
Glutamate Release Induced by Activation of Glycine and GABA Transporters in Spinal Cord is Enhanced...
10
Glutamate Release Induced by Activation of Glycine and GABA Transporters in Spinal Cord is Enhanced in a Mouse Model of Amyotrophic Lateral Sclerosis Luca Raiteri 1 , Simona Zappettini 1 , Sara Stigliani 1 , Silvio Paluzzi 1 , Maurizio Raiteri 1,2 , Giambattista Bonanno 1,2, * 1 Department of Experimental Medicine, Pharmacology and Toxicology Section, University of Genoa, 16148 Genoa, Italy 2 Center of Excellence for Biomedical Research, University of Genoa, 16132 Genoa, Italy Received 12 January 2005; accepted 26 January 2005 Available online 10 May 2005 Abstract Amyotrophic lateral sclerosis is a progressive and fatal neurodegenerative disease, involving both upper and lower motor neurons, the cause of which is obscure, although glutamate (GLU)-induced excitotoxicity has been suggested to play a major role. We studied the release of [ 3 H]D-aspartate ([ 3 H]D-ASP) and endogenous glutamate evoked by glycine (GLY) or GABA from spinal cord synaptosomes in mice expressing a mutant form of human SOD1 with a Gly 93 Ala substitution ([SOD1-G93A(+)]), a transgenic model of amyotrophic lateral sclerosis, in mice expressing the non-mutated form of human SOD1 [SOD1(+)], and in non-transgenic littermates [SOD1()/G93A()]. In parallel experiments, we also studied the release of [ 3 H]GABA evoked by GLY and that of [ 3 H]GLY evoked by GABA. Mutant mice were killed at advanced phase of pathology or during the pre-symptomatic period. In SOD1()/G93A() or SOD1(+) mice GLY evoked [ 3 H]D-ASP and [ 3 H]GABA release, while GABA caused [ 3 H]D-ASP, but not [ 3 H]GLY, release. The GLY-evoked release of [ 3 H]D-ASP, but not that of [ 3 H]GABA, and the GABA-evoked [ 3 H]D-ASP release, but not that of [ 3 H]GLY, were more pronounced in SOD1-G93A(+) than in SOD1(+) or SOD1()/G93A() mice. Furthermore, the excessive potentiation of [ 3 H]D-ASP by GLYor GABA was already present in asymptomatic 30–40 day-old SOD1-G93A(+) mice. The releases of endogenous glutamate and GABA also were enhanced by GLYand the GLY-evoked release of endogenous glutamate, but not of endogenous GABA, was higher in SOD1-G93A(+) than in control animals. Potentiation of the spontaneous amino acid release is likely to be mediated by activation of a GLY or a GABA transporter, since the effect of GLY was counteracted by the GLY transporter blocker glycyldodecylamide but not by the GLY receptor antagonists strychnine and 5,7-dichlorokynurenate while the effect of GABA was diminished by the GABA transporter blocker SKF89976-A but not by the GABA receptor antagonists SR9531 and CGP52432. It is concluded that the glutamate release machinery seems excessively functional in SOD1-G93A(+) animals. # 2005 Elsevier Inc. All rights reserved. Keywords: Amyotrophic lateral sclerosis; SOD1-G93A(+) mice; Glutamate release; Glycine heterotransporters; GABA heterotransporters; Release mechanisms INTRODUCTION Amyotrophic lateral sclerosis (ALS) is a chronic neuromuscular disorder clinically characterized by muscle wasting, weakness and spasticity reflecting a NeuroToxicology 26 (2005) 883–892 * Corresponding author. Tel.: +39 010 3532651; fax: +39 010 3993360. E-mail address: [email protected] (G. Bonanno). 0161-813X/$ – see front matter # 2005 Elsevier Inc. All rights reserved. doi:10.1016/j.neuro.2005.01.015
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Glutamate Release Induced by Activation of Glycine and GABA Transporters in Spinal Cord is Enhanced...

Glutamate Release Induced by Activation ofGlycine and GABA Transporters in SpinalCord is Enhanced in a Mouse Model of
Amyotrophic Lateral SclerosisLuca Raiteri 1, Simona Zappettini 1, Sara Stigliani 1, Silvio Paluzzi 1,
Maurizio Raiteri 1,2, Giambattista Bonanno 1,2,*1Department of Experimental Medicine, Pharmacology and Toxicology Section, University of Genoa, 16148 Genoa,
Italy2Center of Excellence for Biomedical Research, University of Genoa, 16132 Genoa, Italy
Received 12 January 2005; accepted 26 January 2005
Available online 10 May 2005
Abstract
Amyotrophic lateral sclerosis is a progressive and fatal neurodegenerative disease, involving both upper and lower
motor neurons, the cause of which is obscure, although glutamate (GLU)-induced excitotoxicity has been suggested to
play a major role. We studied the release of [3H]D-aspartate ([3H]D-ASP) and endogenous glutamate evoked by glycine
(GLY) or GABA from spinal cord synaptosomes in mice expressing a mutant form of human SOD1 with a Gly93Ala
substitution ([SOD1-G93A(+)]), a transgenic model of amyotrophic lateral sclerosis, in mice expressing the non-mutated
form of human SOD1 [SOD1(+)], and in non-transgenic littermates [SOD1(�)/G93A(�)]. In parallel experiments, we
also studied the release of [3H]GABA evoked by GLYand that of [3H]GLY evoked by GABA. Mutant mice were killed at
advanced phase of pathology or during the pre-symptomatic period. In SOD1(�)/G93A(�) or SOD1(+) mice GLYevoked
[3H]D-ASPand [3H]GABA release, while GABA caused [3H]D-ASP, but not [3H]GLY, release. The GLY-evoked release of
[3H]D-ASP, but not that of [3H]GABA, and the GABA-evoked [3H]D-ASP release, but not that of [3H]GLY, were more
pronounced in SOD1-G93A(+) than in SOD1(+) or SOD1(�)/G93A(�) mice. Furthermore, the excessive potentiation of
[3H]D-ASP by GLYor GABA was already present in asymptomatic 30–40 day-old SOD1-G93A(+) mice. The releases of
endogenous glutamate and GABA also were enhanced by GLYand the GLY-evoked release of endogenous glutamate, but
not of endogenous GABA, was higher in SOD1-G93A(+) than in control animals. Potentiation of the spontaneous amino
acid release is likely to be mediated by activation of a GLY or a GABA transporter, since the effect of GLY was
counteracted by the GLY transporter blocker glycyldodecylamide but not by the GLY receptor antagonists strychnine and
5,7-dichlorokynurenate while the effect of GABA was diminished by the GABA transporter blocker SKF89976-A but not
by the GABA receptor antagonists SR9531 and CGP52432. It is concluded that the glutamate release machinery seems
excessively functional in SOD1-G93A(+) animals.
# 2005 Elsevier Inc. All rights reserved.
Keywords: Amyotrophic lateral sclerosis; SOD1-G93A(+) mice; Glutamate release; Glycine
heterotransporters; GABA heterotransporters; Release mechanisms
NeuroToxicology 26 (2005) 883–892
* Corresponding author. Tel.: +39 010 3532651;
fax: +39 010 3993360.
E-mail address: [email protected] (G. Bonanno).
0161-813X/$ – see front matter # 2005 Elsevier Inc. All rights reserv
doi:10.1016/j.neuro.2005.01.015
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a chronic
neuromuscular disorder clinically characterized by
muscle wasting, weakness and spasticity reflecting a
ed.

