
A Legal, Economic, and Religious Analysis of Selling Kidneys
Upload
independentCategory
view
0download
0
...
Facilitated Diagnosis of the Contiguous Gene Syndrome: Thberous Sc1erosis and Polycystic Kidneys
by Means of Haplotype Studies
Roser Torra, MD, PhD, Celia Badenas, PhD, Alejandro Darnell, MO, PhO, Juan Antonio Camacho, MO,Richard Aspinwall, PhO, Peter C. Harris, PhO, and Xavier Estivill, MO, PhO
• Tuberous sclerosis (TSC) and autosomal dominant polycystic kidney disease (ADPKD) are genetically heterogeneous diseases. The major gene for ADPKD (PKD1) lIes adjacent to Ihe rSC2 gene on chromosome 16p13. Some reports in Ihe IIterature referred to an unusual presenlalion 01 TSC with enlarged cystic kidneys al birth, but it was not until the 10calization of the TSC2 and PKD1 genes that it was possible to analyze Ihe interactlon between both genes. We describe a case 01 a child with TSC and enlarged cystic kidneys. The study of genetic marker segregalion In the family pointed lo the presence of a deletion involvlng the 3' reglon of PKD1. A lurther study of the reglon showed a deletion of 40 kb Involving both PKD1 and TSC2. We suggest Ihat an additive or synerglstic effect between PKD1 and TSC2 may cause thls renal phenolype. A contiguous gene syndrome involving PKD1 and TSC2 should be suspecled in chlldren wilh TSC and enlarged polycystic kidneys al birth. The first approach to identlly a delelion of both genes could be the analysis of Ihe segregatlon of PKD1 and TSC2 markers In Ihe famlly. © 1998 by the Na/lonal Kidney Founda/ion, Ine.
lNDEX WORDS: Polycystic kidneys; tuberous sclerosls type 2; genetics; deletion¡ contiguous gene syndrome; chlldren' mlcrosalelllles; loss 01 helerozigosity.
CHROMOSO E ALTERATIO S involving (ADPKD) and tuberOllS sclerosis (TSC) was the 5contiguous genes usually show a wide spec key to the identification of the PKDI gene
tmm of clinica1 features.! The severity of the TSC is an autosomal dominant disease with a
disease usual1y ret1ects the number of genes high mutation rate (two thirds of al1 cases are
involved. Two contiguous syndromes affecting new mutations6 that appear lO affect cel1 migra
the kidney are Wilms tumor-ani.ridia-genitouri tion and differentiation in a va.riety of organs 7
nary tract malfonnations-mental retardation (de The estimated prevalence of this disease is 1 in
letion 11 p13? and Alport's leiOrrUomatosis (dele 10,000 live births. 8 TSC is genetical1y heteroge
tion COL4A5 O 4A6)3.4 The information neous6 with at leasl two loci causing the disease: 9q34 (TSCl)9 and 16p13.3 (TSC2)W Both lociderived from contiguous gene syndromes perhave been recently clonedll.ll The TS 2 gene mits localization of important disease genes. It encodes a I rotein of 1,784 amino acids that is was precis Iy one of these cases that facilitated called tuberin. the identification of the polycystic disease type I
ADPKD is more frequent than TSC (1 ingene (PKDl). A family with a translocation 1,000 live bitths) and is also a systemic disorder.(breakpoints at 16p 13.3 and 22q 11.21) showing The main complication of this disease is theautosomal dominant polycystic kidney disease progressive enlargement of the cystic lUdneys that eventually may lead to end-stage renal disease13 ADPKD is also a genetically heteroge
From Ihe Nephrology and Genelics Services, Hospiral neous disease caused by mutations either in Clínic; Nephrology Savice, Hospital Sanl loan de Déu,
the PKDI gene (l6p13.3),5 the PKD2 geneBarcelona, Spain; and Ihe Medical Research Council Molecu
(4q13-q23),14.15 or the still unmapped PKD3lar HemalOlogy Unil, InslilLlle oI Molecular Medicine, lohn Radcliffe Hospilal. Oxjord, UK gene. 16. 17 The PKDI and PKD2 genes have been
Receil'ed luly 28, 1997, accepled in rel'isedform Oclober idenlified and sequenced. The PKD1 gene en24, 1997. codes a protein of 4,303 amino acids that is
Supporled by grant no. F/S 9410343 from rhe Fondo de called polycystin. IB-20
Il1\'esligaciones Sanilarias de la Seguridad Social. R. T IS a reripienl offello\Vship 110 Fl 9419.102 from Ihe Direcció The PKDI gene lies immediately adjacent (60 General de Recerca de la Genera/iral de Catalunya. bp) to the rSC2 gene, in tail-to-tail orientation.
