
Electrostatic calculation of linear and non-linear optical properties of ice Ih, II, IX and VIII
16
Electrostatic calculation of linear and non-linear optical properties of ice Ih, II, IX and VIII H. Reis a, * , S.G. Raptis a,b , M.G. Papadopoulos a,1 a Institute of Organic and Pharmaceutical Chemistry, National Hellenic Research Foundation, Vasileos Constantinou 48, GR–11635 Athens, Greece b Chemical Engineering Department, National Technical University of Athens, Zografou Campus, GR–15773 Athens, Greece Received 14 June 2000; in final form 25 October 2000 Abstract Macroscopic first- and third-order susceptibilities of ice Ih, ice II, ice IX and ice VIII are calculated using static and frequency-dependent electronic and static vibrational molecular (hyper)polarizabilities at the MP2 level. The molecular properties are in good agreement with experiment and with high-level ab initio calculations. Intermolecular electrostatic and polarization eects due to induced dipoles are taken into account using a rigorous local-field theory. The electric field due to permanent dipoles is used to calculate eective in-crystal (hyper)polarizabilities. The polarizability depends only weakly on the permanent field, but the dipole moment and the hyperpolarizabilities are strongly aected. The calculated linear susceptibility is in good agreement with available experimental data for ice Ih, and the third-order susceptibility for a third harmonic generation experiment is in reasonable agreement with experimental values for liquid water. The molecular vibrational contributions have a small eect on the susceptibilities. The electric properties of a water tetramer are calculated and used to estimate the eect of non-dipolar interactions on the susceptibilities of ice Ih, which are found to be small. Ó 2001 Elsevier Science B.V. All rights reserved. 1. Introduction The computational prediction and experimental determination of electric properties of a water molecule in various environments has been an active field of research for quite a long time. For the free molecule, accurately determined experi- mental dipole and quadrupole moments and the (hyper)polarizabilities are known and could be reproduced by ab initio calculations, if reasonably high correlation levels and large basis sets were used [1–3]. Experimental determination of the corresponding eective properties in condensed phases, on the other hand, is much more dicult and rests on a number of assumptions and ap- proximations whose limitations are dicult to as- sess. The strong associated character and the extended hydrogen-bonded network formed in liquid water e.g. may raise doubts about the ap- plicability of the usual continuum models to ex- tract (hyper)polarizabilities from corresponding (non)linear experimental measurements. A complementary strategy is oered by com- putational approaches, e.g. classical or ab initio Chemical Physics 263 (2001) 301–316 www.elsevier.nl/locate/chemphys * Corresponding author. Fax: +30-17273831. E-mail addresses: [email protected] (H. Reis), [email protected] (M.G. Papadopoulos). 1 Also corresponding author. 0301-0104/01/$ - see front matter Ó 2001 Elsevier Science B.V. All rights reserved. PII:S0301-0104(00)00367-0
Transcript of Electrostatic calculation of linear and non-linear optical properties of ice Ih, II, IX and VIII

Electrostatic calculation of linear and non-linear opticalproperties of ice Ih, II, IX and VIII
H. Reis a,*, S.G. Raptis a,b, M.G. Papadopoulos a,1
a Institute of Organic and Pharmaceutical Chemistry, National Hellenic Research Foundation, Vasileos Constantinou 48,
GR±11635 Athens, Greeceb Chemical Engineering Department, National Technical University of Athens, Zografou Campus, GR±15773 Athens, Greece
Received 14 June 2000; in ®nal form 25 October 2000
Abstract
Macroscopic ®rst- and third-order susceptibilities of ice Ih, ice II, ice IX and ice VIII are calculated using static and
frequency-dependent electronic and static vibrational molecular (hyper)polarizabilities at the MP2 level. The molecular
properties are in good agreement with experiment and with high-level ab initio calculations. Intermolecular electrostatic
and polarization e�ects due to induced dipoles are taken into account using a rigorous local-®eld theory. The electric
®eld due to permanent dipoles is used to calculate e�ective in-crystal (hyper)polarizabilities. The polarizability depends
only weakly on the permanent ®eld, but the dipole moment and the hyperpolarizabilities are strongly a�ected. The
calculated linear susceptibility is in good agreement with available experimental data for ice Ih, and the third-order
susceptibility for a third harmonic generation experiment is in reasonable agreement with experimental values for liquid
water. The molecular vibrational contributions have a small e�ect on the susceptibilities. The electric properties of a
water tetramer are calculated and used to estimate the e�ect of non-dipolar interactions on the susceptibilities of ice Ih,
which are found to be small. Ó 2001 Elsevier Science B.V. All rights reserved.
1. Introduction
The computational prediction and experimentaldetermination of electric properties of a watermolecule in various environments has been anactive ®eld of research for quite a long time. Forthe free molecule, accurately determined experi-mental dipole and quadrupole moments and the(hyper)polarizabilities are known and could be
reproduced by ab initio calculations, if reasonablyhigh correlation levels and large basis sets wereused [1±3]. Experimental determination of thecorresponding e�ective properties in condensedphases, on the other hand, is much more di�cultand rests on a number of assumptions and ap-proximations whose limitations are di�cult to as-sess. The strong associated character and theextended hydrogen-bonded network formed inliquid water e.g. may raise doubts about the ap-plicability of the usual continuum models to ex-tract (hyper)polarizabilities from corresponding(non)linear experimental measurements.
A complementary strategy is o�ered by com-putational approaches, e.g. classical or ab initio
Chemical Physics 263 (2001) 301±316
www.elsevier.nl/locate/chemphys
* Corresponding author. Fax: +30-17273831.
E-mail addresses: [email protected] (H. Reis), [email protected]
(M.G. Papadopoulos).1 Also corresponding author.
0301-0104/01/$ - see front matter Ó 2001 Elsevier Science B.V. All rights reserved.
PII: S03 0 1-0 1 04 (0 0 )0 03 6 7- 0

molecular dynamics [4,5], supermolecule calcula-tions [6,7] for liquid water and periodic Hartree±Fock [8] or local-®eld methods [2,9±11] for ices.With the latter methods, which in principle arealso applicable to liquids if a corresponding sim-ulated liquid structure is available [12], it is pos-sible to use the molecular properties of an isolatedmolecule in order to calculate the environmentale�ect caused by the surrounding charge distribu-tions. The charge distributions are usually approx-imated by mono-centered or distributed multipoles,and environmental e�ects arising from electro-static and polarization e�ects can be calculated,taking full account of the discrete nature of thecondensed phase [13±15]. In contrast to periodicHartree±Fock methods, it is possible to use highlyaccurate molecular properties as input. On theother hand, no short-range e�ects like quantum-mechanical exchange or charge-transfer e�ects aretaken into account, although they may be con-sidered at least approximately using supermoleculeapproaches [16].
Most applications of local-®eld methods on icehave concentrated on a calculation of the in-crys-tal molecular dipole moment and the permanentelectric ®eld in ice Ih. Recently, Batista et al. havepublished a thorough investigation of these prop-erties [2,9,10], and have concluded that a reliabledescription of the electric ®eld is possible if mul-tipolar expansions up to hexadecapoles are used,although the predicted dipole moments dependvery much on the scheme used for distributing themultipoles over the molecule. However, all theirconclusions rested on purely theoretical calcula-tions and no comparison with experimental datawas done, probably due to the rather indirect in-¯uence of the properties investigated on measur-able macroscopic properties of ice.
Another well-established use of discrete local-®eld methods for molecular crystals is the cal-culation of macroscopic linear and non-linearsusceptibilities [13,14]. Here, the prediction of ex-perimentally measurable quantities, together withthe use of accurate molecular input data allows amore detailed assessment of the reliability of theelectrostatic intermolecular potential used for thecrystal in question. We will apply this methodhere to predict the susceptibilities of several ice
polymorphs. The choice of this system has beenpartly motivated by the smallness of the molecule,which allows the accurate calculation of the mo-lecular properties with reasonable calculationalcost. A further advantage is that the single pointapproximation for the expansion of the chargedistribution can be expected to work well for sucha small system. The dipolar approximation will beused here, which has been found in previous workto yield accurate predictions for other crystals [16±19]. The in¯uence of non-dipolar interactions willbe assessed approximately by applying the local-®eld method to the molecular properties of acluster of four water molecules. For the ®rsttime, we will also take into account the e�ect ofmolecular vibrational contributions on the sus-ceptibilities.
2. Methods
A method for calculating the macroscopic op-tical susceptibilities v�1�, v�2�, and v�3� of molecular
crystals using molecular properties as input hasbeen developed by Hurst and Munn [13,14]. Thetheory has been extensively treated earlier (seeRefs. [13,14,16,20] and references therein) andonly a short summary will be given here. The local®eld at the site of a molecule is calculated bysumming over the ®eld contributions from allsurrounding molecules in the crystal. The inducedcharge density distribution of the molecules is ei-ther approximated by point dipoles or in a morepartitioned way, placing identical point subdipolesat some suitably de®ned submolecules, in order totake into account the size of large molecules [21].For small molecules the point dipole approxima-tion yields essentially the same results as morepartitioned models [16,17]. For the water moleculewe will therefore use the point dipole approxima-tion.
