
ES 2 291 378 T3
31
19 OFICINA ESPAÑOLA DE PATENTES Y MARCAS ESPAÑA 11 Número de publicación: 2 291 378 51 Int. Cl.: C07D 263/32 (2006.01) C07D 277/42 (2006.01) C07D 413/04 (2006.01) C07D 417/04 (2006.01) A61K 31/4439 (2006.01) A61K 31/422 (2006.01) A61K 31/454 (2006.01) A61K 31/496 (2006.01) A61K 31/541 (2006.01) A61K 31/497 (2006.01) A61K 31/4545 (2006.01) A61P 3/10 (2006.01) 12 TRADUCCIÓN DE PATENTE EUROPEA T3 86 Número de solicitud europea: 01994985 .8 86 Fecha de presentación : 21.12.2001 87 Número de publicación de la solicitud: 1354879 87 Fecha de publicación de la solicitud: 22.10.2003 54 Título: Compuestos derivados de dihidronaftaleno y medicamentos que utilizan estos compuestos como ingrediente activo. 30 Prioridad: 25.12.2000 JP 2000-392723 45 Fecha de publicación de la mención BOPI: 01.03.2008 45 Fecha de la publicación del folleto de la patente: 01.03.2008 73 Titular/es: ONO PHARMACEUTICAL Co., Ltd. 1-5, Doshomachi 2-chome Chuo-ku, Osaka-shi, Osaka 541-8526, JP 72 Inventor/es: Tajima, Hisao; Nakayama, Yoshisuke y Fukushima, Daikichi 74 Agente: Ungría López, Javier Aviso: En el plazo de nueve meses a contar desde la fecha de publicación en el Boletín europeo de patentes, de la mención de concesión de la patente europea, cualquier persona podrá oponerse ante la Oficina Europea de Patentes a la patente concedida. La oposición deberá formularse por escrito y estar motivada; sólo se considerará como formulada una vez que se haya realizado el pago de la tasa de oposición (art. 99.1 del Convenio sobre concesión de Patentes Europeas). ES 2 291 378 T3 Venta de fascículos: Oficina Española de Patentes y Marcas. Pº de la Castellana, 75 – 28071 Madrid
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of ES 2 291 378 T3

19© OFICINA ESPAÑOLA DEPATENTES Y MARCAS
ESPAÑA
11© Número de publicación: 2 291 37851© Int. Cl.:
C07D 263/32 (2006.01)
C07D 277/42 (2006.01)
C07D 413/04 (2006.01)
C07D 417/04 (2006.01)
A61K 31/4439 (2006.01)
A61K 31/422 (2006.01)
A61K 31/454 (2006.01)
A61K 31/496 (2006.01)
A61K 31/541 (2006.01)
A61K 31/497 (2006.01)
A61K 31/4545 (2006.01)
A61P 3/10 (2006.01)
12© TRADUCCIÓN DE PATENTE EUROPEA T3
86© Número de solicitud europea: 01994985 .886© Fecha de presentación : 21.12.200187© Número de publicación de la solicitud: 135487987© Fecha de publicación de la solicitud: 22.10.2003
54© Título: Compuestos derivados de dihidronaftaleno y medicamentos que utilizan estos compuestos comoingrediente activo.
30© Prioridad: 25.12.2000 JP 2000-392723
45© Fecha de publicación de la mención BOPI:01.03.2008
45© Fecha de la publicación del folleto de la patente:01.03.2008
73© Titular/es: ONO PHARMACEUTICAL Co., Ltd.1-5, Doshomachi 2-chomeChuo-ku, Osaka-shi, Osaka 541-8526, JP
72© Inventor/es: Tajima, Hisao;Nakayama, Yoshisuke yFukushima, Daikichi
74© Agente: Ungría López, Javier
Aviso: En el plazo de nueve meses a contar desde la fecha de publicación en el Boletín europeo de patentes, dela mención de concesión de la patente europea, cualquier persona podrá oponerse ante la Oficina Europeade Patentes a la patente concedida. La oposición deberá formularse por escrito y estar motivada; sólo seconsiderará como formulada una vez que se haya realizado el pago de la tasa de oposición (art. 99.1 delConvenio sobre concesión de Patentes Europeas).E
S2
291
378
T3
Venta de fascículos: Oficina Española de Patentes y Marcas. Pº de la Castellana, 75 – 28071 Madrid

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
DESCRIPCIÓN
Compuestos derivados de dihidronaftaleno y medicamentos que utilizan estos compuestos como ingrediente activo.
Campo técnico
La presente invención se refiere a un compuesto derivado de dihidronaftaleno.
Más específicamente, la presente invención se refiere a ácido 3-(5-(2-(2-(4-metilfenil)-5-metiloxazol-4-il)etoxi)-3,4-dihidronaftalen-1-il)propanoico, o una sal no tóxica del mismo.
Técnica anterior
Recientemente en el estudio de los factores de transcripción concerniente a la expresión de genes marcadoresen la diferenciación de adipocitos, se ha centrado la atención en el receptor activado por el factor proliferante deperoxisomas (abreviado más adelante como PPAR), que es uno de los receptores intranucleares. Los ADNc de losPPAR se clonaron a partir de diferentes clases de animales, y se encontraron genes de isoformas plurales, se conocenconcretamente tres tipos de isoformas (α, δ, γ) en mamíferos (véase J. Steroid Biochem. Molec. Biol., 51, 157 (1994);Gene Expression., 4, 281 (1995); Biochem Biophys. Res. Commun., 224, 431 (1996); Mol. Endocrinology., 6, 1634(1992)). La isoforma γ de PPAR es expresada predominantemente en tejidos adiposos, células inmunitarias, glándulaadrenal, bazo, intestino delgado. La isoforma α de PPAR es expresada principalmente en tejido adiposo, hígado, retina,y la isoforma δ de PPAR es expresada ampliamente sin especificidad por el tejido (véase Endocrinology., 137, 354(1996)).
Por otra parte, los siguientes derivados de tiazolidina son conocidos como agentes para el tratamiento de la diabe-tes mellitus no insulino-dependiente (NIDDM) y son agentes hipoglucémicos que son utilizados para la mejora de lahiperglucemia en pacientes que sufren diabetes. También son efectivos para la mejora de la hiperinsulinemia, la tole-rancia a la glucosa y el descenso de los lípidos en suero y por tanto se piensa que son considerablemente prometedorescomo agentes para el tratamiento de la resistencia a la insulina.
Una de las proteínas diana en las células de estos derivados de tiazolidina es exactamente PPAR γ y se ha resuel-to que aumentan la actividad de transcripción de PPAR γ (véase Endocrinology., 137, 4189 (1996); Cell., 83, 803(1995); Cell., 83, 813 (1995); J. Biol. Chem., 270, 12953 (1995)). Por consiguiente se piensa que un activador dePPAR γ (agonista) que aumente su actividad de transcripción es prometedor como agente hipoglucémico y/o agentehipolipidémico. Además, puesto que se sabe que un agonista de PPAR γ promueve la expresión de la propia proteínaPPAR γ (Genes & Development., 10, 974 (1996)), también se piensa que puede ser clínicamente útil un agente queincremente la expresión de la propia proteína PPAR γ así como del agente activador de PPAR γ.
PPAR γ está relacionado con la diferenciación de los adipocitos (véase J. Biol. Chem., 272, 5637 (1997) y Cell.,83, 803 (1995)). Se sabe que los derivados de tiazolidina que activan este receptor promueven la diferenciación delos adipocitos. Recientemente se informó que los derivados de tiazolidina aumentan la masa de grasa y hacen que loshombres ganen peso y se vuelvan obesos (véase Lancet., 349, 952 (1997)). Por consiguiente, también se piensa que losagonistas que inhiben la actividad de PPAR γ y los agentes que disminuyen la expresión de la propia proteína PPARγ son también aplicables clínicamente. Por otra parte, se informa de un compuesto que fosforila la proteína PPAR γy disminuye su actividad (Science., 274, 2100 (1996)). Esto implica que también es aplicable clínicamente un agenteque no se une a la proteína PPAR γ como ligando, pero inhibe su actividad.
2

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
De estos, se espera que los activadores de PPAR γ (agonistas) y los reguladores de la expresión de PPAR γ quepueden aumentar la expresión de la propia proteína sean útiles como agentes hipoglucémicos, agentes hipolipidémicosy agentes para la prevención y/o el tratamiento de enfermedades asociadas con trastornos metabólicos tales comola diabetes, la obesidad, el síndrome X, la hipercolesterolemia y la hiperlipoproteinemia etc., la hiperlipidemia, laaterosclerosis, la hipertensión, las enfermedades circulatorias y la sobreingesta etc.
Por otra parte, se espera que los antagonistas que inhiben la actividad de transcripción de PPAR γ o de los regu-ladores de PPAR γ que inhiben la expresión de la propia proteína sean útiles como agentes hipoglucémicos y agentespara la prevención y/o el tratamiento de enfermedades asociadas con trastornos metabólicos tales como la diabetes, laobesidad, el síndrome X etc., la hiperlipidemia, la aterosclerosis, la hipertensión y la sobreingesta etc.
El siguiente compuesto fibrato (por ej. clofibrato) es conocido como agente hipolipidémico.
Y, también se ha resuelto que una de las proteínas diana en las células de los compuestos fibrato es PPAR α (véaseNature., 347, 645 (1990); J. Steroid Biochem. Molec. Biol., 51, 157 (1994); Biochemistry., 32, 5598 (1993)). A partirde estos hechos, se piensa que los reguladores de PPAR α que pueden ser activados por los compuestos fibrato tienenun efecto hipolipidémico, y en ese caso se espera que sean útiles como agentes para la prevención y/o el tratamientode la hiperlipidemia etc.
Además, se ha informado recientemente que PPAR α posee actividad anti-obesidad en la memoria de la publicaciónWO 9736579. Además, se ha informado que la elevación del nivel de colesterol asociado a lipoproteínas de altadensidad (HDL) y la reducción del colesterol asociado a lipoproteínas de baja densidad (LDL), el colesterol asociadoa lipoproteínas de muy baja densidad (VLDL) y los niveles de triglicéridos fueron inducidas por la activación dePPAR α (J. Lipid Res., 39, 17 (1998)). También se ha informado que la composición de los ácidos grasos en sangre,la hipertensión y la resistencia a la insulina fueron mejoradas mediante la administración de bezafibrato que es uno delos compuestos fibrato (Diabetes., 46, 348 (1997)).
Por consiguiente, los agonistas que activan PPAR α y los reguladores de PPAR α que promueven la expresión dela propia proteína PPAR α son útiles como agentes hipolipidémicos y agentes para el tratamiento de la hiperlipidemia,y se espera que tengan un efecto elevador del nivel de colesterol HDL, un efecto reductor de los niveles de colesterolLDL y/o colesterol VLDL, una inhibición del progreso de la aterosclerosis y un efecto anti-obesidad. Por consiguiente,se piensa que son agentes prometedores para el tratamiento y/o la prevención de la diabetes como agentes hipoglucé-micos, para la mejora de la hipertensión, para la supresión del factor de riesgo del síndrome X y para la prevención dela aparición de enfermedades coronarias isquémicas.
Por otra parte, se han encontrado pocos informes sobre ligandos que activan PPAR δ significativamente o sobrelas actividades biológicas asociadas con PPAR δ. PPAR δ es denominado a veces PPAR β, o también es denominadoNUC1 en humanos. Hasta ahora, como para la actividad de PPAR δ, en la memoria de la publicación WO 9601430 sedescribe que hNUC1B (subtipo de PPAR cuya estructura es diferente de la de NUC1 humano en un aminoácido) inhibelas actividades de transcripción de PPAR α humano y del receptor de hormonas tiroideas. Recientemente en la memoriade la publicación WO 9728149, se informó que se han encontrado los compuestos que poseen una alta afinidad por laproteína PPAR δ y que podrían activar PPAR δ significativamente (es decir los agonistas) y que tenían una actividadelevadora del nivel de colesterol asociado a HDL (lipoproteína de alta densidad). Por consiguiente, se espera quelos agonistas que pueden activar PPAR δ tengan un efecto elevador del nivel de colesterol HDL, y así se esperaque sean útiles para la inhibición del progreso de la aterosclerosis y para el tratamiento de la misma, como agenteshipolipidémicos y agentes hipoglucémicos, para el tratamiento de la hiperlipidemia, como agentes hipoglucémicos,para el tratamiento de la diabetes, para el alivio del factor de riesgo del síndrome X, y para la prevención de la apariciónde enfermedades coronarias isquémicas.
Por ejemplo, la memoria de la publicación WO9828254 describe que un compuesto representado por la fórmula(A)
3

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
(donde, AA es un anillo arílico o heterocíclico sustituido opcionalmente, X1A es un enlace, átomo de O, etc., Y1A esalquileno C1-C8 sustituido opcionalmente, X2A es un enlace, átomo de O, etc., W es naftaleno sustituido opcional-mente, etc., BA es carboxilo, etc., X3A es un átomo de O, etc., R3A es alquilo C1-C8 opcionalmente sustituido, etc., nAes un número entero de 1-4.)
o una sal del mismo tiene una actividad hipoglucémica y una actividad hipolipidémica (las partes necesarias seextrajeron de la descripción de los grupos).
La memoria de la publicación W09911255 describe que un compuesto de los representados mediante la fórmula(B)
(donde, R1B es alquilo C1-C8, etc., R2B es -COOR3B (donde R3B es hidrógeno, o alquilo C1-C4.), AB es alquileno C1-C8, etc., GB es un anillo carbocíclico, o heteroanillo (el anillo carbocíclico y el anillo heterocíclico anteriores estánsustituidos opcionalmente con alquilo C1-C8, etc.), E1B es alquileno C1-C8, etc., E2B es -O-, etc., E3B es un enlace,etc., Cyc1B es un anillo carbocíclico saturado, parcialmente saturado o insaturado, etc.) o una sal del mismo tiene unaactividad moduladora del receptor activado por el factor de proliferación de peroxisomas (las partes necesarias fueronextraídas de la descripción de los grupos).
Asimismo, en el Ejemplo 3(35) en la memoria anterior, se describe el compuesto de fórmula (B-1).
Descripción de la invención
Con el fin de encontrar un compuesto que tenga una actividad moduladora de PPAR, los autores de la presenteinvención han dirigido estudios intensivos y han encontrado, como resultado que se pueden completar los objetosmediante los compuestos mencionados más abajo, y de este modo se ha completado la presente invención.
La presente invención se refiere a ácido 3-(5-(2-(2-(4-metilfenil)-5-metiloxazol-4-il)etoxi)-3,4-dihidronaftalen-1-il)propanoico, o una sal no tóxica del mismo.
Descripción detallada de la invención
En la memoria, el grupo alquilo C1-C8 incluye los grupos metilo, etilo, propilo, butilo, pentilo, hexilo, heptilo, yoctilo, y los isómeros de los mismos.
En la memoria, el grupo alquileno C1-C4 incluye metileno, etileno, trimetileno, y tetrametileno groups, y losisómeros de los mismos.
En la memoria, el grupo alquileno C1-C5 incluye los grupos metileno, etileno, trimetileno, tetrametileno, y penta-metileno, y los isómeros de los mismos.
En la memoria, el grupo alquileno C1-C2 incluye los grupos metileno, y etileno, y los isómeros de los mismos.
En la memoria, el grupo alquileno C1-C3 incluye los grupos metileno, etileno, y trimetileno, y los isómeros de losmismos.
En la memoria, el grupo alquileno C2-C3 incluye los grupos etileno, y trimetileno, y los isómeros de los mismos.
En la memoria, el grupo alcoxi C1-C8 incluye los grupos metoxi, etoxi, propoxi, butoxi, pentiloxi, hexiloxi, hep-tiloxi, y octiloxi, y los isómeros de los mismos.
En la memoria, el átomo de halógeno significa un átomo de cloro, bromo, fluoro o yodo.
4

