
Dynamics of the O(3P) + C2H2 reaction from crossed molecular beam experiments with soft electron...
15
This journal is © the Owner Societies 2014 Phys. Chem. Chem. Phys. Cite this: DOI: 10.1039/c3cp54729a Dynamics of the O( 3 P) + C 2 H 2 reaction from crossed molecular beam experiments with soft electron ionization detection Francesca Leonori, Nadia Balucani,* Giovanni Capozza, Enrico Segoloni, Gian Gualberto Volpi and Piergiorgio Casavecchia* The reaction between ground state oxygen atoms, O( 3 P), and the acetylene molecule, C 2 H 2 , has been investigated in crossed molecular beam experiments with mass-spectrometric detection and time-of- flight analysis at three different collision energies, E c = 34.4, 41.1 and 54.6 kJ mol 1 . From product angular and velocity distribution measurements of the HCCO and CH 2 products in the laboratory frame, product angular and translational energy distributions in the center-of-mass frame were determined. Measurements on the CH 2 product were made possible by employing for product detection the recently implemented soft electron-ionization (EI) technique with low-energy, tunable electrons, which has permitted suppressing interference coming from the dissociative ionization of reactants, products and background gases. It has been found that the title reaction leads only to two competing channels: H + HCCO (ketenyl) and CO + CH 2 (triplet methylene). The branching ratio of cross sections between the two competing channels has been determined to be s(HCCO)/[s(HCCO) + s(CH 2 )] = 0.79 0.05, independent of collision energy within the experimental uncertainty. This value is in line with that obtained in the most recent and accurate kinetics determination at room temperature as well as with that predicted from recent theoretical calculations based on statistical rate theory and weak-collision master equation analysis and on dynamics surface-hopping quasiclassical trajectory calculations on-the-fly on coupled triplet/singlet ab initio potential energy surfaces. The firm assessment of the branching ratio as a function of translational energy for this important reaction, besides its fundamental significance, is of considerable relevance for the implementation of theoretical models of hydrocarbon combustion. I. Introduction The characterization of the dominant elementary reactions occurring in combustion and flames is central for the under- standing of these complex chemical processes and may ulti- mately lead to their optimization. 1–3 While kinetics studies provide us with their rate coefficients as a function of tempera- ture, the characterization of the channel yields in kinetics experiments is mostly conducted at room temperature, that is far from the typical conditions of combustion. Nevertheless, measurements of branching ratios (BRs) at the high tempera- tures of interest in combustion are needed, because they can strongly depend on the available energy, especially if new reaction channels open up at high temperature. Product BRs can be provided by dynamics experiments as a function of relative translational energy. These results are especially valuable if all open channels can be investigated under the same experimental conditions and with the same degree of accuracy. Experimentally, this can be achieved using a ‘‘universal’’ detection method such as mass-spectrometry (MS) to interrogate all product channels on the same footing and the crossed molecular beam (CMB) technique. 1,2 An important advantage of CMB experiments is that the reactions are investigated under single collision conditions, which is crucial as primary products are often transient species which can undergo secondary or wall collisions in bulk experiments. 1,2 The reaction between ground state oxygen atoms, O( 3 P), and acetylene, C 2 H 2 (X 1 S g + ), is one of the first steps in the oxidation of acetylene, and plays a key role also in the combus- tion of other hydrocarbons because C 2 H 2 is easily formed both during the combustion of small hydrocarbons, such as methane, and during the combustion of large aliphatic and Dipartimento di Chimica, Biologia e Biotecnologie, Universita ` degli Studi di Perugia, 06123 Perugia, Italy. E-mail: [email protected], [email protected] Received 8th November 2013, Accepted 14th January 2014 DOI: 10.1039/c3cp54729a www.rsc.org/pccp PCCP PAPER Published on 15 January 2014. Downloaded by University of Perugia on 17/03/2014 11:40:57. View Article Online View Journal
-
Upload
vegajournal -
Category
Documents
-
view
2 -
download
0
Transcript of Dynamics of the O(3P) + C2H2 reaction from crossed molecular beam experiments with soft electron...

This journal is© the Owner Societies 2014 Phys. Chem. Chem. Phys.
Cite this:DOI: 10.1039/c3cp54729a
Dynamics of the O(3P) + C2H2 reaction fromcrossed molecular beam experiments withsoft electron ionization detection
Francesca Leonori, Nadia Balucani,* Giovanni Capozza, Enrico Segoloni,Gian Gualberto Volpi and Piergiorgio Casavecchia*
The reaction between ground state oxygen atoms, O(3P), and the acetylene molecule, C2H2, has been
investigated in crossed molecular beam experiments with mass-spectrometric detection and time-of-
flight analysis at three different collision energies, Ec = 34.4, 41.1 and 54.6 kJ mol�1. From product
angular and velocity distribution measurements of the HCCO and CH2 products in the laboratory frame,
product angular and translational energy distributions in the center-of-mass frame were determined.
Measurements on the CH2 product were made possible by employing for product detection the
recently implemented soft electron-ionization (EI) technique with low-energy, tunable electrons, which
has permitted suppressing interference coming from the dissociative ionization of reactants, products
and background gases. It has been found that the title reaction leads only to two competing channels:
H + HCCO (ketenyl) and CO + CH2 (triplet methylene). The branching ratio of cross sections between
the two competing channels has been determined to be s(HCCO)/[s(HCCO) + s(CH2)] = 0.79 � 0.05,
independent of collision energy within the experimental uncertainty. This value is in line with that
obtained in the most recent and accurate kinetics determination at room temperature as well as with
that predicted from recent theoretical calculations based on statistical rate theory and weak-collision
master equation analysis and on dynamics surface-hopping quasiclassical trajectory calculations
on-the-fly on coupled triplet/singlet ab initio potential energy surfaces. The firm assessment of the
branching ratio as a function of translational energy for this important reaction, besides its fundamental
significance, is of considerable relevance for the implementation of theoretical models of hydrocarbon
combustion.
I. Introduction
The characterization of the dominant elementary reactionsoccurring in combustion and flames is central for the under-standing of these complex chemical processes and may ulti-mately lead to their optimization.1–3 While kinetics studiesprovide us with their rate coefficients as a function of tempera-ture, the characterization of the channel yields in kineticsexperiments is mostly conducted at room temperature, that isfar from the typical conditions of combustion. Nevertheless,measurements of branching ratios (BRs) at the high tempera-tures of interest in combustion are needed, because theycan strongly depend on the available energy, especially ifnew reaction channels open up at high temperature. ProductBRs can be provided by dynamics experiments as a function of
relative translational energy. These results are especiallyvaluable if all open channels can be investigated under thesame experimental conditions and with the same degreeof accuracy. Experimentally, this can be achieved using a‘‘universal’’ detection method such as mass-spectrometry(MS) to interrogate all product channels on the same footingand the crossed molecular beam (CMB) technique.1,2 Animportant advantage of CMB experiments is that the reactionsare investigated under single collision conditions, which iscrucial as primary products are often transient specieswhich can undergo secondary or wall collisions in bulkexperiments.1,2
The reaction between ground state oxygen atoms, O(3P),and acetylene, C2H2(X1Sg
+), is one of the first steps in theoxidation of acetylene, and plays a key role also in the combus-tion of other hydrocarbons because C2H2 is easily formedboth during the combustion of small hydrocarbons, such asmethane, and during the combustion of large aliphatic and
Dipartimento di Chimica, Biologia e Biotecnologie, Universita degli Studi di Perugia,
06123 Perugia, Italy. E-mail: [email protected], [email protected]
Received 8th November 2013,Accepted 14th January 2014
DOI: 10.1039/c3cp54729a
www.rsc.org/pccp
PCCP
PAPER
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article OnlineView Journal

Phys. Chem. Chem. Phys. This journal is© the Owner Societies 2014
aromatic hydrocarbons.4–6 The title reaction has three energe-tically and spin-allowed open channels:
O(3P) + C2H2(X1Sg+) - H(2S1/2) + HCCO(X2A00)
DH 00 = �82.4 kJ mol�1 (1a)
- CH2(X3B1) + CO(X1S+) DH 00 = �198.7 kJ mol�1 (1b)
- H2(X1Sg+) + CCO(X3Sg
�) DH 00 = �93.7 kJ mol�1 (1c)
(for the values of the enthalpies of reaction, see ref. 7).Notably, the reaction channel (1a) is alleged to be an
important step in soot formation8–12 because the H and HCCOproducts can further react leading to formation of singletmethylene CH2(a1A1) and CO. CH2(a1A1) in turn reacts readilywith C2H2 to form the propargyl radical (CH2CCH), which isbelieved to be responsible for the formation of the first aro-matic ring and hence of polycyclic aromatic hydrocarbons andsoot. The products of reaction (1a) are also thought to beresponsible for prompt CO2 formation in acetylene flamesthrough the reaction HCCO + O2.13 In addition, reaction (1) isof some relevance in hydrocarbon-rich planetary atmospheres,such as that of Titan, where a little amount of oxygen com-pounds is present,14 as well as in the interstellar medium.15
Because of its paramount practical relevance, reaction (1)has been studied very extensively from the kinetics standpointusing a variety of experimental techniques8,16–22 and over awide temperature range, e.g., from 200 K to 1950 K. While theoverall rate constant has been well established (the recom-mended value of k(T) from 200 K to 2500 K is 1.95 � 10�15 T1.40
exp(�1110/T) cm3 mol�1 s�1),23 the question of the identity ofthe primary reaction products and their relative importance hasbeen a subject of considerable debate over the years. Thehistory of the branching ratio is summarized in Table 1. Allrecent kinetics determinations19–21 agreed on the fact thatonly channels (1a) and (1b) are open and that the productsHCCO + H are predominant over CH2 + CO, with the yield of theH-elimination channel being 80 � 10% and nearly independentof temperature in the range 290–1200 K. In particular, the mostrecent kinetics determination20,21 at room temperature has givenk(1a)/[k(1a) + k(1b)] = 0.83 � 0.08 and k(1b)/[k(1a) + k(1b)] =0.17 � 0.08. These values are in good agreement with theore-tical predictions based on ab initio calculations of the triplet
potential energy surface (PES) by Harding and Wagner,24
who also computed the rate coefficients using transition statetheory. A good agreement was found between theoretical andexperimental rate coefficients at temperatures below 1000 K,but the predicted rates are about a factor of 2 lower at highertemperatures.24 More recently, Nguyen et al.7 have performed adetailed theoretical study of reaction (1) by performing electronicstructure calculations of the two lowest lying triplet PESs. Speci-fically, they have studied the reaction using coupled-clustertheory and the CBS-QCI/APNO combination methods to con-struct the two lowest-lying triplet surfaces, 3A00 and 3A0, and haveused these PESs in high-level theoretical kinetics analysis. The3A00 and 3A0 PESs are depicted schematically in Fig. 1. As can beseen, the reactants correlate adiabatically with the products alongthe ground state 3A00 PES. However, the HCCO radical has a low-lying electronically excited state (located 12.9 kJ mol�1 above theground state) which is formed on the 3A0 state PES. Therefore,a fourth reaction pathway is possible:
O(3P) + C2H2(X1Sg+) - H + HCCO(A2A0) DH 0
0 = �69.5 kJ mol�1
(1d)
The theoretical work of Nguyen et al.7 underscored for thefirst time the role played by the first excited triplet 3A0 PESwhich becomes significant only at high temperatures becauseof the presence of an entrance barrier higher than that asso-ciated with the ground state PES. In this way, Nguyen et al. wereable to better reproduce the experimental values of rate coeffi-cients at high temperatures.7
In addition to the potential role of the 3A0 PES, a contri-bution from intersystem crossing (ISC) from the triplet to thesinglet PES can be present for reaction (1). In O(3P) reactionswith closed-shell substrates, the reactants approach on a tripletPES which intersects a singlet PES usually supporting a stableintermediate. ISC is then possible from the triplet to the singletPES, making the dynamics which involve motion on the under-lying singlet PES different from those involving motion onlyover the triplet PES. Previous measurements of the reactivescattering of O(3P) with various alkyl iodides have indicated theoccurrence of ISC.25–27 Nevertheless, the investigation of atomicoxygen reactions with small molecules containing light atoms,such as O + H2S, clearly excluded the occurrence of ISC,28
Table 1 History of the branching ratio of the O(3P) + C2H2 reaction
HCCO + H (%) CH2 + CO (%)
Early work (1969–1981) (see ref. 39) Dominant productWilliamson and Bayes (1969) (see ref. 39) E25Vinckier et al. (1979) (see ref. 39) E50Harding and Wagner (1986) (up to 3000 K)24 70+10
�35
Peeters et al. (1986) (285–535 K)17 62 � 23Frank et al. (1986) (1500–2500 K)18 64 � 15Schmoltner et al. (1989) (CMB)39 42 � 10 58 � 10Michael and Wagner (1990) (900–1200 K)19 80 � 15Boullart and Peeters (1992) (290 K)20 85+4
�9
Peeters et al. (1994) (290 K)21 83 � 8 17 � 8Nguyen et al. (2006) (200–2000 K) (RRKM)7 E80 E20Rajak and Maiti (2010) (QCT-SH)37 Ec = 39.7 kJ mol�1 79 21This work (CMB) Ec = 34.4, 41.1 and 54.6 kJ mol�1 79 � 5 21 � 5
Paper PCCP
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online

This journal is© the Owner Societies 2014 Phys. Chem. Chem. Phys.
