
Creatine supplementation prevents fatty liver in rats fed choline-deficient diet: a burden of...
7
Creatine supplementation prevents fatty liver in rats fed choline-deficient diet: a burden of one-carbon and fatty acid metabolism Rafael Deminice a,c, ⁎, Gabriela Salim Ferreira de Castro a,1 , Lucas Vieira Francisco a,1 , Lilian Eslaine Costa Mendes da Silva a,1 , João Felipe Rito Cardoso a,1 , Fernando Tadeu Trevisan Frajacomo a,1 , Bruno Gonzaga Teodoro d , Leonardo dos Reis Silveira b,d,1 , Alceu Afonso Jordao a,1 a Nutrition and Metabolism, Faculty of Medicine of Ribeirao Preto, University of Sao Paulo, Av. Bandeirantes 3900, Ribeirao Preto, Sao Paulo, Brazil b School of Physical Education of Ribeirao Preto, University of Sao Paulo, Av. Bandeirantes 3900, Ribeirao Preto, Sao Paulo, Brazil c Department of Physical Education, Faculty of Physical Education and Sport, State University of Londrina. Rodovia Celso Garcia Cid | Pr 445 Km 380 | Campus Universitário, Londrina, Paraná, Brazil d Department of Biochesmtry and Imunology, Faculty of Medicine of Ribeirão Preto Av. Bandeirantes 3900, Ribeirao Preto, Sao Paulo, Brazil Received 4 August 2014; received in revised form 5 November 2014; accepted 19 November 2014 Abstract Aim: To examine the effects of creatine (Cr) supplementation on liver fat accumulation in rats fed a choline-deficient diet. Methods: Twenty-four rats were divided into 3 groups of 8 based on 4 weeks of feeding an AIN-93 control diet (C), a choline-deficient diet (CDD) or a CDD supplemented with 2% Cr. The CDD diet was AIN-93 without choline. Results: The CDD significantly increased plasma homocysteine and TNFα concentration, as well as ALT activity. In liver, the CDD enhanced concentrations of total fat (55%), cholesterol (25%), triglycerides (87%), MDA (30%), TNFα (241%) and decreased SAM concentrations (25%) and the SAM/SAH ratio (33%). Cr supplementation prevented all these metabolic changes, except for hepatic SAM and the SAM/SAH ratio. However, no changes in PEMT gene expression or liver phosphatidylcholine levels were observed among the three experimental groups, and there were no changes in hepatic triglyceride transfer protein (MTP) mRNA level. On the contrary, Cr supplementation normalized expression of the transcription factors PPARα and PPARγ that were altered by the CDD. Further, the downstream targets and fatty acids metabolism genes, UCP2, LCAD and CPT1a, were also normalized in the Cr group as compared to CDD-fed rats. Conclusion: Cr supplementation prevented fat liver accumulation and hepatic injures in rats fed with a CDD for 4 weeks. Our results demonstrated that one- carbon metabolism may have a small role in mitigating hepatic fat accumulation by Cr supplementation. The modulation of key genes related to fatty acid oxidation pathway suggests a new mechanism by which Cr prevents liver fat accumulation. © 2015 Elsevier Inc. All rights reserved. Keywords: Creatine supplementation; Choline-deficient diet; Fatty liver; One-carbon metabolism; Fat oxidation 1. Introduction In the last few years, hepatic fat accumulation and the progression of nonalcoholic steatohepatitis (NASH) have been associated with impairment of hepatic one-carbon metabolism [1]. This has been attributed to the dysfunction of specific enzymes that result in the decreased availability of S-adenosylmethionine (SAM) in the liver and increased homocysteine (Hcy) formation [2, 3]. SAM acts primarily as the universal methyl donor for the methylation of DNA and synthesis of N50-methylated compounds including adrenaline, carnitine, creatine and phosphatidylcholine (PC). A byproduct of these transmethylation reactions is S-adenosylhomocysteine (SAH), which is reversibly hydrolyzed to adenosine and Hcy [4]. Once formed, Hcy can be remethylated by two fashions to reform methionine and support SAM synthesis, thus maintaining the methylation capacity [5]. In the first, 5-methyltetrahydrofolate donates a methyl group to the vitamin B-12 containing enzyme methionine synthase, which is subsequently added to Hcy. Alterna- tively, the remethylation of Hcy is also catalyzed by betaine Hcy methyltransferase (BHMT) that uses betaine as a methyl donor [4]. Because betaine is endogenously synthesized from choline, then it follows that a choline-deficient diet (CDD) impairs remethylation via BHMT and, thus, decreases the availability of SAM [6]. Indeed, it has been demonstrated that a CDD decreased hepatic SAM concentrations and subsequently impaired PC synthesis via phosphatidylethanol- amine N-methyltransferase (PEMT). Furthermore, a CDD diminished secretion of liver triglyceride as very low density lipoprotein (VLDL) and led to the accumulation of liver triglycerides and NASH in rats and mice [7–9]. Available online at www.sciencedirect.com ScienceDirect Journal of Nutritional Biochemistry 26 (2015) 391 – 397 ⁎ Corresponding author at: Department of Physical Education, Faculty of Physical Education and Sport - State University of Londrina. Rodovia Celso Garcia Cid | Pr 445 Km 380 | Campus Universitário, Londrina, Paraná, Brazil. Tel.: +55-16-36024564, +55-43-36024564. E-mail address: [email protected] (R. Deminice). 1 Tel.: +55-16-36024564. http://dx.doi.org/10.1016/j.jnutbio.2014.11.014 0955-2863/© 2015 Elsevier Inc. All rights reserved.
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Creatine supplementation prevents fatty liver in rats fed choline-deficient diet: a burden of...

