
Energy balance regulation by thyroid hormones at central level
10
Energy balance regulation by thyroid hormones at central level Miguel Lo ´ pez 1, 2 , Clara V. Alvarez 1 , Rube ´n Nogueiras 1, 2 , and Carlos Die ´ guez 1, 2 1 Department of Physiology, CIMUS, University of Santiago de Compostela-Instituto de Investigacio ´ n Sanitaria, Santiago de Compostela, 15782, Spain 2 CIBER Fisiopatologı´a de la Obesidad y Nutricio ´ n (CIBERobn), 15706, Santiago de Compostela, Spain Classically, medical textbooks taught that most effects of thyroid hormones (THs) on energy homeostasis are directly exerted in peripheral tissues. However, current evidence is changing (and challenging) our perspective about the role of THs from a ‘peripheral’ to a ‘central’ vision, implying that they affect food intake, energy expenditure, and metabolism by acting, to a large ex- tent, at the central level. Interestingly, effects of THs are interrelated with global energy sensors in the central nervous system (CNS), such as uncoupling protein 2 (UCP2), AMP-activated protein kinase (AMPK; the ‘AMPK–BAT axis’), and mechanistic target of rapamycin (mTOR). Here, we review what is currently known about THs and their regulation of energy balance and metabo- lism in both peripheral and central tissues. The ‘classical view’ of thyroid hormones as modulators of energy balance Thyroid hormones (THs; Box 1) have been known to regu- late basal metabolic rate for more than a century and are now recognized to control a bulk of physiological processes, such as growth, development, and metabolic rate [1–4]. Dysregulation of the thyroid axis leads to marked altera- tions in energy balance, and its potential as a target in the treatment of obesity has been known for decades but treatment efforts have been hampered by side effects [5–8]. The homeostatic significance of THs in energy metabo- lism is exemplified in patients with thyroid dysfunction that results in hyperthyroidism or hypothyroidism. Hyper- thyroidism is a clinical syndrome in which overactive cells within the thyroid gland produce large amounts of THs, namely triiodothyronine (T3) and L-thyroxine (T4), with a resulting excess of circulating free THs and increased metabolic rate. Notably, up to 85% of patients with hyper- thyroidism exhibit weight loss despite increased food in- take, both at mealtimes and between meals, with energy intake failing to meet the increased caloric demands asso- ciated with increased energy expenditure [1,9]. Hypothy- roidism, by contrast, is associated with decreased metabolic rate, yielding weight gain despite reduced food intake [10,11]. Typically, most of these effects have been attributed to the direct actions of THs on metabolically active tissues such as the liver, white and brown adipose tissue (WAT and BAT), heart, and skeletal muscle [1,12,13]. In fact, different molecular pathways have been identified in these tissues that can explain the observed effects at the molecular level, and this evidence is pre- sented below. Peripheral effects of thyroid hormones on thermogenesis Homeotherm (‘warm-blooded’) species generate more heat than poikilotherm (‘cold-blooded’) species, both due to a more active metabolism and lower thermodynamic effi- ciency [14]. These two differences largely depend on THs. The effect of TH is twofold, by promoting the genera- tion of energy and reducing thermodynamic efficiency, leading to heat production and increased body tempera- ture [1,15]. Thus, THs are critical for obligatory thermo- genesis, that is, the heat production automatically caused by the metabolic rate [1,15]. In a thermoneutral environment, obligatory thermogene- sis can maintain body temperature without the participation of any other temperature homeostatic (thermoregulatory) mechanism [1,15,16]. The lowest ambient temperature at which this happens is called the thermoneutrality tempera- ture. Smaller species, such as rodents, as a consequence of their higher surface area-to-volume ratio (which causes them to lose more heat), have a higher thermoneutrality tempera- ture (30 8C in mice and 28 8C in rats) than larger species, such as humans (23 8C) [1,15,16]. When ambient tempera- ture falls below thermoneutrality, the immediate response in mammals is to activate heat-saving mechanisms, such as vasoconstriction, piloerection, rounded positions, and mobil- ity reduction. The heat-saving potential of these strategies is very limited, and additional thermogenic pathways are quickly activated to produce heat ‘on demand’ through a process termed facultative or adaptive thermogenesis [1,15,16]. Shivering is the earliest and most primitive re- sponse to cold. However, homeotherm species have devel- oped more efficient and long-term mechanisms of non- shivering facultative thermogenesis, which allows for the use of metabolic mechanisms to produce heat. The site and mechanism for facultative thermogenesis diverges in the two classes of homoeothermic animals, birds and mammals. In birds, the main site for facultative thermogenesis is skeletal muscle [17], whereas in mammals, the major site is BAT [15,16,18]. Review 1471-4914/$ – see front matter ß 2013 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.molmed.2013.04.004 Corresponding author: Lo ´pez, M. ([email protected]). Keywords: thyroid hormones; central nervous system; hypothalamus; energy balance; lipid metabolism; glucose homeostasis. 418 Trends in Molecular Medicine July 2013, Vol. 19, No. 7
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Energy balance regulation by thyroid hormones at central level

Energy balance regulation by thyroidhormones at central levelMiguel Lopez1,2, Clara V. Alvarez1, Ruben Nogueiras1,2, and Carlos Dieguez1,2
1 Department of Physiology, CIMUS, University of Santiago de Compostela-Instituto de Investigacio n Sanitaria, Santiago de
Compostela, 15782, Spain2 CIBER Fisiopatologıa de la Obesidad y Nutricio n (CIBERobn), 15706, Santiago de Compostela, Spain
Review
Classically, medical textbooks taught that most effectsof thyroid hormones (THs) on energy homeostasis aredirectly exerted in peripheral tissues. However, currentevidence is changing (and challenging) our perspectiveabout the role of THs from a ‘peripheral’ to a ‘central’vision, implying that they affect food intake, energyexpenditure, and metabolism by acting, to a large ex-tent, at the central level. Interestingly, effects of THs areinterrelated with global energy sensors in the centralnervous system (CNS), such as uncoupling protein 2(UCP2), AMP-activated protein kinase (AMPK; the‘AMPK–BAT axis’), and mechanistic target of rapamycin(mTOR). Here, we review what is currently known aboutTHs and their regulation of energy balance and metabo-lism in both peripheral and central tissues.
The ‘classical view’ of thyroid hormones as modulatorsof energy balanceThyroid hormones (THs; Box 1) have been known to regu-late basal metabolic rate for more than a century and arenow recognized to control a bulk of physiological processes,such as growth, development, and metabolic rate [1–4].Dysregulation of the thyroid axis leads to marked altera-tions in energy balance, and its potential as a target in thetreatment of obesity has been known for decades buttreatment efforts have been hampered by side effects [5–8].
The homeostatic significance of THs in energy metabo-lism is exemplified in patients with thyroid dysfunctionthat results in hyperthyroidism or hypothyroidism. Hyper-thyroidism is a clinical syndrome in which overactive cellswithin the thyroid gland produce large amounts of THs,namely triiodothyronine (T3) and L-thyroxine (T4), with aresulting excess of circulating free THs and increasedmetabolic rate. Notably, up to 85% of patients with hyper-thyroidism exhibit weight loss despite increased food in-take, both at mealtimes and between meals, with energyintake failing to meet the increased caloric demands asso-ciated with increased energy expenditure [1,9]. Hypothy-roidism, by contrast, is associated with decreasedmetabolic rate, yielding weight gain despite reduced foodintake [10,11]. Typically, most of these effects have beenattributed to the direct actions of THs on metabolically
1471-4914/$ – see front matter
� 2013 Elsevier Ltd. All rights reserved. http://dx.doi.org/10.1016/j.molmed.2013.04.004
Corresponding author: Lopez, M. ([email protected]).Keywords: thyroid hormones; central nervous system; hypothalamus; energy balance;lipid metabolism; glucose homeostasis.
418 Trends in Molecular Medicine July 2013, Vol. 19, No. 7
active tissues such as the liver, white and brown adiposetissue (WAT and BAT), heart, and skeletal muscle[1,12,13]. In fact, different molecular pathways have beenidentified in these tissues that can explain the observedeffects at the molecular level, and this evidence is pre-sented below.