For frequency-dependent external ®elds thesusceptibilities are given by
v�1��ÿx0; x0� � ��0v�ÿ1X
k
ak�ÿx0; x0�dk
�x0�;
�1�
302 H. Reis et al. / Chemical Physics 263 (2001) 301±316

where the sums run over all Z molecules in the unitcell of volume v, the output frequency x0 is thesum of the respective input frequencies, I123 is aninterchange operator generating all terms obtainedby interchanging x1, x2 and x3, together withtheir associated cartesian indices. The hyperpo-larizabilities are de®ned according to a Taylorexpansion of the dipole moment. L
kk0 are Lorentz-factor tensors, i.e. lattice dipole sums that give the®eld at a particular molecule k due to dipole mo-ments on all molecules of type k0. They are calcu-lated by Ewald summations [22]. The quantityd
k�x� is a local-®eld tensor given by
dk�x� �
Xk0
Dkk0 �x�
�X
k0Ihÿ L � a� ÿ x; x�=��0v�
iÿ1
kk0: �4�
Here the bold-faced matrices L, I, a are of order3Z, I is a generalized unit tensor with 3� 3 sub-tensors 1dkk0 , while L and a�ÿx; x� have subma-trices L
kk0 and ak�ÿx; x�dkk0 , respectively.
The components of the ®rst-order susceptibilityv�1�ii in the main axis system of the indicatrix arerelated to the refractive indices ni by
v�1�ii � n2i ÿ 1: �5�
The molecular (hyper)polarizabilities occurringin Eqs. (1)±(4) are those of a molecule subjected tothe permanent electric crystal ®eld F k0 occurring atthe molecular site in a crystal without applied ex-
ternal ®eld [16]. In the dipole approximation this®eld is given by [23]
F k0 � ��0v�ÿ1Xk0k00
Dkk0 � Lk0k00 � lk00 ; �6�
where lk00 is the permanent dipole moment of the
free molecule. The right-hand side of the equationdepends implicitly on F k0, because the polariz-ability occurring in D
kk0 (compare Eq. (4)) refers toF k0. The ®elds and the molecular properties havetherefore to be determined by a self-consistentprocedure, as explained in Ref. [16]: the molecularproperties without crystal ®eld are used to calcu-late a ®rst approximation to F k0, which is used asadditional electric ®eld in a second round to cal-culate the molecular properties a, b and c. Thisprocess is repeated until the absolute ®eld changebetween successive iterations is lower than a giventhreshold, which was chosen here to be 1%.
The crystal structure necessary for the calcula-tion of crystal susceptibilities is generally takenfrom experiment. A large variety of ice polymor-phs are known, with a degree of proton-orderingranging from complete ordering to complete dis-ordering, while in general the oxygen atoms are®xed in translationally equivalent places. We havechosen to calculate the linear and non-linear op-tical susceptibilities of the two proton-orderedhigh pressure forms ice VIII [24] and ice II [25±27],the nearly proton-ordered low-temperature formice IX [28] and the proton-disordered form ice Ih,which is the stable form at atmospheric pressure
v�2��ÿx0; x1;x2� � �2�0v�ÿ1X
k
bk�ÿx0; x1;x2�..
.d
k�x2�dk
�x1�dk�x0�
h i; �2�
v�3��ÿx0; x1;x2;x3� � v�3�
direct
�ÿx0; x1;x2;x3� � v�3�
cascading
�ÿx0; x1;x2;x3�
� �6�0v�ÿ1X
k
ck
�ÿx0; x1;x2;x3����� dk�x3�dk
�x2�dk�x1�dk
�x0�h i
� �2�0v�ÿ2 I123
Xkk0k00
dTk�x0�dT
k�x1�
h i: b
k�ÿx0; x1;xi�
� Dkk0 �xi� � Lk0k00 � b
k00�ÿxi; x2;x3� : d
k00�x2�dk00
�x3�h i
; �3�
H. Reis et al. / Chemical Physics 263 (2001) 301±316 303

and temperatures below the melting point. Ice IIcrystallizes in the rhombohedral space group R�3and there are 36 molecules per unit cell in thecorresponding hexagonal setting, with dimensionsa � 12:920 �A, c � 6:234 �A at P � 3 kbar andT � 233 K [25], ice VIII crystallizes in the tetra-gonal space group I41=amd, with eight molecules ina unit cell of dimensions a � 4:6779 �A, c � 6:8029�A at P � 28 kbar and T � 269 K [24] and thespace group of ice IX is also tetragonal with spacegroup P41212, and 12 molecules per unit cellof dimensions a � 6:73 �A, c � 6:83 �A at P � 3kbar and T � 77 K [28]. Ice IX has been treatedhere as completely proton ordered, although is hasbeen reported to be slightly disordered [28]. Thebasic unit cell of ice Ih is hexagonal [29], with fourmolecules per unit cell. Two of these unit cells canbe combined to form a eight-molecule unit cellwith orthorhombic symmetry [30]. In order to takeinto account the proton disordering in a more re-alistic way it is necessary to use larger unit cellswhich are then replicated by translation to gener-ate the crystal. Generally, these larger unit cells areconstructed in such a way as to satisfy severalconstraints, such as vanishing total dipole mo-ment, randomness of hydrogen bonding etc. Alarge set of unit cells with di�erent number ofwater molecules and di�erent unit cell dimensionshave been carefully determined by Hayward andReimers [30] according to di�erent constraints.For our calculations, we have used three of theirunit cells, which are available from the world wideweb [30], namely two unit cells with 96 molecules(3� 2� 2-CH and 3� 2� 2-C2 in the notation ofHayward and Reimers), with unit cell parametersa � 13:52 �A, b � 15:61 �A, c � 14:72 �A and oneunit cell with 192 molecules �4� 3� 2� with pa-rameters a � 18:03 �A, b � 23:42 �A, c � 14:72 �A.No attempt has been made to take into accountthat ice Ih is a solid rotator state of ice [31,32] withconsiderable reorientational degrees of freedomgiving rise to an orientational polarization thatincreases the permitivities � in the low-frequencylimit considerably over that expected from pureelectronic polarization. Whenever static suscepti-bilities are reported here, they should be comparedwith experimental susceptibilities at optical fre-quencies extrapolated to zero frequency.
The geometry used for the calculation of theelectronic contributions is the optimized geometryat the MP2 level with the HyPol basis set, and isgiven by rOH � 0:9668 �A and an HOH angle of103.8°. The molecular z-axis lies along the molec-ular dipole moment, pointing from oxygen to-wards the hydrogen atoms, the y-axis lies in theplane of the molecule and the x-axis completes thecartesian axis system. The MP2/HyPol minimumgeometry di�ers from the experimentally deter-mined geometry, especially in ice Ih, where a bondlength of 1.0 �A and an angle of 109.5° hasbeen reported [33]. It has been shown recently thatthe electronic contribution pel to an electric prop-erty at a non-optimum geometry Q0 can be givenapproximately by [34,35]:
pel�Q0� � pel�Qopte � � pgc�Q0�; �7�
where Qopte denotes the optimized geometry, cal-
culated with the speci®c basis set and level ofcorrelation considered, and pgc�Q0� is the so-calledgeometry correction term which involves ®rstgradient terms with respect to the geometry coor-dinates. At the equilibrium geometry in ice theseterms would vanish and the best approximationfor the experimental electronic properties pel;exp
at the experimental minimum Qexpe is therefore
pel;exp�Qexpe � � pel�Qopt
e �.In the Born±Oppenheimer approximation the
total molecular electric property ptot can be ap-proximately given by ptot � pel � pvib, where pvib isthe vibrational contribution to ptot. In previouscalculations of crystal susceptibilities, vibrationalcontributions have been completely ignored, al-though for free molecules they may be far fromnegligible compared with the electronic contri-butions [36]. We will therefore take into accountthe e�ect of the vibrational contributions on thecrystal susceptibilities of ice. To this e�ect weconsider the division of these contributions intothe so-called pure vibrational (PV) part and thepart emanating from the zero point vibrationalaveraging (ZPVA). The PV contributions can bevery large in the static limit, but tend to be verysmall if only optical ®elds are applied, and may beneglected in this case. The ZPVA on the otherhand follows a dispersion behavior that is similar
304 H. Reis et al. / Chemical Physics 263 (2001) 301±316

to that of the electronic properties [36], andtherefore can not be neglected even for all-optical®elds. We will only calculate the static contribu-tions here, and do not attempt to calculate electric®eld e�ects. The di�erent frequency dependence ofthe two contributions is taken approximately intoaccount by adding both the pure contribution andthe ZPVA to the electronic contributions to cal-culate the permanent crystal ®eld, but addingonly the ZPVA contribution for the calculation ofthe susceptibilities. The vibrational contributionswere calculated with the Pol basis set at the MP2level, using the corresponding optimized geome-try, which with rOH � 0:9681, \HOH � 103:5° [35]di�ers only marginally from the minimum ob-tained with MP2/HyPol. All terms up to m� n � 2were considered in the perturbation series forthe properties, where n is the order of electricalanharmonicity and m is the order of mechanicalanharmonicity [36].
The electronic property components were com-puted by employing the ®nite ®eld approach, usinga ®eld value of F � 0:003 a.u., if no additionalpermanent crystal ®elds were applied. The ade-quacy of this value has been checked by comparingthe ®nite ®eld results at the SCF level with resultsobtained at the coupled perturbed Hartree±Focklevel. For the calculation of the properties in thedipolar crystal ®eld, a smaller ®eld value of F �0:001 a.u. had to be used, due to the increasinglynon-linear dependence of the properties with in-creasing applied permanent ®eld.
