
Contributions to catalysis and potential interactions of the three catalytic domains in a contiguous...
9
Contributions to catalysis and potential interactions of the three catalytic domains in a contiguous trimeric creatine kinase Gregg G. Hoffman 1 , Omar Davulcu 2 , Sona Sona 1 and W. Ross Ellington 1,3 1 Department of Biological Science, Florida State University, Tallahassee, FL, USA 2 Department of Chemistry and Biochemistry, Florida State University, Tallahassee, FL, USA 3 Institute of Molecular Biophysics, Florida State University, Tallahassee, FL, USA Creatine kinase (CK) plays a central role in energy homeostasis in cells that display high or variable rates of ATP utilization, such as neurons, muscle fibers, transport epithelia and spermatozoa [1]. The physio- logical roles of the CK reaction are greatly facilitated by the presence of three nuclear gene families, each targeted to and localized in specific intracellular compartments – cytoplasmic (CyCK), mitochondrial (MtCK) and flagellar (FlgCK). Two of these isoforms, CyCK and MtCK, are oligomeric [2]. Both have been the subject of intensive research due to their physiolog- ical importance and their utility as models for under- standing bimolecular catalysis. CyCKs are obligate dimers, while most MtCKs function in an equilibrium of dimers and octamers, with the latter predominating under physiological conditions, at least in higher organisms [2]. This quaternary structure appears to be required for catalysis in both the cytoplasmic and mitochondrial isoforms, and there is compelling evi- dence indicating that the active sites do not function independently within a given oligomer [3–7]. FlgCKs exist as contiguous trimers, with three catalytically complete domains, each with its respective N- and C-domains, fused into a single polypeptide [8,9]. Struc- tural studies have not been conducted on FlgCKs, and the catalytic competence of individual domains and the potential interactions between domains remain unknown. Considerable effort has been focused on determining the physiological and functional importance of the quaternary structure in CyCKs and MtCKs, as oligo- merization is strongly correlated with intracellular localization in both [1,2]. The potential for interaction between adjacent subunits in CKs has been the source of much speculation, but recent X-ray crystallographic [5,6,10] and enzyme kinetics analyses of heterodimers Keywords contiguous trimer; cooperativity; domain interaction; flagellar creatine kinase; kinetics Correspondence W. R. Ellington, Institute of Molecular Biophysics, Florida State University, Tallahassee, FL, USA Fax: +1 850 644 0481 Tel: +1 850 644 5406 E-mail: [email protected] (Received 23 July 2007, revised 26 Novem- ber 2007, accepted 10 December 2007) doi:10.1111/j.1742-4658.2007.06226.x Three separate creatine kinase (CK) isoform families exist in animals. Two of these (cytoplasmic and mitochondrial) are obligate oligomers. A third, flagellar, is monomeric but contains the residues for three complete CK domains. It is not known whether the active sites in each of the contiguous flagellar domains are catalytically competent, and, if so, whether they are capable of acting independently. Here we have utilized site-directed muta- genesis to selectively disable individual active sites and all possible combi- nations thereof. Kinetic studies showed that these mutations had minimal impact on substrate binding and synergism. Interestingly, the active sites were not catalytically equivalent, and were in fact interdependent, a phenomenon that has previously been reported only in the oligomeric CK isoforms. Abbreviations AK, arginine kinase; CK, creatine kinase; CyCK, cytoplasmic CK; FlgCK, flagellar CK; k cat , catalytic turnover; MtCK, mitochondrial CK; PCr, phosphocreatine; TSAC, transition state analog complex. 646 FEBS Journal 275 (2008) 646–654 ª 2008 The Authors Journal compilation ª 2008 FEBS
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Contributions to catalysis and potential interactions of the three catalytic domains in a contiguous...

Contributions to catalysis and potential interactionsof the three catalytic domains in a contiguous trimericcreatine kinaseGregg G. Hoffman1, Omar Davulcu2, Sona Sona1 and W. Ross Ellington1,3
1 Department of Biological Science, Florida State University, Tallahassee, FL, USA
2 Department of Chemistry and Biochemistry, Florida State University, Tallahassee, FL, USA
3 Institute of Molecular Biophysics, Florida State University, Tallahassee, FL, USA
Creatine kinase (CK) plays a central role in energy
homeostasis in cells that display high or variable rates
of ATP utilization, such as neurons, muscle fibers,
transport epithelia and spermatozoa [1]. The physio-
logical roles of the CK reaction are greatly facilitated
by the presence of three nuclear gene families, each
targeted to and localized in specific intracellular
compartments – cytoplasmic (CyCK), mitochondrial
(MtCK) and flagellar (FlgCK). Two of these isoforms,
CyCK and MtCK, are oligomeric [2]. Both have been
the subject of intensive research due to their physiolog-
ical importance and their utility as models for under-
standing bimolecular catalysis. CyCKs are obligate
dimers, while most MtCKs function in an equilibrium
of dimers and octamers, with the latter predominating
under physiological conditions, at least in higher
organisms [2]. This quaternary structure appears to be
required for catalysis in both the cytoplasmic and
mitochondrial isoforms, and there is compelling evi-
dence indicating that the active sites do not function
independently within a given oligomer [3–7]. FlgCKs
exist as contiguous trimers, with three catalytically
complete domains, each with its respective N- and
C-domains, fused into a single polypeptide [8,9]. Struc-
tural studies have not been conducted on FlgCKs,
and the catalytic competence of individual domains
and the potential interactions between domains remain
unknown.