Peripheral effects of thyroid hormones onthermogenesisHomeotherm (‘warm-blooded’) species generate more heatthan poikilotherm (‘cold-blooded’) species, both due to amore active metabolism and lower thermodynamic effi-ciency [14]. These two differences largely depend onTHs. The effect of TH is twofold, by promoting the genera-tion of energy and reducing thermodynamic efficiency,leading to heat production and increased body tempera-ture [1,15]. Thus, THs are critical for obligatory thermo-genesis, that is, the heat production automatically causedby the metabolic rate [1,15].
In a thermoneutral environment, obligatory thermogene-sis can maintain body temperature without the participationof any other temperature homeostatic (thermoregulatory)mechanism [1,15,16]. The lowest ambient temperature atwhich this happens is called the thermoneutrality tempera-ture. Smaller species, such as rodents, as a consequence oftheir higher surface area-to-volume ratio (which causes themto lose more heat), have a higher thermoneutrality tempera-ture (30 8C in mice and 28 8C in rats) than larger species,such as humans (23 8C) [1,15,16]. When ambient tempera-ture falls below thermoneutrality, the immediate response inmammals is to activate heat-saving mechanisms, such asvasoconstriction, piloerection, rounded positions, and mobil-ity reduction. The heat-saving potential of these strategies isvery limited, and additional thermogenic pathways arequickly activated to produce heat ‘on demand’ through aprocess termed facultative or adaptive thermogenesis[1,15,16]. Shivering is the earliest and most primitive re-sponse to cold. However, homeotherm species have devel-oped more efficient and long-term mechanisms of non-shivering facultative thermogenesis, which allows for theuse of metabolic mechanisms to produce heat. The site andmechanism for facultative thermogenesis diverges in the twoclasses of homoeothermic animals, birds and mammals. Inbirds, the main site for facultative thermogenesis is skeletalmuscle [17], whereas in mammals, the major site is BAT[15,16,18].

Box 1. Molecular aspects of thyroid hormone actions
The thyroid gland mainly produces thyroxine (3,30,5,50-tetraio-
dothyronine or T4), which has low biological activity. Intracellular
removal of an outer-ring iodine produces 3,30,5-triiodothyronine
(T3). This critical step in TH action is catalyzed by type 1 and type 2
iodothyronine deiodinases (D1 and D2) and is an essential step in
modulating TH action because T3 possesses a 100-fold higher
affinity for the TH receptor than T4 [3,4,77]. By contrast, type 3
deiodinase (D3) inactivates TH by converting T3 into 3,30-diiodo-L-
thyronine (T2) and T4 into 3,30,50-triiodothyronine (reverse T3 or rT3)
[3,4,77]. D1 is also able to deiodinate both T3 and rT3 into 3,5-T2 or
30,50-T2, respectively [3,4,77]. THs exert their major effects through
nuclear hormone receptors, encoded by two genes: the TR a and b
genes [3]. The transcripts from the TRb gene produce three (TRb1–3)
ligand-binding proteins (nuclear hormone receptors) with a con-
served C-terminal region including DNA and ligand-binding do-
mains but with different N-terminal portions. Regarding the TRa
gene, the situation is more complex. Just one of the two major
product genes, TRa1, gives rise to a ligand receptor; the other three
main isoforms, namely TRa2, TRa3, and TRa4, do not bind to the
ligand but maintain DNA-binding capabilities; their in vivo function
is unclear [3,4,77]. Although all receptors are well known at the
molecular level, convincing evidence for specific functions of each
receptor isoform is lacking. The TR a and b genes are differentially
expressed in a tissue- and temporal-specific pattern. However,
isoform-specific knockout mouse models indicate that receptors can
replace each other in many functions while having few specific
functions [3]. Many of the well-known effects of THs are mediated by
transcriptional regulation of target genes by nuclear TRs [3,4].
Alternative (non-transcriptional or non-genomic) regulatory me-
chanisms have been proposed for some of the metabolic effects of
THs [3,4,77].
Box 2. The hypothalamus, a key modulator of energy
balance and peripheral metabolism
Investigations performed during the past two decades have shown
the molecular and cellular mechanism through which the hypotha-
lamus, a brain region located below the thalamus, regulates an
enormous number of homeostatic functions; among them, regula-
tion of endocrine axes, reproductive function, and energy balance
are of particular importance. The hypothalamus is organized in
anatomically discrete neuronal clusters (nuclei) that form intercon-
nected neuronal circuits via axonal projections. The ARC, PVH,
DMH, and VMH nuclei, as well as the LHA are among the most
relevant hypothalamic sites modulating energy homeostasis (see
Figure 2 in main text) [81–84]. These nuclei receive multiple inputs of
information as diverse as the sensory experience of eating, the
process of ingestion, absorption, metabolism, and levels of energy
storage. The hypothalamus alters the expression of specific signals
(i.e., hypothalamic neuropeptides) with resultant adjustments in
energy balance by integrating afferent signals such as glucose,
amino acids, and lipids, as well as hormones such as leptin, ghrelin,
adiponectin (ADPN), resistin (RSTN), glucagon-like peptide-1 (GLP-
1), insulin, estrogens, and THs [81–84]. The ARC is considered as the
‘master hypothalamic center’ for food intake control. Two distinct
neuronal populations in the ARC integrate those peripheral signals.
One set of neurons express the orexigenic (feeding promoters)
neuropeptides AgRP and NPY. A second population of neurons into
the ARC produces the anorexigenic (feeding inhibitors) products of
POMC, the precursor of alpha-melanocyte stimulating hormone (a-
MSH) and CART. Other hypothalamic sites, such as the LHA express
the orexigenic neuropeptides MCH and OXs. Furthermore, recent
evidence has demonstrated that the hypothalamus plays a major
role major role in the regulation of energy expenditure and also in
the modulation of peripheral glucose and lipid metabolism (see
Figure 2 in main text), through ANS, both the SNA and PSNS. Thus,
the hypothalamus integrates peripheral and central signaling and
modulates: (i) hepatic insulin sensitivity by controlling insulin action
[70]; (ii) the fat depots in the WAT by controlling adipogenesis and
also the storage or oxidation of fatty acids in the adipocytes [69,72];
(iii) fatty acid oxidation in skeletal muscle [85]; and finally (iv) energy
expenditure by modulating the thermogenic program in the BAT
[21,39].
Review Trends in Molecular Medicine July 2013, Vol. 19, No. 7
In muscle, THs increase ATP consumption to maintainion gradients across the cell membranes, such as Na+, K+
[1,19]. This is thought to be largely due to the fact that THscan stimulate both the expression of Na+–K+-ATPase[1,19], as well as its activity by promoting the entranceof Na+ into the cells or the exit of K+ outside the cells,requiring Na+–K+-ATPase activity to restate the gradientsof these two ions through the cell membrane, which leadsto ATP consumption [1,19]. Moreover, THs increase ATPconsumption by acting on the Ca2+ gradient between thesarcoplasmic reticulum and the cytoplasm [20,21]. Thisaction is due to the activity of the sarcoplasmic/endoplas-mic reticulum Ca2+-dependent ATPase (SERCA) [22,23].
