
Frequent epigenetic inactivation of hSRBC in gastric cancer and its implication in attenuated p53...
12
Frequent epigenetic inactivation of hSRBC in gastric cancer and its implication in attenuated p53 response to stresses Jin-Hee Lee 1 , Do-Sun Byun 1 , Min-Goo Lee 1 , Byung-Kyu Ryu 1 , Min-Ju Kang 1 , Kwon-Seok Chae 1,2 , Kil Yeon Lee 3 , Hyo-Jong Kim 4 , Heonyong Park 5 and Sung-Gil Chi 1 * 1 School of Life Sciences and Biotechnology, Korea University, Seoul, Korea 2 Department of Biology Education, Teacher’s College, Kyungpook National University, Daegu, Korea 3 Department of General Surgery, School of Medicine, Kyung Hee University, Seoul, Korea 4 Department of Internal Medicine, School of Medicine, Kyung Hee University, Seoul, Korea 5 Department of Molecular Biology, Dankook University, Seoul, Korea hSRBC is a putative tumor suppressor located at 11p15.4, at which frequent genomic loss has been observed in several human malig- nancies. To explore the candidacy of hSRBC as a suppressor of gastric tumorigenesis, we analyzed the expression and mutation status of hSRBC in gastric tissues and cell lines. hSRBC transcript was expressed in all normal and benign tumor tissues examined, but undetectable or very low in 73% (11/15) cancer cell lines and 41% (46/111) primary tumors. Loss or reduction of hSRBC expression was tumor-specific and correlated with stage and grade of tumors. While allelic loss or somatic mutations of the gene were infrequent, its expression was restored in tumor cells by 5-aza-2 0 - deoxycytidine treatment and aberrant hypermethylation of 23 CpG sites in the promoter region showed a tight association with altered expression. Transient or stable expression of hSRBC led to aG 1 cell cycle arrest and apoptosis of tumor cells, and strongly suppresses colony forming ability and xenograft tumor growth. In addition, hSRBC elevated apoptotic sensitivity of tumor cells to genotoxic agents, such as 5-FU, etoposide and ultraviolet. Interest- ingly, hSRBC increased the protein stability of p53 and expression of p53 target genes, such as p21 Waf1 , PUMA and NOXA, while hSRBC-mediated cell cycle arrest and apoptosis were abolished by blockade of p53 function. Our findings suggest that hSRBC is a novel tumor suppressor whose epigenetic inactivation contributes to the malignant progression of gastric tumors, in part, through attenuated p53 response to stresses. ' 2007 Wiley-Liss, Inc. Key words: hSRBC; promoter hypermethylation; gastric cancer; p53; tumor suppressor gene Gastric cancer is one of the most commonly diagnosed malig- nancies worldwide, and around 1.1 million new cases are expected in the year 2010. 1 Gastric cancer incidence shows large geo- graphic variation with relatively high rates in certain areas such as in Korea, Japan, China, Eastern Europe and South America, par- ticularly Chile. 2–4 Although evidence has accumulated indicating the involvement of a number of molecular abnormalities, includ- ing microsatellite instability, telomerase reactivation, and genetic and epigenetic alterations of genes such as p53, hMLH1, E-cad- herin, K-ras, c-erbB2 and K-sam, the pathogenesis of this disease and the underlying molecular events that contribute to its develop- ment remain largely unclear. 5,6 Chromosomal deletion and allelic loss is a common feature of the malignant progression of human tumors, and a high rate of loss of heterozygosity (LOH) may lead to the identification of putative tumor suppressor genes residing at a particular chromosomal region. The short arm of chromosome 11 is a frequent target of genetic loss in solid tumors. 7–10 In particular, homozygous or hemi- zygous loss at the chromosomal region 11p15 is observed in many cancer types including breast, lung and bladder cancers. 11–16 In gas- tric cancer, 11p15 was identified as a specific region of chromo- somal rearrangement and 37–62% of gastric adenocarcinomas were found to harbor LOH affecting 11p15. 17–20 An extensive alle- lotyping study using high-density polymorphic markers demon- strated that 11p15.5 is a critical region of LOH for chromosome 11 in gastric carcinomas, suggesting the presence of a putative tumor suppressor gene(s) in this region. 21 Recently, the human SRBC [serum deprivation response factor (sdr)-related gene product that binds to c-kinase]] (hSRBC) gene has been mapped to the chromosomal region 11p15.5-p15.4. 22 SRBC belongs to a superfamily of proteins consisting of 3 mem- bers (SDPR, SRBC and PTRF), which have the sequence and structural resemblance. The possible implication of hSRBC in cell cycle control has been suggested by previous observations that mRNA and protein expression of hSRBC is induced by serum starvation and decreased during G 0 -G 1 transition. 23 The hSRBC gene encodes for a protein of 261 amino acids with the N-terminal leucine zipper-like motif and the putative protein kinase C (PKC) phosphorylation site in the middle. 22 hSRBC was initially isolated in a two-hybrid screen for protein interacting with the product of the breast cancer susceptibility gene BRCA1, suggesting that hSRBC may participate in DNA damage response including DNA repair processes. Identification of hSRBC as a BRCA1-interacting protein also raises the possibility that genetic and/or epigenetic inactivation of hSRBC may compromise BRCA1-mediated tumor suppression functions. Interestingly, several frame shift and trun- cation mutations of the hSRBC gene were identified in a few ovar- ian and lung cancer cell lines. 22 Moreover, the aberrant promoter hypermethylation leading to down-regulation of hSRBC expres- sion was observed in a large fraction of breast, lung and ovarian cancer cell lines, whereas strong expression of hSRBC was detected in normal mammary and lung epithelial cells. A recent methylation study using a large collection of primary lung cancer samples also demonstrated that the 5 0 CpG island of the hSRBC gene is methylated in 41% of primary nonsmall cell lung cancers and in 80% of primary small cell lung cancers, but only in 4% cor- responding nonmalignant lung tissues. 24 These observations sug- gest that hSRBC may have a tumor suppressor function and its inactivation would be implicated in the pathogenesis of several types of human cancer. This article contains supplementary material available via the Internet at http://www.interscience.wiley.com/jpages/0020-7136/suppmat. Abbreviations: 5-aza-dC, 5-aza-2-deoxycytidine; PCR, hSRBC, human serum deprivation response factor-related gene product that binds to c-ki- nase; LOH, loss of heterozygosity; PCR, polymerase chain reaction; siRNA, short-interfering RNA; SSCP, single strand conformation poly- morphism; TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling. Grant sponsor: Korea Science and Engineering Foundation; Grant num- ber: R02-2003-000-10031-0. Grant sponsor: Korea Research Foundation; Grant number: 2003-070-C00031. Grant sponsor: National Cancer Center, Korea; Grant number: 0420230. Grant sponsor: Korea University (2005), Republic of Korea. The first two authors contributed equally to this paper. *Correspondence to: School of Life Sciences and Biotechnology, Korea University, 136-701 Seoul, Republic of Korea. Fax: 182-2-927-5458. E-mail: [email protected] Received 17 May 2007; Accepted after revision 16 August 2007 DOI 10.1002/ijc.23166 Published online 4 December 2007 in Wiley InterScience (www.interscience. wiley.com). Int. J. Cancer: 122, 1573–1584 (2008) ' 2007 Wiley-Liss, Inc. Publication of the International Union Against Cancer
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of Frequent epigenetic inactivation of hSRBC in gastric cancer and its implication in attenuated p53...

Frequent epigenetic inactivation of hSRBC in gastric cancer and
its implication in attenuated p53 response to stresses
Jin-Hee Lee1, Do-Sun Byun1, Min-Goo Lee1, Byung-Kyu Ryu1, Min-Ju Kang1, Kwon-Seok Chae1,2,Kil Yeon Lee3, Hyo-Jong Kim4, Heonyong Park5 and Sung-Gil Chi1*
1School of Life Sciences and Biotechnology, Korea University, Seoul, Korea2Department of Biology Education, Teacher’s College, Kyungpook National University, Daegu, Korea3Department of General Surgery, School of Medicine, Kyung Hee University, Seoul, Korea4Department of Internal Medicine, School of Medicine, Kyung Hee University, Seoul, Korea5Department of Molecular Biology, Dankook University, Seoul, Korea
hSRBC is a putative tumor suppressor located at 11p15.4, at whichfrequent genomic loss has been observed in several human malig-nancies. To explore the candidacy of hSRBC as a suppressor ofgastric tumorigenesis, we analyzed the expression and mutationstatus of hSRBC in gastric tissues and cell lines. hSRBC transcriptwas expressed in all normal and benign tumor tissues examined,but undetectable or very low in 73% (11/15) cancer cell lines and41% (46/111) primary tumors. Loss or reduction of hSRBCexpression was tumor-specific and correlated with stage and gradeof tumors. While allelic loss or somatic mutations of the gene wereinfrequent, its expression was restored in tumor cells by 5-aza-20-deoxycytidine treatment and aberrant hypermethylation of 23CpG sites in the promoter region showed a tight association withaltered expression. Transient or stable expression of hSRBC led toa G1 cell cycle arrest and apoptosis of tumor cells, and stronglysuppresses colony forming ability and xenograft tumor growth. Inaddition, hSRBC elevated apoptotic sensitivity of tumor cells togenotoxic agents, such as 5-FU, etoposide and ultraviolet. Interest-ingly, hSRBC increased the protein stability of p53 and expressionof p53 target genes, such as p21Waf1
, PUMA and NOXA, whilehSRBC-mediated cell cycle arrest and apoptosis were abolishedby blockade of p53 function. Our findings suggest that hSRBC is anovel tumor suppressor whose epigenetic inactivation contributesto the malignant progression of gastric tumors, in part, throughattenuated p53 response to stresses.' 2007 Wiley-Liss, Inc.
