
Microviridins DF, serine protease inhibitors from the cyanobacterium Oscillatoria agardhii (NIES-204

International Journal of Mass Spectrometry and Ion Processes 121(1993) 17-26 0168-l 176/93/$06.00 0 1993 - Elsevier Science Publishers B.V. All rights reserved
17
Gas-phase fragmentations of anionic complexes of serine- and threonine-containing peptides
Alex Reiter, Lynn M. Teesch, Hong Zhao, Jeanette Adams*
Department of Chemistry, Emory University, Atlanta, GA 30322, USA
(Received 10 September 1992; accepted 20 April 1993)
Abstract
The (M + Ca - 3H)- and (M - H)- anions of 11 tetra- and larger peptides that contain either Ser or Thr are studied by using fast atom bombardment (FAB) and collision-induced decomposition (CID). The Ser- and Thr-containing negative ions decompose to lose either 30 u (Ser) or 44 u (Thr) in the ion source, metastably, and upon CID. The CID of the FAB- formed (M + Ca - 3H - X)- or (M - H - X)- fragment ions, in which X = 30 or 4lu, is the same as the CID of (M + Ca - 3H)- or (M - H)- anions of peptides in which the Ser and Thr residues are replaced with Gly. Two possible mechanisms for the decompositions are discussed, and theoretical molecular mechanical calculations provide further evidence regarding the mechanisms.
Key words: Peptides; Anions; Calcium; Serine; Threonine.
Introduction
Several investigations of the fundamental gas-phase ion chemistry of cationic complexes between peptides and alkali [l-7] and alkaline earth metal ions [g] have recently been reported. Of even more recent interest is the chemistry of anionic complexes of alkaline earth metal ions [9]. Metastable ion and collision-induced decomposition (CID) of the alkaline earth (M + Cat2+ - 3H)- complexes is strongly depen- dent on the nature of the amino acid residues. For example, the complexes must contain either His, Trp, Tyr, Asp, or Glu to decompose to give N-terminal (c,_, + Cat2+ - 3H)- sequence
* Corresponding author. ’ Publication number seven of the Cherry L. Emerson Center for Scientific Computation.
ions [9(a),(b)]. Molecular mechanical calculations indicate that these sequence ions are structure- specific and contain the metal ion intramolecularly chelated to the deprotonated amino acid side chain and to two deprotonated amide groups [9(b)]. Interestingly, however, (M + Cat2+ - 3H)- com- plexes that contain either Ser (structure 1) or Thr (structure 2) decompose almost exclusively meta- stably and by CID via facile cleavage of the side chains to give losses of 30 u (Ser) and 44u (Thr) [9(c),(d)]. Waugh et al. [lo] also recently reported similar CID of (M - H)- anions of dipeptides that contain either Ser or Thr.
Here, we report a study of both (M + Ca - 3H)- and (M - H)- anions of 11 tetra- and larger peptides that contain either Ser or Thr. We use experimental data and theoretical molecular mechanical calcula- tions to address two possible mechanisms for the fragmentations.

18 A. Reiter et aLlInt. J. Mass Spectrom. Ion Processes 127 (1993) 17-26
NH2-peptide peptide-C02H
Experimental section
Peptides were obtained either from Sigma or from the Emory University Microchemical Facility. They include AYTPAA, AATYPA, AYGPAA, DTHAAA, ADTHAA, YTGLFT, VESSK, VEGGK, VHLTP, VHLGP, and VGSE. The matrix used for fast atom bombardment (FAB) was 2 : 1 thioglycerol/glycerol (T/G) and was obtained from Aldrich. The (M + Ca - 3H)- complexes were prepared by mixing small amounts of the peptides with the FAB matrix, which had been previously saturated with Ca(OH)*.
Mass spectrometric experiments were performed by using a VG 70-S, forward-geometry mass spec- trometer. The mass spectrometer was equipped with an Ion Tech saddle-field FAB gun and a commercial FAB ion source. Precursor ions were produced by bombarding the sample with 7 keV Ar or 8 keV Xe atoms at an atom gun current of 2 mA. Ions produced were accelerated to 8 keV trans- lational energy.
Product ions that were formed by metastable ion or collision-induced decomposition (CID) in the first field-free region between the ion source and the ESA were observed by using linked scans at a constant ratio of B/E. Helium was used as collision gas (= 50% beam reduction). Experiments were performed at a product ion resolution of approxi- mately 1000 (10% valley), and magnet calibration was performed from a mixture of glycerol and CsI.
All spectra were acquired by using VG software, and CID spectra are the result of averaging lo-20 scans. Background spectra were acquired for all experiments in order to eliminate artifact ions that might arise from chemical noise [l 11.
