
G-protein estrogen receptor as a regulator of low-density lipoprotein cholesterol metabolism:...
15
213 T he importance of estrogen as a regulator of plasma lipid metabolism has been most clearly demonstrated in set- tings of estrogen deficiency (eg, menopause). Bilateral ovari- ectomy in premenopausal women causes an increase in plasma levels of low-density lipoprotein (LDL) when compared with premenopausal women with preserved ovaries. 1 Furthermore, LDL cholesterol levels increase after onset of menopause. 2 Multiple mechanisms have been proposed by which estro- gens regulate lipoprotein metabolism in humans, including induction of hepatic LDL receptor expression, 3,4 reduction of hepatic secretion of acyl-coenzyme A: cholesterol acyltrans- ferase 2–derived cholesteryl esters in plasma lipoproteins, 5 reduction of hepatic lipase, 6 and reduction of levels of propro- tein convertase subtilisin kexin type 9 (PCSK9), 7 a key regula- tor of LDL receptor metabolism. 8 The receptor(s) mediating estrogen’s effects on LDL metabolism is unclear. Estrogen’s effects are triggered via 2 main receptor types. Classic estrogen receptors (ERs) have been shown to have both nuclear and cytoplasmic/membrane- associated sites of effect. 9,10 More recently, a G-protein– coupled receptor, G-protein ER (GPER; aka GPR30) has been shown to mediate some of the so-called rapid (nongenomic) effects of estrogen. 11 We have recently shown that GPER is also activated by aldosterone. 12 © 2014 American Heart Association, Inc. Arterioscler Thromb Vasc Biol is available at http://atvb.ahajournals.org DOI: 10.1161/ATVBAHA.114.304326 Objective—Estrogen deficiency is linked with increased low-density lipoprotein (LDL) cholesterol. The hormone receptor mediating this effect is unknown. G-protein estrogen receptor (GPER) is a recently recognized G-protein–coupled receptor that is activated by estrogens. We recently identified a common hypofunctional missense variant of GPER, namely P16L. However, the role of GPER in LDL metabolism is unknown. Therefore, we examined the association of the P16L genotype with plasma LDL cholesterol level. Furthermore, we studied the role of GPER in regulating expression of the LDL receptor and proprotein convertase subtilisin kexin type 9. Approach and Results—Our discovery cohort was a genetically isolated population of Northern European descent, and our validation cohort consisted of normal, healthy women aged 18 to 56 years from London, Ontario. In addition, we examined the effect of GPER on the regulation of proprotein convertase subtilisin kexin type 9 and LDL receptor expression by the treatment with the GPER agonist, G1. In the discovery cohort, GPER P16L genotype was associated with a significant increase in LDL cholesterol (mean±SEM): 3.18±0.05, 3.25±0.08, and 4.25±0.33 mmol/L, respectively, in subjects with CC (homozygous for P16), CT (heterozygotes), and TT (homozygous for L16) genotypes (P<0.05). In the validation cohort (n=339), the GPER P16L genotype was associated with a similar increase in LDL cholesterol: 2.17±0.05, 2.34±0.06, and 2.42±0.16 mmol/L, respectively, in subjects with CC, CT, and TT genotypes (P<0.05). In the human hepatic carcinoma cell line, the GPER agonist, G1, mediated a concentration-dependent increase in LDL receptor expression, blocked by either pretreatment with the GPER antagonist G15 or by shRNA-mediated GPER downregulation. G1 also mediated a GPER- and concentration-dependent decrease in proprotein convertase subtilisin kexin type 9 expression. Conclusions —GPER activation upregulates LDL receptor expression, probably at least, in part, via proprotein convertase subtilisin kexin type 9 downregulation. Furthermore, humans carrying the hypofunctional P16L genetic variant of GPER have increased plasma LDL cholesterol. In aggregate, these data suggest an important role of GPER in the regulation of LDL receptor expression and consequently LDL metabolism. ( Arterioscler Thromb Vasc Biol . 2015;35:213-221. DOI: 10.1161/ATVBAHA.114.304326.) Key Words: cholesterol, LDL ◼ GPER protein, human ◼ receptors, LDL ◼ PCSK9 protein, human Received on: July 25, 2014; final version accepted on: October 28, 2014. From the Robarts Research Institute (Y.H., Q.D., M.R.B., A.D.M., M.W.H., R.G., R.A.H., R.D.F.) and Departments of Medicine (M.W.H., R.G., R.A.H., R.D.F.), Physiology and Pharmacology (R.G., R.A.H., R.D.F.), and Biochemistry (M.W.H.), Western University, London, Ontario, Canada; Department of Medicine, University of Toronto, Toronto, Ontario, Canada (P.W.C.); and Department of Public Health and Social Policy, University of Victoria, Victoria, British Columbia, Canada (J.H.B.). The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.114.304326/-/DC1. Correspondence to Ross D. Feldman, MD, Departments of Medicine and of Physiology and Pharmacology, Schulich School of Medicine and Dentistry, Robarts Research Institute, University of Western Ontario, 1151 Richmond St N, London, Ontario, Canada N6A 5B7. E-mail [email protected] G-Protein Estrogen Receptor as a Regulator of Low-Density Lipoprotein Cholesterol Metabolism Cellular and Population Genetic Studies Yasin Hussain, Qingming Ding, Philip W. Connelly, J. Howard Brunt, Matthew R. Ban, Adam D. McIntyre, Murray W. Huff, Robert Gros, Robert A. Hegele, Ross D. Feldman at UNIVERSITY WESTERN ONTARIO on February 11, 2015 http://atvb.ahajournals.org/ Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015 http://atvb.ahajournals.org/ Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015 http://atvb.ahajournals.org/ Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015 http://atvb.ahajournals.org/ Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015 http://atvb.ahajournals.org/ Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015 http://atvb.ahajournals.org/ Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015 http://atvb.ahajournals.org/ Downloaded from
Transcript of G-protein estrogen receptor as a regulator of low-density lipoprotein cholesterol metabolism:...

213
The importance of estrogen as a regulator of plasma lipid metabolism has been most clearly demonstrated in set-
tings of estrogen deficiency (eg, menopause). Bilateral ovari-ectomy in premenopausal women causes an increase in plasma levels of low-density lipoprotein (LDL) when compared with premenopausal women with preserved ovaries.1 Furthermore, LDL cholesterol levels increase after onset of menopause.2
Multiple mechanisms have been proposed by which estro-gens regulate lipoprotein metabolism in humans, including induction of hepatic LDL receptor expression,3,4 reduction of hepatic secretion of acyl-coenzyme A: cholesterol acyltrans-ferase 2–derived cholesteryl esters in plasma lipoproteins,5
reduction of hepatic lipase,6 and reduction of levels of propro-tein convertase subtilisin kexin type 9 (PCSK9),7 a key regula-tor of LDL receptor metabolism.8
The receptor(s) mediating estrogen’s effects on LDL metabolism is unclear. Estrogen’s effects are triggered via 2 main receptor types. Classic estrogen receptors (ERs) have been shown to have both nuclear and cytoplasmic/membrane- associated sites of effect.9,10 More recently, a G-protein–coupled receptor, G-protein ER (GPER; aka GPR30) has been shown to mediate some of the so-called rapid (nongenomic) effects of estrogen.11 We have recently shown that GPER is also activated by aldosterone.12
© 2014 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol is available at http://atvb.ahajournals.org DOI: 10.1161/ATVBAHA.114.304326
Objective—Estrogen deficiency is linked with increased low-density lipoprotein (LDL) cholesterol. The hormone receptor mediating this effect is unknown. G-protein estrogen receptor (GPER) is a recently recognized G-protein–coupled receptor that is activated by estrogens. We recently identified a common hypofunctional missense variant of GPER, namely P16L. However, the role of GPER in LDL metabolism is unknown. Therefore, we examined the association of the P16L genotype with plasma LDL cholesterol level. Furthermore, we studied the role of GPER in regulating expression of the LDL receptor and proprotein convertase subtilisin kexin type 9.