The computations of the electronic propertieshave been performed with the GAUSSIAN 98GAUSSIAN 98 [37]and the DALTONDALTON [38] packages. For the compu-tation of the vibrational contributions the recentlydeveloped numerical derivative package NUMDERNUMDER
[39] implemented in CADPACCADPAC [40] has been used.The static electronic properties of the water mol-ecule have been calculated with two medium-sizedbasis sets, the Pol basis set, developed by Sadlej[41] for the accurate calculation of polarizabilities,and with the HyPol set, which is an extension ofthe Pol basis set, developed by Pluta and Sadlej[42] for the accurate calculation of hyperpolariz-abilities. The calculations with the HyPol basis sethave been performed using spherical basis func-tions, in order to eliminate large linear dependen-
cies that occur using cartesian function sets. ThePol basis set has been used for the vibrationalcontributions and cartesian basis sets were em-ployed, because CADPACCADPAC does not allow the use ofspherical basis sets. However, the use of cartesianbasis sets leads only to two additional basis func-tions on the oxygen atom, which do not lead toproblems due to linear dependencies among thebasis functions.
Correlation e�ects to the electronic propertieshave been taken into account using the methodsMP2 [43] and CCSD(T) [44], while MP2 was usedfor the vibrational contributions.
3. Results and discussion
The calculated electronic and vibrational con-tributions to the electric properties are given inTable 1. For the electronic polarizability both basissets yield very similar results at all levels of theoryconsidered, but for the ®rst and second hyperpo-larizabilities the two basis sets yield substantiallydi�erent values. The values obtained with theHyPol basis set at the SCF level are much closerto the near Hartree±Fock limit as reported byMaroulis [1]. His values for the quantities bk andcav are ÿ11:07 and 985 a.u., respectively, in goodagreement with our results for the HyPol basisset. As also observed already by Maroulis [1], thevalues of the properties calculated at the MP2 levelare very close to those obtained at the CCSD(T)level. The vibrational contributions calculated byus are in general comparable with other resultsgiven in the literature [35,45±47]. The di�erencescan be explained by di�erences in the order ofmechanical/electrical anharmonicities which weretaken into account, di�erent basis sets and/or dif-ferent level of electronic correlation considered.
In Table 2 we show the average frequency-dependent properties aav�ÿx; x�, bk�ÿ2x; x;x�,cav�ÿ2x; x;x; 0� and cav�ÿ3x; x;x;x�, calculatedat the often used laser frequency corresponding tothe wavelength k � 1064 nm. The frequency de-pendence was computed at the SCF level, em-ploying the random-phase approximation (RPA).In order to incorporate approximately the e�ect of
H. Reis et al. / Chemical Physics 263 (2001) 301±316 305
electronic correlation, we used a simple multipli-cative correction [48]:
aii�MP2;x� � aii�SCF;x�aii�SCF; 0� aii�MP2; 0�; �8�
and similarly for bijj, ciijj. The resulting averagevalues are also shown in Table 2, and are com-pared both with available experimental gas-phasevalues and with results calculated from dispersionequations published by Christiansen et al. [3],
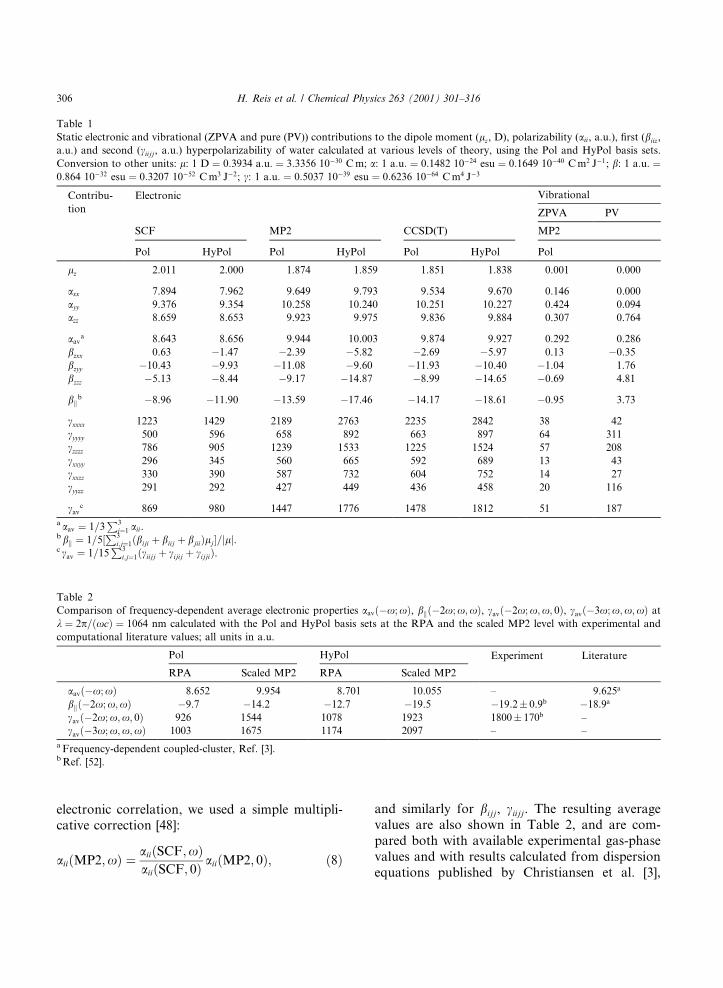
Table 1
Static electronic and vibrational (ZPVA and pure (PV)) contributions to the dipole moment (lz, D), polarizability (aii, a.u.), ®rst (biiz,
a.u.) and second (ciijj, a.u.) hyperpolarizability of water calculated at various levels of theory, using the Pol and HyPol basis sets.
Conversion to other units: l: 1 D � 0:3934 a:u: � 3:3356 10ÿ30 C m; a: 1 a:u: � 0:1482 10ÿ24 esu � 0:1649 10ÿ40 C m2 Jÿ1; b: 1 a:u: �0:864 10ÿ32 esu � 0:3207 10ÿ52 C m3 Jÿ2; c: 1 a:u: � 0:5037 10ÿ39 esu � 0:6236 10ÿ64 C m4 Jÿ3
Contribu-
tion
Electronic Vibrational
ZPVA PV
SCF MP2 CCSD(T) MP2
Pol HyPol Pol HyPol Pol HyPol Pol
lz 2.011 2.000 1.874 1.859 1.851 1.838 0.001 0.000
axx 7.894 7.962 9.649 9.793 9.534 9.670 0.146 0.000
ayy 9.376 9.354 10.258 10.240 10.251 10.227 0.424 0.094
azz 8.659 8.653 9.923 9.975 9.836 9.884 0.307 0.764
aava 8.643 8.656 9.944 10.003 9.874 9.927 0.292 0.286
bzxx 0.63 ÿ1.47 ÿ2.39 ÿ5.82 ÿ2.69 ÿ5.97 0.13 ÿ0.35
bzyy ÿ10.43 ÿ9.93 ÿ11.08 ÿ9.60 ÿ11.93 ÿ10.40 ÿ1.04 1.76
bzzz ÿ5.13 ÿ8.44 ÿ9.17 ÿ14.87 ÿ8.99 ÿ14.65 ÿ0.69 4.81
bkb ÿ8.96 ÿ11.90 ÿ13.59 ÿ17.46 ÿ14.17 ÿ18.61 ÿ0.95 3.73
cxxxx 1223 1429 2189 2763 2235 2842 38 42
cyyyy 500 596 658 892 663 897 64 311
czzzz 786 905 1239 1533 1225 1524 57 208
cxxyy 296 345 560 665 592 689 13 43
cxxzz 330 390 587 732 604 752 14 27
cyyzz 291 292 427 449 436 458 20 116
cavc 869 980 1447 1776 1478 1812 51 187
a aav � 1=3P3
i�1 aii.b bk � 1=5�P3
i;j�1�biji � biij � bjii�lj�=jlj.c cav � 1=15
P3i;j�1�ciijj � cijij � cijji�.
Table 2
Comparison of frequency-dependent average electronic properties aav�ÿx; x�, bk�ÿ2x; x;x�, cav�ÿ2x; x;x; 0�, cav�ÿ3x; x;x;x� at
k � 2p=�xc� � 1064 nm calculated with the Pol and HyPol basis sets at the RPA and the scaled MP2 level with experimental and
computational literature values; all units in a.u.
Pol HyPol Experiment Literature
RPA Scaled MP2 RPA Scaled MP2
aav�ÿx; x� 8.652 9.954 8.701 10.055 ± 9.625a
bk�ÿ2x; x;x� ÿ9.7 ÿ14.2 ÿ12.7 ÿ19.5 ÿ19.2� 0.9b ÿ18.9a
cav�ÿ2x; x;x; 0� 926 1544 1078 1923 1800� 170b ±
cav�ÿ3x; x;x;x� 1003 1675 1174 2097 ± ±
a Frequency-dependent coupled-cluster, Ref. [3].b Ref. [52].
306 H. Reis et al. / Chemical Physics 263 (2001) 301±316

which were obtained using frequency-dependentcoupled cluster calculations. 2 The hyperpolariz-abilities obtained with the HyPol basis set at thescaled MP2 level in general compare well withexperiment and the highly accurate coupled clustervalues of Ref. [3], and perform much better thanthose obtained with the Pol basis set. For the po-larizability aav both basis sets give again nearly thesame values.
As a result of the comparison between ourcalculated values and experimental and other abinitio results, we chose to use the electronic prop-erties calculated with the HyPol basis set at thescaled MP2 level as input values for the permanentcrystal ®eld and the susceptibility calculations.