so leading to the conclusion that ISC could be significant onlyin the presence of heavy atoms because of the larger spin–orbitcoupling. In this respect, it came as a surprise that for thereaction O(3P) + C2H4 ISC accounts for about 50% of the totalreactivity,29–31 and even more for the reactions O(3P) + CH2CCH2
(allene)32 and O(3P) + CH3CCH (methylacetylene).33 The role ofISC in the O(3P) + C2H4 reaction has been confirmed by detailedtheoretical work.29,30,34
In the case of the title reaction the role of ISC has alreadybeen examined regarding the channel
O(3P) + C2H2(X1Sg+) - CH2(a1A1) + CO DH 0
0 =�158.2 kJ mol�1
(1e)
because the formation of singlet methylene requires ISC tothe singlet PES. Several attempts have been done to observesinglet CH2 in kinetics experiments,20,21 but in the cases whereCH2(a1A1) was observed by laser-induced-fluorescence (LIF), thedata analysis proved that it was formed by secondary reactions(such as HCCO + H) rather than being an active primarychannel of reaction (1).8,20,21 In addition, an attempt to detectCH2(a1A1) emission in collision free experiments failed.35
Nevertheless, according to the theoretical study reported byYarkony,36 ISC can occur likely, while recent quasiclassical-trajectory surface-hopping (QCT-SH) results reported by Rajakand Maiti37 suggested that a fraction of about one third of theCH2 + CO channel (which overall is found to account for 21% ofthe reaction yield at Ec = 39.7 kJ mol�1) (see Table 1) originatesvia ISC leading to singlet CH2.
The dynamics of the title reaction was investigated in twoearly CMB studies38,39 carried out in two different laboratoriesat a collision energy of ca. 25 kJ mol�1. While one study38 waslimited to the dynamics of channel (1a), an estimate of thebranching ratio of channels (1a) and (1b) was given by Schmolt-ner et al.39 who used a beam of 18O to detect, in addition toHCCO, also 18CO (at m/z = 30), for which the detector had amuch lower background than at m/z = 28. A branching ratio ofcross sections s(1a)/[s(1a) + s(1b)] = 0.58 � 0.21 was derived,which is somewhat lower than the value obtained from kineticsstudies (see Table 1). More recently, the reaction was also
studied using Fourier transform infrared (FTIR) emissionspectroscopy in a flow system at room temperature, where thevibrational population of CO was determined,40 and in CMBexperiments at hyperthermal collision energies, where otherhighly endothermic channels become open.41
Considering the importance of the reaction of acetylene withatomic oxygen in hydrocarbon combustion and flames, we havere-investigated the dynamics and the branching ratio of thisreaction using an improved CMB technique which exploits(i) an increased instrumental sensitivity, (ii) an improved (withrespect to previous CMB studies) resolution for measuring productangular and time-of-flight (TOF) distributions, and (iii) the newability to detect cleanly the CH2 radical by using soft electron-ionization (EI).42,43 A preliminary account of this work wasreported in a Communication44 showing some data at onecollision energy. In addition, by exploiting the new capabilityof tuning the kinetic energy of the ionizing electrons down tovery low values, we have determined the electron ionizationefficiency curve of the reaction product HCCO and estimatedexperimentally the ionization energy of the HCCO radical.42
In this paper we report on product angular and velocity distri-bution measurements of the HCCO and CH2 products, generatedin channels (1a) and (1b), respectively, at three different collisionenergies, Ec (Ec = 34.4, 41.1 and 54.6 kJ mol�1). The reactionmechanism for both channels is elucidated and the branchingratio is derived at the investigated collision energies. Thedynamic results are discussed in light of the most recentelectronic structure calculations of the relevant PESs.7,41,45
II. Experimental
The basics of our CMB apparatus have been described elsewhere,46
while critical improvements, such as the soft electron-ionizationand variable crossing beam set-ups, have been detailedrecently.42,43 Briefly, two supersonic beams of the reactants arecrossed at 901 or 1351 under single collision conditions in a largescattering chamber kept at about 2� 10�6 mbar under operatingconditions. Two experiments (at Ec = 34.4 and 41.1 kJ mol�1)have been performed at a beam crossing angle g of 901,
Fig. 1 Schematic potential energy surfaces for the O(3P) + C2H2 reaction (adapted from ref. 7).
PCCP Paper
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online

Phys. Chem. Chem. Phys. This journal is© the Owner Societies 2014
while that at the highest Ec of 54.6 kJ mol�1 has beenperformed at a beam crossing angle of 1351. The angular andvelocity distributions of the reaction products are recordedby a triply differentially pumped, ultra-high-vacuum (UHV)(10�11 mbar) detector equipped with a tunable electron impactionizer followed by a quadrupole mass filter and an off-axiselectron multiplier. The whole detector unit can be rotatedin the plane of the two beams around their intersection axis(Y = 01 represents the direction of the atomic oxygen beam).The velocity of reactants and products is derived using single-shot and pseudo-random, respectively, TOF analysis. Thesupersonic beams of O atoms were produced by means of aradio-frequency (RF) discharge source operating at a RF powerof about 300 Watt and a pressure of 200–300 mbar of a dilute(5%) mixture of O2 in He carrier gas.47–49 This source producesatomic oxygen mainly in the ground 3PJ state, but a smallconcentration in the excited 1D2 state (lying B2 eV above theground state) is also present.47 The contribution of O(1D) to thereactive signal is expected to be negligible in the presentexperiments because at the experimental Ecs the reactive crosssections of the triplet and singlet reactions are similar (the rateconstant at room temperature of O(1D) + C2H2 has beendetermined to be (3.1 � 0.2) � 10�10 cm3 s�1),50 and thefraction of O(1D) in the beam is very small (r5%) with respectto O(3P); furthermore, the O(1D) reaction is expected to producemainly OH + CCH,51 so that we do not expect any interferencein the study of the reaction channels (1a) and (1b).
A supersonic beam of acetylene was produced by expandingneat gas through a stainless steel nozzle kept at room tempera-ture. An acetone/dry ice slush trap (�78 1C) was used on theC2H2 gas line to trap acetone impurities for the experiment atEc = 54.6 kJ mol�1. Because the effect of the impurities turnedout to be almost negligible, no trap was used for the experi-ments at the other Ecs. In a first series of experiments, wheng = 901 the O(3P) peak velocity, vpeak, and the speed ratio, S, were2762 m s�1 and 5.1, respectively, while the C2H2 beam hadvpeak = 817 m s�1 and S = 6.5, resulting in a canonical collisionenergy of 41.1 kJ mol�1.52 When g = 1351 the O(3P) beam hadvpeak = 2702 m s�1 and S = 6.9, while the C2H2 beam had vpeak =805 m s�1 and S = 6.9, resulting in Ec = 54.6 kJ mol�1. In the lastmeasurements with g = 901 the O-beam peak velocity was2499 m s�1 and S = 5.6, while the C2H2 beam had vpeak =839 m s�1 and S = 6.1, resulting in Ec = 34.4 kJ mol�1. Note thatin the first series of experiments, despite the nearly identicalbeam characteristics, the collision energy is significantlyincreased by simply changing the beam intersection angle g.42,43
The slight differences in the beam characteristics are due toslightly different experimental conditions in the two experi-ments: at Ec = 41.1 kJ mol�1, the C2H2 nozzle diameter was70 mm and the backing pressure was 0.75 bar, while 100 micronnozzle and 0.4 bar were used at Ec = 54.6 kJ mol�1; in addition,two slightly different nozzles and backing pressure and RFpower were used in the O beam (similar arguments hold for theexperiment at Ec = 34.4 kJ mol�1). Under these expansionconditions C2H2 clustering was negligible. Because of thesignificant cooling during supersonic expansion, the acetylene
molecules in the beam are expected to be in the lowest rota-tional states of essentially the ground vibrational level, andtherefore the internal energy of the molecular reactant contri-butes negligibly to the total available energy.
