
Technological approaches to linguistic documentation and meta-documentation
Upload
khangminh22Category
view
3download
0
GaudiMM DocumentationRelease 0.0.8+1.g7374b62
Jaime Rodríguez-Guerra, Jean-Didier Maréchal
Dec 03, 2021


User guide
1 How to install 3
2 Quick usage 5
3 Input files 7
4 Output files 9
5 FAQ & Known issues 11
6 GaudiMM basics (start here!) 13
7 Tutorial: Your first GaudiMM calculation 17
8 Tutorial: Visualizing your first results 23
9 Developers guide 27
10 API documentation 31
11 Indices and tables 75
Python Module Index 77
Index 79
i

ii

GaudiMM Documentation, Release 0.0.8+1.g7374b62
GaudiMM, for Genetic Algorithms with Unrestricted Descriptors for Intuitive Molecular Modeling, helps to sketchnew molecular designs that require complex interactions.
User guide 1

GaudiMM Documentation, Release 0.0.8+1.g7374b62
2 User guide

CHAPTER 1
How to install
Quick steps:
1 - Download the latest stable copy of UCSF Chimera and install it with:
chmod +x chimera-*.bin && sudo ./chimera-*.bin
2 - Install Miniconda Python 2.7 Distribution for your platform and install it with:
bash Miniconda2*.sh
3 - Install gaudi with conda in a new environment called insilichem (or whatever name you prefer after the -nflag), using these custom channels (-c flags):
conda create -n insilichem -c omnia -c salilab -c insilichem -c conda-forge -c→˓bioconda -c tpeulen gaudi
4 - Activate the new environment as proposed:
conda activate insilichem
or
source activate insilichem
5 - Run it!
gaudi
1.1 Check everything is OK
If everything went OK, you will get the usage screen:
3

GaudiMM Documentation, Release 0.0.8+1.g7374b62
Usage: gaudi [OPTIONS] COMMAND [ARGS]...
GaudiMM: Genetic Algorithms with Unrestricted Descriptors for IntuitiveMolecular Modeling
(C) 2017, InsiliChemhttps://github.com/insilichem/gaudi
Options:--version Show the version and exit.-h, --help Show this message and exit.
Commands:prepare Create or edit a GAUDI input file.run Launch a GAUDI input file.view Analyze the results in a GAUDI output file.
4 Chapter 1. How to install

CHAPTER 2
Quick usage
Running GAUDI jobs is quite easy with gaudi.cli.gaudi_run:
gaudi run /path/to/some_file.gaudi-input
To learn how to create input files, go to Input files, and make sure to check the tutorials!
• GaudiMM basics (start here!)
• Tutorial: Your first GaudiMM calculation
After the job is completed, you can check the results with gaudi.cli.gaudi_view:
gaudi view /path/to/some_file.gaudi-output
You can choose between two molecular visualization tools: Chimera itself (using GaudiView extension), or our in-house GUI, GAUDInspect.
5
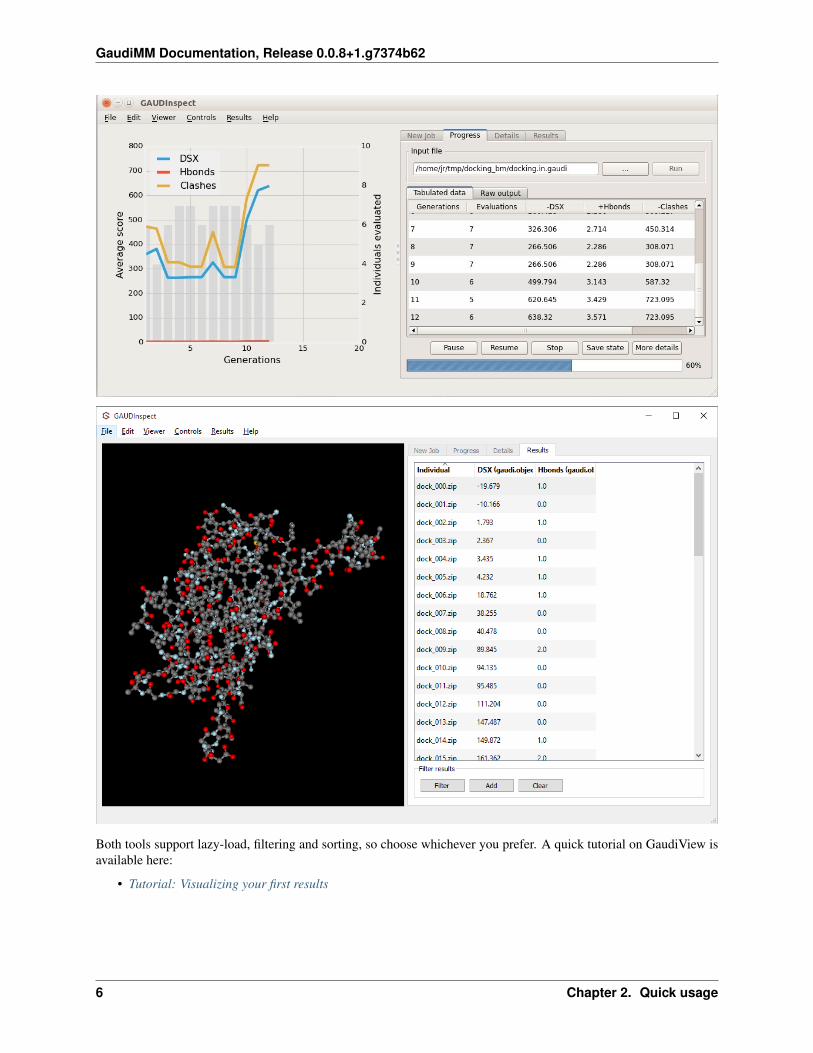
GaudiMM Documentation, Release 0.0.8+1.g7374b62
Both tools support lazy-load, filtering and sorting, so choose whichever you prefer. A quick tutorial on GaudiView isavailable here:
• Tutorial: Visualizing your first results
6 Chapter 2. Quick usage

CHAPTER 3
Input files
GaudiMM uses YAML-formatted files for both input and output files. YAML is a human-readable serialization format,already implemented in a broad range of languages. Input files must contain these five sections:
• output. Project options. Configure it to your liking
• ga. Genetic algorithm configuration. Normally, you don’t have to touch this, except maybe the number ofgenerations and population size.
• similarity. The similarity function to compare potentially redundant solutions.
• genes. List of descriptors used to define an individual
• objectives. The list of functions that will evaluate your individuals.
You can check some sample input files in the examples directory.
3.1 How to create GaudiMM input files
If you don’t mind installing GAUDInspect, it provides a full GUI to create GAUDI input files, step by step. Add genesand objectives, configure the paths, number of generations and population size, and run it. Simple and easy.
Note: The development of GAUDInspect is currently stalled.
However, you can also edit them manually, since they are just plain text files. Create a copy of one of the examplesand edit them to your convenience. While you can check the API documentation of gaudi.genes and gaudi.objectivesto find a list of available components, we really recommend checking the beginners tutorial:
• Tutorial: Your first GaudiMM calculation.
7

GaudiMM Documentation, Release 0.0.8+1.g7374b62
8 Chapter 3. Input files

CHAPTER 4
Output files
GAUDI output files are also YAML-formatted, so you can just open the file and read it in your favourite editor.However, that’s not probably what you were expecting to do.
For the time being, your best bet to understand how to analyze the results is to read the visualization tutorial:
• Tutorial: Visualizing your first results
Todo:
• Multi-objective solutions: Pareto front vs lexicographically sorted elite
9

GaudiMM Documentation, Release 0.0.8+1.g7374b62
10 Chapter 4. Output files

CHAPTER 5
FAQ & Known issues
5.1 Known issues
Most problems we have faced have to do with the installation and Chimera-Conda incompatibilities. Those are coveredin the pychimera documentation, so please check that list before raising an issue in this repository!
5.2 How can I cite GaudiMM? What licenses apply?
GaudiMM is scientific software, funded by public research grants. If you make use of GaudiMM in scientific publica-tions, please cite it. It will help measure the impact of our research and future funding!
@preamble{ " \newcommand{\noop}[1]{} " }@article{GaudiMM2017,
title = {GaudiMM: A Modular Multi-Objective Platform for Molecular Modeling},author = {Rodr{\'i}guez-Guerra Pedregal, Jaime and Sciortino, Giuseppe and Guasp,
→˓Jordi and Municoy, Mart{\'i} and Mar{\'e}chal, Jean-Didier},year = {\noop{2017}submitted},
}
GaudiMM itself is licensed under Apache License 2.0, but includes work from other developers, whose licenses apply.Please check the LICENSE file in the root directory for further details.
5.3 How many generations / Which population size should I pick?
This depends on the complexity of your search space (related to the number and type of genes in use), and theevaluation power of the chosen objectives. A simple job would work OK with 50 generations and populations of 100individuals, while more complex ones would require 500 generations for populations of 1000 indivuals. This alsodepends on the values of mu and lambda parameters.
11

GaudiMM Documentation, Release 0.0.8+1.g7374b62
5.4 The output produces a lot of similar results and I want to focuson diversity
You should play with the cutoff value of the RMSD similarity section. If the RMSD of two potentially similarsolutions is under the cutoff, one of them is discarded. However, this is only applied if the score of two solutions arethe same; ie, if there is a draw.
To force draws, one can reduce the number of decimal positions returned by every objective, which is controlled bythe precision parameter. It is setup globally in the output section, but can also be overriden by any objective.By default, precision is 3. Also, if you are using the gaudi.genes.search.Search gene, make sure itsprecision parameter is small enough so you don’t lose exploration efforts in too similar positions.
5.5 The output produces very different results and I am interested inclustered solutions
In this case, one should increase the precision value (both globally and in the gaudi.genes.search.Searchgene, if it applies) and reduce the RMSD similarity cutoff.
5.6 What is the difference between atom_names and atom_types insome objectives or genes?
atom_names refers to the name attribute of chimera.Atom objects, while atom_types is applied withidatmType attributes. Normally, the name attribute is picked directly from the molecule file (PDB, mol2), whilethe idatmType is assigned algorithmically by UCSF Chimera.
Tip: Any further questions? Feel free to submit your inquiries to our issues page!
12 Chapter 5. FAQ & Known issues

CHAPTER 6
GaudiMM basics (start here!)
The main strength of GaudiMM is, possibly, its flexible approach to solve molecular modeling problems. However,flexibility does not come without a price: the learning curve can be somewhat steep for beginners. In this tutorial wewill try to smooth that curve out.
GaudiMM makes a big effort to clearly separate exploration from evaluation. As such, both stages are configured indifferent sections of the input file: genes and objectives.
6.1 Exploration and genes
Most molecular modeling problems can be explained using search space analogies. This is, given a problem, onecan identify the dimensions it must explore to find adequate solutions for that problem. That n-dimensional spacecan be comprised of one or more (bio)chemical structures and their respective spatial variability. Modifying thetopology and/or composition of such structures would be equivalent to moving along the (bio)chemical axes of suchsearch space, while modifying their atomic positions would be equivalent to moving along the cartesian and/or internalcoordinates axes. Thus, visiting different regions of the whole search space means creating new candidate solutions tothe problem. . . We will call this stage exploration, and it is detailed in the genes section of the input file. If you wantto know why they are called genes, check the Genetic Algorithms Primer in the Developers guide.
The more important gene is, almost always, the gaudi.genes.molecule.Molecule gene. This is used to load(bio)chemical structures, such as proteins, peptides and small cofactors. Normally they are straight PDB or Mol2 files,but can also be more complex constructs. As of now, it is the only one capable of providing topologies. The othergenes are responsible of applying changes to the coordinates of that initial structure. Some examples include torsionangles of rotatable bonds (gaudi.genes.torsion.Torsion gene), translating and rotating a Molecule in the3D space (gaudi.genes.search.Search gene), or importing the atomic positions of a molecular dynamicstrajectory frame (gaudi.genes.trajectory.Trajectory gene).
6.2 Evaluation and objectives
However, there is no guarantee that, given a new position on such search space, that hypothetical solution is actuallygood enough to solve the problem. To assess the quality of a random solution, it must be evaluated with some criteria
13

GaudiMM Documentation, Release 0.0.8+1.g7374b62
or objectives. All objectives have something in common: they take a candidate solution (ie, a point of the searchspace) and measure some property to return a numeric score. Some examples include the distance between two givenatoms (gaudi.objectives.distance.Distance objective), the steric clashes of the molecules (gaudi.objectives.contacts.Contacts objective) or the forcefield energy of the system (gaudi.objectives.energy.Energy objective).
6.3 Examples of application
Now we got the basics covered, we can start to think of how we could solve some standard (and not so standard)molecular modeling problems with GaudiMM.
6.3.1 Docking
Docking problems devote to finding the correct orientation and position of a small molecule (the ligand) within thecavity of a bigger one (normally, a protein). For the exploration stage, at least three genes must be defined:
• A gaudi.genes.molecule.Molecule gene to load the file corresponding to the protein structure.
• Another gaudi.genes.molecule.Molecule gene to load the ligand itself.
• A gaudi.genes.search.Search gene to translate and rotate the ligand around the protein surroundings.
This is all you need to move a rigid ligand around a protein. However, most of the time you want to implement somekind of internal flexibility, such as torsions in the ligand or some residues sidechains. To do that, add these to yourgenes section:
• A gaudi.genes.torsion.Torsion gene with the ligand as target to implement dihedral angle torsionsalong the rotatable bonds of the ligand.
• A gaudi.genes.rotamers.Rotamers gene with some residues of the protein as target. This will ran-domly modify the dihedral torsions of the sidechains of the specified residues according to the angles providedby the rotamer library (defaults to Dunbrack’s).
There is also a gaudi.genes.mutamers.Mutamers gene that allows you to mutate residues in addition torotameric exploration. This will provide some traversal along the biochemical axis of your protein, but careful, becausethe current implementation is RAM hungry!
The evaluation stage can be comprised of several objectives, but normally you’d want to:
• Minimize steric clashes with gaudi.objectives.contacts.Contacts.
• Maximize hydrophobic patches with another gaudi.objectives.contacts.Contacts entry.
• Use a proper docking scoring function, such as gaudi.objectives.ligscore.LigScore or gaudi.objectives.dsx.DSX. Depending on the one you choose, it should be minimized (the usual case) or max-imized.
6.3.2 Covalent docking
Not all docking studies consider a free ligand. Sometimes the ligand is anchored to some part of the protein. Whilethere’s no specific gene to implement a covalent bond (for now, at least), you can mimick it with a Search genewith radius=0 and rotate=True. The origin of this null search sphere should be set to the atom that is partof the covalent bond, so you would have add an extra atom in one of the molecules. Also, we recommend settingan gaudi.objectives.angle.Angle objective between the involved atoms in the covalent bond so that theresulting rotation matches the expected geometry of the new bond (109.5º for sp3, 120º for sp2, 180º for sp1).
14 Chapter 6. GaudiMM basics (start here!)

GaudiMM Documentation, Release 0.0.8+1.g7374b62
6.3.3 Naked metal ions docking
If instead a small organic molecule you choose to load a naked metal ion. . . would it work? Well, in most dockingprograms probably not, but with GaudiMM the answer is. . . it depends!
It depends on the objectives you choose. Since we ship a gaudi.objectives.coordination.Coordination objective that can deal with coordination geometries, you can dock naked metal ions in any proteinor peptide.
6.3.4 Competitive docking
Since you can instantiate as many gaudi.genes.molecule.Molecule genes as you want, nothing preventsyou from adding more ligand molecules at the same time. They will compete to find its place in the protein(s).Just remember to replicate any dependent genes (gaudi.genes.search.Search, gaudi.genes.torsion.Torsion and so on) accordingly. Also, why not two proteins which the ligand should choose from (but it’s true thereare finer ways to assess that)?
6.3.5 Hacking Molecule genes for complex studies
In addition to loading molecular structure files, the gaudi.genes.molecule.Molecule does a couple of extrathings. The path parameter can be set to two different values:
• The path to a PDB or Mol2 file (or any format that Chimera can open). This is the standard behaviour. It willload the structure and that’s it.
• The path to a directory, whose contents determine the final behaviour:
A) If the directory contains molecule files, GaudiMM will choose one of them randomly for each case.
B) If the directory contains subdirectories which, in turn, contain molecules files, GaudiMM will sort thosesubdirectories by name and then pick one molecule from each, in that order. The chosen molecules willbe chained linearly as specified in the accompanying *.attr files. Perfect for drug design, since you canfill a couple of directories with different alkanes to link a cofactor
Case A allows GaudiMM to test a library of compounds against certain criteria: ie, virtual screening!
Case B makes drug design studies possible. If you want to test different linker molecules to anchor a cofactor to aprotein, you can use a two-directories scheme: one directory would contain the aforementioned linkers, and the otherone just the cofactor (it’s fine it there’s only one molecule in one directory).
6.4 Peptide folding & Conformational analysis
Loading an unfolded peptide with gaudi.genes.molecule.Molecule is pretty easy. Just specify the path.Then, if you apply a gaudi.genes.torsion.Torsion gene on such peptide, but considering only the bondsinvolving an alpha-carbon, you can effectively explore the conformational space of its folding. Use gaudi.genes.rotamers.Rotamers to implement sidechain flexibility.
For the evaluation, you can use the gaudi.objectives.energy.Energy objective to assess the forcefieldenergy as provided by OpenMM GPU calculations. This type of study could be considered some sort of highlyexplosive metadynamics.
6.4. Peptide folding & Conformational analysis 15

GaudiMM Documentation, Release 0.0.8+1.g7374b62
6.4.1 Homology modeling
In the same fashion, given an unfolded peptided or protein segment, you could apply the same Torsion scheme toexplore different conformations. A hypothetical objective could be devised to calculate the RMS deviation of the newfragment and a reference one, which then the GA would minimize to obtain a similar structure.
6.4.2 Conformational analysis
Another possible variation of this scheme is to impose geometric guides as additional objectives, like gaudi.objectives.distance.Distance, gaudi.objectives.angle.Angle or gaudi.objectives.coordination.Coordination. The resulting calculation could be regarded as a restrained conformationalanalysis, very useful for finding initial structures of unparametrized small molecules you want to study with higherlevels of theory, such as QM.
6.4.3 Trajectory analysis
GaudiMM features a gaudi.genes.trajectory.Trajectory gene capable of importing frames of MDmovies and apply them to the corresponding gaudi.genes.molecule.Molecule instance. This way, all theobjectives are available as trajectory analysis tools, maybe not present in other specific software. For example, findingcoordination geometries of a given metal ion along a molecular mechanics simulation.
16 Chapter 6. GaudiMM basics (start here!)