L. Raiteri et al. / NeuroToxicology 26 (2005) 883–892884
progressive degeneration of upper and lower motor
neurons (Brown, 1995). The prevalence of the disease
is three to five patients per 100,000 individuals, making
it the most frequent paralytic disease in adults (Przed-
borski et al., 2003). To date only a few approved
treatments prolong survival in ALS patients of some
extent (Louvel et al., 1997). Development of more
effective neuroprotective therapies is impeded by lack
of understanding of the mechanisms of neuronal death
in ALS and how the disease propagates.
ALS occurs both in sporadic and familial forms
(Horton et al., 1976). Approximately, 10% of cases
are inherited, usually as an autosomal dominant trait
(Mulder et al., 1986). In about 25%of familiar cases, the
disease is caused by mutation in the gene encoding
cytosolic copper–zinc superoxide dismutase (SOD1)
(Rosen et al., 1993). Consistently, transgenic mice
over-expressing familialALS-associatedSOD1mutants
(G93A,G37R, andG85R) develop a rapidly progressive
neuromuscular disease resembling humanALS (Gurney
et al., 1994; Brown, 1995;Wong et al., 1995). Since both
sporadic and familial ALS patients show very similar
pathological traits, there is a general consensus that
clarification of the mechanisms underlying neurotoxi-
city in familialALS should help the understandingof the
pathogenic determinants in sporadic ALS.
There is increasing evidence that glutamate-
mediated excitotoxicity is implicated in ALS (see,
for reviews: Leigh and Meldrum, 1996; Morrison
and Morrison, 1999; Shaw and Eggett, 2000; Cluskey
and Ramsden, 2001). An abnormality in the function of
the glial glutamate transporter GLT-1 (EAAT-2), which
is largely responsible for removing extracellular glu-
tamate, has been proposed as one major cause of
excessive glutamate receptor activation in both spora-
dic and familiar ALS (Rothstein et al., 1995). On the
other hand, alterations in the glial glutamate transporter
may not alone account for the excitotoxic degeneration
(see Morrison and Morrison, 1999). Other reasons for
the gains of glutamatergic function present in mutant
forms of SOD1, including increase of glutamate
release, should therefore be considered.
Transporters for different transmitters often are coex-
pressed on the same neuron, particularly on the same
terminal button; that is, transporters for the reuptake of
the transmitter just released into the synapse (homo-
transporters) and transporters able to recognize and
capture transmitters/modulators coming from neigh-
bouring structures (heterotransporters) often coexist
on the same membrane. Interestingly, activation of
heterotransporters can elicit release of the transmitter
previously taken up through the coexisting homotran-
sporters or of the endogenous counterpart (Bonanno and
Raiteri, 1994; Raiteri et al., 2002; Sepkuty et al., 2002).
Heterotransporters have also been found to exist on
neuronal cell bodies/dendrites; for example, the excita-
tory amino acid transporter 4 (EAAT4) is present on
GABAergic Purkinje cells in the cerebellum (Yamada
et al., 1996; Furuta et al., 1997; Nagao et al., 1997).
Studying the heterotransporter-mediated modula-
tion of glutamate release in the spinal cord of a
transgenic mouse model of familial ALS carrying a
human SOD1 cDNA with the G93A-associated muta-
tion (Gurney et al., 1994), we here found a potential
novel mechanism by which the extracellular glutamate
concentration can be increased.
METHODS
Animals
B6SJL-TgN SOD1-G93A(+)1Gur mice expressing
high copy number of mutant human SOD1 with a
Gly93Ala substitution [SOD1-G93A(+)] and B6SJL-
TgN (SOD1)2Gur mice expressing wild-type human
SOD1 [SOD1(+)] were obtained from Jackson Labora-
tories (Bar Harbor, ME, USA) and bred at the animal
facility of the Pharmacology and Toxicology Section,
Department of Experimental Medicine in Genoa.
Transgenic mice were originally produced by micro-
injection of the transgene in fertilized eggs obtained
from hybrid mice: the G1 line was used for G93A and
the N1029 line for SOD1 (Gurney et al., 1994). The
transgene number is different in the two lines, with G1
havingmore than double the number of gene copies. As
estimated by Gurney et al. (1994), G1 mice have a gene
copy (per diploid gene) = 18, while N1029 mice have a
gene copy (per diploid gene) = 7.2; however, it was
reported that the N1029 line expressed comparable or
even greater amounts of total brain human SOD1 (Dal
Canto and Gurney, 1994; Gurney et al., 1994). Selec-
tive breeding maintained each transgene in the hemi-
zigous state on an F1 hybrid C57Bl6 � SJL genetic
background (Gurney et al., 1994). Nontransgenic lit-
termates of SOD1-G93A(+) and SOD1(+) mice were
used as controls [SOD1(�)/G93A(�)]. All transgenic
mice were identified analyzing extracts from tail tips
(homogenized in phosphate-buffer saline, freeze/
thawed twice and centrifuged at 23,000 � g for
15 min at + 4 8C) by staining for SOD1 after Laemm-
li’s polyacrylamide gel electrophoresis (10% resolving
and 4% stacking). In the SOD1-G93A(+) strain, the
first signs of the disease appeared between 120 and 140

L. Raiteri et al. / NeuroToxicology 26 (2005) 883–892 885
days of life. Progression of the disease was then rapid
so that after 7–9 days, the animals were killed because
of their ingestion disability, due to paralysis of the
posterior limbs. Animals were housed at constant
temperature (22 � 1 8C) and relative humidity (50%)
under a regular dark-light schedule (light on 7 a.m. to 7
p.m.). Food and water were freely available. All
experiments were carried out in accordance with the
European Community Council Directive of 24 Novem-
ber 1986 (86/609/EEC). All efforts were made to
minimize animal suffering and to use only the number
of animals necessary to produce reliable results.
Preparation of Synaptosomes
Animals were sacrificed and the spinal cord rapidly
removed after exposure of the spinal column. Synapto-
somes were prepared essentially as described pre-
viously (Raiteri et al., 1984). Briefly, the tissue was
homogenized in 40 volumes of 0.32 M sucrose, buf-
fered at pH 7.4 with phosphate, using a glass-teflon
tissue grinder (clearance 0.25 mm, 12 up and down
strokes in about 1 min). The homogenate was centri-
fuged (5 min, 1000 � g) to remove nuclei and debris,
and synaptosomes were isolated from the supernatant
by centrifugation at 12,000 � g for 20 min. The synap-
tosomal pellet was then resuspended in a physiological
medium having the following composition (mM):
NaCl, 125; KCl, 3; MgSO4, 1.2; CaCl2, 1.2; NaH2PO4,
1; NaHCO3, 22; glucose, 10 (aeration with 95%O2 and
5% CO2); pH 7.2–7.4. All the above procedures were
performed at 0–4 8C. Protein was measured according
to Bradford (1976) using BSA as a standard.
Experiments of Release
Synaptosomes were incubated at 37 8C for 15 min in
the presence of 0.02 mM [3H]GABA and 50 mM of the
GABA transaminase inhibitor aminooxyacetic acid, to
avoid [3H]GABA metabolism, or of 0.02 mM [3H]D-
ASP, a non-metabolized compound often used as a
marker to mimic glutamate in release studies, or of
0.3 mM [3H]glycine. Aliquots (about 50 mg of protein)
of the synaptosomal suspension were distributed on
microporous filters placed at the bottom of a set of
parallel superfusion chambers maintained at 37 8C(Raiteri and Raiteri, 2000). Superfusion was then
started with standard medium (containing aminooxya-
cetic acid in experiments of [3H]GABA release) at a
rate of 0.5 ml/min and continued for 48 min. After
33 min of superfusion to equilibrate the system, five 3-
min fractions were collected. Synaptosomes were
exposed to glycine (1–1000 mM) or GABA (0.1–
1000 mM) at the end of the second fraction collec-
ted (t = 39 min). Strychnine, 5,7-dichlorokynurenate
(5,7-DCK), glycyldodecylamide (GDA), N[3-(40-fluorophenyl)-3-(40-phenylphenoxy)propyl]sarcosine(NFPS), amoxapine, 2-(30-carbethoxy-20-propenyl)-3-amino-6-paramethoxy-phenyl-pyridazinium bro-
mide (SR95531), [3-[[(3,4-dichlorophenyl)methyl]a-
mino]propyl](diethoxymethyl)phosphinic acid (CGP
52432), and N-(4,4-diphenyl-3-butenyl)nipecotic acid
(SKF89976A) were introduced at t = 30 min. Radio-
activity was measured in each fraction collected and in
the superfused filters. In some experiments, fractions
were analyzed for their endogenous glutamate and
GABA content.
