Relativistic Methods for Calculating Electron Paramagnetic ...
Upload
independentCategory
view
5download
0
Appl. Magn. Reson. 30, 103-120 (2006) AppUed Magnetlc Resonance �9 Springer-Verlag 2006 Printed in Austria
Evidence for the Deve lopment of Persistent Carbon-Centered Radicals from a Benzyl Phenol ic Antioxidant: an Electron
Paramagnet ic Resonance Study
S. N. Mendiara and M. E. J. Coronel
Departamento de QuŸ Universidad Nacional de Mar del Plata, Mar del Plata, Argentina
Received December 15, 2005; revised February 8, 2006
Abstract. Radicals have been generated from the benzyl phenolic antioxidant 1,3,5-trimethyl-2,4,6- tris(3,5-di-tert-butyl-4-hydroxybenzyl) benzene, carrying out oxidative and hydrogen abstraction reac- tions. Transient phenoxyl radicals were directly visualized but only persistent carbon-centered radi- cals were monitored by electron paramagnetic resonance (EPR). The experimental EPR results let us rationalize our analysis as the sum of two different radicals. One, called the methylene radical, de- veloped from the loss of a benzylic hydrogen gave place to a doublet of triplets with ac~ ~ 2.7 mT anda H = 0.165 mT. Besides, the methyl radical, developed after an intramolecular hydrogen trans- fer involving a methyl group on the central aromatic ring of the molecule, formed a triplet of trip- lets, with a~ H around 0.060 mT and a2 H = 0.169 mT. AII the co~tact interactions were tested by EPR simulation of the experimental data.
1 Introduction
The compound 1,3,5-tri-methyl-2,4,6-tris(3,5,-di-tert-butyl-4-hydroxybenzyl) ben- zene (Fig. 1), commercially available as Irganox 1330 and Ethanox 330, trade names of Ciba and Albemarle, respectively, is a phenolic antioxidant, currently added as a stabilizer to polymers and other materials that can undergo thermo- oxidative degradation [1].
Phenols and aromatic amines are recognized antioxidants and have been sys- tematically studied for decades. Moreover, several natural and synthetic antioxi- dants have been investigated with the purpose of explaining the high antioxi- dant power of tocopherols, both from a chemical a n d a biochemical point of view [2, 3].
The industrial achievements in the manufacture of synthetic organic polymers also induce investigations on antioxidants, in order to improve the stability of their products [4].
Thermal, mechanical, photochemical, high-energy-induced, and biochemical processes taking place in the presence of oxygen, generally show a free radical

104 S. N. Mendiara and M. E. J. Coronel
H3C q ~ C -.. CH3
CH 3 H 3 C ~ CH3 CH 3
H3C.~ C H Ÿ CH2 >C / CH3 H~C / ~ CH, ~ ~CH,
HO" [ T "OH
C C H3C ~ I ~CH3 H 3 C / I ~ C H 3
CH3 CH 3
Fig. 1. Compound 1,3,5-tri-methyl-2,4,6-tris(3,5,-di-tert-butyl-4-hydroxybenzyl) benzene, molecule 1.
character. The primary carbon-centered free radical species, R o, develop oxygen- centered radicals and hydroperoxides, then chain propagation steps settle down, and so carbon-centered radicals may be regenerated.
A chain propagation reaction can be summarized as follows
R - + 0 2 - - RO2- ,
RO2o + RH -- RO2H + R"
A chain-breaking antioxidant can stop the peroxidation reaction. If ArOH stands for a phenolic antioxidant, we may describe the following reactions
ArOH + R.
ArOH + RO2-
ArO ~ + ArO-
-- RH + ARO.,
-- ArO. + ROzH,
- - nonradical products.
The antioxidant behavior of phenols is well correlated with their O-H group reactivity through the O-H bond dissociation enthalpy (BDE(o_H)) [2, 3, 5-7]. Also, the reactivity is evaluated by the rate of the hydrogen transfer to peroxyl and alkyl radicals [3, 8]. The occurrence of electron followed by proton transfer is also supported by experimental work [9, 10].
Molecule 1 has been the subject of a recent study where the quinone type products of its antioxidant activity, in synergistic interaction with an aromatic amine, have been identified [11]. For sterically hindered phenols like molecule 1, it is generally assumed that in the first step the phenolic hydrogen is donated to radical species. In the second step, a benzylic hydrogen is donated leading to a quinone strucmre. The oxidation sequence may take place over each phenolic

