
Enhancement of the catalytic oxidation of hydrogen-lean chlorinated VOCs in the presence of...
11
Applied Catalysis B: Environmental 24 (2000) 33–43 Enhancement of the catalytic oxidation of hydrogen-lean chlorinated VOCs in the presence of hydrogen-supplying compounds J.R. González-Velasco * , A. Aranzabal, R. López-Fonseca, R. Ferret, J.A. González-Marcos Department of Chemical Engineering, Faculty of Sciences, Universidad del Pa´ ıs Vasco/EHU, P.O. Box 644, E-48080 Bilbao, Spain Received 20 February 1999; received in revised form 12 July 1999; accepted 12 July 1999 Abstract The complete catalytic oxidation of trichloroethylene (TCE) over alumina-supported noble metal catalysts (Pt and Pd) and in the presence of hydrogen-rich compounds, i.e. water, hexane and toluene was evaluated. Experiments were performed at conditions of lean TCE concentration (around 1000 ppm) in air, between 250 and 550 ◦ C in a conventional fixed-bed reactor. Hexane and toluene were added to the feedstream in a concentration of around 1000 ppm and water concentration varied from 1000 to 15 000 ppm. TCE oxidation occurred faster in the presence of hexane and toluene over both catalysts. Over palladium catalysts, water did not alter catalytic activity, whereas over platinum catalysts water enhanced TCE oxidation at low temperatures (<400 ◦ C) but inhibited it at higher temperatures (>400 ◦ C). Selectivity to HCl was much improved by feeding water as a hydrogen-supplying reactant; 7500 ppm of water enhanced HCl outputs from 39.4 to 78.0% with Pd, and from 37.5 to 58.9% with Pt. Selectivities to C 2 Cl 4 , formed by chlorination of the feed, and Cl 2 were greatly reduced. On the other hand water promoted complete oxidation of TCE to CO 2 , and thus reduced selectivity to CO. In the presence of hexane and toluene, formation of HCl was also enhanced. Hexane showed higher inhibition ability than toluene over both catalysts for the C 2 Cl 4 and Cl 2 formation. Unlike in the presence of water, selectivity to CO increased, as a consequence of partial oxidation of both hydrocarbons. ©2000 Elsevier Science B.V. All rights reserved. Keywords: Catalytic oxidation; Selectivity; Trichloroethylene; Noble metal catalysts; VOC 1. Introduction Chlorinated volatile organic compounds (CVOCs) are known to be hazardous to the environment and public health. Catalytic oxidation of CVOCs is cur- rently receiving increased attention to keep their emissions in compliance with the increasingly strin- gent environmental regulations (US 1990 Clean Air Act), since chlorinated compounds, including toxic * Corresponding author. Tel.: +34-94-6012-681; fax: +34-94- 4648-500 E-mail address: [email protected] (J.R. Gonz´ alez-Velasco) by-products can be destroyed into CO 2 ,H 2 O and HCl at relatively low temperatures (<550 ◦ C) compared with conventional thermal incineration. Among the large number of CVOCs which are discharged into the environment, those containing more chlorine atoms than hydrogen atoms within their molecules, cannot be totally converted by air to the most desirable HCl, which is easily scrubbed in an alkaline medium, and therefore even more toxic products such as Cl 2 and COCl 2 are formed. To im- prove selectivity to HCl, either a hydrogen-rich fuel or water vapour may be added to the feedstream. Indeed, in the real flue gases, CVOCs are present 0926-3373/00/$ – see front matter ©2000 Elsevier Science B.V. All rights reserved. PII:S0926-3373(99)00087-9
-
Upload
cicenergigune -
Category
Documents
-
view
0 -
download
0
Transcript of Enhancement of the catalytic oxidation of hydrogen-lean chlorinated VOCs in the presence of...

Applied Catalysis B: Environmental 24 (2000) 33–43
Enhancement of the catalytic oxidation of hydrogen-lean chlorinatedVOCs in the presence of hydrogen-supplying compounds
J.R. González-Velasco∗, A. Aranzabal, R. López-Fonseca, R. Ferret, J.A. González-MarcosDepartment of Chemical Engineering, Faculty of Sciences, Universidad del Paıs Vasco/EHU, P.O. Box 644, E-48080 Bilbao, Spain
Received 20 February 1999; received in revised form 12 July 1999; accepted 12 July 1999
Abstract
The complete catalytic oxidation of trichloroethylene (TCE) over alumina-supported noble metal catalysts (Pt and Pd) andin the presence of hydrogen-rich compounds, i.e. water, hexane and toluene was evaluated. Experiments were performed atconditions of lean TCE concentration (around 1000 ppm) in air, between 250 and 550◦C in a conventional fixed-bed reactor.Hexane and toluene were added to the feedstream in a concentration of around 1000 ppm and water concentration variedfrom 1000 to 15 000 ppm. TCE oxidation occurred faster in the presence of hexane and toluene over both catalysts. Overpalladium catalysts, water did not alter catalytic activity, whereas over platinum catalysts water enhanced TCE oxidationat low temperatures (<400◦C) but inhibited it at higher temperatures (>400◦C). Selectivity to HCl was much improved byfeeding water as a hydrogen-supplying reactant; 7500 ppm of water enhanced HCl outputs from 39.4 to 78.0% with Pd, andfrom 37.5 to 58.9% with Pt. Selectivities to C2Cl4, formed by chlorination of the feed, and Cl2 were greatly reduced. On theother hand water promoted complete oxidation of TCE to CO2, and thus reduced selectivity to CO. In the presence of hexaneand toluene, formation of HCl was also enhanced. Hexane showed higher inhibition ability than toluene over both catalystsfor the C2Cl4 and Cl2 formation. Unlike in the presence of water, selectivity to CO increased, as a consequence of partialoxidation of both hydrocarbons. ©2000 Elsevier Science B.V. All rights reserved.
