
EGFRvIII undergoes activation-dependent downregulation mediated by the Cbl proteins
13
ORIGINAL ARTICLE EGFRvIII undergoes activation-dependent downregulation mediated by the Cbl proteins GC Davies 1 , PE Ryan 1,2 , L Rahman 3 , M Zajac-Kaye 3 and S Lipkowitz 1 1 Laboratory of Cellular and Molecular Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA 2 George Washington University Institute of Biomedical Sciences, Washington, DC, USA and 3 Molecular Therapeutics Program, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA The overexpression or mutation of tyrosine kinases (TKs), such as the epidermal growth factor receptor (EGFR), can lead to the development of cancer. The most common mutation of the EGFR in glioblastomas is the deletion of exons 2–7 known as the EGFRvIII. This mutant receptor cannot bind EGF but, instead, is constitutively active. The Cbl family of ubiquitin ligases (Cbl, Cbl-b, and Cbl-c) targets the activated EGFR for degradation. As the EGFRvIII is transforming, we investigated whether it could be downregulated by the Cbl proteins. The over- expression of all three Cbl proteins resulted in the ubiquitination and degradation of the EGFRvIII. As with the wild-type EGFR, the TK-binding domain and the RING finger of Cbl-b are sufficient for the downregula- tion of the EGFRvIII. Also, we found that Cbl-b is recruited to the EGFRvIII and inhibits the transformation of NIH 3T3 cells by the EGFRvIII. Mutation of the Cbl- binding site (Y1045F) in the EGFRvIII inhibits its ubiquitination and downregulation by Cbl-b and enhances its ability to transform. Furthermore, the EGFR TK inhibitor, AG 1478, prevents the downregulation of the EGFRvIII by the Cbl proteins and antagonizes the ability of an immunotoxin directed against the EGFRvIII to kill cells expressing this receptor. In conclusion, the EGFR- vIII does not transform by escaping regulation by Cbl proteins and this activation-induced downregulation of the EGFRvIII has an important role in mediating the toxicity of anti-EGFRvIII immunotoxins. Oncogene (2006) 25, 6497–6509. doi:10.1038/sj.onc.1209662; published online 15 May 2006 Keywords: EGFRvIII; Cbl proteins; ubiquitin; cancer Introduction The epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein that plays an important role in the growth and differentiation of normal cells. The inappropriate activity of the EGFR tyrosine kinase (TK) is associated with the development of a wide range of cancers (Blume-Jensen and Hunter, 2001). Amplifica- tion of the EGFR gene occurs in approximately 50% of glioblastoma multiformes (reviewed in Kuan et al., 2001). In many cases, the amplification of the EGFR gene is associated with its genomic rearrangement. The most common of these rearrangements is known as the EGFR variant III (EGFRvIII, de2–7 EGFR, or DEGFR) and is characterized by the genomic deletion of exons 2–7. This deletion results in the loss of the amino-terminal amino-acid residues 6–273 in the extra- cellular domain of the EGFR. The expression of the EGFRvIII in glioblastoma multiforme is a negative prognostic indicator (Shinojima et al., 2003; Heimberger et al., 2005). However, it has been reported recently that the co-expression of the EGFRvIII and the tumor-suppre- ssor protein PTEN predicts response of patients with glioblastoma to the EGFR TK inhibitors (Mellinghoff et al., 2005). In addition, the expression of the EGFRvIII has been reported in breast, non-small-cell lung, ovarian, and prostate carcinomas, but not in normal tissues (Garcia de Palazzo et al., 1993; Moscatello et al., 1995; Wikstrand et al., 1995). Therefore, the EGFRvIII is a tumor-specific target for cancer therapy (reviewed in Kuan et al., 2001; Pedersen et al., 2001). The ectopic expression of the EGFRvIII in a glioblastoma cell line increased the speed with which these cells formed tumors in nude mice (Nishikawa et al., 1994; Nagane et al., 1996). The EGFRvIII is also capable of significantly enhancing the tumorgenicity of immortalized murine fibroblasts (Batra et al., 1995; Moscatello et al., 1996) and the breast cancer cell line MCF-7 (Tang et al., 2000). Also, the EGFRvIII increases the in vitro invasiveness of a small-cell lung cancer cell line (Damstrup et al., 2002). Using the murine hematopoietic 32D cell line, Tang et al. (2000) demonstrated that the EGFRvIII is capable of directly transforming a non-tumorigenic cell line. Unlike the wild-type (WT) EGFR, the EGFRvIII is unable to bind to EGF or transforming growth factor-a (Ekstrand et al., 1994; Nishikawa et al., 1994; Moscatello et al., 1996) but, instead, it can dimerize spontaneously (Moscatello et al., 1996; Fernandes et al., 2001; Montgomery, 2002). The spontaneous dimerization and ensuing TK activation of the EGFRvIII is necessary Received 3 February 2006; revised 23 March 2006; accepted 23 March 2006; published online 15 May 2006 Correspondence: Dr S Lipkowitz, LCMB/CCR/NCI/NIH, Bldg 37/Rm 2066, 37 Convent Drive, Bethesda, MD 20892-4255, USA. E-mail: [email protected] Oncogene (2006) 25, 6497–6509 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of EGFRvIII undergoes activation-dependent downregulation mediated by the Cbl proteins

ORIGINAL ARTICLE
EGFRvIII undergoes activation-dependent downregulation mediated
by the Cbl proteins
GC Davies1, PE Ryan1,2, L Rahman3, M Zajac-Kaye3 and S Lipkowitz1
1Laboratory of Cellular and Molecular Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health,Bethesda, MD, USA 2George Washington University Institute of Biomedical Sciences, Washington, DC, USA and 3MolecularTherapeutics Program, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
The overexpression or mutation of tyrosine kinases (TKs),such as the epidermal growth factor receptor (EGFR), canlead to the development of cancer. The most commonmutation of the EGFR in glioblastomas is the deletion ofexons 2–7 known as the EGFRvIII. This mutant receptorcannot bind EGF but, instead, is constitutively active. TheCbl family of ubiquitin ligases (Cbl, Cbl-b, and Cbl-c)targets the activated EGFR for degradation. As theEGFRvIII is transforming, we investigated whether itcould be downregulated by the Cbl proteins. The over-expression of all three Cbl proteins resulted in theubiquitination and degradation of the EGFRvIII. As withthe wild-type EGFR, the TK-binding domain and theRING finger of Cbl-b are sufficient for the downregula-tion of the EGFRvIII. Also, we found that Cbl-b isrecruited to the EGFRvIII and inhibits the transformationof NIH 3T3 cells by the EGFRvIII. Mutation of the Cbl-binding site (Y1045F) in the EGFRvIII inhibits itsubiquitination and downregulation by Cbl-b and enhancesits ability to transform. Furthermore, the EGFR TKinhibitor, AG 1478, prevents the downregulation of theEGFRvIII by the Cbl proteins and antagonizes the abilityof an immunotoxin directed against the EGFRvIII to killcells expressing this receptor. In conclusion, the EGFR-vIII does not transform by escaping regulation by Cblproteins and this activation-induced downregulation of theEGFRvIII has an important role in mediating the toxicityof anti-EGFRvIII immunotoxins.Oncogene (2006) 25, 6497–6509. doi:10.1038/sj.onc.1209662;published online 15 May 2006
Keywords: EGFRvIII; Cbl proteins; ubiquitin; cancer
Introduction
The epidermal growth factor receptor (EGFR) is atransmembrane glycoprotein that plays an importantrole in the growth and differentiation of normal cells.