Approach and Results—Our discovery cohort was a genetically isolated population of Northern European descent, and our validation cohort consisted of normal, healthy women aged 18 to 56 years from London, Ontario. In addition, we examined the effect of GPER on the regulation of proprotein convertase subtilisin kexin type 9 and LDL receptor expression by the treatment with the GPER agonist, G1. In the discovery cohort, GPER P16L genotype was associated with a significant increase in LDL cholesterol (mean±SEM): 3.18±0.05, 3.25±0.08, and 4.25±0.33 mmol/L, respectively, in subjects with CC (homozygous for P16), CT (heterozygotes), and TT (homozygous for L16) genotypes (P<0.05). In the validation cohort (n=339), the GPER P16L genotype was associated with a similar increase in LDL cholesterol: 2.17±0.05, 2.34±0.06, and 2.42±0.16 mmol/L, respectively, in subjects with CC, CT, and TT genotypes (P<0.05). In the human hepatic carcinoma cell line, the GPER agonist, G1, mediated a concentration-dependent increase in LDL receptor expression, blocked by either pretreatment with the GPER antagonist G15 or by shRNA-mediated GPER downregulation. G1 also mediated a GPER- and concentration-dependent decrease in proprotein convertase subtilisin kexin type 9 expression.
Conclusions—GPER activation upregulates LDL receptor expression, probably at least, in part, via proprotein convertase subtilisin kexin type 9 downregulation. Furthermore, humans carrying the hypofunctional P16L genetic variant of GPER have increased plasma LDL cholesterol. In aggregate, these data suggest an important role of GPER in the regulation of LDL receptor expression and consequently LDL metabolism. (Arterioscler Thromb Vasc Biol. 2015;35:213-221. DOI: 10.1161/ATVBAHA.114.304326.)
Key Words: cholesterol, LDL ◼ GPER protein, human ◼ receptors, LDL ◼ PCSK9 protein, human
Received on: July 25, 2014; final version accepted on: October 28, 2014.From the Robarts Research Institute (Y.H., Q.D., M.R.B., A.D.M., M.W.H., R.G., R.A.H., R.D.F.) and Departments of Medicine (M.W.H., R.G., R.A.H.,
R.D.F.), Physiology and Pharmacology (R.G., R.A.H., R.D.F.), and Biochemistry (M.W.H.), Western University, London, Ontario, Canada; Department of Medicine, University of Toronto, Toronto, Ontario, Canada (P.W.C.); and Department of Public Health and Social Policy, University of Victoria, Victoria, British Columbia, Canada (J.H.B.).
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.114.304326/-/DC1.Correspondence to Ross D. Feldman, MD, Departments of Medicine and of Physiology and Pharmacology, Schulich School of Medicine and Dentistry,
Robarts Research Institute, University of Western Ontario, 1151 Richmond St N, London, Ontario, Canada N6A 5B7. E-mail [email protected]
G-Protein Estrogen Receptor as a Regulator of Low-Density Lipoprotein Cholesterol Metabolism
Cellular and Population Genetic Studies
Yasin Hussain, Qingming Ding, Philip W. Connelly, J. Howard Brunt, Matthew R. Ban, Adam D. McIntyre, Murray W. Huff, Robert Gros, Robert A. Hegele, Ross D. Feldman
at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from
214 Arterioscler Thromb Vasc Biol January 2015
The role of GPER in hepatic lipid metabolism is unknown. GPER is expressed in normal hepatocytes.13 Notably, mice with genetic deletion of GPER have increased cholesterol levels.14 In contrast, total cholesterol levels have not been reported to be elevated in ERα knockout mice.15
To examine the relationship between GPER func-tion and lipid metabolism in humans, we have studied the effect of carrying a common hypofunctional GPER genetic variant. The P16L GPER genetic variant is com-mon with an allelic frequency in the general population of ≈20%. Recent studies from our laboratory have shown that this proline to leucine change at amino acid residue 16 of GPER (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs=11544331) results in a hypofunctional response to the GPER agonist G1.16
In these studies, we evaluate the association of GPER P16L genotype with plasma LDL cholesterol in a previously characterized,17 genetically isolated population of Northern European descent from Alberta (n=415). To validate our find-ings, we examine a London, Ontario-based population of healthy women (n=339).18
We found that in both population samples, the hypofunc-tional GPER P16L variant was associated with increased LDL concentrations in women, following a recessive model. Furthermore, in cellular studies, we demonstrate that G1 ago-nist activation of GPER in HepG2 cells, which endogenously express GPER,19,20 leads to reduced PCSK9 expression and increased LDL receptor expression. Overall, these studies support an important role of GPER in regulation of plasma lipoprotein metabolism.
Materials and MethodsMaterials and Methods are available in the online-only Data Supplement.
ResultsFemale Carriers of GPER P16L Have Higher Plasma LDL CholesterolIn the discovery cohort, the GPER P16L allele frequency was 12.3%, with no significant difference between men (13.1%) and women (11.7%). The frequencies of CC, CT, and TT genotypes were 77.1%, 21.2%, and 1.7%, respectively; these did not significantly deviate from the Hardy–Weinberg distribution. The body mass index and waist circumference were not different between geno-types (Table 1). The plasma LDL and total cholesterol concentrations in this sample were significantly higher in homozygotes for the P16L allele, and followed an appar-ent recessive model (P<0.05; Table 2). After adjusting for covariates, there was no change in the significance of the associations (Table 3).
Subgroup analysis showed that the increase in LDL choles-terol levels in carriers of the GPER P16L variant was sex spe-cific; in particular, women but not men showed the increase in plasma LDL and total cholesterol levels (Table 2). After adjusting for covariates in women, there was no change in the significance of the associations (Table 3).
In the validation cohort of 339 women, the allele fre-quency of the P16L GPER variant was 25.2%. The frequen-cies of GPER variant genotype were 56.3%, 36.9%, and 6.8%, respectively, for wild type (CC), heterozygote (CT), and P16L homozygote (CT) and did not significantly deviate from the Hardy–Weinberg distribution. The plasma LDL cholesterol lev-els were significantly higher in those carrying the P16L GPER variant (TT>CT>CC; P<0.05; Table 4). The body mass index and waist circumference did not vary significantly between genotypes (Table 5). The increase in plasma LDL cholesterol levels associated with the expression of the P16L GPER vari-ant remained significant after adjusting for covariates (Table 6).
Nonstandard Abbreviations and Acronyms
GPER G-protein estrogen receptor
LDL low-density lipoprotein
PCSK9 proprotein convertase subtilisin kexin type 9
Table 1. Subject Demographics: Alberta Population
CC CT TT P Value
Whole population (n=415) n=320 n=88 n=7 ...
Age, y 36.32±0.83 38.44±1.62 40.92±5.13 NS
BMI, kg/m2 28.25±0.31 27.89±0.56 29.48±2.38 NS
Waist circumference, cm 85.97±0.77 87.99±1.45 88.29±3.62 NS
Women (n=235) n=185 n=45 n=5 ...
Age, y 37.52±1.13 37.15±2.44 47.23±4.52 NS
BMI, kg/m2 28.64±0.47 27.43±1.00 31.06±3.33 NS
Waist circumference, cm 81.43±0.99 81.27±1.86 89.00±5.15 NS
Men (n=180) n=135 n=43 n=2 ...
