
The high-conductance channel of porin-less yeast mitochondria
Upload
u-bordeauxCategory
view
1download
0
From calcium signaling to cell death: two conformations for themitochondrial permeability transition pore.
Switching from low- to high-conductance state
Franc°ois Ichas a;b;*, Jean-Pierre Mazat a
a INSERM-CJF 9705, Integrated Biological Systems Study Group, Victor Segalen-Bordeaux 2 University, 146 rue Leo Saignat,F-33076 Bordeaux Cedex, France
b CNR Center for the Study of Biomembranes, Department of Biomedical Sciences, Laboratory of Biophysics and Membrane Biology,University of Padua, Viale G. Colombo 3, I-35121 Padua, Italy
Received 27 January 1998; revised 30 March 1998; accepted 31 March 1998
Abstract
The permeability transition pore (PTP) is a channel of the inner mitochondrial membrane that appears to operate at thecrossroads of two distinct physiological pathways, i.e., the Ca2� signaling network during the life of the cell, and the effectorphase of the apoptotic cascade during Ca2�-dependent cell death. Correspondingly, two open conformations of the PTP canalso be observed in isolated organelles. A low-conductance state, that allows the diffusion of small ions like Ca2�, is pH-operated, promoting spontaneous closure of the channel. A high-conductance state, that allows the unselective diffusion ofbig molecules, stabilizes the channel in the open conformation, disrupting in turn the mitochondrial structure and causing therelease of proapoptotic factors. Our current results indicate that switching from low- to high-conductance state is anirreversible process that is strictly dependent on the saturation of the internal Ca2�-binding sites of the PTP. Thus, the high-conductance state of the PTP, which was shown to play a pivotal role in the course of excitotoxic and thapsigargin-inducedcell death, might result from a Ca2�-dependent conformational shift of the low-conductance state, normally participating inthe regulation of cellular Ca2� homeostasis as a pH-operated channel. These observations lead us to propose a simplebiophysical model of the transition between Ca2� signaling and Ca2�-dependent apoptosis. ß 1998 Elsevier Science B.V.All rights reserved.
Keywords: Apoptosis ; Bcl-2; Calcium; Cyclosporin A; Cytochrome c ; Excitotoxicity; Glutamate; Inositol 1,4,5-trisphosphate; Mito-chondria; Neuron; N-Methyl-D-aspartate; Permeability transition pore; Thapsigargin
1. Introduction
For some obscure reasons, and in spite of an ex-haustive number of clues, it took nearly 40 yearsafter Mitchell proposed his chemosmotic model ofoxidative phosphorylation [1] to realize that mito-chondria are not simply cellular power plants as-signed to ATP production. As a matter of fact, themitochondrial transmembrane proton gradient main-
0005-2728 / 98 / $19.00 ß 1998 Elsevier Science B.V. All rights reserved.PII: S 0 0 0 5 - 2 7 2 8 ( 9 8 ) 0 0 1 1 9 - 4
Abbreviations: CICR, Ca2�-induced Ca2� release; CsA, cyclo-sporin A; v8, mitochondrial membrane potential ; ER, endoplas-mic reticulum; IP3, inositol 1,4,5-trisphosphate; NMDA, N-methyl-D-aspartate; PTP, permeability transition pore; RaM,rapid mode of mitochondrial Ca2� uptake
* Corresponding author, at address b. Fax: +39 (49) 8276361;E-mail : [email protected]
BBABIO 44663 28-7-98
Biochimica et Biophysica Acta 1366 (1998) 33^50

tained by respiration is used by the organelle as aversatile driving force for a variety of tasks thathave little to do with ATP synthesis [2]. Amongthese, two purely dissipative tasks, i.e. that do notcouple the dissipation of the proton gradient to anykind of thermodynamic work, have recently beenproposed to play pivotal roles in the life and in thedeath of the cell. Both these mitochondrial tasks cor-respond to signal transduction processes, and as suchinvolve the downhill release of signaling ions and/ormolecules from the organelle. Both these signalingevents have been proposed to depend on a Ca2�-ac-tivated channel of the inner mitochondrial mem-brane ^ the permeability transition pore (PTP) ^characterized by Hunter and Haworth in the 1970s[3^8] (see [9] for a comprehensive review), and theopening of which results in the dissipation of severaltransmembrane electrochemical gradients. Indeed,while evidence indicates that the PTP is a channelthat mitochondria operate to release Ca2� and par-ticipate in the intracellular Ca2� signaling networkduring the normal life of the cell [10^14], severalreports concerning neuronal excitotoxicity [15^19]and thapsigargin-induced apoptosis [20] give alsocredit to the idea that the Ca2�-dependent operationof the PTP is a key event committing the cell to anapoptotic fate, probably by causing swelling of themitochondrial matrix, rupture of the outer mem-brane, and release of proapoptotic factors containedin the former intermembrane space [21^23]. We dis-cuss here this dual function, and analyze it in termsof variability of PTP open conformation [7,9,14,24^26].
2. Low-conductance state of the PTP and Ca2+
signaling
The idea that the PTP might be relevant to cellularCa2� homeostasis as a mitochondrial Ca2�-activatedCa2� release channel has been hypothesized for along time, mainly on the basis of phenomenologicand thermodynamic considerations [3,7,27]. The ¢rstexperimental report giving credit to this view camefrom the group of Halestrap and concerned the reg-ulation of mitochondrial volume in hepatocytes chal-lenged with the Ca2�-mobilizing hormone vasopres-sin [10]. In their study, the authors found that after
vasopressin challenge, mitochondria were undergoingin situ a swelling process that could be partially pre-vented by cyclosporin A (CsA). Since CsA is a po-tent inhibitor of the PTP [28^31], these results couldbe interpreted in terms of PTP opening triggered byCa2� signaling events [10]. A few years later, a keyreport concerning Ca2� homeostasis in intact cardi-omyocytes at rest provided further evidence of PTP£ickering in situ [11]. In this report, Altschuld andcolleagues showed that a signi¢cant amount of thetotal cellular Ca2� transits through mitochondria,entering the organelle in a ruthenium red-sensitivemanner, i.e., via the Ca2� uniporter, and leaving itthrough a CsA-sensitive pathway, i.e., the PTP. Us-ing the same type of approach in hepatocytes, thegroup of Orrenius reported the same year observa-tions compatible with the latter view [12]. However,with regard to the models of the PTP that were cur-rent at that time, few people in the ¢eld were open tothe idea that a channel apparently solely capable ofcausing a global and nearly irremediable disruptionof mitochondrial homeostasis in vitro could repre-sent a structure of any relevance to the normal phys-iology of the cell.
2.1. Low-conductance state of the PTP andmitochondrial Ca 2+-induced Ca 2+
release (CICR)
Yet, it became rapidly evident that the PTP isendowed with the characteristics of a fully reversibleCICR channel capable of transient opening[13,14,32,33], preserving other mitochondrial func-tions such as volume homeostasis [14]. Signs ofsuch behavior had been observed in vitro for yearsby the group of Evtodienko (reviewed in [32^34]),but were only recently attributed to the operationof a speci¢c narrow conformation of the pore [14].It is important to note that as early as 1979, Ha-worth and Hunter detected and discussed the processof PTP £ickering [6], as well as the existence of aPTP subconductance state [7] (see also Section 6).
In intact organelles, we observed that mitochon-drial CICR is promoted by the latter type of PTPconformation [14] (i) with an apparent molecularcuto¡ 6 300 Da, i.e. 5 times smaller than the high-conductance state [6], and permeable to small ionslike protons, Ca2�, or K�, (ii) that does not cause
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^5034

mitochondrial swelling, (iii) and principally operatedby matrix pH [14,35] (see also Table 1). Other au-thors have recently proposed an even narrower con-formation of the PTP that would be selective forprotons [25,26], and that could promote mitochon-drial CICR as well by behaving as a pure depolariz-ing channel (see explanation below).
In electrophysiological studies, several subconduc-tance levels of the PTP have been described by Szabo©and Zoratti [9], and by the group of Kinnally [24].One of these, corresponding to a half-conductancestate of approx. 500 pS, promotes pore £ickering,at variance from the full-conductance state thatmost often develops from the 500 pS state and sub-sequently blocks the pore in the open conformationat 1200 pS [9]. It thus seems likely that the £ickering500 pS state of the PTP observed in patch clampstudies [9,24] corresponds to the low-conductanceconformation of the PTP evidenced in intact organ-elles [7,14].
The proposed mechanism by which the PTP pro-motes mitochondrial CICR is based on the observa-tion that matrix pH seems to be the major regulatorof the low-conductance conformation [14,35] (seealso Section 4), and can be described as follows: (i)a Ca2� signal reaches the organelle, (ii) Ca2� entersthe mitochondrion due to v8 which is negative in-side, (iii) charge compensation by the respiratorychain raises matrix pH, (iv) high matrix pH triggersPTP opening, (v) PTP opening collapses the protongradient causing decline of v8, outward redistribu-tion of Ca2�, and matrix acidi¢cation, (vi) matrixacidi¢cation closes the PTP, (vii) respiratory chain
Table 1Modulators of the two conductance states of the PTP
Channel modulator Low-conductance PTP High-conductance PTP
E¡ect Ref. E¡ect Ref.
Ca2� u [13,14,32,33] u [3^8]Sr2� v [34] ;[b] t [3,5^8]Mg2� s [a] s [3,5^8]H� s [14] ;[b] s [6,44,65]ADP s [14,32,33] s [3,5^8]Pi t [a] v [3]CsA s [13,14,32,33] s [28^31]SDZ-PSC833 t [13] a [a]
Up arrow, activator; down arrow, inhibitor; circle, no e¡ect; [a] Ichas et al. unpublished; [b] present manuscript; single arrows, com-mon modulators; double arrows, modulators having di¡erent e¡ects on the two conductance states. The list is limited to the modula-tors that have been investigated on both conductance states.
Fig. 1. Schematic model of the transmembrane ion £uxes asso-ciated with triggering, opening, and closure of the PTP in itslow-conductance conformation. After [13,14,35]. In the car-toons: left, Ca2� uniporter (white) ; central, PTP (black); right,respiratory chain (grey). Phase 1: the respiratory chain main-tains the proton gradient that is responsible for membrane po-larization (v8). Ca2� enters mitochondria due to v8 throughthe uniporter, while the respiratory chain electrically compen-sates the resulting in£ux of positive charges by extruding moreprotons. As a result, matrix pH increases. Phase 2: high matrixpH triggers PTP opening. The latter provides an inward path-way for protons that shunts the activity of the respiratorychain, and collapses v8. As a result, Ca2� undergoes an out-ward redistribution, and matrix pH decreases. Note that Ca2�
e¥ux takes place through both the uniporter and the PTP.Phase 3: low matrix pH closes the PTP. PTP closure stops pro-ton in£ow, and the respiratory chain restores the proton gra-dient and corresponding v8. Ca2� is reaccumulated due to re-stored v8, and matrix pH increases again.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^50 35

