophic lateral sclerosis Review
www.future-drugs.com 253
Another methodology for uncovering protein biomarkers uti-lizes antibody microarrays. This assay determines levels of pro-tein expression with antibodies spotted onto a matrix or surfaceto capture specific proteins. This approach has been used tomonitor protein expression in spinal muscular atrophy andmore recent studies are beginning to examine cytokine expres-sion in ALS patients [32,33]. However, since altered cytokineexpression occurs during inflammation, a common event inneurodegenerative diseases, it may be difficult to uncover acytokine expression profile unique to ALS.
An alternative approach for biomarker discovery is to searchfor differentially expressed peptides in the very complex peptidepools generated if total proteins from biofluids are depleted forthe most abundant proteins and then enzymatically digested. Insuch approaches, the number of detected and identified proteinscan be greatly increased by using techniques such as multidimen-sional protein identification technology (MudPIT) [34]. A typicalLC-MS system separates peptides using a reverse-phase column,but in MudPIT the sample is first loaded on to a strong cationexchange column and fractions are eluted online throughincreasing salt concentrations. Each fraction is then further sepa-rated on a reverse-phase column and peptides directly analyzedby MS. Sample analysis requires several hours, limiting the totalnumber of samples used in any one experiment. Another plat-form suitable for this type of analysis is online capillary LC cou-pled to Fourier transform ion cyclotron resonance MS. The highresolution together with high sensitivity makes it possible todetect a huge number of peptides. In one study, this methodol-ogy was used in concert with a pattern recognition strategy todetect protein patterns in CSF specific to ALS [35]. Quantifica-tion of peptides can be performed using isotope labeling tech-niques, such as isotope-coded affinity tags and iTraq™, or soft-ware-based label-free differential expression techniques [36,37].Biomarker discovery at the peptide level has the advantage thattime-consuming protein separation can be avoided, but the maindisadvantage is that the interpretation of results sometimes can
be very complex, as peptides can be differently expressed due toalternative splicing or post-translational modifications. Incom-plete enzymatic digestion of the starting material can also induceartifacts and lead to false-positive biomarker discoveries. The val-idation of findings on peptide levels can be difficult and oftenrequires antibody-based methods such as immunoblot or ELISA.Antipeptide-specific antibodies are often not available and mustbe generated and characterized prior to validation studies.
Lessons from genome-wide association studies
Rapid technologic developments in whole genome-wide associ-ation screens have made possible comparisons of single nucleo-tide polymorphisms (SNPs) across the complete genome ofindividuals [38]. Recently, five whole genome-wide SNP screen-ings were preformed for sporadic ALS [39–43]. These studieswere performed to identify putative genetic polymorphismsassociated with sporadic ALS (SALS) (TABLE 3).
Two of the first genome screenings could not find an associa-tion for any of the SNPs analyzed to ALS. One study analyzedapproximately 500,000 SNPs in 276 SALS and 271 controls [40].The other study analyzed 1277 SNPs in 1402 SALS and 1233controls [41]. Three larger studies were also preformed, one ana-lyzed 300,000 SNPs using 1337 SALS and 1356 controls [39],the second nearly 800,000 SNPs with 1287 SALS and 1567controls [42] and the third 312,000 SNPs using 1767 ALS and1916 control subjects [43]. The first of these larger studies identi-fied inositol 1,4,5-triphosphate receptor 2 gene as associatedwith SALS, and the second study identified five different genes,of which the FLJ10986 gene correlated best to SALS. The lateststudy using both American and European ancestry subjects iden-tified the DPP6 gene, a dipeptidyl-peptidase-like protein, as adisease susceptibility gene. The complete lack of overlap betweenthe potential susceptibility genes within these studies highlightsthe likely heterogeneous nature of the disease and the possibilitythat all genome-wide association screens performed to date have
Table 3. Summary of five different whole genome-wide association screens correlating genetic polymorphisms to sporadic amyotrophic lateral sclerosis.
Subjects(ALS/Con)
Ethnicity Platform SNPs analyzed Genes found Ref.
1287/1567 US White and non-White Affymetrix GeneChip Human Mapping 500K andIllumina Infinium II HumanHap300K
766,955 FLJ10986; LOXHD1; PTPRT; MAGI2; IL18RAP
[42]
1402/1233 British and German 1536-plex Golden Gate 1277 None [41]
276/271 US White non-Hispanic Illumina Infinium II HumanHap550K 553,352 None [40]
1337/1356 Dutch, Belgian and Swedish Illumina Infinium II HumanHap300K 300,000 ITPR2 [39]
1767/1916 US, Dutch, Belgian and Swedish
Illumina Infinium II HumanHap300K 311,946 DPP6 [43]
ALS: Amyotrophic lateral sclerosis; Con: Control; DPP6: Dipeptidyl aminopeptidase-like protein 6; FLJ10986: Hypothetical protein with unknown function; LOXHD1: Lipoxygenase homology domains 1; IL18RAP: IL-18 receptor accessory protein; ITPR2: Inositol 1,4,5-triphosphate receptor, type 2; MAGI2: Membrane-associated guanylate kinase, WW and PDZ domain containing 2; PTPRT: Protein tyrosine phosphatase, receptor type T; SNP: Single nucleotide polymorphism.

254 Expert Rev. Proteomics 5(2), (2008)
Review Ryberg & Bowser
been underpowered. Differences in subject ethnicity between thestudy groups may also contribute to the inability to discovercommon genetic polymorphisms associated with SALS.
From these studies we suggest that no single genetic factoroverwhelmingly contributes to the onset of SALS, indicating thatmultiple biochemical pathways contribute to the disease patho-genesis. Proteomic biomarker studies may, therefore, requirelarge numbers of test subjects during the discovery phase in orderto uncover potential biomarkers that will ultimately be validatedin subsequent studies.
Protein biomarkers in plasma & serum
Levels in either plasma or serum from ALS and control subjectshave been described for a number of proteins. TABLE 4 lists indi-vidual proteins examined in each published report and the totalnumber of test subjects used for the analysis. The vast majorityof these have been described for serum, possibly reflecting apreference for serum sampling in a clinical setting.
A number of the potential blood-based biomarker proteinsfunction as growth factors or hormones. Of these, three may bedifferentially expressed. Insulin [44] and insulin-like growth factorsbinding protein 1 (IGFBP-1) [45] have been described as decreasedin ALS, whereas basic fibroblast GF-2 has been reported asincreased [46]. Growth hormone [44], brain-derived neurotrophicfactor [47] and erythropoietin (EPO) [48,49] have all been reportedto be unchanged. However, conflicting results have been reportedfor VEGF [50–53] and IGF-1 [44,45]. These studies used differentcommercially available immunoassays for VEGF and IGF-1, onepotential confounding factor generating the conflicting results.
