
Enhancer of Zeste Homolog 2 Silences MicroRNA-218 in Human Pancreatic Ductal Adenocarcinoma Cells by...
21
Enhancer of Zeste Homolog 2 Silences MicroRNA-218 in Human Pancreatic Ductal Adenocarcinoma Cells by Inducing Formation of Heterochromatin CHI HAN LI, 1 KA–FAI TO, 2 JOANNA HUNG–MAN TONG, 2 ZHANGANG XIAO, 1 TIAN XIA, 1 PAUL BS LAI, 3 SHEUNG CHING CHOW, 1 YIN–XIN ZHU, 1 STEPHEN L CHAN, 4 VICTOR E MARQUEZ, 5 and YANGCHAO CHEN 1,6 1 School of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, Hong Kong; 2 Department of Anatomical and Cellular Pathology, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong; 3 Department of Surgery, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong; 4 Department of Clinical Oncology, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong; 5 Chemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, Frederick, Maryland; and 6 Shenzhen Research Institute, The Chinese University of Hong Kong, Shenzhen, China BACKGROUND & AIMS: Enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase that is overex- pressed by pancreatic ductal adenocarcinoma (PDAC) cells and increases their aggressiveness. We identified mi- croRNAs (miRs) that are regulated by EZH2 and studied their functions in PDAC cells. METHODS: We per- formed miR profile analysis of PDAC cells incubated with EZH2 inhibitor 3-deazaneplanocin A, and pancreatic duc- tal epithelial cells that overexpressed EZH2. Expression levels of miRs and the targets of miRs were analyzed by quantitative reverse transcription polymerase chain reac- tion and immunohistochemistry. We expressed different forms of EZH2 to analyze functional domains and used small interfering RNAs to reduce its level in PDAC cells. RESULTS: Expression of miR-218 was repressed by EZH2 in PDAC cells. Levels of miR-218 were significantly re- duced in primary PDAC tumor samples compared with paired, adjacent nontumor tissue. Overexpression of miR- 218 in SW1990 cells reduced their proliferation and tu- mor formation and metastasis in nude mice. Loss of miR-218 from SW1990 cells increased levels of UDP- glycosyltransferase 8 and miR-218 was found to bind to its 3=-UTR. Levels of UDP-glycosyltransferase protein and messenger RNA were associated with the metastatic po- tential of PDAC cell lines and progression of tumors in patients. EZH2 was found to silence miR-218 by binding to its promoter, promoting heterochromatin formation, and recruiting the DNAs methyltransferase 1, 3A, and 3B. CONCLUSIONS: EZH2 is up-regulated in PDAC sam- ples from patients and silences miR-218. MicroRNA-218 prevents proliferation of PDAC cells in culture, and tu- mor growth and metastasis in nude mice. MicroRNA- 218 reduces levels of UDP-glycosyltransferase, which is associated with the metastatic potential of PDAC tumors in mice and progression of human PDAC. Keywords: Pancreatic Cancer; Gene Regulation; Epigenetic; Messenger RNA Processing. P ancreatic cancer is the fourth leading cause of cancer- related death in the United States, with 5% 5-year survival rate and little improvement over 30 years. 1,2 Eighty-five percent of all incidences of pancreatic cancer are pancreatic ductal adenocarcinoma (PDAC) and the poor outcomes of PDAC are attributed to its highly ag- gressive nature in that the tumor cells readily undergo invasion and dissemination. 3,4 Recently, it was shown that the gene expression regulator enhancer of zeste homolog (EZH)2 is frequently overexpressed in diverse malignan- cies, including PDAC, and it possesses important roles in cancer development and progression. 5,6 MicroRNAs (miRs) belong to a class of small noncod- ing RNAs and primarily function to regulate gene expres- sion based on sequence complementation and contribute immensely to cancer biology. 7–9 In 2011, Cao et al first demonstrated EZH2’s ability to regulate miRs along the PRC2PRC1 oncoprotein axis, 10 suggesting the feasibility for EZH2-induced miR dysregulation. Here, we further rein- force the importance of EZH2 in suppressing miR-218 by demonstrating the carcinogenic roles, delineating the onco- genic axis, and revealing the involvement of de novo hetero- chromatin formation in EZH2-mediated gene silencing. Materials and Methods Clinical Samples, Cell Lines, and Drug Treatment Sixty pairs of PDAC tumor and surrounding normal tissues were obtained from patients who underwent pancreatic resection at the Prince of Wales Hospital. All specimens were fixed and embedded into paraffin. Cell lines were obtained from American Type Culture Collection (Manassas, VA). Human pan- creatic ductal epithelial (HPDE) cell line was a gift from Dr Ming-Sound Tsao (University Health Network, Ontario Cancer Institute and Princess Margaret Hospital Site, Toronto) and was maintained according to the publications. 11,12 In drug treat- Abbreviations used in this paper: 3=-UTR, 3=-untranslated region; 5AZAC, 5-azacytidine; DNMT, DNA methyltransferase; DZnep, 3-deazaneplanocin A; EZH2, enhancer of zeste 2 homolog; H3K27me3, histone 3 lysine 27 trimethylation; HPDE, human pancreatic ductal epithelium; miR, microRNA; PDAC, pancreatic ductal adenocarcinoma; PRC2, polycomb repressive complex 2; qRT-PCR, quantitative reverse transcription polymerase chain reaction; shRNA, short-hairpin RNA; siRNA, small interfering RNA; UGT8, UDP-glycosyltransferase 8; VOPP1, vesicular, overexpressed in cancer, prosurvival protein 1. © 2013 by the AGA Institute 0016-5085/$36.00 http://dx.doi.org/10.1053/j.gastro.2013.01.058 BASIC AND TRANSLATIONAL PANCREAS GASTROENTEROLOGY 2013;144:1086 –1097
Transcript of Enhancer of Zeste Homolog 2 Silences MicroRNA-218 in Human Pancreatic Ductal Adenocarcinoma Cells by...

P
BK
idp2mmgimtptaC
M
s
BA
SICA
ND
TRA
NSLA
TION
AL
PA
NCREA
S
GASTROENTEROLOGY 2013;144:1086–1097
Enhancer of Zeste Homolog 2 Silences MicroRNA-218 in HumanPancreatic Ductal Adenocarcinoma Cells by Inducing Formation ofHeterochromatinCHI HAN LI,1 KA–FAI TO,2 JOANNA HUNG–MAN TONG,2 ZHANGANG XIAO,1 TIAN XIA,1 PAUL BS LAI,3
SHEUNG CHING CHOW,1 YIN–XIN ZHU,1 STEPHEN L CHAN,4 VICTOR E MARQUEZ,5 and YANGCHAO CHEN1,6
1School of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, Hong Kong; 2Department of Anatomical and Cellular Pathology,rince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong; 3Department of Surgery, Prince of Wales Hospital, The Chinese University of
Hong Kong, Shatin, Hong Kong; 4Department of Clinical Oncology, Prince of Wales Hospital, The Chinese University of Hong Kong, Shatin, Hong Kong; 5Chemical6Shenzhen Research Institute, The Chinese University of Hong
iology Laboratory, Center for Cancer Research, National Cancer Institute, Frederick, Maryland; andong, Shenzhen, China
t(cc
BACKGROUND & AIMS: Enhancer of zeste homolog 2(EZH2) is a histone methyltransferase that is overex-pressed by pancreatic ductal adenocarcinoma (PDAC)cells and increases their aggressiveness. We identified mi-croRNAs (miRs) that are regulated by EZH2 and studiedtheir functions in PDAC cells. METHODS: We per-formed miR profile analysis of PDAC cells incubated withEZH2 inhibitor 3-deazaneplanocin A, and pancreatic duc-tal epithelial cells that overexpressed EZH2. Expressionlevels of miRs and the targets of miRs were analyzed byquantitative reverse transcription polymerase chain reac-tion and immunohistochemistry. We expressed differentforms of EZH2 to analyze functional domains and usedsmall interfering RNAs to reduce its level in PDAC cells.RESULTS: Expression of miR-218 was repressed by EZH2n PDAC cells. Levels of miR-218 were significantly re-uced in primary PDAC tumor samples compared withaired, adjacent nontumor tissue. Overexpression of miR-18 in SW1990 cells reduced their proliferation and tu-or formation and metastasis in nude mice. Loss ofiR-218 from SW1990 cells increased levels of UDP-
lycosyltransferase 8 and miR-218 was found to bind tots 3=-UTR. Levels of UDP-glycosyltransferase protein and
essenger RNA were associated with the metastatic po-ential of PDAC cell lines and progression of tumors inatients. EZH2 was found to silence miR-218 by bindingo its promoter, promoting heterochromatin formation,nd recruiting the DNAs methyltransferase 1, 3A, and 3B.ONCLUSIONS: EZH2 is up-regulated in PDAC sam-
ples from patients and silences miR-218. MicroRNA-218prevents proliferation of PDAC cells in culture, and tu-mor growth and metastasis in nude mice. MicroRNA-218 reduces levels of UDP-glycosyltransferase, which isassociated with the metastatic potential of PDAC tumorsin mice and progression of human PDAC.
Keywords: Pancreatic Cancer; Gene Regulation; Epigenetic;essenger RNA Processing.