In the study of the O(3P) + C2H2 reaction with the CMBtechnique it is very easy to detect the heavy HCCO (ketenyl)fragment corresponding to channel (1a), because HCCO iskinematically constrained in a narrow angular region aroundthe center-of-mass. The detection of any of the two productsfrom the CH2 + CO forming channel is much more problematic.In particular, detection of CO is plagued by the high inherentdetector background at m/z = 28 due to residual CO in any UHVchamber. Detection of the CH2 counter-fragment at m/z = 14 isalso problematic in classic CMB experiments employing hard EI(70–200 eV), because of the high inherent background at thismass (though lower than at m/z = 28) due to dissociativeionization of residual CH4 to CH2
+, of residual N2 to N+, butespecially due to the interference from the dissociative ioniza-tion to 13CH+ as well as CH2
+ of the intense HCCO productsignal and of the elastically scattered, intense C2H2 beam.Although 13C represents only about 2% of the total C in HCCOand C2H2, the resulting m/z = 14 signal from the aboveprocesses is comparable to the reactive scattering signal dueto CH2 product formation. For all these reasons, detection ofCH2 from reaction (1b) was never attempted in the past.
In our laboratory, after having reduced to a very low value thepartial pressure of methane in the detector (and consequently itscontribution to m/z = 14 via dissociative ionization), we wereready to also tackle all the other contributions to m/z = 14. Thecontribution arising from dissociative ionization of residualN2 was suppressed by reducing the electron energy below theappearance energy (AE = 24.3 eV) of N+ from N2.53 To eliminatethe contribution from elastically scattered C2H2 to 13CH+ andCH2
+ the electron energy was lowered below the AE of CH+
(AE = 20.9 eV) and CH2+ (AE = 18.2–19.7 eV) from C2H2.53
To eliminate the contribution from dissociative ionization ofHCCO to m/z = 14 would require to lower the electron energybelow about 14 eV. However, since only a very small fraction ofCH+ appears at m/z = 14, all the final measurements at m/z = 14were performed at 17 eV, which was sufficient to suppress allthe interferences (see Fig. 1 in ref. 44).
The product laboratory (LAB) angular distributions, N(Y),were recorded at m/z = 41 (HCCO+) and 14 (CH2
+) by countingfor 30 s and 100 s, respectively, at each angle and averaging overat least five scans. The C2H2 beam was modulated at 160 Hz bya tuning-fork chopper for background subtraction. The HCCOand CH2 LAB angular distribution are reported in Fig. 2–4 forthe experiments at Ec = 34.4, 41.1 and 54.6 kJ mol�1, respec-tively, together with the corresponding velocity vector (Newton)diagram of the experiment. Signal-to-noise (S/N) at the center-of-mass (CM) angle was for m/z = 41 about 470 at 60 eV (S/N =200 at 17 eV) and 35 for m/z = 14 at 17 eV at Ec = 41.1 kJ mol�1
and 54.6 kJ mol�1, while it was somewhat lower at the lowest Ec
of 34.4 kJ mol�1. Angular and TOF distributions at m/z = 41were also recorded using 17 eV electron energy for signalcalibration purposes with respect to the m/z = 14 distributions
Paper PCCP
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online
This journal is© the Owner Societies 2014 Phys. Chem. Chem. Phys.
which were always measured at 17 eV. Angular distributionsrecorded at m/z = 40 with 60 eV electron energy proved at all Ecsto be identical to those recorded at m/z = 41 (see next section).
TOF distributions of the products were obtained at selectedlaboratory angles using typically the cross-correlation TOFtechnique with four 127-bit pseudorandom sequences. High-time resolution was achieved by spinning the pseudorandomTOF disk, located at the entrance of the detector, at 393.7 Hzcorresponding to a dwell time of 5 ms per channel; typicalcounting times were 5–20 minutes per angle. The flight lengthwas 24.3 cm. However, in the experiments at Ec = 41.1 and54.6 kJ mol�1, because of the excellent signal-to-noise, them/z = 41 TOF spectra (obtained using 60 eV electron energy)could also be recorded using the single-shot method at 3 ms perchannel employing a TOF wheel with 4 equally spaced slits(width 1 mm) spinning at 300 Hz; typical counting times were15–60 minutes per angle, depending on signal intensity. Alsothe m/z = 14 TOF spectra (obtained using 17 eV electron energy)were also recorded with the single-shot technique becauseof the negligible background at this mass when using 17 eV
ionizing electrons (a TOF wheel with 8 equally spaced slits ofwidth 1.2 mm was used in this case); typical counting timeswere 10–120 minutes per angle, depending on signal intensity.In order to obtain a somewhat higher time resolution the finalTOF spectra shown in this paper at these two Ecs were allobtained with the single-shot method at 3 ms per channel. Incontrast, the product TOF data at Ec = 34.4 kJ mol�1 wererecorded only using the pseudorandom TOF method with a dwelltime of 6 ms per channel (corresponding to a wheel frequency of328.1 Hz); counting times varied from 10 to 60 min dependingupon product mass and signal intensity.
III. Results and analysis
Fig. 2–4 show comparatively the LAB angular distributionsof the HCCO (top panel) and CH2 (bottom panel) products atEc = 34.4, 41.1 and 54.6 kJ mol�1, respectively. The TOF spectrarecorded at selected LAB angles are shown in Fig. 5–7 form/z = 41 and Fig. 8–10 for m/z = 14 (for this mass only oneTOF spectrum at the CM angle was recorded at the lowest Ec,
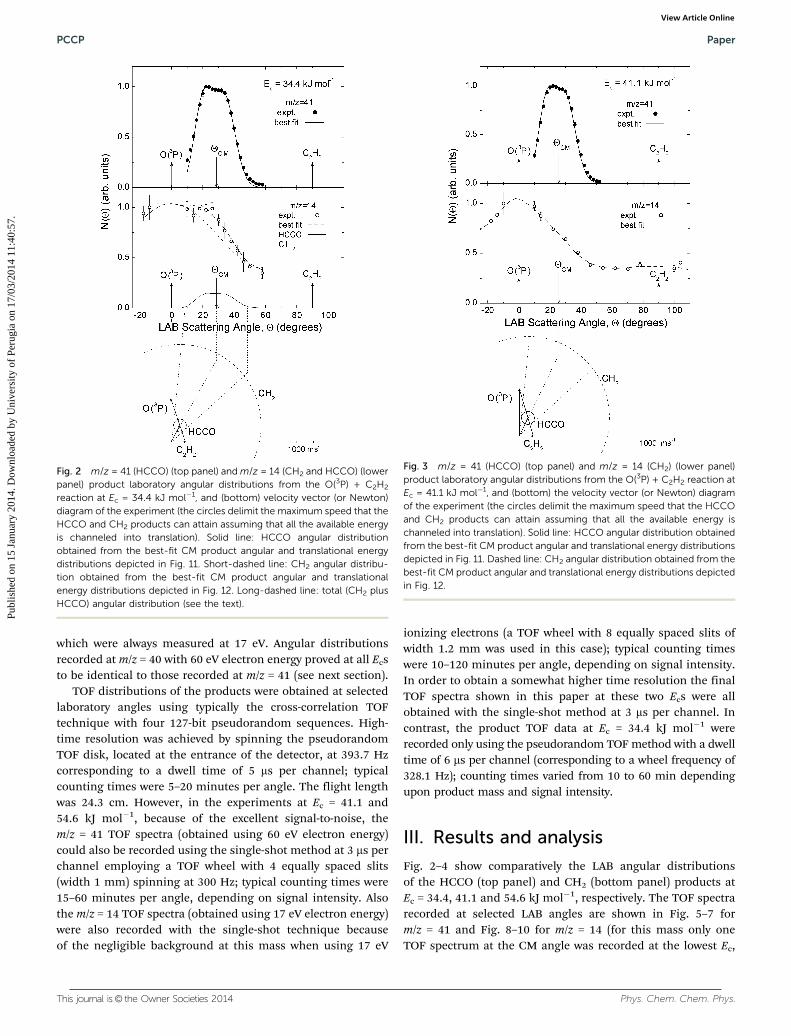
Fig. 2 m/z = 41 (HCCO) (top panel) and m/z = 14 (CH2 and HCCO) (lowerpanel) product laboratory angular distributions from the O(3P) + C2H2
reaction at Ec = 34.4 kJ mol�1, and (bottom) velocity vector (or Newton)diagram of the experiment (the circles delimit the maximum speed that theHCCO and CH2 products can attain assuming that all the available energyis channeled into translation). Solid line: HCCO angular distributionobtained from the best-fit CM product angular and translational energydistributions depicted in Fig. 11. Short-dashed line: CH2 angular distribu-tion obtained from the best-fit CM product angular and translationalenergy distributions depicted in Fig. 12. Long-dashed line: total (CH2 plusHCCO) angular distribution (see the text).
Fig. 3 m/z = 41 (HCCO) (top panel) and m/z = 14 (CH2) (lower panel)product laboratory angular distributions from the O(3P) + C2H2 reaction atEc = 41.1 kJ mol�1, and (bottom) the velocity vector (or Newton) diagramof the experiment (the circles delimit the maximum speed that the HCCOand CH2 products can attain assuming that all the available energy ischanneled into translation). Solid line: HCCO angular distribution obtainedfrom the best-fit CM product angular and translational energy distributionsdepicted in Fig. 11. Dashed line: CH2 angular distribution obtained from thebest-fit CM product angular and translational energy distributions depictedin Fig. 12.
PCCP Paper
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online

Phys. Chem. Chem. Phys. This journal is© the Owner Societies 2014
and three spectra at the highest Ec). It should be noted that,while at Ec = 41.1 and 54.6 kJ mol�1 the CH2 product has beendetected cleanly using 17 eV electron energy, at the lowest Ec
the energy of the ionizing electrons was somewhat higher than17 eV (about 18.5 eV) and consequently some contribution atm/z = 14 from dissociative ionization of the HCCO mainproduct to 13CH+ is discernable, both in the angular and TOFdistributions (see Fig. 2 and 8). This has been taken intoaccount in the data analysis (see below).