Considerable effort has been focused on determining
the physiological and functional importance of the
quaternary structure in CyCKs and MtCKs, as oligo-
merization is strongly correlated with intracellular
localization in both [1,2]. The potential for interaction
between adjacent subunits in CKs has been the source
of much speculation, but recent X-ray crystallographic
[5,6,10] and enzyme kinetics analyses of heterodimers
Keywords
contiguous trimer; cooperativity; domain
interaction; flagellar creatine kinase; kinetics
Correspondence
W. R. Ellington, Institute of Molecular
Biophysics, Florida State University,
Tallahassee, FL, USA
Fax: +1 850 644 0481
Tel: +1 850 644 5406
E-mail: [email protected]
(Received 23 July 2007, revised 26 Novem-
ber 2007, accepted 10 December 2007)
doi:10.1111/j.1742-4658.2007.06226.x
Three separate creatine kinase (CK) isoform families exist in animals. Two
of these (cytoplasmic and mitochondrial) are obligate oligomers. A third,
flagellar, is monomeric but contains the residues for three complete CK
domains. It is not known whether the active sites in each of the contiguous
flagellar domains are catalytically competent, and, if so, whether they are
capable of acting independently. Here we have utilized site-directed muta-
genesis to selectively disable individual active sites and all possible combi-
nations thereof. Kinetic studies showed that these mutations had minimal
impact on substrate binding and synergism. Interestingly, the active sites
were not catalytically equivalent, and were in fact interdependent, a
phenomenon that has previously been reported only in the oligomeric
CK isoforms.
Abbreviations
AK, arginine kinase; CK, creatine kinase; CyCK, cytoplasmic CK; FlgCK, flagellar CK; kcat, catalytic turnover; MtCK, mitochondrial CK;
PCr, phosphocreatine; TSAC, transition state analog complex.
646 FEBS Journal 275 (2008) 646–654 ª 2008 The Authors Journal compilation ª 2008 FEBS

of wild-type and inactive CK subunits [3,4,11] convinc-
ingly show that intra-oligomer interactions modulate
catalytic activity in a manner that has been described
as ‘flip-flop cooperativity’ in the case of chicken cyto-
plasmic CK [3,4,11].
Numerous approaches, including X-ray crystallogra-
phy [5,6], hydrogen ⁄deuterium exchange–mass spec-
trometry [12], small angle X-ray scattering [13] and
site-directed mutagenesis [14] have demonstrated that
CyCKs and MtCKs undergo substantial conforma-
tional changes upon transition from the open, sub-
strate-free state to the closed transition state analog
complex (TSAC) that is seen when MgADP, creatine
and nitrate are bound to CKs. This transition involves
the movement of two flexible loops (residues 60–72
and 323–333 in both Torpedo and rabbit CyCKs) and
at the N-terminus, over distances up to 19 A as the
molecule responds to occupancy of the active sites
[5,6]. The homodimeric apo-crystal structure of rabbit
muscle CK consists of two identical conformational
states for the monomeric subunits in the dimer [15]. In
contrast, the recently published crystal structure of the
TSAC of rabbit muscle CK [6] (and the TSAC struc-
ture for Torpedo [5]) is highly asymmetrical, with only
one of the monomers in the closed configuration.
When superimposed, these asymmetrical monomers
reveal significant movement of five structural elements,
which may explain the difference between the apo and
closed states [6].
These clear, large-scale and widely dispersed confor-
mational changes pose unique constraints upon any
tertiary structure that functionally competent contigu-
ous trimers of FlgCK may potentially adopt. This pos-
sibility raises some fundamental questions regarding
the connections between structure and catalysis in this
relatively unstudied molecule, i.e. how can loop move-
ment and intra-subunit communication be accommo-
dated in a contiguous trimer, and, if there are
constraints, do they have an impact on catalysis in
other domains or do the domains function indepen-
dently across the molecule?
To address the above issues, we have cloned and
expressed a 1167 residue FlgCK from the marine
worm Chaetopterus variopedatus (referred to here as
CVFlgCK), and utilized site-directed mutagenesis of
the active-site cysteine residue(s) to selectively eliminate
catalysis in each of the individual domains and in all
possible combinations of domains. Inactivation of this
cysteine has been shown to reduce catalytic turnover
(kcat) by > 99% compared with wild-type in sev-
eral CKs [16–18]. Our results show that the mutations,
with a few exceptions, had no significant effect on sub-
strate binding and synergism. Interestingly, while all
three CK domains were shown to be catalytically com-
petent, they were not equivalent in terms of catalytic
turnover rates. More importantly, the relative contri-
bution of any given active site depended on the cata-
lytic state of the active site within the remaining
domains. Both CyCK and MtCK have been shown to
undergo substantial conformational changes upon sub-
strate binding, and it is reasonable to expect that simi-
lar movements and interactions also occur in FlgCKs.
The catalytic non-equivalence reported here clearly
indicates that this is indeed the case, and that these
interactions may be representative of a suite of inter-
actions and structural changes that are required for
catalysis across this entire enzyme family.
Results and Discussion
FlgCKs lack quaternary structure and are monomers
that contain three apparently complete CK domains.
Recently, a number of other enzymes with multiple
catalytic domains have been identified – two-domain
arginine kinases [19–21], a two-domain carbonic anhy-
drase [22], a three-domain luciferase [23] and a three-
domain adenylate kinase [24]. The present study
provides insight into the inter-dependent functional
properties of the three domains of FlgCK, and lays
the groundwork for study of the relationship between
these functional properties and the structural interac-
tions that potentially mediate them.
Analysis of the primary structures of the three
FlgCK domains
Two CK TSAC crystal structures have been published
(Torpedo and rabbit muscle). Both have one subunit in
a quasi-open, binary complex with MgADP and one in
a closed TSAC with MgADP, creatine and nitrate
[5,6]. This active-site asymmetry occurs even though
the crystals for both Torpedo and rabbit muscle CK
were grown under conditions that would strongly
favor TSAC formation, indicating that, at least in mul-
timeric CKs, only one monomer within a given dimer
can form the TSAC, or that formation of this TSAC
somehow stabilizes the open state of the adjoining
active site or precludes binding of all components to
form a TSAC. Comparison of the two monomers
within a given isoform reveals that two sets of confor-
mational changes are potentially important for cataly-
sis and inter-subunit communication; the first involves
movements within the two loops that act to control
access to the active site(s), and the second involves a
significant structural change within the first 20 N-ter-
minal residues.