Pancreatic cancer is the fourth leading cause of cancer-related death in the United States, with �5% 5-year
urvival rate and little improvement over 30 years.1,2
Eighty-five percent of all incidences of pancreatic cancer
are pancreatic ductal adenocarcinoma (PDAC) and thepoor outcomes of PDAC are attributed to its highly ag-gressive nature in that the tumor cells readily undergoinvasion and dissemination.3,4 Recently, it was shown thathe gene expression regulator enhancer of zeste homologEZH)2 is frequently overexpressed in diverse malignan-ies, including PDAC, and it possesses important roles inancer development and progression.5,6
MicroRNAs (miRs) belong to a class of small noncod-ing RNAs and primarily function to regulate gene expres-sion based on sequence complementation and contributeimmensely to cancer biology.7–9 In 2011, Cao et al firstdemonstrated EZH2’s ability to regulate miRs along thePRC2�PRC1 oncoprotein axis,10 suggesting the feasibilityfor EZH2-induced miR dysregulation. Here, we further rein-force the importance of EZH2 in suppressing miR-218 bydemonstrating the carcinogenic roles, delineating the onco-genic axis, and revealing the involvement of de novo hetero-chromatin formation in EZH2-mediated gene silencing.
Materials and MethodsClinical Samples, Cell Lines, and DrugTreatmentSixty pairs of PDAC tumor and surrounding normal
tissues were obtained from patients who underwent pancreaticresection at the Prince of Wales Hospital. All specimens werefixed and embedded into paraffin. Cell lines were obtained fromAmerican Type Culture Collection (Manassas, VA). Human pan-creatic ductal epithelial (HPDE) cell line was a gift from DrMing-Sound Tsao (University Health Network, Ontario CancerInstitute and Princess Margaret Hospital Site, Toronto) and wasmaintained according to the publications.11,12 In drug treat-
Abbreviations used in this paper: 3=-UTR, 3=-untranslated region;5AZAC, 5-azacytidine; DNMT, DNA methyltransferase; DZnep,3-deazaneplanocin A; EZH2, enhancer of zeste 2 homolog; H3K27me3,histone 3 lysine 27 trimethylation; HPDE, human pancreatic ductalepithelium; miR, microRNA; PDAC, pancreatic ductal adenocarcinoma;PRC2, polycomb repressive complex 2; qRT-PCR, quantitative reversetranscription polymerase chain reaction; shRNA, short-hairpin RNA;siRNA, small interfering RNA; UGT8, UDP-glycosyltransferase 8; VOPP1,vesicular, overexpressed in cancer, prosurvival protein 1.
© 2013 by the AGA Institute0016-5085/$36.00
http://dx.doi.org/10.1053/j.gastro.2013.01.058

Aat
a
T(1atfi
ha(tn
BA
SIC
AN
DTR
AN
SLA
TIO
NA
LPA
NCREA
S
May 2013 EZH2 SILENCES miR-218 IN PANCREATIC CANCER 1087
ments, 5 �M 3-deazaneplanocin A (DZnep), 500 nM trichostatin, and 1 �M 5-azacytidine (5AZAC) (Sigma, St Louis, MO) werepplied to cells seeded overnight, followed by 24 –72 h incuba-ion.
Plasmids, Lentivirus Production, andTransductionMyc-tagged full-length human EZH2 and various mu-
tant constructs were cloned into a lentiviral vector coexpressingenhanced green fluorescent protein. Lentiviral vector containingeither short-hairpin RNA (shRNA) targeting EZH2, or the stemloop of miR-218-2 (retrieved from miRBase) was cloned into H1promoter containing lentiviral vector.13 The VSV-G–pseu-dotyped lentiviruses were produced by co-transfecting 293T cellswith the transfer vectors and 3 other packaging vectors: pMDLg/pRRE, pRSV-REV, and pCMV-VSVG.14 The 5 � 104 cancer cellsor HPDE cells were plated in 24-well plates and transduced withlentivirus in the presence of 8 �g/mL hexadimethrine bromide(Sigma).
Promoter and 3=-UTR Luciferase ReporterAssayTo validate that UDP-glycosyltransferase 8 (UGT8) was a
direct target of miR-218, and determine the effect of variousEZH2 domains on miR-218-2 promoter activity,15 luciferase
ssays were performed (see Supplementary Materials).
MicroRNA-218 In Situ Hybridization inTissue MicroarrayIn situ hybridization of mature miR-218 was conducted
on a freshly sectioned pancreatic cancer tissue microarray con-sisting of 87 PDAC cases and 10 normal cases (PA2082; USBiomax Inc, Rockville, MD). Hybridization was performed usingmiRCURY LNA detection probe for hsa-miR-218 (30 nM) la-beled with DIG at both 5= and 3= end (Exiqon). The section wasapplied with alkaline phosphatase– conjugated anti-DIG (Roche,San Francisco, CA) and the signal was developed by 5-bromo-4-chloro-3-inodolyl phosphate/nitroblue tetrazolium. Cells werecounterstained with nuclear fast red before mounting.
Cell Proliferation, Migration, and InvasionAssayA 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide assay was performed to measure cell proliferation.Chamber migration and invasion assay were performed to de-termine cell migration and invasion rate (see SupplementaryMaterials).
In Vivo Tumor Growth and Metastasis AssayMale BALB/c nude mice aged 4 to 6 weeks were acquired
from Laboratory Animal Services Centre of the Chinese Univer-sity of Hong Kong. Animal handling and experimental proce-dures were approved by the Animal Experimental Ethics Com-mittee of the institute. For tumor growth assay, SW1990 cells(5 � 106) were injected subcutaneously (5 mice per group).
umor volume was calculated by the equation: volume �length � width2)/2. For metastasis study, SW1990 cells (1 �06) were injected orthotopically into the major pancreatic duct,nd the tumor was allowed to grow for 3 months. The mice werehen sacrificed, dissected, and speculated metastatic tissues were
xed for H&E staining.Chromatin ImmunoprecipitationChromatin immunoprecipitation was performed using
the SimpleChIP Enzymatic Chromatin IP Kit, Magnetic Beads(Cell Signaling Technology, Danvers, MA). Cross-linked chroma-tin was incubated overnight at 4°C with anti-H3 (�ve ctrl),anti-IgG (�ve ctrl), anti-EZH2, anti–DNA methyltransferase(DNMT) 1, anti-DNMT3A, anti-DNMT3B, anti-HP1�, anti-HP1�, and anti-H3K9me2 (Cell Signaling Technology), anti–
istone 3 lysine 27 trimethylation (H3K27me3), anti–polymer-se II, and anti-EED (Millipore, Billerica, MA), anti-SUV39H1Abcam, Cambridge, MA). The precipitated DNA were quanti-ated absolutely by real-time polymerase chain reaction andormalized by respective 2% input.
Results
EZH2 Inhibits miR-218 Expression in PDACWe performed a locked nucleic acid–based human
global miR quantitative reverse transcription polymerasechain reaction (qRT-PCR) profiling in EZH2-overex-pressed HPDE and its corresponding vector control. Here,231 (approximately 30%) miRs were down-regulated �1.5-fold. In parallel, we used DZnep to treat PDAC cell lineSW1990. DZnep is an inhibitor that showed potent de-pletion effect on EZH2, PRC2 components, andH3K27me3 (Supplementary Figure 1B). The miR profilingshowed 96 (approximately 13%) miRs were up-regulated�1.5-fold after DZnep treatment (Supplementary Figure1A). When combining both studies, 23 miRs were foundboth down-regulated in EZH2-overexpressed HPDE cellsand up-regulated in DZnep-treated SW1990 cells (Figure1A). In PDAC, it is highly possible that EZH2 directlysuppressed these miRs and the loss of miR expressioncontributed to malignant development or progression.
Additional qRT-PCR validation (Supplementary Figure1C and D) showed miR-218 was a promising target be-cause its expression was increased on DZnep treatmentsin various PDAC cell lines SW1990, PANC1, and CAPAN2(Figure 1B), and reduced in EZH2-overexpressed HPDE(Figure 1C). The miR-218 level was consistently up-regu-lated after EZH2 knockdown by either shRNA or smallinterfering RNA (siRNA) (Figure 1D, Supplementary Fig-ure 1E). Loss of miR-218 is critical in PDAC, as we ob-served that all PDAC cell lines had a low miR-218 expres-sion compared with HPDE (Figure 1E). To demonstratethe clinical relevance of miR-218, we conducted in situhybridization analysis of miR-218 in a human pancreaticcancer tissue microarray. The intensity of miR-218 stain-ing was quantified, and statistical analysis showed thatmiR-218 signal was reduced in tumor compared withnormal tissues (P � .0001) (Figure 1F, left panel, Supple-mentary Table 1). In addition, a reduction of miR-218level was observed in PDAC tumor compared with adja-cent nontumor tissues with statistical significance (P �.0004) (Figure 1F, right panel; Supplementary Table 2) asmeasured by qRT-PCR normalized with RNU6B (Supple-
mentary Figure 1F).