Molecular mechanical calculations were performed by using the program SYBYL, version 6.0 from Tripos Associations, Inc. (Matthew et al. [12]). Calculations were performed for (M + Ca - 3H)- and (M - H)- ions of SSS, VTL, LVT, TVL, VESSK, and AYTPAA. The strain energy is described as the summation of the bond length deformation (E,,,), valence angle deformation (E bend), torsion angle deformation (Et,,,), out-of-plane deformation (E,,,,), non- bonded interaction (Evdw), 1,4 interactions (E,,J,
electrostatic non-bonded interactions (E&, and electrostatic 1,4 interactions (El+&, according to Eq. (1):
+ c 4,4e1e (1)
To prevent the aromatic ring of the Tyr residue in AYTPAA from puckering upon coordination of the metal to the Tyr oxygen, the Tyr residue was treated as an aggregate, which required the addition of an extra term, (E,,), (Eq. (2)):
Gotal = c Estr + c bend + c Ems + c Eoop
+ c -%w + c El,4 + c he
(2)
The strain energies were minimized by using the Powell method [13], and charge was calculated using the Pullman method [14].
The force field used to calculate strain energy for the (M + Ca - 3H)- complexes is a combination of the parameters described by Raos et al. [15] for copper(I1) chelation by amino acids and of the parameters of Tripos [12]. The parameters for the bond length deformation, valence angle deformation, and torsion angle deformation have
A. Reiter et al./Int. J. Mass Spectrom. Ion Processes 127 (1993) 17-26 19
basis in those described by Raos et al. [ 151, whereas the remaining parameters are Tripos parameters. The bond length used for the Ca-0 bond is 2.55& an average of the bond lengths reported by Izac et al. [16] for calcium-oxygen interac- tions. The bond lengths for the Ca-N bonds are those described by Tripos. This combination of the Raos and Tripos parameters was used because it gives better agreement with the X-ray crystal struc- tures of nickel(I1) and copper(I1) tetraglycine [ 171 complexes than the parameters of Tripos alone.
To ensure that a global minimum for the metal ion-peptide complexes was obtained, the com- plexes were varied until the strain energies agreed to within 2.1 kJ mol-’ after an entire cycle through the complexes. The torsion angles of the backbone and bulky side chains, which are not included in the chelate rings, were varied from 0 to 360” in 60” increments, whereas the torsion angles in the chelate rings were varied from - 15 to + 15” in 6” increments. The variations of the torsion angles were performed sequentially beginning with the C-terminus and ending with the N-terminus, fol- lowed by the side chains of the amino acids. The structure with the lowest strain energy from the previous set of conformations that were created by varying the torsion angles, as described above, was used as the starting structure for evaluating the next set of conformations.
To model the hydrogen-bonding interactions in
a 2 1 s AYTPAA
(M-H-44).
:
541
FAB 2 .- 3 - z I
the metal ion complexes, a formal 2+ charge was placed on the calcium ion, a - 1 charge was placed on one of the C-terminal carboxylate oxygens, and the rest of the molecular charge was calculated by the Pullman method [14]. The Ca*+ was also bound to two specific deprotonated sites in the peptide. The complexes were then minimized using the above method.
The force field that was used in the molecular mechanical calculations of the (M - H)- ions was AMBER [18], included in the SYBYL package. To ensure that a global minimum was obtained, the lowest energy structures were those that were obtained after 1000 cycles using a random search of all possible structures [19].
Results and discussion
Eleven tetra- through hexapeptides that contain at least one Ser (1) or Thr (2) residue were studied by negative ion fast atom bombardment (FAB) and metastable ion and collision-induced decompo- sition (CID). All of the peptides also contain one protic amino acid (His, Trp, Tyr, Asp, or Glu). Analogs of three of these peptides in which Gly replaced Ser or Thr were also studied.