CHAPTER 7
Tutorial: Your first GaudiMM calculation
Running a GaudiMM calculation is easy. All you need is a simple command:
gaudi run input_file.yaml
The question is. . . how do I create that input_file.yaml.
7.1 How to create an input file from scratch
Input files in GaudiMM are formatted with YAML, which follows readable conventions that are still parseable bycomputers. To easily edit YAML files, we recommend using a text editor that supports syntax highlighting, such asSublime Text 3, Atom or Visual Studio Code. If you use .yaml as the extension of the input filename, the editor willcolorize the text automatically. Else, you can always configure it manually. Check the docs of your editor to do so. Ifyou don’t want syntax highlighting it’s fine, GaudiMM will work the same.
Input files must contain the sections mentioned above: output, ga, similarity, genes, and objectives. Each of thissections it’s defined with a colon and a newline, and its contents will be inside an indented block. For readability, Iusually insert a blank line between sections.
output:contents of output
ga:contents of ga
similarity:contents of similarity
genes:contents of genes
objectives:contents of objectives
17

GaudiMM Documentation, Release 0.0.8+1.g7374b62
Note: The API documentation of gaudi.parse.Settings contains the full list of parameters for outputand ga sections. Programmatically defined default values are always defined in gaudi.parse.Settings.default_values. The appropriate types and whether they are required or not are defined in gaudi.parse.Settings.schema. GaudiMM will check if the submitted values conform to these rules and report any possiblemistakes.
7.1.1 The output section
This section governs how the results and reports will be created. Everything is optional, since each key has a defaultvalue, but it is preferrable to at least specify the name of the job (otherwise, it will be set to five random characters),and the path where the result files will be written. Like this:
output:name: some_examplepath: results
Note: All the relative paths in GaudiMM are relative to the location of the input file, not the working directory. Toease this difference, we recommend running the jobs from the same folder where the input file is located. Of course,you can always use absolute paths.
7.1.2 The ga section
This section hosts the parameters of the Genetic Algorithm GaudiMM uses. Unless you know what you are doing, theonly values you should modify are population and generations. To see the appropriate values, refer to FAQ& Known issues. For example:
ga:population: 200generations: 100
7.1.3 The similarity section
This section contains the parameters to the similarity operator, which, given two individuals with the same fitness,whether they can be considered the same solution or not. This section is deliberatedly loose: you define the Pythonfunction to call, together with its positional and keyword arguments.
For the time being, the only similarity function we ship is based on the RMSD of the two structures: gaudi.similarity.rmsd(). The arguments are which Molecule genes should be compared and the RMSD thresholdto consider whether they are equivalent or not.
similarity:module: gaudi.similarity.rmsdargs: [[Ligand], 1.0]kwargs: {}
18 Chapter 7. Tutorial: Your first GaudiMM calculation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
7.1.4 The genes section
This section describes the components of the exploration stage of the algorithm; ie, the features of each Individual inthe population. While the previous sections were dictionaries (this is, a collection key-value pairs), the genes andobjectives section is actually a list of dictionaries. As a result, you need to specify them like this:
genes:- name: Protein
module: gaudi.genes.moleculepath: /path/to/protein.mol2
- name: Torsionmodule: gaudi.genes.torsiontarget: Ligandflexibility: 360
Notice the dash - next to name. This, and the extra indentation, define a list. Each element of this list is a new gene.Each gene must include two compulsory values:
• name. A unique identifier for this gene. If you add two genes with the same name, GaudiMM will complain.
• module. The Python import path to the module that contains the gene. All GaudiMM builtin genes are locatedat gaudi.genes.
All other parameters are determined by the chosen gene. Check the corresponding documentation for each one!
Note:
How do I know which genes to use?Unless you code a gene of your own to replace it, you will always need one or moregaudi.genes.molecule.Molecule genes. Then, choose the flexibility models you want to implement on topof such molecule. Several examples are provided in GaudiMM basics (start here!).
7.1.5 The objectives section
Like the genes section, the objectives section is also a list of dictionaries, so they follow the same syntax:
- name: Clashesmodule: gaudi.objectives.contactswhich: clashesweight: -1.0probes: [Ligand]radius: 5.0
- name: LigScoremodule: gaudi.objectives.ligscoreweight: -1.0proteins: [Protein]ligands: [Ligand]method: pose
In addition to the required name and module parameters, each objective needs a weight parameter. If set to 1.0,the algorithm will maximize the score returned by the objective; if set to -1.0, it will be minimized. Theoretically,any other positive or negative float will work, but stick to the convention of using 1.0 or -1.0.
7.1. How to create an input file from scratch 19

GaudiMM Documentation, Release 0.0.8+1.g7374b62
Any other parameters present in an objective are responsibility of that objective, and are specified in its correspondingdocumentation.
That’s it! Now save it with a memorable filename and run it!
7.2 How to run your input file
Let’s get back to the beginning of the tutorial: all you need to do is typing:
gaudi run input_file.yaml
If everything is fine, you’ll see the following output in the console:
$> gaudi run input_file.yaml
.g8"""bgd db `7MMF' `7MF'`7MM"""Yb. `7MMF'.dP' `M ;MM: MM M MM `Yb. MMdM' ` ,V^MM. MM M MM `Mb MM `7MMpMMMb.pMMMb. `7MMpMMMb.→˓pMMMb.MM ,M `MM MM M MM MM MM MM MM MM MM MM→˓MMMM. `7MMF' AbmmmqMA MM M MM ,MP MM MM MM MM MM MM→˓MM`Mb. MM A' VML YM. ,M MM ,dP' MM MM MM MM MM MM→˓MM`"bmmmdPY .AMA. .AMMA.`bmmmmd"' .JMMmmmdP' .JMML..JMML JMML JMML..JMML JMML
→˓JMML.--------------------------------------------------------------------------------------→˓----GaudiMM: Genetic Algorithms with Unrestricted Descriptors for Intuitive Molecular→˓Modeling2017, InsiliChem · v0.0.2+251.g122cdf0.dirty
Loaded input input_file.yamlLaunching job with...
Genes: Protein, Ligand, Rotamers, Torsion, SearchObjectives: Clashes, Contacts, HBonds, LigScore
After the first iteration is complete, the realtime report data will kick in:
gen progress nevals speed eta avg→˓ std min→˓ max0 4.76% 20 1.25 ev/s 0:16:34 [ 3.080e+03 -2.690e+02 7.500e-01 1.→˓179e+03] [ 1.027e+03 7.200e+01 8.292e-01 4.964e+02] [ 584.881 -398.517→˓ 0. 144.78 ] [ 4.753e+03 -1.053e+02 3.000e+00 2.066e+03]1 9.52% 60 1.28 ev/s 0:16:32 [ 2.787e+03 -2.659e+02 1.400e+00 9.→˓142e+02] [ 1198.699 98.675 1.393 484.309] [ 415.912 -398.517→˓ 0. 16.45 ] [ 4538.766 -71.562 5. 1854.63 ]
The first time you see this it might result too confusing, especially if the terminal wraps long lines. Let’s describe eachtab-separated column:
• gen. The current generation.
• progress. Percentage of completion of the job. This is estimated with the expected number of operations:(𝑔𝑒𝑛𝑒𝑟𝑎𝑡𝑖𝑜𝑛𝑠+ 1) * 𝑙𝑎𝑚𝑏𝑑𝑎_ * (𝑐𝑥𝑝𝑏+𝑚𝑢𝑡𝑝𝑏)
20 Chapter 7. Tutorial: Your first GaudiMM calculation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
• nevals. Number of evaluations performed in current generation.
• speed. Estimated number of evaluations per second. This does not take into account the time spent in thevariation stage.
• eta. Estimated time left.
• avg. Average of all the fitness values reported by each objective in the current generation. They are listed in theorder given in the input file, and also reflected above, after the Launching job with. . . line.
• std. Same as avg, but for the standard deviation.
• max. The maximum fitness value reported by each objective in the current generation.
• min. Same as above, but for the minimum value.
If the setting check_every in the output section is greater than zero, GaudiMM will dump the current populationevery check_every generations. That way, you can assess the progress visually along the simulation.
Also, if you feel that the algorithm has progressed enough to satisfy your needs, you can cancel it prematurely withCtrl+C. GaudiMM will detect the interruption and offer to dump the current state of the simulation:
^C[!]
Interruption detected. Write results so far? (y/N):
Answer y and wait a couple of seconds while GaudiMM writes the results. To analyze them, check the followingtutorial:
• Tutorial: Visualizing your first results
7.2. How to run your input file 21

GaudiMM Documentation, Release 0.0.8+1.g7374b62
22 Chapter 7. Tutorial: Your first GaudiMM calculation

CHAPTER 8
Tutorial: Visualizing your first results
When a GaudiMM job finishes correctly, you will find the results in the directory specified at output.path. Thereyou will find:
• *.gaudi-input. An (updated) copy of the input file used in this job, for reproducibility purposes.
• *.gaudi-log. A detailed log of the simulation, for debugging purposes.
• *.gaudi-output. A YAML file with the scores and paths of the solutions.
• (Potentially lots of) *.zip. As many as the elite population size. Each zip contains an Individual, which meansthat you will find molecule files (normally, mol2 files), as well as the allele metadata of the rest of the genes.
To analyze the results, currently the best tool is our own extension for UCSF Chimera, GaudiView. After installing it,you can type the following:
gaudi view *.gaudi-output
, and hopefully it will open Chimera and GaudiView to load the output file. If it doesn’t work (please, if that’s thecase, report it in the issues page!), you can always UCSF Chimera, fire up GaudiView from Tools> InsiliChem>GAUDIView and browse the file manually.
23
GaudiMM Documentation, Release 0.0.8+1.g7374b62
One way or another, you will end up seeing the following dialog:
24 Chapter 8. Tutorial: Visualizing your first results
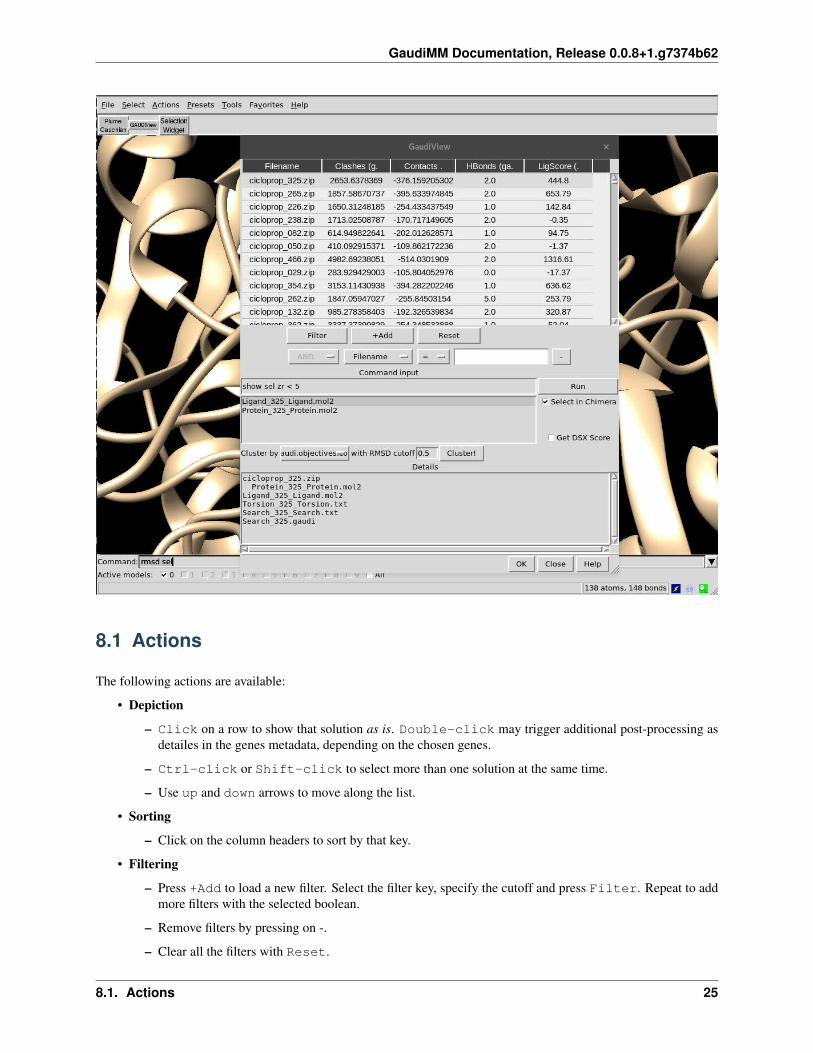
GaudiMM Documentation, Release 0.0.8+1.g7374b62
8.1 Actions
The following actions are available:
• Depiction
– Click on a row to show that solution as is. Double-click may trigger additional post-processing asdetailes in the genes metadata, depending on the chosen genes.
– Ctrl-click or Shift-click to select more than one solution at the same time.
– Use up and down arrows to move along the list.
• Sorting
– Click on the column headers to sort by that key.
• Filtering
– Press +Add to load a new filter. Select the filter key, specify the cutoff and press Filter. Repeat to addmore filters with the selected boolean.
– Remove filters by pressing on -.
– Clear all the filters with Reset.
8.1. Actions 25

GaudiMM Documentation, Release 0.0.8+1.g7374b62
• Commands
– Every time you change the selected row, the command specified in the command input panel will be run.You can force it by pressing Enter key or click on Run.
– Combine the command line with the dynamic selection panel to run commands like show sel zr <5 to depict the surrounding residues of docking poses.
– Multiple commands can be chained with ;.
• Clustering
– First, choose the column to use as energy criteria.
– Select the RMSD cutoff. If two solutions have RMSD bigger than this value, they won’t be consideredsimilar and thus not clustered together.
– Press Cluster!. Since this method requires to load all solutions at once, it can take some time if youhave a lot of solutions.
– When it’s done, a new column Cluster will be available at the end of the table. Sort by that column togroup them together and Shift-click to depict the full cluster.
When you are done with the analysis, you can exit GaudiView in different ways:
• Click OK. The depicted solutions at that moment won’t be closed, and thus available for further post-processingin Chimera. Every other solution previously loaded will be removed from the canvas and closed.
• Click Close or upper-corner X. All solutions, visible or not, will be removed from canvas and closed.
Error: There’s a known bug in GaudiView that may trigger a non-critical exception if you filter the solutions andthen try to sort them again without selecting a new row beforehand. You can dismiss the dialog and simply clickin any row to avoid this bug. It will be fixed soon, promise!
26 Chapter 8. Tutorial: Visualizing your first results

CHAPTER 9
Developers guide
9.1 Introduction to Genetic Algorithms
The GA in GaudiMM stands for Genetic Algorithm, a search heuristic inspired by natural selection that is used foroptimization processes.
Genetic Algorithms use a biologicist terminology. Each candidate solution to the problem is considered an individual,which is part of the so-called population (the set of all candidate solutions).
The initial population is generated from scratch, almost always randomly. These individuals also comprise the firstgeneration of the evolution process. As in nature, only the fittest survive. The survival process is simulated with anevaluation function, that tests them against the optimization variable(s). This is called selection.
• How do we create an individual? With one or more genes, naturally. Genes describe how an individual shouldlook like, of course!
• How do we evaluate that individual? With one or more objectives.
Also, as in nature, the fittest individuals are allowed to mate (exchange their defining values), and mutate (sponta-neously modify their own defining values). This adds some more variability to the process, and that is key to survival.
After a number of generations repeating the process of selection, choosing the fittest over the weakest, we will obtainbetter and better solutions to our problem. Since it’s all heuristics, it’s up to us to stop at some point. We won’tprobably get the solution, but we can live with pretty good ones, right?
9.1.1 The One Max Problem
Ok, that was a lot of biology and we are trying to code, I get it. Let’s explain a classical GA example, the trivial OneMax Problem, adapted from the original deap documentation.
In this problem, we have a list of integers that can be either 0 or 1, and we want to obtain a list full of 1s. So, in thisexample, we have individuals defined by a single gene and evaluated with a single objective.
We build individuals with the gene onemax:
27

GaudiMM Documentation, Release 0.0.8+1.g7374b62
import random.randintdef onemax(size=5):
return [random.randint(0, 1) for i in range(size)]
adam = onemax(5) # returns [0, 0, 0, 1, 0]eve = onemax(5) # returns [0, 1, 1, 0, 0]
The objective is also trivial. We have to maximize the sum of the numbers inside a given individual:
def evaluate(individual):return sum(individual)
So. . . which is one is fittest, adam or eve? Obviously, eve:
evaluate(adam) # returns 1evaluate(eve) # returns 2
Of course, an initial population is usually larger! At least, a hundred individuals. With such a trivial case, given a bigenough population, we may obtain the solution in the first generation by pure change. However, we must not rely onthe initial population as the only diversity source.
Additional diversity is achieved with the mutation and mating operations, implemented as additional functions:
def mate(a, b):""" Let a and b mate, in hope of fitter children """i = crossover_point = random.random() * min(len(a), len(b))c, d = a[:], b[:]c[:i], d[i:] = d[:i], c[i:]return c, d
def mutate(individual, probability):""" Spontaneous mutation at random places can result in a fitter individual """return [random.randint(0, 1) for i in individual if random.random() < probability]
Let’s see how this is useful:
cain, abel = mate(adam, eve)# cain = [ 0, 1, 1, 1, 0 ]# abel = [ 0, 0, 0, 0, 0 ]evaluate(cain) # returns 3evaluate(abel) # returns 0
See? adam and eve gave birth to cain and abel. cain had luck and inherited the good parts, while abel. . .Well, he was not that lucky. In the next selection process, cain will be selected over abel, and probably over itsown father adam. Now, the population (cain and eve) as a whole is fitter, with an average fitness of 2.5. That’shigher than the average in the previous generation (1.5). Evolution!
Mutation works similarly:
enoch = mutate(cain)# enoch = [ 1, 1, 1, 1, 0]seth = mutate(eve)# seth = [ 0, 0, 1, 0, 0]
Take into account that mutations can be beneficial, like in the case of enoch, but also detrimental, as in the case ofseth. Some of them will contribute to evolution, and some of them not. Lucky ones will be selected, the others,discarded.
28 Chapter 9. Developers guide