n
BA
SICA
ND
TRA
NSLA
TION
AL
PA
NCREA
S
1088 LI ET AL GASTROENTEROLOGY Vol. 144, No. 5
MicroRNA-218 Inhibits Tumor Growth andMetastasis by Targeting VOPP1 and UGT8,RespectivelyCell proliferation assay demonstrated that in-
creased miR-218 level by mimics transfection (Supple-mentary Figure 2A) inhibited cell growth in PDAC celllines (Figure 2A and B, Supplementary Figure 2B). Thenegative regulatory effect of miR-218 on PDAC cellgrowth was also observed in the in vivo study. SW1990transduced with miR-218 expressing lentivirus showedreduced tumor growth in nude mice compared with thecontrol lentivirus transduced group (n � 5) (Figure 2C).In addition, highly vascularized subcutaneous tumorswere observed macroscopically (Supplementary Figure2C). Endothelial marker mouse CD31 staining showedtumor developed from miR-218 overexpressed SW1990had reduced microvessel formation (Figure 2D). In addi-
Figure 1. MicroRNA-218 expression is inhibited in PDAC cell lines andPDAC. (A) Twenty-three miRs were concurrently up-regulated in DZnepcells. (B, C) Validation of the profiling was performed. DZnep treatmentSW1990. (C) MicroRNA-218 was suppressed in HPDE cells transducedusing both siEZH2 and shEZH2 increased the miR-218 level in SW1990 cHPDE. (F, left panel) There was positive miR-218 signal in normal ductDown-regulation of miR-218 with statistical significance (P � .0001, N �ormal tissues. *P � .05; **P � .01.
tion to tumor growth, miR-218 inhibited cell migration
and invasion in PDAC in vitro (Figure 2E). Nude micemetastasis study showed 4 of 5 (80%) nude mice orthoto-pically injected with control SW1990 had either lung orliver metastasis, but none injected with miR-218 overex-pressing SW1990 developed distal metastasis (Figure 2F).Taken together, miR-218 negatively regulates PDACgrowth and metastasis by inhibiting PDAC cell prolifera-tion, tumor microvessel formation, and cell migrationand invasion.
To delineate the EZH2-miR-218 pathway, we aimed toidentify the functional targets of miR-218. By qRT-PCR(Supplementary Figure 3A), we showed vesicular, overex-pressed in cancer, prosurvival protein (VOPP) 1, andUGT8 messenger RNA levels were reduced after transfec-tion of miR-218 mimics into SW1990 (Figure 3A). Knock-down of VOPP1 by siRNAs attenuated cell growth in-duced by miR-218 inhibitors in SW1990 (Figure 3B).
or tissues by EZH2. MicroRNA-218 was regulated by EZH2 in humanated SW1990 cells and down-regulated in EZH2-overexpressed HPDE-regulated miR-218 in PDAC cell lines PANC1, CAPAN2 in addition toEZH2 expressing lentivirus. (D) RNA interference knockdown of EZH2. (E) MicroRNA-218 level was lower in all PDAC cell lines compared withs, but low or no miR-218 signal in malignant duct cells. (F, right panel)) was observed in PDAC tumor tissues compared with paired adjacent
tum-treupbyellscell
60
Measurement of VOPP1 messenger RNA level showed

iiit(Ft
� .
BA
SIC
AN
DTR
AN
SLA
TIO
NA
LPA
NCREA
S
May 2013 EZH2 SILENCES miR-218 IN PANCREATIC CANCER 1089
that the majority of PDAC lines had high VOPP1 expres-sion compared with HPDE (Figure 3C). In addition, dif-ferential EZH2 expression in PDAC tumor and adjacentnormal pairs were highly correlated with that of VOPP1 (P �.01) (Supplementary Figure 3B), which reinforced the va-lidity of the EZH2-miR-218 dysregulation pathway.
Next, we showed that increased expression of anothermiR-218 target, UGT8, was associated with PDAC cellmigration and invasion. By 3=-UTR reporter assay, theluciferase activity of the reporter containing WT-UGT8-3=-UTR, but not the seed-region removed mutant, was re-duced after transfection of miR-218 mimics in SW1990(Figure 3D, left panel). Overexpression of miR-218 alsodecreased UGT8 protein expression as determined byWestern blot (Figure 3D, right panel). Measurement ofboth UGT8 messenger RNA and protein levels showed acell-line–specific expression pattern, and that the expres-
Figure 2. MicroRNA-218 negatively regulates tumor growth and metascell proliferation, and conversely transfection of miR-218 inhibitors incrattenuated the antiproliferative effect of shEZH2 in SW1990 cells. (C) SW218) showed reduced subcutaneous tumor growth in nude mice (n �reduced microvessel density from tissues derived from miR-218 overemiR-218 mimics reduced SW1990 cell migration and invasion rate, as mAround 3 months post injection, no metastasis was observed in nude mlentiviral control (Lenti-CTRL) SW1990 developed distal metastases. *P
sion of UGT8 was highly associated with the metastatic
potential of the cell lines (Figure 3E). BxPC3, CFPAC1,and SW1990, which more readily undergo metastasis, hadhigher UGT8 expression compared with other cell lineswith no or low metastatic potential.16 –18 It is worth not-ng that SW1990, having the highest abundance of UGT8,s the PDAC cell line derived from metastatic organ. Mostmportantly, knockdown of UGT8 by siRNAs counteractedhe pro-migrating effect of miR-218 inhibitors in SW1990Figure 3F), and impaired wound healing (Supplementaryigure 3C), showing that UGT8 was a critical functionalarget of miR-218 in promoting PDAC metastasis.
UGT8 Is Frequently Overexpressed in PDACTumor TissuesImmunohistochemical staining of EZH2 and UGT8 in
60 pairs of PDAC tumor and adjacent normal tissues showedthat the majority of normal duct cells had no positive EZH2 or
s both in vitro and in vivo. (A) Transfection of miR-218 mimics decreaseded cell growth in SW1990 cells. (B) Transfection of miR-218 inhibitors90 cells transduced with miR-218 overexpressing lentivirus (Lenti-miR-(D) Endothelial cells CD31 staining of the tumors showed there was aessing SW1990 cells. Original magnification �200. (E) Transfection ofsured by chamber migration assay and invasion assay, respectively. (F)grafted with Lenti-miR-218 SW1990 cells (n � 5), while 80% (4 of 5) of05; **P � .01; ***P � .001.
tasieas19
5).xpreaice
UGT8 staining, while nucleus EZH2 and membranous UGT8

sic
uat
BA
SICA
ND
TRA
NSLA
TION
AL
PA
NCREA
S
1090 LI ET AL GASTROENTEROLOGY Vol. 144, No. 5
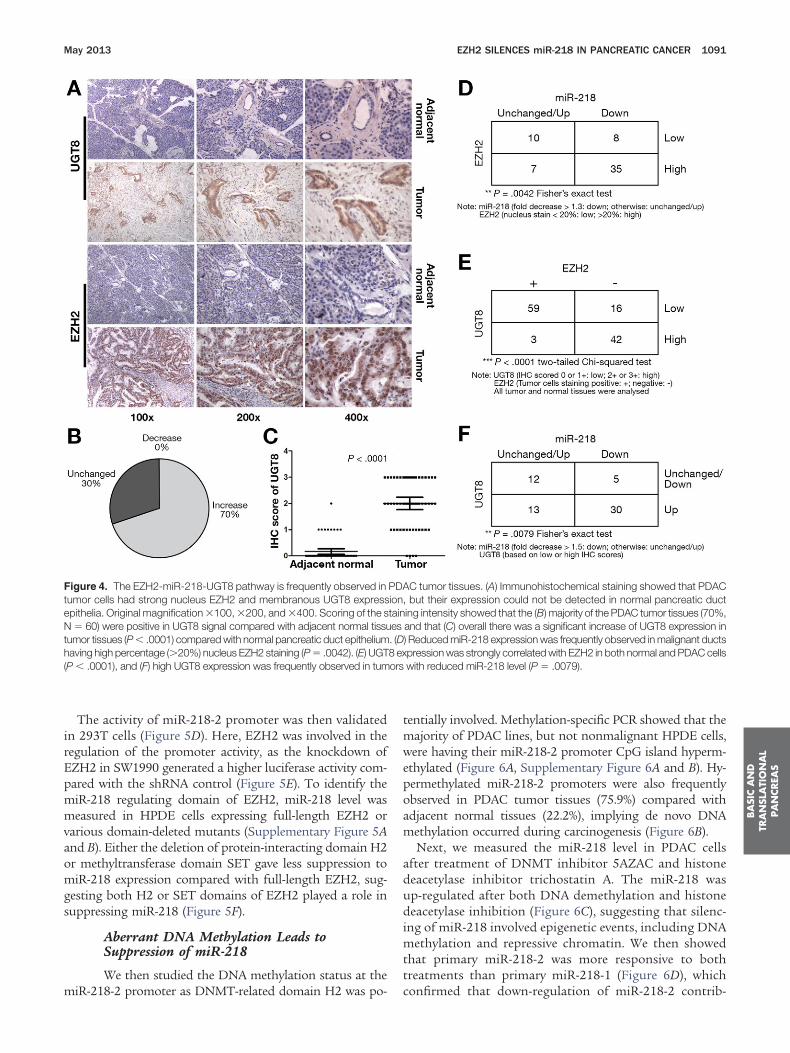
expression were frequently observed in tumor cells (Figure 4A).After scoring of the UGT8 intensity, it was observed that 70% ofthe tumor tissues had increased UGT8 expression relative totheir adjacent normal duct cells (Figure 4B), and overall therewas a higher UGT8 level in PDAC tumor cells (P � .0001)(Figure 4C).