The inappropriate activity of the EGFR tyrosine kinase(TK) is associated with the development of a wide rangeof cancers (Blume-Jensen and Hunter, 2001). Amplifica-tion of the EGFR gene occurs in approximately 50% ofglioblastoma multiformes (reviewed in Kuan et al.,2001). In many cases, the amplification of the EGFRgene is associated with its genomic rearrangement. Themost common of these rearrangements is known as theEGFR variant III (EGFRvIII, de2–7 EGFR, orDEGFR) and is characterized by the genomic deletionof exons 2–7. This deletion results in the loss of theamino-terminal amino-acid residues 6–273 in the extra-cellular domain of the EGFR. The expression of theEGFRvIII in glioblastoma multiforme is a negativeprognostic indicator (Shinojima et al., 2003; Heimbergeret al., 2005). However, it has been reported recently thatthe co-expression of the EGFRvIII and the tumor-suppre-ssor protein PTEN predicts response of patients withglioblastoma to the EGFR TK inhibitors (Mellinghoffet al., 2005). In addition, the expression of the EGFRvIIIhas been reported in breast, non-small-cell lung, ovarian,and prostate carcinomas, but not in normal tissues (Garciade Palazzo et al., 1993; Moscatello et al., 1995; Wikstrandet al., 1995). Therefore, the EGFRvIII is a tumor-specifictarget for cancer therapy (reviewed in Kuan et al., 2001;Pedersen et al., 2001).The ectopic expression of the EGFRvIII in a
glioblastoma cell line increased the speed with whichthese cells formed tumors in nude mice (Nishikawaet al., 1994; Nagane et al., 1996). The EGFRvIII is alsocapable of significantly enhancing the tumorgenicity ofimmortalized murine fibroblasts (Batra et al., 1995;Moscatello et al., 1996) and the breast cancer cell lineMCF-7 (Tang et al., 2000). Also, the EGFRvIIIincreases the in vitro invasiveness of a small-cell lungcancer cell line (Damstrup et al., 2002). Using themurine hematopoietic 32D cell line, Tang et al. (2000)demonstrated that the EGFRvIII is capable of directlytransforming a non-tumorigenic cell line. Unlike thewild-type (WT) EGFR, the EGFRvIII is unable to bindto EGF or transforming growth factor-a (Ekstrandet al., 1994; Nishikawa et al., 1994; Moscatello et al.,1996) but, instead, it can dimerize spontaneously(Moscatello et al., 1996; Fernandes et al., 2001;Montgomery, 2002). The spontaneous dimerizationand ensuing TK activation of the EGFRvIII is necessary
Received 3 February 2006; revised 23 March 2006; accepted 23 March2006; published online 15 May 2006
Correspondence: Dr S Lipkowitz, LCMB/CCR/NCI/NIH, Bldg37/Rm 2066, 37 Convent Drive, Bethesda, MD 20892-4255, USA.E-mail: [email protected]
Oncogene (2006) 25, 6497–6509& 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00
www.nature.com/onc

to transform cells (Han et al., 1996; Nagane et al., 1996;Huang et al., 1997; Antonyak et al., 1998; O’Rourkeet al., 1998; Montgomery, 2002; Johns et al., 2003;Abulrob et al., 2004; Luwor et al., 2004; Pedersen et al.,2004).The Cbl proteins (Cbl, Cbl-b, and Cbl-c) are negative
regulators of WT EGFR signaling (Ettenberg et al.,1999a, b, 2001; Keane et al., 1999; Levkowitz et al.,1999; Waterman et al., 1999a; Yokouchi et al., 1999;Duan et al., 2003). The Cbl proteins all contain anamino-terminal TK-binding (TKB) domain, a RINGfinger domain, and a region of proline-rich sequences intheir carboxy-terminus (Nau and Lipkowitz, 2003).They are recruited to the activated EGFR either bythe direct binding of their TKB domain to thephosphotyrosine residue at position 1045 in the EGFR(Levkowitz et al., 1999) or by an indirect mechanismmediated by Grb2 – the SH3 domains of Grb2 bind theproline-rich region of the Cbl proteins and the SH2domain of Grb2 binds the phosphorylated EGFR(Waterman et al., 2002; Jiang et al., 2003). The RINGfinger domain of the Cbl proteins allows them tofunction as ubiquitin ligases (E3s) and so to target theEGFR signaling complex for internalization and sub-sequent degradation in the lysosome (Ettenberg et al.,1999b, 2001; Levkowitz et al., 1999; Waterman et al.,1999a; Yokouchi et al., 1999; Duan et al., 2003). Thus,the Cbl proteins mediate the downregulation of theEGFR following stimulation with EGF.It has been hypothesized that a failure to down-
regulate the EGFRvIII once it becomes activatedcontributes to its capacity to transform (Huang et al.,1997). Supporting this, a recent study reported that theEGFRvIII does not interact with either Cbl or Cbl-band is not downregulated (Schmidt et al., 2003).Mutations in the Cbl-binding site of several receptortyrosine kinases (RTKs) result in transforming forms ofthese RTKs (Peschard et al., 2001; Mancini et al., 2002;Peschard and Park, 2003). However, the intracellulardomain of the EGFRvIII is not mutated and thus theCbl protein-binding sites are intact. The Cbl proteinsbind the phosphorylated EGFR (Levkowitz et al., 1999)and the phosphorylation pattern of active EGFRvIII issimilar to that of the activated WT EGFR (Fernandeset al., 2001). The inability of the Cbl proteins to interactwith or downregulate the EGFRvIII suggested a novelmechanism regulating the interaction between theEGFRvIII and the Cbl proteins. Therefore, we investi-gated the interaction between the Cbl proteins and theEGFRvIII further. In contrast to the published data, wefound that the overexpression of all three Cbl proteinscaused the ubiquitination and downregulation of theEGFRvIII. Also, we demonstrated that Cbl-b binds tothe EGFRvIII, that the requirements for Cbl-b-mediated degradation of the EGFRvIII are identicalto that of the WT EGFR, and that only activeEGFRvIII is downregulated. Consequently, Cbl-binhibits the transformation of NIH 3T3 fibroblasts bythe EGFRvIII and the mutation of the Cbl-binding sitein the EGFRvIII (Y1045F) enhances the ability of theEGFRvIII to transform. Finally, we demonstrated that
the inhibition of the EGFRvIII TK by AG 1478, whichabrogates Cbl-mediated downregulation of the EGFR-vIII, antagonizes the ability of an immunotoxin directedagainst the EGFRvIII to kill cells expressing thisreceptor. Thus, the EGFRvIII undergoes activation-dependent downregulation mediated by the Cbl pro-teins.
Results
Cbl proteins ubiquitinate and downregulate theconstitutively active EGFRvIIIThe overexpression of Cbl proteins enhances EGF-induced ubiquitination and downregulation of the WTEGFR (Ettenberg et al., 1999b, 2001; Levkowitz et al.,1999; Waterman et al., 1999b; Yokouchi et al., 1999).Therefore, we investigated whether the Cbl proteins alsoregulate the constitutively active mutant EGFRvIII in acell line Chinese hamster ovary (CHO) that does notexpress the WT EGFR. The co-transfection of CHOcells with the EGFRvIII and either Cbl, Cbl-b, or Cbl-cresulted in a decrease in EGFRvIII protein levels(Figure 1a–c; compare lanes 2 and 3). Also, the co-transfection of Cbl, Cbl-b, or Cbl-c increased theamount of ubiquitinated proteins seen in immunopreci-pitates of the EGFRvIII (Figure 1d–f; compare lanes 2and 3). These ubiquitinated species represent ubiquiti-nated forms of the EGFRvIII suggesting that, like theactive WT EGFR, Cbl proteins are capable ofubiquitinating and downregulating the EGFRvIII.As all three Cbl proteins caused the degradation of
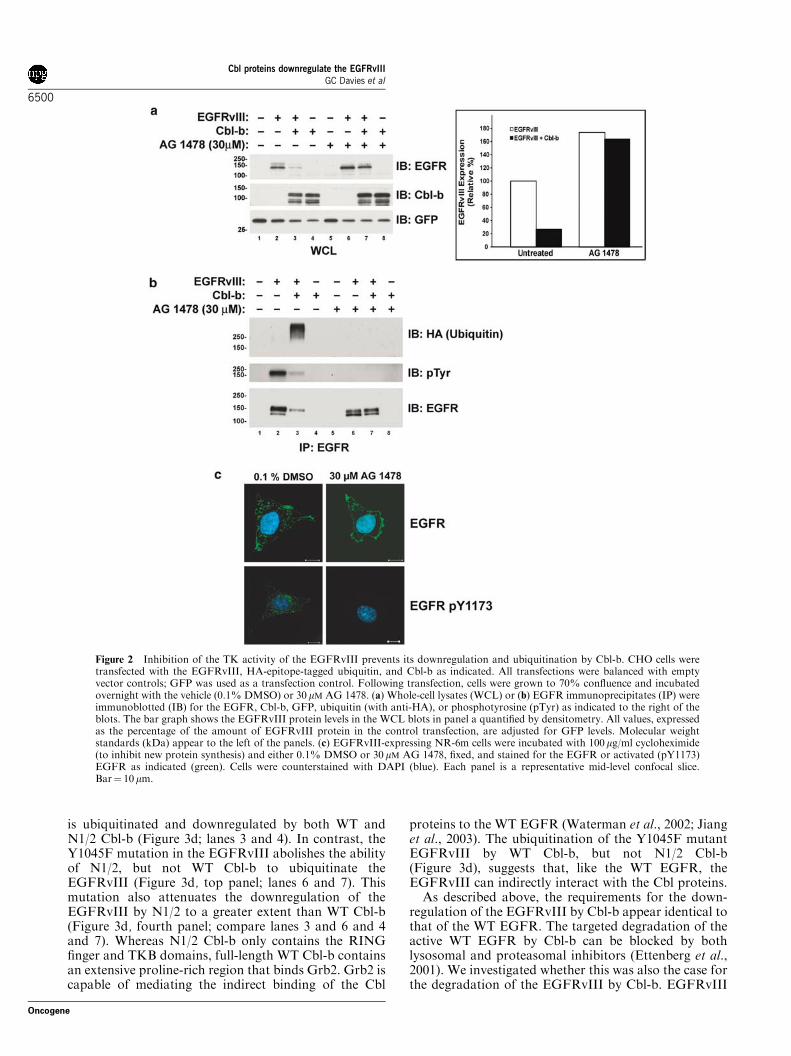