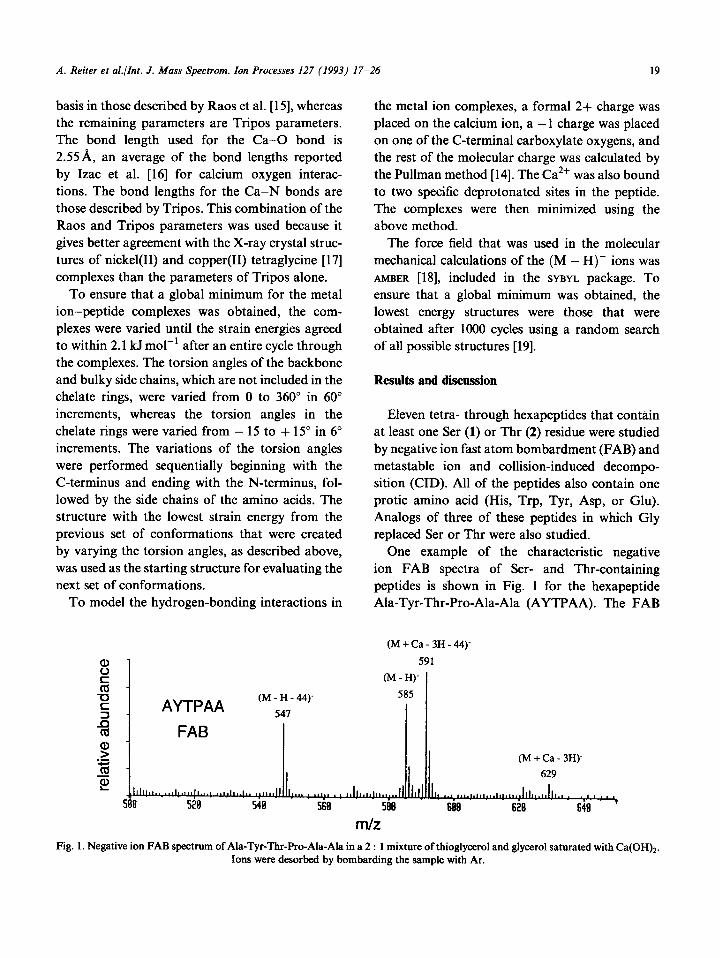
One example of the characteristic negative ion FAB spectra of Ser- and Thr-containing peptides is shown in Fig. 1 for the hexapeptide Ala-Tyr-Thr-Pro-Ala-Ala (AYTPAA). The FAB
(M+Ca-3H-44)‘
591
(M - H)-
585
(M + Ca - 3H)-
629
m/z Fig. 1. Negative ion FAB spectrmn of Ala-Tyr-Thr-Pro-Ala-Ala in a 2 : 1 mixture of thioglycerol and glycerol saturated with Ca(OH)2.
Ions were desorbed by bombarding the sample with Ar.
20 A. Reiter et al./Int. J. Mass Spectrom. Ion Processes 127 (1993) 17-26
. a (AYTPAA + Ca - 3H)-
- CID
(AYTPAA - H)-
$ _ CID
s z 21s 388 -
(VESSK + Ca - 3H)- < NH2
CID -60
T.‘-- 1.. I ..~*.h.
388 488 58E I
-30 517
Id _ (VESSK H)- -18 -
. CID
288 488 588
m/z
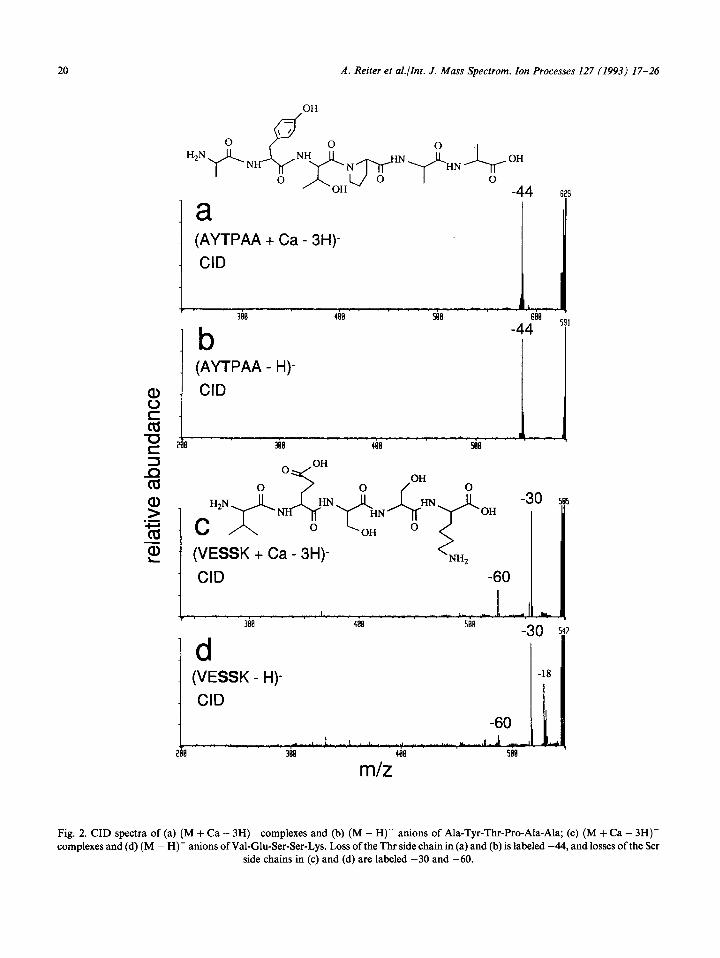
Fig. 2. CID spectra of (a) (M + Ca - 3H)- complexes and (b) (M - H)- anions of Ala-Tyr-Thr-Pro-Ala-Ala; (c) (M + Ca - 3H)- complexes and (d) (M - H)- anions of Val-Glu-Ser-Ser-Lys. Loss of the Thr side chain in (a) and (b) is labeled -44, and losses of the Ser
side chains in (c) and (d) are labeled -30 and -60.