rebuilds the proton gradient, thus restoring v8, (vii)restored v8 drives Ca2� re-uptake (Fig. 1) [14,35]. Itshould be noted that phase (vii) is equivalent tophase (i), and thus, that the whole cycle might repeatinde¢nitely, leading to pore £ickering and oscillatoryion £uxes [32,35] (Fig. 2). Regarding conformationalconsiderations, it should also be noted that a proton-selective conformation of the PTP [25,26] could ac-count for phase (v) as well. Indeed, the Ca2� uni-porter present in the inner mitochondrial membraneis a fast and symmetric Ca2� channel that can trans-port Ca2� in both directions [27]. Thus, transientcollapse of v8 by a proton-selective PTP wouldalso result in the outward redistribution of Ca2�,due to reverse £ow through the Ca2� uniporter.
The dependence of the low-conductance PTP con-formation on matrix pH ^ rather than on matrixCa2� ^ is a notable characteristic. Owing to suchcontrol, triggering of mitochondrial CICR does notdepend on the absolute amount of Ca2� taken up bymitochondria, but rather on the rate at which Ca2� isprovided to the organelles [13,14,34,35]. Indeed, forthe same amount of Ca2� provided, mitochondriacan behave either as Ca2� storage organelles whenCa2� is delivered slowly, or undergo CICR whenCa2� is delivered at a fast rate (see also Fig. 8).Thus, triggering of the narrow conformation ofthe PTP by matrix pH rather than by Ca2� itselfallows the organelles to integrate and processCa2� inputs on a temporal/frequency-dependent ba-
sis [13,14,34,35]. We will come back to this feature inSection 4 and Section 5 regarding cell death.
2.2. PTP and Ca 2+ signaling: theoretical sketch
The features of the low-conductance state of thePTP, and of its function of CICR channel deter-mined in vitro, can be used to determine whetherits participation in the intracellular Ca2� network isfeasible in situ, at least on theoretical grounds. Thein vitro experiments tell us that signi¢cant amountsof Ca2� need to be translocated inside mitochondria(30^50 nmol mg31 [13,14,32^34]) at a fast rate (s 90nmol mg31Umin31 [14]), in order to raise matrix pHhigh enough to trigger mitochondrial CICR. If weassume that theses quantitative values are directlyrelevant to the in situ context, we can predict thatonly a subset of the cellular mitochondrial popula-tion is susceptible to operate the PTP as a CICRchannel.
If we consider mitochondria sensing the averageheight of a standard cytosolic Ca2� transient, i.e.with a peak amplitude of 500^1500 nM and lastingfor 5 s, we can deduce from the kinetics of the Ca2�
uniporter [27] that a maximum of 0.5^4 nmol mg31
can be transferred through the uniporter, at a ratevarying between 10 and 80 nmol mg31Umin31 [27].However, operation of the rapid uptake mode(RaM) must also be taken into account [36]. Consid-ering that RaM-mediated Ca2� uptake occurs duringthe initial 100 ms of the peak, at a rate two orders ofmagnitude higher than uniport-mediated uptake(T.E. Gunter, personal communication; see also[36]), then an additional amount of 1.7^13 nmolmg31 must be considered. In this case, we come upwith a total Ca2� transfer of 2.3^17 nmol mg31, at amean rate varying between 46 and 340 nmolmg31Umin31. While the upper value exceeds ^ interms of rate ^ the threshold of mitochondrialCICR, the amounts translocated do not meet the invitro requirements.
Let us now consider mitochondria tightly associ-ated with the intracellular Ca2� release channels ofthe endoplasmic reticulum (ER) [37]. When Ca2�
transients are generated, these particular mitochon-dria sense local Ca2� `hot spots' and take up thecation at an extremely fast rate due to the high localCa2� concentration (s 20 WM according to recent
Fig. 2. Heuristic model of mitochondrial CICR. After [14,35].For details see text and Fig. 1.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^5036

direct measurements; R. Rizzuto, personal commu-nication). This results in free matrix [Ca2�] transientspeaking at 5^10 WM above the baseline [37,38]. Sincethe bound/free Ca2� ratio in mitochondria in vivo isapprox. 6000 [39], the amplitude of the free matrix[Ca2�] transients observed at the level of these par-ticular mitochondria re£ects a quantity of Ca2�
translocated of 30^60 nmol mg31. Moreover, thistransfer occurs within approx. 5 s [37,38], giving arate between 600 and 1200 nmol mg31Umin31.These values, deduced from matrix parameters, aresimilar to the ones that can be calculated from thekinetics of the uniporter. Indeed, for extramitochon-drial Ca2� concentrations exceeding 20 WM, the uni-porter transports Ca2� at a rate that varies between600 and 1200 nmol mg31Umin31 [27], which, in 5 s,results in the transfer of 50^100 nmol mg31. Thus,whatever the mode of calculation, these values allexceed the requirements of mitochondrial CICR op-eration observed in vitro.
Thus, this analysis suggests that mitochondrialCICR is most likely (i) to occur speci¢cally at thelevel of the mitochondria tightly associated with theCa2� release channels of the ER (like the IP3 recep-tor), and (ii) to be triggered immediately after, andeach time Ca2� is released from the ER (Fig. 3).
2.3. PTP and Ca 2+ signaling: experimental results
Experimentally, three studies con¢rm the idea thatPTP operation occurs in situ, and is dependent onsuch a Ca2� cross-talk between the ER and theneighboring mitochondria. Davidson and Halestrap
reported as early as 1990 that in hepatocytes chal-lenged with vasopressin ^ which mobilizes Ca2� fromthe ER ^ mitochondria undergo a swelling process insitu that can be partially inhibited by CsA [10]. Thisresult can be interpreted in terms of PTP openingtriggered by the transfer of Ca2� from the ER tomitochondria. Four years later, while we were exam-ining the di¡erential sensitivity of mitochondrialCICR to two distinct CsA derivatives in vitro, weobserved in parallel experiments that the amplitudeof IP3-dependent cytosolic Ca2� signals triggered inintact Ehrlich carcinoma cells exhibited a similar dif-ferential sensitivity to the two CsA derivatives understudy [13]. In other words, the cyclosporin that wasthe more potent at inhibiting mitochondrial CICR,appeared to be also the more e¤cient at decreasingthe size of the cytosolic Ca2� transients in intactcells. In addition, the inhibiting e¡ects of CsA onthe cytosolic Ca2� signals could be mimicked by pre-treating the cells with antimycin A that collapses v8,and prevents all mitochondrial Ca2� £uxes. We thusproposed that mitochondrial CICR, i.e., transientPTP operation, is triggered shortly after Ca2� is re-leased from the ER, and contributes to a signi¢cantpart of the ¢nal cytosolic Ca2� signal [13]. By mon-itoring both intramitochondrial Ca2� and v8 duringthe early phase of IP3-dependent Ca2� signals, welater obtained results con¢rming that the PTP is aCa2� release channel relevant to the regulation ofcellular Ca2� homeostasis. We found that shortlyafter Ca2� is released from the ER by IP3, mitochon-dria undergo a transient membrane depolarization,as well as a transient decrease in their Ca2� contents,
Fig. 3. Schematic model of mitochondrial CICR participation in the Ca2� signaling network. After [13,14,37]. The reticular structurerepresents the ER, and the organelles with cristae are mitochondria. The right-hand mitochondrion is located in a Ca2� microdomain,close to an IP3 receptor of the ER (empty square). (A) During the initial mobilization of Ca2� from the ER, the right-hand mito-chondrion takes up Ca2� via the uniporter (closed circle), while the left-hand mitochondrion is too far away from the Ca2� releasesite to do so. (B) Mitochondrial CICR occurs at the level of the right-hand mitochondrion due to PTP opening (empty circle), whichampli¢es the cytosolic signal, and the left-hand mitochondrion starts only now accumulating Ca2�. (C) All the organelles accumulateCa2� (uniporter: closed circle; Ca2�-ATPase: closed square). The transmembrane arrows indicate the direction of the net Ca2� £uxes.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^50 37