the EGFRvIII, we chose to use Cbl-b to investigate themechanism by which they regulate this oncogenicEGFR mutant. Given that the TK activity andautophosphorylation of the WT EGFR are necessaryfor its ubiquitination and degradation by the Cblproteins (Levkowitz et al., 1999), we examined whetherthis is also the case with the EGFRvIII. Although theWT EGFR is regulated by ligand binding, the EGFR-vIII is spontaneously active. Therefore, we used theEGFR TK inhibitor AG 1478 to inhibit the activity ofthe EGFRvIII. Treatment of CHO cells overexpressingthe EGFRvIII with AG 1478 prevented tyrosineautophosphorylation of the EGFRvIII (Figure 2b,middle panel; compare lanes 2 and 3 to lanes 6 and 7).Inactivation of the EGFRvIII TK by AG 1478attenuated its downregulation by Cbl-b (Figure 2a, toppanel; compare lanes 2 and 3 to lanes 6 and 7). Co-expression of Cbl-b resulted in downregulation of theEGFRvIII by 73% in the absence of AG 1478(Figure 2a, bar graph). In the presense of AG 1478,the level of the EGFRvIII was higher and co-expressionof Cbl-b only resulted in 5% downregulation (Figure 2a,bar graph). AG 1478 completely abolished ubiquitina-tion of EGFRvIII by Cbl-b (Figure 2b, top panel;compare lanes 3 and 7). Also, AG 1478 treatmentinhibited the ubiquitination and downregulation of theEGFRvIII by Cbl (data not shown). Therefore, the TKactivity of the EGFRvIII is necessary for its down-regulation by the Cbl proteins. As AG 1478 inhibits the
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6498
Oncogene

activation-induced downregulation of the EGFRvIII bythe Cbl proteins, we examined the effects of AG 1478upon the subcellular localization of the EGFRvIII in themurine fibroblast cell line NR-6m (Figure 2c). The NR-6m cell line is a variant of Swiss 3T3 cells which has beenstably transfected with the EGFRvIII, resulting intransformation of the cells (Batra et al., 1995). It waschosen for the localization studies because the cell linedoes not express endogenous WT EGFR, thus allowingthe use of anti-EGFR and anti-phospho-EGFR anti-bodies. In these cells, the EGFRvIII was localized inboth the plasma membrane and intracellular vesicles.The majority of active EGFRvIII, as detected by EGFRphosphotyrosine 1173 staining, appears to be localizedin intracellular vesicles. Inhibition of the TK activity ofthe EGFRvIII by AG 1478 treatment abolishedphosphotyrosine 1173 staining and resulted in a reduc-tion of the amount of EGFRvIII in intracellular vesiclesand an increase in the proportion of the EGFRvIIIlocated at the plasma membrane compared to intracel-lular vesicles. This is consistent with AG 1478 treatmentpreventing activation-induced internalization and down-regulation of the EGFRvIII from the plasma mem-brane.
Requirements for Cbl-b-mediated downregulation of theEGFRvIIIWe mapped the regions of Cbl-b necessary for thedownregulation of the EGFRvIII by transfecting CHOcells with the EGFRvIII and various constructs of Cbl-b(Figure 3a). As described above (Figure 1), WT Cbl-bdownregulates the EGFRvIII (Figure 3b, lane 3). Thedeletion of the proline-rich, carboxy-terminal half ofCbl-b did not inhibit its ability to downregulate theEGFRvIII (N1/2 Cbl-b; Figure 3b, lane 4). In contrast,
the deletion of the TKB domain containing the amino-terminus of Cbl-b (C2/3 Cbl-b) prevented the down-regulation of the EGFRvIII by Cbl-b (Figure 3b, lane5). Finally, a RING finger mutant of Cbl-b (C373A)that has been shown to lack E3 activity (Ettenberg et al.,2001) was unable to downregulate the EGFRvIII(Figure 3b, lane 6). Quantification of the downregula-tion of the EGFRvIII by the various constructs of Cbl-brevealed that N1/2 and WT Cbl-b downregulate theEGFRvIII to a similar extent, that the overexpression ofC2/3 Cbl-b did not affect EGFRvIII levels, and that theRING finger mutant of Cbl-b tended to increase theamount of the EGFRvIII protein (Figure 3c). There-fore, like the WT EGFR (Ettenberg et al., 2001), theTKB and RING finger domains of Cbl-b are sufficientfor the downregulation of the EGFRvIII. Also, the E3activity of Cbl-b is necessary for the downregulation ofthe EGFRvIII by Cbl-b.The TKB domain of the Cbl proteins has been shown
to mediate a specific binding to a phosphotyrosineresidue (Y1045) in the activated WT EGFR (Levkowitzet al., 1999; Waterman et al., 2002). The mutation of thisresidue attenuates the downregulation of the EGFR. Wetested the ability of the equivalent mutation (Y1045F) inthe EGFRvIII to affect its regulation by Cbl-b(Figure 3d). Using an antibody against phosphotyrosine1045 EGFR, we detected phosphorylation of theEGFRvIII at this residue that was abolished by itsmutation to phenylalanine (Figure 3d, second panel;compare lanes 2 and 5). As in the WT EGFR, Y1045appears to be a minor phosphotyrosine residue (Levko-witz et al., 1999), as the loss of Y1045 phosphorylation(Figure 3d, second panel) by mutation of this residuedoes not decrease significantly the content of EGFRvIIIphosphotyrosine (Figure 3d, third panel; compare lanes2 and 5). As described above (Figure 3b), the EGFRvIII
Figure 1 The constitutively active mutant EGFRvIII is ubiquitinated and downregulated by Cbl proteins. CHO cells were transfectedwith the EGFRvIII, HA-epitope-tagged ubiquitin, and either Cbl, Cbl-b, or Cbl-c as indicated. All transfections were balanced withempty vector controls; GFP was used as a transfection control. Cells were serum starved overnight and protein lysates were prepared.(a–c) Whole-cell lysates (WCL) or (d–f ) EGFR immunoprecipitates (IP) were immunoblotted (IB) for the EGFR, Cbl, Cbl-b, Cbl-c,ubiquitin (with anti-HA), or GFP as indicated to the right of the blots. An asterisk indicates the band corresponding to Cbl-c.Immunoblots of the WCL were used to assess the levels of the transfected proteins. Immunoblots of the precipitated EGFRvIII wereused to assess ubiquitination. Molecular weight standards (kDa) appear to the left of the panels.
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6499
Oncogene
is ubiquitinated and downregulated by both WT andN1/2 Cbl-b (Figure 3d; lanes 3 and 4). In contrast, theY1045F mutation in the EGFRvIII abolishes the abilityof N1/2, but not WT Cbl-b to ubiquitinate theEGFRvIII (Figure 3d, top panel; lanes 6 and 7). Thismutation also attenuates the downregulation of theEGFRvIII by N1/2 to a greater extent than WT Cbl-b(Figure 3d, fourth panel; compare lanes 3 and 6 and 4and 7). Whereas N1/2 Cbl-b only contains the RINGfinger and TKB domains, full-length WT Cbl-b containsan extensive proline-rich region that binds Grb2. Grb2 iscapable of mediating the indirect binding of the Cbl
proteins to the WT EGFR (Waterman et al., 2002; Jianget al., 2003). The ubiquitination of the Y1045F mutantEGFRvIII by WT Cbl-b, but not N1/2 Cbl-b(Figure 3d), suggests that, like the WT EGFR, theEGFRvIII can indirectly interact with the Cbl proteins.As described above, the requirements for the down-
regulation of the EGFRvIII by Cbl-b appear identical tothat of the WT EGFR. The targeted degradation of theactive WT EGFR by Cbl-b can be blocked by bothlysosomal and proteasomal inhibitors (Ettenberg et al.,2001). We investigated whether this was also the case forthe degradation of the EGFRvIII by Cbl-b. EGFRvIII
Figure 2 Inhibition of the TK activity of the EGFRvIII prevents its downregulation and ubiquitination by Cbl-b. CHO cells weretransfected with the EGFRvIII, HA-epitope-tagged ubiquitin, and Cbl-b as indicated. All transfections were balanced with emptyvector controls; GFP was used as a transfection control. Following transfection, cells were grown to 70% confluence and incubatedovernight with the vehicle (0.1% DMSO) or 30 mM AG 1478. (a) Whole-cell lysates (WCL) or (b) EGFR immunoprecipitates (IP) wereimmunoblotted (IB) for the EGFR, Cbl-b, GFP, ubiquitin (with anti-HA), or phosphotyrosine (pTyr) as indicated to the right of theblots. The bar graph shows the EGFRvIII protein levels in the WCL blots in panel a quantified by densitometry. All values, expressedas the percentage of the amount of EGFRvIII protein in the control transfection, are adjusted for GFP levels. Molecular weightstandards (kDa) appear to the left of the panels. (c) EGFRvIII-expressing NR-6m cells were incubated with 100mg/ml cycloheximide(to inhibit new protein synthesis) and either 0.1% DMSO or 30 mM AG 1478, fixed, and stained for the EGFR or activated (pY1173)EGFR as indicated (green). Cells were counterstained with DAPI (blue). Each panel is a representative mid-level confocal slice.Bar¼ 10mm.