G. G. Hoffman et al. Catalysis in a contiguous trimeric creatine kinase
FEBS Journal 275 (2008) 646–654 ª 2008 The Authors Journal compilation ª 2008 FEBS 647

Figure 1 shows a multiple sequence alignment in
which the sequences of the three contiguous domains
of FlgCK (ChaetFlgD1–3) are aligned with the
sequences of Torpedo and rabbit muscle CK mono-
mers. The flexible loops, key catalytic residues and a
conserved proline that seems in the rabbit crystal
structure to act as a hinge point when the N-terminal
undergoes conformational changes upon conversion to
the TSAC are indicated (many of the N-terminal resi-
dues in the Torpedo structure were not well resolved
[5] and were excluded from the final model). The speci-
ficity loop (creatine binding pocket, residues 60–72 in
Torpedo) is nearly identical in all five CK domains,
and the nucleotide binding loop (323–335 in Torpedo)
is quite similar (shown in blue in Fig. 1). The key cata-
lytic residues identified in Torpedo CK are conserved
in all three FlgCK domains (shown in red in Fig. 1),
as is the ‘hinge’ proline (position 21 in Torpedo, show
in pink in Fig. 1).
Based on the above comparisons, it appears that all
three FlgCK domains have the requisite elements for
catalysis and are at least capable of the same types of
structural interactions described for oligomeric CK iso-
forms. It is important to note that these isoforms have
Fig. 1. Multiple sequence alignment of the
sequences for Torpedo [5] and rabbit mus-
cle [6] CKs and each of the three FlgCK
domains (ChaetFlgCKD1–3). Residues
directly implicated in catalysis are shown in
red, the flexible loops that have been shown
to undergo conformational changes upon
substrate binding are shown in blue, and
the N-terminal ‘hinge’ proline is shown in
pink. The highly conserved reactive cysteine
residues that were the mutagenic target of
this study are shown in green.
Catalysis in a contiguous trimeric creatine kinase G. G. Hoffman et al.
648 FEBS Journal 275 (2008) 646–654 ª 2008 The Authors Journal compilation ª 2008 FEBS

conserved this sequence similarity for as long as
675 million years, when Chaetopterus (a lophotrocozo-
an invertebrate) last shared a common ancestor with
the deuterostomes [25]. This suggests that these struc-
tural elements play an important functional role in this
enzyme system.
Expression of wild-type and mutant FlgCKs
Seven mutant constructs were engineered using the
wild-type C. variopedatus FlgCK as the platform. All
mutations involved conversion of the reactive cysteine
residue within a domain (C299, C667 and C1052 in
CVFlgCK; see Fig. 1), or a combination of domains,
to serine. In this context, each FlgCK domain will be
referred to as D1, D2 and D3, respectively. Previous
work has shown that this cysteine to serine mutation
dramatically reduces enzyme activity in the reverse cat-
alytic direction, especially at low Cl) concentrations
[4,16,26,27]. The following combinations of mutated
domains were constructed: D1SD2D3, D1D2SD3,
D1D2D3S, D1D2SD3S, D1SD2 D3S, D1SD2SD3 and
D1SD2SD3S (where the subscript S corresponds to the
C fi S mutant and no subscript corresponds to a wild-
type domain). Expression of wild-type FlgCK and sin-
gle and double C fi S FlgCK mutants yielded large
amounts of soluble, recombinant protein that was eas-
ily purified to homogeneity by low-pressure chroma-
tography. As expected, expression of the triple mutant
(D1SD2SD3S) yielded CK with dramatically reduced
activity. In fact, it was necessary to significantly con-
centrate the purified protein from 2 L of bacterial
culture to obtain sufficient recombinant triple
mutant CK for kinetic analyses.
Kinetic analysis of wild-type and mutant flgCKs
Binary (KS) and ternary (KM) substrate-binding con-
stants for both ADP and phosphocreatine (PCr), as
well as the substrate-binding synergism (KS ⁄KM), were
determined for the wild-type and the seven C fi S
mutant constructs. With only a few exceptions, muta-
tion of the reactive cysteine had no significant impact
on KS or KM values for the recombinant flgCKs
(Table 1). There was a significant decrease of KS(PCr) in
the D1D2SD3S mutant as well as of the KS(ADP) and
KM(ADP) values for the triple mutant. The wild-type
and all mutant constructs demonstrated very limited
substrate-binding synergism as evidenced by KS ⁄KM
values slightly above unity. Synergy values for the
mutants were not significantly different from those of
the wild-type. Overall, our results show that C fi S
mutations in the FlgCK domains, individually and in
combination, had little impact on substrate binding in
the reverse catalytic direction. This has also been
observed for chicken [4] and human [27] cytoplas-
mic CKs. Interestingly, this is not the case for octa-
meric mitochondrial CK, where an 11-fold increase in
KM(PCr) was reported for the C fi S mutant [16]. Our
values for ADP binding in the wild-type CVFlgCK
contigious trimer are similar to but somewhat lower
than those reported for the oligomeric CK isoforms.
KM(ADP) values range from 150 lm for MtCK octa-
mers [2] to between 190 and 440 lm for Cy CK
dimers. This trend is more pronounced for PCr bind-
ing. Tombes and Shapiro reported a KM(PCr) value that
is twice that reported here [28].
In contrast to our substrate-binding parameters,
the cysteine mutations in the domains of FlgCK were
observed to have a profound impact on catalytic
rates and relative efficiency (Table 2 and Fig. 2). Pre-
vious work has shown that inactivation of the reac-
tive cysteine produces a dramatic reduction in Vmax
and kcat for a variety of CKs [4,17,26,27], and our
triple mutant, as expected, displayed very limited
activity as evidenced by very low Vmax and kcat val-
ues (Table 2 and Fig. 2). If each CK domain of the
FlgCK has an equal potential for catalytic rate
enhancement, then it might be anticipated
that C fi S mutations in individual domains and
Table 1. Kinetic parameters for wild-type and mutant FlgCK constructs. Values represent mean ± 1 SD (n = 3).