Available online at www.sciencedirect.com
ScienceDirect
Journal of Nutritional Biochemistry 26 (2015) 391–397
Creatine supplementation prevents fatty liver in rats fed choline-deficient diet:a burden of one-carbon and fatty acid metabolism
Rafael Deminicea,c,⁎, Gabriela Salim Ferreira de Castroa,1, Lucas Vieira Franciscoa,1,Lilian Eslaine Costa Mendes da Silvaa,1, João Felipe Rito Cardosoa,1, Fernando Tadeu Trevisan Frajacomoa,1,
Bruno Gonzaga Teodorod, Leonardo dos Reis Silveirab,d,1, Alceu Afonso Jordaoa,1
aNutrition and Metabolism, Faculty of Medicine of Ribeirao Preto, University of Sao Paulo, Av. Bandeirantes 3900, Ribeirao Preto, Sao Paulo, BrazilbSchool of Physical Education of Ribeirao Preto, University of Sao Paulo, Av. Bandeirantes 3900, Ribeirao Preto, Sao Paulo, Brazil
cDepartment of Physical Education, Faculty of Physical Education and Sport, State University of Londrina. Rodovia Celso Garcia Cid | Pr 445 Km 380 | Campus Universitário, Londrina,Paraná, Brazil
dDepartment of Biochesmtry and Imunology, Faculty of Medicine of Ribeirão Preto Av. Bandeirantes 3900, Ribeirao Preto, Sao Paulo, Brazil
Received 4 August 2014; received in revised form 5 November 2014; accepted 19 November 2014
Abstract
Aim: To examine the effects of creatine (Cr) supplementation on liver fat accumulation in rats fed a choline-deficient diet.Methods: Twenty-four rats were divided into 3 groups of 8 based on 4 weeks of feeding an AIN-93 control diet (C), a choline-deficient diet (CDD) or a CDDsupplemented with 2% Cr. The CDD diet was AIN-93 without choline.Results: The CDD significantly increased plasma homocysteine and TNFα concentration, as well as ALT activity. In liver, the CDD enhanced concentrations of totalfat (55%), cholesterol (25%), triglycerides (87%), MDA (30%), TNFα (241%) and decreased SAM concentrations (25%) and the SAM/SAH ratio (33%). Crsupplementation prevented all these metabolic changes, except for hepatic SAM and the SAM/SAH ratio. However, no changes in PEMT gene expression or liverphosphatidylcholine levels were observed among the three experimental groups, and there were no changes in hepatic triglyceride transfer protein (MTP)mRNA level. On the contrary, Cr supplementation normalized expression of the transcription factors PPARα and PPARγ that were altered by the CDD. Further, thedownstream targets and fatty acids metabolism genes, UCP2, LCAD and CPT1a, were also normalized in the Cr group as compared to CDD-fed rats.Conclusion: Cr supplementation prevented fat liver accumulation and hepatic injures in rats fed with a CDD for 4 weeks. Our results demonstrated that one-carbon metabolism may have a small role in mitigating hepatic fat accumulation by Cr supplementation. The modulation of key genes related to fatty acidoxidation pathway suggests a new mechanism by which Cr prevents liver fat accumulation.© 2015 Elsevier Inc. All rights reserved.
Keywords: Creatine supplementation; Choline-deficient diet; Fatty liver; One-carbon metabolism; Fat oxidation
1. Introduction
In the last few years, hepatic fat accumulation and the progressionof nonalcoholic steatohepatitis (NASH) have been associated withimpairment of hepatic one-carbon metabolism [1]. This has beenattributed to the dysfunction of specific enzymes that result in thedecreased availability of S-adenosylmethionine (SAM) in the liver andincreased homocysteine (Hcy) formation [2, 3]. SAM acts primarily asthe universal methyl donor for the methylation of DNA and synthesisof N50-methylated compounds including adrenaline, carnitine,creatine and phosphatidylcholine (PC). A byproduct of these
⁎ Corresponding author at: Department of Physical Education, Faculty ofPhysical Education and Sport - State University of Londrina. Rodovia CelsoGarcia Cid | Pr 445 Km 380 | Campus Universitário, Londrina, Paraná, Brazil.Tel.: +55-16-36024564, +55-43-36024564.
E-mail address: [email protected] (R. Deminice).1 Tel.: +55-16-36024564.
http://dx.doi.org/10.1016/j.jnutbio.2014.11.0140955-2863/© 2015 Elsevier Inc. All rights reserved.
transmethylation reactions is S-adenosylhomocysteine (SAH),which is reversibly hydrolyzed to adenosine and Hcy [4]. Onceformed, Hcy can be remethylated by two fashions to reformmethionine and support SAM synthesis, thus maintaining themethylation capacity [5]. In the first, 5-methyltetrahydrofolatedonates a methyl group to the vitamin B-12 containing enzymemethionine synthase, which is subsequently added to Hcy. Alterna-tively, the remethylation of Hcy is also catalyzed by betaine Hcymethyltransferase (BHMT) that uses betaine as a methyl donor [4].Because betaine is endogenously synthesized from choline, then itfollows that a choline-deficient diet (CDD) impairs remethylation viaBHMT and, thus, decreases the availability of SAM [6]. Indeed, it hasbeen demonstrated that a CDD decreased hepatic SAM concentrationsand subsequently impaired PC synthesis via phosphatidylethanol-amine N-methyltransferase (PEMT). Furthermore, a CDD diminishedsecretion of liver triglyceride as very low density lipoprotein (VLDL)and led to the accumulation of liver triglycerides and NASH in rats andmice [7–9].

392 R. Deminice et al. / Journal of Nutritional Biochemistry 26 (2015) 391–397
Creatine synthesis consumes a considerable amount of hepatic SAMand thus produces an equivalent amount of Hcy [10]. Previous studieshave shown that creatine supplementation (CR) down-regulates theendogenous formation of creatine from its substrate guanidinoaceticacid (GAA), via Guanidinoacetate methyltransferase (GAMT), whichreduces Hcy synthesis [11–14]. Because both PEMT and GAMT drawfrom the same pool of hepatic SAM, CR may also increase SAMavailability for PC synthesis (via PEMT) and, moreover, increase VLDLsecretion and prevent the accumulation of fat in the liver during CDDfeeding (Fig. 2). Previous data fromour grouphas demonstrated that CRprevents fatty liver after 3 weeks of feeding a high-fat diet [15].However, feeding a high-fat diet was unable to explain the specificcontribution of one-carbon metabolism to hepatic fat accumulation.Therefore, we proposed that the induction of NASH by feeding a CDDand supplementing with creatine would clarify the relationshipbetween one-carbon metabolism and hepatic fat accumulation. Theaim of the present study was to examine the effects of CR on liver fataccumulation and one-carbon metabolism in rats fed a CDD.