that can both be inhibited by CsA. We again ob-served that the release of Ca2� from mitochondriathat occurs during this phase ampli¢es the cytosolicCa2� signal, further indicating that the PTP is achannel that mitochondria operate transiently totake an active part in Ca2� signaling processes [14].
Two other studies, published in 1992, also evi-denced that PTP £ickering is a process contributingto the regulation of cellular Ca2� homeostasis[11,12]. However, in these reports, the possible de-pendence of PTP opening on an initial release ofCa2� from the ER was not investigated. Altschuldand colleagues [11] reported that the net uptake of45Ca2� by isolated cardiomyocytes at rest is dramat-ically increased when the cells are incubated in thepresence of CsA, and that the e¡ects of CsA can befully reversed by the introduction of ruthenium redin the extracellular medium (ruthenium red blocksmitochondrial Ca2� uptake [27]). The authors logi-cally concluded that in the presence of CsA mito-chondria are continuously accumulating Ca2� with-out possible release through the PTP, and that suchmitochondrial overaccumulation is prevented whenthe in£ux pathway ^ the mitochondrial Ca2� uni-porter ^ is also blocked with ruthenium red [11].The group of Orrenius observed in the same yearthat in the presence of CsA the mitochondria of in-tact hepatocytes become progressively overloadedwith Ca2� in situ [12], again suggesting that thePTP is a physiological pathway for mitochondrialCa2� e¥ux.
Finally, a recent communication by Di Lisa andcolleagues, based on a di¡erent and original method,gives further credit to the idea that the PTP is achannel operated almost constantly by mitochondriain situ (Di Lisa, personal communication, and [40]).However, in this work, the correlation of PTP func-tion with Ca2� homeostasis was not investigated. Intheir study, the authors loaded hepatoma cells withcalcein-AM, and subsequently quenched the cyto-solic signal by perfusing the cells with cobalt. Aftercobalt washout, mitochondria (which did not take upcobalt during the perfusion) could be imaged as £u-orescent calcein-containing organelles over a darkcellular background. By recording the time courseof calcein £uorescence at the level of individual mi-tochondria, the authors could evidence a spontane-ous and constant £uorescence decrease that could be
abolished by CsA. Since mitochondria maintain ahigh mean v8 value under these conditions, the re-sults are indicative of constant £ickering of the PTP(i) under a conformation that does not impair mito-chondrial functions, and (ii) allowing gradual calceinrelease and/or cobalt in£ux.
2.4. Summary
The PTP can adopt di¡erent open conformations.Under its low-conductance conformation, the PTP (i)does not impair mitochondrial functions, like volumehomeostasis, and can £icker in vitro and in situ; (ii)is operated by the changes in matrix pH accompany-ing mitochondrial Ca2� uptake; and (iii) is involvedas a mitochondrial Ca2� release channel in the reg-ulation of cellular Ca2� homeostasis.
3. High-conductance state of the PTP and cell death
Historically, the high-conductance state of the PTPwas the ¢rst to be characterized by Hunter and Ha-worth in isolated organelles [3^8]. The process ofpermeability transition was described as a pathwayfor mitochondrial Ca2� e¥ux [7], dependent on theopening of a Ca2�-activated hydrophilic channel ofthe inner membrane [6,8], later characterized electro-physiologically and termed `megachannel' by Sza-bo© and Zoratti [9,41,43,44,46,47], or `multiple con-ductance channel' by the group of Kinnally [24,42,45,48,49]. In its high-conductance state, the PTP ren-ders the inner mitochondrial membrane unselectivelypermeable to solutes of molecular mass 6 1500 Da[6] and remains open, unless Ca2� is chelated byaddition of EGTA [3,6]. Since mitochondria withan open high-conductance pore can no longer main-tain a proton gradient, and thus no longer sustainoxidative phosphorylation [1], it was proposed thatthe cellular toxicity of several exogenous/arti¢cialagents (chemical, or physical like ischemia/reperfu-sion cycles) known to promote PTP opening in vitro,might be accounted for by an in situ activation of thePTP causing ^ among other things ^ an arrest ofaerobic ATP synthesis [50^60] (see also [9] and otherreviews in this issue).
However, as demonstrated by Hunter and Ha-worth [3^8] (see also Section 6) and several other
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^5038

groups, the high-conductance state of the PTP is notonly operated by Ca2�, and the toxic e¡ects of avariety of exogenous compounds are in fact revealinga few key built-in regulatory mechanisms of thechannel [3^9,24^31,41^49,61^73]. To these built-insensors correspond clearly identi¢ed endogenous ef-fectors, suggesting that beyond its role in accidental/toxic cell death [50^60], the high-conductance stateof the PTP might be physiologically triggered duringprogrammed cell death. This view is reinforced bythe observations that (i) the antiapoptotic e¡ects ofthe Bcl-2 gene product seem to correlate with itsmitochondrial location [74,75], and that (ii) in vitro,Bcl-2 seems to inhibit operation of the PTP in itshigh-conductance state [21,76,77].
3.1. High-conductance state of the PTP
In its high-conductance state, the PTP was charac-terized as a channel activated by the cooperativebinding of two Ca2� ions to its matrix domain[6,8], with a ¢xed molecular cuto¡ of approx. 1500Da [6], and an electrical conductance of approx. 1.2nS [9,24]. In this conformation, opening of the PTPinduces in vitro a complete and sustained collapse ofthe transmembrane proton gradient, allows the e¥uxof a variety of other ions (like Ca2�) [7] and of smallmolecules (like pyrimidylic and adenylic nucleotides)[3,6], and promotes the di¡usion of components fromthe incubation medium into the matrix (like sucrose)[3^8]. The high-conductance state of the PTP wasfound to be highly regulated, and exhibits the fea-tures of a Ca2�-, voltage-, and pH-gated channel [3^8,44,64,65], modulated by the redox and phosphatepotentials [3^8] (see Fig. 4 and Section 6). Moreover,opening of the PTP appears to be regulated by directbinding of a mitochondrial cyclophilin (cyclophilin
D) to its matrix domain, accounting for the inhibi-tory e¡ect of CsA [62,63,66,68^71]. More recently,the PTP has been proposed to be also directly regu-lated by the reactive oxygen species leaking from therespiratory chain [67], and by the rate of electrontransfer across the respiratory chain complex I [73].
From a structural point of view, the PTP was pro-posed as soon as 1979 to be formed by the ADP/ATP translocator [5]. This proposal was made byHunter and Haworth when they discovered that thePTP was regulated by the binding of ADP [5^8], andthat the two inhibitors of the ADP/ATP transloca-tor, i.e. atractyloside and bongkrekic acid, respec-tively activated and inhibited the PTP [5]. Thisview recently gained further support [31], especiallywhen it was reported that the translocator reconstit-uted into liposomes exhibits a channel activity, withan electrical conductance, a Ca2� sensitivity, and avoltage dependence that resemble those of the high-conductance PTP conformation [78]. Still concerningstructural considerations, Haworth and Hunter alsoproposed in 1980 that the PTP might participate inmultiprotein complexes bridging the two mitochon-drial membranes, which they called `transient gapjunctions' [8]. Again, this pioneering view gainedsupport in electrophysiological studies indicatingthat the PTP might be interacting with the outermembrane porins [46,47], and with the mitochondrialbenzodiazepine receptor [45]. Moreover, isolated mi-tochondrial contact sites constituted of cyclophilin D(matrix), ADP/ATP translocator (inner membrane),creatine kinase (facultative and tissue-dependent, in-termembrane space), porin (outer membrane), andhexokinase (facultative and tissue-dependent, extra-mitochondrial space) exhibit, once integrated intoliposomes, a behavior reminiscent of the high-con-ductance state of the PTP [79,80].
3.2. PTP and cell death: a mechanism for the releaseof proapoptotic factors
As mentioned before, PTP opening in its high-con-ductance conformation induces unselective solute£uxes that dissipate the concentration gradients ofrelatively big molecules, but since most proteins re-main trapped in the matrix, the resulting oncoticimbalance (at least in vitro) causes high amplitudeswelling of the organelle. During swelling, the cristae
Fig. 4. Bioenergetic regulation of the high-conductance state ofthe PTP. After [3^8,44,65]. The dashed arrows represent regula-tive interactions. For explanation see text.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^50 39