GaudiMM Documentation, Release 0.0.8+1.g7374b62
By the way, deap already defines some mutation and mating operators for you that will work in most cases. So,hopefully, this part will be trivial.
And that’s it! Deap does the rest! So, to sum up, you only need to worry about:
• How to define your individuals.
• How to evaluate them.
• How to implement mutation and mating (normally, with deap built-in operators).
If you want to know more about Deap and Genetic Algorithms, go check their documentation. It’s great!
9.2 Our implementation
GaudiMM is built as an extensible and highly modular Python platform. Although the main focus is Chemistry andmolecular design, you can use your own genes and objectives. You can think of GaudiMM as a new API for deap thatprovides an object-oriented interface to easily create new individuals and objectives.
In deap an individual can be any Python object (check their overview and GA examples), which is a very versatileapproach, but it tends to be very limited when your individual gets complex. For example, if an individual needs to bedefined by several genes with different mutation strategies.
In GaudiMM, each individual is a gaudi.base.Individual, which is a very (bio)fancy name for a list ofgenes. To create a gene, you just subclass gaudi.genes.GeneProvider and define the needed methods:express, unexpress, mutate, and mate. The gaudi.base.Individual class then provides some wrappermethods that call the respective counterparts in each gene.
To evaluate the fitness of an individual, you must first define the set of evaluation functions. Each function is calledobjective, and you keep them inside a gaudi.base.Environment.
To create a new objective, you have to subclass gaudi.objectives.ObjectiveProvider, which pro-vides a very simple interface: evaluate. Define your function there, and that’s it!
Todo:
• Tutorial: How to create your own gene
• Tutorial: How to create your own objective
9.2. Our implementation 29

GaudiMM Documentation, Release 0.0.8+1.g7374b62
30 Chapter 9. Developers guide

CHAPTER 10
API documentation
The GaudiMM package is comprised of several core modules that establish the base architecture to build an extensibleplatform of molecular design.
The main module is gaudi.base, which defines the gaudi.base.Individual, whose instances represent thepotential solutions to the proposed problem. Two plugin packages allow easy customization of how individuals aredefined (gaudi.genes) and how they are evaluated (gaudi.objectives). Additionally:
• gaudi.algorithms is the place to look for the actual GA implementation
• gaudi.box is a placeholder for several small functions that are used across GaudiMM.
• gaudi.exceptions defines custom exceptions.
• gaudi.parse contains parsing utilities to retrieve the configuration files.
• gaudi.parallel contains helpers to deal with parallel GaudiMM jobs.
• gaudi.plugin holds some magic to make the plugin system work.
• gaudi.similarity defines the diversity enhancers.
10.1 gaudi.cli
This package holds all the CLI scripts and wrappers provided by GAUDI.
10.1.1 gaudi.cli.gaudi_cli
gaudi.cli.gaudi_cli is the CLI entry point for all GaudiMM scripts.
Available commands:
• run
• view
• prepare
31

GaudiMM Documentation, Release 0.0.8+1.g7374b62
gaudi.cli.gaudi_cli.echo_banner()
gaudi.cli.gaudi_cli.load_chimera(nogui=True)
gaudi.cli.gaudi_cli.test_import(command, module)
gaudi.cli.gaudi_cli.timeit(func, *args, **kwargs)
10.1.2 gaudi.cli.gaudi_run
gaudi.cli.gaudi_run is the main hub for launching GAUDI jobs.
It sets up the configuration environment needed by DEAP (responsible for the GA) and ties it up to the GAUDI customclasses that shape up the invididuals and objectives. All in a loosely-coupled approach based on Python modules calledon-demand.
Usage. Simply, type:
gaudi run /path/to/job.gaudi-input
gaudi.cli.gaudi_run.enable_logging(path=None, name=None, debug=False)Register loggers and handlers for both stdout and file
gaudi.cli.gaudi_run.launch(cfg)Runs a GAUDI job
Parameters cfg (gaudi.parse.Settings) – Parsed YAML dict with attribute-like access
gaudi.cli.gaudi_run.main(cfg, debug=False)Starts a GAUDI job
Parameters
• cfg (str or gaudi.parse.Settings) – Path to YAML input file or an alreadyparsed YAML file via gaudi.parse.Settings class
• debug (bool, optional, default=False) – Whether to enable verbose loggingor not.
gaudi.cli.gaudi_run.unbuffer_stdout()
10.2 gaudi.genes
These are the built-in genes in GAUDI. You can also build your own, but these are ready to use.
10.2.1 Molecule gene
This gene implements a wrapper around Chimera.molecule objects to expand its original features, such as appendingnew molecules.
This allows to build new structures with a couple of building blocks as a starting point, as well as keeping severalligands as different potential solutions to the essay (think about multi-molecule alternative docking). The user canalso request more gaudi.genes.molecule instances for the genome of the individual, resulting in a competitivemulti-docking essay.
To handle all this diversity, each construction is cached the first time is built.
This class is a dependency of most of the other genes (and even objectives), so it will be requested almost always.
32 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
class gaudi.genes.molecule.Compound(molecule=None, origin=None, seed=0.0, **kwargs)Bases: object
Wraps chimera.Molecule instances and allows to perform copies, appending new fragments, free placement andextended attributes.
Parameters
• molecule (chimera.Molecule or str, optional) – The chimera.Moleculeobject to wrap, or
’dummy’ (for a empty molecule), or
The path to a mol2 file that will be parsed by Chimera to return a valid chimera.Moleculeobject.
• origin (3-tuple of float, optional) – Coordinates to the place where themolecule should be placed
• seed (float, optional) – Random seed, used by the vertex chooser on appending.
• optional (kwargs,) – Additional parameters that should be injected as attributes
mol
Type chimera.Molecule
donorIf self were to be bonded to another Compound, this atom would be the one involved in the bond. Bydefault, first element in chimera.Molecule.atoms
Type chimera.Atom
acceptorIf another Compound wanted to be bonded to self, this atom would be the one involved in the bond. Bydefault, last element in chimera.Molecule.atoms
Type chimera.Atom
originDefault location of mol in the 3D canvas.
Type 3-tuple of float
seedRandomness seed for vertex computation.
Type float
rotatable_bondsBonds that can be torsioned
Type list of chimera.Bond
nonrotatableBonds that, although possible, should not be torsiond. Ie, fixed bonds.
Type list of chimera.Bond
built_atomsMemo of already built_atoms
Type list of chimera.Atom
10.2. gaudi.genes 33

GaudiMM Documentation, Release 0.0.8+1.g7374b62
Notes
Todo: Instead of a molecule parameter overload, think of using equivalent classmethods.
add_dummy_atom(where, name=’dum’, element=None, residue=None, bonded_to=None, se-rial=None)
Adds a placeholder atom at the coordinates specified by where
Parameters
• where (chimera.Atom or 3-tuple of float) – Coordinates of target loca-tion. A chimera.Atom can be supplied, in which case its coordinates will be used (via.coord())
• name (str, optional) – Name for the new atom
• element (chimera.Element, optional) – Element of the new atom
• residue (chimera.Residue, optional) – Residue that will incorporate the newatom
• bonded_to (chimera.Atom, optional) – Atom that will form a bond with newatom
• serial (int) – Serial number that will be assigned to atom
add_hydrogens()Add missing hydrogens to current molecule
append(molecule)Wrapper around attach to add a new molecule to self, using self.acceptor as bonding atom.
Parameters molecule (Compound) –
apply_pdbfix(pH=7.0)Run PDBFixer and replace original molecule with new one
attach(molecule, acceptor, donor)Call join to bond molecule to self.mol and updates attributes.
Parameters
• molecule (Compound) – The molecule that will be attached to self
• acceptor (chimera.Atom) – Atom of self that will participate in the new bond
• donor (chimera.Atom) – Atom of molecule that will participate in the new bond
Notes
Note: After joining the two molecules together, we have to update the attributes of self to match the newmolecular reality. For example, the atom participating in the bond will not be available for new bonds (weare forcing linear joining for now), so the new donor will be inherited from molecule, before deleting theobject.
destroy()Removes mol from canvas, deletes it and then, deletes itself
34 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
join(molecule, acceptor, donor, newres=False)Take a molecule and bond it to self.mol, copying the atoms in the process so they’re contained in the samechimera.Molecule object.
Parameters
• molecule (Compound) – The molecule that will be attached to self
• acceptor (chimera.Atom) – Atom of self that will participate in the new bond
• donor (chimera.Atom) – Atom of molecule that will participate in the new bond
• newres (bool) – If True, don’t reuse acceptor.residue for the new molecule, and createa new blank one instead.
Returns built_atoms – Maps original atoms in molecules to their new counterparts in self.mol.
Return type dict
Notes
Note: Chimera does not allow bonds between different chimera.Molecule objects, so firstly, we have tocopy the atoms of molecule to self.mol and, only then, make the joining bond.
It traverses the atoms of molecule and adds a copy of each of them to self.mol using chimera.molEdit.addAtom in the same spot of space. All the bonds are preserved and, finally, bond the twomolecules.
The algorithm starts by adding the bonding atom of molecule (donor), to the sprouts list. Then, theloop starts:
while sprouts contains atoms:sprout = sprouts.pop(0)copy sprout to self.molfor each neighbor of sprout
copy neighbor to self.molif neighbor itself has more than one neighbor (ie, sprout)
add neighbor to sprouts
Along the way, we have to take care of already processed sprouts, so we don’t repeat ourselves. That’swhat the built_atoms dict is for.
Also, instead of letting addAtom guess new serial numbers, we calculate them beforehand by computingthe highest serial number in self.mol prior to the additions and then incremeting one by one on aper-element basis.
Todo: This code is UGLY. Find a better way!
parse_attr()
place(where, anchor=None)Convenience wrapper around translate, supplying default
Parameters
• where (3-tuple of float, or chimera.Atom) – Coordinates of destination.If chimera.Atom, use supplied coord()
10.2. gaudi.genes 35

GaudiMM Documentation, Release 0.0.8+1.g7374b62
• anchor (chimera.Atom) – The atom that will guide the translation.
Notes
Note: Anchor atoms are called that way in the API because I picture them as the one we pick with ourhands to drag the molecule to the desired place.
place_for_bonding(target, anchor=None)Translate self.mol to a covalent distance of target atom, with an adequate orientation.
Parameters
• target (chimera.Atom) – The atom we are willing to bond later on.
• anchor (chimera.Atom, optional) –
prepend(molecule)Wrapper around attach to add a new molecule to self, using self.donor as bonding atom.
Parameters molecule (Compound) –
set_vdw_radii(vdw_radii)
update_attr(d)
class gaudi.genes.molecule.Molecule(path=None, symmetry=None, hydrogens=False, pdb-fix=False, vdw_radii=None, **kwargs)
Bases: gaudi.genes.GeneProvider
Interface around the gaudi.genes.molecule.Compound to handle the GAUDI protocol and cachingfeatures.
Parameters
• path (str, optional) – Path to a molecule file or a directory containing dirs ofmolecule files.
• symmetry (str, optional) – If path is a directory, list of pairs of directories whosechosen mocule must be the same, thus enabling symmetry.
• hydrogens (bool, optional) – Add hydrogens to Molecule (True) or not (False).
• pdbfix (bool, optional) – Only for testing and debugging. Better run pdbfixer priorto GAUDI. Fix potential issues that may cause troubles with OpenMM forcefields.
• vdw_radii (dict {str: float} , optional) – Set a specific vdw_radius fora particular element (instead of standard Chimera VdW table). It can be useful in particularcases together with a contacts objective. Example of use in the .yaml file: vdw_radii: {
Fe: 2.00, Cu: 2.16}
Defaults to None
allelePaths to every fragment that composes a given Compound. It will consist of a single value tuple if there’sno dynamic building involved; ie, a mol2 or pdb file as is.
Type tuple of str
_CATALOGClass attribute (shared among all Molecule instances) that holds all the possible molecules GAUDI canbuild given current path.
36 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
If path is a single molecule file, that’s the only possibility, but if it’s set to a directory, the engine canpotentially build all the combinations of molecule blocks found in those subdirectories.
Normally, it is accessed via the catalog property.
Type dict
Notes
Use of ‘_cache‘
Molecule class uses _cache to store already built molecules. Its entry in the _cache directory is aboltons.cacheutils.LRU cache (least recently used) set to a maximum size of 300 entries.
Todo: The LRU cache size should be proportional to job size, depending on the population size and number ofgenerations, but also taking available memory into account (?).
SUPPORTED_FILETYPES = ('mol2', 'pdb')
build(key, where=None)Builds a Compound following the recipe contained in key through Compound.append methods.
Parameters key (tuple of str) – Paths to the molecule blocks that comprise the finalmolecule. A single molecule is just a one-block recipe.
Returns The final molecule result of the sequential appending.
Return type Compound
classmethod clear_cache()
compoundGet expressed allele on-demand (read-only attribute)
express()Adds Chimera molecule object to the viewer canvas.
It also converts pseudobonds (used by Chimera to depict coordinated ligands to metals) to regular bonds.
find_atom(serial)
find_atoms(serial, only_one=False)
find_residue(position)
find_residues(position, only_one=False)
get(key)Looks for the compound corresponding to key in _cache. If found, return it. Else, build it on demand, storeit on cache and return it.
Parameters key (str) – Path (or combination of) to the requested molecule. It should beextracted from catalog.
Returns The result of building the requested molecule.
Return type gaudi.genes.molecule.Compound
mate(mate)
10.2. gaudi.genes 37

GaudiMM Documentation, Release 0.0.8+1.g7374b62
Todo: Allow mating while preserving symmetry
mutate(indpb)VERY primitive. It only gets another compound.
unexpress()Removes the Chimera molecule from the viewer canvas (without deleting the object).
write(path=None, name=None, absolute=None, combined_with=None, filetype=’mol2’)Writes full mol2 to disk.
Todo: It’d be preferable to get a string instead of a file
xyz(transformed=True)
gaudi.genes.molecule.enable(**kwargs)
10.2.2 Mutamers gene
This modules allows to explore side chains flexibility in proteins, as well as mutation.
It needs that at least a gaudi.genes.rotamers.molecule.Molecule has been requested in the input file.Residues of those are referenced in the residues argument.
It also allows mutations in the selected residues. However, the resulting structure keeps the same backbone, whichmay not be representative of the in-vivo behaviour. Use with caution.
class gaudi.genes.mutamers.Mutamers(residues=None, library=’Dunbrack’,avoid_replacement=False, mutations=[], liga-tion=False, hydrogens=False, **kwargs)
Bases: gaudi.genes.GeneProvider
Mutamers class
Parameters
• residues (list of str) – Residues that can mutate. This has to be in the form:
[ Protein/233, Protein/109 ]
where the first element (before slash) is the gaudi.genes.molecule name and the secondelement (after slash) is the residue position number in that molecule.
This list of str is later parsed to the proper chimera.Residue objects
• library ({'Dunbrack', 'Dynameomics'}) – The rotamer library to use.
• mutations (list of str, required) – Aminoacids (in 3-letter codes) residuescan mutate to.
• ligation (bool, optional) – If True, all residues will mutate to the same type ofaminoacid.
• hydrogens (bool, optional) – If True, add hydrogens to replacing residues (buggy)
alleleFor i residues, it contains i tuples with two values each: residue type and a float within [0, 1), which willbe used to pick one of the rotamers for that residue type.
Type list of 2-tuple (str, float)
38 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
static add_hydrogens_to_isolated_rotamer(rotamers)
choice(l)Overrides random.choice with custom one so we can reuse a previously obtained random number.This helps dealing with the ligation parameter, which forces all the requested residues to mutate to thesame type
express()Compile the gene to an evaluable object.
get_rotamers(mol, pos, restype)Gets the requested rotamers out of cache and if not found, creates the library and stores it in the cache.
Parameters
• mol (str) – gaudi.genes.molecule name that contains the residue
• pos – Residue position in mol
• restype – Get rotamers of selected position with this type of residue. It does not needto be the original type, so this allows mutations
Returns
Return type List of rotamers returned by Rotamers.getRotamers.
mate(mate)Perform a crossover with another gene of the same kind.
mutate(indpb)Perform a mutation on the gene.
unexpress()Revert expression.
static update_rotamer_coords(residue, rotamer)
gaudi.genes.mutamers.enable(**kwargs)
10.2.3 NormalModes gene
This module allows to explore molecular folding through normal modes analysis.
It works by calculating normal modes for the input molecule and moving along a combination of normal modes.
It needs at least a gaudi.genes.rotamers.molecule.Molecule.
class gaudi.genes.normalmodes.NormalModes(method=’prody’, target=None, modes=None,n_samples=10000, rmsd=1.0, group_by=None,group_lambda=None, path=None,write_modes=False, **kwargs)
Bases: gaudi.genes.GeneProvider
NormalModes class
Parameters
• method (str, optional, default=prody) – Either: - prody : calculate normalmodes using prody algorithms - gaussian : read normal modes from a gaussian output file
• target (str) – Name of the Gene containing the actual molecule
• modes (list, optional, default=range(12)) – Modes to be used to move themolecule
10.2. gaudi.genes 39