The results for the permanent local electric ®eldin dipole approximation at a water molecule forthe di�erent ice polymorphs investigated are givenin Table 3, expressed in the molecular frame. Amaximum of two iterations was necessary to yieldconverged values for the ®eld. In the case of ice IIand ice IX there are two symmetry-independenttypes of water molecules in non-equivalent envi-ronments (identi®ed by Mol. 1 and Mol. 2 in Table3 and the following tables), which consequentlyexperience di�erent local ®elds and acquire di�er-ent molecular properties. Those di�erences have
been fully taken into account for the two ices. Forproton-disordered ice Ih the ®elds in general aredi�erent for each molecule, and the ®eld given inTable 3 is the local ®eld averaged over the mole-cules in the unit cell. The results for the three largeunit cells used for ice Ih di�ered only slightly, asindicated by the standard deviation given in Table3. Therefore we used only one cell in the subse-quent calculations, namely the cell 3� 2� 2-C2 inthe notation of Hayward and Reimers [30]. The®eld components in the molecular x and y direc-tion were considered to be not signi®cantly di�erentfrom zero and were neglected for the recalcula-tion of the molecular in-®eld properties for ice Ih.Generally, the calculated ®elds increase with in-creasing mass density of the investigated poly-morphs, except for the polymorph with the highestdensity, ice VIII, where the dipolar ®eld is with0.49 GV mÿ1 very small compared to the dipolar®eld in the other polymorphs. This shows thatfor ice VIII the dipole±dipole interactions playa negligible role for the energetic stability of thecrystal.
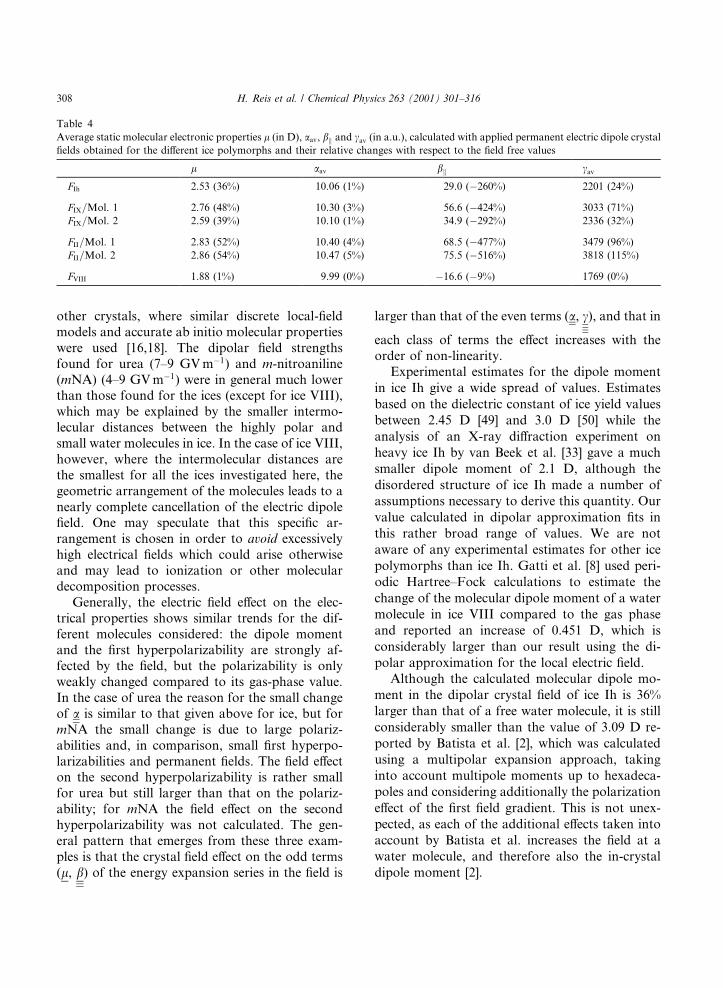
The vibrational contributions always lead to anenhancement of the ®eld, although the e�ect isquite small: using only the electronic contributionsleads to �5% smaller ®eld values.
The e�ect of the dipolar ®elds on the averagemolecular electric properties is shown in Table 4.The ®eld e�ect especially on the ®rst hyperpolar-izability, but also on the dipole moment and thesecond hyperpolarizability is seen to be quite large,while the e�ect on the polarizability is rathersmall. The small ®eld dependence of a can be un-derstood in terms of a series expansion in theexternal ®eld F :
a�F � � a0� b
0
F � 1=2c0
: F F ; �9�
where the subscript 0 denotes the respectivequantities at zero ®eld. The second term and thethird term are of opposite signs at the electric ®eldsoccurring here, and are of the same order ofmagnitude due to the small absolute values of b
0
for water, thus roughly canceling each other.
The permanent crystal ®eld and its e�ects onthe molecular in-crystal electric properties in icesmay be compared to those reported previously for
Table 3
Local electric crystal ®eld (F , GV mÿ1) at a water molecule,
expressed in the molecular frame, calculated in dipolar ap-
proximation
Fx Fy Fz
Ice Ih Aver.a ;b ÿ0.1� 0.2 0.2� 0.2 12.3� 0.4
Mol. 1 3.49 1.40 17.93
Ice IX
Mol. 2 0.00 0.00 14.87
Mol. 1 3.63 ÿ2.56 19.32
Ice II
Mol. 2 5.04 1.09 19.71
Ice VIII 0.00 0.00 0.49
a The average ®eld over the molecules in the unit cell.b The values for Ice Ih are averages for three di�erent simulated
unit cells with the corresponding standard deviation, see text.
2 The factor ÿ21:38 in Eq. (7) of Ref. [3] was changed to
ÿ17:38.
H. Reis et al. / Chemical Physics 263 (2001) 301±316 307
other crystals, where similar discrete local-®eldmodels and accurate ab initio molecular propertieswere used [16,18]. The dipolar ®eld strengthsfound for urea (7±9 GV mÿ1) and m-nitroaniline(mNA) (4±9 GV mÿ1) were in general much lowerthan those found for the ices (except for ice VIII),which may be explained by the smaller intermo-lecular distances between the highly polar andsmall water molecules in ice. In the case of ice VIII,however, where the intermolecular distances arethe smallest for all the ices investigated here, thegeometric arrangement of the molecules leads to anearly complete cancellation of the electric dipole®eld. One may speculate that this speci®c ar-rangement is chosen in order to avoid excessivelyhigh electrical ®elds which could arise otherwiseand may lead to ionization or other moleculardecomposition processes.
Generally, the electric ®eld e�ect on the elec-trical properties shows similar trends for the dif-ferent molecules considered: the dipole momentand the ®rst hyperpolarizability are strongly af-fected by the ®eld, but the polarizability is onlyweakly changed compared to its gas-phase value.In the case of urea the reason for the small changeof a is similar to that given above for ice, but formNA the small change is due to large polariz-abilities and, in comparison, small ®rst hyperpo-larizabilities and permanent ®elds. The ®eld e�ecton the second hyperpolarizability is rather smallfor urea but still larger than that on the polariz-ability; for mNA the ®eld e�ect on the secondhyperpolarizability was not calculated. The gen-eral pattern that emerges from these three exam-ples is that the crystal ®eld e�ect on the odd terms(l, b) of the energy expansion series in the ®eld is
larger than that of the even terms (a, c), and that in
each class of terms the e�ect increases with theorder of non-linearity.
Experimental estimates for the dipole moment
in ice Ih give a wide spread of values. Estimates
based on the dielectric constant of ice yield values
between 2.45 D [49] and 3.0 D [50] while the
analysis of an X-ray di�raction experiment on
heavy ice Ih by van Beek et al. [33] gave a much
smaller dipole moment of 2.1 D, although the
disordered structure of ice Ih made a number of
assumptions necessary to derive this quantity. Our
value calculated in dipolar approximation ®ts in
this rather broad range of values. We are not
aware of any experimental estimates for other ice
polymorphs than ice Ih. Gatti et al. [8] used peri-
odic Hartree±Fock calculations to estimate the
change of the molecular dipole moment of a water
molecule in ice VIII compared to the gas phase
and reported an increase of 0.451 D, which is
considerably larger than our result using the di-
polar approximation for the local electric ®eld.Although the calculated molecular dipole mo-
ment in the dipolar crystal ®eld of ice Ih is 36%
larger than that of a free water molecule, it is still
considerably smaller than the value of 3.09 D re-
ported by Batista et al. [2], which was calculated
using a multipolar expansion approach, taking
into account multipole moments up to hexadeca-
poles and considering additionally the polarization
e�ect of the ®rst ®eld gradient. This is not unex-
pected, as each of the additional e�ects taken into
account by Batista et al. increases the ®eld at a
water molecule, and therefore also the in-crystal
dipole moment [2].
Table 4
Average static molecular electronic properties l (in D), aav, bk and cav (in a.u.), calculated with applied permanent electric dipole crystal
®elds obtained for the di�erent ice polymorphs and their relative changes with respect to the ®eld free values
l aav bk cav
FIh 2.53 (36%) 10.06 (1%) 29.0 (ÿ260%) 2201 (24%)
FIX=Mol. 1 2.76 (48%) 10.30 (3%) 56.6 (ÿ424%) 3033 (71%)
FIX=Mol. 2 2.59 (39%) 10.10 (1%) 34.9 (ÿ292%) 2336 (32%)
FII=Mol. 1 2.83 (52%) 10.40 (4%) 68.5 (ÿ477%) 3479 (96%)
FII=Mol. 2 2.86 (54%) 10.47 (5%) 75.5 (ÿ516%) 3818 (115%)
FVIII 1.88 (1%) 9.99 (0%) ÿ16.6 (ÿ9%) 1769 (0%)
308 H. Reis et al. / Chemical Physics 263 (2001) 301±316

Most of the reports on electric (hyper)polariz-abilities of a water molecule in the condensedphase have been made on liquid water. A signchange of bk in going from the gas to the liquidphase has been reported from EFISH measure-ments [51,52]. This is also observed here for thesolid phases, except for ice VIII. Ab initio calcu-lations on clusters of water molecules simulatingthe structure of liquid water [7] and moleculardynamics simulations using a quantum-mechani-cal method that accounts for the electronic struc-ture variation of the bulk [5] both yield a decrease
of the molecular mean polarizability in liquidwater compared to the gas-phase value, whiletaking into account only the e�ect of the electric®eld arising from the surrounding molecules leadsgenerally to an increase of the polarizability [7], ashas been found here for the ices, too. Only in therange 0±10 GV mÿ1 the mean polarizability isslightly smaller than that of the free molecule [7].This is responsible for the small decrease of aav forice VIII.