A second group of proteins investigated as potential bio-markers are various cytokines, chemokines and other immune-related proteins. Of these proteins, endoglin [54] and thrombo-spondin [55] were shown to be downregulated in ALS, whereascaspase-1 [56], tumor necrosis factor (TNF) receptor I [57],TNF receptor II [57], RANTES [58] and fragment of thrombo-spondin [55] were upregulated. The levels for intercellular adhe-sion molecule (ICAM)-1 [59], endothelial leukocyte adhesionmolecule (ELAM)-1 [59], Fas receptor (CD95) [56] and Fms-like tyrosine kinase receptor-3 (Flt3 or CD135) [60] werereported to be unchanged.
For chemokine (C-C motif ) ligand 2 (monocyte chemo-attractant protein [MCP]-1 or CCL2) [61,62], TNF-α [57,63] andIL-6 [63,64] have inconsistent results have been reported. Further-more, different immunoassays have been used, and a possibilityis that the different antibodies recognize different variants ofMCP-1, TNF-α and IL-6. However, for TNF-α, the sizes of thestudy groups are an important factor as one larger study [57]
showed an upregulation while a smaller study showed no differ-ence [63]. TGF-β1 has been described as upregulated in ALSplasma [65], but in another study the protein levels weredescribed as unchanged in serum [66]. This inconsistency may bedue to the different samples types used but could also be due toassay differences.
Besides growth factors, hormones, interleukins and immunesystem-related proteins a number of other proteins have alsobeen measured. Creatine kinase [67], angiogenin [50] and matrixmetallopeptidase (MMP)-9 [68] have been reported to be upregu-lated. Fibronectin has been reported as downregulated [69]. TIMPmetallopeptidase inhibitor 1 (TIMP-1) [68], collagen IV [70],S100β [71] and SOD [72] have been reported to be unchanged.
All of these studies utilized small study group sizes, with10–20 individuals in either disease or control groups. As notedpreviously, genome-wide association screens indicate that nodominant genetic pathway contributes to the development ofsporadic ALS. This suggests that multiple biochemical pathwayslikely contribute to the development and progression of ALS.Therefore, protein biomarker discovery efforts using small sam-ple groups of 10–20 individuals are probably underpowered andshould be replicated using a much larger number of ALS andcontrol subjects. Moreover, many of the results have never beenvalidated using separate subject cohorts. Independent studieshave often failed to confirm prior findings for the same protein.
Protein biomarkers in cerebrospinal fluid
Protein levels within the CSF of ALS and control subjects havebeen described for approximately 40 different proteins (TABLE 4).Similar to plasma and serum, one large group of identified puta-tive protein biomarkers are hormone and growth factors.Included in this group of proteins are insulin [44], growth hor-mone [44] and EPO [48,49], which were reported to be decreased inALS CSF. On the other hand, FGF-2 [46], glial cell-line-derivedneurotrophic factor (GDNF) [73], pigment epithelium-derivedfactor (PEDF) [74] and hepatocyte growth factor (HGF) [75,76]
were reported to be increased in ALS CSF. The levels for IGFbinding protein 2 (IGFBP-2) [77] and brain-derived neurotrophicfactor [47,73] were reported to be unchanged. For IGF-1 [44,77] andVEGF [52,53,78,79] conflicting results have been reported, whichcould be due to the fact that different immunoassays were usedin these studies.
Amongst interleukins and other immune-related proteins,RANTES [58], Flt3 [60] and MCP-1 [61,62,80] have been shown tobe upregulated in ALS CSF, whereas the levels of TGF-β1 [66]
and CD95 [56] have been shown to be unchanged. Caspase-1 hasbeen shown to be downregulated [56]. Inconsistent results beenreported for IL-6 [63,81], which could be due to the differentimmunoassays used.
Two different neuron-specific proteins have been investigatedin the CSF of ALS and control subjects. The first neuron-specificprotein, microtubule-associated protein tau, has failed to exhibitaltered CSF levels during ALS in three different studies [49,82,83],whereas two reports showed an increase for tau [67,84]. In addi-tion, one of these studies demonstrated that the phosphorylatedform of tau (p-tau) was unchanged in ALS [82]. The reasons forthese discrepancies are not clear, as the same research group hasshowed both upregulated tau levels [84] and small, but nonsignifi-cant, decreased levels in separate studies [49]. The neuron-specific

Protein biomarkers for amyotrophic lateral sclerosis Review
www.future-drugs.com 255
Table 4. Summary of putative amyotrophic lateral sclerosis protein biomarkers in cerebrospinal fluid, plasma and serum.
Protein Study Subjects (ALS/Con)
CSF Serum Plasma Ref.
Hormones and growth factors
Insulin 24/40 ↓ ↓ [44]
IGF-1 Bilic (2006) 24/40 ↓ No change [44]
Hosback (2007) 28/28 ↑ [45]
Pirttila (2004) 14/25 No change [77]
IGFBP-1 28/28 ↓ [45]
IGFBP-2 14/25 No change [77]
Growth hormone 24/40 ↓ No change [44]
EPO Brettschneider (2007) 60/33 ↓ No change [48]
Brettschneider( 2006) 30/49 ↓ No change [49]
FGF-2 15/10–15 ↑ ↑ [46]
VEGF Devos (2004) 24/19 ↓ No change No change [52]
Ilzecka (2004) 30/30 No change [78]
Moreau (2006) 10/10 No change No change [53]
Nagata (2007) 42/16 No change [80]
Nygren (2002) 13/13 ↑ [51]
Cronin (2006) 79/72 No change [50]
HGF Kern (2001) 12/11 ↑ [75]
Tsuboi (2002) 10/32 ↑ [76]
BDNF Grundström (2000) 15/11 No change [73]
Ilzecka (2002) 20/15 No change No change [47]
GDNF 15/11 ↑ [73]
PEDF 15/22 ↑ [74]
Inflammatory system related
Endoglin 25/25 ↓ [54]
Caspase-1 25/15 ↓ ↑ [56]
CD95 25/15 No change No change [56]
IL-6 Sekizawa (1998) 27/20 ↑ [81]
Ono (2001) 11/11 ↑ [64]
Moreau (2005) 10/10 No change No change [63]
TNF-α Moreau (2005) 10/10 No change [63]
Poloni (2000) 51/36 ↑ [57]
The number of ALS and control subjects are listed for each investigation. An up arrow (↑) denotes that the investigated protein was reported to be upregulated in ALS in that particular biofluid, down arrow (↓) denotes a downregulation and (No change) denotes that no changes were reported for the protein. Furthermore, the potential biomarkers were stratified into groups according to their reported function.ALS: Amyotrophic lateral sclerosis; BDNF: Brain-derived neurotrophic factor; Con: Control; CSF: Cerebrospinal fluid; ELAM: Endothelial leukocyte adhesion molecule; EPO: Erythropoietin; Flt: Fms-like tyrosine kinase receptor; GDNF: Glial cell line-derived neurotrophic factor; ICAM: Intercellular adhesion molecule; IGFBP: IGF-binding protein; MCP: Monocyte chemoattractant protein; MMP: Matrix metallopeptidase; n.d.: Not determined; NFH: Neurofilament heavy subunit; NFL: Neurofilament light subunit; PEDF: Pigment epithelium-derived factor; SOD: Superoxide dismutase; TSP: Thrombospondin; TTR: Transthyretin.