Key words: hSRBC; promoter hypermethylation; gastric cancer;p53; tumor suppressor gene
Gastric cancer is one of the most commonly diagnosed malig-nancies worldwide, and around 1.1 million new cases are expectedin the year 2010.1 Gastric cancer incidence shows large geo-graphic variation with relatively high rates in certain areas such asin Korea, Japan, China, Eastern Europe and South America, par-ticularly Chile.2–4 Although evidence has accumulated indicatingthe involvement of a number of molecular abnormalities, includ-ing microsatellite instability, telomerase reactivation, and geneticand epigenetic alterations of genes such as p53, hMLH1, E-cad-herin, K-ras, c-erbB2 and K-sam, the pathogenesis of this diseaseand the underlying molecular events that contribute to its develop-ment remain largely unclear.5,6
Chromosomal deletion and allelic loss is a common feature ofthe malignant progression of human tumors, and a high rate of lossof heterozygosity (LOH) may lead to the identification of putativetumor suppressor genes residing at a particular chromosomalregion. The short arm of chromosome 11 is a frequent target ofgenetic loss in solid tumors.7–10 In particular, homozygous or hemi-zygous loss at the chromosomal region 11p15 is observed in manycancer types including breast, lung and bladder cancers.11–16 In gas-tric cancer, 11p15 was identified as a specific region of chromo-somal rearrangement and �37–62% of gastric adenocarcinomaswere found to harbor LOH affecting 11p15.17–20 An extensive alle-lotyping study using high-density polymorphic markers demon-strated that 11p15.5 is a critical region of LOH for chromosome 11in gastric carcinomas, suggesting the presence of a putative tumorsuppressor gene(s) in this region.21
Recently, the human SRBC [serum deprivation response factor(sdr)-related gene product that binds to c-kinase]] (hSRBC) genehas been mapped to the chromosomal region 11p15.5-p15.4.22
SRBC belongs to a superfamily of proteins consisting of 3 mem-bers (SDPR, SRBC and PTRF), which have the sequence andstructural resemblance. The possible implication of hSRBC in cellcycle control has been suggested by previous observations thatmRNA and protein expression of hSRBC is induced by serumstarvation and decreased during G0-G1 transition.23 The hSRBCgene encodes for a protein of 261 amino acids with the N-terminalleucine zipper-like motif and the putative protein kinase C (PKC)phosphorylation site in the middle.22 hSRBC was initially isolatedin a two-hybrid screen for protein interacting with the product ofthe breast cancer susceptibility gene BRCA1, suggesting thathSRBC may participate in DNA damage response including DNArepair processes. Identification of hSRBC as a BRCA1-interactingprotein also raises the possibility that genetic and/or epigeneticinactivation of hSRBC may compromise BRCA1-mediated tumorsuppression functions. Interestingly, several frame shift and trun-cation mutations of the hSRBC gene were identified in a few ovar-ian and lung cancer cell lines.22 Moreover, the aberrant promoterhypermethylation leading to down-regulation of hSRBC expres-sion was observed in a large fraction of breast, lung and ovariancancer cell lines, whereas strong expression of hSRBC wasdetected in normal mammary and lung epithelial cells. A recentmethylation study using a large collection of primary lung cancersamples also demonstrated that the 50CpG island of the hSRBCgene is methylated in 41% of primary nonsmall cell lung cancersand in 80% of primary small cell lung cancers, but only in 4% cor-responding nonmalignant lung tissues.24 These observations sug-gest that hSRBC may have a tumor suppressor function and itsinactivation would be implicated in the pathogenesis of severaltypes of human cancer.
This article contains supplementary material available via the Internet athttp://www.interscience.wiley.com/jpages/0020-7136/suppmat.Abbreviations: 5-aza-dC, 5-aza-2-deoxycytidine; PCR, hSRBC, human
serum deprivation response factor-related gene product that binds to c-ki-nase; LOH, loss of heterozygosity; PCR, polymerase chain reaction;siRNA, short-interfering RNA; SSCP, single strand conformation poly-morphism; TUNEL, terminal deoxynucleotidyltransferase-mediateddUTP-biotin nick end labeling.Grant sponsor: Korea Science and Engineering Foundation; Grant num-
ber: R02-2003-000-10031-0. Grant sponsor: Korea Research Foundation;Grant number: 2003-070-C00031. Grant sponsor: National Cancer Center,Korea; Grant number: 0420230. Grant sponsor: Korea University (2005),Republic of Korea.The first two authors contributed equally to this paper.*Correspondence to: School of Life Sciences and Biotechnology, Korea
University, 136-701 Seoul, Republic of Korea. Fax: 182-2-927-5458.E-mail: [email protected] 17 May 2007; Accepted after revision 16 August 2007DOI 10.1002/ijc.23166Published online 4 December 2007 in Wiley InterScience (www.interscience.
wiley.com).
Int. J. Cancer: 122, 1573–1584 (2008)' 2007 Wiley-Liss, Inc.
Publication of the International Union Against Cancer

In an attempt to explore the candidacy of hSRBC as a suppres-sor of gastric carcinogenesis, we investigated the expression andmutation status of the gene in a series of gastric tissue specimensand cell lines. Our study demonstrates that hSRBC is inactivatedin a considerable fraction of gastric cell lines and primary tumorsby aberrant promoter CpG sites hypermethylation. We also foundthat hSRBC induces a G1 arrest of the cell cycle and apoptosis,and suppresses in vitro cellular growth and xenograft tumorgrowth by enhancing the protein stability of p53. These resultsestablished hSRBC as a novel tumor suppressor whose epigeneticinactivation contributes to the malignant progression of humangastric epithelial cells though attenuated p53 response to variousstresses.
Material and methods
Tissue specimens and cancer cell lines
Total 187 gastric tissues including 111 primary adenocarcino-mas, 3 adenomas, 6 hamartomas, 7 hyperplastic polyps, and 60normal gastric tissues were obtained from 111 cancer patients and76 noncancer patients by surgical resection in the Kyung Hee Uni-versity Medical Center (Seoul, Korea). Signed informed consentwas obtained from each patient. Tissue specimens were snap-frozen in liquid N2 and stored at 270�C. Bits of tumors and adja-cent portions of each tumor were fixed and used for hematoxylinand eosin staining for histopathological evaluation. Tumor speci-mens composed of at least 70% carcinoma cells and adjacenttissues found not to contain tumor cells were chosen for mole-cular analysis. Fifteen human gastric cancer cell lines (SNU1,SNU5, SNU16, SNU216, SNU484, SNU601, SNU620, SNU638,SNU719, MKN1, MKN28, MKN45, MKN74, AGS and KATO-III) were obtained from Korea Cell Line Bank (Seoul, Korea) orAmerican Type Culture Collection (Rockville, MD).
Semi-quantitative polymerase chain reaction analysis
Extraction of total cellular RNA and synthesis of cDNA wereperformed as described previously.25,26 Genomic DNA wasextracted from the same cells of the tissues from the DNA phaseafter RNA was extracted. Briefly, 1 lg of DNase1-treated RNAwas converted to cDNA by reverse transcription using randomhexamer primers and M-MLV reverse transcriptase (InvitrogenCorporation, Carlsbad, CA). Polymerase chain reaction (PCR)was initially performed over a range of cycles (24, 26, 28, 30, 32,34, 36, and 38 cycles) and 2 ll of 1:4 diluted cDNA (12.5 ng/50 llPCR reaction) undergoing 28–36 cycles was observed to be withinthe logarithmic phase of amplification with primers hSRBC-4(sense; 50-TTCTGCTCTTCAAGGAGGAG-30) and hSRBC-7(antisense; 50-CCAAGGCGAGGCGGCTTGAC-30) for hSRBCand G2 (sense; 50-CATGTGGGCCATGAGGTCCACCAC-30)and G3 (antisense; 50-AACCATGAGAAGTATGACAACAGC-30) for an endogenous expression standard gene GAPDH. PCRwas done for 32–36 cycles at 95�C (1 min), 56–60�C (0.5 min),and 72�C (1 min) in 1.5 mM MgCl2-containing reaction buffer(PCR buffer II, Perkin Elmer). For genomic PCR, intron 2 regionof hSRBC, exon 8 of p53, and intron 5 of GAPDH were amplifiedwith intron-specific primers hSRBC-IN1 (sense; 50-CGTCCGCAGAATTTGGTCTG-30) and hSRBC-5 (antisense; 50-AAGGGCTCTGGTGCCTTCTG-30), SG85 (sense; 50-TCCTTACTGCCTCTTGCTTCTCTTT-30) and SG83 (antisense: 50- TCTCCTCCACCGCTTCTTGT-30), and G3 (see above) and G5 (antisense; 50-GAGTCCTTCCACGATACCAAAG-30), respectively. About 10ll of PCR products were resolved on 2% agarose gels. Quantita-tion was achieved by densitometric scanning of the ethidium bro-mide-stained gels. Absolute area integrations of the curves repre-senting each specimen were then compared after adjustment forGAPDH. Integration and analysis was performed using MolecularAnalyst software program (Bio-Rad, Hercules, CA). QuantitativePCR was repeated at least 3 times for each specimen and the meanwas obtained.
Northern blot analysis
A total of 20 lg of total RNA was separated on formaldehydeagarose gel. RNA was transferred to Nylon membrane using Tur-boBlotter (Scleicher & Schulell) and the membrane was baked inthe CL1000 UV crosslinker (UVP). Dioxigenin-labeled probe forhSRBC was prepared using PCR. Hybridization was conductedwith Dig-labeled probes in the high SDS hybridization solution at50�C for 12 hr. Detection was performed using DIG chemilumi-nescent detection kit (Roche) according to the manufacturer’s pro-tocol.