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
En la memoria, el grupo 1H-tetrazol-5-ilo significa
En la memoria, el grupo 3,5-dioxoisooxazolidin-4-ilo
En la memoria, el grupo arilo mono- o bi-carbocíclico C3-C10 opcionalmente parcialmente o totalmente saturadorepresentado por el anillo 1 y R6, significa, por ejemplo, ciclopropano, ciclobutano, ciclohexano, ciclohexano, ciclo-heptano, ciclooctano, ciclononano, ciclodecano, ciclopropeno, ciclobuteno, ciclopenteno, ciclohexeno, ciclohepteno,cicloocteno, ciclopentadieno, ciclohexadieno, cicloheptadieno, ciclooctadieno, benceno, pentaleno, azuleno, perhi-droazuleno, perhidropentaleno, indeno, perhidroindeno, indano, naftaleno, tetrahidronaftaleno, perhidronaftaleno, etc.
En la memoria, los grupos arilo mono- o bi-heterocíclicos C3-C10 opcionalmente parcialmente o totalmente sa-turados que contienen 1-4 heteroátomos seleccionados entre un átomo de oxígeno, nitrógeno o azufre, el grupo arilomono- o bi-heterocíclico de 3 a 10 miembros que contiene 1-4 heteroátomos seleccionados entre un átomo de oxígeno,nitrógeno o azufre, significan, por ejemplo, pirrol, imidazol, triazol, tetrazol, pirazol, piridina, pirazina, pirimidina, pi-ridazina, azepina, diazepina, furano, pirano, oxepina, tiofeno, tiina, tiepina, oxazol, isoxazol, tiazol, isotiazol, furazano,oxadiazol, oxazina, oxadiazina, oxazepina, oxadiazepina, tiadiazol, tiazina, tiadiazina, tiazepina, tiadiazepina, indol,isoindol, indolizina, benzofurano, isobenzofurano, benzotiofeno, isobenzotiofeno, ditianaftaleno, indazol, quinolina,isoquinolina, quinolizina, purina, ftalazina, pteridina, naftiridina, quinoxalina, quinazolina, cinolina, benzoxazol, ben-zotiazol, benzimidazol, cromeno, benzofurazano, benzotiadiazol, benzotriazol, etc.
Asimismo, el grupo arilo mono- o bi-heterocíclico de 3-10 miembros parcialmente o totalmente saturado quecontiene 1-4 heteroátomos seleccionados entre un átomo de oxígeno, nitrógeno o azufre, significa, aziridina, azeti-dina, pirrolina, pirrolidina, imidazolina, imidazolidina, triazolina, triazolidina, tetrazolina, tetrazolidina, pirazolina,pirazolidina, dihidropiridina, tetrahidropiridina, piperidina, dihidropirazina, tetrahidropirazina, piperazina, dihidro-pirimidina, tetrahidropirimidina, perhidropirimidina, dihidropiridazina, tetrahidropiridazina, perhidropiridazina, di-hidroazepina, tetrahidroazepina, perhidroazepina, dihidrodiazepina, tetrahidrodiazepina, perhidrodiazepina, oxirano,oxetano, dihidrofurano, tetrahidrofurano, dihidropirano, tetrahidropirano, dihidrooxepina, tetrahidrooxepina, perhi-drooxepina, tiirano, tietano, dihidrotiofeno, tetrahidrotiofeno, dihidrotiina (dihidrotiopirano), tetrahidrotiina (tetrahi-drotiopirano), dihidrotiepina, tetrahidrotiepina, perhidrotiepina, dihidrooxazol, tetrahidrooxazol (oxazolidina), dihi-droisoxazol, tetrahidroisoxazol (isooxazolidina), dihidrotiazol, tetrahidrotiazol (tiazolidina), dihidroisotiazol, tetrahi-droisotiazol (isotiazolidina), dihidrofurazano, tetrahidrofurazano, dihidrooxadiazol, tetrahidrooxadiazol (oxadiazolidi-na), dihidrooxazina, tetrahidrooxazina, dihidrooxadiazina, tetrahidrooxadiazina, dihidrooxazepina, tetrahidrooxazepi-na, perhidrooxazepina, dihidrooxadiazepina, tetrahidrooxadiazepina, perhidrooxadiazepina, dihidrotiadiazol, tetrahi-drotiadiazol (tiadiazolidina), dihidrotiazina, tetrahidrotiazina, dihidrotiadiazina, tetrahidrotiadiazina, dihidrotiazepina,tetrahidrotiadiazepina, perhidrotiazepina, dihidrotiadiazepina, tetrahidrotiadiazepina, perhidrotiadiazepina, morfoli-na, tiomorfolina, oxatiano, indolina, isoindolina, dihidrobenzofurano, perhidrobenzofurano, dihidroisobenzofurano,perhidroisobenzofurano, dihidrobenzotiofeno, perhidrobenzotiofeno, dihidroisobenzotiofeno, perhidroisobenzotiofe-no, dihidroindazol, perhidroindazol, dihidroquinolina, tetrahidroquinolina, perhidroquinolina, dihidroisoquinolina, te-trahidroisoquinolina, perhidroisoquinolina, dihidroftalazina, tetrahidroftalazina, perhidroftalazina, dihidronaftiridina,tetrahidronaftiridina, perhidronaftiridina, dihidroquinoxalina, tetrahidroquinoxalina, perhidroquinoxalina, dihidroqui-nazolina, tetrahidroquinazolina, perhidroquinazolina, dihidrocinolina, tetrahidrocinolina, perhidrocinolina, benzoxa-tiano, dihidrobenzoxazina, dihidrobenzotiazina, pirazinomorfolina, dihidrobenzoxazol, perhidrobenzoxazol, dihidro-benzotiazol, perhidrobenzotiazol, dihidrobenzimidazol, perhidrobenzimidazol, dioxolano, dioxano, ditiolano, ditiano,dioxaindano, benzodioxano, cromano, benzoditiolano, benzoditiano, etc.
En la presente invención, el regulador de PPAR incluye todos los reguladores de PPAR α, γ, δ, α+γ, α+δ, γ+δy α+γ+δ. La manera reguladora preferible es, regulador de PPAR α, regulador de PPAR γ, regulador de PPAR δ,regulador de PPAR α+γ, regulador de PPAR α+δ, más preferiblemente regulador de PPAR α+γ. El regulador dePPAR también incluye al agonista de PPAR y el antagonista de PPAR, preferiblemente el agonista de PPAR, máspreferiblemente el agonista de PPAR α, el agonista de PPAR γ, el agonista de PPAR δ, el agonista de PPAR α+γ o elagonista de PPAR α+δ, particularmente preferiblemente el agonista de PPAR α+γ.
A no ser que se especifique lo contrario, todos los isómeros están incluidos en la presente invención. Por ejemplo,los grupos alquilo, alcoxi y alquileno incluyen los lineales y los ramificados. Además, los isómeros del enlace doble,el anillo, el anillo fusionado (isómero E, Z, cis, trans), los isómeros generados a partir de los átomos de carbono
5

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
asimétricos (isómero R, S, α, β, enantiómero, diastereómero), los isómeros ópticamente activos (isómero D, L, d,l), los compuestos polares generados mediante separación cromatográfica (compuesto más polar, compuesto menospolar), los compuestos en equilibrio, las mezclas de los mismos en proporciones voluntarias y las mezclas racémicastambién están incluidas en la presente invención.
Según la presente invención, a no ser que se indique de otro modo y como resulta evidente para los expertos en latécnica, el símbolo
indica que está unido al lado opuesto de la lámina (es decir configuración α), el símbolo
indica que está unido al lado frontal de la lámina (es decir configuración β), el símbolo
indica que es α-, β- o una mezcla de las mismas, y el símbolo
indica que es una mezcla de la configuración α y la configuración β.
El compuesto de la presente invención puede ser convertido en una sal no tóxica mediante métodos conocidos.
Una sal no tóxica es preferiblemente farmacéuticamente aceptable y soluble en agua.
Una sal no tóxica significa, por ejemplo, las sales de metales alcalinos (por ej., potasio, sodio, litio, etc.), lassales de metales alcalinotérreos (por ej., calcio, magnesio, etc.), las sales de amonio (por ej., tetrametilamonio, te-trabutilamonio, etc.), las sales de aminas orgánicas (por ej., trietilamina, metilamina, dimetilamina, ciclopentilamina,bencilamina, fenetilamina, piperidina, monoetanolamina, dietanolamina, tris(hidroximetil)metilamina, lisina, argini-na, N-metil-D-glucamina, etc.), las sales de adición de ácido (por ej., sales de ácidos inorgánicos (por ej., hidrocloruro,hidrobromato, hidroyodato, sulfato, fosfato, y nitrato, etc.), sales de ácidos orgánicos (por ej., acetato, trifluoroaceta-to, lactato, tartrato, oxalato, fumarato, maleato, benzoato, citrato, metanosulfonato, etanosulfonato, bencenosulfonato,toluenosulfonato, isetionato, glucuronato, gluconato, etc.), etc.
Además, un solvato del compuesto de la presente invención, y los metales alcalino(térreos), el amonio, las aminasorgánicas y las sales de adición de ácido del mismo anteriores, están incluidos en la presente invención.
El solvato es preferiblemente no tóxico y soluble en agua. Los solvatos apropiados significan, por ejemplo, solvatostales como agua, un disolvente alcohólico (por ej., etanol, etc.), etc.
(1) Los compuestos representados mediante la fórmula (IA)
(donde R5−1 representa un grupo alquilo C1-C8,
X representa (1) un enlace, o (2) alquileno C1-C4,
Y representa (1) -O-, o (2) -S-,
6

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
Z representa alquileno C1-C4,
A representa (1) -O-, o (2) -S-,
R2 y R3 representa cada uno independiente (1) hidrógeno, (2) alquilo C1-C8, (3) alcoxi C1-C8, o (4) alcoxi C1-C8sustituido con fenilo,
R4 representa (1) hidrógeno, o (2) alquilo C1-C8,
D representa D1, D2 o D3
D1 representa
anillo 1 representa un grupo arilo mono- o bicarbocíclico C3-10 opcionalmente parcialmente o totalmente saturado,
D2 representa
anillo 2 representa un grupo arilo mono- o biheterocíclico de 3-10 miembros opcionalmente parcialmente o totalmentesaturado que contiene 1-4 heteroátomos seleccionados entre un átomo de oxígeno, nitrógeno o azufre,
D3 representa alquilo C1-C8,
R6 representa (1) hidrógeno, (2) alquilo C1-C8, (3) nitro, (4) NR7R8, (5) halógeno, (6) alcoxi C1-C8, (7) alquil(C1-C8)tio, (8) CF3, (9) CF3O, (10) arilo mono- o bicarbocíclico C3-10 opcionalmente parcialmente o totalmentesaturado, o (11) arilo mono- o biheterocíclico de 3-10 miembros opcionalmente parcialmente o totalmente saturadoque contiene 1-4 heteroátomos seleccionados entre un átomo de oxígeno, nitrógeno o azufre,
R7 y R8 representan cada uno independiente (1) un átomo de hidrógeno, o (2) alquilo C1,
m representa 1-3, se puede preparar mediante los siguientes métodos.
El compuesto representado mediante la fórmula (IA) se puede preparar haciendo reaccionar un compuesto repre-sentado mediante la fórmula (II)
7