As can be seen the heavy HCCO product, scattered by thevery light H counterpart, is confined in a narrow angular rangearound the CM angle, YCM, (see Fig. 2–4) and is quite slow withpeaks in the TOF spectra at around 200–250 ms (see Fig. 5–7),that is somewhat faster than the center of mass. In contrast,the much lighter CH2 product, scattered by the heavy COco-fragment, is characterized by very broad angular distribu-tions (see Fig. 2–4) and is very fast, peaking at about 60–70 ms(see Fig. 8–10), which is at times much shorter than that of thecenter of mass, because of linear momentum conservation andalso, to a minor extent, because of a larger reaction exoergicity.It should be noted that the time-of-flight of the center-of-massin Fig. 5–10 is 224, 208 and 319 ms for the experiment atEc = 34.4, 41.1 and 54.6 kJ mol�1, respectively. The HCCOLAB angular distributions show some bimodality with respectto YCM, more evident at the higher Ec (because the 1351 cross-ing beam configuration affords a higher angular and TOFresolution, as inspection of the Newton diagrams in Fig. 2–4indicates), that is, product is scattered both in the forward and
backward directions. This, together with the bimodality of theTOF spectra at angles close to YCM, indicates that the CMangular distribution of HCCO extends over all CM angularranges and that the product translational energy distributionpeaks away from zero, reflecting a significant amount of energybeing released into translation (see below).
Notably, N(Y) and N(Y, t) at m/z = 41 (HCCO+) and 40 (CCO+)(not shown) were found to be identical, indicating that the signalat m/z = 40 is all coming from the dissociative ionization ofHCCO, and not from the dynamically and energetically differentH2 elimination channel (1c). This suggests that the H2 elimina-tion pathway is closed at these Ecs. Recent QCT calculations forreaction (1) at very high (hyperthermal) collision energies havepointed out that the H2-elimination channel can give a smallcontribution only starting from a collision energy of about190 kJ mol�1, thus corroborating our experimental findings.41
For quantitative and physically meaningful information on thereaction dynamics, the product angular and velocity distributions
Fig. 4 As in Fig. 3, but at Ec = 54.6 kJ mol�1.
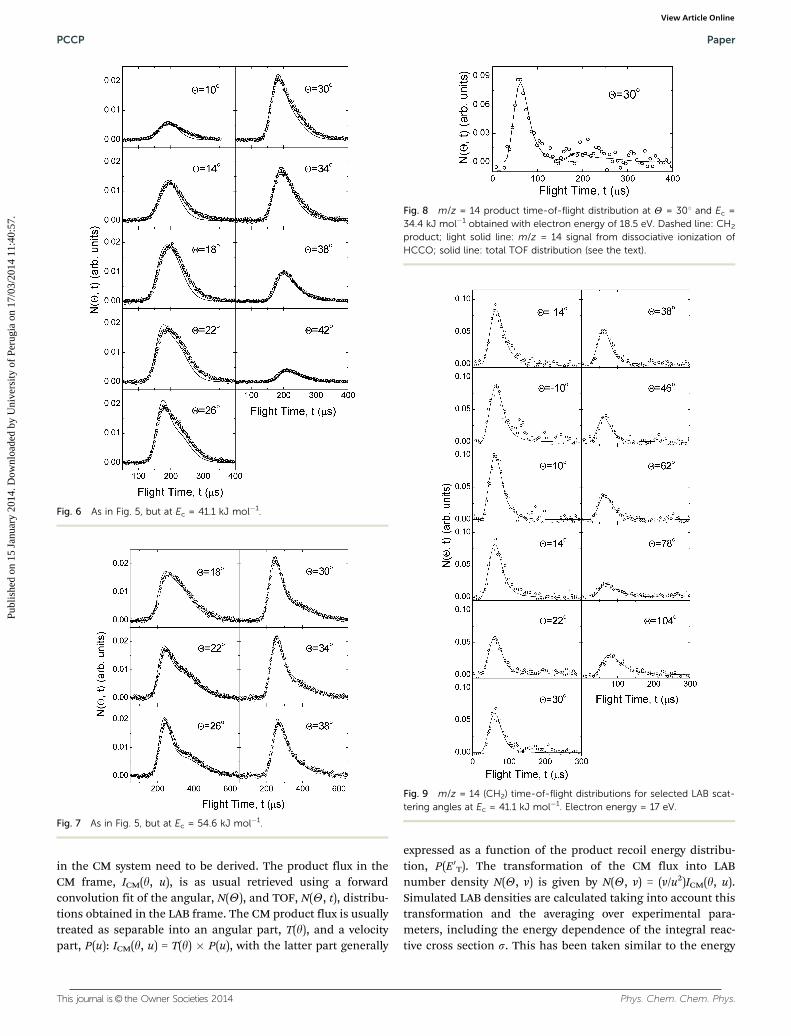
Fig. 5 m/z = 41 (HCCO) time-of-flight distributions for selected LABscattering angles at Ec = 34.4 kJ mol�1. Electron energy = 60 eV. Solidlines: TOF distributions calculated from the best-fit CM product angularand translational energy distributions depicted in Fig. 11.
Paper PCCP
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online
This journal is© the Owner Societies 2014 Phys. Chem. Chem. Phys.
in the CM system need to be derived. The product flux in theCM frame, ICM(y, u), is as usual retrieved using a forwardconvolution fit of the angular, N(Y), and TOF, N(Y, t), distribu-tions obtained in the LAB frame. The CM product flux is usuallytreated as separable into an angular part, T(y), and a velocitypart, P(u): ICM(y, u) = T(y) � P(u), with the latter part generally
expressed as a function of the product recoil energy distribu-tion, P(E0T). The transformation of the CM flux into LABnumber density N(Y, v) is given by N(Y, v) = (v/u2)ICM(y, u).Simulated LAB densities are calculated taking into account thistransformation and the averaging over experimental para-meters, including the energy dependence of the integral reac-tive cross section s. This has been taken similar to the energy
Fig. 6 As in Fig. 5, but at Ec = 41.1 kJ mol�1.
Fig. 7 As in Fig. 5, but at Ec = 54.6 kJ mol�1.
Fig. 8 m/z = 14 product time-of-flight distribution at Y = 301 and Ec =34.4 kJ mol�1 obtained with electron energy of 18.5 eV. Dashed line: CH2
product; light solid line: m/z = 14 signal from dissociative ionization ofHCCO; solid line: total TOF distribution (see the text).
Fig. 9 m/z = 14 (CH2) time-of-flight distributions for selected LAB scat-tering angles at Ec = 41.1 kJ mol�1. Electron energy = 17 eV.
PCCP Paper
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online

Phys. Chem. Chem. Phys. This journal is© the Owner Societies 2014
dependence of the cross section for HCCO + H formation asmeasured by Clemo et al.38 from the threshold up to 24 kJ mol�1.The same functional form s p (Ec � E0)s/Ec, where E0 is thethreshold energy of 8 kJ mol�1, was used with s = 1.5. The fittingprocedure is repeated until a satisfying fit of the experimentalLAB densities, N(Y) and N(Y, t), is achieved and the CMfunctions, T(y) and P(E0T), so determined are the best-fit func-tions. For the experiment at Ec = 34.4 kJ mol�1 the m/z = 14 datahave been analyzed using for the total flux ICM(y, u) the relationICM(y, u) = [T(y) � P(u)]CH2
+ a � [T(y) � P(u)]HCCO, where areflect the contribution of the HCCO channel at m/z = 14 and isa best-fit parameter. A similar approach has been used to fit theTOF data at 23 eV electron energy (see below).
A coupling may exist between the product angular andvelocity distribution in the CM frame. Such a coupling maybe subtle and usually high S/N is needed to disentangle it. Inthe present study this coupling was observed for the HCCO + Hchannel, but it proved to be small. When taken into account,the resulting angle dependent P(E0T) functions fell within theerror bars of the P(E0T) obtained in the best-fit analysis withoutangle dependence (see below). Therefore, the sensitivity of thedata did not warrant the use of P(E0T, y) functions and this wasnot pursued further in the data analysis.
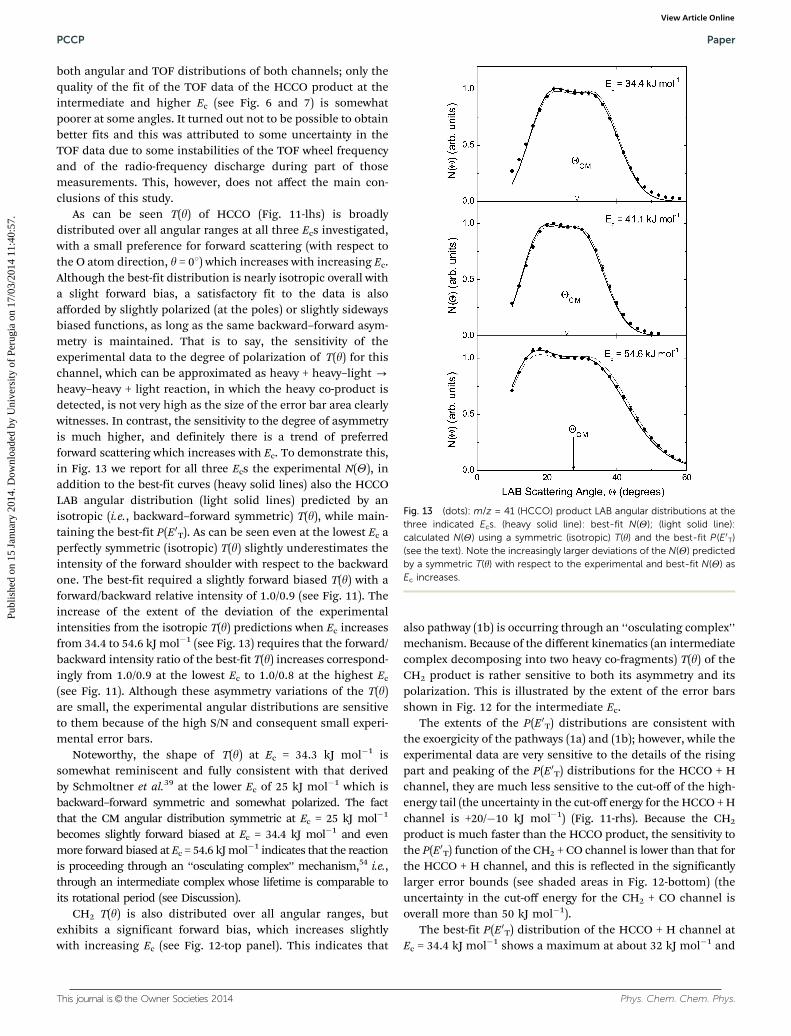
From LAB product angular and TOF distributions at m/z = 41and 14, CM product angular, T(y), and translational energy,P(E0T), distributions were derived for both channels (1a) and(1b). The best-fit T(y) functions of the HCCO and CH2 productsat the three Ecs are depicted in the left-hand-side (lhs) of Fig. 11and in the top panel of Fig. 12, respectively. The correspondingbest-fit P(E0T) for the HCCO + H and CH2 + CO channels arereported in the right-hand-side (rhs) and in the bottom panel ofthe same two figures, respectively. While for the best-fit CMfunctions of the HCCO + H channel the error bars are shown as
shaded area for all Ecs investigated (see Fig. 11), for those of theCH2 + CO channel they are indicated only at the intermediate Ec
(similar uncertainties hold for the other two Ecs) (see Fig. 12).CM functions within the shaded areas still produce a satis-factory fit of the LAB data. Overall the best-fits are very good for
Fig. 10 As in Fig. 9, but at Ec = 54.6 kJ mol�1.