Keywords:Catalytic oxidation; Selectivity; Trichloroethylene; Noble metal catalysts; VOC
1. Introduction
Chlorinated volatile organic compounds (CVOCs)are known to be hazardous to the environment andpublic health. Catalytic oxidation of CVOCs is cur-rently receiving increased attention to keep theiremissions in compliance with the increasingly strin-gent environmental regulations (US 1990 Clean AirAct), since chlorinated compounds, including toxic
∗ Corresponding author. Tel.: +34-94-6012-681; fax: +34-94-4648-500E-mail address:[email protected] (J.R. Gonzalez-Velasco)
by-products can be destroyed into CO2, H2O and HClat relatively low temperatures (<550◦C) comparedwith conventional thermal incineration.
Among the large number of CVOCs which aredischarged into the environment, those containingmore chlorine atoms than hydrogen atoms withintheir molecules, cannot be totally converted by air tothe most desirable HCl, which is easily scrubbed inan alkaline medium, and therefore even more toxicproducts such as Cl2 and COCl2 are formed. To im-prove selectivity to HCl, either a hydrogen-rich fuelor water vapour may be added to the feedstream.Indeed, in the real flue gases, CVOCs are present
0926-3373/00/$ – see front matter ©2000 Elsevier Science B.V. All rights reserved.PII: S0926-3373(99)00087-9

34 J.R. Gonzalez-Velasco et al. / Applied Catalysis B: Environmental 24 (2000) 33–43
in a matrix of non-chlorinated organic compounds(VOC) [1,2], where inhibition [1,3] or enhancement[4,5] of oxidation of a given compound may appear;i.e. the catalytic reaction of a compound in a mixturecannot be predicted solely from the behaviour of theindividual compound [1].
This work is a continuation of a previous paper [6],in which the activity and product distribution in theCVOCs oxidation catalysed by Pd and Pt ong-aluminawas reported. HCl, Cl2 and C2Cl4 were found to be thechlorine containing products in trichloroethylene cat-alytic oxidation [6–9]. The noble metals seemed to beresponsible for the formation of C2Cl4 by chlorinationof the adsorbed TCE, through metal (oxy)chlorides.In this context palladium showed higher chlorinationability than platinum [10] resulting in higher selectiv-ity to C2Cl4 in the former. CO was also detected atlow temperatures as a consequence of incompleteoxidation of CVOCs.
The objective of the present study is to evaluatethe effect of some hydrogen-rich compounds, namelywater, hexane and toluene, in the trichloroethylene(TCE) oxidation over supported low-loading platinumand palladium catalysts as a way of providing hydro-gen in order to convert all the chlorine to hydrogenchloride and to minimise undesirable by-product for-mation. TCE was chosen as a suitable hydrogen-leanmodel compound present in many off-gases, such asgroundwater stripping emissions, where TCE is ad-mixtured with hydrogen-rich hydrocarbons (aromat-ics, alkenes, alkanes, etc.) and high water vapour con-centration (0.8–3%) [11].
2. Experimental
SCM-129Xg-alumina spheres supplied by Rhône-Poulenc were calcined in air at 700◦C for 4 h andthen crushed and sieved to the desired particle size. Itscharacterisation led to the following properties: BETsurface area, 114 m2 g−1; pore volume, 0.5 cm3 g−1;average pore radius, 8.7 nm; predominant pore radius,8.6 nm; isoelectric point, 9.
The active phases (Pt and Pd) were incorporated byadsorption from aqueous solution of their respectivesalts — H2PtCl6·nH2O and PdCl2— using 40 cm3 ofsolution per gram of alumina. The nominal compo-sitions of the prepared catalysts were 0.5 wt%. After
drying for 1 h at 150◦C, final activation of the pre-cursors was made by calcining at 550◦C in air streamfor 4 h and reducing at 550◦C in a N2/H2 = 3/1 streamfor 2 additional hours. The final catalysts resulted in0.44 wt% Pt and 0.42 wt% Pd, measured by atomicabsorption spectroscopy (AAS); the metal dispersionwas determined by hydrogen chemisorption, resultingin 53 and 43% dispersion, respectively.
Oxidation reactions were carried out in a conven-tional fixed-bed flow reactor under atmospheric pres-sure. It consisted in a 12 mm i.d. stainless steel tubelocated inside an electrical furnace. At the lower partof the reactor, 0.85 g (around 2 cm3) of catalyst wereplaced on a plug of glass wool. In order to preheat thegas feedstream crushed quartz glass (10 cm3, 2 mmo.d.) was placed above the catalyst bed. The temper-ature at the inlet was controlled automatically with aK-type thermocouple.
The flow rate through the reactor was set at500 cm3 min−1 that produced a space velocity of15 000 h−1. The feedstream to the reactor wasprepared by delivering the liquid hydrocarbons(1000 ppmv TCE, 1000 ppm hexane and 1000 ppmtoluene) and water by syringe pumps into a dry,oil-free compressed air, which was purified using acommercial PSA drier and metered by a mass flowcontroller. The injection point was electrically heatedto ensure complete evaporation. Before entering thereactor, the feedstream went through a 2 dm3 staticmixer to dampen out possible concentration oscilla-tions associated with the running of the syringe pump.After the reactor teflon tubing was employed becauseof its inertness and insensitivity to corrosion by hy-drogen chloride. Negligible corrosion of the stainlesssteel reactor was expected above 200◦C [12].