GaudiMM Documentation, Release 0.0.8+1.g7374b62
• group_by (str or callable, optional, default=None) – Available strnames: residues, mass. This is, consider only pseudoatoms that group a whole residue,groups of contiguous atoms that amount for certain mass, or only alpha carbons. If residuesor mass, the group_lambda parameter can customize the behaviour.
• group_lambda (int, optional) – Either: number of residues per group (default=7),or number of groups with same mass (default=100).
• path (str) – Gaussian or prody modes output path. Required if method is gaussian.
• write_modes (bool, optional) – write a molecule_modes.nmd file with theProDy modes
• n_samples (int, optional, default=10000) – number of conformations togenerate
• rmsd (float, optional, default=1.0) – RMSD (in angstrom) that the confor-mations will have with respect to the initial conformation. In some cases, it can be slightlyhigher than the requested threshold (e.g. 1.07A instead of 1A)
alleleRandomly picked coordinates from NORMAL_MODE_SAMPLES
Type slice of prody.ensemble
NORMAL_MODESnormal modes calculated for the molecule or readed from the gaussian frequencies output file stored in aprody modes class (ANM or RTB)
Type prody.modes
NORMAL_MODE_SAMPLESconfigurations applying modes to molecule
Type prody.ensemble
_original_coordsParent coordinates
Type numpy.array
_chimera2prody_chimera2prody[chimera_index] = prody_index
Type dict
NORMAL_MODES
NORMAL_MODES_SAMPLES
calculate_prody_normal_modes()calculate normal modes, creates a diccionary between chimera and prody indices and calculate n_confsnumber of configurations using this modes
express()Apply new coords as provided by current normal mode
mate(mate)
Todo: Combine coords between two samples in NORMAL_MODES_SAMPLES? Or two sam-ples between diferent NORMAL_MODES_SAMPLES? Or combine samples between two NOR-MAL_MODES_SAMPLES?
40 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
For now : pass
molecule
mutate(indpb)(mutate to/get) another SAMPLE with probability = indpb
read_gaussian_normal_modes()read normal modes, creates a diccionary between chimera and prody indices and calculate n_confs numberof configurations using this modes
read_prody_normal_modes()
unexpress()Undo coordinates change
gaudi.genes.normalmodes.alg3(moldy, max_bonds=3, **kwargs)TESTS PENDING!
Coarse Grain Algorithm 3: Graph algorithm.
New group when a vertice: have more than n, have 0 edges new chain
Parameters
• moldy (prody.AtomGroup) –
• n (int, optional, default=2) – maximum bonds number
Returns moldy – New Betas added
Return type prody.AtomGroup
gaudi.genes.normalmodes.chimeracoords2numpy(molecule)
Parameters molecule (chimera.molecule) –
Returns
Return type numpy.array with molecule.atoms coordinates
gaudi.genes.normalmodes.chunker(end, n)divide end integers in closed groups of n
gaudi.genes.normalmodes.convert_chimera_molecule_to_prody(molecule)Function that transforms a chimera molecule into a prody atom group
Parameters molecule (chimera.Molecule) –
Returns
• prody_molecule (prody.AtomGroup())
• chimera2prody (dict) – dictionary: chimera2prody[chimera_atom.coordIndex] = i-thm el-ement prody getCoords() array
gaudi.genes.normalmodes.enable(**kwargs)
gaudi.genes.normalmodes.gaussian_modes(path)Read the modes Create a prody.modes instance
Parameters path (str) – gaussian frequencies output path
Returns modes
Return type ProDy modes ANM or RTB
10.2. gaudi.genes 41

GaudiMM Documentation, Release 0.0.8+1.g7374b62
gaudi.genes.normalmodes.group_by_mass(molecule, n=100)Coarse Grain Algorithm 2: groups per mass percentage
Parameters
• molecule (prody.AtomGroup) –
• n (int, optional, default=100) – Intended number of groups. The mass of thesystem will be divided by this number, and each group will have the corresponding propor-tional mass. However, the final number of groups can be slightly different.
Returns molecule – New Betas added
Return type prody.AtomGroup
gaudi.genes.normalmodes.group_by_residues(molecule, n=7)Coarse Grain Algorithm 1: groups per residues
Parameters
• molecule (prody.AtomGroup) –
• n (int, optional, default=7) – number of residues per group
Returns molecule – New betas added
Return type prody.AtomGroup
gaudi.genes.normalmodes.prody_modes(molecule, max_modes, algorithm=None, **options)
Parameters
• molecule (prody.AtomGroup) –
• nax_modes (int) – number of modes to calculate
• algorithm (callable, optional, default=None) – coarseGrain(prm) wichmake molecule.select().setBetas(i) where i is the index Coarse Grain group Where prm isprody AtomGroup
• options (dict, optional) – Parameters for algorithm callable
Returns modes
Return type ProDy modes ANM or RTB
10.2.4 Rotamers gene
This modules allows to explore side chains flexibility in proteins, as well as mutation.
It needs that at least a gaudi.genes.rotamers.molecule.Molecule has been requested in the input file.Residues of those are referenced in the residues argument.
It also allows mutations in the selected residues. However, the resulting structure keeps the same backbone, whichmay not be representative of the in-vivo behaviour. Use with caution.
class gaudi.genes.rotamers.Rotamers(residues=None, library=’Dunbrack’,with_original=True, **kwargs)
Bases: gaudi.genes.GeneProvider
Rotamers class
Parameters
• residues (list of str) – Residues that should be analyzed. This has to be in theform:
42 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
[ Protein/233, Protein/109 ]
where the first element (before slash) is the gaudi.genes.molecule name and the secondelement (after slash) is the residue position number in that molecule.
This list of str is later parsed to the proper chimera.Residue objects
• library ({'Dunbrack', 'Dynameomics'}) – The rotamer library to use.
• with_original (bool, defaults to True) – Whether to include the original setof chi angles as part of the rotamer library.
alleleFor i residues, it contains i floats within [0, 1), that will point to the selected rotamer torsions for eachresidue.
Type list of float
static all_chis(residue)
express()Compile the gene to an evaluable object.
mate(mate)Perform a crossover with another gene of the same kind.
mutate(indpb)Perform a mutation on the gene.
static patch_residue(residue)
retrieve_rotamers(molecule, position, residue, library=’Dunbrack’)
unexpress()Revert expression.
static update_rotamer(residue, chis)
gaudi.genes.rotamers.enable(**kwargs)
10.2.5 Search gene
This module provides spatial exploration of the environment.
It works by creating a sphere with radius self.radius and origin at self.origin. The movement is achieved with threematrices that contain a translation, a rotation, and a reference position.
It depends on gaudi.genes.molecule.Molecule, since these are the ones that will be moved around. Com-bined with the adequate objectives, this module can be used to implement docking experiments.
class gaudi.genes.search.Search(target=None, center=None, radius=None, rotate=True, preci-sion=0, interpolation=0.5, **kwargs)
Bases: gaudi.genes.GeneProvider
Parameters
• target (namedtuple or Name of gaudi.genes.molecule) – Can be either:- The anchor atom of the molecule we want to move, with syntax <molecule_name>/<index>. For example, if we want to move Ligand using atom with serial number = 1 aspivot, we would specify Ligand/1. It’s parsed to the actual chimera.Atom later on. - Aname of gaudi.genes.molecule instance. In this case, the anchor atom for the movement ofthe molecule will be set to its nearest atom to the geometric center of the molecule.
10.2. gaudi.genes 43

GaudiMM Documentation, Release 0.0.8+1.g7374b62
• center (3-item list or tuple of float, optional) – Coordinates to thecenter of the desired search sphere
• radius (float) – Maximum distance from center that the molecule can move
• rotate (bool, bool) – If False, don’t rotatate the molecule - only translation
• precision (int, bool) – Rounds the decimal part of the 3D search matrix to get acoarser model of space. Ie, less points can be accessed, the search is less exhaustive, morevariability in less runs.
alleleA 4x3 matrix of float, as explained in Notes.
Type 3-tuple of 4-tuple of floats
originThe initial position of the requested target molecule. If we don’t take this into account, we can’t move themolecule around was not originally in the center of the sphere.
Type 3-tuple of float
Notes
How matricial translation and rotation takes place
A single movement is summed up in a 4x3 matrix:
( (R1, R2, R3, T1), (R4, R5, R6, T2), (R7, R8, R9, T3) )
R-elements contain the rotation information, while T elements account for the translation movement.
That matrix can be obtained from multipying three different matrices with this expression:
multiply_matrices(translation, rotation, to_zero)
To understand the operation, it must be read from the right:
1. First, translate the molecule the origin of coordinates 0,0,0
2. In that position, the rotation can take place.
3. Then, translate to the final coordinates from zero. There’s no need to get back to the original position.
How do we get the needed matrices?
• to_zero. Record the original position (origin) of the molecule and multiply it by -1. Done with methodto_zero().
• rotation. Obtained directly from FitMap.search.random_rotation
• translation. Check docstring of random_translation() in this module.
center
express()Multiply all the matrices, convert the result to a chimera.CoordFrame and set that as the xform for thetarget molecule. If precision is set, round them.
mate(mate)Interpolate the matrices and assign them to each individual. Ind1 gets the rotated interpolation, while Ind2gets the translation.
molecule
44 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
mutate(indpb)Perform a mutation on the gene.
origin
random_transform()Wrapper function to provide translation and rotation in a single call
to_zeroReturn a translation matrix that takes the molecule from its original position to the origin of coordinates(0,0,0).
Needed for rotations.
unexpress()Reset xform to unity matrix.
gaudi.genes.search.center(mol)
gaudi.genes.search.distance(a, b)
gaudi.genes.search.enable(**kwargs)
gaudi.genes.search.nearest_atom(mol, position)
gaudi.genes.search.parse_origin(origin, individual=None)The center of the sphere can be given as an Atom, or directly as a list of three floats (x,y,z). If it’s an Atom, findit and return the xyz coords. If not, just turn the list into a tuple.
Parameters
• origin (3-item list of coordinates, or chimera.Atom) –
• genes (gaudi.parse.Settings.genes) – List of gene-dicts to look for moleculesthat may contain the referred atom in origin
Returns The x,y,z coordinates
Return type Tuple of float
gaudi.genes.search.rand_xform(origin, destination, r, rotate=True)
gaudi.genes.search.random_translation(center, r)Get a random point from the cube built with l=r and test if it’s within the sphere. Most of the points will be, butnot all of them, so get another one until that criteria is met.
Parameters
• center (3-tuple of float) – Coordinates of the center of the search sphere
• r (float) – Radius of the search sphere
Returns
Return type A translation matrix to a random point in the search sphere, with no rotation.
gaudi.genes.search.rotate(molecule, at, alpha)
gaudi.genes.search.translate(molecule, anchor, target)
10.2.6 Torsions gene
This module helps explore small molecules flexibility.
It does so by performing bond rotations in the selected gaudi.genes.molecule.Molecule objects, if they exhibit freebond rotations.
10.2. gaudi.genes 45

GaudiMM Documentation, Release 0.0.8+1.g7374b62
class gaudi.genes.torsion.Torsion(target=None, flexibility=360.0, max_bonds=None, an-chor=None, rotatable_atom_types=(’C3’, ’N3’, ’C2’, ’N2’,’P’), rotatable_atom_names=(), rotatable_elements=(),non_rotatable_bonds=(), rotatable_bonds=(), precision=1,**kwargs)
Bases: gaudi.genes.GeneProvider
Parameters
• target (str) – Name of gaudi.genes.molecule instance to perform rotation on
• flexibility (int or float) – Maximum number of degrees a bond can rotate
• max_bonds – Expected number of free rotations in molecule. Needed to store arbitraryrotations.
• anchor (str) – Molecule/atom_serial_number of reference atom for torsions
• rotatable_atom_types (list of str) – Which type of atom types (as inchimera.Atom.idatmType) should rotate. Defaults to (‘C3’, ‘N3’, ‘C2’, ‘N2’, ‘P’).
• rotatable_atom_names (list of str) – Which type of atom names (as inchimera.Atom.name) should rotate. Defaults to ().
• rotatable_bonds (list of [SerialNumberAtom1,SerialNumberAtom2, SerialNumberAnchor]) – Concrete bonds that areallowed to rotate. Atoms have to be designated using their chimera serial number. IM-PORTANT: if set, these will be the ONLY bonds allowed to rotate, ignoring other possibleconditions (e.g. rotatable_atom_types, rotatable_atom_names. . . ).
alleleFor i rotatable bonds in molecule, it contains i floats which correspond to each torsion angle. As such,each falls within [-180.0, 180.0).
Type tuple of float
Notes
Todo: max_bonds is now automatically computed, but probably won’t deal nicely with block-built ligands.
BONDS_ROTS = {}
anchorGet the molecular anchor. Ie, the root of the rotations, the fixed atom of the molecule.
Usually, this is the target atom in the Search gene, but if we can’t find it, get the nearest atom to thegeometric center of the molecule, and if it’s not possible, the donor atom of the molecule.
clear_cache()
express()Apply rotations to rotatable bonds
mate(mate)Perform a crossover with another gene of the same kind.
molecule
mutate(indpb)Perform a mutation on the gene.
46 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
random_angle()Returns a random angle within flexibility limits
rotatable_bondsGets potentially rotatable bonds in molecule
First, it retrieves all the atoms. Then, the bonds are filtered, discarding coordination (pseudo)bonds andsort them by atom serial.
For each bond, try to retrieve it from the cache. If unavailable, request a bond rotation object tochimera.BondRot.
In this step, we have to discard non rotatable atoms (as requested by the user), and make sure the involvedatoms are of compatible type. Namely, one of them must be either C3, N3, C2 or N2, and both of them,non-terminal (more than one neighbor).
If the bond is valid, get the BondRot object. Chimera will complain if we already have requested that bondpreviously, or if the bond is in a cycle. Handle those exceptions silently, and get the next bond in that case.
If no exceptions are raised, then store the rotation anchor in the BondRot object (that’s the nearest atom inthe bond to the molecular anchor), and store the BondRot object in the rotations cache.
unexpress()Undo the rotations
gaudi.genes.torsion.center(mol)
gaudi.genes.torsion.distance(a, b)
gaudi.genes.torsion.enable(**kwargs)
gaudi.genes.torsion.nearest_atom(mol, position)
10.2.7 Trajectory gene
This module provides spatial exploration of the environment through a MD trajectory file.
class gaudi.genes.trajectory.Trajectory(target=None, path=None, max_frame=None,stride=1, preload=False, **kwargs)
Bases: gaudi.genes.GeneProvider
Parameters
• target (str) – The Molecule that contains the topology of the trajectory.
• path (str) – Path to a MD trajectory file, as supported by mdtraj.
• max_frame (int) – Last frame of the trajectory that can be loaded.
• stride (int, optional) – Only load one in every stride frames
• preload (bool, optional) – Load the full trajectory in memory to accelerate expres-sion. Not recommended for large files!
alleleThe index of a frame in the MD trajectory.
Type int
_trajAlias to the frames cache
Type dict
10.2. gaudi.genes 47

GaudiMM Documentation, Release 0.0.8+1.g7374b62
express()Load the frame requested by the current allele into a new CoordSet object (always at index 1) and set thatas the active one.
load_frame(n)
mate(mate)Simply exchange alleles. Can’t try to interpolate an intermediate structure because the result wouldn’tprobably belong to the original trajectory!
moleculeThe target Molecule gene
mutate(indpb)Perform a mutation on the gene.
random_frame_number()
topologyReturns the equivalent mdtraj Topology object of the currently expressed Chimera molecule
unexpress()Set the original coordinates (stored at mol.coordSets[0]) as the active ones.
gaudi.genes.trajectory.enable(**kwargs)
10.2.8 Base class for all genes
Genes are modules that reside in the gaudi.genes package, and have a certain class structure.
class gaudi.genes.GeneProvider(parent=None, name=None, cx_eta=5.0, mut_eta=5.0,mut_indpb=0.75, **kwargs)
Bases: object
Base class that every genes plugin MUST inherit.
The methods listed here are compulsory for all subclasses, since the individual will be using them anyway.If it’s not relevant in your plugin, just define them with a single pass statement. Also, don’t forget to callGeneProvider.__init__ in your overriden __init__ function; it registers compulsory
— From (M.A. itself)[http://martyalchin.com/2008/jan/10/simple-plugin-framework/]: Now that we have amount point, we can start stacking plugins onto it. As mentioned above, individual plugins will subclass themount point. Because that also means inheriting the metaclass, the act of subclassing alone will suffice as pluginregistration. Of course, the goal is to have plugins actually do something, so there would be more to it than justdefining a base class, but the point is that the entire contents of the class declaration can be specific to the pluginbeing written. The plugin framework itself has absolutely no expectation for how you build the class, allowingmaximum flexibility. Duck typing at its finest.
classmethod clear_cache()
express()Compile the gene to an evaluable object.
mate(gene)Perform a crossover with another gene of the same kind.
mutate()Perform a mutation on the gene.
plugins = [<class 'gaudi.genes.search.Search'>, <class 'gaudi.genes.molecule.Molecule'>, <class 'gaudi.genes.mutamers.Mutamers'>, <class 'gaudi.genes.normalmodes.NormalModes'>, <class 'gaudi.genes.rotamers.Rotamers'>, <class 'gaudi.genes.torsion.Torsion'>, <class 'gaudi.genes.trajectory.Trajectory'>]
48 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
unexpress()Revert expression.
classmethod validate(data, schema=None)
classmethod with_validation(**kwargs)
write(path, name, *args, **kwargs)Write results of expression to a file representation.
10.3 gaudi.objectives
These are the built-in objectives in GaudiMM. You can also build your own, but these are ready to use.
10.3.1 Angle objective
This objective calculates the angle formed by three given atoms (or the dihedral, if four atoms are given) and returnsthe absolute difference of that angle and the target value.
class gaudi.objectives.angle.Angle(threshold=None, probes=None, *args, **kwargs)Bases: gaudi.objectives.ObjectiveProvider
Angle class
Parameters
• threshold (float) – Optimum angle
• probes (list of str) – Atoms that make the angle, expressed as a series of<molecule_name>/<serial_number> strings
Returns Deviation from threshold angle, in degrees
Return type float
evaluate(ind)Return the score of the individual under the current conditions.
probes(ind)
gaudi.objectives.angle.enable(**kwargs)
10.3.2 Contacts objective
This objective provides a wrapper around Chimera’s DetectClash that detects clashes and contacts. Clashes are un-derstood as steric conflicts that increases the energy of the system. They are evaluated as the sum of volumetricoverlapping of the Van der Waals’ spheres of the implied atoms. Contacts are considered as stabilizing, and they areevaluated with a Lennard-Jones 12-6 like function.
class gaudi.objectives.contacts.Contacts(probes=None, radius=5.0, which=’hydrophobic’,clash_threshold=0.6, hydrophobic_threshold=-0.4, cutoff=0.0, hydrophobic_elements=(’C’,’S’), bond_separation=4, same_residue=True,only_internal=False, *args, **kwargs)
Bases: gaudi.objectives.ObjectiveProvider
Contacts class :param probes: Name of molecule gene that is object of contacts analysis :type probes: str :paramradius: Maximum distance from any point of probes that is searched
10.3. gaudi.objectives 49