Fig. 11 Center-of-mass best-fit HCCO angular (lhs) and product transla-tional energy (rhs) distributions for the channel HCCO + H. (top): Ec =34.4 kJ mol�1; (middle) Ec = 41.1 kJ mol�1; (bottom) Ec = 54.6 kJ mol�1.Shaded areas indicate the limits of the error bars (see text). ETOT indicatesthe total available energy.
Fig. 12 Center-of-mass best-fit CH2 angular (top) and product transla-tional energy (bottom) distributions for the channel CH2 + CO. (——): Ec =34.4 kJ mol�1; (------): 41.1 kJ mol�1; (-.-.-.-) Ec = 54.6 kJ mol�1. Shadedareas indicate the limits of the error bars for Ec = 41.1 kJ mol�1 (error barsat the lowest and highest Ec span similar ranges as at the intermediate Ec).ETOT indicates the total available energy.
Paper PCCP
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online
This journal is© the Owner Societies 2014 Phys. Chem. Chem. Phys.
both angular and TOF distributions of both channels; only thequality of the fit of the TOF data of the HCCO product at theintermediate and higher Ec (see Fig. 6 and 7) is somewhatpoorer at some angles. It turned out not to be possible to obtainbetter fits and this was attributed to some uncertainty in theTOF data due to some instabilities of the TOF wheel frequencyand of the radio-frequency discharge during part of thosemeasurements. This, however, does not affect the main con-clusions of this study.
As can be seen T(y) of HCCO (Fig. 11-lhs) is broadlydistributed over all angular ranges at all three Ecs investigated,with a small preference for forward scattering (with respect tothe O atom direction, y = 01) which increases with increasing Ec.Although the best-fit distribution is nearly isotropic overall witha slight forward bias, a satisfactory fit to the data is alsoafforded by slightly polarized (at the poles) or slightly sidewaysbiased functions, as long as the same backward–forward asym-metry is maintained. That is to say, the sensitivity of theexperimental data to the degree of polarization of T(y) for thischannel, which can be approximated as heavy + heavy–light -heavy–heavy + light reaction, in which the heavy co-product isdetected, is not very high as the size of the error bar area clearlywitnesses. In contrast, the sensitivity to the degree of asymmetryis much higher, and definitely there is a trend of preferredforward scattering which increases with Ec. To demonstrate this,in Fig. 13 we report for all three Ecs the experimental N(Y), inaddition to the best-fit curves (heavy solid lines) also the HCCOLAB angular distribution (light solid lines) predicted by anisotropic (i.e., backward–forward symmetric) T(y), while main-taining the best-fit P(E0T). As can be seen even at the lowest Ec aperfectly symmetric (isotropic) T(y) slightly underestimates theintensity of the forward shoulder with respect to the backwardone. The best-fit required a slightly forward biased T(y) with aforward/backward relative intensity of 1.0/0.9 (see Fig. 11). Theincrease of the extent of the deviation of the experimentalintensities from the isotropic T(y) predictions when Ec increasesfrom 34.4 to 54.6 kJ mol�1 (see Fig. 13) requires that the forward/backward intensity ratio of the best-fit T(y) increases correspond-ingly from 1.0/0.9 at the lowest Ec to 1.0/0.8 at the highest Ec
(see Fig. 11). Although these asymmetry variations of the T(y)are small, the experimental angular distributions are sensitiveto them because of the high S/N and consequent small experi-mental error bars.
Noteworthy, the shape of T(y) at Ec = 34.3 kJ mol�1 issomewhat reminiscent and fully consistent with that derivedby Schmoltner et al.39 at the lower Ec of 25 kJ mol�1 which isbackward–forward symmetric and somewhat polarized. The factthat the CM angular distribution symmetric at Ec = 25 kJ mol�1
becomes slightly forward biased at Ec = 34.4 kJ mol�1 and evenmore forward biased at Ec = 54.6 kJ mol�1 indicates that the reactionis proceeding through an ‘‘osculating complex’’ mechanism,54 i.e.,through an intermediate complex whose lifetime is comparable toits rotational period (see Discussion).
CH2 T(y) is also distributed over all angular ranges, butexhibits a significant forward bias, which increases slightlywith increasing Ec (see Fig. 12-top panel). This indicates that
also pathway (1b) is occurring through an ‘‘osculating complex’’mechanism. Because of the different kinematics (an intermediatecomplex decomposing into two heavy co-fragments) T(y) of theCH2 product is rather sensitive to both its asymmetry and itspolarization. This is illustrated by the extent of the error barsshown in Fig. 12 for the intermediate Ec.
The extents of the P(E0T) distributions are consistent withthe exoergicity of the pathways (1a) and (1b); however, while theexperimental data are very sensitive to the details of the risingpart and peaking of the P(E0T) distributions for the HCCO + Hchannel, they are much less sensitive to the cut-off of the high-energy tail (the uncertainty in the cut-off energy for the HCCO + Hchannel is +20/�10 kJ mol�1) (Fig. 11-rhs). Because the CH2
product is much faster than the HCCO product, the sensitivity tothe P(E0T) function of the CH2 + CO channel is lower than that forthe HCCO + H channel, and this is reflected in the significantlylarger error bounds (see shaded areas in Fig. 12-bottom) (theuncertainty in the cut-off energy for the CH2 + CO channel isoverall more than 50 kJ mol�1).
The best-fit P(E0T) distribution of the HCCO + H channel atEc = 34.4 kJ mol�1 shows a maximum at about 32 kJ mol�1 and
Fig. 13 (dots): m/z = 41 (HCCO) product LAB angular distributions at thethree indicated Ecs. (heavy solid line): best-fit N(Y); (light solid line):calculated N(Y) using a symmetric (isotropic) T(y) and the best-fit P(E0T)(see the text). Note the increasingly larger deviations of the N(Y) predictedby a symmetric T(y) with respect to the experimental and best-fit N(Y) asEc increases.
PCCP Paper
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online
Phys. Chem. Chem. Phys. This journal is© the Owner Societies 2014
this corresponds to an average product translational energy hE0Ti,defined as hE0Ti =
PP(E0T)E0T/
PP(E0T), of about 49 kJ mol�1,
that is, the average fraction of total available energy ETOT (ETOT =Ec� DH0
0) channelled into translation, fT, defined as fT = hE0Ti/ETOT,is 0.42. The remaining fraction (0.58) will be channelled into theinternal degrees of freedom of the HCCO product. The fraction fT
is found to decrease slightly with increasing Ec (to fT = 0.38 at Ec =41.1 kJ mol�1 and to fT = 0.35 at Ec = 54.6 kJ mol�1), and this factsuggests that when rising Ec the extra collision energy is mostlyconverted into internal excitation of the HCCO product. Thiscould well indicate also some electronic excitation (besidesro-vibrational excitation), with channel (1d) becoming moresignificant as Ec is raised, as predicted by the theoretical workof Nguyen et al.7
An attempt was made to verify whether the experimentaldata were sensitive to the distinct contributions of the twolowest triplet PESs. However, the analysis of the scattering datacould not distinguish the two contributions without someambiguity, perhaps because the dynamics of formation ofHCCO is similar in the two cases. In fact, the decomposingHCCHO transition states on both surfaces are similar, with theH atoms departing at an angle close to 901 with respect to theplane of the heavy CCO atoms (see Fig. 3 in ref. 7).
The best-fit P(E0T) distribution of the CH2 + CO channelpeaks at a much higher energy (ca. 65–70 kJ mol�1 at all Ecs)(see Fig. 12-bottom panel) and this is consistent with adissociation process characterized by an exit barrier, as intriplet-CH2CO - CH2(3B1) + CO. In contrast, since the processsinglet-CH2CO - CH2(1A1) + CO occurs without an exitpotential barrier,55 we would observe a P(E0T) distributionpeaking near zero were this channel dominant. Perhaps thisdynamic feature is the only argument supporting the conclu-sion that CH2 is mainly formed in its ground triplet state. As amatter of fact, because of the large exoergicity of channel (1b)and of the small energy gap (37.6 kJ mol�1) between triplet andsinglet methylene, it is not possible to infer from the P(E0T)distribution whether singlet CH2 was formed to any significantextent (see below). The average fraction of energy in translationis about 0.42 at all Ecs for the CH2 + CO channel, indicatingthat about 58% of the total available energy goes into internal(ro-vibrational) excitation of the CH2 and CO products.