Loss of heterozygosity analysis
Loss of heterozygosity (LOH) of the hSRBC gene was deter-mined using an intraexonic SNP (50-AGCGCT/A-30) located at1690 nucleotide (T690A in exon 2). PCR amplification was per-formed on each tumor and normal DNA sample pair obtainedfrom 48 patients using primers hSRBC-15 (sense; 50-CTCGGACGAGGAGCCGGTGG-30) and hSRBC-12 (antisense; 50-GGCTACACTCTCCATTTGGAG-30). Total of 5 ll of the PCR productswere used for cutting with the endonuclease AfeI and enzyme-digested PCR products were electrophoresed on 2% agarose gels.If the 2 alleles appeared in the normal tissue DNA, the patient wasconsidered an informative case for the marker. Signal intensity offragments and the relative ratio of both tumor and normal alleleintensities were determined by scanning densitometry. Becausecertain numbers of noncancerous cells might be present in tumortissues, LOH was assigned when the intensity ratio of the 2 tumoralleles differed by at least 50% from that observed on its corre-sponding normal DNA.
Nonisotopic RT-PCR-single strand conformationpolymorphism analysis
To screen the presence of somatic mutations, RT-PCR-singlestrand conformation polymorphism (SSCP) analysis of hSRBCwas performed. The hSRBC transcript was amplified with 4 sets ofprimers that were designed to cover the entire coding region of thegene. Sequences of the primers used will be obtained uponrequest. About 20 ll of PCR products mixed with 10 ll of 0.5 NNaOH, 10 mM EDTA, and 15 ll of denaturing loading buffer(95% formamide, 20 mM EDTA, 0.05% bromophenol blue, and0.05% xylene cyanol). After heating at 95�C for 5 min, sampleswere loaded in wells precooled to 4�C and run using 8% nondena-turating acrylamide gels containing 10% glycerol at 4–8�C and18–22�C.
5-aza-20-deoxycytidine treatmentTo assess re-activation of hSRBC expression, cell lines showing
no or low levels of hSRBC transcript were plated in 6-well tissueplates 48 h before treatment. 5-Aza-dC (Sigma) was added to thefresh medium at concentrations of 5 lM in duplicate and cellswere harvested after 4 days of treatment. Expression level ofhSRBC transcript before and after 5-Aza-dC treatment were ana-lysed by RT-PCR.
Methylation specific PCR (MSP) and bisulfite DNAsequencing analysis
Totally 1 lg of genomic DNA was incubated with 3M sodiumbisulfite (pH 5.0) and 200 ng of bisulfite-modified DNA was sub-jected to PCR amplification of the hSRBC promoter region usingmethylation-specific primers MSP-M04 (sense; 50-GAAATAAAAATTTTCGTGATTC-30) and MSP-M03 (antisense; 50-CTTAAAAACGTTTCGCCTTCCG-30) and unmethylation-specific primersMSP-U04 (sense; 50-GTTGTGTTAATATAGTTTTTGT-30) andMSP-U03 (antisense; 50-AAAATCTCTTAAAAACATTTCA-30).PCR was performed for 38 cycles at 95�C (1 min), 60�C (1 min)and 72�C (1 min) and 15 ll of the PCR products were resolved on
1574 LEE ET AL.

a 2% agarose gel. For bisulfite sequencing analysis, 50 ng of bisul-fite-modified DNA was subjected to PCR amplification of thehSRBC promoter region using primers MS-seq-1 (sense; 50-CCATCTTCACTAATATAAAAAA-30) and MS-seq-2 (antisense;50-GTTTTAGTTGTGATTTAGGTAG-30). The PCR productswere cloned into pCRII vectors (Invitogen Corporation, Carlsbad,CA). Five clones of each specimen were sequenced by automatedfluorescence-based DNA sequencing to determine the methylationstatus.
Immunoblot assay
Cells were lysed in a lysis buffer containing 20 mM Tris (pH7.4), 300 mM NaCl, 10 mM EDTA, 0.2% SDS, 2% Triton X-100,2% sodium deoxycholate and 1 mM PMSF. The cell lysate wasclarified by centrifugation and 20 lg of total protein were supple-mented with Laemmli buffer and loaded on a 10% SDS-polyacryl-amide gel for electrophoresis. The polyclonal antibody againsthSRBC was generated by immunizing rabbit with a GST-fusionprotein containing amino acids 164–180 of hSRBC by standardfusion. Western analyses were performed using antibodies specificfor p53 (Santa Cruz Biotechnology, CA), phospho-p53 (Cell Sig-naling, Bevery, MA), p21Waf1 (Santa Cruz Biotechnology), and b-tubulin (Sigma, St. Louis, MO). Antibody binding was detectedby enhanced chemiluminescence (Amersham Biosciences, Piscat-away, NJ) using a secondary antibody conjugated to horseradishperoxidase. For stripping, the blots were incubated in a strippingbuffer [0.2 M glycine (pH 2.2), 0.1% SDS, 1% Tween-20] at roomtemperature for 60 min.
Expression plasmids, siRNA and transfection
hSRBC expression vector was constructed by PCR using spe-cific oligonucleotide primers hSRBC-11 (50-ATGAGGGAGAGTGCGTTGGAGCCG-30) and hSRBC-12 (50-GGTACCTCAGGCTACACTCTCCATTTGGAG-30). The PCR products wereligated to a pCR2.1 vector (Invitrogen Corporation) and subse-quently subcloned into pcDNA3.1 (Invitrogen Corporation). Tocreate N-terminal GFP-tagged hSRBC construct, PCR was per-formed using primers SRBC-GFP-C1-S (50-CTCGAGAAATGAGGGAGAGTGCGTTGGAGC-30) and hSRBC-GFP-C1-AS1(50-GAATTCGTCAGGCTACACTCTCCATTTGGA-30) and theproducts were ligated to a linearized pCR2.1 vector. EcoR1/XhoIfragment of pCR2.1 were subcloned into pEGFP-C1 (ClontechLaboratories, Mountain view, CA). Correct directionality and in-frame sequences in pCMVTaqII and pcDNA3.1 were verified byDNA sequencing. Transfection of constructs was performed usingLipofectamine 2000 (Invitrogen Corporation) or ExGen500 (Fer-mentas Life Sciences) according to the manufacturer’s protocol.Each transfection experiment was carried out in triplicate. Thetransfection efficiency was monitored using a fluorescence micros-copy for GFP or CAT assay (Boehringer Mannheim, Indianapolis,IN) according to the instruction of manufacturer. To generatehSRBC-expressing cell lines, AGS cells were transfected with4 lg of hSRBC expression vectors and colonies were isolated byG418 selection (1,600 lg/ml). After 6 weeks selection, 2 AGSsublines expressing different levels of hSRBC were chosen formolecular biological assays. As a control subline, a pool of emptyvector-transfected cells was used. siRNA duplex against hSRBC(siGENOME SMART pool reagent, M-016416-00-0005, HumanPRKCDBP) and control siRNA were synthesized by DharmaconResearch (Lafayette, CO). For transfection, 1 3 105 cells wereplated on 60-mm-diameter dishes 24 hr before transfection. Thecells were incubated with a siRNA-Oligofectamine mixture at37�C for 4 hr, followed by addition of fresh medium containing10% fetal bovine serum.
Flow cytometric analysis of cell cycle and apoptosis
To measure in vitro cellular growth, cells were seeded in 6-wellplate at the density of 1 3 105 cells per well in duplicate and weremaintained in the presence of 10% FBS. Cell numbers were
counted using a hemocytometer for 8 days at 48-hr intervals. Forflow cytometry analysis, cells transfected with GFP-hSRBC orGFP-control vectors were treated with etoposide (10 lM) for 24,48 and 72 hr. Cells were fixed with 70% ethanol and resuspendedin 1 ml of PBS containing 50 mg/ml RNase and 50 mg/ml propi-dium iodide (Sigma). The assay was performed on a FACScanflow cytometer (Becton Dickinson, San Jose, CA), and thecell cycle profile was analyzed using Modfit software (BectonDickinson).
BrdU immunofleurocence assay
AGS cells seeded at a density of 1000 cells/2-well on slideglassplate were incubated with BrdU (10 lM) for 24 hr. The cells weretreated with 2 N HCl at 37�C for 30 min to denature DNA. Afterblocking in normal goat serum with 0.3% Triton X-100, sectionswas reacted with rat anti-BrdU antibody (abcam, Cambridge, MA)and incubated in Alexa 488-conjugated goat anti-rat IgG (1:200,Vector Laboratories). After washing, slides were reacted withmouse anti-PCNA (Chemicon, Billerica, MA) and incubated withAlexa 546-conjugated goat anti mouse IgG (1:200, Vector Labora-tories). The slides were counterstained with DAPI (Biotian, Hay-ward, CA). Fluorescence image was viewed using a fluorescencemicroscope equipped with a Spot digital camera.
TUNEL assay
TUNEL assay was performed to evaluate apoptotic effect ofhSRBC. Briefly, AGS were transfected with control or hSRBCexpression vectors, and the cells were grown for 12–72 hr. Thecells were then fixed with 4% paraformaldehyde in PBS, and thebuffer containing 3% bovine serum albumin and 0.1% Triton X-100 was added and incubated for 15 min at 4�C. The cells werelabeled by the terminal deoxynucleotidyltransferase-mediateddUTP-biotin nick end labeling (TUNEL) reaction mixture usingthe In Situ Cell Death Detection kit (Roche). TUNEL signals ofcells were visualized directly under microscopy.