Endogenous Glutamate and GABADetermination
Endogenous glutamate and GABA were measured
by high performance liquid chromatography analysis
following pre-column derivatization with o-phthalal-
dehyde and separation on a C18 reverse-phase chro-
matographic column (10 � 4.6 mm, 3 mm; at 30 8C;Chrompack, Middleburg, The Netherlands) coupled
with fluorometric detection (excitation wavelength
350 nm; emission wavelength 450 nm; Raiteri et al.,
2000). Buffers and the gradient program were as
follows: solvent A, 0.1 M sodium acetate (pH 5.8)/
methanol, 80:20; solvent B, 0.1 M sodium acetate (pH
5.8)/methanol, 20:80; solvent C, sodium acetate (pH
6.0)/methanol, 80:20; gradient program, 100% C for
4 min from the initiation of the program; 90% A and
10% B in 1 min; 42% A and 58% B in 14 min; 100% B
in 1 min; isocratic flow 2 min; 100% C in 3 min; flow
rate 0.9 ml/min. Homoserine was used as an internal
standard.
Calculations
Tritium released in each fraction collected was
calculated as fractional rate �100. The endogenous
glutamate and GABA released in each fraction was
expressed as pmol/mg of synaptosomal protein. Drug
effects were evaluated by performing the ratio between
the efflux in the fourth fraction collected (in which the
maximum effect of glycine or GABA was generally
reached) and that of the second fraction. This ratio was
compared to the corresponding ratio obtained under
control conditions. Appropriate controls were always
run in parallel. The concentration–response curves
shown in Figs. 1, 2 and 4 were fitted to the experimental
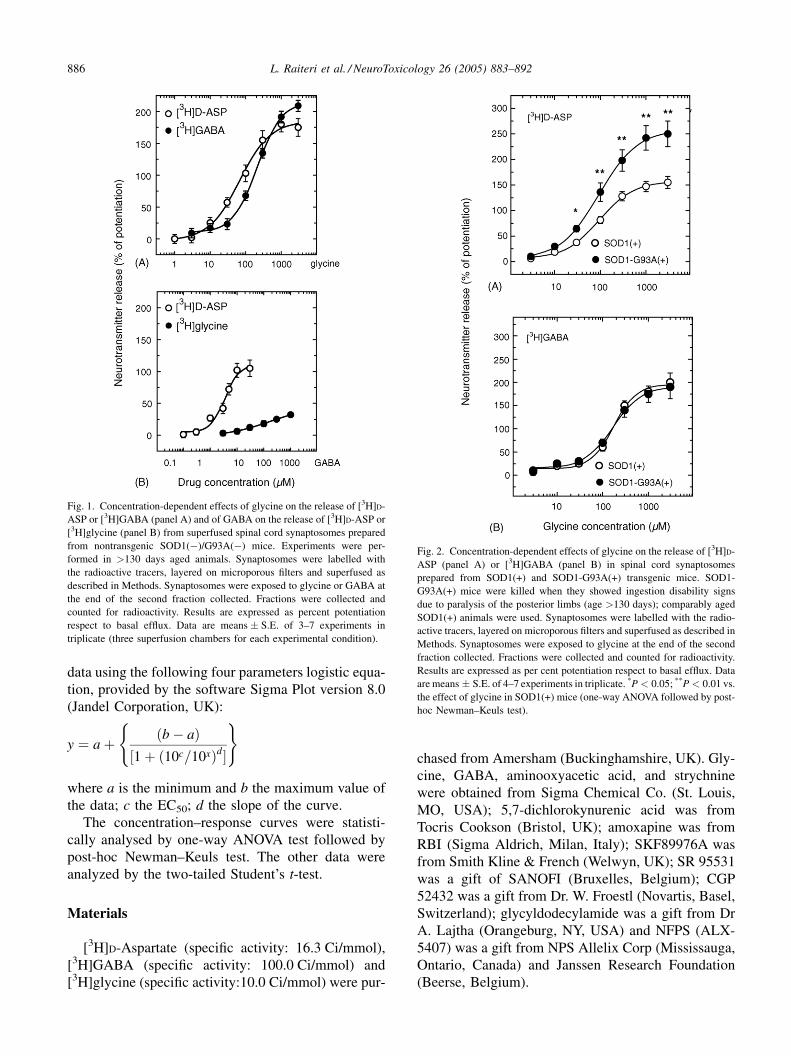
L. Raiteri et al. / NeuroToxicology 26 (2005) 883–892886
Fig. 1. Concentration-dependent effects of glycine on the release of [3H]D-
ASP or [3H]GABA (panel A) and of GABA on the release of [3H]D-ASP or
[3H]glycine (panel B) from superfused spinal cord synaptosomes prepared
from nontransgenic SOD1(�)/G93A(�) mice. Experiments were per-
formed in >130 days aged animals. Synaptosomes were labelled with
the radioactive tracers, layered on microporous filters and superfused as
described in Methods. Synaptosomes were exposed to glycine or GABA at
the end of the second fraction collected. Fractions were collected and
counted for radioactivity. Results are expressed as percent potentiation
respect to basal efflux. Data are means � S.E. of 3–7 experiments in
triplicate (three superfusion chambers for each experimental condition).
Fig. 2. Concentration-dependent effects of glycine on the release of [3H]D-
ASP (panel A) or [3H]GABA (panel B) in spinal cord synaptosomes
prepared from SOD1(+) and SOD1-G93A(+) transgenic mice. SOD1-
G93A(+) mice were killed when they showed ingestion disability signs
due to paralysis of the posterior limbs (age >130 days); comparably aged
SOD1(+) animals were used. Synaptosomes were labelled with the radio-
active tracers, layered on microporous filters and superfused as described in
Methods. Synaptosomes were exposed to glycine at the end of the second
fraction collected. Fractions were collected and counted for radioactivity.
Results are expressed as per cent potentiation respect to basal efflux. Data
are means � S.E. of 4–7 experiments in triplicate. *P < 0.05; **P < 0.01 vs.
the effect of glycine in SOD1(+) mice (one-way ANOVA followed by post-
hoc Newman–Keuls test).
data using the following four parameters logistic equa-
tion, provided by the software Sigma Plot version 8.0
(Jandel Corporation, UK):
y ¼ aþ ðb� aÞ½1þ ð10c=10xÞd�
( )
where a is the minimum and b the maximum value of
the data; c the EC50; d the slope of the curve.
The concentration–response curves were statisti-
cally analysed by one-way ANOVA test followed by
post-hoc Newman–Keuls test. The other data were
analyzed by the two-tailed Student’s t-test.