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
(donde R9 representa un grupo eliminable (por ej., un átomo de halógeno, un grupo mesiloxi o un grupo tosiloxi,etc.), D4 tiene el mismo significado que D, con la condición de que el grupo amino incluido en el grupo representadopor D4 está protegido si fuera necesario. Otros símbolos tienen los mismos significados que se han descrito antes.) conun compuesto representado mediante la fórmula (III)
(donde R10 representa un grupo OH o un grupo SH, y los otros símbolos tienen los mismos significados que se handescrito antes.), si fuera necesario sometiéndolo después si fuera necesario a una reacción de desprotección del grupoprotector.
Esta reacción es conocida. Por ejemplo, se lleva a cabo de 0 a 80ºC en un disolvente orgánico (por ej., tetrahidrofu-rano (THF), éter dietílico, cloruro de metileno, cloroformo, tetracloruro de carbono, hexano, hexano, benceno, tolueno,dimetilformamida (DMF), dimetilsulfóxido (DMSO), hexametilfosforamida (HMPA), etc.) en presencia de una base(por ej., hidruro de sodio, carbonato de potasio, trietilamina, piridina, yoduro de sodio, carbonato de cesio, etc.).
La reacción de desprotección de estos grupos protectores se puede llevar a cabo mediante los siguientes métodos.
La reacción de desprotección de estos grupos protectores de un grupo amino es conocida, y los ejemplos incluyen
(1) una reacción de desprotección en condiciones ácidas,
(2) una reacción de desprotección mediante hidrogenolisis, y similares.
Estos métodos se describen específicamente más abajo.
(1) La reacción de desprotección en condiciones ácidas se lleva a cabo, por ejemplo, en un disolvente orgánico(por ej., cloruro de metileno, cloroformo, dioxano, acetato de etilo, anisol, metanol, etanol, alcohol isopropílico, etc.)o en ausencia de un disolvente orgánico o una solución acuosa del mismo, utilizando un ácido orgánico (por ej., ácidoacético, ácido trifluoroacético, ácido metanosulfónico, etc.), un ácido inorgánico (por ej., ácido clorhídrico, ácidosulfúrico, etc.) o una mezcla de los mismos (por ej., bromuro de hidrógeno/ácido acético, etc.) de 0 a 100ºC.
(2) La reacción de desprotección mediante hidrogenolisis se lleva a cabo, por ejemplo, en un disolvente, (por ej.,un sistema etérico (por ej., tetrahidrofurano, dioxano, dimetoxietano, éter dietílico, etc.), un sistema alcohólico (porej., metanol, etanol), un sistema bencénico (por ej., benceno, tolueno, etc.), un sistema cetónico (por ej., acetona,metiletilcetona, etc.), un sistema nitrílico (por ej., acetonitrilo, etc.), un sistema amídico (por ej., dimetilformamida,etc.), agua, acetato de etilo, ácido acético o una mezcla disolvente de dos o más de ellos, etc.), en presencia de uncatalizador (por ej., paladio-carbono, negro de paladio, hidróxido de paladio, óxido de platino, níquel Raney, etc.) apresión normal o forzada en una atmósfera de hidrógeno o en presencia de formiato de amonio de 0 a 200ºC.
Los ejemplos del grupo protector de un grupo amino incluyen un grupo benciloxicarbonilo, un grupo t-butoxicar-bonilo, un grupo trifluoroacetilo, y un grupo 9-fluorenilmetoxicarbonilo.
Los grupos protectores de un grupo amino no están particularmente limitados a los anteriores, y también se puedenutilizar otros grupos, con tal que pueden ser liberados fácilmente y selectivamente. Por ejemplo, se pueden utilizar losdescritos por T.W. Greene en Protective Groups in Organic Synthesis, 3a edicion, Wiley, New York, 1999.
Si bien los expertos en la técnica lo pueden entender fácilmente, se puede preparar fácilmente un compuestoobjetivo de la presente invención utilizando apropiadamente estas reacciones de desprotección.
Además, entre los compuestos representados mediante la fórmula (IA), se puede preparar un compuesto donde Yrepresenta un grupo -O-, es decir, un compuesto representado mediante la fórmula (IA-1)
8

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
(donde todos los símbolos tienen los mismos significados que se han descrito antes.) haciendo reaccionar uncompuesto representado mediante la fórmula (IV)
(donde todos los símbolos tienen los mismos significados que se han descrito antes.) con un compuesto represen-tado mediante la fórmula (III-1)
(donde todos los símbolos tienen los mismos significados que se han descrito antes.), si fuera necesario sometiéndoloa una reacción de desprotección del grupo protector.
Esta reacción es conocida. Por ejemplo, se lleva a cabo mediante reacción con un compuesto alcohólico correspon-diente en un disolvente orgánico (por ej., diclorometano, éter dietílico, tetrahidrofurano, acetonitrilo, benceno, tolueno,etc.) en presencia de un compuesto azoico (por ej., azodicarboxilato de dietilo, azodicarboxilato de diisopropilo, 1,1’-(azodicarbonil)dipiperidina, 1,1’-azobis(N,N-dimetilformamida), etc.) y un compuesto fosfina (por ej., trifenilfosfina,tributilfosfina, trimetilfosfina, etc.).
La reacción de desprotección de un grupo protector se puede llevar a cabo mediante los métodos descritos antes.
(2) Los compuestos representados mediante la fórmula (IB)
(donde todos los símbolos tienen los mismos significados que se han descrito antes.) se pueden preparar mediante lossiguientes métodos.
El compuesto representado mediante la fórmula (IB) se puede preparar sometiendo el compuesto anterior repre-sentado mediante la fórmula (IA) a una reacción de hidrólisis.
Dicha reacción de hidrólisis es conocida. Se lleva a cabo, por ejemplo,
(1) en un disolvente orgánico miscible con agua (por ej., THF, dioxano, etanol, metanol etc.) o una mezcla disol-vente de los mismos, utilizando una solución acuosa de un álcali (por ej., hidróxido de potasio, hidróxido de sodio,hidróxido de litio, carbonato de potasio, carbonato de sodio etc.), o
(2) en un alcanol (por ej., metanol, etanol etc.), utilizando el álcali anterior en condiciones anhidras. Estas reaccio-nes se pueden llevar a cabo a 0 ∼ 100ºC normalmente.
Los compuestos representados mediante las fórmulas (II) y (IV) son compuestos conocidos o se pueden prepararfácilmente mediante métodos conocidos o métodos descritos en los Ejemplos.
Por ejemplo, entre los compuestos de fórmula (IV), el 2-(5-metil-2-feniloxazol-4-il)etanol se puede preparar me-diante los métodos descritos en J. Med. Chem., 35, 1853-1864 (1992).
Por ejemplo, entre los compuestos de fórmula (IV), el 2-(5-metil-2-(morfolin-4-il)oxazol-4-il)etanol se puedepreparar mediante los métodos descritos in J. Med. Chem., 41, 5037-5054(1998).
9
5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
Los compuestos representados mediante las fórmulas (II), (III), (III-1), y (IV) son compuestos conocidos o sepueden preparar fácilmente mediante métodos conocidos o métodos descritos en los Ejemplos.
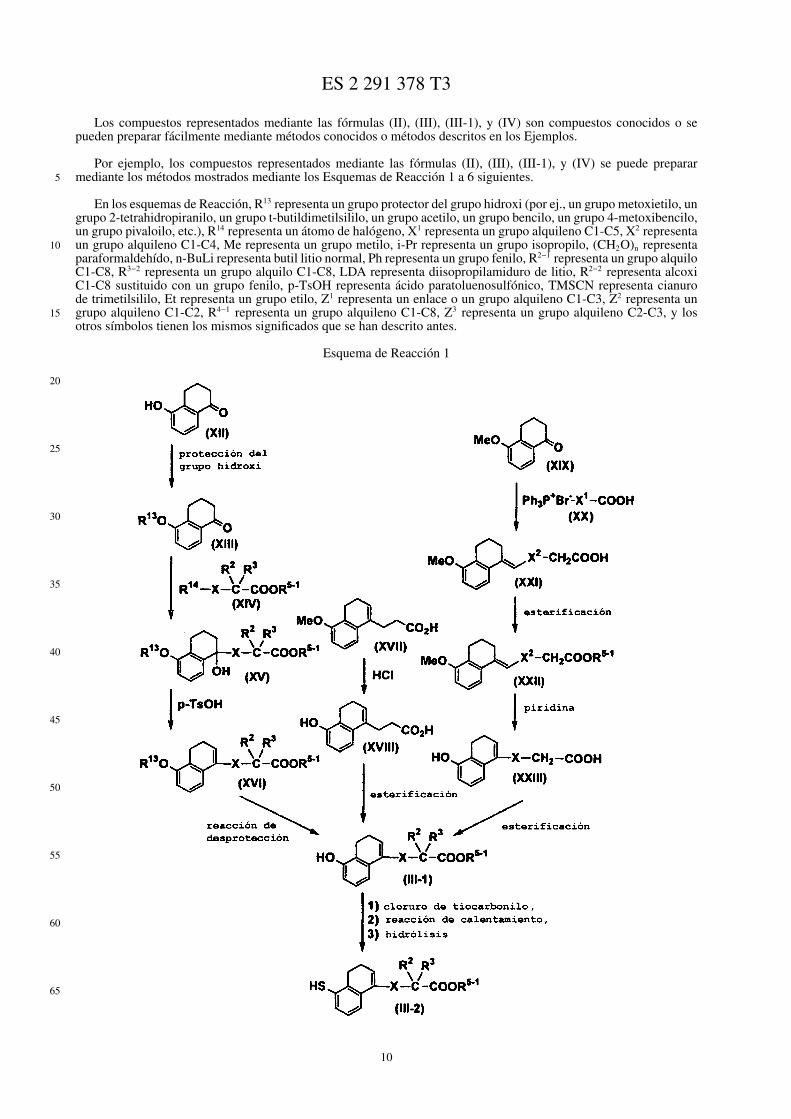
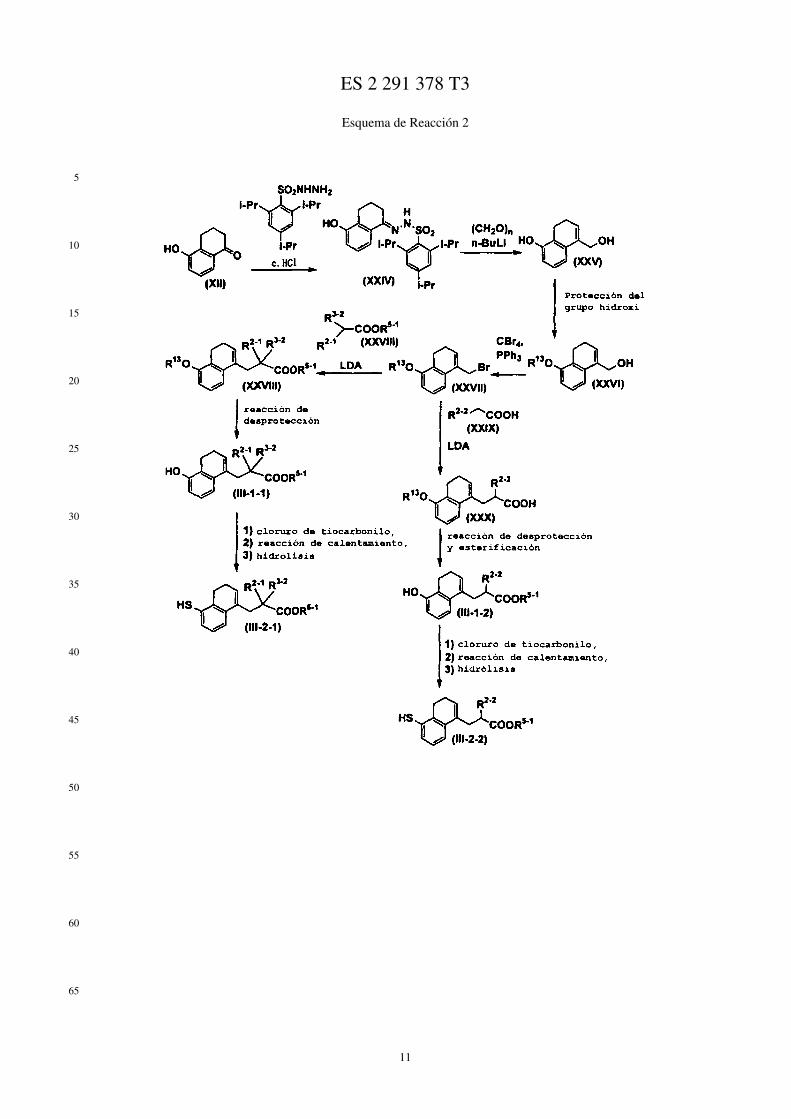
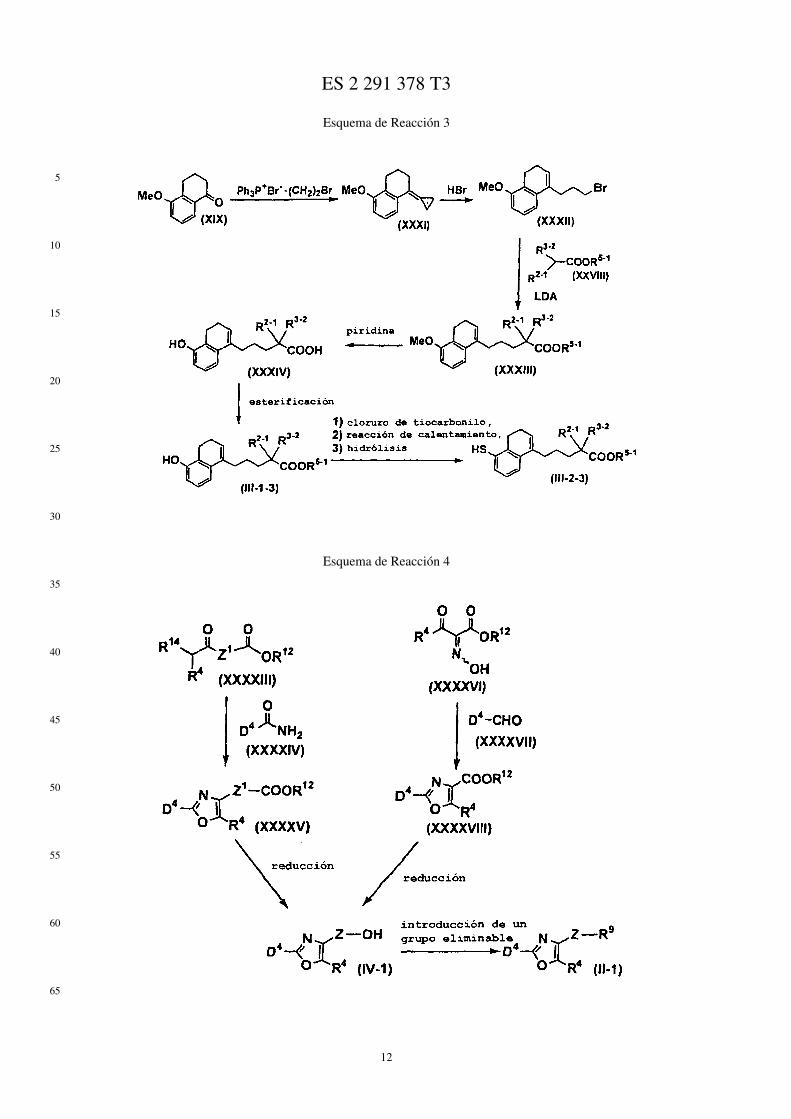
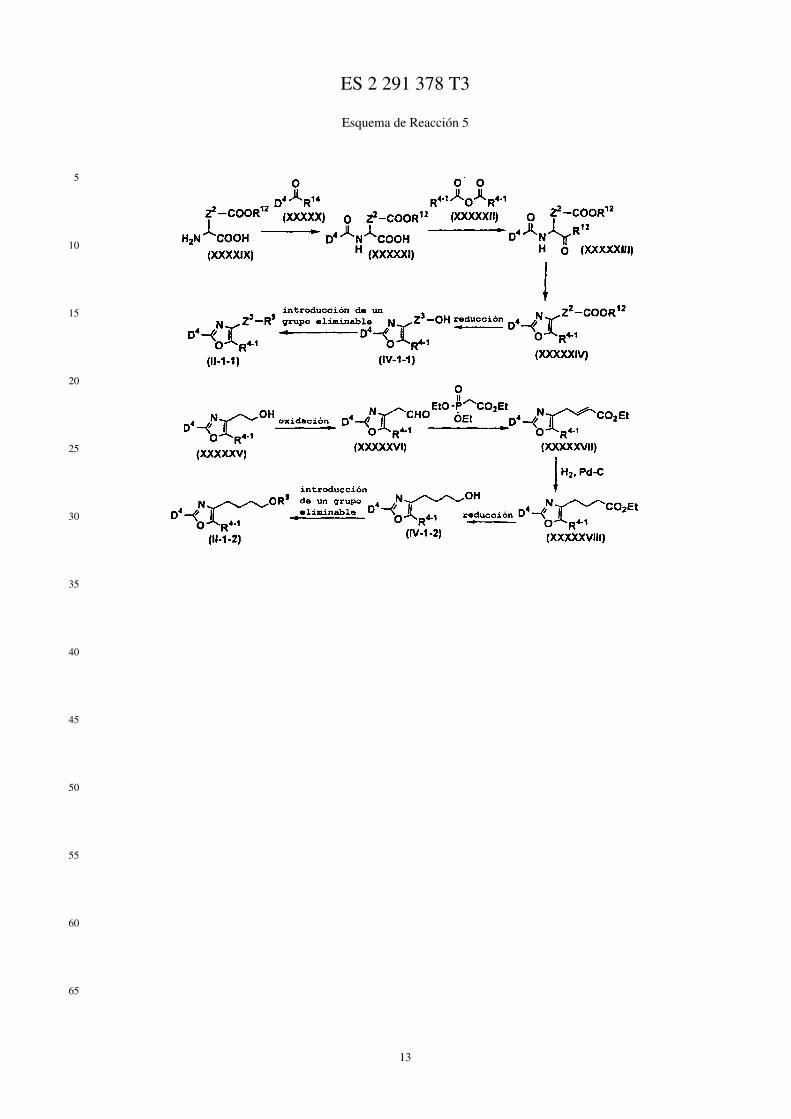
Por ejemplo, los compuestos representados mediante las fórmulas (II), (III), (III-1), y (IV) se puede prepararmediante los métodos mostrados mediante los Esquemas de Reacción 1 a 6 siguientes.
En los esquemas de Reacción, R13 representa un grupo protector del grupo hidroxi (por ej., un grupo metoxietilo, ungrupo 2-tetrahidropiranilo, un grupo t-butildimetilsililo, un grupo acetilo, un grupo bencilo, un grupo 4-metoxibencilo,un grupo pivaloilo, etc.), R14 representa un átomo de halógeno, X1 representa un grupo alquileno C1-C5, X2 representaun grupo alquileno C1-C4, Me representa un grupo metilo, i-Pr representa un grupo isopropilo, (CH2O)n representaparaformaldehído, n-BuLi representa butil litio normal, Ph representa un grupo fenilo, R2−1 representa un grupo alquiloC1-C8, R3−2 representa un grupo alquilo C1-C8, LDA representa diisopropilamiduro de litio, R2−2 representa alcoxiC1-C8 sustituido con un grupo fenilo, p-TsOH representa ácido paratoluenosulfónico, TMSCN representa cianurode trimetilsililo, Et representa un grupo etilo, Z1 representa un enlace o un grupo alquileno C1-C3, Z2 representa ungrupo alquileno C1-C2, R4−1 representa un grupo alquileno C1-C8, Z3 representa un grupo alquileno C2-C3, y losotros símbolos tienen los mismos significados que se han descrito antes.
Esquema de Reacción 1
10
5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
Esquema de Reacción 2
11
5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
Esquema de Reacción 3
Esquema de Reacción 4
12
5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
Esquema de Reacción 5
13

