
Creatine as a compatible osmolyte in muscle cells exposed to hypertonic stress
Upload
independentCategory
view
0download
0
H
NHA
moCu
Neurobiology of Disease 8, 479–491 (2001)doi:10.1006/nbdi.2001.0406, available online at http://www.idealibrary.com on
A
Creatine Increases Survival and Delays MotorSymptoms in a Transgenic Animal Modelof Huntington’s Disease
Ole A. Andreassen,* Alpaslan Dedeoglu,* Robert J. Ferrante,†
Bruce G. Jenkins,‡ Kimberly L. Ferrante,* ,§ Melissa Thomas,¶
Avi Friedlich,§ Susan E. Browne,§ Gabriele Schilling,\
David R. Borchelt,\ Steven M. Hersch,** Christopher A. Ross,††
and M. Flint Beal* ,§
*Neurochemistry Laboratory, Neurology Service, ‡MGH-NMR Center, Department ofRadiology, and ¶Molecular Endocrinology Laboratory, Massachusetts General Hospital and
arvard Medical School, Boston, Massachusetts; †Geriatric Research Education and ClinicalCenter, Bedford VA Medical Center, Bedford, Massachusetts and Department of Neurology,Pathology, and Psychiatry, Boston University School of Medicine, Boston, Massachusetts;§Department of Neurology and Neuroscience, Weill Medical College of Cornell University,
ew York Presbyterian Hospital, New York, New York; Department of Pathology, Johnsopkins University, Baltimore, Maryland; **Department of Neurology, Emory University,tlanta, Georgia; and ††Departments of Psychiatry and Neuroscience, Johns Hopkins
University, Baltimore, Maryland
Received August 4, 2000; revised November 30, 2000; accepted March 5, 2000
There is substantial evidence for bioenergetic defects in Huntington’s disease (HD). Creatine admin-istration increases brain phosphocreatine levels and it stabilizes the mitochondrial permeabilitytransition. We examined the effects of creatine administration in a transgenic mouse model of HDproduced by 82 polyglutamine repeats in a 171 amino acid N-terminal fragment of huntingtin (N171-82Q). Dietary supplementation of 2% creatine significantly improved survival, slowed the developmentof motor symptoms, and delayed the onset of weight loss. Creatine lessened brain atrophy and theformation of intranuclear inclusions, attenuated reductions in striatal N-acetylaspartate as assessedby NMR spectroscopy, and delayed the development of hyperglycemia. These results are similar tothose observed using dietary creatine supplementation in the R6/2 transgenic mouse model of HD andprovide further evidence that creatine may exert therapeutic effects in HD. © 2001 Academic Press
Key Words: Huntington’s; transgenic; creatine; N-acetylaspartate; diabetes; NMR spectroscopy.
3srphes
INTRODUCTION
Huntington’s disease (HD) is an autosomal domi-nant inherited neurodegenerative disorder character-ized by a progressive development of motor, psychi-atric, and cognitive symptoms (Hersch and Ferrante,1997; Kowall et al., 1999). The onset of HD is usually in
id-life with a mean survival of 15–20 years afternset. The HD gene mutation is an expanded unstableAG/polyglutamine repeat in huntingtin, a protein ofnknown function with a relative molecular weight of
0969-9961/01 $35.00Copyright © 2001 by Academic Press
ll rights of reproduction in any form reserved. 479
50,000 (The Huntington’s Disease Collaborative Re-earch Group, 1993). Normal individuals possess aepeat length of 6–35 glutamines, whereas HD isresent when alleles of more than 36 repeats are in-erited. The age of onset and the severity of the dis-ase are correlated with the length of the CAG expan-ion (Andrew et al., 1993; Duyao et al., 1993; Snell et al.,
1993).Although the discovery of the HD gene defect has
played an important role in understanding the patho-genesis of the disease, the pathogenetic mechanism is

G
ri1at
480 Andreassen et al.
A
not yet known. No effective treatment is presentlyavailable. It is now widely accepted that there is aselective pattern of neuronal degeneration in HD suchthat the neostriatum is the primary site of involve-ment, with medium-sized spiny striatal neurons dis-proportionately affected early and most severely andrelative sparing of striatal aspiny NADPH-d and cho-line acetyltransferase positive neurons (Kowall andFerrante, 1998). Recent studies have reported the pres-ence of intranuclear and cytosolic aggregates, usingn-terminal huntingtin antibodies (DiFiglia et al., 1997;
utekunst et al., 1999). While it is unclear what rolenuclear huntingtin aggregation may play in the patho-genesis of HD, the intracellular consequences of theseaggregates has been the object of intense investigation(Sisodia, 1998). Expansion of trinucleotide repeats isnow recognized as a major cause of neurological dis-ease, including several forms of spinocerebellar ataxia(SCA), spinobulbar muscular atrophy (SBMA), anddentatorubropallidoluysian atrophy (DRPLA) (Ross,1997; Ross et al., 1998). These diseases share severalcommon features, such as selective neuronal loss, re-peat length instability, and anticipation.
The pathogenetic mechanism of HD is presumed tobe a toxic gain of function. Several lines of evidencesuggest that impaired energy metabolism is a majorfactor in the development of HD (Beal, 2000). We andothers have shown that lactate is elevated in the cortexof HD patients (Jenkins et al., 1993), that there is aeduced phosphocreatine/inorganic phosphate ration resting muscle of HD patients (Koroshetz et al.,997), that mitochondrial toxins produce striatal dam-ge, which resembles the histopathology of HD pa-ients (Beal et al., 1993; Brouillet et al., 1995), and that
there are reduced activity of the mitochondrial en-zymes in the respiratory chain in HD patients (Browneet al., 1997; Gu et al., 1996). Recent studies showed thatmitochondria in lymphoblasts from HD patients aremore susceptible to depolarization and that this di-rectly correlates with CAG repeat length (Sawa et al.,1999). The maximal rate of mitochondrial ATP gener-ation in muscle is significantly reduced in both symp-tomatic HD patients and in presymptomatic HD genecarriers (Lodi et al., 2000).
A major advance in HD research was the develop-ment of transgenic mouse models. We have shownthat R6/2 mice develop marked decreases in NAA(Jenkins et al., 2000) and that they are more susceptibleto mitochondrial toxins (Bogdanov et al., 1998). Othersfound increased markers of oxidative stress and de-creased activity of mitochondrial enzymes in thesemice, which further supports the involvement of im-
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
paired energy metabolism (Tabrizi et al., 2000). Re-cently, we developed another transgenic mouse modelof HD in which transgenic mice (N171-82Q) express acDNA encoding a 171 amino acid N-terminal frag-ment of huntingtin containing 82 CAG repeats (Schill-ing et al., 1999). These transgenic mice develop a pro-gressive neurological phenotype beginning at about90 d of age with a mean death at 135 days of age. Thephenotype consists of behavioral abnormalities in-cluding loss of coordination, tremors, ataxia, hypoki-nesia and abnormal gait, and premature death. Dif-fuse nuclear immunolabeling, neuronal intranuclearinclusions, and neuritic aggregates are observed usingantibodies against huntingtin (Schilling et al., 1999).
If energy impairment plays a major role in thepathogenesis of HD, then compounds that improveenergy metabolism may reduce neuronal dysfunctionand loss. Creatine has several potential neuroprotec-tive effects, including buffering of intracellular energyreserves, stabilizing intracellular calcium concentra-tions, and inhibiting the activation of the mitochon-drial permeability transition pore (Brdiczka et al., 1998;Hemmer and Wallimann, 1993; Leist and Nicotera,1998; Nicotera and Lipton, 1999; O’Gorman et al.,1996). We previously showed that creatine is protec-tive in mitochondrial toxin models of HD (Matthewset al., 1998), and reported a significant neuroprotectiveeffect of creatine in the R6/2 transgenic mouse modelof HD (Ferrante et al., 2000). In the present study, weexamined whether dietary creatine supplementationdelays the behavioral and neuropathological pheno-type and extends survival in N171-82Q mice.