Construct KS(ADP) (lM) KM(ADP) (lM) KS(PCr) (mM) KM(PCr) (mM) KS ⁄ KM
Wild-type 151 ± 49.3 100 ± 0.9 3.1 ± 0.6 2.2 ± 0.5 1.4 ± 0.4
D1SD2D3 178 ± 79.8 123 ± 28.4 2.9 ± 0.8 2.1 ± 0.5 1.4 ± 0.7
D1D2SD3 109 ± 26.7 96 ± 3.3 2.1 ± 0.4 1.9 ± 0.2 1.1 ± 0.3
D1D2D3S 163 ± 13.3 104 ± 11.5 2.5 ± 0.4 1.6 ± 0.2 1.6 ± 0.3
D1SD2D3S 138 ± 41.7 90 ± 12.5 3.0 ± 0.7 2.0 ± 0.2 1.5 ± 0.2
D1D2SD3S 137 ± 33.5 112 ± 15.8 1.6 ± 0.2a 1.4 ± 0.4 1.2 ± 0.4
D1SD2SD3 173 ± 9.7 123 ± 23.2 2.5 ± 0.1 1.7 ± 0.3 1.4 ± 0.1
D1SD2SD3S 54 ± 4.5a 52 ± 4.1a 3.0 ± 0.4 2.9 ± 0.3 1.0 ± 0.1
a Values that are significantly different from wild-type (P < 0.05).
G. G. Hoffman et al. Catalysis in a contiguous trimeric creatine kinase
FEBS Journal 275 (2008) 646–654 ª 2008 The Authors Journal compilation ª 2008 FEBS 649
combinations of domains will produce proportionate
decreases in catalytic turnover.
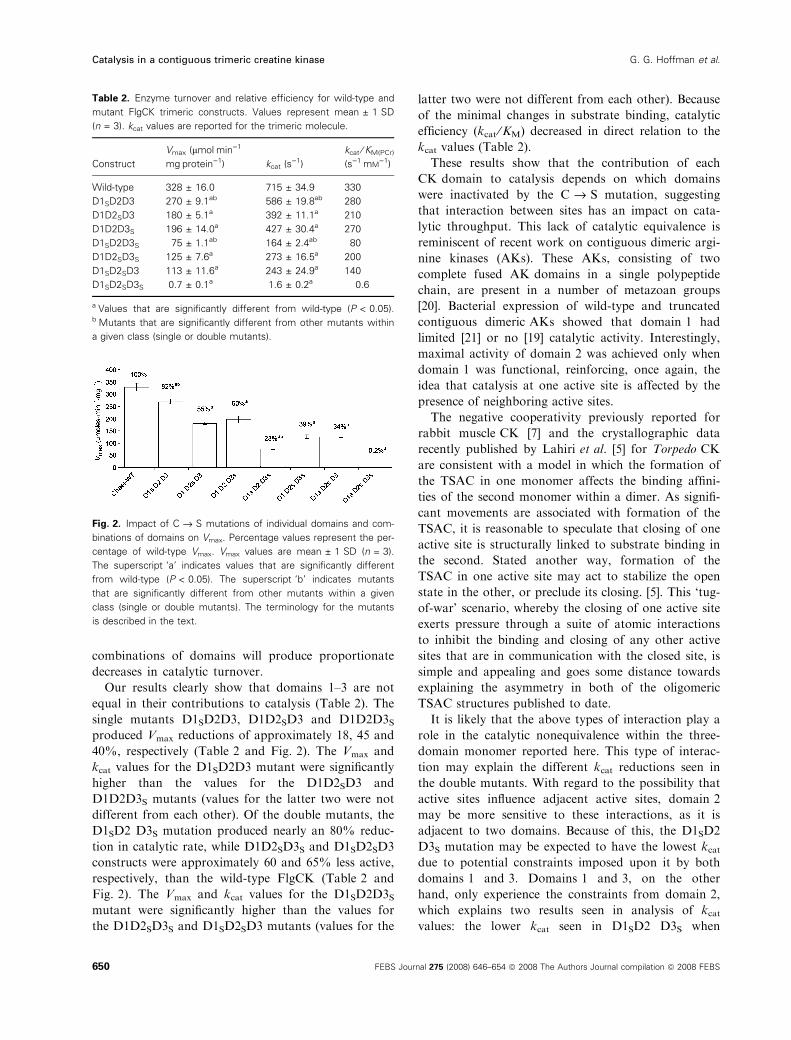
Our results clearly show that domains 1–3 are not
equal in their contributions to catalysis (Table 2). The
single mutants D1SD2D3, D1D2SD3 and D1D2D3Sproduced Vmax reductions of approximately 18, 45 and
40%, respectively (Table 2 and Fig. 2). The Vmax and
kcat values for the D1SD2D3 mutant were significantly
higher than the values for the D1D2SD3 and
D1D2D3S mutants (values for the latter two were not
different from each other). Of the double mutants, the
D1SD2 D3S mutation produced nearly an 80% reduc-
tion in catalytic rate, while D1D2SD3S and D1SD2SD3
constructs were approximately 60 and 65% less active,
respectively, than the wild-type FlgCK (Table 2 and
Fig. 2). The Vmax and kcat values for the D1SD2D3Smutant were significantly higher than the values for
the D1D2SD3S and D1SD2SD3 mutants (values for the
latter two were not different from each other). Because
of the minimal changes in substrate binding, catalytic
efficiency (kcat ⁄KM) decreased in direct relation to the
kcat values (Table 2).