Age, y 34.68±1.20 39.78±2.11 25.15±0.08 NS
BMI, kg/m2 27.70±0.32 28.29±0.58 26.33±1.76 NS
Waist circumference, cm 92.23±1.00 94.88±1.67 86.50±2.50 NS
BMI indicates body mass index; and NS, not significant.
at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from

Hussain et al GPER Regulation of LDL Cholesterol 215
Role of GPER in the Regulation of LDL Receptor Protein ExpressionTo determine whether the increase in LDL cholesterol lev-els in carriers of the GPER P16L variant might indicate a role for GPER in the regulation of lipoprotein metabolism, we studied whether GPER activation regulated LDL recep-tor protein expression, which is an important determinant of plasma LDL levels.21 In HepG2 cells, the GPER agonist, G122, induced a concentration-dependent increase in LDL receptor protein expression with a maximal effect of a 36±6%
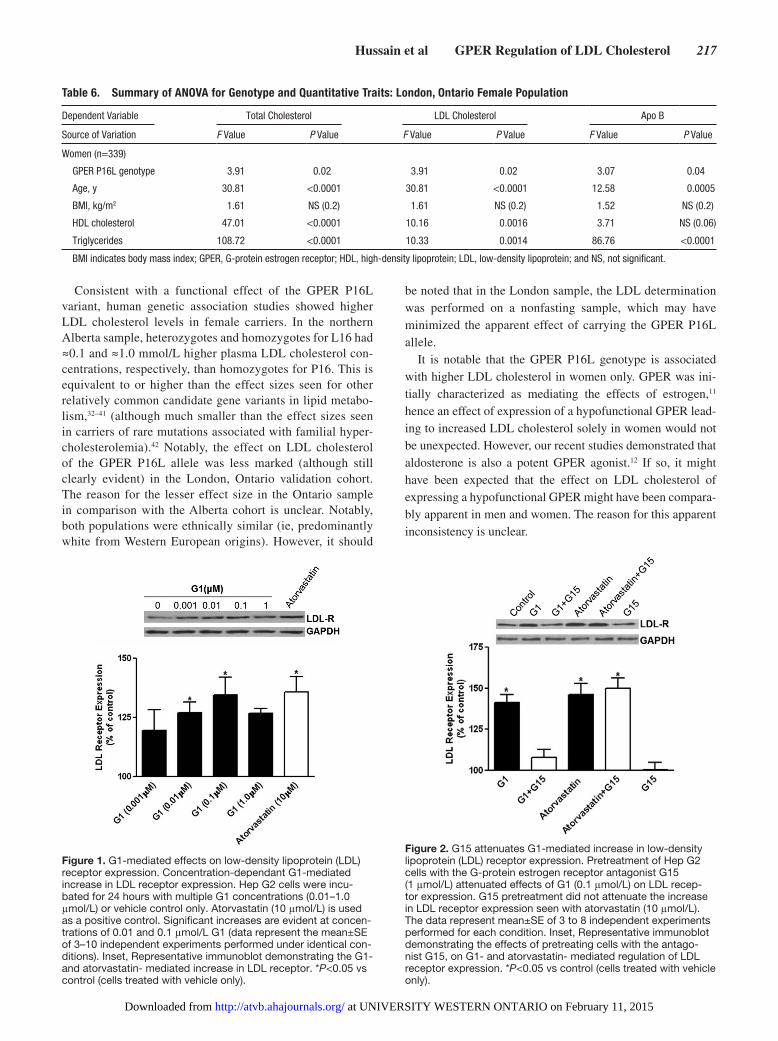
(mean±SE; n=10) increase in LDL receptor protein above baseline at a G1 concentration of 0.1 μmol/L (Figure 1); this was comparable with the increase in LDL receptor protein induced by atorvastatin (33±9%, n=3; Figure 1).23
To confirm the GPER dependence of the observed effect of G1, we examined the effect of the GPER antagonist, G1524 (1 μmol/L) on G1-mediated stimulation of LDL receptor pro-tein expression. Pretreatment of HepG2 cells with G15 inhib-ited the G1-mediated increase in LDL receptor (Figure 2). G15 alone had no significant effect on LDL receptor protein
Table 2. Lipid Profiles by Genotype (Least Squares Means Shown): Alberta Population
P Value
CC CT TT CC vs CT CC vs TT CT vs TT
Whole population (n=415) n=320 n=88 n=7 ... ... ...
Total cholesterol, mmol/L 5.28 5.28 6.25 NS (0.96) 0.0031 0.0038
LDL cholesterol, mmol/L 3.27 3.21 4.30 NS (0.52) 0.0011 0.0008
Apo B, g/L 1.20 1.19 1.41 NS(0.89) 0.02 0.02
Women (n=235) n=185 n=45 n=5 ... ... ...
Total cholesterol, mmol/L 5.17 5.04 6.51 NS (0.32) 0.0005 0.0002
LDL cholesterol, mmol/L 3.09 2.96 4.47 NS (0.28) 0.0002 <0.0001
Apo B, g/L 1.12 1.09 1.45 NS (0.43) 0.0015 0.0009
Men (n=180) n=135 n=43 n=2 ... ... ...
Total cholesterol, mmol/L 5.34 5.46 5.56 NS (0.43) NS (0.72) NS (0.87)
LDL cholesterol, mmol/L 3.37 3.40 3.67 NS (0.80) 0.60 NS (0.66)
Apo B, g/L 1.28 1.29 1.28 NS (0.75) 0.98 NS (0.95)
LDL indicates low-density lipoprotein.
Table 3. Summary of ANOVA for Genotype and Quantitative Traits: Alberta Population
Dependent Variable Total Cholesterol LDL Cholesterol Apo B
Source of Variation F Value P Value F Value P Value F Value P Value
Whole population (n=415)
GPER P16L genotype 4.46 0.01 5.72 0.0035 2.91 NS (0.06)
Age, y 24.93 <0.0001 13.88 0.0002 32.37 <0.0001
Sex 9.17 0.002 14.01 0.0002 3.91 0.04
BMI, kg/m2 16.26 <0.0001 11.96 0.0006 13.06 0.0003
HDL cholesterol 71.24 <0.0001 3.51 NS (0.06) 3.29 NS (0.07)
Triglycerides 68.66 <0.0001 3.36 NS (0.06) 59.22 <0.0001
Women (n=235)
GPER P16L genotype 7.05 0.0001 8.01 0.0004 5.65 0.004
Age, y 15.43 0.0001 9.49 0.0023 21.04 <0.0001
BMI, kg/m2 12.56 0.0005 7.76 0.0058 8.31 0.004
HDL cholesterol 54.41 <0.0001 2.84 NS (0.09) 1.83 NS (0.2)
Triglycerides 35.87 <0.0001 3.86 0.05 26.55 <0.0001
Men (n=180)
GPER P16L genotype 0.37 NS (0.69) 0.17 NS (0.84) 0.05 NS (0.9)
Age, y 7.36 0.007 3.43 NS (0.07) 8.39 0.004
BMI, kg/m2 4.22 0.04 4.04 0.05 5.72 0.02
HDL cholesterol 18.16 <0.0001 0.86 NS (0.35) 1.16 NS (0.3)
Triglycerides 24.82 <0.0001 0.21 NS (0.66) 1.96 NS (0.16)
BMI indicates body mass index; GPER, G-protein estrogen receptor; HDL, high-density lipoprotein; LDL, low-density lipoprotein; and NS, not significant.
at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from

216 Arterioscler Thromb Vasc Biol January 2015
expression. In addition, G15 had no significant effect on atorvastatin-mediated upregulation of LDL receptor protein (Figure 2).
In addition, we confirmed the GPER-dependent effect of G1 on LDL receptor protein expression using a GPER-targeted shRNA to downregulate GPER expression. Infection of HepG2 cells with the adenoshGPER construct resulted in ≈50% downregulation of GPER mRNA con-tent when compared with the control construct (adenosh-GFP; Figure 3A). GPER downregulation, using shGPER, attenuated G1-mediated increase in LDL receptor protein expression (Figure 3B). Atorvastatin-mediated LDL recep-tor upregulation was not affected by shRNA-induced GPER downregulation.
Role of GPER on Regulation of LDL Uptake and LDL Receptor mRNATo confirm the functional effect of GPER activation on LDL receptor protein expression, we assessed G1-mediated regula-tion of LDL uptake. G1 induced a concentration-dependent increase in LDL uptake with a maximal response of 27±2% above control at G1 concentrations >0.1 μmol/L (n=4; P<0.05; Figure 4A).
To determine whether the increase in LDL receptor pro-tein expression and LDL uptake was related to an increase in LDL receptor mRNA, we performed quantitative polymerase chain reaction studies. No alterations in LDLR mRNA levels were apparent at G1 concentrations ranging from 1 nmol/L to 1 μmol/L (n=3). In contrast, atorvastatin increased LDL receptor mRNA levels 57±14% above control (n=4; P<0.05; Figure 4B).