MATERIALS AND METHODS
Animals
As previously described, transgenic mice express-ing a cDNA encoding an N-terminal fragment (171amino acids) of huntingtin with 82 or 18 glutamineswere used in these studies (Schilling et al., 1999). Maletransgenic mice from the N171-82Q line (line 81),which have 82 CAG repeats (82Q) were bred withfemale B6C3F1 mice (Jackson Laboratories, Bar Har-bor, ME) for 5 generations. As controls, we used wild-type littermates and N171-18Q mice. The N171-18Qline has 18 CAG repeats (18Q) and no underlyingclinical or neuropathological phenotype. They werebred with female B6C3F1 mice (Jackson Laboratories)for 1 generation. The offspring were genotyped with aPCR assay on tail DNA (Schilling et al., 1999). The mice

(8uCaamrw
S
empc
H
sd4awsa
1
Oba2ddsa
titaNmt
481Creatine Effects in Huntington’s Disease Mice
were housed under standard conditions with free ac-cess to water and food.
Treatment
At 4 weeks of age, N171-82Q animals were placedon a diet supplemented with 2% creatine (AvicenaGroup, Cambridge, MA). The amount of food intakeper mouse was found to be 4–5 g per day, with nosignificant difference between treated and untreatedmice. Survival data was obtained from a total of 118unsupplemented and 43 creatine 2% supplementedN171-82Q mice.
Behavior and Weight Assessment
Motor performance was assessed from 90 d of age inunsupplemented N171-82Q (n 5 18) and N171-18Qn 5 8) mice, and in 2% creatine supplemented N171-2Q (n 5 10) mice. The mice were trained for one weeksing a rotarod apparatus (Columbus Instruments,olumbus, OH) and subsequently tested twice a weekt 12 rpm. The time elapsed on the rotarod was useds the measure of the competency to the task. Eachouse was given three 60-s trials, with the best result
ecorded. Mice in each group were weighed once aeek.
urvival
Mice were observed each morning. The mice wereuthanized when they were no longer able to initiateovement after being gently prodded for 2 min. This
oint was the measure of time of death and wasonfirmed by two independent observers.
istology
Groups of 10 2% creatine supplemented and un-upplemented N171-82Q and N171-18Q mice wereeeply anesthetized and transcardially perfused with% paraformaldehyde at 90, 100, 110, 120, 130 days ofge. The brains and pancrei were removed, postfixedith the perfusant for 2 h, cryoprotected in a graded
eries of 10% and 20% glycerol/2% DMSO, and seri-lly frozen sectioned at 50 mm. Brain tissue sections
were subsequently stained for Nissl substance (cresylviolet) and immunostained with an antibody recog-nizing the first 256 amino acids of human huntingtin(EM48, dilution, 1:1000), using a previously describedimmunohistochemical method (Kuemmerle et al.,999). An antibody to ubiquitin (dilution, 1:200, Dako
Corp. Carpintena, CA) was used in tissue sections toconfirm the presence of aggregates.
Stereology/Quantitation
Serial-cut coronal tissue-sections from the rostralsegment of the neostriatum to the level of the anteriorcommissure (interaural 5.34 mm/Bregma 1.54 mm tointeraural 3.7 mm/Bregma 20.10 mm) (Franklin andPaxinos, 1997), including the primary motor cortex,were used for huntingtin aggregate analysis. Unbiasedstereologic counts of huntingtin-positive aggregates($1.0 mm) were obtained from the neostriatum andlayer 2 of the neocortex in 10 animals from unsupple-mented and 2% creatine supplemented N171-82Qmice at 90, 100, 110, 120, and 130 days using Neurolu-cida Stereo Investigator software (Microbrightfield,Colchester, VT). The total areas of the neostriatum andmotor cortex were defined in serial sections in whichcounting frames were randomly sampled. The dissec-tor counting method was employed in which hunting-tin-positive aggregates were counted in an unbiasedselection of serial sections in a defined volume of theneostriatum and neocortex. All computer identifiedprofiles were manually verified as aggregates, ex-ported to Microsoft Excel and analyzed using Stat-view. Striatal neuron areas were analyzed by micro-scopic videocapture using a Windows-based imageanalysis system for area measurement (Optimas, Bio-scan Incorporated, Edmonds, WA). The software au-tomatically identifies and measures profiles.
NMR Spectroscopy
We performed in vivo spectroscopy at 4.7T (GEmega CSI, GE, Freemont, CA) using a sinusoidalird cage coil as described in detail earlier (Jenkins etl., 2000). Magnetic resonance spectroscopy was run in5 N171-82Q mice (13 normal diet and 12 fed with 2%ietary creatine supplementation) between 90 and 105ays of age. Spectra were collected using a PRESSequence with TR 2s and TE 136 ms at 4.7T. Theverage voxel size was 60 ml (6.5 3 3 3 3 mm) cover-
ing both striata and the lateral septum. Mice wereimaged under halothane/N2O2 anesthesia (1.5% halo-hane) and were maintained at 38°C using a circulat-ng water blanket. The data were analyzed by integra-ion of peak areas fitting the peaks to mixed Lorenzi-n–Gaussian lineshapes. Values were determined forAA and creatine using choline as a standard. Nor-alization of the choline values to water spectra from
he same voxel (TR/TE 10 s/10 ms) indicated no sig-
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
bFoNTt
482 Andreassen et al.
A
nificant difference between the choline values in thecreatine supplemented and nonsupplemented mice.
Plasma Glucose Measurements
The mice were lightly anesthetized with isofluranegas and tail vein blood was collected. Glucose wasmeasured with a Lifescan One Touch Basic GlucoseMonitoring System (Lifescan Inc., CA) and validatedby semiautomatic glucose oxidase enzyme assay(Beckman, Fullerton, CA). The glucose measurementswere performed at 2:00–4:00 pm after 6–7 h of fasting.A diabetic glucose was defined as a fasting bloodglucose of more than 188 mg/dl, which was 2 S.D.above those in N171-18Q mice.
Glucose Tolerance Test (GTT)
After 6–7 h of fasting, baseline levels of glucosewere measured. The mice were subsequently given abolus injection of glucose (1.5 g/kg i.p.), and plasmaglucose levels were measured 30 and 60 min later.
Insulin Levels
Insulin levels at 75, 100, and 125 days of age (n 55–8) were measured in duplicate with a rat insulinELISA kit normalized with mouse insulin standards(Chrystal Chem. Inc., Chicago, IL).
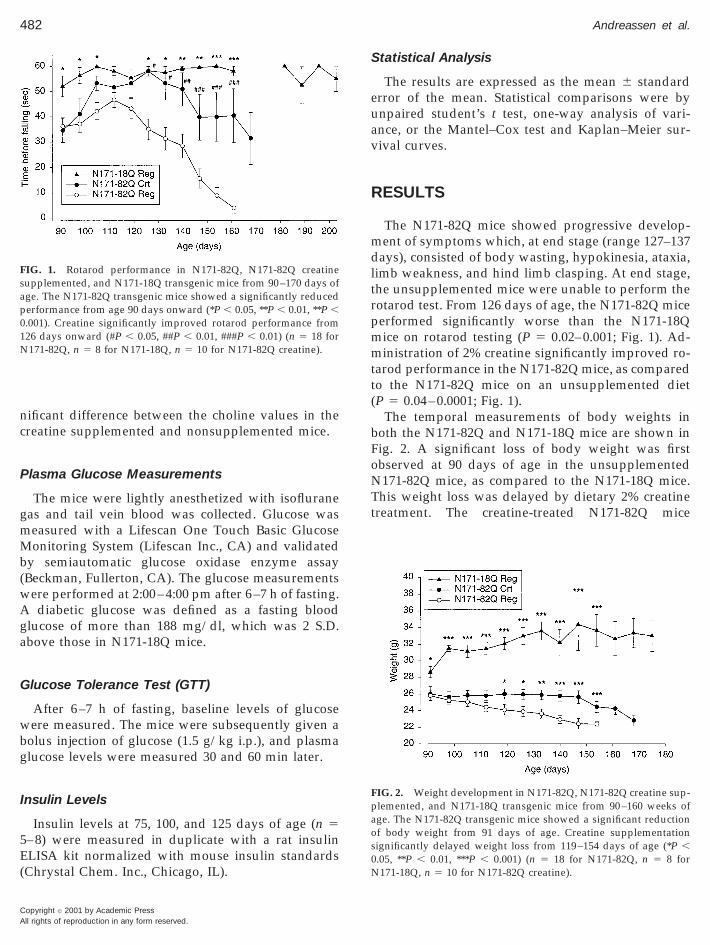
FIG. 1. Rotarod performance in N171-82Q, N171-82Q creatinesupplemented, and N171-18Q transgenic mice from 90–170 days ofage. The N171-82Q transgenic mice showed a significantly reducedperformance from age 90 days onward (*P , 0.05, **P , 0.01, **P ,0.001). Creatine significantly improved rotarod performance from126 days onward (#P , 0.05, ##P , 0.01, ###P , 0.01) (n 5 18 forN171-82Q, n 5 8 for N171-18Q, n 5 10 for N171-82Q creatine).