These results show that the contribution of each
CK domain to catalysis depends on which domains
were inactivated by the C fi S mutation, suggesting
that interaction between sites has an impact on cata-
lytic throughput. This lack of catalytic equivalence is
reminiscent of recent work on contiguous dimeric argi-
nine kinases (AKs). These AKs, consisting of two
complete fused AK domains in a single polypeptide
chain, are present in a number of metazoan groups
[20]. Bacterial expression of wild-type and truncated
contiguous dimeric AKs showed that domain 1 had
limited [21] or no [19] catalytic activity. Interestingly,
maximal activity of domain 2 was achieved only when
domain 1 was functional, reinforcing, once again, the
idea that catalysis at one active site is affected by the
presence of neighboring active sites.
The negative cooperativity previously reported for
rabbit muscle CK [7] and the crystallographic data
recently published by Lahiri et al. [5] for Torpedo CK
are consistent with a model in which the formation of
the TSAC in one monomer affects the binding affini-
ties of the second monomer within a dimer. As signifi-
cant movements are associated with formation of the
TSAC, it is reasonable to speculate that closing of one
active site is structurally linked to substrate binding in
the second. Stated another way, formation of the
TSAC in one active site may act to stabilize the open
state in the other, or preclude its closing. [5]. This ‘tug-
of-war’ scenario, whereby the closing of one active site
exerts pressure through a suite of atomic interactions
to inhibit the binding and closing of any other active
sites that are in communication with the closed site, is
simple and appealing and goes some distance towards
explaining the asymmetry in both of the oligomeric
TSAC structures published to date.
It is likely that the above types of interaction play a
role in the catalytic nonequivalence within the three-
domain monomer reported here. This type of interac-
tion may explain the different kcat reductions seen in
the double mutants. With regard to the possibility that
active sites influence adjacent active sites, domain 2
may be more sensitive to these interactions, as it is
adjacent to two domains. Because of this, the D1SD2
D3S mutation may be expected to have the lowest kcatdue to potential constraints imposed upon it by both
domains 1 and 3. Domains 1 and 3, on the other
hand, only experience the constraints from domain 2,
which explains two results seen in analysis of kcatvalues: the lower kcat seen in D1SD2 D3S when
Table 2. Enzyme turnover and relative efficiency for wild-type and
mutant FlgCK trimeric constructs. Values represent mean ± 1 SD
(n = 3). kcat values are reported for the trimeric molecule.
Construct
Vmax (lmolÆmin)1Æ
mgÆprotein)1) kcat (s)1)
kcat ⁄ KM(PCr)
(s)1ÆmM)1)
Wild-type 328 ± 16.0 715 ± 34.9 330
D1SD2D3 270 ± 9.1ab 586 ± 19.8ab 280
D1D2SD3 180 ± 5.1a 392 ± 11.1a 210
D1D2D3S 196 ± 14.0a 427 ± 30.4a 270
D1SD2D3S 75 ± 1.1ab 164 ± 2.4ab 80
D1D2SD3S 125 ± 7.6a 273 ± 16.5a 200
D1SD2SD3 113 ± 11.6a 243 ± 24.9a 140
D1SD2SD3S 0.7 ± 0.1a 1.6 ± 0.2a 0.6
a Values that are significantly different from wild-type (P < 0.05).b Mutants that are significantly different from other mutants within
a given class (single or double mutants).
Fig. 2. Impact of C fi S mutations of individual domains and com-
binations of domains on Vmax. Percentage values represent the per-
centage of wild-type Vmax. Vmax values are mean ± 1 SD (n = 3).
The superscript ‘a’ indicates values that are significantly different
from wild-type (P < 0.05). The superscript ‘b’ indicates mutants
that are significantly different from other mutants within a given
class (single or double mutants). The terminology for the mutants
is described in the text.
Catalysis in a contiguous trimeric creatine kinase G. G. Hoffman et al.
650 FEBS Journal 275 (2008) 646–654 ª 2008 The Authors Journal compilation ª 2008 FEBS

compared with D1D2SD3S and D1SD2SD3, and the
similar kcat values seen in D1D2SD3S and D1SD2SD3.
Given the wealth of structural data available, it is
surprising that little evidence for a structural network
such as that described above exists for CK. An intrigu-
ing alternative to the classical model of multidomain
interactions has been proposed by Hawkins and
McLeish [29]. They present a model in which allostery
arises from coupling of changes in local vibrational
modes to changes in global entropy, in which altera-
tions in protein flexibility upon ligand binding at one
site affect the entropic cost of binding at neighboring
sites. This idea stems from the fact that proteins exist as
dynamic ensembles of conformational states, and ligand
binding redistributes the population within the ensem-
ble, leading to altered conformations at other, some-
times distant, sites [29,30]. These potentially distal sites
may also experience an increase in flexibility, which,
together with enthalpic contributions such as hydrogen
bond formation between substrate and enzyme, may
serve to partially offset the loss in entropy that accom-
panies substrate binding. This increase in flexibility,
however, may also have the side effect of impeding
binding in adjacent active sites, essentially allowing only
one of a set of interacting active sites to complete a
catalytic cycle at a time. Further understanding of
catalysis and the interaction of active sites in these
unique contiguous trimeric FlgCKs will depend on the
outcome of on-going studies of expressed truncated
contiguous dimers and monomers, as well as X-ray
crystallographic determination of the atomic structure.