formed by the inner membrane progressively unfold,causing eventually a rupture of the outer membranethat brings into direct communication the formerintermembrane space and the extramitochondrialmedium. Thus, mitochondria with an open high-con-ductance pore undergo an extensive structural im-pairment that causes the release in the outer medium(provided it is saline) of the soluble components nor-mally trapped in the intermembrane space. Cyto-chrome c and apoptosis inducing factor (AIF) aretwo of these [21^23].
Cytochrome c is a small heme-containing proteinnormally adsorbed at the outer aspect of the innermitochondrial membrane, which shuttles electronsbetween the respiratory chain complexes III and IV[2]. When released from mitochondria, cytochrome cinduces the activation of caspase 3 which is a keycytosolic cell death protease [81^83]. AIF is a 50kDa protein ^ presumably resident of the intermem-brane space ^ that, once released, can promote directactivation of caspase 3 and endonucleases [21]. Insummary, the release of cytochrome c and/or AIFfrom mitochondria can be identi¢ed with a key eventcommitting the cell to death. Thus, because of theouter membrane alterations it induces, PTP openingin its high-conductance conformation appears as agood candidate mechanism for the release of apopto-genic factors from mitochondria [21^23].
Such a mechanism has been proposed to takeplace in many forms of apoptosis, suggesting thatthe high-conductance state of the PTP could play auniversal role of central executioner of the cell deathprogram [21]. This generalization was probably ex-cessive, since in some key cellular models the releaseof apoptogenic factors from mitochondria appears tooccur independently of PTP opening [82^85].
However, in some speci¢c cases ^ all concerningCa2�-dependent cell death ^ the PTP was unambig-uously shown to play a pivotal role in the sequenceof events committing the cell to death [15^20]. In thenext section, we summarize these models for which aconsensus seems to have been reached.
3.3. PTP and Ca 2+-dependent cell death:experimental results
A ¢rst indication that PTP opening in its high-conductance state might act as an apoptotic trigger
came from key studies published by Vayssie©re et al.in 1994 [86], and Petit et al. in 1995 [87], where theauthors showed that early commitment to apoptosisin di¡erent cell types is associated with both v8collapse and alterations of the mitochondrial struc-ture. Concordant observations were then reported ina study where cells were challenged with proapop-totic stimuli in vivo, and subsequently sorted accord-ing to the value of mitochondrial v8 [88]. After ashort-term culture, cells that had retained a high v8after the proapoptotic treatment did not undergoapoptosis, while those with a low v8 did. The au-thors could thus establish a correlation between mi-tochondrial v8 collapse and early commitment to anapoptotic fate. At the same time, numerous reportswere suggesting that the early v8 dissipation ob-served in various types of toxic cell death were de-pendent on PTP opening [50^60]. Thus, the PTP im-mediately became the subject of a close scrutiny inthe ¢eld of apoptotic cell death (see other reviews inthis issue).
For the time being, excitotoxic neuronal cell deathobserved after sustained glutamate stimulation re-mains the principal model where several groupsclearly identi¢ed opening of the PTP with an earlycausative event in the cell death cascade [15^19]. Glu-tamate neurotoxicity is triggered by massive Ca2�
in£ux arising from overstimulation of the N-meth-yl-D-aspartate (NMDA) receptors. Such mode ofneuronal cell death is associated with neurodegener-ative disorders and cerebral ischemia. In 1994,Thayer and coworkers showed that glutamate-in-duced Ca2� overloads in neurons are sequesteredby mitochondria, causing `uncoupling' of respirationand intracellular acidosis, and proposed that mito-chondria might be responsible for glutamate-inducedcell death [89^91]. At the same time, the group ofMiller observed that neuronal excitotoxic degenera-tion is initiated at speci¢c sites enriched in mitochon-dria, and that local Ca2� in£ux in these regions re-sults in mitochondrial v8 collapse [92]. In 1996 and1997 the groups of Reynolds [15,19], Montal [16],Nicotera [17], as well as Nieminen and coworkers[18] all reported that in response to sustained stim-ulation of NMDA receptors (i) mitochondria take upCa2�, (ii) v8 collapses, and (iii) cell death occurs.Strikingly, CsA was shown to be able to allow com-plete recovery of v8, and to prevent cell death in all
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^5040

of these studies [15^19]. Based on these ¢ndings, asimple `cell death sequence' was proposed: (i) pro-longed stimulation of the NMDA receptors of theneuronal plasma membrane causes a sustained in£uxof Ca2� from the extracellular medium; (ii) mito-chondria take up the Ca2� entering the cell ; (iii)opening of the PTP in its high-conductance state istriggered; (iv) mitochondrial v8 collapses; and (v)cell death occurs. Since the morphological features ofexcitotoxic cell death associate necrotic and apop-totic signs, it seems likely that PTP opening resultsin cell death because it causes both (i) an arrest ofaerobic ATP synthesis ^ promoting necrotic signs[93] ; and (ii) mitochondrial swelling, outer mem-brane rupture, and release of proapoptotic factors ^promoting apoptotic signs [93] (for a recent reviewsee [94]).
Another model of Ca2�-dependent cell deathwhere the PTP might play a role corresponds tothe induction of apoptosis by thapsigargin [20,95](a model introduced by the group of Distelhorst[96^100]). Thapsigargin inhibits the ER Ca2�-AT-Pase, blocking the uptake leg of the ER Ca2� cycle.As a result, the ER Ca2� stores become progressivelyand irreversibly emptied, causing in turn opening ofthe store-operated Ca2� entry channels in the plasmamembrane, and massive in£ow of Ca2� from the ex-tracellular medium. Under such conditions (i) mito-chondria take up Ca2� [97,101]; (ii) mitochondrialv8 collapses [20]; and (iii) apoptosis occurs [20,95^100]. In general mechanistic terms, this situation isvery reminiscent of excitotoxic neuronal cell death.Strikingly, here also CsA was reported to be e¤cientat preventing both mitochondrial v8 decrease [20],and signs of apoptosis [20,95]. It should be noted,however, that there is a controversy on whether thecapacitative Ca2� in£ux taking place after thapsigar-gin treatment is or is not necessary for apoptosis tooccur [102,103]. Since operation of the PTP in itshigh-conductance state is di¤cult to conceive in theabsence of mitochondrial Ca2� uptake caused by asustained store-operated Ca2� entry [101], furtherstudies will be needed to clarify the latter point.
Ca2�-dependent mechanisms involving the PTPmight also be responsible for the cases of apoptosisobserved (i) after interleukin withdrawal from inter-leukin-dependent cell lines [104], and (ii) in L-amy-loid neurotoxicity [105]. Indeed, (i) in the case of
interleukin withdrawal, progressive emptying of theER Ca2� pool concomitant with mitochondrial Ca2�
uptake was reported to be correlated with the onsetof apoptosis [104]; and (ii) in the case of L-amyloidneurotoxicity, neuronal cell death was shown to beassociated with mitochondrial v8 collapse, whileboth v8 collapse and cell death could be inhibitedby Ca2� bu¡ers and antioxidants [105]. Thus, itshould be interesting to test the e¡ects of CsA inthese models.
Strikingly, in all four cases of Ca2�-dependent celldeath cited above, Bcl-2 was found to be an e¤cientblocker of the apoptotic program [96^100,103^107].However, in spite of the prominent mitochondriallocalization of Bcl-2 [74,75], and of possible directinhibitory e¡ects of Bcl-2 on the PTP [21,76,77], dur-ing Ca2�-dependent apoptosis, Bcl-2 seems to act onother targets and/or stages of the cell death pathway.In the case of glutamate neurotoxicity, the protectiveaction of Bcl-2 seems to take place far downstreamfrom PTP operation (compare the time courses ofPTP operation in [15^19], and of Bcl-2 protectionin [107]). On the contrary, in thapsigargin-inducedcell death [96^100], or during apoptosis caused byinterleukin withdrawal [104], Bcl-2 seems to act up-stream from PTP operation by preventing the initial(non-mitochondrial) steps of Ca2� mobilization. Fi-nally, in L-amyloid neurotoxicity, Bcl-2 might actupstream from and/or directly at the mitochondriallevel [105]. These examples demonstrate that the Bcl-2 sensitivity of a process is not a proof of PTP in-volvement, and that CsA remains the main availabletool to test the PTP dependence of a sequence ofevents.
3.4. Summary
Opening of the PTP in its high conductance statecauses irreversible dissipation of v8, colloidosmoticswelling of the matrix, rupture of the outer mito-chondrial membrane, and release of proapoptoticfactors from the intermembrane space. Even this `le-thal' conformation of the channel is endowed withseveral built-in control points of regulation, suggest-ing a participation in the physiological pathways toprogrammed cell death. Persistent v8 collapse com-patible with PTP opening in its high-conductancestate has consistently been observed during Ca2�-de-
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^50 41