256 Expert Rev. Proteomics 5(2), (2008)
Review Ryberg & Bowser
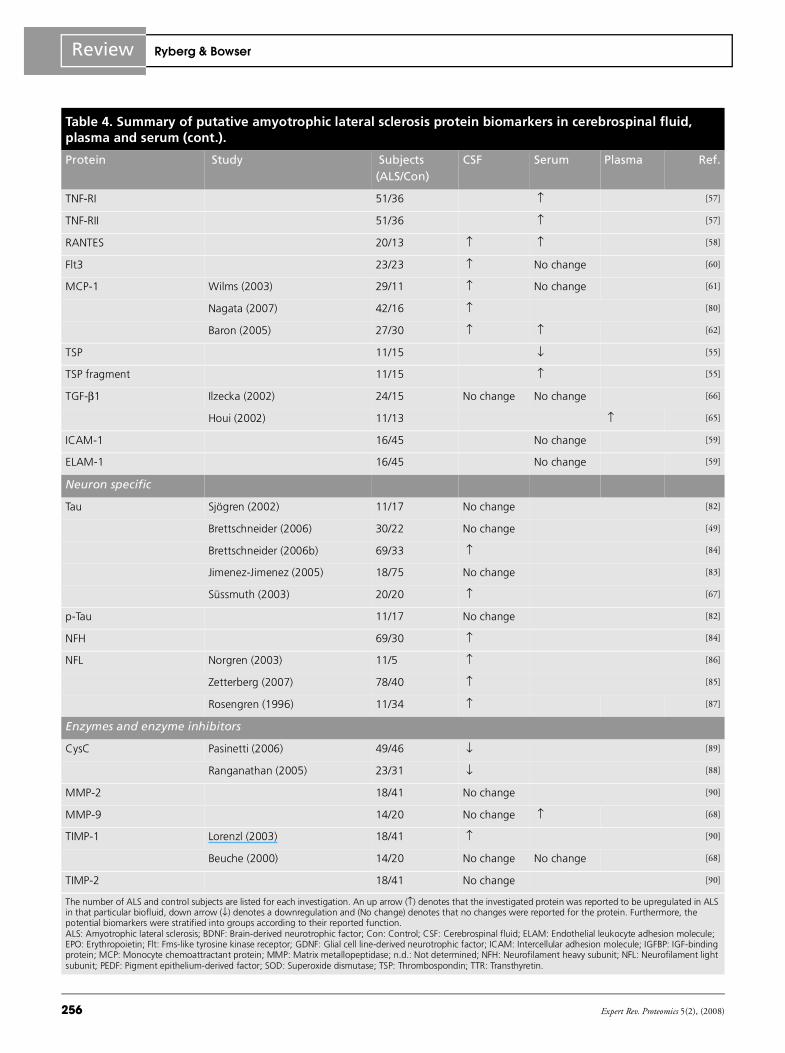
Table 4. Summary of putative amyotrophic lateral sclerosis protein biomarkers in cerebrospinal fluid, plasma and serum (cont.).
Protein Study Subjects (ALS/Con)
CSF Serum Plasma Ref.
TNF-RI 51/36 ↑ [57]
TNF-RII 51/36 ↑ [57]
RANTES 20/13 ↑ ↑ [58]
Flt3 23/23 ↑ No change [60]
MCP-1 Wilms (2003) 29/11 ↑ No change [61]
Nagata (2007) 42/16 ↑ [80]
Baron (2005) 27/30 ↑ ↑ [62]
TSP 11/15 ↓ [55]
TSP fragment 11/15 ↑ [55]
TGF-β1 Ilzecka (2002) 24/15 No change No change [66]
Houi (2002) 11/13 ↑ [65]
ICAM-1 16/45 No change [59]
ELAM-1 16/45 No change [59]
Neuron specific
Tau Sjögren (2002) 11/17 No change [82]
Brettschneider (2006) 30/22 No change [49]
Brettschneider (2006b) 69/33 ↑ [84]
Jimenez-Jimenez (2005) 18/75 No change [83]
Süssmuth (2003) 20/20 ↑ [67]
p-Tau 11/17 No change [82]
NFH 69/30 ↑ [84]
NFL Norgren (2003) 11/5 ↑ [86]
Zetterberg (2007) 78/40 ↑ [85]
Rosengren (1996) 11/34 ↑ [87]
Enzymes and enzyme inhibitors
CysC Pasinetti (2006) 49/46 ↓ [89]
Ranganathan (2005) 23/31 ↓ [88]
MMP-2 18/41 No change [90]
MMP-9 14/20 No change ↑ [68]
TIMP-1 Lorenzl (2003) 18/41 ↑ [90]
Beuche (2000) 14/20 No change No change [68]
TIMP-2 18/41 No change [90]
The number of ALS and control subjects are listed for each investigation. An up arrow (↑) denotes that the investigated protein was reported to be upregulated in ALS in that particular biofluid, down arrow (↓) denotes a downregulation and (No change) denotes that no changes were reported for the protein. Furthermore, the potential biomarkers were stratified into groups according to their reported function.ALS: Amyotrophic lateral sclerosis; BDNF: Brain-derived neurotrophic factor; Con: Control; CSF: Cerebrospinal fluid; ELAM: Endothelial leukocyte adhesion molecule; EPO: Erythropoietin; Flt: Fms-like tyrosine kinase receptor; GDNF: Glial cell line-derived neurotrophic factor; ICAM: Intercellular adhesion molecule; IGFBP: IGF-binding protein; MCP: Monocyte chemoattractant protein; MMP: Matrix metallopeptidase; n.d.: Not determined; NFH: Neurofilament heavy subunit; NFL: Neurofilament light subunit; PEDF: Pigment epithelium-derived factor; SOD: Superoxide dismutase; TSP: Thrombospondin; TTR: Transthyretin.

Protein biomarkers for amyotrophic lateral sclerosis Review
www.future-drugs.com 257
proteins neurofilament heavy subunit and neurofilament lightsubunit (NFL) have been shown to be significantly upregulatedin ALS CSF in four different studies [84–87]. Increased levels ofneurofilament proteins are not unique to ALS and can be seen forother neurodegenerative diseases, including Alzheimer’s disease(AD) and multiple sclerosis. However, the levels show a close cor-relation to disease duration and could also potentiallydifferentiate SOD1 familial forms from other types of ALS [85].