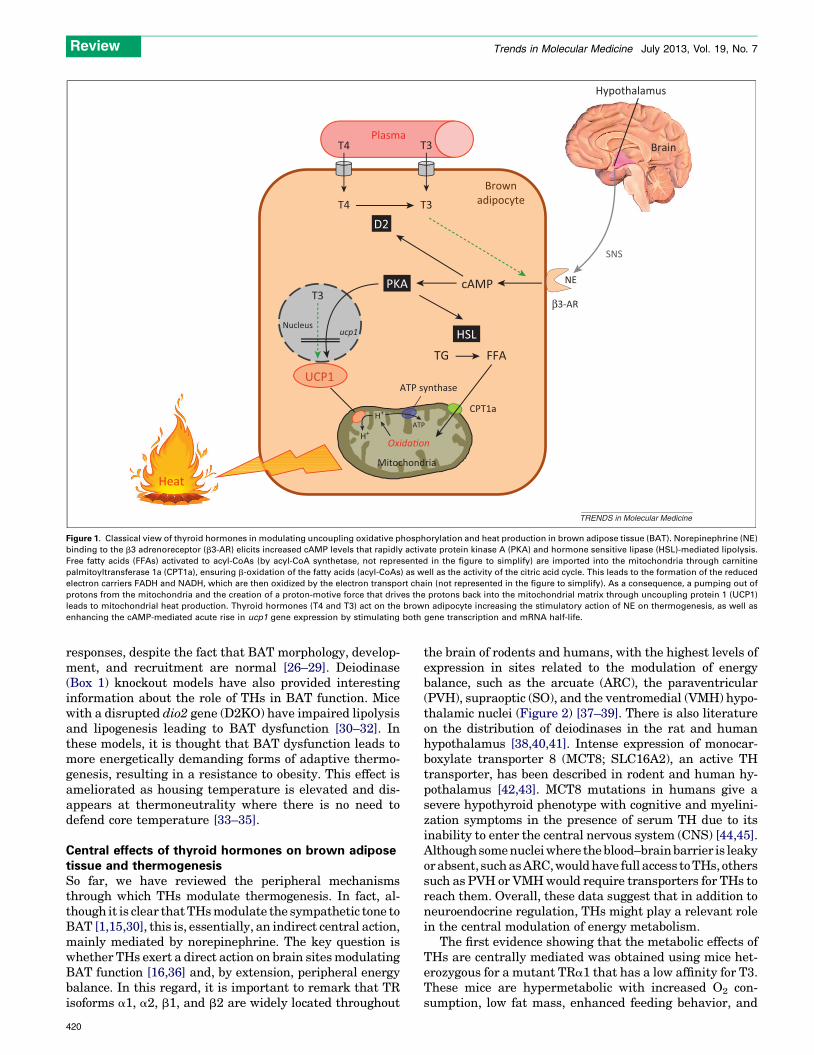
In BAT, the inner mitochondrial membrane contains aprotein termed uncoupling protein 1 (UCP1) that providesan alternative pathway for the protons back into themitochondrial matrix, circumventing ATP synthase andproducing heat [1,15,16]. The role of THs in modulatinguncoupling oxidative phosphorylation and heat productionin BAT has been historically intriguing. Classically, BATthermogenesis is centrally activated by the hypothalamus(a brain area important for energy balance, Box 2) via thesympathetic nervous system (SNS), with norepinephrineas the key neurotransmitter mediating activation(Figure 1). In mature brown adipocytes, norepinephrineinteracts with all types of adrenergic receptors: b1, b2, b3,a1, and a2, with b3 adrenergic stimulation of thermogen-esis as the most significant pathway [15,16]. Norepineph-rine binding to the b3 adrenoreceptor (b3-AR) increasescAMP levels that rapidly activate protein kinase A (PKA)-and hormone-sensitive lipase (HSL)-mediated lipolysis.
Free fatty acids are imported into the mitochondriathrough carnitine palmitoyltransferase 1a (CPT1a), wherethey are oxidized (via b-oxidation and further citric acidcycle), leading to the formation of NADH and FADH, whichare then oxidized by the electron transport chain. Thisresults in a pumping out of protons from the mitochondriaand the creation of a proton-motive force that drives theprotons back into the mitochondrial matrix through UCP1.The energy stored in the proton-motive force is then re-leased, starting mitochondrial heat production (Figure 1)[15,16].
In addition to this SNS-mediated activation, BAT is adirect target of THs and possesses a large number of a1 andb1 TH receptors (TRs). THs are essential to maintain thenorepinephrine signaling in BAT [24], and the TRa1 iso-form is required to maintain the normal adrenergic re-sponsiveness of brown adipocytes, whereas TRb mediatesT3-induced UCP1 gene expression [13,25]. Studies of dif-ferent TR knockout mouse models and the use of isoform-selective agonists have confirmed the role of the differentTH isoforms in BAT thermogenesis. Global TRa1 and TR-a1/b deficiency, as well as deletion of all isoforms of TRa
(Thra-0/0), lead to hypothermia and cold intolerance asso-ciated with reduced BAT thermogenesis (but not obligatorythermogenesis) due to blunted norepinephrine-induced
419
T4 T3
NE
D2
cAMP
TG FFA
Oxida�on H+
H+
SNS
Brownadipocyte
ATP
β3-AR T3
UCP1
PKA
HSL
ATP synthase
CPT1a
HeatMitochondria
Nucleusucp1
Hypothalamus
T3Plasma
T4 Brain
TRENDS in Molecular Medicine
Figure 1. Classical view of thyroid hormones in modulating uncoupling oxidative phosphorylation and heat production in brown adipose tissue (BAT). Norepinephrine (NE)
binding to the b3 adrenoreceptor (b3-AR) elicits increased cAMP levels that rapidly activate protein kinase A (PKA) and hormone sensitive lipase (HSL)-mediated lipolysis.
Free fatty acids (FFAs) activated to acyl-CoAs (by acyl-CoA synthetase, not represented in the figure to simplify) are imported into the mitochondria through carnitine
palmitoyltransferase 1a (CPT1a), ensuring b-oxidation of the fatty acids (acyl-CoAs) as well as the activity of the citric acid cycle. This leads to the formation of the reduced
electron carriers FADH and NADH, which are then oxidized by the electron transport chain (not represented in the figure to simplify). As a consequence, a pumping out of
protons from the mitochondria and the creation of a proton-motive force that drives the protons back into the mitochondrial matrix through uncoupling protein 1 (UCP1)
leads to mitochondrial heat production. Thyroid hormones (T4 and T3) act on the brown adipocyte increasing the stimulatory action of NE on thermogenesis, as well as
enhancing the cAMP-mediated acute rise in ucp1 gene expression by stimulating both gene transcription and mRNA half-life.
Review Trends in Molecular Medicine July 2013, Vol. 19, No. 7
responses, despite the fact that BAT morphology, develop-ment, and recruitment are normal [26–29]. Deiodinase(Box 1) knockout models have also provided interestinginformation about the role of THs in BAT function. Micewith a disrupted dio2 gene (D2KO) have impaired lipolysisand lipogenesis leading to BAT dysfunction [30–32]. Inthese models, it is thought that BAT dysfunction leads tomore energetically demanding forms of adaptive thermo-genesis, resulting in a resistance to obesity. This effect isameliorated as housing temperature is elevated and dis-appears at thermoneutrality where there is no need todefend core temperature [33–35].
Central effects of thyroid hormones on brown adiposetissue and thermogenesisSo far, we have reviewed the peripheral mechanismsthrough which THs modulate thermogenesis. In fact, al-though it is clear that THs modulate the sympathetic tone toBAT [1,15,30], this is, essentially, an indirect central action,mainly mediated by norepinephrine. The key question iswhether THs exert a direct action on brain sites modulatingBAT function [16,36] and, by extension, peripheral energybalance. In this regard, it is important to remark that TRisoforms a1, a2, b1, and b2 are widely located throughout
420
the brain of rodents and humans, with the highest levels ofexpression in sites related to the modulation of energybalance, such as the arcuate (ARC), the paraventricular(PVH), supraoptic (SO), and the ventromedial (VMH) hypo-thalamic nuclei (Figure 2) [37–39]. There is also literatureon the distribution of deiodinases in the rat and humanhypothalamus [38,40,41]. Intense expression of monocar-boxylate transporter 8 (MCT8; SLC16A2), an active THtransporter, has been described in rodent and human hy-pothalamus [42,43]. MCT8 mutations in humans give asevere hypothyroid phenotype with cognitive and myelini-zation symptoms in the presence of serum TH due to itsinability to enter the central nervous system (CNS) [44,45].Although some nuclei where the blood–brain barrier is leakyor absent, such as ARC, would have full access to THs, otherssuch as PVH or VMH would require transporters for THs toreach them. Overall, these data suggest that in addition toneuroendocrine regulation, THs might play a relevant rolein the central modulation of energy metabolism.