Address repril1/ requesls lO Roser Torra. MD, PhD, Sen'i The proximily of [he two genes suggested a role cio de Nefrologia, Hosplial C/(nic, Vil/arroel 170, 08036 for the PKDI gene in cases of rSC2 with severe Barcelona, Spain. E-mail: [email protected]
cystic involvement. Brook-Carter et al2 identi,',) 1998 b\' rhe Naliollal Kidl1ey Foundalion, Il1e. 0272-638619813106-0020$3 ODIO fied six rSC2 children with very severe polycys-
American Journal af Kidney Diseases, Vol 31, No 6 (June), 1998: pp 1038-1043 1038

1039 POLYCYSTIC KIDNEYS AND TUBEROUS SCLEROSIS
tic disease showing deletions that involved both genes. These children, as well as others reported in the literature, present with enlarged polycystic kidneys recognizable at birth or shortly thereafter. Their kidneys are filJed by a multitude of variably sized cysts, closely resembling those seen in advanced stages of A PKD. These children usually enter end-stage renal disease before the second decade of ¡ife. They al so present symptoms corresponding to TSC, such as seizures, hamartomas, psychomotor retardation, and skin lesions (hypomelanotic macules and facial angiofibromas). ;Jther typical lesions of T e, such as angiomyolipomas, Koenen's tumors, and welJ-established facial angiofibromas, only appeal' later in life. Frequently, if the stigmata of TSe are overlooked, this entity may be diagnosed as early-onset ADPKD or autosomal recessive polycystic .kidney disease.
We present a case of TSC2 and PKDJ deletion that was suspected because of hemizigosity for microsatellite markers at the 3' end of PKD J. The deletion was confirmed by pulse field gel clectrophoresis (PFGE) showing a deletion of 40 kb disrupting both genes. We suggest that the first approach to diagnose this contiguous gene syndrome could be to search for hemizigosity by the use of genetic markers from PKD l and TSC2.
MATERIALS ANO METHOOS
Blood samples were extracted from the parents and the probando Genomic DNA was extracted with the salting-out method22 A cel] line from the proband was constructed. D A was anaJyzed for the microsalellite KG8, which lies in the 3' UTR of the PKDl gene, clase to che TSC2 genc.5
polymorphism A4091 A (exon 45),23 and CW2 and l6AC2.5, which are proximal lO che 5' end of che PKDl gene. 14
Analysis of these microsatellites was performed as previously described25 and detected with the silver-staining technique 26 PFGE \Vas pelformed as previously described21 with DNA probes within and flanking che TSC2 and PKDl genes.