Colony formation assay
AGS cells were transfected with expression vectors encodinghSRBC or empty vector (pcDNA3.1) and maintained in the pres-ence of G418 (1600 lg/ml) for 4–6 weeks. Colonies were fixedwith methanol for 15 min and stained with 0.05% crystal violet in20% ethanol.
Xenograft tumor growth assay
Five-week-old female SCID mice (NOD. CB17-Prkdc, AnimalResources Centre, Australia) were maintained in pressurized ven-tilated cages. AGS-pcDNA and AGS-hSRBC-1 cells (1 3 107)were injected subcutaneously into the right flank (200 ll/mouse)of 5 mice, respectively. After 3 days, 2D tumor sizes were meas-ured every 3 days. Tumor growth was monitored regularly andvolume (V) was calculated using the formula V 5 1/2 3 length 3(width)2.
Statistical analysis
The results of apoptosis and colony forming assays wereexpressed as mean 6 SD. A student’s t-test was used to determinethe statistical significance of the difference. The v2 test was usedto determine the statistical significance of expression and methyla-tion levels between tumor and normal tissues. A p value of lessthan 0.05 was considered significant.
Results
Loss or reduction of hSRBC expression in human gastric cancers
We initially examined hSRBC expression in normal gastric tis-sues and benign tumors using semi-quantitative RT-PCRapproach. hSRBC transcript was easily detectable in all 60 normaland 16 benign tumor tissues (3 adenomas, 6 hamartomas and 7hyperplastic polyps) we tested. No significant variations were
1575hSRBC INACTIVATION IN GASTRIC CANCER

observed in its expression levels (hSRBC/GAPDH) among speci-mens [normal; 0.87–1.50 (mean; 1.21) and benign; 0.92–1.47(mean; 1.18)] (Fig. 1a). By contrast, 15 gastric cancer cell linesexhibited variable expression levels of hSRBC (0.00–1.34, mean;0.66) (Fig. 1b). hSRBC transcript was undetectable in 4 cell lines(SNU1, SNU484, SNU620, and AGS) and very low in 5 cell lines(SNU16, SNU601, SNU719, MKN28, and MKN74). An immuno-blot assay revealed that protein expression of hSRBC in cancercell lines was fairly consistent with its transcript expression (Fig.1b). However, hSRBC protein products were undetectable from 2
cell lines (SNU638 and MKN45) which express normal level oftranscript, suggesting the post-transcriptional down-regulation ofhSRBC in these tumor cells. Therefore, 11 of 15 (73.3%) gastriccancer cell lines displayed no or extremely low expression ofhSRBC protein.
In contrast to normal and benign tissues, a substantial fractionof primary carcinomas showed a marked reduction of hSRBC tran-script (Fig. 1c). Moreover, among 48 matched sets, 32 (66.7%)cases showed detectable reduction of hSRBC in cancerous tissuescompared to its adjacent noncancerous tissues, which frequently
FIGURE 1 – Expression status of hSRBC in gastric tissues and cell lines. (a) Semi-quantitative RT-PCR analysis of hSRBC in normal and be-nign tumor tissues. PCR was performed using exon-specific primers and 10 ll of the PCR products were resolved on a 2% agarose gel. GAPDHwas used as an endogenous control. N, normal tissues; Ha, harmatomas; Hp, hyperplastic polyps; Ad, adenomas. (b) Expression of hSRBC tran-script and protein in cancer cell lines. For immunoblot assay, 30 lg of total protein was fractionated using 10% SDS-PAGE and hSRBC wasdetected using an anti-hSRBC polyclonal antibody and enhanced chemiluminescence. (c) hSRBC expression in primary carcinomas. N, normaltissues; T, primary carcinomas. (d) Tumor- specific reduction of hSRBC. hSRBC expression was compared between cancer and adjacent noncan-cerous tissues obtained from the same cancer patients. (e) Northern blot assay of hSRBC. Expression levels of hSRBC were verified by Northernblot analysis in cell lines and primary tumors. (f) Expression levels of hSRBC in tissues and cell lines. Data represent means of triplicate assays.Bar indicates the mean expression level of each specimen group. (g) Comparison of hSRBC levels between early (E) and advanced (A) tumors,well differentiated (WD), moderately differentiated (MD), and poorly differentiated (PD) tumors, and intestinal (I) and diffused (D) types oftumors.
1576 LEE ET AL.
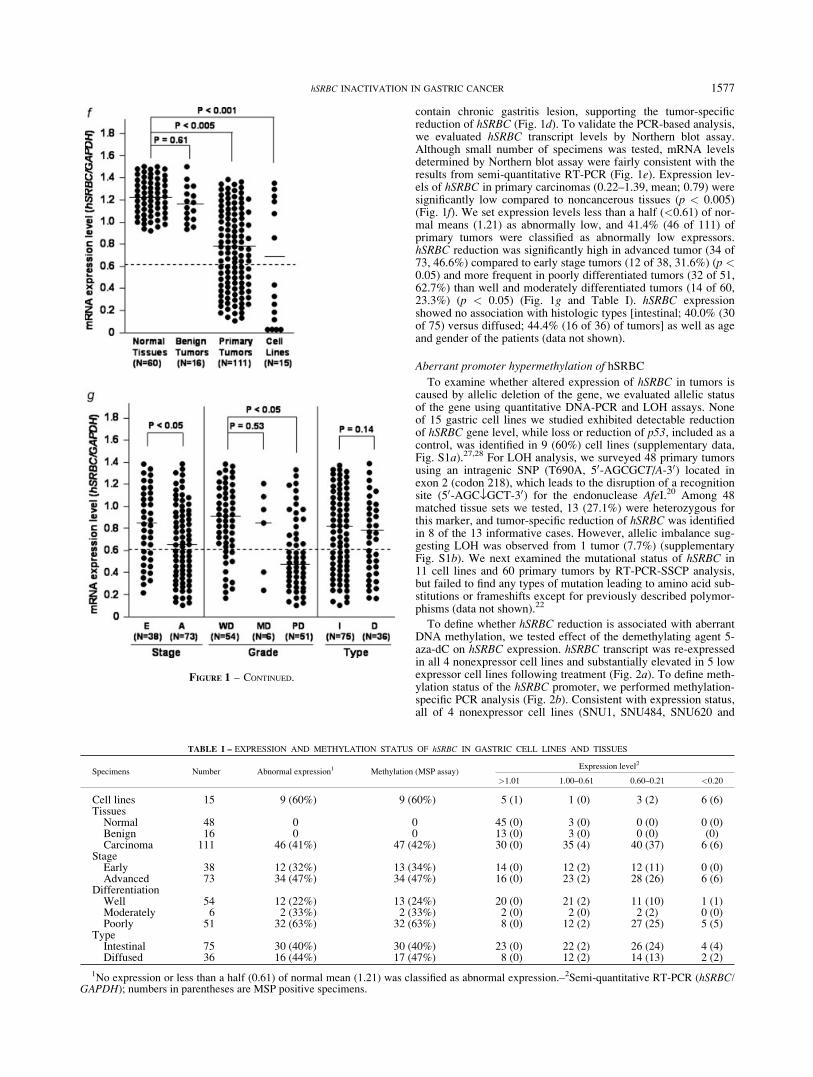
contain chronic gastritis lesion, supporting the tumor-specificreduction of hSRBC (Fig. 1d). To validate the PCR-based analysis,we evaluated hSRBC transcript levels by Northern blot assay.Although small number of specimens was tested, mRNA levelsdetermined by Northern blot assay were fairly consistent with theresults from semi-quantitative RT-PCR (Fig. 1e). Expression lev-els of hSRBC in primary carcinomas (0.22–1.39, mean; 0.79) weresignificantly low compared to noncancerous tissues (p < 0.005)(Fig. 1f). We set expression levels less than a half (<0.61) of nor-mal means (1.21) as abnormally low, and 41.4% (46 of 111) ofprimary tumors were classified as abnormally low expressors.hSRBC reduction was significantly high in advanced tumor (34 of73, 46.6%) compared to early stage tumors (12 of 38, 31.6%) (p <0.05) and more frequent in poorly differentiated tumors (32 of 51,62.7%) than well and moderately differentiated tumors (14 of 60,23.3%) (p < 0.05) (Fig. 1g and Table I). hSRBC expressionshowed no association with histologic types [intestinal; 40.0% (30of 75) versus diffused; 44.4% (16 of 36) of tumors] as well as ageand gender of the patients (data not shown).