pendent cell death. Moreover, both mitochondrialdepolarization and progression into the apoptoticprogram can be prevented by CsA that blocks thePTP. In these apoptotic models, PTP opening ap-pears to depend on an initial phase of mitochondrialCa2� uptake.
4. Two conformations for the PTP: switching fromlow- to high-conductance state
Based on the observations summarized in the twolast sections, it appears that either during normalCa2� signaling events, or during Ca2�-dependentcell death, a similar process is responsible for trigger-ing PTP opening: Ca2� uptake by mitochondria.How then to explain that di¡erent open conforma-tions of the pore can arise from a similar type oftrigger signal? In other words, what are the factorsthat divert the PTP from its Ca2� release channelfunction (low-conductance state) to transform itinto a cell death-promoting channel (high-conduct-ance state)?
4.1. Conductance switch: pH versus Ca 2+
Fig. 5 shows isolated mitochondria challengedwith Ca2� undergoing the transient membrane per-meability change that we termed mitochondrialCICR [13,14]. As shown previously, mitochondrialCICR takes the form of a Ca2� transient detectablein the extramitochondrial medium due to transitoryPTP opening [13,14] (Fig. 5A). This process is notaccompanied by swelling and, instead, mitochondriaundergo a phase of shrinkage (Fig. 5A,B). Thisshrinkage is due the net solute loss associated withK� release through the PTP [14], while mitochondriaremain impermeable to sucrose. Thus, the PTPadopts here a low-conductance conformation selec-tive for small ions (see Section 2). This is con¢rmedby the experiments of Fig. 5B, where mitochondriawere submitted to osmotic pressure steps by addingsucrose at di¡erent stages of the CICR spike. Therate of mitochondrial shrinkage observed after su-crose addition is independent of the stage at whichthe osmotic pressure is raised (Fig. 5B), con¢rmingthat mitochondria operate the PTP in low-conduct-ance state during mitochondrial CICR [7,14].
Strikingly, if the size of the Ca2� load is increased,the response no longer takes the appearance of aspike, but rather exhibits a CsA-sensitive triphasicpattern (Fig. 6A). The initial phase of the responseis similar to a CICR spike, but before Ca2� re-up-take is completed, a new cycle of Ca2� release takesplace. The latter, however, is irreversible and associ-ated with large amplitude swelling of mitochondria(Fig. 6A). Comparative analysis of K� £uxes andvolume changes rules out the possibility that thePTP sequentially opens under di¡erent conforma-tions in di¡erent mitochondrial subpopulations. In-deed, (i) during the initial phase of the response,mitochondria remain impermeable to sucrose (Fig.6B), while all of their K� contents is released (Fig.6A); and (ii) during the subsequent phase of irrever-
Fig. 5. Opening of the PTP under its low-conductance stateduring mitochondrial CICR in vitro. See also [13,14,32^35]. Ratliver mitochondria were isolated, suspended in a hypotonic me-dium, and stimulated with Ca2� (40 nmol mg31) as in [13]. (A)Typical mitochondrial CICR response. The simultaneouschanges in extramitochondrial Ca2�, mitochondrial volume, andextramitochondrial potassium were monitored as in [14]. Notethe mitochondrial contraction concomitant with potassium ef-£ux. (B) Osmometric demonstration of the low-conductance(sucrose-impermeable) conformation of the PTP during mito-chondrial CICR. For explanation see text and [14].
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^5042
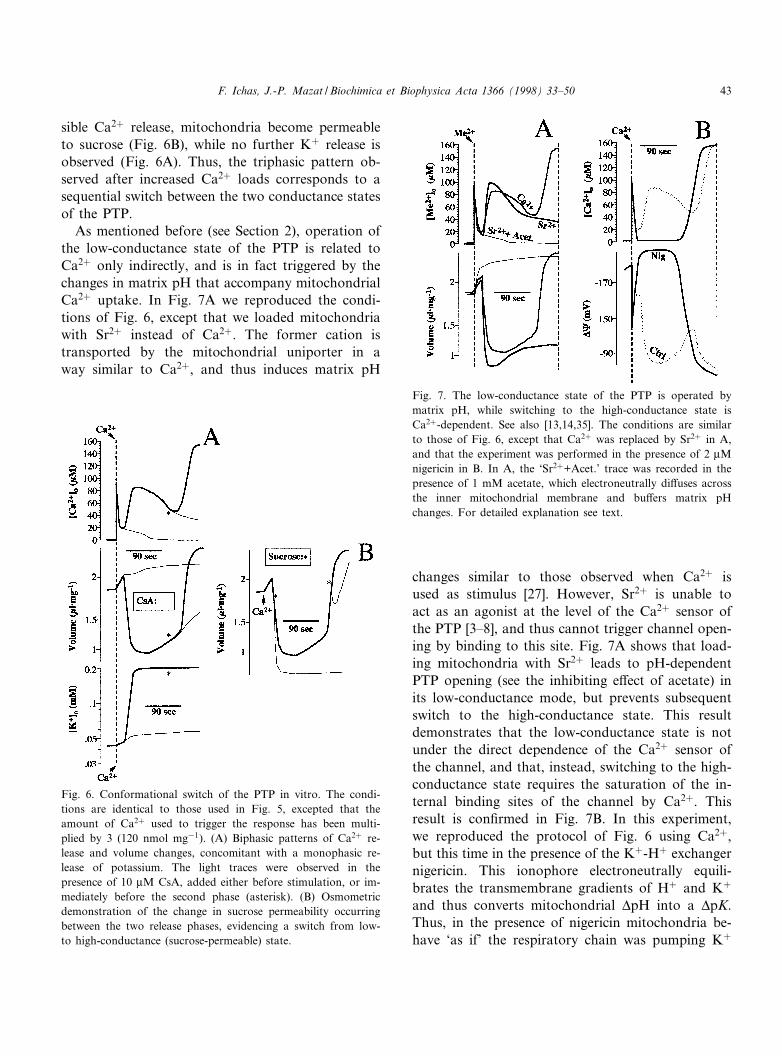
sible Ca2� release, mitochondria become permeableto sucrose (Fig. 6B), while no further K� release isobserved (Fig. 6A). Thus, the triphasic pattern ob-served after increased Ca2� loads corresponds to asequential switch between the two conductance statesof the PTP.
As mentioned before (see Section 2), operation ofthe low-conductance state of the PTP is related toCa2� only indirectly, and is in fact triggered by thechanges in matrix pH that accompany mitochondrialCa2� uptake. In Fig. 7A we reproduced the condi-tions of Fig. 6, except that we loaded mitochondriawith Sr2� instead of Ca2�. The former cation istransported by the mitochondrial uniporter in away similar to Ca2�, and thus induces matrix pH
changes similar to those observed when Ca2� isused as stimulus [27]. However, Sr2� is unable toact as an agonist at the level of the Ca2� sensor ofthe PTP [3^8], and thus cannot trigger channel open-ing by binding to this site. Fig. 7A shows that load-ing mitochondria with Sr2� leads to pH-dependentPTP opening (see the inhibiting e¡ect of acetate) inits low-conductance mode, but prevents subsequentswitch to the high-conductance state. This resultdemonstrates that the low-conductance state is notunder the direct dependence of the Ca2� sensor ofthe channel, and that, instead, switching to the high-conductance state requires the saturation of the in-ternal binding sites of the channel by Ca2�. Thisresult is con¢rmed in Fig. 7B. In this experiment,we reproduced the protocol of Fig. 6 using Ca2�,but this time in the presence of the K�-H� exchangernigericin. This ionophore electroneutrally equili-brates the transmembrane gradients of H� and K�
and thus converts mitochondrial vpH into a vpK.Thus, in the presence of nigericin mitochondria be-have `as if' the respiratory chain was pumping K�
Fig. 6. Conformational switch of the PTP in vitro. The condi-tions are identical to those used in Fig. 5, excepted that theamount of Ca2� used to trigger the response has been multi-plied by 3 (120 nmol mg31). (A) Biphasic patterns of Ca2� re-lease and volume changes, concomitant with a monophasic re-lease of potassium. The light traces were observed in thepresence of 10 WM CsA, added either before stimulation, or im-mediately before the second phase (asterisk). (B) Osmometricdemonstration of the change in sucrose permeability occurringbetween the two release phases, evidencing a switch from low-to high-conductance (sucrose-permeable) state.
Fig. 7. The low-conductance state of the PTP is operated bymatrix pH, while switching to the high-conductance state isCa2�-dependent. See also [13,14,35]. The conditions are similarto those of Fig. 6, except that Ca2� was replaced by Sr2� in A,and that the experiment was performed in the presence of 2 WMnigericin in B. In A, the `Sr2�+Acet.' trace was recorded in thepresence of 1 mM acetate, which electroneutrally di¡uses acrossthe inner mitochondrial membrane and bu¡ers matrix pHchanges. For detailed explanation see text.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^50 43

instead of protons. As a result, matrix pH is clampedto low values. Fig. 7B shows that after Ca2� loading,operation of the PTP in its low-conductance state istotally prevented by nigericin. However, opening ofthe PTP in its high-conductance state is still observedafter a lag time, at a distance from the Ca2� stimulus(Fig. 7B).
4.2. Conductance switch: fast versus slow Ca 2+
loading
These experiments show that conversion betweenthe two open conformations of the PTP can be ob-served in vitro by an increased mitochondrial Ca2�
load su¤cient to saturate the Ca2�-binding sites ofthe channel, and to override the control exerted bypH on the pore. As noted before (see Section 2), thechanges in matrix pH consecutive to mitochondrialCa2� uptake that are responsible for triggering PTPopening in its low-conductance state result from theactivity of the respiratory chain [14,35]. The respira-tory chain is a complex set of proton pumps thatcontinuously expel protons from the matrix, andwhose function is regulated by the value of the trans-membrane proton electrochemical gradient, i.e. thesum of v8 and vpH (expressed in electrical terms)[1,2]. For the non-specialist reader, we can comparewhat the respiratory chain does with protons in mi-tochondria, to what a voltage-clamp device does withelectrons in electrophysiological studies. When themitochondrial membrane becomes depolarized, e.g.,when Ca2� (positive charges) enters the matrix, therespiratory chain `senses' the v8 drop, and immedi-ately increases its proton extrusion rate until theoriginal `resting' v8 (plus vpH) value is restored.This roughly corresponds to a voltage-clamp activity.However, at major variance from arti¢cial voltage-clamp devices, the respiratory chain pumps out pro-tons, and thus the voltage corrections performed areaccompanied by changes in matrix pH. Thesechanges in matrix pH, however, are bu¡ered intime by the electroneutral redistribution of a varietyof permeant weak acids, such as, for instance, phos-phate or oxidizable substrates [14,35] (Fig. 8). Thus,when the rate of Ca2� in£ux causing a v8 drop isfaster than the rate of electroneutral redistribution ofweak acids, voltage correction by the respiratorychain is accompanied by a pronounced matrix alka-
linization. On the contrary, if a similar amount ofCa2� is accumulated, but this time at a slow rate,the redistribution of weak acid can keep pace, andvoltage correction by the respiratory chain is no lon-ger accompanied by signi¢cant increases in matrixpH [14,35] (Fig. 8). Thus, a fast rate of Ca2� deliverycan trigger PTP opening under its low-conductance,pH-dependent conformation, while a slow rate pre-vents pH-dependent channel opening, and results inprogressive mitochondrial Ca2� overloading (Figs. 8and 9). In the latter case, the Ca2� sensor of the PTPbecomes progressively saturated, and the channel
Fig. 8. pH-dependent operation of the PTP is sensitive to therate of mitochondrial Ca2� uptake. After [13,14,35]. The condi-tions are similar to those of Fig. 1 [14]. The simultaneouschanges in extramitochondrial Ca2� and matrix pH were moni-tored as in [14]. (A) Slow Ca2� stimulation of mitochondriawith three Ca2� additions (3U10 nmol mg31). (B) Rapid stimu-lation with three Ca2� additions using the same amount ofCa2� as in A. AH, permeant weak acids di¡using electroneu-trally down the vpH. In the cartoons, the thickness of the ar-rows schematically re£ects the rates of transport. For explana-tion see text.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^5044

