
Ionization Equilibria of Acids and Bases under Hydrothermal Conditions
52
Chapter 13 Ionization equilibria of acids and bases under hydrothermal conditions Peter Tremaine, a, * Kai Zhang, a Pascale Be ´ne ´zeth b and Caibin Xiao c a Department of Chemistry, University of Guelph, Guelph, Ont., Canada N1G 2W1 b Chemical Sciences Division, Oak Ridge National Laboratory, Building 4500S, P.O. Box 2008, Oak Ridge, TN 37831-6110, USA c GE Water Technologies, 4636 Somerton Road, P.O. Box 3002, Trevose, PA 19053-6783, USA 13.1. Introduction 13.1.1. Acids and Bases Under Hydrothermal Conditions The properties of acids and bases control much of the aqueous chemistry of geochemical, industrial and biological systems. Ionization constants for simple acids and bases at 25 8C are tabulated in many sources, including all under- graduate chemistry textbooks. The behavior of acids and bases, including the ionization of water itself, under extremes of temperature and pressure is much less widely known. Our purpose in this chapter is to present a practical discussion and compilation of the effects of temperature, pressure, and in some cases, ionic strength on the ionization constants of simple acids and bases, from room temperature to hydrothermal conditions. The first measurements of the ionization constants of water and aqueous acids and bases were made by Noyes (1907), who used the change in conductance associated with ionization to measure equilibrium constants up to about 300 8C at steam saturation. Only modest research on hydrothermal effects was carried out until the 1950s, when interest in nuclear reactor coolant chemistry led national laboratories in several countries to develop experimental methodologies suitable for the corrosive conditions encountered at elevated temperatures. Complementary studies by geochemists investigating geothermal systems and ore body formation have led to the development of additional experi- mental techniques suitable for near critical and supercritical conditions * Corresponding author. E-mail: [email protected] Aqueous Systems at Elevated Temperatures and Pressures: Physical Chemistry in Water, Steam and Hydrothermal Solutions D.A. Palmer, R. Ferna ´ndez-Prini and A.H. Harvey (editors) q 2004 Elsevier Ltd. All rights reserved
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Ionization Equilibria of Acids and Bases under Hydrothermal Conditions

Chapter 13
Ionization equilibria of acids and basesunder hydrothermal conditions
Peter Tremaine,a,* Kai Zhang,a Pascale Benezethb and Caibin Xiaoc
a Department of Chemistry, University of Guelph, Guelph, Ont., Canada N1G 2W1b Chemical Sciences Division, Oak Ridge National Laboratory, Building 4500S, P.O. Box 2008,
Oak Ridge, TN 37831-6110, USAc GE Water Technologies, 4636 Somerton Road, P.O. Box 3002, Trevose, PA 19053-6783, USA
13.1. Introduction
13.1.1. Acids and Bases Under Hydrothermal Conditions
The properties of acids and bases control much of the aqueous chemistry of
geochemical, industrial and biological systems. Ionization constants for simple
acids and bases at 25 8C are tabulated in many sources, including all under-
graduate chemistry textbooks. The behavior of acids and bases, including the
ionization of water itself, under extremes of temperature and pressure is much less
widely known. Our purpose in this chapter is to present a practical discussion and
compilation of the effects of temperature, pressure, and in some cases, ionic
strength on the ionization constants of simple acids and bases, from room
temperature to hydrothermal conditions.
The first measurements of the ionization constants of water and aqueous acids
and bases were made by Noyes (1907), who used the change in conductance
associated with ionization to measure equilibrium constants up to about 300 8C at
steam saturation. Only modest research on hydrothermal effects was carried out
until the 1950s, when interest in nuclear reactor coolant chemistry led national
laboratories in several countries to develop experimental methodologies
suitable for the corrosive conditions encountered at elevated temperatures.
Complementary studies by geochemists investigating geothermal systems and
ore body formation have led to the development of additional experi-
mental techniques suitable for near critical and supercritical conditions
*Corresponding author. E-mail: [email protected]
Aqueous Systems at Elevated Temperatures and Pressures:Physical Chemistry in Water, Steam and Hydrothermal SolutionsD.A. Palmer, R. Fernandez-Prini and A.H. Harvey (editors)q 2004 Elsevier Ltd. All rights reserved

(see, e.g., Mesmer et al., 1997; Ulmer and Barnes, 1983). Academic research has
been spurred by these applications and by a desire to use wide variations in
temperature and pressure as a probe to understand ion–water and ion–ion
interactions.
The first two sections of this chapter consist of a short review of the
underlying chemical thermodynamics, experimental methods and the substituent
and hydration effects that determine the magnitude of the ionization constants of
acids and bases at elevated temperatures and pressures. The remaining four
sections describe the behavior of several classes of inorganic and organic acids
and bases. The chapter includes practical tables for use by non-specialists, based
on the equations and database for the dissociation of water developed at Oak
Ridge National Laboratory (Mesmer et al., 1970; Busey and Mesmer, 1978;
Palmer and Drummond, 1988).
13.1.2. Thermodynamic Relations
13.1.2.1. Equations for Pressure and Temperature Dependence of DGo
and log10 K
Here we are concerned with the Bronsted definition of acids and bases, as solutes
capable of releasing hydrogen ions and hydroxide ions, respectively.
HAðaqÞO HþðaqÞ þ A2ðaqÞ ð13:1Þ
and
BðaqÞ þ H2OðlÞO BHþðaqÞ þ OH2ðaqÞ ð13:2Þ
The ionization product A2(aq) is the conjugate base of HA(aq), because it
behaves as a base in reacting with water to form HA(aq).
A2ðaqÞ þ H2OðlÞO HAðaqÞ þ OH2ðaqÞ ð13:3Þ
Similarly, BHþ(aq) is the conjugate acid of B(aq).
The equilibrium quotient of the acid ionization reaction, reaction 13.1, is
defined as
Q1a ¼ mA2mH2=mHA ð13:4Þ
where mA2 ; mHþ and mHA are molalities of the species A2(aq), Hþ(aq) and
HA(aq), respectively. Molalities (mol·kg21) are generally used instead of concen-
tration (mol·dm23) because molalities are not affected by the expansion or
contraction of water with temperature and pressure. The equilibrium quotient Q1a
is a function of temperature, pressure and ionic strength.
The equilibrium constant of reaction 13.1 refers to infinitely dilute solutions
in the hypothetical 1 mol·kg21 standard state, and thus it is not a function
P. Tremaine et al.442

of ionic strength:
K1a ¼ aA2aHþ=aHA ð13:5Þ
where aA2 ; aHA and aHþ are activities in molality units. The corresponding
expressions may be written for the ionization of bases, which we define as Qb and
Kb. The ratio between molalities and activities of solutes is defined as the activity
coefficient, g ¼ a=m; so that
log10 K1a ¼ log10 Q1a þ log10ðgA2gHþ=gHAÞ ð13:6Þ
where gA2 ; gHþ and gHA are activity coefficients of A2(aq), Hþ(aq) and HA(aq),
respectively. Since the last term in Eq. 13.6 vanishes as ionic strength approaches
zero, log10 K1a is usually obtained from experimentally determined log10 Q1a
values by extrapolation to infinite dilution.
Values for log10 K can also be calculated from the Gibbs energy change of the
reaction through the relationship:
DGo ¼ 2RT ln K ð13:7Þ
where T is the temperature in kelvin and R is the molar gas constant. The Gibbs
energy change is not usually determined experimentally at elevated temperature
but rather is calculated from experimental values of K measured by means of
potentiometric titration or other techniques. The temperature dependence of
log10 K is described by the following relationships.
Starting with two basic thermodynamic equations, DGo ¼ DHo 2 TDSo and
DSo ¼ 2ð›DGo=›TÞp; we have
ðdðDGo=TÞ ¼ 2DHo=T2 dT ð13:8Þ
If DH o of a reaction is independent of temperature and pressure, then
log10 KT ;p ¼ log KTr;prþ DHo
Tr;prð1=Tr 2 1=TÞ=ð2:303RÞ ð13:9Þ
where r is the reference state ðTr ¼ 298:15 KÞ: The above equation does not
provide a satisfactory estimation for log10 K at elevated temperatures, especially
for reactions with an asymmetric charge distribution.
More accurate estimations for log10 K require the addition of standard partial
molar heat capacity and volume functions for the reaction. Adding DV ¼
ð›DG=›pÞT and DCp ¼ Tð›DS=›TÞp into the exact differential equation yields the
expression:
dDG ¼ ð›DG=›TÞpdT þ ð›DG=›pÞTdp ð13:10Þ
Ionization equilibria of acids and bases 443

which can be integrated from the reference state ðTr; prÞ to state ðT ; pÞ; yielding the
expression:
DGoT ;p ¼ DGo
Tr;prþ
ðpath
ðpath
DCop=T dT þ DSo
Tr;pr
� �dT þ DVo dp
� �ð13:11Þ
The above line integral is independent of the path chosen. However, the path for
the first part (heat capacity) must be the same as for the second (volume) part. For
T , critical temperature, integration along the saturation curve yields
log10 KT ;p ¼ ðlog10 KTr;prþ DHo
Tr;prð1=Tr 2 1=TÞÞ=ð2:303RÞ
þððDCo
p=TÞ dT 2 ð1=TÞðDCo
p dT 2ððDVo=TÞ dp ð13:12Þ
Appropriate expressions for DCop and DVo can be used as a fitting expression in
Eq. 13.12, to represent the temperature and pressure dependence of experimental
values for log10 K. Alternatively, if DCop and DV o are known as a function of
temperature, Eq. 13.12 can be used to calculate log10 K vs. T and p (Pitzer, 1995;
Mesmer et al., 1988). The effects of temperature and pressure on activity
coefficients are discussed below.
13.1.3. Acid–Base Equilibria to 300 8C
13.1.3.1. Factors Controlling the Ionization of Acids and Bases at Elevated
Temperatures and Pressures
The principles governing the ionization of acids and bases at elevated
temperatures and pressures have been discussed by Mesmer et al. (1988, 1991)
and others (e.g., Fernandez-Prini et al., 1992; Levelt Sengers, 1991), using
interpretations based on hard-won experimental data for about 20 systems,
described in subsequent sections. At ambient temperatures, liquid water consists
of long-range hydrogen-bonded networks, roughly tetrahedral, that extend on a
time-averaged basis to three or more nearest neighbors, with a considerable degree
of thermal motion and inter-penetration (Svishchev and Kusalik, 1995). As the
temperature is raised along the saturation pressure curve, long-range hydrogen
bonding breaks down and water becomes more compressible until, at the critical
temperature and pressure, the compressibility of water becomes infinite.
The degree of ionization, i.e., the magnitude of the ionization constant in
reaction 13.1 or 13.2, is governed by the thermodynamic relationship that defines
Gibbs energy:
DGo ¼ DHo 2 TDSo ¼ DUo þ pDVo 2 TDSo ð13:13Þ
At ambient temperatures and pressures, the hydration of the species HA(aq),
Hþ(aq) and A2(aq) reflects hydrogen bonding effects associated with both
P. Tremaine et al.444

short-range and long-range interactions with water. Strong hydrogen bonding to
the acid or conjugate base that minimizes energetic effects (DU o) may cause the
entropic term (DS o) and volumetric term (DV o) to also be reduced. The difficulties
in modeling hydration effects that have occupied researchers for more than 100
years result from the subtle balance between these three effects that exists at
temperatures near 25 8C.
Raising the temperature and pressure causes the equilibrium of ionization
reactions 13.1 and 13.2 to shift in the direction that favors smaller volumes
( pDV o , 0) and greater entropies (TDS o . 0). The effect is illustrated
schematically in Fig. 13.1, which depicts the ionization process as the insertion
of uncharged and charged spheres into liquid water. At ambient temperatures,
short-range and long-range interactions around the ionized and neutral acids and
bases are species-specific so that DG o can shift in either a positive or negative
direction with modest increases in temperature and pressure, depending on the
number of hydrogen-bond acceptors and donors, the charge, and the size and shape
of the species in question. At temperatures above about 200 8C, however, long-
range solute–water interactions begin to dominate as a result of the decreased
hydrogen bonding in water itself and the resulting increased compressibility of
liquid water. Figure 13.2 shows that the standard partial molar volumes of
morpholine and its chloride salt, morpholinium chloride up to 300 8C (Tremaine
et al., 1997) deviate towards þ and 2 infinity at the critical point of water. The
result is that we can draw the following general conclusions:
† Increasing the temperature above about 250 8C along the steam saturation
pressure curve towards the critical point causes the ionization constants of
neutral acids and bases to decrease.
† Increasing the pressure at temperatures above about 250 8C causes the
ionization constant to increase.
Fig. 13.1. The solvation of ions and non-electrolytes in high-temperature water. Elevated
temperatures favor ion-pairing and long-range interactions dominate as the compressibility of
liquid water increases.
Ionization equilibria of acids and bases 445

† At temperatures below 100 8C, ionization behavior is species-specific.
† Ionization constants in the range 100 , t , 250 8C display intermediate
behavior.
Here and elsewhere, the symbol t is used for temperature in 8C. Typical values
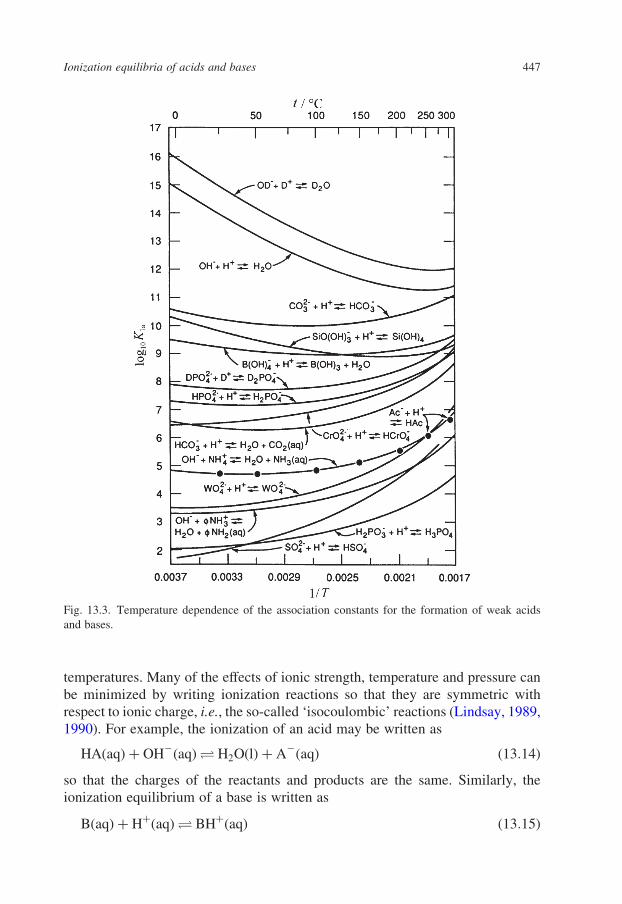
of log10 K for ionization equilibria below 300 8C are plotted as a function of
temperature in Fig. 13.3.
Experimental ionization and association constants have been measured under
supercritical conditions, primarily using the conductivity, heat capacity and
density methods described in a later section and by Mesmer et al. (1991). The
major factors controlling ionization above the critical point of water continue to be
temperature and solvent density, for the reasons described above.
13.1.3.2. Isocoulombic Extrapolations
Experimental measurements of log10 K and the other thermodynamic constants
used in Eq. 13.12 at elevated temperatures and pressures are extremely difficult.
As a result, the standard Gibbs energies of formation, standard enthalpies of
formation and partial molar entropies for many species are known only at ambient
0 50 100 150 200 250 300 3500
25
50
75
100
125
150
Neutral SpeciesC4H8ONH
Ionic SpeciesC4H8ONH2
+Cl–
t / ˚C
V2/(
cm3 .
mol
–1)
Fig. 13.2. Standard partial molar volumes of neutral morpholine and its salt, morpholinium chloride,
showing increasingly large positive and negative deviations as the temperature is raised along the
steam saturation curve (Tremaine et al., 1997). The solid line is a fit to an extended version of the
‘density’ model discussed in the text.
P. Tremaine et al.446
temperatures. Many of the effects of ionic strength, temperature and pressure can
be minimized by writing ionization reactions so that they are symmetric with
respect to ionic charge, i.e., the so-called ‘isocoulombic’ reactions (Lindsay, 1989,
1990). For example, the ionization of an acid may be written as
HAðaqÞ þ OH2ðaqÞO H2OðlÞ þ A2ðaqÞ ð13:14Þ
so that the charges of the reactants and products are the same. Similarly, the
ionization equilibrium of a base is written as
BðaqÞ þ HþðaqÞO BHþðaqÞ ð13:15Þ
Fig. 13.3. Temperature dependence of the association constants for the formation of weak acids
and bases.
Ionization equilibria of acids and bases 447