2. Methods
Twenty-four male Wistar rats (initial weight ~120 g) wereobtained from Central Animal Care at the University of São Paulo,Faculty of Medicine of Ribeirão Preto. All procedures were approvedby the Ethics Committee for Animal Use at the same institutionand were in accordance with the Guidelines of the COBEA (BrazilianCollege of experiments with animals). The rats were housed inindividual cages on a 12-h-light/-dark cycle at a mean temperature of22°C andwere randomly assigned to three dietary groups of eight ratseach: control (C); CDD; CDD plus CR. Group C was fed standard AIN-93 maintenance diet as proposed by Reeves et al. [16] which contains0.25% choline. CDD groups received AIN-93 standard diet withoutcholine. CR was performed by adding 2% (wt:vol) creatine mono-hydrate to the CDD diet. The rats had free access to food throughoutthe 4 weeks, and food intake was measured daily to assessconsumption of fat, creatine and total energy. Body weight wasmeasured twice a week.
2.1. Tissue preparation
After 4 weeks of experimental feeding, rats were anesthetisedwith an intraperitoneal injection of sodium pentobarbital (65 mg/kgip) and killed by decapitation. Animals were sacrificed between 9 and11 a.m. At necropsy, blood was collected into heparinized tubes andcentrifuged. The plasma fraction was separated and stored at −80°Cuntil analysis. Livers were extracted, weighed and freeze-clampedwith aluminum tongs. Weighed samples were and stored at −80°C.All procedures were performed under standard RNase-free conditionsto avoid exogenous RNase contamination. A portion of fresh livertissue was weighed and cut in small cubes of approximately 5×5×5mm and embedded using 10% buffered formalin for histopathologicevaluation. Left kidney was removed and immediately homogenizedin ice-cold 50 mmol/L potassium phosphate buffer (pH 7.4) forapproximately 30 s. This homogenate was used for the analysis ofAGAT activity. Epididymal fat pads were removed and weighed as anindication of visceral fat.
2.2. Hcy and related metabolites
Plasma Hcy and other sulfur-containg amino acids were deriva-tized with commercially available kit EZ:Faast Amino Acid Analysis(Phenomenex®) and assayed by gas-chomatography (GC-FID, GC-17A Shimadzu®, Kyoto, Japan). For SAM and SAH determinations,freeze-clamped liver samples were homogenized in ice-cold 8%(wt/vol) trichloroacetic acid. Homogenates were centrifuged at
13,000 g for 5 min at 4°C. The supernatants were removed andanalyzed by HPLC using a Phenomenex® C18 column equilibratedwith 96% of buffer A (50-mmol/L NaH2PO4 containing 10-mmol/Lheptanesulfonic acid at pH 3.2) and 4% acetonitrile. SAM and SAHwere separated by means of a gradient of 96–80% of buffer A and 4–20% of acetonitrile for 15 min. SAM and SAH peaks were detected at258 nm and quantified using Millennium32 (Version 2) software(Waters, Milford, MA, USA). PC concentrations in liver and plasmawere assayed using a commercially available kit from CaymanChemical Company catalogue #10009926 (Ann Harbor, MI, USA).
Creatine concentrations in plasma and liver were determinedusing previously described methods [13]. Kidney AGAT activity wasdetermined as described by Van Pilsum et al. [17], and protein wasassayed using the Biuret method.
2.3. Hepatic histology and lipid analysis
To examine liver morphology, the tissue was preserved in 10%phosphate-buffered formalin, pH 7.0. Formalin-preserved liversamples were exposed to hematoxylin and eosin (H&E) staining.Slides were scored for fat droplets using a conventional lightmicroscope (200×). Total liver fat was determined by homogenizing0.5 g of liver in 1.0 ml of distilled water. Subsequently, 5 ml ofchloroform-methanol (2:1) was added, and the tubes were thor-oughly mixed and centrifuged to separate fats. After centrifugation,the organic phase was transferred to a preweighed tube. Thischloroform:methanol extraction was repeated twice and the organicphases combined. The extracts were evaporated to dryness under astreamof nitrogen and re-weighed. Thehepatic fatwas re-suspended in1ml of 1-propanol for thequantification of triglycerides and cholesterol,using commercially available kits from Labtest (Lagoa Santa, MinasGerais, Brazil).
2.4. Inflammatory and oxidative stress markers
Plasma alanine aminotransferase (ALT) activity was performedusing a commercially available kit (Labtest, Lagoa Santa, Minas Gerais,Brazil). Liver malondialdehyde (MDA) was determined on an HPLC(LC-20A Shimadzu®, Kyoto, Japan) according to Spirlandeli et al. [18].Hepatic reduced (GSH) and oxidized glutathione (GSSG) wereperformed according to described by Rahman et al. [19]. Plasmaand liver TNF-α were determined using a commercially available kit(eBioscience®, USA).