Different enzymes and enzyme inhibitors have also been iden-tified as potential CSF-based biomarkers for ALS. Cys C has intwo different studies been reported to be present at lower levels inALS CSF [88,89]. Both of these studies used the SELDI-TOF-MSplatform and validated the findings using immunoassays specificto Cys C. TIMP metallopeptidase inhibitor 2 (TIMP-2) [90],MMP-2 and MMP-9 [68,90] have been reported as unchanged inALS, whereas for TIMP metallopeptidase inhibitors 1 (TIMP-1)conflicting results have been reported [68,90]. Increased levels ofSOD1 have been reported [91]. These investigations suggest thatproteases and protease inhibitors within the CSF exhibit alteredactivities and may contribute to the disease pathogenesis.
In addition to the proteins discussed previously, a number ofother proteins have been reported to exhibit altered levels in theCSF of ALS patients. Cytochrome C [92], neurosecretory proteinVGF (VGF) [89], transthyretin (TTR) [88] and β-amyloid 42(Aβ42) [82] have been reported as downregulated. A C-terminalfragment of the neuroendocrine protein 7B2 (7B2) [88] and thelevels of substance P (Sub-P) have been shown to be increasedwithin the CSF [93]. Levels of of S100β were not altered in theCSF of ALS patients [67].
In two different studies the levels of EPO, a hormone regulat-ing red blood cell production, were shown to be decreased in theCSF of ALS patients [48,49]. The levels were not altered in otherneurodegenerative diseases investigated, such as AD, vasculardementia and frontotemporal dementia. This suggests EPOmay be able to differentiate ALS from other neurodegenerativediseases and dementia.
In two independent studies, HGF has been shown to be upreg-ulated in ALS CSF [75,76], but HGF was also increased in otherneurodegenerative diseases including AD and Parkinson’s disease.Therefore, this potential biomarker is not specific to ALS.
Table 4. Summary of putative amyotrophic lateral sclerosis protein biomarkers in cerebrospinal fluid, plasma and serum (cont.).
Protein Study Subjects (ALS/Con)
CSF Serum Plasma Ref.
CK 20/20 ↑ [67]
Angiogenin 79/72 ↑ [50]
SOD Jacobsson (2001) 66/37 ↑ [91]
Ihara (1995) 28/16 No change [72]
Others
Cyt C 40/40 ↓ [92]
Fibronectin 28/39 ↓ [69]
S100β Süssmuth (2003) 20/20 No change [67]
Otto (1998) 41/32 No change [71]
VGF 49/46 ↓ [89]
Sub-P 11/16 ↑ [93]
TTR 23/31 ↓ [88]
Aβ42 11/17 ↓ [82]
7B2 23/31 ↑ [88]
Collagen IV 30/30 No change [70]
Apolipoprotein E 403/0 n.d. n.d. n.d. [94]
The number of ALS and control subjects are listed for each investigation. An up arrow (↑) denotes that the investigated protein was reported to be upregulated in ALS in that particular biofluid, down arrow (↓) denotes a downregulation and (No change) denotes that no changes were reported for the protein. Furthermore, the potential biomarkers were stratified into groups according to their reported function.ALS: Amyotrophic lateral sclerosis; BDNF: Brain-derived neurotrophic factor; Con: Control; CSF: Cerebrospinal fluid; ELAM: Endothelial leukocyte adhesion molecule; EPO: Erythropoietin; Flt: Fms-like tyrosine kinase receptor; GDNF: Glial cell line-derived neurotrophic factor; ICAM: Intercellular adhesion molecule; IGFBP: IGF-binding protein; MCP: Monocyte chemoattractant protein; MMP: Matrix metallopeptidase; n.d.: Not determined; NFH: Neurofilament heavy subunit; NFL: Neurofilament light subunit; PEDF: Pigment epithelium-derived factor; SOD: Superoxide dismutase; TSP: Thrombospondin; TTR: Transthyretin.

258 Expert Rev. Proteomics 5(2), (2008)
Review Ryberg & Bowser
Levels of the monocyte chemotactic protein MCP-1 havebeen reported to be upregulated in the CSF of ALS patients bythree independent studies. One study reported that the CSFfrom 21 of 29 ALS patients exhibited higher levels of MCP-1than any control subject [61]. Similar protein levels were alsomeasured in another study [62]. A third study reported that ALSpatients had significantly higher MCP-1 levels than controlsand spinocerebellar ataxia patients, but levels similar to Parkin-son’s patients [80]. Taken together these data suggest MCP-1 as abiomarker in ALS that warrants further investigation.
Similar to the potential ALS biomarkers reported in serumand plasma, many of the CSF-based studies utilize small subjectnumbers within the sample groups and the results require fur-ther validation studies to confirm the initial findings. However,within the CSF-based proteomic studies there appears moreconcordance between laboratories with respect to protein alter-ations. The reduced overall complexity of the CSF proteomewith respect to serum and plasma may contribute to improvedconsistency between the laboratories.
These proteins, EPO, HGF, MCP-1, NFL, neurofilamentheavy subunit, Cys C and TTR, represent the best describedprotein biomarkers for ALS in CSF to date. From thesebiomarkers, it is not possible to highlight any particular molec-ular pathway that may be especially important for the patho-genesis in ALS, perhaps due to the fact that many differentpathways are involved. Instead, it might be that all or a numberof these proteins could be used in a panel of biomarkers for anaccurate diagnosis of ALS.
Disease progression biomarkers
The ultimate goal for much ALS research is to find therapeuticinterventions that can slow or reverse disease progression. Inorder to evaluate such interventions, disease progressionbiomarkers or surrogate markers would be extremely valuabletools. Unfortunately, very little has been done in the field ofALS disease progression biomarkers. There is a notable lack oflongitudinal studies, which is probably a consequence of therapid disease progression.
However, a number of recent studies have reported potentialdisease progression biomarkers. In a large study, the survivaltime for 403 ALS patients correlated to apolipoprotein E(APOE) phenotype and APOE plasma levels [94]. The plasmalevels of APOE were not determined for the controls. However,approximately one quarter of the ALS patients had levels abovethe maximum reference level and for those patients the diseaseprogression rate was significantly more rapid. This study sug-gests that APOE could be a progression biomarker for ALS, butfurther longitudinal studies that correlate the levels to clinicalmeasures are needed.