EPR Evidence of Carbon-Centered Radicals 105
group available. However, the possibility of finding carbon-centered radicals from benzyl phenolic antioxidants has been proposed [12].
During current work in our laboratory, persistent radicals built up from molecule 1, when applying oxidative techniques and also with hydrogen abstrac- tion reactions [ 13, 14]. Electron paramagnetic resonance (EPR) spectra have been recorded and tested by EPR simulation. In order to understand the recorded spec- tra we carried out a detailed analysis dealing with the possible contributions of selected radical species.
2 Experimental
All the solvents (Carlo Erba) were freshly distilled. Phenol molecules (Figs. 1 and 2) were crystallized from hexane. Solid gal-
vinoxyl radical (Aldrich) was used as received; its solutions were obtained by weighing, dissolution and further dilutions in benzene or benzene-toluene (1:1). The purity of molecule 1 was assessed by thin-layer chromatography, high-pres- sure liquid chromatography and 'H nuclear magnetic resonance (NMR) spectros- copy. Proton NMR spectra, 200 MHz, in deuterochloroform, were recorded on a Bruker AC-200 NMR spectrometer at the spectroscopy service unit UMYMFOR (CONICET-FCEN, University of Buenos Aires).
The solutions of molecule 1 were prepared in benzene, toluene, 1,4-dioxane, and in a mixture of benzene-l,4-dioxane (1:1).
Persistent radicals were obtained from molecule 1 by applying three types of reactions:
i. Oxidation with lead dioxide. This reaetion was accomplished by addition of 8 mg of lead dioxide to 0.5 or 1 ml of a 10 -3 M solution of molecule 1. Air was previously removed by argon bubbling through the solution.
ii. Oxidation with alkaline solution of potassium ferricyanide. 0.5 ml of the reagent was stirred with 0.5 or 1 ml of 10 -3 M solution of molecule 1 under argon bubbling [14].
100 gl aliquots of the solutions of either i or ii were transferred to EPR sample tubes under argon atmosphere. Degassing was carried out by argon bub- bling or by freeze-and-pump cycles.
iii. Hydrogen abstraction with galvinoxyl radical [15]. This reaction was car- ried out in EPR tubes by mixing 100 gl of a solution of 0.05 M molecule 1 with 20 gl of 2 .10 -4 M galvinoxyl radical in benzene or benzene-toluene (1:1). Degassing was achieved by freezing and pumping in a cyclic way. Galvinoxyl radical was totally quenched and the signal of persistent radicals from molecute 1 was obtained.
H O - ~ C H 2 - ~ OH
Fig. 2. Phenol molecule, molecule 2.

106 S. N. Mendiara and M. E. J. Coronel
All EPR spectra were monitored and recorded on an X-band Bruker ER 200D EPR spectrometer equipped with an ER 4111VT variable-temperature control system unit of the type BVT1000. Radical concentrations were estimated by tak- ing galvinoxyl radical a s a reference. Typical first-derivative spectra for persis- tent radicals obtained out of molecule 1 are shown in Fig. 3, the intensity of the signal is measured in arbitrary units and the magnetic field in gauss, 1 G = 0.1 mT. The experimental general instrumental settings for acquiring EPR spectra are given in the legend of Fig. 3.
In order to carry out variable-temperature studies the solutions were prepared in benzene-toluene (I:1). The alkaline-ferricyanide method was used and the system was carefully degassed with argon. The EPR sample tubes were sealed with an intermediate latex tubing in order to maintain the argon atmosphere and minimize pressure changes over the range from 198 to 354 K. EPR spectra were recorded at 198, 220, 230, 242, 274, 304, 318, and 354 K.
For geometry optimization the Gaussian98 software and the semiempiricat AM1 method were applied to phenol motecule 1 and the radicals existence of which could be postulated [16]. The results for isomer radicals can be reliably compared.
The simulation of the experimental spectra was accomptished by using the ESR2 Simulator software [17]. The standard Origin plotting program was used in order to develop the processing of the results provided by the simulations.
3 Results
3.1 Experimental Spectra
Persistent radicals were obtained by three different techniques as it was pointed out in Sect. 2.
In the oxidation with lead dioxide the reaction yielded radical concentrations around 10 -4 M.
In the oxidation with the alkaline solution of potassium ferricyanide, also con- centrations in the order of 10 -4 M were reached. In our work, in the course of this type of oxidation the organic phase developed an intense blue color, repre- sentative of the presence of phenoxyl radicals. Cook and Norcross [14] have al- ready described the deep blue due to the presence of phenoxyl radicals; they also observed the fast fading out of the deep blue and the generation of a yel- low color. In our experiments the blue color persisted in the organic phase un- der stirring but disappeared when the stirring was interrupted, giving rise to a brilliant yellow. So, our observations agree with the formation of transient phe- noxyt radicals followed by the formation of quinone structures, j u s t a s it is re- ported in the literature [11, 14]. We could not monitor these transient phenoxyl radicals, they were short-lived intermediates and only typical spectra similar to those shown in Fig. 3 were available.
In the hydrogen abstraction reaction with galvinoxyl radical, we measured persistent radicals concentrations ranging from 10 -5 to 10 -6 M.

EPR Evidence of Carbon-Centered Radicals 107
Expe¡ Spectra Sirnulated Spectra
Spectrum car¡ out in benzene
0.34 mT 1.0
~ 0.5 A ._~ o.o
-o.5
-1.0 i
3460
~ t ~ ~ ~ , ~ 1"0 -4 0.5
-- ~ 0.0
-o.5
-1.0 Spectrum carried out in toluene
3460
J
3i70 ' 3i80 ' 3i90 ' Magnetie Field (G)
i i i
3270 ' 3280 ' 34'9o ' Magnetie Field (G)
3500
3500
_)
Spectrum car¡ out in benzene-dioxane
1.0
"~" 0.5
.~ o.o
-0.5
-1.0
3460 3470 3480 3490 3500 Magnetie Field (G)
Fig. 3. Experimental EPR spectra and simulated speetra of the persistent radicals obtained from moleeule 1, in different solvents and at room temperature. Simulated spectra are the sum of two proposed radicals: methylene radical and methyl radical. The hyperfine coupling constants are shown in Table 1. EPR general instrumental settings: microwave frequency, 9.7 GHz; modulation amplitude, 0.125 G; modulation frequency, 100 kHz; attenuation, 10 dB; reeeiver gain, 1.25. 104;
time constant, 0.2 s; sean rate, 200 s; persistent radical eoneentration, 10 -4 M.
In all the cases the in tens i ty o f the s igna l r e m a i n s p rac t i ca l ly c o n s t a n t throughout the exper imenta l measuremen t . Hence, we m a y deduce that the sys- tem has a t ta ined the s tat ionary state.
3.2 Analysis of Experimental Spectral Hyperfine Constants
The spectra o f the persis tent radicals keep the same pat tem, no matter the me thod used for their generat ion. In Fig. 3, we show the spectra recorded after apply-