A portion of the effluent stream was analysedon-line by a Hewlett Packard 5890 Series II gas chro-matograph equipped with an electron capture detector(ECD) and a thermal conductivity detector (TCD).CO and CO2 were separated by both a 13X molecularsieve column and a Hayesep N column interconnectedto a gas sampling loop by a single 10-port valve andwere analysed on the TCD. The concentration ofTCE as well as any other chlorinated hydrocarbonsformed in the reaction were determined on the ECDafter being separated in a HP-VOC column. Analy-sis of both Cl2 and HCl was performed by bubblingthe effluent stream through a 0.0125 N NaOH solu-

J.R. Gonzalez-Velasco et al. / Applied Catalysis B: Environmental 24 (2000) 33–43 35
tion [13]. Then, Cl2 concentration was determinedby titration with ferrous ammonium sulphate (FAS)using N,N-diethyl-p-phenylenediamine (DPD) as in-dicator [14]. The concentration of chloride ions inthe bubbled solution was determined by using an ionselective electrode.
The selectivity values for the reaction products re-ported in this paper have been calculated as
SC2Cl4 = 4C2Cl42Cl2 + HCl + 4C2Cl4
× 100 (1)
SCl2 = 2Cl22Cl2 + HCl + 4C2Cl4
× 100 (2)
SHCl = HCl
2Cl2 + HCl + 4C2Cl4× 100 (3)
SCO = CO
CO+ CO2× 100 (4)
where C2Cl4, Cl2, HCl and CO are the concentra-tions of tetrachloroethlylene, chlorine, hydrogen chlo-ride and carbon monoxide, respectively, in the effluentstream.
3. Results and discussion
3.1. TCE destruction activity
Fig. 1 shows the effect of water concentration onthe destruction of TCE over both 0.42 wt% Pd/Al2O3and 0.44 wt% Pt/Al2O3 catalysts expressed as conver-sion versus temperature. It must be noted that prior tothe analysis of the following results some experiments,specially for the conditions where conversion temper-ature curves are close between themselves, were madethree times in order to check experimental error, whichwas lower than 2% in all the temperature ranges, andeven lower than 0.1% for conversions higher than 99%.A ‘white test’ placing just 0.3–0.5 mm crushed quartzglass into the reactor was also carried out, in order tocheck whether some reaction under thermal combus-tion conditions could take place. It was found that ho-mogeneous reaction only occurred above 425◦C at alower extent than the catalytic reactions. These resultsare discussed elsewhere [6].
With Pd/Al2O3 (empty points), the reaction pro-ceeded to almost the same extent either in the ab-
Fig. 1. TCE oxidation light-off curves over 0.42 wt% Pd/Al2O3
and 0.44 wt% Pt/Al2O3 in the presence of water.
sence or presence of water, even when increasingits concentration 10 times. However, TCE oxidationlight-off curves over Pt/Al2O3 (solid points) showedan interesting feature: catalyst activity was enhancedby water at the lower temperatures (<400◦C); at400◦C conversion was nearly the same in dry andhumid streams and above 400◦C the presence of wa-ter seemed to inhibit the activity for TCE oxidation.Those enhancements and inhibitions were larger asthe water content in the feedstream was increased,pointing out that water played an important role inTCE destruction. This was confirmed by an additionalexperiment carried out in the absence of oxygen andin the presence of 1.5% water, which is referred to ashydrolysis in Fig. 1. The conversion values for thisrun were practically the same to those obtained inthe presence of oxygen, concluding that, at the con-ditions of our study, over Pt/Al2O3, oxidation is themajor pathway for TCE destruction in dry air and lowwater contents (1000 ppm), whereas hydrolysis is thefavoured pathway in excess of water (1.5%). Bondand Rosa-Calzadilla [15] reported that water, and notoxygen, was responsible for the conversion of CH2Cl2and CCl4 over Pt/g-alumina catalysts, through the alu-minium hydroxyl groups [16], CO and HCl being theonly detected products. Nevertheless, oxygen plays
36 J.R. Gonzalez-Velasco et al. / Applied Catalysis B: Environmental 24 (2000) 33–43
an important role in the complete oxidation of TCEto CO2, since in pure hydrolysis conditions (in theabsence of oxygen) TCE destruction showed muchhigher selectivity to CO in the overall studied rangeof temperature, whereas in the presence of oxygenselectivity to CO2 was promoted. Agarwal et al. [17]and Lester and Marinangeli [18] also concluded thatboth oxidation and hydrolysis accounted for CNCldestruction in the presence of water vapour, and Aidaet al. [19] reported that the oxidation of CH3Cl wasinitiated by the substitution of Cl by superficial OH,followed by hydrolysis of chlorinated alumina andthen, oxidation of CH3OH proceeding on the metalsites.
The degree of destruction for a VOC may dependon whether the compound is alone in the feed or aspart of a mixture of VOCs. Earlier studies have foundthat some mixture effect exists in the oxidation of hy-drocarbons, mostly as inhibition of the oxidation ofone hydrocarbon in the presence of others due to com-petition for adsorption sites. It is known that halo-genated VOCs are difficult to be destroyed over anoble metal catalyst supported on alumina [20] andthey also play an inhibiting effect in the oxidation ofnon-halogenated hydrocarbons. Cullis et al. [21] ob-served that methane oxidation was inhibited by thepresence of methylene chloride, although oxidationactivity and selectivity to CO2 and water were com-pletely restored when methylene chloride was elimi-nated from the feed.