Aberrant promoter hypermethylation of hSRBC
To examine whether altered expression of hSRBC in tumors iscaused by allelic deletion of the gene, we evaluated allelic statusof the gene using quantitative DNA-PCR and LOH assays. Noneof 15 gastric cell lines we studied exhibited detectable reductionof hSRBC gene level, while loss or reduction of p53, included as acontrol, was identified in 9 (60%) cell lines (supplementary data,Fig. S1a).27,28 For LOH analysis, we surveyed 48 primary tumorsusing an intragenic SNP (T690A, 50-AGCGCT/A-30) located inexon 2 (codon 218), which leads to the disruption of a recognitionsite (50-AGCflGCT-30) for the endonuclease AfeI.20 Among 48matched tissue sets we tested, 13 (27.1%) were heterozygous forthis marker, and tumor-specific reduction of hSRBC was identifiedin 8 of the 13 informative cases. However, allelic imbalance sug-gesting LOH was observed from 1 tumor (7.7%) (supplementaryFig. S1b). We next examined the mutational status of hSRBC in11 cell lines and 60 primary tumors by RT-PCR-SSCP analysis,but failed to find any types of mutation leading to amino acid sub-stitutions or frameshifts except for previously described polymor-phisms (data not shown).22
To define whether hSRBC reduction is associated with aberrantDNA methylation, we tested effect of the demethylating agent 5-aza-dC on hSRBC expression. hSRBC transcript was re-expressedin all 4 nonexpressor cell lines and substantially elevated in 5 lowexpressor cell lines following treatment (Fig. 2a). To define meth-ylation status of the hSRBC promoter, we performed methylation-specific PCR analysis (Fig. 2b). Consistent with expression status,all of 4 nonexpressor cell lines (SNU1, SNU484, SNU620 and
TABLE I – EXPRESSION AND METHYLATION STATUS OF hSRBC IN GASTRIC CELL LINES AND TISSUES
Specimens Number Abnormal expression1 Methylation (MSP assay)Expression level2
>1.01 1.00–0.61 0.60–0.21 <0.20
Cell lines 15 9 (60%) 9 (60%) 5 (1) 1 (0) 3 (2) 6 (6)Tissues
Normal 48 0 0 45 (0) 3 (0) 0 (0) 0 (0)Benign 16 0 0 13 (0) 3 (0) 0 (0) (0)Carcinoma 111 46 (41%) 47 (42%) 30 (0) 35 (4) 40 (37) 6 (6)
StageEarly 38 12 (32%) 13 (34%) 14 (0) 12 (2) 12 (11) 0 (0)Advanced 73 34 (47%) 34 (47%) 16 (0) 23 (2) 28 (26) 6 (6)
DifferentiationWell 54 12 (22%) 13 (24%) 20 (0) 21 (2) 11 (10) 1 (1)Moderately 6 2 (33%) 2 (33%) 2 (0) 2 (0) 2 (2) 0 (0)Poorly 51 32 (63%) 32 (63%) 8 (0) 12 (2) 27 (25) 5 (5)
TypeIntestinal 75 30 (40%) 30 (40%) 23 (0) 22 (2) 26 (24) 4 (4)Diffused 36 16 (44%) 17 (47%) 8 (0) 12 (2) 14 (13) 2 (2)
1No expression or less than a half (0.61) of normal mean (1.21) was classified as abnormal expression.–2Semi-quantitative RT-PCR (hSRBC/GAPDH); numbers in parentheses are MSP positive specimens.
FIGURE 1 – CONTINUED.
1577hSRBC INACTIVATION IN GASTRIC CANCER

AGS) showed only methylation-specific products and 4 of 5 lowexpressors (SNU16, SNU719, MKN28 and MKN74) showedboth methylation- and unmethylation-specific products, whereas5 of 6 normal expressors (SNU5, SNU601, MKN1, MKN45 andKATOIII) displayed only unmethylation-specific products (Fig.2c). Methylation-specific products were also detected in 47 of 111(42%) primary tumors. While 93% (43 of 46) of primary tumorswith low expression level displayed methylation, only 6% (4 of65) of tumors with normal expression level showed promotermethylation (Table I). Next, the methylation status of 23 CpGssites located in the promoter region (nucleotides –71 to –394) wasevaluated using bisulfite PCR and DNA sequencing analysis (Fig.2d). Five PCR clones were sequenced to determine methylationfrequency at individual CpG sites (complete methylation; 80–100%, partial methylation; 20–60%, and unmethylation; 0%).Among 23 CpG sites, 20–23 sites (87–100%) and 8–13 sites (35–57%) were completely methylated in 4 nonexpressor and 5 lowexpressor cell lines, respectively, whereas less than 9% (0–2 sites)were methylated in 6 normal expressor cell lines (Fig. 2e). Partialmethylation at the remaining sites was also more frequent in 4nonexpressors (4 of 6 sites; 67%) and 5 low expressors (26 of 64sites; 41%) than 6 normal expressors (24 of 128 sites; 19%), indi-cating that overall methylation extent correlates with transcriptlevel. Consistent with cell lines, primary tumors with low tran-script level displayed complete or partial methylation at 9–14CpG sites (39–61%) while adjacent noncancerous tissues or pri-mary tumors with normal expression level showed partial methyl-
ation at 1–4 sites (4–13%) (Fig. 2f). In particular, methylation sta-tus of 10 CpG sites (numbers 14–23 in Fig. 2b) within nucleotides2201 to 2394 was most tightly associated with expression level.Approximately 30–50% (3–5 sites) and 90–100% (9–10 sites) ofthese 10 CpG sites were completely methylated in 5 low- and 4non-expressor cell lines, respectively whereas none of the sitesshowed complete methylation in 6 hSRBC-expressing cell lines,suggesting that hypermethylation of CpG sites within this regionmight be critical for the transcriptional silencing of hSRBC.
Induction of G1 cell cycle arrest and apoptosis by hSRBCexpression
To understand hSRBC effect on cell proliferation and apoptosis,we generated 2 AGS sublines (AGS-hSRBC-1 and -2), whichexpress different levels of hSRBC (Fig. 3a). Compared to vector-transfected control (AGS-pcDNA), AGS-hSRBC sublines dis-played �51–70% decrease in cellular growth (supplementarydata, Fig. S2a). Flow cytometry analysis revealed that AGS-hSRBC sublines have more G1 phase cells compared to AGS-pcDNA cells (Fig. 3b). Cell proliferation assays using BrdU incor-poration and PCNA expression also showed that AGS-hSRBCsublines are �25–65% low in BrdU uptake and PCNA expressioncompared to AGS-pcDNA cells (Fig. 3c and Fig. S2b). Consis-tently, transient expression of hSRBC led to an accumulation ofthe G1 phase cells in a dose-dependent manner (Fig. 3d). Together,these results indicate that hSRBC exerts anti-prolferative effect. It
FIGURE 2 – Promoter hypermethylation of hSRBC. (a) Re-expression of hSRBC by 5-aza-dC treatment. Nine cancer cell lines with no or lowhSRBC expression were treated with 5-aza-dC (5 lM) for 4 days and hSRBC expression was evaluated by semi-quantitative RT-PCR. C, control;T, treated. (b) Nucleotide sequences of the 50 upstream region of the hSRBC gene. CpGs (nucleotides –71 to –394) are underlined and numbered.Oligonucleotide primers used for methylation-specific PCR (M04/M03 and U04/U03) and bisulfite sequencing analysis (MS-Seq1/MS-Seq2) wereindicated by arrows. (c) Methylation-specific PCR analysis of hSRBC. About 200 ng of bisulfite-modified DNA was subjected to PCR amplificationof the hSRBC promoter sequences using unmethylation-specific (U) and methylation-specific (M) primer sets (U04/U03 for unmethylation-specificPCR and M04/M03 for methylation specific PCR). NL, normal lymphocyte DNA; P, positive control (SssI methyltransferase-treated lymphocyteDNA); BS (-), bisulfite-untreated lymphocyte DNA. (d) A map of the CpG sites of the 50 upstream region of hSRBC. Twenty-three CpGs analyzedare represented by vertical lines and numbered 1–23. The transcription start site is indicated by an arrow at 11. (e) Methylation status of 23 CpGsites in 15 cancer cell lines. The region comprised of 23 CpGs was amplified by PCR. The PCR products were cloned and 5 plasmid clones weresequenced for each cell line. Percent methylation was determined from the number of alleles containing a methylated CpG at each position relativeto the total number of alleles analyzed. Black, gray and white squares represent complete methylation (4–5 clones), partial methylation (1–3clones), and unmethylation, respectively. (f) Comparison of promoter CpG sites methylation between primary tumors and its adjacent noncanceroustissues. N, adjacent noncancerous tissue; T, tumor tissue.
1578 LEE ET AL.
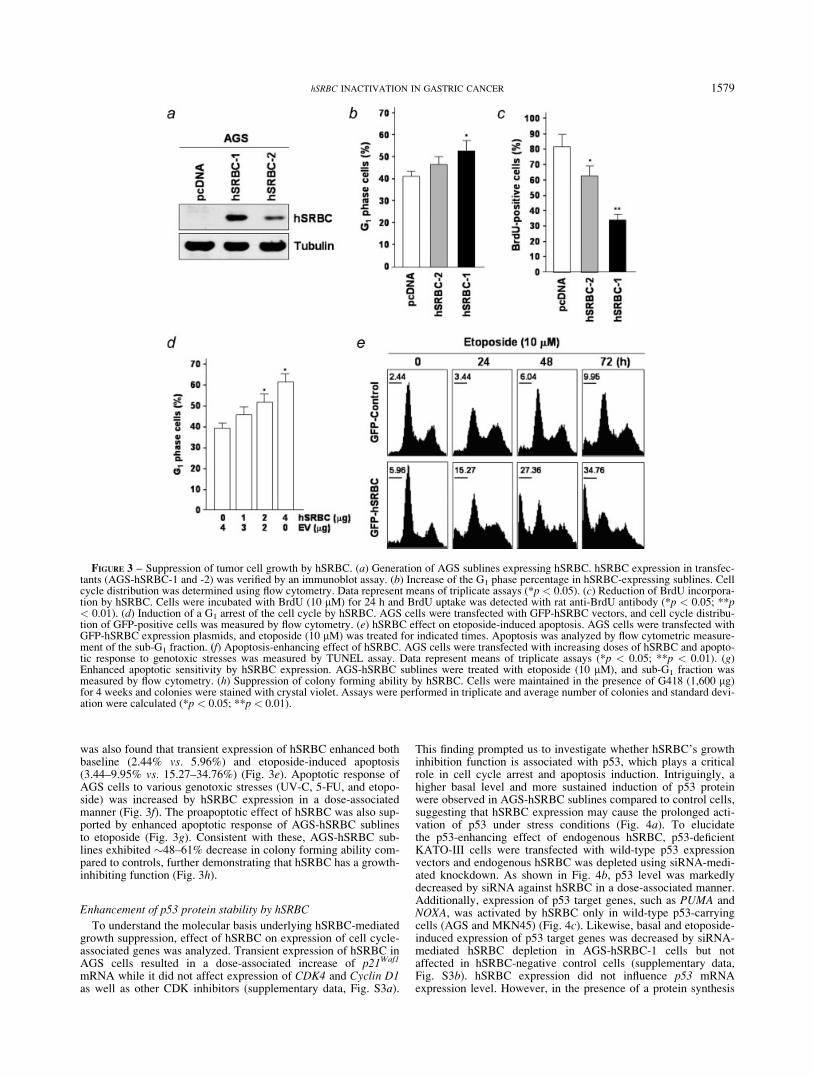
was also found that transient expression of hSRBC enhanced bothbaseline (2.44% vs. 5.96%) and etoposide-induced apoptosis(3.44–9.95% vs. 15.27–34.76%) (Fig. 3e). Apoptotic response ofAGS cells to various genotoxic stresses (UV-C, 5-FU, and etopo-side) was increased by hSRBC expression in a dose-associatedmanner (Fig. 3f). The proapoptotic effect of hSRBC was also sup-ported by enhanced apoptotic response of AGS-hSRBC sublinesto etoposide (Fig. 3g). Consistent with these, AGS-hSRBC sub-lines exhibited �48–61% decrease in colony forming ability com-pared to controls, further demonstrating that hSRBC has a growth-inhibiting function (Fig. 3h).