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
Esquema de Reacción 6
En los Esquemas de Reacción, los compuestos que se van a utilizar como las sustancias de partida representadasmediante las fórmulas (XII), (XIV), (XVII), (XIX), (XX), (XXVIII), (XXXXIII), (XXXXIV), (XXXXVI), (XXXX-VII), (XXXXIX), (XXXXX), (XXXXXII), (XXXXXIX), (XXXXXXI), (XXXXXXII) y (XXXXXXIV) son com-puestos conocidos o se pueden preparar fácilmente mediante métodos conocidos.
En cada reacción descrita aquí, el producto de reacción se puede purificar mediante técnicas de purificación ge-nerales tales como destilación a presión ordinaria o a presión reducida, cromatografía líquida de alto rendimiento,cromatografía en capa fina o cromatografía en columna utilizando gel de sílice o silicato de magnesio, lavado y recris-talización. La purificación se puede llevar a cabo en cada reacción o después de completar varias reacciones.
Actividad Farmacológica
Se confirmó que los compuestos de la presente invención tienen actividades reguladoras de PPAR mediante lossiguientes experimentos.
Medición de las actividades agonística de PPAR α y agonística de PPAR γ
(1) Preparación de materiales en el análisis con luciferasa utilizando PPAR α o γ humano
Todas las operaciones se llevaron a cabo mediante los métodos básicos de las técnicas de ingeniería genética y losmétodos convencionales de los sistemas de Un híbrido o Dos híbridos en levaduras.
Como vector de expresión del gen de la luciferasa bajo el control del promotor de la timidina quinasa (TK), seescindió el gen estructural de la luciferasa de PicaGene Basic Vector 2 (nombre comercial, Toyo Ink Inc., Núm. decatálogo 309-04821), para preparar el vector de expresión del gen de la luciferasa pTK-Luc. bajo el control del pro-motor TK (-105/+51) como una actividad promotora esencial mínima de pTKβ que tenía el promotor TK (ChrontechInc., Núm. de catálogo 6179-1). En la corriente superior del promotor TK, se insertó repetida cuatro veces la secuencia
14

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
UAS, que es el elemento de respuesta de la proteína Gal4, un factor de transcripción básico en levaduras, para construir4 X UAS-TK-Luc. como gen informador. La siguiente es la secuencia intensificadora utilizada (Secuencia No. 1).
Secuencia No. 1: Secuencia intensificadora que repite el elemento de respuesta Gal4 cuatro veces en tándem.
5’-T(CGACGGAGTACTGTCCTCCG)x4 AGCT-3’
Se preparó un vector como se describe más adelante que expresa la proteína receptora quimérica donde en elextremo carboxi de la proteína Gal4 de levadura se fusionó el dominio de unión al ADN al dominio de unión alligando de PPAR α o γ humano. Es decir, PicaGene Basic Vector 2 (nombre comercial, Toyo Ink Inc., Núm. decatálogo 309-04821) se utilizó como vector de expresión básico, el gen estructural se cambió por el de la proteínareceptora quimérica, mientras que los dominios del promotor y del intensificador se mantuvieron tal cual.
El ADN que codifica una proteína fusionada compuesta por el dominio de unión al ADN de Gal4, la secuencia deaminoácidos del 1º al 147º conectada al dominio de unión al ligando de PPAR α o γ humano en marco se insertó aguasabajo del promotor/intensificador en PicaGene Basic Vector 2 (nombre comercial, Toyo Ink Inc., Núm. de catálogo309-04821). Aquí el ADN se alineó como sigue; en el extremo amino del dominio de unión al ligando de PPAR α oγ humano, se añadió la señal de traslocación nuclear originada a partir del antígeno T de SV-40, Ala Pro Lys Lys LysArg Lys Val Gly (secuencia No. 2) para hacer que la proteína de fusión se localice intranuclearmente. Por otra parte,en sus extremos carboxi, se añadió el epítopo de la hemaglutinina de la influenza, Tyr Pro Tyr Asp Val Pro Asp TyrAla (secuencia No. 3) y un codón de terminación de la traducción por este orden, para detectar una secuencia marcadacon una etiqueta epitópica de la proteína fusionada expresada.
Según la comparación de las estructuras de los PPAR humanos descritas en las publicaciones por R. Mukherjeeet al. (Véase J. Steroid Biochem. Molec. Biol., 51, 157 (1994)), M. E. Green et al., (Véase Gene Expression., 4, 281(1995)), A. Elbrecht et al. (Véase Biochem Biophys. Res. Commun., 224, 431 (1996)) o A. Schmidt et al. (Véase Mol.Endocrinology., 6, 1634 (1992)), la porción del gen estructural utilizada como dominio de unión al ligando de PPARα o γ humano fue ADN que codifica el siguiente péptido:
dominio de unión al ligando de PPAR α humano: Ser167-Tyr468
dominio de unión al ligando de PPAR γ humano: Ser176- Tyr478
(cada dominio de unión al ligando de PPAR γ1 humano y dominio de unión al ligando de PPAR γ2 humano esSer204-Tyr506 que son secuencias idénticas entre sí).
Con el fin de medir el nivel basal de transcripción, se preparó también un vector de expresión que contenía eldominio de unión al ADN de la proteína Gal4 que carecía del dominio de unión al ligando de PPAR, que es codificadoexclusivamente por la secuencia de aminoácidos del 1º al 147º en la proteína Gal4.
(2) Análisis con Luciferasa utilizando PPAR α o γ humano
Las células CV-1 utilizadas como células anfitrionas se cultivaron mediante una técnica convencional. Es decir,se utilizaron medio de Eagle modificado de Dulbecco (DMEM) con un suplemento de suero bovino fetal al 10%(GIBCO BRL Inc., Núm. de catálogo 26140-061) y 50 U/ml de penicilina G y 50 µg/ml de sulfato de estreptomicinapara cultivar las células CV-1 en una atmósfera con gas dióxido de carbono al 5% a 37ºC.
Se sembraron 2 x 106 células en una placa de 10 cm, y se lavaron una vez con el medio sin suero, seguido dela adición del medio (10 ml). Se mezclaron bien el gel informador (10 µg), el vector de expresión de Gal4-PPAR(0,5 µg) y 50 µl de LipofectAMINE (GIBRO BRL Inc., Núm. de catálogo 18324-012) y se añadieron al cultivo paraintroducir estos ADN en las células anfitrionas. Se cultivaron a 37ºC durante 5-6 horas, y a esto se le añadieron 10ml de medio que contenía 20% de suero bovino fetal sometido a diálisis (GIBRO BRL Inc., Núm. de catálogo 26300-061), y después se cultivaron a 37ºC durante la noche. Las células se dispersaron con tripsina, y se sembraron de nuevoen placas de 96 pocillos a una densidad de 8.000 células/100 ml de DMEM-10% suero sometido a diálisis/pocillo.Varias horas después del cultivo, cuando las células se anclaron al soporte de plástico, se añadieron a esto 100 µl deDMEM-suero sometido a diálisis al 10% que contenía los compuestos de la presente invención, cuya concentraciónes dos veces tan alta como sus concentraciones finales. El cultivo se sedimentó a 37ºC durante 42 horas y las célulasse disolvieron para medir la actividad luciferasa según las instrucciones del fabricante.
En cuanto a la actividad agonística de PPAR α, la actividad relativa de los compuestos de la presente invención(10 µM) se mostró en la Tabla 14, bajo la condición de que la actividad luciferasa se definió como 1.0 en el casode la carbaciclina (10 µM) como compuesto de control positivo, lo que podría activar la transcripción del gen de laluciferasa significativamente para PPAR α (Véase Eur. J. Biochem., 233, 242 (1996); Genes & Development., 10, 974(1996)).
En cuanto a la actividad agonística de PPAR γ, la actividad relativa de los compuestos de la presente invención(10 µM) se mostró en la Tabla 15, bajo la condición de que la actividad luciferasa se definió como 1.0 en el casode la troglitazona (10 µM) como compuesto de control positivo, lo que podría activar la transcripción del gen de la
15

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
luciferasa significativamente para PPAR γ (Véase Cell., 83, 863 (1995); Endocrinology., 137, 4189 (1996) y J. Med.Chem., 39, 665 (1996)) y se ha introducido en el mercado como agente hipoglucémico.
Además, se llevó a cabo el análisis de cada compuesto tres veces para examinar su reproducibilidad y para confir-mar la actividad dependiente de la dosis.
Asimismo, el siguiente compuesto descrito en el Ejemplo 3(35) en la memoria de la publicación WO9911255 seutilizó como compuesto comparativo.
compuesto descrito en el Ejemplo 3(35) en la memoria de la publicación WO9911255
TABLA 14
TABLA 15
Por ejemplo, los efectos hipoglucémico e hipolipidémico de los compuestos de la presente invención se puedenmedir mediante los siguientes métodos.
Efecto Hipoglucémico e hipolipidémico (1)
Ratones macho KKAy/TA jcl de 8 semanas de edad (cinco ratones por grupo) se crían previamente individualmenteen jaulas sencillas durante aproximadamente una semana y se les proporciona dieta granulada y agua del grifo desdeuna botella de agua de alimentación ad libitum. Los ratones se aclimatan al cambio con dieta molida durante tres días.El primer día del experimento (Día 0), se mide el peso corporal de los ratones. Se recogen muestras de sangre de la venacoccígea utilizando un microcapilar para medir la concentración de glucosa en plasma. Basándose en la concentraciónde glucosa en plasma, los ratones se dividen en varios grupos (cinco ratones por grupo) utilizando un método dealeatorización estratificado. El peso corporal de los ratones se mide la mañana del día siguiente, y a partir del díasiguiente durante seis días se les proporcionan los compuestos mediante una mezcla alimentaria que contenía 0,03%(p/p), 0,01% (p/p) o 0,003% (p/p) del compuesto de la presente invención o sólo mediante dieta molida. La mañanadel cuarto y séptimo día, se determinaron sus pesos corporales y sus tomas de alimento para calcular la dosis mediaadministrada. La mañana del sexto día, se recogieron muestras de sangre de la vena coccígea para medir los nivelesde glucosa y triglicéridos (TG). El séptido día después de medir el peso corporal, se recogieron muestras de sangrede la vena cava abdominal bajo condiciones de anestesia con éter para determinar los niveles de insulina en plasma,ácidos grasos no esterificados (NEFA), GOT y GPT utilizando kits adquiribles comercialmente. Y, el hígado se retiray se pesa. Los ARN totales se preparan a partir del lóbulo izquierdo del hígado y se mide el nivel de expresión génica
16