We observed that miR-218 expression was frequently re-duced in tumors with high percentage EZH2-positive nucleus(P � .0042) (Figure 4D). The post-surgery survival of patientswith reduced miR-218 level in primary tumor tissues was sig-nificantly shorter than patients without loss of miR-218 (Sup-plementary Figure 4). In addition, level of UGT8 was stronglyassociated with both EZH2 and miR-218 expression. UGT8expression was highly correlated with EZH2 expression in bothnormal and malignant pancreatic duct cells (P � .0001) (Figure4E), and high UGT8 level was negatively correlated with expres-ion of miR-218 (P � .0079) (Figure 4F). Reduced miR-218 leveln PDAC tumor tissues was frequently accompanied by in-
Figure 3. MicroRNA-218 inhibits cell proliferation and invasion by targeand UGT8 was observed after transfection of miR-218 mimics. (B) Knockrate. (C) VOPP1 transcript level was higher in the majority of PDAC lineshowed that miR-218 inhibited the activity of luciferase reporter contamiR-218 binding site were deleted. (D, right panel) The protein level of UUGT8 was positively correlated with the metastatic potential of PDACinhibitors increased invasion rate of PDAC cells, and the effect was atten
reased UGT8 expression, giving 85.7% specificity.
EZH2 Suppresses miR-218 by Targeting Locus2 of miR-218 Precursor
The mature form of miR-218 is generated from 2separate loci—miR-218-1 and miR-218-2—and it is reportedthat they are coexpressed with their host genes SLIT2 andSLIT3, respectively.19 We measured SLIT2 and SLIT3 levelsin pancreatic cell lines, and showed that only expression ofSLIT3, not SLIT2, was low in most malignant cell lines(Figure 5A). Consistently, the primary form, miR-218-2, wasup-regulated on DZnep treatment of SW1990, in contrast tomiR-218-1, which was less responsive (Figure 5B). Knock-down of EZH2 resulted in up-regulation of miR-218 andSLIT3 simultaneously (Figure 5C). Measurement of miR-218precursors showed that miR-218-2 was frequently reduced inPDAC tissues (Supplementary Figure 5A). To conclude, weshowed that reduction of miR-218 level was heavily attrib-uted to the reduced expression of primary miR-218-2 and its
VOPP1 and UGT8, respectively. (A) Reduced transcript level of VOPP1n of VOPP1 attenuated the effect of miR-218 to reduce cell proliferation
ompared with HPDE cells. (D, left panel) UGT8-3=-UTR reporter assayg the wildtype UGT8-3=-UTR, but not the mutant in which 8-nt of the8 was reduced after transfection of miR-218 mimics. (E) Expression ofl lines in both transcript and protein level. (F) Transfection of miR-218ed by UGT8 knockdown concurrently. *P � .05; **P � .01; ***P � .001.
tingdows cininGTcel
host gene SLIT3.
omgs
poam
th( ors
BA
SIC
AN
DTR
AN
SLA
TIO
NA
LPA
NCREA
S
May 2013 EZH2 SILENCES miR-218 IN PANCREATIC CANCER 1091
The activity of miR-218-2 promoter was then validatedin 293T cells (Figure 5D). Here, EZH2 was involved in theregulation of the promoter activity, as the knockdown ofEZH2 in SW1990 generated a higher luciferase activity com-pared with the shRNA control (Figure 5E). To identify themiR-218 regulating domain of EZH2, miR-218 level wasmeasured in HPDE cells expressing full-length EZH2 orvarious domain-deleted mutants (Supplementary Figure 5Aand B). Either the deletion of protein-interacting domain H2
r methyltransferase domain SET gave less suppression toiR-218 expression compared with full-length EZH2, sug-
esting both H2 or SET domains of EZH2 played a role inuppressing miR-218 (Figure 5F).
Aberrant DNA Methylation Leads toSuppression of miR-218
We then studied the DNA methylation status at the
Figure 4. The EZH2-miR-218-UGT8 pathway is frequently observed intumor cells had strong nucleus EZH2 and membranous UGT8 expressepithelia. Original magnification �100, �200, and �400. Scoring of the sN � 60) were positive in UGT8 signal compared with adjacent normal tissuumor tissues (P � .0001) compared with normal pancreatic duct epitheliumaving high percentage (�20%) nucleus EZH2 staining (P � .0042). (E) UGT
P � .0001), and (F) high UGT8 expression was frequently observed in tum
miR-218-2 promoter as DNMT-related domain H2 was po-
tentially involved. Methylation-specific PCR showed that themajority of PDAC lines, but not nonmalignant HPDE cells,were having their miR-218-2 promoter CpG island hyperm-ethylated (Figure 6A, Supplementary Figure 6A and B). Hy-
ermethylated miR-218-2 promoters were also frequentlybserved in PDAC tumor tissues (75.9%) compared withdjacent normal tissues (22.2%), implying de novo DNAethylation occurred during carcinogenesis (Figure 6B).Next, we measured the miR-218 level in PDAC cells
after treatment of DNMT inhibitor 5AZAC and histonedeacetylase inhibitor trichostatin A. The miR-218 wasup-regulated after both DNA demethylation and histonedeacetylase inhibition (Figure 6C), suggesting that silenc-ing of miR-218 involved epigenetic events, including DNAmethylation and repressive chromatin. We then showedthat primary miR-218-2 was more responsive to bothtreatments than primary miR-218-1 (Figure 6D), which
C tumor tissues. (A) Immunohistochemical staining showed that PDAC, but their expression could not be detected in normal pancreatic ducting intensity showed that the (B) majority of the PDAC tumor tissues (70%,and that (C) overall there was a significant increase of UGT8 expression in) Reduced miR-218 expression was frequently observed in malignant ductspression was strongly correlated with EZH2 in both normal and PDAC cellswith reduced miR-218 level (P � .0079).
PDAiontaines. (D8 ex
confirmed that down-regulation of miR-218-2 contrib-

petiamSccEi(scdkD
stm
Ms ZH
BA
SICA
ND
TRA
NSLA
TION
AL
PA
NCREA
S
1092 LI ET AL GASTROENTEROLOGY Vol. 144, No. 5
uted to the loss of miR-218 expression. Co-transfectingmiR-218-2 promoter reporter with various EZH2 express-ing construct showed EZH2 lacking domain H2 lost theinhibition effect (Figure 6E) to miR-218-2 promoter,which validates the involvement of EZH2 domain H2during miR-218 suppression. Lastly, depletion of eitherDNMT1, DNMT3A, or DNMT3B by siRNAs elevated themiR-218 expression in SW1990 (Figure 6F and Supple-mentary Figure 6C). The involvement of DNMTs is directevidence supporting de novo methylation at the miR-218-2 promoter.
EZH2 Induces Histone Methylation andHeterochromatin Formation at miR-218-2By chromatin immunoprecipitation, EZH2 and
H3K27me3 were frequently found at the miR-218-2 pro-moter of PDAC cells, but not in HPDE cells (Figure 7A, leftpanels, Supplementary Figure 6D). The occupancy ofH3K27me3 at the promoter in HPDE cells was elevated
Figure 5. SLIT3/miR-218-2 precursor is silenced by EZH2 at the transchowed that SLIT3, but not SLIT2, was generally lower in expression ofhat of miR-218-1, was up-regulated in PDAC cell lines on DZnep treaiR-218 and SLIT3 mRNA. (D) Transfection of reporter containing miR-21
compared with blank reporter, and (E) knockdown of EZH2 further increasicroRNA-218 expression was measured in HPDE cells transduced wit
uppression was observed in cells expressing H2 or SET domain-deleted E
when EZH2 was ectopically expressed (Figure 7A, right a
anels). To investigate the effect of EZH2 to the chromatinnvironment of miR-218-2 promoter, we co-precipitatedhe chromatin containing miR-218-2 promoter with var-ous factors, including PRC2 component EED, polymer-se II, DNMTs, and heterochromatin-related factors orarkers. Here, the amount of precipitated DNA in
W1990 with EZH2 knockdown was determined andompared with shRNA control. Quantitation of the pre-ipitated DNA showed there was a reduced occupancy ofZH2 at miR-218-2 promoter, accompanied by a decrease
n EED, and simultaneous enrichment of polymerase IIFigure 7B). Knockdown of either EED or SUZ12 byiRNAs up-regulated the level of miR-218 in SW1990ells, which confirmed the involvement of PRC2 complexuring the silencing of miR-218 (Figure 7C). Similarly,nockdown of EZH2 resulted in reduced occupancies ofNMT1, DNMT3A, and DNMT3B (Figure 7D).Finally, we proved that expression of miR was also
ional level. (A) Measurement of SLIT2 and SLIT3 among PDAC cell lineslignant cells compared with HPDE. (B) Precursor of miR-218-2, but notnt. (C) Knockdown of EZH2 simultaneously increased the expression ofpromoter showed there was an increase in luciferase activity in 293T cellshe activity of miR-218-2 promoter containing reporter in SW1990 cells. (F)ntivirus containing full-length and mutant EZH2. Attenuation of miR-2182 compared with full-length EZH2. *P � .05; **P � .01; ***P � .001.
riptmatme8-2ed th le
ffected by altered chromatin configuration. Depletion

ptfrEc2pm
BA
SIC
AN
DTR
AN
SLA
TIO
NA
LPA
NCREA
S
May 2013 EZH2 SILENCES miR-218 IN PANCREATIC CANCER 1093
of EZH2 caused a reduced miR-218-2 promoter occu-pancy of heterochromatin-associated factors, such assilencing marker H3K9me2, H3K9 methyltransferaseSUV39H1, and HP1-� and HP1-� at the miR-218-2
romoter (Figure 7E). All these factors, together withhe presence of H3K27me3, were heavily linked to theormation of facultative heterochromatin as a means toegulate gene expression. Collectively, we showed thatZH2 played a critical role in recruiting repressivehromatin-related factors, and they suppressed miR-18 expression via heterochromatin formation at theromoter of miR-218-2 in addition to DNA and H3K27ethylation (Figure 7F).