IV. Determination of the branching ratios
It is of high interest for the modelling of combustion systemsto know accurately the branching ratio of the O(3P) + C2H2
reaction. To estimate the absolute yields of each channel in CMBexperiments one should know (i) the absolute beam intensities,(ii) the exact size of the collision volume, and (iii) the detectionefficiency. These quantities are not easy to determine accurately;however, since the first two are constant and the third can bereasonably estimated, we can easily determine relative cross sec-tions. We have followed the procedure outlined by Schmoltneret al.39 and have taken advantage of the soft EI approach. Althoughfrom the best-fit procedure it is possible to derive the total
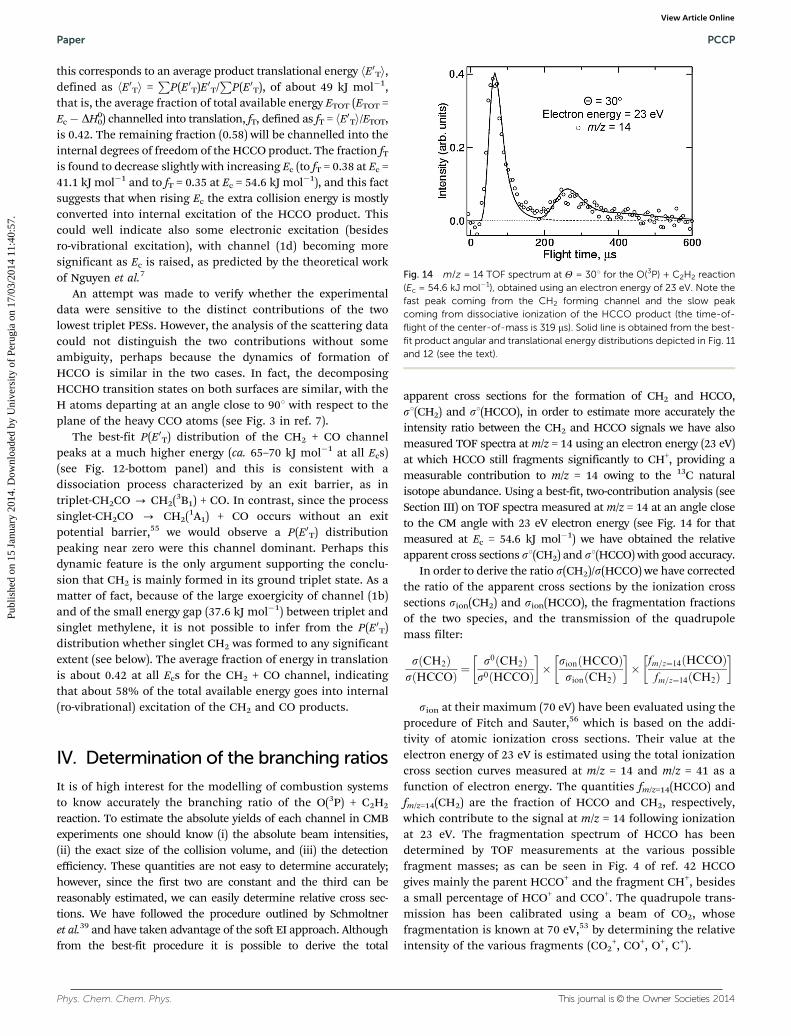
apparent cross sections for the formation of CH2 and HCCO,s1(CH2) and s1(HCCO), in order to estimate more accurately theintensity ratio between the CH2 and HCCO signals we have alsomeasured TOF spectra at m/z = 14 using an electron energy (23 eV)at which HCCO still fragments significantly to CH+, providing ameasurable contribution to m/z = 14 owing to the 13C naturalisotope abundance. Using a best-fit, two-contribution analysis (seeSection III) on TOF spectra measured at m/z = 14 at an angle closeto the CM angle with 23 eV electron energy (see Fig. 14 for thatmeasured at Ec = 54.6 kJ mol�1) we have obtained the relativeapparent cross sections s1(CH2) and s1(HCCO) with good accuracy.
In order to derive the ratio s(CH2)/s(HCCO) we have correctedthe ratio of the apparent cross sections by the ionization crosssections sion(CH2) and sion(HCCO), the fragmentation fractionsof the two species, and the transmission of the quadrupolemass filter:
s CH2ð ÞsðHCCOÞ ¼
s0 CH2ð Þs0ðHCCOÞ
� �� sionðHCCOÞ
sion CH2ð Þ
� ��
fm=z¼14ðHCCOÞfm=z¼14 CH2ð Þ
� �
sion at their maximum (70 eV) have been evaluated using theprocedure of Fitch and Sauter,56 which is based on the addi-tivity of atomic ionization cross sections. Their value at theelectron energy of 23 eV is estimated using the total ionizationcross section curves measured at m/z = 14 and m/z = 41 as afunction of electron energy. The quantities fm/z=14(HCCO) andfm/z=14(CH2) are the fraction of HCCO and CH2, respectively,which contribute to the signal at m/z = 14 following ionizationat 23 eV. The fragmentation spectrum of HCCO has beendetermined by TOF measurements at the various possiblefragment masses; as can be seen in Fig. 4 of ref. 42 HCCOgives mainly the parent HCCO+ and the fragment CH+, besidesa small percentage of HCO+ and CCO+. The quadrupole trans-mission has been calibrated using a beam of CO2, whosefragmentation is known at 70 eV,53 by determining the relativeintensity of the various fragments (CO2
+, CO+, O+, C+).
Fig. 14 m/z = 14 TOF spectrum at Y = 301 for the O(3P) + C2H2 reaction(Ec = 54.6 kJ mol�1), obtained using an electron energy of 23 eV. Note thefast peak coming from the CH2 forming channel and the slow peakcoming from dissociative ionization of the HCCO product (the time-of-flight of the center-of-mass is 319 ms). Solid line is obtained from the best-fit product angular and translational energy distributions depicted in Fig. 11and 12 (see the text).
Paper PCCP
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online

This journal is© the Owner Societies 2014 Phys. Chem. Chem. Phys.
From the fragmentation pattern of HCCO at 23 eV, correct-ing for the quadrupole transmission, we derive that the frac-tion of HCCO appearing as CH+ (m/z = 13) is 0.08. Since thenatural isotopic abundance of 13C is 0.011, the fraction ofHCCO appearing at m/z = 14 is 0.08 � 0.011 = 8.8 � 10�4.The fraction of CH2 appearing as parent ion CH2
+ at 23 eV is0.79, according to the work of Deutsch et al.57 Becauses1(CH2) = 1, s1(HCCO) = 0.01, fm/z=14(HCCO)/fm/z=14(CH2) =8.8 � 10�4/0.79, sion(CH2) = 1.5 Å2, sion(HCCO) = 3.5 Å2, weobtain s(CH2)/s(HCCO) = 0.26� 0.05 and therefore the followingbranching ratios (at Ec = 54.6 kJ mol�1):
s CH2ð Þs CH2ð Þ þ sðHCCOÞ ¼ 0:21� 0:05
sðHCCOÞs CH2ð Þ þ sðHCCOÞ ¼ 0:79� 0:05
The uncertainty is attributable to three main reasons. Firstof all, the ionization cross sections have an uncertainty of about20%, since for their determination we have employed anempirical procedure whose applicability to polyatomic radicalshas not been verified yet. However, it should be noted that therecently developed ‘‘modified additivity rules’’ appear to givesatisfactory results also for polyatomic radicals;57 furthermore,the total and partial ionization cross sections of CH2 are experi-mentally known and this has permitted to test the additivityrules.57 Assuming that the overestimate of sion obtained follow-ing Fitch and Sauter56 is similar for HCCO and CH2, their ratioshould be reliable to within 20%. Secondly, the fragmentationof the HCCO and CH2 products has been measured with anuncertainty to within 20%. Finally, the apparent cross sectionsderived from the best-fit analysis of the experimental data havean accuracy of the order of 10%.
The value of the branching ratio (BR) estimated at the lowerEcs of 41.1 kJ mol�1 and 34.4 kJ mol�1 is within the error barsthe same as at Ec = 54.6 kJ mol�1 (see Fig. 15). The presentestimate of the BR (see Table 1) for the two reactive channels ofthe O + C2H2 reaction is more accurate than that performed by
Schmoltner et al.39 at Ec = 25 kJ mol�1 because of the higherquality of the present experimental product angular and TOFdistributions, which has permitted an improved derivation ofthe CM differential cross sections for the two channels, andof a better determination of the HCCO fragmentation pattern(at 23 eV HCCO fragments much less extensively than at 200 eV,which is the value of electron energy used in the previous CMBexperiments at 25 kJ mol�1).
V. Discussion
The shape of the CM angular distribution for a bimolecularreaction can be related to the micromechanism of the reactivecollision. In particular, forward-peaked or backward-peakedT(y)s can be attributed to a direct type mechanism, while abackward–forward symmetric T(y) can generally be related toan indirect mechanism occurring through the formation of along-lived complex, i.e., a complex whose lifetime is longer(>6–7 times) than its rotational period.54 The detailed shapeof T(y), in particular its degree of polarization, is linked to thepartitioning of the total angular momentum. The possiblelimiting forms of T(y) depend on the correlation between theinitial orbital angular momentum Li and the final orbitalangular momentum Lf (usually in CMB experiments, becauseof the strong rotational cooling occurring in supersonic beams,the initial rotational angular momentum associated with thereactants ji is small and can be assumed to be E0). When one ofthe two co-products is a H-atom, the initial angular momentumis mostly converted into jf (Li B jf) because Lf is very small due tothe small reduced mass of the products (L = mvb). This impliesthat the molecular products are rotationally excited and thatthere is no correlation between Lf and Li. The resulting T(y) has asmall degree of polarization and is usually isotropic. Thesearguments well account for the best-fit almost isotropic T(y)derived for the HCCO + H channel. The small bias for forwardscattering (that increases with the available energy) can beexplained by invoking the osculating model of chemical reactions.54
Therefore, at low collision energy the O(3P) + C2H2 reaction onthe ground state surface is likely to proceed through a long-livedcomplex which starts to osculate as Ec increases. Fig. 11-lhsshows that T(y) is slightly forward biased even at the lowest Ec
investigated in this work, but the same channel is characterizedby a backward–forward symmetric T(y) at the lower Ec of theexperiments by Schmoltner et al.39
The best fit T(y) for channel (1b) is, instead, significantlymore polarized. In addition, the backward–forward asymmetryis enhanced, possibly because the increased exothermicity ofthis channel shortens the intermediate lifetime. The observeddegree of polarization suggests that the three heavy atomsremain coplanar during the decomposition of the transitionstate. In this respect, it is interesting to note that Huang et al.,22
following the experimental observation that the CO productsare rotationally cold, suggested that the decomposing transi-tion state for this channel should actually be collinear with analmost C2v symmetry. Theoretical prediction, however, did not
Fig. 15 Branching ratio sHCCO/(sHCCO + sCH2) for the reaction O(3P) +
C2H2 as a function of collision energy. The points at Ec = 34.4, 41.1 and54.6 kJ mol�1 are from the present work, while that at Ec = 3.7 kJ mol�1 istaken from kinetics work at room temperature (see the text). The dashedline is drawn through the points only to guide the eye.