The equilibrium quotients of neutralization reactions, such as reactions 13.14
and 13.15, are defined as
Q1a;OH ¼ Q1a=Qw ¼mA2
mHAmOH2
ð13:16Þ
and
Q1b;H ¼ Q1b=Qw ¼mBHþ
mBmHþ
ð13:17Þ
with the analogous equations for K1a;OH and K1b;H:For several aqueous systems, both K1a;OH (or K1b;H) and functions for DV o or
DCop have been independently measured. As an example to illustrate the usefulness
of Eq. 13.12, Tremaine et al. (1997) have used the experimental values of V o for
morpholine and the morpholinium ion shown in Fig. 13.2, and values of Cop
obtained below 55 8C, to estimate DCop for the morpholine ionization equilibrium
at high temperatures using the semi-empirical Helgeson–Kirkham–Flowers
(HKF) model. Combining these contributions with that from the first term in
Eq. 13.12 yields log10 K ¼ 24:843 at 300 8C, which is in excellent agreement
with the values 24.79 ^ 0.06 and 24.69 ^ 0.06 measured in KCl media by
Mesmer and Hitch (1977), and in sodium trifluoromethanesulfonate (NaCF3SO3)
by Ridley et al. (2000), respectively.
As a consequence of the symmetry of isocoulombic reactions, plots of
log10 Q1a,OH vs. 1/T are almost linear over a very wide range of temperatures, and
the temperature dependence can be described quite accurately by assuming a
constant mean value of DCop between some condition of T and p, and a reference
condition, Tr and pr:
log10 K1a;OH;T ;p ¼ log10 K1a;OH;Tr;prþ {DHo
Tr;prð1=Tr 2 1=TÞ
þ DCop½ln ðT=TrÞ þ Tr=T 2 1�2 ðDVo=TÞ½p 2 pr�}=ð2:303RÞ
ð13:18Þ
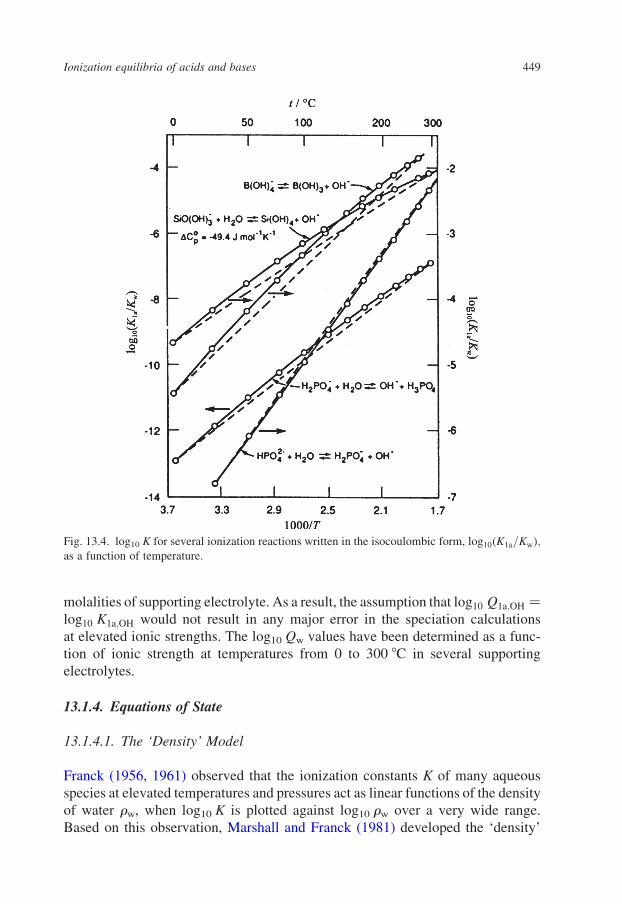
Typical examples are plotted in Fig. 13.4 in their isocoulombic forms. Figure
13.5 shows the temperature dependence of DCop for several of these reactions when
written in the non-isocoulombic and isocoulombic forms. For isocoulombic
equilibria at temperatures below 300 8C, the contribution of DV o to log10 K1a,OH,T
is often less than the experimental uncertainty.
Values of log10 Q1a for ionization reaction 13.1 can be calculated from the
experimentally determined value for the isocoulombic ionization equilibrium (Eq.
13.14), by using the value for the ionization of water at ionic strength I, log10 Qw:
log10 Q1a ¼ log10 Qw þ log10 K1a;OH ðIsocoulombicÞ ð13:19Þ
It has been shown in a number of experimental studies (Mesmer et al., 1988,
1991; Lindsay, 1989, 1990) that for most reactions log10 Q1a,OH is almost
independent of ionic strength for most isocoulombic equilibria at moderate
P. Tremaine et al.448
molalities of supporting electrolyte. As a result, the assumption that log10 Q1a;OH ¼
log10 K1a;OH would not result in any major error in the speciation calculations
at elevated ionic strengths. The log10 Qw values have been determined as a func-
tion of ionic strength at temperatures from 0 to 300 8C in several supporting
electrolytes.
13.1.4. Equations of State
13.1.4.1. The ‘Density’ Model
Franck (1956, 1961) observed that the ionization constants K of many aqueous
species at elevated temperatures and pressures act as linear functions of the density
of water rw, when log10 K is plotted against log10 rw over a very wide range.
Based on this observation, Marshall and Franck (1981) developed the ‘density’
Fig. 13.4. log10 K for several ionization reactions written in the isocoulombic form, log10ðK1a=KwÞ;as a function of temperature.
Ionization equilibria of acids and bases 449

model to represent the ionization constant Kw of water at temperatures up to
1273 K and at pressures up to 1000 MPa. Equations of this form have been used
for representing K of general ionization reactions by Mesmer et al. (1988):
log10 K ¼ a þb
Tþ
c
T2þ
d
T3
� �þ k log10 rw ð13:20Þ
k ¼ e þf
Tþ
g
T2
� �ð13:21Þ
where a, b, c, d, e, f and g are adjustable parameters (a number of these parameters
may be set to zero for many reactions) and rw is the density of pure water
(in g·cm23).
Fig. 13.5. The behavior of DCop for the ionization reactions written in both the non-isocoulombic and
isocoulombic forms, as a function of temperature.
P. Tremaine et al.450

Other thermodynamic quantities can be derived from the above equations. The
Gibbs energy of ionization DG o is related to K by Eq. 13.7, so that
DGo ¼ 22:303RT a þb
Tþ
c
T2þ
d
T3
� �þ e þ
f
Tþ
g
T2
� �log10 rw
� �
ð13:22Þ
The enthalpy of ionization DH o can be obtained from the identity:
›
›T
DGo
T
� �p¼ 2
DHo
T2ð13:23Þ
to yield
DHo ¼ 22:303R b þ2c
Tþ
3d
T2þ f þ
2g
T
� �log10 rw
� �2 RT2kaw ð13:24Þ
where aw ¼ 2ð1=rwÞð›rw=›TÞp is the thermal expansion coefficient of water and
k is the fitted function given in Eq. 13.21.
Similarly, the entropy of ionization DS o, the standard partial molar heat
capacity of ionization DrCpo, and the standard partial molar volume of ionization
DV o can be derived from DG o using standard thermodynamic identities (Mesmer
et al., 1988) so that
DSo ¼ 2:303R a 2c
T22
2d
T3þ e 2
g
T2
� �log10 rw
� �2 RTkaw ð13:25Þ
DCop ¼2 2:303R 2
2c
T22
6d
T32
2g
T2
� �log10 rw
� �
2 Raw 2eT 22g
T
� �2 RT2k
›aw
›T
� �p
ð13:26Þ
DVo ¼ 2RTkbw ð13:27Þ
Here bw ¼ ð1=rwÞð›rw=›pÞT is the compressibility of water.
Equation 13.22 can be further simplified over a restricted region, and the
simplified form has fewer parameters (Anderson et al., 1991). More complex
versions have been adopted to describe DV o accurately at low temperatures
(Clarke et al., 2000). Figure 13.6 shows the behavior of log10 K, DSo, DCop and
DV o for the ionization of ammonia over a wide range of temperature and pressure,
according to the density model fit reported by Mesmer et al. (1988). The functions
for DCop and DV o clearly show the very large electrostriction effects that arise from
the ability of ions to attract increasing numbers of water molecules as the
compressibility of water increases under near critical conditions.
Ionization equilibria of acids and bases 451

Fig. 13.6.
P. Tremaine et al.452

13.1.4.2. The Revised Helgeson–Kirkham–Flowers Model
Helgeson and co-workers (Helgeson et al., 1981; Tanger and Helgeson, 1988)
have developed an equation of state, based on the Born equation for ionic
hydration, which is widely used by geochemists (see Chapter 4 for more details).
Briefly, the HKF model consists of expressions for standard partial heat capacity
and volume functions in Eq. 13.12, and assumes that the standard molar Gibbs
energy and enthalpy of formation of each species at 298.15 K and 0.1 MPa are
known properties.
In this model, the standard molar properties, Y o, of aqueous ions are considered
to have two contributions: an electrostatic term based on the Born equation DYoBorn;
and a non-electrostatic term Yon :
Yo ¼ Yon þ DYo
Born ð13:28Þ
The Born equation describes the Gibbs energy of ionic hydration, i.e., the
transfer of an ion from the ideal gas state to liquid water, by representing the ion as
Fig. 13.6. The behavior of (a) log10 K, (b)DS o and (c)DV o for the association of ammonia over a wide
range of temperature and pressure, according to the density model fit reported by Mesmer et al. (1988).
Ionization equilibria of acids and bases 453

a charged conducting sphere and water as a continuous dielectric medium without
molecular structure. The Born equation takes the form:
DGoBorn ¼ 2veffð1=1r 2 1Þ ð13:29Þ
with
veff ¼NAðzeÞ2
8p10reff
ð13:30Þ
where 1r is the solvent dielectric constant; veff is a term that includes the ionic
charge z, electron charge e, ionic radius reff, permittivity of free space 10 and
Avogadro’s number NA.
The HKF model employs an effective ionic radius, where reff is a linear function
of crystallographic radius rx and charge z, reff ¼ rx þ 0:94lzl (with r in angstroms)
for cations and reff ¼ rx for anions. The revised HKF model (Tanger and
Helgeson, 1988; Shock and Helgeson, 1988) also considered reff to be a function of
T and p. The appropriate temperature and pressure derivatives of DGoBorn yield
expressions for DCop;Born and DVo
Born in terms of the same parameters.
The non-electrostatic term Yon includes three contributions: (i) the intrinsic gas
phase property of the solute, (ii) the change arising from the difference in standard
states between the gas phase and solution, and (iii) short-range hydration effects
(Fernandez-Prini et al., 1992). In the HKF treatment, Yon is used as an empirical
fitting equation with the following form:
Vo ¼ a1 þ a2
1
›2V
›v2þ p
0BBB@
1CCCAþ a3 þ a4
1
Cþ p
� �� �1
T 2Q
� �þ Vo
Born ð13:31Þ
Here Q is a solvent parameter equal to 228 K, which corresponds to the
temperature at which supercooled liquid water may undergo anomalous behavior
(Angell, 1983); C is a similar solvent parameter equal to 2600 bar; and a1, a2, a3
and a4 are temperature- and pressure-independent, but species-dependent, fitting
parameters.
The non-electrostatic contribution DCop;n can be represented by a temperature-
dependent function similar to that used for V o. The pressure dependence of DCop;n
can be derived from the V o expression based on the thermodynamic identity
ð›Cp=›pÞT ¼ 2Tð›2V=›T2Þp to yield the following expression for the entire
standard partial molar heat capacity:
Cop ¼ c1 þ c2
1
T 2Q
� �2
22T1
T 2Q
� �3
a3ðp2 prÞþ a4 lnCþ p
Cþ pr
� �� �þCo
p;Born
ð13:32Þ
P. Tremaine et al.454

Here c1 and c2 are temperature- and pressure-independent, but species-
dependent, parameters; Q is again a parameter with the value of 228 K; pr is
the reference pressure (1 bar); and a3 and a4 are determined by the fits to V o
from Eq. 13.31.
Standard partial molar volumes and heat capacities of aqueous ions and
electrolytes typically exhibit an inverted U-shape as a function of temperature
(Fig. 13.2). This is consistent with the singularity at water’s critical temperature
of 647 K where second-derivative thermodynamic parameters approach þ1,
and with the behavior in supercooled water where these properties also show a
large increase (which may or may not be associated with a singularity near 228 K).
The parameters in the revised HKF model have been selected so that the
electrostatic contribution dominates at high temperatures where the Born model is
most satisfactory, and the non-electrostatic contribution to V o and Cop dominates at
low temperatures. The revised HKF model has been used widely for the
extrapolation of low-temperature standard partial molar properties of aqueous ions
and electrolytes to elevated temperatures and pressures.
The revised HKF model has also been used by Shock and Helgeson (1990)
for the prediction of the standard partial molar properties of neutral aqueous
organic species up to 1273 K and 500 MPa. It was fitted to the available
experimental data for neutral aqueous organic species at elevated temperatures
and pressures. The fitted parameters were then used to develop correlations with
other low-temperature thermodynamic constants. In contrast to the negative
Cop;Born and Vo
Born for aqueous ions and electrolytes, as required by theory, the fitted
Born terms for neutral species can be either positive or negative. According to Eq.
13.30, positive values of Cop;Born and Vo
Born correspond to negative values of veff, so
that z, the effective charge, is an imaginary number. Clearly, the expression for the
electrostatic contribution has no physical meaning, and the validity of the revised
HKF model for neutral species is questionable. The predictive capability stated in
the paper is also limited by the rather sparse experimental data available at the
time the correlations were derived.
13.1.4.3. Fluctuation Solution Theory Models
O’Connell, Wood and their co-workers (Plyasunov et al., 2000a,b; Sedlbauer et al.,
2000) have developed equations of state for aqueous electrolytes and non-
electrolytes based on the approach proposed by O’Connell et al. (1996). This
approach makes use of the dimensionless Krichevskii parameter
A12 ¼Vo
2
kRT¼ lim
n2!0
›ðpV=RTÞ
›n2
� �T ;V;n2
ð13:33Þ
which is a smooth, continuous and finite function, even at the critical point. Here
n2 is the number of moles of solute. The equations are constructed so that they
Ionization equilibria of acids and bases 455