Experimental procedures
Amplification of full-length FlgCK cDNA
Chaetopterus variopedatus mRNA previously isolated by
our group [8] was used to amplify, clone and sequence the
FlgCK cDNA full-length transcript. Briefly, single-stranded
cDNA was reverse-transcribed using Ready-to-Go You
Prime beads (GE Healthcare, Piscataway, NY, USA) and a
lock-docking oligo(dT) reverse primer [31] according to the
manufacturer’s instructions. The full-length cDNA was
produced and PCR-amplified in a Hybaid PCR Sprint
thermocycler (Ashford, UK) using gene-specific primers
designed to amplify the full-length coding sequence from
the start to the stop codon using PfuTurbo Hotstart DNA
polymerase (Stratagene, La Jolla, CA, USA). PCR amplifi-
cation was carried out using a 1.5 min incubation at 95 �C,followed by 17 cycles of 95 �C for 40 s, 60 �C for 40 s, and
68 �C for 16 min. A single PCR product was produced,
and this was gel-purified using a QiaQuick spin kit (Qiagen,
Valencia, CA, USA). This product was subcloned into a
puC19 TA (TOPO) cloning vector (Invitrogen, Carlsbad,
CA, USA), and plasmids from two independent clones were
completely sequenced in both directions on an automated
Applied Biosystems model 3100 genetic analyzer (Foster
City, CA, USA).
Expression and purification of recombinant
protein
The sequence-verified full-length CVFlgCK cDNA was
ligated into the pETBlue1 vector system (EMD Bioscienc-
es ⁄Novagen, La Jolla, CA, USA), and used to transform
BL21 Tuner(DE3)-pLacI expression hosts (Novagen)
according to the manufacturer’s instructions. Recombinant
FlgCK was expressed according to the protocol used for
other invertebrate CKs [32,33]. Bacteria were harvested by
centrifugation at 4 �C for 15 min at 17 000 g. The pelleted
cells were resuspended in lysis buffer (50 mm Tris, 300 mm
NaCl, 5 mm EDTA, pH 7.8) using a Polytron homogenizer
(Brinkman, Westbury, NY, USA), and then lysed using 100
cycles of microfluidization (Microfluidics, Newton, MA) in
N2 gas. Cellular debris was pelleted by centrifugation at
23 000 g for 20 min at 4 �C. CK expression was verified
using a reverse-direction (PCr fi ATP) spectrophotomet-
ric assay as previously described [34]. Expression of recom-
binant wild-type FlgCK yielded substantial levels of soluble
enzyme activity.
Wild-type and mutant constructs of CVFlgCK were all
easily purified from cellular lysates using two rounds of low-
pressure chromatography. Lysates were exhaustively dia-
lyzed against DEAE running buffer (10 mm Tris, 0.5 mm
EDTA, 1 mm DTT at pH 8.1), briefly centrifuged at 4 �Cfor 15 min at 23 000 g, and then applied to a 40 mL DEAE–
Sepharose Fast Flow column(GE Biotech, Piscataway, NJ,
USA) equilibrated with running buffer. After washing, pro-
teins were eluted with a 400 mL linear gradient of NaCl
(from 0 to 250 mm in running buffer). Fractions showing
CK activity were pooled, exhaustively dialyzed against
hydroxyapatite running buffer (5 mm potassium phosphate,
1 mm DTT at pH 7.0), and applied to an 80 mL Bio-Gel HT
hydroxyapatite column (Bio-Rad Laboratories, Hercules,
CA, USA). After washing, proteins were eluted with a
400 mL linear gradient of 5–400 m potassium phosphate
(pH 7.0). For each construct, active hydroxyapatite fractions
were analyzed by SDS–PAGE [35]. FlgCK fractions were
pooled and concentrated using pressure filtration. Protein
content was determined using a Bio-Rad protein assay kit
based on the Bradford method [36], using bovine serum
albumin as the standard. The resulting FlgCK preparations
were essentially homogeneous.
Site-directed mutagenesis
As Fig. 2 clearly shows, the residues surrounding the reac-
tive cysteines are highly conserved in all three FlgCK
G. G. Hoffman et al. Catalysis in a contiguous trimeric creatine kinase
FEBS Journal 275 (2008) 646–654 ª 2008 The Authors Journal compilation ª 2008 FEBS 651

domains; therefore, they could not be directly mutated in
the full-length expression vector as the mutagenic primers
would not be domain-specific. Thus, each of the domains
was excised using restriction enzymes, ligated into TOPO
cloning vectors and mutated. The mutated construct was
then excised and re-ligated back into the original expression
vector containing the two non-mutated domains. The fol-
lowing restriction enzymes were used to separate individual
domains: D1, MfeI and XhoI; D2, XhoI and AatII; D3,
AatII and AvrII. PCR using Ex Taq HS polymerase (Taka-
ra USA, Santa Ana, CA, USA) was performed to fill in the
sticky ends and add adenine nucleotide overhangs before
ligating the individual domains into the TOPO vectors
using the primers listed in Table 3.
Mutations were carried out using the QuikChange muta-
genesis kit (Stratagene) according to the manufacturer’s
protocol. The specific primers used for the mutation(s) are
listed in Table 3. Briefly, the template plasmid was ampli-
fied using PfuUltra Hotstart DNA polymerase (Stratagene)
with a forward primer and its reverse complement, both
coding for the target mutation. The original methylated
template plasmid was digested using the restriction enzyme
DpnI by incubating at 37 �C for 1 h. The amplified plasmid
was then transformed into Escherichia coli XL1-Blue super-
competent cells (Stratagene) according to the manufac-
turer’s protocol. Carbenicillin-resistant transformed cells
were plated, and plasmids were isolated from overnight cul-
tures grown from single colonies. These plasmids were iso-
lated using a Qiagen QIAprep Spin Miniprep kit. The
mutant inserts were verified by sequencing and manipulated
as described above. The site-directed mutants were
expressed and purified to homogeneity as for the wild-type.
The purity and protein content of the mutant FlgCKs were
determined as for the wild-type preparation. All mutant
constructs yielded active soluble protein, although the
D1SD2SD3S mutant had minimal catalytic activity.
Enzyme kinetics
Kinetic assays were run on a Cary 100 UV–visible spectro-
photometer (Varian, Walnut Creek, CA, USA) using the
manufacturer’s software. Initial velocity values were deter-
mined for the reverse reaction by varying the concentration
of one substrate versus six fixed concentrations of the sec-
ond substrate and vice versa, resulting in a 6 · 6 matrix.