Enhancement of p53 protein stability by hSRBC
To understand the molecular basis underlying hSRBC-mediatedgrowth suppression, effect of hSRBC on expression of cell cycle-associated genes was analyzed. Transient expression of hSRBC inAGS cells resulted in a dose-associated increase of p21Waf1
mRNA while it did not affect expression of CDK4 and Cyclin D1as well as other CDK inhibitors (supplementary data, Fig. S3a).
This finding prompted us to investigate whether hSRBC’s growthinhibition function is associated with p53, which plays a criticalrole in cell cycle arrest and apoptosis induction. Intriguingly, ahigher basal level and more sustained induction of p53 proteinwere observed in AGS-hSRBC sublines compared to control cells,suggesting that hSRBC expression may cause the prolonged acti-vation of p53 under stress conditions (Fig. 4a). To elucidatethe p53-enhancing effect of endogenous hSRBC, p53-deficientKATO-III cells were transfected with wild-type p53 expressionvectors and endogenous hSRBC was depleted using siRNA-medi-ated knockdown. As shown in Fig. 4b, p53 level was markedlydecreased by siRNA against hSRBC in a dose-associated manner.Additionally, expression of p53 target genes, such as PUMA andNOXA, was activated by hSRBC only in wild-type p53-carryingcells (AGS and MKN45) (Fig. 4c). Likewise, basal and etoposide-induced expression of p53 target genes was decreased by siRNA-mediated hSRBC depletion in AGS-hSRBC-1 cells but notaffected in hSRBC-negative control cells (supplementary data,Fig. S3b). hSRBC expression did not influence p53 mRNAexpression level. However, in the presence of a protein synthesis
FIGURE 3 – Suppression of tumor cell growth by hSRBC. (a) Generation of AGS sublines expressing hSRBC. hSRBC expression in transfec-tants (AGS-hSRBC-1 and -2) was verified by an immunoblot assay. (b) Increase of the G1 phase percentage in hSRBC-expressing sublines. Cellcycle distribution was determined using flow cytometry. Data represent means of triplicate assays (*p < 0.05). (c) Reduction of BrdU incorpora-tion by hSRBC. Cells were incubated with BrdU (10 lM) for 24 h and BrdU uptake was detected with rat anti-BrdU antibody (*p < 0.05; **p< 0.01). (d) Induction of a G1 arrest of the cell cycle by hSRBC. AGS cells were transfected with GFP-hSRBC vectors, and cell cycle distribu-tion of GFP-positive cells was measured by flow cytometry. (e) hSRBC effect on etoposide-induced apoptosis. AGS cells were transfected withGFP-hSRBC expression plasmids, and etoposide (10 lM) was treated for indicated times. Apoptosis was analyzed by flow cytometric measure-ment of the sub-G1 fraction. (f) Apoptosis-enhancing effect of hSRBC. AGS cells were transfected with increasing doses of hSRBC and apopto-tic response to genotoxic stresses was measured by TUNEL assay. Data represent means of triplicate assays (*p < 0.05; **p < 0.01). (g)Enhanced apoptotic sensitivity by hSRBC expression. AGS-hSRBC sublines were treated with etoposide (10 lM), and sub-G1 fraction wasmeasured by flow cytometry. (h) Suppression of colony forming ability by hSRBC. Cells were maintained in the presence of G418 (1,600 lg)for 4 weeks and colonies were stained with crystal violet. Assays were performed in triplicate and average number of colonies and standard devi-ation were calculated (*p < 0.05; **p < 0.01).
1579hSRBC INACTIVATION IN GASTRIC CANCER
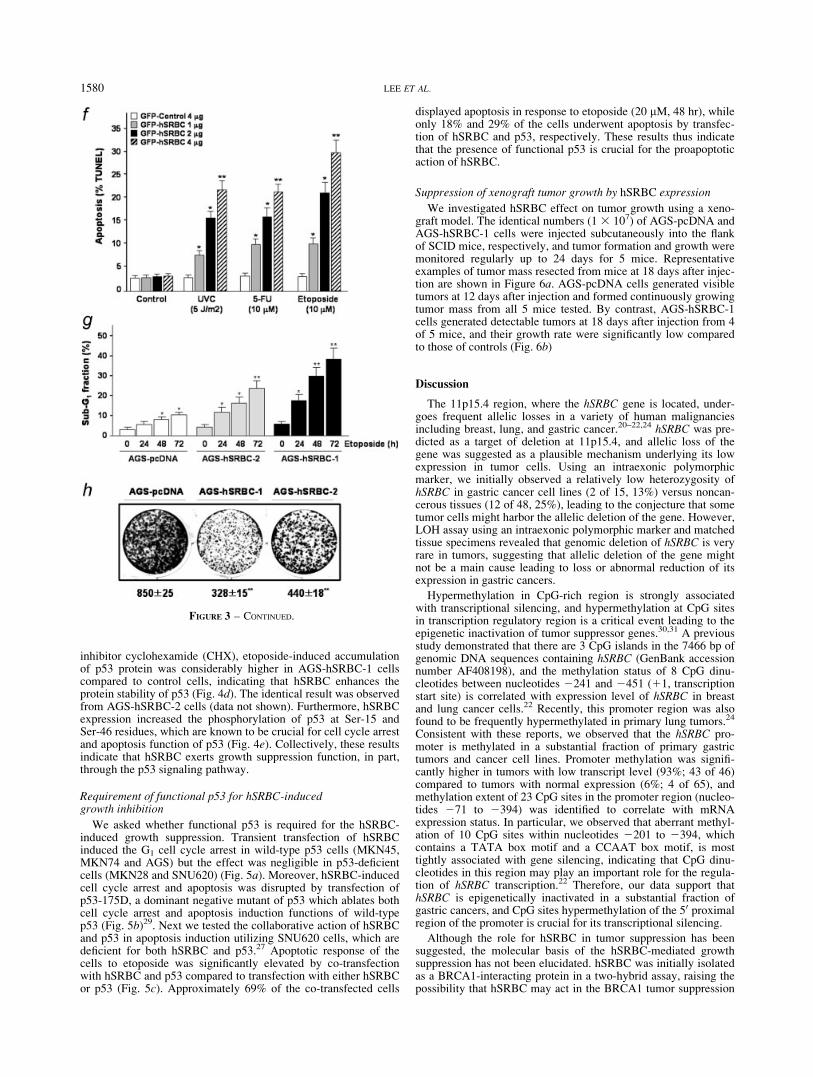
inhibitor cyclohexamide (CHX), etoposide-induced accumulationof p53 protein was considerably higher in AGS-hSRBC-1 cellscompared to control cells, indicating that hSRBC enhances theprotein stability of p53 (Fig. 4d). The identical result was observedfrom AGS-hSRBC-2 cells (data not shown). Furthermore, hSRBCexpression increased the phosphorylation of p53 at Ser-15 andSer-46 residues, which are known to be crucial for cell cycle arrestand apoptosis function of p53 (Fig. 4e). Collectively, these resultsindicate that hSRBC exerts growth suppression function, in part,through the p53 signaling pathway.
Requirement of functional p53 for hSRBC-inducedgrowth inhibition
We asked whether functional p53 is required for the hSRBC-induced growth suppression. Transient transfection of hSRBCinduced the G1 cell cycle arrest in wild-type p53 cells (MKN45,MKN74 and AGS) but the effect was negligible in p53-deficientcells (MKN28 and SNU620) (Fig. 5a). Moreover, hSRBC-inducedcell cycle arrest and apoptosis was disrupted by transfection ofp53-175D, a dominant negative mutant of p53 which ablates bothcell cycle arrest and apoptosis induction functions of wild-typep53 (Fig. 5b)29. Next we tested the collaborative action of hSRBCand p53 in apoptosis induction utilizing SNU620 cells, which aredeficient for both hSRBC and p53.27 Apoptotic response of thecells to etoposide was significantly elevated by co-transfectionwith hSRBC and p53 compared to transfection with either hSRBCor p53 (Fig. 5c). Approximately 69% of the co-transfected cells
displayed apoptosis in response to etoposide (20 lM, 48 hr), whileonly 18% and 29% of the cells underwent apoptosis by transfec-tion of hSRBC and p53, respectively. These results thus indicatethat the presence of functional p53 is crucial for the proapoptoticaction of hSRBC.