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
Statistical Analysis
The results are expressed as the mean 6 standarderror of the mean. Statistical comparisons were byunpaired student’s t test, one-way analysis of vari-ance, or the Mantel–Cox test and Kaplan–Meier sur-vival curves.
RESULTS
The N171-82Q mice showed progressive develop-ment of symptoms which, at end stage (range 127–137days), consisted of body wasting, hypokinesia, ataxia,limb weakness, and hind limb clasping. At end stage,the unsupplemented mice were unable to perform therotarod test. From 126 days of age, the N171-82Q miceperformed significantly worse than the N171-18Qmice on rotarod testing (P 5 0.02–0.001; Fig. 1). Ad-ministration of 2% creatine significantly improved ro-tarod performance in the N171-82Q mice, as comparedto the N171-82Q mice on an unsupplemented diet(P 5 0.04–0.0001; Fig. 1).
The temporal measurements of body weights inoth the N171-82Q and N171-18Q mice are shown inig. 2. A significant loss of body weight was firstbserved at 90 days of age in the unsupplemented171-82Q mice, as compared to the N171-18Q mice.his weight loss was delayed by dietary 2% creatine
reatment. The creatine-treated N171-82Q mice
FIG. 2. Weight development in N171-82Q, N171-82Q creatine sup-plemented, and N171-18Q transgenic mice from 90–160 weeks ofage. The N171-82Q transgenic mice showed a significant reductionof body weight from 91 days of age. Creatine supplementationsignificantly delayed weight loss from 119–154 days of age (*P ,0.05, **P , 0.01, ***P , 0.001) (n 5 18 for N171-82Q, n 5 8 forN171-18Q, n 5 10 for N171-82Q creatine).
5wm(
piaN2
1
483Creatine Effects in Huntington’s Disease Mice
weighed significantly more than the unsupplementedN171-82Q mice from 119 days of age (P 5 0.04–0.0007,Fig. 2).
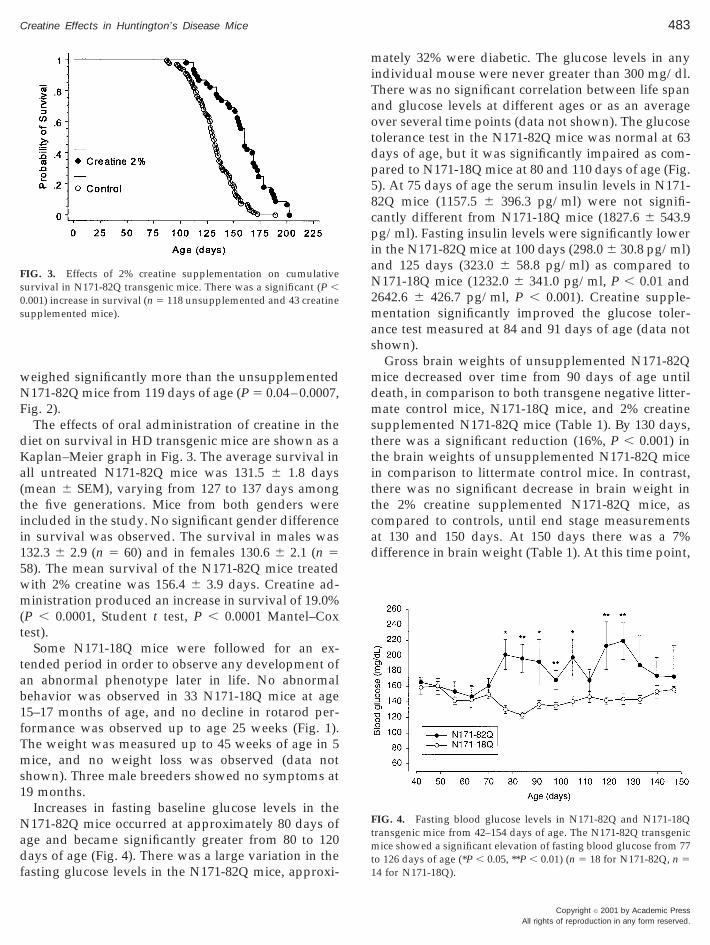
The effects of oral administration of creatine in thediet on survival in HD transgenic mice are shown as aKaplan–Meier graph in Fig. 3. The average survival inall untreated N171-82Q mice was 131.5 6 1.8 days(mean 6 SEM), varying from 127 to 137 days amongthe five generations. Mice from both genders wereincluded in the study. No significant gender differencein survival was observed. The survival in males was132.3 6 2.9 (n 5 60) and in females 130.6 6 2.1 (n 58). The mean survival of the N171-82Q mice treatedith 2% creatine was 156.4 6 3.9 days. Creatine ad-inistration produced an increase in survival of 19.0%
P , 0.0001, Student t test, P , 0.0001 Mantel–Coxtest).
Some N171-18Q mice were followed for an ex-tended period in order to observe any development ofan abnormal phenotype later in life. No abnormalbehavior was observed in 33 N171-18Q mice at age15–17 months of age, and no decline in rotarod per-formance was observed up to age 25 weeks (Fig. 1).The weight was measured up to 45 weeks of age in 5mice, and no weight loss was observed (data notshown). Three male breeders showed no symptoms at19 months.
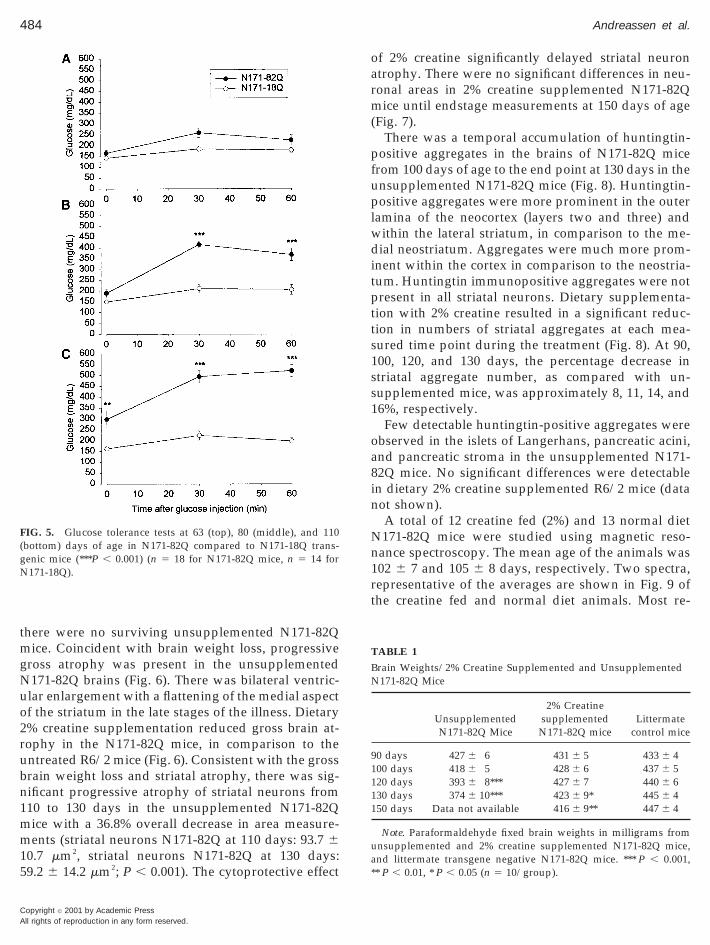
Increases in fasting baseline glucose levels in theN171-82Q mice occurred at approximately 80 days ofage and became significantly greater from 80 to 120days of age (Fig. 4). There was a large variation in thefasting glucose levels in the N171-82Q mice, approxi-
FIG. 3. Effects of 2% creatine supplementation on cumulativesurvival in N171-82Q transgenic mice. There was a significant (P ,0.001) increase in survival (n 5 118 unsupplemented and 43 creatinesupplemented mice).