The first evidence showing that the metabolic effects ofTHs are centrally mediated was obtained using mice het-erozygous for a mutant TRa1 that has a low affinity for T3.These mice are hypermetabolic with increased O2 con-sumption, low fat mass, enhanced feeding behavior, and

Brain Hypothalamus
Liver BATWAT Beta-cell(pancreas)
Food intake
Autonomicnervous system
Glucose and lipid metabolism Energy expenditure
Hormonal andnutri�onal signals
PVH PVHLHALHA
DMH DMH
VMH VMH
ARC ARC3V
MCHOXs
AgRPNPY
CARTPOMC
(α-MSH)
Skeletalmuscle
TRENDS in Molecular Medicine
Figure 2. Hypothalamic regulation of whole-body energy balance and metabolism. There exists a complex and dynamic interplay between the peripheral metabolic
environment and the central brain mechanisms that regulate it. Specific nuclei in the hypothalamus respond to alterations in food availability, energy stores, and nutritional
requirements and communicate hormonally and via the autonomous nervous system (ANS) to elicit functional changes in a range of tissues including the liver, pancreatic
beta cell, muscle, white adipose tissue (WAT), and brown adipose tissue (BAT). This homeostatic loop modulates whole-body energy homeostasis and metabolism.
Review Trends in Molecular Medicine July 2013, Vol. 19, No. 7
resistance to diet-induced obesity [33]. In addition, thesemutant mice show enhanced insulin sensitivity and in-creased lipid utilization. The hypermetabolism of thesemutant mice was explained by high BAT activity, increas-ing thermogenesis, and energy expenditure. These meta-bolic alterations are blunted after the functionaldenervation of sympathetic signaling to BAT, indicatingthat the CNS controlled the metabolism of these mice viathe autonomic nervous system (ANS). It was proposed thatthe aporeceptor TRa1 may have a part in controllinghypothalamic T3 concentration and subsequently affectingsympathetic outflow to BAT, contributing to the hypermet-abolic phenotype [33].
Despite this key evidence, the molecular mechanismmediating the central effect of THs on BAT thermogenesisremained elusive until a couple of years ago, when apreviously unrecognized homeostatic link between thecentral effects of THs on AMP-activated protein kinase(AMPK), a cellular sensor [46,47], and BAT function wasfirst identified. Specifically, it was shown that activation ofthe thermogenic program in BAT through the SNSdepends on T3-mediated activation of de novo lipogenesisin the hypothalamus by inhibiting AMPK (Figure 3) [39].The relevance of these results is substantial for severalreasons. THs were already known to modulate lipid me-tabolism in peripheral tissues [48–50], but no previous
evidence had demonstrated a regulatory action of THson CNS lipid metabolism. These data first demonstratedthat central THs increase de novo lipogenesis in the hypo-thalamus but not in other brain regions, such as the cortexor the cerebellum. These prolipogenic changes are mediat-ed by the inactivation of AMPK and subsequent activationof its downstream targets acetyl-CoA carboxylase (ACC)and fatty acid synthase (FAS), as well as inhibition of CPT1[39]. The physiological relevance of this effect is unclear,but the data indicate that resultant accumulation of mal-onyl-CoA and complex lipids in the hypothalamus plays arole in signaling, as demonstrated by the fact that theirincrease is correlated with increased sympathetic stimula-tion of BAT [39]. Notably, targeted inhibition of TH signal-ing in the VMH, using adenoviral injection of a dominantnegative TR, totally reverses the effects of hyperthyroid-ism on energy balance, inactivating the thermogenic pro-gram in BAT, and leading to weight gain despiteunaffected feeding [39]. In addition, the effects of THson AMPK signaling are specific to the VMH, given thatstereotaxical treatment with adenoviruses harboring con-stitutively active isoforms of AMPK (AMPK-CA) reducesthe activation of BAT, preventing the weight loss associat-ed with hyperthyroidism without increased feeding. Al-though the VMH was known as a key hypothalamicnucleus largely involved in controlling metabolic activity
421

Thyroid gland
↑ Energy expenditure
Body weight
BAT
↑ Food intake
T3 Hyperthyroidism
AMPK
VMH
3V
ARC mTOR
NPY AgRP
POMC
β3-AR
PVH
RPa IO
SNS
Complex lipids? Malonyl-CoA?
Liver
Glucose produc�onInsulin resistance↑
?
Lipid metabolism?
DMV
PSNS (vagus nerve)SNS
IML
AMPK? mTOR?
UCP2
T4
T3
T4
T3
Plasma Plasma
TRENDS in Molecular Medicine
Figure 3. Central actions of thyroid hormones (THs): energy balance and peripheral metabolism. THs exert dissociated actions on hypothalamic metabolic sensors. While
central T3 regulates feeding through mammalian target of rapamycin (mTOR) and uncoupling protein 2 (UCP2) in the arcuate nucleus of the hypothalamus (ARC), it
modulates the thermogenic program in the brown adipose tissue (BAT) via AMP-activated protein kinase (AMPK) in the ventromedial nucleus of the hypothalamus (VMH).
Central T3 acting in the paraventricular nucleus of the hypothalamus (PVH) also modulates hepatic glucose homeostasis in the liver, although the molecular details of this
interaction at this level remain unknown. Functional changes in those hypothalamic energy sensors (i.e., impaired lipid metabolism in the VMH) are associated with
activation of the parasympathetic and the sympathetic nervous system (PSNS and SNS) through changes in brainstem nuclei, such as the dorsal motor nucleus of the
vagus (DMV), the inferior olive (IO), and the raphe pallidus (RPa), as well as the intermediolateral column (IML) in the spinal cord. While both sympathetic and
parasympathetic outflow from the PVH modulate hepatic glucose metabolism, BAT function is modulated in the VMH only through the SNS. If central T3 modulates hepatic
lipid metabolism is currently unknown.
Review Trends in Molecular Medicine July 2013, Vol. 19, No. 7
of BAT [15,16], the molecular mechanism controlling thataction was unknown until this report. We have named thisphysiological pathway as the ‘AMPK–BAT axis’ [21,39],which seems to be a canonical circuit, non-exclusive forTHs, because other molecules such as bone morphogeneticprotein 8b (BMP8b) [51] and nicotine [52] can also modu-late this molecular pathway.
The significance of central THs on BAT thermogenesismay be interesting with regard to treatment efforts forobesity. In contrast to previous paradigms, during the pastfew years it has been revealed that humans possess meta-bolically active BAT [18,53]. Importantly, thyroid statushas been shown to affect the activity of BAT in humans. Ithas been observed using positron emission tomography-computed tomography (PET-CT) with 18F-fluoro-deoxyglu-cose (18F-FDG) that THs activate BAT, including increasedexpression of UCP1 and deiodinase 2 (D2) expression [54].BAT activation ameliorates diabetes in patients with
422
severe insulin resistance [54], and it would be highlyinteresting to investigate whether such mechanismsmay be relevant in pathological states characterized bynegative energy balance, such as cachexia, wasting dis-eases, or life-threatening conditions such as ‘thyroidstorm’, all of which are situations where selective modula-tion of AMPK activity in the VMH could theoreticallyprovide a useful therapeutic strategy.
Central effects of thyroid hormones on food intakeIt is well known that alterations in thyroid status areassociated with changes in feeding in both humans androdents. Hyperphagia is observed in 85% of hyperthyroidpatients, despite associated weight loss [9]. Similarly,hyperthyroid rats display a hyperphagic phenotype andleanness [39,55,56]. A bulk of data has demonstrated thathyperthyroid-induced hyperphagia is associated with amarked dysregulation of hypothalamic neuropeptide

Review Trends in Molecular Medicine July 2013, Vol. 19, No. 7
systems (Box 2 and Figure 2). mRNAs for agouti-relatedprotein (AgRP) and neuropeptide Y (NPY) are upregulatedand proopiomelanocortin (POMC) mRNA is decreased inthe ARC of hyperthyroid rats [39] probably in order tocompensate the alterations in energy expenditure. Of note,this hyperthyroidism-induced pattern of impairment inneuropeptide expression does not affect other hypothalam-ic areas, and therefore appears to be highly specific becauseno changes have been found in the expression of otherhypothalamic neuropeptides (Box 2 and Figure 2), such asthe cocaine and amphetamine regulated transcript (CART)in the ARC and in the lateral hypothalamic area (LHA) ormelanin-concentrating hormone (MCH) and orexin (OX) inthe LHA [39,55,57].