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6500
Oncogene

protein levels were stabilized by both proteasomal(ALLN, MG 132, and lactacystin) and lysosomal(leupeptin, chloroquine, folimycin, and ammoniumchloride) inhibitors in CHO cells co-transfected withthe EGFRvIII and Cbl-b (Figure 3e; compare lane 1 tolanes 2–8). Therefore, it appears that the degradation ofthe WT EGFR and the EGFRvIII by Cbl-b share asimilar mechanism.
The EGFRvIII and Cbl-b associateThe ligand-induced downregulation of the WT EGFRby the Cbl proteins requires their binding to thereceptor. We examined the ability of Cbl-b to bind tothe EGFRvIII. In contrast to the WT EGFR followingEGF stimulation, only a small proportion of the
EGFRvIII is active at any given time (Huang et al.,1997; Fernandes et al., 2001). As Cbl-b targets thisactive pool of the EGFRvIII for degradation, theEGFRvIII bound to Cbl-b would be predicted to be avery small fraction of total EGFRvIII protein. UnlikeWT Cbl-b, Cbl-b with a mutation in its RING finger(C373A) does not downregulate the EGFRvIII (seeFigure 3b), thereby increasing the likelihood of obser-ving an interaction between the EGFRvIII and Cbl-b.Indeed, when CHO cells were transfected with acombination of the EGFRvIII and a RING fingermutant of Cbl-b, we observed an association betweenthe EGFRvIII and Cbl-b when either Cbl-b (Figure 4a,lane 3) or the EGFRvIII (Figure 4b, lane 3) wereprecipitated. We were also able to co-precipitate WTCbl-b along with the EGFRvIII (data not shown).
Figure 3 The RING finger of Cbl-b is necessary for the downregulation of the EGFRvIII. (a) WT Cbl-b (aa 1–938), N1/2 Cbl-b (aa1–483), C2/3 Cbl-b (aa 327–938), or Cbl-b RING finger mutant (RFmt Cbl-b; C373A) expression constructs were used to map theregions of Cbl-b necessary for the downregulation of the EGFRvIII. (b) CHO cells were transfected with the EGFRvIII alone or incombination with various Cbl-b constructs as indicated. Whole-cell lysates (WCL) were immunoblotted (IB) for the EGFR, Cbl-b, orGFP as specified to the right of the blots. (c) EGFRvIII protein levels were then quantified by densitometry. All values, expressed asthe percentage of the amount of EGFRvIII protein in the control transfection, are adjusted for GFP levels and are the mean and s.e.m.of n¼ 3 experiments. (d) Mutation of Y1045 in the EGFRvIII abrogates its downregulation by Cbl-b. CHO cells were transfected withthe EGFRvIII, the Y1045F EGFRvIII, HA-epitope-tagged ubiquitin, WT Cbl-b, and N1/2 Cbl-b (aa 1–483) as indicated. WCL orEGFR immunoprecipitates (IP) were immunoblotted for the EGFR, Cbl-b, or ubiquitin (with anti-HA) as indicated. The positions ofWT Cbl-b (white arrow) and N1/2 Cbl-b (black arrow) are indicated along the right-hand side of the blot. Note that a non-specificartefact band migrates at the size of N1/2 Cbl-b in all lanes, a darker band corresponding to N1/2 Cbl-b can be seen in the relevantlanes. (e) Proteasomal and lysosomal inhibitors prevent Cbl-b-mediated degradation of the EGFRvIII. CHO cells were transfectedwith the EGFRvIII and Cbl-b. Cells were serum starved overnight and then incubated with 0.1% DMSO, 20 mM ALLN, 20 mM MG132, 20mM Lactacystin, 200mM Leupeptin, 200mM Chloroquine, 200mM Folimycin, or 20mM ammonium chloride (NH4Cl) for 8 h.WCL were immunoblotted for the EGFR or GFP as indicated. All transfections were balanced with empty vector controls; GFP wasused as a transfection control. Molecular weight standards (kDa) appear to the left of the panels.
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6501
Oncogene

As in CHO cells (Figures 2 and 3d), the co-transfection of the EGFRvIII and Cbl-b into humanembryonic kidney (HEK) 293T cells decreased EGFR-vIII protein levels and tyrosine phosphorylation(Figure 4c). In addition, we were also able to co-precipitate the EGFRvIII and WT Cbl-b from thelysates of HEK 293T cells transfected with theseproteins (Figure 4d). Activation of the endogenousEGFR by EGF (100 ng/ml; 30min) did not affectsignificantly the downregulation of the EGFRvIII byCbl-b, nor did it affect the association between theseproteins. Similarly, the co-expression of the WT EGFRwith the EGFRvIII in CHO cells did not appear toaffect the regulation of EGFRvIII by Cbl-b (data notshown).
Cbl-b prevents the ability of the EGFRvIII to inducetransformation of NIH 3T3 fibroblastsThe EGFRvIII has been shown to mediate celltransformation as a consequence of its constituti-vely active TK (Nagane et al., 1996; Huang et al.,1997). As Cbl-b downregulates active EGFRvIII,
we tested the ability of Cbl-b to inhibit EGFRvIII-induced transformation using a cell focus formingassay. Immortalized NIH 3T3 cells were transfectedwith either the EGFRvIII, Cbl-b, RING fingermutant Cbl-b, or a combination of the EGFRvIII andCbl-b or RING finger mutant Cbl-b. All transfectionswere balanced with empty control vectors.Stable Zeocin- and G-418-resistant clones were pooledand a focus-forming assay was performed. We foundthat cells ectopically expressing the EGFRvIII gaverise to foci 10–14 days after inoculation (Figure 5a, plate2). The overexpression of Cbl-b alone did not inducefoci formation (Figure 5a, plate 3), instead it inhibitedthe formation of foci by the EGFRvIII (Figure 5a,plate 4). Western blotting of the pooled Zeocin- andG-418-resistant clones indicated that Cbl-b down-regulates the EGFRvIII in NIH 3T3 cells (data notshown). In contrast, a RING finger mutant of Cbl-b(C373A) failed to suppress the induction of foci by theEGFRvIII (Figure 5a, plate 6). Therefore, Cbl-b inhibitsthe ability of the EGFRvIII to transform andthis inhibition is dependent upon the E3 activity ofCbl-b.
Figure 4 Cbl-b associates with the EGFRvIII. CHO cells were transfected with the EGFRvIII and HA-epitope-tagged RING fingermutant (RFmt; C373A) Cbl-b. Cells were serum starved overnight, lysed, and either (a) Cbl-b (with anti-HA) or (b) the EGFRvIII wasimmunoprecipitated. Whole-cell lysates (WCL) or the immunoprecipitates (IP) were immunoblotted (IB) for the EGFR, Cbl-b, orGFP as indicated to the right-hand of the blots. (c, d) Cbl-b associates with and downregulates the EGFRvIII in HEK 293T cells. HEK293T cells were transfected with the EGFRvIII and HA-epitope-tagged Cbl-b as indicated. The cells were serum starved overnight andincubated with or without 100 ng/ml EGF for 30min. (c) WCL or (d) EGFR IP were immunoblotted for the EGFR, Cbl-b (with anti-HA), phosphotyrosine (pTyr), or GFP as indicated to the right-hand side of the blots. The positions of the tyrosine-phosphorylatedEGFRvIII (white arrow) and HA-Cbl-b (black arrow) proteins are indicated along the right-hand side of the blot. All transfectionswere balanced with empty vector controls; GFP was used as a transfection control. Molecular weight standards (kDa) appear to theleft of the panels.