Role of GPER in the Regulation on PCSK9 ExpressionPCSK9 is recognized as a critical regulator of overall LDL receptor functional capacity.8 Furthermore, the regulation of PCSK9 levels has been shown to be cAMP dependent,25 which is the second messenger pathway regulated by GPER activation.26 To determine whether GPER plays a role in PCSK9 protein expression, which in turn could explain the effects of GPER on the regulation of LDL receptor pro-tein, we examined the effect of G1 on PCSK9 levels as assessed in HepG2 conditioned media samples. G1 medi-ated a concentration-dependent decrease in PCSK9 protein
expression in HepG2 cells, with a maximal decrease to 72±8% (n=8) of control with G1 at a concentration of 1.0 μmol/L (Figure 5A). Atorvastatin, in contrast, increased PCSK9 protein content by 40% (P<0.05). Comparable findings were obtained when PCSK9 content was assessed in samples taken from cell lysates (Figure 5B).
To determine whether the decrease in PCSK9 protein expression was related to a decrease in PCSK9 receptor mRNA, we performed quantitative polymerase chain reaction studies. G1 mediated a concentration-dependent decrease in PCSK9 mRNA content (Figure 5C). In contrast (and as expected), atorvastatin treatment increased PCSK9 mRNA content.
To confirm the GPER dependence of the observed effect of G1, we examined the effect of G15 (1 μmol/L). Pretreatment of HepG2 cells with G15 significantly attenuated the decrease in PCSK9 protein induced by G1 (Figure 6). G15 alone had no significant effect on PCSK9 protein expression. In addi-tion, G15 had no significant effect on atorvastatin-mediated upregulation of PCSK9 protein expression. Furthermore, we confirmed the GPER-dependent effect of G1 on PCSK9 levels using the GPER-targeted shRNA. GPER down-regulation almost completely attenuated the G1-mediated decrease in PCSK9 expression (Figure 7). The atorvastatin-mediated PCSK9 upregulation was not affected by GPER downregulation.
DiscussionThe importance of the G-protein–coupled receptor, GPER, in mediating the vascular effects of estrogen (and of aldo-sterone) has been increasingly appreciated.27–31 However, the importance of GPER in regulation of lipoprotein metab-olism in humans has been unexplored to date. Using a com-bination of human genetics and functional cellular biology, we found that the relatively common missense GPER P16L had a significant influence on LDL cholesterol concentra-tions. Specifically, the GPER P16L genotype was associated with higher plasma LDL cholesterol levels in women, but not in men. Furthermore, in HepG2 cells expressing GPER, we demonstrate that GPER activation (1) increased LDL receptor protein expression, without change in mRNA con-tent and (2) decreased PCSK9 mRNA content and protein levels. These independent lines of experimental evidence are directionally consistent and in aggregate support the importance of GPER as a regulator of LDL metabolism, probably via regulation of PCSK9.
Table 4. Lipid Profiles by Genotype (Least Squares Means Shown): London, Ontario Female Population
Women (n=339) P Value
CC (n=191)
CT (n=125)
TT (n=23) CC vs CT
CC vs TT CT vs TT
Total cholesterol, mmol/L
4.23 4.39 4.53 0.02 0.03 NS (0.33)
LDL cholesterol, mmol/L
2.17 2.34 2.42 0.03 0.03 NS (0.33)
Apo B, g/L 0.69 0.72 0.76 NS (0.10) 0.03 NS (0.21)
LDL indicates low-density lipoprotein; and NS, not significant.
Table 5. Subject Demographics: London, Ontario Female Population
Women (n=339) CC (n=191) CT (n=125) TT (n=23) P Value
Age, y 27.51±0.72 28.33±0.93 24.83±1.56 NS
BMI, kg/m2 22.91±0.28 23.35±0.40 22.26±0.59 NS
Waist circumference (cm)
76.26±0.63 77.31±0.89 75.65±1.61 NS
BMI indicates body mass index; and NS, not significant.
at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from
Hussain et al GPER Regulation of LDL Cholesterol 217
Consistent with a functional effect of the GPER P16L variant, human genetic association studies showed higher LDL cholesterol levels in female carriers. In the northern Alberta sample, heterozygotes and homozygotes for L16 had ≈0.1 and ≈1.0 mmol/L higher plasma LDL cholesterol con-centrations, respectively, than homozygotes for P16. This is equivalent to or higher than the effect sizes seen for other relatively common candidate gene variants in lipid metabo-lism,32–41 (although much smaller than the effect sizes seen in carriers of rare mutations associated with familial hyper-cholesterolemia).42 Notably, the effect on LDL cholesterol of the GPER P16L allele was less marked (although still clearly evident) in the London, Ontario validation cohort. The reason for the lesser effect size in the Ontario sample in comparison with the Alberta cohort is unclear. Notably, both populations were ethnically similar (ie, predominantly white from Western European origins). However, it should
be noted that in the London sample, the LDL determination was performed on a nonfasting sample, which may have minimized the apparent effect of carrying the GPER P16L allele.
It is notable that the GPER P16L genotype is associated with higher LDL cholesterol in women only. GPER was ini-tially characterized as mediating the effects of estrogen,11 hence an effect of expression of a hypofunctional GPER lead-ing to increased LDL cholesterol solely in women would not be unexpected. However, our recent studies demonstrated that aldosterone is also a potent GPER agonist.12 If so, it might have been expected that the effect on LDL cholesterol of expressing a hypofunctional GPER might have been compara-bly apparent in men and women. The reason for this apparent inconsistency is unclear.
Table 6. Summary of ANOVA for Genotype and Quantitative Traits: London, Ontario Female Population
Dependent Variable Total Cholesterol LDL Cholesterol Apo B
Source of Variation F Value P Value F Value P Value F Value P Value
Women (n=339)
GPER P16L genotype 3.91 0.02 3.91 0.02 3.07 0.04
Age, y 30.81 <0.0001 30.81 <0.0001 12.58 0.0005
BMI, kg/m2 1.61 NS (0.2) 1.61 NS (0.2) 1.52 NS (0.2)
HDL cholesterol 47.01 <0.0001 10.16 0.0016 3.71 NS (0.06)
Triglycerides 108.72 <0.0001 10.33 0.0014 86.76 <0.0001
BMI indicates body mass index; GPER, G-protein estrogen receptor; HDL, high-density lipoprotein; LDL, low-density lipoprotein; and NS, not significant.
Figure 1. G1-mediated effects on low-density lipoprotein (LDL) receptor expression. Concentration-dependant G1-mediated increase in LDL receptor expression. Hep G2 cells were incu-bated for 24 hours with multiple G1 concentrations (0.01–1.0 μmol/L) or vehicle control only. Atorvastatin (10 μmol/L) is used as a positive control. Significant increases are evident at concen-trations of 0.01 and 0.1 μmol/L G1 (data represent the mean±SE of 3–10 independent experiments performed under identical con-ditions). Inset, Representative immunoblot demonstrating the G1- and atorvastatin- mediated increase in LDL receptor. *P<0.05 vs control (cells treated with vehicle only).
Figure 2. G15 attenuates G1-mediated increase in low-density lipoprotein (LDL) receptor expression. Pretreatment of Hep G2 cells with the G-protein estrogen receptor antagonist G15 (1 μmol/L) attenuated effects of G1 (0.1 μmol/L) on LDL recep-tor expression. G15 pretreatment did not attenuate the increase in LDL receptor expression seen with atorvastatin (10 μmol/L). The data represent mean±SE of 3 to 8 independent experiments performed for each condition. Inset, Representative immunoblot demonstrating the effects of pretreating cells with the antago-nist G15, on G1- and atorvastatin- mediated regulation of LDL receptor expression. *P<0.05 vs control (cells treated with vehicle only).
at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from

218 Arterioscler Thromb Vasc Biol January 2015
We considered why this genetic variant has not been previously identified as a significant determinant on prior genome-wide association studies of plasma LDL cholesterol levels. However, as far as we have been able to determine, none of the commonly used microarrays in the catalogue of published genome-wide association studies carried the single nucleotide polymorphism of interest (rs11544331) (https://www.genome.gov/page.cfm?pageid=26525384#searchForm).43–56 In this context, the findings in the present study emphasize that the previous genome-wide association studies may be incomplete about potential candidate genes in dyslipidemia because the associations are limited to the content on the microarray. A recent genome-wide association study57 did identify a variant of GPR146 (viz., rs1997243) as being associated with cholesterol levels (Table II in the online-only Data Supplement). Notably, this genetic variant was also reported to be in linkage disequilibrium with P16L GPER (r2=0.554), supporting our present findings.