mately 32% were diabetic. The glucose levels in anyindividual mouse were never greater than 300 mg/dl.There was no significant correlation between life spanand glucose levels at different ages or as an averageover several time points (data not shown). The glucosetolerance test in the N171-82Q mice was normal at 63days of age, but it was significantly impaired as com-pared to N171-18Q mice at 80 and 110 days of age (Fig.5). At 75 days of age the serum insulin levels in N171-82Q mice (1157.5 6 396.3 pg/ml) were not signifi-cantly different from N171-18Q mice (1827.6 6 543.9
g/ml). Fasting insulin levels were significantly lowern the N171-82Q mice at 100 days (298.0 6 30.8 pg/ml)nd 125 days (323.0 6 58.8 pg/ml) as compared to171-18Q mice (1232.0 6 341.0 pg/ml, P , 0.01 and
642.6 6 426.7 pg/ml, P , 0.001). Creatine supple-mentation significantly improved the glucose toler-ance test measured at 84 and 91 days of age (data notshown).
Gross brain weights of unsupplemented N171-82Qmice decreased over time from 90 days of age untildeath, in comparison to both transgene negative litter-mate control mice, N171-18Q mice, and 2% creatinesupplemented N171-82Q mice (Table 1). By 130 days,there was a significant reduction (16%, P , 0.001) inthe brain weights of unsupplemented N171-82Q micein comparison to littermate control mice. In contrast,there was no significant decrease in brain weight inthe 2% creatine supplemented N171-82Q mice, ascompared to controls, until end stage measurementsat 130 and 150 days. At 150 days there was a 7%difference in brain weight (Table 1). At this time point,
FIG. 4. Fasting blood glucose levels in N171-82Q and N171-18Qtransgenic mice from 42–154 days of age. The N171-82Q transgenicmice showed a significant elevation of fasting blood glucose from 77to 126 days of age (*P , 0.05, **P , 0.01) (n 5 18 for N171-82Q, n 54 for N171-18Q).
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
484 Andreassen et al.
A
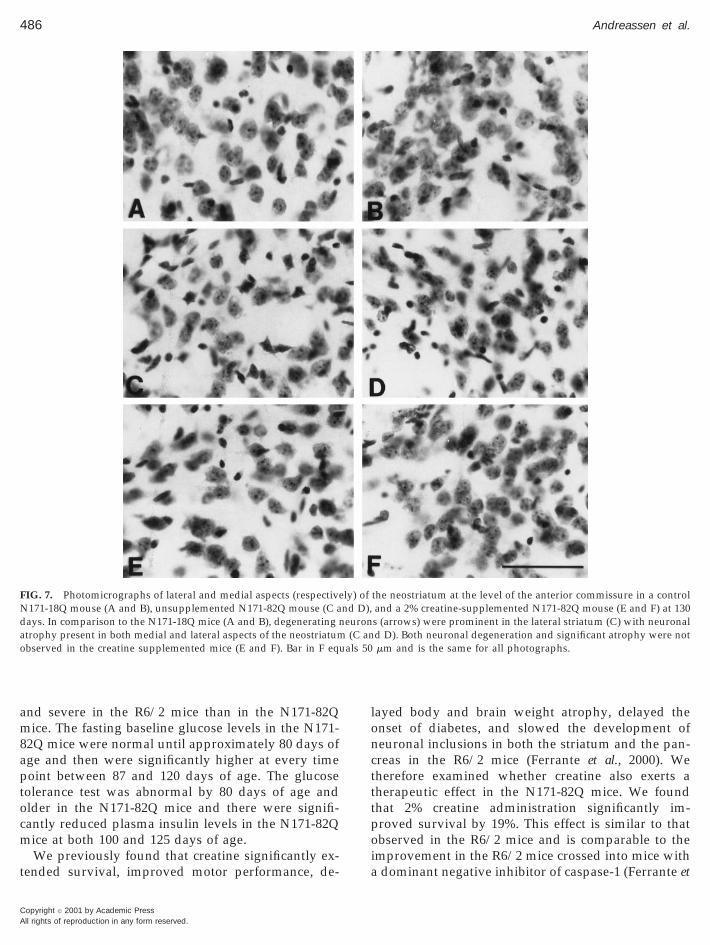
there were no surviving unsupplemented N171-82Qmice. Coincident with brain weight loss, progressivegross atrophy was present in the unsupplementedN171-82Q brains (Fig. 6). There was bilateral ventric-ular enlargement with a flattening of the medial aspectof the striatum in the late stages of the illness. Dietary2% creatine supplementation reduced gross brain at-rophy in the N171-82Q mice, in comparison to theuntreated R6/2 mice (Fig. 6). Consistent with the grossbrain weight loss and striatal atrophy, there was sig-nificant progressive atrophy of striatal neurons from110 to 130 days in the unsupplemented N171-82Qmice with a 36.8% overall decrease in area measure-ments (striatal neurons N171-82Q at 110 days: 93.7 610.7 mm2, striatal neurons N171-82Q at 130 days:59.2 6 14.2 mm2; P , 0.001). The cytoprotective effect
FIG. 5. Glucose tolerance tests at 63 (top), 80 (middle), and 110(bottom) days of age in N171-82Q compared to N171-18Q trans-genic mice (***P , 0.001) (n 5 18 for N171-82Q mice, n 5 14 forN171-18Q).
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
of 2% creatine significantly delayed striatal neuronatrophy. There were no significant differences in neu-ronal areas in 2% creatine supplemented N171-82Qmice until endstage measurements at 150 days of age(Fig. 7).
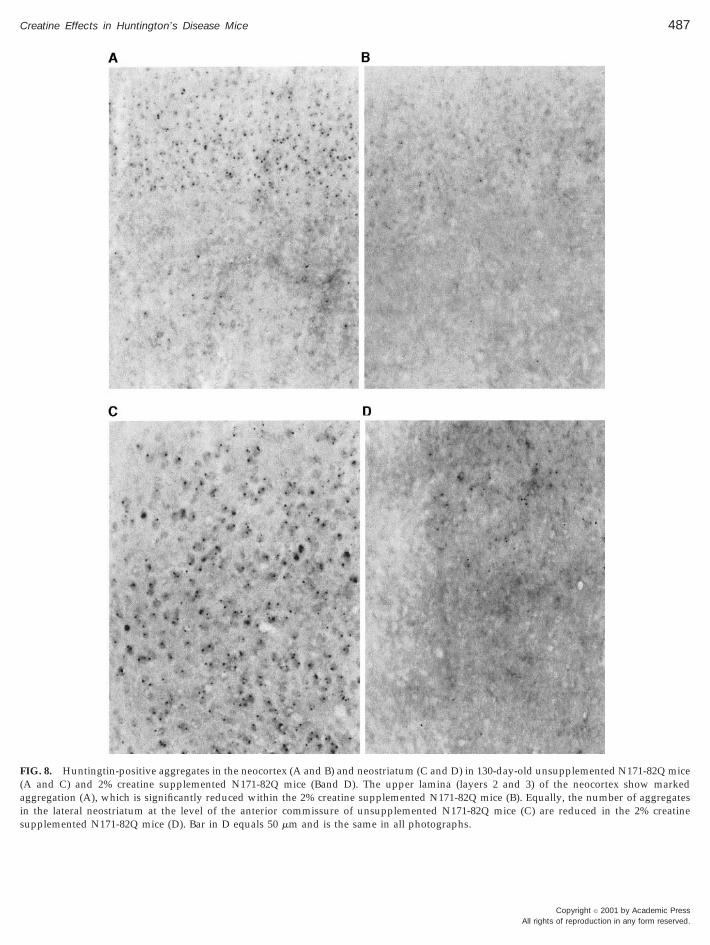
There was a temporal accumulation of huntingtin-positive aggregates in the brains of N171-82Q micefrom 100 days of age to the end point at 130 days in theunsupplemented N171-82Q mice (Fig. 8). Huntingtin-positive aggregates were more prominent in the outerlamina of the neocortex (layers two and three) andwithin the lateral striatum, in comparison to the me-dial neostriatum. Aggregates were much more prom-inent within the cortex in comparison to the neostria-tum. Huntingtin immunopositive aggregates were notpresent in all striatal neurons. Dietary supplementa-tion with 2% creatine resulted in a significant reduc-tion in numbers of striatal aggregates at each mea-sured time point during the treatment (Fig. 8). At 90,100, 120, and 130 days, the percentage decrease instriatal aggregate number, as compared with un-supplemented mice, was approximately 8, 11, 14, and16%, respectively.