In Table 5 we show the average frequency-
dependent electric properties of a water molecule
calculated in the di�erent dipolar crystal ®elds
at the scaled MP2 level, together with the changes
of the frequency-dependent properties compared
to the static properties. Comparing with the ®eld
strengths given in Table 3 it can be seen that with
increasing ®eld strength the frequency dependence
becomes more pronounced, which in turn indicates
a ®eld-induced lowering of the transition energies
�hxgj for dipole-allowed electronic transitions be-
tween the ground state g and one or several excitedstates j.
In Table 6 the calculated ®rst-order suscepti-bility v�1� in the static limit and at the frequenciescorresponding to k � 1064 and 532 nm and, for iceIh, k � 623:9 nm are shown, together with avail-able experimental results taken from the literature.The values have been calculated with and withoutinclusion of the permanent electric crystal ®eld F k0
in dipolar approximation. Comparing the di�erentphases for each kind of ®eld approximation, thetrends in magnitude of the ®rst susceptibilitiesfollow closely the trend in the mass density q,which increases in the order ice Ih > Ice IX J IceII > Ice VIII [53].
Generally, consideration of the local-®eld e�ectleads to modest enhancements of the componentsv�1�ii . The largest increases occur for ice II and donot exceed 8%.
To our knowledge, experimental values for v�1�
or for the refractive indices ni (see Eq. (5)), of theices investigated here are only available for ice Ih.Minton [54] reports values for the refractive indi-ces ni at k � 623:9 nm and ÿ65°C, which corre-sponds to a density of q � 0:9237 g cmÿ3 andcoincides very well with that of the crystal struc-ture used for our calculations, q � 0:9234 g cmÿ3.The corresponding ®rst susceptibilities di�er by5% and 6% from our calculated values withoutand with the e�ect of the dipolar ®eld, respec-tively. A more recent compilation of refractive datafrom polycrystalline ice Ih at 266 K has been pub-lished by Warren [55], collecting data from several
Table 5
Average frequency-dependent electronic properties aav�ÿx; x�, aav�ÿ2x; 2x�, aav�ÿ3x; 3x�, bk�ÿ2x; x;x� and cav�ÿ3x; x;x;x�in a:u:��, �k � 2pc=x � 1064 nm� at the scaled MP2 level, calculated with applied permanent electric dipole crystal ®elds obtained for
the di�erent ice polymorphs and their relative changes with respect to the static values
aav�ÿx; x� aav�ÿ2x; 2x� aav�ÿ3x; 3x� bk�ÿ2x; x;x� cav�ÿ3x; x;x;x�FIh 10.11 (0%) 10.28 (2%) 10.58 (5%) 31.5 (9%) 2691 (22%)
FIX=Mol. 1 10.36 (1%) 10.54 (2%) 10.88 (6%) 63.9 (13%) 3994 (32%)
FIX=Mol. 2 10.16 (1%) 10.33 (2%) 10.64 (5%) 37.7 (8%) 2887 (24%)
FII=Mol. 1 10.46 (1%) 10.66 (3%) 11.02 (6%) 77.3 (13%) 4770 (37%)
FII=Mol. 2 10.53 (1%) 10.73 (2%) 11.11 (6%) 85.7 (14%) 5403 (42%)
FVIII 10.05 (1%) 10.21 (2%) 10.50 (5%) ÿ17.9 (8%) 2087 (18%)
H. Reis et al. / Chemical Physics 263 (2001) 301±316 309
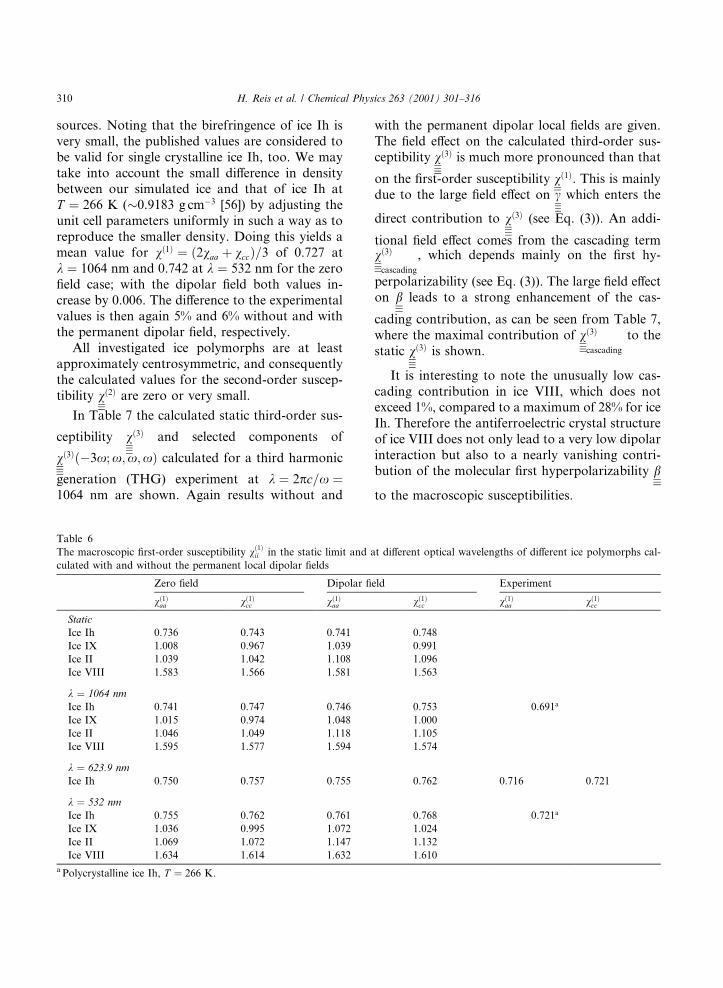
sources. Noting that the birefringence of ice Ih isvery small, the published values are considered tobe valid for single crystalline ice Ih, too. We maytake into account the small di�erence in densitybetween our simulated ice and that of ice Ih atT � 266 K (�0.9183 g cmÿ3 [56]) by adjusting theunit cell parameters uniformly in such a way as toreproduce the smaller density. Doing this yields amean value for v�1� � �2vaa � vcc�=3 of 0.727 atk � 1064 nm and 0.742 at k � 532 nm for the zero®eld case; with the dipolar ®eld both values in-crease by 0.006. The di�erence to the experimentalvalues is then again 5% and 6% without and withthe permanent dipolar ®eld, respectively.
All investigated ice polymorphs are at leastapproximately centrosymmetric, and consequentlythe calculated values for the second-order suscep-tibility v�2� are zero or very small.
In Table 7 the calculated static third-order sus-
ceptibility v�3� and selected components of
v�3��ÿ3x; x;x;x� calculated for a third harmonic
generation (THG) experiment at k � 2pc=x �1064 nm are shown. Again results without and
with the permanent dipolar local ®elds are given.The ®eld e�ect on the calculated third-order sus-ceptibility v�3� is much more pronounced than that
on the ®rst-order susceptibility v�1�. This is mainlydue to the large ®eld e�ect on c which enters the
direct contribution to v�3� (see Eq. (3)). An addi-
tional ®eld e�ect comes from the cascading termv�3�
cascading
, which depends mainly on the ®rst hy-
perpolarizability (see Eq. (3)). The large ®eld e�ecton b leads to a strong enhancement of the cas-
cading contribution, as can be seen from Table 7,where the maximal contribution of v�3�
cascading
to thestatic v�3� is shown.
It is interesting to note the unusually low cas-cading contribution in ice VIII, which does notexceed 1%, compared to a maximum of 28% for iceIh. Therefore the antiferroelectric crystal structureof ice VIII does not only lead to a very low dipolarinteraction but also to a nearly vanishing contri-bution of the molecular ®rst hyperpolarizability b
to the macroscopic susceptibilities.
Table 6
The macroscopic ®rst-order susceptibility v�1�ii in the static limit and at di�erent optical wavelengths of di�erent ice polymorphs cal-
culated with and without the permanent local dipolar ®elds
Zero ®eld Dipolar ®eld Experiment
v�1�aa v�1�cc v�1�aa v�1�cc v�1�aa v�1�cc
StaticIce Ih 0.736 0.743 0.741 0.748
Ice IX 1.008 0.967 1.039 0.991
Ice II 1.039 1.042 1.108 1.096
Ice VIII 1.583 1.566 1.581 1.563
k � 1064 nm
Ice Ih 0.741 0.747 0.746 0.753 0.691a
Ice IX 1.015 0.974 1.048 1.000
Ice II 1.046 1.049 1.118 1.105
Ice VIII 1.595 1.577 1.594 1.574
k � 623.9 nm
Ice Ih 0.750 0.757 0.755 0.762 0.716 0.721
k � 532 nm
Ice Ih 0.755 0.762 0.761 0.768 0.721a
Ice IX 1.036 0.995 1.072 1.024
Ice II 1.069 1.072 1.147 1.132
Ice VIII 1.634 1.614 1.632 1.610
a Polycrystalline ice Ih, T � 266 K.