RESULTS
Clinical Data
A 7-month-old girl was admitted for the study of psychomotor retardation and petit mal seizures. In a prenatal sonography sean, she was diagnosed with polyeystic kidneys, and when she was bom, bilateral ftank masses were noted. There was no family history of cystjc kidneys or renal, cutaneous, 01' neurologic disease. Her blood
pressure was 160/1 00 mm Hg. he physical examination showed depigmented areas of 2 cm in diameter on the trunk and lower extremities and enlarged kidney . An elcctroencephalogram showed multifocal paroxysms predominantly at the right hemispheriulll On ophthalmologic evaluation, a phacoma was detected in the lower part of the right macula. Laboratory data included a creatinine level of O.S mg/dJ, and a glomerular filtration rate of 49 mUmin! 1.73 m2
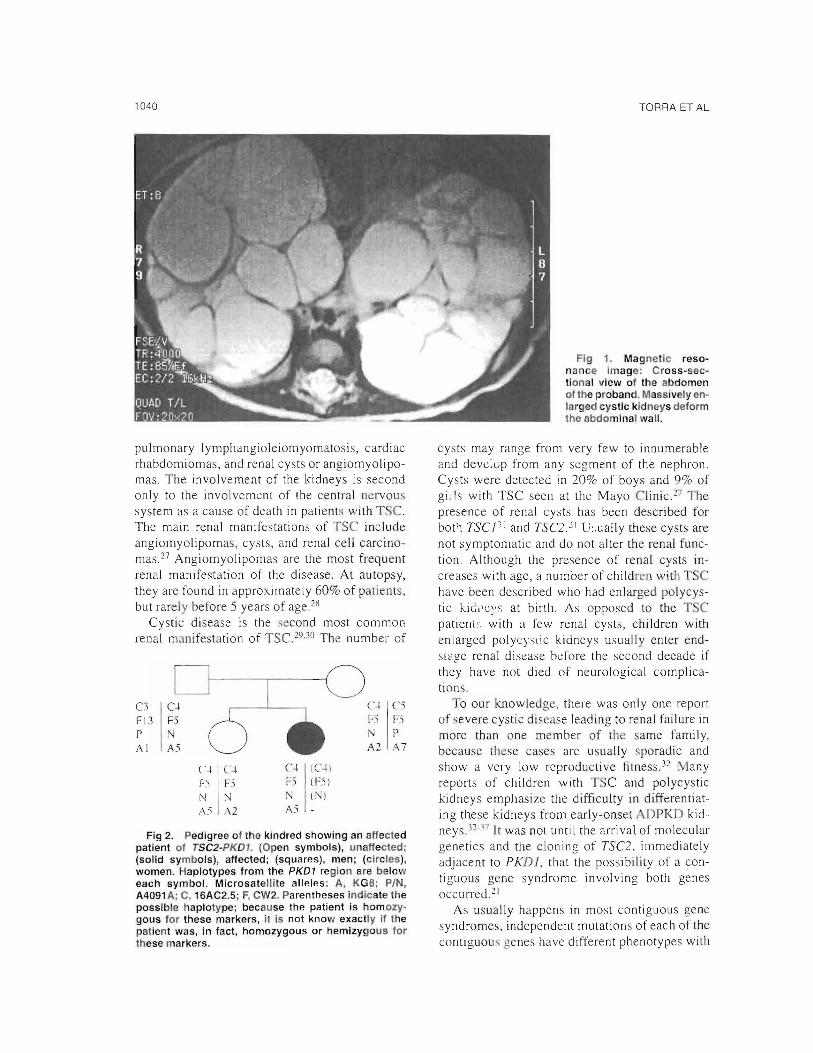
Gchocardiogram showed a concentric myocardic hypertrophy. A cranial magnetic resonance imaging (MRI) disclosed mulLiplc subependimary nodules. An abdominal MRI showed massively enlarged cystic kidneys (Fig 1). Renal arteriography did not reveal any angiomiolypoma. t present, the patient is 14 months old and is being treated with valproic acid and captopril; both the seizures and the high blood pressure are under control. Her renal function remains stable.
Molecular Analysis
Analysis of the family with the KGS CA repeat marker showed that the proband was hemizyguus for this marker and noninformative for CW2, AC2.5, and the polymorphism A4091A (Fig 2). The hernizigosity for KG8 pointed to a deletion involving the 3' region of the PKDJ gene. For this reasan, a further study of that region was performed by PFGE. The DNA probes JHI and ew10 shawed a deleted D A fragment :::!:40 kb smal1er than the normal product deleted in Nrul or Mlul digests (Fig 3). Further analysis with the probes BFSS and lli8 showed no breakpoint fragment by a lower signal intensity, indjcating that both pro bes were deleted. The estimated area deleted in this patient is shown in Fig 3.