Discussion
Our study demonstrates a pivotal role of EZH2
Figure 6. Aberrant methylation at miR-218-2 promoter contributes to mmiR-218-2 was hypermethylated in majority of PDAC cell lines but not inpromoter was frequently observed in PDAC tumor tissues (75.9%) comsignificantly increased in SW1990 cells treated with either trichostatinprominent than that of miR-218-1 precursor on TSA and 5AZAC treaco-transfected with miR-218-2 promoter containing reporter into 293Tdomain showed a reduced inhibition of reporter activity. (F) Knockdown oexpression of miR-218 significantly. **P � .01.
during the suppression of miR-218 in PDAC and re-
veals a previously underemphasized silencing mecha-nism involving EZH2-induced de novo heterochroma-tinization in cancer biology. Our finding that EZH2directly suppressed miR-218 revealed several significantaspects. First, EZH2-miR-218 pathway dysregulationincreased cell aggressiveness via up-regulation ofVOPP1 and UGT8. Second, heterochromatin inductionat miR-218 promoter added an extra dimension toEZH2-mediated aberration that explained a persistentsilencing effect. Third, targeting EZH2 alone suffi-ciently abolished aberrant epigenetic effects in PDAC.Finally, the frequent association of EZH2-miR-218-UGT8 pathway with metastatic phenotypes highlightedits clinical values in terms of novel therapeutic strategyor prognostic markers development.
To dissect the functional roles of miR-218 in PDAC
218 down-regulation. (A) Methylation-specific PCR showed promoter ofPDE. M, methylated; U, unmethylated. (B) Hypermethylated miR-218-2ed with adjacent normal tissues (22.2%). (C) miR-218 expression wasTSA) or 5AZAC. (D) Up-regulation of miR-218-2 precursor was morents. (E) Full-length EZH2 or various mutants expressing vector were
lls. While full-length EZH2 inhibited the reporter activity, deletion of H2NMT1, DNMT3A, or DNMT3B by siRNAs in SW1990 cells increased the
iR-H
parA (tmecef D
progression, we identified VOPP1 and UGT8 as both the

sr2tbwowbtU
tc
E
BA
SICA
ND
TRA
NSLA
TION
AL
PA
NCREA
S
1094 LI ET AL GASTROENTEROLOGY Vol. 144, No. 5
regulating and functional targets of miR-218 that con-ferred EZH2-miR-218 oncogenic effects. Previous studiesshowed VOPP1 was involved in the regulation of nuclearfactor �B transcription activity, and contributed to apo-ptosis resistance,20 and here we reaffirmed its cell growthinduction role in PDAC. In addition, we demonstratedthat UGT8 promoted PDAC cell migration. Taken to-gether, we suggest that loss of miR-218 gave a growthadvantage among the tumor cell population attributed toVOPP1, and were readily undergone disseminationthrough the effect of promigration protein UGT8. Inaddition to the in vivo studies mentioned previously, thetherapeutic effect of miR-218 was further proven in thesubcutaneous tumor growth study with intratumoral in-jection of miR-218 mimics. Consistently, restoring miR-218 within the tumor cells inhibited tumor growth (Sup-
Figure 7. EZH2 recruits diverse silencing partners to the miR-218-2 pation using anti-EZH2 and anti-H3K27me3 antibody showed that EZHompared with HPDE (upper panels), and ectopic EZH2 expression in H
(B) Knockdown of EZH2 in SW1990 cells reduced the occupancy of boRNA knockdown of EED and SUZ12 increased the expression of miR-miR-218-2 promoter was observed in shEZH2 expressing SW1990 coincluding H3K27me3, H3K9me2, HP1-�, HP1-�, and SUV39H1 were o
ZH2-mediated heterochromatinization of the miR-218-2 promoter.
plementary Figure 7). The miR-218 restoration therapy is f
potentiated with our in-depth molecular basis and thera-peutic effect evaluation.
The functional characterization of UGT8 provided newinsight into the highly aggressive phenotype of PDACwith high EZH2 level.5 As reflected by frequent coexpres-ion of EZH2 and UGT8 in clinical samples, UGT8 up-egulation was a common consequence of the EZH2-miR-18 pathway dysregulation. Previous studies have linkedhe expression of UGT8 with prognosis of breast cancery showing a strong positive correlation of UGT8 levelith lung metastasis,21,22 but they lack the investigationf UGT8 functional roles during this progression. Here,e proved that up-regulation of UGT8 was not only ayproduct in EZH2-miR-218 dysregulation, but was func-ional in promoting metastasis. UGT8 belongs to theDP-glycosyltransferase family, and it catalyzes the trans-
oter that leads to heterochromatization. (A) Chromatin immunoprecipi-nd H3K27me3 were enriched in PDAC cell lines PANC1 and SW1990
cells increased both EZH2 and H3K27me3 enrichment (lower panels).ZH2 and EED, and increased that of polymerase II. (C) Small interfering. (D) A reduced enrichment of DNMT1, DNMT3A, and DNMT3B at theared with negative control. (E) Reduction of heterochromatin markers,erved in shEZH2 expressing SW1990 cells. (F) A hypothetical model of
rom2 aPDEth E218mpbs
er of galactose to ceramide, which is a key enzymatic step

P
t
2f
mpinmmndmgt
dnhartIC
ac
Poam
BA
SIC
AN
DTR
AN
SLA
TIO
NA
LPA
NCREA
S
May 2013 EZH2 SILENCES miR-218 IN PANCREATIC CANCER 1095
in the biosynthesis of sphingolipid subtype. In PDAC,overexpression of UGT8 possibly affected the ceramide/sphingolipid rheostat, and this changes the proportionamong sphingolipid subtypes. It was reported that alteredsphingolipid metabolism affected the onset, dissemina-tion, and formation of metastasis in different cancertypes.23 Having a crucial role in PDAC progression, it isworth investigating the principle underlying UGT8-cer-amide mediated promotion of metastasis.
The miR-218 expression is lost in multiple solid tu-mors, but the underlying mechanism is unexplored. Here,all 3 DNMT members contributed to miR-218 silencing,despite having specific roles in DNA methylation.DNMT3A and DNMT3B catalyze de novo DNA methyl-ation,24 and their expression was recently correlated with
DAC aggressiveness,25 but any participation during pre-cancerous transformation is elusive. Hence, aberrant denovo DNA methylation during early tumorigenesis is un-likely a result of DNMT3A or DNMT3B up-regulation.Instead, redirecting them within genomic region becauseof cofactor dysregulation should be the possible scenarioduring PDAC carcinogenesis. Here, ectopically expressedEZH2 guided both DNMTs to miR-218 promoter, in-duced CpG island methylation, and silenced miR-218. Asfor DNMT1, it maintained the methylation induced byDNMT3A and DNMT3B during cancerous clonal expan-sion. In addition, the active recruitment of DNMT1 byEZH2 to miR-218 promoter suggested DNMT1 mightalso de novo methylate miR-218 promoter, despite havingweaker activity.26,27
We demonstrated that EZH2 is a central player promot-ing epigenetic suppression to miR-218 by establishingsuppressive histone marker H3K27me3 and promoterDNA methylation, but not exclusively. We showed chro-matin-modulating factors were involved at the suppressedmiR-218-2 promoter, which gave a new angle to under-standing the role of EZH2. Other than H3K27me3,H3K9me2, linked with silenced region of euchromatin,28
was enriched at miR-218-2 promoter. The occurrence ofH3K9me2 might be a result of the hyperactivity of PRC2complex on EZH2 overexpression, as it is reported thatthe PRC2 components mediated the methylation ofH3K9. EZH2 and SUZ12 physically interact with factorUHRF1, which mediates the methylation of H3K9through the recruitment of methyltransferaseSUV39H1.29 It also explains why SUV39H1 was colocallyenriched with EZH2. SUV39H1 is responsible for trim-ethylation of H3K9, which leads to the formation ofconstitutive heterochromatin.30 Studies showed thatSUV39H1 silenced gene expression through heterochro-matin formation,31 and here dimethylated H3K9 mightprovide a platform for SUV39H1 for the induction ofH3K9me3 and subsequent heterochromatin at the miR-218-2 promoter. Lastly, the enrichment of HP1 memberswas attributed to H3K9 hypermethylation, as the chro-modomain of HP1 proteins specifically recognizes themethyl marks of H3K9.32 Commonly associated with
ranscriptional inactive region, HP1-� and HP1-� at miR-18-2 promoter provided critical evidence supporting theormation of repressive chromatin structure.