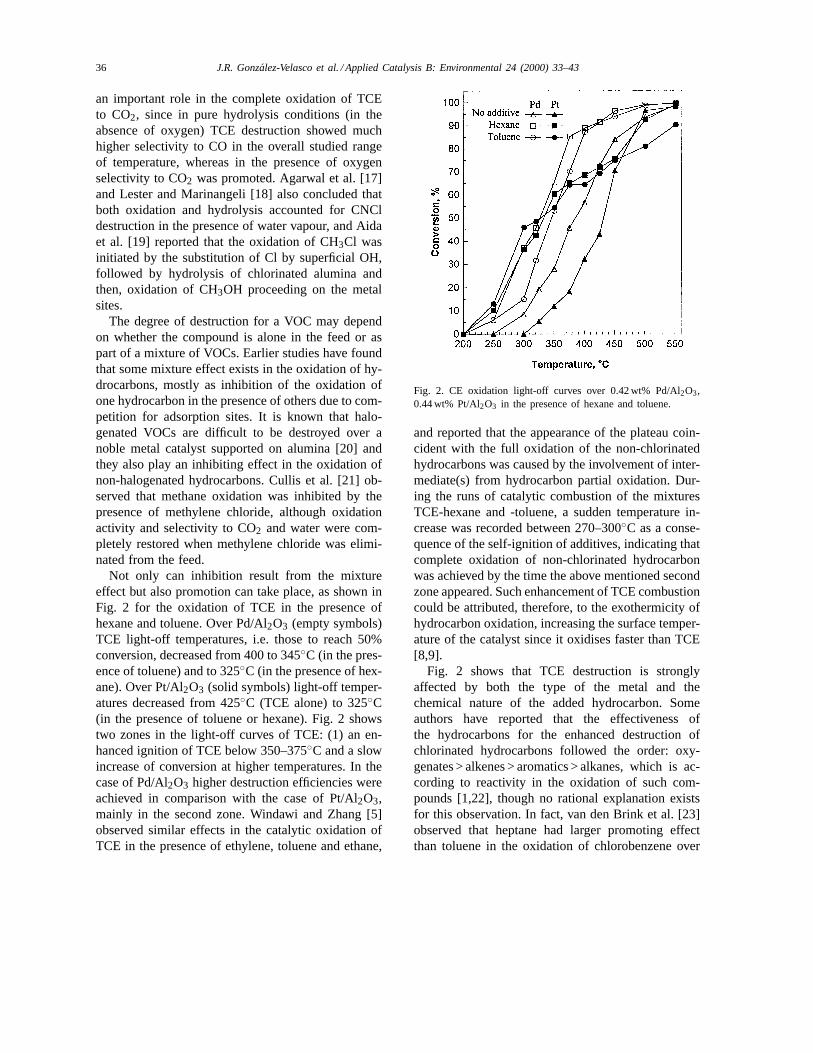
Not only can inhibition result from the mixtureeffect but also promotion can take place, as shown inFig. 2 for the oxidation of TCE in the presence ofhexane and toluene. Over Pd/Al2O3 (empty symbols)TCE light-off temperatures, i.e. those to reach 50%conversion, decreased from 400 to 345◦C (in the pres-ence of toluene) and to 325◦C (in the presence of hex-ane). Over Pt/Al2O3 (solid symbols) light-off temper-atures decreased from 425◦C (TCE alone) to 325◦C(in the presence of toluene or hexane). Fig. 2 showstwo zones in the light-off curves of TCE: (1) an en-hanced ignition of TCE below 350–375◦C and a slowincrease of conversion at higher temperatures. In thecase of Pd/Al2O3 higher destruction efficiencies wereachieved in comparison with the case of Pt/Al2O3,mainly in the second zone. Windawi and Zhang [5]observed similar effects in the catalytic oxidation ofTCE in the presence of ethylene, toluene and ethane,
Fig. 2. CE oxidation light-off curves over 0.42 wt% Pd/Al2O3,0.44 wt% Pt/Al2O3 in the presence of hexane and toluene.
and reported that the appearance of the plateau coin-cident with the full oxidation of the non-chlorinatedhydrocarbons was caused by the involvement of inter-mediate(s) from hydrocarbon partial oxidation. Dur-ing the runs of catalytic combustion of the mixturesTCE-hexane and -toluene, a sudden temperature in-crease was recorded between 270–300◦C as a conse-quence of the self-ignition of additives, indicating thatcomplete oxidation of non-chlorinated hydrocarbonwas achieved by the time the above mentioned secondzone appeared. Such enhancement of TCE combustioncould be attributed, therefore, to the exothermicity ofhydrocarbon oxidation, increasing the surface temper-ature of the catalyst since it oxidises faster than TCE[8,9].
Fig. 2 shows that TCE destruction is stronglyaffected by both the type of the metal and thechemical nature of the added hydrocarbon. Someauthors have reported that the effectiveness ofthe hydrocarbons for the enhanced destruction ofchlorinated hydrocarbons followed the order: oxy-genates > alkenes > aromatics > alkanes, which is ac-cording to reactivity in the oxidation of such com-pounds [1,22], though no rational explanation existsfor this observation. In fact, van den Brink et al. [23]observed that heptane had larger promoting effectthan toluene in the oxidation of chlorobenzene over

J.R. Gonzalez-Velasco et al. / Applied Catalysis B: Environmental 24 (2000) 33–43 37
a 2% Pt/g-alumina catalyst, in agreement with ourresults with hexane (the same molecule-type thanheptane) and toluene.
In addition to these changes in TCE destruction ac-tivity, the presence of water, hexane, and toluene inthe TCE feedstream also has an important effect onreaction product distribution, the production of CO2,H2O and HCl being desirable , but inhibition of CO,Cl2 and C2Cl4.