These studies suggest a mechanism whereby GPER activa-tion has beneficial effects on LDL cholesterol metabolism—and thus specify a mechanism by which carrying a hypofunc-tional GPER variant may lead to increased LDL cholesterol levels. LDL receptor expression is significantly increased by GPER activation with effects comparable with those of HMG (3-hydroxy-3-methyl-glutaryl) CoA reductase inhibition.
In summary, our studies demonstrate that the expression of a common GPER genetic variant parallels increased LDL lev-els and that GPER activation increases LDL receptor protein expression, probably via downregulation of PCSK9 levels. The findings could explain, at least in part, the relationship between functional estrogen deficiency and increased LDL cholesterol. They also show that the GPER P16L variant has a clearly dis-tinct effect on LDL cholesterol, without any apparent associ-ated or concomitant effect on HDL cholesterol concentrations,
Figure 3. G-protein estrogen receptor (GPER) downregulation and its effect on G1-mediated increase in low-density lipoprotein (LDL) receptor expression. A, Infection of cells with adenoshGPER construct resulted in a significant downregulation of GPER RNA expression. Infection with adenoshGFP had no significant effect. Representative reverse transcriptase polymerase chain reac-tion. B, shGPER-mediated downregulation of GPER expression caused a decrease in the G1-mediated increase (0.1 μmol/L) in LDL receptor expression. The atorvastatin-mediated increase in LDL receptor expression was not attenuated by GPER downregu-lation. The data represent the mean±SE of 3 to 5 independent experiments performed for each condition. Inset, Representative immunoblot demonstrating the G1- and atorvastatin-mediated increase in LDL receptor expression vs control in both shGFP- and shGPER-infected cells. *P<0.05 vs its respective control (ie, cells infected with the same adenosh construct, treated with vehicle only).
Figure 4. G-protein estrogen receptor–mediated upregulation of low-density lipoprotein (LDL) uptake but not of LDL recep-tor translation. A, G1-mediated LDL uptake. Concentration-dependent increase in G1-mediated LDL uptake as assessed by the extent of uptake of LDL labeled with the fluorescent probe 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI-LDL). Data represent the mean±SE of 4 inde-pendent experiments performed under identical conditions (*P<0.05 vs control, ie, cells treated with vehicle only). B, G1 does not increase LDL receptor mRNA content. LDL recep-tor mRNA content was not increased by G1, as assessed by real-time polymerase chain reaction analysis of the level of LDL receptor mRNA expression In contrast, atorvastatin treat-ment upregulated LDL receptor mRNA content. Data represent the mean±SE of 4 independent experiments performed under identical conditions (*P<0.05 vs control, ie, cells treated with vehicle only).
at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from

Hussain et al GPER Regulation of LDL Cholesterol 219
which are also well known to be associated with estrogen sta-tus. Overall, these findings support an important role of GPER in the regulation of LDL metabolism in humans.
AcknowledgmentsWe are grateful for the expert technical assistance of Cynthia Sawyez in performing the biochemical analyses for these studies.
Figure 5. G1-mediated effects on proprotein convertase subtilisin kexin type 9 (PCSK9) expression. Concentration-dependent G1-mediated decrease in PCSK9 expression assessed in supernatants (A) and in cell lysates (B). Hep G2 cells were incubated for 24 hours with multiple G1 concentrations (0.01–1.0 μmol/L). Atorvastatin (10 μmol/L) is used as a positive control. A statistically sig-nificant decrease in PCSK9 expression is evident at concentration of G1>0.1 μmol/L. Data represent the mean±SE of 3 to 10 inde-pendent experiments performed under identical conditions in supernatants (A) and cell lysates (B). Note: The control condition in A was medium from cells treated with vehicle only. The control condition in B was lysates from cells treated with vehicle only. Insets, Representative immunoblots demonstrating the G1-mediated decrease and atorvastatin-mediated increase in PCSK9 expression. *P<0.05 vs control. C, G1 increases PCSK9 mRNA content. PCSK9 mRNA content was decreased by G1, as assessed by real-time polymerase chain reaction analysis. In contrast, atorvastatin treatment upregulated PCSK9 mRNA content. Data repre-sent the mean±SE of 4 independent experiments performed under identical conditions (*P<0.05 vs control, ie, cells treated with vehicle only).
Figure 6. G15 attenuates G1-mediated decrease in proprotein convertase subtilisin kexin type 9 (PCSK9) expression. Pretreat-ing Hep G2 cells with the G-protein estrogen receptor antagonist G15 (1 μmol/L) attenuated the effects on PCSK9 expression of G1 (0.1 μmol/L). Atorvastatin-mediated increase in PCSK9 expression was not altered with G15 pretreatment. Data repre-sent mean±SE of 3 to 5 independent experiments performed for each condition. Inset, Representative immunoblot demonstrat-ing the effects of pretreating cells with the antagonist G15, on G-1 and atorvastatin-mediated regulation of PCSK9 expression. *P<0.05 vs control (cells treated with vehicle only).
Figure 7. G-protein estrogen receptor (GPER) downregulation and its effect on G1-mediated decrease in proprotein convertase subtilisin kexin type 9 (PCSK9) expression. shGPER-mediated downregulation of GPER expression caused an attenuation in the G1-mediated decrease (0.1 μmol/L) in PCSK9 expression (data represent the mean±SE of 3 independent experiments performed for each condition) Inset, Representative immunoblot demonstrat-ing the G1-mediated decrease and atorvastatin-mediated increase in PCSK9 expression vs control in both shGFP- and shGPER-infected cells. *P<0.05 vs its respective control (ie, cells infected with the same adenosh construct, treated with vehicle only).
at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from

220 Arterioscler Thromb Vasc Biol January 2015
Sources of FundingR.D. Feldman and R. Gros had support from Heart and Stroke Foundation for the submitted work.
DisclosuresNone.
References 1. Yoshida T, Takahashi K, Yamatani H, Takata K, Kurachi H. Impact
of surgical menopause on lipid and bone metabolism. Climacteric. 2011;14:445–452.
2. Kannel WB. Metabolic risk factors for coronary heart disease in women: perspective from the Framingham Study. Am Heart J. 1987;114:413–419.
3. Inukai T, Takanashi K, Takebayashi K, Tayama K, Aso Y, Takiguchi Y, Takemura Y. Estrogen markedly increases LDL-receptor activity in hyper-cholesterolemic patients. J Med. 2000;31:247–261.
4. Parini P, Angelin B, Rudling M. Importance of estrogen receptors in hepatic LDL receptor regulation. Arterioscler Thromb Vasc Biol. 1997;17:1800–1805.
5. Kavanagh K, Davis MA, Zhang L, Wilson MD, Register TC, Adams MR, Rudel LL, Wagner JD. Estrogen decreases atherosclerosis in part by reducing hepatic acyl-CoA:cholesterol acyltransferase 2 (ACAT2) in mon-keys. Arterioscler Thromb Vasc Biol. 2009;29:1471–1477.
6. Jones DR, Schmidt RJ, Pickard RT, Foxworthy PS, Eacho PI. Estrogen receptor-mediated repression of human hepatic lipase gene transcription. J Lipid Res. 2002;43:383–391.
7. Persson L, Henriksson P, Westerlund E, Hovatta O, Angelin B, Rudling M. Endogenous estrogens lower plasma PCSK9 and LDL cholesterol but not Lp(a) or bile acid synthesis in women. Arterioscler Thromb Vasc Biol. 2012;32:810–814.
8. Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32:71–77.
9. Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu Rev Physiol. 2005;67:335–376.
10. Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7:715–726.
11. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A trans-membrane intracellular estrogen receptor mediates rapid cell signaling. Science (New York, N.Y.). 2005;307:1625–1630.
12. Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J, Feldman RD. GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension. 2011;57:442–451.
13. Hsieh YC, Yu HP, Frink M, Suzuki T, Choudhry MA, Schwacha MG, Chaudry IH. G protein-coupled receptor 30-dependent protein kinase A pathway is critical in nongenomic effects of estrogen in attenuating liver injury after trauma-hemorrhage. Am J Pathol. 2007;170:1210–1218.