Recently, extensive work has focused on the detailedassessment of the mechanism underlying thyroid-inducedchanges in neuropeptide expression, particularly in theARC. TH action has been linked to other TH targets inthe same hypothalamic nucleus, such as D2 (Box 1) [58],uncoupling protein 2 (UCP2) [59], and mechanistic target ofrapamycin (mTOR) [57]. D2-expressing glial cells are indirect contact with AgRP/NPY neurons, which also expressUCP2 [59]. Diano and Horvath elegantly demonstrated thatfood deprivation increases D2 activity in the ARC, leading toa local rise in T3 levels and subsequent UCP2-mediatedmitochondrial uncoupling and proliferation, with increasedexcitability of AgRP/NPY neurons and resultant increase inappetite [59]. Remarkably, expression levels and activity ofUCP2 are also increased after central injection of T3 alone,but reduced in hypothyroidism, indicating direct action of T3on the UCP2–AgRP/NPY axis (Figure 3) [59].
Besides the role of AMPK and UCP2 in the modulationof energy homeostasis by THs at the central level, currentevidence indicates that hypothalamic mTOR is under TH-mediated control of feeding. mTOR is an evolutionarilyconserved serine–threonine kinase that modulates cell-cycle progression and growth by sensing changes in energybalance, growth factors, nutrients, and oxygen [60]. Cur-rent literature has reported that hypothalamic mTORsignaling plays a main role in modulating energy balance[57,61]. The mTOR pathway is highly regulated in specifichypothalamic nuclei, such as the ARC, where it co-localizeswith AgRP, NPY, and POMC [61]. At this level, mTORsignaling plays a pivotal role in the hypothalamic networksthat respond to nutrient availability (i.e., leucine levels)and the hormonal milieu. Thus, leptin, ghrelin, insulin,ciliary neurotrophic factor (CNTF), and bone morphoge-netic protein 7 (BMP7) all act on hypothalamic mTORsignaling to regulate energy homeostasis [61–63].
In line with the above evidence, recent data have demon-strated that hyperphagia induced by acute central T3 ad-ministration or by chronic hyperthyroidism is mediated byspecific upregulation of the mTOR pathway in the ARC,where mTOR highly co-localizes with TRa, but not in theVMH [57]. In fact, the orexigenic state characterizing hyper-thryroidism is totally reversed by central treatment withrapamycin, a specific mTOR inhibitor, as a result of normal-ized AgRP and NPY expression in the ARC [57]. The rele-vance of these results in terms of central actions of THs isfairly remarkable. Thus, this evidence indicates that T3exerts dissociated actions on hypothalamic metabolic
sensors: T3 regulates feeding through mTOR in the ARC,while modulating energy expenditure via AMPK in theVMH (Figure 3) [39,57]. Overall, these data are in agree-ment with previous data that point to AgRP and NPYneuronal populations as critical mediators of the orexigeniceffect of T3 [39,57]. They are also in agreement with the well-defined role of glia as generator of T3 for the neurons,through the D2 enzyme expressed solely by the glia. Wheth-er mTOR and UCP2 crosstalk downstream of THs in theARC is currently unresolved, but considering that both ofthem share molecular pathways with AMPK [57,61,64], thishypothesis may provide new cues about the central role ofTHs in the modulation of energy balance.
Central effects of thyroid hormones on hepatic glucosemetabolismTHs appear to influence almost all aspects of glucosemetabolism [65], including the following: (i) promotingintestinal glucose absorption and uptake of glucose byWAT and muscle; (ii) influencing hepatic glycogen synthe-sis and glycogenolysis; (iii) modulating the responsivenessof liver, WAT, and muscle to other hormones such asinsulin and catecholamines; and (iv) increasing insulindegradation in hyperthyroid patients. In keeping with this,patients with thyrotoxicosis usually exhibit impaired oralglucose tolerance or overt diabetes [65,66].
Elegant data from Kalsbeek and Fliers have shown thatTHs modulate glucose production and insulin sensitivityvia a sympathetic pathway from the PVH to the liver,independent of circulating glucoregulatory hormones[67,68]. They performed a specific functional denervationof the hepatic sympathetic or parasympathetic nervoussystem (SNS and PSNS, respectively) in euthyroid andthyrotoxic rats. In euthyroid rats, selective hepatic sympa-thetic or parasympathetic denervation did not affect glu-cose concentration, insulin concentration, or glucoseproduction. As expected, thyrotoxicosis increased hepaticglucose production and reduced insulin sensitivity in theliver. Sympathectomy attenuated the increased hepaticglucose production observed in thyrotoxicosis, whereashepatic parasympathectomy did not affect hepatic glucoseproduction but induced a marked increase in basal insulinconcentration, indicating hepatic insulin resistance, sug-gesting a centrally mediated effect of THs on glucosemetabolism [67]. In a further study, the same groupattempted to investigate the specific neuronal populationmodulating brain TH control on hepatic glucose productionand because TRs are abundantly expressed in the PVH,they administered T3 directly to this nucleus and assessedhepatic glucose production in euthyroid rats. The specificinjection of T3 into the PVH increased hepatic glucoseproduction (without changes in glucoregulatory hormones)and selective hepatic sympathectomy blunted this effect[68]. Therefore, this study concluded that central THsrequire an intact SNS to act through the PVH–liver axisto control glucose production (Figure 3).
Overall, these data unequivocally demonstrate the keyrole of central THs in the regulation of hepatic glucosehomeostasis. This is a very challenging concept. Currentdata suggest that peptide hormones such as leptin, insulin,and ghrelin, which bind to extracellular receptors, may act
423

Box 3. Thyroid hormone derivatives and energy balance
TH derivatives could also have metabolic actions in the body.
Derivatives include thyronines with less than three iodines. Some of
these molecules are present in the body but have been considered
byproducts of TH metabolism. However, recent data suggest
potential physiological effects, such as ligands of non-TRs either
at physiological concentrations or injected at pharmacological
concentrations [86]. One of the most interesting candidates is 3,5
diiodothyronine (T2). Acute injection of pharmacological doses of
T2 in rats with high-fat diet (HFD) activated mitochondrial metabo-
lism in the liver, significantly reduced the weight gain observed in
these rats and ameliorated their unfavorable serum lipid profile [87].
T2 did not alter T3, T4, or TSH and did not seem to have any
thyrotoxic effect. It has recently been shown that long-term
treatment with T2 prevents lipid accumulation in the liver and
muscle of rats fed a HFD by increasing fatty acid oxidation, which
leads to prevention of insulin resistance without any side effect
associated with T3 actions [88,89]. T2 actions seem mediated by a
nuclear co-activator pathway including sirtuin (SIRT1) and de-
acetylation of transcription factors [90]. To date, human studies
are lacking, but a recent publication described the injection of T2
during 3 weeks in two healthy volunteers, with a moderate weight
loss without apparent side effects or alteration of TH levels [91].
Thyronamines (TAMs) are derivatives lacking the N-terminal
carboxyl group characteristic of thyronines, but maintaining the
presence of one, two, or more iodines [86]. The physiological serum
concentrations of the different TAMs are currently under discussion,
but it seems that 3-iodothyronamine (T1AM) could be present at
high enough concentrations to be considered an endogenous
hormone ligand [86]. Independent of the physiology or pharmacol-
ogy of their actions, TAMs are very interesting candidates to
consider in the metabolic portfolio. Intraperitoneal injection of
T1AM in rodents induces acute bradycardia, increase in plasma
corticosterone, hyperglycemia with hyperglucagonemia and slight
hypoinsulinemia, reduction of the metabolic rate, marked hypother-
mia, and a shift from glucose to lipids as the main cell energy
source, with the subsequent loss of adipose mass [92,93]. Globally,
it seems that TAM actions are partially opposite to T3. Hyperglyce-
mia and hyperglucagonemia with an absence of changes in insulin
levels are also achieved through central injection of low T1AM
doses, suggesting central effects on regulation of pancreatic islets
[94]. This injection also stimulated hepatic glucose production,
whereas circulating insulin levels decreased [94]. Recent data have
highlighted a central action of TAMs on energy metabolism,
independently of TH levels. Direct intra-ARC injections of T1AM
induced an orexigenic effect even at low doses not altering oxygen
consumption or locomotor activity [95].