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
de la proteína bi-funcional (hidrasa-deshidrogenasa, HD) mediante el método de transferencia Northern. Realmente,no existe una diferencia significativa en la toma de alimento entre el grupo de control (sólo dieta molida) y el grupotratado con los compuestos (dieta molida que contenía 0,03%, 0,01% o 0,003% de los compuestos). La dosis calculadaes de aproximadamente 40 mg/kg/día en el grupo al que se administró la dieta que contenía 0,03% del compuesto.
Se sugiere la posibilidad como agente para la prevención y/o el tratamiento de la diabetes mellitus, la hiperlipide-mia, la aterosclerosis etc., a partir de los efectos mejoradores de los niveles de glucosa en plasma, insulina en plasma,NEFA o TG en ratones KKAy/Ta bien alimentados. Es probable que este efecto esté mediado por la activación dePPAR γ in vivo. Adicionalmente, es probable que un aumento de peso del hígado y de la expresión del ARNm de HDdependa de la activación de PPAR α in vivo.
Efecto Hipoglucémico e hipolipidémico (2)
Ratas Zucker fa/fa (Cepa: Crj-[ZUC]-fa/fa) de 8 semanas de edad y ratas delgadas Zucker sanas (Cepa: Crj-[ZUC]-lean) para ser contrastadas se crían previamente individualmente en jaulas sencillas durante aproximadamente dossemanas y se les proporciona dieta granulada y agua del grifo desde un equipo automático para el suministro deagua ad libitum. Durante cinco días antes del tratamiento, las ratas se aclimatan a la administración mediante gavageoral. Durante este período, se observan sus condiciones generales, y se utilizan para el experimento ratas sanas de10 semanas de edad. Se mide el peso corporal de cada rata la mañana del primer día del experimento (Día 0) yse recogen muestras de sangre de la vena coccígea utilizando un microcapilar para medir las concentraciones deglucosa, TG, NEFA en plasma y HbA1c. Basándose en HbA1c y en el peso corporal, las ratas se asignan a gruposcompuestos por cinco animales cada uno utilizando un método de aleatorización estratificado. Adicionalmente, lasratas se intercambian opcionalmente para evitar la desviación de otras medias de los parámetros entre grupos. El pesocorporal de cada animal se mide todas las mañanas de los días posteriores al agrupamiento. Los volúmenes que sevan a administrar se calculan sobre la base del peso corporal medido el día de la administración, y se lleva a cabola administración mediante gavage oral del compuesto de la presente invención o de vehículo solo (metilcelulosa al0,5%) una vez al día durante 13 días. A los animales sanos (ratas delgadas) se les administra solamente vehículo.
Se mide el consumo de alimentos durante la mañana de los Días 1, 4, 7, 10 y 13 para calcular las tomas medias dealimento. El séptimo día, se recogieron muestras de sangre de la vena coccígea utilizando un microcapilar para medirlas concentraciones de glucosa, TG, NEFA en plasma y HbA1c. Y el día 14º, se realiza el ensayo de tolerancia oral ala glucosa (OGTT) para evaluar el efecto mejorador sobre la intolerancia a la glucosa. Se hace ayunar a las ratas el díaprevio (Día 13) para realizar el OGTT. Después de recoger las muestras de sangre al día siguiente (Día 14), se cargauna solución de glucosa al 40% a un volumen de 2 g/5 ml/kg por administración oral. Sesenta y 120 minutos despuésde la carga, se recogen muestras de sangre de la vena coccígea utilizando un microcapilar para determinar los nivelesde glucosa en plasma.
A los animales se les proporciona alimento después del OGTT y se les administra el compuesto de la presenteinvención en Día 15. La mañana del día 16º después de medir el peso corporal, se recogen muestras de sangre de lavena cava abdominal en condiciones de anestesia con éter para determinar los niveles de glucosa en plasma, insulinaen plasma, TG, NEFA, GOT y GPT. Y el hígado se retira y se pesa.
Se sugiere la posibilidad como agente para la prevención y/o el tratamiento de la diabetes mellitus, la hiperlipi-demia, la aterosclerosis etc., para mejorar los efectos de los niveles de glucosa en plasma, insulina en plasma, TG,NEFA o de HbA1c en ratas Zucker fa/fa bien alimentadas. Asimismo, un efecto de descenso de la glucosa en plasmaen ayunas y un efecto mejorador de la intolerancia a la glucosa durante OGTT sugieren la posibilidad como agentepara la prevención y/o el tratamiento de la diabetes mellitus. Estos efectos están mediados probablemente a travésde la activación de PPAR γ in vivo. Adicionalmente, se sugiere que un aumento del peso del hígado depende de laactivación de PPAR α in vivo.
Efecto Hipoglucémico e hipolipidémico (3)
Se realiza una inspección médica a monos macho cynomolgus de 3 a 4 años de edad (peso corporal medio: aproxi-madamente 3 kg) para que tengan una inspección médica real y se aclimatan para proporcionarles aproximadamente100 g de dieta granulada una vez al día y agua del grifo desde un equipo de suministro de agua automático ad libitum,individualmente en jaulas para monos sencillas durante más de un mes. Después de eso, los animales volvieron a tomaruna dieta en una hora. Adicionalmente, los animales se criaron previamente durante 14 días. Catorce y 7 días antes deltratamiento, se mide el peso corporal, y después se recogen muestras de sangre de la vena safena del miembro pos-terior para medir los parámetros hematológicos (glóbulos rojos, hematocrito, hemoglobina, plaquetas y leucocitos) ybioquímicos (GOT, GPT, fosfatasa alcalina, proteína total, nitrógeno de urea en sangre, creatinina, creatinina quinasa,bilirrubina total, glucosa, colesterol total, HDL, LDL y TG). Adicionalmente, se observa la condición general de losanimales durante la aclimatación y la crianza previa, y se utilizan los animales sanos para el experimento. Asimismo,se mide cada día el consumo de alimentos.
Sobre la base del peso corporal medido el día final del período de aclimatación, los animales se dividen en variosgrupos (tres animales por grupo) utilizando un método de aleatorización estratificado. La mañana del Día 1, 3, 7,10 y 14, se mide el peso corporal. Los volúmenes que se van a administrar se calculan basándose en el último pesocorporal, y se lleva a cabo la administración mediante gavage oral del compuesto de la presente invención (3-100
17

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
mg/kg/día) o vehículo solo (solución diluida) una vez al día durante 14 días. 1, 7 y 14 días después del tratamiento,se recogen muestras de sangre para medir los parámetros hematológicos y bioquímicos anteriormente mencionadosantes de la administración del compuesto de la presente invención. Se confirma que la glucosa en sangre no cambiacon el compuesto de la presente invención. Al cabo de tres semanas, y 14 días después del comienzo del tratamiento,se recogen muestras de sangre de la vena safena del miembro posterior o de la vena antebraquial 1, 2 y 4 horasdespués del gavage oral, y también 1, 2 y 3 horas después de proporcionar una dieta, para medir la glucosa y los TG enplasma.
Se sugiere la posibilidad de un agente para la prevención y/o el tratamiento de la hiperlipidemia y la aterosclerosisetc., para mejorar los efectos de los niveles de TG en plasma en los monos en ayunas. Estos efectos están mediadosprobablemente por la activación de PPAR α in vivo. También se observa un efecto supresor del aumento de TG post-prandial. Adicionalmente, se puede estimar si el compuesto tiene riesgo de toxicidad a partir de otros parámetrosbioquímicos.
Toxicidad
La toxicidad del compuesto representado mediante la fórmula (I) de la presente invención es muy baja de maneraque se considera que el compuesto es suficientemente seguro para su uso como fármaco.
Aplicabilidad industrial
Aplicación a fármacos
Puesto que el compuesto representado mediante la fórmula (I) de la presente invención y las sales no tóxicas delmismo tienen una actividad moduladora de PPAR, se espera su aplicación como agente hipoglucémico, agente hipoli-pidémico, agente para la prevención y/o el tratamiento de las enfermedades asociadas con trastornos metabólicos talescomo la diabetes, la obesidad, el síndrome X, la hipercolesterolemia y la hiperlipoproteinemia etc., la hiperlipidemia,la aterosclerosis, la hipertensión, las enfermedades circulatorias, la sobreingesta, las enfermedades cardíacas corona-rias etc., agentes que elevan el colesterol HDL, los agentes que rebajan el colesterol LDL y/o el colesterol VLDL y losagentes para suprimir los factores de riesgo de la diabetes o el síndrome X.
Asimismo, puesto que el compuesto representado mediante la fórmula (I) de la presente invención, y las sales notóxicas del mismo, tienen un efecto agonista de PPARα y/o un efecto agonista de PPAR γ, se espera que sean aplicadoscomo agentes hipoglucémicos, agentes hipolipidémicos, agentes para la prevención y/o el tratamiento de enfermeda-des asociadas con trastornos metabólicos tales como la diabetes, la obesidad, el síndrome X, la hipercolesterolemia,la hiperlipoproteinemia etc., la hiperlipidemia, la aterosclerosis, la hipertensión, las enfermedades circulatorias y lasobreingesta etc., el efecto elevador del colesterol HDL, el efecto reductor del colesterol LDL y/o el colesterol VLDL,la inhibición del progreso de la aterosclerosis y su tratamiento, y el efecto inhibidor de la obesidad. También se esperaque sean útiles para el tratamiento y/o la prevención de la diabetes como agentes hipoglucémicos, para la mejora dela hipertensión, para la supresión de los factores de riesgo del síndrome X, y como agentes para la prevención de laaparición de enfermedades cardíacas coronarias.
Para los fines anteriormente descritos, los compuestos de la presente invención de fórmula (I) y las sales no tóxicasde los mismos se pueden administrar normalmente sistémicamente o localmente, usualmente mediante administraciónoral o parenteral.
El compuesto representado mediante la fórmula (I) de la presente invención, y una sal no tóxica del mismo se ad-ministra generalmente sistémicamente o tópicamente y oralmente o parenteralmente cuando se utiliza para los objetosanteriores.
Las dosis se determinan dependiendo de la edad, el peso corporal, los síntomas, el efecto terapéutico, la ruta deadministración, la duración del tratamiento y similares. Generalmente, se administran oralmente de 1 mg a 1.000 mgpor adulto de una a varias veces al día, o se administran parenteralmente de 1 mg a 100 mg por adulto (preferiblementemediante administración intravenosa) de una a varias veces al día, o se administra continuamente en vena durante 1 a24 horas al día.
Puesto que los cambios de dosis dependen de las diferentes condiciones descritas antes, existen casos donde sepueden utilizar dosis mayores o menores que los intervalos anteriores.
El compuesto representado mediante la fórmula (I) de la presente invención se puede administrar en forma decomposiciones sólidas, composiciones líquidas y otras composiciones para la administración oral, e inyectables, lini-mentos, supositorios y similares para la administración parenteral.
Las composiciones sólidas para la administración oral incluyen comprimidos, píldoras, cápsulas, polvos dispersa-bles, gránulos y similares.
Las cápsulas incluyen cápsulas duras y cápsulas blandas.
18

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
En tales composiciones sólidas, uno o más compuestos activos se mezclan con al menos un diluyente inerte talcomo lactosa, manitol, glucosa, hidroxipropilcelulosa, celulosa microcristalina, almidón, polivinilpirrolidona, o meta-silicato aluminato de magnesio. La composición puede contener también sustancias adicionales distintas de un dilu-yente inerte, por ej., lubricantes tales como estearato de magnesio, agentes disgregantes tales como glicolato cálcico decelulosa, agentes estabilizantes tales como lactosa, y agentes para ayudar a la disolución tales como ácido glutámicoy ácido aspártico según los métodos habituales. Si fuera necesario, están incluidas las tabletas o píldoras pueden estarrecubiertas con agentes de recubrimiento de película gástrica o entérica tales como azúcar, gelatina, hidroxipropilce-lulosa y ftalato de hidroxipropilcelulosa, o estar recubiertas de materiales absorbibles tales como gelatina.
Las composiciones líquidas para la administración oral incluyen emulsiones, soluciones, jarabes, elixires y simi-lares farmacéuticamente aceptables. En tales composiciones líquidas, están contenidos uno o más compuestos activosen un diluyente inerte utilizado comúnmente (por ej., agua purificada, etanol). Además, tales composiciones puedencontener también sustancias auxiliares tales como agentes humectantes o agentes suspensores, agentes edulcorante,agentes aromatizantes, agentes aromatizantes, y agentes conservantes.
Otras composiciones para la administración oral incluyen pulverizaciones que contienen uno o más compuestosactivos que se preparan mediante métodos conocidos. Tales composiciones pueden contener agentes estabilizantestales como hidrogenosulfato de sodio, agentes tamponadores para conferir isotonicidad, soluciones isotónicas talescomo cloruro de sodio, citrato de sodio o ácido cítrico, además de diluyentes inertes. El procedimiento para prepararpulverizaciones se describen en las Patentes de los Estados Unidos Núms. 2.868.691 y 3.095.355.
Los inyectables para la administración parenteral en la presente invención incluyen soluciones, suspensiones yemulsiones acuosas o no acuosas estériles. Las soluciones y suspensiones acuosas incluyen agua destilada para inyec-tables y solución salina fisiológica. Las soluciones y suspensiones no acuosas incluyen propilenglicol, polietilenglicol,aceites vegetales tales como aceite de oliva, alcoholes tales como etanol, POLYSORBATE80 (marca registrada), ysimilares. Las soluciones, suspensiones y emulsiones acuosas y no acuosas estériles se pueden utilizar en forma demezcla. Tales composiciones pueden contener adicionalmente agentes conservantes, agentes humectantes, agentesemulsionantes, agentes dispersantes, agentes estabilizantes (por ej., lactosa), agentes auxiliares tales como agentesauxiliares solubilizantes (por ej., ácido glutámico, ácido aspártico). Se pueden esterilizar mediante filtración a travésde un filtro de retención de bacterias, incorporación de un agente esterilizador o irradiación. Por ejemplo, también sepueden elaborar en forma de composiciones sólidas estériles que se pueden disolver en agua estéril u otro diluyenteestéril para inyectables antes del uso del producto liofilizado.
Otras composiciones para la administración parenteral incluyen líquidos para el uso externo, linimentos endérmi-cos, pomadas, supositorios para la administración intrarrectal, pesarios para la administración intravaginal y similaresque contienen uno o más compuestos activos que se pueden preparar mediante métodos conocidos.
Mejor modo de llevar a cabo la invención
La presente invención se explica más abajo en detalle basándose en los Ejemplos de Referencia y los Ejemplos, noobstante, la presente invención no está limitada a estos.
Los disolventes entre paréntesis muestran los disolventes de desarrollo o elución y las proporciones de los disol-ventes utilizado están en volumen en las separaciones cromatográficas o la TLC. Los disolventes entre paréntesis enel RMN muestran los disolventes para la medición.
Ejemplo de Referencia 1
Ácido 3-(5-hidroxi-3,4-dihidronaftalen-1-il)propanoico
A hidrocloruro de piridina (200 g), se le añadió ácido 3-(5-metoxi-3,4-dihidronaftalen-1-il)propanoico (25,1 g;compuesto conocido (Véase J. Chem. Soc. Perkin Trans. I., 1739-1742(1987)), seguido de agitación a 180ºC durante3 horas. La mezcla de reacción se enfrió a temperatura ambiente, y se diluyó con agua. La capa acuosa se acidulócon ácido clorhídrico concentrado. La capa acuosa se extrajo con acetato de etilo. El extracto se extrajo con unasolución acuosa saturada de hidrogenocarbonato de sodio. La capa acuosa combinada se aciduló con ácido clorhídricoconcentrado, seguido extracción con acetato de etilo. La capa orgánica combinada se lavó con solución salina saturada,se secó con sulfato de sodio anhidro, y se concentró a presión reducida para obtener de ese modo el compuesto deltítulo (11,8 g) que tiene los siguientes datos físicos.
TLC: Rf 0,42 (cloroformo : metanol = 6 : 1);
19