have the correct limiting behavior in dilute solutions of low-pressure steam, i.e.,
they converge to the second cross virial coefficient between the solute and water.
Details are discussed in Chapter 4.
13.1.4.4. Propagation of Error
Uncertainties associated with log10DCop in Eq. 13.9 lead to an uncertainty in the
estimated values for log10 KT,p:
s2log10 K ¼
Xð› log10 K=›xÞ2s2
x ð13:34Þ
where s2x is the variance of independent variable x and s2
log10 K is the variance of
the dependent variable log10 KT,p. For the special case of Eq. 13.12, where DCop is
temperature independent and DV o is small:
s2log10 K ¼ s2
log10 K;298 þ ½ð1=298:15 2 1=TÞ=R�s2H
þ ½{lnðT=298:15Þ þ 298:15=T 2 1}=R�s2Cp
ð13:35Þ
If the uncertainty in DH o at 25 8C is assumed to be 4 kJ·mol21, this leads to an
uncertainty of 0.34 in log10 K at 300 8C. If the uncertainty in DCop is 2 J·K21·mol21
and 10 J·K21·mol21 at 25 and 300 8C, respectively, and linear with respect to
temperature in this temperature range, these uncertainties result in an error of 0.05
in log10 K at 300 8C. This analysis reveals that accurate DH o values for the heat
capacity function at the reference temperature (usually 25 8C) are essential for the
accurate estimation of log10 K using the equations given above.
13.1.5. Activity Coefficients
Equilibrium quotients Q are usually measured in a solution in which ionic strength
is dictated by the addition of supporting electrolytes such as NaCl, KCl or
NaCF3SO3. Clearly, activity coefficient models are needed to extrapolate the Q
values to infinite dilution for such equilibria. A detailed discussion of models that
incorporate pressure and temperature effects has been given by Millero (1979) and
Pitzer (1991). As an example, the following semi-empirical equation has been
widely used to analyze high-temperature potentiometric titration data by the
ORNL group:
log10 Q ¼ log10 K 2 ðDz2Aw=2:303Þ{ffiffiI
p=ð1 þ 1:2
ffiffiI
pÞ þ 1:667 lnð1 þ 1:2
ffiffiI
pÞ}
þ a1I þ a2I2 þ a3FðIÞ þ 0:0157fI ð13:36Þ
Here I is the ionic strength (I ¼ ð1=2ÞP
miz2i ; where the summation extends to
all ions in solution of molality m and charge z), Dz2 ¼P
z2ðproductsÞ2Pz2ðreactantsÞ is related to the coulombic asymmetry of the equilibrium and Aw
represents the Debye–Huckel limiting slope for the osmotic coefficient. The term
P. Tremaine et al.456

f is the osmotic coefficient of the solution, which is only needed for equilibria in
which water is involved. The values of f for NaCl reported by Archer (1992) as a
function of temperature and ionic strength are used as an approximation. The
Debye–Huckel limiting law term in the above equation was proposed by Pitzer
(1991); the function F(I), takes the form:
FðIÞ ¼ ½1 2 ð1 þ 2ffiffiI
p2 2IÞ expð22
ffiffiI
pÞ�=ð4IÞ ð13:37Þ
The quadratic term a2I 2 is not always needed. Because solute ions undergo
specific interactions with the highly charged anions and cations in the supporting
electrolyte, activity coefficients cannot be described solely by the ionic strength of
the medium. However, it has been found that the nature of the supporting
electrolyte (KCl, NaCl or NaCF3SO3) barely affects the value of log10 K.
Practically, Eq. 13.36 with the coefficients reported by the ORNL group can be
used in speciation calculations at ionic strengths up to 5 mol·kg21 (Baes and
Mesmer, 1986).
Concentrated aqueous media containing more than one pair of ions can be
treated with the Pitzer ion interaction theory for activity coefficients. The Pitzer
equation contains many terms that arise from the binary and ternary interactions of
the ions. These parameters are usually determined by fitting the Pitzer equation
to the experimental activity coefficient of a single electrolyte or a common-ion
mixed electrolyte system and can be used to calculate activity coefficients for
more complicated systems. Although the binary and ternary interaction parameters
for many ions are reported at ambient conditions, these parameters for several
important electrolyte systems such as sulfate and phosphate are still not available
at elevated temperatures. Extensive databases for the Pitzer ion interaction model,
as well as its application to modeling industrial and geochemical systems, have
been presented in several reviews (see, e.g., Pitzer, 1991).
† It is very important to use the same activity coefficient model as that used to
treat the original extrapolation of log10 Q to infinite dilution, or errors will
arise from loss of self-consistency.
When no data are available, empirical and semi-empirical approaches can be
used to estimate activity coefficients for concentrated, mixed electrolytes at
elevated temperatures. For many engineering applications (e.g., bulk properties of
steam condensate and boiler water), the aqueous media are rather dilute
(I p 0.01 mol·kg21), so that the Debye–Huckel limiting law provides sufficiently
accurate activity coefficients. The most practical method at low to moderate ionic
strengths (I & 2.0 mol·kg21) is to avoid using any activity coefficient model if
possible by writing each weak acid/base ionization equilibrium in an isocoulombic
fashion and using Eq. 13.18.
When the isocoulombic approximation is not feasible, an approach suggested
by Lindsay (1989, 1990) can be useful in engineering calculations involving acid–
base equilibria. Here, the activity coefficient of a single ion is estimated by using
Ionization equilibria of acids and bases 457

NaCl(aq) as a model system, through the expression:
glzl ¼ g z2
^ðNaClÞ ð13:38Þ
where gjzj is the single-ion activity coefficient for an ion with charge z, and g^(NaCl)
is the activity coefficient of NaCl(aq) at the same temperature and ionic strength.
Within this approximation, the activity coefficient quotient, log10ðgHPO224=
gH2PO4gOH2Þ for H2PO2
4 ðaqÞ þ OH2ðaqÞO HPO224 ðaqÞ; is estimated to be
2 log10 g^(NaCl). It has been shown that this approximation could provide a
reasonable estimation for activity coefficients of electrolytes at ionic strengths up
to about 1.0 mol·kg21 at temperatures in the range 200 , t , 300 8C (Lindsay,
1989, 1990).
13.2. Experimental Methods
13.2.1. Electrical Conductance
The use of electric conductance measurements to determine the degree of
association in aqueous solutions at high temperature was pioneered by Noyes
(1907) and a detailed description of the conductance technique is given in Chapter
10. Throughout the 1950s and 1960s, Franck and Marshall carried out electrical
conductance studies of a number of electrolytes, mostly in the temperature–
pressure ranges of 400–800 8C and 1–400 MPa, using a platinum-lined cell
described by Franck (1956, 1961), Franck et al. (1962) and Quist and Marshall
(1968a). The aqueous electrolytes studied include the alkali metal halides, K2SO4,
KHSO4, HBr and NH3. A modified version of this apparatus was described by Ho
et al. (1994). Much of our knowledge about ion association at temperatures above
200 8C was obtained from these investigations, which were made before other
methods became available.
Measuring the conductance of aqueous solutions under ambient conditions is
straightforward, but specialized techniques are required to extend these techniques
to high temperature and pressure conditions. Experimental challenges include the
need to use corrosion-resistant metals (usually platinum and its alloys), accurate
temperature and pressure control, and electrically insulated high-pressure seals for
the electrodes. Experience has shown that a static apparatus, of the type used by
Quist and Marshall, does not allow accurate measurements for very dilute
solutions, especially under conditions close to the critical temperature of water.
Accurate conductance values for dilute solutions (,1025 mol·kg21) are essential
if one wants to calculate ion association constants, which are independent of
the activity coefficient model chosen. To overcome this problem, Wood and his
co-workers at the University of Delaware and Ho and Palmer at Oak Ridge
P. Tremaine et al.458

National Laboratory have developed flow-through conductance apparatus for
high-temperature applications (Zimmerman et al., 1995; Ho et al., 2000a). The
flow-through cells allow rapid and accurate electric conductance measurements to
be made on aqueous solutions with concentration as low as 4 £ 1028 mol·kg21,
even in the vicinity of the critical point of water. Since then, a number of acids
(HCl), bases (LiOH, KOH and NaOH) and salts (Na2SO4, alkali metal halides)
have been studied (Zimmerman et al., 1995; Gruszkiewicz and Wood, 1997;
Sharygin et al., 2001; Ho et al., 1994, 2000b, 2001; Ho and Palmer, 1995, 1996,
1997, 1998).
Calculating ion association constants from conductivity data is tedious and
difficult, because modern conductance equations for simple electrolyte solutions
often contain several dozen terms (Fernandez-Prini, 1969). Recently, Wood and
co-workers have evaluated several data interpretation strategies for electrolyte
mixtures that use various combinations of mixing rules, theoretical conductance
equations and activity coefficient models (Sharygin et al., 2001). It was found that
the latest conductance equation developed by Turq et al. (1995), together with the
constant-ionic-strength mixing rule, was suitable for treating high-temperature
conductance data for Na2SO4(aq) solutions containing six ionic species, to
calculate ion association constants for species such as the Naþ·SO422(aq) ion pair.
This method should allow rapid and accurate determination of the equilibrium
constant for any association reaction, which changes the concentration of ions in
solution. Conductance techniques are treated in detail in Chapter 10.
13.2.2. The Hydrogen-Electrode Concentration Cell (HECC)
The use of hydrogen electrodes in a concentration cell configuration was
pioneered at Oak Ridge 30 years ago (Mesmer et al., 1970). The design and
function of the HECC have been described in numerous publications (e.g.,
Mesmer et al., 1970; Kettler et al., 1991; Benezeth et al., 1997) and have been
used in a large number of studies of reactions such as acid–base ionization, metal
ion hydrolysis and complexation, solubility measurements and adsorption studies.
A detailed discussion of this cell is given in Chapter 11, but briefly, it consists of a
300 mL or 1 L capacity pressure vessel containing two concentric Teflon cups
separated by a porous Teflon plug, which acts as a liquid junction completing the
electric circuit. Teflon-insulated platinum wires coated with platinum black
protrude into each cup and serve as electrodes. The solutions in each cup are
stirred magnetically. The solution in the inner cup serves as the reference of
known hydrogen ion molality (usually a strong acid or base), whereas the outer
cup contains the test solution in which a titration can be performed. Both solutions
are thoroughly purged with hydrogen at ambient temperature prior to placing the
vessel in the aluminum block tube furnace or oil bath for equilibration at
temperature.
Ionization equilibria of acids and bases 459

The initial configuration of the cell in a typical study of the ionization of a weak
acid, HA, is as follows:
Pt;H2lmHA;mNaCl;mNaATest
llmNaCl;mHClReference
lH2; Pt ð13:39Þ
where NaCl represents a supporting, non-complexing electrolyte, which is ideally
50–100 times more abundant than the other components so that gHþ,test < gHþ,ref
and the liquid junction potential is minimized. Note that the working definition of
pH is pHm ¼ 2log10 mHþ in stoichiometric molal concentration units. The
convention used here is that Hþ is not complexed by the medium ions and ion
pairing is treated implicitly by the activity coefficient model employed.
Each platinum–hydrogen electrode responds to the half-cell reaction:
H2ðgÞ! 2HþðaqÞ þ 2e2 ð13:40Þ
and the difference in potential between the electrodes is described by the Nernst
equation:
DE ¼ 2RT
FlnðmHþ;t=mHþ;rÞ2 Elj ð13:41Þ
where mHþ;t and mHþ;r refer to the stoichiometric molalities of hydrogen ions in the
test and reference compartments, respectively. The stoichiometric molal activity
coefficients of Hþ in the test and reference compartments are assumed to be equal
at all points in the titration. The ideal gas and Faraday constants are designated by
R and F, respectively; T denotes the temperature in kelvin; and Elj represents the
liquid junction potential based on the full Henderson equation (Baes and Mesmer,
1986), which involves the limiting conductivities of the individual ions.
From the measured mHþ together with a solution of known pHm
(; 2 log10 mHþ in molal units) used in the reference cup, and mass and charge
balance constraints, the molal dissociation constant (QHA) for the acid at the ionic
strength of interest can be calculated, typically with an accuracy of about ^0.01 -
log10 units. By varying the total ionic strength from ca. 0.1–5 molal, the pK
value and activity coefficient ratio for the dissociation reaction can be obtained
by regressing the equation log10 QHA ¼ log10 KHA 2 log10ðgHþgA2=gHAÞ using
an appropriate activity coefficient model such as the Pitzer ion interaction
treatment.
13.2.3. Other pH Sensing Electrodes and Reference Electrodes
Over the few past decades, numerous efforts have been made to develop instru-
ments suitable for pH measurements in aqueous fluids at elevated temperatures
and pressures other than the HECC described above. These include:
† yttria-stabilized zirconia (YSZ) membrane electrodes (150–500 8C) (Mac-
Donald et al., 1988; Hettiarachchi et al., 1992; Ding and Seyfried, 1995, 1996;
Lvov et al., 1999);
P. Tremaine et al.460

† metal–metal oxide electrodes (100–300 8C) such as Pt–PtO2, Ir–IrO2, Zr–
ZrO2, Rh–Rh2O3, W/WO3 (e.g., Kriksunov et al., 1994);
† glass electrode (25–200 8C) (Diakonov et al., 1996).
The principles, development, application and limitations of pH-sensing
electrodes and reference electrodes are discussed in detail in Chapter 11.
13.2.4. Spectroscopic Methods
Over the years, there have been a series of attempts to use UV–visible and
Raman spectroscopy to measure equilibrium constants at elevated temperatures,
usually at steam saturation. The availability of stable UV–visible spectrometers
with fast data acquisition has allowed several workers to develop high-pressure
flow systems with on-line injection, using sapphire or quartz windows.
Suleimenov and Seward (1997) have used these to determine ionization and
complexation constants of species where the spectra of the acid and conjugate
base differ in the visible or near-UV. An alternative approach has been taken
by Johnston’s group, who have developed several thermally stable colorimetric
pH indicators for hydrothermal applications (Johnston et al., 1997; Xiang et al.,
1996).
Several researchers have used Raman spectroscopy to study the speciation
of hydrothermal solutions (see, e.g., Rudolph et al., 1997). Because of the small
diameter of the exciting laser beam, cell construction can make use of small
sapphire tubes or diamond windows, thereby simplifying construction and
extending the temperature range. Recent developments in hydrothermal diamond
anvil cells (Bassett et al., 1993), the use of high-pressure flow injection systems
and the availability of Raman microscopes promise to increase the versatility of
this technique as a tool for obtaining ionization constant data under extreme
conditions.
13.2.5. Flow Calorimetry
Flow calorimetry is an important tool for determining the thermodynamic
properties of aqueous solutes under hydrothermal conditions. Two kinds of flow
calorimeters have been used to determine ionization constants. High-pressure
heat-of-mixing calorimeters have been used at Brigham Young University to
determine DrHo of ionization reactions as a function of temperature, up to about
325 8C. These yield DrCop by differentiation. In favorable cases, the instruments
can be operated as titration calorimeters to obtain equilibrium constants directly at
elevated temperature and pressure (Oscarson et al., 1992).
The second method uses heat capacity flow micro-calorimeters and vibrating
tube densimeters to determine the standard partial molar heat capacities and
Ionization equilibria of acids and bases 461