A. Reiter et aLlInt. J. Mass Spectrom. Ion Processes 127 (1993) 17-26 21
spectra contain abundant fragment ions that are the result of loss of 3Ou (Ser) or 44u (Thr) from both (M + Ca - 3H)- and (M - H)- precursors. The fragment ions for AYTPAA are labeled (M + Ca - 3H - 44)- and (M - H - 44)- in
Fig. 1. For most peptides, the (M + Ca - 3H)- complexes are observed in the FAB spectra at very low abundances (ion of m/z 629 in Fig. 1). The (M-H)- ions are more abundant, as are the fragment ions formed via losses of either the Ser (-30~) or Thr (-44 u) side chains even though the matrix is saturated with Ca(OH)2.
Waugh et al. [lo] reported previously that (M - H)- ions of Ser- and Thr-containing dipep- tides fragment by CID to lose 30 or 44u, respec- tively. We find that tetra- and larger peptide (M + Ca - 3H)- complexes and (M - H)- ions also undergo these same losses by both metastable ion and CID. For example, CID spectra of (M + Ca i 3H)- complexes (Fig. 2(a)) and (M - H)- ions (Fig. 2(b)) of AYTPAA only show product ions that arise from cleavage of the Thr side chain (-44 u). Similarly, CID of precursor anions of Val-Glu-Ser-Ser-Lys (VESSK) (Fig. 2(c) and 2(d)) show product ions that result from cleav- ages of a single Ser (-30 u) and of both Ser (-60 u) side chains. No informative sequence ions are observed. These results hold for all peptides studied. Hu and Gross [9(d)] also recently reported these same losses for tripeptides.
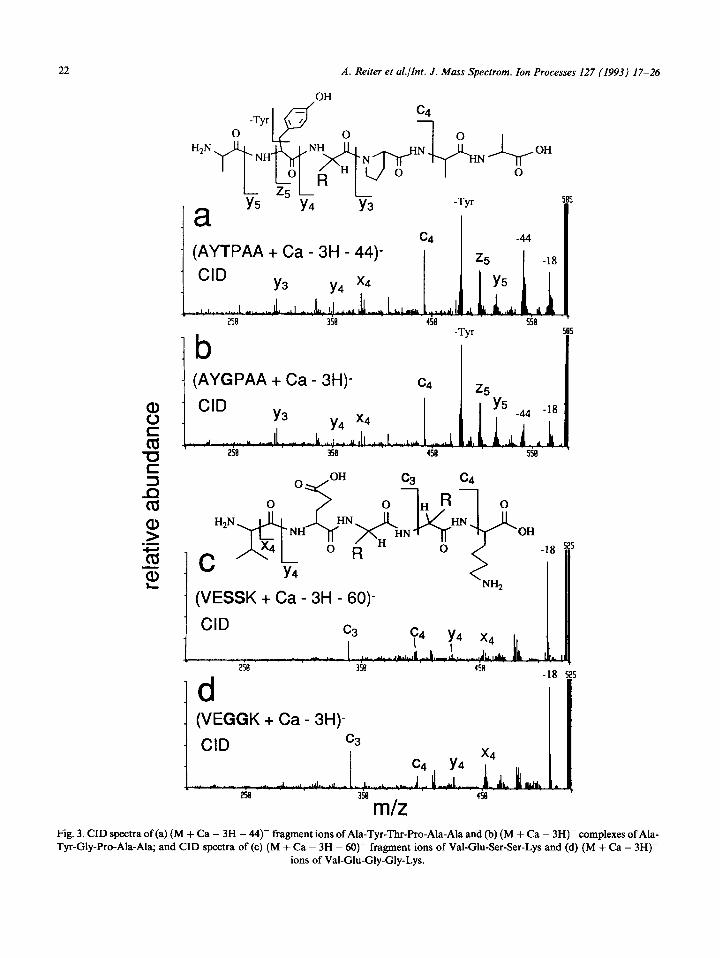
Results from our previously described study of (M + Cat2+ - 3H)- complexes [9(a),(b)] suggested that losses of the side chains could involve a cleav- age in which the side chain is cleaved at the o-carbon and a hydrogen is transferred to the -CH- group of the ionic fragment. The ionic frag- ment would simply be a Gly-containing peptide complex [9(c)]. Hu and Gross [9(d)] also recently presented this same idea. This hypothesis was tested here by comparing the CID spectra of the FAB-formed (M + Ca - 3H - X)- fragment ions, in which X = 30 or 44u, to CID spectra of (M + Ca - 3H)- complexes of analogous Gly- containing peptide complexes. For example, CID of FAB-formed (M + Ca - 3H - 44)- fragment
ions of AYTPAA (Fig. 3(a)) are virtually identical to CID of (M + Ca - 3H)- ions of Ala-Tyr-Gly- Pro-Ala-Ala (AYGPAA) (Fig. 3(b)). Similarly, CID of FAB-formed (M + Ca - 3H - 60)- frag- ment ions of VESSK (Fig. 3(c)) are virtually identical to CID of (M + Ca - 3H)- ions of Val-Glu-Gly-Gly-Lys (VEGGK) (Fig. 3(d)).