Most importantly, depletion of EZH2 effectively re-oved all of these suppressive factors from the miR-218-2
romoter, which indicated EZH2 was indispensible dur-ng heterochromatin formation. EZH2 induces perma-ent repression of miR-218 by induction of heterochro-atin around the miR-218-2 promoter in a progressiveanner. This persistent effect is essential to alter gene
etwork homeostasis that provides an advantageous con-ition for cell growth and dissemination. Expression ofiR-218 is also reduced in other cancer types, such as
astric cancer, lung squamous cell carcinoma, glioblas-oma,19,33,34 human papillomavirus–positive cells of both
cervical cancer, and head and neck cancer.35,36 Coinci-ently, EZH2 overexpression is a common molecular phe-omenon observed in these malignancies, so EZH2 mayave a universal effect on miR-218 in these cancer types,s shown in our hypothetical model of EZH2-mediatedepression to miR-218. In humans, EZH2 is directed to itsarget via recruitment by long noncoding RNA HOTAIR.t is revealed that the consensus sequence of HOTAIR isG-rich,37 so its affinity to CG-rich region (ie, CpG island)
is high. We speculated that EZH2 enrichment at the CpGisland of miR-218-2 promoter may be mediated byHOTAIR. Interestingly, HOTAIR is recently shown to behighly expressed in pancreatic cancer tissue and celllines.38 Abrogation of the recruitment machinery may be
n alternative approach to attenuate EZH2 aberration inancer.
This work adds essential elements to the currentDAC progression model that depicts an accumulationf both somatic mutation and epigenetic aberrationlong the progression of PDAC from ductal lesion toalignant neoplasm.39,40 In alignment with most mod-
els, we emphasized the abnormality of chromatin dy-namics within PDAC cell, in this case mediated byEZH2, and unveiled a unique dysregulation pathwayattributed to the invasive phenotype of PDAC. We dem-onstrated the unprecedented linkage between poly-comb complex and H3K9me-HP1: 2 independent sys-tems predominate for instituting a repressivechromatin landscape in controlling gene expression.Here, we showed there was crosstalk, in between whichEZH2 indirectly induced H3K9 methylation and thesubsequent HP1 recruitment. The cooperative effect ofthe 2 systems contributed to aberrant gene silencing inPDAC. Now it is widely accepted that level of EZH2strongly associates with severity of malignant progres-sion, but the role of HP1 in PDAC is still largelyunexplored. Future works should focus on the tempo-ral and spatial distribution of HP1 during PDAC pro-gression, and distinguished origins of various HP1 pat-terns (ie, EZH2-mediated or canonical H3K9me-induced). The investigation of HP1 dynamics onchromatin during PDAC progression can provide the
puzzle pieces missing in the current model.
dHromm
d
1
1
1
1
1
1
1
1
1
1
2
2
2
2
2
2
2
2
2
2
3
3
3
3
3
3
BA
SICA
ND
TRA
NSLA
TION
AL
PA
NCREA
S
1096 LI ET AL GASTROENTEROLOGY Vol. 144, No. 5
This study provided several molecular bases to supportadditional evaluation on potential clinical values of EZH2inhibitor. We showed that DZnep inhibited cell prolifera-tion, promoted apoptosis, and induced cell cycle arrest inPDAC cells (Supplementary Figure 8A–C). DZnep effectively
epleted PRC2 components, decreased the global3K27me3 level in PDAC cells, and subsequently caused
e-expression of a set of miRs, including miR-218. The abilityf DZnep to restore the expression of tumor suppressoriRs should be more strong evidence to apply this smallolecule in clinical usage.
Supplementary Materials
Note: To access the supplementary materialaccompanying this article, visit the online version ofGastroenterology at www.gastrojournal.org, and at http://
x.doi.org/10.1053/j.gastro.2013.01.058.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CACancer J Clin 2012;62:10–29.
2. Edwards BK, Brown ML, Wingo PA, et al. Annual report to thenation on the status of cancer, 1975�2002, featuring population-based trends in cancer treatment. J Natl Cancer Inst 2005;97:1407–1427.
3. Warshaw AL, Fernandez-del Castillo C. Pancreatic carcinoma.N Engl J Med 1992;326:455–465.
4. Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics.Nat Rev Cancer 2002;2:897–909.
5. Chen Y, Xie D, Yin Li W, et al. RNAi targeting EZH2 inhibits tumorgrowth and liver metastasis of pancreatic cancer in vivo. CancerLett 2010;297:109–116.
6. Grzenda A, Ordog T, Urrutia R. Polycomb and the emerging epige-netics of pancreatic cancer. J Gastrointest Cancer 2011;42:100–111.
7. Li A, Omura N, Hong SM, et al. Pancreatic cancers epigeneticallysilence SIP1 and hypomethylate and overexpress miR-200a/200bin association with elevated circulating miR-200a and miR-200blevels. Cancer Res 2010;70:5226–5237.
8. Li Y, Vandenboom TG 2nd, Wang Z, et al. miR-146a suppressesinvasion of pancreatic cancer cells. Cancer Res 2010;70:1486–1495.
9. Volinia S, Calin GA, Liu CG, et al. A microRNA expression signatureof human solid tumors defines cancer gene targets. Proc NatlAcad Sci U S A 2006;103:2257–2261.
0. Cao Q, Mani RS, Ateeq B, et al. Coordinated regulation of poly-comb group complexes through microRNAs in cancer. Cancer Cell2011;20:187–199.
1. Ouyang H, Mou LJ, Luk C, et al. Immortal human pancreatic ductepithelial cell lines with near normal genotype and phenotype.Am J Pathol 2000;157:1623–1631.
2. Furukawa T, Duguid WP, Rosenberg L, et al. Long-term culture andimmortalization of epithelial cells from normal adult human pan-creatic ducts transfected by the E6E7 gene of human papillomavirus 16. Am J Pathol 1996;148:1763–1770.
3. Chen Y, Lin MC, Wang H, et al. Proteomic analysis of EZH2downstream target proteins in hepatocellular carcinoma. Proteom-ics 2007;7:3097–3104.
4. Chen Y, Lin MC, Yao H, et al. Lentivirus-mediated RNA interfer-ence targeting enhancer of zeste homolog 2 inhibits hepatocellu-lar carcinoma growth through down-regulation of stathmin. Hepa-tology 2007;46:200–208.
5. Marson A, Levine SS, Cole MF, et al. Connecting microRNA genesto the core transcriptional regulatory circuitry of embryonic stem
cells. Cell 2008;134:521–533.6. Sakurai Y, Sawada T, Chung YS, et al. Identification and charac-terization of motility stimulating factor secreted from pancreaticcancer cells: role in tumor invasion and metastasis. Clin ExpMetastasis 1997;15:307–317.
7. Miyazawa Y, Uekita T, Hiraoka N, et al. CUB domain-containingprotein 1, a prognostic factor for human pancreatic cancers,promotes cell migration and extracellular matrix degradation. Can-cer Res 2010;70:5136–5146.
8. Duxbury MS, Ito H, Zinner MJ, et al. CEACAM6 gene silencingimpairs anoikis resistance and in vivo metastatic ability of pan-creatic adenocarcinoma cells. Oncogene 2004;23:465–473.
9. Tie J, Pan Y, Zhao L, et al. miR-218 inhibits invasion and metas-tasis of gastric cancer by targeting the Robo1 receptor. PLoSGenet 2010;6:e1000879.
0. Gao C, Zhang Z, Liu W, et al. Reduced microRNA-218 expressionis associated with high nuclear factor kappa B activation in gastriccancer. Cancer 2010;116:41–49.
1. Dziegiel P, Owczarek T, Plazuk E, et al. Ceramide galactosyltrans-ferase (UGT8) is a molecular marker of breast cancer malignancyand lung metastases. Br J Cancer 2010;103:524–531.
2. Culhane AC, Quackenbush J. Confounding effects in “a six-genesignature predicting breast cancer lung metastasis.” Cancer Res2009;69:7480–7485.
3. Ruckhäberle E, Rody A, Engels K, et al. Microarray analysis ofaltered sphingolipid metabolism reveals prognostic significance ofsphingosine kinase 1 in breast cancer. Breast Cancer Res Treat2008;112:41–52.
4. Okano M, Bell DW, Haber DA, et al. DNA methyltransferasesDnmt3a and Dnmt3b are essential for de novo methylation andmammalian development. Cell 1999;99:247–257.
5. Zhang JJ, Zhu Y, Zhu Y, et al. Association of increased DNAmethyltransferase expression with carcinogenesis and poor prog-nosis in pancreatic ductal adenocarcinoma. Clin Transl Oncol2012;14:116–124.
6. Pradhan S, Bacolla A, Wells RD, et al. Recombinant human DNA(cytosine-5) methyltransferase. I. Expression, purification, andcomparison of de novo and maintenance methylation. J Biol Chem1999;274:33002–33010.
7. Peng DF, Kanai Y, Sawada M, et al. Increased DNA methyltrans-ferase 1 (DNMT1) protein expression in precancerous conditionsand ductal carcinomas of the pancreas. Cancer Sci 2005;96:403–408.
8. Rice JC, Briggs SD, Ueberheide B, et al. Histone methyltrans-ferases direct different degrees of methylation to define distinctchromatin domains. Mol Cell 2003;12:1591–1598.
9. Babbio F, Pistore C, Curti L, et al. The SRA protein UHRF1 pro-motes epigenetic crosstalks and is involved in prostate cancerprogression. Oncogene 2012;31:4878–4887.
0. Peters AH, O’Carroll D, Scherthan H, et al. Loss of the Suv39hhistone methyltransferases impairs mammalian heterochromatinand genome stability. Cell 2001;107:323–337.