3.2. Effect of water on product distribution
Fig. 3 shows the effect of temperature and waterconcentration on the formation of C2Cl4. The amountof C2Cl4, under dry conditions, increased with temper-ature, reached a maximum at 450◦C and was reducedpractically to zero at 550◦C [6]. The C2Cl4 concentra-tion was three times higher over Pd/Al2O3 than overPt/Al2O3. In the presence of 1000 ppm of water C2Cl4formation was reduced by a factor of 4 over Pd/Al2O3and by a factor of 2 over Pt/Al2O3. The addition ofhigher water amounts (7500 and 15 000 ppm) led tosimilar C2Cl4 concentrations (around 25 and 15 ppm)on Pd/Al2O3 and a slightly higher concentrations overPt/Al2O3 (34 and 26 ppm).
Similar effects were found when measuring HCl/Cl2outlet concentration, as shown in Fig. 4. The presenceof water vapour in the feedstream greatly lowered theproduction of molecular chlorine, in favour of increas-ing HCl formation. In the runs over Pd/Al2O3, when1000 ppm water were added, chlorine production washalf of that produced under dry conditions and was10 times lower in excess of water (1.5%). Platinumshowed higher selectivity to chlorine, hence the effectof water was slightly lower. When the reaction pro-ceeded in the absence of oxygen and in the presenceof 1.5% water (referred to as hydrolysis), no chlorinewas observed, suggesting that Cl2 measured in thepresence of both water (1.5%) and oxygen (21%) isformed by the Deacon reaction:
2HCl + 12O2 ⇔ H2O + Cl2 (5)
in which HCl formation is thermodynamicallyfavoured with temperature. Nevertheless, the mea-sured Cl2 concentration at the reaction conditionsis significantly lower than that corresponding to thefull equilibrium [6], which is displaced by formation
Fig. 3. C2Cl4 concentration profiles in the presence of water.
of Cl2, because of the high concentration of oxygen(20%) compared to that of water (1.5%). The increasein Cl2 concentration shown in Fig. 4 is explainedbecause the Deacon reaction rate is enhanced withtemperature [24,25].
The improved HCl selectivity in the presence of wa-ter can be explained as an interaction between waterand the chlorine atoms liberated from the TCE oxida-tion on Pd/Al2O3 and Pt/Al2O3 catalysts. The adsorp-tion and subsequent oxidative decomposition of TCEon palladium and platinum involves C–Cl bond disso-

38 J.R. Gonzalez-Velasco et al. / Applied Catalysis B: Environmental 24 (2000) 33–43
Fig. 4. HCl and Cl2 concentration profiles in the presence of water.
ciation, by a chemical interaction of the halogen withthe precious metal and the support, resulting in pre-cious metal (oxide) chloride species, [PtIV (Ox)Cly] , onalumina [26–28], and aluminium chloride [7,29]. Themetal (oxide) chloride species can directly decomposeto molecular chlorine (Cl2) and also react with the feedTCE by transferring chlorine (Cl2) to the double bond.The intermediate pentachloroethane is spontaneouslydehydrochlorinated by HCl elimination, resulting inthe more stable tetrachlorethylene. The chlorination
of TCE is catalysed by the metal [6,30], especially bypalladium, which showed higher chlorination power.When water is present in the feed, it is dissociated overthe platinum or palladium surface precovered by oxy-gen, into H+ and OH− species. The protons recom-bine with chloride interacting with the metal, whichaids in desorbing HCl from the catalyst surface, andthus it reduces the formation of Cl2 (Fig. 4, solid sym-bols) and C2Cl4 (Fig. 3) and improves HCl selectivity(Fig. 4, empty symbols).

J.R. Gonzalez-Velasco et al. / Applied Catalysis B: Environmental 24 (2000) 33–43 39
Fig. 5. CO concentration profiles in the presence of water.
The interaction of chlorine atoms with alumina sup-port was also reported by Bickle [7] and Castro [29].The interaction with the metal in the form of Pt/Pdchlorides was observed by Forger and Jaeger [26] andMiyake and Hanaya [10], whereas Pt/Pd oxychlorideswere observed by Lieske et al. [27,28] at temperaturesup to 600◦C, using TPR and UV-Vis spectroscopy,and by Barbier et al. [31] for high Cl/Pt (≥6) ratio un-der an oxidising atmosphere of up to 730◦C. Miyakeand Hanaya [10] also reported higher activity for pal-ladium than platinum in the chlorination of propene,which agrees with the observed higher selectivity ofpalladium to tetrachloroethylene, by chlorination ofthe feed.
The role of water in removing chlorine from thesurface of noble metal catalysts was also reported byCastro et al. [29] and Marecot et al. [32], conclud-ing that the water content in the atmosphere and tem-perature were the main factors controlling the chlo-rine loss rate. Likewise, van den Brink et al. [33] ob-served lower formation of polychlorinated benzenesin the catalytic combustion of chlorobenzene over aPt/g-alumina catalyst in the presence of water.
The presence of CO in the TCE oxidation outletstream is a consequence of the incomplete combus-tion of TCE [6]. Its formation followed a similar trendto C2Cl4, but peaked at 400◦C (Fig. 5), afterwards
Fig. 6. C2Cl4 concentration profiles in the presence of hexane andtoluene.
decreased and finally at 500◦C increased again as aresult of C2Cl4 decomposition to CO or gas phaseTCE reaction, since homogeneous reaction was de-tected above 450◦C, as reported elsewhere [6]. OverPd/Al2O3 (empty symbols) the amount of CO de-creased by a factor of 4 between 350 and 400◦C when1000 ppm of water were added, and with higher wa-ter concentration CO was not formed. Over Pt/Al2O3under both dry and humid conditions CO was notdetected.