eventually opens in its high-conductance conforma-tion (Fig. 9). Thus, both conformations of the PTPare triggered by Ca2�, but by di¡erent mechanisms,and it is mainly the rate of the extramitochondrial[Ca2�] rise and the rate of mitochondrial Ca2� up-take that determine whether the PTP will open in alow-conductance state after limited Ca2� loading(fast rates), or in a high-conductance state after mas-sive Ca2� overloading (slow rates).
4.3. Summary
The PTP can switch from low- to high-conduct-ance state. While the low-conductance conformationis mainly pH-operated, the conformational switchappears to be strictly dependent on the saturationof the internal Ca2� binding of the channel. Fastmitochondrial Ca2� uptake promotes signi¢cantchanges in matrix pH for modest Ca2� loads, andresults in operation of the PTP in its low-conduc-tance, pH-dependent conformation. Slow mitochon-drial Ca2� uptake results in massive Ca2� overload-ing without signi¢cant changes in matrix pH, andeventually causes opening of the PTP in its high-con-ductance, Ca2�-dependent conformation.
5. From Ca2+ signaling to Ca2+-dependent cell death:a biophysical model
The latter observations demonstrate that operationof either pore conformation simply depends on therate of the extramitochondrial [Ca2�] increases, andon the rate of mitochondrial Ca2� uptake. We showbelow that the tight dependence of the PTP openconformational state on such kinetic parameters pro-vides a simple reading frame to predict the outcome(survival or death) of the Ca2� signals in living cells.This leads us to propose a model of cell death wherethe trigger apoptotic signal is simply encoded in theshape and the origin of the cytosolic Ca2� signals.
5.1. Shape of Ca 2+ signals and rate of mitochondrialCa 2+uptake : fromsignalingtocelldeath
During physiological signaling events triggered byCa2� mobilizing hormones, the cytosolic Ca2� sig-nals are organized in time as single or repetitivespikes of variable frequency, and in space as propa-gating wavefronts [108^113]. In non-excitable cells,these elementary spikes result in ¢rst instance fromthe coordinated operation of Ca2� release from theER, Ca2� extrusion by the plasma membrane Ca2�-ATPases, Ca2� entry through the store-operatedCa2� channels, and Ca2� re-uptake by the ERCa2�-ATPases (reviewed in [114]). In this context,the mitochondria located near the IP3-dependentCa2� release channels of the ER periodically senseintense and short-lived Ca2� transients [37]. Duringthe generation of such localized Ca2� hot spots, theneighboring mitochondria take up Ca2� at an ex-tremely fast rate, due to the high local Ca2� concen-tration that activates the mitochondrial Ca2� uni-porter [37] (see also Section 2). Since the rate ofmitochondrial Ca2� uptake is probably su¤cient totransiently override matrix pH bu¡ering, the PTPopens in its low-conductance conformation, and mi-tochondrial CICR occurs, contributing to the ampli-¢cation of the mean cytosolic Ca2� spike [13,14].Note that in this case, no mitochondrial Ca2� over-loading occurs since PTP opening is triggered after avery moderate ^ but fast ^ transient increase in mi-tochondrial Ca2� [14]. As a result, mitochondria par-ticipate in Ca2� signaling, do not swell, and maintaintheir normal functions (Fig. 10A).
Fig. 9. The rate of mitochondrial Ca2� uptake determines themode of PTP operation. The conditions are similar to those ofFig. 8. The simultaneous changes in extramitochondrial Ca2�
and mitochondrial volume were monitored as in [14], but thelight scattering signal was not calibrated. (A) Rapid Ca2� stim-ulation as in Fig. 8; low-conductance PTP opening is observed(shrinkage). (B) Slow Ca2� stimulation using pulses of individu-al height as in A, and continued until Ca2� release occurred;high-conductance PTP opening is observed (swelling). For ex-planation see text.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^50 45

Let us now consider the case of Ca2� signals suchas those observed after thapsigargin challenge. Asnoted before, thapsigargin irreversibly empties theER Ca2� pool, and the monotonic persistent cyto-solic Ca2� elevation observed in treated cells resultsfrom the establishment of a steady state betweenCa2� entry through the store-operated channels ofthe plasma membrane, Ca2� extrusion by the plasmamembrane Ca2�-ATPases, and Ca2� uptake by mi-tochondria [101]. In this case, the localized Ca2� hotspots cited above are not generated, and mitochon-
dria face a steady submicromolar plateau of cytosolicCa2� concentration. Under these conditions, RaM-mediated Ca2� uptake spontaneously inactivatesafter a few milliseconds [36,101], and the mitochon-drial Ca2� uniporter remains the sole operating up-take pathway. At those submicromolar cytosolicCa2� concentrations, uniporter-mediated Ca2� up-take is slow [27], and mitochondria get progressivelyoverloaded with Ca2� at an extremely slow rate[101]. The massive mitochondrial Ca2� overloadingobserved under these conditions indicates that the
Fig. 10. Model of the transition between Ca2� signaling and Ca2�-dependent apoptosis. The model elements are schematized as inFig. 3. (A) Normal Ca2� signaling as in Fig. 3; the PTP is transiently operated (phase 2) in its low-conductance, pH-dependent con-formation. (B) Pathological Ca2� signaling, like in thapsigargin-treated cells ; mitochondrial Ca2� overloading leads to PTP opening inits high-conductance, Ca2�-dependent conformation (phase 3); as a result, mitochondria swell and the outer membrane is disrupted.For explanation see text.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^5046

PTP remains closed for long periods. This resultis expected since slow mitochondrial Ca2� uptakeprobably occurs without signi¢cant change in matrixpH as observed in vitro, thus precluding operationof the PTP under its pH-dependent conformation,i.e., preventing mitochondrial CICR from takingplace. As the Ca2� load further increases, however,the PTP eventually opens in its high-conductancestate, causing irreversible mitochondrial depolar-ization, and committing the cell to death [20] (Fig.10B).
A very similar scenario can be proposed for exci-totoxic neuronal cell death [15^19]. In neurons over-stimulated with glutamate or its agonists, a steady-state increase in cytosolic Ca2� takes place that re-sults from the concerted operation of Ca2� in£uxthrough the NMDA channels of the plasma mem-brane, Ca2� extrusion by the plasma membraneCa2�-ATPases, and Ca2� uptake by mitochondria[89^92] (for a recent review see [94]). Since the cyto-solic Ca2� concentration is maintained at near micro-molar steady values, Ca2� in£ux through the Ca2�
uniporter is ^ here also ^ slow due to the low extra-mitochondrial Ca2� concentration. As a result, (i)pH-dependent PTP opening is prevented, (ii) mito-chondria become progressively overloaded withCa2�, and (iii) the PTP eventually opens in itshigh-conductance state, committing the neuron todeath [15^19].
5.2. General conclusion
Our results indicate that switching from the low-conductance state of the PTP to the high-conduc-tance open conformation is an irreversible processthat is strictly dependent on the saturation of theinternal Ca2�-binding sites of the channel. Thus,the high-conductance state of the PTP, which wasshown to play a pivotal role in the course of excito-toxic and thapsigargin-induced cell death, might re-sult from a Ca2�-dependent conformational shift ofthe low-conductance state, normally participating inthe regulation of cellular Ca2� homeostasis as a pH-operated channel. These observations have led us topropose a simple biophysical model of the transitionbetween Ca2� signaling and Ca2�-dependent apopto-sis, where, depending on the shape of the cytosolicCa2� signals, the di¡erences in the kinetics of mito-
chondrial Ca2� uptake determine the type of PTPconformation operated (Fig. 10).
6. Tribute to Hunter and Haworth
By 1980, Hunter and Haworth had already discov-ered and characterized many of the key features ofthe PTP [3^8]. However, few people were ready tobelieve in the existence of mitochondrial channels atthis time. As a result, their studies went unheeded for10 years (for notable exceptions, see [29,30,61]), andwhen new groups ¢nally decided to develop theirwork, Hunter had left research, and Haworth hadchanged his ¢eld of investigation. In this annex, wesummarize the striking contribution of these two au-thors, with the hope that the reader will feel stimu-lated to read the ¢ve key papers (plus one abstract)that pioneered the ¢eld [3^8].
While the process of Ca2�-induced mitochondrialswelling, referred to as `mitochondrial damage', or`mitochondrial aging', had been known for years,Hunter and Haworth were the ¢rst to clearly showthat the progressive global swelling, loss of respira-tory control, and Ca2� release observed in mitochon-drial suspensions after Ca2� addition were in factdue to an `all-or-nothing' abrupt increase in the per-meability of the mitochondrial membrane occurringat the level of individual mitochondria, and progres-sively recruiting increasing fractions of the mitochon-drial population [3^8]. They thus introduced the con-cept of a variable threshold for activation of theprocess of permeability transition in di¡erent individ-ual mitochondria, and applied electron microscopy,respiratory control assays, calcium release assays,and light scattering methods to the quantitative de-termination of the fraction of mitochondria with anopen pore within a whole mitochondrial population[3^8]. In 1976, they showed that the PTP was unse-lectively permeable to H�, Ca2�, Mg2�, choline, glu-cose, sucrose, and NAD(P)H [3,6]. Using dextrans ofdi¡erent molecular weight, they determined the mo-lecular mass cuto¡ of the PTP (6 1500 Da) [6]. Theyalso showed that PTP opening is a reversible process,since mitochondria with an open pore could be com-pletely resealed by simply adding ATP plus Mg2�, orby chelating Ca2� with EGTA [3,6].
They showed that the PTP is a channel activated
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^50 47