DISCUSSION
Tse is an autosomal dominant disease that affects all tissues wlth the possible exception of the peripheral nervous system, meninges, and . keletal muscles 7 The main chnical findings leading to the diagnosis of TSC are cortical tubers, subependimal glial nodules, astrocytomas, retinal hamartomas, facial angiofibromas, fibrous forehead plaque, ungueal fibroma, and shagreen pateh. Apan from these neurological and dermatologic findings, some visceral features are also presumptive of TSC, which are
1040 TORRA ET AL
pulmonary Iymphi:lngioleiomyomatosis, cardiac rhabdomíomas, and renal cysts or angiomyolipomas. The involvement of the kidneys is second only to the involvcment of the central ncrvous system as a cause of death in patients with T, C. The main renal manifestations of TSC include angiomyolipomas, cysts, and renal cell carcinomas 27 Angiomyolipomas are the most frequent renal manifestatíon of the disease. At autopsy, they are found in approxjmately 60% ofpatienrs, but rarely before 5 years of age. 28
Cystie dísease is the second most common renal manifestation of TSC. H111 The number of
C3 C-J. ("~
Fl3 F5 1".1 P P
Al A2 A7
C-J. C-J. C-+II(-1-1 F5 F) ¡-) I (F5)
N N [\·1(.'1A) .A.1 A2
Fig 2. Pedigree 01 the kindred showing an ar1ected patient 01 rSC2-PKD1. (Open symbols), unafteC1ed; (solid symbols), affected; (squares), men; (c1rcles), women. Haplotypes trom the PKDl region are below each symbol. Microsatellite alleles: A, KG8; PIN, A4091 A; e, 16AC2.5; F, CW2. Parentheses indicate the possible haplotype; because lhe patient is homozygous lor these markers, it is not know exactly il lhe palienl was, in tact, homozygous or hemizygous tor lhese markers.
Flg 1. Magnetíc reso· nance Image: Cross-seclional view 01 the abdomen 01 the probando Massively enlarged cystic kidneys delorm lhe abdominal wall.
cysts may range from very fcw to innumerable and devc:up from any segment of the nephron. Cysts were detected in 20% of boys and 9% of gí:-ls with TSC seen at the Mayo Clínic n The presence of renal cysts has been described for both TSC ¡1! and TSC2." 1 U:,ually these cysts are not symptomatic and do not alter the renal function. Although the presence of renal cysts íncreases with agc, a number of childrcn wi h TSe have been described who had enlarged polycystic kü.:"cy<; at birth. As opposed to the T e patieni': with a few renal cysts, children with enlarged P01YCYSlic kidneys usui:llly en ter endstq!e renal disease before the second decade if they have not died of neurological complicatians.
'o our knowledge, there was only one report uf severe cystic discase leading to renal failure in more than one member of lh same family, because these cases are usually sporadic and show a very low reproductive fitness. 32 l\Ilany reports of children with TSC and polycystic kidneys emphasizc the difficulty in differentiating these kidneys from early-onsel. DPKD kidneys.'2-:1.7 It was not until the arrival of molecular genetics and the cloning of TSC2. ill1mediately adjacent to PKDJ, that the possibility 01' a contiguous gene syndrome involving both genes occurred. 21
As usually happens in most contiguous gene syndromes, independent mutations of each ol' the contíguOllS genes have different phenotypes wilb

1041
8
POLYCYSTIC KIDNEYS AND TUBEROUS SCLEROSIS
A JH1 JH8 CW10
N P kb N P kb N P kb
-145 -145 -145
- 97 - 97 - 97
.... deletian
lükb
R ~.-\S.,....:...." ....,"- ~'PK"9-~""""""""""""""""" ..'''''':'-''''''''''''':''\
r= c::J U -pie' = JHI 81-'55 JH8 CWIO
Fig 3. (A) PFGE anaJysis of DNA from a normal control (N) and the patienl (P), digested with Nvul and hybridized wlth JHI (lefl), JHB (center), and CW O(right). A deleted fragment :±: 40 kb smaller than normal is seen with JHI and CW10 but not JHB, which is detected. (B) Map of the TSC2IPKD1 region showlng the locations of the genomic probes used (open boxes), the Nrul sites (Al flanklng the genes, the positions 01 PKD1 and TSC2 genes, and the directlons of the transcriptlon. The area deleted In the patient ls shown aboye the map.