1. Murayama A, Ohmori K, Fujimura A, et al. Epigenetic control ofrDNA loci in response to intracellular energy status. Cell 2008;133:627–639.
2. Lachner M, O’Carroll D, Rea S, et al. Methylation of histone H3lysine 9 creates a binding site for HP1 proteins. Nature 2001;410:116–120.
3. Wald AI, Hoskins EE, Wells SI, et al. Alteration of microRNAprofiles in squamous cell carcinoma of the head and neck celllines by human papillomavirus. Head Neck 2010;33:504–512.
4. Davidson MR, Larsen JE, Yang IA, et al. MicroRNA-218 is deletedand downregulated in lung squamous cell carcinoma. PLoS One2010;5:e12560.
5. Song L, Huang Q, Chen K, et al. miR-218 inhibits the invasiveability of glioma cells by direct downregulation ofr IKK-�. BiochemBiophys Res Commun 2010;402:135–140.
36. Martinez I, Gardiner AS, Board KF, et al. Human papillomavirustype 16 reduces the expression of microRNA-218 in cervical car-
cinoma cells. Oncogene 2008;27:2575–2582.
3
3
4
May 2013 EZH2 SILENCES miR-218 IN PANCREATIC CANCER 1097
37. Tsai MC, Manor O, Wan Y, et al. Long noncoding RNA as modularscaffold of histone modification complexes. Science 2010;329(5992):689–693.
8. Kim K, Jutooru I, Chadalapaka G, et al. HOTAIR is a negativeprognostic factor and exhibits pro-oncogenic activity in pancreaticcancer. Oncogene 2012 May 21. [Epub ahead of print]
9. Hruban RH, Goggins M, Parsons J, et al. Progression model forpancreatic cancer. Clin Cancer Res 2000;6:2969–2972.
0. Lomberk G, Urrutia R. Epigenetics and its applications to a revisedprogression model of pancreatic cancer. In: Neoptolemos JP, Urrutia R,Abbruzzese JL, et al, eds. Pancreatic Cancer. New York: Springer; 2009;143–170.
Received May 17, 2012. Accepted January 9, 2013.
Reprint requestsAddress requests for reprints to: Yangchao Chen, PhD, School of
Biomedical Sciences, Faculty of Medicine, the Chinese University of
Hong Kong, Shatin, Hong Kong. e-mail: [email protected];fax: �852 26035123.
Conflicts of interestThe authors disclose no conflicts.
FundingThis work was supported by grants from the Research Grants
Council-General Research Fund of Hong Kong SpecialAdministrative Region, China (CUHK462109 and CUHK462211),National Natural Science Foundation of China (81101888),Shenzhen Basic Research Program (JC201105201092A), DirectGrant from CUHK to YC. This research was supported in part bythe Intramural Research Program of the National Institutes ofHealth, National Cancer Institute, Center for Cancer Research.
The study sponsor played no role in the study sponsor in thestudy design in the collection, analysis, and interpretation
of date.BA
SIC
AN
DTR
AN
SLA
TIO
NA
LPA
NCREA
S

m
opffmtd
mvg
Cyu
e
1097.e1 LI ET AL GASTROENTEROLOGY Vol. 144, No. 5
Supplementary Materials and Methods
MicroRNA-218 Mimics, Inhibitors, andsiRNAs TransfectionMicroRNA-218 mimics, miR-218 inhibitors, and
siRNAs targeting EZH2, UGT8, VOPP1, EED, SUZ12,DNMT1, DNMT3A, or DNMT3B (Shanghai GenePharmaCo., Ltd, Shanghai, China) were transfected into cells usingDharmaFECT 1 siRNA transfection reagent (Thermo Sci-entific, Lafayette, CO).
Total RNA and DNA ExtractionTotal RNA and DNA from cell cultures were ex-
tracted using Trizol (Invitrogen, Carlsbad, CA) and QIAampDNA Mini Kit (Qiagen, Valencia, CA), respectively. ForPDAC patient tissue samples, 10-um thick specimens weresectioned from paraffin block and mounted on noncoatedslides. Regions containing either tumor or normal tissueswere identified under microscope, and were isolated fortotal RNA and DNA extraction by miRNeasy FFPE Kit andQIAamp DNA FFPE Tissue Kit (Qiagen), respectively.
Quantitative RT-PCRMicroRNA profilings were conducted using mi-
croRNA Ready-to-Use PCR, Human panel I�II, V2.M (Exiqon,Vedbaek, Denmark). For messenger RNA and primary miR,ccomplementary DNA was converted from total RNA usingHigh-Capacity cDNA Reverse Transcription Kit (Applied Bio-systems, Foster City, CA). Taqman Pri-miRNA assays wereperformed to measure primary miR-218-1 and miR-218-2 (As-say ID Hs03302986_pri and Hs03302992_pri). In both assays,glyceraldehyde-3-phosphate dehydrogenase was measuredas the reference gene. For miR expression level measure-ment, Ncode First Strand cDNA Synthesis and qRT-PCRkit was used during validation and candidates screening,while Taqman MicroRNA assay was used to measure miR-218 expression. For measurement of mature miRs, RNU6Bwas used as the reference gene. Quantitative PCR was per-formed by ABI 7900HT Real-Time PCR system using eitherSYBR Green PCR Master Mix or Taqman Universal PCRMaster Mix (Applied Biosystems). Comparative CT method(2���CT) was acquired to measure fold changes.
Western Blot AnalysisWestern blottings were conducted under standard
procedures. After blocking, the membrane was incubatedat 4°C overnight with anti-UGT8 (Sigma), anti-EZH2,anti–myc-tag (Cell Signaling Technology), or Actin(Santa Cruz Biotechnology, Santa Cruz, CA) diluted innonfat dry milk (Santa Cruz Biotechnology), followed byhorseradish peroxidase–labeled secondary antibody incu-bation, and chemiluminescence signal was develop byECL Plus Western Blotting Detection Reagents (GEHealthcare Life Sciences, Piscataway, NJ).
Migration and Invasion AssaysPDAC cells resuspended in unsupplemented Dul-
becco’s modified Eagle medium were seeded into a chamber t
containing an 8-�m pore size polyethylene terephthalateembrane either with or without 100 �L Matrigel (BD
Biosciences, San Jose, CA). All assays were performed understandard procedures. For chamber migration assay, crystal vi-olet staining was redissolved by acetic acid and 550 nm absor-bance was measured. For invasion assay, 6 fields on the mem-brane were randomly captured and number of cells on eachfield was counted.
Promoter and 3=-UTR Luciferase Reporter AssaysPromoter region of miR-218-2, 2.7-kbp upstream
of transcriptional start site reported by Marson et al15
was cloned into pGL3 basic vector (Promega, Madison,WI). The 3=-UTR of UGT8 (approximately 500 bp), and amutant with 8mer miR-218 binding sited removed, wascloned into pmiR-Report (Ambion, Austin, TX). Accuracy
f the constructs was confirmed by DNA sequencing. Re-orter vectors and Renilla luciferase vector were co-trans-ected into either 293T or SW1990 cells using Lipo-ectamine 2000 (Invitrogen). For 3=-UTR reporter assay,
iR-218 mimics was co-transfected with the luciferase vec-ors using Lipofectamine 200. After 24–48 h incubation,ual-luciferase reporter assay (Promega) was performed.
H&E and Immunohistochemical StainingH&E staining of the nude mice lung metastasis
tissues was performed under standard procedures. For pri-mary antibody incubation, subcutaneous tumor was incu-bated with anti-CD31 (Abcam), and PDAC clinical tissueswere incubated with anti-EZH2 (Cell Signaling Technology) oranti-UGT8 (Sigma) overnight at 4°C. Scoring of the immuno-histochemical staining was based on the percentage of positivecells and staining intensity under a light microscope. Threerandom fields at 200� magnification were captured per sec-tions for evaluation. During scoring for UGT8 IHC staining, 4categories (0, 1�, 2�, and 3�) were denoted as negative, weak,
oderate, and strong, respectively, and were further subdi-ided into high-expression (2� and 3�) and low-expressionroups (0 and 1�).
Methylation-Specific PCRBisulfite treatment of genomic DNA was performed
by EZ DNA Methylation-Gold Kit (Zymo Research, Irvine,A). Bisulfite-treated DNA were used as the template in meth-lation-specific PCR using primers specific to methylated ornmethylated miR-218-2 promoter, and bisulfite-converted
�-actin allele. Universal methylated DNA was used as a hyper-methylated control to optimize the methylation-specific PCRcondition. The accuracy of the methylation-specific PCR reac-tion was confirmed by DNA sequencing.
Statistical AnalysisGraphPad Prism 5 (GraphPad Software, La Jolla,
CA) was used for statistical analysis. Statistical tests for dataanalysis included 2-tailed Student t test, �2 test, and Fisher’sxact test. Data were presented as mean � standard devia-
ion. P values �.05 were considered statistically significant.