volumes of the acid and conjugate base, Cop and V o, from which Co
p and DrVo
can be calculated for use in Eq. 13.12 (Sedlbauer et al., 2000; Clarke et al., 2000).
The method is particularly attractive because it yields standard partial molar
properties of individual species, so that equations of state can be derived.
13.3. Ionization of Water
13.3.1. log10 Kw as a Function of Temperature and Pressure
A number of research groups have used EMF methods, conductivity, calorimetry
and spectroscopy to determine values of log10 Qw, which corresponds to the self-
ionization reaction:
H2OðlÞO HþðaqÞ þ OH2ðaqÞ ð13:42Þ
at elevated temperatures, as a function of ionic strength. The critical review
by Marshall and Franck (1981) contains most of the modern values, and
a comprehensive, weighted fit of the infinite-dilution values, log10 Kw, to the
density model (Eq. 13.21):
log10 Kw ¼ 24:098 2 3245:2=ðT=KÞ þ 2:2362 £ 105=ðT=KÞ2
2 3:984 £ 107=ðT=KÞ3 þ ½13:957 2 1262:3=ðT=KÞ
þ 8:5641 £ 105=ðT=KÞ2� log10 rw ð13:43Þ
The temperature- and pressure-dependence of log10 Kw yields values of DH o,
DS o, DCop and DV o up to 1000 8C and 1 GPa. IAPWS has adopted the Marshall–
Franck formulation as its ‘best’ values for log10 Kw vs. T and p for densities
.0.35 g·cm23.
Mesmer, Baes and others at Oak Ridge National Laboratory have measured
ionization constants for water, log10 Qw, vs. I in KCl, NaCl and NaCF3SO3 media
from 0 to 300 8C, and the corresponding values of DH o, DS o, DCop and DV o
(Sweeton et al., 1974; Busey and Mesmer, 1978; Palmer and Drummond, 1988).
These are not entirely consistent with the IAPWS selection of Marshall and
Franck’s values for the ion product of water, but they are complete, internally
consistent with each other, and have been used as the de facto standard by most
other workers. Values of log10 Qw vs. I are listed in Table 13.1. Olofsson and
Hepler (1975) reported a critical evaluation of the consistency of the available data
with the calorimetric results, and found them to be in very good agreement. Values
of log10 Kw from Sweeton et al. (1974) are compared with those of Marshall and
Franck (1981) in Table 13.2.
P. Tremaine et al.462
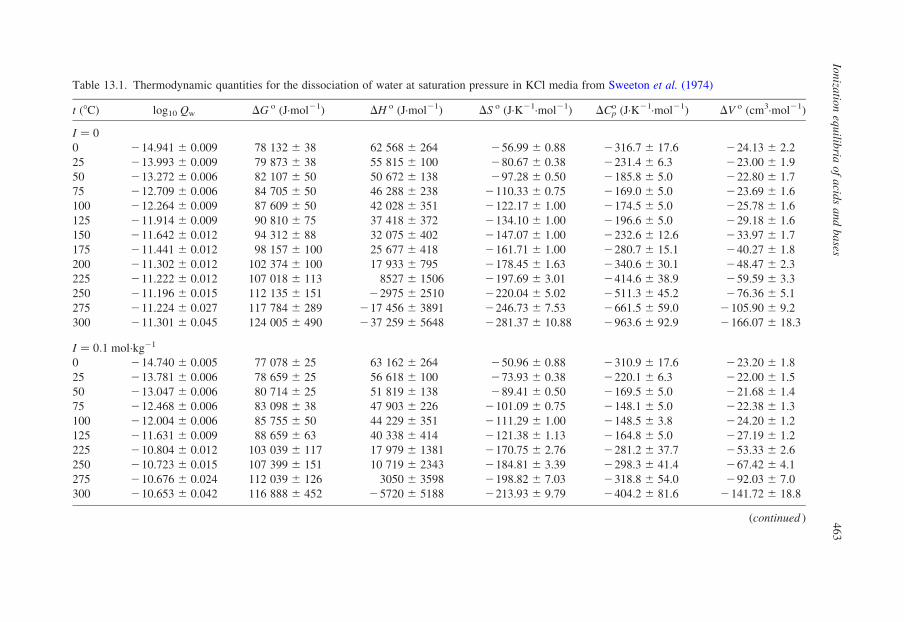
Table 13.1. Thermodynamic quantities for the dissociation of water at saturation pressure in KCl media from Sweeton et al. (1974)
t (8C) log10 Qw DG o (J·mol21) DH o (J·mol21) DS o (J·K21·mol21) DCpo (J·K21·mol21) DV o (cm3·mol21)
I ¼ 0
0 214.941 ^ 0.009 78 132 ^ 38 62 568 ^ 264 256.99 ^ 0.88 2316.7 ^ 17.6 224.13 ^ 2.2
25 213.993 ^ 0.009 79 873 ^ 38 55 815 ^ 100 280.67 ^ 0.38 2231.4 ^ 6.3 223.00 ^ 1.9
50 213.272 ^ 0.006 82 107 ^ 50 50 672 ^ 138 297.28 ^ 0.50 2185.8 ^ 5.0 222.80 ^ 1.7
75 212.709 ^ 0.006 84 705 ^ 50 46 288 ^ 238 2110.33 ^ 0.75 2169.0 ^ 5.0 223.69 ^ 1.6
100 212.264 ^ 0.009 87 609 ^ 50 42 028 ^ 351 2122.17 ^ 1.00 2174.5 ^ 5.0 225.78 ^ 1.6
125 211.914 ^ 0.009 90 810 ^ 75 37 418 ^ 372 2134.10 ^ 1.00 2196.6 ^ 5.0 229.18 ^ 1.6
150 211.642 ^ 0.012 94 312 ^ 88 32 075 ^ 402 2147.07 ^ 1.00 2232.6 ^ 12.6 233.97 ^ 1.7
175 211.441 ^ 0.012 98 157 ^ 100 25 677 ^ 418 2161.71 ^ 1.00 2280.7 ^ 15.1 240.27 ^ 1.8
200 211.302 ^ 0.012 102 374 ^ 100 17 933 ^ 795 2178.45 ^ 1.63 2340.6 ^ 30.1 248.47 ^ 2.3
225 211.222 ^ 0.012 107 018 ^ 113 8527 ^ 1506 2197.69 ^ 3.01 2414.6 ^ 38.9 259.59 ^ 3.3
250 211.196 ^ 0.015 112 135 ^ 151 22975 ^ 2510 2220.04 ^ 5.02 2511.3 ^ 45.2 276.36 ^ 5.1
275 211.224 ^ 0.027 117 784 ^ 289 217 456 ^ 3891 2246.73 ^ 7.53 2661.5 ^ 59.0 2105.90 ^ 9.2
300 211.301 ^ 0.045 124 005 ^ 490 237 259 ^ 5648 2281.37 ^ 10.88 2963.6 ^ 92.9 2166.07 ^ 18.3
I ¼ 0:1 mol·kg21
0 214.740 ^ 0.005 77 078 ^ 25 63 162 ^ 264 250.96 ^ 0.88 2310.9 ^ 17.6 223.20 ^ 1.8
25 213.781 ^ 0.006 78 659 ^ 25 56 618 ^ 100 273.93 ^ 0.38 2220.1 ^ 6.3 222.00 ^ 1.5
50 213.047 ^ 0.006 80 714 ^ 25 51 819 ^ 138 289.41 ^ 0.50 2169.5 ^ 5.0 221.68 ^ 1.4
75 212.468 ^ 0.006 83 098 ^ 38 47 903 ^ 226 2101.09 ^ 0.75 2148.1 ^ 5.0 222.38 ^ 1.3
100 212.004 ^ 0.006 85 755 ^ 50 44 229 ^ 351 2111.29 ^ 1.00 2148.5 ^ 3.8 224.20 ^ 1.2
125 211.631 ^ 0.009 88 659 ^ 63 40 338 ^ 414 2121.38 ^ 1.13 2164.8 ^ 5.0 227.19 ^ 1.2
225 210.804 ^ 0.012 103 039 ^ 117 17 979 ^ 1381 2170.75 ^ 2.76 2281.2 ^ 37.7 253.33 ^ 2.6
250 210.723 ^ 0.015 107 399 ^ 151 10 719 ^ 2343 2184.81 ^ 3.39 2298.3 ^ 41.4 267.42 ^ 4.1
275 210.676 ^ 0.024 112 039 ^ 126 3050 ^ 3598 2198.82 ^ 7.03 2318.8 ^ 54.0 292.03 ^ 7.0
300 210.653 ^ 0.042 116 888 ^ 452 25720 ^ 5188 2213.93 ^ 9.79 2404.2 ^ 81.6 2141.72 ^ 18.8
(continued )
Ion
izatio
neq
uilib
riao
fa
cids
an
db
ases
46
3
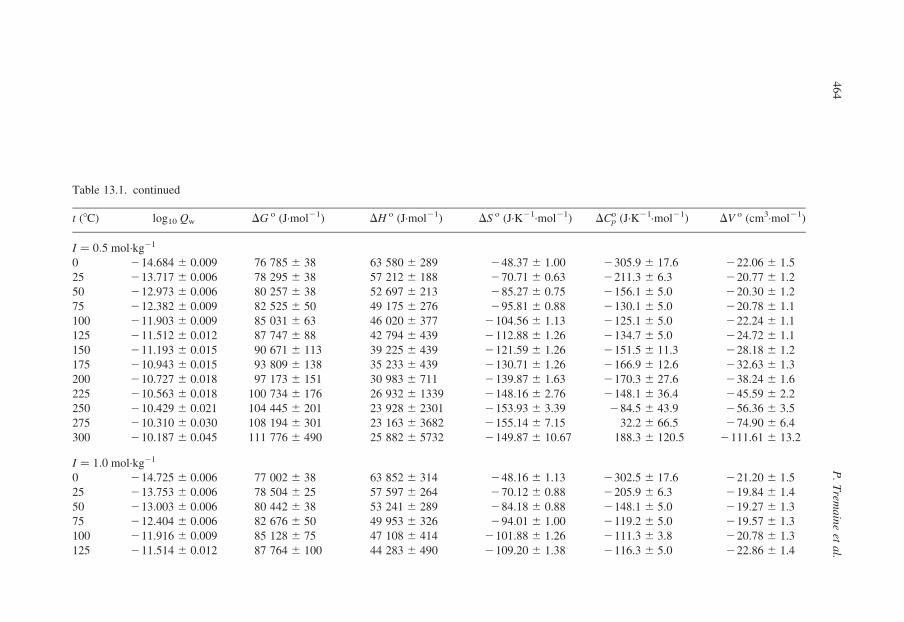
Table 13.1. continued
t (8C) log10 Qw DG o (J·mol21) DH o (J·mol21) DS o (J·K21·mol21) DCpo (J·K21·mol21) DV o (cm3·mol21)
I ¼ 0:5 mol·kg21
0 214.684 ^ 0.009 76 785 ^ 38 63 580 ^ 289 248.37 ^ 1.00 2305.9 ^ 17.6 222.06 ^ 1.5
25 213.717 ^ 0.006 78 295 ^ 38 57 212 ^ 188 270.71 ^ 0.63 2211.3 ^ 6.3 220.77 ^ 1.2
50 212.973 ^ 0.006 80 257 ^ 38 52 697 ^ 213 285.27 ^ 0.75 2156.1 ^ 5.0 220.30 ^ 1.2
75 212.382 ^ 0.009 82 525 ^ 50 49 175 ^ 276 295.81 ^ 0.88 2130.1 ^ 5.0 220.78 ^ 1.1
100 211.903 ^ 0.009 85 031 ^ 63 46 020 ^ 377 2104.56 ^ 1.13 2125.1 ^ 5.0 222.24 ^ 1.1
125 211.512 ^ 0.012 87 747 ^ 88 42 794 ^ 439 2112.88 ^ 1.26 2134.7 ^ 5.0 224.72 ^ 1.1
150 211.193 ^ 0.015 90 671 ^ 113 39 225 ^ 439 2121.59 ^ 1.26 2151.5 ^ 11.3 228.18 ^ 1.2
175 210.943 ^ 0.015 93 809 ^ 138 35 233 ^ 439 2130.71 ^ 1.26 2166.9 ^ 12.6 232.63 ^ 1.3
200 210.727 ^ 0.018 97 173 ^ 151 30 983 ^ 711 2139.87 ^ 1.63 2170.3 ^ 27.6 238.24 ^ 1.6
225 210.563 ^ 0.018 100 734 ^ 176 26 932 ^ 1339 2148.16 ^ 2.76 2148.1 ^ 36.4 245.59 ^ 2.2
250 210.429 ^ 0.021 104 445 ^ 201 23 928 ^ 2301 2153.93 ^ 3.39 284.5 ^ 43.9 256.36 ^ 3.5
275 210.310 ^ 0.030 108 194 ^ 301 23 163 ^ 3682 2155.14 ^ 7.15 32.2 ^ 66.5 274.90 ^ 6.4
300 210.187 ^ 0.045 111 776 ^ 490 25 882 ^ 5732 2149.87 ^ 10.67 188.3 ^ 120.5 2111.61 ^ 13.2
I ¼ 1:0 mol·kg21
0 214.725 ^ 0.006 77 002 ^ 38 63 852 ^ 314 248.16 ^ 1.13 2302.5 ^ 17.6 221.20 ^ 1.5
25 213.753 ^ 0.006 78 504 ^ 25 57 597 ^ 264 270.12 ^ 0.88 2205.9 ^ 6.3 219.84 ^ 1.4
50 213.003 ^ 0.006 80 442 ^ 38 53 241 ^ 289 284.18 ^ 0.88 2148.1 ^ 5.0 219.27 ^ 1.3
75 212.404 ^ 0.006 82 676 ^ 50 49 953 ^ 326 294.01 ^ 1.00 2119.2 ^ 5.0 219.57 ^ 1.3
100 211.916 ^ 0.009 85 128 ^ 75 47 108 ^ 414 2101.88 ^ 1.26 2111.3 ^ 3.8 220.78 ^ 1.3
125 211.514 ^ 0.012 87 764 ^ 100 44 283 ^ 490 2109.20 ^ 1.38 2116.3 ^ 5.0 222.86 ^ 1.4
P.
Trem
ain
eet
al.
46
4
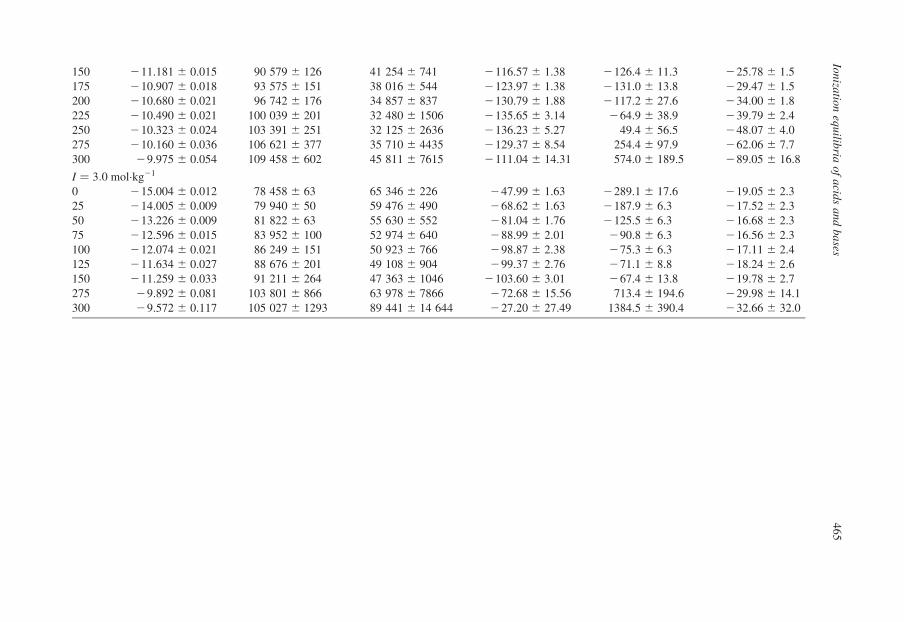
150 211.181 ^ 0.015 90 579 ^ 126 41 254 ^ 741 2116.57 ^ 1.38 2126.4 ^ 11.3 225.78 ^ 1.5
175 210.907 ^ 0.018 93 575 ^ 151 38 016 ^ 544 2123.97 ^ 1.38 2131.0 ^ 13.8 229.47 ^ 1.5
200 210.680 ^ 0.021 96 742 ^ 176 34 857 ^ 837 2130.79 ^ 1.88 2117.2 ^ 27.6 234.00 ^ 1.8
225 210.490 ^ 0.021 100 039 ^ 201 32 480 ^ 1506 2135.65 ^ 3.14 264.9 ^ 38.9 239.79 ^ 2.4
250 210.323 ^ 0.024 103 391 ^ 251 32 125 ^ 2636 2136.23 ^ 5.27 49.4 ^ 56.5 248.07 ^ 4.0
275 210.160 ^ 0.036 106 621 ^ 377 35 710 ^ 4435 2129.37 ^ 8.54 254.4 ^ 97.9 262.06 ^ 7.7
300 29.975 ^ 0.054 109 458 ^ 602 45 811 ^ 7615 2111.04 ^ 14.31 574.0 ^ 189.5 289.05 ^ 16.8
I ¼ 3:0 mol·kg21
0 215.004 ^ 0.012 78 458 ^ 63 65 346 ^ 226 247.99 ^ 1.63 2289.1 ^ 17.6 219.05 ^ 2.3
25 214.005 ^ 0.009 79 940 ^ 50 59 476 ^ 490 268.62 ^ 1.63 2187.9 ^ 6.3 217.52 ^ 2.3
50 213.226 ^ 0.009 81 822 ^ 63 55 630 ^ 552 281.04 ^ 1.76 2125.5 ^ 6.3 216.68 ^ 2.3
75 212.596 ^ 0.015 83 952 ^ 100 52 974 ^ 640 288.99 ^ 2.01 290.8 ^ 6.3 216.56 ^ 2.3
100 212.074 ^ 0.021 86 249 ^ 151 50 923 ^ 766 298.87 ^ 2.38 275.3 ^ 6.3 217.11 ^ 2.4
125 211.634 ^ 0.027 88 676 ^ 201 49 108 ^ 904 299.37 ^ 2.76 271.1 ^ 8.8 218.24 ^ 2.6
150 211.259 ^ 0.033 91 211 ^ 264 47 363 ^ 1046 2103.60 ^ 3.01 267.4 ^ 13.8 219.78 ^ 2.7
275 29.892 ^ 0.081 103 801 ^ 866 63 978 ^ 7866 272.68 ^ 15.56 713.4 ^ 194.6 229.98 ^ 14.1
300 29.572 ^ 0.117 105 027 ^ 1293 89 441 ^ 14 644 227.20 ^ 27.49 1384.5 ^ 390.4 232.66 ^ 32.0
Ion
izatio
neq
uilib
riao
fa
cids
an
db
ases
46
5
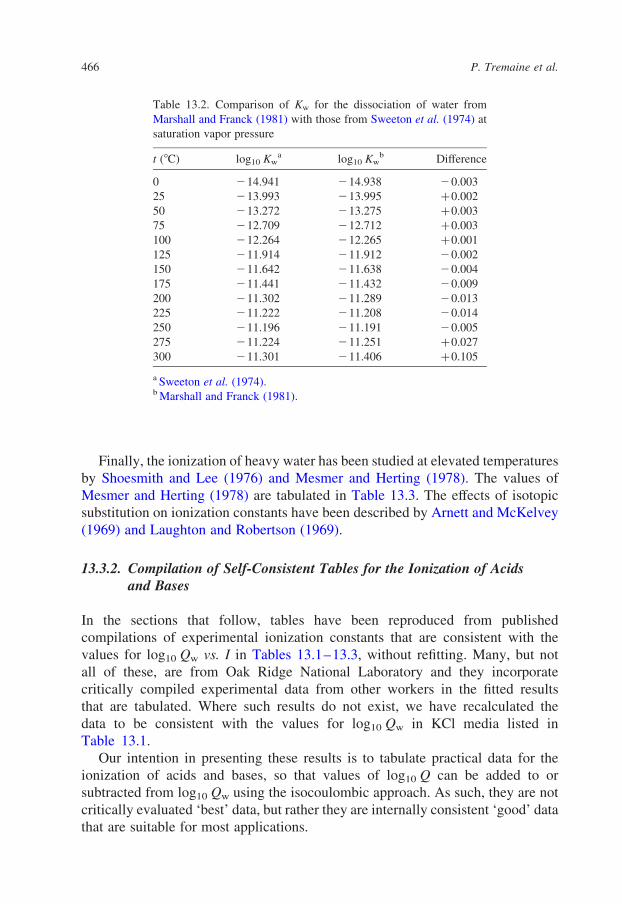
Finally, the ionization of heavy water has been studied at elevated temperatures
by Shoesmith and Lee (1976) and Mesmer and Herting (1978). The values of
Mesmer and Herting (1978) are tabulated in Table 13.3. The effects of isotopic
substitution on ionization constants have been described by Arnett and McKelvey
(1969) and Laughton and Robertson (1969).
13.3.2. Compilation of Self-Consistent Tables for the Ionization of Acidsand Bases
In the sections that follow, tables have been reproduced from published
compilations of experimental ionization constants that are consistent with the
values for log10 Qw vs. I in Tables 13.1–13.3, without refitting. Many, but not
all of these, are from Oak Ridge National Laboratory and they incorporate
critically compiled experimental data from other workers in the fitted results
that are tabulated. Where such results do not exist, we have recalculated the
data to be consistent with the values for log10 Qw in KCl media listed in
Table 13.1.
Our intention in presenting these results is to tabulate practical data for the
ionization of acids and bases, so that values of log10 Q can be added to or
subtracted from log10 Qw using the isocoulombic approach. As such, they are not
critically evaluated ‘best’ data, but rather they are internally consistent ‘good’ data
that are suitable for most applications.
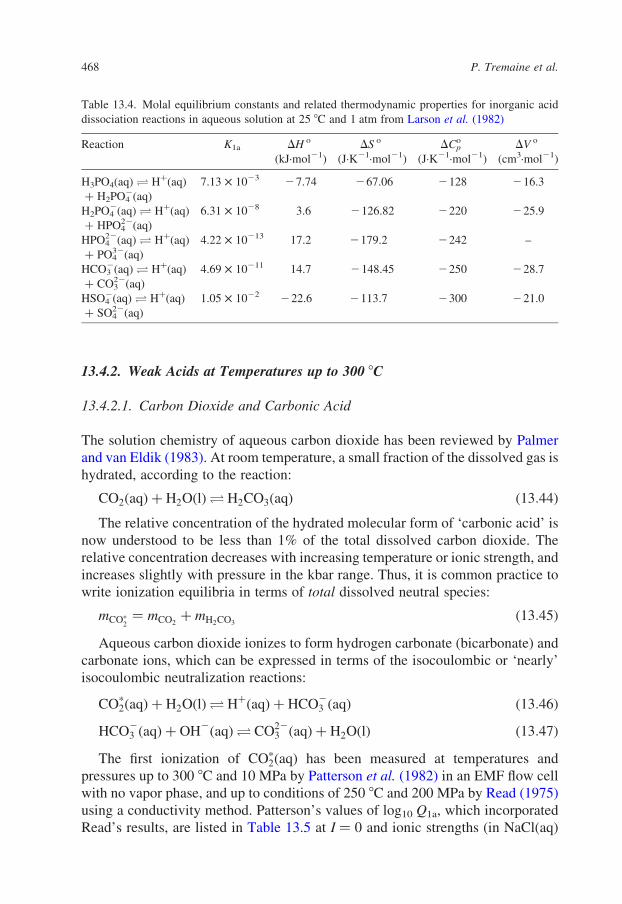
Table 13.2. Comparison of Kw for the dissociation of water from
Marshall and Franck (1981) with those from Sweeton et al. (1974) at
saturation vapor pressure
t (8C) log10 Kwa log10 Kw
b Difference
0 214.941 214.938 20.003
25 213.993 213.995 þ0.002
50 213.272 213.275 þ0.003
75 212.709 212.712 þ0.003
100 212.264 212.265 þ0.001
125 211.914 211.912 20.002
150 211.642 211.638 20.004
175 211.441 211.432 20.009
200 211.302 211.289 20.013
225 211.222 211.208 20.014
250 211.196 211.191 20.005
275 211.224 211.251 þ0.027
300 211.301 211.406 þ0.105
a Sweeton et al. (1974).b Marshall and Franck (1981).
P. Tremaine et al.466