108 S. N. Mendiara and M. E. J. Coronel
Table 1. Hyperfine coupling constants at room temperature.
Solvent Viscosity (mPa. s) Methylene radical (mT) a Methyl radical (mT) b
ac~ a H a2 a a~
1,4-Dioxane 1.20 2.691 0.169 0.169 0.091 Benzene-dioxane ~ 1.07 2.680 0.163 0.169 0.084 Benzene 0.60 2.679 0.165 0.169 0.068 Toluene 0.55 2.644 0.169 0.169 0.062
a EPR data. b Simulation assignments. They fit very well the experimental pattems. We may observe in Fig. 3
that the values assigned to a~ in benzene, toluene and benzene-l,4-dioxane reproduce quite differ- ent experimental patterns corresponding to the central part of the spectra.
r The viscosity of the mixture was calculated with the relationship r/, b = (r/a)X~ , where r/a bis the viscosity of the mixture of solvents a and b; Z~ and Zb ate their respective molar fractions.
ing the lead dioxide technique in benzene and in toluene, at room temperature. We apprecia ted that the spectra did not show the pattern expected for phenoxyl radicals. Moreover, we observed that it was quite beneficial to carry out our mea- surements in different solvents, the central part o f the spectra showed vis ib le variat ions with them.
In order to interpret our exper imenta l results, we per formed a de ta i l ed s tudy throughout our exper imenta l EPR spect ra set. At the same time, we cons idered every poss ib le generat ion o f radicals , including those from the qu inone prod- ucts.
We deduced ana ly t ica l ly some hyper f ine coupl ing constants and p r o p o s e d others. To assess the qual i ty o f our approach we per formed a ser ies o f spec- tral compute r s imulat ions. Our inves t iga t ions let us ra t ional ize our ana lys i s as the sum o f two different radicals . We can apprec ia te dis t inct ive sets o f l ines: the external ones are well def ined a s a double t o f tr iplets and the centra l pat- tern fo l lows the representa t ion o f a t r iplet o f tr iplets. The external sys tem o f l ines can be at t r ibuted to a radical result ing f rom the loss o f a me thy l ene hy- d rogen and we wil l call it me thy lene radical . The central group o f s ignals can have its or igin in the loss o f the hydrogen from a methyl group bound to the central r ing o f the molecular structure and it wil l be cal led methyl radical . Both spec t ra were s imula ted and added with very good results. Our in te rpre ta t ion can be enl ightened by examining carefu l ly the react ions outl ined in Figs . 7 and 9, (Sects. 4.2 and 4.3).
In benzene solution, the methy lene radical is recognized by two hyperf ine contact interactions due to one methylene hydrogen, ac~ = 2.679 mT, and two hydrogen atoms on the phenolic ring, a H = 0.165 mT. In toluene solution, this radical shows sl ightly different values for those splittings: 2.644 and 0.169 mT, respectively. In benzene - l , 4 -d ioxane those splittings are 2.680 and 0.163 mT. This information is completed in Table 1.

EPR Evidence of Carbon-Centered Radicals 109
In Table 1 we present the hyperfine coupling constants deduced analytically or estimated by simulation. Spectra were run in different solvents and at room temperature.
Notice that even for the short differences between the values assigned to a~ in benzene and toluene, we can observe quite different expe¡ pattems cor- responding to the lines of the central part of the spectra (Fig. 3). The solvent effects wilI be discussed in Sect. 4.4. We assigned to the methyl radical the following hyperfine interactions: two methylic and two methylenic hydrogen atoms with a~ = 0.068 mT and a~ = 0.169 mT in benzene and a~ = 0.062 mT and a2 H =.0.169 mT in toluene. In benzene-l ,4-dioxane those values are 0.084 and 0.169, respectively. A hyperconjugative effect is taking place [18]. The av ~ constant was evaluated over a range from 0.05 to 0.10 mT, but only the results given above fit the simulations. As the line width affects the shape of the sig- nal, we tested different values ranging from 0.025 to 0.050 mT. The best fit was achieved with the line width of 0.038 mT.
Both sets of signals are coincident at the same central field, due to similar values of the factor g for carbon-centered radicals that share a chemical envi- ronment.
The external doublet of triplets could only be provided in this molecule by a radical centered in a benzylic carbon (methylene radical) resulting from the loss of one of its hydrogen atoms. This signal cannot be contributed by a spin centered on the oxygen of the phenolic group. A phenoxyl radical should give a triptet of triplets with relative intensities 1:2:1 for the external and central lines, which should arise by the hyperconjugation in the methylene group, together with the interactions of two meta-hydrogen atoms in the phenolic ring. Pannell [19] has studied the pattem of" lines for the phenoxyl radical derived from bis(3,5-di- tert-butyl-4-hydroxyphenyl)methane, molecule 2. He calculated the hyperfine con-
Fig. 4. EPR spectrum of the phenoxyl radical obtained from molecule 2 in benzene. Hyperfine coup- ling constants deduced: acHn~~ = 0.91 toT, amo~H _- 0.16 mT.