40 J.R. Gonzalez-Velasco et al. / Applied Catalysis B: Environmental 24 (2000) 33–43
Fig. 7. HCl and Cl2 concentration profiles in the presence of hexane and toluene.
Lower selectivity to CO is also associated with wa-ter dissociation on the surface metal (Pt, Pd), so thatthe resulting OH− species promotes CO oxidation toCO2 by the water gas shift reaction (WGSR).
This statement agrees with that reported by Herasand Viscido [34] when water is adsorbed overoxygen-precovered noble metals and with Bose andSenkan [35] in gas phase TCE oxidation. Alterna-tively, as reported by Narayanan and Greene [36],surface gas reaction of Cl anions with water resultsin HCl and OH−. Any of these two mechanisms ex-
plains our results, and also those observed by otherauthors, e.g. Agarwal et al. [17] who reported a dropin CO selectivity from 60 to 0% when adding 7800ppm water in the catalytic oxidation of cyanogenchloride over 2.5% Pt/a-alumina.
3.3. Effect of hydrocarbons on product distribution
Without water but in the presence of 1000 ppm hex-ane and toluene, as additional hydrogen source, C2Cl4
J.R. Gonzalez-Velasco et al. / Applied Catalysis B: Environmental 24 (2000) 33–43 41
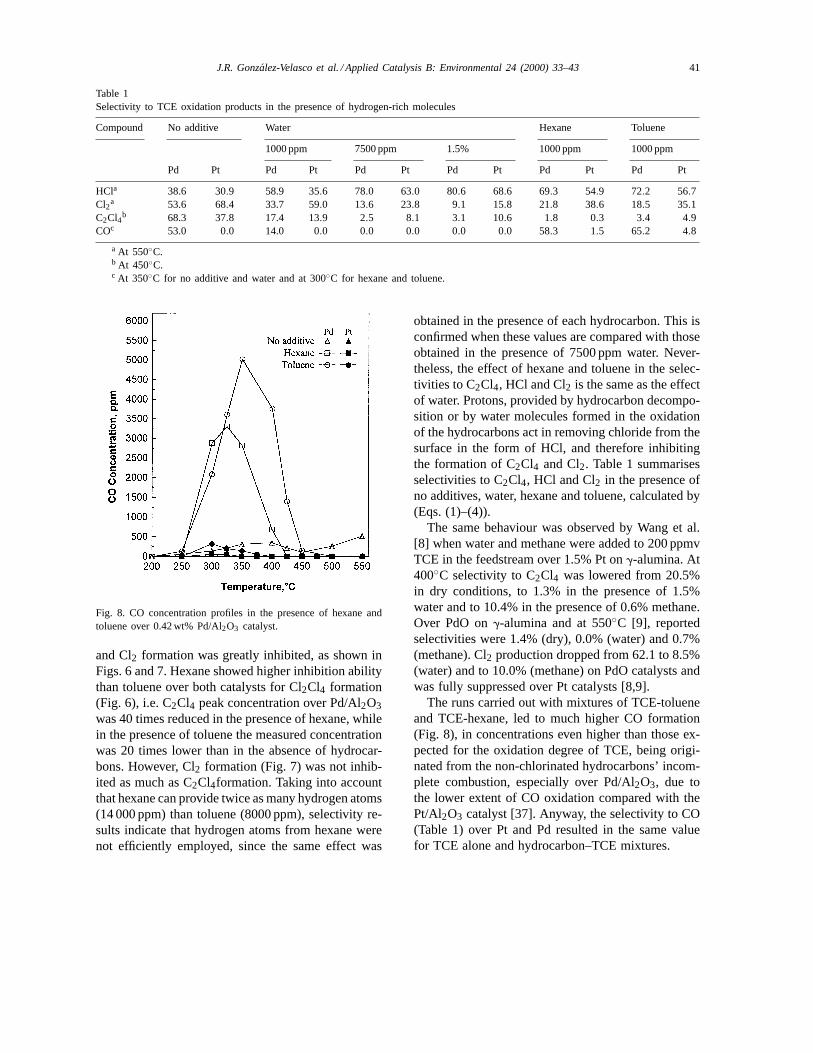
Table 1Selectivity to TCE oxidation products in the presence of hydrogen-rich molecules
Compound No additive Water Hexane Toluene
1000 ppm 7500 ppm 1.5% 1000 ppm 1000 ppm
Pd Pt Pd Pt Pd Pt Pd Pt Pd Pt Pd Pt
HCla 38.6 30.9 58.9 35.6 78.0 63.0 80.6 68.6 69.3 54.9 72.2 56.7Cl2a 53.6 68.4 33.7 59.0 13.6 23.8 9.1 15.8 21.8 38.6 18.5 35.1C2Cl4b 68.3 37.8 17.4 13.9 2.5 8.1 3.1 10.6 1.8 0.3 3.4 4.9COc 53.0 0.0 14.0 0.0 0.0 0.0 0.0 0.0 58.3 1.5 65.2 4.8
a At 550◦C.b At 450◦C.c At 350◦C for no additive and water and at 300◦C for hexane and toluene.