no COlnmon chnical features. but in the case of PKDI and 7SC2, both enlilies can be associated with renal cysts. Thus, lhe presence of a single mutation in any of these genes is enough to cause cystic kidneys, making it difficuJt to determine the contribution of each gene in this contiguous gene syndrome. Although Peral et ap8 described a nonsense mutation in the 3' region ofthe PKDl gene associated with a case of infantile-onset polycystic kidney disease, most of the mutations found in this region are usuaUy associated with late-onset ADPKD. Thus, it ma ' be an additive or synergistic effect between PKD 1 and TSC2, rather than an inactivation of PKD1, that causes the severe cystic involvement in this syndrome. Although it is attractive to consider that DNA changes in the TSC2 gene may be involved in early-onset PKD1, the cases described so far did not have any clinical features ofTSC and usually had a family history of adult-onset disease, whereas the six patients described by Brook~'arter et al21 and the patient reported here showed definite features of TSC. Up to now, the mechanism of cystogenesis in both di eases and aIso in this contiguous gene syndrome remained unknown. Although the two-hit model has been well shown in TSC hamartomas,39 it remains to be elucidated ifrenal cy t show loss ofheterozygosity (LOH) in rSC2-PKD1. Recently, LOH has been shown for PKD1:"oAI raising the possi
bílity that a two-hit mechanism is involved in the TSC2-PKDI cases. The t\Vo-hit model was first reported by Knudson42 in 1971 in the retinoblastoma and has been applied to many types of cancel' since then. It implies that the disease would only occur when a germinal mutaríon inherited from a parent 01' as a result of a new mutaríon coexisted witll a somaríc mutatíon. When this somatic mutation is a deletion, it is called an LOH.
We have highlighted lh fact that children presenting with enlarged cystic kidneys at birth, in the absence of an ADPKD familial history or clínical signs of autosomal recessive polycystic kidney disease, may have a deh::tion of TSC2 and PKD1. The renal prognosis is much worse in these patients than in typical TSC cases. A nrst approach to diagnose this syndrome, after the su picion of TSC arises, may be the study of the segregation of genetic markers of PKDl and TSC2 in the family. The loss of one allele in the proband wouId suggest the presence of a deletion that couId be fLll1her studied through molecular means.
REFERENCES
I Schmicke! RD: Contiguous gene syndromes: A compo· nem of recognizable syndromes. J Pedíatr 109:231·241, 1986
2. Riccardi V. Sujansky E. Smith A. Francke U: Chromo·

1042
somal imbalance in the aniridia-Wilm's tumor association II p intcrstitial delelion, Pediatric> 61 :604·6 j O. 1978
3, Antignac C, Zhou J, San~lk: M, Cochat P. ROllSsel P. Oeschenes G. Grns ¡:, K.nebelman 8, Hors·Cayla MC. Tryg· gvason K. Gubler MC: Alpon syndrome and diffuse leiomyo· matosis: Oeletions in Ihe ('OL4AS collagen gene. Kidney Int421178·IIX3.1992
4. Zhou J. Moehizuki T. Smeets H, Antignac C. L~llrila P, de Paepe A. Tryggavson K. Reeders ST: Oeletion oC the pajred aS (IV) and a6 l i Y) collagen genes in inherited smooth muscle lumors Science 261' J j 67·1169. 1993
S. European Polyeystic Kidney Disease Consortium: The polycystic kidney disease 1 gene cncodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 77:881·894,1994