Materials
[3H]D-Aspartate (specific activity: 16.3 Ci/mmol),
[3H]GABA (specific activity: 100.0 Ci/mmol) and
[3H]glycine (specific activity:10.0 Ci/mmol) were pur-
chased from Amersham (Buckinghamshire, UK). Gly-
cine, GABA, aminooxyacetic acid, and strychnine
were obtained from Sigma Chemical Co. (St. Louis,
MO, USA); 5,7-dichlorokynurenic acid was from
Tocris Cookson (Bristol, UK); amoxapine was from
RBI (Sigma Aldrich, Milan, Italy); SKF89976A was
from Smith Kline & French (Welwyn, UK); SR 95531
was a gift of SANOFI (Bruxelles, Belgium); CGP
52432 was a gift from Dr. W. Froestl (Novartis, Basel,
Switzerland); glycyldodecylamide was a gift from Dr
A. Lajtha (Orangeburg, NY, USA) and NFPS (ALX-
5407) was a gift from NPS Allelix Corp (Mississauga,
Ontario, Canada) and Janssen Research Foundation
(Beerse, Belgium).
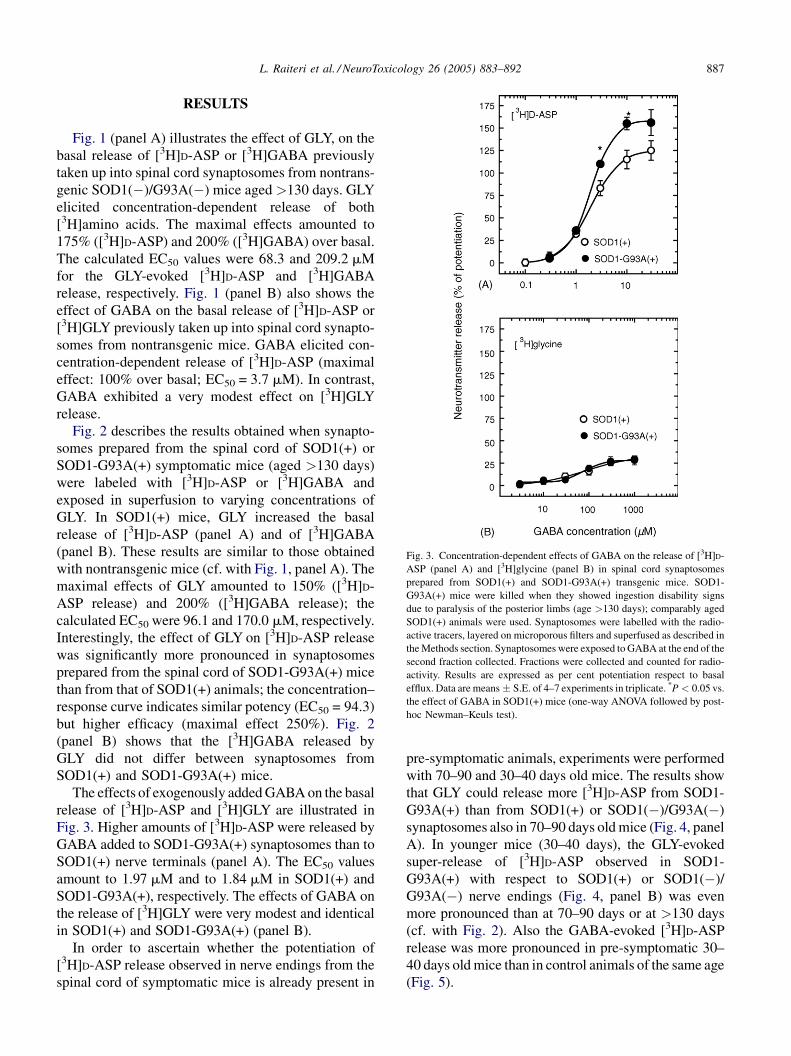
L. Raiteri et al. / NeuroToxicology 26 (2005) 883–892 887
Fig. 3. Concentration-dependent effects of GABA on the release of [3H]D-
ASP (panel A) and [3H]glycine (panel B) in spinal cord synaptosomes
prepared from SOD1(+) and SOD1-G93A(+) transgenic mice. SOD1-
G93A(+) mice were killed when they showed ingestion disability signs
due to paralysis of the posterior limbs (age >130 days); comparably aged
SOD1(+) animals were used. Synaptosomes were labelled with the radio-
active tracers, layered on microporous filters and superfused as described in
theMethods section. Synaptosomes were exposed to GABA at the end of the
second fraction collected. Fractions were collected and counted for radio-
activity. Results are expressed as per cent potentiation respect to basal
efflux. Data are means � S.E. of 4–7 experiments in triplicate. *P < 0.05 vs.
the effect of GABA in SOD1(+) mice (one-way ANOVA followed by post-
hoc Newman–Keuls test).
RESULTS
Fig. 1 (panel A) illustrates the effect of GLY, on the
basal release of [3H]D-ASP or [3H]GABA previously
taken up into spinal cord synaptosomes from nontrans-
genic SOD1(�)/G93A(�) mice aged >130 days. GLY
elicited concentration-dependent release of both
[3H]amino acids. The maximal effects amounted to
175% ([3H]D-ASP) and 200% ([3H]GABA) over basal.
The calculated EC50 values were 68.3 and 209.2 mM
for the GLY-evoked [3H]D-ASP and [3H]GABA
release, respectively. Fig. 1 (panel B) also shows the
effect of GABA on the basal release of [3H]D-ASP or
[3H]GLY previously taken up into spinal cord synapto-
somes from nontransgenic mice. GABA elicited con-
centration-dependent release of [3H]D-ASP (maximal
effect: 100% over basal; EC50 = 3.7 mM). In contrast,
GABA exhibited a very modest effect on [3H]GLY
release.
Fig. 2 describes the results obtained when synapto-
somes prepared from the spinal cord of SOD1(+) or
SOD1-G93A(+) symptomatic mice (aged >130 days)
were labeled with [3H]D-ASP or [3H]GABA and
exposed in superfusion to varying concentrations of
GLY. In SOD1(+) mice, GLY increased the basal
release of [3H]D-ASP (panel A) and of [3H]GABA
(panel B). These results are similar to those obtained
with nontransgenic mice (cf. with Fig. 1, panel A). The
maximal effects of GLY amounted to 150% ([3H]D-
ASP release) and 200% ([3H]GABA release); the
calculated EC50 were 96.1 and 170.0 mM, respectively.
Interestingly, the effect of GLY on [3H]D-ASP release
was significantly more pronounced in synaptosomes
prepared from the spinal cord of SOD1-G93A(+) mice
than from that of SOD1(+) animals; the concentration–
response curve indicates similar potency (EC50 = 94.3)
but higher efficacy (maximal effect 250%). Fig. 2
(panel B) shows that the [3H]GABA released by
GLY did not differ between synaptosomes from
SOD1(+) and SOD1-G93A(+) mice.
The effects of exogenously addedGABAon the basal
release of [3H]D-ASP and [3H]GLY are illustrated in
Fig. 3. Higher amounts of [3H]D-ASP were released by
GABA added to SOD1-G93A(+) synaptosomes than to
SOD1(+) nerve terminals (panel A). The EC50 values
amount to 1.97 mM and to 1.84 mM in SOD1(+) and
SOD1-G93A(+), respectively. The effects of GABA on
the release of [3H]GLY were very modest and identical
in SOD1(+) and SOD1-G93A(+) (panel B).