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6502
Oncogene

The mutation of the Cbl-binding site (Y1045F) in theEGFRvIII attenuates its downregulation by Cbl-b(Figure 3c). This mutation increased the number of fociformed by the EGFRvIII (Figure 5b, compare plates 2and 3). In NIH 3T3 cells, the EGFRvIII is localized inboth the plasma membrane and in intracellular vesicles(Figure 5c). However, the proportion of EGFRvIIIlocated at the plasma membrane compared to intracel-lular vesicles is increased by mutation of Y1045F
(Figure 5c). In cells, the only proteins known to bindY1045 when it is phosphorylated are the Cbl proteins.As both Cbl and Cbl-b are endogenous to NIH 3T3 cellsthis change in localization – similar to that seen with theinhibition of the EGFRvIII TK activity (Figure 2c) – isconsistent with the Y1045F EGFRvIII being defectivein Cbl-mediated downregulation. Although the Y1045Fmutation affected the localization of the EGFRvIII andmarkedly enhanced foci formation in NIH 3T3 cells, this
Figure 5 The transformation of NIH 3T3 cells by the EGFRvIII is regulated by Cbl proteins. (a) NIH 3T3 cells were transfected with2mg of the EGFRvIII, HA-epitope-tagged WT Cbl-b, HA-epitope-tagged RING finger mutant (RFmt; C373A) Cbl-b, or empty vectorcontrols as indicated before selection in medium containing 600mg/ml Zeocin and 600mg/ml G-418. At passage 3, foci assays wereperformed. (b) NIH 3T3 cells were transfected with 1 mg of the EGFRvIII, EGFRvIII Y1045F, or empty vector before selection inmedium containing 600mg/ml Zeocin. At passage 3, foci assays were performed. (c) EGFRvIII- or Y1045 EGFRvIII-expressing NIH3T3 cells were fixed and stained for the EGFR (green). Each panel is a representative mid-level confocal slice. Bar¼ 10 mm.
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6503
Oncogene

mutation had a relatively modest effect upon thedownregulation of the EGFRvIII by Cbl-b in CHOcells (Figure 3d). This is likely due to the lowendogenous levels of the Cbl proteins present in theNIH 3T3 cells used in the focus-forming assaycompared to the levels of Cbl-b when it is overexpressedin CHO cells. Similarly, Waterman et al. (2002) reportedthat mitogenic signaling from the WT EGFR wasincreased significantly by the Y1045F mutation in thecontext of endogenous Cbl proteins.As the formation of foci is increased by the mutation
of the Cbl-binding site in the EGFRvIII and decreasedby the overexpression of Cbl-b (Figure 5), the ability ofthe EGFRvIII to transform is regulated by the Cblproteins.
The cytotoxicity of an EGFRvIII-specific immunotoxin isantagonized by an EGFRvIII TK inhibitorTo confirm further that the EGFRvIII undergoesactivation-dependent downregulation, we investigatedthe effects of an EGFR TK inhibitor, AG 1478, uponthe activity of an anti-EGFRvIII immunotoxin (MR1-1(scFv)-PE38) (Beers et al., 2000). Immunotoxins mustbe internalized upon binding to their receptor in order tokill cells (Pastan et al., 1992). As we have shown above(Figure 2), AG 1478 treatment inhibits the activation-induced downregulation of the EGFRvIII by the Cblproteins. Therefore, the inhibition of the EGFRvIII TKwould be expected to decrease the efficacy of the anti-EGFRvIII immunotoxin MR1-1(scFv)-PE38. The effectof MR1-1(scFv)-PE38 treatment upon the viability of a
murine fibroblast cell line (NR-6) and a subclone thatstably expresses the EGFRvIII (NR-6m) (Batra et al.,1995) was measured using an MTS dye reduction assay(Figure 6a). Previously, we have shown that this indirectmeasurement of cytotoxicity correlates with cell death(Keane et al., 1996). A 24 h incubation with MR1-1(scFv)-PE38 causes a concentration-dependent decreasein the viability of NR-6m cells. In contrast, the viabilityof the parental cell line (NR-6), which does not expressthe EGFRvIII, is not affected by treatment with thefusion toxin. Treatment with 30 mM AG 1478 attenuatedthe decrease in viability of NR-6m cells caused by MR1-1(scFv)-PE38 (Figure 6b). The concentration of MR1-1(scFv)-PE38 necessary to reduce cell viability by 50%was approximately 1000-fold higher when cells wereincubated with 30 mM AG 1478 (IC50E10 mg/ml) thanwhen they were incubated with the vehicle (IC50E10 ng/ml). Therefore, the TK activity of the EGFRvIII has animportant role in mediating the toxicity of anti-EGFRvIII immunotoxins. In addition, this result isconsistent with the EGFRvIII undergoing activation-induced downregulation.
Discussion
The ability of all three members of the Cbl family of E3s(Cbl, Cbl-b, and Cbl-c) to ubiquitinate and down-regulate the EGFR following stimulation with EGFis well-characterized (Ettenberg et al., 1999b, 2001;Levkowitz et al., 1999; Waterman et al., 1999a; Yokouchi
Figure 6 The EGFR TK inhibitor, AG 1478, prevents MR1-1(scFv)-PE38-mediated cell death of EGFRvIII-expressing cells. (a) Themurine fibroblast cell lines NR-6 (�) and the EGFRvIII-expressing NR-6m (’) were incubated with increasing concentrations (0.01–10 000ng/ml) of MR1-1(scFv)-PE38 for 24 h. The cell viability, measured by an MTS dye reduction assay, is expressed as a percentageof maximum. Values are the mean71 s.d. of a representative experiment. (b) EGFRvIII-expressing NR-6m cells were pretreated witheither the vehicle (0.1%DMSO;’) or 30mMAG 1478 (m) for 4 h. The cells were then incubated with a combination of 0.01–10 000ng/ml MR1-1(scFv)-PE38 in the continued presence of either the vehicle or 30 mM AG 1478. The cell viability, measured by an MTS dyereduction assay, is expressed as a percentage of maximum. Values are the mean7s.e.m. of n¼ 3 experiments.
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6504
Oncogene

et al., 1999; Duan et al., 2003). In this study, we establishthat the Cbl proteins can downregulate the constitu-tively active mutant of the EGFR known as theEGFRvIII. The overexpression of Cbl, Cbl-b, or Cbl-ccaused a decrease in the level of EGFRvIII proteinin CHO cells (Figure 1a–c). We observed also that theco-expression of the Cbl proteins enhanced the ubiqui-tination of the EGFRvIII (Figure 1d–f). This down-regulation of the EGFRvIII by Cbl-b was blocked bythe use of an EGFR TK inhibitor, AG 1478 (Figure 2),and by the Y1045F mutation of the EGFRvIII (Figures3 and 5). As in the active WT EGFR, Y1045 isphosphorylated in the EGFRvIII and the Y1045Fmutation prevents phosphorylation of this residue(Figure 3d). This prevents the direct binding of theCbl proteins, the only proteins known to interact withthis phosphotyrosine residue in cells. The abrogation ofthe interaction of the EGFRvIII with endogenous Cblproteins by either EGFRvIII Y1045F mutation (Figure5b and c) or TK inhibition (Figures 2 and 6b) blocksEGFRvIII downregulation. Therefore, it appears thatthe Cbl proteins mediate the activation-induced down-regulation of the EGFRvIII.The ligand-induced activation of the WT EGFR
results in its autophosphorylation and the subsequentrecruitment of Cbl-b (Ettenberg et al., 1999a). There-fore, we investigated the interaction between theEGFRvIII and Cbl-b using a cell line that expressesendogenous EGFR (HEK 293T) and a cell line that doesnot (CHO). We observed an association between theEGFRvIII and Cbl-b in both of these cell lines(Figure 4). The interaction between the EGFRvIII andCbl-b in HEK 293T cells appears to be unaffected by theactivation of WT EGFR by EGF. In addition, the co-transfection of the WT EGFR and the EGFRvIII intoCHO cells did not appear to prevent the downregulationof either of these proteins by Cbl-b (data not shown).Therefore, it appears that the constitutive associationbetween Cbl-b and the EGFRvIII is independent of theWT EGFR. Like the WT EGFR, we found that therecruitment of Cbl-b to the EGFRvIII involves twomechanisms: one that involves the TKB domain of Cbl-b, the other that involves the proline-rich carboxy-terminus of Cbl-b. Using the end point of receptordegradation, we found that the EGFRvIII is down-regulated by both WT Cbl-b and a truncated form ofCbl-b (N1/2) that contains its TKB and RING fingerdomains, but not its extensive proline-rich carboxy-terminus (Figure 3). Mutation of the Cbl TKB-bindingsite in the WT EGFR (Y1045F) impairs the ligand-induced ubiquitination and downregulation of theEGFR (Levkowitz et al., 1999). When we mutated theequivalent residue in the EGFRvIII, we prevented theubiquitination and downregulation of this receptor byN1/2 Cbl-b (Figure 3d). However, the mutation of thisresidue does not appear to have as significant an effectupon the interaction between the EGFRvIII and WTCbl-b. As the proline-rich region of the Cbl proteins canindirectly bind to the WT EGFR via Grb2 (Watermanet al., 2002; Jiang et al., 2003), this is likely also the casewith the EGFRvIII. The EGFRvIII has been shown to