13.4. Inorganic Acids and Bases
13.4.1. Weak Acids at 25 8C and 100 kPa
The properties of aqueous inorganic acids and bases at 25 8C are well understood,
and they are discussed in depth in several authoritative texts, reviews and
monographs, many of which date from the 1960s (see, e.g., Cotton and Wilkinson,
1988; Hepler and Hopkins, 1979). The introduction of modern titration calori-
meters, flow micro-calorimeters and vibrating-tube densimeters resulted in a
major increase in the number and quality of data for DH o, DS o, DCop and DV o.
Many of these derivative properties and the ‘best’ values for log10 K have been
tabulated by Pettit and Powell (1997) and Smith and Martell (1997). Table 13.4
lists values at 25 8C from Larson et al. (1982).
Table 13.3. Thermodynamic quantities for the ionization of D2O at saturation pressure in KCl media
from Mesmer and Herting (1978)
t (8C) log10 Qw DH o (J·mol21) DS o (J·K21·mol21) DCpo (J·K21·mol21)
I ¼ 0
0 215.972 ^ 0.021 67 488 ^ 1632 258.58 ^ 5.86 2347.3 ^ 46.0
25 214.951 ^ 0.008 60 040 ^ 879 284.89 ^ 2.97 2257.3 ^ 27.6
50 214.176 ^ 0.011 54 308 ^ 628 2103.39 ^ 2.01 2207.1 ^ 20.1
75 213.574 ^ 0.015 49 455 ^ 711 2117.86 ^ 2.22 2185.4 ^ 13.4
100 213.099 ^ 0.020 44 852 ^ 795 2130.62 ^ 2.26 2185.8 ^ 11.3
125 212.725 ^ 0.024 39 999 ^ 753 2143.13 ^ 2.34 2202.9 ^ 16.7
150 212.434 ^ 0.027 34 602 ^ 837 2156.31 ^ 2.47 2233.5 ^ 25.5
175 212.215 ^ 0.028 28 242 ^ 1297 2170.83 ^ 2.97 2276.6 ^ 33.1
200 212.060 ^ 0.030 20 669 ^ 2134 2187.02 ^ 4.60 2331.0 ^ 46.0
225 211.964 ^ 0.036 11 590 ^ 3305 2205.85 ^ 7.11 2399.6 ^ 54.4
250 211.923 ^ 0.049 418 ^ 4602 2227.19 ^ 10.04 2489.5 ^ 66.9
275 211.933 ^ 0.070 213 389 ^ 6694 2252.71 ^ 12.97 2636.0 ^ 71.1
300 211.992 ^ 0.098 232 635 ^ 8368 2286.19 ^ 16.74 2933.0 ^ 87.9
I ¼ 1:0 mol·kg21
0 215.591 ^ 0.068 59 036 ^ 3096 282.42 ^ 10.04 2330.1 ^ 50.6
25 214.612 ^ 0.033 52 049 ^ 2134 2105.14 ^ 6.69 2235.6 ^ 36.4
50 213.849 ^ 0.023 46 903 ^ 1464 2120.08 ^ 4.60 2180.7 ^ 31.8
75 213.231 ^ 0.025 42 760 ^ 1004 2130.42 ^ 3.10 2154.4 ^ 28.5
100 212.717 ^ 0.028 38 995 ^ 962 2138.91 ^ 2.72 2149.8 ^ 27.2
125 212.282 ^ 0.029 35 146 ^ 1255 2146.90 ^ 3.26 2162.3 ^ 30.1
150 211.910 ^ 0.031 30 794 ^ 1841 2155.27 ^ 4.52 2187.9 ^ 35.6
175 211.590 ^ 0.036 25 648 ^ 2636 2164.85 ^ 6.28 2225.5 ^ 41.8
200 211.312 ^ 0.048 19 414 ^ 3640 2175.73 ^ 8.37 2276.1 ^ 50.2
225 211.069 ^ 0.066 11 799 ^ 5021 2188.28 ^ 10.88 2338.9 ^ 58.6
250 210.852 ^ 0.089 2385 ^ 6276 2203.34 ^ 13.81 2418.4 ^ 71.1
275 210.650 ^ 0.120 29623 ^ 8368 2221.33 ^ 17.15 2539.7 ^ 79.5
300 210.430 ^ 0.150 225 522 ^ 10042 2243.93 ^ 20.50 2748.9 ^ 92.0
Ionization equilibria of acids and bases 467
13.4.2. Weak Acids at Temperatures up to 300 8C
13.4.2.1. Carbon Dioxide and Carbonic Acid
The solution chemistry of aqueous carbon dioxide has been reviewed by Palmer
and van Eldik (1983). At room temperature, a small fraction of the dissolved gas is
hydrated, according to the reaction:
CO2ðaqÞ þ H2OðlÞO H2CO3ðaqÞ ð13:44Þ
The relative concentration of the hydrated molecular form of ‘carbonic acid’ is
now understood to be less than 1% of the total dissolved carbon dioxide. The
relative concentration decreases with increasing temperature or ionic strength, and
increases slightly with pressure in the kbar range. Thus, it is common practice to
write ionization equilibria in terms of total dissolved neutral species:
mCOp2¼ mCO2
þ mH2CO3ð13:45Þ
Aqueous carbon dioxide ionizes to form hydrogen carbonate (bicarbonate) and
carbonate ions, which can be expressed in terms of the isocoulombic or ‘nearly’
isocoulombic neutralization reactions:
COp2ðaqÞ þ H2OðlÞO HþðaqÞ þ HCO2
3 ðaqÞ ð13:46Þ
HCO23 ðaqÞ þ OH2ðaqÞO CO22
3 ðaqÞ þ H2OðlÞ ð13:47Þ
The first ionization of COp2ðaqÞ has been measured at temperatures and
pressures up to 300 8C and 10 MPa by Patterson et al. (1982) in an EMF flow cell
with no vapor phase, and up to conditions of 250 8C and 200 MPa by Read (1975)
using a conductivity method. Patterson’s values of log10 Q1a, which incorporated
Read’s results, are listed in Table 13.5 at I ¼ 0 and ionic strengths (in NaCl(aq)
Table 13.4. Molal equilibrium constants and related thermodynamic properties for inorganic acid
dissociation reactions in aqueous solution at 25 8C and 1 atm from Larson et al. (1982)
Reaction K1a DH o
(kJ·mol21)
DS o
(J·K21·mol21)
DCpo
(J·K21·mol21)
DV o
(cm3·mol21)
H3PO4(aq) O Hþ(aq)
þ H2PO42(aq)
7.13 £ 1023 27.74 267.06 2128 216.3
H2PO42(aq) O Hþ(aq)
þ HPO422(aq)
6.31 £ 1028 3.6 2126.82 2220 225.9
HPO422(aq) O Hþ(aq)
þ PO432(aq)
4.22 £ 10213 17.2 2179.2 2242 –
HCO32(aq) O Hþ(aq)
þ CO322(aq)
4.69 £ 10211 14.7 2148.45 2250 228.7
HSO42(aq) O Hþ(aq)
þ SO422(aq)
1.05 £ 1022 222.6 2113.7 2300 221.0
P. Tremaine et al.468
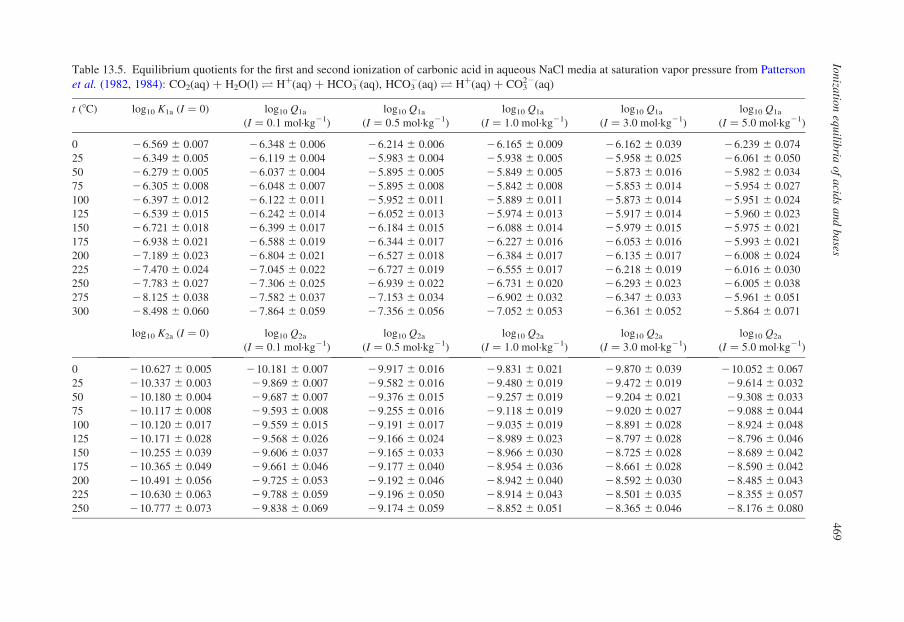
Table 13.5. Equilibrium quotients for the first and second ionization of carbonic acid in aqueous NaCl media at saturation vapor pressure from Patterson
et al. (1982, 1984): CO2(aq) þ H2O(l) O Hþ(aq) þ HCO32(aq), HCO3
2(aq) O Hþ(aq) þ CO322(aq)
t (8C) log10 K1a (I ¼ 0) log10 Q1a
(I ¼ 0:1 mol·kg21)
log10 Q1a
(I ¼ 0:5 mol·kg21)
log10 Q1a
(I ¼ 1:0 mol·kg21)
log10 Q1a
(I ¼ 3:0 mol·kg21)
log10 Q1a
(I ¼ 5:0 mol·kg21)
0 26.569 ^ 0.007 26.348 ^ 0.006 26.214 ^ 0.006 26.165 ^ 0.009 26.162 ^ 0.039 26.239 ^ 0.074
25 26.349 ^ 0.005 26.119 ^ 0.004 25.983 ^ 0.004 25.938 ^ 0.005 25.958 ^ 0.025 26.061 ^ 0.050
50 26.279 ^ 0.005 26.037 ^ 0.004 25.895 ^ 0.005 25.849 ^ 0.005 25.873 ^ 0.016 25.982 ^ 0.034
75 26.305 ^ 0.008 26.048 ^ 0.007 25.895 ^ 0.008 25.842 ^ 0.008 25.853 ^ 0.014 25.954 ^ 0.027
100 26.397 ^ 0.012 26.122 ^ 0.011 25.952 ^ 0.011 25.889 ^ 0.011 25.873 ^ 0.014 25.951 ^ 0.024
125 26.539 ^ 0.015 26.242 ^ 0.014 26.052 ^ 0.013 25.974 ^ 0.013 25.917 ^ 0.014 25.960 ^ 0.023
150 26.721 ^ 0.018 26.399 ^ 0.017 26.184 ^ 0.015 26.088 ^ 0.014 25.979 ^ 0.015 25.975 ^ 0.021
175 26.938 ^ 0.021 26.588 ^ 0.019 26.344 ^ 0.017 26.227 ^ 0.016 26.053 ^ 0.016 25.993 ^ 0.021
200 27.189 ^ 0.023 26.804 ^ 0.021 26.527 ^ 0.018 26.384 ^ 0.017 26.135 ^ 0.017 26.008 ^ 0.024
225 27.470 ^ 0.024 27.045 ^ 0.022 26.727 ^ 0.019 26.555 ^ 0.017 26.218 ^ 0.019 26.016 ^ 0.030
250 27.783 ^ 0.027 27.306 ^ 0.025 26.939 ^ 0.022 26.731 ^ 0.020 26.293 ^ 0.023 26.005 ^ 0.038
275 28.125 ^ 0.038 27.582 ^ 0.037 27.153 ^ 0.034 26.902 ^ 0.032 26.347 ^ 0.033 25.961 ^ 0.051
300 28.498 ^ 0.060 27.864 ^ 0.059 27.356 ^ 0.056 27.052 ^ 0.053 26.361 ^ 0.052 25.864 ^ 0.071
log10 K2a (I ¼ 0) log10 Q2a
(I ¼ 0:1 mol·kg21)
log10 Q2a
(I ¼ 0:5 mol·kg21)
log10 Q2a
(I ¼ 1:0 mol·kg21)
log10 Q2a
(I ¼ 3:0 mol·kg21)
log10 Q2a
(I ¼ 5:0 mol·kg21)
0 210.627 ^ 0.005 210.181 ^ 0.007 29.917 ^ 0.016 29.831 ^ 0.021 29.870 ^ 0.039 210.052 ^ 0.067
25 210.337 ^ 0.003 29.869 ^ 0.007 29.582 ^ 0.016 29.480 ^ 0.019 29.472 ^ 0.019 29.614 ^ 0.032
50 210.180 ^ 0.004 29.687 ^ 0.007 29.376 ^ 0.015 29.257 ^ 0.019 29.204 ^ 0.021 29.308 ^ 0.033
75 210.117 ^ 0.008 29.593 ^ 0.008 29.255 ^ 0.016 29.118 ^ 0.019 29.020 ^ 0.027 29.088 ^ 0.044
100 210.120 ^ 0.017 29.559 ^ 0.015 29.191 ^ 0.017 29.035 ^ 0.019 28.891 ^ 0.028 28.924 ^ 0.048
125 210.171 ^ 0.028 29.568 ^ 0.026 29.166 ^ 0.024 28.989 ^ 0.023 28.797 ^ 0.028 28.796 ^ 0.046
150 210.255 ^ 0.039 29.606 ^ 0.037 29.165 ^ 0.033 28.966 ^ 0.030 28.725 ^ 0.028 28.689 ^ 0.042
175 210.365 ^ 0.049 29.661 ^ 0.046 29.177 ^ 0.040 28.954 ^ 0.036 28.661 ^ 0.028 28.590 ^ 0.042
200 210.491 ^ 0.056 29.725 ^ 0.053 29.192 ^ 0.046 28.942 ^ 0.040 28.592 ^ 0.030 28.485 ^ 0.043
225 210.630 ^ 0.063 29.788 ^ 0.059 29.196 ^ 0.050 28.914 ^ 0.043 28.501 ^ 0.035 28.355 ^ 0.057
250 210.777 ^ 0.073 29.838 ^ 0.069 29.174 ^ 0.059 28.852 ^ 0.051 28.365 ^ 0.046 28.176 ^ 0.080
Ion
izatio
neq
uilib
riao
fa
cids
an
db
ases
46
9