14. Sharma G, Hu C, Brigman JL, Zhu G, Hathaway HJ, Prossnitz ER. GPER deficiency in male mice results in insulin resistance, dyslipidemia, and a proinflammatory state. Endocrinology. 2013;154:4136–4145.
15. Villablanca AC, Tenwolde A, Lee M, Huck M, Mumenthaler S, Rutledge JC. 17beta-estradiol prevents early-stage atherosclerosis in estrogen recep-tor-alpha deficient female mice. J Cardiovasc Transl Res. 2009;2:289–299.
16. Feldman RD, Gros R, Ding Q, Hussain Y, Ban MR, McIntyre AD, Hegele RA. A common hypofunctional genetic variant of GPER is associated with increased blood pressure in women. Br J Clin Pharmacol. 2014. doi: 10.1111/bcp.12471
17. Hegele RA, Evans AJ, Tu L, Ip G, Brunt JH, Connelly PW. A gene-gender interaction affecting plasma lipoproteins in a genetic isolate. Arterioscler Thromb. 1994;14:671–678.
18. Hodges GJ, Gros R, Hegele RA, Van Uum S, Shoemaker JK, Feldman RD. Increased blood pressure and hyperdynamic cardiovascular responses in carriers of a common hyperfunctional variant of adenylyl cyclase 6. J Pharmacol Exp Ther. 2010;335:451–457.
19. Santolla MF, Lappano R, De Marco P, Pupo M, Vivacqua A, Sisci D, Abonante S, Iacopetta D, Cappello AR, Dolce V, Maggiolini M. G pro-tein-coupled estrogen receptor mediates the up-regulation of fatty acid synthase induced by 17β-estradiol in cancer cells and cancer-associated fibroblasts. J Biol Chem. 2012;287:43234–43245.
20. Ikeda Y, Tajima S, Izawa-Ishizawa Y, Kihira Y, Ishizawa K, Tomita S, Tsuchiya K, Tamaki T. Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes. PLoS One. 2012;7:e40465.
21. Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science (New York, N.Y.). 1986;232:34–47.
22. Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and biomolecular screening converge on a selec-tive agonist for GPR30. Nat Chem Biol. 2006;2:207–212.
23. Moon JH, Kang SB, Park JS, Lee BW, Kang ES, Ahn CW, Lee HC, Cha BS. Up-regulation of hepatic low-density lipoprotein receptor-related pro-tein 1: a possible novel mechanism of antiatherogenic activity of hydroxy-methylglutaryl-coenzyme A reductase inhibitor Atorvastatin and hepatic LRP1 expression. Metabolism. 2011;60:930–940.
24. Dennis MK, Burai R, Ramesh C, et al. In vivo effects of a GPR30 antago-nist. Nat Chem Biol. 2009;5:421–427.
25. Mayer G, Hamelin J, Asselin MC, Pasquato A, Marcinkiewicz E, Tang M, Tabibzadeh S, Seidah NG. The regulated cell surface zymogen activation of the proprotein convertase PC5A directs the processing of its secretory substrates. J Biol Chem. 2008;283:2373–2384.
26. Filardo EJ, Thomas P. GPR30: a seven-transmembrane-spanning estro-gen receptor that triggers EGF release. Trends Endocrinol Metab. 2005;16:362–367.
27. Feldman RD, Gros R. Vascular effects of aldosterone: sorting out the receptors and the ligands. Clin Exp Pharmacol Physiol. 2013;40:916–921.
28. Filardo EJ, Thomas P. Minireview: G protein-coupled estrogen receptor-1, GPER-1: its mechanism of action and role in female reproductive cancer, renal and vascular physiology. Endocrinology. 2012;153:2953–2962.
29. Lindsey SH, Chappell MC. Evidence that the G protein-coupled mem-brane receptor GPR30 contributes to the cardiovascular actions of estro-gen. Gend Med. 2011;8:343–354.
30. Lindsey SH, Liu L, Chappell MC. Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids. 2014;81:99–102.
31. Prabhushankar R, Krueger C, Manrique C. Membrane estrogen receptors: their role in blood pressure regulation and cardiovascular disease. Curr Hypertens Rep. 2014;16:408.
32. Acton S, Osgood D, Donoghue M, Corella D, Pocovi M, Cenarro A, Mozas P, Keilty J, Squazzo S, Woolf EA, Ordovas JM. Association of polymorphisms at the SR-BI gene locus with plasma lipid levels and body mass index in a white population. Arterioscler Thromb Vasc Biol. 1999;19:1734–1743.
33. Benn M, Stene MC, Nordestgaard BG, Jensen GB, Steffensen R, Tybjaerg-Hansen A. Common and rare alleles in apolipoprotein B contrib-ute to plasma levels of low-density lipoprotein cholesterol in the general population. J Clin Endocrinol Metab. 2008;93:1038–1045.
34. Bennet AM, Di Angelantonio E, Ye Z, Wensley F, Dahlin A, Ahlbom A, Keavney B, Collins R, Wiman B, de Faire U, Danesh J. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. 2007;298:1300–1311.
35. Boright AP, Connelly PW, Brunt JH, Morgan K, Hegele RA. Association and linkage of LDLR gene variation with variation in plasma low density lipoprotein cholesterol. J Hum Genet. 1998;43:153–159.
36. Clee SM, Zwinderman AH, Engert JC, Zwarts KY, Molhuizen HO, Roomp K, Jukema JW, van Wijland M, van Dam M, Hudson TJ, Brooks-Wilson A, Genest J Jr, Kastelein JJ, Hayden MR. Common genetic variation in ABCA1 is associated with altered lipoprotein levels and a modified risk for coronary artery disease. Circulation. 2001;103:1198–1205.
37. Kotowski IK, Pertsemlidis A, Luke A, Cooper RS, Vega GL, Cohen JC, Hobbs HH. A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet. 2006;78:410–422.
38. Larson IA, Ordovas JM, Barnard JR, Hoffmann MM, Feussner G, Lamon-Fava S, Schaefer EJ. Effects of apolipoprotein A-I genetic variations on plasma apolipoprotein, serum lipoprotein and glucose levels. Clin Genet. 2002;61:176–184.
39. Tomaszewski M, Charchar FJ, Barnes T, Gawron-Kiszka M, Sedkowska A, Podolecka E, Kowalczyk J, Rathbone W, Kalarus Z, Grzeszczak W, Goodall AH, Samani NJ, Zukowska-Szczechowska E. A common vari-ant in low-density lipoprotein receptor-related protein 6 gene (LRP6) is associated with LDL-cholesterol. Arterioscler Thromb Vasc Biol. 2009;29:1316–1321.
40. Wang J, Freeman DJ, Grundy SM, Levine DM, Guerra R, Cohen JC. Linkage between cholesterol 7alpha-hydroxylase and high plasma low-density lipoprotein cholesterol concentrations. J Clin Invest. 1998;101:1283–1291.
41. Zhu H, Tucker HM, Grear KE, Simpson JF, Manning AK, Cupples LA, Estus S. A common polymorphism decreases low-density lipoprotein
at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from

Hussain et al GPER Regulation of LDL Cholesterol 221
receptor exon 12 splicing efficiency and associates with increased choles-terol. Hum Mol Genet. 2007;16:1765–1772.
42. Burnett JR, Hooper AJ. Common and rare gene variants affecting plasma LDL cholesterol. The Clinical biochemist. Reviews / Australian Association of Clinical Biochemists. 2008;29:11–26.
43. Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D et al. Loci influenc-ing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009;41(1):47–55.
44. Burkhardt R, Kenny EE, Lowe JK, Birkeland A, Josowitz R, Noel M, Salit J, Maller JB, Pe’er I, Daly MJ, Altshuler D, Stoffel M, Friedman JM, Breslow JL. Common SNPs in HMGCR in micronesians and whites asso-ciated with LDL-cholesterol levels affect alternative splicing of exon13. Arterioscler Thromb Vasc Biol. 2008;28:2078–2084.
45. Coram MA, Duan Q, Hoffmann TJ, et al. Genome-wide characteriza-tion of shared and distinct genetic components that influence blood lipid levels in ethnically diverse human populations. Am J Hum Genet. 2013;92:904–916.
46. Kathiresan S, Melander O, Guiducci C, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cho-lesterol or triglycerides in humans. Nat Genet. 2008;40:189–197.