bind to Grb2 in NIH 3T3 fibroblasts (Moscatello et al.,1996). Interestingly, stable clones of NIH 3T3 cellsexpressing high levels of the EGFRvIII have decreasedlevels of Grb2 protein (Moscatello et al., 1996). This isconsistent with the ability of the Cbl proteins todownregulate the EGFR signaling complex, includingGrb2 (Ettenberg et al., 2001).In contrast to the present study, Schmidt et al. (2003)
reported that the EGFRvIII does not interact witheither Cbl or Cbl-b. In their investigation, HEK 293 cellswere transfected with EGFRvIII and either Cbl or Cbl-b. Then the EGFRvIII was precipitated with an anti-EGFRvIII-specific antibody. Although they observedthe co-precipitation of both Cbl and Cbl-b with theEGFRvIII, the WT EGFR was also precipitated in theirexperiments. They concluded that the anti-EGFRvIIIantibody was crossreacting with the WT receptor, so insubsequent experiments they precleared the lysate withan anti-EGFR antibody before the precipitation of theEGFRvIII. Following preclearing of the lysates, theyfailed to observe either Cbl or Cbl-b when theEGFRvIII was precipitated. In addition, they were alsounable to observe any ubiquitination of the EGFRvIIIfollowing this preclearing. As the EGFRvIII and theWT EGFR are capable of heterodimerizing (Luworet al., 2004), it is possible that this preclearing stepremoved any of the EGFRvIII that is bound to the WTEGFR. As this heterodimerized protein may be theactive pool of the EGFRvIII, this could account for anydifferences between the two studies. Our experiments inCHO cells, which do not express the WT EGFR,allowed us to investigate the interaction between theEGFRvIII and the Cbl proteins in the absence of theWT receptor. In addition, we used a mutant (C373A) ofCbl-b deficient in E3 activity to test an interactionbetween the EGFRvIII and Cbl-b in CHO cells. As thismutant cannot target the complex of Cbl-b and theEGFRvIII for lysosomal degradation, the amount ofactive EGFRvIII bound to Cbl-b should be increasedrelative to cells transfected with WT Cbl-b. Therefore,any association between these proteins should bedetected with a greater sensitivity than if WT Cbl-bwas used. Only a small fraction (approximately 10%) ofthe EGFRvIII protein is active at any given timecompared to the WT EGFR that has been stimulated byEGF (Huang et al., 1997; Fernandes et al., 2001). Thus,it is possible that the interaction between the EGFRvIIIand the Cbl proteins was below the level of sensitivity ofthe immunoprecipitation and immunoblotting proce-dure used by Schmidt et al. (2003).The constitutive TK activity of the EGFRvIII results
in the malignant transformation of cells (Han et al.,1996; Nagane et al., 1996; Huang et al., 1997; Antonyaket al., 1998; Montgomery, 2002; Johns et al., 2003;Abulrob et al., 2004; Luwor et al., 2004; Pedersen et al.,2004). In this study, we found that the EGFRvIII isregulated by the Cbl proteins in an identical manner tothe WT EGFR. This is unsurprising given that theactivity and phosphorylation pattern of the dimerizedEGFRvIII is similar to that of the WT EGFR followingEGF stimulation (Fernandes et al., 2001). Indeed, we
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6505
Oncogene

were able to detect phosphorylation of the Cbl TKB-binding site (Y1045) on the EGFRvIII using a specificantibody (Figure 3d). In addition, Reist et al. (1995)reported that the EGFRvIII is internalized rapidly fromthe surface of fibroblasts transfected with the EGFR-vIII, suggesting that it is downregulated. Conversely, ina study using glioblastoma cells transfected with eitherthe WT EGFR or the EGFRvIII, Huang et al. (1997)reported that, while the EGF-stimulated WT EGFR israpidly endocytosed, the EGFRvIII is internalized at asimilar rate to that of the unstimulated WT EGFR. Thissuggests that the EGFRvIII is not downregulated.However, only a small proportion of the total EGFR-vIII protein is active when compared to the ligandbound EGFR (Huang et al., 1997; Fernandes et al.,2001). It is likely that, compared to the spontaneousendocytosis of the overexpressed WT EGFR, theenhanced internalization of the small amount of activeEGFRvIII does not significantly affect the overall rateof endocytosis.Our work indicates that active EGFRvIII is degraded
by a Cbl protein-dependent mechanism. However,cancer cells with amplification of the EGFRvIIIconstitutively synthesize new inactive EGFRvIII pro-tein. Experiments using the EGFR inhibitor AG 1478demonstrate that the Cbl proteins do not mediateubiquitination or degradation of inactive EGFRvIII(Figure 2). The amplification and overexpression of theEGFRvIII creates a large pool of inactive receptor, asmall fraction of which spontaneously activates toreplenish the pool of downregulated active EGFRvIII.Thus, at steady-state equilibrium, there always will beactive EGFRvIII and this results in the transformationof cells. The overexpression of Cbl-b inhibits thetransformation of fibroblasts by the EGFRvIII byenhancing the degradation of the active EGFRvIII.Conversely, the mutation of the Cbl-binding site in theEGFRvIII increases its capacity to transform bypreventing degradation of the active EGFRvIII.The anti-EGFRvIII immunotoxin, MR1-1(scFv)-
PE38, kills glioblastoma cells that ectopically expressthe EGFRvIII (Beers et al., 2000). In this study, we usedan MTS dye reduction assay to test the ability of thisimmunotoxin to kill a Swiss 3T3-derived cell line (NR-6)that does not express the WT EGFR (Pruss andHerschman, 1977). Although MR1-1(scFv)-PE38 didnot effect the growth of NR-6 cells, it caused aconcentration-dependent death of EGFRvIII-expressingNR-6m cells (Figure 6a). This finding confirmed theprevious report (Beers et al., 2000) that MR1-1(scFv)-PE38 specifically kills EGFRvIII-expressing cells. TheIC50 of MR1-1(scFv)-PE38 in this study (approximately10 ng/ml) is similar to previously reported values (Beerset al., 2000). To function, immunotoxins must beinternalized upon binding to their receptors (Pastanet al., 1992); indeed anti-EGFRvIII monoclonal anti-bodies — including MR1-1(scFv)-PE38 — are rapidlyinternalized by EGFRvIII-expressing cells (Reist et al.,1995; Kuan et al., 2000). These internalized antibodiesbecome localized to vesicles in the perinuclear Golgiregion and are rapidly catabolized, suggesting that the
internalized EGFRvIII:monoclonal antibody complex istrafficked to the lysosome. The Cbl proteins are criticalregulators of the trafficking of the WT EGFR to thelysosome (Duan et al., 2003) and this study hasestablished that they regulate the constitutively activeEGFRvIII. Furthermore, the inhibition of the TKactivity of the EGFRvIII prevents its downregulationby the Cbl proteins and decreases the amount ofEGFRvIII located in intracellular vesicles (Figure 2).Therefore, we tested whether inhibition of the EGFR-vIII TK affects the efficacy of MR1-1(scFv)-PE38.Consistent with the ability of the EGFRvIII to undergoactivation-induced downregulation, we found thattreatment with AG 1478 caused an approximately1000-fold increase in the IC50 of MR1-1(scFv)-PE38(Figure 6b). Thus, the inhibition of the TK activity ofthe EGFRvIII appears to antagonize MR1-1(scFv)-PE38 in vitro. Like the WT EGFR, the EGFRvIII alsocan be spontaneously endocytosed in an activation-independent manner. Thus, MR1-1(scFv)-PE38 is stillcapable of killing cells in the presence of AG1478, albeitwith an IC50 1000-fold higher than untreated cells. Thisfinding suggests that TK inhibitors and immunotoxinsmay be antagonistic if used together for the treatment ofEGFRvIII-expressing tumors.This study has demonstrated that the EGFRvIII
undergoes activation-induced downregulation by theCbl proteins. This suggests that the ability of theEGFRvIII to transform cells is not a consequence ofunattenuated signaling from this mutant, but is duerather to the spontaneous activity of this TK. The abilityof the EGFRvIII to be regulated by the Cbl proteins hasimplications for the treatment of malignancies. Thera-pies, such as immunotoxins, that exploit the down-regulation of the EGFRvIII or therapies aimed atenhancing the activation-induced degradation of thismutant offer a promising approach to the treatment ofEGFRvIII-expressing tumors. However, the use of TKinhibitors in conjunction with these therapies maydecrease their efficacy.