Fig. 8. CO concentration profiles in the presence of hexane andtoluene over 0.42 wt% Pd/Al2O3 catalyst.
and Cl2 formation was greatly inhibited, as shown inFigs. 6 and 7. Hexane showed higher inhibition abilitythan toluene over both catalysts for Cl2Cl4 formation(Fig. 6), i.e. C2Cl4 peak concentration over Pd/Al2O3was 40 times reduced in the presence of hexane, whilein the presence of toluene the measured concentrationwas 20 times lower than in the absence of hydrocar-bons. However, Cl2 formation (Fig. 7) was not inhib-ited as much as C2Cl4formation. Taking into accountthat hexane can provide twice as many hydrogen atoms(14 000 ppm) than toluene (8000 ppm), selectivity re-sults indicate that hydrogen atoms from hexane werenot efficiently employed, since the same effect was
obtained in the presence of each hydrocarbon. This isconfirmed when these values are compared with thoseobtained in the presence of 7500 ppm water. Never-theless, the effect of hexane and toluene in the selec-tivities to C2Cl4, HCl and Cl2 is the same as the effectof water. Protons, provided by hydrocarbon decompo-sition or by water molecules formed in the oxidationof the hydrocarbons act in removing chloride from thesurface in the form of HCl, and therefore inhibitingthe formation of C2Cl4 and Cl2. Table 1 summarisesselectivities to C2Cl4, HCl and Cl2 in the presence ofno additives, water, hexane and toluene, calculated by(Eqs. (1)–(4)).
The same behaviour was observed by Wang et al.[8] when water and methane were added to 200 ppmvTCE in the feedstream over 1.5% Pt ong-alumina. At400◦C selectivity to C2Cl4 was lowered from 20.5%in dry conditions, to 1.3% in the presence of 1.5%water and to 10.4% in the presence of 0.6% methane.Over PdO ong-alumina and at 550◦C [9], reportedselectivities were 1.4% (dry), 0.0% (water) and 0.7%(methane). Cl2 production dropped from 62.1 to 8.5%(water) and to 10.0% (methane) on PdO catalysts andwas fully suppressed over Pt catalysts [8,9].
The runs carried out with mixtures of TCE-tolueneand TCE-hexane, led to much higher CO formation(Fig. 8), in concentrations even higher than those ex-pected for the oxidation degree of TCE, being origi-nated from the non-chlorinated hydrocarbons’ incom-plete combustion, especially over Pd/Al2O3, due tothe lower extent of CO oxidation compared with thePt/Al2O3 catalyst [37]. Anyway, the selectivity to CO(Table 1) over Pt and Pd resulted in the same valuefor TCE alone and hydrocarbon–TCE mixtures.

42 J.R. Gonzalez-Velasco et al. / Applied Catalysis B: Environmental 24 (2000) 33–43
Poor chlorine balances have been specially notedin HCl production, which resulted below that ex-pected from reaction stoichiometry. As no reactionbetween HCl and the reactor and teflon was checkedat the operational conditions, the poor balances canonly be explained by some interaction between HCland the catalyst. We must mention that this work hasbeen performed with fresh catalysts and the activityof long-term exposed catalysts was not examined.Though it is well known that Al-containing catalystsare gradually converted into AlCl3 during halocar-bon destruction by HCl under moisture conditions[38–41] and that the catalyst temperature may causevapourisation of AlCl3 (boiling point, 178◦C). Thispoint, that deserves further attention in the future,could explain the fact that in all the runs carriedout, chlorine balance decreased from 80 to 60% asreaction temperature increased.
4. Conclusions
The oxidative destruction of TCE admixtured withwater, hexane and toluene over 0.42 wt% Pd/Al2O3and 0.44 wt% Pt/Al2O3 catalysts has been experimen-tally studied under oxygen-rich conditions.
The presence of water in the feedstream hardlyinfluences the catalytic destruction of TCE overPd/Al2O3. Over Pt/Al2O3 TCE destruction in thepresence of water is enhanced at low tempera-tures but inhibited at higher temperatures. Com-parison of destruction data in the presence and ab-sence of oxygen in the environment suggests thatTCE destruction occurred via both hydrolysis andoxidation.
The light-off temperatures, at which 50% of TCEis converted, are shifted to lower values in the pres-ence of non-halogenated VOC admixtures with TCEin the feedstream. The magnitude of this shift dependson the VOC nature: hexane produced a stronger ef-fect towards the enhancement of TCE conversion thantoluene, especially over Pd/Al2O3.
HCl selectivity was much improved with the addi-tion of hydrogen-supplying compounds to the feed-stream by removing chlorine attached to the catalystsurface. Increasing water concentration resulted inlower selectivity to Cl2 and C2Cl4. Both hexane andtoluene showed similar effectiveness in promoting
selectivity to HCl, and hence inhibiting Cl2 and C2Cl4formation.
Water was also found to promote complete oxida-tion of CO, by the water-shift reaction, resulting in100% selectivity to CO2. Selectivity to CO was notinfluenced by the presence of non-halogenated VOC,as a consequence of their partial oxidation to CO. Inany case, at temperatures around 550◦C, where TCEwas almost completely destroyed, selectivity to CO2was 100%.
Acknowledgements
The authors wish to thank Universidad del PaısVasco/EHU (UPV069.310-0111/94) Gobierno Vasco(GV069.310-0111/94) and Ministerio de Educación yCultura (QUI96-0471) for the financial support. Oneof the authors (A.A.) acknowledges Gobierno Vascofor the grant BF194.010.
References
[1] B.A. Tichenor, M.A. Palazzolo, Environ. Progress 6 (1987)172.
[2] S.K. Agarwal, J.J. Spivey, J.B. Butt, Appl. Catal. A 82 (1992)259.
[3] S.K. Gangwal, M.E. Mullins, J.J. Spivey, P.R. Caffrey, B.A.Tichenor, Appl. Catal. A 36 (1988) 231.
[4] B. Mendyka, A. Musialik-Piotrowska, K. Syczewska, Catal.Today 11 (1992) 597.