110 S. N. Mendiara and M. E. J. Coronel
stants aHH,_ = 0.91 mT and ametaH ~-0.16 mT, in good agreement with its EPR spectrum. We also recorded this spectrum in our laboratory (Fig. 4).
3.3 Effects of Temperature on the Spectra
The series of spectra run at different temperatures from 198 to 354 K are gath- ered in Fig. 5. The simulations developed to fit these spectra indicate that
3 .3G
274 K
J
220 K
�91 304 K
/? 354 K
Fig. 5. Persistent radical spectra. They were recorded after molecule 1 oxidation was car¡ out by the alkaline-ferricyanide method, at different temperatures. The solvent was a mixture of benzene and toluene (1:1). That appropriate mixture allows good solubility all through the experiments.

EPR Evidence of Carbon-Centered Radicals I I I
a~ = 0.068 mT from 198 to 274 K and it changes to 0.062 mT between 304 and 354 K. In the latter range of temperature, the coalescence into a seemingly broad triplet becomes apparent. The same sets of values can be applied for the spectra shown in Fig. 3, the first value for the spectrum obtained in benzene and the second value to that obtained in toluene.
On the other hand, the spectra recorded from 274 to 198 K progressively undergo a dipolar broadening expectable for lower temperature and higher vis- cosity of the solutions [18]. This effect is very clear because the molecule 1, molecular weight about 775, and the radicals derived from it are slowly tum- bling species.
We may compare those radical records with the EPR signal of a smaller- size persistent radical, galvinoxyl radical, which contains one quinonic and one benzenic ring bond to a methyne group and its spectrum is a doublet of quin- tets [19]. This radical, molecular weight 421, can be taken a s a reference for a shorter correlation time, i.e., more rapid tumbling in solution that allows a bet- ter resolution of the quintets at lower temperature (Fig. 6).
Another fact of interest can be seen in the spectra of radicals coming from molecule 1 registered between 354 and 198 K. A lesser peak-to-peak amplitude is observed in the external lines of the methylene radical doublet at lower field, towards the left-hand part of the spectra. These results are discussed in Sect. 4.4. Since the derivative amptitude is inversely proportional to the square of the line width, small changes in line widths cause appreciable changes in the rela- tive amplitudes of the lines [18]. The a H hyperfine constant denotes a little rate c. of change from 354 to 198 K.
4 Discussion
4.1 Hydrogen Transfer in the Genesis of the Radicals
In a recent paper, Scaiano and coworkers [20] have demonstrated in a work dealing with antioxidants that hydrogen transfer is dominant with respect to
_J
318K
b a
?
'7198 K
Fig. 6. Galvinoxyl spectra in benzene-toluene (1:1) at two different temperatures, a 198 K, b 318 K. Hyperfine coupling constants deduced: aCHH = 0.579 mT; am,,,-H - 0.132 mT.

112 S. N. Mendiara and M. E. J. Coronel
electron transfer in the hydrogen abstraction by phenoxyl radicals. Since galvi- noxyl is a phenoxyl radical, our work clearly shows that both hydrogen ab- straction with galvinoxyl radical and the two oxidative procedures render the same signal for persistent radicals. The simultaneous probable formation of phe- noxyl and methylene radicals must be taken into account, concerning specially the hydrogen abstraction with galvinoxyl. Since the BDE(o_H) in substituted phe- nols is currently evaluated from 348.2 to 334.4 kJ/mol and the BDE(H_cH ) is about 340.6 kJ/mol, we may assume that both dissociations can take place [21, 22]. In Fig. 4, we can appreciate the spectrum of the radical of molecule 2 that rep- resents the corresponding phenoxyl radical. The radical was obtained by oxida- tion with lead dioxide in benzene. This is in accord with a higher rate con- stant for the oxidation of the O-H group of phenols in aromatic solvents [23]. The steric hindrance on the phenolic O-H group is a factor that favors the loss of the hydrogen [24]. The methylene groups that connect the phenolic moiety to the central ring in molecule 1 are also hindered, however this is not the case in molecule 2, where the methylene hydrogen atoms are not subjected to the same degree of hindering forces. Then, the presence of carbon-centered persistent radicals derived from a methylene group in molecule 1, the methyl- ene radical, can be supported by stabilizing interactions with the molecular surroundings.
The possibility of the formation of peroxyl radicals is not considered for our samples because we worked under air-free conditions. That kind of radicals can be long lived but their spectra do not have hyperfine structure [25].
4.2 Hyperfine Constant Patterns in the Radicals
The methylene radical shows hyperfine coupling with the a-hydrogen (a dou- blet), and with two equivalent ortho hydrogen nuclei located at the aromatic ring (a triplet). In the literature, the et-hydrogen has ah ac~ ranging around 1.3-1.7 mT [26, 27]. In our spectra this interaction is about 2.68 mT in benzene. The triplet substructure originating from the two ortho hydrogen nuclei in the aro- matic phenolic ring can take values close to 0.5 mT in the benzyl radical de- rived from toluene [26, 28, 29]. In molecule 1, the presence of a phenolic O-H group should decrease this constant due to partial spin localization on the oxy- gen atom. Its value in our spectra is about 0.16 mT, and the contact interaction with the O-H hydrogen has not become apparent.
The high value we have observed for the ac~ can have its origin in an in- tramolecular interaction within the surrounding atoms that can stabilize the me- thylene radical, in addition to the steric hindrance. A hydrogen transfer might involve one of the methyl substituting groups attached to the central ring of the molecule. For example, the possibility of an intramolecular hydrogen transfer has been studied for an aromatic molecule. In that case, the spin density is mainly localized on the carbon-centered primary radical, being very low at the carbon that bears the migrating hydrogen [30, 31]. The hydrogen transfer could justify