Review Trends in Molecular Medicine July 2013, Vol. 19, No. 7
through the hypothalamus to fine-tune metabolic control inperipheral organs, such as liver, WAT, and BAT [69–72].However, the notion that small non-peptide hormones exert-ing their effects in target tissues by binding to nuclearreceptors act at the central level to regulate peripheralmetabolism is fairly remarkable. Furthermore, this idearaises another relevant question: is this a specific effect ofcentral THs on liver and BAT metabolism or are central THsa global modulator of the whole peripheral metabolism?Bearing in mind the strong effects exerted by alterationsin thyroid status on lipid metabolism in liver, muscle, WAT,and BAT, as well as the lipid uptake and processing by thoseorgans [39,48,57] and the autonomic innervations of thosetissues [15,39,68,73], it is tempting to speculate that, besidestheir actions on hepatic glucose metabolism, central THsmight be a global metabolic regulator. Supporting this no-tion, it has been demonstrated that central THs activate CNScenters modulating both the SNS and PSNS. T3 elicits amarked increase in neuronal activity in the dorsal motornucleus of the vagus (DMV), the main nucleus modulatingvagus nerve firing to the liver [39], as well as the inferior olive(IO) and the raphe pallidus (RPa) nuclei, which modulatesympathetic nerves innervating BAT (Figure 3)[15,16,39,51]. Notably, all these nuclei receive neuronalprojections from the VMH [15,39], which would reinforcethe view of this nucleus as a key site globally regulatingenergy balance.
Clinical implicationsEpidemiological studies have shown an incidence of up to20% of subclinical hypothyroidism in obese subjects [5,7,49].In addition, the pronounced effects of THs in energy andmetabolic homeostasis have put TH-related compounds atthe forefront of developing a drug-based therapy for obesity[5,7,49]. The coexistence of hyperphagia and decreased bodyweight in patients with hyperthyroidism leaves this ap-proach especially attractive. By contrast, the phenotype ofpatients with mutations in the TRa or TRb gene suggeststhat many actions are specific of one of the two receptors.Thus, patients with TRb mutations present pituitary resis-tance to THs with high thyroid-stimulating hormone (TSH)in the presence of high TH levels, goiter, and tachycardia.But otherwise, they generally present a mild cognitive andgrowth phenotype [74]. However, the two recently describedpatients with TRa inactivating mutation present clinicalmanifestations of severe hypothyroidism (constipation,growth retardation, myelinization defects, low IQ) withoutalteration of TSH levels [75,76].
Drug development is, however, hampered by the need toensure that these molecules do not lead to undesirableeffects on cardiac function, skeletal tissue, muscle, or theCNS. Data gleaned in recent years uncovering the differen-tial distribution at the tissue level of TRs [38,40,41] andtheir mechanism of action [3,4,77] have facilitated the de-velopment of TH analogs with selective actions for thetreatment of dyslipidemia and obesity [7,8]. Among theseveral selective agonists developed and tested in eitherrodents and/or humans, GC-1, GC-24, KB-141, which areTRb-high affinity ligands, as well as other TH derivatives,such as 3,5-diiodothyroproprionic acid (DITPA), have beenfound to decrease body weight largely through an increase in
424
energy expenditure, whereas KB-2115 did not alter bodyweight [7,78]. Of note, all of them led to a significantimprovement in circulating levels of cholesterol and triglyc-erides, showing that the effects on energy homeostasis anddyslipidemia can be targeted selectively [7,78], becauseplasma cholesterol levels have been shown to be moresensitive to some thyromimetics, such as KB-2115, thaneffects on the cardiovascular system, metabolic rate, andbody weight reduction [79]. In conclusion, these data dem-onstrate the potentially beneficial actions of thyromimetic-based drugs in patients with dyslipidemia and obesity.However, it is currently unknown whether those beneficialmetabolic actions are due to a direct effect on key metabolicorgans or whether central effects may be involved.
Concluding remarks and future perspectives: centralactions of thyroid hormones, not just a metaboliclandscapeTHs and probably TH derivatives (Box 3) are importantmodulators of both energy balance and metabolism [1–4].

Review Trends in Molecular Medicine July 2013, Vol. 19, No. 7
Textbooks used to convey that THs primarily exert theireffects through direct action in peripheral organs, and thegeneral consensus assumes that most effects of THs onenergy homeostasis are exerted directly at the peripherallevel, that is, through the binding of circulating THs to TRsin metabolically active peripheral tissues. Current data,reviewed here, are changing and challenging this perspec-tive from a ‘peripheral vision’ to a ‘central vision’, showingthat THs also exert dissociated actions on hypothalamicnuclei, such as the ARC, the PVH, and the VMH, bymodulating specific metabolic sensors to regulate energyhomeostasis (Figure 3) [39,57,59]. Importantly, this newconcept for the central actions of THs seems not to betotally constrained to the modulation of whole-body me-tabolism and energy balance. In fact, current evidenceextends the relevance of central actions of THs to cardio-vascular function. Very recently, it was demonstrated thatTHs are necessary for the proper development of a previ-ously unknown population of parvalbuminergic neurons inthe anterior hypothalamus (AHA), which plays a majorrole in central autonomic control of blood pressure andheart rate, indicating that developmental hypothyroidismmay represent a previously unknown risk factor for car-diovascular disorders [80].
Overall, the evidence presented here reinforces the ideathat THs are critical modulators of peripheral systemsthrough the CNS. However, the key question will be toaddress the relevance of these central actions on humanpathology, not just in syndromes related to alterations isthyroid status per se but also revealing new moleculartargets and hopefully designing molecular analogs to treatenergy balance disorders, such as obesity. In light of theever-growing obese population, this will be a new excitingavenue that biomedical research will need to address inupcoming years.
AcknowledgmentsWe emphatically thank Dr Silje Skrede (University of Bergen) for hercomments and criticisms. The research leading to these results hasreceived funding from the European Community’s Seventh FrameworkProgram (FP7/2007–2013) under grant agreement no. 281854 – theObERStress project (ML), 245009 – the Neurofast project (R.N., C.D., andM.L.), Xunta de Galicia (CAV: 09CSA011208PR and XUGA CN2012/142;M.L.: 10PXIB208164PR and 2012-CP070; R.N.: EM 2012/039 and 2012-CP069), Fondo Investigationes Sanitarias (M.L.: PI12/01814), MINECOco-funded by the FEDER Program of EU (C.A.V.: BFU2010-16652; R.N.:RyC-2008-02219 and BFU2012-35255; C.D.: BFU2011-29102), CIBER deFisiopatologıa de la Obesidad y Nutricion is an initiative of ISCIII.
References1 Silva, J.E. (2006) Thermogenic mechanisms and their hormonal
regulation. Physiol. Rev. 86, 435–4642 Hollenberg, A.N. and Forrest, D. (2008) The thyroid and metabolism:
the action continues. Cell Metab. 8, 10–123 Cheng, S.Y. et al. (2010) Molecular aspects of thyroid hormone actions.
Endocr. Rev. 31, 139–1704 Brent, G.A. (2012) Mechanisms of thyroid hormone action. J. Clin.
Invest. 122, 3035–30435 Weaver, J.U. (2008) Classical endocrine diseases causing obesity.