47. Kathiresan S, Willer CJ, Peloso GM, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009;41:56–65.
48. Rasmussen-Torvik LJ, Pacheco JA, Wilke RA, et al. High density GWAS for LDL cholesterol in African Americans using electronic medi-cal records reveals a strong protective variant in APOE. Clin Transl Sci. 2012;5:394–399.
49. Sabatti C, Service SK, Hartikainen AL, et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet. 2009;41:35–46.
50. Sandhu MS, Waterworth DM, Debenham SL, et al.; Wellcome Trust Case Control Consortium. LDL-cholesterol concentrations: a genome-wide association study. Lancet. 2008;371:483–491.
51. Shen H, Damcott CM, Rampersaud E, et al. Familial defective apolipoprotein B-100 and increased low-density lipoprotein cholesterol and coronary artery calcification in the old order amish. Arch Intern Med. 2010;170:1850–1855.
52. Teslovich TM, Musunuru K, Smith AV, et al. Biological, clinical and pop-ulation relevance of 95 loci for blood lipids. Nature. 2010;466:707–713.
53. Trompet S, de Craen AJ, Postmus I, Ford I, Sattar N, Caslake M, Stott DJ, Buckley BM, Sacks F, Devlin JJ, Slagboom PE, Westendorp RG, Jukema JW; PROSPER Study Group. Replication of LDL GWAs hits in PROSPER/PHASE as validation for future (pharmaco)genetic analyses. BMC Med Genet. 2011;12:131.
54. Wallace C, Newhouse SJ, Braund P, et al. Genome-wide association study identifies genes for biomarkers of cardiovascular disease: serum urate and dyslipidemia. Am J Hum Genet. 2008;82:139–149.
55. Waterworth DM, Ricketts SL, Song K, et al.; Wellcome Trust Case Control Consortium. Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler Thromb Vasc Biol. 2010;30:2264–2276.
56. Willer CJ, Sanna S, Jackson AU, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–169.
57. Willer CJ, Schmidt EM, Sengupta S et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283.
G-protein estrogen receptor (GPER) is a newly recognized G-protein–coupled receptor linked to the actions of estrogen (and to a lesser extent aldosterone). We have identified that women carrying a common hypofunctional missense genetic variant of GPER, P16L GPER, have increased low-density lipoprotein cholesterol. Furthermore, GPER activation in vitro leads to increased low-density lipoprotein receptor ex-pression mediated at least, in part, by inhibition of PCSK9 expression, an important regulator of low-density lipoprotein receptor degradation. In total, these studies support an important role of GPER in low-density lipoprotein cholesterol regulation.
Significance
at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from

1
Materials and Methods Cell culture. HepG2 cells (obtained from ATCC, Manassas, Virginia, USA) were cultured in
DMEM (Invitrogen, Mississauga, ON) containing penicillin, streptomycin and 10% fetal calf
serum (FBS) in humidified atmosphere (37°C, 5% CO2). Previous studies have shown that these
cells express GPER1,2
as well as ERα and ERβ3.
Assessment of LDL receptor and PCSK9 protein. The effect of the GPER agonist G1 4 on
PCSK9 and LDL receptor protein was assessed by immunoblotting. Cultured HepG2 cells were
serum starved for 24 hours in phenol red-free DMEM and then incubated with increasing
concentration of G1 for an additional 24 hours. In experiments using the GPER antagonist, G155,
cells were pretreated with G15 for 15 min prior to addition of the agonist. Cells were
sequentially washed with phosphate buffered saline (PBS) and lysed in ice-cold buffer
containing 20mM Tris, pH 8.0, 1%NP-40.0, 0.1% SDS, 140mM NaCl and 1mM
phenylmethylsulfonyl fluoride. Conditioned media (for secreted PCSK924,25
) or cell lysates (for
LDL receptor) were resolved by 8 %SDS-PAGE and transferred electrophoretically onto
Immun-Blot polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA) and blocked for 1
hour at room temperature in a buffer containing 20 mM Tris (pH 7.4), 0.5 M NaCl, and 0.1%
Tween 20 and 5% skim milk. The membranes were then incubated with anti-LDL receptor
antibody or anti-PCSK9 (Abcam, Toronto ON; 1:1000 dilutions for both). Blots were washed in
Tris-buffered saline for 1 h and then incubated in HRP conjugated secondary anti-rabbit (1:5,000
dilution) antibody for 1 h at room temperature. GAPDH was used as a loading control for
immunoblots of cell lysates. In immunoblots of supernatant samples for PCSK9 expression,
samples were normalized for protein content
Proteins were detected by chemiluminescence, as described by the manufacturer's
protocol (DuPont NEN, Boston, MA). Bands corresponding to PCSK9 or LDL receptor were
quantified by densitometry.
Assessment of LDL uptake. LDL uptake was determined by using LDL labeled with the
fluorescent probe 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI-LDL,
Alfa Aesar, Ward Hill, MA) as previously described6. Briefly, HepG2 cells in 6-well (35mm)
plates were serum-starved for 24 hours in phenol red-free DMEM and then were incubated with
increasing concentration of GPER agonist G1(0.001-1μM) for 24 hour. Subsequently, cells were
incubated in fresh media in the presence of 10 μg/mL DiI-LDL for 5 h. Cells were trypsinized
and resuspended in phosphate-buffered saline for FACS analysis with BD FACSCalibur (BD
Biosciences, Franklin Lakes, NJ) Data were collected and analyzed with BD Cell Quest Pro
Software
Real time PCR analysis of the level of LDL receptor and PCSK9 mRNA expression. HepG2
cells were serum- starved in phenol red-free DMEM for 24 hours, and then incubated with
increasing concentration of G1 (0.001-1μM) for 24 hours. Total RNA was isolated by using
RNeasy Mini Kit (Qiagen, Toronto, ON). RNA (2 μg) was then reverse-transcribed using high
capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) as per
manufacturer instruction, and mRNA abundances were determined by a 2-step quantitative real-
time reverse transcription PCR method on an ABI Prism (model 7900HT) Sequence Detection

2
System (Applied Biosystems, Foster City, CA) as previously described6. Briefly, cDNA (20 ng)
generated by reverse transcription as described above was analyzed in triplicate for LDLR or
PCSK9 mRNA expression which was normalized to the abundance of glyceraldehyde 3-
phosphate dehydrogenase (GAPDH) utilizing standard curve method. Quantities of mRNA were
interpolated from a standard curve plotted as cycle threshold versus the log of the quantity of
serial dilutions of known GAPDH and LDLR or PCSK9 cDNA standards. Primers for LDLR,
PCSK9 and GAPDH were obtained from Taqman Assays-on-Demand (Applied Biosystems,
Foster City, CA).
Gene transfer studies. The AdMaxTM
adenovirus creation kit was used to generate the
adenoviral vectors used in these studies as per manufacturer’s instructions (Microbix BioSystems
Inc., Toronto, Canada), as we have previously described7. Hep G2 cells were cultured for 24
hours and then infected with adenovirus expressing GPER shRNA (shGPER) or shGFP (control)
for 16 hours, followed by incubation in phenol red-free DMEM for 24 hours. Finally, cells were
treated with receptor agonists for 24 hours.
Human genetic studies
Discovery cohort: Alberta population of Northern European descent: 415 healthy subjects
were studied from 21 adjacent Hutterite colonies in Alberta8. Both females and males were
included. Subjects were 17-73 years of age, with the following demographic characteristics:
56.6% were women; age, body mass index (BMI) and waist circumference were (mean+
standard deviation) 36.9±14.9 years, 28.2±5.3 kg/m2 and 86.6±13.7 cm, respectively. Fasting
blood samples (12 hours) were drawn to determine the biochemical profile and genotype of the
subjects. Exclusion criteria were restricted to blood samples inadequate for genetic and/or
biochemical profiling. Informed consent was obtained for all analyses, with approval from the
Western University Research Ethics Board.
Validation cohort: London Ontario based population: We studied 339 normal, healthy females,
18-56 years of age. Recruitment was based on local advertising and email invitations for
volunteers within the Robarts Research Institute and the University of Western Ontario.