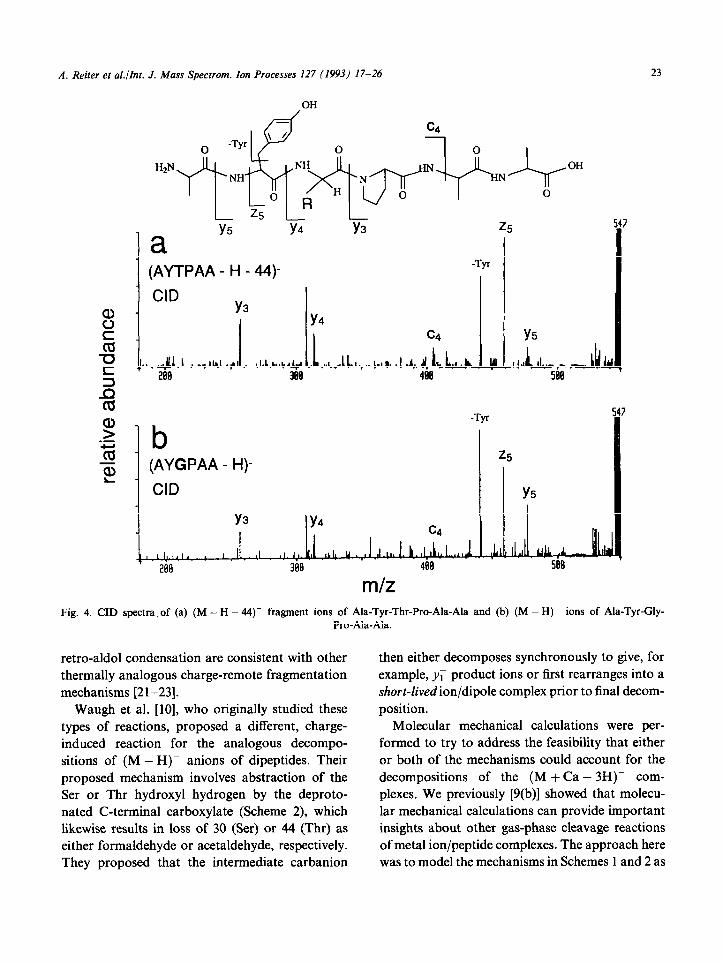
Repeating these same experiments for the (M - H)- anions reveals that the FAB-formed (M - H - X)- fragment ions, in which X = 30 or 44u, likewise have the structure of (M - H)- ions of Gly-containing peptides. This is shown in Fig. 4 for CID of the FAB-formed (M - H - 44- ions of AYTPAA (Fig. 4(a)) versus CID of the (M - H)- ions of AYGPAA (Fig. 4(b)). It should be noted that minor differences in relative product ion abundances in Figs. 3 and 4 may simply be the result of differences in experimental conditions such as collision gas pressure and ion internal energy.
One mechanism that could account for losses of 30 and 44 u from both (M + Ca - 3H)- complexes and (M - H)- anions involves a six-membered transition state in which hydrogen is transferred from the hydroxyl group of the side chain to the adjacent amide carbonyl oxygen (Scheme 1) [9(c)]. The resulting ion is an enolate that can then rearrange to its keto form to give a complex that now contains Gly. The neutral loss for Thr (Scheme 1) is acetaldehyde, and for Ser, the neutral loss is formaldehyde. Hu and Gross [9(d)] also recently suggested this mechanism. We point out that in addition, the mechanism in Scheme 1 is an exact analogy to the thermal retro-aldol conden- sation of the P-hydroxy ketones to give acetone and an enolate, which likewise isomerizes to its keto form [20]. The thermal retro-aldol reaction has an activation energy of 1.4eV and an Arrhen- ius A factor of x 1012 s-‘. An important feature of the mechanism and structure shown in Scheme 1 is that the anionic charge site(s) of the precursor ions is(are) not involved in the transition state of the reaction. Consequently, the reaction would be charge-remote [21]. Furthermore, the activation energy and Arrhenius A factor for the thermal
22 A. Reiter et al./Int. J. Mass Spectrom. Ion Processes 127 (1993) 17-26
OH
. (AYTPAA + Ca - 3H - 44)-
rb -Tyr
- (AYGPAA + Ca - 3H)- c4 z5
- (VESSK + Ca - 3H - 60)-
CID
-r 2se
1 d (VEGGK + Ca - 3H)-
. CID
Fig. 3. CID spectra of (a) (M + Ca - 3H - 44- fragment ions of Ala-Tyr-Thr-Pro-Ala-Ala and (b) (M + Ca - 3H)- complexes of Ala- Tyr-Gly-Pro-Ala-Ala; and CID spectra of(c) (M + Ca - 3H - 60- fragment ions of Val-Glu-Ser-Ser-Lys and (d) (M + Ca - 3H)-
ions of Val-Glu-Gly-Gly-Lys.