Few detectable huntingtin-positive aggregates wereobserved in the islets of Langerhans, pancreatic acini,and pancreatic stroma in the unsupplemented N171-82Q mice. No significant differences were detectablein dietary 2% creatine supplemented R6/2 mice (datanot shown).
A total of 12 creatine fed (2%) and 13 normal dietN171-82Q mice were studied using magnetic reso-nance spectroscopy. The mean age of the animals was102 6 7 and 105 6 8 days, respectively. Two spectra,representative of the averages are shown in Fig. 9 ofthe creatine fed and normal diet animals. Most re-
TABLE 1
Brain Weights/2% Creatine Supplemented and UnsupplementedN171-82Q Mice
UnsupplementedN171-82Q Mice
2% Creatinesupplemented
N171-82Q miceLittermate
control mice
90 days 427 6 6 431 6 5 433 6 4100 days 418 6 5 428 6 6 437 6 5120 days 393 6 8*** 427 6 7 440 6 6130 days 374 6 10*** 423 6 9* 445 6 4150 days Data not available 416 6 9** 447 6 4
Note. Paraformaldehyde fixed brain weights in milligrams fromunsupplemented and 2% creatine supplemented N171-82Q mice,and littermate transgene negative N171-82Q mice. *** P , 0.001,** P , 0.01, * P , 0.05 (n 5 10/group).
erav
igmavstmtTddv
485Creatine Effects in Huntington’s Disease Mice
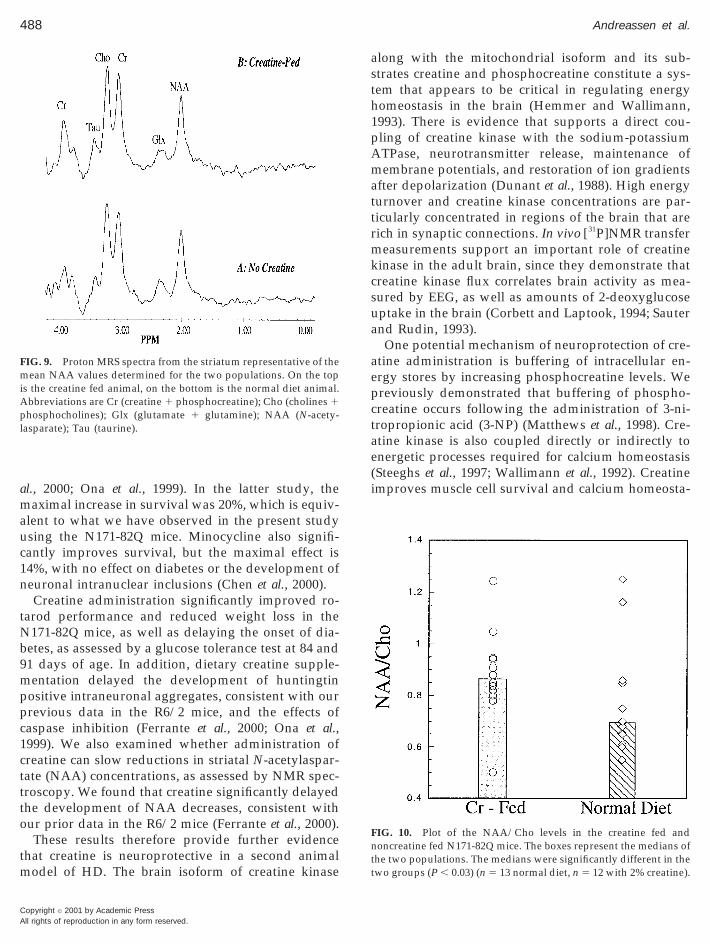
markable in the spectra, compared to the N171-18Qanimals, is a decrease in the NAA. The mean NAA/Cho ratio was 0.88 6 0.17, 0.76 6 0.2, and 1.15 6 0.2 inthe creatine fed, normal diet, and N171-18Q normaldiet mice, respectively. Creatine has no effects onNAA/Cho in normal mice (data not shown). Shown inFig. 10 are the NAA/Cho values in the creatine fedand normal diet animals. An appropriate nonparamet-ric test (Komolgorov–Smirnov test) showed the twogroups were significantly different (P , 0.02). Themedians of the two groups showed a larger differencethan did the means (0.87 vs 0.69 for the creatine fedand normal diet, respectively). A distribution-free per-mutation test of the medians also showed the twogroups were significantly different (P , 0.03). Themean increase in the creatine levels in the brains of thecreatine fed mice based upon mean increases in thecreatine peaks at 3.03 and 3.96 ppm was 13.3 6 5.9%.
DISCUSSION
A major development in studying the pathogenesisof neurodegenerative diseases has been the develop-ment of transgenic mouse models. They are particu-larly valuable in studying the effects of experimental
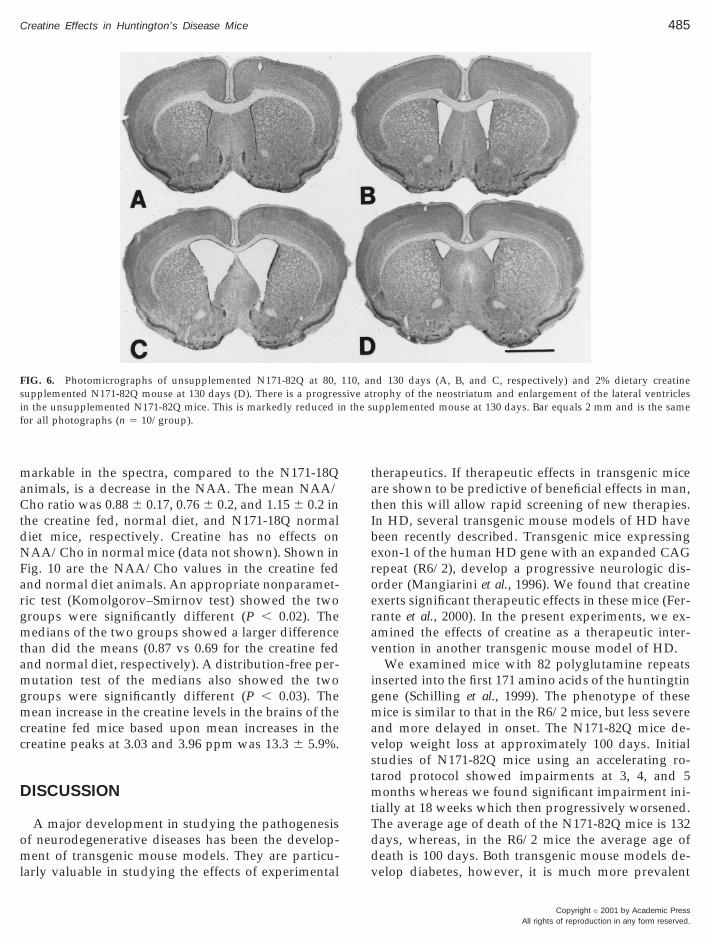
FIG. 6. Photomicrographs of unsupplemented N171-82Q at 80,supplemented N171-82Q mouse at 130 days (D). There is a progresin the unsupplemented N171-82Q mice. This is markedly reduced ifor all photographs (n 5 10/group).