In order to ascertain whether the potentiation of
[3H]D-ASP release observed in nerve endings from the
spinal cord of symptomatic mice is already present in
pre-symptomatic animals, experiments were performed
with 70–90 and 30–40 days old mice. The results show
that GLY could release more [3H]D-ASP from SOD1-
G93A(+) than from SOD1(+) or SOD1(�)/G93A(�)
synaptosomes also in 70–90 days oldmice (Fig. 4, panel
A). In younger mice (30–40 days), the GLY-evoked
super-release of [3H]D-ASP observed in SOD1-
G93A(+) with respect to SOD1(+) or SOD1(�)/
G93A(�) nerve endings (Fig. 4, panel B) was even
more pronounced than at 70–90 days or at >130 days
(cf. with Fig. 2). Also the GABA-evoked [3H]D-ASP
release was more pronounced in pre-symptomatic 30–
40 days oldmice than in control animals of the same age
(Fig. 5).
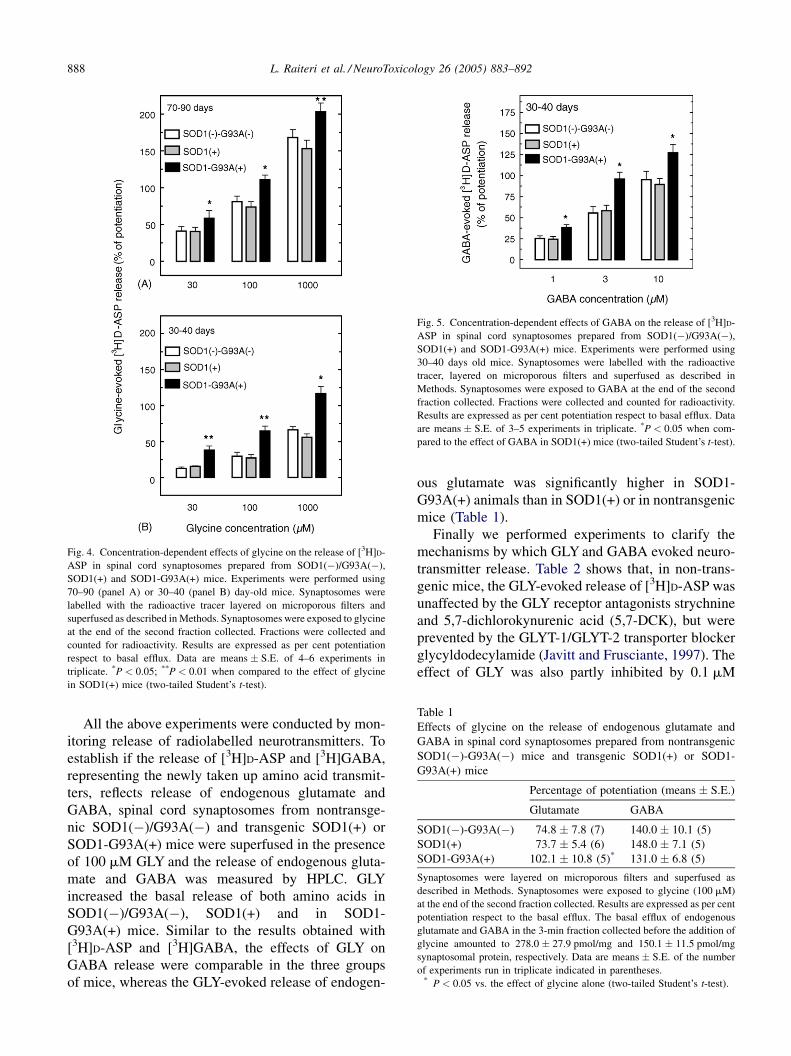
L. Raiteri et al. / NeuroToxicology 26 (2005) 883–892888
Fig. 4. Concentration-dependent effects of glycine on the release of [3H]D-
ASP in spinal cord synaptosomes prepared from SOD1(�)/G93A(�),
SOD1(+) and SOD1-G93A(+) mice. Experiments were performed using
70–90 (panel A) or 30–40 (panel B) day-old mice. Synaptosomes were
labelled with the radioactive tracer layered on microporous filters and
superfused as described in Methods. Synaptosomes were exposed to glycine
at the end of the second fraction collected. Fractions were collected and
counted for radioactivity. Results are expressed as per cent potentiation
respect to basal efflux. Data are means � S.E. of 4–6 experiments in
triplicate. *P < 0.05; **P < 0.01 when compared to the effect of glycine
in SOD1(+) mice (two-tailed Student’s t-test).
Fig. 5. Concentration-dependent effects of GABA on the release of [3H]D-
ASP in spinal cord synaptosomes prepared from SOD1(�)/G93A(�),
SOD1(+) and SOD1-G93A(+) mice. Experiments were performed using
30–40 days old mice. Synaptosomes were labelled with the radioactive
tracer, layered on microporous filters and superfused as described in
Methods. Synaptosomes were exposed to GABA at the end of the second
fraction collected. Fractions were collected and counted for radioactivity.
Results are expressed as per cent potentiation respect to basal efflux. Data
are means � S.E. of 3–5 experiments in triplicate. *P < 0.05 when com-
pared to the effect of GABA in SOD1(+) mice (two-tailed Student’s t-test).
Table 1
Effects of glycine on the release of endogenous glutamate and
GABA in spinal cord synaptosomes prepared from nontransgenic
SOD1(�)-G93A(�) mice and transgenic SOD1(+) or SOD1-
G93A(+) mice
Percentage of potentiation (means � S.E.)
Glutamate GABA
SOD1(�)-G93A(�) 74.8 � 7.8 (7) 140.0 � 10.1 (5)
SOD1(+) 73.7 � 5.4 (6) 148.0 � 7.1 (5)
SOD1-G93A(+) 102.1 � 10.8 (5)* 131.0 � 6.8 (5)
Synaptosomes were layered on microporous filters and superfused as
described in Methods. Synaptosomes were exposed to glycine (100 mM)
at the end of the second fraction collected. Results are expressed as per cent
potentiation respect to the basal efflux. The basal efflux of endogenous
glutamate and GABA in the 3-min fraction collected before the addition of
glycine amounted to 278.0 � 27.9 pmol/mg and 150.1 � 11.5 pmol/mg
synaptosomal protein, respectively. Data are means � S.E. of the number
of experiments run in triplicate indicated in parentheses.* P < 0.05 vs. the effect of glycine alone (two-tailed Student’s t-test).
All the above experiments were conducted by mon-
itoring release of radiolabelled neurotransmitters. To
establish if the release of [3H]D-ASP and [3H]GABA,
representing the newly taken up amino acid transmit-
ters, reflects release of endogenous glutamate and
GABA, spinal cord synaptosomes from nontransge-
nic SOD1(�)/G93A(�) and transgenic SOD1(+) or
SOD1-G93A(+) mice were superfused in the presence
of 100 mM GLY and the release of endogenous gluta-
mate and GABA was measured by HPLC. GLY
increased the basal release of both amino acids in
SOD1(�)/G93A(�), SOD1(+) and in SOD1-
G93A(+) mice. Similar to the results obtained with
[3H]D-ASP and [3H]GABA, the effects of GLY on
GABA release were comparable in the three groups
of mice, whereas the GLY-evoked release of endogen-
ous glutamate was significantly higher in SOD1-
G93A(+) animals than in SOD1(+) or in nontransgenic
mice (Table 1).
Finally we performed experiments to clarify the
mechanisms by which GLY and GABA evoked neuro-
transmitter release. Table 2 shows that, in non-trans-
genic mice, the GLY-evoked release of [3H]D-ASP was
unaffected by the GLY receptor antagonists strychnine
and 5,7-dichlorokynurenic acid (5,7-DCK), but were
prevented by the GLYT-1/GLYT-2 transporter blocker
glycyldodecylamide (Javitt and Frusciante, 1997). The
effect of GLY was also partly inhibited by 0.1 mM

L. Raiteri et al. / NeuroToxicology 26 (2005) 883–892 889
Table 2
Effects of glycine receptor antagonists and of glycine uptake
inhibitors on the glycine-evoked release of [3H]D-ASP and effects
of GABA receptor antagonists and of GABA uptake inhibitors on
the GABA-evoked release of [3H]D-ASP in spinal cord synapto-
somes prepared from nontransgenic SOD1(�)-G93A(�) mice and
transgenic SOD1(+) or SOD1-G93A(+) mice
Drugs Percentage of
potentiation
(means � S.E.)