EPR Evidence of Carbon-Centered Radicals
\ /~CH. + --CH 3 = q 2 + --CH 2.
methylene methyl radical radical
Fig. 7. Hydrogen transfer.
113
the stabilization of the methylene radical and methyl radical. The exchange re- action is sketched in Fig. 7.
Methylene radical, in first place, is originated from the benzyl group. Me'- thyl radical is generated by hydrogen migration from a methyl group attached to the central ring. The radical center is the methylene with greater probability than the methyl because the latter group has a higher BDE, 367.6 kJ/mol. That value has been determined for the hydrogen at the methyl group of the pen- tamethyl substituted toluene [32]. The hydrogen transfer equilibrium should sta- bilize both radicals and the methyl radical could only show hyperconjugation with the resultant methylene involved in the transfer, giving rise to a triplet of trip- lets with poor spin density.
4.3 Details of the Intramolecular Hydrogen Exchange
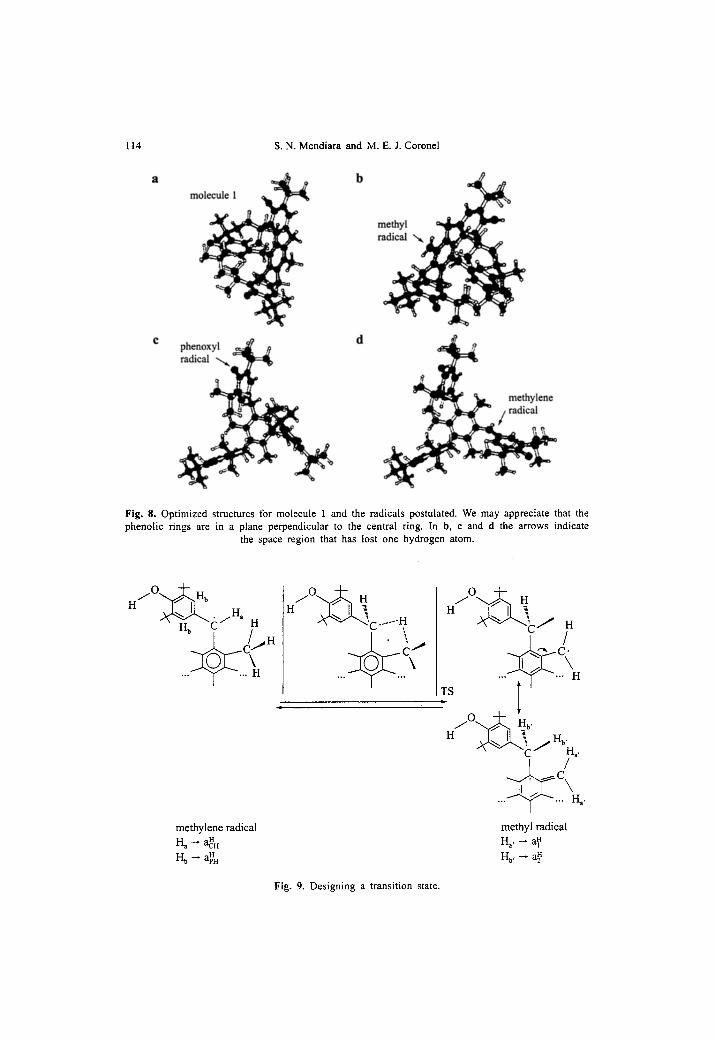
In Fig. 8, we present the optimized structures for the radicals proposed. Methylene radical has the most stable conformation when the phenolic rings
are in a plane perpendicular to the central ring. Then, the overtapping of the single occupied molecular orbital with the central r~ system is prevented. At the same time, the interactions with the methyl groups are favored. In Fig. 8d an arrow indicates the region where the hydrogen atom was lost. The distance from the methylene radical carbon to the methyl hydrogen atoms is 0.212 nm. The same value was found for the distance between the carbon in the methyl radical and the hydrogens in the methylene radical. We may also observe that phenoxyl, methyl and methylene radicals and molecule 1 display similar optimized geom- etries.
On the other hand, the phenoxyl radical from molecule 2 can only exhibit hyperconjugation with the nearby methylene group. This enables the fast con- version of phenoxyl radical into the quinone structures already identified [11].
Early in the development of magnetic resonance methods it was possible to determine the existence of short-lived complexes between reagents that involve hydrogen transfer [33]. The geometry of a transition state and the energy bar- rier for the formation of hydrogen transfer complexes are still demanding atten- tion [34]. Intermediate complexes also should allow an electron transfer, followed by a proton transfer, but hydrogen exchange seems to be the preferential way for the reaction that involves toluene and benzyl radical [35, 36].
114 S. N. Mendiara and M. E. J. Coronel
molecule 1 b me.thyl
r phenoxyl ~=~ /~ d ~ ) l l .
methylene ~ /radical
Fig. 8. Optimized structures for molecule 1 and the radicals postulated. We may appreciate that the phenolic rings are in a plane perpendicular to the central ¡ In la, e and d the arrows indicate
the space region that has lost one hydrogen atom.
h/O'-.v• H~
H b tS H , / i . ...~..c,.
methylene radical H a -- aH
H b ~ a~H
i / 0 ~ . , H
H ~ C ...... g / o . . . ~ H
H ~ C / H
H q
...~.r..~ Ha '
methyl radical H~, - a~
Hb, - - a~
Fig. 9. Designing a transition state.