6. Sampson JR. Yates JR. Pirrit LA, Fleury P. Winship 1, Beighton P, Coonor JM: Evideoee COl geoetic heterogeneity io tuberolls >clerosis J Med Genet 26:511·516. 1989
7. Torres VE: Tuberolls sclerosis complex. in Walson ML, Torres VE (eds) Polycystic Kidney Oisease. OxCord. England, Oxford Univ~rsity Press, 1996. pp 283·308
8. Wiederholt WC. Gomez MR. Kurland LT: Incidencc and prevalence 01' tuberousclcrosis in Rochesler, Minnesota, 1950 through 1982. Neurology 35600·603, 1985
9. Fryer AE, Chalmers A. Connor JM, Fraser l. Povey S, Yates AO, Yates JRW, Osbome JP: Evidence that the gene Cor tllberous sclerosis is in chromosome 9. Lancct 1:659·661. 1987
10. Kandt RS, Haincs JL, Smith M, Norrhrup H, ardner RJM. Short :vIP, Oumars K, Roach ES, Steingold S. WaJl S, Blanton SH, Flodman P, Kwiatkowskj OJ, Jcwell A. Weber JL, Roses AO, Pericak-Vallce MA: Linkage 01' an important gene loclls for tub rOlls sclerosis to a chromosome 16 marker for polycystic kidncy dlsease Nature Genet 2:37-41. 1992
ti. European Chromosome 16 Tuberous Selerosis Con· sortium: Identificatíon and characterization 01' the tuberous sclerosis gene on chIomosome 16 Cel! 75 1305·1315. 1993
12. van Slegtenhorst M. de Hoogt R. Herman, C. I ellist M. Janssen B, Verhoef S, Lindhoul O, van den Ouweland A, Halley O, Young J, Burley M. Jeremiah S, Woodward K. Nahmias J. Fox M, Ekong R, sborne J. Wolfe J, Povey S, Snell RG, Cheadle JP, Jon~s AC, Tachataki M. Ravine O, Sampson JR. Reeve MP, Richardson P. Wj]mer F, Munro C, llawkins TL. Sepp T, Ali mM, Ward S. Grecn AJ, Yates JRW, Kwiatkowska J, Hensk~ EP, Short MP. Haines JH. J07.wiak S. Kwiatkowski DJ: Identification of the tuberous sclerosis gene TSC j on chromosome 9q34. Sciencc 277:805· 808. 1997
13. Gabow PA. Autosomal dominant polycystic kidney disease. N Eng J Me<.l 329332·342. 1993
14. Kimberling WJ, Kumar S, Gabow P. Kenyon JB. Conolly CJ, Somlo S: Autosomal dominant polycystic kid· ney disease: Localization 01" the second gene to chromosome 4q 13·q23. Genomil's 18:467-472, 1993
15. Peters OJM, Spnlit L, Saris J1. Ravine D, Sandkuijl LA, Fossdal R, Boersma J, Van Eijk R, Norby S, Constanti· nou-Oeltas CO, Picrides A. BI"l~senden J •• Frants RR. Van Ommen G J B, Breuning MH: Chromosome 4 10laJization of a second gene for autosomal dominant polycystic kidney discasc. Nature Genet S 359-362. 1993
TORRA ET AL
16. Oaoust MC. Bichet OG. Somlo S A French-Canadian family with autosomal dominanl polyeystiekidney disease (AOPKD) unlinkecl lo AOPKDI 0[' AOPKD2 J Am Soc ;--';ephrol 4:262, 1993 (abstr)
17 de Almeida S, de Almeida E. Peters O, Pinto JR, [avora 1, Lavioha J, Breuning M. Prata MM: Autosomal dominant polycystic kidney disease: Evidencc ror the existenee of a third locus in a Porluguese family. Hum Genet 96 83-88, 1995
18. Hughes J, Warcl el, Peral B, /\spinwall R, Clark K, San i'"íillán JL, Gamble V, Harris PC: ','he polycystie kidney disease 1 (PKD/) gene encodcs a novel protein wíth mul· tiple cell recognition domains. Nature Genet 10: 151-160, 1995
19. American AOPKDI Consonium: Analysis of the genomic sequence for lhe autosomal dominanl polycyslic kidney disease (PKDI) gene prediels lhe presence of a leucine-rich repear. Ilurn Molec Genet 4:575·582. 1995
20. International ADPKO Consortium: PolycYSlic kidney <.lisease: The complete structure of lhe PKD I gene and its prolein. Cell 81289-298, 1995