GaudiMM Documentation, Release 0.0.8+1.g7374b62
for possible interactions
Parameters
• which ({'hydrophobic', 'clashes'}) – Type of interactions to measure
• clash_threshold (float, optional) – Maximum overlap of van-der-Waalsspheres that is considered as a contact (attractive). If the overlap is greater, it’s considered aclash (repulsive)
• hydrophobic_threshold (float, optional) – Maximum overlap for hydropho-bic patches.
• hydrophobic_elements (list of str, optional, defaults to [C,S]) – Which elements are allowed to interact in hydrophobic patches
• cutoff (float, optional) – If the overlap volume is greater than this, a penalty isapplied. Useful to filter bad solutions.
• bond_separation (int, optional) – Ignore clashes or contacts between atomswithin n bonds.
• only_internal (bool, optional) – If set to True, take into account only in-tramolecular interactions, defaults to False
Returns Lennard-Jones-like energy when which‘=‘hydrophobic, and volumetric overlap of VdWspheres in A3 if which‘=‘clashes.
Return type float
evaluate_clashes(ind)
evaluate_hydrophobic(ind)
find_interactions(ind)
molecules(ind)
probes(ind)
gaudi.objectives.contacts.enable(**kwargs)
10.3.3 Coordination objective
This objective performs rough estimations of good orientations of ligating residues in a protein to coordinate a givenmetal or small molecule. The geometry is approximated by computing average distances from ligating atoms the metalcentre (self.probe) as well as the angles formed by the probe, the ligating atom and its immediate neighbor. Goodplanarity is assured by a dihedral check.
class gaudi.objectives.coordination.Coordination(probe=None, radius=3.0,atom_types=(), atom_elements=(),atom_names=(), residues=(),geometry=’tetrahedral’, dis-tance=0, min_atoms=1, pre-vent_intruders=True, en-force_all_residues=False,only_one_ligand_per_residue=False,center_of_mass_correction=False,distance_correction=False, *args,**kwargs)
Bases: gaudi.objectives.ObjectiveProvider
50 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
Coordination class
Parameters
• probe (tuple) – The atom that acts as the metal center, expressed as<molecule_name>/<atom serial>. This will be parsed later on.
• residues (list of str) – Residues that must coordinate to probe, expressed as<molecule_name>/<residue position>. Position can be *.
• radius (float, optional, default=3.0) – Distance from probe where ligatingatoms must be found
• atom_types (list of str, optional) – Types of atoms that are considered lig-ands to probe
• atom_names (list of str, optional) – Names of atoms that are considered lig-ands to probe
• atom_elements (list of str, optional) – Elements of atoms that are consid-ered ligands to probe
• distance (float, optional) – Perfect distance a ligand atom should be from target.
• geometry (str or list of 3-tuple floats, optional) – Which geome-try should be fitted. Choose from GEOMETRIES dict or specify a set of vectors.
• enforce_all_residues (bool, optional) – Whether to force or not if all speci-fied residues should coordinate.
• only_one_ligand_per_residue (bool, optional) – Enforce that only one lig-and for each residue should coordinate.
• prevent_intruders (bool, optional) – Don’t let non-ligand atoms to be closerto the target than the selected ligand atoms.
• center_of_mass_correction (bool, optional) – If True, calculate the dis-tance between the metal center and the center of mass of the ligand atoms, and sum thatto the final score.
• distance_correction (bool, optional) – If True, report the deviation of theexperimental coordination bond length and the ideal one, as tabulated by chimera.Element, and sum that to the final score.
Returns Sum of RMSD of vertices from ideal RMSD and average cosine of angle deviation fromideal orientation of ligand neighbors. A perfect match should report 0.0.
Return type float
coordination_sphere(ind)1. Get atoms and residues found within self.radius angstroms from self.probe. Found residuesMUST include self.residues. Otherwise, apply penalty.
2. Sort atoms by absolute difference of self.distance and distance to self.probe. That way,nearest atoms are computed first. If found atoms do not include some of the requested types, apply penalty.
evaluate(ind)
1. Get requested atoms sorted by distance
2. If they meet the minimum quantity, return the rmsd for that geometry
3. If that’s not possible of they are not enough, return penalty
molecules(ind)
10.3. gaudi.objectives 51

GaudiMM Documentation, Release 0.0.8+1.g7374b62
probe(ind)
residues(ind)
gaudi.objectives.coordination.enable(**kwargs)
gaudi.objectives.coordination.ideal_bond_deviation(metal, ligand, other_ligands=())Assess if the current bond vector is well oriented with respect to the ideal bond vector.
Parameters
• metal (chimera.Atom) – The ion ligands are coordinating to
• ligand (chimera.Atom) – Potential ligand atoms to metal
Returns Absolute sine of the angle between the ideal vector and the ligand-metal one.
Return type float
gaudi.objectives.coordination.ideal_bonded_positions(atom, element, geome-try=None)
10.3.4 Distance objective
This objective calculates the distance between two given atoms. It returns the absolute difference between the calcu-lated distance and the target value.
class gaudi.objectives.distance.Distance(threshold=None, tolerance=None, target=None,probes=None, center_of_mass=False, *args,**kwargs)
Bases: gaudi.objectives.ObjectiveProvider
Distance class
Parameters
• threshold (float) – Optimum distance to meet
• tolerance (float) – Maximum deviation from threshold that is not penalized
• target (str) – The atom to measure the distance to, expressed as <moleculename>/<atom serial>
• probes (list of str) – The atoms whose distance to target is being measured, ex-pressed as <molecule name>/<atom serial>. If more than one is provided, the average of allof them is returned
• center_of_mass (bool) –
Returns (Mean of) absolute deviation from threshold distance, in A.
Return type float
atoms(ind, *targets)
evaluate_center_of_mass(ind)
evaluate_distances(ind)Measure the distance
gaudi.objectives.distance.enable(**kwargs)
52 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
10.3.5 DrugScoreX objective
This objective is a wrapper around the binaries provided by Neudert and Klebe and calculates the score of the currentpose.
The lower, the better, so usually you will use a -1.0 weight.
class gaudi.objectives.dsx.DSX(binary=None, potentials=None, proteins=(’Protein’, ), lig-ands=(’Ligand’, ), terms=None, sorting=1, cofactor_mode=0,with_covalent=False, with_metals=True, *args, **kwargs)
Bases: gaudi.objectives.ObjectiveProvider
DSX class
Parameters
• protein (str) – The molecule name that is acting as a protein
• ligand (str) – The molecule name that is acting as a ligand
• binary (str, optional) – Path to the DSX binary. Only needed if drugscorex isnot in PATH.
• potentials (str, optional) – Path to DSX potentials. Only needed ifDSX_POTENTIALS env var has not been set by the installation process (condainstall -c insilichem drugscorex normally takes care of that).
• terms (list of bool, optional) – Enable (True) or disable (False) certain termsin the score function in this order: distance-dependent pair potentials, torsion potentials,intramolecular clashes, sas potentials, hbond potentials
• sorting (int, defaults to 1) – Sorting mode. An int between 0-6, read binaryhelp for -S:
-S int : Here you can specify the mode that affects how the→˓results
will be sorted. The default mode is '-S 1', which sorts theligands in the same order as they are found in the lig_
→˓file.The following modes are possible::
0: Same order as in the ligand file1: Ordered by increasing total score2: Ordered by increasing per-atom-score3: Ordered by increasing per-contact-score4: Ordered by increasing rmsd5: Ordered by increasing torsion score6: Ordered by increasing per-torsion-score
• cofactor_mode (int, defaults to 0) – Cofactor handling mode. An int between0-7, read binary help for -I:
-I int : Here you can specify the mode that affects how cofactors,waters and metals will be handeled.The default mode is '-I 1', which means, that all moleculesare treated as part of the protein. If a structure shouldnot be treated as part of the protein you have supply aseperate file with seperate MOLECULE entries correspondingto each MOLECULE entry in the ligand_file (It is assumedthat the structure, e.g. a cofactor, was kept flexible indocking, so that there should be a different geometry
(continues on next page)
10.3. gaudi.objectives 53

GaudiMM Documentation, Release 0.0.8+1.g7374b62
(continued from previous page)
corresponding to each solution. Otherwise it won't makesense not to treat it as part of the protein.).The following modes are possible:
0: cofactors, waters and metals interact with protein,ligand and each other1: cofactors, waters and metals are treated as part ofthe protein2: cofactors and metals are treated as part of the
→˓protein(waters as in mode 0)3: cofactors and waters are treated as part of the
→˓protein4: cofactors are treated as part of the protein5: metals and waters are treated as part of the protein6: metals are treated as part of the protein7: waters are treated as part of the protein
Please note: Only those structures can be treatedindividually, which are supplied in seperate files.
• with_covalent (bool, defaults to False) – Whether to deal with covalentlybonded atoms as normal atoms (False) or not (True)
• with_metals (bool, defaults to True) – Whether to deal with metal atoms asnormal atoms (False) or not (True)
Returns Interaction energy as reported by DSX output logs.
Return type float
clean()
evaluate(ind)Run a subprocess calling DSX binary with provided options, and parse the results. Clean tmp files at exit.
get_molecule_by_name(ind, *names)Get a molecule gene instance of individual by its name
parse_output(stream)
prepare_command()
prepare_ligands(ligands)
prepare_proteins(proteins)
gaudi.objectives.dsx.enable(**kwargs)
10.3.6 Energy (OpenMM) objective
This objective is a wrapper around OpenMM, providing a GPU-accelerated energy calculation of the system with asimple forcefield evaluation.
class gaudi.objectives.energy.Energy(targets=None, forcefields=(’amber99sbildn.xml’, ),auto_parametrize=None, parameters=None, plat-form=None, *args, **kwargs)
Bases: gaudi.objectives.ObjectiveProvider
Calculate the energy of a system
Parameters
54 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
• targets (list of str, default=None) – If set, which molecules should be eval-uated. Else, all will be evaluated.
• forcefields (list of str, default=('amber99sbildn.xml',)) –Which forcefields to use
• auto_parametrize (list of str, default=None) – List of Molecule in-stances GAUDI should try to auto parametrize with antechamber.
• parameters (list of 2-item list of str) – List of (gaff.mol2, .frcmod) filesto use as parametrization source.
• platform (str) – Which platform to use for calculations. Choose between CPU, CUDA,OpenCL.
Returns The estimated potential energy, in kJ/mol
Return type float
calculate_energy(coordinates)Set up an OpenMM simulation with default parameters and return the potential energy of the initial state
Parameters coordinates (simtk.unit.Quantity) – Positions of the atoms in the sys-tem
Returns potential_energy – Potential energy of the system, in kJ/mol
Return type float
static chimera_molecule_to_openmm_positions(*molecules)
static chimera_molecule_to_openmm_topology(*molecules)Convert a Chimera Molecule object to OpenMM structure, providing topology and coordinates.
Parameters molecule (chimera.Molecule) –
Returns
• topology (simtk.openmm.app.topology.Topology)
• coordinates (simtk.unit.Quantity)
evaluate(individual)Calculates the energy of current individual
Notes
For static calculations, where molecules are essentially always the same, but with different coordinates, weonly need to generate topologies once. However, for dynamic jobs, with potentially different moleculesinvolved each time, we cannot guarantee having the same topology. As a result, we generate it again foreach evaluation.
molecules(individual)
simulationBuild a new OpenMM simulation if not yet defined and return it
Notes
self.topology must be defined previously! Use self.chimera_molecule_to_openmm_topology to set it.
10.3. gaudi.objectives 55

GaudiMM Documentation, Release 0.0.8+1.g7374b62
gaudi.objectives.energy.calculate_energy(filename, forcefields=None)Calculate energy from PDB file with desired forcefields. If not specified, amber99sbildn will be used. Returnspotential energy in kJ/mol.
gaudi.objectives.energy.enable(**kwargs)
10.3.7 GOLD objective
This objective is a wrapper around the scoring functions provided by CCDC’s GOLD.
It will use the rescoring abilities in GOLD to extract the fitness corresponding to any of the available scoring functions:
• GoldScore (goldscore)
• ChemScore (chemscore)
• Astex Statistical Potential (asp)
• CHEMPLP (chemplp)
Since GOLD is commercial software, you will need to install it separately and provide a valid license! This isjust a wrapper. Make sure to set all the needed environment variables, such as CCDC_LICENSE_FILE, and that‘gold_auto’ is in $PATH. Check tests/test_objectives_gold.py for an example; make sure to have GOLDXX/bin beforeGOLDXX/GOLD/bin!
class gaudi.objectives.gold.Gold(protein=’Protein’, ligand=’Ligand’, scoring=’chemscore’,score_component=’Score’, radius=10, *args, **kwargs)
Bases: gaudi.objectives.ObjectiveProvider
Gold class
Parameters
• protein (str) – The name of molecule acting as protein
• ligand (str) – The name of molecule acting as ligand
• scoring (str, optional, defaults to chemscore) – Fitness function touse. Choose between chemscore, chemplp, goldscore and asp.
• score_component (str, optional, defaults to 'Score') – Scoringfields to parse out of the rescore.log file, such as Score, DG, S(metal), etc.
• radius (float, optional, defaults to 10.0) – Radius (in A) of binding sitesphere, the origin of which is automatically centered at the ligand’s center of mass.
Returns Interaction energy as reported by GOLD’s chosen scoring function
Return type float
clean()
evaluate(ind)Run a subprocess calling LigScore binary with provided options, and parse the results. Clean tmp files atexit.
get_molecule_by_name(ind, *names)Get a molecule gene instance of individual by its name
origin(molecule)
parse_output(filename)Get last word of first line (and unique) and parse it into float
prepare_command(protein_path, ligand_path, origin)
56 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
prepare_ligands(ligands)
prepare_proteins(proteins)
gaudi.objectives.gold.enable(**kwargs)
10.3.8 Hydrogen bonds objective
This objective is a wrapper around Chimera’s FindHBond. It returns the number of hydrogen bonds that can be formedbetween the target molecule and its environment. .. todo:
Evaluate the possible HBonds with some kind of function thatgives a rough idea of the strength (energy) of each of them.
class gaudi.objectives.hbonds.Hbonds(probes=None, radius=5.0, distance_tolerance=0.4,angle_tolerance=20.0, only_intermolecular=True,only_probes=False, *args, **kwargs)
Bases: gaudi.objectives.ObjectiveProvider
Hbonds class :param probes: Names of molecules being object of analysis :type probes: list of str :param radius:Maximum distance from any point of probe that is searched
for a possible interaction
Parameters
• distance_tolerance (float, optional) – Allowed deviation from ideal dis-tance to consider a valid H bond.
• angle_tolerance (float, optional) – Allowed deviation from ideal angle toconsider a valid H bond.
• only_intermolecular (boolean, optional) – Only intermolecular interactionsare considered (defaults to True)
• only_probes (boolean, optional) – Only interactions between probe moleculesare considered, excluding other molecule genes. (defaults to False)
Returns Number of detected Hydrogen bonds.
Return type int
display(bonds)Mock method to show a graphical depiction of the found H Bonds.
evaluate(ind)Find H bonds within self.radius angstroms from self.probes, and return only those that interact with probe.Ie, discard those hbonds in that search space whose none of their atoms involved are not part of self.probe.
molecules(ind)
probes(ind)
gaudi.objectives.hbonds.enable(**kwargs)
10.3.9 Inertia objective
This objective calculates the alignment between the axes of inertia of the given molecules.
10.3. gaudi.objectives 57

GaudiMM Documentation, Release 0.0.8+1.g7374b62
class gaudi.objectives.inertia.AxesOfInertia(reference=None, targets=None,only_primaries=False, threshold=0.84,*args, **kwargs)
Bases: gaudi.objectives.ObjectiveProvider
Calculates the axes of inertia of given molecules and returns their alignment deviation.
Parameters
• reference (str) – Molecule name targets should align to.
• targets (list of str) – Names of molecules to be aligned to reference
• threshold (float) – Target average of cosine of angle of alignment between targetsand reference.
• only_primaries (bool) – Consider only the largest inertia vectors.
Returns Mean absolute difference of threshold alignment and mean of all the cosines involved foreach axis.
Return type float
evaluate(individual)Return the score of the individual under the current conditions.
reference(individual)The reference molecule. Usually, the biggest in size
targets(individual)
gaudi.objectives.inertia.calculate_alignment(reference_axis, *probes_axes)
gaudi.objectives.inertia.calculate_axes_of_inertia(molecule)
gaudi.objectives.inertia.calculate_inertial_matrix(coordinates, masses)
gaudi.objectives.inertia.centroid(coordinates, masses)
gaudi.objectives.inertia.enable(**kwargs)
10.3.10 LigScore objective
This objective is a wrapper around the scoring fuction provided by IMP’s ligand_score.
The lower, the better, so usually you will use a -1.0 weight.
class gaudi.objectives.ligscore.LigScore(proteins=(’Protein’, ), ligands=(’Ligand’, ),method=’pose’, binary=None, library=None,*args, **kwargs)
Bases: gaudi.objectives.ObjectiveProvider
LigScore class
Parameters
• proteins (list of str) – The name of molecules that are acting as proteins
• ligands (list of str) – The name of molecules that are acting as ligands
• binary (str, optional) – Path to ligand_score executable
• library (str, optional) – Path to LigScore lib file
Returns Interaction energy as reported by IMP’s ligand_score.
58 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
Return type float
clean()
evaluate(ind)Run a subprocess calling LigScore binary with provided options, and parse the results. Clean tmp files atexit.
get_molecule_by_name(ind, *names)Get a molecule gene instance of individual by its name
parse_output(stream)Get last word of first line (and unique) and parse it into float
prepare_command(protein_path, ligand_path)
prepare_ligands(ligands)
prepare_proteins(proteins)
gaudi.objectives.ligscore.enable(**kwargs)
10.3.11 NWChem objective
This objective is a wrapper around NWChem.
It expects an additional input template with the keyword $MOLECULE, which will be replaced by the currentlyexpressed molecule(s). See TEMPLATE for an example, which works as a default template if none is provided.
A ~/.nwchemrc file should be present. If you installed NWChem with our conda recipe, you will find the file in$CONDA_PREFIX/etc/default.nwchemrc. Copy it to your $HOME.
class gaudi.objectives.nwchem.NWChem(template=None, targets=(’Ligand’, ), parser=None, ti-tle=None, executable=None, basis_library=None, pro-cessors=None, *args, **kwargs)
Bases: gaudi.objectives.ObjectiveProvider
NWChem class
Parameters
• targets (list of str) – Molecule name(s) to be processed with NWChem. Smallones!
• template (str, optional) – NWChem input template (or path to a file with suchcontents) containing a $MOLECULE placeholder to be replaced by the currently expressedmolecule(s) requested in targets, and optionally, a $TITLE placeholder to be replacedby the job name. If not provided, it will default to the TEMPLATE example (single-point dftenergy).
• parser (str, optional) – Path to a Python script containing a top-level functioncalled parse_output which will parse the NWChem output and return a float. This replacesthe default parser, which looks for the last ‘Total <whatever> energy’ value.
• processors (int, optional=None) – Number of physical processors to use withopenmpi
Returns Any numeric value as reported by the parser routines. By default, last ‘Total <whatever>energy’ value.
Return type float
clean()
10.3. gaudi.objectives 59