In another study, EPO levels were measured in the serumand CSF from 60 ALS patients [49]. These patients were fur-ther stratified as either slowly progressing (n = 30) or rapidlyprogressing (n = 30). The investigators demonstrated that
CSF EPO levels in the rapidly progressing group wereapproximately half of those in the slowly progressing group.The levels in serum were similar for the two groups. Thesedata suggest that EPO might also be a useful progressionbiomarker, but further studies are needed to correlate EPOlevels to clinical measures such as the ALS functional ratingscale or forced vital capacity.
A recent study reported a negative correlation between diseaseprogression and CSF levels of NFL in 78 ALS patients [85]. ALSpatients with NFL levels over the median had considerablyshorter disease duration. A similar relationship has also beenshown for neurofilament heavy protein in the CSF from 20 ALSpatients [84]. This same study also followed changes for CSF taulongitudinally in a smaller number of patients. In another studyserum collagen IV protein was found to be slightly elevated inALS patients with disease duration longer than 3 years (n = 19)as compared with patients with shorter duration (n = 11) [70].This longitudinal study also followed changes for CSF tau in asmaller number of patients. Furthermore, it has been reportedthat TGFβ1 levels in plasma showed a correlation to diseaseduration, although this study only included 11 ALS patients [65]
and in another study CSF levels of VEGF in 30 ALS patientswere correlated to disease duration [78].
Additional studies are needed to find definitive biomarkersthat function as surrogate markers of disease progression in ALS,as well as to validate already published potential disease progres-sion biomarkers. Longitudinal studies that correlate the surrogatemarker to clinical measures would be especially valuable.
Potential of panels of protein biomarkers
The heterogeneity of ALS as indicated by whole genome-wideassociation screens and the multitude of potential proteinbiomarkers suggests that no single protein biomarker will iden-tify the disease with high specificity and selectivity. In a studyusing online capillary LC and Fourier transform ion cyclotronresonance MS, close to 4000 peptides were detected in peptidepools from an enzymatic digest generated from ALS and controlCSF [35]. In this experiment it was concluded that no single pro-tein could identify ALS, although patterns of proteins in the ALSCSF existed that could accurately separate ALS cases from con-trol cases. This is also in accordance with two protein biomarkerstudies performed with ALS and control CSF using the SELDIplatform [88,89]. In both these studies, at least three different pro-tein biomarkers had to be used to achieve satisfactory selectivityand sensitivity in the separation of ALS and control cases.
There are currently no reasons to believe that ALS is a lesscomplex disease than other major neurodegenerative diseases.For example, in AD the combination of protein biomarkers inthe CSF has been proposed to increase sensitivity and selectivityof the diagnosis, especially in early diagnosis of the disease [95]. Inblood, the situation is probably much more complex. In a recentstudy using plasma samples, 18 different potential proteinbiomarkers were used for accurate identification of AD [96].

Protein biomarkers for amyotrophic lateral sclerosis Review
www.future-drugs.com 259
Thus, it is likely that a panel of biomarkers must be used forthe selective diagnosis of ALS. A potential problem is that a verylarge biomarker panel may be difficult to use in a clinical setting.It would be advantageous if the number of proteins could bekept to minimum so that more traditional technologies, such asELISA, could be used.
Expert commentary & five-year view
ALS is likely a much more heterogeneous disease than believedonly a decade ago. The genetics behind familial ALS are com-plex and we have not yet identified all genes that induce familialforms of ALS [4,5]. Genome-wide association screens indicatethat genetic contributions to sporadic ALS are more complexthan first believed, and environmental factors may contribute tothe onset of disease [39–43]. In addition, some ALS patients havefrontotemporal dementia. These patients have TDP-43 positiveinclusions, which can also be seen in sporadic ALS cases, but notin SOD1 familial ALS [13]. The identification of proteins withinaggregates that often appear during ALS will lead to newinsights into the disease pathogenesis. The presence of cognitivedeficits in some ALS patients suggests that common mecha-nisms exist between ALS and other neurodegenerative diseasessuch as AD. The likely heterogeneous nature of the disease sug-gests that no single biomarker will be able to detect or differenti-ate ALS from control subjects and a panel of biomarkers mustbe generated for a specific and sensitive diagnostic test. It willalso be interesting to correlate protein biomarkers to alteredmRNA expression levels detected in tissue microarray studies forALS [97–100].
The disease heterogeneity also suggests that stratifyingpatients into subgroups based on site of disease onset, age, gen-der and ethnicity might be very important for successful identi-fication of biomarkers for ALS. This must also be considered
when validating potential biomarker candidates. Diseaseheterogeneity is an important factor to consider when design-ing a biomarker investigation, requiring sufficient sample sizeand proper patient subclassifications. Conflicting results pub-lished for protein biomarkers in CSF and blood (TABLE 4) mayindicate that many studies have been underpowered. Alterna-tively, it could mean that ethnicity, environmental factors andlifestyle factors are important contributors to ALS.
Increased collaboration between research centers will beimportant for biomarker discovery and validation for ALS.Standardization of sample acquisition and sample storage con-ditions will, therefore, be increasingly important for theexchange of samples without introducing artifacts. Collectionof patient clinical information should also be uniform, so thatthe correct subclassifications and matching of control subjectscould be achieved.
We predict that during the next 5 years, an array of new bio-markers will be identified and validated, both for blood and CSF.Blood biomarkers could aid a primary care physician to identifypotential ALS cases and refer those to a specialist, while CSFbiomarkers will aid the specialist in an accurate and early diagno-sis of the disease. Surrogate biomarkers will be available to helpthe physician to monitor the efficiency of treatment and could beused in clinical trials to find more effective interventions.
Financial & competing interests disclosure
The corresponding author (Robert Bowser) is a cofounder of KnoppNeurosciences Inc., a biotechnology company focused on the development oftherapeutics for ALS and other neurodegenerative diseases. The authors haveno other relevant affiliations or financial involvement with any organizationor entity with a financial interest in or financial conflict with the subjectmatter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Key issues
• Standardized acquisition, processing and storage methods for biofluids are required to obtain reliable proteomics data during the discovery phase.
• Owing to the heterogeneous nature of amyotrophic lateral sclerosis (ALS), panels of protein biomarkers derived from cerebrospinal fluid and blood may be most valuable for generating an accurate diagnostic test for ALS. Protein-based biomarkers may help determine the overall heterogeneity within the disease.
• Additional biofluids, such as urine and saliva, should also be pursued with respect to biomarker discovery efforts.
• Longitudinal studies are required to identify surrogate biomarkers for ALS disease progression. Such biomarkers may identify protein alterations that correlate to slow or more rapid disease progression.
• Most biomarker discovery efforts have utilized too few subjects in the study design. Increased collaboration between academic and industry investigators is required to generate larger experimental sample sizes in order to both discover and validate ALS-specific biomarkers.