Front. Horm. Res. 36, 212–2286 Kaptein, E.M. et al. (2009) Thyroid hormone therapy for obesity and
nonthyroidal illnesses: a systematic review. J. Clin. Endocrinol. Metab.94, 3663–3675
7 Baxter, J.D. and Webb, P. (2009) Thyroid hormone mimetics: potentialapplications in atherosclerosis, obesity and type 2 diabetes. Nat. Rev.Drug Discov. 8, 308–320
8 Pearce, E.N. (2012) Thyroid hormone and obesity. Curr. Opin.Endocrinol. Diabetes Obes. 19, 408–413
9 Ingbar, S.H. (1985) The thyroid gland. In Williams Textbook ofEndocrinology (Wilson, J.D. and Foster, D.W., eds), pp. 975–1170,Philadelphia, PA, USA, W.B. Saunders Company
10 Kim, B. (2008) Thyroid hormone as a determinant of energyexpenditure and the basal metabolic rate. Thyroid 18, 141–144
11 Sainsbury, A. and Zhang, L. (2012) Role of the hypothalamus in theneuroendocrine regulation of body weight and composition duringenergy deficit. Obes. Rev. 13, 234–257
12 Bianco, A.C. et al. (2005) Adaptive activation of thyroid hormone andenergy expenditure. Biosci. Rep. 25, 191–208
13 Ribeiro, M.O. et al. (2010) Expression of uncoupling protein 1 in mousebrown adipose tissue is thyroid hormone receptor-b isoform specificand required for adaptive thermogenesis. Endocrinology 151, 432–440
14 Hulbert, A.J. and Else, P.L. (2004) Basal metabolic rate: history,composition, regulation, and usefulness. Physiol. Biochem. Zool. 77,869–876
15 Cannon, B. and Nedergaard, J. (2004) Brown adipose tissue: functionand physiological significance. Physiol. Rev. 84, 277–359
16 Whittle, A.J. et al. (2011) Using brown adipose tissue to treat obesity –the central issue. Trends Mol. Med. 17, 405–411
17 Duchamp, C. and Barre, H. (1993) Skeletal muscle as the major site ofnonshivering thermogenesis in cold-acclimated ducklings. Am. J.Physiol. 265, R1076–R1083
18 Nedergaard, J. et al. (2007) Unexpected evidence for active brownadipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab.293, E444–E452
19 Ismail-Beigi, F. (1992) Regulation of Na+,K+-ATPase expression bythyroid hormone. Semin. Nephrol. 12, 44–48
20 Block, B.A. (1994) Thermogenesis in muscle. Annu. Rev. Physiol. 56,535–577
21 Cannon, B. and Nedergaard, J. (2010) Thyroid hormones: ignitingbrown fat via the brain. Nat. Med. 16, 965–967
22 Simonides, W.S. et al. (2001) Mechanism of thyroid-hormone regulatedexpression of the SERCA genes in skeletal muscle: implications forthermogenesis. Biosci. Rep. 21, 139–154
23 van den Berg, S.A. et al. (2011) Skeletal muscle mitochondrialuncoupling, adaptive thermogenesis and energy expenditure. Curr.Opin. Clin. Nutr. Metab. Care 14, 243–249
24 Bianco, A.C. et al. (1988) Triiodothyronine amplifies norepinephrinestimulation of uncoupling protein gene transcription by a mechanismnot requiring protein synthesis. J. Biol. Chem. 263, 18168–18175
25 Martinez de, M.R. et al. (2010) The T3 receptor b1 isoform regulatesUCP1 and D2 deiodinase in rat brown adipocytes. Endocrinology 151,5074–5083
26 Wikstrom, L. et al. (1998) Abnormal heart rate and body temperaturein mice lacking thyroid hormone receptor a1. EMBO J. 17, 455–461
27 Golozoubova, V. et al. (2004) Depressed thermogenesis but competentbrown adipose tissue recruitment in mice devoid of all hormone-binding thyroid hormone receptors. Mol. Endocrinol. 18, 384–401
28 Marrif, H. et al. (2005) Temperature homeostasis in transgenic micelacking thyroid hormone receptor-a gene products. Endocrinology 146,2872–2884
29 Ramadan, W. et al. (2011) Type-2 iodothyronine 50deiodinase (D2) inskeletal muscle of C57Bl/6 mice. II. Evidence for a role of D2 in thehypermetabolism of thyroid hormone receptor a-deficient mice.Endocrinology 152, 3093–3102
30 De Jesus, L.A. et al. (2001) The type 2 iodothyronine deiodinase isessential for adaptive thermogenesis in brown adipose tissue. J. Clin.Invest. 108, 1379–1385
31 Christoffolete, M.A. et al. (2004) Mice with targeted disruption of theDio2 gene have cold-induced overexpression of the uncoupling protein 1gene but fail to increase brown adipose tissue lipogenesis and adaptivethermogenesis. Diabetes 53, 577–584
32 Castillo, M. et al. (2011) Disruption of thyroid hormone activation intype 2 deiodinase knockout mice causes obesity with glucoseintolerance and liver steatosis only at thermoneutrality. Diabetes60, 1082–1089
425

Review Trends in Molecular Medicine July 2013, Vol. 19, No. 7
33 Sjogren, M. et al. (2007) Hypermetabolism in mice caused by thecentral action of an unliganded thyroid hormone receptor a1. EMBOJ. 26, 4535–4545
34 Pelletier, P. et al. (2008) Mice lacking the thyroid hormone receptor-agene spend more energy in thermogenesis, burn more fat, and are lesssensitive to high-fat diet-induced obesity. Endocrinology 149, 6471–6486
35 Marsili, A. et al. (2011) Mice with a targeted deletion of the type 2deiodinase are insulin resistant and susceptible to diet induced obesity.PLoS ONE 6, e20832
36 Nedergaard, J. et al. (1997) The interaction between thyroid andbrown-fat thermogenesis. Central or peripheral effects? Ann. N. Y.Acad. Sci. 813, 712–717
37 Lechan, R.M. et al. (1993) Immunocytochemical delineation of thyroidhormone receptor b 2-like immunoreactivity in the rat central nervoussystem. Endocrinology 132, 2461–2469
38 Alkemade, A. et al. (2005) Neuroanatomical pathways for thyroidhormone feedback in the human hypothalamus. J. Clin. Endocrinol.Metab. 90, 4322–4334
39 Lopez, M. et al. (2010) Hypothalamic AMPK and fatty acid metabolismmediate thyroid regulation of energy balance. Nat. Med. 16, 1001–1008
40 Friesema, E.C. et al. (2012) Thyroid hormone transporters anddeiodinases in the developing human hypothalamus. Eur. J.Endocrinol. 167, 379–386
41 Freitas, B.C. et al. (2010) Paracrine signaling by glial cell-derivedtriiodothyronine activates neuronal gene expression in the rodentbrain and human cells. J. Clin. Invest. 120, 2206–2217
42 Visser, W.E. et al. (2011) Thyroid hormone transporters: the knownsand the unknowns. Mol. Endocrinol. 25, 1–14
43 Alkemade, A. et al. (2011) Expression of thyroid hormone transportersin the human hypothalamus. J. Clin. Endocrinol. Metab. 96, E967–E971
44 Friesema, E.C. et al. (2004) Association between mutations in a thyroidhormone transporter and severe X-linked psychomotor retardation.Lancet 364, 1435–1437
45 Dumitrescu, A.M. et al. (2004) A novel syndrome combining thyroidand neurological abnormalities is associated with mutations in amonocarboxylate transporter gene. Am. J. Hum. Genet. 74, 168–175
46 Lage, R. et al. (2008) AMPK: a metabolic gauge regulating whole-bodyenergy homeostasis. Trends Mol. Med. 14, 539–549
47 Lopez, M. et al. (2008) Hypothalamic fatty acid metabolism mediatesthe orexigenic action of ghrelin. Cell Metab. 7, 389–399
48 Klieverik, L.P. et al. (2009) Thyroid hormone effects on whole-bodyenergy homeostasis and tissue-specific fatty acid uptake in vivo.Endocrinology 150, 5639–5648
49 Pearce, E.N. (2012) Update in lipid alterations in subclinicalhypothyroidism. J. Clin. Endocrinol. Metab. 97, 326–333