2.5. Gene expression and western blotting
RNA was isolated from 50 mg of frozen liver using a RiboPure Kit(Ambion, part number AM 1924, USA) according to the manufacturer'sinstructions. Total RNA was quantified by spectrophotometry at OD260/280 (NanoDrop2000c, ThermoScientific, USA). The quality andintegrity of the isolated RNAwere assessed using a 1.2% agarose gel andelectrophoresis. An additional DNase I treatment (DNA-free Kit,Ambion, part number AM1906, USA) was performed to removecontaminating DNA from the isolated RNA. cDNA was synthesizedfrom 1000 ηg of RNA using a high-capacity cDNA Reverse TranscriptionKit (Applied Biosystems, part number 4374966, USA). Quantitative PCRwas performed using the 7500 Fast Real-time PCR System (AppliedBiosystems, USA). The following Taqman® Gene Expression probes(Applied Biosystems, USA) were used in this study: Rn01644299_m1(ChDh), Rn00567492_m1 (Chka), Rn00755199_g1 (Chkb),Rn00589584_m1 (Pcyt 1a) , Rn00564517_m1 (PEMT) ,Rn00578255_m1 (BHMT), Rn00567215_m1 (GNMT), Rn00560948_m1(CBS), Rn00566193_m1 (Ppara), Rn01754856_m1 (Ucp2),Rn00580241_m1 (Ppargc1a) Rn00690933_m1 (cyclophilin A).SYBRgreen master mix was used for qPCR of the following primers:
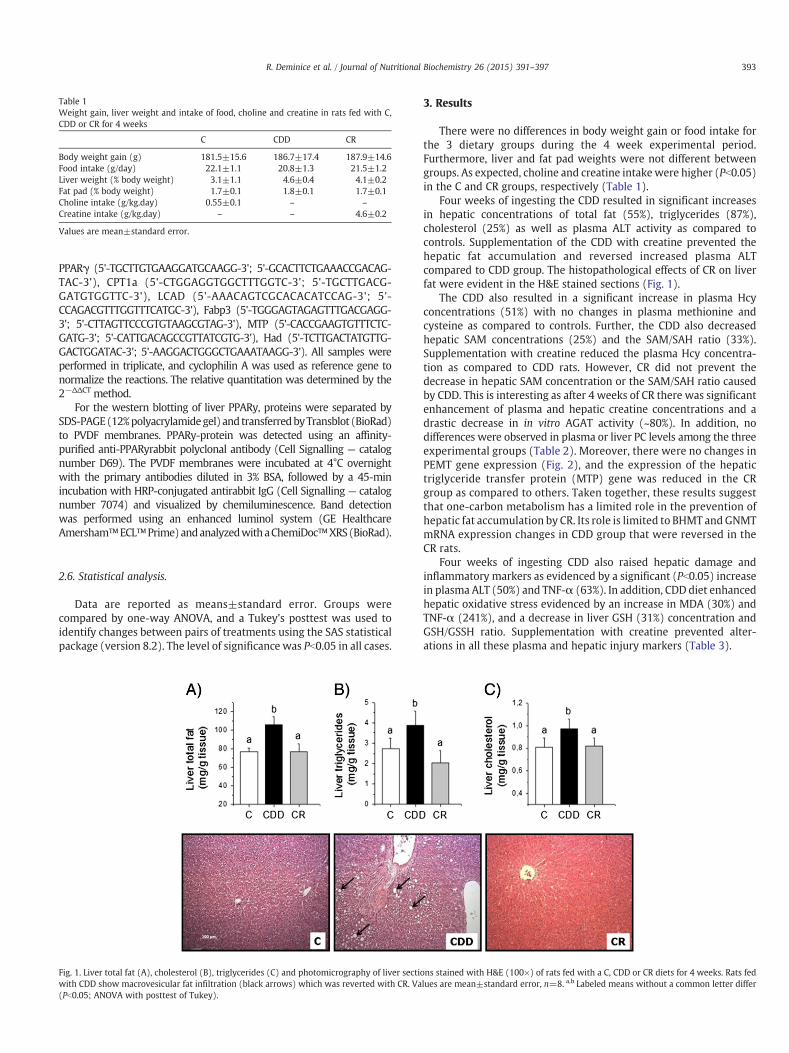
Table 1Weight gain, liver weight and intake of food, choline and creatine in rats fed with C,CDD or CR for 4 weeks
C CDD CR
Body weight gain (g) 181.5±15.6 186.7±17.4 187.9±14.6Food intake (g/day) 22.1±1.1 20.8±1.3 21.5±1.2Liver weight (% body weight) 3.1±1.1 4.6±0.4 4.1±0.2Fat pad (% body weight) 1.7±0.1 1.8±0.1 1.7±0.1Choline intake (g/kg.day) 0.55±0.1 – –
Creatine intake (g/kg.day) – – 4.6±0.2
Values are mean±standard error.
393R. Deminice et al. / Journal of Nutritional Biochemistry 26 (2015) 391–397
PPARγ (5'-TGCTTGTGAAGGATGCAAGG-3'; 5'-GCACTTCTGAAACCGACAG-TAC-3'), CPT1a (5'-CTGGAGGTGGCTTTGGTC-3'; 5'-TGCTTGACG-GATGTGGTTC-3'), LCAD (5'-AAACAGTCGCACACATCCAG-3'; 5'-CCAGACGTTTGGTTTCATGC-3'), Fabp3 (5'-TGGGAGTAGAGTTTGACGAGG-3'; 5'-CTTAGTTCCCGTGTAAGCGTAG-3'), MTP (5'-CACCGAAGTGTTTCTC-GATG-3'; 5'-CATTGACAGCCGTTATCGTG-3'), Had (5'-TCTTGACTATGTTG-GACTGGATAC-3'; 5'-AAGGACTGGGCTGAAATAAGG-3'). All samples wereperformed in triplicate, and cyclophilin A was used as reference gene tonormalize the reactions. The relative quantitation was determined by the2−ΔΔCT method.
For the western blotting of liver PPARy, proteins were separated bySDS-PAGE(12%polyacrylamidegel) and transferredbyTransblot (BioRad)to PVDF membranes. PPARy-protein was detected using an affinity-purified anti-PPARyrabbit polyclonal antibody (Cell Signalling — catalognumber D69). The PVDF membranes were incubated at 4°C overnightwith the primary antibodies diluted in 3% BSA, followed by a 45-minincubation with HRP-conjugated antirabbit IgG (Cell Signalling — catalognumber 7074) and visualized by chemiluminescence. Band detectionwas performed using an enhanced luminol system (GE HealthcareAmersham™ECL™Prime)andanalyzedwithaChemiDoc™XRS(BioRad).