therapeutics. If therapeutic effects in transgenic miceare shown to be predictive of beneficial effects in man,then this will allow rapid screening of new therapies.In HD, several transgenic mouse models of HD havebeen recently described. Transgenic mice expressingexon-1 of the human HD gene with an expanded CAGrepeat (R6/2), develop a progressive neurologic dis-order (Mangiarini et al., 1996). We found that creatinexerts significant therapeutic effects in these mice (Fer-ante et al., 2000). In the present experiments, we ex-mined the effects of creatine as a therapeutic inter-ention in another transgenic mouse model of HD.We examined mice with 82 polyglutamine repeats
nserted into the first 171 amino acids of the huntingtinene (Schilling et al., 1999). The phenotype of theseice is similar to that in the R6/2 mice, but less severe
nd more delayed in onset. The N171-82Q mice de-elop weight loss at approximately 100 days. Initialtudies of N171-82Q mice using an accelerating ro-arod protocol showed impairments at 3, 4, and 5
onths whereas we found significant impairment ini-ially at 18 weeks which then progressively worsened.he average age of death of the N171-82Q mice is 132ays, whereas, in the R6/2 mice the average age ofeath is 100 days. Both transgenic mouse models de-elop diabetes, however, it is much more prevalent
d 130 days (A, B, and C, respectively) and 2% dietary creatinerophy of the neostriatum and enlargement of the lateral ventriclesupplemented mouse at 130 days. Bar equals 2 mm and is the same
110, ansive atn the s
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
tttpoia
486 Andreassen et al.
A
and severe in the R6/2 mice than in the N171-82Qmice. The fasting baseline glucose levels in the N171-82Q mice were normal until approximately 80 days ofage and then were significantly higher at every timepoint between 87 and 120 days of age. The glucosetolerance test was abnormal by 80 days of age andolder in the N171-82Q mice and there were signifi-cantly reduced plasma insulin levels in the N171-82Qmice at both 100 and 125 days of age.
We previously found that creatine significantly ex-tended survival, improved motor performance, de-
FIG. 7. Photomicrographs of lateral and medial aspects (respectiveN171-18Q mouse (A and B), unsupplemented N171-82Q mouse (C adays. In comparison to the N171-18Q mice (A and B), degenerating natrophy present in both medial and lateral aspects of the neostriatumobserved in the creatine supplemented mice (E and F). Bar in F eq
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
layed body and brain weight atrophy, delayed theonset of diabetes, and slowed the development ofneuronal inclusions in both the striatum and the pan-creas in the R6/2 mice (Ferrante et al., 2000). Weherefore examined whether creatine also exerts aherapeutic effect in the N171-82Q mice. We foundhat 2% creatine administration significantly im-roved survival by 19%. This effect is similar to thatbserved in the R6/2 mice and is comparable to the
mprovement in the R6/2 mice crossed into mice withdominant negative inhibitor of caspase-1 (Ferrante et
the neostriatum at the level of the anterior commissure in a controland a 2% creatine-supplemented N171-82Q mouse (E and F) at 130s (arrows) were prominent in the lateral striatum (C) with neuronald D). Both neuronal degeneration and significant atrophy were notmm and is the same for all photographs.
ly) ofnd D),euron(C an
uals 50
487Creatine Effects in Huntington’s Disease Mice
FIG. 8. Huntingtin-positive aggregates in the neocortex (A and B) and neostriatum (C and D) in 130-day-old unsupplemented N171-82Q mice(A and C) and 2% creatine supplemented N171-82Q mice (Band D). The upper lamina (layers 2 and 3) of the neocortex show markedaggregation (A), which is significantly reduced within the 2% creatine supplemented N171-82Q mice (B). Equally, the number of aggregatesin the lateral neostriatum at the level of the anterior commissure of unsupplemented N171-82Q mice (C) are reduced in the 2% creatinesupplemented N171-82Q mice (D). Bar in D equals 50 mm and is the same in all photographs.
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
1cttto
tm
asth1pAmattr
ae(i
miApl
488 Andreassen et al.
A
al., 2000; Ona et al., 1999). In the latter study, themaximal increase in survival was 20%, which is equiv-alent to what we have observed in the present studyusing the N171-82Q mice. Minocycline also signifi-cantly improves survival, but the maximal effect is14%, with no effect on diabetes or the development ofneuronal intranuclear inclusions (Chen et al., 2000).
Creatine administration significantly improved ro-tarod performance and reduced weight loss in theN171-82Q mice, as well as delaying the onset of dia-betes, as assessed by a glucose tolerance test at 84 and91 days of age. In addition, dietary creatine supple-mentation delayed the development of huntingtinpositive intraneuronal aggregates, consistent with ourprevious data in the R6/2 mice, and the effects ofcaspase inhibition (Ferrante et al., 2000; Ona et al.,999). We also examined whether administration ofreatine can slow reductions in striatal N-acetylaspar-ate (NAA) concentrations, as assessed by NMR spec-roscopy. We found that creatine significantly delayedhe development of NAA decreases, consistent withur prior data in the R6/2 mice (Ferrante et al., 2000).These results therefore provide further evidence
hat creatine is neuroprotective in a second animalodel of HD. The brain isoform of creatine kinase
FIG. 9. Proton MRS spectra from the striatum representative of theean NAA values determined for the two populations. On the top
s the creatine fed animal, on the bottom is the normal diet animal.bbreviations are Cr (creatine 1 phosphocreatine); Cho (cholines 1hosphocholines); Glx (glutamate 1 glutamine); NAA (N-acety-
asparate); Tau (taurine).
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
long with the mitochondrial isoform and its sub-trates creatine and phosphocreatine constitute a sys-em that appears to be critical in regulating energyomeostasis in the brain (Hemmer and Wallimann,993). There is evidence that supports a direct cou-ling of creatine kinase with the sodium-potassiumTPase, neurotransmitter release, maintenance ofembrane potentials, and restoration of ion gradients
fter depolarization (Dunant et al., 1988). High energyurnover and creatine kinase concentrations are par-icularly concentrated in regions of the brain that areich in synaptic connections. In vivo [31P]NMR transfer
measurements support an important role of creatinekinase in the adult brain, since they demonstrate thatcreatine kinase flux correlates brain activity as mea-sured by EEG, as well as amounts of 2-deoxyglucoseuptake in the brain (Corbett and Laptook, 1994; Sauterand Rudin, 1993).
One potential mechanism of neuroprotection of cre-atine administration is buffering of intracellular en-ergy stores by increasing phosphocreatine levels. Wepreviously demonstrated that buffering of phospho-creatine occurs following the administration of 3-ni-tropropionic acid (3-NP) (Matthews et al., 1998). Cre-tine kinase is also coupled directly or indirectly tonergetic processes required for calcium homeostasisSteeghs et al., 1997; Wallimann et al., 1992). Creatinemproves muscle cell survival and calcium homeosta-
FIG. 10. Plot of the NAA/Cho levels in the creatine fed andnoncreatine fed N171-82Q mice. The boxes represent the medians ofthe two populations. The medians were significantly different in thetwo groups (P , 0.03) (n 5 13 normal diet, n 5 12 with 2% creatine).

B
B
489Creatine Effects in Huntington’s Disease Mice
sis of dystrophic myotubes in culture (Pulido et al.,1998). Creatine also protects against glutamate andb-amyloid toxicity in cultured hippocampal neurons(Brewer and Wallimann, 2000). In addition, creatinehas direct effects on glutamate uptake, and therebycan reduce extracellular glutamate concentrations,which may contribute to its neuroprotective effects(Xu et al., 1996).
Another neuroprotective mechanism of creatinemay be by inhibiting activation of the mitochondrialpermeability transition (MPT). A critical role of theMPT has been suggested for both apoptotic and ne-crotic cell death (Bernardi et al., 1998). Mitochondrialcreatine kinase has been implicated as interacting be-tween the outer membrane voltage dependent anionchannel and the adenine nucleotide translocator in theinner membrane, which are thought to be componentsof the MPT (Brdiczka et al., 1998). Creatine adminis-tration can stabilize the mitochondrial creatine kinasein an octomeric form, which then inhibits activation ofthe MPT (O’Gorman et al., 1996). Creatine administra-tion also results in increased ADP concentrationswhich inhibit the activation of the MPT (Bernardi et al.,1998).