Suppression of xenograft tumor growth by hSRBC expression
We investigated hSRBC effect on tumor growth using a xeno-graft model. The identical numbers (1 3 107) of AGS-pcDNA andAGS-hSRBC-1 cells were injected subcutaneously into the flankof SCID mice, respectively, and tumor formation and growth weremonitored regularly up to 24 days for 5 mice. Representativeexamples of tumor mass resected from mice at 18 days after injec-tion are shown in Figure 6a. AGS-pcDNA cells generated visibletumors at 12 days after injection and formed continuously growingtumor mass from all 5 mice tested. By contrast, AGS-hSRBC-1cells generated detectable tumors at 18 days after injection from 4of 5 mice, and their growth rate were significantly low comparedto those of controls (Fig. 6b)
Discussion
The 11p15.4 region, where the hSRBC gene is located, under-goes frequent allelic losses in a variety of human malignanciesincluding breast, lung, and gastric cancer.20–22,24 hSRBC was pre-dicted as a target of deletion at 11p15.4, and allelic loss of thegene was suggested as a plausible mechanism underlying its lowexpression in tumor cells. Using an intraexonic polymorphicmarker, we initially observed a relatively low heterozygosity ofhSRBC in gastric cancer cell lines (2 of 15, 13%) versus noncan-cerous tissues (12 of 48, 25%), leading to the conjecture that sometumor cells might harbor the allelic deletion of the gene. However,LOH assay using an intraexonic polymorphic marker and matchedtissue specimens revealed that genomic deletion of hSRBC is veryrare in tumors, suggesting that allelic deletion of the gene mightnot be a main cause leading to loss or abnormal reduction of itsexpression in gastric cancers.
Hypermethylation in CpG-rich region is strongly associatedwith transcriptional silencing, and hypermethylation at CpG sitesin transcription regulatory region is a critical event leading to theepigenetic inactivation of tumor suppressor genes.30,31 A previousstudy demonstrated that there are 3 CpG islands in the 7466 bp ofgenomic DNA sequences containing hSRBC (GenBank accessionnumber AF408198), and the methylation status of 8 CpG dinu-cleotides between nucleotides 2241 and 2451 (11, transcriptionstart site) is correlated with expression level of hSRBC in breastand lung cancer cells.22 Recently, this promoter region was alsofound to be frequently hypermethylated in primary lung tumors.24
Consistent with these reports, we observed that the hSRBC pro-moter is methylated in a substantial fraction of primary gastrictumors and cancer cell lines. Promoter methylation was signifi-cantly higher in tumors with low transcript level (93%; 43 of 46)compared to tumors with normal expression (6%; 4 of 65), andmethylation extent of 23 CpG sites in the promoter region (nucleo-tides 271 to 2394) was identified to correlate with mRNAexpression status. In particular, we observed that aberrant methyl-ation of 10 CpG sites within nucleotides 2201 to 2394, whichcontains a TATA box motif and a CCAAT box motif, is mosttightly associated with gene silencing, indicating that CpG dinu-cleotides in this region may play an important role for the regula-tion of hSRBC transcription.22 Therefore, our data support thathSRBC is epigenetically inactivated in a substantial fraction ofgastric cancers, and CpG sites hypermethylation of the 50 proximalregion of the promoter is crucial for its transcriptional silencing.
Although the role for hSRBC in tumor suppression has beensuggested, the molecular basis of the hSRBC-mediated growthsuppression has not been elucidated. hSRBC was initially isolatedas a BRCA1-interacting protein in a two-hybrid assay, raising thepossibility that hSRBC may act in the BRCA1 tumor suppression
FIGURE 3 – CONTINUED.
1580 LEE ET AL.

pathway.22 The possible involvement of hSRBC in cell cycle con-trol has been also suggested by observations that mRNA and pro-tein expression of hSRBC is induced on serum starvation anddown-regulated during G0-G1 transition, and that SRBC expres-sion is increased in response to dithiolethione, a chemopreventiveagent in the livers of rats.23,32 The hSRBC protein has the putativePKC phosphorylation site in the middle (172–194 amino acids),and rat SRBC with 81% amino acid identity to hSRBC wasreported to bind and phosphorylated by PKC-d that is involved ingrowth inhibition and tumor suppression.22,33,34 These findingsalso led to the conjecture that hSRBC might be engaged in thePKC signaling pathway. In the present study, we demonstrate firstthat hSRBC leads to a G1 arrest of the cell cycle and enhances theapoptotic sensitivity of tumor cells to genotoxic stresses, such asetoposide, 5-FU and UV-C. Consistently, hSRBC expressiondecreased in vitro growth, colony forming ability and xenograftgrowth of tumor cells. Interestingly, we also found that hSRBCenhances the protein stability of p53 and expression of its targetgenes, including p21Waf1, PUMA and NOXA. Compared tohSRBC-negative control cells, hSRBC-expressing sublines exhib-ited higher steady-state level of p53 as well as prolonged activa-tion under stress condition. Furthermore, both anti-proliferativeand pro-apoptotic activity of hSRBC was more strongly evoked in
wild-type p53 versus p53-deficient cells, and abolished by p53blockade. Our data thus suggest that p53 might play an importantrole for the hSRBC-mediated tumor suppression although the p53-independent role for hSRBC can not be excluded. In fact, the exis-tence of p53-independent function of hSRBC was predicted byour observation that cell cycle arrest and apoptosis of p53-defi-cient cells is increased by hSRBC expression. Collectively, thesefindings support that hSRBC would be a novel effector of p53 andits growth suppression function stems partially from the ability topotentiate the p53 signaling. In this context, it is conceivable thatdysregulation of hSRBC may result in the attenuated p53 responseto stress conditions, and thus contribute to survival advantages ofpreneoplastic or malignant cells.
Recently, it was demonstrated that PKC-d induces multiple an-tineoplastic effects in human colon cancer cells that depend on theactivities of p21Waf1 and p53.33 Interestingly, p21Waf1 and p53were transiently up-regulated after PKC-d transfection, and PKC-d expression did not affect tumorigenicity or differentiation inp21Waf1-null or p53-deficient cells, suggesting that p21Waf1 andp53 are essential mediators of the cellular effects of PKC-d. More-over, cellular accumulation of p53 protein in response to geno-toxic stresses was dramatically inhibited by treatment with PKC-downregulating concentrations of TPA, or by the PKC-d inhibitor
FIGURE 4 – hSRBC induction of p53 and its target gene expression. (a) hSRBC induction of p53 protein. hSRBC-expressing AGS sublines(hSRBC-1 and hSRBC-2) and control (AGS-pcDNA) were treated with etoposide (10 lM, 0-12 hr) and p53 level was analyzed by an immuno-blot assay. (b) Effect of endogenous hSRBC blockade on p53 level. KATO-III cells (p53-null) were co-transfected with wild-type p53 (WTp53)expression vector and increasing doses of hSRBC siRNA. The cells were treated with etoposide (10 lM) for 24 hr, and p53 and hSRBC levelswere measured at 48 hr after transfection. (c) Activation of p53 target gene expression by hSRBC. Expression of p21Waf1, PUMA and NOXAwas examined by semi-quantitative RT-PCR. (d) hSRBC enhancement of p53 protein stability. AGS-hSRBC-1 and AGS-pcDNA cells were pre-incubated with cyclohexamide (CHX, 100 lM) for 1 hr before etoposide treatment (10 lM) and p53 protein level was determined 12–48 hr aftertreatment. (e) Increased p53 phosphorylation by hSRBC. AGS and MKN45 cells were transfected with increasing does of hSRBC expressionvectors. Phosphorylated p53 proteins were detected using phospho-specific antibodies.
1581hSRBC INACTIVATION IN GASTRIC CANCER

rottlerin.35 Based on these, it is plausible that p53 induction byPKC-d might be mediated by hSRBC or its collaboration withhSRBC. It is also conceivable that hSRBC could play a crucial
role in the PKC-d tumor suppression pathway, and antineoplasticeffects of PKC-d might be mediated in part via hSRBC-mediatedactivation of p53 and p21Waf1.
FIGURE 5 – Involvement of p53 in hSRBC-mediated growth suppression. (a) Induction of the G1 cell cycle arrest by hSRBC. GFP-hSRBC (4lg) or GFP-control (4 lg) vectors were transfected into p53-deficient cells (MKN28 and SNU620) and wild-type p53 cells (MKN45, MKN74and AGS), and its effect on cell cycle progression was determined by flow cytometry (*p < 0.05). (b) Inhibition of hSRBC-induced G1 arrestand apoptosis by p53 blockade. AGS cells were transfected with GFP-hSRBC and/or p53-175D expression vectors and percentages of the G1
and sub-G1 fraction were measured by flow cytometry (*p < 0.05; **p < 0.01). (c) Collaborative action of hSRBC and p53 in apoptosis induc-tion. SNU620 cells were transfected with hSRBC and p53 alone or both and its effect on cellular sensitivity to etoposide was measured byTUNEL assay (*p < 0.05; **p < 0.01).