2.6. Statistical analysis.
Data are reported as means±standard error. Groups werecompared by one-way ANOVA, and a Tukey's posttest was used toidentify changes between pairs of treatments using the SAS statisticalpackage (version 8.2). The level of significance was Pb0.05 in all cases.
Fig. 1. Liver total fat (A), cholesterol (B), triglycerides (C) and photomicrography of liver sectiwith CDD show macrovesicular fat infiltration (black arrows) which was reverted with CR. Va(Pb0.05; ANOVA with posttest of Tukey).
3. Results
There were no differences in body weight gain or food intake forthe 3 dietary groups during the 4 week experimental period.Furthermore, liver and fat pad weights were not different betweengroups. As expected, choline and creatine intakewere higher (Pb0.05)in the C and CR groups, respectively (Table 1).
Four weeks of ingesting the CDD resulted in significant increasesin hepatic concentrations of total fat (55%), triglycerides (87%),cholesterol (25%) as well as plasma ALT activity as compared tocontrols. Supplementation of the CDD with creatine prevented thehepatic fat accumulation and reversed increased plasma ALTcompared to CDD group. The histopathological effects of CR on liverfat were evident in the H&E stained sections (Fig. 1).
The CDD also resulted in a significant increase in plasma Hcyconcentrations (51%) with no changes in plasma methionine andcysteine as compared to controls. Further, the CDD also decreasedhepatic SAM concentrations (25%) and the SAM/SAH ratio (33%).Supplementation with creatine reduced the plasma Hcy concentra-tion as compared to CDD rats. However, CR did not prevent thedecrease in hepatic SAM concentration or the SAM/SAH ratio causedby CDD. This is interesting as after 4 weeks of CR there was significantenhancement of plasma and hepatic creatine concentrations and adrastic decrease in in vitro AGAT activity (~80%). In addition, nodifferences were observed in plasma or liver PC levels among the threeexperimental groups (Table 2). Moreover, there were no changes inPEMT gene expression (Fig. 2), and the expression of the hepatictriglyceride transfer protein (MTP) gene was reduced in the CRgroup as compared to others. Taken together, these results suggestthat one-carbon metabolism has a limited role in the prevention ofhepatic fat accumulation by CR. Its role is limited to BHMT and GNMTmRNA expression changes in CDD group that were reversed in theCR rats.
Four weeks of ingesting CDD also raised hepatic damage andinflammatory markers as evidenced by a significant (Pb0.05) increasein plasma ALT (50%) and TNF-α (63%). In addition, CDD diet enhancedhepatic oxidative stress evidenced by an increase in MDA (30%) andTNF-α (241%), and a decrease in liver GSH (31%) concentration andGSH/GSSH ratio. Supplementation with creatine prevented alter-ations in all these plasma and hepatic injury markers (Table 3).
ons stained with H&E (100×) of rats fed with a C, CDD or CR diets for 4 weeks. Rats fedlues are mean±standard error, n=8. a,b Labeled means without a common letter differ

Table 2One-carbon metabolites determined in plasma, liver and kidney of rats fed with C, CDDor CR for 4 weeks
C CDD CR
PlasmaCreatine (μmol/L) 83.6±7.6a 70.1±6.9a 340.4±30.8b
Hcy (μmol/L) 11.2±0.7a 16.9±1.3b 10.2±1.3a
Methionine (μmol/L) 50.5±4.4 48.3±5.5 41.7±6.3Cysteine (μmol/L) 207.5±11.4 252.3±33.5 234.7±25.6PC (mg/dl) 114.1±11.3 102.1±13.1 104.4±10.1
LiverCreatine (μmol/g tissue) 0.5±0.1a 0.5±0.1a 2.6±0.2b
SAM (nmol/g tissue) 123.5±7.6a 92.2±10.1b 107.8±10.6a,b
SAH (nmol/g tissue) 17.7±1.2a 20.5±1.1a 27.2±0.8b
SAM/SAH 7.1±0.6a 4.7±0.7b 3.9±0.4b
PC (mg/g tissue) 139.6±8.7 155.2±10.2 158.9±10.7Kidney
AGAT activity (nom/mg/min) 978.5±70.6a 896.7±65.3a 203.3±22.3b
Values are mean±standard error, n=8. a,b Means in a row without a common letterdiffer (Pb0.05; ANOVA with posttest of Tukey).
394 R. Deminice et al. / Journal of Nutritional Biochemistry 26 (2015) 391–397
In order to determine the preventive effects of CR on fatty liver, weexamined an abundance of genes and proteins involved in fatty acidmetabolism (Fig. 3). CDD significantly increased expression of thetranscription factors PPARα and PPARγ. The translation of PPARγ intoprotein was confirmed by western blotting. The changes in thesetranscription factors corresponded to the modulation of some of theirdownstream target genes which are putatively involved in fatty acidoxidation. These included UCP2, PGC1a, LCAD and CPT1a. Interest-ingly, CR normalized the expression of the UCP2, LCAD and CPT1atranscripts to that of control rats.
Fig. 2. Creatine, Hcy and PCmetabolic interactions (A); methionine metabolism (B); phospholipweeks. Values are mean±standard error, n=8. a,b Labeled means without a common letterexpression which results in decreased hepatic SAM availability and increased Hcy. CR reversconcentration. AGAT, arginine:glycine amidinotransferase; GAMT, SAM:guanidinoacetatecytidylyltransferase; BHMT, betaine-homocysteine S-methyltransferase; GNMT, Glycine N-me
4. Discussion
We have previously shown that CR prevents hepatic fat accumu-lation in rats fed with high-fat diet [15]. More recently, da Silva et al.[20] demonstrated that creatine treatment reduced lipid accumula-tion in rat hepatoma cultured cells. Our data provide novel evidencethat CR prevented hepatic fat accumulation, decreased inflammatoryand oxidative stress markers and plasma ALT in rats fed with a CDDfor 4 weeks.