by the allosteric binding of two Ca ions at the matrixside of the channel [3,6,8], and that ADP and Mg2�
act as competitive inhibitors [5,7,8]. They found thatthe two redox couples NAD(P)�/NAD(P)H werealso regulating the pore, due to a direct interactionof the nucleotides with the channel [3^5,8]. Theyshowed that pH was modulating the PTP [6], andrecognized that energization per se, independentlyof its origin ^ substrate oxidation by the respiratorychain [7], or ATP hydrolysis by the mitochondrialATPase [5] ^ was a pore modulator, since in bothcases FCCP was causing PTP opening [5,7].
They showed that phosphate and fatty acids werepore inducers [3], and that Sr2� was an inhibitor ofthe PTP acting at the level of the internal Ca2�-bind-ing sites of the channel [3^8]. They characterized theinhibiting e¡ects of N-ethylmaleimide (NEM) andthe activating e¡ects of arsenate [3], and showedthat atractyloside and bongkrekic acid were respec-tively activating and inhibiting the PTP [5].
Based on the e¡ects of ADP [3^8], atractyloside[5], and bongkrekic acid [5], they proposed that theATP/ADP translocator might be part of the PTP [5].Strikingly, they also made the hypothesis that thePTP might participate in mitochondrial contact sites:`the channel which is most similar to the inner mem-brane channel, in being both nonspeci¢c and chemi-cally gated, is perhaps the channel which spans theintercellular gap junction ... It is tempting to specu-late that opening of the inner membrane channelcould entail fusion with the outer membrane, pro-ducing a transient gap junction' [8].
Haworth and Hunter discovered PTP £ickering in1979: `the Ca2�-induced permeability is discontinu-ous in time: the treated mitochondria must be con-tinuously switching between a permeable state andan impermeable state' [6]. Finally, they also detectedand analyzed the subconductance state of the PTP [7]as they found that mitochondria impermeable to su-crose could still release Ca2� at a fast rate, in aruthenium red-insensitive manner, and in the absenceof added Na� : `Do the mitochondria which remainin the aggregated con¢guration release Ca2� by amechanism totally unrelated to the transition, or isthe release still transitional (in the sense of having alag period followed by a sudden release) withoutchange in permeability to sucrose? This remains anopen question. Some involvement of the transition
mechanism is suggested by the observation that therelease at low levels of Ca2� is still insensitive toruthenium red' [7].
Acknowledgements
We are deeply indebted to Dr. P. Bernardi foradvice during manuscript preparation. We also thankthe following colleagues for scienti¢c interactionsthat contributed to our current conception of PTPfunction: Drs. L. Buntinas, F. Di Lisa, O. Eriksson,Yu.V. Evtodienko, E. Fontaine, K.D. Garlid, K.K.Gunter, T.E. Gunter, E.L. Holmuhamedov, K.W.Kinnally, V.A. Selivanov, S.S. Sidash, V.V. Teplova,A.P. Thomas, and M. Zoratti. We thank F. De Gior-gi for critical reading of the manuscript. F.I. is fellowof the FEBS (1997) and Human Frontier ScienceProgram (1998) organizations at the University ofPadua. The original experimental data presentedwere obtained with the support of research grantsto J.-P.M. from l'AFM, la FRM, la Ligue Contrele Cancer, and la Region Aquitaine.
References
[1] P. Mitchell, Nature 191 (1961) 144^148.[2] V.P. Skulatchev, Membrane Bioenergetics, Springer Verlag,
Berlin, 1988.[3] D.R. Hunter, R.A. Haworth, J.H. Southard, J. Biol. Chem.
251 (1976) 5069^5077.[4] D.R. Hunter, Fed. Proc. 36 (1977) 903.[5] D.R. Hunter, R.A. Haworth, Arch. Biochem. Biophys. 195
(1979) 453^459.[6] R.A. Haworth, D.R. Hunter, Arch. Biochem. Biophys. 195
(1979) 460^467.[7] D.R. Hunter, R.A. Haworth, Arch. Biochem. Biophys. 195
(1979) 468^477.[8] R.A. Haworth, D.R. Hunter, J. Membr. Biol. 54 (1980) 231^
236.[9] M. Zoratti, I. Szabo© , Biochim. Biophys. Acta 1241 (1995)
139^176.[10] A.M. Davidson, A.P. Halestrap, Biochem. J. 268 (1990)
147^152.[11] R.A. Altschuld, C.M. Hohl, L.C. Castillo, A.A. Garleb,
R.C. Starling, G.P. Brierley, Am. J. Physiol. 262 (1992)H1699^H1704.
[12] G.E.N. Kass, M.J. Juedes, S. Orrenius, Biochem. Pharma-col. 44 (1992) 1995^2003.
[13] F. Ichas, L.S. Jouaville, S.S. Sidash, J.-P. Mazat, E.L. Hol-muhamedov, FEBS Lett. 348 (1994) 211^215.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^5048

[14] F. Ichas, L.S. Jouaville, J.-P. Mazat, Cell 89 (1997) 1145^1153.
[15] R.J. White, I.J. Reynolds, J. Neurosci. 16 (1996) 5688^5697.[16] A.F. Schinder, E.C. Olson, N.C. Spitzer, M. Montal, J. Neu-
rosci. 16 (1996) 6125^6133.[17] M. Ankarcrona, J.M. Dypbukt, S. Orrenius, P. Nicotera,
FEBS Lett. 394 (1996) 321^324.[18] A.L. Nieminen, T.G. Petrie, J.J. Lemasters, W.R. Selman,
Neuroscience 75 (1996) 993^997.[19] K.R. Hoyt, T.A. Sharma, I.J. Reynolds, Br. J. Pharmacol.
122 (1997) 803^808.[20] P. Waring, J. Beaver, Exp. Cell Res. 227 (1996) 264^276.[21] N. Zamzami, S.A. Susin, P. Marchetti, T. Hirsch, I. Gomez-
Monterrey, M. Castedo, G. Kroemer, J. Exp. Med. 183(1996) 1533^1544.
[22] S.P. Kantrow, C.A. Piantadosi, Biochem. Biophys. Res.Commun. 232 (1997) 669^671.
[23] J.L. Scarlett, M.P. Murphy, FEBS Lett. 418 (1997) 282^286.
[24] D.B. Zorov, K.W. Kinnally, S. Perini, H. Tedeschi, Biochim.Biophys. Acta 1105 (1992) 263^270.
[25] S.A. Novgorodov, T.I. Gudz, J. Bioenerg. Biomembr. 28(1996) 139^146.
[26] K.M. Brokemeier, D.R. Pfei¡er, in: New Perspectives inMitochondrial Research Workshop, Sept. 1997, Padua,1997, pp. 43^44.
[27] T.E. Gunter, D.R. Pfei¡er, Am. J. Physiol. 258 (1990) C755^C786.
[28] N. Fournier, G. Ducet, A. Crevat, J. Bioenerg. Biomembr.19 (1987) 297^303.
[29] M. Crompton, H. Ellinger, A. Costi, Biochem. J. 255 (1988)357^360.
[30] K.M. Brokemeier, M.E. Dempsey, D.R. Pfei¡er, J. Biol.Chem. 264 (1989) 7826^7830.
[31] A.P. Halestrap, A.M. Davidson, Biochem. J. 268 (1990)153^160.
[32] Yu.V. Evtodienko, V.V. Teplova, J. Khavaja, N.E. Saris,Cell Calcium 15 (1994) 142^152.
[33] V.V. Teplova, S.S. Sidash, P.R. Makarov, Yu.V. Evtodien-ko, Biokhimiia 60 (1995) 944^952.
[34] E.L. Holmuhamedov, V.V. Teplova, E.A. Chukhlova, Yu.V.Evtodienko, R.G. Ulrich, Biochem. Mol. Biol. Int 36 (1995)39^49.
[35] V.A. Selivanov, F. Ichas, E.L. Holmuhamedov, L.S. Joua-ville, Yu.V. Evtodienko, J.-P. Mazat, Biophys. Chem. 72(1998) 111^121.
[36] G.C. Sparagna, K.K. Gunter, S.S. Sheu, T.E. Gunter, J. Biol.Chem. 270 (1995) 27510^27515.
[37] R. Rizzuto, M. Brini, M. Murgia, T. Pozzan, Science 262(1993) 744^747.
[38] G.A. Rutter, P. Burnett, R. Rizzuto, M. Brini, M. Murgia,T. Pozzan, J.M. Tavare, R.M. Denton, Proc. Natl. Acad.Sci. USA 93 (1996) 5489^5494.
[39] Y. Horikawa, A. Goel, A.P. Somlyo, A.V. Somlyo, Biophys.J. 74 (1998) 1579^1590.
[40] V. Petronilli, P. Bernardi, F. Di Lisa, in: New Perspectives
in Mitochondrial Research Workshop, Sept. 1997, Padua,1997, pp. 39^40.
[41] I. Szabo©, M. Zoratti, J. Biol. Chem. 266 (1991) 3376^3379.[42] K.W. Kinnally, D. Zorov, Y. Antonenko, S. Perini, Bio-
chem. Biophys. Res. Commun. 176 (1991) 1183^1188.[43] I. Szabo©, M. Zoratti, J. Bioenerg. Biomembr. 24 (1992) 111^
117.[44] I. Szabo© , P. Bernardi, M. Zoratti, J. Biol. Chem. 267 (1992)
2940^2946.[45] K.W. Kinnally, D.B. Zorov, Y.N. Antonenko, S.H. Snyder,
M.W. McEnery, H. Tedeschi, Proc. Natl. Acad. Sci. USA 90(1993) 1374^1378.
[46] I. Szabo©, M. Zoratti, FEBS Lett. 330 (1993) 201^205.[47] I. Szabo©, M. Zoratti, FEBS Lett. 330 (1993) 206^210.[48] T.A. Lohret, K.W. Kinnally, J. Biol. Chem. 270 (1995)
15950^15953.[49] T.A. Lohret, R.E. Jensen, K.W. Kinnally, J. Cell Biol. 137
(1997) 377^386.[50] W. Nazareth, N. Yafei, M. Crompton, J. Mol. Cell. Cardiol.
23 (1991) 1351^1354.[51] M. Crompton, O. McGuinness, W. Nazareth, Biochim. Bio-
phys. Acta 1101 (1992) 214^217.[52] M.R. Duchen, O. McGuinness, L.A. Brown, M. Crompton,
Cardiovasc. Res. 27 (1993) 1790^1794.[53] E.J. Gri¤ths, A.P. Halestrap, J. Mol. Cell. Cardiol. 25
(1993) 1461^1469.[54] J.G. Pastorino, J.W. Snyder, J.B. Hoek, J.L. Farber, Am. J.
Physiol. 268 (1995) C676^C685.[55] E.J. Gri¤ths, A.P. Halestrap, Biochem. J. 307 (1995) 93^98.[56] A.L. Nieminen, A.K. Saylor, S.A. Tesfai, B. Herman, J.J.
Lemasters, Biochem. J. 307 (1995) 99^106.[57] J.G. Pastorino, G. Simbula, K. Yamamoto, P.A. Glascott,
R.J. Rothman, J.L. Farber, J. Biol. Chem. 271 (1996) 29792^29798.
[58] A.L. Nieminen, A.M. Byrne, B. Herman, J.J. Lemasters,Am. J. Physiol. 272 (1997) C1286^C1294.
[59] T. Qian, A.L. Nieminen, B. Herman, J.J. Lemasters, Am. J.Physiol. 273 (1997) C1783^C1792.
[60] L.C. Trost, J.J. Lemasters, Toxicol. Appl. Pharmacol. 147(1997) 431^441.
[61] M. Crompton, A. Costi, Eur. J. Biochem. 178 (1988) 489^501.
[62] E.J. Gri¤ths, A.P. Halestrap, Biochem. J. 274 (1991) 611^614.
[63] A.P. Halestrap, Biochem. J. 278 (1991) 715^719.[64] P. Bernardi, S. Vassanelli, P. Veronese, R. Colonna, I. Sza-
bo© , M. Zoratti, J. Biol. Chem. 267 (1992) 2934^2939.[65] P. Bernardi, J. Biol. Chem. 267 (1992) 8834^8839.[66] C.P. Connern, A.P. Halestrap, Biochem. J. 302 (1994) 321^
324.[67] A.J. Kowaltowski, R.F. Castiho, A.E. Vercesi, Am. J. Phys-
iol. 269 (1995) C141^C147.[68] A. Nicolli, E. Basso, V. Petronilli, P. Bernardi, J. Biol.
Chem. 271 (1996) 2185^2192.[69] C.P. Connern, A.P. Halestrap, Biochemistry 35 (1996) 8172^
8180.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^50 49