media) up to 5.0 mol·kg21. The second ionization constant, which has been
measured to 250 8C by Patterson et al. (1984), decreases with increasing tempera-
ture for reasons outlined earlier.
13.4.2.2. Phosphoric Acid
The sodium salts of aqueous phosphoric acid are widely used as pH buffers in the
boiler water of thermal electric power stations. Phosphoric acid ionizes according
to the reactions:
H3PO4ðaqÞ þ OH2ðaqÞO H2PO24 ðaqÞ þ H2OðlÞ ð13:48Þ
H2PO24 ðaqÞ þ OH2ðaqÞO HPO22
4 ðaqÞ þ H2OðlÞ ð13:49Þ
HPO224 ðaqÞ þ OH2ðaqÞO PO32
4 ðaqÞ þ H2OðlÞ ð13:50Þ
The first and second dissociation equilibria, reactions 13.48 and 13.49, have
been determined potentiometrically at temperatures up to 300 8C by Mesmer and
Baes (1974). Their values of log10 Q1a,OH and log10 Q2a,OH at ionic strengths in
KCl(aq) up to 1.0 mol·kg21 are listed in Table 13.6. The third ionization constant
is expected to decrease with increasing temperature. Values have been estimated
by Lindsay (1990) and Shock and Helgeson (1988).
13.4.2.3. Hydrogen Sulfate Ion
Sulfuric acid, H2SO4(aq), is considered to be a strong acid, which ionizes
according to the reaction:
H2SO4ðaqÞO HSO24 ðaqÞ þ HþðaqÞ ð13:51Þ
Colorimetric measurements of the pH of ammonia/sulfuric acid mixtures by
Xiang et al. (1996) yielded values for the first dissociation constant of H2SO4(aq)
from 350 to 400 8C.
The hydrogen sulfate (‘bisulfate’) ion is a moderately strong acid, which is
found in boilers and in the feed-train of nuclear and thermal electric power stations
as an impurity from condenser leaks. The HSO24 ion is iso-electronic with
perchlorate ClO24 and, as a result, it can be used as a non-complexing ion for high-
temperature experiments (Rudolph et al., 1997). The ionization of hydrogen
sulfate has been studied as the reaction:
HSO24 ðaqÞO HþðaqÞ þ SO22
4 ðaqÞ ð13:52Þ
up to 250 8C by Dickson et al. (1990). The values for log10 Q2a at ionic strengths in
NaCl(aq) up to 5.0 mol·kg21 are tabulated in Table 13.7.
P. Tremaine et al.470
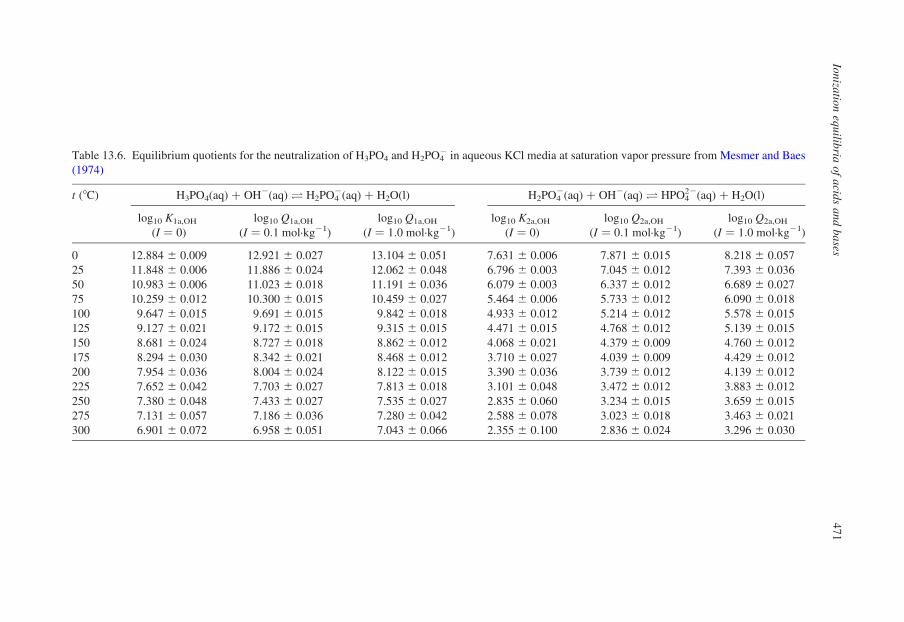
Table 13.6. Equilibrium quotients for the neutralization of H3PO4 and H2PO42 in aqueous KCl media at saturation vapor pressure from Mesmer and Baes
(1974)
t (8C) H3PO4(aq) þ OH2(aq) O H2PO42(aq) þ H2O(l) H2PO4
2(aq) þ OH2(aq) O HPO422(aq) þ H2O(l)
log10 K1a,OH
(I ¼ 0)
log10 Q1a,OH
(I ¼ 0:1 mol·kg21)
log10 Q1a,OH
(I ¼ 1:0 mol·kg21)
log10 K2a,OH
(I ¼ 0)
log10 Q2a,OH
(I ¼ 0:1 mol·kg21)
log10 Q2a,OH
(I ¼ 1:0 mol·kg21)
0 12.884 ^ 0.009 12.921 ^ 0.027 13.104 ^ 0.051 7.631 ^ 0.006 7.871 ^ 0.015 8.218 ^ 0.057
25 11.848 ^ 0.006 11.886 ^ 0.024 12.062 ^ 0.048 6.796 ^ 0.003 7.045 ^ 0.012 7.393 ^ 0.036
50 10.983 ^ 0.006 11.023 ^ 0.018 11.191 ^ 0.036 6.079 ^ 0.003 6.337 ^ 0.012 6.689 ^ 0.027
75 10.259 ^ 0.012 10.300 ^ 0.015 10.459 ^ 0.027 5.464 ^ 0.006 5.733 ^ 0.012 6.090 ^ 0.018
100 9.647 ^ 0.015 9.691 ^ 0.015 9.842 ^ 0.018 4.933 ^ 0.012 5.214 ^ 0.012 5.578 ^ 0.015
125 9.127 ^ 0.021 9.172 ^ 0.015 9.315 ^ 0.015 4.471 ^ 0.015 4.768 ^ 0.012 5.139 ^ 0.015
150 8.681 ^ 0.024 8.727 ^ 0.018 8.862 ^ 0.012 4.068 ^ 0.021 4.379 ^ 0.009 4.760 ^ 0.012
175 8.294 ^ 0.030 8.342 ^ 0.021 8.468 ^ 0.012 3.710 ^ 0.027 4.039 ^ 0.009 4.429 ^ 0.012
200 7.954 ^ 0.036 8.004 ^ 0.024 8.122 ^ 0.015 3.390 ^ 0.036 3.739 ^ 0.012 4.139 ^ 0.012
225 7.652 ^ 0.042 7.703 ^ 0.027 7.813 ^ 0.018 3.101 ^ 0.048 3.472 ^ 0.012 3.883 ^ 0.012
250 7.380 ^ 0.048 7.433 ^ 0.027 7.535 ^ 0.027 2.835 ^ 0.060 3.234 ^ 0.015 3.659 ^ 0.015
275 7.131 ^ 0.057 7.186 ^ 0.036 7.280 ^ 0.042 2.588 ^ 0.078 3.023 ^ 0.018 3.463 ^ 0.021
300 6.901 ^ 0.072 6.958 ^ 0.051 7.043 ^ 0.066 2.355 ^ 0.100 2.836 ^ 0.024 3.296 ^ 0.030
Ion
izatio
neq
uilib
riao
fa
cids
an
db
ases
47
1

Table 13.7. Equilibrium quotients for the hydrogen sulfate ionization in aqueous NaCl media at saturation vapor pressure from Dickson et al. (1990):
HSO42(aq) O Hþ(aq) þ SO4
22(aq)
t
(8C)
log10 K2a
(I ¼ 0)
log10 Q2a
(I ¼ 0:1 mol·kg21)
log10 Q2a
(I ¼ 0:5 mol·kg21)
log10 Q2a
(I ¼ 1:0 mol·kg21)
log10 Q2a
(I ¼ 3:0 mol·kg21)
log10 Q2a
(I ¼ 5:0 mol·kg21)
0 21.659 ^ 0.030 21.198 ^ 0.030 20.900 ^ 0.030 20.788 ^ 0.031 20.737 ^ 0.032 20.806 ^ 0.032
25 21.964 ^ 0.018 21.487 ^ 0.017 21.178 ^ 0.017 21.055 ^ 0.018 20.971 ^ 0.019 21.010 ^ 0.019
50 22.316 ^ 0.012 21.817 ^ 0.010 21.493 ^ 0.009 21.358 ^ 0.010 21.238 ^ 0.011 21.250 ^ 0.011
75 22.686 ^ 0.009 22.161 ^ 0.007 21.817 ^ 0.005 21.669 ^ 0.005 21.511 ^ 0.007 21.495 ^ 0.006
100 23.061 ^ 0.008 22.504 ^ 0.006 22.135 ^ 0.004 21.972 ^ 0.004 21.775 ^ 0.006 21.730 ^ 0.005
125 23.436 ^ 0.007 22.840 ^ 0.005 22.442 ^ 0.003 22.261 ^ 0.003 22.020 ^ 0.005 21.946 ^ 0.005
150 23.809 ^ 0.007 23.167 ^ 0.005 22.735 ^ 0.003 22.533 ^ 0.003 22.244 ^ 0.005 22.140 ^ 0.004
175 24.182 ^ 0.007 23.488 ^ 0.006 23.015 ^ 0.004 22.788 ^ 0.004 22.446 ^ 0.005 22.309 ^ 0.005
200 24.561 ^ 0.008 23.804 ^ 0.006 23.282 ^ 0.004 23.027 ^ 0.004 22.624 ^ 0.005 22.450 ^ 0.005
225 24.951 ^ 0.009 24.118 ^ 0.007 23.537 ^ 0.005 23.248 ^ 0.005 22.775 ^ 0.006 22.560 ^ 0.006
250 25.355 ^ 0.012 24.432 ^ 0.010 23.778 ^ 0.009 23.447 ^ 0.009 22.892 ^ 0.011 22.629 ^ 0.012
P.
Trem
ain
eet
al.
47
2