In the current study, the CDD impaired methionine metabolism bydecreasing hepatic SAM availability and the SAM/SAH ratio. Further,CDD reduced BHMT and GNMT gene expression. Together theseresults suggest that feeding a CDD reduces the remethylation of Hcyto methionine by BHMT and restricts available methyl groups forSAM formation and subsequent transmethylation (Fig. 2). This is inagreement with that shown elsewhere [6]. In contrast, CR has beenwidely demonstrated to modulate methylation demand [11] anddecrease plasma Hcy concentration [11–14]. In the present study, wefound 4 weeks of CR increased plasma (303%) and liver (478%)creatine concentrations. As consequence, a remarkable (~80%) down-regulation of renal AGAT enzyme activity was found, showing areduction in the endogenous formation of creatine (Table 2). Ascreatine synthesis is responsible for a considerable portion of methylgroups and Hcy formation [10], the suppression of creatine synthesisthrough supplementation led to decreased plasma Hcy in the CRgroup compared to CDD.
Despite a reduction in Hcy levels, CR was not able to restorehepatic SAM concentrations and the SAM/SAH ratio that wasdiminished by CDD. SAM is required for synthesis of PC via PEMT,and PC is an essential component of VLDL [21]. This results in ahypothetical mechanism for the development of NASH by CDD.
ids metabolism (C); andMTP (D) genes mRNA of rats fed with a C, CDD or CR diets for 4differ (Pb0.05; ANOVA with posttest of Tukey). CDD impaired ChDh and BHMT geneed BHMT and GNMT egen expression, however, was unable to increase hepatica SAMN-methyltransferase; PE, phosphatidlyethanolamine; CT, CTP:phosphorylcholine
thyltransferase.

Table 3Plasma and liver inflammatory and oxidative stress markers in rats fed with C, CDD orCR for 4 weeks
C CDD CR
PlasmaALT (U/L) 45.6±3.5a 68.7±4.7b 52.9±3.9a
TNF-α (pg/ml) 10.3±2.2a 16.8±3.1b 11.5±2.7a
LiverMDA (nmol/g tissue) 3.6±0.4a 4.7±0.3b 3.0±0.4a
GSH (mmol/g tissue) 5.2±0.2a 4.0±0.2b 5.0±0.1a
GSH/GSSG 19.1±3.1a 7.3±1.0b 17.0±1.8a
TNF-α (pg/g tissue) 156.7±17.4a 378.1±31.8b 186.3±21.4a
Values are mean±standard error, n=8. a,b Means in a row without a common letterdiffer (Pb0.05; ANOVA with posttest of Tukey).
395R. Deminice et al. / Journal of Nutritional Biochemistry 26 (2015) 391–397
Studies have shown that CDD impairs SAM availability and PCsynthesis which reduces the secretion of liver triglyceride as VLDL,resulting in accumulation of liver triglycerides [7]. PC and creatinesynthesis are quantitatively themost prominent consumers of hepaticSAM. Indeed, hepatic PC synthesis by PEMT is responsible forapproximately 50% of hepatic Hcy formation [7, 21]. Creatinesynthesis also consumes a considerable portion of hepatic SAM,perhaps as much as 40% [9]. Because both PEMT and GAMT share thesame hepatic SAM pool, we originally hypothesized that CR could leadto a sparing effect on SAM, resulting in increased PC formation andhepatic VLDL secretion. However, CR could not restore SAM levels orthe SAM/SAH ratio in rats fed CDD. Further, no change in PEMT geneexpression or liver PC levels occurred among the three experimentalgroups (Table 2). Fig. 2 also demonstrated reduced MTP geneexpression in CR group compared to others. MTP codes formicrosomal triglyceride transfer protein, which catalyzes the transferof triglycerides and PC between phospholipid surfaces which is
Fig. 3. Hepatic transcription factors (A); fatty acid oxidation genes mRNA (B); and PPARγ prstandard error, n=8. a,b Labeled means without a common letter differ (Pb0.05; ANOVA withrelated to fatty acid b-oxidation pathway, which was reversed by CR.
required for the secretion of VLDL in the liver [22]. These resultstogether partially refute our initial hypothesis.
BHMT has a major role in the overall conversion of Hcy tomethionine in the remethylation pathway. GNMT is also important tomaintain the homeostasis of SAM, which utilizes glycine as themethyl receptor to consume excess of SAM to form a nontoxic product[23]. Studies have shown that BHMT and GNMT knockout mice hadreduction in liver SAM and severe steatosis [23–25]. Our data revealedthat, although not enough to increase SAM concentration and SAM/SAH ratio to the control levels, CR restored the remethylation byBHMT enough to return GNMT to control levels in rats fed CDD. Theseresults showed that the normalization of BHMT and GNMT geneexpression by creatine may have a critical role in CDD mediated fatliver accumulation; however, we are unable to exactly define thecontribution of BHMT and GNMT pathway to the preventive effects ofcreatine on fatty liver disease. These data are important and must beconsidered in future studies.
In addition to its effects on one-carbon metabolism, recent studieshave demonstrated that creatine treatment may stimulate fattyacid oxidation [15, 20]. The up-regulation in LCAD found in CR-supplemented group compared to CDD provides an evidence that CRcan stimulates fatty acid oxidation. LCAD is a mitochondrial matrixenzyme responsible for the first step of long-chain fatty acyl-CoAsoxidation, and its abnormality or deletion has been related tomitochondrial dysfunction and hepatic steatosis [26, 27]. A recentstudy from da Silva et al. [20] revealed that creatine treatmentreduced lipid accumulation in oleate-treated McArdle RH-7777 rathepatoma cells by the stimulation of fatty acid oxidation andtriglycerides secretion. These authors reported increased PPARαgene expression as well as several of its downstream targets linkedto fatty acid oxidation such as CPT1a, ACOX1, LCAD, MCAD and LCADin creatine treated cells. These data are consistent with previously
otein expression of rats fed with a C, CDD or CR diets for 4 weeks. Values are mean±posttest of Tukey). CDD modulated substantially transcription factors and key genes

396 R. Deminice et al. / Journal of Nutritional Biochemistry 26 (2015) 391–397
results that demonstrated increased LCAD gene expression andprevention of accumulation of fat in the liver after 3 weeks of CR inrats fed high-fat diet [15].