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
RMN (CDCl3): δ 9,21 (s, 1H), 6,98 (dd, J = 7,8, 7,6 Hz, 1H), 6,71 (d, J = 7,6 Hz, 1H), 6,70 (d, J = 7,8 Hz, 1H),5,82 (t, J = 4,4 Hz, 1H), 2,68-2,50 (m, 4H), 2,36 (m, 2H), 2,12 (m, 2H).
Ejemplo de Referencia 2
Éster metílico de ácido 3-(5-hidroxi-3,4-dihidronaftalen-1-il)propanoico
Se enfrió metanol anhidro (40 ml) a -10ºC, y a esto se le añadió gota a gota cloruro de tionilo (5,92 ml) enatmósfera de argón, seguido de agitación a -10ºC durante 20 minutos. A esta solución, se le añadió el compuesto (11,8g) preparado en el Ejemplo de Referencia 1, seguido de agitación a temperatura ambiente durante 1 hora. La mezcla dereacción se concentró a presión reducida, sometiéndola después a la formación del azeotropo con tolueno (dos veces).El residuo se purificó mediante cromatografía en columna de gel de sílice (cloroformo a cloroformo : metanol = 50 :1) para obtener de ese modo el compuesto del título (10,6 g) que tiene los siguientes datos físicos.
TLC: Rf 0,72 (cloroformo : metanol = 10 : 1);
RMN (CDCl3): δ 7,06 (dd, J = 7,8, 7,6 Hz, 1H), 6,88 (d, J = 7,6 Hz, 1H), 6,70 (d, J = 7,8 Hz, 1H), 5,88 (t, J = 4,4Hz, 1H), 4,93 (s, 1H), 3,68 (s, 3H), 2,82-2,62 (m, 4H), 2,58-2,49 (m, 2H), 2,26 (m, 2H).
Ejemplo de Referencia 3
5-Pivaloiloxi-1,2,3,4-tetrahidronaftalen-1-ona
A una solución en piridina (180 ml) de 5-hidroxi-1-tetralona (30,0 g), se le añadió 4-dimetilaminopiridina (1,13 g),y a esto se le añadió cloruro de pivaloilo (25,0 ml) enfriando con hielo, seguido de agitación a temperatura ambientedurante la noche. La mezcla de reacción se enfrió con hielo, y a esto se le añadió ácido clorhídrico concentrado,seguido extracción con acetato de etilo. El extracto se lavó con agua y solución salina saturada en este orden, se secócon sulfato de magnesio anhidro, y se concentró. El residuo se purificó mediante cromatografía en columna de gel desílice (hexano : acetato de etilo = 9 : 1 a 5 : 1) para obtener de ese modo el compuesto del título (45,4 g) que tiene lossiguientes datos físicos.
TLC: Rf 0,42 (hexano : acetato de etilo =5 : 1);
RMN (CDCl3): δ 7,95 (dd, J = 7,8, 1,4 Hz, 1H), 7,33 (t, J = 7,8 Hz, 1H), 7,19 (dd, J = 7,8, 1,4 Hz, 1H), 2,79 (t, J =6,0 Hz, 2H), 2,65 (dd, J = 7,6, 6,0 Hz, 2H), 2,19-2,05 (m, 2H), 1,40 (s, 9H).
Ejemplo de Referencia 4
Éster etílico de ácido 2-(1-hidroxi-5-pivaloiloxi-1,2,3,4-tetrahidronaftalen-1-il)acético
A una suspensión en benceno anhidro (60 ml) de zinc (16,9 g), se le añadió yodo (cantidad catalítica), seguido dereflujo calentando, y a esto se le añadió gota a gota una solución en benceno anhidro (120 ml) del compuesto (45,4 g)preparado en el Ejemplo de Referencia 3 y a esto se le añadió gota a gota éster etílico de ácido bromoacético (25,0 ml),seguido de reflujo calentando durante la noche. La mezcla de reacción se enfrió a temperatura ambiente. La mezcla dereacción se añadió a agua helada, y a esto se le añadió ácido clorhídrico concentrado, seguido extracción con acetatode etilo. El extracto se lavó con agua y solución salina saturada en este orden, se secó con sulfato de magnesio anhidro,y se concentró. El residuo se purificó mediante cromatografía en columna de gel de sílice (hexano : acetato de etilo =8 : 1 a 5 : 1) para obtener de ese modo el compuesto del título (33,5 g) que tiene los siguientes datos físicos.
20

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
TLC: Rf 0,52 (hexano : acetato de etilo = 85 : 15);
RMN (CDCl3): δ 7,42 (dd, J = 8,0, 2,0 Hz, 1H), 7,17 (t, J = 8,0 Hz, 1H), 6,85 (dd, J = 8,0, 2,0 Hz, 1H), 4,16 (c, J =7,0 Hz, 2H), 4,10-3,90 (ancho, 1H), 2,80 (d, J = 14,0 Hz, 1H), 2,76 (d, J =14,0 Hz, 1H), 2,68-2,40 (m, 2H), 2,12-1,44(m, 4H), 1,35 (s, 9H), 1,24 (t, J = 7,0 Hz, 3H).
Ejemplo de Referencia 5
Éster etílico de ácido 2-(5-pivaloiloxi-3,4-dihidronaftalen-1-il)acético
A una solución en tolueno (80 ml) del compuesto (33,5 g) preparado en el Ejemplo de Referencia 4, se le añadiómonohidrato de ácido p-toluenosulfónico (1,52 g), seguido de reflujo calentando durante la noche. La mezcla de reac-ción se enfrió a temperatura ambiente, se diluyó con acetato de etilo, se lavó con agua, una solución acuosa saturada dehidrogenocarbonato de sodio, agua y solución salina saturada en este orden, se secó con sulfato de magnesio anhidro,y se concentró. El residuo se purificó mediante cromatografía en columna de gel de sílice (hexano : acetato de etilo =20 : 1 a 10 : 1) para obtener de ese modo el compuesto del título (13,2 g) que tiene los siguientes datos físicos.
TLC: Rf 0,56 (hexano : acetato de etilo = 3 : 1);
RMN (CDCl3): δ 7,18 (t, J = 8,0 Hz, 1H), 7,08 (dd, J = 8,0, 1,0 Hz, 1H), 6,86 (dd, J = 8,0, 1,0 Hz, 1H), 6,01 (t, J= 4,5 Hz, 1H), 4,14 (c, J = 7,0 Hz, 2H), 3,44-3,40 (m, 2H), 2,63 (t, J = 8,0 Hz, 2H), 2,36-2,23 (m, 2H), 1,38 (s, 9H),1,22 (t, J = 7,0 Hz, 3H).
Ejemplo de Referencia 6
Éster etílico de ácido 2-(5-hidroxi-3,4-dihidronaftalen-1-il)acético
Enfriando con hielo, a una solución etanólica (50 ml) del compuesto (13,2 g) preparado en el Ejemplo de Referencia5, se le añadió gota a gota una solución etanólica de etilato de sodio (20 ml, 2,6 M), seguido de agitación a temperaturaambiente durante 3 horas. La mezcla de reacción se añadió a una mezcla de ácido clorhídrico 2 N y hielo, seguidoextracción con acetato de etilo. El extracto se lavó con solución salina saturada, se secó con sulfato de magnesioanhidro, y se concentró. El residuo se purificó mediante cromatografía en columna de gel de sílice (hexano : acetatode etilo = 4 : 1 a 2 : 1). El aceite obtenido se sometió a cristalización mediante una mezcla disolvente de hexano yacetato de etilo. Además, los cristales obtenidos se sometieron a recristalización en una mezcla disolvente de hexanoy acetato de etilo para obtener de ese modo el compuesto del título (7,73 g) que tiene los siguientes datos físicos.
TLC: Rf 0,34 (hexano : acetato de etilo = 3 : 1);
RMN (CDCl3): δ 7,00 (t, J = 8,0 Hz, 1H), 6,78 (d, J = 8,0, 1,0 Hz, 1H), 6,63 (dd, J = 8,0, 1,0 Hz, 1H), 5,98 (t, J =4,5 Hz, 1H), 5,25 (s ancho, 1H), 4,15 (c, J = 7,0 Hz, 2H), 3,44-3,41 (m, 2H), 2,74 (t, J = 8,0 Hz, 2H), 2,36-2,23 (m,2H), 1,23 (t, J = 7,0 Hz, 3H).
Ejemplo de Referencia 7
Ácido 5-(5-metoxi-3,4-dihidronaftalen-1(2H)ilideno)hexanoico
A una solución en tetrahidrofurano anhidro (200 ml) de bromuro de (4-carboxibutil)trifenilfosfonio (25,0 g), se leañadió t-butóxido de potasio (12,7 g), seguido de agitación a 30ºC durante 1 hora. A la mezcla de reacción se le añadió
21

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
una solución en tetrahidrofurano (20 ml) de 5-metoxi-1-tetralona (5,0 g), seguido de agitación a temperatura ambientedurante la noche. La mezcla de reacción se añadió a una mezcla de una solución acuosa saturada de cloruro de amonioy hielo, seguido extracción con acetato de etilo. El extracto se concentró para obtener de ese modo el compuesto deltítulo bruto que tiene los siguientes datos físicos. El compuesto obtenido se utilizó sin purificación en la reacciónsubsiguiente.
TLC: Rf 0,34 (hexano : acetato de etilo = 2 : 1).
Ejemplo de Referencia 8
Éster metílico de ácido 5-(5-metoxi-3,4-dihidronaftalen-1(2H)ilideno)hexanoico
A una solución en dimetilformamida anhidra (40 ml) del compuesto preparado en el Ejemplo de Referencia 7, sele añadieron yoduro de metilo (5,3 ml) y carbonato de potasio (17,6 g), seguido de agitación a temperatura ambientedurante la noche. La mezcla de reacción se añadió a agua helada, seguido extracción con acetato de etilo. El extractose lavó con solución salina saturada, se secó con sulfato de magnesio anhidro, y se concentró. El residuo se purificómediante cromatografía en columna de gel de sílice (hexano : acetato de etilo = 5 : 1) para obtener de ese modo elcompuesto del título (6,80 g) que tiene los siguientes datos físicos.
TLC: Rf 0,72 (hexano : acetato de etilo = 2 : 1);
RMN (CDCl3): δ 7,18 (dd, J = 7,6, 1,2 Hz, 1H), 7,10 (t, J = 7,6 Hz, 1H), 6,70 (dd, J = 7,6, 1,2 Hz, 1H), 5,96 (tancho, J = 7,2 Hz, 1H), 3,81 (s, 3H), 3,66 (s, 3H), 2,71 (t, J = 6,4 Hz, 2H), 2,48-2,18 (m, 6H), 1,89-1,71 (m, 4H).
Ejemplo de Referencia 9
5-(5-hidroxi-3,4-dihidronaftalen-1-il)hexanoico acid
La mezcla del compuesto (6,83 g) preparado en el Ejemplo de Referencia 8 y piridina-ácido clorhídrico (39 g) seagitó at 180ºC durante 2 horas. La mezcla de reacción se enfrió a temperatura ambiente, y a esto se le añadió agua,seguido extracción con acetato de etilo. El extracto se lavó con ácido clorhídrico 2 N y solución salina saturada en esteorden, se secó con sulfato de magnesio anhidro, y se concentró para obtener de ese modo el compuesto del título brutoque tiene los siguientes datos físicos. El compuesto obtenido se utilizó sin purificación en la reacción subsiguiente.
TLC: Rf 0,12 (hexano : acetato de etilo = 2:1).
Ejemplo de Referencia 10
Éster metílico de ácido 5-(5-hidroxi-3,4-dihidronaftalen-1-il)hexanoico
Se añadió cloruro de tionilo (1,9 ml) a metanol (25 ml) a -30ºC, seguido de agitación a -20ºC durante 15 minutos.A la solución de reacción, se le añadió la solución metanólica (10 ml) del compuesto preparado en el Ejemplo deReferencia 9, seguido de agitación a temperatura ambiente durante 30 minutos. La mezcla de reacción se concentró.El residuo se diluyó con acetato de etilo. La solución diluida se lavó con una solución acuosa saturada de hidroge-nocarbonato de sodio, agua y solución salina saturada en este orden, se secó con sulfato de magnesio anhidro, y seconcentró. El residuo se purificó mediante cromatografía en columna de gel de sílice (hexano : acetato de etilo = 4 :1) para obtener de ese modo el compuesto del título (4,91 g) que tiene los siguientes datos físicos.
22