GaudiMM Documentation, Release 0.0.8+1.g7374b62
evaluate(ind)Run a subprocess calling DSX binary with provided options, and parse the results. Clean tmp files at exit.
get_molecule_by_name(ind, *names)Get a molecule gene instance of individual by its name
get_xyz(*molecules)
parse_output(stream)
prepare_nwfile(*molecules)
gaudi.objectives.nwchem.enable(**kwargs)
10.3.12 Solvation objective
This objective calculates SASA for the given system (or region).
class gaudi.objectives.solvation.Solvation(targets=None, threshold=0.0, radius=5.0,method=’area’, *args, **kwargs)
Bases: gaudi.objectives.ObjectiveProvider
Solvation class
Parameters
• targets ([str]) – Names of the molecule genes being analyzed
• threshold (float, optional, default=0) – Optimize the difference to thisvalue
• radius (float, optional, default=5.0) – Max distance to search for neighboratoms from targets.
• method (str, optional, default=area) – Which method should be used. Bothmethods compute the surface of the solvated molecule. area returns the surface area of suchsurface, while volume returns the volume occuppied by the model.
Returns Surface area of solvated shell, in A2 (if method=area), or volume of solvated shell, in A3
(if method=volume).
Return type float
evaluate_area(ind)
evaluate_volume(ind)
molecules(ind)
surface(ind)
targets(ind)
zone_atoms(probes, molecules)
gaudi.objectives.solvation.enable(**kwargs)
gaudi.objectives.solvation.grid_sas_surface(atoms, probe_radius=1.4,grid_spacing=0.5)
Stripped from Chimera’s Surface.gridsurf
60 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
10.3.13 Vina objective
This objective is a wrapper around the scoring functions provided by AutoDock Vina.
class gaudi.objectives.vina.Vina(receptor=’Protein’, ligand=’Ligand’, prepare_each=False,*args, **kwargs)
Bases: gaudi.objectives.ObjectiveProvider
Vina class
Parameters
• receptor (str) – Key of the gene containing the molecule acting as receptor (protein)
• ligand (str) – Key of the gene containing the molecule acting as ligand
• prepare_each (bool) – Whether to prepare receptors and ligands in every evaluationor try to cache the results for faster performance.
Returns Interaction energy in kcal/mol, as reported by AutoDock Vina –score-only.
Return type float
Notes
• AutoDock scripts prepare_ligand4.py and prepare_receptor4.py are
used to prepare the corresponding .pdqt files that will be used as input for AutoDock Vina scorer. - No repairs norcleanups will be performed on ligand/receptor molecules, so the user has to take into account that provided .mol2or .pdb files have correct atom types and correct structure (including Hydrogen atoms that will be taken into ac-count in the docking evaluation). Otherwise, AutoDock errors/warnings could appear (e.g. ValueError:Could not find atomic number for Lp Lp) - Gasteiger charges will be added during the prepara-tion of the .pdbqt files. - All torsions of the ligand will be marked as inactive for AutoDock, because torsionchanges are part of GaudiMM genes.
clean()
evaluate(ind)Run a subprocess calling Vina binary with provided options, and parse the results. Clean tmp files at exit.
parse_output(stream)
prepare_ligand(molecule)
prepare_receptor(molecule)
tmpfile
gaudi.objectives.vina.enable(**kwargs)
10.3.14 Volume objective
This objective calculates the volume occupied by the requested Molecule gene instance.
Note: Volume is calculated from the surfacePiece created by a new experimental method found in Chimera’sSurface.gridsurf. This could be used as an objective for SES, instead of Solvation.
10.3. gaudi.objectives 61

GaudiMM Documentation, Release 0.0.8+1.g7374b62
class gaudi.objectives.volume.Volume(threshold=0.0, target=None, cavities=False, *args,**kwargs)
Bases: gaudi.objectives.ObjectiveProvider
Volume class
Parameters
• threshold (float or 'auto') – Final volume to target. If ‘auto’, it will calculatethe sum of VdW volumes of all requested atoms in probes. (Unimplemented!)
• target (list of str) – Molecule gene name to calculate volume over
• cavities (boolean, optional, default=False) – If True, evaluate cavitiesvolume creating a convex hull and calculating the difference between convex hull volumeand molecule volume
Returns Calculated volume in A3
Return type float
evaluate_convexhull(ind)
evaluate_volume(ind)
target(ind)
gaudi.objectives.volume.convexhull_volume(surface)This function gets a surface, creates the convex hull and calculates its volume
Parameters surface (Surface.gridsurf.ses_surface(molecule.atoms)) –
Returns volume – Convex hull volume
Return type float
Notes
Some systems may produce small volume blobs, resulting in a number of different surface pieces.This should be discussed in the future. # points = surface.surfacePieces[0].geometry[0] # convexhull =scipy.spatial.ConvexHull(points)
gaudi.objectives.volume.enable(**kwargs)
10.3.15 Base class for all objectives
Objectives are modules that reside in the gaudi.objectives package, and have a certain class structure
class gaudi.objectives.ObjectiveProvider(environment=None, name=None, weight=None,zone=None, precision=3, **kwargs)
Bases: object
Base class that every objectives plugin MUST inherit.
Mount point for plugins implementing new objectives to be evaluated by DEAP. The objective resides withinthe Fitness attribute of the individual. Do whatever you want, but use an evaluate() function to return the results.Apart from that, there’s no requirements.
The base class includes some useful attributes, so don’t forget to call ObjectiveProvider.__init__ in your over-riden __init__. For example, self.zone is a Chimera.selection.ItemizedSelection object which is shared amongall objectives. Use that to get atoms in the surrounding of the target gene, and remember to self.zone.clear() itbefore use.
62 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
— From (M.A. itself)[http://martyalchin.com/2008/jan/10/simple-plugin-framework/]: Now that we have amount point, we can start stacking plugins onto it. As mentioned above, individual plugins will subclass themount point. Because that also means inheriting the metaclass, the act of subclassing alone will suffice as pluginregistration. Of course, the goal is to have plugins actually do something, so there would be more to it than justdefining a base class, but the point is that the entire contents of the class declaration can be specific to the pluginbeing written. The plugin framework itself has absolutely no expectation for how you build the class, allowingmaximum flexibility. Duck typing at its finest.
classmethod clear_cache()
evaluate(individual)Return the score of the individual under the current conditions.
plugins = [<class 'gaudi.objectives.angle.Angle'>, <class 'gaudi.objectives.contacts.Contacts'>, <class 'gaudi.objectives.coordination.Coordination'>, <class 'gaudi.objectives.distance.Distance'>, <class 'gaudi.objectives.dsx.DSX'>, <class 'gaudi.objectives.energy.Energy'>, <class 'gaudi.objectives.gold.Gold'>, <class 'gaudi.objectives.hbonds.Hbonds'>, <class 'gaudi.objectives.inertia.AxesOfInertia'>, <class 'gaudi.objectives.ligscore.LigScore'>, <class 'gaudi.objectives.nwchem.NWChem'>, <class 'gaudi.objectives.solvation.Solvation'>, <class 'gaudi.objectives.vina.Vina'>, <class 'gaudi.objectives.volume.Volume'>]
classmethod validate(data, schema=None)
classmethod with_validation(**kwargs)
10.4 gaudi.algorithms
This module implements evolutionary algorithms as seen in DEAP, and extends their functionality to make use ofGAUDI goodies.
Todo:
• Genealogy
gaudi.algorithms.dump_population(population, cfg, subdir=None)
gaudi.algorithms.ea_mu_plus_lambda(population, toolbox, mu, lambda_, cxpb, mutpb, ngen,cfg, stats=None, halloffame=None, verbose=True,prompt_on_exception=True)
This is the (𝜇+ 𝜆) evolutionary algorithm.
Parameters
• population – A list of individuals.
• toolbox – A Toolbox that contains the evolution operators.
• mu – The number of individuals to select for the next generation.
• lambda_ – The number of children to produce at each generation.
• cxpb – The probability that an offspring is produced by crossover.
• mutpb – The probability that an offspring is produced by mutation.
• ngen – The number of generation.
• stats – A Statistics object that is updated inplace, optional.
• halloffame – A HallOfFame object that will contain the best individuals, optional.
• verbose – Whether or not to log the statistics.
Returns The final population.
10.4. gaudi.algorithms 63

GaudiMM Documentation, Release 0.0.8+1.g7374b62
First, the individuals having an invalid fitness are evaluated. Then, the evolutionary loop begins by producinglambda_ offspring from the population, the offspring are generated by a crossover, a mutation or a reproductionproportionally to the probabilities cxpb, mutpb and 1 - (cxpb + mutpb). The offspring are then evaluated andthe next generation population is selected from both the offspring and the population. Briefly, the operators areapplied as following
evaluate(population)for i in range(ngen):
offspring = varOr(population, toolbox, lambda_, cxpb, mutpb)evaluate(offspring)population = select(population + offspring, mu)
This function expects toolbox.mate(), toolbox.mutate(), toolbox.select() and toolbox.evaluate() aliases to be registered in the toolbox. This algorithm uses the varOr() variation.
10.5 gaudi.base
Contains the core classes we use to build individuals (potential solutions of the optimization process).
class gaudi.base.BaseIndividual(cfg=None, cache=None, **kwargs)Bases: object
Base class for individual objects that are evaluated by DEAP.
Each individual is a potential solution. It contains all that is needed for an evaluation. With multiprocessing inmind, individuals should be self-contained so it can be passed between threads.
The defined methods are only wrapper calls to the respective methods of each gene.
Parameters
• cfg (gaudi.parse.Settings) – The full parsed object from the configuration YAMLfile.
• cache (dict or dict-like) – A mutable object that can be used to store valuesacross instances.
• dummy (bool) – If True, create an uninitialized Individual, only containing the cfg at-tribute. If false, call __ready__ and complete initialization.
__CACHEClass attribute that caches gene data across instances
Type dict
__CACHE_OBJClass attribute that caches objectives data across instances
Type dict
Todo: write() should use Pickle and just save the whole object, but Chimera’s inmutable objects (Atoms,Residues, etc) get in the way. A workaround may be found if we take a look a the session saving code.
clear_cache()
evaluate(environment)Express individual, evaluate it and unexpress it.
Parameters environment (Environment) – Objectives that will evaluate the individual
64 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
express()Express genes in this environment. Very much like ‘compiling’ the individual to a chimera.Molecule.
mate(other)Recombine genes of self with other. It simply calls mate on each gene instance
Parameters other (Individual) – Another individual to mate with.
mutate(indpb)Trigger a round of possible mutations across all genes
Parameters indpb (float) – Probability of suffering a mutation
post_express()
post_unexpress()
pre_express()
pre_unexpress()
similar(other)Compare self and other with a similarity function.
Returns
Return type bool
unexpress()Undo .express()
write(i, path=None)Export the individual to a mol2 file
Parameters
• i (int) – Individual identificator in current generation or hall of fame
• note :: (.) – Maybe someday we can pickle it all :/ >>> filename = os.path.join(path,‘{}_{}.pickle.gz’.format(name,i)) >>> with gzip.GzipFile(filename, ‘wb’) as f: >>>cPickle.dump(self, f, 0) >>> return filename
class gaudi.base.Environment(cfg=None, *args, **kwargs)Bases: object
Objective container and helper to evaluate an individual. It must be instantiated with a gaudi.parse.Settingsobject.
Parameters cfg (gaudi.parse.Settings) – The parsed configuration YAML file that con-tains objectives information
clear_cache()
evaluate(individual)individual : Individual
class gaudi.base.Fitness(weights)Bases: deap.base.Fitness
wvalues = ()
class gaudi.base.MolecularIndividual(*args, **kwargs)Bases: gaudi.base.BaseIndividual
find_molecule(name)
post_express()
10.5. gaudi.base 65

GaudiMM Documentation, Release 0.0.8+1.g7374b62
xyz(gene=None)
gaudi.base.expressed(*args, **kwds)
10.6 gaudi.box
This module is a messy collection of useful functions used all along GAUDI.
Todo: Some of these functions are hardly used, so maybe we should clean it a little in the future. . .
gaudi.box.atoms_between(atom1, atom2)Finds all connected atoms between two given atoms
gaudi.box.atoms_by_serial(*serials, **kw)Find atoms in kw[‘atoms’] with serialNumber = serials.
Parameters
• serials (int) – List of serial numbers to match
• atoms (list of chimera.Atom, optional) – List of atoms to be traversed whilelooking for serial numbers
Returns
Return type list of chimera.Atom
gaudi.box.create_single_individual(path)Create an individual within Chimera. Convenience method for Chimera IDLE.
gaudi.box.do_cprofile(func)Decorator to cProfile a certain function and output the results to cprofile.out
gaudi.box.draw_interactions(interactions, startCol=’FF0000’, endCol=’FFFF00’, key=None,name=’Custom pseudobonds’)
Draw pseudobonds depicting atoms relationships.
Parameters
• interactions (list of tuples) – Each tuple contains an interaction, defined, atleast, by the two atoms involved.
• startCol (str, optional) – Hex code for the initial color of the pseudobond (closerto the first atom of the pair).
• endCol (str, optional) – Hex code for the final color of the pseudobond. (closer tothe second atom of the pair)
• key (int, optional) – The index of an interaction tuple that represent the alpha chan-nel in the color used to depict the interaction.
• name (str, optional) – Name of the pseudobond group created.
Returns
Return type chimera.pseudoBondGroup
gaudi.box.files_in(path, ext=None)Returns all the files in a given directory, filtered by extension if desired.
Parameters
66 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
• path (str) –
• ext (list of str, optional) – File extension(s) to filter on.
Returns
Return type List of absolute paths
gaudi.box.find_nearest(anchor, atoms)Find the atom of atoms that is closer to anchor, in terms of number of atoms in between.
gaudi.box.highest_atom_indices(r)Returns a dictionary with highest atom indices in given residue Key: value -> element.name: highest index inresidue
gaudi.box.incremental_existing_path(path, separator=’__’)
gaudi.box.open_models_and_close(*args, **kwds)
gaudi.box.pseudobond_to_bond(molecule, remove=False)Transforms every pseudobond in molecule to a covalent bond
Parameters
• molecule (chimera.Molecule) –
• remove (bool) – If True, remove original pseudobonds after actual bonds have been cre-ated.
gaudi.box.rmsd(a, b)
gaudi.box.sequential_bonds(atoms, s)Returns bonds in atoms in sequential order, beginning at atom s
gaudi.box.silent_stdout(*args, **kwds)
gaudi.box.stdout_to_file(workspace, stderr=True)
gaudi.box.suppress_ksdssp(trig_name, my_data, molecules)Monkey-patch Chimera triggers to disable KSDSSP computation
gaudi.box.write_individuals(inds, outpath, name, evalfn, remove=True)Write an individual to disk.
Note: Deprecated since an Individual object is able to write itself to disk.
10.7 gaudi.exceptions
This module collects more meaningful exceptions than builtins.
exception gaudi.exceptions.AtomsNotFoundBases: exceptions.Exception
exception gaudi.exceptions.MoleculesNotFoundBases: exceptions.Exception
exception gaudi.exceptions.ResiduesNotFoundBases: exceptions.Exception
exception gaudi.exceptions.TooManyAtomsBases: exceptions.Exception
10.7. gaudi.exceptions 67

GaudiMM Documentation, Release 0.0.8+1.g7374b62
exception gaudi.exceptions.TooManyResiduesBases: exceptions.Exception
10.8 gaudi.parallel
Helper functions to deal with parallel execution of GaudiMM jobs.abs
Useful for benchmarks.
gaudi.parallel.run_parallel(fn, args=(), processes=None, initializer=None, initargs=(), max-tasksperchild=1, map_timeout=9999999, map_chunksize=1,map_callback=None)
Create a pool instance with built-in exception handling.
10.9 gaudi.parse
This module parses and validates YAML input files into convenient objects that allow per-attribute access to configu-ration parameters.
gaudi.parse.AssertList(*validators, **kwargs)Make sure the value is contained in a list
gaudi.parse.Coordinates(v)
gaudi.parse.Degrees(v)
gaudi.parse.ExpandUserPathExists(v)
gaudi.parse.Importable(v)
gaudi.parse.MakeDir(validator)
class gaudi.parse.MoleculeAtom(molecule, atom)Bases: tuple
atomAlias for field number 1
moleculeAlias for field number 0
class gaudi.parse.MoleculeResidue(molecule, residue)Bases: tuple
moleculeAlias for field number 0
residueAlias for field number 1
gaudi.parse.Molecule_name(v)Ideal implementation:
def fn(v):valid = [i['name'] for i in items if i['module'] == 'gaudi.genes.molecule']if v not in valid:
raise Invalid("{} is not a valid Molecule name".format(v))return v
return fn
68 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
However, I must figure a way to get the gene list beforehand
gaudi.parse.Named_spec(*names)Assert that str is formatted like “Molecule/123”, with Molecule being a valid name of a Molecule gene and 123a positive int or *
gaudi.parse.RelPathToInputFile(inputpath=None)
gaudi.parse.ResidueThreeLetterCode(v)
class gaudi.parse.Settings(path=None, validation=True)Bases: munch.Munch
Parses a YAML input file with PyYAML, validates it with voluptuous and builds a attribute-accessible dict withMunch.
Hence, all the attributes in this class are generated automatically from the default values, and the updated withthe contents of the YAML file.
Parameters path (str) – Path to YAML file
outputContains the parameters to determine how to write and report results.
Type dict
output.pathDirectory that will contain the result files. If it does not exist, it will be created. If it does, the contentscould be overwritten. Better change this between different attempts.
Type str, optional, defaults to . (current dir)
output.nameA small identifier for your calculation. If not set, it will use five random characters.
Type str, optional
output.precisionHow many decimals should be used along the simulation. This won’t only affect reports, but also thereported scores by the objectives during the selection process.
Type int, optional, defaults to 3
output.compressWhether to apply compression to the individual ZIP files or not.
Type bool, optional, defaults to True
output.historyWhether to save all the genealogy of the individuals created along the simulation or not. Only for advancedusers.
Type bool, optional, defaults to False
output.paretoIf True, the elite population will be the Pareto front of the population. If False, the elite population will bethe best solutions, according to the lexicographic sorting of the fitness values.
Type bool, optional, defaults to True
output.verboseWhether to realtime report the progress of the simulation or not.
Type bool, optional, defaults to True
10.9. gaudi.parse 69