PCCP Paper
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online

Phys. Chem. Chem. Phys. This journal is© the Owner Societies 2014
confirm that as far as the triplet PES is concerned. A signifi-cantly polarized T(y) for channel (1b) was also derived bySchmoltner et al.39
The relatively large fraction of energy released as producttranslational energy can be rationalized by the presence of anexit barrier for both channels (1a) and (1b). In the case ofchannel (1b) a very small fraction of energy has been observedto be channelled into vibrational and rotational excitation ofCO.22,40 A significant internal excitation of CH2, possiblyincluding electronic excitation, should account for the remain-ing available energy. The observation of a fraction of vibrationaland rotational energy even smaller than that predicted bystatistical methods leads to the conclusion that channel (1b)does not behave statistically and that its reaction mechanism isdominated by dynamic effects.22,40
It is of interest to comment the present results for channel(1a) in light of other CMB studies, even though a directcomparison is not possible because of the different experi-mental Ec. Clemo et al.38 derived a P(E0T) for the HCCO + Hchannel at Ec = 25 kJ mol�1 which has an abrupt cut-off at about80 kJ mol�1 and a very peculiar shape; Schmoltner et al.39
derived P(E0T) similar in shape to that obtained in this study,but exhibiting a maximum at about 45 kJ mol�1, which isslightly higher than that obtained here. The P(E0T) tail ofSchmoltner et al.39 extended up to 138 kJ mol�1, implying anexoergicity of the HCCO + H channel of about 113 kJ mol�1. Thebest fit P(E0T) obtained in the present work extends to about117 kJ mol�1 at Ec = 34.4 kJ mol�1, consistent with an exoergi-city of 82.4 kJ mol�1, which is today a well established valuefollowing the rather accurate determination of DHo
f (HCCO) =176.6 � 2.9 kJ mol�1 (1.83 � 0.03 eV).58 The discrepancy withregard to the P(E0T) peak position with respect to the work ofSchmoltner et al. can be attributed to the lower data quality ofthe previous study. As far as T(y) is concerned, our results aremore in line with those of Schmoltner et al. who derived abackward–forward symmetric T(y) with some polarization atEc = 25 kJ mol�1, while Clemo et al. derived a T(y) somewhatsideways peaked with a slightly preferred backward bias.Lahankar et al.41 have recently investigated channel (1a) forthe isotopic variant O(3P) + C2D2 at hyperthermal collisionenergies (from 255 up to 420 kJ mol�1). At the lowest Ec
investigated (255 kJ mol�1) the product angular distributionis almost backward–forward symmetric with a small preferencefor backward scattering. The preference for backward scatter-ing with respect to the direction of the reagent O atomsincreases with increasing collision energy (see Fig. S2a–d ofref. 41, which show distributions for Ec = 255, 337, 387 and420 kJ mol�1, respectively). Since at our much lower Ecs we haveobserved some preference for forward scattering, which isassociated with the shortening of the intermediate lifetimewith increasing Ec, we suggest that the almost symmetric T(y)observed41 at Ec = 255 kJ mol�1 is already the result of the twodifferent reaction mechanisms.
The present experimental results can be interpreted in moredetail by considering the available information on the under-lying potential energy surface as determined in electronic
structure calculations by Nguyen et al.7 and Garashchuket al.45 According to their calculations, the O(3P) + C2H2 reac-tion is initiated by electrophilic O-addition onto a C-atom ofacetylene, with the formation of the bound triplet additiontrans-HCCHO intermediate. Because of the large internalenergy content, the trans-HCCHO can either convert to itsrotamer cis, undergo a C–H bond fission with the formation ofH + HCCO(X2A00) or rearrange to triplet ketene by a 1,2-H shift.The cis-HCCHO isomer has a similar destiny, as it can isomerizeback to trans-HCCHO, dissociate into H + HCCO(X2A00) orrearrange to triplet ketene. Triplet ketene easily dissociatesinto CH2(X3B1) + CO. Trans- and cis-HCCHO(3A00) lead toHCCO(X2A00) + H via two transition states located at 44.8 and17.2 kJ mol�1 above the product asymptote, respectively. Sincethe two transition states for H-migration from trans/cis-HCCHOare tighter and lie higher in energy than the two transitionstates for H-elimination, the H + HCCO(X2A00) product yield ispredicted to be the favoured channel. This is in agreement withour and previous experimental results.
A very interesting aspect of the theoretical study of Nguyenet al.7 is the conclusion that the statistical yield of CH2 + CO is7–10% when considering only the ground state triplet PES. Thisvalue is significantly smaller than that derived experimentally.To reconcile experimental and theoretical results, it wassuggested that the O + C2H2 reaction on the 3A00 PES is non-statistical. More specifically, it was argued that trans-HCCHO -
cis-HCCHO isomerisation is less efficient than what predictedby RRKM as a consequence of a non-statistical energy partition-ing in trans-HCCHO. If this is the case, a larger fraction of theproducts would be formed from the trans-HCCCHO isomer,which needs to surmount a lower barrier than its cis isomer toyield CH2 + CO. An alternative explanation for the too lowpredicted CH2 yield is that triplet HCCHO (trans or cis) mayundergo competitive ISC to singlet HCCHO, which shouldrapidly isomerize to the low-lying singlet ketene CH2CO anddissociate into singlet CH2(1A1) and CO. A 10% yield of singletmethylene would explain the discrepancy between theory andexperiment. The recent QCT-SH calculations by Rajak andMaiti,37 which explicitly considered the coupling betweentriplet and singlet PES, derived a global BR of 0.20 for theCH2 + CO channel, of which about 1/3 (BR = 0.07) is given by theformation of singlet CH2. However, the experimental confirma-tion of CH2(1A1) formation is still lacking.
In any case, the absence of a significant triplet - singlet ISCin the O(3P) + C2H2 reaction contrasts with the observation ofvery significant triplet - singlet ISC in the homologous O(3P) +C2H4 reaction30,31 and in the related C(3P) + C2H2 reaction.59,60
According to Nguyen et al.,7 the lack of an efficient ISC can beattributed to (i) the faster (with respect to O + C2H4) chemicaldecomposition of the chemically activated triplet trans-HCCHOadduct and (ii) the narrow geometry range of the HCCHO tripletand singlet surface crossing seams.
Electrophilic addition of oxygen onto a C-atom of acetylenecan also proceed on the excited 3A0 PES via a transition statewhich is calculated to lie 25.5 kJ mol�1 above the reactants.Electronically excited trans-HCCHO(3A0) readily isomerizes to
Paper PCCP
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online

This journal is© the Owner Societies 2014 Phys. Chem. Chem. Phys.
the cis form. In this case, only the cis form can undergo aC–H bond cleavage to electronically excited products H +HCCO(A2A0), located about 12 kJ mol�1 above the ground stateproducts. Internal conversion of the excited triplet adduct tothe lowest lying triplet adduct is theoretically predicted to bevery unlikely because the lifetime of the excited adduct isestimated to be only B1 ps at room temperature.7 This sug-gests that the excited triplet adduct can only fragment toexcited H + HCCO products, thus increasing the H + HCCOproduct yield. It is interesting to note that the excited HCCHOlifetime is much shorter than that predicted by the sametheoretical method for the ground state addition intermediate(the lifetime of the triplet trans-HCCHO is predicted to beB15 ps at 1000 K and B4 ps at 2000 K). Because of the muchlower stability of the excited triplet HCCHO adduct and con-sequent much shorter lifetime (o1 ps), the reaction on theexcited triplet PES is envisaged to be more direct, i.e., proceed-ing through a complex which starts to osculate at much lowerEcs than the complex in the ground state triplet PES. Indeed,our experimental observation that the T(y) of HCCO exhibits anincreased T(01)/T(1801) ratio with increasing Ec (see Fig. 11-lhs)could also be rationalized in terms of contributions fromscattering on the two lowest triplet PES, one long-lived andone osculating. Our lowest Ec is, in fact, higher than theentrance barrier, while that of Schmoltner et al.’s experimentis slightly lower. Unfortunately, as already commented on, wehave not been able to discriminate between the formation ofHCCO in its ground or first electronically excited states becauseof the similar reaction exothermicity of channel (1a) and (1d)and because of their expected similar mechanisms.
Nguyen et al.7 have compared their statistical predictionswith our previous CMB results at the collision energy ofca. 40 kJ mol�1. In doing so, they have converted entirely thecollision energy to additional internal vibrational energy of theinitially formed triplet adduct HCCHO. A similar energy con-tent would be achieved for a thermal energy of the reactantcorresponding to about 750 K. Solving the master equation withonly the ground state 3A00 PES yields 76% H + HCCO(X2A00) and24% CH2(X3B1) + CO. When these numbers are combined withan estimated 16% population for the excited 3A0 state PES and a84% population for the 3A00 state PES, an overall BR of 80% forH + HCCO and 20% for CH2(X3B1) + CO was derived. Thesenumbers compare very well with our CMB determination of79 � 5% and 21 � 5% (see Table 1).
In conclusion, the present experimental results confirm thetheoretical prediction that channels (1a) and (1b) are thedominant ones for the reaction between O(3P) and acetylene,with the HCCO + H channel being about four times moreimportant. Interestingly, the BR value obtained at the highestcollision energy (54.6 kJ mol�1) of this study is still very similarto the room temperature (corresponding to about 4 kJ mol�1)recommended value. The substantial invariance of the productyield in such a wide energy/temperature range confirms whatanticipated by the calculations of Harding and Wagner24 andNguyen et al.7 In light of the recent theoretical results,7,37
however, this apparent invariance is actually the result of a
compensation of varying factors, such as the contribution ofthe excited triplet PES and/or ISC to the singlet PES. Moretheoretical work is needed to address this point and experi-mental evidence of HCCO and CH2 formation in their excitedstates is desirable. Clearly, a significant presence of excitedradicals in combustion environments can affect the final out-come and should be considered in combustion models as wellexemplified by the important role of CH2(a1A1) as opposed toCH2(X3B1) in soot formation.