50 Duntas, L.H. and Brenta, G. (2012) The effect of thyroid disorders onlipid levels and metabolism. Med. Clin. North Am. 96, 269–281
51 Whittle, A.J. et al. (2012) BMP8B increases brown adipose tissuethermogenesis through both central and peripheral actions. Cell149, 871–885
52 Martinez de Morentin, P.B. et al. (2012) Nicotine induces negativeenergy balance through hypothalamic AMP-activated protein kinase.Diabetes 61, 807–817
53 Cypess, A.M. et al. (2009) Identification and importance of brownadipose tissue in adult humans. N. Engl. J. Med. 360, 1509–1517
54 Skarulis, M.C. et al. (2010) Thyroid hormone induced brown adiposetissue and amelioration of diabetes in a patient with extreme insulinresistance. J. Clin. Endocrinol. Metab. 95, 256–262
55 Lopez, M. et al. (2001) Prepro-orexin mRNA levels in the rathypothalamus, and orexin receptors mRNA levels in the rathypothalamus and adrenal gland are not influenced by the thyroidstatus. Neurosci. Lett. 300, 171–175
56 Lopez, M. et al. (2002) Thyroid status regulates CART but not AgRPmRNA levels in the rat hypothalamus. Neuroreport 13, 1775–1779
57 Varela, L. et al. (2012) Hypothalamic mTOR pathway mediates thyroidhormone-induced hyperphagia in hyperthyroidism. J. Pathol. 227,209–222
58 Coppola, A. et al. (2005) Suppression of hypothalamic deiodinase typeII activity blunts TRH mRNA decline during fasting. FEBS Lett. 579,4654–4658
426
59 Coppola, A. et al. (2007) A central thermogenic-like mechanism infeeding regulation: an interplay between arcuate nucleus T3 andUCP2. Cell Metab. 5, 21–33
60 Wullschleger, S. et al. (2006) TOR signaling in growth and metabolism.Cell 124, 471–484
61 Cota, D. et al. (2006) Hypothalamic mTOR signaling regulates foodintake. Science 312, 927–930
62 Mayer, C.M. and Belsham, D.D. (2010) Central insulin signaling isattenuated by long-term insulin exposure via insulin receptorsubstrate-1 serine phosphorylation, proteasomal degradation, andlysosomal insulin receptor degradation. Endocrinology 151, 75–84
63 Townsend, K.L. et al. (2012) Bone morphogenetic protein 7 (BMP7)reverses obesity and regulates appetite through a central mTORpathway. FASEB J. 26, 2187–2196
64 Andrews, Z.B. et al. (2008) UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 454, 846–851
65 Fliers, E. et al. (2010) Novel neural pathways for metabolic effects ofthyroid hormone. Trends Endocrinol. Metab. 21, 230–236
66 Dimitriadis, G. et al. (2011) Effect of hyperthyroidism on clearance andsecretion of glucagon in man. Exp. Clin. Endocrinol. Diabetes 119, 214–217
67 Klieverik, L.P. et al. (2008) Effects of thyrotoxicosis and selectivehepatic autonomic denervation on hepatic glucose metabolism inrats. Am. J. Physiol. Endocrinol. Metab. 294, E513–E520
68 Klieverik, L.P. et al. (2009) Thyroid hormone modulates glucoseproduction via a sympathetic pathway from the hypothalamicparaventricular nucleus to the liver. Proc. Natl. Acad. Sci. U.S.A.106, 5966–5971
69 Theander-Carrillo, C. et al. (2006) Ghrelin action in the brain controlsadipocyte metabolism. J. Clin. Invest. 116, 1983–1993
70 Ono, H. et al. (2008) Activation of hypothalamic S6 kinase mediatesdiet-induced hepatic insulin resistance in rats. J. Clin. Invest. 118,2959–2968
71 Buettner, C. et al. (2008) Leptin controls adipose tissue lipogenesis viacentral, STAT3-independent mechanisms. Nat. Med. 14, 667–675
72 Sangiao-Alvarellos, S. et al. (2009) Central ghrelin regulatesperipheral lipid metabolism in a growth hormone-independentfashion. Endocrinology 150, 4562–4574
73 Bartness, T.J. et al. (2010) Sensory and sympathetic nervous systemcontrol of white adipose tissue lipolysis. Mol. Cell. Endocrinol. 318, 34–43
74 Refetoff, S. and Dumitrescu, A.M. (2007) Syndromes of reducedsensitivity to thyroid hormone: genetic defects in hormone receptors,cell transporters and deiodination. Best Pract. Res. Clin. Endocrinol.Metab. 21, 277–305
75 Bochukova, E. et al. (2012) A mutation in the thyroid hormone receptora gene. N. Engl. J. Med. 366, 243–249
76 van Mullem, A. et al. (2012) Clinical phenotype and mutant TRa1. N.Engl. J. Med. 366, 1451–1453
77 Gereben, B. et al. (2008) Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr. Rev. 29, 898–938
78 Lin, J.Z. et al. (2012) Thyroid hormone receptor agonists reduce serumcholesterol independent of the LDL receptor. Endocrinology 153, 6136–6144
79 Berkenstam, A. et al. (2008) The thyroid hormone mimetic compoundKB2115 lowers plasma LDL cholesterol and stimulates bile acidsynthesis without cardiac effects in humans. Proc. Natl. Acad. Sci.U.S.A. 105, 663–667
80 Mittag, J. et al. (2013) Thyroid hormone is required for hypothalamicneurons regulating cardiovascular functions. J. Clin. Invest. 123, 509–516
81 Coll, A.P. et al. (2007) The hormonal control of food intake. Cell 129,251–262
82 Lopez, M. et al. (2007) Peripheral tissue–brain interactions in theregulation of food intake. Proc. Nutr. Soc. 66, 131–155
83 Williams, K.W. and Elmquist, J.K. (2012) From neuroanatomy tobehavior: central integration of peripheral signals regulating feedingbehavior. Nat. Neurosci. 15, 1350–1355
84 Yeo, G.S. and Heisler, L.K. (2012) Unraveling the brain regulation ofappetite: lessons from genetics. Nat. Neurosci. 15, 1343–1349
85 Minokoshi, Y. and Kahn, B.B. (2003) Role of AMP-activated proteinkinase in leptin-induced fatty acid oxidation in muscle. Biochem. Soc.Trans. 31, 196–201

Review Trends in Molecular Medicine July 2013, Vol. 19, No. 7
86 Piehl, S. et al. (2011) Thyronamines – past, present, and future.Endocr. Rev. 32, 64–80
87 Lanni, A. et al. (2005) 3,5-Diiodo-L-thyronine powerfully reducesadiposity in rats by increasing the burning of fats. FASEB J. 19,1552–1554
88 de Lange, P. et al. (2011) Nonthyrotoxic prevention of diet-inducedinsulin resistance by 3,5-diiodo-L-thyronine in rats. Diabetes 60, 2730–2739
89 Moreno, M. et al. (2011) 3,5-Diiodo-L-thyronine prevents high-fat-diet-induced insulin resistance in rat skeletal muscle through metabolicand structural adaptations. FASEB J. 25, 3312–3324
90 Nogueiras, R. et al. (2012) Sirtuin 1 and sirtuin 3: physiologicalmodulators of metabolism. Physiol. Rev. 92, 1479–1514
91 Antonelli, A. et al. (2011) 3,5-Diiodo-L-thyronine increases restingmetabolic rate and reduces body weight without undesirable sideeffects. J. Biol. Regul. Homeost. Agents 25, 655–660
92 Scanlan, T.S. et al. (2004) 3-Iodothyronamine is an endogenous andrapid-acting derivative of thyroid hormone. Nat. Med. 10, 638–642
93 Braulke, L.J. et al. (2008) 3-Iodothyronamine: a novel hormonecontrolling the balance between glucose and lipid utilisation. J.Comp. Physiol. B 178, 167–177
94 Klieverik, L.P. et al. (2009) Central effects of thyronamines on glucosemetabolism in rats. J. Endocrinol. 201, 377–386
95 Dhillo, W.S. et al. (2009) The thyroid hormone derivative 3-iodothyronamine increases food intake in rodents. Diabetes Obes.Metab. 11, 251–260
427




