SOD1(�)-G93A(�)
Glycine (100 mM) 91.0 � 7.4 (5)
Glycine (100 mM) + strychnine (0.1 mM) 81.6 � 12.1 (4)
Glycine (100 mM) + 5,7-DCK (1 mM) 93.1 � 11.5 (4)
Glycine (100 mM) + GDA (50 mM) 21.0 � 4.2 (5)**
Glycine (100 mM) + NFPS (0.1 mM) 37.5 � 1.7 (4) **
Glycine (100 mM) + amoxapine (50 mM) 50.5 � 8.6 (4)*
GABA (10 mM) 73.9 � 8.0 (5)
GABA(10 mM) + SR95531 64.9 � 8.2 (4)
GABA (10 mM) + CGP 52432 57.6 � 6.1 (4)
GABA (10 mM) + SKF89976A 21.5 � 4.6 (5) *
SOD1(+)
Glycine (100 mM) 81.1 � 7.4 (6)
Glycine (100 mM) + strychnine (0.1 mM) 72.1 � 11.1 (3)
Glycine (100 mM) + 5,7-DCK (1 mM) 75.5 � 12.5 (3)
Glycine (100 mM) + GDA (50 mM) 18.7 � 4.2 (3)**
Glycine (100 mM) + NFPS (0.1 mM) 28.3 � 8.5 (4)**
Glycine(100 mM) + amoxapine (50 mM) 36.5 � 5.2 (3)**
SOD1-G93A(+)
Glycine (100 mM) 115.0 � 14.1 (4)
Glycine (100 mM) + strychnine (0.1 mM) 100.3 � 14.5 (3)
Glycine (100 mM) + 5,7-DCK (1 mM) 112.0 � 12.0 (3)
Glycine (100 mM) + GDA (50 mM) 28.7 � 7.8 (3)**
Synaptosomes were labelled with the radioactive tracer, layered on micro-
porous filters and superfused as described in Methods. Synaptosomes were
exposed to glycine (100 mM) or GABA (10 mM) at the end of the second
fraction collected. Receptor antagonists and uptake inhibitors were added to
the superfusion medium 9 min before glycine or GABA. Results are
expressed as per cent potentiation respect to the basal efflux. Data are
means � S.E. of the number of experiments run in triplicate indicated in
parentheses.* P < 0.05 vs. the effect of glycine or GABA alone (two-tailed Student’s
t-test).** P < 0.01 vs. the effect of glycine or GABA alone (two-tailed Student’s
t-test).
NFPS, a potent and selective non-transportable blocker
ofGLY transporters of theGLYT-1 type (Atkinson et al.,
2001; Herdon et al., 2001) or by amoxapine, added at
50 mM, a concentration reported to be selective for the
GLYT-2 transporter (Nunez et al., 2000). Similar results
were obtained with transgenic SOD1-G93A(+) or
SOD1(+) (Table 2). To note, also the GLY-evoked
[3H]GABA release was insensitive to GLY receptor
antagonists but it was abolished by GLY transport
blockers (data not shown). Table 2 also shows that
the release of [3H]D-ASP evoked by GABAwas insen-
sitive to the GABAA receptor antagonist SR95531 or the
GABAB receptor antagonist CGP 52432, but was pre-
vented by the GABA transport blocker SKF89976A, a
selective GAT-1 inhibitor (Borden et al., 1994; Zhou
et al., 2004) in SOD1(�)/G93A(�) mice.
DISCUSSION
The aim of this work has been to study on the effects
of glycine and GABA on the release of glutamate in a
transgenic mouse model of familial ALS, using spinal
cord synaptosomes in superfusion. Synaptosomal pre-
parations contain the different nerve ending families
present in the mammalian CNS. To study the effects of
a modulatory compound on the release of a given
neurotransmitter, indirect effects have to be minimized.
This can be obtained by stratifying synaptosomes on
microporous filters (diameter 2.5 cm) in such an
amount (<100 mg protein/filter) that, based on pre-
vious estimates (Raiteri et al., 1986), the particles
constitute no more than a monolayer. Many studies
have demonstrated that, when such a synaptosomal
layer is up–down superfused, any compound released
is removed before it can activate synaptosomal targets
(transporters, presynaptic receptors and so on). These
targets remain therefore ligand-free and can be selec-
tively activated by ligands added to the superfusion
medium (Raiteri and Raiteri, 2000). Under such con-
ditions, changes in the release of a neurotransmitter A
produced by a compound B can be attributed to direct
action of B on the particles releasing A.
Due to the above considerations, the augmentation
of glutamate release provoked by GLY or by GABA
indicates that GLY or GABA directly acts on gluta-
mate-releasing terminals of SOD1(�)/G93A(�),
SOD1(+) and SOD1-G93A(+) mice. Since the GLY
effect was strychnine-insensitive, GLY does not acti-
vate strychnine-sensitive receptors, which sometimes
mediate stimulation of transmitter release (Darstein
et al., 2000). GLY does not act as glutamate co-agonist
on NMDA receptors either, because its releasing effect
was insensitive to 5,7-DCK, a selective antagonist at
the NMDA receptor glycine site. Instead, the GLY
effect was prevented by the GLY transporter blocker
glycyldodecylamide (Javitt and Frusciante, 1997) sug-
gesting that GLYuptake into glutamatergic terminals is
the primary trigger for glutamate release. We can
conclude that axon terminals able to take up [3H]D-
ASP through glutamate transporters also possess GLY
transporters, the activation of which elevates basal
glutamate release. The GLY effect has an EC50 of
about 90 mM, comparable with reported Km values
for GLYuptake (Lopez-Corcuera et al., 1998; Morrow

L. Raiteri et al. / NeuroToxicology 26 (2005) 883–892890
et al., 1998). Two GLY transporter subtypes, termed
GLYT-1 and GLYT-2, have been reported to exist (see,
for a review, Lopez-Corcuera et al., 2001) and non-
transportable selective blockers have become recently
available. The GLY-evoked glutamate release was
partly sensitive to NFPS, a potent GLYT-1 inhibitor
(Atkinson et al., 2001; Herdon et al., 2001), and partly
to amoxapine, which exhibits GLYT-2 selectivity
(Nunez et al., 2000; see Table 2), thus indicating that
GLYT-1 and GLYT-2 are present on glutamatergic
nerve terminals of mouse spinal cord. Considering that
GLYT-1 and GLYT-2 display different expression pat-
terns in the spinal cord (Adams et al., 1995; Zafra et al.,
1995a,b), the possibility that some glutamate nerve
terminals express either GLYT-1 or GLYT-2 appears
more likely than the existence of glutamatergic axon
terminals endowed with both GLYT-1 and GLYT-2.
The GABA-evoked glutamate release was insensi-
tive to GABAA and GABAB receptor antagonists, but it
was blocked by a GABA transporter inhibitor, indicat-
ing involvement of GABA transporters coexisting with
glutamate transporters in spinal cord glutamatergic
nerve terminals. The EC50 of GABA amounted to
about 3 mM, in line with Km values previously reported
in uptake studies (Levi, 1970; Debler and Lajtha,
1987). The finding that the GABA releasing effect
was blocked by the selective GAT-1 inhibitor
SKF89976-A (Borden et al., 1994; Zhou et al.,
2004) indicates that the GABA heterotransporters
involved belongs to the GAT-1 subtype.