A. Reiter et aLlInt. J. Mass Spectrom. Ion Processes 127 (1993) 17-26 23
OH
b (AYGPAA - H)-
CID
G
Y5
m/z Fig. 4. CID spectra, of (a) (M - H - 44)- fragment ions of Ala-Tyr-Thr-Pro-Ala-Ala and (b) (M - H)- ions of Ala-Tyr-Gly-
Pro-Ala-Ala.
retro-aldol condensation are consistent with other thermally analogous charge-remote fragmentation mechanisms [2 l-231.
Waugh et al. [lo], who originally studied these types of reactions, proposed a different, charge- induced reaction for the analogous decompo- sitions of (M - H)- anions of dipeptides. Their proposed mechanism involves abstraction of the Ser or Thr hydroxyl hydrogen by the deproto- nated C-terminal carboxylate (Scheme 2), which likewise results in loss of 30 (Ser) or 44 (Thr) as either formaldehyde or acetaldehyde, respectively. They proposed that the intermediate carbanion
then either decomposes synchronously to give, for example, r; product ions or first rearranges into a shorr-livedionldipole complex prior to final decom- position.
Molecular mechanical calculations were per- formed to try to address the feasibility that either or both of the mechanisms could account for the decompositions of the (M + Ca - 3H)- com- plexes. We previously [9(b)] showed that molecu- lar mechanical calculations can provide important insights about other gas-phase cleavage reactions of metal ion/peptide complexes. The approach here was to model the mechanisms in Schemes 1 and 2 as
A. Reiter et aLlInt. J. Mass Spectrom. Ion Processes 127 (1993) 17-26
-3H+Ca I -H
[peptide-CO,Hl 1
I OH l- \ -3H+Cal-H +
[peptide-CO,H] 1
I ,P
1 lHzN-pep~del y$, -3H+Cal-H r
l-
L fi ‘H -@eptide-COZHI -I
Scheme 1.
first requiring either a precursor ion structure in which the carbonyl oxygen was hydrogen bonded to the side-chain hydroxyl hydrogen (Scheme 1) or in which the deprotonated C-terminus was hydro- gen-bonded to the hydroxyl hydrogen (Scheme 2). Calculations for all the peptide/metal ion com- plexes studied here gave the same results: The low- est energy structures all have the deprotonated C- terminus bound to the Ca2+ ion, not interacting with the side chain or any other structure in the peptide. This is not unexpected, but the calcula- tions provide evidence that the mechanism shown
N-terminus 0 bo
C-terminus
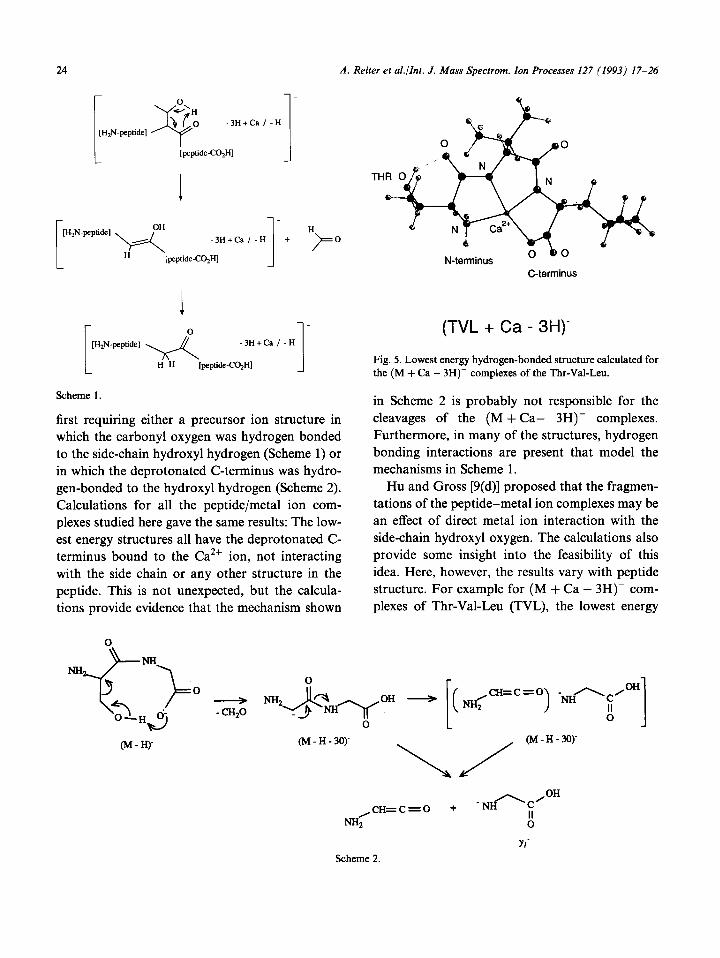
(TVL + Ca - 3H)-
Fig. 5. Lowest energy hydrogen-bonded structure calculated for the (M + Ca - 3H)- complexes of the Thr-Val-Leu.