Materials and methods
MaterialsDulbecco’s modified Eagle’s medium (DMEM), fetal bovineserum (FBS), penicillin, streptomycin sulfate, and Zeocin wereobtained from Invitrogen (Carlsbad, CA, USA). Dulbecco’sphosphate-buffered saline (DPBS) and G-418 sulfate werepurchased from Mediatech Inc. (Herndon, VA, USA). AG1478, ALLN (MG 101), cycloheximide, MG 132, lactacystin,and folimycin (concanamycin A) were acquired from EMDBiosciences Inc. (San Diego, CA, USA). Leupeptin hemisulfatewas bought from MP Biomedicals (Irvine, CA, USA).Chloroquine, ammonium chloride, and DMSO were obtainedfrom Sigma-Aldrich Corp. (St Louis, MO, USA). Recombi-nant human EGF was purchased from BD Biosciences, Inc.(San Jose, CA, USA). A recombinant immunotoxin generatedfrom an EGFRvIII-specific single chain Fv domain fused todomains I and II of the Pseudomonas exotoxin (MR1-1(scFv)-PE38) was provided by Dr Ira Pastan (Beers et al., 2000).Tissue culture plastic ware and other laboratory consumableswere purchased from commercial sources.
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6506
Oncogene

Expression constructsThe expression plasmids for full-length WT and HA-epitope-tagged Cbl, Cbl-b, and Cbl-c along with HA-epitope-taggedfull-length RING finger mutant (C373A) Cbl-b, C2/3 Cbl-b(aa 327–938), N1/2 Cbl-b (aa 1–483), and the control vector(pCEFL) have been described previously (Ettenberg et al.,1999a; Ettenberg et al., 2001). The cDNA for the EGFRvIIIwas a gift from Dr Gordon N Gill and was cloned intopSVZeo(�) (Invitrogen, Carlsbad, CA, USA). Site-directedmutagenesis of EGFRvIII (Y1045F) was performed using theQuick Change Kit (Stratagene, La Jolla, CA, USA). All of theconstructs were confirmed by DNA sequencing. The GFPexpression plasmid (pCDNA3.1/zeo) was obtained fromInvitrogen (Carlsbad, CA, USA). The HA-epitope-taggedubiquitin expression plasmid was provided by Dr DirkBohmann (Treier et al., 1994).
Cell culture, transfections, and foci assaysCHO, HEK 293T, and NIH 3T3 cells were maintained inculture in DMEM supplemented with 10% FBS, 100U/mlpenicillin, and 100mg/ml streptomycin sulfate. NR-6 cells weremaintained in DMEM supplemented with 5% FBS, 100U/mlpenicillin, and 100 mg/ml streptomycin sulfate. NR-6m cells, asubclone of NR-6 that stably expresses the EGFRvIII, wereprovided by Dr Darrel Bigner and were maintained in DMEMsupplemented with 10% FBS, 100U/ml penicillin, 100 mg/mlstreptomycin sulfate, and 750mg/ml G-418. CHO cells weretransfected with various constructs using FuGENE 6 (RocheDiagnostics Corp., Indianapolis, IN, USA), whereas HEK293T cells were transfected using calcium phosphate (Profec-tion; Promega Corp., Madison, WI, USA). Following trans-fection, cells were grown to 70% confluence and starvedovernight in DMEM supplemented with 0.5% FBS. Then,cells were treated as described in the figure legends before thepreparation of cell lysates.NIH 3T3 cells were transfected with the EGFRvIII, Y1045F
EGFRvIII, HA-Cbl-b, C373A HA-Cbl-b, or empty vectorcontrols as indicated using Effectine (Qiagen Inc., Valencia,CA, USA). A day after the transfection, the cells were split 1:3and grown for 14 days in selection medium containing either600mg/ml Zeocin alone or a combination of 600mg/ml Zeocinand 600 mg/ml G-418. Stable clones were pooled and fociassays were performed at passage 3 by plating 1� 106 cells per100mm tissue culture dish. Cells were incubated 1–2 weeks,fixed with 10% methanol, 10% acetic acid solution for 15min,and stained with 20% ethanol, 0.4% crystal violet for 5min.
Immunoblotting and immunoprecipitationTo harvest proteins, cells were washed twice in ice-cold DPBScontaining 200 mM sodium orthovanadate (Fisher Chemicals,Fairlawn, NJ, USA) and then lysed in ice-cold lysis buffer(10mM Tris-HCl pH 7.5, 150mM NaCl, 5mM EDTA, 1%Triton X-100, 10% glycerol, 100mM iodoacetamide (Sigma-Aldrich Corp., St Louis, MO, USA), 2mM sodium orthova-nadate, and protease inhibitors (Complete tabss, RocheDiagnostics Corp., Indianapolis, IN, USA)). The lysates werecleared of debris by centrifugation at 16 000 g for 10min at41C. Supernatant protein concentrations were determinedusing a BioRad protein assay (BioRad, Hercules, CA, USA).For immunoblotting, lysates (2 mg protein/ml) were boiled inloading buffer (62.5mM Tris-HCl pH 6.8, 10% glycerol, 2%SDS, 1mg/ml bromphenol blue, 0.3573M b-mercaptoethanol)for 5min. For immunoprecipitation, lysates containing 500 mgprotein were incubated with either a mouse monoclonal anti-EGFR antibody (528; Santa Cruz Biotechnology, Santa Cruz,CA, USA) and Protein A/Gþ agarose beads (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) or HA-affinity matrix(Roche Diagnostics Corp., Indianapolis, IN, USA) overnightat 41C with tumbling. Immune complexes were washed fivetimes in cold lysis buffer, resuspended in 2� loading bufferand boiled for 5min. The proteins were resolved by SDS–PAGE and transferred to PVDF membranes (Immobilon P;Millipore, Billerica, MA, USA). Membranes were probed witheither rabbit polyclonal anti-EGFR (2232; Cell SignalingTechnology Inc., Beverly, MA, USA), rabbit polyclonal anti-phosphotyrosine 1045 EGFR (2237; Cell Signaling Technol-ogy Inc., Beverly, MA, USA), rabbit polyclonal anti-Cbl (C15;Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbitpolyclonal anti-Cbl-b (H121 or H454; Santa Cruz Biotechnol-ogy, Santa Cruz, CA, USA), goat polyclonal anti-Cbl-c (C15;Santa Cruz Biotechnology, Santa Cruz, CA, USA), mousemonoclonal anti-HA (12CA5; Roche Diagnostics Corp.,Indianapolis, IN, USA), mouse monoclonal anti-GFP (C163;Zymed, San Francisco, CA, USA), mouse monoclonal anti-a-Tubulin (DM 1A; Sigma-Aldrich Corp., St Louis, MO, USA),or peroxidase-linked anti-phosphotyrosine (4G10; UpstateBiotechnology Inc., Waltham, MA, USA) antibodies. Horse-radish peroxidase-linked donkey anti-rabbit (GE Healthcare,Piscataway, NJ, USA), donkey anti-mouse (GE Healthcare,Piscataway, NJ, USA), or rabbit anti-goat (Santa CruzBiotechnology, Santa Cruz, CA, USA) immunoglobulin wasused with SuperSignal (Pierce Biotechnology Inc., Rockford,IL, USA) to visualize the blots. Immunoblots were quantifiedon a PC computer using the public domain NIH Imageprogram (developed at the US National Institutes of Healthand available on the Internet at http://rsb.info.nih.gov/nih-image/).
Immunofluorescence and confocal microscopyNR-6m cells or pooled clones of NIH 3T3 cells expressing theEGFRvIII or Y1045F EGFRvIII were plated at 2� 104 cells/well in four-well chambered coverslips (Nalgene Nunc Inter-national, Rochester, NY, USA) and incubated overnight.Then, the NR-6m cells were incubated for 3 h with 100 mg/mlcycloheximide and either 30 mM AG 1478 or 0.1% DMSO.Following a rinse with PBS, both NR-6m and NIH 3T3 cellswere fixed with 2% paraformaldehyde in PBS for 30min atroom temperature. The chambers were rinsed three times withPBS, washed three times with PFNS buffer (PBS, containing10% FBS, 0.02% sodium azide (Sigma-Aldrich Corp., StLouis, MO, USA), and 10% saponin (Sigma-Aldrich Corp., StLouis, MO, USA)) and blocked with PFNS-G (PFNS buffersupplemented with 2% normal goat serum) for 30min at roomtemperature. Blocked chambers were then incubated overnightat 41C with either mouse monoclonal anti-EGFR (528; SantaCruz Biotechnology, Santa Cruz, CA, USA) or mousemonoclonal anti-phosphotyrosine 1173 EGFR (9H2; UpstateBiotechnology Inc., Waltham, MA, USA) antibodies dilutedin PFNS-G, washed three times with PFNS, and incubatedwith Alexa-Fluor 488-conjugated goat anti-mouse antibodydiluted in PFNS-G for 1 h at room temperature. The chamberswere then washed three times with PBS containing 2%saponin, stained with 300 nM DAPI in PBS for 3min, andrinsed three times with PBS. All images were collected using aZiess 510 META confocal microscope with a � 63 (NA 1.4)Plan-Apochromat oil immersion objective (Carl Zeiss Micro-imaging Inc., Thornwood, NY, USA). Alexa-Fluor 488staining was imaged using a 488 nm Argon Laser line inconjunction with a HFT 405/488/543/633 multiple beamsplitter, NFT 545 dichroic, and a BP 505–570 emission filter.DAPI was imaged using a 405 nm laser diode line, HFT 405/488/543/633 multiple beam splitter, NFT 505 dichroic, and a
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6507
Oncogene

BP 420–480 emission filter. The laser power was set to 4%transmission with the pinhole opened to 1 Airy unit. Confocalimage series were recorded with a frame size of 512� 512pixels and a pixel size of 110–140 nm. Images were processedwith Zeiss LSM Image Browser (version 3.5). Adobe Photo-shop (version 7.0) was used to prepare composite images.