GaudiMM Documentation, Release 0.0.8+1.g7374b62
output.check_everyDump the elite population every n generations. Switched off if set to 0.
Type int, optional, defaults to 10
output.prompt_on_exceptionWhen an exception is raised, GaudiMM tries to dump the current population to disk as an emergencyrescue plan. This includes pressing Ctrl+C. If this happens, it prompts the user whether to dump it or not.For interactive sessions this is desirable, but no so much for unsupervised cluster jobs. If set to False, thisbehaviour will be disabled.
Type bool, optional, defaults to True
gaContains the genetic algorithm parameters.
Type dict
ga.populationSize of the starting population, in number of individuals.
Type int
ga.generationsNumber of generations to simulate.
Type float
ga.muThe number of children to select at each generation, expressed as a multiplier of ga.population.
Type float, optional, defaults to 1.0
ga.lambda_The number of children to produce at each generation, expressed as a multiplier of ga.population.
Type float, optional, defaults to 3.0
ga.mut_etaCrowding degree of the mutation. A high eta will produce a mutant resembling its parent, while a smalleta will produce a solution much more different.
Type float, optional, defaults to 5
ga.mut_pbThe probability that an offspring is produced by mutation.
Type float, optional, defaults to 0.5
ga.mut_indpbIndependent probability for each gene to be mutated.
Type float, optional, defaults to 0.75
ga.cx_etaCrowding degree of the crossover. A high eta will produce children resembling to their parents, while asmall eta will produce solutions much more different.
Type float, optional, defaults to 5
ga.cx_pbThe probability that an offspring is produced by crossover.
Type float, optional, defaults to 0.5
70 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
similarityContains the parameters to the similarity operator, which, given two individuals with the same fitness,whether they can be considered the same solution or not.
Type dict
similarity.moduleThe function to call when a fitness draw happens. It should be expressed as Python importable path; ie,separated by dots: gaudi.similarity.rmsd.
Type str
similarity.argsPositional arguments to the similarity function.
Type list
similarity.kwargsOptional arguments to the similarity function.
Type dict
genesContains the list of genes that each Individual will have.
Type list of dict
objectivesContains the list of objectives that will make the Environment object to evaluate the Individuals.
Type list of dict
default_values = {'ga': {'cx_eta': 5, 'cx_pb': 0.5, 'generations': 3, 'lambda_': 3, 'mu': 1, 'mut_eta': 5, 'mut_indpb': 0.75, 'mut_pb': 0.5, 'population': 10}, 'genes': [{}], 'objectives': [{}], 'output': {'check_every': 10, 'compress': True, 'history': False, 'name': 'dLKBW', 'pareto': True, 'path': '.', 'precision': 3, 'prompt_on_exception': True, 'verbose': True}, 'similarity': {'args': [['Ligand'], 2.5], 'kwargs': {}, 'module': 'gaudi.similarity.rmsd'}}
name_objectives
schema = {'genes': All(Length(min=1, max=None), [<type 'dict'>], msg=None), 'objectives': All(Length(min=1, max=None), [<type 'dict'>], msg=None), 'output': {'check_every': All(Coerce(int, msg=None), Range(min=0, max=None, min_included=True, max_included=True, msg=None), msg=None), 'compress': Coerce(bool, msg=None), 'history': Coerce(bool, msg=None), 'name': All(<type 'basestring'>, Length(min=1, max=255), msg=None), 'pareto': Coerce(bool, msg=None), 'path': <function fn>, 'precision': All(Coerce(int, msg=None), Range(min=-3, max=6, min_included=True, max_included=True, msg=None), msg=None), 'prompt_on_exception': Coerce(bool, msg=None), 'verbose': Coerce(bool, msg=None)}, 'similarity': {'args': <type 'list'>, 'kwargs': <type 'dict'>, 'module': <type 'basestring'>}, '_path': <function ExpandUserPathExists>, 'ga': {'cx_eta': All(Coerce(int, msg=None), Range(min=0, max=None, min_included=True, max_included=True, msg=None), msg=None), 'cx_pb': All(Coerce(float, msg=None), Range(min=0, max=1, min_included=True, max_included=True, msg=None), msg=None), 'generations': All(Coerce(int, msg=None), Range(min=0, max=None, min_included=True, max_included=True, msg=None), msg=None), 'lambda_': All(Coerce(float, msg=None), Range(min=0, max=None, min_included=True, max_included=True, msg=None), msg=None), 'mu': All(Coerce(float, msg=None), Range(min=0, max=1, min_included=True, max_included=True, msg=None), msg=None), 'mut_eta': All(Coerce(int, msg=None), Range(min=0, max=None, min_included=True, max_included=True, msg=None), msg=None), 'mut_indpb': All(Coerce(float, msg=None), Range(min=0, max=1, min_included=True, max_included=True, msg=None), msg=None), 'mut_pb': All(Coerce(float, msg=None), Range(min=0, max=1, min_included=True, max_included=True, msg=None), msg=None), 'population': All(Coerce(int, msg=None), Range(min=2, max=None, min_included=True, max_included=True, msg=None), msg=None)}}
validate(data=None)
weights
gaudi.parse.deep_update(source, overrides)Update a nested dictionary or similar mapping.
Modify source in place.
gaudi.parse.parse_rawstring(s)It parses reference strings contained in some fields.
These strings contain references to genes.molecule instances, and one of its atoms
gaudi.parse.validate(schema, data)
10.10 gaudi.plugin
This module provides the basic functionality for the plugin system of genes and objectives.
class gaudi.plugin.PluginMount(name, bases, attrs)Bases: type
Base class for plugin mount points.
10.10. gaudi.plugin 71

GaudiMM Documentation, Release 0.0.8+1.g7374b62
Metaclass trickery obtained from Marty Alchin’s blog Each mount point (ie, genes and objectives),MUST inherit this one.
gaudi.plugin.import_plugins(*pluginlist)Import requested modules, only once, when launch.py is called and the configuration is parsed successfully.
Parameters pluginlist (list of gaudi.parse.Param) – Usually, the genes or objec-tives list resulting from the configuration parsing.
gaudi.plugin.load_plugins(plugins, container=None, **kwargs)Requests an instance of the class that resides in each plugin. For genes, each individual has its own instance,but objectives are treated like a singleton. So, they are only instantiated once. That’s the reason behind usen amutable container.
Parameters
• plugins (list of gaudi.parse.Param) – Modules to load. Each Param must have a moduleattr with a full import path.
• container (dict or dict-like) – If provided, use this container to retain instancesacross individuals.
• kwargs – Everything else will be passed to the requested plugin instances.
10.11 gaudi.similarity
This module contains the similarity functions that are used to discard individuals that are not different enough.
gaudi.similarity.rmsd(ind1, ind2, subjects, threshold, *args, **kwargs)Returns the RMSD between two individuals
Parameters
• ind1 (gaudi.base.Individual) –
• ind2 (gaudi.base.Individual) –
• subjects (list of str) – Name of gaudi.genes.molecule instances to measure
• threshold (float) – Maximum RMSD value to consider two individuals as similar. Ifrmsd > threshold, they are considered different.
Returns True if rmsd is within threshold, False otherwise. It will always return False if number ofatoms is not equal in the two Individuals.
Return type bool
10.12 gaudi._cpdrift
Coherent Point Drift (affine and rigid) Python2/3 implementation, adapted from kwohlfahrt’s.
Only 3D points are supported in this version.
Depends on:
• Python 2.7, 3.4+
• Numpy
• Matplotlib (plotting only)
72 Chapter 10. API documentation

GaudiMM Documentation, Release 0.0.8+1.g7374b62
class gaudi._cpdrift.Quaternion(s, i, j, k)Bases: object
axis_angle
conjugate(other=None)
classmethod fromAxisAngle(v, theta)
matrix()
norm()
reciprocal()
unit()
vector
gaudi._cpdrift.RMSD(X, Y)
gaudi._cpdrift.affine_cpd(X, Y, w=0.0, B=None)
gaudi._cpdrift.affine_xform(X, B=<Mock name=’mock.array()’ id=’140309713689552’>, t=0)
gaudi._cpdrift.coherent_point_drift(X, Y, w=0.0, B=None, guess_steps=5,max_iterations=20, method=’affine’)
gaudi._cpdrift.common_steps(X, Y, Y_, w, sigma_sq)
class gaudi._cpdrift.frange(start, stop, step)Bases: object
gaudi._cpdrift.last(sequence)
gaudi._cpdrift.pairwise_sqdist(X, Y)
gaudi._cpdrift.plot(x, y, t)Plot the initial datasets and registration results.
Parameters
• x (ndarray) – The static shape that y will be registered to. Expected array shape is[n_points_x, n_dims]
• y (ndarray) – The moving shape. Expected array shape is [n_points_y, n_dims]. Notethat n_dims should be equal for x and y, but n_points does not need to match.
• t (ndarray) – The transformed version of y. Output shape is [n_points_y, n_dims].
gaudi._cpdrift.rigid_cpd(X, Y, w=0.0, R=None)
gaudi._cpdrift.rigid_xform(X, R=<Mock name=’mock.array()’ id=’140309713689552’>, t=0.0,s=1.0)
gaudi._cpdrift.rotation_matrix(*angles)
gaudi._cpdrift.spaced_rotations(N)
gaudi._cpdrift.std(x)
10.12. gaudi._cpdrift 73

GaudiMM Documentation, Release 0.0.8+1.g7374b62
74 Chapter 10. API documentation

CHAPTER 11
Indices and tables
• genindex
• modindex
• search
75

GaudiMM Documentation, Release 0.0.8+1.g7374b62
76 Chapter 11. Indices and tables

Python Module Index
ggaudi, 31gaudi._cpdrift, 72gaudi.algorithms, 63gaudi.base, 64gaudi.box, 66gaudi.cli, 31gaudi.cli.gaudi_cli, 31gaudi.cli.gaudi_run, 32gaudi.exceptions, 67gaudi.genes, 48gaudi.genes.molecule, 32gaudi.genes.mutamers, 38gaudi.genes.normalmodes, 39gaudi.genes.rotamers, 42gaudi.genes.search, 43gaudi.genes.torsion, 45gaudi.genes.trajectory, 47gaudi.objectives, 62gaudi.objectives.angle, 49gaudi.objectives.contacts, 49gaudi.objectives.coordination, 50gaudi.objectives.distance, 52gaudi.objectives.dsx, 53gaudi.objectives.energy, 54gaudi.objectives.gold, 56gaudi.objectives.hbonds, 57gaudi.objectives.inertia, 57gaudi.objectives.ligscore, 58gaudi.objectives.nwchem, 59gaudi.objectives.solvation, 60gaudi.objectives.vina, 61gaudi.objectives.volume, 61gaudi.parallel, 68gaudi.parse, 68gaudi.plugin, 71gaudi.similarity, 72
77

GaudiMM Documentation, Release 0.0.8+1.g7374b62
78 Python Module Index

Index
Symbols_CATALOG (gaudi.genes.molecule.Molecule attribute),
36__CACHE (gaudi.base.BaseIndividual attribute), 64__CACHE_OBJ (gaudi.base.BaseIndividual attribute),
64_chimera2prody (gaudi.genes.normalmodes.NormalModes
attribute), 40_original_coords (gaudi.genes.normalmodes.NormalModes
attribute), 40_traj (gaudi.genes.trajectory.Trajectory attribute), 47
Aacceptor (gaudi.genes.molecule.Compound attribute),
33add_dummy_atom() (gaudi.genes.molecule.Compound
method), 34add_hydrogens() (gaudi.genes.molecule.Compound
method), 34add_hydrogens_to_isolated_rotamer()
(gaudi.genes.mutamers.Mutamers staticmethod), 38
affine_cpd() (in module gaudi._cpdrift), 73affine_xform() (in module gaudi._cpdrift), 73alg3() (in module gaudi.genes.normalmodes), 41all_chis() (gaudi.genes.rotamers.Rotamers static
method), 43allele (gaudi.genes.molecule.Molecule attribute), 36allele (gaudi.genes.mutamers.Mutamers attribute), 38allele (gaudi.genes.normalmodes.NormalModes at-
tribute), 40allele (gaudi.genes.rotamers.Rotamers attribute), 43allele (gaudi.genes.search.Search attribute), 44allele (gaudi.genes.torsion.Torsion attribute), 46allele (gaudi.genes.trajectory.Trajectory attribute), 47anchor (gaudi.genes.torsion.Torsion attribute), 46Angle (class in gaudi.objectives.angle), 49append() (gaudi.genes.molecule.Compound method),
34
apply_pdbfix() (gaudi.genes.molecule.Compoundmethod), 34
args (gaudi.parse.Settings.similarity attribute), 71AssertList() (in module gaudi.parse), 68atom (gaudi.parse.MoleculeAtom attribute), 68atoms() (gaudi.objectives.distance.Distance method),
52atoms_between() (in module gaudi.box), 66atoms_by_serial() (in module gaudi.box), 66AtomsNotFound, 67attach() (gaudi.genes.molecule.Compound method),
34AxesOfInertia (class in gaudi.objectives.inertia), 57axis_angle (gaudi._cpdrift.Quaternion attribute), 73
BBaseIndividual (class in gaudi.base), 64BONDS_ROTS (gaudi.genes.torsion.Torsion attribute),
46build() (gaudi.genes.molecule.Molecule method), 37built_atoms (gaudi.genes.molecule.Compound at-
tribute), 33
Ccalculate_alignment() (in module
gaudi.objectives.inertia), 58calculate_axes_of_inertia() (in module
gaudi.objectives.inertia), 58calculate_energy()
(gaudi.objectives.energy.Energy method),55
calculate_energy() (in modulegaudi.objectives.energy), 55
calculate_inertial_matrix() (in modulegaudi.objectives.inertia), 58
calculate_prody_normal_modes()(gaudi.genes.normalmodes.NormalModesmethod), 40
center (gaudi.genes.search.Search attribute), 44
79

GaudiMM Documentation, Release 0.0.8+1.g7374b62
center() (in module gaudi.genes.search), 45center() (in module gaudi.genes.torsion), 47centroid() (in module gaudi.objectives.inertia), 58check_every (gaudi.parse.Settings.output attribute),
69chimera_molecule_to_openmm_positions()
(gaudi.objectives.energy.Energy static method),55
chimera_molecule_to_openmm_topology()(gaudi.objectives.energy.Energy static method),55
chimeracoords2numpy() (in modulegaudi.genes.normalmodes), 41
choice() (gaudi.genes.mutamers.Mutamers method),39
chunker() (in module gaudi.genes.normalmodes), 41clean() (gaudi.objectives.dsx.DSX method), 54clean() (gaudi.objectives.gold.Gold method), 56clean() (gaudi.objectives.ligscore.LigScore method),
59clean() (gaudi.objectives.nwchem.NWChem method),
59clean() (gaudi.objectives.vina.Vina method), 61clear_cache() (gaudi.base.BaseIndividual method),
64clear_cache() (gaudi.base.Environment method), 65clear_cache() (gaudi.genes.GeneProvider class
method), 48clear_cache() (gaudi.genes.molecule.Molecule
class method), 37clear_cache() (gaudi.genes.torsion.Torsion
method), 46clear_cache() (gaudi.objectives.ObjectiveProvider
class method), 63coherent_point_drift() (in module
gaudi._cpdrift), 73common_steps() (in module gaudi._cpdrift), 73Compound (class in gaudi.genes.molecule), 32compound (gaudi.genes.molecule.Molecule attribute),
37compress (gaudi.parse.Settings.output attribute), 69conjugate() (gaudi._cpdrift.Quaternion method), 73Contacts (class in gaudi.objectives.contacts), 49convert_chimera_molecule_to_prody() (in
module gaudi.genes.normalmodes), 41convexhull_volume() (in module
gaudi.objectives.volume), 62Coordinates() (in module gaudi.parse), 68Coordination (class in
gaudi.objectives.coordination), 50coordination_sphere()
(gaudi.objectives.coordination.Coordinationmethod), 51
create_single_individual() (in module
gaudi.box), 66cx_eta (gaudi.parse.Settings.ga attribute), 70cx_pb (gaudi.parse.Settings.ga attribute), 70
Ddeep_update() (in module gaudi.parse), 71default_values (gaudi.parse.Settings attribute), 71Degrees() (in module gaudi.parse), 68destroy() (gaudi.genes.molecule.Compound method),
34display() (gaudi.objectives.hbonds.Hbonds method),
57Distance (class in gaudi.objectives.distance), 52distance() (in module gaudi.genes.search), 45distance() (in module gaudi.genes.torsion), 47do_cprofile() (in module gaudi.box), 66donor (gaudi.genes.molecule.Compound attribute), 33draw_interactions() (in module gaudi.box), 66DSX (class in gaudi.objectives.dsx), 53dump_population() (in module gaudi.algorithms),
63
Eea_mu_plus_lambda() (in module
gaudi.algorithms), 63echo_banner() (in module gaudi.cli.gaudi_cli), 32enable() (in module gaudi.genes.molecule), 38enable() (in module gaudi.genes.mutamers), 39enable() (in module gaudi.genes.normalmodes), 41enable() (in module gaudi.genes.rotamers), 43enable() (in module gaudi.genes.search), 45enable() (in module gaudi.genes.torsion), 47enable() (in module gaudi.genes.trajectory), 48enable() (in module gaudi.objectives.angle), 49enable() (in module gaudi.objectives.contacts), 50enable() (in module gaudi.objectives.coordination),
52enable() (in module gaudi.objectives.distance), 52enable() (in module gaudi.objectives.dsx), 54enable() (in module gaudi.objectives.energy), 56enable() (in module gaudi.objectives.gold), 57enable() (in module gaudi.objectives.hbonds), 57enable() (in module gaudi.objectives.inertia), 58enable() (in module gaudi.objectives.ligscore), 59enable() (in module gaudi.objectives.nwchem), 60enable() (in module gaudi.objectives.solvation), 60enable() (in module gaudi.objectives.vina), 61enable() (in module gaudi.objectives.volume), 62enable_logging() (in module gaudi.cli.gaudi_run),
32Energy (class in gaudi.objectives.energy), 54Environment (class in gaudi.base), 65evaluate() (gaudi.base.BaseIndividual method), 64evaluate() (gaudi.base.Environment method), 65
80 Index