EPR Evidence of Carbon-Centered Radicals 115
Another aspect of the hydrogen transfer shown in Fig. 7 is that the reac- tion takes place in a molecular cage, which gives the frame to postulate the existente of a transition state (TS). We designed a representative TS in Fig. 9. For example, Mayer et al. [36] describe the degenerate hydrogen exchange reaction between benzyl radical and toluene postulating a transition structure that allows the 2p-Ÿ orbital on the benzylic carbon of the radical to approach to a benzylic hydrogen of toluene. This can be the case of the hydrogen transfer between the methylene radical and the methyl radical in our molecule 1. The geometry results obtained by the optimization calculations in Fig. 8 are in agreement with that postulation.
We formulate an interpretation of the central lines, the overlapped triptet of triplets.
The TS leads to the origin of the methyl radical and the hyperconjugative structure that delocalize the electron spin in the circuir of the hydrogen trans- fer, so giving rise to a triplet of triplets that fits by simulation the central lines of the spectra, as it can be seen in Fig. 3. In Fig. 9 we may observe that the following correspondence holds: H a generates the hyperfine interaction aCHH and the two H b generate the hyperfine interaction a~. In methyl radical, both H a, generate the hyperfine interaction a~ and both H b, generate the hyperfine in- teraction a~.
We may now put forward some comments about the rate constant. The time- scale of the EPR experiment is such that the spectra representan averaged pic- ture of the whole set of radicals formed. The two molecular structures involved in the hydrogen transfer can give rise to a well resolved spectrum due to the shorter spin lifetime of the EPR compared with earlier NMR expe¡ [33].
Otherwise, if the rate of interconversion was increased, the first consequence observed should be a broadening of the lines, because of the reduced lifetime of a given radical.
From the Heisenberg uncertainty principle, AW. St ~- h(2rt) -1 (AW= h - A v is the energy uncertainty), the mean lifetime At would have to be ~ 5. 10 -7 S or less, for line broadening to be significant, if we considered a line width close to 0.01 toT [18]. Then, if the interconversion in the hydrogen exchange is slow enough, i.e., less than (At) -I, 2.106 s -l, both spectra can be seen with good definition and relatively thin lines. This applies to the methylene radical and methyl radical signals in our spectra.
4.4 Effects o f Solvent and Temperature on the Spectra
Not only chemical but also physical processes affect the line width. The spectra in Fig. 3 clearly expresses changes of the central set of lines when the solvent is changed: the solvent is benzene (viscosity, 0.60 mPa. s), toluene (viscosity, 0.55 roPa-s) a n d a mixture (1:1) of 1,4-dioxane and benzene (viscosity, 1.07 ropa. s). The spectrum in toluene shows the change of the central lines into a seemingly broad triplet. Spectra were also obtained in 1,4-dioxane.

116 S. N. Mendiara and M. E. J. Coronel
The resultant values of the contact interactions are gathered in Table 1. We observe that the simulation of the respective spectra rendered small differences for some contact interactions. A tendency is shown for the ac~ and a~ to be enlarged as the viscosity increased, from pure toluene to 1,4-dioxane.
The mixture of benzene and toluene (1:1) was chosen for carrying out the va¡ study in order to obtain stable solutions and to prevent freez- ing and boiling over the range from 198 to 354 K. Also, benzene-toluene mix- tures are considered quasi ideal solutions that obey Raoult's law [37].
We attfibute the detected effects to subtle changes in the rate of hydrogen exchange.
As it was commented in Sect. 3.3, spectral effects of slow molecular tum- bling rates can be observed for the records obtained from 198 to 274 K. For free radicals in solutions with low viscosity, anisotropic interactions are averaged to zero. This statement is not always true, especially when the molecular struc- ture that contains the radical is considerably large and/or the solvent has a moderate viscosity.
For the records obtained between 198 and 274 K, the tumbling rate is rapid enough so that the line positions correspond to the completely averaged spectra. However, the line widths due to the methylene radical are not uniform. The low- field lines, on the left-hand side, are broader than the high-field ones, on the right-hand region of the recorded spectra.
In general, the line widths in spectra that exhibit the effects of slow tum- bling rates can be approximated by the following relation:
F = A + Z BiMe + Z CeMi z + Z EijMiMj i i i<j
In this expression, F is the addition of all the contributions to the line width. M i and Mj are the total z component of the nuclear spin quantum numbers for sets i and j of equivalent nuclei. The coefficient A is a constant term that includes all line broadening effects which are the same for all hyperfine components. The coefficient B e causes the spectra to be asymmetric. It arises from the product of the g-tensor and the hyperfine tensors; ir the p-electron spin density is negative and the g-tensor component parallel to the field is less than the g-tensor compo- nent perpendicular to the field, then the low-field lines will be broader than the high-field ones. This effect should be present in the spectra recorded between 198 and 274 K. The C e coefficient is only dependent on the hyperfine anisotropy; broad- ening due to anisotropic hyperfine effects generally varies as h/T, hence C e coef- ficients add broadening to all the line widths. The E U coefficient arises from the products of the hyperfine tensors of nuclei from different equivalent sets [18].
The calculation of the values of the anisotropic tensors and consequentty the calculus of the coefficients is out of the scope of this work, but a qualitative insight is useful to understand the several sources of the so-called dipolar broad- ening, due to slow tumbling rate.
Temperature effects on the contact interaction have been the matter of a considerable number of studies. The earlier EPR works about the effect of tem-