Moreover, LCAD is a target of PPARα, important regulators of lipidmetabolism involved in NASH progression and pathophysiology[28–30]. PPARα-null mice exhibited both hepatic steatosis anddecreased expression of transcripts related to fatty acid oxidation[28]. Matsusue et al. [29] demonstrated that liver-specific knockoutPPARγ mice presented decreased hepatic lipid stores. Our datarevealed that 4 weeks of feeding with a CDD promoted elevatedPPARα and PPARγ gene expression and PPARγ protein content.Curiously, CR decreased to control levels the expression of PPARα,PPARγ, and CPT1a when feeding with a CDD. It seems controversialthat CDD should increase the expression of both PPARα and PPARγand some of their targets. However, rodent studies have demonstrat-ed high-fat diets up-regulated the expression of of both PPARα andPPARγ [31, 32]. In ob/ob mice, increased rates of fatty acid oxidationhave been found [33, 34].While there is some divergence in literature,early in the course of fatty liver disease as found in CDD, an effort tocompensate the increased fat deposition may increase the fattyoxidation by liver mitochondria [35, 36]; it is consistent to increasedavailability of the pool of free fatty acids to be used as substrates [37].In contrast, our data demonstrated that CR was able to inhibits bothPPARα and PPARγ up-regulation induced by CDD; reflecting theabsence of liver fat deposition.
Finally, our results demonstrated reduced UCP2 gene expression,reduced liver and plasma TNFα and restored hepatic GSH/GSSG ratio;it may also be considered as protective effects of creatine. Whileantioxidant capacity of creatine has been well documented [38], tothe best of our knowledge, this is the first study demonstrating thatcreatine may prevent liver injure markers in fatty liver disease.Inflammation and oxidative stress have been considered importantdysfunctions related to NASH progression. Hepatic fat liver accumu-lation increases liver susceptibility to lipid peroxidation and alde-hydes formation; it has been shown to increase UCP2 activity [39].Our data suggest that CR may prevent fatty liver progression,characterized to lipid peroxidation and inflammation.
In conclusion, CR prevented fat liver accumulation and hepaticinjures in rats fed with a CDD for 4 weeks. Changes in BHMT andGNMT gene expression by creatine have a critical role in CDD-mediated fat liver accumulation; however, creatine protection effectgoes beyond modulation in one-carbon metabolism. Our resultssuggest that CR modifies fatty acid oxidation, especially transcriptionfactors (PPARα and PPARγ) and key genes related to fatty acidmetabolism as LCAD that were impaired by 4 weeks of CDD.
This study is supported by grants from Capes (Coordenação deAperfeiçoamento de Pessoal de Nível Superior, Brazil) and RUSP(Reitoria da Universidade de São Paulo, Brazil).
References
[1] Kim SK, Choi KH, Kim YC. Effect of acute betaine administration on hepaticmetabolism of S-amino acids in rats and mice. Biochem Pharmacol 2003;65:1565–74.
[2] Kim SK, Kim YC. Effects of betaine supplementation on hepatic metabolism ofsulfur-containing amino acids in mice. J Hepatol 2005;42:907–13.
[3] Kwon do Y, Jung YS, Kim SJ, Park HK, Park JH, Kim YC. Impaired sulfur-amino acidmetabolism and oxidative stress in nonalcoholic fatty liver are alleviated bybetaine supplementation in rats. J Nutr 2009;139:63–8.
[4] Brosnan JT, da Silva R, Brosnan ME. Amino acids and the regulation of methylbalance in humans. Curr Opin Clin Nutr Metab Care 2007;10(1):52–7.
[5] da Silva RP, Kelly KB, Al Rajabi A, Jacobs RL. Novel insights on interactions betweenfolate and lipid metabolism. Biofactors 2014;40(3):277–83.
[6] Setoue M, Ohuchi S, Morita T, Sugiyama K. Choline deprivation induceshyperhomocysteinemia in rats fed low methionine diets. J Nutr Sci Vitaminol(Tokyo) 2008;54(6):483–90.
[7] Li Z, Vance DE. Phosphatidylcholine and choline homeostasis. J Lipid Res 2008;49(6):1187–94.
[8] Raubenheimer PJ, Nyirenda MJ, Walker BR. A choline-deficient diet exacerbatesfatty liver but attenuates insulin resistance and glucose intolerance in mice fed ahigh-fat diet. Diabetes 2006;55(7):2015–20.
[9] Kashireddy PR, Rao MS. Sex differences in choline-deficient diet-inducedsteatohepatitis in mice. Exp Biol Med (Maywood) 2004;229(2):158–62.
[10] Stead LM, Brosnan JT, Brosnan ME, Vance DE, Jacobs RL. Is it time to reevaluatemethyl balance in humans? Am J Clin Nutr 2006;83(1):5–10.
[11] Edison EE, Brosnan ME, Meyer C, Brosnan JT. Creatine synthesis: production ofguanidinoacetate by the rat and human kidney in vivo. Am J Physiol Renal Physiol2007;293(6):F1799–804.
[12] Stead LM, Au KP, Jacobs RL, Brosnan ME, Brosnan JT. Methylation demand andhomocysteine metabolism: effects of dietary provision of creatine and guanidi-noacetate. Am J Physiol Endocrinol Metab 2001;281:E1095–100.
[13] Deminice R, Portari GV, Vannucchi H, Jordao AA. Effects of creatine supplemen-tation on homocysteine levels and lipid peroxidation in rats. Br J Nutr 2009;102(1):110–6.