Cytotoxicity assaysNR-6 or NR-6m cells were plated at 1.5� 104 cells/well in 96-well microtiter plates and incubated overnight. Then, NR-6mcells were incubated for 4 h with either 30 mM AG 1478 or 0.1%DMSO. NR-6m cells were then incubated for a further 24 hwith either 30 mM AG 1478 or 0.1% DMSO alone or incombination with 0.1–10 000 ng/ml MR1-1(scFv)-PE38. NR-6cells were incubated with MR1-1(scFv)-PE38 (0.1–10 000 ng/ml) or mock-treated for 24 h. Cell viability was assessed by theMTS dye reduction assay (Cell Titer 96 AQueous One SolutionCell Proliferation assay; Promega Corp., Madison, WI, USA).Six wells were used in each experimental group. The cell
viability in each experimental group was expressed as the meanpercentage of maximum viability. Values are the mean71 s.d.of a representative experiment or the mean7s.e.m. for n¼ 3experiments.
Acknowledgements
We thank Marion M Nau for helpful discussion andcritical review of the manuscript. We thank Jeremy Karlinfor helpful discussion and for help with quantitation ofthe immunoblots. Also, we thank Valarie A Barr for helpwith the confocal microscopy. Philip E Ryan is a graduatestudent in the Genetics Program of the George WashingtonUniversity Institute of Biomedical Sciences. The workpresented here is in partial fulfillment for the degree of PhD.This research was supported by the Intramural ResearchProgram of the NIH, National Cancer Institute, Center forCancer Research.
References
Abulrob A, Giuseppin S, Andrade MF, McDermid A, MorenoM, Stanimirovic D. (2004). Oncogene 23: 6967–6979.
Antonyak MA, Moscatello DK, Wong AJ. (1998). J BiolChem 273: 2817–2822.
Batra SK, Castelino-Prabhu S, Wikstrand CJ, Zhu X,Humphrey PA, Friedman HS et al. (1995). Cell GrowthDiffer 6: 1251–1259.
Beers R, Chowdhury P, Bigner D, Pastan I. (2000). ClinCancer Res 6: 2835–2843.
Blume-Jensen P, Hunter T. (2001). Nature 411: 355–365.Damstrup L, Wandahl Pedersen M, Bastholm L, Elling F,Skovgaard Poulsen H. (2002). Int J Cancer 97: 7–14.
Duan L, Miura Y, Dimri M, Majumder B, Dodge IL, ReddiAL et al. (2003). J Biol Chem 278: 28950–28960.
Ekstrand AJ, Longo N, Hamid ML, Olson JJ, Liu L, CollinsVP et al. (1994). Oncogene 9: 2313–2320.
Ettenberg SA, Keane MM, Nau MM, Frankel M, Wang LM,Pierce JH et al. (1999a). Oncogene 18: 1855–1866.
Ettenberg SA, Magnifico A, Cuello M, Nau MM, RubinsteinYR, Yarden Y et al. (2001). J Biol Chem 276: 27677–27684.
Ettenberg SA, Rubinstein YR, Banerjee P, Nau MM, KeaneMM, Lipkowitz S. (1999b). Mol Cell Biol Res Commun 2:111–118.
Fernandes H, Cohen S, Bishayee S. (2001). J Biol Chem 276:5375–5383.
Garcia de Palazzo IE, Adams GP, Sundareshan P, Wong AJ,Testa JR, Bigner DD et al. (1993). Cancer Res 53:3217–3220.
Han Y, Caday CG, Nanda A, Cavenee WK, Huang HJ.(1996). Cancer Res 56: 3859–3861.
Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J,Gilbert M et al. (2005). Clin Cancer Res 11: 1462–1466.
Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, JiXD et al. (1997). J Biol Chem 272: 2927–2935.
Jiang X, Huang F, Marusyk A, Sorkin A. (2003).Mol Biol Cell14: 858–870.
Johns TG, Luwor RB, Murone C, Walker F, Weinstock J,Vitali AA et al. (2003). Proc Natl Acad Sci USA 100: 15871–15876.
Keane MM, Ettenberg SA, Lowrey GA, Russell EK,Lipkowitz S. (1996). Cancer Res 56: 4791–4798.
Keane MM, Ettenberg SA, Nau MM, Banerjee P, Cuello M,Penninger J et al. (1999). Oncogene 18: 3365–3375.
Kuan CT, Wikstrand CJ, Archer G, Beers R, Pastan I,Zalutsky MR et al. (2000). Int J Cancer 88: 962–969.
Kuan CT, Wikstrand CJ, Bigner DD. (2001). Endocr RelatCancer 8: 83–96.
Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygan-kov AY, Alroy I et al. (1999). Mol Cell 4: 1029–1040.
Luwor RB, Zhu HJ, Walker F, Vitali AA, Perera RM, BurgessAW et al. (2004). Oncogene 23: 6095–6104.
Mancini A, Koch A, Wilms R, Tamura T. (2002). J Biol Chem277: 14635–14640.
Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, ZhuS, Dia EQ et al. (2005). N Engl J Med 353: 2012–2024.
Montgomery RB. (2002). Int J Cancer 101: 111–117.Moscatello DK, Holgado-Madruga M, Godwin AK, RamirezG, Gunn G, Zoltick PW et al. (1995). Cancer Res 55:5536–5539.
Moscatello DK, Montgomery RB, Sundareshan P,McDanel H, Wong MY, Wong AJ. (1996). Oncogene 13:85–96.
Nagane M, Coufal F, Lin H, Bogler O, Cavenee WK, HuangHJ. (1996). Cancer Res 56: 5079–5086.
Nau MM, Lipkowitz S. (2003). Gene 308: 103–113.Nishikawa R, Ji XD, Harmon RC, Lazar CS, Gill GN,Cavenee WK et al. (1994). Proc Natl Acad Sci USA 91:7727–7731.
O’Rourke DM, Nute EJ, Davis JG, Wu C, Lee A, Murali Ret al. (1998). Oncogene 16: 1197–1207.
Pastan I, Chaudhary V, FitzGerald DJ. (1992). Annu RevBiochem 61: 331–354.
Pedersen MW, Meltorn M, Damstrup L, Poulsen HS. (2001).Ann Oncol 12: 745–760.
Pedersen MW, Tkach V, Pedersen N, Berezin V, Poulsen HS.(2004). Int J Cancer 108: 643–653.
Peschard P, Fournier TM, Lamorte L, Naujokas MA, BandH, Langdon WY et al. (2001). Mol Cell 8: 995–1004.
Peschard P, Park M. (2003). Cancer Cell 3: 519–523.Pruss RM, Herschman HR. (1977). Proc Natl Acad Sci USA74: 3918–3921.
Reist CJ, Archer GE, Kurpad SN, Wikstrand CJ, Vaidya-nathan G, Willingham MC et al. (1995). Cancer Res 55:4375–4382.
Schmidt MH, Furnari FB, Cavenee WK, Bogler O. (2003).Proc Natl Acad Sci USA 100: 6505–6510.
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6508
Oncogene

Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M,Nakamura H et al. (2003). Cancer Res 63: 6962–6970.
Tang CK, Gong XQ, Moscatello DK, Wong AJ, LippmanME. (2000). Cancer Res 60: 3081–3087.
Treier M, Staszewski LM, Bohmann D. (1994). Cell 78: 787–798.
Waterman H, Alroy I, Strano S, Seger R, Yarden Y. (1999a).EMBO J 18: 3348–3358.
Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, ElsonA et al. (2002). EMBO J 21: 303–313.
Waterman H, Levkowitz G, Alroy I, Yarden Y. (1999b). J BiolChem 274: 22151–22154.
Wikstrand CJ, Hale LP, Batra SK, Hill ML, Humphrey PA,Kurpad SN et al. (1995). Cancer Res 55: 3140–3148.
Yokouchi M, Kondo T, Houghton A, Bartkiewicz M, HorneWC, Zhang H et al. (1999). J Biol Chem 274: 31707–31712.
Cbl proteins downregulate the EGFRvIIIGC Davies et al
6509
Oncogene




