Actual concentrations of both substrates were empirically
determined by enzymatic standardization (for PCr) and
spectrophotometric standardization (for ADP). Magnesium
acetate was added to a concentration of 1 mm above the
concentration of ADP to ensure full saturation of ADP
by Mg2+. Assay buffer (100 mm Na-HEPES, pH 7) was
added to each 3 mL cuvette to bring the total reaction
volume to 2.5 mL. All assays were run at 25 �C and were
nominally Cl)-free to maximize the inhibitory impact of
the C fi S mutation. Kinetic rate measurements were fit to
the following rate equation for a random order, sequential,
bimolecular–bimolecular reaction mechanism using non-
linear least-squares regression [37]:
m¼Vmax½PCr�½ADP�
aKSðPCrÞKSðADPÞ þaKSðPCrÞ½ADP�þaKSðADPÞ½PCr�þ ½PCr�½ADP�
Vmax, KS(PCr), KS(ADP) and a were simultaneously deter-
mined. KS(PCr) and KS(ADP) are the dissociation constants of
phosphocreatine and ADP binary complexes, respectively.
KM, the dissociation constant for the Michaelis complex
with both phosphocreatine and ADP bound, was deter-
mined from the relationship KM = a(KS). Vmax is expressed
as specific activity, and kcat is calculated from Vmax using
molecular mass and a conversion from minutes to seconds.
Errors of mean values for each parameter were determined
as the standard deviation of the triplicate set. Data analyses
were performed using sigmaplot (SPSS, Chicago, IL,
USA).
Acknowledgments
This research was supported by National Science
Foundation grants IOB-0130024 and IOB-0542236 to
WRE and National Institutes of Health grant R01-
GM077643 to OD. We thank the staff of the DNA
Sequencing and Molecular Cloning facilities for their
assistance.
References
1 Ellington WR (2001) Evolution and physiological roles
of phosphagen systems. Annu Rev Physiol 63, 289–325.
2 Wallimann T, Wyss M, Brdiczka D, Nicolay K &
Eppenberger HM (1992) Intracellular compartmenta-
tion, structure and function of creatine kinase
Table 3. Primers used for filling in and for C fi S mutation of indi-
vidual FlgCK domains.
Primer name Sequence (5¢- to 3¢)
PCR primers
D1 forward GAG CAC AAC AAT TGG ATG GCCD1 reverse CCT TTC TCG AGT CTC TTC TCCD2 forward GAG ACT CGA GAA AGG AGA GGD2 reverse GGC AGA CGT CAG CAG TGGD3 forward CCA CTG CTG ACG TCT GCCD3 reverse ATC AGC CTA GGC CCT TTC GTC
QuikChange mutagenic primers
D1 forward CAT CCA CAC GTC CCC CAG TAA CTT AGGD1 reverse CCT AAG TTA CTG GGG GAC GTG TGG ATGD2 forward CGT GCT GAC ATC CCC CAG CAA CCT GGGD2 reverse CCC AGG TTG CTG GGG GAT GTC AGC ACGD3 forward CAT CCT GAC CTC CCC TAG CAA CCT GGGD3 reverse CCC AGG TTG CTA GGG GAG GTC AGG ATG
Catalysis in a contiguous trimeric creatine kinase G. G. Hoffman et al.
652 FEBS Journal 275 (2008) 646–654 ª 2008 The Authors Journal compilation ª 2008 FEBS

isoenzymes in tissues with high and fluctuating energy
demands: the ‘phosphocreatine circuit’ for cellular
energy homeostasis. Biochem J 281, 21–40.
3 Grossman SH & Sellers DS (1998) Subunit conforma-
tion and dynamics in a heterodimeric protein: studies of
the hybrid isozyme of creatine kinase. Biochim Biophys
Acta 1387, 447–453.
4 Hornemann T, Rutishauser D & Wallimann T (2000)
Why is creatine kinase a dimer? Evidence for cooper-
ativity between the two subunits. Biochim Biophys Acta
1480, 365–373.
5 Lahiri SD, Wang PF, Babbitt PC, McLeish MJ,
Kenyon GL & Allen KN (2002) The 2.1 A structure of
Torpedo californica creatine kinase complexed with the
ADP-Mg2+-NO3)-creatine transition-state analogue
complex. Biochemistry 41, 13861–13867.
6 Ohren JF, Kundracik ML, Borders CL Jr, Edmiston P
& Viola RE (2007) Structural asymmetry and intersub-
unit communication in muscle creatine kinase. Acta
Crystallogr D Biol Crystallogr 63, 381–389.
7 Price NC & Hunter MG (1976) Non-identical behaviour
of the subunits of rabbit muscule creatine kinase. Bio-
chim Biophys Acta 445, 364–376.
8 Suzuki T, Mizuta C, Uda K, Ishida K, Mizuta K, Sona
S, Compaan DM & Ellington WR (2004) Evolution
and divergence of the genes for cytoplasmic, mito-
chondrial, and flagellar creatine kinases. J Mol Evol 59,
218–226.
9 Wothe DD, Charbonneau H & Shapiro BM (1990) The
phosphocreatine shuttle of sea urchin sperm: flagellar
creatine kinase resulted from a gene triplication. Proc
Natl Acad Sci USA 87, 5203–5207.
10 Eder M, Schlattner U, Becker A, Wallimann T, Kabsch
W & Fritz-Wolf K (1999) Crystal structure of brain-
type creatine kinase at 1.41 A resolution. Protein Sci 8,
2258–2269.
11 Lin L, Perryman MB, Friedman D, Roberts R & Ma
TS (1994) Determination of the catalytic site of creatine
kinase by site-directed mutagenesis. Biochim Biophys
Acta 1206, 97–104.
12 Mazon H, Marcillat O, Forest E & Vial C (2003)
Changes in MM-CK conformational mobility upon for-
mation of the ADP-Mg2+-NO3)-creatine transition
state analogue complex as detected by hydrogen ⁄ deute-rium exchange. Biochemistry 42, 13596–13604.