[14] Deminice R, Vannucchi H, Simões-Ambrosio LM, Jordao AA. Creatine supplemen-tation reduces increased homocysteine concentration induced by acute exercisein rats. Eur J Appl Physiol 2011;111(11):2663–70.
[15] Deminice R, da Silva RP, Lamarre SG, Brown C, Furey GN, McCarter SA, et al.Creatine supplementation prevents the accumulation of fat in the livers of rats feda high-fat diet. J Nutr 2011;141(10):1799–804.
[16] Reeves PG, Nielsen FH, Fahey Jr GC. AIN-93 purified diets for laboratory rodents:final report of the American Institute of Nutrition ad hoc writing committee on thereformulation of the AIN-76A rodent diet. J Nutr 1993;123:1939–51.
[17] Van Pilsum JF, Taylor D, Zakis B, McCormick P. Simplified assay for transamidinaseactivities of rat kidney homogenates. Anal Biochem 1970;35:277–86.
[18] Spirlandeli AL, Deminice R, Jordao AA. Plasma malondialdehyde as biomarker oflipid peroxidation: effects of acute exercise. Int J Sports Med 2014;35:14–8.
[19] Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathioneand glutathione disulfide levels using enzymatic recycling method. Nat Protoc2006;1:3159–65.
[20] da Silva RP, Kelly KB, Leonard KA, Jacobs RL. Creatine reduces hepatic TGaccumulation in hepatocytes by stimulating fatty acid oxidation. Biochim BiophysActa 1841;2014:1639–46.
[21] Cole LK, Vance JE, Vance DE. Phosphatidylcholine biosynthesis and lipoproteinmetabolism. Biochim Biophys Acta 2012;1821(5):754–61.
[22] Yi-qiang L, Maeda T, Yuko Fujimaki Y, Kojima K, Fujita M, Kinoshita M, et al. Role ofmicrosomal triglyceride transfer protein in induction of fatty liver in suncun.Hepatol Res 1999;16(1):19–25.
[23] Martínez-Chantar ML, Vázquez-Chantada M, Ariz U, Martínez N, Varela M, Luka Z,et al. Loss of the glycine N-methyltransferase gene leads to steatosis andhepatocellular carcinoma in mice. Hepatology 2008;47:1191–9.
[24] Collinsova M, Strakova J, Jiracek J, Garrow TA. Inhibition of betaine-homocysteineS-methyltransferase in mice causes hyperhomocysteinemia. J Nutr 2006;136:1493–7.
[25] Varela-Rey M, Martínez-López N, Fernández-Ramos D, Embade N, Calvisi DF,Woodhoo A, et al. Fatty liver and fibrosis in glycine N-methyltransferase knockoutmice is prevented by nicotinamide. Hepatology 2010;52:105–14.
[26] Zhang D, Liu ZX, Choi CS, Tian L, Kibbey R, Dong J, et al. Mitochondrialdysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causeshepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci U S A 2007;104:17075–80.
[27] Kurtz DM, Rinaldo P, Rhead WJ, Tian L, Millington DS, Vockley J, et al. Targeteddisruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucialroles for fatty acid oxidation. Proc Natl Acad Sci U S A 1998;95:15592–7.
[28] Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, et al. Alteredconstitutive expression of fatty acid-metabolizing enzymes in mice lacking theperoxisome proliferator-activated receptor alpha (PPARalpha). J Biol Chem 1998;273:5678–84.
[29] Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, et al. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liverbut aggravates diabetic phenotypes. J Clin Invest 2003;111(5):737–47.
[30] Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ.PPAR&alpha expression protects male mice from high fatinduced nonalcoholicfatty liver. J Nutr 2011;141:603–10.
[31] Schultz A, Neil D, Aguila MB, Mandarim-de-Lacerda CA. Hepatic adverse effects offructose consumption independent of overweight/obesity. Int J Mol Sci 2013;14(11):21873–86.
[32] Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, et al. PPARgamma mediates high-fat diet-induced adipocyte hypertrophy and insulinresistance. Mol Cell 1999;4(4):597–609.
[33] García-Ruiz I, Rodríguez-Juan C, Díaz-Sanjuan T, del Hoyo P, Colina F, Muñoz-Yagüe T, et al. Uric acid and anti-TNF antibody improvemitochondrial dysfunctionin ob/ob mice. Hepatology 2006;44:581–91.
[34] García-Ruiz I, Rodríguez-Juan C, Díaz-Sanjuán T, Martínez MA, Muñoz-Yagüe T,Solís-Herruzo JA. Effects of rosiglitazone on the liver histology and mitochondrialfunction in ob/ob mice. Hepatology 2007;46:414–23.
[35] Ciapaite J, van den Broek NM, Te Brinke H, Nicolay K, Jeneson JA, Houten SM, et al.Differential effects of short- and long-term high-fat diet feeding on hepatic fattyacid metabolism in rats. Biochim Biophys Acta 1811;2011:441–51.
[36] Lazarin Mde O, Ishii-Iwamoto EL, Yamamoto NS, Constantin RP, Garcia RF, daCosta CE, et al. Liver mitochondrial function and redox status in an experimentalmodel of non-alcoholic fatty liver disease induced by monosodium L-glutamate inrats. Exp Mol Pathol 2011;91:687–94.

397R. Deminice et al. / Journal of Nutritional Biochemistry 26 (2015) 391–397
[37] Carmiel-Haggai M, Cederbaum AI, Nieto N. A high-fat diet leads to the progressionof non-alcoholic fatty liver disease in obese rats. FASEB J 2005;19:136–8.
[38] Sestili P, Martinelli C, Colombo E, Barbieri E, Potenza L, Sartini S, et al. Creatine asan antioxidant. Amino Acids 2011;40:1385–96.
[39] Serviddio G, Bellanti F, Tamborra R, Rollo T, Capitanio N, Romano AD, et al.Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increasessusceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut 2008;57:957–65.




