ReferencesPapers of special note have been highlighted as:
• of interest
•• of considerable interest
1 Mitchell JD, Borasio GD. Amyotrophic lateral sclerosis. Lancet 369, 2031–2041 (2007).
2 Baek WS, Desai NP. ALS: pitfalls in the diagnosis. Pract. Neurol. 7, 74–81 (2007).
3 Schymick JC, Talbot K, Traynor BJ. Genetics of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 16, 233–242 (2007).

260 Expert Rev. Proteomics 5(2), (2008)
Review Ryberg & Bowser
4 Majoor-Krakauer D, Willems PJ, Hofman A. Genetic epidemiology of amyotrophic lateral sclerosis. Clin. Genet. 63, 83–101 (2003).
5 Kunst CB. Complex genetics of amyotrophic lateral sclerosis. Am. J. Hum. Genet. 75, 933–947 (2004).
6 Valentine JS, Doucette PA, Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu. Rev. Biochem. 74, 563–593 (2005).
7 Bowser R, Cudkowicz M, Kaddurah-Daouk R. Biomarkers for amyotrophic lateral sclerosis. Expert Rev. Mol. Diagn. 6, 387–398 (2006).
8 Cudkowicz M, Qureshi M, Shefner J. Measures and markers in amyotrophic lateral sclerosis. NeuroRx 1, 273–283 (2004).
9 Ince P, Stout N, Shaw P et al. Parvalbumin and calbindin D-28k in the human motor system and in motor neuron disease. Neuropathol. Appl. Neurobiol. 19, 291–299 (1993).
10 Alexianu ME, Ho BK, Mohamed AH et al. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 36, 846–858 (1994).
11 Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N. Engl. J. Med. 344, 1688–1700 (2001).
12 Neumann M, Sampathu DM, Kwong LK et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006).
•• Identified TAR-DNA binding protein 43 as a component of inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis (ALS).
13 Mackenzie IR, Bigio EH, Ince PG et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61, 427–434 (2007).
14 Neumann M, Kwong LK, Sampathu DM et al. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch. Neurol. 64, 1389–1394 (2007).
15 Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J. 24, 1311–1317 (2005).
16 Gruzman A, Wood WL, Alpert E et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc. Natl Acad. Sci. USA 104, 12524–12529 (2007).
17 Rakhit R, Robertson J, Vande Velde C et al. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nature Med. 13, 754–759 (2007).
18 Sunderland T, Gur RE, Arnold SE. The use of biomarkers in the elderly: current and future challenges. Biol. Psychiatry 58, 272–276 (2005).
19 Shaw LM, Korecka M, Clark CM et al. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat. Rev. Drug Discovery 6, 295–303 (2007).
20 Gil J, Preux P-M, Alioum A et al. Disease progression and survivial in ALS: first multi-state model approach. Amyotroph. Lateral Scler. 8, 224–229 (2007).
21 O’toole O, Traynor BJ, Brennan P et al. Epidemiology and clinical features of amyotrophic lateral sclerosis in Ireland between 1995 and 2004. J. Neurol. Neurosurg. Psychiatry 79, 30–32 (2008).
22 Kolarcik C, Bowser R. Plasma and cerebrospinal fluid-based protein biomarkers for motor neuron disease. Mol. Diagn. Ther. 10, 281–292 (2006).
23 Katz R. Biomarkers and surrogate markers: an FDA perspective. NeuroRx 1, 189–195 (2004).
24 Ranganathan S, Nicoll GC, Henry S et al. Comparative proteomic profiling of cerebrospinal fluid between living and post mortem ALS and control subjects. Amyotroph. Lateral Scler. 3, 1–7 (2007).
25 Leigh PN, Simmons A, Williams S et al. Imaging: MRS/MRI/PET/SPECT: summary. Amyotroph. Lateral Scler. Other Motor Neuron. Disord. 3(Suppl. 1), S75–S80 (2002).
26 Hu S, Loo JA, Wong DT. Human body fluid proteome analysis. Proteomics 6, 6326–6353 (2006).
27 Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 7, 41–53 (2006).
28 Adachi J, Kumar C, Zhang Y et al. The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. Genome Biol. 7, R80 (2006).
29 West-Nielsen M, Hogdall EV, Marchiori E et al. Sample handling for mass spectrometric proteomic investigations of human sera. Anal. Chem. 77, 5114–5123 (2005).
30 Ranganathan S, Polshyna A, Nicholl G et al. Assessment of protein stability in cerebrospinal fluid using surface-enhanced
laser desorption/ionization time-of-flight mass spectrometry protein profiling. Clin. Proteom. 2, 91–101 (2006).
31 Marouga R, David S, Hawkins E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Chim. Acta 382, 669–678 (2005).
32 Anderson K, Potter A, Baban D et al. Protein expression changes in spinal muscular atrophy revealed with a novel antibody array technology. Brain 126, 2052–2064 (2003).
33 Connor JR, Mitchell RM, Lee SY et al. Biomarkers in ALS patients stratified by HFE genotype. Soc. Neurosci. Abstr. (2007).
34 McDonald WH, Yates JR III. Shotgun proteomics and biomarker discovery. Dis. Markers 18, 99–105 (2002).
35 Ramstrom M, Ivonin I, Johansson A et al. Cerebrospinal fluid protein patterns in neurodegenerative disease revealed by liquid chromatography–Fourier transform ion cyclotron resonance mass spectrometry. Proteomics 4, 4010–4018 (2004).
36 Comuzzi B, Sadar MD. Proteomic analyses to identify novel therapeutic targets for the treatment of advanced prostate cancer. Cellscience 3, 61–81 (2006).
37 Wong JW, Sullivan MJ, Cagney G. Computational methods for the comparative quantification of proteins in label-free LCn-MS experiments. Brief. Bioinform. (2007).
38 Elahi E, Kumm J, Ronaghi M. Global genetic analysis. J. Biochem. Mol. Biol. 37, 11–27 (2004).
39 van Es MA, van Vaught PW Blauw HM et al. ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol. 6, 869–877 (2007).
40 Schymick JC, Scholz SW, Fung HC et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 6, 322–328 (2007).
41 Kasperaviciute D, Weale ME, Shianna KV et al. Large-scale pathways-based association study in amyotrophic lateral sclerosis. Brain 130, 2292–2301 (2007).
42 Dunckley T, Huentelman MJ, Craig DW et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N. Engl. J. Med. 357, 775–788 (2007).
43 van Es MA, van Vught PW, Blauw HM et al. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 40, 29–31 (2008).

Protein biomarkers for amyotrophic lateral sclerosis Review
www.future-drugs.com 261
• Whole-genome-wide association screening in sporadic ALS that identified DPP6.
44 Bilic E, Bilic E, Rudan I et al. Comparison of the growth hormone, IGF-1 and insulin in cerebrospinal fluid and serum between patients with motor neuron disease and healthy controls. Eur. J. Neurol. 13, 1340–1345 (2006).