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
TLC: Rf 0,48 (hexano : acetato de etilo = 2 : 1);
RMN (CDCl3): δ 7,08 (t, J = 7,8 Hz, 1H), 6,86 (d ancho, J = 7,8 Hz, 1H), 6,68 (dd, J = 7,8, 1,0 Hz, 1H), 5,85 (t, J= 4,4 Hz, 1H), 4,96 (s ancho, 1H), 3,66 (s, 3H), 2,70 (t, J = 8,0 Hz, 2H), 2,49-2,17 (m, 6H), 1,89-1,45 (m, 4H).
Ejemplo de Referencia 11
N’-((1E)-5-hidroxi-3,4-dihidronaftalen-1(2H)ilideno)-2,4,6-triisopropilbencenosulfonohidrazida
A una solución metanólica (250 ml) de 2,4,6-triisopropilbencenosulfonilhidrazida (36,8 g) y 5-hidroxi-1,2,3,4-tetrahidronaftalen-1-ona (20,0 g), se le añadió ácido clorhídrico concentrado (4,3 ml) a temperatura ambiente, seguidode agitación a 40ºC durante 2 horas. Enfriando con hielo, la mezcla de reacción se agitó durante 1 hora. Los cristalesdepositados se separaron. La separación se lavó con metanol frío, se secó a presión reducida para obtener de ese modoel compuesto del título (49,2 g) que tiene los siguientes datos físicos.
TLC: Rf 0,28 (hexano : acetato de etilo = 3 : 1);
RMN (CDCl3): δ 7,57 (ancho, 1H), 7,56 (d, J = 8,1 Hz, 1H), 7,16 (s, 2H), 6,99 (t, J = 8,1 Hz, 1H), 6,71 (d, J = 8,1Hz, 1H), 4,37 - 4,24 (m, 2H), 2,88 (m, 1H), 2,69 (t, J = 6,0 Hz, 2H), 2,43 (t, J = 6,6 Hz, 2H), 1,96 - 1,85 (m, 2H), 1,30(d, J = 6,9 Hz, 12H), 1,23 (d, J = 6,9 Hz, 6H).
Ejemplo de Referencia 12
5-hidroxi-3,4-dihidronaftalen-1-ilmetanol
A una solución en tetrahidrofurano anhidro (510 ml) del compuesto (49,2 g) preparado en el Ejemplo de Referencia11, se le añadió n-butil litio (221 ml, 1,56 M en hexano) a -78ºC, seguido de agitación a -78ºC durante 30 minutos. Lamezcla de reacción se calentó a 0ºC, seguido de agitación a 0ºC durante 30 minutos. Enfriando con hielo, se añadióparaformaldehído (11,7 g) a la mezcla de reacción, seguido de calentamiento a temperatura ambiente, y la mezcla dereacción se agitó durante 1 hora. Enfriando con hielo, se añadió una solución acuosa saturada de cloruro de amonio ala mezcla de reacción para la separación del líquido. La capa acuosa se extrajo con acetato de etilo. La capa orgánicacombinada se lavó con solución salina saturada, se secó con sulfato de sodio anhidro, y se concentró. El residuo sepurificó mediante cromatografía en columna de gel de sílice (hexano : acetato de etilo = 3 : 1 a 1 : 1) para obtener deese modo el compuesto del título (15,7 g) que tiene los siguientes datos físicos.
TLC: Rf 0,28 (hexano : acetato de etilo = 2 : 1);
RMN (CDCl3): δ 9,17 (s ancho, 1H), 6,94 (t, J = 8,1 Hz, 1H), 6,74 (d, J = 8,1 Hz, 1H), 6,68 (dd, J = 8,1, 1,2 Hz,1H), 6,01 (t, J = 4,8 Hz, 1H), 4,50 (s ancho, 1H), 4,25 (d, J = 1,2 Hz, 2H), 2,60 (t, J = 7,8 Hz, 2H), 2,22 - 2,09 (m, 2H).
Ejemplo de Referencia 13
5-metoximetoxi-3,4-dihidronaftalen-1-ilmetanol
Enfriando con hielo, a una solución en tetrahidrofurano anhidro (135 ml) del compuesto (15,7 g) preparado en elEjemplo de Referencia 12, se le añadió hidruro de sodio (3,75 g, 63,1%), seguido de agitación a temperatura ambiente
23

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
durante 30 minutos. A la mezcla de reacción se le añadió gota a gota, clorometil metil éter (7,41 ml) enfriando conhielo, seguido de agitación a temperatura ambiente durante 13 horas. A la mezcla de reacción se le añadieron, aguahelada y a una solución acuosa saturada de cloruro de amonio, seguido extracción con acetato de etilo. El extracto selavó con agua y solución salina saturada en este orden, se secó con sulfato de sodio anhidro, y se concentró. El residuose purificó mediante cromatografía en columna de gel de sílice (hexano : acetato de etilo = 4 : 1 a 2 : 1) para obtenerde ese modo el compuesto del título (15,1 g) que tiene los siguientes datos físicos.
TLC: Rf 0,38 (hexano : acetato de etilo = 2:1);
RMN (CDCl3): δ 7,16 (t, J = 7,8 Hz, 1H), 7,05 (d, J = 7,8 Hz, 1H), 7,00 (d, J = 7,8 Hz, 1H), 6,14 (t, J = 4,5 Hz,1H), 5,20 (s, 2H), 4,51 (s ancho, 2H), 3,49 (s, 3H), 2,82 (t, J = 8,1 Hz, 2H), 2,30 (td, J = 8,1, 4,5 Hz, 2H), 1,46 (sancho, 1H).
Ejemplo de Referencia 14
1-bromometil-5-metoximetoxi-3,4-dihidronaftaleno
A una solución en cloruro de metileno (110 ml) del compuesto (7,53 g) preparado en el Ejemplo de Referencia 13y trifenilfosfina (9,59 g), se le añadió tetrabromometano (12,1 g) enfriando con hielo, seguido de agitación enfriandocon hielo durante 40 minutos. La mezcla de reacción se concentró. Al residuo, se le añadió una mezcla disolvente deéter dietílico y hexano (5 : 1) para excluir el óxido de trifenilfosfina. El producto bruto obtenido se purificó mediantecromatografía en columna de gel de sílice (hexano : acetato de etilo = 20 : 1 a 10 : 1) para obtener de ese modo elcompuesto del título (6,32 g) que tiene los siguientes datos físicos.
TLC: Rf 0,76 (hexano : acetato de etilo = 2 : 1);
RMN (CDC13): δ 7,20 (t, J = 8,1 Hz. 1H), 7,12 (d, J = 8,1 Hz, 1H), 7,03 (d, J = 8,1 Hz, 1H), 6,30 (t, J = 4,8 Hz,1H), 5,20 (s, 2H), 4,36 (s, 2H), 3,49 (s, 3H), 2,83 (t, J = 8,7 Hz, 2H), 2,31 (td, J = 8,7, 4,8 Hz, 2H).
Ejemplo de Referencia 15
Éster metílico de ácido 2,2-dimetil-3-(5-metoximetoxi-3,4-dihidronaftalen-1-il)propanoico
Enfriando con hielo, a una solución en tetrahidrofurano anhidro (30 ml) de éster metílico de ácido 2-metilpropa-noico (5,11 ml), se le añadió gota a gota diisopropilamiduro de litio (22,3 ml), seguido de agitación a 30ºC durante30 minutos. Enfriando con hielo, a la mezcla de reacción, se le añadió gota a gota una solución en tetrahidrofuranoanhidro (20 ml) del compuesto (6,32 g) preparado en el Ejemplo de Referencia 14, seguido de agitación a temperaturaambiente durante 2 horas. A la mezcla de reacción se le añadió una solución acuosa saturada de cloruro de amonio,seguido extracción con acetato de etilo. El extracto se lavó con solución salina saturada, se secó con sulfato de magne-sio anhidro, y se concentró. El residuo se purificó mediante cromatografía en columna de gel de sílice (hexano:acetatode etilo = 8 : 1 a 6 : 1) para obtener de ese modo el compuesto del título (7,02 g) que tiene los siguientes datosfísicos.
TLC: Rf 0,55 (hexano : acetato de etilo = 5 : 1);
RMN (CDCl3): δ 7,10 (t, J = 8,1 Hz, 1H), 6,97 (d, J = 8,1 Hz, 1H), 6,94 (d, J = 8,1 Hz, 1H), 5,85 (t, J = 4,5 Hz,1H), 3,49 (s, 3H), 3,47 (s, 3H), 2,74 (t, J = 7,8 Hz, 2H), 2,72 (s, 2H), 2,17 (td, J = 7,8, 4,5 Hz, 2H), 1,15 (s, 6H).
24

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
Ejemplo de Referencia 16
Éster metílico de ácido 2,2-dimetil-3-(5-hidroxi-3,4-dihidronaftalen-1-il)propanoico
A una solución metanólica (110 ml) del compuesto (6,78 g) preparado en el Ejemplo de Referencia 15, se le añadióuna solución 4 N de cloruro de hidrógeno-dioxano (8,4 ml), seguido de agitación a temperatura ambiente durante 15horas. La mezcla de reacción se concentró. El residuo se extrajo con acetato de etilo. El extracto se lavó con unasolución acuosa saturada de hidrogenocarbonato de sodio y solución salina saturada en este orden, se secó con sulfatode sodio anhidro, y se concentró para obtener de ese modo el compuesto del título (5,78 g) que tiene los siguientesdatos físicos.
TLC: Rf 0,30 (hexano:acetato de etilo = 5:1);
RMN (CDCl3): δ 7,03 (t, J = 8,1 Hz, 1H), 6,89 (d, J = 8,1 Hz, 1H), 6,65 (dd, J = 1,2, 8,1 Hz, 1H), 5,85 (t, J = 4,5Hz, 1H), 4,71 (s, 1H), 3,46 (s, 3H), 2,71 (d, J = 1,2 Hz, 2H), 2,67 (t, J = 8,1 Hz, 2H), 2,20 (td, J = 8,1, 4,5 Hz, 2H),1,56 (s, 6H).
Ejemplo de Referencia 17
1-Ciclopropiliden-5-metoxi-1,2,3,4-tetrahidronaftaleno
Enfriando con hielo, a una solución en tetrahidrofurano anhidro (200 ml) de bromuro de (3-bromopropil)trifenil-fosfinio (19,8 g), se le añadió t-butóxido de potasio (9,58 g), seguido de agitación a temperatura ambiente durante 1,5horas. A la mezcla de reacción se le añadió, 5-metoxi-1-tetralona (5,0 g), seguido de agitación a temperatura ambientedurante 5 horas. La mezcla de reacción se vertió en una solución acuosa saturada de cloruro de amonio. La capa acuosase extrajo con acetato de etilo. La capa orgánica combinada se lavó con solución salina saturada, se secó con sulfato demagnesio anhidro, y se concentró. El residuo se purificó mediante cromatografía en columna de gel de sílice (hexano :acetato de etilo = 10 : 1) para obtener de ese modo el compuesto del título (5,66 g) que tiene los siguientes datosfísicos.
TLC: Rf 0,86 (hexano : acetato de etilo = 10 : 1);
RMN (CDCl3): δ 7,56 (d, J = 7,8 Hz, 1H), 7,13 (t, J = 7,8 Hz, 1H), 6,70 (d, J = 7,8 Hz, 1H), 3,83 (s, 3H), 2,76 (t, J= 6,4 Hz, 2H), 2,66-2,56 (m, 2H), 1,94-1,80 (m, 2H), 1,51-1,40 (m, 2H), 1,12-1,02 (m, 2H).
Ejemplo de Referencia 18
1-(3-Bromopropil)-5-metoxi-3,4-dihidronaftaleno
A una solución en ácido acético (60 ml) del compuesto (5.00 g) preparado en el Ejemplo de Referencia 17, se leañadió una solución acuosa de bromuro de hidrógeno al 47% (20 ml), seguido de agitación a temperatura ambientedurante 2 horas. A la mezcla de reacción se le añadió agua helada, seguido extracción con acetato de etilo. El extractose lavó con solución salina saturada, se secó con sulfato de magnesio anhidro, y se concentró. El residuo se purificómediante cromatografía en columna de gel de sílice (hexano : acetato de etilo = 10 : 1) para obtener de ese modo elcompuesto del título (7,05 g) que tiene los siguientes datos físicos.
TLC: Rf 0,69 (hexano : acetato de etilo =10 : 1);
25