310 H. Reis et al. / Chemical Physics 263 (2001) 301±316
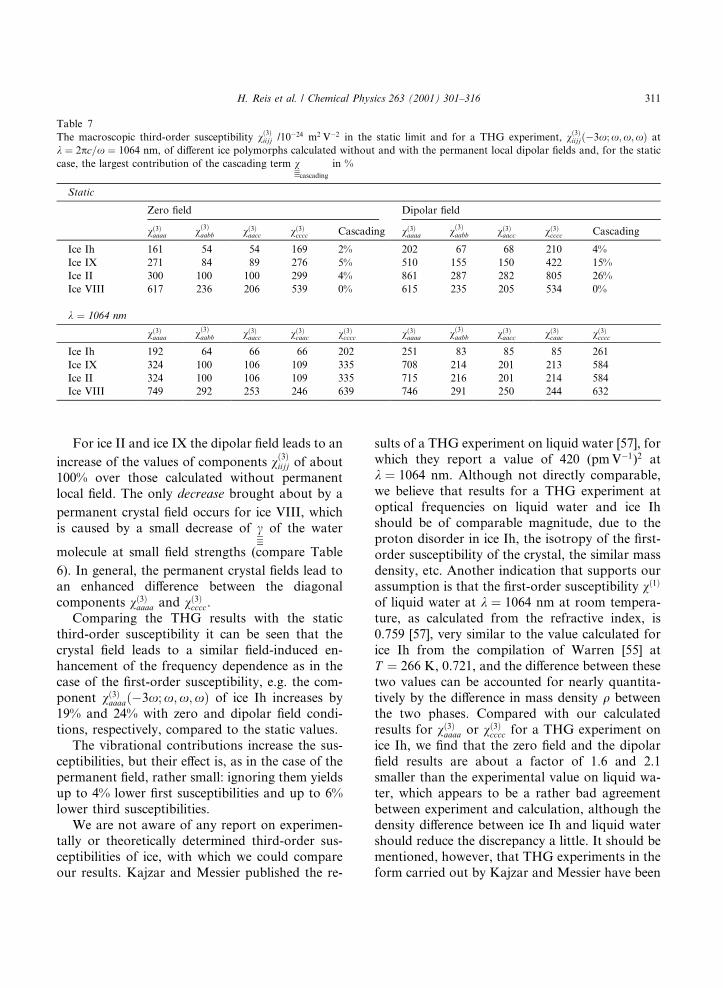
For ice II and ice IX the dipolar ®eld leads to an
increase of the values of components v�3�iijj of about100% over those calculated without permanentlocal ®eld. The only decrease brought about by a
permanent crystal ®eld occurs for ice VIII, whichis caused by a small decrease of c of the water
molecule at small ®eld strengths (compare Table
6). In general, the permanent crystal ®elds lead toan enhanced di�erence between the diagonalcomponents v�3�aaaa and v�3�cccc.
Comparing the THG results with the staticthird-order susceptibility it can be seen that thecrystal ®eld leads to a similar ®eld-induced en-hancement of the frequency dependence as in thecase of the ®rst-order susceptibility, e.g. the com-ponent v�3�aaaa�ÿ3x; x;x;x� of ice Ih increases by19% and 24% with zero and dipolar ®eld condi-tions, respectively, compared to the static values.
The vibrational contributions increase the sus-ceptibilities, but their e�ect is, as in the case of thepermanent ®eld, rather small: ignoring them yieldsup to 4% lower ®rst susceptibilities and up to 6%lower third susceptibilities.
We are not aware of any report on experimen-tally or theoretically determined third-order sus-ceptibilities of ice, with which we could compareour results. Kajzar and Messier published the re-
sults of a THG experiment on liquid water [57], forwhich they report a value of 420 (pm Vÿ1)2 atk � 1064 nm. Although not directly comparable,we believe that results for a THG experiment atoptical frequencies on liquid water and ice Ihshould be of comparable magnitude, due to theproton disorder in ice Ih, the isotropy of the ®rst-order susceptibility of the crystal, the similar massdensity, etc. Another indication that supports ourassumption is that the ®rst-order susceptibility v�1�
of liquid water at k � 1064 nm at room tempera-ture, as calculated from the refractive index, is0.759 [57], very similar to the value calculated forice Ih from the compilation of Warren [55] atT � 266 K, 0.721, and the di�erence between thesetwo values can be accounted for nearly quantita-tively by the di�erence in mass density q betweenthe two phases. Compared with our calculatedresults for v�3�aaaa or v�3�cccc for a THG experiment onice Ih, we ®nd that the zero ®eld and the dipolar®eld results are about a factor of 1.6 and 2.1smaller than the experimental value on liquid wa-ter, which appears to be a rather bad agreementbetween experiment and calculation, although thedensity di�erence between ice Ih and liquid watershould reduce the discrepancy a little. It should bementioned, however, that THG experiments in theform carried out by Kajzar and Messier have been
Table 7
The macroscopic third-order susceptibility v�3�iijj /10ÿ24 m2 Vÿ2 in the static limit and for a THG experiment, v�3�iijj�ÿ3x; x;x;x� at
k � 2pc=x � 1064 nm, of di�erent ice polymorphs calculated without and with the permanent local dipolar ®elds and, for the static
case, the largest contribution of the cascading term vcascading
in %
Static
Zero ®eld Dipolar ®eld
v�3�aaaa v�3�aabb v�3�aacc v�3�cccc Cascading v�3�aaaa v�3�aabb v�3�aacc v�3�cccc Cascading
Ice Ih 161 54 54 169 2% 202 67 68 210 4%
Ice IX 271 84 89 276 5% 510 155 150 422 15%
Ice II 300 100 100 299 4% 861 287 282 805 26%
Ice VIII 617 236 206 539 0% 615 235 205 534 0%
k � 1064 nm
v�3�aaaa v�3�aabb v�3�aacc v�3�caac v�3�cccc v�3�aaaa v�3�aabb v�3�aacc v�3�caac v�3�cccc
Ice Ih 192 64 66 66 202 251 83 85 85 261
Ice IX 324 100 106 109 335 708 214 201 213 584
Ice II 324 100 106 109 335 715 216 201 214 584
Ice VIII 749 292 253 246 639 746 291 250 244 632
H. Reis et al. / Chemical Physics 263 (2001) 301±316 311

considered to overestimate the third-order suscep-tibility substantially [58], due to uncertainties con-cerning the calibration process used to obtain thevalue for the reference standard, fused silica [59,60].Kajzar and Messier used the value reported byMeredith et al. [58], 2:79� 1014 esu. Using anotherreference value obtained by Mito et al. [60], 0:95�1014 esu the experimental value of Kajzar andMessier for THG of liquid water would dropto 140� 10ÿ24 (pm Vÿ1)2, which is smaller than ourvalues. As long as this uncertainty in the correctreference values remains, we only can state thatour values agree with the experimental values inthe rather large error limits of the latter.
As mentioned above, the electrostatic modelused does only take into account long-rangedipolar interactions. No short-range e�ects asquantum-mechanical exchange and intermolecularcharge-transfer e�ects are included, which mayarise e.g. in ices due to the extended hydrogen-bonded network formed by the molecules. Furthere�ects arising from multipoles higher than dipolehave been neglected, too. Attempts to take intoaccount these additional e�ects e.g. for the mo-lecular polarizability in liquid water have beenreported using a supermolecule approach [7], andhave found a substantial decrease of about 15%for the mean polarizability aav. Similarly, a largee�ect on the ®rst hyperpolarizability has beenfound by Mikkelsen et al. [6], using the super-molecule approach.
We have tried to get an estimate of the in¯uenceof these e�ects on the susceptibilities of ice bycalculating the electric properties of a cluster ofwater molecules and then using the electrostaticmodel to calculate the macroscopic susceptibilities.Comparing with the susceptibilities computed withthe electrostatic model applied to the properties ofa single molecule we can assess the in¯uence of theshort-range and non-dipolar e�ects in the clusteron the susceptibilities. We constructed an hypo-thetical ice Ih crystal, using the ®rst four moleculesof the 3� 2� 2-C2 unit cell of Hayward andReimers [30] as the cluster, for which the proper-ties were calculated. In order to get the eightmolecules necessary for the smallest possible or-thorhombic unit cell, the cluster was replicated bya mirror plane in the bc plane at a=2 and translated
by b=2 along b. In order to calculate the propertiesof the cluster at the MP2 level, we had to choosethe Pol basis set, because the HyPol basis setwould have been outside of our computationalresources. Vibrational properties were ignored.The diagonal components and the average valuesof the static electronic properties of the cluster atthe MP2 level are given in Table 8, together withthe properties of the same cluster composed offour non-interacting molecules, calculated bysumming up the molecular properties of a freemolecule. In general, the deviations caused by theintermolecular interactions for the single compo-nents are much larger than those of the averagevalues, except for b, where all changes are large,while the di�erences for the polarizabilities arequite small. The single components enter in theformulae for the susceptibilities and not the aver-age values. Therefore, larger di�erences for thepredicted susceptibilities than suggested by thesmall di�erences in the average values may stillarise.