in Scheme 2 is probably not responsible for the cleavages of the (M + Ca- 3H)- complexes. Furthermore, in many of the structures, hydrogen bonding interactions are present that model the mechanisms in Scheme 1.
Hu and Gross [9(d)] proposed that the fragmen- tations of the peptide-metal ion complexes may be an effect of direct metal ion interaction with the side-chain hydroxyl oxygen. The calculations also provide some insight into the feasibility of this idea. Here, however, the results vary with peptide structure. For example for (M + Ca - 3H)- com- plexes of Thr-Val-Leu (TVL), the lowest energy
,CH=C=O + NH2 6
Scheme 2. Yl’

A. Reiter et al./Int. J. Mass Spectrom. Ion Processes 127 (1993) 17-26 25
hydrogen-bonded structure does not contain the side chain interacting with the CaZf ion, but the side chain is involved in a hydrogen-bonded struc- ture that instead models the mechanism in Scheme 1 (Fig. 5). Other calculated low-energy structures of some of the peptides, in contrast, reveal that interactions between the side-chain oxygen and the metal ion are possible. We wish to point out, however, that having a binding interaction between the metal ion and the side-chain oxygen would reduce the likelihood that the cleavage reactions would occur as low-energy (metastable ion) reac- tions because an extra bond, the Ca-0 dative bond, would have to be broken in the transition state.
We also performed similar calculations to address the chemistry of the (M - H)- anions. The approach here was to evaluate possible hydrogen bonding interactions between the free C-terminal carboxylate anion and the different available hydrogens in the peptide chain. For all the peptides studied except Val-Thr-Leu (VTL), the lowest energy structures contain the deproto- nated C-terminal carboxylate involved in hydrogen bonding with the side-chain hydroxyl hydrogen. One example is shown in Fig. 6 for the (M - H)- anions of TVL. This is even the case for the hexa- peptide AYTPAA in which the Thr side chain is four amino acids distant from the C-terminal car-
THR 0 N-terminus
(TVL - H)-
Fig. 6. Lowest energy hydrogen-bonded structure calculated for the (M - H)- ions of Thr-Val-Leu.
boxylate. Thus, it is quite feasible that the reaction shown in Scheme 2 can account for decompositions of the (M - H)- anions. We wish to point out, however, that the product ions that arise from cleavages of the side chains of the tetra- and larger peptides are clearly long-lived, relatively low-
energy, stable species because they do not detectably decompose further by CID in the same field-free region (refer to Fig. 2(b) and 2(d)). Furthermore, these stable “intermediate” species only decompose by subsequent CID as do their Gly-containing peptide analogues (refer to Fig. 4). Thus, if the intermediate product ions from losses of the Ser and Thr side chains were ion/dipole complexes, as suggested in Scheme 2, they would either have to have relatively strong ion/dipole bonds or have to be close to being in their ground states. Perhaps it is more reasonable that the intermediate ions instead have the depro- tonated Gly-like structure also suggested in Scheme 2.
The molecular mechanical calculations provide evidence for possible precursor ion structures that could or could not decompose via the two mechan- isms shown in Schemes 1 and 2. The metal ion complexes would fragment via the mechanism in Scheme 1 because there would not be a free deprotonated C-terminal carboxylate available for the transition state shown in Scheme 2. In con- trast, the (M - H)- anions could decompose via either reaction. The decompositions from both types of precursor ions, however, appear to be similar in energetics, both being metastable ion decompositions. A true charge-remote decompo- sition (Scheme 1) should be higher in energy, how- ever, than a charge-induced reaction (Scheme 2). This seems to point toward two possibilities. One is that the metal ion-containing precursor isomers that fragment via Scheme 1 contain the metal ion in close proximity to the Ser or Thr side chain so that the bonds are sufficiently polarized so that the transition state energy is reduced and is compar- able to that in Scheme 2. The other possibility is that both types of precursor anions decompose via a true charge-remote decomposition (Scheme 1) so

26 A. Reiter et al./Int. J. Mass Spectrom. Ion Processes 127 (1993) 17-26
that the energetics are comparable. One way to address these two possibilities is to acquire the metastable ion kinetic energy release distribu- tions, which should be identical if the transition states and precursor ion internal energies are com- parable. This is an avenue of our future research.