Our results show for the first time that glutamate
release can be abnormally enhanced in an animal model
of familial ALS.As shown in Figs. 1–3, GLYandGABA
are more efficacious in releasing glutamate when added
to nerve endings from SOD1-G93A(+) than from
SOD1(+) or SOD1(�)/G93A(�) mice. In contrast,
the GLY-evoked GABA release exhibits identical pat-
tern in SOD1-G93A(+) and in SOD1(+) or SOD1(�)/
G93A(�) animals. Furthermore, GABA was almost
ineffective on the release of GLY. If these effects take
place also in vivo, they could induce unbalances
between inhibitory and excitatory transmitters.
The mechanisms underlying the increased release of
glutamate may be manifold including augmented
expression of GLY and GABA heterotransporters in
the plasmalemmal membrane of glutamatergic axon
terminals, possibly consequent to improved transporter
trafficking. Another possibility is represented by an
augmented efficiency of the glutamate translocation
processes throughout the terminal membrane following
heterotransporter activation. Accordingly, it has been
reported that GLY- or GABA-evoked glutamate release
occurs by reversal of the homotransporter or by the
opening of anion channels (Raiteri et al., 2005). Finally,
the excessive release of glutamate could be triggered by
modifications of the intracellular pathways involved in
heterotransporter-induced neurotransmitter release, pos-
sibly linked to the co-transport of Na+ occurring during
the process of heterotransporter activation. Clarification
of these mechanisms deserves further investigation
because their precise understanding is crucial to the
development of appropriate therapeutic interventions.
The observation that the release of glutamate eli-
cited by GLYor GABAwas higher in SOD1-G93A(+)
mice than in SOD1(+) animals also in experiments
with presymptomatic mice (30–40 or 70–90 days old)
gives support to the idea that the observed effects are
related to the mutation and are not an aspecific con-
sequence of the loss of motor neurons, which is not
present in 8-week-old (Bendotti et al., 2001) or 12-
week-old (Howland et al., 2002) transgenic mice.
Particularly relevant is the observation that the poten-
tiation of GLY-evoked glutamate release was most
pronounced in asymptomatic 30–40 days old SOD1-
G93A(+) mice (Fig. 4). In fact, no changes in GLT-1
(EAAT2) were observed at 60 days of age, before the
appearance of clinical symptoms (Bendotti et al.,
2001). Similarly, glutamate uptake was not decreased
between 8 and 17 weeks in transgenic mice, but was
significantly reduced only at 21 weeks (Canton et al.,
1998). Therefore, the increase of glutamate release
evoked by GLY and by GABA here observed seems
to represent the earliest sign of disease in SOD1-
G93A(+) animals. If the decrease in the glial EAAT2
transporter observed in ALS is a consequence of the
increased glutamate release remains to be established.
Interestingly, elevation of glutamate concentrations
in plasma and cerebrospinal fluid has been documented
in ALS patients (Perry et al., 1990; Rothstein et al.,
1990). Since in our superfused synaptosomes indirect
effects are prevented (Raiteri and Raiteri, 2000), it
seems reasonable to propose that SOD1 mutation
causes modifications in glutamatergic nerve endings,
particularly in the glutamate release machinery. If this
is the case, a thorough examination of the mechanisms
involved in glutamate release should clarify, at least in
part, the relations between SOD1 mutation and
enhanced release of glutamate.
Although the concentrations of GLY and GABA in
the synaptic cleft remain unknown, they could be
inferred from the Km values of the GLY and GABA
uptake processes. The EC50 for the GABA-evoked
glutamate release amounts to about 3 mM, quite com-
patible with the Km values reported for the high affinity

L. Raiteri et al. / NeuroToxicology 26 (2005) 883–892 891
uptake by different authors (Levi, 1970; Debler and
Lajtha, 1987). As to the GLY-evoked glutamate
release, the Km values obtained in uptake studies with
transiently or stably expressed GLYT-1 and GLYT-2
transporters (Smith et al., 1992; Lopez-Corcuera et al.,
1998; Morrow et al., 1998) are well in the range of the
EC50 calculated from the present concentration–
response curves. Based on the information available,
the possibility therefore exists that, in particular con-
ditions, GLY and GABA activate their respective het-
erotransporters on glutamatergic terminals causing
excessive glutamate release. The GLY- and the
GABA-evoked release of glutamate may have obvious
pathophysiological significance in presence of a defi-
ciency in the astroglial glutamate transporter EAAT2.
In this scenario, selective GLY and GABA transporter
subtype blockers could be useful not only to limit the
GLY and GABA heterotransporter-induced release of
glutamate, but also to increase the extracellular avail-
ability of the two inhibitory amino acids, thus offering
substantial benefits as future treatments.
ACKNOWLEDGEMENTS
This work was supported by grants from Italian
MIUR (COFIN 2002) and from Italian Ministry of
Health (1% Project). The authors wish to thank Mrs.
Maura Agate for her skilful secretarial assistance.
REFERENCES
Adams RH, Sato K, Shimada S, Tohyama M, Puschel AW, Betz H.
Gene structure and glial expression of the glycine transporter
GlyT1 in embryonic and adult rodents. J Neurosci
1995;15:2524–32.
Atkinson BN, Bell SC, De Vivo M, Kowalski LR, Lechner SM,
Ognyanov VI, et al. ALX 5407: a potent, selective inhibitor of
the hGlyT1 glycine transporter. Mol Pharmacol 2001;60:1414–
20.
Bendotti C, Tortarolo M, Suchak SK, Calvaresi N, Carvelli L,
Bastone A, et al. Transgenic SOD1 G93A mice develop
reduced GLT-1 in spinal cord without alterations in cerebrosp-
inal fluid glutamate levels. J Neurochem 2001;79:737–46.
Bonanno G, Raiteri M. Release-regulating presynaptic heterocar-
riers. Prog Neurobiol 1994;44:451–62.
Borden LA, Dhar TGM. Smith KF, Weinshank RL, Branchek TA,
Gluchowski C. Tiagabine, SK&F 89976-A, CT-966, and NNC-
711 are selective for the cloned GABA transporter GAT-1. Eur J
Pharmacol Mol Pharmacol Sect 1994;269:219–24.
Bradford MM. A rapid and sensitive method for the quantitation of
microgram quantities of protein utilizing the principle of protein
dye binding. Anal Biochem 1976;72:248–54.
Brown RH. Amyotrophic lateral sclerosis: recent insights from
genetic and transgenic mice. Cell 1995;80:687–92.
Canton T, Pratt J, Stutzmann J-M, Imperato A, Boireau A. Glu-
tamate uptake is decreased tardively in the spinal cord of FALS
mice. Neuroreport 1998;9:775–8.
Cluskey S, Ramsden DB. Mechanisms of neurodegeneration in
amyotrophic lateral sclerosis. J Clin Pathol Mol Pathol
2001;54:386–92.
Dal Canto MC, Gurney ME. Development of central nervous
system pathology in a murine transgenic model of human
amyotrophic lateral sclerosis. Am J Pathol 1994;145:1271–9.
Darstein M, Landwehrmeyer GB, Klig C, Becker C-M, Feuerstein
TJ. Strychnine-sensitive glycine receptors in rat caudatoputa-
men are expressed by cholinergic interneurons. Neuroscience
2000;96:33–9.
Debler EA, Lajtha A. High-affinity transport of gamma-aminobu-
tyric acid, glycine, taurine, L-aspartic acid, and L-glutamic acid
in synaptosomal (P2) tissue: a kinetic and substrate specificity. J
Neurochem 1987;48:1851–6.