VI. Conclusion
By combining our experimental results and theoretical infor-mation on the relevant potential energy surfaces we have now acomplete picture of reaction (1). The mechanism of the O(3P) +acetylene reaction sees the initial electrophilic attack of the Oatom to the triple bond of the C2H2 molecule with formation ofa triplet diradical adduct (HCCHO) that under single collisionconditions undergoes competitively C–H bond cleavage to formHCCO + H and isomerization to form triplet ketene (H2CCO)followed by C–C bond cleavage to form 3CH2 + CO. The HCCO + Hchannel accounts for ca. 80% of the overall yield while CH2 + CO forthe rest (20%) at all collision energies investigated (see Fig. 15). Thepresent results agree well with recent kinetics determination of thebranching ratio at room temperature, with statistical calculationsbased on the two lowest triplet potential energy surfaces and withQCT-SH predictions explicitly considering ISC between the tripletand the singlet PESs. Nguyen et al.7 predict that about 30% of thereaction proceeds on the excited triplet surface at 2000 K, and thiswas shown to be able to rationalize quantitatively the temperaturedependence of the rate constants up to very high values (2000 K).Rajak and Maiti37 predicted an ISC contribution of B7% via theformation of singlet CH2. Our dynamics results are fully consistentwith these findings, but do not allow us to disentangle the relativecontributions to the reaction dynamics of the two lowest tripletsurfaces and possible (minor) ISC contribution. The firm assess-ment of the branching ratio as a function of translational energy forthis important reaction, besides its fundamental significance, is ofconsiderable interest for the implementation of theoretical modelsof hydrocarbon combustion.
Acknowledgements
Financial support by Italian MIUR (PRIN 2010-2011, grant2010ERFKXL) and EC COST Action CM0901 ‘‘Detailed ChemicalModels for Cleaner Combustion’’ is gratefully acknowledged.We thank Marta Suriani and Simone Famiani for their contri-bution to taking some data.
References
1 N. Balucani, F. Leonori and P. Casavecchia, Energy, 2012,43, 47.
2 N. Balucani, F. Leonori and P. Casavecchia, in Cleaner Com-bustion, Green Energy and Technology, ed. F. Battin-Leclerc,
PCCP Paper
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online

Phys. Chem. Chem. Phys. This journal is© the Owner Societies 2014
J. M. Simmie and E. Blurok, Springer-Verlag, London, 2013,ch. 22, pp. 577–606.
3 J. A. Miller, M. J. Pilling and J. Troe, Proc. Combust. Inst.,2005, 30, 43.
4 A. Williams and D. B. Smith, Chem. Rev., 1970, 70, 267.5 J. Peeters, Bull. Soc. Chim. Belg., 1997, 106, 357, and refer-
ences therein.6 D. J. Hucknall, Chemistry of Hydrocarbon Combustion,
Chapman Hall, New York, 1985.7 T. L. Nguyen, L. Vereecken and J. Peeters, J. Phys. Chem. A,
2006, 110, 6696.8 J. Peeters, S. Vanhaelemeersch, J. Van Hoeymissen,
R. Borms and D. J. Vermeylen, J. Phys. Chem., 1989, 93,3892.
9 W. Hack, M. Koch, H. Gg. Wagner and H. Wilms, Ber.Bunsen-Ges. Phys. Chem., 1988, 92, 674.
10 U. Alkemade and K. H. Homann, Z. Phys. Chem., 1989,19, 161.
11 P. R. Westmoreland, A. M. Dean, J. B. Howard and P. J.Longwell, J. Phys. Chem., 1989, 93, 8171.
12 S. A. Carl, L. Vereecken and J. Peeters, Phys. Chem. Chem.Phys., 2007, 9, 4071.
13 D. L. Osborn, J. Phys. Chem. A, 2003, 107, 3728.14 M. Dobrijevic, E. Hebrard, J. C. Loison and K. M. Hickson,
Icarus, 2014, 228, 324.15 A. Occhiogrosso, S. Viti and N. Balucani, Mon. Not. R. Astron.
Soc., 2013, 432, 3423.16 K. Mahmud and A. Fontijn, J. Phys. Chem., 1987, 91, 1918,
and refs. therein.17 J. Peeters, M. Schaekers and C. Vinckier, J. Phys. Chem.,
1986, 90, 6552.18 P. Frank, K. A. Bhaskaran and T. H. Just, 21th Symp. (Int.)
Combustion, 1986, p. 885.19 J. V. Michael and A. F. Wagner, J. Phys. Chem., 1990,
94, 2453.20 W. Boullart and J. Peeters, J. Phys. Chem., 1992, 96,
9810.21 J. Peeters, W. Boullart and I. Langhans, Int. J. Chem. Kinet.,
1994, 26, 869.22 X. Huang, G. Xing and R. Bershon, J. Chem. Phys., 1994,
101, 5818.23 D. L. Baulch, C. T. Bowman, C. J. Cobos, R. A. Cox, Th. Just,
J. A. Kerr, M. J. Pilling, D. Stocker, J. Troe, W. Tsang,R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data,2005, 34, 757.
24 L. B. Harding and A. F. Wagner, J. Phys. Chem., 1986,90, 2974.
25 M. Alagia, N. Balucani, L. Cartechini, P. Casavecchia,M. van Beek, G. G. Volpi, L. Bonnet and J. C. Rayez, FaradayDiscuss., 1999, 113, 133.
26 X. Gao, J. Essex-Lopresti, S. Munro, M. P. Hall, D. J.Smith and R. Grice, J. Chem. Soc., Faraday Trans., 1998,93, 641.
27 M. Alagia, N. Balucani, P. Casavecchia, A. Lagana, G. Ochoade Aspuru, E. H. van Kleef, G. G. Volpi and G. Lendvay,Chem. Phys. Lett., 1996, 258, 1.
28 N. Balucani, D. Stranges, P. Casavecchia and G. G. Volpi,J. Chem. Phys., 2004, 120, 9571.
29 P. Casavecchia, G. Capozza, E. Segoloni, F. Leonori,N. Balucani and G. G. Volpi, J. Phys. Chem. A, 2005,109, 3527.
30 B. Fu, Y.-C. Han, J. M. Bowman, F. Leonori, N. Balucani,L. Angelucci, A. Occhiogrosso, R. Petrucci and P. Casavecchia,J. Chem. Phys., 2012, 137, 22A532.
31 B. Fu, Y.-C. Han, J. M. Bowman, L. Angelucci, N. Balucani,F. Leonori and P. Casavecchia, Proc. Natl. Acad. Sci. U. S. A.,2012, 109, 9733.
32 F. Leonori, A. Occhiogrosso, N. Balucani, A. Bucci,R. Petrucci and P. Casavecchia, J. Phys. Chem. Lett., 2012,3, 75.
33 N. Balucani, F. Leonori, V. Nevrly, S. Falcinelli, D. Stranges,A. Bergeat and P. Casavecchia, Phys. Chem. Chem. Phys.,2014, submitted.
34 W. Hu, G. Lendvay, B. Maiti and G. C. Schatz, J. Phys.Chem. A, 2008, 112, 2093.
35 G. Xing, X. Huang, X. Wang and R. Bersohn, J. Chem. Phys.,1996, 105, 488.
36 D. R. Yarkony, J. Phys. Chem. A, 1998, 102, 5305.37 K. Rajak and B. Maiti, J. Chem. Phys., 2010, 133, 011101.38 A. R. Clemo, G. L. Duncan and R. Grice, J. Chem. Soc.,
Faraday Trans. 2, 1982, 78, 1231.39 A. M. Schmoltner, P. M. Chu and Y. T. Lee, J. Chem. Phys.,
1989, 91, 5365.40 V. Chikan and S. R. Leone, J. Phys. Chem. A, 2005, 109, 2525.41 S. A. Lahankar, J. M. Zhang, S. Garashchuk, G. C. Schatz and
T. K. Minton, J. Phys. Chem. Lett., 2012, 3, 75.42 P. Casavecchia, F. Leonori, N. Balucani, R. Petrucci,
G. Capozza and E. Segoloni, Phys. Chem. Chem. Phys.,2009, 11, 46.
43 N. Balucani, G. Capozza, F. Leonori, E. Segoloni andP. Casavecchia, Int. Rev. Phys. Chem., 2006, 25, 109.
44 G. Capozza, E. Segoloni, F. Leonori, G. G. Volpi andP. Casavecchia, J. Chem. Phys., 2004, 120, 4557.
45 S. Garashchuk, V. A. Rassolov and B. J. Braams, Chem. Phys.Lett., 2013, 588, 22.
46 M. Alagia, N. Balucani, P. Casavecchia, D. Stranges andG. G. Volpi, J. Chem. Soc., Faraday Trans., 1995, 91, 575.
47 M. Alagia, V. Aquilanti, D. Ascenzi, N. Balucani,D. Cappelletti, L. Cartechini, P. Casavecchia, F. Pirani,G. Sanchini and G. G. Volpi, Isr. J. Chem., 1997, 37, 329.
48 F. Leonori, K. M. Hickson, S. D. Le Picard, X. Wang,R. Petrucci, P. Foggi, N. Balucani and P. Casavecchia,Mol. Phys., 2010, 108, 1097.
49 S. J. Sibener, R. J. Buss, C. Y. Ng and Y. T. Lee, Rev. Sci.Instrum., 1980, 51, 167.
50 S. A. Carl, Phys. Chem. Chem. Phys., 2005, 7, 4051.51 Y. Girard and P. Chaquin, J. Phys. Chem. A, 2003, 107, 10462.52 In the early experiments, while the reported beam velocities
were correct, the collision energy was erroneously indicatedto be 39.7 kJ mol�1 (9.5 kcal mol�1) (see ref. 44). Here wereport the correct value of 41.1 kJ mol�1. This does notaffect the conclusions of the early work.
Paper PCCP
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online

This journal is© the Owner Societies 2014 Phys. Chem. Chem. Phys.
53 NIST Chemistry WebBook, 2002.54 G. A. Fisk, J. D. McDonald and D. R. Herschbach, Discuss.
Faraday Soc., 1967, 44, 228.55 C. C. Hayden, D. M. Neumark, K. Shobatake, R. K. Sparks
and Y. T. Lee, J. Chem. Phys., 1982, 76, 3607.56 W. L. Fitch and A. D. Sauter, Anal. Chem., 1983, 55, 832.57 H. Deutsch, K. Becker, D. Matt and T. D. Maerk, Int. J. Mass
Spectrom., 2000, 197, 37.
58 D. L. Osborn, D. H. Mordaunt, H. Choi, R. T. Bise,D. M. Neumark and C. McMichael Rohlfing, J. Chem. Phys.,1997, 106, 10087.
59 F. Leonori, R. Petrucci, E. Segoloni, A. Bergeat, K. H.Hickson, N. Balucani and P. Casavecchia, J. Phys. Chem. A,2008, 112, 1363.
60 A. M. Mebel, V. V. Kislov and M. Hayashi, J. Chem. Phys.,2007, 126, 204310.
PCCP Paper
Publ
ishe
d on
15
Janu
ary
2014
. Dow
nloa
ded
by U
nive
rsity
of
Peru
gia
on 1
7/03
/201
4 11
:40:
57.
View Article Online




