45 Hosback S, Hardiman O, Nolan CM et al. Circulating insulin-like growth factors and related binding proteins are selectively altered in amyotrophic lateral sclerosis and multiple sclerosis. Growth Horm. IGF Res. 17, 472–479 (2007).
46 Johansson A, Larsson A, Nygren I et al. Increased serum and cerebrospinal fluid FGF-2 levels in amyotrophic lateral sclerosis. Neuroreport 14, 1867–1869 (2003).
47 Ilzecka J, Stelmasiak Z. Brain-derived neurotrophic factor is not altered in the serum and cerebrospinal fluid of amyotrophic lateral sclerosis patients. Neurol. Sci. 22, 473–474 (2002).
48 Brettschneider J, Widl K, Schattauer D et al. Cerebrospinal fluid erythropoietin (EPO) in amyotrophic lateral sclerosis. Neurosci. Lett. 416, 257–260 (2007).
49 Brettschneider J, Widl K, Ehrenreich H et al. Erythropoietin in the cerebrospinal fluid in neurodegenerative diseases. Neurosci. Lett. 404, 347–351 (2006).
50 Cronin S, Greenway MJ, Ennis S et al. Elevated serum angiogenin levels in ALS. Neurology 67, 1833–1836 (2006).
• Larger study that identified angiogenin as a potential biomarker for ALS in serum.
51 Nygren I, Larsson A, Johansson A et al. VEGF is increased in serum but not in spinal cord from patients with amyotrophic lateral sclerosis. Neuroreport 13, 2199–2201 (2002).
52 Devos D, Moreau C, Lassalle P et al. Low levels of the vascular endothelial growth factor in CSF from early ALS patients. Neurology 62, 2127–2129 (2004).
53 Moreau C, Devos D, Brunaud-Danel V et al. Paradoxical response of VEGF expression to hypoxia in CSF of patients with ALS. J. Neurol. Neurosurg. Psychiatry 77, 255–257 (2006).
54 Ilzecka J. Decreased serum endoglin level in patients with amyotrophic lateral sclerosis: a preliminary report. Scand. J. Clin. Lab. Invest. 12, 1–5 (2007).
55 Smirnova IV, Festoff BW. Alterations in serum thrombospondin in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 127, 207–213 (1994).
56 Ilzecka J, Stelmasiak Z, Dobosz B. Interleukin-1β converting enzyme/caspase-1 (ICE/caspase-1) and soluble APO-1/Fas/CD 95 receptor in amyotrophic lateral sclerosis patients. Acta Neurol. Scand. 103, 255–258 (2001).
57 Poloni M, Facchetti D, Mai R et al. Circulating levels of tumour necrosis factor-alpha and its soluble receptors are increased in the blood of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 287, 211–214 (2000).
58 Rentzos M, Nikolaou C, Rombos A et al. RANTES levels are elevated in serum and cerebrospinal fluid in patients with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron. Disord. 8, 283–287 (2007).
59 Rentzos M, Michalopoulou M, Nikolaou C et al. Serum levels of soluble intercellular adhesion molecule-1 (s-ICAM-1) and soluble endothelial leukocyte adhesion molecule-1(s-ELAM-1) in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron. Disord. 6, 118–121 (2005).
60 Ilzecka J. Cerebrospinal fluid Flt3 ligand level in patients with amyotrophic lateral sclerosis. Acta Neurol. Scand. 114, 205–209 (2006).
61 Wilms H, Sievers J, Dengler R et al. Intrathecal synthesis of monocyte chemoattractant protein-1 (MCP-1) in amyotrophic lateral sclerosis: further evidence for microglial activation in neurodegeneration. J. Neuroimmunol. 144, 139–142 (2003).
62 Baron P, Bussini S, Cardin V et al. Production of monocyte chemoattractant protein-1 in amyotrophic lateral sclerosis. Muscle Nerve 32, 541–544 (2005).
63 Moreau C, Devos D, Brunaud-Danel V et al. Elevated IL-6 and TNF-α levels in patients with ALS: inflammation or hypoxia? Neurology 65, 1958–1960 (2005).
64 Ono S, Hu J, Shimizu N et al. Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis. J. Neurol. Sci. 187, 27–34 (2001).
65 Houi K, Kobayashi T, Kato S et al. Increased plasma TGF-β1 in patients with amyotrophic lateral sclerosis. Acta Neurol. Scand. 106, 299–301 (2002).
66 Ilzecka J, Stelmasiak Z, Dobosz B. Transforming growth factor-β 1 (TGF-β 1) in patients with amyotrophic lateral sclerosis. Cytokine 20, 239–243 (2002).
67 Sussmuth SD, Tumani H, Ecker D et al. Amyotrophic lateral sclerosis: disease stage related changes of tau protein and S100 β in cerebrospinal fluid and creatine kinase in serum. Neurosci. Lett. 353, 57–60 (2003).
• One of the first attempts to correlate protein alternations in ALS to disease progression.
68 Beuche W, Yushchenko M, Mader M et al. Matrix metalloproteinase-9 is elevated in serum of patients with amyotrophic lateral sclerosis. Neuroreport 11, 3419–3422 (2000).
69 Ono S, Imai T, Shimizu N et al. Decreased plasma levels of fibronectin in amyotrophic lateral sclerosis. Acta Neurol. Scand. 101, 391–394 (2000).
70 Houi K, Kobayashi T, Kato S et al. Serum type IV collagen increases with duration of amyotrophic lateral sclerosis. Muscle Nerve 23, 430–432 (2000).
71 Otto M, Bahn E, Wiltfang J et al. Decrease of S100 β protein in serum of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 240, 171–173 (1998).
• Larger study that identified S100-β as a potential biomarker for ALS in serum.
72 Ihara Y, Mori A, Hayabara T et al. Superoxide dismutase and free radicals in sporadic amyotrophic lateral sclerosis: relationship to clinical data. J. Neurol. Sci. 134, 51–56 (1995).
73 Grundstrom E, Lindholm D, Johansson A et al. GDNF but not BDNF is increased in cerebrospinal fluid in amyotrophic lateral sclerosis. Neuroreport 11, 1781–1783 (2000).
74 Kuncl RW, Bilak MM, Bilak SR et al. Pigment epithelium-derived factor is elevated in CSF of patients with amyotrophic lateral sclerosis. J. Neurochem. 81, 178–184 (2002).
75 Kern MA, Friese M, Grundstrom E et al. Amyotrophic lateral sclerosis: evidence for intact hepatocyte growth factor/MET signalling axis. Cytokine 15, 315–319 (2001).
76 Tsuboi Y, Kakimoto K, Akatsu H et al. Hepatocyte growth factor in cerebrospinal fluid in neurologic disease. Acta Neurol. Scand. 106, 99–103 (2002).