We previously found neuroprotective effects of cre-atine against 3-NP, malonate and MPTP induced neu-rotoxicity (Matthews et al., 1999; Matthews et al., 1998).We also found that creatine significantly improvedsurvival and delayed loss of anterior horn motor neu-rons in a transgenic mouse model of ALS (Klivenyi etal., 1999). In the present study, we found that creatineimproves survival, delays the onset of diabetes, delaysmotor deficits, and delays the development of neuro-pathologic damage in the N171-82Q transgenic mousemodel of HD. These studies demonstrate that creatineadministration exerts neuroprotective effects in twodifferent transgenic mouse models of HD, the resultsof which are remarkably congruent. These findingsfurther suggest that treatment with creatine may be auseful therapeutic strategy to slow or halt the progres-sion of neurodegeneration in HD.
ACKNOWLEDGMENTS
This work was supported by The Hereditary Disease Foundation,The Huntington Disease Society of America, The Department ofDefense and NIH Grants NS38180 (M.F.B.), NS35255 (R.J.F. andS.M.H.), NS37102, and AG13846 (R.J.F.), and AG12992 (M.F.B. andR.J.F.), AT00613 (R.J.F., S.M.H., and M.F.B.), NS16375 (C.A.R. andD.R.B.), and NS38144 (C.A.R.) the Veterans Administration (R.J.F.),and the Norwegian Research Council (O.A.A.). The secretarial as-sistance of Sharon Melanson and technical support of Karen Smithand John R. Schleicher are gratefully acknowledged.
REFERENCES
Andrew, S. E., Goldberg, Y. P., Kremer, B., Telenius, H., Theilmann,J., Adam, S., Starr, E., Squitieri, F., Lin, B., Kalchman, M. A.,Graham, R. K., & Hayden, M. R. (1993) The relationship betweentrinucleotide (CAG) repeat length and clinical features of Hun-tington’s disease. Nature Genet. 4, 398–403.
Beal, M. F. (2000) Energetics in the pathogenesis of neurodegenera-tive diseases. Trends Neurosci. 23, 294–300.
Beal, M. F., Hyman, B. T., & Koroshetz, W. J. (1993) Do defects inmitochondrial energy metabolism underlie the pathology of neu-rodegenerative diseases. Trends Neurosci. 16, 125–131.
Bernardi, P., Basso, E., Colonna, R., Costantini, P., Di Lisa, F.,Eriksson, O., Fontaine, E., Forte, M., Ichas, F., Massari, S., Nicolli,A., Petronilli, V., & Scorrano, L. (1998) Perspectives of the mito-chondrial permeability transition. Biochim. Biophys. Acta 1365,200–206.
Bogdanov, M. B., Ferrante, R. J., Kuemmerle, S., Klivenyi, P., & Beal,M. F. (1998) Increased vulnerability to 3-nitropropionic acid in ananimal model of Huntington’s disease. J. Neurochem. 71, 2642–2644.
rdiczka, D., Beutner, G., Ruck, A., Dolder, M., & Wallimann, T.(1998) The molecular structure of mitochondrial contact sites.Their role in regulation of energy metabolism and permeabilitytransition. Biofactors 8, 235–242.
rewer, G. J., & Wallimann, T. W. (2000) Protective effect of theenergy precursor creatine against toxicity of glutamate andb-amyloid in rat hippocampal neurons. J. Neurochem. 74, 1968–1978.
Brouillet, E., Hantraye, P., Ferrante, R. J., Dolan, R., Leroy-Willig, A.,Kowall, N. W., & Beal, M. F. (1995) Chronic mitochondrial energyimpairment produces selective striatal degeneration and abnor-mal choreiform movements in primates. Proc. Natl. Acad. Sci. USA92, 7105–7109.
Browne, S. E., Bowling, A. C., MacGarvey, U., Baik, M. J., Berger,S. C., Muqit, M. M. K., Bird, E. D., & Beal, M. F. (1997) Oxidativedamage and metabolic dysfunction in Huntington’s disease: Se-lective vulnerability of the basal ganglia. Ann. Neurol. 41, 646–653.
Chen, M., Ona, V. O., Li, M., Ferrante, R. J., Fink, K. B., Zhu, S., Bian,J., Guo, L., Farrell, L. A., Hersch, S. M., Hobbs, W., Vonsattel, J. P.,Cha, J. H., & Friedlander, R. M. (2000) Minocycline inhibitscaspase-1 and caspase-3 expression and delays mortality in atransgenic mouse model of huntington disease. Nature Med. 6,797–801.
Corbett, R. J. T., & Laptook, A. R. (1994) Age-related changes inswine brain creatine kinase-catalyzed 31P exchange measured invivo using 31P NMR magnetization transfer. J. Cereb. Blood FlowMetab. 14, 1070–1077.
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P.,Vonsattel, J. P., & Aronin, N. (1997) Aggregation of huntingtin inneuronal intranuclear inclusions and dystrophic neurites in brain.Science 277, 1990–1993.
Dunant, Y., Loctin, F., Marsal, J., Muller, D., Parducz, A., & Ra-basseda, X. (1988) Energy metabolism and quantal acetylcholinerelease: Effects of botulinum toxin, 1-fluoro-2,4-dinitrobenzene,and diamide in the Torpedo electric organ. J. Neurochem. 50, 431–439.
Duyao, M., Ambrose, C., Myers, R., Novelletto, A., Persichetti, F.,Frontali, M., Folstein, S., Ross, C., Franz, M., Abbott, M., Gray, J.,Conneally, P., Young, A., Penney, J., Hollingsworth, Z., Shoulson,I., Lazzarini, A., Falek, A., Koroshetz, W., Sax, D., Bird, E., Von-
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.

F
G
G
H
H
J
J
K
K
K
K
K
L
L
M
M
M
N
O
O
P
R
R
S
S
S
S
S
S
490 Andreassen et al.
A
sattel, J., Bonilla, E., Alvir, J., Bickham Gonde, J., Cha, J.-H., Dure,L., Gomez, F., Ramos, M., Sanchez-Ramos, J., Snodgrass, S., deYoung, M., Wexler, N., Moscowitz, C., Penchaszadeh, G., Mac-Farlane, H., Anderson, M., Jenkins, B., Srinidhi, J., Barnes, G.,Gusella, J., & MacDonald, M. (1993) Trinucleotide repeat lengthinstability and age of onset in Huntington’s disease. Nature Genet.4, 387–392.
Ferrante, R. J., Andreassen, O. A., Jenkins, B. G., Dedeoglu, A.,Kuemmerle, S., Kubilus, J. K., Kaddurah-Daouk, R., Hersch, S. M.,& Beal, M. F. (2000) Neuroprotective effects of creatine in atransgenic mouse model of Huntington’s disease. J. Neurosci. 20,4389–4397.
ranklin, K. B. J., & Paxinos, G. (1997) The Mouse Brain in StereotaxicCoordinates. Academic Press.u, M., Gash, M. T., Mann, V. M., Javoy-Agid, F., Cooper, J. M., &Schapira, A. H. V. (1996) Mitochondrial defect in Huntington’sdisease caudate nucleus. Ann. Neurol. 39, 385–389.utekunst, C. A., Li, S. H., Yi, H., Mulroy, J. S., Kuemmerle, S.,Jones, R., Rye, D., Ferrante, R. J., Hersch, S. M., & Li, X. J. (1999)Nuclear and neuropil aggregates in Huntington’s disease: Rela-tionship to neuropathology. J. Neurosci. 19, 2522–2534.emmer, W., & Wallimann, T. (1993) Functional aspects of creatinekinase in brain. Dev. Neurosci. 15, 249–260.ersch, S. M., & Ferrante, R. J. (1997) Neuropathology and patho-physiology of Huntington’s disease. In: Movement Disorders: Neu-rologic Principles and Practice (R. L. Watts and W. C. Koller, Eds.),pp. 503–518. McGraw-Hill, New York.
enkins, B. G., Klivenyi, P., Kustermann, E., Andreassen, O. A.,Ferrante, R. J., Rosen, B. R., & Beal, M. F. (2000) Exponentialdecrease over time in n-acetylaspartate levels in the absence ofneuronal loss and increases in glutamine and glucose in trans-genic Huntington’s disease mice. J. Neurochem. 74, 2108 –2119.