[70] A. Tanveer, S. Virji, L. Andreeva, N.F. Totty, J.J. Hsuan,J.M. Ward, M. Crompton, Eur. J. Biochem. 238 (1996) 166^172.
[71] A.P. Halestrap, K.Y. Wood¢eld, C.P. Connern, J. Biol.Chem. 272 (1997) 3346^3354.
[72] A.J. Kowaltowski, R.F. Castilho, Biochim. Biophys. Acta1322 (1997) 221^229.
[73] E. Fontaine, O. Eriksson, F. Ichas, P. Bernardi, J. Biol.Chem. 273 (1998) 12662^12668.
[74] D.M. Hockenberry, G. Nunez, C.L. Milliman, R.D. Schreib-er, S.J. Korsmeyer, Nature 348 (1990) 334^338.
[75] D.M. Hockenberry, Z.N. Oltvai, X.-M. Yin, C.L. Milliman,S.J. Korsmeyer, Cell 75 (1993) 241^251.
[76] S. Shimizu, Y. Egushi, W. Kamiike, Y. Funahashi, A.Mignon, V. Lacronique, H. Matsuda, Y. Tsujimoto, Proc.Natl. Acad. Sci. USA 95 (1998) 1455^1459.
[77] A. Moodie, R.C. Murphy, R. Song, P. Fox, M.L. Campo,D. Lawrence, E. Schneider, K.W. Kinnally, (1998) Lectureat the 42nd Biophysical Meeting, Kansas City, KS, Feb.1998.
[78] N. Brustovetsky, M. Klingenberg, Biochemistry 35 (1996)8483^8488.
[79] G. Beutner, A. Ru«ck, B. Riede, W. Welte, D. Brdiczka,FEBS Lett. 396 (1996) 189^195.
[80] E. O'Gorman, G. Beutner, M. Dolder, A.P. Koretsky, D.Brdiczka, T. Wallimann, FEBS Lett. 414 (1997) 253^257.
[81] X. Liu, C.N. Kim, J. Yang, R. Jemmerson, X. Wang, Cell 86(1996) 147^157.
[82] R.M. Kluck, E. Bossy-Wetzel, D.R. Green, D.D. Newmeyer,Science 275 (1997) 1132^1136.
[83] J. Yang, X. Liu, K. Bhalla, C.N. Kim, A.M. Ibrado, J. Cai,T.I. Peng, D.P. Jones, X. Wang, Science 275 (1997) 1129^1132.
[84] M.G. Vander Heiden, N.S. Chandel, E.K. Williamson, P.T.Schumacker, G.B. Thompson, Cell 91 (1997) 627^637.
[85] E. Bossy-Wetzel, D.D. Newmeyer, D.R. Green, EMBO J. 17(1998) 37^49.
[86] J.L. Vayssie©re, P.X. Petit, Y. Risler, B. Mignotte, Proc. Natl.Acad. Sci. USA 91 (1994) 11752^11756.
[87] P.X. Petit, H. LecEur, E. Zorn, C. Dauguet, B. Mignotte,M.L. Gougeon, J. Cell Biol. 130 (1995) 157^167.
[88] N. Zamzami, P. Marchetti, M. Castedo, C. Zanin, J.L. Vays-sie©re, P.X. Petit, G. Kroemer, J. Exp. Med. 181 (1995) 1661^1672.
[89] G.J. Wang, R.D. Randall, S.A. Thayer, J. Neurophysiol. 72(1994) 2563^2569.
[90] S.A. Thayer, G.J. Wang, Clin. Exp. Pharmacol. Physiol. 22(1995) 303^304.
[91] G.J. Wang, S.A. Thayer, J. Neurophysiol. 76 (1996) 1611^1621.
[92] V.P. Bindokas, R.J. Miller, J. Neurosci. 15 (1995) 6999^7011.
[93] M. Leist, B. Single, A.F. Castoldi, S. Ku«hnle, P. Nicotera,J. Exp. Med. 185 (1997) 1481^1486.
[94] R.J. Miller, Trends Neurosci. 21 (1998) 95^96.[95] Q.Q. Huang, M. Fang, H.Q. Zhang, S.B. Xue, Acta Phar-
macol. Sin. 18 (1997) 262^266.[96] M. Lam, G. Dubyak, L. Chen, G. Nunen, R.L. Miesfeld,
C.W. Distelhorst, Proc. Natl. Acad. Sci. USA 91 (1994)6569^6573.
[97] C.W. Distelhorst, T.S. McCormick, Cell Calcium 19 (1996)473^483.
[98] C.W. Distelhorst, M. Lam, T.S. McCormick, Oncogene 12(1996) 2051^2055.
[99] H. He, M. Lam, T.S. McCormick, C.W. Distelhorst, J. CellBiol. 138 (1997) 1219^1228.
[100] X.M. Qi, M. Simonson, C.W. Distelhorst, Oncogene 15(1997) 2849^2853.
[101] B.V. Chernyak, FEBS Lett. 418 (1997) 131^134.[102] S. Jiang, S.C. Chow, P. Nicotera, S. Orrenius, Exp. Cell
Res. 212 (1994) 84^92.[103] X. Bian, F.M. Hughes, Y. Huang, J.A. Cidlowski, J.W.
Putney Jr., Am. J. Physiol. 272 (1997) C1241^C1249.[104] G. Ba¡y, T. Miyashita, J.R. Williamson, J.C. Reed, J. Biol.
Chem. 268 (1993) 6511^6519.[105] J.H. Prehn, V.P. Bindokas, J. Jordan, M.F. Galindo, G.D.
Ghadge, R.P. Roos, L.H. Boise, C.B. Thompson, S. Kra-jewski, J.C. Reed, R.J. Miller, Mol. Pharmacol. 49 (1996)319^328.
[106] J.H. Prehn, V.P. Bindokas, C.J. Marcuccilli, S. Krajewski,J.C. Reed, R.J. Miller, Proc. Natl. Acad. Sci. USA 91(1994) 12599^12603.
[107] Y. Wang, W. Jia, M. Cynader, Neuroreport 8 (1997) 3323^3326.
[108] N.M. Woods, K.S. Cuthbertson, P.H. Cobbold, Nature 319(1986) 600^602.
[109] M.J. Berridge, P.H. Cobbold, K.S. Cuthbertson, Phil.Trans. R. Soc. Lond. B 320 (1988) 325^343.
[110] T.A. Rooney, E.J. Sass, A.P. Thomas, J. Biol. Chem. 264(1989) 17131^17141.
[111] T.A. Rooney, E.J. Sass, A.P. Thomas, J. Biol. Chem. 265(1990) 10792^10796.
[112] J.D. Lechleiter, D.E. Clapham, Cell 69 (1992) 283^294.[113] L.D. Robb-Gaspers, A.P. Thomas, J. Biol. Chem. 270
(1995) 8102^8107.[114] S. Miyazaki, Curr. Opin. Cell Biol. 7 (1995) 190^196.
BBABIO 44663 28-7-98
F. Ichas, J.-P. Mazat / Biochimica et Biophysica Acta 1366 (1998) 33^5050
Copyright © 2022 FDOKUMEN




