13.4.2.4. Hydrogen Sulfide
It is not possible to study the ionization of hydrogen sulfide in electrochemical or
conductivity cells that contain platinum electrodes. Suleimenov and Seward
(1997) have circumvented this problem by determining the degree of ionization of
H2S/HS2 buffers by UV–visible spectrophotometry at temperatures up to 350 8C.
The resulting values for log10 K1a are listed in Table 13.8, for the reaction:
H2SðaqÞO HþðaqÞ þ HS2ðaqÞ ð13:53Þ
The further ionization of HS2(aq) to form the sulfide ion:
HS2ðaqÞO S22ðaqÞ þ HþðaqÞ ð13:54Þ
has been shown to be negligible, even in very concentrated solutions of base (Rao
and Hepler, 1977; Giggenbach, 1971). The destabilization of polyvalent ions in
high-temperature water is expected to make S22(aq) even more unstable.
13.4.2.5. Nitric Acid
The ionization constant of nitric acid has been determined at temperatures from
250 to 320 8C by Oscarson et al. (1992) by using flow calorimetry to determine
enthalpies of dilution as a function of nitric acid molality for the ionization
reaction:
HNO3ðaqÞO NO23 ðaqÞ þ HþðaqÞ ð13:55Þ
Table 13.8. First ionization constant of H2S (molal) and ionization constant of HNO3 from
Suleimenov and Seward (1997) and Oscarson et al. (1992)
t (8C) H2S(aq) O Hþ(aq) þ HS2(aq) HNO3(aq) O Hþ(aq) þ NO32(aq)
p (MPa) log10 K1a log10 K1a (p ¼ psata) p (MPa) log10 K1a
25 10 26.96 ^ 0.022 26.99
50 10 26.66 ^ 0.018 26.68
100 10 26.46 ^ 0.013 26.49
150 10 26.47 ^ 0.019 26.49
200 10 26.70 ^ 0.023 26.73
250 10 27.16 ^ 0.076 27.19 10.3 21
275 11 21.42
300 11 27.87 ^ 0.076 27.89 11 21.92
319 12.8 22.39
350 20 28.77 ^ 0.051 28.89
a Saturation vapor pressure.
Ionization equilibria of acids and bases 473
The results are consistent with earlier high-temperature conductance (Noyes,
1907) and solubility (Marshall and Slusher, 1975a,b) studies, which show that
nitric acid becomes an increasingly weak acid at elevated temperature as do all
acids, and that undissociated HNO3 is an important species above about 250 8C.
Values for log10 K1a are reported in Table 13.8. Chlistunoff et al. (1999) have used
UV–visible spectroscopy to determine equilibrium constants for the reactions by
which HNO3(aq) converts to the more reduced species HNO2(aq) and NO(aq) in
supercritical water.
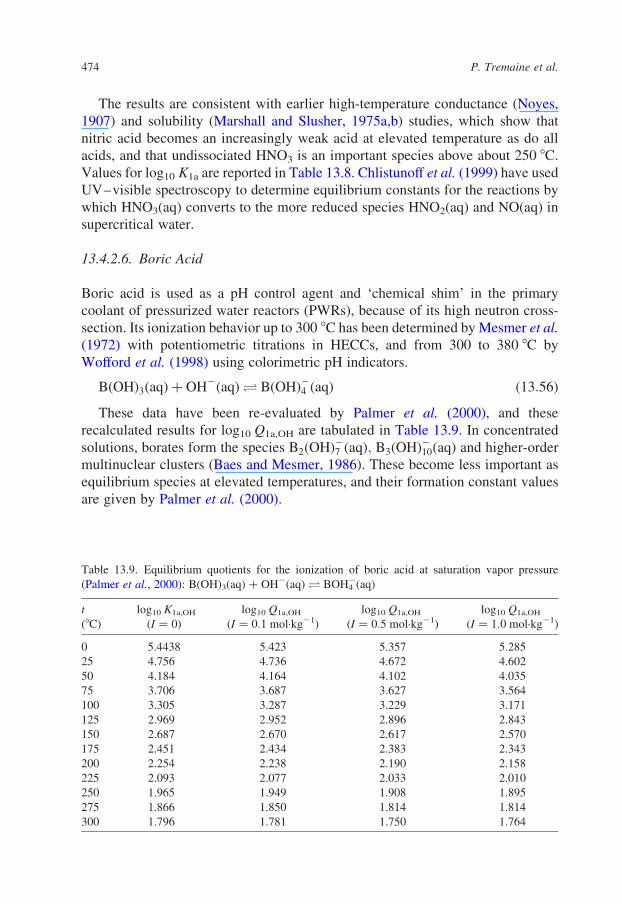
13.4.2.6. Boric Acid
Boric acid is used as a pH control agent and ‘chemical shim’ in the primary
coolant of pressurized water reactors (PWRs), because of its high neutron cross-
section. Its ionization behavior up to 300 8C has been determined by Mesmer et al.
(1972) with potentiometric titrations in HECCs, and from 300 to 380 8C by
Wofford et al. (1998) using colorimetric pH indicators.
BðOHÞ3ðaqÞ þ OH2ðaqÞO BðOHÞ24 ðaqÞ ð13:56Þ
These data have been re-evaluated by Palmer et al. (2000), and these
recalculated results for log10 Q1a,OH are tabulated in Table 13.9. In concentrated
solutions, borates form the species B2ðOHÞ27 ðaqÞ; B3ðOHÞ210ðaqÞ and higher-order
multinuclear clusters (Baes and Mesmer, 1986). These become less important as
equilibrium species at elevated temperatures, and their formation constant values
are given by Palmer et al. (2000).
Table 13.9. Equilibrium quotients for the ionization of boric acid at saturation vapor pressure
(Palmer et al., 2000): B(OH)3(aq) þ OH2(aq) O BOH42(aq)
t
(8C)
log10 K1a,OH
(I ¼ 0)
log10 Q1a,OH
(I ¼ 0:1 mol·kg21)
log10 Q1a,OH
(I ¼ 0:5 mol·kg21)
log10 Q1a,OH
(I ¼ 1:0 mol·kg21)
0 5.4438 5.423 5.357 5.285
25 4.756 4.736 4.672 4.602
50 4.184 4.164 4.102 4.035
75 3.706 3.687 3.627 3.564
100 3.305 3.287 3.229 3.171
125 2.969 2.952 2.896 2.843
150 2.687 2.670 2.617 2.570
175 2.451 2.434 2.383 2.343
200 2.254 2.238 2.190 2.158
225 2.093 2.077 2.033 2.010
250 1.965 1.949 1.908 1.895
275 1.866 1.850 1.814 1.814
300 1.796 1.781 1.750 1.764
P. Tremaine et al.474

13.4.2.7. Silicic Acid
The ionization of silicic acid is one of the most important reactions in geo-
chemistry, as it is responsible for the enhanced solubility of quartz and other
silicate minerals at high pH. Busey and Mesmer (1977) used potentiometric
titrations to determine log10 Q1a,OH for the reaction:
SiðOHÞ4ðaqÞ þ OH2ðaqÞO SiOðOHÞ23 ðaqÞ þ H2OðlÞ ð13:57Þ
The results are listed in Table 13.10 at ionic strengths in NaCl(aq) up to
5.0 mol·kg21. Silicates also form the multimeric clusters in concentrated solutions
(Baes and Mesmer, 1986). Like the polynuclear borates, polysilicates are less
stable at elevated temperatures.
13.4.3. Weak and ‘Almost Strong’ Acids and Bases at Temperaturesto Supercritical Conditions
13.4.3.1. Hydrochloric Acid
Like nitric acid, HCl(aq) becomes a weak acid at elevated temperatures and
pressures. Heat of dilution studies (Holmes et al., 1987) have shown that
the association of hydrochloric acid under subcritical conditions becomes
important at temperatures above about 250 8C. The degree of ionization of
HCl(aq) in the supercritical region has been studied by a number of authors using
conductance methods. Recent high-resolution conductance measurements by Ho
et al. (2001) have resolved earlier discrepancies, and values of 2 log10 K1a for the
association reaction:
HþðaqÞ þ Cl2ðaqÞO HClðaqÞ ð13:58Þ
from this work are presented in Table 13.11. Mesmer et al. (1988, 1991) have used
a treatment based on a fit of the density model to earlier experimental data to
describe the factors that affect the association of HCl(aq) in the sub- and super-
critical regions.
13.4.3.2. Alkali Metal Hydroxides
The ionization properties of the alkali metal hydroxides LiOH, NaOH and KOH
are of much importance because LiOH(aq) is used under subcritical conditions to
control the pH of the primary coolant circuits in nuclear reactors, while NaOH(aq)
and KOH(aq) are common components of geologic fluids that formed under
supercritical conditions. Conductance measurements by a number of groups have
shown that association increases at elevated temperatures for reasons similar to
those for HCl(aq) (Mesmer et al., 1991). Tables 13.12 and 13.13 list values for the
Ionization equilibria of acids and bases 475
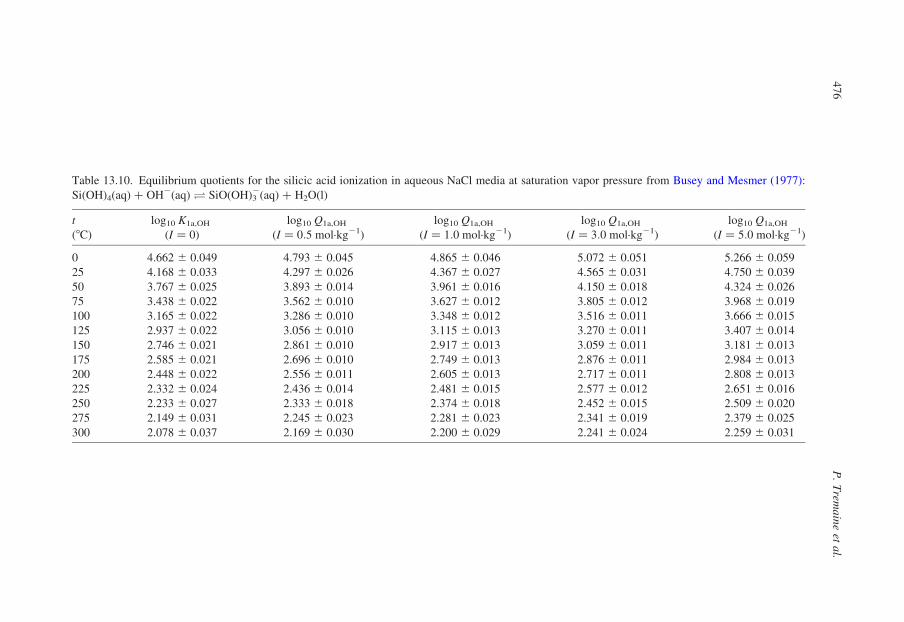
Table 13.10. Equilibrium quotients for the silicic acid ionization in aqueous NaCl media at saturation vapor pressure from Busey and Mesmer (1977):
Si(OH)4(aq) þ OH2(aq) O SiO(OH)32(aq) þ H2O(l)
t
(8C)
log10 K1a,OH
(I ¼ 0)
log10 Q1a,OH
(I ¼ 0:5 mol·kg21)
log10 Q1a,OH
(I ¼ 1:0 mol·kg21)
log10 Q1a,OH
(I ¼ 3:0 mol·kg21)
log10 Q1a,OH
(I ¼ 5:0 mol·kg21)
0 4.662 ^ 0.049 4.793 ^ 0.045 4.865 ^ 0.046 5.072 ^ 0.051 5.266 ^ 0.059
25 4.168 ^ 0.033 4.297 ^ 0.026 4.367 ^ 0.027 4.565 ^ 0.031 4.750 ^ 0.039
50 3.767 ^ 0.025 3.893 ^ 0.014 3.961 ^ 0.016 4.150 ^ 0.018 4.324 ^ 0.026
75 3.438 ^ 0.022 3.562 ^ 0.010 3.627 ^ 0.012 3.805 ^ 0.012 3.968 ^ 0.019
100 3.165 ^ 0.022 3.286 ^ 0.010 3.348 ^ 0.012 3.516 ^ 0.011 3.666 ^ 0.015
125 2.937 ^ 0.022 3.056 ^ 0.010 3.115 ^ 0.013 3.270 ^ 0.011 3.407 ^ 0.014
150 2.746 ^ 0.021 2.861 ^ 0.010 2.917 ^ 0.013 3.059 ^ 0.011 3.181 ^ 0.013
175 2.585 ^ 0.021 2.696 ^ 0.010 2.749 ^ 0.013 2.876 ^ 0.011 2.984 ^ 0.013
200 2.448 ^ 0.022 2.556 ^ 0.011 2.605 ^ 0.013 2.717 ^ 0.011 2.808 ^ 0.013
225 2.332 ^ 0.024 2.436 ^ 0.014 2.481 ^ 0.015 2.577 ^ 0.012 2.651 ^ 0.016
250 2.233 ^ 0.027 2.333 ^ 0.018 2.374 ^ 0.018 2.452 ^ 0.015 2.509 ^ 0.020
275 2.149 ^ 0.031 2.245 ^ 0.023 2.281 ^ 0.023 2.341 ^ 0.019 2.379 ^ 0.025
300 2.078 ^ 0.037 2.169 ^ 0.030 2.200 ^ 0.029 2.241 ^ 0.024 2.259 ^ 0.031
P.
Trem
ain
eet
al.
47
6
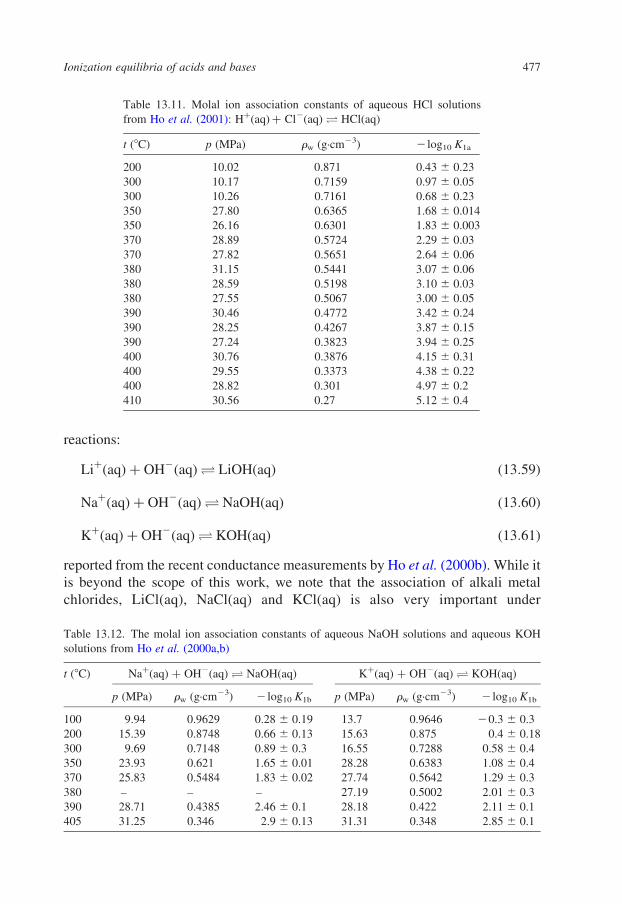
reactions:
LiþðaqÞ þ OH2ðaqÞO LiOHðaqÞ ð13:59Þ
NaþðaqÞ þ OH2ðaqÞO NaOHðaqÞ ð13:60Þ
KþðaqÞ þ OH2ðaqÞO KOHðaqÞ ð13:61Þ
reported from the recent conductance measurements by Ho et al. (2000b). While it
is beyond the scope of this work, we note that the association of alkali metal
chlorides, LiCl(aq), NaCl(aq) and KCl(aq) is also very important under
Table 13.12. The molal ion association constants of aqueous NaOH solutions and aqueous KOH
solutions from Ho et al. (2000a,b)
t (8C) Naþ(aq) þ OH2(aq) O NaOH(aq) Kþ(aq) þ OH2(aq) O KOH(aq)
p (MPa) rw (g·cm23) 2 log10 K1b p (MPa) rw (g·cm23) 2 log10 K1b
100 9.94 0.9629 0.28 ^ 0.19 13.7 0.9646 20.3 ^ 0.3
200 15.39 0.8748 0.66 ^ 0.13 15.63 0.875 0.4 ^ 0.18
300 9.69 0.7148 0.89 ^ 0.3 16.55 0.7288 0.58 ^ 0.4
350 23.93 0.621 1.65 ^ 0.01 28.28 0.6383 1.08 ^ 0.4
370 25.83 0.5484 1.83 ^ 0.02 27.74 0.5642 1.29 ^ 0.3
380 – – – 27.19 0.5002 2.01 ^ 0.3
390 28.71 0.4385 2.46 ^ 0.1 28.18 0.422 2.11 ^ 0.1
405 31.25 0.346 2.9 ^ 0.13 31.31 0.348 2.85 ^ 0.1
Table 13.11. Molal ion association constants of aqueous HCl solutions
from Ho et al. (2001): Hþ(aq)þ Cl2(aq) O HCl(aq)
t (8C) p (MPa) rw (g·cm23) 2 log10 K1a
200 10.02 0.871 0.43 ^ 0.23
300 10.17 0.7159 0.97 ^ 0.05
300 10.26 0.7161 0.68 ^ 0.23
350 27.80 0.6365 1.68 ^ 0.014
350 26.16 0.6301 1.83 ^ 0.003
370 28.89 0.5724 2.29 ^ 0.03
370 27.82 0.5651 2.64 ^ 0.06
380 31.15 0.5441 3.07 ^ 0.06
380 28.59 0.5198 3.10 ^ 0.03
380 27.55 0.5067 3.00 ^ 0.05
390 30.46 0.4772 3.42 ^ 0.24
390 28.25 0.4267 3.87 ^ 0.15
390 27.24 0.3823 3.94 ^ 0.25
400 30.76 0.3876 4.15 ^ 0.31
400 29.55 0.3373 4.38 ^ 0.22
400 28.82 0.301 4.97 ^ 0.2
410 30.56 0.27 5.12 ^ 0.4
Ionization equilibria of acids and bases 477