262 Expert Rev. Proteomics 5(2), (2008)
Review Ryberg & Bowser
77 Pirttila T, Vanhatalo S, Turpeinen U et al. Cerebrospinal fluid insulin-like growth factor-1, insulin growth factor binding protein-2 or nitric oxide are not increased in MS or ALS. Acta Neurol. Scand. 109, 337–341 (2004).
78 Ilzecka J. Cerebrospinal fluid vascular endothelial growth factor in patients with amyotrophic lateral sclerosis. Clin. Neurol. Neurosurg. 106, 289–293 (2004).
79 Nagata T, Nagano I, Shiote M et al. Elevation of MCP-1 and MCP-1/VEGF ratio in cerebrospinal fluid of ALS patients Neurol. Res. 29, 772–776 (2007).
80 Nagata T, Nagano I, Shiote M et al. Elevation of MCP-1 and MCP-1/VEGF ratio in cerebrospinal fluid of ALS patients Neurol. Res. 29, 772–776 (2007).
81 Sekizawa T, Openshaw H, Ohbo K et al. Cerebrospinal fluid interleukin 6 in amyotrophic lateral sclerosis: immunological parameter and comparison with inflammatory and non-inflammatory central nervous system diseases. J. Neurol. Sci. 154, 194–199 (1998).
82 Sjogren M, Davidsson P, Wallin A et al. Decreased CSF-β-amyloid 42 in Alzheimer’s disease and amyotrophic lateral sclerosis may reflect mismetabolism of β-amyloid induced by disparate mechanisms. Dement. Geriatr. Cogn. Disord. 13, 112–118 (2002).
83 Jimenez-Jimenez FJ, Hernanz A, Medina-Acebron S et al. Tau protein concentrations in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neurol. Scand. 111, 114–117 (2005).
84 Brettschneider J, Petzold A, Sussmuth SD et al. Axonal damage markers in cerebrospinal fluid are increased in ALS. Neurology 66, 852–856 (2006).
• Larger study that describes the levels of heavy neurofilament protein and tau protein in cerebospinal fluid (CSF).
85 Zetterberg H, Jacobsson J, Rosengren L et al. Cerebrospinal fluid neurofilament light levels in amyotrophic lateral sclerosis: impact of SOD1 genotype. Eur. J. Neurol. 14, 1329–1333 (2007).
•• Larger study showing that the light neurofilament protein is strongly elevated in ALS CSF and that levels correspond to disease duration. Levels were considerably higher in SOD1 wild-type patients as compared with those with SOD1 mutations.
86 Norgren N, Rosengren L, Stigbrand T. Elevated neurofilament levels in neurological diseases. Brain Res. 987, 25–31 (2003).
87 Rosengren LE, Karlsson JE, Karlsson JO et al. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neurofilament protein in CSF. J. Neurochem. 67, 2013–2018 (1996).
88 Ranganathan S, Williams E, Ganchev P et al. Proteomic profiling of cerebrospinal fluid identifies biomarkers for amyotrophic lateral sclerosis. J. Neurochem. 95, 1461–1471 (2005).
•• First article to use mass spectrometry to determine the protein profile in CSF for ALS. Some of the protein biomarkers were identified and validated.
89 Pasinetti GM, Ungar LH, Lange DJ et al. Identification of potential CSF biomarkers in ALS. Neurology 66, 1218–1222 (2006).
90 Lorenzl S, Albers DS, LeWitt PA et al. Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative diseases. J. Neurol. Sci. 207, 71–76 (2003).
91 Jacobsson J, Jonsson PA, Andersen PM et al. Superoxide dismutase in CSF from amyotrophic lateral sclerosis patients with and without CuZn-superoxide dismutase mutations. Brain 124, 1461–1466 (2001).
92 Ilzecka J. Decreased cerebrospinal fluid cytochrome c levels in patients with amyotrophic lateral sclerosis. Scand. J. Clin. Lab. Invest. 67, 264–269 (2007).
93 Matsuishi T, Nagamitsu S, Shoji H et al. Increased cerebrospinal fluid levels of substance P in patients with amyotrophic lateral sclerosis. Short communication. Neural. Transm. 106, 943–948 (1999).
94 Lacomblez L, Doppler V, Beucler I et al. APOE: a potential marker of disease progression in ALS. Neurology 58, 1112–1114 (2002).
95 Borroni B, Premi E, Di Luca M et al. Combined biomarkers for early Alzheimer disease diagnosis. Curr. Med. Chem. 14, 1171–1178 (2007).
96 Ray S, Britschgi M, Herbert C et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nature Med. 13, 1359–1362 (2007).
97 Jiang YM, Yamamoto M, Kobayashi Y et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann. Neurol. 57, 236–251 (2005).
98 Malaspina A, Kaushik N, de Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J. Neurochem. 77, 132–145 (2001).
99 Wang XS, Simmons Z, Liu W et al. Differential expression of genes in amyotrophic lateral sclerosis revealed by profiling the post mortem cortex. Amyotroph. Lateral Scler. 7, 201–210 (2006).
100 Dangond F, Hwang D, Camelo S et al. Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol. Genomics 16, 229–239 (2004).
101 Kay A, Petzold A, Kerr M et al. Decreased cerebrospinal fluid apolipoprotein E after subarachnoid hemorrhage: correlation with injury severity and clinical outcome. Stroke 34, 637–642 (2003).
102 Russman AN, Lederman RJ, Calabrese LH et al. Multifocal varicella–zoster virus vasculopathy without rash. Arch. Neurol. 60, 1607–1609 (2003).
103 Gruener N, Gozlan O, Goldstein T et al. Iron, transferrin, and ferritin in cerebrospinal fluid of children. Clin. Chem. 37, 263–265 (1991).
104 Lejon V, Robays J, N’Siesi FX et al. Treatment failure related to intrathecal immunoglobulin M (IgM) synthesis, cerebrospinal fluid IgM, and interleukin-10 in patients with hemolymphatic-stage sleeping sickness. Clin. Vaccine Immunol. 14, 732–737 (2007).
Affiliations
• Henrik Ryberg, PhDUniversity of Pittsburgh School of Medicine, Department of Pathology, Center for ALS Research; Pittsburgh Institute for Neurodegenerative Diseases, Pittsburgh, PA 15261, USATel.: +1 412 648 9655Fax: +1 412 648 [email protected]
• Robert Bowser, PhDUniversity of Pittsburgh School of Medicine, Department of Pathology, Center for ALS Research; Pittsburgh Institute for Neurodegenerative Diseases, Pittsburgh, PA 15261, USATel.: +1 412 383 7819Fax: +1 412 648 [email protected]




