Acknowledgments
Funding for this research was provided by The Emory University Research Fund, the National Science Foundation (CHE-9 113272 and CHE- 9209637), and the Petroleum Research Fund (25280-AC5), which is administered by the Amer- ican Chemical Society. A grant-in-aid of research from Sigma Xi, The Scientific Research Society, also provided partial support. A preliminary report of these results was presented at the 40th ASMS Conference on Mass Spectrometry and Allied Topics, June, 1992, Washington, DC.
References
R.B. Cody, I.J. Amster and F.W. McLtierty, Proc. Natl. Acad. Sci., USA, 82 (1985) 6367. (a) L.M. Mallis and D.H. Russell, Anal. Chem., 58 (1986) 1076. (b) D.H. Russell, Mass Spectrom. Rev., 5 (1986) 167. (c) D.H. Russell, ES. McGlohon and L.M. Mallis, Anal. Chem., 60 (1988) 1818. X. Tang, W. Ens, KG. Standing and J.B. Westmore, Anal. Chem., 60 (1988) 1791. D. Renner and G. Spiteller, Biomed. Environ. Mass Spec- trom., 15 (1988) 75. (a) R.P. Grese, R.L. Cemy and M.L. Gross, J. Am. Chem. Sot., 111 (1989) 2835. (b) R.P. Grese and M.L. Gross, J. Am. Chem. Sot., 112 (1990) 5098. (a) J.A. Leary, T.D. Williams and G. Bott, Rapid Com- mun. Mass Spectrom., 3 (1989) 192.
7
8
9
10
11
12
13 14
15
16
17
18
19
20 21 22
23
(b) J.A. Leary, Z. Zhou, S.A. Ogden and T.D. Williams, J. Am. Sot. Mass Spectrom., 1 (1990) 473. (a) L.M. Teesch and J. Adams, J. Am. Chem. Sot., 113 (1991) 812. (b) L.M. Tees&, R.C. Orlando and J. Adams, J. Am. Chem. Sot., 113 (1991) 3668. (c) H. Zhao and J. Adams, Int. J. Mass Spectrom. Ion Processes, 125 (1993) 195. L.M. Teesch and J. Adams, J. Am. Chem. Sot., 112 (1990) 4110. (a) L.M. Teesch and J. Adams, Proc. 39th ASMS Confer- ence on Mass Spectrometry and Allied Topics (1991) 128. (b) H. Zhao, A. Reiter, L.M. Teesch and J. Adams, J. Am. Chem. Sot., 115 (1993) 2854. (c) L.M. Teesch and J. Adams, Proc. 40th ASMS Confer- ence on Mass Spectrometry and Allied Topics (1992) 45. (d) P. Hu and M.L. Gross, J. Am. Chem. Sot., 114 (1992) 9153. (e) P. Hu and M.L. Gross, J. Am. Chem. Sot., 114 (1992) 9161. R.J. Waugh, M. Eckersley, J.H. Bowie and R.N. Hayes, Int. J. Mass Spectrom. Ion Processes, 98 (1990) 135. M.J. Contado and J. Adams, Anal. Chim. Acta, 246 (1991) 187. C. Matthew, R.D. Cramer and N. Van Opendosch, J. Comput. Chem., 10 (1989) 982. M.J.D. Powell, Math. Program., 12 (1977) 241. (a) H. Berthod and A. Pullman, J. Chem. Phys., 62 (1965) 942. (b) H. Berthod and A. Pullman, Theor. Chim. Acta, 8 (1967) 212. N. Raos, S.R. Niketic and V. Simeon, J. Inorg. Biochem., 16 (1982) 1. P. Izac, J. Libman, A. Shanzer, C.E. Felder and S. Lifson, J. Am. Chem. Sot., 113 (1991) 3431. (a) H.C. Freeman, J.M. Guss and R.L. Sinclair, Acta Crys- tallogr. Sect. B, 34 (1970) 2459. (b) H.C. Freeman and M.R. Taylor, Acta Crystallogr., 18 (1965) 939. S.J. Weiner, P.A. Kollman, D.T. Nguyen and D.A. Case, J. Comput. Chem., 7 (1986) 230. D.M. Ferguson and D.J. Raber, J. Am. Chem. Sot., 111 (1989) 4371. G.G. Smith and B.L. Yates, J. Org. Chem., 30 (1965) 2067. J. Adams, Mass Spectrom. Rev., 9 (1990) 141. J. Adams and M.L. Gross, J. Am. Chem. Sot., 111 (1989) 435. M.J. Contado, J. Adams, N.J. Jensen and M.L. Gross, J. Am. Sot. Mass Spectrom., 2 (1991) 180.
Copyright © 2022 FDOKUMEN




