ASb
May 2013 EZH2 SILENCES miR-218 IN PANCREATIC CANCER 1097.e2
Supplementary Figure 1. EZH2 plays a role in regulating miR expresup-regulated in SW1990 after DZnep treatment, and down-regulatedcomparative CT method, and change �1.5-fold was considered significEZH2, EED, and SUZ12, as well as the global H3K27me3 level in SWmiR-218, miR-302a, miR-363, miR-378, miR-516a-5p, and miR-616)
mong the 8 miRs, miR-218 gave the most significant increase in exprcatter plot of the normalize RNU6B in qRT-PCR measurement of maetween tumor and nontumor groups.
sion. (A) By qRT-PCR-based global human miR profiling, 21 miRs were bothafter overexpression of EZH2 in HPDE. Fold change was measured by
ant. (B) Western blots showed DZnep treatment reduced the protein level of1990. (C, D) Validation by qRT-PCR identified 8 miRs (miR-7, miR-206,that had consistent trend of changes with that of the profiling results. (E)
ession in SW1990 after knockdown of EZH2 by either siRNA or shRNA. (F)ture miR-218 in PDAC paired tissues, and there is no significant difference

A
P
P
1097.e3 LI ET AL GASTROENTEROLOGY Vol. 144, No. 5
Supplementary Table 1. Statistics of MicroRNA-218 In SituHybridization in Pancreatic Normaland Tumor Tissue Microarray
No. ofcases P value
PDAC tissues 87 �.0001a
Normal tissues 10Sex
Male 51 .3967Female 42
ge groupsAge �50 y 34 .686850 � Age �60 y 33 (one-way ANOVA)60 � Age �70 y 28
athological typesWell to moderately differentiated 50 �.0001a
Poorly differentiated 19 �.001a
(Test against normal)rimary tumor grades (TNM
system)�.0001a
T1 and T2 27 �.0001a
T3 and T4 49 (Test against normal)
ANOVA, analysis of variance.
aStatistically significant.
May 2013 EZH2 SILENCES miR-218 IN PANCREATIC CANCER 1097.e4
Supplementary Figure 2. MicroRNA-218 inhibits PDAC cell proliferation, in vivo tumor growth, and cell migration. (A) Significant increase ofmiR-218 level resulted after miR-218 mimic transfection in SW1990. (B) Other than SW1990, the cell proliferation rate of PANC1 was also decreasedafter miR-218 mimic transfection, and increased after miR-218 inhibitor transfection. (C) In nude mice, SW1990 xenograft, reduced subcutaneousgrowth, and vascularization were observed macroscopically in lentiviral overexpression of miR-218. (D) Wound healing assay was performed to study
the effect of miR-218 on cell migration. Reduced wound healing rate was observed in CAPAN2 after miR-218 mimic transfection. *P � .05.
frUma
1097.e5 LI ET AL GASTROENTEROLOGY Vol. 144, No. 5
Supplementary Figure 3. MicroRNA-218 regulates UGT8 level and inhibits PDAC cell migration. (A) The mRNA level of potential miR-218unctional targets was measured after transfection of miR-218 mimic in SW1990. Functional targets of miR-218 were either putative miR-218 targetseported in other studies, or they were commonly predicted in Silico by 3 different miR target prediction algorithms. It was observed that ROBO1,GT8, and VOPP1 were significantly reduced after the transfection of miR-218 mimic. (B) Messenger RNA levels of EZH2 and VOPP1 wereeasured in PDAC tumor and adjacent normal tissues by qRT-PCR. Pearson’s correlation showed that the changes in EZH2 expression in tumor
gainst respective normal tissues were positively correlated with the changes in VOPP1 expression (n � 26; P � .01). (C) Wound healing assay wasperformed to study the functional association between miR-218 and UGT8. Although transfection of miR-218 inhibitor increased the wound healing
rate of CAPAN2, simultaneous knockdown of UGT8 by siRNA attenuated such an effect of miR-218 inhibitor. *P � .05; **P � .01.
ciPdwu
aTac
May 2013 EZH2 SILENCES miR-218 IN PANCREATIC CANCER 1097.e6
Supplementary Figure 4. Kaplan–Meier plot of 28 PDAC patients.The Kaplan–Meier survival curve of PDAC patients with reduced miR-218 level was significantly different (P � .0376) from patients with un-hanged/up-regulated miR-218 level. Patients were grouped accord-
ng to the miR-218 level changes in paired normal and tumor tissues.atients with miR-218 fold decrease �1.5 were denoted as miR-218own-regulated (n � 15), and those with miR-218 fold decrease �1.5ere denoted as miR-218 unchanged/up-regulated. Log-rank test was
sed for the statistical analysis of the Kaplan-Meier survival curve.Supplementary Figure 5. Lentivirus-mediated overexpression of full-length EZH2 and various domain deleted mutants. (A) Precursor miR-218-2was frequently reduced in 60 pairs of PDAC tissues (P � .0076). (B) After transduction of full-length or mutant EZH2 containing lentivirus into HPDE,Western blotting was performed to confirm their expression. For full-length EZH2 and �SET, �cys � SET, �H1, and �H2 mutants, eithernti–myc-tag or anti-EZH2 was used during the primary antibody incubation. It showed that all EZH2 constructs were expressed in HPDE cells. (C)he �H1 � H2 EZH2 expression construct could not be detected by either anti-myc or anti-EZH2 antibody, as the construct was not myc-taggednd lacked the carboxyl terminal epitope of the EZH2 antibody. Hence, expression of the untagged �H1 � H2 mutant as well as the blank vector
ontrol was confirmed by coexpression of green fluorescent protein.
1097.e7 LI ET AL GASTROENTEROLOGY Vol. 144, No. 5
Supplementary Figure 6. Various epigenetic mechanisms regulate miR-218 expression. (A) Schematic diagram illustrates the CpG island andCpG dinucleotides distribution at the promoter region of miR-218-2. (B) MSP products of miR-218-2 promoter, using the template methylatedgenomic DNA control Universal Methylated DNA (UMD), was first cloned into pMD-18T vector before subjected to DNA sequencing. It showed thatthe reaction correctly distinguished the bisulfate-treated DNA from untreated, and accurately reflected the hypermethylation status of the miR-218-2promoter. (C) The transcript level of DNMT1, DNMT3A, DNMT3B, EED, and SUZ12 were significantly reduced after transfection of siRNAs. (D)
Chromatin immunoprecipitation showed the enrichment of EZH2 and H3K27me3 marker at the promoter region of miR-218-2.
owomiddmptn2(
May 2013 EZH2 SILENCES miR-218 IN PANCREATIC CANCER 1097.e8
Supplementary Figure 7. Intratumoral injection of miR-218 mimicsinhibits in vivo tumor growth. SW1990 cells (5 � 106) were subcutane-usly inoculated into both flanks of the nude mice (n � 5). The tumorsere allowed to grow around 5 mm � 5 mm in size before the injectionf miR-218 mimics and negative control duplex; 10 nmol miR-218imics or negative control RNA duplex (NC) was mixed with transfect-
ng agent Dharmafect 1 (Fisher Scientific). MicroRNA-218 mimics wereirectly injected into the right flank tumor, and the negative control RNAuplex was injected directly into the left flank tumor of the same nudeice. The intratumoral injections were carried out every 3 days for aeriod of 2 weeks, and the tumor size was measured regularly duringhe RNA duplex injection. After 2 weeks injection, a reduced subcuta-eous tumor growth was observed for the tumors injected with miR-18 mimics compared with those injected with negative control duplex
P � .05).

(aCrsD
NS
AP
R
1097.e9 LI ET AL GASTROENTEROLOGY Vol. 144, No. 5
Supplementary Figure 8. DZnep shows various anticancer properties, including cell growth inhibition, induction of apoptosis, and cell cycle arrest.A) HPDE and all PDAC cell lines were treated with 5 �M DZnep, and they were allowed to grow for 48 h. DZnep treatment reduced cell growth inll tested cell lines that ranged from 20%–60% growth inhibition. (B) Flow cytometry analysis of DZnep effect on the apoptosis of PDAC cell lines.ultures were stained with Annexin V-allophycocyanin and propidium iodide. Increased number of cells had undergone both early apoptosis (lower
ight area) and late apoptosis (upper right area) in SW1990, CAPAN2, and CFPAC1 on DZnep treatment. (C) Cell cycle assay by propidium iodidetaining was carried out to analyze the effect of DZnep to cell cycle. Cell cycle distribution before and after DZnep treatment was illustrated. After
Znep treatment, increased population of cells at the G0/G1 phase was observed in both SW1990 and CAPAN2 cells.Supplementary Table 2. Characteristics of PDAC in 60Patients
MicroRNA-218down-regulated
�1.5-fold
MicroRNA-218unchanged/up-regulated
Pvalue
34 26ex, n (%) .9999Male 17 (50.0) 17 (50.0)Female 16 (61.5) 10 (38.5)
ge, y, median (range) 66 (45–79) 61 (45–75) .0878athological type, n (%) .2033Well differentiated 4 (15.4) 4 (19.0)Moderately
differentiated21 (80.8) 13 (62.0)
Poorly differentiated 1 (3.8) 4 (19.0)egional lymph node
metastasis, n (%).7808
No 13 (41) 15 (65)
Yes 19 (59) 8 (35)



![57] Heterogeneity of heterochromatin in six species of Ctenomys (Rodentia: Octodontoidea: Ctenomyidae) from Argentina revealed by a combined analysis of C-and RE-banding](https://static.fdokumen.com/doc/165x107/631d1a5e5a0be56b6e0e8711/57-heterogeneity-of-heterochromatin-in-six-species-of-ctenomys-rodentia-octodontoidea.jpg)