enkins, B. G., Koroshetz, W. J., Beal, M. F., & Rosen, B. R. (1993)Evidence for impairment of energy metabolism in vivo in Hun-tington’s disease using localized 1H NMR spectroscopy. Neurol-ogy 43, 2689–2695.
livenyi, P., Ferrante, R. J., Matthews, R. T., Bogdanov, M. B., Klein,A. M., Andreassen, O. A., Mueller, G., Werner, M., Kaddurah-Daouk, R., & Beal, M. F. (1999) Neuroprotective effects of creatinein a transgenic animal model of amyotrophic lateral sclerosis.Nature Med. 5, 347–350.
oroshetz, W. J., Jenkins, B. G., Rosen, B. R., & Beal, M. F. (1997)Energy metabolism defects in Huntington’s disease and effects ofcoenzyme Q10. Ann. Neurol. 41, 160–165.
owall, N. W., & Ferrante, R. J. (1998) Huntington’s disease. In:Neuropathology of Dementing Disorders (M. Markesbery, Ed.), pp.219–256. Edward Arnold, London, England.
owall, N. W., Kuemmerle, S., & Ferrante, R. J. (1999) The geneticbasis and molecular pathogenesis of Huntington’s disease. In:Advances in Cell Aging and Gerontology (M. Mattson, Ed.), Vol 3,pp. 81–92. JAI Press, Stamford, CT.
uemmerle, S., Gutekunst, C. A., Klein, A. M., Li, X. J., Li, S. H.,Beal, M. F., Hersch, S. M., & Ferrante, R. J. (1999) Huntingtinaggregates may not predict neuronal death in Huntington’s dis-ease. Ann. Neurol. 46, 842–849.
eist, M., & Nicotera, P. (1998) Calcium and neuronal death. Rev.Physiol. Biochem. Pharmacol. 132, 79–125.
odi, R., Schapira, A. H., Manners, D., Styles, P., Wood, N. W.,Taylor, D. J., & Warner, T. T. (2000) Abnormal in vivo skeletalmuscle energy metabolism in Huntington’s disease and denta-torubropallidoluysian atrophy. Ann. Neurol. 48, 72–76.
Copyright © 2001 by Academic Pressll rights of reproduction in any form reserved.
angiarini, L., Sathasivam, K., Seller, M., Cozens, B., Harper, A.,Hetherington, C., Lawton, M., Trottier, Y., Lehrach, H., Davies,S. W., & Bates, G. P. (1996) Exon 1 of the HD gene with anexpanded CAG repeat is sufficient to cause a progressive neu-rological phenotype in transgenic mice. Cell 87, 493–506.atthews, R. T., Ferrante, R. J., Klivenyi, P., Yang, L., Klein, A. M.,Mueller, G., Kaddurah-Daouk, R., & Beal, M. F. (1999) Creatineand cyclocreatine attenuate MPTP neurotoxicity. Exp. Neurol. 157,142–149.atthews, R. T., Yang, L., Jenkins, B. G., Ferrante, R. J., Rosen, B. R.,Kaddurah-Daouk, R., & Beal, M. F. (1998) Neuroprotective effectsof creatine and cyclocreatine in animal models of Huntington’sdisease. J. Neurosci. 18, 156–163.icotera, P., & Lipton, S. A. (1999) Excitotoxins in neuronal apopto-sis and necrosis. J. Cereb. Blood Flow Metab. 19, 583–591.’Gorman, E., Beutner, G., Wallimann, T., & Brdiczka, D. (1996)Differential effects of creatine depletion on the regulation of en-zyme activities and on creatine-stimulated mitochondrial respira-tion in skeletal muscle, heart, and brain. Biochim. Biophys. Acta1276, 161–170.na, V. O., Li, M., Vonsattel, J. P., Andrews, L. J., Khan, S. Q.,Chung, W. M., Frey, A. S., Menon, A. S., Li, X. J., Stieg, P. E.,Yuan, J., Penney, J. B., Young, A. B., Cha, J. H., & Friedlander,R. M. (1999) Inhibition of caspase-1 slows disease progressionin a mouse model of Huntington’s disease. Nature 399, 263–267.
ulido, S. M., Passaquin, A. C., Leijendekker, W. J., Challet, C.,Wallimann, T., & Ruegg, U. T. (1998) Creatine supplementationimproves intracellular Ca21 handling and survival in mdx skel-etal muscle cells. FEBS Lett. 439, 357–362.
oss, C. A. (1997) Intranuclear neuronal inclusions: A commonpathogenic mechanism for glutamine-repeat neurodegenerativediseases? Neuron 19, 1147–1150.
oss, C. A., Margolis, R. L., Becher, M. W., Wood, J. D., Enge-lender, S., Cooper, J. K., & Sharp, A. H. (1998) Pathogenesis ofneurodegenerative diseases associated with expanded glu-tamine repeats: New answers, new questions. Prog. Brain Res.117, 397– 419.
auter, A., & Rudin, M. (1993) Determination of creatine kinaseparameters in rat brain by NMR magnetization transfer: Correla-tion with brain function. J. Biol. Chem. 268, 13166–13171.
awa, A., Wiegand, G. W., Cooper, J., Margolis, R. L., Sharp, A. H.,Lawler, J. F., Jr., Greenamyre, J. T., Snyder, S. H., & Ross, C. A.(1999) Increased apoptosis of Huntington disease lymphoblastsassociated with repeat length-dependent mitochondrial depolar-ization. Nature Med. 5, 1194–1198.
chilling, G., Becher, M. W., Sharp, A. H., Jinnah, H. A., Duan, K.,Kotzuk, J. A., Slunt, H. H., Ratovitski, T., Cooper, J. K., Jenkins,N. A., Copeland, N. G., Price, D. L., Ross, C. A., & Borchelt, D. R.(1999) Intranuclear inclusions and neuritic aggregates in trans-genic mice expressing a mutant N-terminal fragment of hunting-tin. Hum. Mol. Genet. 8, 397–407.
isodia, S. S. (1998) Nuclear inclusions in glutamine repeat disor-ders: Are they pernicious, coincidental, or beneficial? [Comment].Cell 95, 1–4.
nell, R. G., MacMillan, J. C., Cheadle, J. P., Fenton, I., Lazarou, L. P.,Davies, P., MacDonald, M. E., Gusella, J. F., Harper, P. S., & Shaw,D. J. (1993) Relationship between trinucleotide repeat expansionand phenotypic variation in Huntington’s disease. Nature Genet. 4,393–397.
teeghs, K., Benders, A., Oerlemans, F., de Haan, A., Heerschap, A.,Ruitenbeek, W., Jost, C., van Deursen, J., Perryman, B., Pette, D.,Bruckwilder, M., Koudijs, J., Jap, P., Veerkamp, J., & Wieringa, B.

W
X
491Creatine Effects in Huntington’s Disease Mice
(1997) Altered Ca21 responses in muscles with combined mitochon-drial and cytosolic creatine kinase deficiencies. Cell 89, 93–103.
Tabrizi, S. J., Workman, J., Hart, P. E., Mangiarini, L., Mahal, A.,Bates, G., Cooper, J. M., & Schapira, A. H. (2000) Mitochondrialdysfunction and free radical damage in the Huntington R6/2transgenic mouse. Ann. Neurol. 47, 80–86.
The Huntington’s Disease Collaborative Research Group (1993) Anovel gene containing a trinucleotide repeat that is expanded andunstable on Huntington’s disease chromosomes. Cell 72, 971–983.
allimann, T., Wyss, M., Brdiczka, D., Nicolay, K., & Eppen-berger, H. M. (1992) Intracellular compartmentation, structureand function of creatine kinase isoenzymes in tissues with highand fluctuating energy demands: The ‘phosphocreatine circuit’for cellular energy homeostasis. Biochem. J. 281, 21– 40.
u, C. J., Klunk, W. E., Kanfer, J. N., Xiong, Q., Miller, G.,& Pettegrew, J. W. (1996) Phosphocreatine-dependent gluta-mate uptake by synaptic vesicles. J. Biol. Chem. 271, 13435–13440.
Copyright © 2001 by Academic PressAll rights of reproduction in any form reserved.
Copyright © 2022 FDOKUMEN




