1582 LEE ET AL.

In conclusion, the data presented here clearly show that hSRBCundergoes frequent epigenetic inactivation due to aberrant pro-moter hypermethylation in human gastric cancers, and its alteredexpression is associated with tumors with high stage and grade,supporting the implication of hSRBC abnormality in the malig-nant progression of human gastric tumors. Furthermore, our studyraises the possibility that down-regulation of hSRBC may provide
tumor cells a survival advantage by attenuating the apoptoticresponse of p53 under stress conditions. This finding thus suggeststhat restoration of functional hSRBC could be effective in over-coming therapeutic resistance by sensitization of tumor cells to ap-optotic stimuli. It will be valuable to explore the possible applica-tion of hSRBC as a clinically useful marker for detection andtreatment of human gastric malignancies.
References
1. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002.CA Cancer J Clin 2005;55:74–108.
2. Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A,Feuer EJ, Thun MJ. Cancer statistics, 2005. CA Cancer J Clin 2005;55:10–30.
3. Lynch HT, Grady W, Suriano G, Huntsman D. Gastric cancer: newgenetic developments. J Surg Oncol 2005;90:114–33.
4. Choi Y, Gwack J, Kim Y, Bae J, Jun JK, Ko KP, Yoo KY. Long termtrends and the future gastric cancer mortality in Korea: 1983~2013.Cancer Res Treat 2006;38:7–12.
5. Vauhkonen M, Vauhkonen H, Sipponen P. Pathology and molecularbiology of gastric cancer. Best Practice Res Clin Gastroenterol2006;20:651–74.
6. Tamura G. Alterations of tumor suppressor and tumor-related genesin the development and progression of gastric cancer. World J Gastro-enterol 2006;12:192–8.
7. Wittmann S, Zirn B, Alkassar M, Ambros P, Graf N, Gessler M. Lossof 11q and 16q in Wilms tumors is associated with anaplasia, tumorrecurrence, and poor prognosis. Genes Chromosomes Cancer 2007;46:163–70.
8. Attiyeh EF, London WB, Mosse YP, Wang Q, Winter C, Khazi D,McGrady PW, Seeger RC, Look AT, Shimada H, Brodeur GM, CohnSL, et al. Chromosome 1p and 11q deletions and outcome in neuro-blastoma. N Engl J Med 2005;353:2243–53.
9. Chunder N, Mandal S, Roy A, Roychoudhury S, Panda CK. Analysisof different deleted regions in chromosome 11 and their interrelationsin early- and late-onset breast tumors: association with cyclin D1amplification and survival. Diagn Mol Pathol 2004;13:172–82.
10. Nakadate H, Yokomori K, Watanabe N, Tsuchiya T, Namiki T,Kobayshi H, Suita S, Tsunematsu Y, Horikoshi Y, Hatae Y, Endo M,Komada Y, et al. Mutations/deletions of the WT1 gene, loss of hetero-zygosity on chromosome arms 11p and 11q, chromosome ploidy andhistology in Wilms’ tumors in Japan. Int J Cancer 2001;94:396–400.
11. Roy D, Calaf G, Hei TK. Allelic imbalance at 11p15.5–15.4 corre-lated with c-Ha-ras mutation during radiation-induced neoplastictransformation of human breast epithelial cells. Int J Cancer 2003;103:730–7.
12. Zhao B, Bepler G. Transcript map and complete genomic sequencefor the 310 kb region of minimal allele loss on chromosome segment11p15.5 in non-small-cell lung cancer. Oncogene 2001;20:8154–64.
13. Tran YK, Newsham IF. High-density marker analysis of 11p15.5 innon-small cell lung carcinomas reveals allelic deletion of one sharedand one distinct region when compared to breast carcinomas. CancerRes 1996;56:2916–21.
14. Panani AD, Ferti AD, Raptis SA, Roussos C. Novel recurrent struc-tural chromosomal aberrations in primary bladder cancer. AnticancerRes 2004;24:2967–74.
15. Shaw ME, Knowles MA. Deletion mapping of chromosome 11 in car-cinoma of the bladder. Genes Chromosomes Cancer 1995;13:1–8.
16. Lichy JH, Zavar M, Tsai MM, O’Leary TJ, Taubenberger JK. Loss ofheterozygosity on chromosome 11p15 during histological progressionin microdissected ductal carcinoma of the breast. Am J Pathol1998;153:271–8.
17. Moskaluk CA, Rumpel CA. Allelic deletion in 11p15 is a commonoccurrence in esophageal and gastric adenocarcinoma. Cancer 1998;83:232–9.
18. Rodriguez E, Rao PH, Ladanyi M, Altorki N, Albino AP, Kelsen DP,Jhanwar SC, Chaganti RS. 11p13–15 is a specific region of chromo-somal rearrangement in gastric and esophageal adenocarcinomas.Cancer Res 1990;50:6410–16.
19. Ranzani GN, Renault B, Pellegata NS, Fattorini P, Magni E, Bacci F,Amadori D. Loss of heterozygosity and K-ras gene mutations in gas-tric cancer. Hum Genet 1993;92:244–9.
20. Yustein AS, Harper JC, Petroni GR, Cummings OW, Moskaluk CA,Powell SM. Allelotype of gastric adenocarcinoma. Cancer Res 1999;59:1437–41.
21. Baffa R, Negrini M, Mandes B, Rugge M, Ranzani GN, Hirohashi S,Croce CM. Loss of heterozygosity for chromosome 11 in adenocarci-noma of the stomach. Cancer Res 1996;56:268–72.
22. Xu XL, Wu LC, Du F, Davis A, Peyton M, Tomizawa Y, Maitra A,Tomlinson G, Gazdar AF, Weissman BE, Bowcock AM, Baer R,Minna JD. Inactivation of human SRBC, located within the 11p15.5-p15.4 tumor suppressor region, in breast and lung cancers. Cancer Res2001;61:7943–9.
23. Gustincich S, Schneider C. Serum deprivation response gene isinduced by serum starvation but not by contact inhibition. CellGrowth Differ 1993;4:753–60.
24. Zochbauer-Muller S, Fong KM, Geradts J, Xu X, Seidl S, End-Pfut-zenreuter A, Lang G, Heller G, Zielinski CC, Gazdar AF, Minna JD.Expression of the candidate tumor suppressor gene hSRBC is
FIGURE 6 – Suppression of xenograft tumor growth by hSRBC. (a) Representative examples of xenograft tumors derived from AGS-pcDNAand AGS-hSRBC-1 cells. AGS-pcDNA and AGS-hSRBC-1 cells (1 3 107) were injected subcutaneously into the right flank of 5-week-oldfemale SCID mice. The mice were sacrificed at 18 days after injection and tumor size was measured. (b) Inhibition of tumor growth by hSRBC.AGS-pcDNA and AGS-hSRBC-1 cells injected into 5 SCID mice and 2D tumor sizes were measured every 3 days. Tumor volume was calcu-lated using the formula V 5 1/2 3 length 3 (width)2 (*p < 0.05; **p < 0.01: ***p < 0.001). [Color figure can be viewed in the online issue,which is available at www.interscience.wiley.com.]
1583hSRBC INACTIVATION IN GASTRIC CANCER

frequently lost in primary lung cancers with and without DNA meth-ylation. Oncogene 2005;24:6249–55.
25. Chi SG, Kim HJ, Park BJ, Min HJ, Park JH, Kim YW, Dong SH, KimBH, Lee JI, Chang YW, Chang R, Kim WK, et al. Mutational abroga-tion of the PTEN/MMAC1 gene in gastrointestinal polyps in patientswith Cowden disease. Gastroenterology 1998;115:1084–9.
26. Gumerlock PH, Chi SG, Shi XB, Voeller HJ, Jacobson JW, GelmannEP, deVere White RW. p53 abnormalities in primary prostate cancer:single-strand conformation polymorphism analysis of complementaryDNA in comparison with genomic DNA. J Natl Cancer Inst 1997;89:66–71.
27. Kim JH, Takahashi T, Chiba I, Park JG, Birrer MJ, Roh JK, De LeeH, Kim JP, Minna JD, Gazdar AF. Occurrence of p53 gene abnormal-ities in gastric carcinoma tumors and cell lines. J Natl Cancer Inst1991;83:938–43.
28. Yokozaki H. Molecular characteristics of eight gastric cancer celllines established in Japan. Pathol Int 2000;50:767–77.
29. Ryan KM, Vousden KH. Characterization of structural p53 mutantswhich show selective defects in apoptosis but not cell cycle arrest.Mol Cell Biol 1998;18:3692–8.
30. Ehrlich M, Jiang G, Fiala E, Dome JS, Yu MC, Long TI, Youn B,Sohn OS, Widschwendter M, Tomlinson GE, Chintagumpala M,Champagne M, et al. Hypomethylation and hypermethylation of DNAin Wilms tumors. Oncogene 2002;21:6694–702.
31. Sato F, Meltzer SJ. CpG island hypermethylation in progression ofesophageal and gastric cancer. Cancer 2006;106:483–93.
32. Primiano T, Gastel JA, Kensler TW, Sutter TR. Isolation of cDNAsrepresenting dithiolethione-responsive genes. Carcinogenesis 1996;17:2297–303.
33. Perletti G, Marras E, Dondi D, Osti D, Congiu T, Ferrarese R, deEguileor M, Tashjian AH, Jr. p21Waf1/Cip1 and p53 are downstreameffectors of protein kinase C delta in tumor suppression and dif-ferentiation in human colon cancer cells. Int J Cancer 2005;113:42–53.
34. Gschwendt M. Protein kinase Cd. Eur J Biochem 1999;259:555–64.
35. Abbas T, White D, Hui L, Yoshida K, Foster DA, Bargonetti J. Inhibi-tion of human p53 basal transcription by down-regulation of proteinkinase Cd. J Biol Chem 2004;279:9970–7.
1584 LEE ET AL.




