[5] H. Windawi, Z.C. Zhang, Catal. Today 30 (1996) 99.[6] J.R. González-Velasco, A. Aranzabal, J.I. Gutiérrez Ortiz,
R. López-Fonseca, M.A. Gutiérrez-Ortiz, Appl. Catal. B 19(1998) 189.
[7] G.M. Bickle, T. Suzuki, Y. Mitarai, Appl. Catal. B 4 (1994)141.
[8] Y. Wang, H. Shaw, R.J. Farrauto, Catalytic oxidation of traceconcentrations of trichloroethylene over 1.5% platinum ong-alumina, in: Catalytic Control of Air Pollution, Mobile andStationary Sources; ACS Symposium Series 495; AmericanChemical Society, Washington DC, 1992, pp. 125–140.
[9] T-Ch. Yu, H. Shaw, R.J. Farrauto, Catalytic oxidation oftrichloroethylene over PdO catalyst ong-Al2O3, in: CatalyticControl of Air Pollution, Mobile and Stationary Sources;ACS Symposium Series 495; American Chemical Society,Washington DC, 1992, pp. 141–152.
[10] T. Miyake, M. Hanaya, Appl. Catal. A 121 (1995) L13.[11] M. Kosusko, M.E. Mullins, K. Ramanathan, T.N. Rogers,
Environ. Progress 7 (1988) 136.[12] C.M. Schillmoller, Chem. Eng. 39 (1980) 161.

J.R. Gonzalez-Velasco et al. / Applied Catalysis B: Environmental 24 (2000) 33–43 43
[13] Guıa para la valoración de la contaminación del aire(Colección Senda Ambiental, vol. 2), Ministerio de Sanidady Consumo, Madrid, 1985 (in Spanish).
[14] A.E. Greenberg, L.S. Clesceri, A.D. Eaton (Eds.), StandardMethods for the Examination of Water and Wastewater,American Public Health Association 18th edn., WashingtonDC, 1992, p. 4.43.
[15] G.C. Bond, F. Rosa Calzadilla, Catal. Letters 39 (1996) 261.[16] J. Haber, T. Machej, M. Derewinski, J. Janik, J. Krysciak, H.
Sadowska, Zeitschrift. Phys. Chem. 197 (1996) 97.[17] S.K. Agarwal, J.J. Spivey, D.E. Tevault, Appl. Catal. B 5
(1995) 389.[18] G.R. Lester, R.E. Marinangeli, Development and characteri-
zation of oxidation catalyst for air purification, Reportto the U.S. Army by Signal Research Center Inc.CRDEC-CR-87050, 1987.
[19] T. Aida, R. Higuchi, H. Niiyama, Chem. Lett. (1990) 2247.[20] J.J. Spivey, Ind. Eng. Chem. Res. 26 (1987) 2165.[21] C.F. Cullis, D.E. Keene, D.L. Trimm, J. Catal. 19 (1970) 378.[22] J. Hermia, S. Vigneron, Catal. Today 17 (1993) 349.[23] R.W. Van den Brink, P. Mulder, R. Louw, Catalytic
combustion of chlorobenzene in absence and presence ofhydrocarbons, in: Abstracts of 2nd World Congress onEnvironmental Catalysis, AIChE, Miami Beach, FL, 15–20November 1998.
[24] S. Chatterjee, H.L. Greene, Appl. Catal. 98 (1993) 139.[25] B. Ramachandran, H.L. Greene, S. Chatterjee, Appl. Catal.
B 8 (1996) 157.[26] K. Foger, H. Jaeger, J. Catal. 92 (1985) 64.
[27] H. Lieske, G. Lietz, H. Spindler, J. Voelter, J. Catal. 81 (1983)8–16.
[28] G. Lietz, H. Lieske, H. Spindler, W. Hanke, J. Voelter, J.Catal. 81 (1983) 17.
[29] A.A. Castro, O.A. Scelza, G.T. Baronetti, M.A. Fritzler, J.M.Parera, Appl. Catal. 6 (1983) 347.
[30] L. Becker, H. Föster, J. Catal. 170 (1997) 200.[31] J. Barbier, D. Bahloul, P. Marecot, Catal. Lett. 8 (1991) 327.[32] P. Marecot, A. Falche, B. Kellali, G. Mabilon, M. Prigent, J.
Barbier, Appl. Catal. B 3 (1994) 283.[33] R.W. Van den Brink, R. Louw, P. Mulder, Appl. Catal. B 16
(1998) 219.[34] J.M. Heras, L. Viscido, Catal. Rev.–Sci. Eng. 30 (1988) 281.[35] N. Bose, S.M. Senkan, Combust. Sci. Technol. 35 (1983) 187.[36] S. Narayanan, H.L. Greene, Deactivation by H2S of Cr2O3
emission control catalyst for chlorinated VOC destruction,Technical Report Data for Presentation at 83rd AnnualMeeting, AWMA, Pittsburg, PA, 24–29 June 1990.
[37] G.W. Cordonna, M. Kosanovich, E.R. Becker, PlatinumMetals Rev. 32 (1989) 46.
[38] M.W.M. Hisham, S.W. Benson, J. Phys. Chem. 99 (1995)6194.
[39] M. Tajima, M. Niwa, Y. Fujji, Y. Koinuma, K. Aizawa,S. Kashiyama, S. Kobayashi, K. Mizuno, H. Ohûchi, Appl.Catal. B 12 (1997) 263.
[40] S. Karmakar, H.L. Greene, J. Catal. 148 (1994) 524.[41] M. Briend-Faure, O. Cornu, D. Delafosse, R. Monque, M.J.
Pelter, Appl. Catal. 38 (1988) 71.




