EPR Evidence of Carbon-Centered Radicals 117
perature were carried out to obtain vibrational information. An increase in tem- peramre augments the population in excited vibrational levels. So, the "s" char- acter of the radical and hence the hyperfine splitting due to the ct-C increase, as it has been demonstrated in 13C-labeled radicals [38]. The opposite or comple- mentary behavior has been found for the hyperfine splitting due to the ct-H [39, 40]. This feature can account for very small variations in the contact interac- tion, for example, 0.05 mT over a temperature range of 221 K [39].
In our study, the highest contact interaction, aCH,H shows a change of 0.17 mT over a 156 K amplitude range of temperature, going from lower to higher values as the temperature is decreased. The effect above mentioned for the ct-H applied to the methylene radical contact interaction ac~ and may be included in this rather large value of 0.17 mT, but would not be the only origin of this experimental fact.
In Fig. 10 we may appreciate the representation of the experimental data, deduced from the recorded spectra in Fig. 5. The least-squares fit yields the fol- lowing quadratic equation:
y = 3.17 - 0.0039x + 4.45. l O - 6 x 2 ,
R = 0.9873.
I f the value obtained for this interaction at 198 K is neglected, because of the uncertainty due to the strong anisotropic broadening at that temperature, it is possible to obtain a linear fit with a slope - 8 . 6 2 . 10 -4 mT/K (R = 0.9756).
The contraction of the sample volume and consequently the increase in the density of the solutions along the decrease in temperature can explain the rate of variation of the a H [41, 42]. The increment in solvent density enhances its solvation capability. A more effective solvation with a better stabilization of the
2.74
2.72
2.70
2.68 E ~" 2.66
2.64
2.62
2.60
180
' i , i . t , I , i ' i ' i . i , i
220 260 300 340 T (K)
Fig. 10. Analysis of the temperature effect on the hyperfine constant ac• Representation of the experi- mental data shown in Fig. 5.

118 s. N. Mendiara and M. E. J. Coronel
radical and its surroundings can take place as the temperature is lowered. Simi- lar experiments carried out by Griller [42] with di-tert-butylnitroxide in cyclo- pentane at constant pressure have demonstrated the increment of the contact in- teraction along the decrease in temperature, yielding a variation of - 7 . 7 - 1 0 -5 mT/K. In our study, the stabilization of the radical centered on the benzyl car- bon and hence the increment of the ac~ contact interaction involves the solva- tion of a molecular stmcture much larger than the di-tert-butyl nitroxide radical. Then, the rate of increment of the ac~ contact interaction when the temperature is decreased should be greater than the rate obtained by Griller [42] for the di- tert-butylnitroxide radical.
Finally, it must be said that Griller [42] summarized the effects operating, on the "a N'' in the di-tert-butylnitroxide radical in the following form
OV (~aN 1 =(c3a~ 1 +(~aSl (-~le. t. ~ r .),> t. a r .)~ t. av .)~
This equation takes into account the whole effect of the temperature on a con- tact interaction [42].
5 Conclusions
In the course of our investigation unexpected spectra of persistent carbon-cen- tered radicals, obtained by means of hydrogen abstraction or oxidative reactions, arose from molecule 1. This molecule is a particular case of the hindered phenols currently used as polymer and food stabilizers. Molecule 1 has also methylene groups connecting the phenolic groups to a central benzenic ring. We have suc- ceeded in justifying the formation of methylene radicals. The persistence of methylene radicals could be achieved through stabilizing interactions with their neighbor partners, in particular by means of hydrogen exchange with the methyl groups bond to the central benzene ring, giving rise to methyl radicals.
The analysis of persistent EPR radical spectra and the simulations proved to be useful to postulate the stmctures of the radicals involved. The study devel- oped on solvent and temperature effects throughout spectra have contributed to strengthen the existence of the stmctures proposed.
Oxidation and radical processes are highly complex and involve various mecha- nisms. The activity of many antioxidant compounds is still incompletely charac- te¡ Surely, the studies conceming the persistent radicals generated in the re- actions under investigation will be profitable. Also, all the knowledge acquired about those compounds may aid to establish their real potential as antioxidants.
Acknowledgments
We thank our colleague M. A. Grela for helping us with the simulation scale representation. We are especially grateful to L. L. Perissinotti (University of