Table 8
Diagonal components and average values of the electric prop-
erties of a water tetramer, calculated with full consideration of
intermolecular interactions and with complete neglect of inter-
actions; units as in Table 1
Interaction Zero Full
lx 1.202 0.469
ly ÿ1.202 ÿ0.832
lz ÿ1.700 ÿ2.241
l 2.404 2.436
axx 39.97 40.77
ayy 39.35 36.23
azz 39.50 41.36
aav 39.61 39.45
bxxx ÿ23.0 ÿ5.9
byyy 17.7 15.7
bzzz 35.9 14.2
bk ÿ44.8 ÿ15.1
cxxxx 4899 6010
cyyyy 6220 5580
czzzz 6053 7911
cav 5711 6352
312 H. Reis et al. / Chemical Physics 263 (2001) 301±316
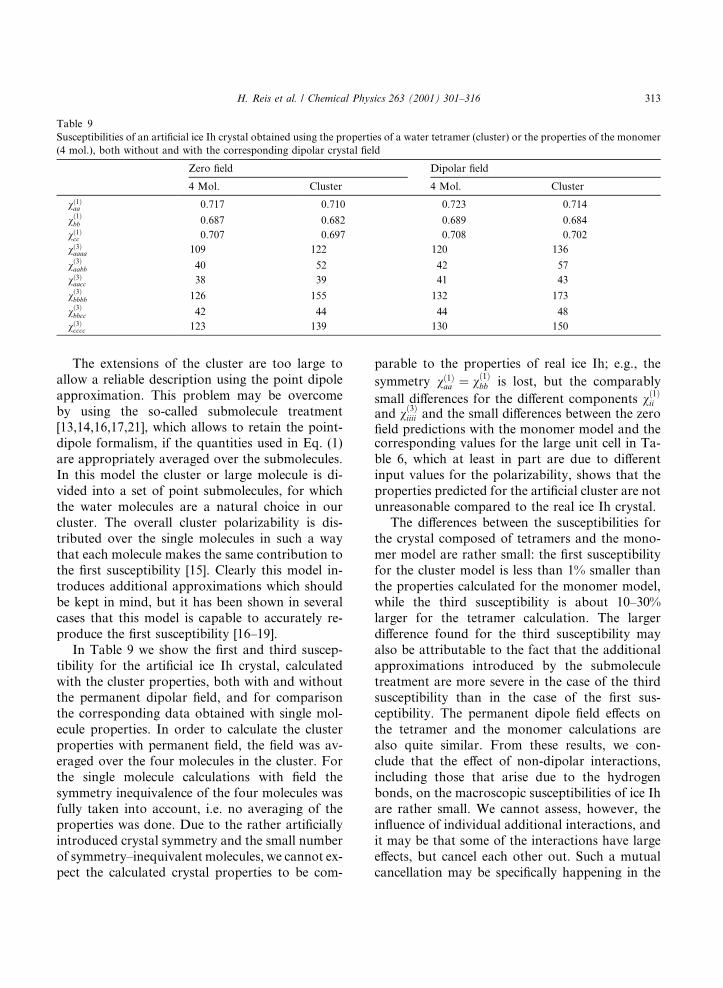
The extensions of the cluster are too large toallow a reliable description using the point dipoleapproximation. This problem may be overcomeby using the so-called submolecule treatment[13,14,16,17,21], which allows to retain the point-dipole formalism, if the quantities used in Eq. (1)are appropriately averaged over the submolecules.In this model the cluster or large molecule is di-vided into a set of point submolecules, for whichthe water molecules are a natural choice in ourcluster. The overall cluster polarizability is dis-tributed over the single molecules in such a waythat each molecule makes the same contribution tothe ®rst susceptibility [15]. Clearly this model in-troduces additional approximations which shouldbe kept in mind, but it has been shown in severalcases that this model is capable to accurately re-produce the ®rst susceptibility [16±19].
In Table 9 we show the ®rst and third suscep-tibility for the arti®cial ice Ih crystal, calculatedwith the cluster properties, both with and withoutthe permanent dipolar ®eld, and for comparisonthe corresponding data obtained with single mol-ecule properties. In order to calculate the clusterproperties with permanent ®eld, the ®eld was av-eraged over the four molecules in the cluster. Forthe single molecule calculations with ®eld thesymmetry inequivalence of the four molecules wasfully taken into account, i.e. no averaging of theproperties was done. Due to the rather arti®ciallyintroduced crystal symmetry and the small numberof symmetry±inequivalent molecules, we cannot ex-pect the calculated crystal properties to be com-
parable to the properties of real ice Ih; e.g., the
symmetry v�1�aa � v�1�bb is lost, but the comparably
small di�erences for the di�erent components v�1�iiand v�3�iiii and the small di�erences between the zero®eld predictions with the monomer model and thecorresponding values for the large unit cell in Ta-ble 6, which at least in part are due to di�erentinput values for the polarizability, shows that theproperties predicted for the arti®cial cluster are notunreasonable compared to the real ice Ih crystal.
The di�erences between the susceptibilities forthe crystal composed of tetramers and the mono-mer model are rather small: the ®rst susceptibilityfor the cluster model is less than 1% smaller thanthe properties calculated for the monomer model,while the third susceptibility is about 10±30%larger for the tetramer calculation. The largerdi�erence found for the third susceptibility mayalso be attributable to the fact that the additionalapproximations introduced by the submoleculetreatment are more severe in the case of the thirdsusceptibility than in the case of the ®rst sus-ceptibility. The permanent dipole ®eld e�ects onthe tetramer and the monomer calculations arealso quite similar. From these results, we con-clude that the e�ect of non-dipolar interactions,including those that arise due to the hydrogenbonds, on the macroscopic susceptibilities of ice Ihare rather small. We cannot assess, however, thein¯uence of individual additional interactions, andit may be that some of the interactions have largee�ects, but cancel each other out. Such a mutualcancellation may be speci®cally happening in the
Table 9
Susceptibilities of an arti®cial ice Ih crystal obtained using the properties of a water tetramer (cluster) or the properties of the monomer
(4 mol.), both without and with the corresponding dipolar crystal ®eld
Zero ®eld Dipolar ®eld
4 Mol. Cluster 4 Mol. Cluster
v�1�aa 0.717 0.710 0.723 0.714
v�1�bb 0.687 0.682 0.689 0.684
v�1�cc 0.707 0.697 0.708 0.702
v�3�aaaa 109 122 120 136
v�3�aabb 40 52 42 57
v�3�aacc 38 39 41 43
v�3�bbbb 126 155 132 173
v�3�bbcc 42 44 44 48
v�3�cccc 123 139 130 150
H. Reis et al. / Chemical Physics 263 (2001) 301±316 313

rather small cluster considered, and it may notoccur if larger clusters are used, but this could notbe explored in this work due to restrictions incomputer power.
4. Conclusions
Using accurately calculated molecular proper-ties and a rigorous electrostatic local-®eld method,we have calculated the macroscopic ®rst and third-order susceptibilities of four di�erent ice polymor-phs. The procedure employed allows to comparepurely computationally obtained macroscopic pro-perties using molecular electric properties directlywith experimentally accessible data, although thelack of experimental data especially for the highpressure polymorphs limits our conclusions for thetime being to ice Ih. The use of the dipolar ap-proximation only, has been found to yield accurate®rst and acceptable third susceptibilities for ice Ih,but the e�ect of the dipolar ®eld on the predictedsusceptibilities is quite small, and the completeneglect of the permanent ®eld yields susceptibilitiesfor ice Ih that compare equally well with the ex-perimental values. The calculated ®eld e�ect onthe susceptibilities of ice IX and ice II is muchlarger, and experimental values for these poly-morphs may be able to establish the reliability ofthe dipolar approximation more clearly. The verysmall dipolar interaction in ice VIII shows that forthis polymorph other intermolecular interactions,or multipolar higher than dipole and/or short-range interactions are dominant. Their in¯uenceon the susceptibilities, however, remains to beinvestigated.
The e�ect of the permanent ®eld on the mo-lecular properties follows a similar pattern asfound previously for urea and mNA, and themagnitude of the changes Dp of the propertiesp � l, a, b, c can roughly be given by the followingsequence: Db� DcJ Dl� Da. The trend of the®eld e�ect on the susceptibilities, on the otherhand, cannot be predicted easily from this pattern,as it depends strongly on the crystal structure: inurea, the crystal symmetry leads to a nearly com-plete cancellation of the strong ®eld e�ect on b in
the second-order susceptibility v�2� [16], and for ice
IX and ice II, the structure dependent cascadingcontribution to the third-order susceptibility leadsa much stronger ®eld dependence of v�3� than the
®eld dependence of the second hyperpolarizabilityalone would suggest.
The e�ect of the molecular vibrational contri-butions both on the permanent ®eld and the sus-ceptibilities is found to be rather small, well below10% in both cases.
The consequences of a multipolar description ofthe charge distribution of the water molecule thatgoes beyond the dipole approximation are di�cultto assess. A consequent description of the ®elde�ects requires the consideration of ®rst andhigher ®eld gradients and requires therefore alsothe knowledge of a large number of higher orderresponse functions, if e.g. the ®eld gradient e�ectson the (hyper)polarizabilities are sought by way ofusing Taylor expansions as in Eq. (9), similar tothe method used by Batista et al. [2,9,10]. Takinginto account higher multipoles leads to higherpermanent ®elds [2], which in turn increase the(hyper)polarizabilities (Table 3) and would there-fore tend to lead to higher susceptibilities. This inturn would worsen the predicted v�1� of ice Ihcompared to experiment, although the in¯uence ofthe ®eld gradients still remains to be considered.On the other hand, the small di�erences found forthe properties of an ice Ih similar crystal composedof tetramers, for which all intermolecular interac-tions had been taken into account, and those ofthe same crystal, taking into account only dipolarinteractions, shows that the e�ect of non-dipolarinteractions on the susceptibilities of ice Ih areprobably small. We believe, therefore, that themacroscopic susceptibilities of the ice polymorphsconsidered, once measured, would be acceptablywell approximated by the results given here.