GaudiMM Documentation, Release 0.0.8+1.g7374b62
evaluate() (gaudi.objectives.angle.Angle method),49
evaluate() (gaudi.objectives.coordination.Coordinationmethod), 51
evaluate() (gaudi.objectives.dsx.DSX method), 54evaluate() (gaudi.objectives.energy.Energy method),
55evaluate() (gaudi.objectives.gold.Gold method), 56evaluate() (gaudi.objectives.hbonds.Hbonds
method), 57evaluate() (gaudi.objectives.inertia.AxesOfInertia
method), 58evaluate() (gaudi.objectives.ligscore.LigScore
method), 59evaluate() (gaudi.objectives.nwchem.NWChem
method), 59evaluate() (gaudi.objectives.ObjectiveProvider
method), 63evaluate() (gaudi.objectives.vina.Vina method), 61evaluate_area() (gaudi.objectives.solvation.Solvation
method), 60evaluate_center_of_mass()
(gaudi.objectives.distance.Distance method),52
evaluate_clashes()(gaudi.objectives.contacts.Contacts method),50
evaluate_convexhull()(gaudi.objectives.volume.Volume method),62
evaluate_distances()(gaudi.objectives.distance.Distance method),52
evaluate_hydrophobic()(gaudi.objectives.contacts.Contacts method),50
evaluate_volume()(gaudi.objectives.solvation.Solvation method),60
evaluate_volume()(gaudi.objectives.volume.Volume method),62
ExpandUserPathExists() (in modulegaudi.parse), 68
express() (gaudi.base.BaseIndividual method), 64express() (gaudi.genes.GeneProvider method), 48express() (gaudi.genes.molecule.Molecule method),
37express() (gaudi.genes.mutamers.Mutamers method),
39express() (gaudi.genes.normalmodes.NormalModes
method), 40express() (gaudi.genes.rotamers.Rotamers method),
43
express() (gaudi.genes.search.Search method), 44express() (gaudi.genes.torsion.Torsion method), 46express() (gaudi.genes.trajectory.Trajectory
method), 47expressed() (in module gaudi.base), 66
Ffiles_in() (in module gaudi.box), 66find_atom() (gaudi.genes.molecule.Molecule
method), 37find_atoms() (gaudi.genes.molecule.Molecule
method), 37find_interactions()
(gaudi.objectives.contacts.Contacts method),50
find_molecule() (gaudi.base.MolecularIndividualmethod), 65
find_nearest() (in module gaudi.box), 67find_residue() (gaudi.genes.molecule.Molecule
method), 37find_residues() (gaudi.genes.molecule.Molecule
method), 37Fitness (class in gaudi.base), 65frange (class in gaudi._cpdrift), 73fromAxisAngle() (gaudi._cpdrift.Quaternion class
method), 73
Gga (gaudi.parse.Settings attribute), 70gaudi (module), 31gaudi._cpdrift (module), 72gaudi.algorithms (module), 63gaudi.base (module), 64gaudi.box (module), 66gaudi.cli (module), 31gaudi.cli.gaudi_cli (module), 31gaudi.cli.gaudi_run (module), 32gaudi.exceptions (module), 67gaudi.genes (module), 48gaudi.genes.molecule (module), 32gaudi.genes.mutamers (module), 38gaudi.genes.normalmodes (module), 39gaudi.genes.rotamers (module), 42gaudi.genes.search (module), 43gaudi.genes.torsion (module), 45gaudi.genes.trajectory (module), 47gaudi.objectives (module), 62gaudi.objectives.angle (module), 49gaudi.objectives.contacts (module), 49gaudi.objectives.coordination (module), 50gaudi.objectives.distance (module), 52gaudi.objectives.dsx (module), 53gaudi.objectives.energy (module), 54gaudi.objectives.gold (module), 56
Index 81

GaudiMM Documentation, Release 0.0.8+1.g7374b62
gaudi.objectives.hbonds (module), 57gaudi.objectives.inertia (module), 57gaudi.objectives.ligscore (module), 58gaudi.objectives.nwchem (module), 59gaudi.objectives.solvation (module), 60gaudi.objectives.vina (module), 61gaudi.objectives.volume (module), 61gaudi.parallel (module), 68gaudi.parse (module), 68gaudi.plugin (module), 71gaudi.similarity (module), 72gaussian_modes() (in module
gaudi.genes.normalmodes), 41GeneProvider (class in gaudi.genes), 48generations (gaudi.parse.Settings.ga attribute), 70genes (gaudi.parse.Settings attribute), 71get() (gaudi.genes.molecule.Molecule method), 37get_molecule_by_name()
(gaudi.objectives.dsx.DSX method), 54get_molecule_by_name()
(gaudi.objectives.gold.Gold method), 56get_molecule_by_name()
(gaudi.objectives.ligscore.LigScore method),59
get_molecule_by_name()(gaudi.objectives.nwchem.NWChem method),60
get_rotamers() (gaudi.genes.mutamers.Mutamersmethod), 39
get_xyz() (gaudi.objectives.nwchem.NWChemmethod), 60
Gold (class in gaudi.objectives.gold), 56grid_sas_surface() (in module
gaudi.objectives.solvation), 60group_by_mass() (in module
gaudi.genes.normalmodes), 41group_by_residues() (in module
gaudi.genes.normalmodes), 42
HHbonds (class in gaudi.objectives.hbonds), 57highest_atom_indices() (in module gaudi.box),
67history (gaudi.parse.Settings.output attribute), 69
Iideal_bond_deviation() (in module
gaudi.objectives.coordination), 52ideal_bonded_positions() (in module
gaudi.objectives.coordination), 52import_plugins() (in module gaudi.plugin), 72Importable() (in module gaudi.parse), 68incremental_existing_path() (in module
gaudi.box), 67
Jjoin() (gaudi.genes.molecule.Compound method), 34
Kkwargs (gaudi.parse.Settings.similarity attribute), 71
Llambda_ (gaudi.parse.Settings.ga attribute), 70last() (in module gaudi._cpdrift), 73launch() (in module gaudi.cli.gaudi_run), 32LigScore (class in gaudi.objectives.ligscore), 58load_chimera() (in module gaudi.cli.gaudi_cli), 32load_frame() (gaudi.genes.trajectory.Trajectory
method), 48load_plugins() (in module gaudi.plugin), 72
Mmain() (in module gaudi.cli.gaudi_run), 32MakeDir() (in module gaudi.parse), 68mate() (gaudi.base.BaseIndividual method), 65mate() (gaudi.genes.GeneProvider method), 48mate() (gaudi.genes.molecule.Molecule method), 37mate() (gaudi.genes.mutamers.Mutamers method), 39mate() (gaudi.genes.normalmodes.NormalModes
method), 40mate() (gaudi.genes.rotamers.Rotamers method), 43mate() (gaudi.genes.search.Search method), 44mate() (gaudi.genes.torsion.Torsion method), 46mate() (gaudi.genes.trajectory.Trajectory method), 48matrix() (gaudi._cpdrift.Quaternion method), 73module (gaudi.parse.Settings.similarity attribute), 71mol (gaudi.genes.molecule.Compound attribute), 33MolecularIndividual (class in gaudi.base), 65Molecule (class in gaudi.genes.molecule), 36molecule (gaudi.genes.normalmodes.NormalModes
attribute), 41molecule (gaudi.genes.search.Search attribute), 44molecule (gaudi.genes.torsion.Torsion attribute), 46molecule (gaudi.genes.trajectory.Trajectory attribute),
48molecule (gaudi.parse.MoleculeAtom attribute), 68molecule (gaudi.parse.MoleculeResidue attribute), 68Molecule_name() (in module gaudi.parse), 68MoleculeAtom (class in gaudi.parse), 68MoleculeResidue (class in gaudi.parse), 68molecules() (gaudi.objectives.contacts.Contacts
method), 50molecules() (gaudi.objectives.coordination.Coordination
method), 51molecules() (gaudi.objectives.energy.Energy
method), 55molecules() (gaudi.objectives.hbonds.Hbonds
method), 57
82 Index

GaudiMM Documentation, Release 0.0.8+1.g7374b62
molecules() (gaudi.objectives.solvation.Solvationmethod), 60
MoleculesNotFound, 67mu (gaudi.parse.Settings.ga attribute), 70mut_eta (gaudi.parse.Settings.ga attribute), 70mut_indpb (gaudi.parse.Settings.ga attribute), 70mut_pb (gaudi.parse.Settings.ga attribute), 70Mutamers (class in gaudi.genes.mutamers), 38mutate() (gaudi.base.BaseIndividual method), 65mutate() (gaudi.genes.GeneProvider method), 48mutate() (gaudi.genes.molecule.Molecule method), 38mutate() (gaudi.genes.mutamers.Mutamers method),
39mutate() (gaudi.genes.normalmodes.NormalModes
method), 41mutate() (gaudi.genes.rotamers.Rotamers method), 43mutate() (gaudi.genes.search.Search method), 44mutate() (gaudi.genes.torsion.Torsion method), 46mutate() (gaudi.genes.trajectory.Trajectory method),
48
Nname (gaudi.parse.Settings.output attribute), 69name_objectives (gaudi.parse.Settings attribute),
71Named_spec() (in module gaudi.parse), 69nearest_atom() (in module gaudi.genes.search), 45nearest_atom() (in module gaudi.genes.torsion), 47nonrotatable (gaudi.genes.molecule.Compound at-
tribute), 33norm() (gaudi._cpdrift.Quaternion method), 73NORMAL_MODE_SAMPLES
(gaudi.genes.normalmodes.NormalModesattribute), 40
NORMAL_MODES (gaudi.genes.normalmodes.NormalModesattribute), 40
NORMAL_MODES_SAMPLES(gaudi.genes.normalmodes.NormalModesattribute), 40
NormalModes (class in gaudi.genes.normalmodes), 39NWChem (class in gaudi.objectives.nwchem), 59
OObjectiveProvider (class in gaudi.objectives), 62objectives (gaudi.parse.Settings attribute), 71open_models_and_close() (in module gaudi.box),
67origin (gaudi.genes.molecule.Compound attribute), 33origin (gaudi.genes.search.Search attribute), 44, 45origin() (gaudi.objectives.gold.Gold method), 56output (gaudi.parse.Settings attribute), 69
Ppairwise_sqdist() (in module gaudi._cpdrift), 73
pareto (gaudi.parse.Settings.output attribute), 69parse_attr() (gaudi.genes.molecule.Compound
method), 35parse_origin() (in module gaudi.genes.search), 45parse_output() (gaudi.objectives.dsx.DSX method),
54parse_output() (gaudi.objectives.gold.Gold
method), 56parse_output() (gaudi.objectives.ligscore.LigScore
method), 59parse_output() (gaudi.objectives.nwchem.NWChem
method), 60parse_output() (gaudi.objectives.vina.Vina
method), 61parse_rawstring() (in module gaudi.parse), 71patch_residue() (gaudi.genes.rotamers.Rotamers
static method), 43path (gaudi.parse.Settings.output attribute), 69place() (gaudi.genes.molecule.Compound method), 35place_for_bonding()
(gaudi.genes.molecule.Compound method), 36plot() (in module gaudi._cpdrift), 73PluginMount (class in gaudi.plugin), 71plugins (gaudi.genes.GeneProvider attribute), 48plugins (gaudi.objectives.ObjectiveProvider at-
tribute), 63population (gaudi.parse.Settings.ga attribute), 70post_express() (gaudi.base.BaseIndividual
method), 65post_express() (gaudi.base.MolecularIndividual
method), 65post_unexpress() (gaudi.base.BaseIndividual
method), 65pre_express() (gaudi.base.BaseIndividual method),
65pre_unexpress() (gaudi.base.BaseIndividual
method), 65precision (gaudi.parse.Settings.output attribute), 69prepare_command() (gaudi.objectives.dsx.DSX
method), 54prepare_command() (gaudi.objectives.gold.Gold
method), 56prepare_command()
(gaudi.objectives.ligscore.LigScore method),59
prepare_ligand() (gaudi.objectives.vina.Vinamethod), 61
prepare_ligands() (gaudi.objectives.dsx.DSXmethod), 54
prepare_ligands() (gaudi.objectives.gold.Goldmethod), 56
prepare_ligands()(gaudi.objectives.ligscore.LigScore method),59
Index 83

GaudiMM Documentation, Release 0.0.8+1.g7374b62
prepare_nwfile() (gaudi.objectives.nwchem.NWChemmethod), 60
prepare_proteins() (gaudi.objectives.dsx.DSXmethod), 54
prepare_proteins() (gaudi.objectives.gold.Goldmethod), 57
prepare_proteins()(gaudi.objectives.ligscore.LigScore method),59
prepare_receptor() (gaudi.objectives.vina.Vinamethod), 61
prepend() (gaudi.genes.molecule.Compound method),36
probe() (gaudi.objectives.coordination.Coordinationmethod), 51
probes() (gaudi.objectives.angle.Angle method), 49probes() (gaudi.objectives.contacts.Contacts method),
50probes() (gaudi.objectives.hbonds.Hbonds method),
57prody_modes() (in module
gaudi.genes.normalmodes), 42prompt_on_exception (gaudi.parse.Settings.output
attribute), 70pseudobond_to_bond() (in module gaudi.box), 67
QQuaternion (class in gaudi._cpdrift), 72
Rrand_xform() (in module gaudi.genes.search), 45random_angle() (gaudi.genes.torsion.Torsion
method), 46random_frame_number()
(gaudi.genes.trajectory.Trajectory method), 48random_transform() (gaudi.genes.search.Search
method), 45random_translation() (in module
gaudi.genes.search), 45read_gaussian_normal_modes()
(gaudi.genes.normalmodes.NormalModesmethod), 41
read_prody_normal_modes()(gaudi.genes.normalmodes.NormalModesmethod), 41
reciprocal() (gaudi._cpdrift.Quaternion method),73
reference() (gaudi.objectives.inertia.AxesOfInertiamethod), 58
RelPathToInputFile() (in module gaudi.parse),69
residue (gaudi.parse.MoleculeResidue attribute), 68residues() (gaudi.objectives.coordination.Coordination
method), 52
ResiduesNotFound, 67ResidueThreeLetterCode() (in module
gaudi.parse), 69retrieve_rotamers()
(gaudi.genes.rotamers.Rotamers method),43
rigid_cpd() (in module gaudi._cpdrift), 73rigid_xform() (in module gaudi._cpdrift), 73RMSD() (in module gaudi._cpdrift), 73rmsd() (in module gaudi.box), 67rmsd() (in module gaudi.similarity), 72Rotamers (class in gaudi.genes.rotamers), 42rotatable_bonds (gaudi.genes.molecule.Compound
attribute), 33rotatable_bonds (gaudi.genes.torsion.Torsion at-
tribute), 47rotate() (in module gaudi.genes.search), 45rotation_matrix() (in module gaudi._cpdrift), 73run_parallel() (in module gaudi.parallel), 68
Sschema (gaudi.parse.Settings attribute), 71Search (class in gaudi.genes.search), 43seed (gaudi.genes.molecule.Compound attribute), 33sequential_bonds() (in module gaudi.box), 67set_vdw_radii() (gaudi.genes.molecule.Compound
method), 36Settings (class in gaudi.parse), 69silent_stdout() (in module gaudi.box), 67similar() (gaudi.base.BaseIndividual method), 65similarity (gaudi.parse.Settings attribute), 70simulation (gaudi.objectives.energy.Energy at-
tribute), 55Solvation (class in gaudi.objectives.solvation), 60spaced_rotations() (in module gaudi._cpdrift), 73std() (in module gaudi._cpdrift), 73stdout_to_file() (in module gaudi.box), 67SUPPORTED_FILETYPES
(gaudi.genes.molecule.Molecule attribute),37
suppress_ksdssp() (in module gaudi.box), 67surface() (gaudi.objectives.solvation.Solvation
method), 60
Ttarget() (gaudi.objectives.volume.Volume method),
62targets() (gaudi.objectives.inertia.AxesOfInertia
method), 58targets() (gaudi.objectives.solvation.Solvation
method), 60test_import() (in module gaudi.cli.gaudi_cli), 32timeit() (in module gaudi.cli.gaudi_cli), 32tmpfile (gaudi.objectives.vina.Vina attribute), 61
84 Index

GaudiMM Documentation, Release 0.0.8+1.g7374b62
to_zero (gaudi.genes.search.Search attribute), 45TooManyAtoms, 67TooManyResidues, 67topology (gaudi.genes.trajectory.Trajectory attribute),
48Torsion (class in gaudi.genes.torsion), 45Trajectory (class in gaudi.genes.trajectory), 47translate() (in module gaudi.genes.search), 45
Uunbuffer_stdout() (in module
gaudi.cli.gaudi_run), 32unexpress() (gaudi.base.BaseIndividual method), 65unexpress() (gaudi.genes.GeneProvider method), 48unexpress() (gaudi.genes.molecule.Molecule
method), 38unexpress() (gaudi.genes.mutamers.Mutamers
method), 39unexpress() (gaudi.genes.normalmodes.NormalModes
method), 41unexpress() (gaudi.genes.rotamers.Rotamers
method), 43unexpress() (gaudi.genes.search.Search method), 45unexpress() (gaudi.genes.torsion.Torsion method),
47unexpress() (gaudi.genes.trajectory.Trajectory
method), 48unit() (gaudi._cpdrift.Quaternion method), 73update_attr() (gaudi.genes.molecule.Compound
method), 36update_rotamer() (gaudi.genes.rotamers.Rotamers
static method), 43update_rotamer_coords()
(gaudi.genes.mutamers.Mutamers staticmethod), 39
Vvalidate() (gaudi.genes.GeneProvider class
method), 49validate() (gaudi.objectives.ObjectiveProvider class
method), 63validate() (gaudi.parse.Settings method), 71validate() (in module gaudi.parse), 71vector (gaudi._cpdrift.Quaternion attribute), 73verbose (gaudi.parse.Settings.output attribute), 69Vina (class in gaudi.objectives.vina), 61Volume (class in gaudi.objectives.volume), 61
Wweights (gaudi.parse.Settings attribute), 71with_validation() (gaudi.genes.GeneProvider
class method), 49
with_validation()(gaudi.objectives.ObjectiveProvider classmethod), 63
write() (gaudi.base.BaseIndividual method), 65write() (gaudi.genes.GeneProvider method), 49write() (gaudi.genes.molecule.Molecule method), 38write_individuals() (in module gaudi.box), 67wvalues (gaudi.base.Fitness attribute), 65
Xxyz() (gaudi.base.MolecularIndividual method), 65xyz() (gaudi.genes.molecule.Molecule method), 38
Zzone_atoms() (gaudi.objectives.solvation.Solvation
method), 60
Index 85
Copyright © 2022 FDOKUMEN




