13 Forstner M, Muller A, Rognan D, Kriechbaum M &
Wallimann T (1998) Mutation of cis-proline 207 in
mitochondrial creatine kinase to alanine leads to
increased acid stability. Protein Eng 11, 563–568.
14 Uda K & Suzuki T (2004) Role of amino acid residues
on the GS region of Stichopus arginine kinase and
Danio creatine kinase. Protein J 23, 53–64.
15 Rao JK, Bujacz G & Wlodawer A (1998) Crystal struc-
ture of rabbit muscle creatine kinase. FEBS Lett 439,
133–137.
16 Furter R, Furter-Graves EM & Wallimann T (1993)
Creatine kinase: the reactive cysteine is required for
synergism but is nonessential for catalysis. Biochemistry
32, 7022–7029.
17 Reddy S, Jones AD, Cross CE, Wong PS & Van Der
Vliet A (2000) Inactivation of creatine kinase by
S-glutathionylation of the active-site cysteine residue.
Biochem J 347, 821–827.
18 Tanaka N, Tonai T & Kunugi S (1997) Site-specific
modification of rabbit muscle creatine kinase with sulf-
hydryl-specific fluorescence probe by use of hydrostatic
pressure. Biochim Biophys Acta 1339, 226–232.
19 Compaan DM & Ellington WR (2003) Functional con-
sequences of a gene duplication and fusion event in an
arginine kinase. J Exp Biol 206, 1545–1556.
20 Suzuki T, Kawasaki Y, Unemi Y, Nishimura Y, Soga
T, Kamidochi M, Yazawa Y & Furukohri T (1998)
Gene duplication and fusion have occurred frequently
in the evolution of phosphagen kinases – a two-domain
arginine kinase from the clam Pseudocardium sachalin-
ensis. Biochim Biophys Acta 1388, 253–259.
21 Suzuki T, Tomoyuki T & Uda K (2003) Kinetic proper-
ties and structural characteristics of an unusual two-
domain arginine kinase of the clam Corbicula japonica.
FEBS Lett 533, 95–98.
22 Leggat W, Dixon R, Saleh S & Yellowlees D (2005) A
novel carbonic anhydrase from the giant clam Tridacna
gigas contains two carbonic anhydrase domains. FEBS
J 272, 3297–3305.
23 Liu L, Wilson T & Hastings JW (2004) Molecular evo-
lution of dinoflagellate luciferases, enzymes with three
catalytic domains in a single polypeptide. Proc Natl
Acad Sci USA 101, 16555–16560.
24 Kinukawa M, Nomura M & Vacquier VD (2007) A sea
urchin sperm flagellar adenylate kinase with triplicated
catalytic domains. J Biol Chem 282, 2947–2955.
25 Doolittle RF, Feng DF, Tsang S, Cho G & Little E
(1996) Determining divergence times of the major king-
doms of living organisms with a protein clock. Science
271, 470–477.
26 Buechter DD, Medzihradszky KF, Burlingame AL &
Kenyon GL (1992) The active site of creatine kinase.
Affinity labeling of cysteine 282 with N-(2,3-epoxy-
propyl)-N-amidinoglycine. J Biol Chem 267, 2173–
2178.
27 Wang PF, McLeish MJ, Kneen MM, Lee G & Kenyon
GL (2001) An unusually low pK(a) for Cys282 in the
active site of human muscle creatine kinase. Biochemis-
try 40, 11698–11705.
28 Tombes RM & Shapiro MM (1987) Enzyme termini of
a phosphocreatine shuttle. J Biol Chem 262, 16011–
16019.
29 Hawkins RJ & McLeish TC (2006) Coupling of global
and local vibrational modes in dynamic allostery of
proteins. Biophys J 91, 2055–2062.
G. G. Hoffman et al. Catalysis in a contiguous trimeric creatine kinase
FEBS Journal 275 (2008) 646–654 ª 2008 The Authors Journal compilation ª 2008 FEBS 653

30 Gunasekaran K, Ma B & Nussinov R (2004) Is allo-
stery an intrinsic property of all dynamic proteins?
Proteins 57, 433–443.
31 Borson ND, Salo WL & Drewes LR (1992) A lock-
docking oligo(dT) primer for 5’ and 3’ RACE PCR.
PCR Methods Appl 2, 144–148.
32 Hoffman GG, Sona S, Bertin M & Ellington WR (2006)
The role of an absolutely conserved tryptophan residue
in octamer formation and stability in mitochondrial crea-
tine kinases. Biochim Biophys Acta 1764, 1512–1517.
33 Sona S, Suzuki T & Ellington WR (2004) Cloning and
expression of mitochondrial and protoflagellar creatine
kinases from a marine sponge: implications for the ori-
gin of intracellular energy transport systems. Biochem
Biophys Res Commun 317, 1207–1214.
34 Strong SJ & Ellington WR (1996) Expression of
horseshoe crab arginine kinase in Escherichia coli and
site-directed mutations of the reactive cysteine peptide.
Comp Biochem Physiol B Biochem Mol Biol 113,
809–816.
35 Laemmli UK (1970) Cleavage of structural proteins
during the assembly of the head of bacteriophage T4.
Nature 227, 680–685.
36 Bradford MM (1976) A rapid and sensitive method for
the quantitation of microgram quantities of protein
utilizing the principle of protein–dye binding. Anal
Biochem 72, 248–254.
37 Segel IH (1975) Enzyme Kinetics – Behavior and
Analysis of Rapid Equilibrium and Steady-State Enzyme
Systems. Wiley, New York, NY.
Catalysis in a contiguous trimeric creatine kinase G. G. Hoffman et al.
654 FEBS Journal 275 (2008) 646–654 ª 2008 The Authors Journal compilation ª 2008 FEBS




