Acknowledgements
We gratefully acknowledge ®nancial supportfrom the European Commission in the form ofa TMR Network Grant (contract no. ERB-FMRXCT960047).
314 H. Reis et al. / Chemical Physics 263 (2001) 301±316

References
[1] G. Maroulis, Chem. Phys. Lett. 289 (1998) 403.
[2] E.R. Batista, S.S. Xantheas, H. J�onsson, J. Chem.
Phys.109 (1998) 4546.
[3] O. Christiansen, J. Gauss, J.F. Stanton, Chem. Phys. Lett.
305 (1999) 147.
[4] P.L. Silvestrelli, M. Parrinello, J. Chem. Phys. 111 (1999)
3572.
[5] B.D. Bursulaya, J. Jeon, D.A. Zichi, H.J. Kim, J. Chem.
Phys. 108 (1998) 3286.
[6] K.V. Mikkelsen, Y. Luo, H. �Agren, P. Jùrgensen, J. Chem.
Phys. 102 (1995) 9362.
[7] A. Morita, S. Kato, J. Chem. Phys. 110 (1999) 11987.
[8] C. Gatti, B. Silvi, F. Colonna, Chem. Phys. Lett. 247
(1995) 135.
[9] E.R. Batista, S.S. Xantheas, H. J�onsson, J. Chem. Phys.
111 (1999) 6011.
[10] E.R. Batista, S.S. Xantheas, H. J�onsson, J. Chem. Phys.
112 (2000) 3285.
[11] C. Coulson, D. Eisenberg, Proc. Roy. Soc. Lond. Ser. A
291 (1966) 445.
[12] R.H.C. Janssen, J.-M. Bomont, D.N. Theodorou, S.
Raptis, M.G. Papadopoulos, J. Chem. Phys. 110 (1999)
6463.
[13] M. Hurst, R.W. Munn, J. Mol. Electron. 2 (1983) 35.
[14] M. Hurst, R.W. Munn, J. Mol. Electron. 2 (1983) 43.
[15] R.W. Munn, Mol. Phys. 64 (1988) 1.
[16] H. Reis, M.G. Papadopoulos, R.W. Munn, J. Chem. Phys.
109 (1998) 6828.
[17] H. Reis, S. Raptis, M.G. Papadopoulos, R.H.C. Janssen,
D.N. Theodorou, R.W. Munn, Theor. Chem. Acc. 99
(1998) 384.
[18] H. Reis, M.G. Papadopoulos, P. Calaminici, A. K�oster, K.
Jug, Chem. Phys. 261 (2000) 359.
[19] H. Reis, M.G. Papadopoulos, C. H�attig, J.G. �Angy�an,
R.W. Munn, J. Chem. Phys. 112 (2000) 6161.
[20] M. Malagoli, R.W. Munn, J. Chem. Phys. 107 (1997) 7926.
[21] T. Luty, Chem. Phys. Lett. 44 (1976) 335.
[22] P.G. Cummins, D.A. Dunmur, R.W. Munn, R.J. Ne-
wham, Acta Cryst. A 32 (1976) 847.
[23] M. Hurst, R.W. Munn, in: R.A. Hann, D. Bloor (Eds.),
Organic Materials for Non-linear Optics, Royal Society of
Chemistry Special Publication No. 69, Royal Society of
Chemistry, London, 1989, p. 3.
[24] J.D. Jorgensen, R.A. Beyerlein, N. Watanabe, T.G.
Worlton, J. Chem. Phys. 81 (1984) 3211.
[25] B. Kamb, Acta Cryst. 17 (1964) 1437.
[26] E.D. Finch, S.W. Radibeau, R.G. Wenzel, N.G. Nereson,
J. Chem. Phys. 49 (1968) 4361.
[27] B. Kamb, W.C. Hamilton, S.J. LaPlaca, A. Prakash,
J. Chem. Phys. 55 (1971) 1934.
[28] S.J. LaPlaca, W.C. Hamilton, B. Kamb, A. Prakash,
J. Chem. Phys. 58 (1973) 567.
[29] J.D. Bernal, R.H. Fowler, J. Chem. Phys. 1 (1933) 515.
[30] J.A. Hayward, J.R. Reimers, J. Chem. Phys. 106 (1997)
1518.
[31] C.J.F. Boettcher, P. Bordewijk, Theory of Electric Polar-
ization, vol. II, Elsevier, Amsterdam, 1978.
[32] G.P. Johari, Contemp. Phys. 22 (1981) 613.
[33] C.G. van Beek, J. Overeem, J.R. Ruble, B.M. Craven, Can.
J. Chem. 74 (1996) 943.
[34] V.E. Ingamells, M.G. Papadopoulos, A.J. Sadlej, J. Chem.
Phys. 112 (2000) 1645.
[35] V.E. Ingamells, M.G. Papadopoulos, A.J. Sadlej, Chem.
Phys. Lett. 316 (2000) 541.
[36] D.M. Bishop, Adv. Chem. Phys. 104 (1998) 1.
[37] GAUSSIAN 98GAUSSIAN 98 (Revision A.2), M.J. Frisch, G.W. Trucks,
H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman,
V.G. Zakrzewski, J.A. Montgomery, R.E. Stratmann, J.C.
Burant, S. Dapprich, J.M. Millam, A.D. Daniels, K.N.
Kudin, M.C. Strain, O. Farkas, J. Tomasi, V. Barone, M.
Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S.
Cli�ord, J. Ochterski, G.A. Petersson, P.Y. Ayala, Q. Cui,
K. Morokuma, D.K. Malick, A.D. Rabuck, K. Raghav-
achari, J.B. Foresman, J. Cioslowski, J.V. Ortiz, B.B.
Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi,
R. Gomperts, R.L. Martin, D.J. Fox, T. Keith, M.A. Al-
Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez, M.
Challacombe, P.M.W. Gill, B.G. Johnson, W. Chen, M.W.
Wong, J.L. Andres, M. Head-Gordon, E.S. Replogle, J.A.
Pople, Gaussian, Inc., Pittsburgh PA, 1998.
[38] T. Helgaker, H.J. Aa. Jensen, P. Jorgensen, J. Olsen, K.
Ruud, H. Agren, T. Andersen, K.L. Bak, V. Bakken, O.
Christiansen, P. Dahle, E.K. Dalskov, T. Enevoldsen, B.
Fernandez, H. Heiberg, H. Hettema, D. Jonsson, S.
Kirpekar, R. Kobayashi, H. Koch, K.V. Mikkelsen, P.
Norman, M.J. Packer, T. Saue, P.R. Taylor, O. Vahtras
(Ed.), DALTONDALTON, an ab initio electronic structure program,
Release 1.0, 1997.
[39] V.E. Ingamells, NUMDERNUMDER, a numerical derivative algo-
rithm, National Hellenic Research Foundation, Athens,
Greece, (2000).
[40] D. Amos, I.L. Alberts, J.S. Andrews, S.M. Colwell, N.C.
Handy, D. Jayatilaka, P.J. Knowles, R. Kobayashi, G.L.
Laming, A.M. Lee, P.E. Maslen, C.W. Murray, P. Palm-
ieri, J.E. Rice, E.M. Simandiras, A.J. Stone, M.D. Su, D.J.
Tozer, CADPACCADPAC 6.06.0, The Cambridge Analytic Derivatives
Package, Cambridge, UK (1995).
[41] A.J. Sadlej, Col. Cz. Chem. Commun. 53 (1988) 1995.
[42] T. Pluta, A.J. Sadlej, Chem. Phys. Lett. 297 (1998) 391.
[43] J.A. Pople, S. Binkley, R. Seeger, Int. J. Quant. Chem. 10
(1976) 1.
[44] J.A. Pople, M. Head-Gordon, K. Raghavachari, J. Chem.
Phys. 87 (1987) 5968.
[45] J.M. Luis, M. Duran, J.L. Andr�es, J. Chem. Phys. 107
(1997) 1501.
[46] J.M. Luis, M. Duran, J.L. Andr�es, B. Champagne, B.
Kirtman, J. Chem. Phys. 111 (1999) 875.
[47] D.M. Bishop, B. Kirtman, H.A. Kurtz, J.E. Rice, J. Chem.
Phys. 98 (1993) 8024.
[48] H. Sekino, R.J. Bartlett, J. Chem. Phys. 94 (1991)
3665.
[49] E. Whalley, Chem. Phys. Lett. 53 (1978) 449.
H. Reis et al. / Chemical Physics 263 (2001) 301±316 315

[50] D. Adams, Nature 293 (1981) 447.
[51] B.F. Levine, C.G. Bethea, J. Chem. Phys. 65 (1976) 2429.
[52] P. Kaatz, E.A. Donley, D.P. Shelton, J. Chem. Phys. 108
(1998) 849.
[53] M.D. Morse, S.A. Rice, J. Chem. Phys. 76 (1982) 650.
[54] A.P. Minton, J. Phys. Chem. 76 (1972) 886.
[55] S.G. Warren, Appl. Opt. 23 (1984) 1206.
[56] S. LaPlaca, B. Post, Acta Cryst. 13 (1960) 503.
[57] F. Kajzar, J. Messier, Phys. Rev. A 32 (1985) 2352.
[58] G.R. Meredith, B. Buchalter, C. Hanzlik, J. Chem. Phys.
78 (1982) 1543.
[59] B. Buchalter, G.R. Meredith, Appl. Opt. 21 (1982) 3221.
[60] A. Mito, K. Hagimoto, C. Takahashi, Nonlinear Opt. 13
(1995) 3.
316 H. Reis et al. / Chemical Physics 263 (2001) 301±316




