Furuta A, Martin LJ, Lin CLG. Dykes-Hoberg M, Rothstein JD.
Cellular and synaptic localization of the neuronal glutamate
transporters excitatory amino acid transporters 3 and 4. Neu-
roscience 1997;81:1031–42.
GurneyME, Pu H, Chiu A, Dal CantoMC, Polchow CY, Alexander
DD, et al. Motor neuron degeneration in mice that express a
human Cu, Zn superoxide dismutase mutation. Science
1994;264:1772–5.
Herdon HJ, Godfrey FM, Brown AM, Coulton S, Evans JR, Cairns
WJ. Pharmacological assessment of the role of the glycine
transporter GLYT1 in mediating high-affinity glycine uptake
by rat cerebral cortex and cerebellum synaptosomes. Neuro-
pharmacology 2001;41:88–96.
Horton WA, Eldridge R, Brody JA. Familial motor neuron disease.
Evidence for at least three different types. Neurology
1976;26:460–5.
Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, et al.
Focal loss of the glutamate transporter EAAT2 in a transgenic
rat model of SOD1mutant-mediated amyotrophic lateral sclero-
sis (ALS). Proc Natl Acad Sci 2002;99:1604–9.
Javitt DC, Frusciante M. Glycyldodecylamide, a phencyclidine
behavioral antagonist, blocks cortical glycine uptake: implica-
tions for schizophrenia and substance abuse. Psychopharmacol-
ogy 1997;129:96–8.
Leigh PN, Meldrum BS. Excitotoxicity in ALS. Neurology
1996;47:S221–7.
Levi G. Cerebral amino acid transport in vitro during development:
a kinetic analysis. Arch Biochem Bioph 1970;138:347–9.
Lopez-Corcuera B, Geerlings A, Aragon C. Glycine neurotrans-
mitter transporters: an update. Mol Membr Biol 2001;18:13–20.
Lopez-Corcuera B, Martinez-Maza R, Nunez E, Roux M, Suppli-
sson S, Aragon C. Differential properties of two stably
expressed brain-specific glycine transporters. J Neurochem
1998;71:2211–9.
Louvel E, Hugon J, Double A. Therapeutic advances in amyo-
trophic lateral sclerosis. Trends Pharmacol Sci 1997;18:196–
203.
Morrison BM, Morrison JH. Amyotrophic lateral sclerosis asso-
ciated with mutations in superoxide dismutase: a putative
mechanism of degeneration. Brain Res Rev 1999;29:121–35.
Morrow JA, Collie IT, Dunbar DR,Walker GB, Shahid M, Hill DR.
Molecular cloning and functional expression of the human

L. Raiteri et al. / NeuroToxicology 26 (2005) 883–892892
glycine transporter GlyT2 and chromosomal localisation of the
gene in the human genome. FEBS Lett 1998;439:334–40.
Mulder D, Kurland L, Offord K, Beard C. Familial adult motor
neuron disease: amyotrophic lateral sclerosis. Neurology
1986;36:511–7.
Nagao S, Kwak S, Kanazawa I. EAAT4, a glutamate transporter
with properties of a chloride channel, is predominantly loca-
lized in Purkinje cell dendrites, and forms parasagittal compart-
ments in rat cerebellum. Neuroscience 1997;78:929–33.
Nunez E, Lopez-Corcuera B, Vazquez J, Gimenez C, Aragon C.
Differential effects of the tricyclic antidepressant amoxapine on
glycine uptake mediated by the recombinant GLYT1 and
GLYT2 glycine transporters. Br J Pharmacol 2000;129:200–6.
Perry TL, Krieger C, Hansen S, Eisen A. Amyotrophic lateral
sclerosis amino acid levels in plasma and cerebrospinal fluid.
Ann Neurol 1990;28:12–7.
Przedborski S, Vila M, Jackson Lewis V. Neurodegeneration: what
is it and where are we? J Clin Invest 2003;111:3–10.
Raiteri L, Raiteri M. Synaptosomes still viable after 25 years of
superfusion. Neurochem Res 2000;25:1265–74.
Raiteri L, Raiteri M, Bonanno G. Coexistence and function of
different neurotransmitter transporters in the plasma membrane
of CNS neurons. Prog Neurobiol 2002;68:287–309.
Raiteri L, Stigliani S, Siri A, Passalacqua M, Melloni E, Raiteri M,
et al. Glycine taken up through GLYT1 and GLYT2 hetero-
transporters into glutamatergic axon terminals of mouse spinal
cord elicits release of glutamate by homotransporter reversal
and through anion channels. Biochem Pharmacol 2005;69:159–
68.
Raiteri M, Bonanno G, Marchi M, Maura G. Is there a functional
linkage between neurotransmitter uptake mechanisms and pre-
synaptic receptors? J Pharmacol Exp Ther 1984;231:671–7.
Raiteri M, Marchi M, Caviglia A. Studies on a possible functional
coupling between presynaptic acetylcholinesterase and high-
affinity choline uptake in the rat brain. J Neurochem
1986;47:1696–9.
Raiteri M, Sala R, Fassio A, Rossetto O, Bonanno G. Entrapping of
impermeant probes of different size into non-permeabilized
synaptosomes as a method to study presynaptic mechanisms.
J Neurochem 2000;74:423–31.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati
A, et al. Mutations in Cu/Zn superoxide dismutase gene are
associated with familial amyotrophic lateral sclerosis. Nature
1993;362:59–62.
Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR,
Drachman DB, et al. Abnormal excitatory amino acid meta-
bolism in amyotrophic lateral sclerosis. Ann Neurol 1990;28:
18–25.
Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW.
Selective loss of glial glutamate transporter GLT-1 in amyo-
trophic lateral sclerosis. Ann Neurol 1995;38:73–84.
Sepkuty JP, Cohen AS, Eccles C, Rafiq A, Behar K, Ganel R, et al.
A neuronal glutamate transporter contributes to neurotransmit-
ter GABA synthesis and epilepsy. J Neurosci 2002;22:
6372–9.
Shaw PJ, Eggett CJ. Molecular factors underlying selective vulner-
ability of motor neurons to neurodegeneration in amyotrophic
lateral sclerosis. J Neurol 2000;247:17–27.
Smith KE, Borden LA, Hartig PR, Branchek T, Weinshank RL.
Cloning and expression of a glycine transporter reveal coloca-
lization with NMDA receptors. Neuron 1992;8:927–35.
Wong PC, Pardo CA, Borchelt DR, LeeMK, Copeland NG, Jenkins
NA, et al. An adverse property of a familial ALS-linked SOD1
mutation causes motor neuron disease characterized by
vacuolar degeneration of mitochondria. Neuron 1995;14:
1105–16.
Yamada K, Watanabe M, Shibata T, Tanaka K, Wada K, Inoue Y.
EAAT4 is a post-synaptic glutamate transporter at Purkinje cell
synapses. Neuroreport 1996;7:2013–7.
Zafra F, Aragon C, Olivares L, Danbolt NC, Gimenez C, Storm-
Mathisen J. Glycine transporters are differentially expressed
among CNS cells. J Neurosci 1995;15:3952–69.
Zafra F, Gomeza J, Olivares L, Aragon C, Gimenez C. Regional
distribution and developmental variation of the glycine trans-
porters GLYT1 and GLYT2 in the rat CNS. Eur J Neurosci
1995;7:1342–52.
Zhou YG, Bennett ER, Kanner BI. The aqueous accessibility in the
external half of transmembrane domain I of the GABA trans-
porter GAT-1 is modulated by its ligands. J Biol Chem
2004;279:13800–8.




