Informed consent was obtained for all analyses, with approval from the Western University
Research Ethics Board. This included 251 subjects from a previously reported cohort study9,
which comprises all of the female subjects in whom non-fasting plasma lipids/lipoproteins were
determined. The cohort is predominantly white (87%) of predominantly Anglo-Saxon origin with
small subsets of East Asians (9%), south Asians (3%) and blacks (1%). Exclusion criteria
included: history of cardiovascular events, average alcohol intake over 2 units per day,
pregnancy and use of anti-hypertensive drugs or anticoagulants. Age, BMI and waist
circumference were 27.6±0.5 years; 23.0±0.2 kg/m2 and 76.6±0.5 cm, respectively.
Biochemical analysis: Plasma concentrations of total cholesterol, high density lipoprotein
(HDL) cholesterol, triglyceride and apolipoprotein (apo) B were determined in the J. Alick Little
Lipid Research Laboratory at St. Michael’s Hospital (for the discovery cohort)8,10
and in the
laboratory of Dr. Murray Huff, Robarts Institute (for the validation cohort)11
. The low density
lipoprotein (LDL) value was calculated.

3
Genotyping: Genomic DNA was extracted from whole blood and genotyped for GPER by
London Regional Genomic Centre (www.lrgc.ca) as previously described12
.
Data analysis: Cellular studies: Initial analysis by one-way ANOVA was followed by Dunnett’s
multiple comparison tests for multiple group comparisons. The significance of difference
between paired groups was determined by Students test for paired data. P < 0.05 on a two-sided
test was taken as a minimum level of significance.
Human genetic studies: Statistical significance of difference in quantitative variables between
wild type P16/P16 homozygotes (CC) and carriers of P16L GPER variant groups (CT plus TT)
was determined by non-parametric one way analysis of variance (Prism 4.0, GraphPad Software,
San Diego, CA). P<0.05 was taken as the nominal significance level. The chi-square test was
used to compare the allele frequencies in the males vs. females of each population. As previously
reported, we used a general linear model ANOVA to test for the association of GPER genotype
with plasma levels of LDL and total cholesterol after adjusting for covariates13
. The GPER
genotype was introduced into the ANOVA under an additive (co-dominant) model (CC, wild
type; CT, heterozygote carrier; and TT, homozygote for the variant). The dependent variable
introduced was either LDL cholesterol, total cholesterol or apo B and the independent variables
were GPER genotype, age, BMI, sex, HDL cholesterol and triglyceride. Genotype frequencies
deviation from the Hardy-Weinberg equilibrium predicted frequencies were evaluated using the
conditional chi-square goodness of fit test using one degree of freedom for both populations
14.
This analysis was done for the Alberta and London Ontario based samples using SAS statistical
software, version 9.1 (SAS institute, Inc., Cary, NC).

4
References
1. Santolla MF, Lappano R, De Marco P, Pupo M, Vivacqua A, Sisci D, Abonante S,
Iacopetta D, Cappello AR, Dolce V, Maggiolini M. G protein-coupled estrogen receptor
mediates the up-regulation of fatty acid synthase induced by 17beta-estradiol in cancer
cells and cancer-associated fibroblasts. The Journal of biological chemistry.
2012;287:43234-43245.
2. Ikeda Y, Tajima S, Izawa-Ishizawa Y, Kihira Y, Ishizawa K, Tomita S, Tsuchiya K,
Tamaki T. Estrogen regulates hepcidin expression via GPR30-BMP6-dependent
signaling in hepatocytes. PLoS One. 2012;7:e40465.
3. Solakidi S, Psarra AM, Sekeris CE. Differential subcellular distribution of estrogen
receptor isoforms: localization of ERalpha in the nucleoli and ERbeta in the mitochondria
of human osteosarcoma SaOS-2 and hepatocarcinoma HepG2 cell lines. Biochimica et
biophysica acta. 2005;1745:382-392.
4. Bologa CG, Revankar CM, Young SM, Edwards BS, Arterburn JB, Kiselyov AS, Parker
MA, Tkachenko SE, Savchuck NP, Sklar LA, Oprea TI, Prossnitz ER. Virtual and
biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol.
2006;2:207-212.
5. Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, Bologa CG, Leitao
A, Brailoiu E, Deliu E, Dun NJ, Sklar LA, Hathaway HJ, Arterburn JB, Oprea TI,
Prossnitz ER. In vivo effects of a GPR30 antagonist. Nat Chem Biol. 2009;5:421-427.
6. Beyea MM, Reaume S, Sawyez CG, Edwards JY, O'Neil C, Hegele RA, Pickering JG,
Huff MW. The oxysterol 24(s),25-epoxycholesterol attenuates human smooth muscle-
derived foam cell formation via reduced low-density lipoprotein uptake and enhanced
cholesterol efflux. J Am Heart Assoc.1:e000810.
7. Gros R, Ding Q, Chorazyczewski J, Pickering JG, Limbird LE, Feldman RD. Adenylyl
cyclase isoform-selective regulation of vascular smooth muscle proliferation and
cytoskeletal reorganization. Circulation research. 2006;99:845-852.
8. Hegele RA, Evans AJ, Tu L, Ip G, Brunt JH, Connelly PW. A gene-gender interaction
affecting plasma lipoproteins in a genetic isolate. Arterioscler Thromb. 1994;14:671-678.
9. Hodges GJ, Gros R, Hegele RA, Van Uum S, Shoemaker JK, Feldman RD. Increased
blood pressure and hyperdynamic cardiovascular responses in carriers of a common
hyperfunctional variant of adenylyl cyclase 6. The Journal of pharmacology and
experimental therapeutics. 2010;335:451-457.
10. MacLean DR, Petrasovits A, Nargundkar M, Connelly PW, MacLeod E, Edwards A,
Hessel P. Canadian heart health surveys: a profile of cardiovascular risk. Survey methods

5
and data analysis. Canadian Heart Health Surveys Research Group. CMAJ : Canadian
Medical Association journal = journal de l'Association medicale canadienne.
1992;146:1969-1974.
11. Cheung R, Lewanczuk RZ, Rodger NW, Huff MW, Oddou-Stock P, Botteri F, Pecher E,
Muirhead N. The effect of valsartan and captopril on lipid parameters in patients with
type II diabetes mellitus and nephropathy. International journal of clinical practice.
1999;53:584-592.
12. Cao H, van der Veer E, Ban MR, Hanley AJ, Zinman B, Harris SB, Young TK, Pickering
JG, Hegele RA. Promoter polymorphism in PCK1 (phosphoenolpyruvate carboxykinase
gene) associated with type 2 diabetes mellitus. The Journal of clinical endocrinology and
metabolism. 2004;89:898-903.
13. Hegele RA, Cao H, Huff MW, Anderson CM. LMNA R482Q mutation in partial
lipodystrophy associated with reduced plasma leptin concentration. The Journal of
clinical endocrinology and metabolism. 2000;85:3089-3093.
14. Hegele RA, Plaetke R, Lalouel JM. Linkage disequilibrium between DNA markers at the
low-density lipoprotein receptor gene. Genet Epidemiol. 1990;7:69-81.

D. McIntyre, Murray W. Huff, Robert Gros, Robert A. Hegele and Ross D. FeldmanYasin Hussain, Qingming Ding, Philip W. Connelly, J. Howard Brunt, Matthew R. Ban, Adam
Metabolism: Cellular and Population Genetic StudiesG-Protein Estrogen Receptor as a Regulator of Low-Density Lipoprotein Cholesterol
Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2014 American Heart Association, Inc. All rights reserved.
Greenville Avenue, Dallas, TX 75231is published by the American Heart Association, 7272Arteriosclerosis, Thrombosis, and Vascular Biology
doi: 10.1161/ATVBAHA.114.3043262014;
2015;35:213-221; originally published online November 13,Arterioscler Thromb Vasc Biol.
http://atvb.ahajournals.org/content/35/1/213World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://atvb.ahajournals.org/content/suppl/2014/11/13/ATVBAHA.114.304326.DC1.htmlData Supplement (unedited) at:
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
at UNIVERSITY WESTERN ONTARIO on February 11, 2015http://atvb.ahajournals.org/Downloaded from




