21. Brook-Carter P, Peral B, Ward CJ, Thompson P, Hllghes J, Mahesshwar MM. i'iellist M, Gamble ,Harris PC, Sampson JR: Oeletion of lhe TSC2 and PKDI genes associated with sevcre inCantilc polycystic kidney disease-A contiguous syndrome. Nature Genet 8:328-332, 1994
22. Miller SA, Oykes DD. Polesky f;¡ : A simple saltingout procedure for extracting [);>.JA from human nucleated cells. NueJcie Acid Res 16: 12 l S, 1988
23. Peral B. Ward CJ. Jan Millán JL, Thomas S, Stallings RL. Moreno F. Harris pe: Evidence of linkage disequilibrium in the Spanish polyeystic kidney disease I population. Am J Hum Genet 54:899-908, 1994
24. Peral B, San Millán JL, Ong A M. Gamble V, Ward C1. Strong C. Harris PC: Screening ¡he 3' region 01" the polycystic kidney disease I (PKDI) gene reveals six novel mutations. Am J Hum Gcnet 58:86·96, 1996
25. Torra R. Badenas \ . Darnel1 A, Nieolau C, Volpini Y, Reverl L. Estivill X: Linkage, clinical features and prognosis of.\I)PKD types 1 and 2. J Am Soc Nephrol 7:2142-2151, 1996
26. Bassam 8J, Caetano·Anollcs G, Grcsshoff PM: Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal Rioehem 196:80-83. J991
27 Torres VE. King BE Holley KE, Blute \>{!., Gome7. MR: The kidney in tuberous sclerosis complex. Adv Nephrol 23:43-70. J994
28. Van Baal JG. FJeury P, Brllmrnelkamp WH: Tuberous sclerosis and the relation with [(~nal angiomyolipoma. A genetic study on the cl i!lieal aspeets. Clin Genet 35: 167·173. 1989
29. Bemstein J. Robbins TU: Renal involvement inlubcr· OllS ,cl~rosis. Ann N Y Acad Sci 61536-49, 1991
30. Stillwell TJ, Gomez MR. Kelalis PP Rena\ lesions in tuberous sclerosis. J Urol 138:477-481, 1987
31 "\ellist M, Broo~·Carter PT, Connor JM. Kwiat· kowski DJ, Johnson P, S~l1lpson J: Identification oC markers Ranking the Illberous sclerosi, locus on ehromosomc 9 (TSC!). J ¡'vled Genet 30:224·227. 1993
32. O'Callaghan TJ. Edwards JA. Tobin IV!. Mookerjee

1043 POLYCYSTIC KIONEYS ANO TUBEROUS SCLEROSIS
BK: Tuberousclerosis with striking renaJ invo]vement in a family. Arch Intern Med 135:1082-1087, 1975
33. Schillinger F, Montagnac R: Chronic renal failure and its treatment in ruberous sclerosis. Nephrol Dial Transplant II :481-485, 1996
34. Wenzl JE, Largos JC, Albers DD: Tuberous sclerosis presenting as polycystic kidneys in an infan!. J Pediatr 77:673-676, 1970
35. Webb DW, Super M, Normand CS, Osbome JP: Tuberousclerosis and polycystic kidney disease. Br Med .1 306: 1258-1259, 1993
36. Yu DT, Sheth KJ: Cystic renal involvement in tuberous sclerosis. Clin Pediatr 24:36-39, 19R5
37. Stapleton FB, .Iohnson D, Kaplan GW, Griswold W The cystic renallesions in ruberousclerosis J Pediatr 97:574579, 1980
38. Peral B, Ong ACM, San Millán JL, Gamble V, Rees
L, Harris PC: A stable nonsense mutation associated with a case of infantile onset poIycystic kidney disease 1 (PKDI). Hum Mol Genet 5:539-542,1996
39. Green AJ, Smith M, Yates JRW: Loss of heterozygosity on chromosome 16p 13.3 in hamartomes from tuberous sclerosis patients. Nature Gene! 6: 193- ¡96, 1994
40. Qian F, Watnick TJ, Onuchic LF, Germino GG: The molecular bases of cyst formation in human autosomal dominant polycystic kidney disease type 1. CeI187:979-987, J996
41. Brasier JL, Henske EP: Los> of the polycystic kidney dísease (PKDl) region of chromosome 16pl3 in renal cysts cells SupporlS a loss-of-function model for cyst pathogenesis. J Clín Invest 99: 194-199, 1997
42. Knudson AG: Mutution and cancer: Statistical study of retinoblastoma Proc Natl Acad Sci U S A 68:820-823, 1971
Copyright © 2022 FDOKUMEN




