EPR Evidence o f Carbon-Centered Radicals 119
Buenos Aires) for her generous and helpful guidance in the development of the geometrical optimizations.
References
1. EPA High Production Volume Program 201-15483 a. Test Plan for Irganox 1330/ Ethanox 330. 1,3,5-trimethyl-2,4,6-tris (3,5-di-t-butyl-4-hydroxybenzyl) benzene. CAS No 1709-70-02. July 30, 2004. Submitted by Ciba Specialty Chemicals and Albemarle Corporation.
2. Burlon G.W., Doba T., Gabe E.J., Hughes L., Lee F.L., Prasad L., Ingold K.U.: J. Am. Chem. Soc. 107, 7053-7064 (1985)
3. McFaul EA., Ingold K.U., Lusztyk J.: J. Org. Chem. 61, 1316-1321 (1996) 4. Pospisil J., Nespurek S.: Polym. Degrad. Stab. 49, 99-110 (1995) 5. Mulder P., Saastad O.W., Griller D.: J. Am Chem. Soc. 110, 4090-4092 (1988) 6. Wright J.S., Carpenter D.J., McKay D.J., Ingold K.U.J.: Am. Chem. Soc. 119, 4245-4252 (1997) 7. Wright J.S., Johnson E.R., DiLabio G.A.: J. Am. Chem. Soc. 123, 1173-1183 (2001) 8. Franchi P., Lucarini M., Pedulli G.F., Valgimigli L., Lunelli B.: J. Am. Chem. Soc. 121, 507-514
(1999) 9. Nagaoka S., Kuranaka A., Tsuboi H., Nagashima U., Mukai K.: J. Phys. Chem. 96, 2754-2761
(1992) 10. Zang H.Y., Wang L.F.: J. Phys. Chem. A 107, 11258-11259 (2003) 11. Barret J., Gijsman P., Swatgen J., Lange R.M.F.: Polym. Degrad. Stab. 75, 367-374 (2002) 12. Matsuura T., Ohkatsu Y.: Polym. Degrad. Stab. 70, 59-63 (2000) 13. Bennett J.E.: Nature 186, 385-386 (1960) 14. Cook C.D., Norcross B.E.: J. Am. Chem. Soc. 78, 3797-3799 (1956) 15. Lucarini M., Pedulli G.F., Cipollone M.: J. Org. Chem. 59, 5063-5070 (1994) 16. Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Zakr'zewski
V.G., Montgomery J.A.Jr., Stratmann R.E., Burant J.C., Dapprich S., Millam J.M., Daniels A.D., Kudin K.N., Strain M.C., Farkas O., Tomasi J., Barone V., Cossi M., Cammi R., Mennucci B., Pomelli C., Adamo C., Clifford S., Ochterski J.W., Petersson G.A., Ayala P.Y., Cui Q., Morokuma K., Malick D.K., Rabuck A.D., Raghavachafi K., Foresman J.B., Cioslowski J., Ortiz J.V., Baboul A.G., Stefanov B.B., Liu G., Liashenko A., Piskorz E, Komaromi I., Gompertz R., Martin R.L., Fox D.J., Keith T., AI-Laham M.A., Peng C.Y., Nanayakkara A., Gonzalez C., Challacombe M., Gill P.M.W., Johnson B.G., Chen W., Wong M.W., Andres J.L., Head-Gordon M., Replogle E.S., Pople J.A.: Gaussian 98, Rev. A7, Gaussian, Inc., Pittsburgh PA, 1998.
17. Mckelvey, Ronald D. ESR Simulator. Department of Chemistry. University of Wisconsin-LaCrosse, USA. 1986.
18. Wertz J.E., Bolton J.R.: Electron Spin Resonance. Elementary Theory and Practical Applications. New York: McGraw-Hill 1972.
19. Pannell J.: Chem. Ind. (London) 1797-1800 (1962) 20. Aliaga C., Asp› A., Scaiano J.C.: Org. Lett. 5, 4145-4148 (2003) 21. Borges dos Santos R.M., Martinho Simoes J.A.: J. Phys. Chem. Ref. Data 27, 707-739 (1998) 22. Lide D.R., Frederikse H.ER.: CRC Handbook of Chemistry and Physics, 75th edn., pp. 9-64.
Boca Raton, Fla.: CRC Press 1994-1995. 23. Valgimigli L., Banks J.T., Ingold K.U., Lusztyk J.: J. Aro. Chem. Soci 117, 9966-9971 (1995) 24. Wayner D.D.M., Lusztyk E., Ingold K.U., Mulder P.: J. Org. Chem. 61, 6430-6433 (1996) 25. Howard A.J.: Free Radicals (Kochi J.K., ed.), p. 42, Vol. 2, chapt. 12, New York: Wiley 1973. 26. Alberti A., Benaglia M., Macciantelli D.: Org. Lett. 2, 1553-1555 (2000) 27. Yamada B., Sakamoto K.: Polymer 41, 5619-5623 (2000) 28. Kochi J.K. in: Free Radicals (Kochi J.K., ed.), vol. 2, chapt. 23. New York: Wiley 1973. 29. Konkin A.L., Roth H.K., Schroedner M., Nazmutdinova G.A., Aganov A.V., Ida T., Garipov R.R.:
Chem. Phys. 287, 377-389 (2003) 30. Sordo T.L., Dannenberg J.J.: J. Org. Chem. 64, 1922-1924 (1999) 31. Karady S., Abramson N.L., Dolling U.-H., Douglas G.J., McManemin G.J., Marcune B.: J. Am.
Chem. Soc. 117, 5425-5426 (1995) 32. Wu Y-D., Wong C.-L., Chan K.W.K., Ji G.Z., Jiang X.-K.: J. Org Chem. 61, 746-750 (1996)

120 Mendiara and Coronel: EPR Evidence of Carbon Centered Radicals
33. Kreilick R.W., Weissman S.L.: J. Am. Chem. Soc. 88, 2645-2652 (1966) 34. Isbom C., Hrovat D.A., Borden W.T., Mayer J.M., Carpenter B.C.: J. Am. Chem. Soc. 127, 5794-
5795 (2005) 35. Foti M., Ingold K.U., Lusztyk J.: J. Am. Chem. Soc. 116, 9440-9447 (1994) 36. Mayer J.M., Hrovat D.A., Thomas J.L., Borden W.T.: J. Am. Chem. Soc. 124, 11142-11147 (2002) 37. Atkins P.W.: Physical Chemistry, 5th edn., pp 216-217. Oxford: Oxford University Press 1994. 38. Griller D. Marriot P.R., Preston K.F.: J. Chem. Phys. 71, 3703-3707 (1979) 39. Griller D., Ingold K.U., Krusic P.J., Fischer H.: J. Am. Chem. Soc. 100, 6750-6752 (1978) 40. G¡ D., Preston K.F.: J. Am. Chem. Soc. 101, 1975-1979 (1979) 41. Griller D., Ingold K.U.: Acc. Chem. Res. 13, 194-200 (1980) 42. Griller D.: J. Am. Chem. Soc. 100, 5240-5241 (1978)
Authors' address: Sara N. Mendiara, Departamento de QuŸ Facultad de Ciencias Exactas y Naturales, Universidad Nacional de Mar del Plata, Funes 3350, 7600 Mar del Plata, Argentina. E-mail:[email protected]
Copyright © 2022 FDOKUMEN