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
RMN (CDCl3): δ 7,16 (t, J = 7,8 Hz, 1H), 6,90 (d, J = 7,8 Hz, 1H), 6,79 (d, J = 7,8 Hz, 1H), 5,92 (t, J = 4,6 Hz,1H), 3,83 (s, 3H), 3,44 (t, J = 6,6 Hz, 2H), 2,74 (t, J = 7,6 Hz, 2H), 2,65-2,55 (m, 2H), 2,28-2,15 (m, 2H), 2,13-1,98(m, 2H).
Ejemplo de Referencia 19
Éster metílico de ácido 2,2-dimetil-5-(5-metoxi-3,4-dihidronaftalen-1-il)hexanoico
Enfriando con hielo, a una solución en tetrahidrofurano anhidro (15 ml) de éster metílico de ácido 2-metilpro-panoico (3,30 g), se le añadió diisopropilamiduro de litio (16,5 ml, 2,0 M), seguido de agitación a 30ºC durante 30minutos. La mezcla de reacción se enfrió a temperatura ambiente, y a esto se le añadió una solución una solución entetrahidrofurano anhidro (5 ml) del compuesto (3,00 g) preparado en el Ejemplo de Referencia 18, seguido de agita-ción a temperatura ambiente durante 3 horas. La mezcla de reacción se vertió en a una solución acuosa saturada decloruro de amonio, seguido extracción con acetato de etilo. El extracto se lavó con solución salina saturada, se secócon sulfato de magnesio anhidro, y se concentró. El residuo se purificó mediante cromatografía en columna de gel desílice (hexano : acetato de etilo = 100 : 1 a 20 : 1) para obtener de ese modo el compuesto del título bruto (3,68 g) quetiene los siguientes datos físicos. El compuesto obtenido se utilizó sin purificación en la reacción subsiguiente.
TLC: Rf 0,84 (hexano : acetato de etilo = 2 : 1).
Ejemplo de Referencia 20
Ácido 2,2-dimetil-5-(5-hidroxi-3,4-dihidronaftalen-1-il)hexanoico
La mezcla del compuesto (3,68 g) preparado en el Ejemplo de Referencia 19 y piridina-ácido clorhídrico (17 g)se agitó a 180ºC durante la noche. La mezcla de reacción se enfrió a temperatura ambiente, y a esto se le añadióagua, seguido extracción con acetato de etilo. El extracto se lavó con solución salina saturada, se secó con sulfato demagnesio anhidro, y se concentró para obtener de ese modo el compuesto del título bruto que tiene los siguientes datosfísicos. El compuesto obtenido se utilizó sin purificación en la reacción subsiguiente.
TLC: Rf 0,30 (hexano : acetato de etilo = 2 : 1).
Ejemplo de Referencia 21
Éster metílico de ácido 2,2-dimetil-5-(5-hidroxi-3,4-dihidronaftalen-1-il)hexanoico
Se añadió cloruro de tionilo (0,86 ml) a metanol frío (11 ml), seguido de agitación a -20ºC durante 15 minutos. A lasolución de reacción, se le añadió una solución metanólica (5 ml) del compuesto preparado en el Ejemplo de Referencia20, seguido de agitación a temperatura ambiente durante 1 hora. La mezcla de reacción se concentró. El residuo sediluyó con acetato de etilo. La solución diluida se lavó con una solución acuosa saturada de hidrogenocarbonato desodio y solución salina saturada en este orden, se secó con sulfato de magnesio anhidro, y se concentró. El residuose purificó mediante cromatografía en columna de gel de sílice (hexano : acetato de etilo = 8 : 1) para obtener de esemodo el compuesto del título (2,02 g) que tiene los siguientes datos físicos.
TLC: Rf 0,66 (hexano : acetato de etilo = 2 : 1);
26

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
RMN (CDCl3): δ 7,05 (t, J = 8,0 Hz, 1H), 6,84 (d, J = 8,0 Hz, 1H), 6,68 (dd, J = 8,0, 1,0 Hz, 1H), 5,84 (t, J = 4,8Hz, 1H), 5,01 (s ancho, 1H), 3,60 (s, 3H), 2,70 (t, J = 8,0 Hz, 2H), 2,44-2,32 (m, 2H), 2,30-2,17 (m, 2H), 1,75-1,34(m, 4H), 1,15 (s, 6H).
Ejemplo de Referencia 22
Ácido 2-benciloxi-3-(5-metoximetoxi-3,4-dihidronaftalen-1-il)propanoico
A una solución en tetrahidrofurano (7 ml) de ácido 2-benciloxiacético (0,30 ml), se le añadió gota a gota diisopro-pilamiduro de litio (2,4 ml) a -78ºC en atmósfera de argón, seguido de agitación a 0ºC durante 10 minutos. La soluciónobtenida antes se añadió a una solución en tetrahidrofurano (3 ml) del compuesto (600 mg) preparado en el Ejemplo deReferencia 14 a -78ºC, seguido de agitación a temperatura ambiente durante 12 horas. La mezcla de reacción se vertióen una solución acuosa saturada de cloruro de amonio para la separación líquida. La capa acuosa se extrajo con acetatode etilo. La capa orgánica combinada se lavó con solución salina saturada, se secó con sulfato de magnesio anhidro, yse concentró. El residuo se purificó mediante cromatografía en columna de gel de sílice (cloroformo : metanol = 50 :1) para obtener de ese modo el compuesto del título (99 mg) que tiene los siguientes datos físicos.
TLC: Rf 0,33 (hexano : acetato de etilo = 4 : 1).
Ejemplo de Referencia 23
Éster metílico de ácido 2-benciloxi-3-(5-hidroxi-3,4-dihidronaftalen-1-il)propanoico
A una solución metanólica (4 ml) del compuesto (99 mg) preparado en el Ejemplo de Referencia 22, se le añadióuna solución 4N de cloruro de hidrógeno-dioxano (0,1 ml), seguido de agitación a temperatura ambiente durante15 horas. La mezcla de reacción se concentró para obtener de ese modo el compuesto del título bruto que tiene lossiguientes datos físicos. El compuesto obtenido se utilizó sin purificación en la reacción subsiguiente.
TLC: Rf 0,48 (hexano : acetato de etilo = 2 : 1).
Ejemplo de Referencia 24
Ácido 3-metoxicarbonil-2-(4-metilbenzoilamino)propanoico
Se disolvió en agua (1,3 L) hidrocloruro de éster β-metílico de ácido aspártico (184 g), y a esto se le añadióhidrogenocarbonato de sodio (277 g), y a esto se le añadieron gota a gota tetrahidrofurano (450 ml) y una solución entetrahidrofurano (50 ml) de cloruro de 4-metilbenzoilo (146 ml), seguido de agitación a temperatura ambiente durante15 horas. La mezcla de reacción se lavó con acetato de etilo. La capa acuosa se neutralizó con ácido clorhídrico 2 N
27

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
a pH de 2 a 3, y se extrajo con acetato de etilo. El extracto se lavó con solución salina saturada, se secó con sulfatode magnesio anhidro, y se concentró para obtener de ese modo el compuesto del título bruto (255 g) que tiene lossiguientes datos físicos. El compuesto obtenido se utilizó sin purificación en la reacción subsiguiente.
TLC: Rf 0,28 (cloroformo : metanol = 5 : 1);
RMN (CDCl3): δ 7,71 (d, J = 8,1 Hz, 2H), 7,34 (d, J = 7,8 Hz, 1H), 7,24 (d, J = 8,1 Hz, 2H), 5,08 (ddd, J = 7,5,4,5, 4,5 Hz, 1H), 3,73 (s, 3H), 3,18 (dd, J = 17,1, 4,5 Hz, 1H), 3,00 (dd, J = 17,1, 4,5 Hz, 1H), 2,40 (s, 3H).
Ejemplo de Referencia 25
Éster metílico de ácido 3-acetil-3-(4-metilbenzoilamino)propanoico
A una solución en piridina (480 ml) del compuesto (255 g) preparado en el Ejemplo de Referencia 24, se leañadieron anhídrido acético (453 ml) y 4-dimetilaminopiridina (3,52 g), seguido de agitación a 90ºC durante 1 hora.La mezcla de reacción se enfrió a temperatura ambiente, y se concentró. El residuo se vertió en agua helada, seguidoextracción con acetato de etilo. El extracto se lavó con agua, ácido clorhídrico 2 N y solución salina saturada en esteorden, se secó con sulfato de magnesio anhidro, y se concentró para obtener de ese modo el compuesto del título brutoque tiene los siguientes datos físicos.
TLC: Rf 0,23 (hexano : acetato de etilo = 2 : 1).
Ejemplo de Referencia 26
Éster metílico de ácido 2-(2-(4-metilfenil)-5-metiloxazol-4-il)acético
A una solución en anhídrido acético (450 ml) del compuesto preparado en el Ejemplo de Referencia 25, se le añadióácido sulfúrico concentrado (86 ml), seguido de agitación a 90ºC durante 1 hora. La mezcla de reacción se enfrió atemperatura ambiente, y se vertió en hielo. La capa acuosa se neutralizó con una solución acuosa 5N de hidróxido desodio, y se extrajo con acetato de etilo. El extracto se lavó con una solución acuosa 1 N de hidróxido de sodio, agua ysolución salina saturada en este orden, se secó con sulfato de magnesio anhidro, y se concentró. El aceite obtenido sedejó estar durante la noche. El sólido obtenido se lavó con hexano, y se separó por filtración mediante aspiración paraobtener de ese modo el compuesto del título (183 g) que tiene los siguientes datos físicos.
TLC: Rf 0,61 (hexano : acetato de etilo = 2 : 1);
RMN (CDCl3): δ 7,87 (d, J = 8,1 Hz, 2H), 7,23 (d, J = 8,1 Hz, 2H), 3,73 (s, 3H), 3,57 (s, 2H), 2,38 (s, 3H), 2,35(s, 3H).
Ejemplo de Referencia 27
2-(2-(4-metilfenil)-5-metiloxazol-4-il)etanol
28

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
Enfriando con hielo, se suspendió hidruro de litio y aluminio (18,6 g) en tetrahidrofurano anhidro (250 ml), y aesto se le añadió gota a gota una solución en tetrahidrofurano anhidro (250 ml) del compuesto (120 g) preparado en elEjemplo de Referencia 26, seguido de agitación durante 30 minutos enfriando con hielo. A la mezcla de reacción sele añadió gota a gota una solución acuosa saturada de sulfato de sodio para la separación líquida. La capa orgánica sesecó con sulfato de magnesio anhidro, y se filtró con celite. El producto filtrado se concentró. El residuo se dejó estardurante la noche. Los cristales obtenidos se lavaron con una mezcla disolvente de hexano y acetato de etilo (10 : 1)para obtener de ese modo el compuesto del título (80,0 g) que tiene los siguientes datos físicos.
TLC Rf 0,18 (hexano : acetato de etilo = 2 : 1);
RMN (CDCl3): δ 7,86 (m, 2H), 7,23 (m, 2H), 3,92 (ancho, 2H), 2,71 (t, J = 6,0 Hz, 2H), 2,39 (s, 3H), 2,32 (s, 3H).
Ejemplo 1
Éster metílico de ácido 3-(5-(2-(2-(4-metilfenil)-5-metiloxazol-4-il)etoxi)-3,4-dihidronaftalen-1-il)propanoico
A una solución en cloruro de metileno (15 ml) del compuesto (600 mg) preparado en el Ejemplo de Referencia2, se le añadieron el compuesto (617 mg) preparado en el Ejemplo de Referencia 27, trifenilfosfina (1,02 g) y 1,1’-(azodicarbonil)dipiperidina (978 mg), seguido de agitación a temperatura ambiente durante 3 horas. La mezcla dereacción se diluyó con éter dietílico, y se filtró con celite. El producto filtrado se concentró a presión reducida. Elresiduo se purificó mediante cromatografía en columna de gel de sílice (hexano : acetato de etilo = 9 : 1 a 7 : 1 a 5 : 1a 7 : 2) para obtener de ese modo el compuesto de la presente invención (1,00 g) que tiene los siguientes datos físicos.
TLC: Rf 0,59 (hexano : acetato de etilo = 2 : 1);
RMN (CDCl3): δ 7,86 (d, J = 8,1 Hz, 2H), 7,23 (d, J = 8,1 Hz, 2H), 7,13 (dd, J = 8,0, 8,0 Hz, 1H), 6,94-6,74 (m,2H), 5,87 (dd, J = 4,6, 4,6 Hz, 1H), 4,25 (t, J = 6,6 Hz, 2H), 3,67 (s, 3H), 2,99 (t, J = 6,6 Hz, 2H), 2,85-2,63 (m, 4H),2,60-2,45 (m, 2H), 2,39 (s, 3H), 2,36 (s, 3H), 2,30-2,10 (m, 2H).
Ejemplo 2
Ácido 3-(5-(2-(2-(4-metilfenil)-5-metiloxazol-4-il)etoxi)-3,4-dihidronaftalen-1-il)propanoico
El compuesto (950 mg) preparado en el Ejemplo 1 se disolvió en metanol (8,0 ml) y tetrahidrofurano (8,0 ml), y aesto se le añadió una solución acuosa 2N de hidróxido de sodio (3,3 ml), seguido de agitación a temperatura ambientedurante la noche. La mezcla de reacción se aciduló con ácido clorhídrico 1 N, y se extrajo con una mezcla disolventede acetato de etilo y tetrahidrofurano. El extracto se lavó con solución salina saturada, se secó con sulfato de magnesioanhidro, y se concentró a presión reducida. El residuo se recristalizó con una mezcla disolvente de acetato de etilo ytetrahidrofurano para obtener de ese modo el compuesto de la presente invención (745 mg) que tiene los siguientesdatos físicos.
TLC: Rf 0,63 (cloroformo : metanol = 8 : 1);
RMN (DMSO-d6): δ 7,79 (d, J = 8,2 Hz, 2H), 7,29 (d, J = 8,2 Hz, 2H), 7,14 (dd, J = 8,0, 8,0 Hz, 1H), 6,97-6,78(m, 2H), 5,84 (t ancho, 1H), 4,19 (t, J = 5,8 Hz, 2H), 2,91 (t, J = 5,8 Hz, 2H), 2,75-2,20 (m, 6H), 2,33 (s, 3H), 2,36 (s,3H), 2,20-1,94 (m, 2H).
29

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
Ejemplo de Formulación
Ejemplo de Formulación 1
Los siguientes componentes se mezclaron mediante un método convencional y se troquelaron para obtener 100comprimidos que contenían cada uno 50 mg del ingrediente activo.
* Ácido 3-(5-(2-(2-(4-metilfenil)-5-metiloxazol-4-il)etoxi)-3,4- 5,0 gdihidronaftalen-1-il)propanoico
* Carboximetil celulosa cálcica(agente disgregante) 0,2 g
* Estearato de magnesio (lubricante) 0,1 g* Celulosa microcristalina 4,7 g
Ejemplo de Formulación 2
Los siguientes componentes se mezclaron mediante un método convencional, y la solución se esterilizó medianteun método convencional, se colocó en ampollas de 5 ml y se liofilizó mediante un método convencional para obtenerde ese modo 100 ampollas que contenían cada una 20 mg del ingrediente activo.
* Ácido 3-(5-(2-(2-(4-metilfenil)-5-metiloxazol-4-il)etoxi)-3,4- 2,0 gdihidronaftalen-1-il)propanoico
* Manitol 20 g* Agua destilada 1000 ml
30

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 291 378 T3
REIVINDICACIONES
1. Un compuesto químico seleccionado del grupo que consiste en ácido 3-(5-(2-(2-(4-metilfenil)-5-metiloxazol-4-il)etoxi)-3,4-dihidronaftalen-1-il)propanoico y una sal no tóxica del mismo.
2. El compuesto químico según la reivindicación 1 que es ácido 3-(5-(2-(2-(4-metilfenil)-5-metiloxazol-4-il)etoxi)-3,4-dihidronaftalen-1-il)propanoico.
31




