supercritical conditions and must be considered in speciation calculations
(Mesmer et al., 1991).
13.4.4. Representation of Inorganic Acid–Base Ionization Constantsby the ‘Density’ Model
Many of the ionization constants for inorganic acids and bases have been
represented by fitting an expanded form of the ‘density’ model, Eq. 13.20. Table
13.14 lists parameters for the general equation:
log10 K ¼ q1 þ q2=T þ q3 ln T þ q4T þ q5T2 þ q6=T2
þ ðq7 þ q8T þ q9=TÞ log10 rw ð13:62Þ
as reported in the original papers cited. Here T is the temperature in kelvin and qn
are fitting parameters for each acid. Where fits were not reported we have fitted
Eq. 13.62 to the reported values of log10 K as tabulated.
13.5. Carboxylic Acids and Phenols
13.5.1. Background
Many techniques such as those described earlier have been used to determine
the ionization of aliphatic and aromatic carboxylic acids, and the thermodynamic
properties of the reaction ðDrCp;DrHÞ: It is not the task of this chapter to
Table 13.13. The molal ion association constants of aqueous LiOH
solutions from Ho et al. (2000a,b): Liþ(aq) þ OH2(aq) O LiOH(aq)
t (8C) p (MPa) rw (g·cm23) 2 log10 K1b
50 4.40 0.9899 0.53 ^ 0.17
100 4.88 0.9606 1.03 ^ 0.17
150 9.40 0.9220 1.29 ^ 0.19
200 9.91 0.8709 1.28 ^ 0.1
250 11.02 0.8069 1.41 ^ 0.15
300 9.52 0.7144 1.36 ^ 0.04
300 10.39 0.7164 1.56 ^ 0.1
300 12.87 0.7216 1.60 ^ 0.02
350 23.86 0.6206 2.00 ^ 0.03
350 27.44 0.6352 1.81 ^ 0.07
370 28.79 0.5715 2.49 ^ 0.06
380 29.85 0.5323 2.67 ^ 0.15
390 28.88 0.4433 3.18 ^ 0.03
400 32.49 0.4328 3.10 ^ 0.23
400 30.57 0.3794 3.48 ^ 0.03
410 32.20 0.3273 3.75 ^ 0.2
P. Tremaine et al.478
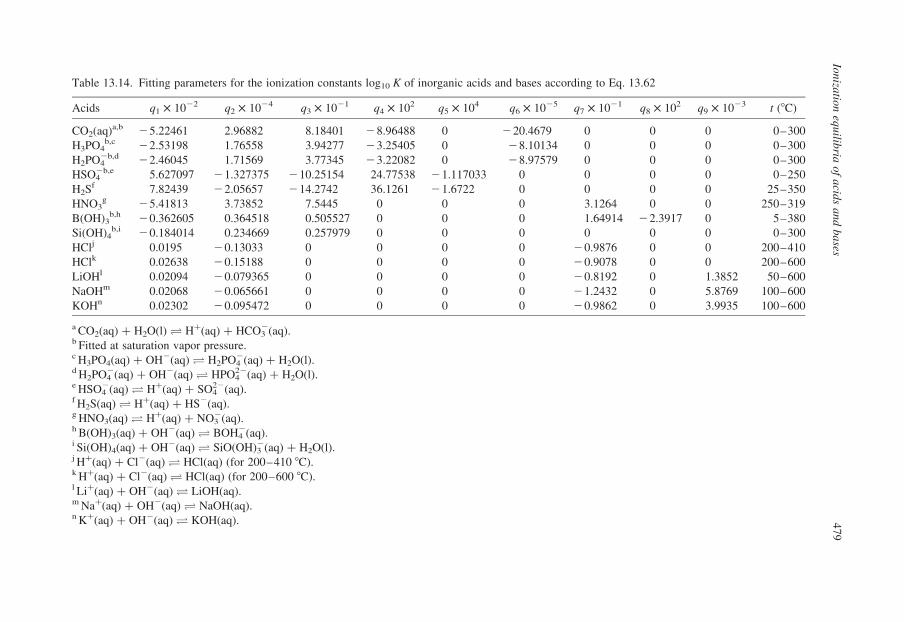
Table 13.14. Fitting parameters for the ionization constants log10 K of inorganic acids and bases according to Eq. 13.62
Acids q1 £ 1022 q2 £ 1024 q3 £ 1021 q4 £ 102 q5 £ 104 q6 £ 1025 q7 £ 1021 q8 £ 102 q9 £ 1023 t (8C)
CO2(aq)a,b 25.22461 2.96882 8.18401 28.96488 0 220.4679 0 0 0 0–300
H3PO4b,c 22.53198 1.76558 3.94277 23.25405 0 28.10134 0 0 0 0–300
H2PO42b,d 22.46045 1.71569 3.77345 23.22082 0 28.97579 0 0 0 0–300
HSO42b,e 5.627097 21.327375 210.25154 24.77538 21.117033 0 0 0 0 0–250
H2Sf 7.82439 22.05657 214.2742 36.1261 21.6722 0 0 0 0 25–350
HNO3g 25.41813 3.73852 7.5445 0 0 0 3.1264 0 0 250–319
B(OH)3b,h 20.362605 0.364518 0.505527 0 0 0 1.64914 22.3917 0 5–380
Si(OH)4b,i 20.184014 0.234669 0.257979 0 0 0 0 0 0 0–300
HClj 0.0195 20.13033 0 0 0 0 20.9876 0 0 200–410
HClk 0.02638 20.15188 0 0 0 0 20.9078 0 0 200–600
LiOHl 0.02094 20.079365 0 0 0 0 20.8192 0 1.3852 50–600
NaOHm 0.02068 20.065661 0 0 0 0 21.2432 0 5.8769 100–600
KOHn 0.02302 20.095472 0 0 0 0 20.9862 0 3.9935 100–600
a CO2(aq) þ H2O(l) O Hþ(aq) þ HCO32(aq).
b Fitted at saturation vapor pressure.c H3PO4(aq) þ OH2(aq) O H2PO4
2(aq) þ H2O(l).d H2PO4
2(aq) þ OH2(aq) O HPO422(aq) þ H2O(l).
e HSO42(aq) O Hþ(aq) þ SO4
22(aq).f H2S(aq) O Hþ(aq) þ HS2(aq).g HNO3(aq) O Hþ(aq) þ NO3
2(aq).h B(OH)3(aq) þ OH2(aq) O BOH4
2(aq).i Si(OH)4(aq) þ OH2(aq) O SiO(OH)3
2(aq) þ H2O(l).j Hþ(aq) þ Cl2(aq) O HCl(aq) (for 200–410 8C).k Hþ(aq) þ Cl2(aq) O HCl(aq) (for 200–600 8C).l Liþ(aq) þ OH2(aq) O LiOH(aq).m Naþ(aq) þ OH2(aq) O NaOH(aq).n Kþ(aq) þ OH2(aq) O KOH(aq).
Ion
izatio
neq
uilib
riao
fa
cids
an
db
ases
47
9
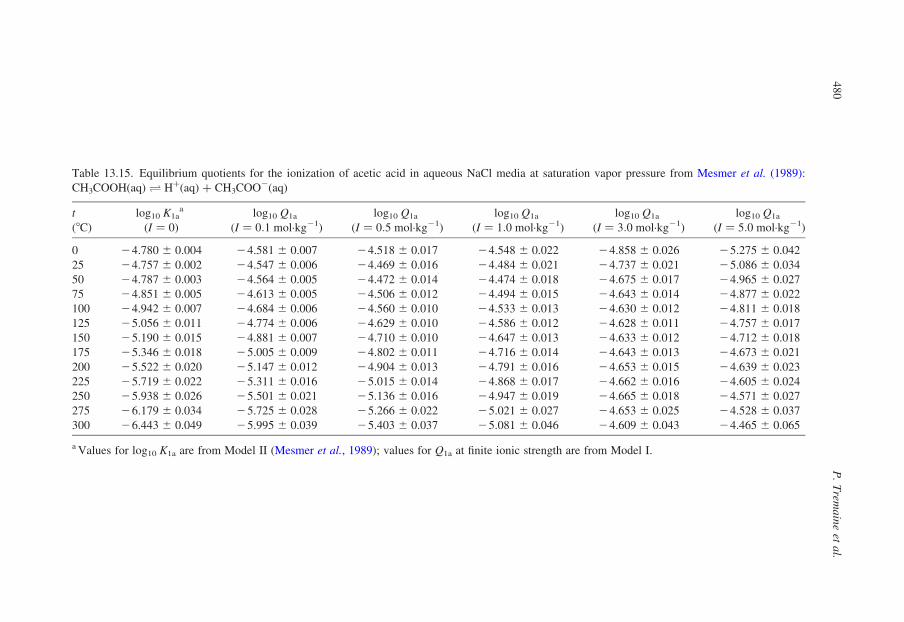
Table 13.15. Equilibrium quotients for the ionization of acetic acid in aqueous NaCl media at saturation vapor pressure from Mesmer et al. (1989):
CH3COOH(aq) O Hþ(aq) þ CH3COO2(aq)
t
(8C)
log10 K1aa
(I ¼ 0)
log10 Q1a
(I ¼ 0:1 mol·kg21)
log10 Q1a
(I ¼ 0:5 mol·kg21)
log10 Q1a
(I ¼ 1:0 mol·kg21)
log10 Q1a
(I ¼ 3:0 mol·kg21)
log10 Q1a
(I ¼ 5:0 mol·kg21)
0 24.780 ^ 0.004 24.581 ^ 0.007 24.518 ^ 0.017 24.548 ^ 0.022 24.858 ^ 0.026 25.275 ^ 0.042
25 24.757 ^ 0.002 24.547 ^ 0.006 24.469 ^ 0.016 24.484 ^ 0.021 24.737 ^ 0.021 25.086 ^ 0.034
50 24.787 ^ 0.003 24.564 ^ 0.005 24.472 ^ 0.014 24.474 ^ 0.018 24.675 ^ 0.017 24.965 ^ 0.027
75 24.851 ^ 0.005 24.613 ^ 0.005 24.506 ^ 0.012 24.494 ^ 0.015 24.643 ^ 0.014 24.877 ^ 0.022
100 24.942 ^ 0.007 24.684 ^ 0.006 24.560 ^ 0.010 24.533 ^ 0.013 24.630 ^ 0.012 24.811 ^ 0.018
125 25.056 ^ 0.011 24.774 ^ 0.006 24.629 ^ 0.010 24.586 ^ 0.012 24.628 ^ 0.011 24.757 ^ 0.017
150 25.190 ^ 0.015 24.881 ^ 0.007 24.710 ^ 0.010 24.647 ^ 0.013 24.633 ^ 0.012 24.712 ^ 0.018
175 25.346 ^ 0.018 25.005 ^ 0.009 24.802 ^ 0.011 24.716 ^ 0.014 24.643 ^ 0.013 24.673 ^ 0.021
200 25.522 ^ 0.020 25.147 ^ 0.012 24.904 ^ 0.013 24.791 ^ 0.016 24.653 ^ 0.015 24.639 ^ 0.023
225 25.719 ^ 0.022 25.311 ^ 0.016 25.015 ^ 0.014 24.868 ^ 0.017 24.662 ^ 0.016 24.605 ^ 0.024
250 25.938 ^ 0.026 25.501 ^ 0.021 25.136 ^ 0.016 24.947 ^ 0.019 24.665 ^ 0.018 24.571 ^ 0.027
275 26.179 ^ 0.034 25.725 ^ 0.028 25.266 ^ 0.022 25.021 ^ 0.027 24.653 ^ 0.025 24.528 ^ 0.037
300 26.443 ^ 0.049 25.995 ^ 0.039 25.403 ^ 0.037 25.081 ^ 0.046 24.609 ^ 0.043 24.465 ^ 0.065
a Values for log10 K1a are from Model II (Mesmer et al., 1989); values for Q1a at finite ionic strength are from Model I.
P.
Trem
ain
eet
al.
48
0

enumerate all the reported experimental data, but rather to give reliable values
of the dissociation constants and related thermodynamic properties of major
carboxylic acids at elevated temperatures. Experimental values above 100 8C
have been reported for several aliphatic carboxylic acids (acetic, formic, oxalic,
malonic, succinic and citric acid) and one aromatic acid (benzoic). Most of these
data were measured in the last decade at Oak Ridge National Laboratory by using
the HECC. In each case presented here, the fits to their data and the equations
generated for log10 K included other critically evaluated thermodynamic data
(such as DrCop; DrH
o, etc.). Details are described in the original literature. The
values are reported in Tables 13.15–13.17 at saturation vapor pressure, as a
function of temperature. The ionic strength corresponds to infinite dilution, except
for acetic acid and formic acid, which are listed at several ionic strengths. These
data were generated from fits of the general Eq. 13.62 to the experimental data,
with values of the fitting parameters reported in Table 13.18. None of the
measurements cited in Table 13.18 extended to temperatures high enough to fit
statistically significant values for the ‘density’ terms q7, q8 and q9. We also note
the results of Ellis (1963), who measured the acid dissociation constants for acetic,
propionic, n-butyric and benzoic acids using conductance methods, at tempera-
tures up to 225 8C.
13.5.1.1. Acetic and Formic Acids
Acetic and formic acids are the most thoroughly studied carboxylic acids at
elevated temperatures, both because of their simplicity and thermal stability and
because of their importance to the electric power industry as corrosive trace
contaminants in boiler water. For acetic acid, the results from Mesmer et al. (1989)
represent the first potentiometric study of an organic acid at high temperatures.
The ionization constants of the reaction:
CH3COOHðaqÞO CH3COO2ðaqÞ þ HþðaqÞ ð13:63Þ
were carefully determined in NaCl media to 5 mol·kg21 from 50 to 250 8C by
potentiometry. The values reported in Table 13.15 were generated from their
Table 13.16. Equilibrium quotients for the dissociation of formic acid in aqueous NaCl media at
saturation vapor pressure from Bell et al. (1993): HCOOH(aq) O Hþ(aq) þ HCOO2(aq)
t (8C) log10 K1a
(I ¼ 0)
log10 Q1a
(I ¼ 0:1 mol·kg21)
log10 Q1a
(I ¼ 0:5 mol·kg21)
log10 Q1a
(I ¼ 1:0 mol·kg21)
log10 Q1a
(I ¼ 5:0 mol·kg21)
25 23.755 ^ 0.002 23.544 ^ 0.003 23.465 ^ 0.007 23.480 ^ 0.008 23.980 ^ 0.015
50 23.781 ^ 0.003 23.556 ^ 0.003 23.457 ^ 0.007 23.461 ^ 0.008 23.879 ^ 0.011
75 23.849 ^ 0.003 23.607 ^ 0.003 23.485 ^ 0.006 23.477 ^ 0.007 23.809 ^ 0.009
100 23.951 ^ 0.005 23.690 ^ 0.004 23.554 ^ 0.005 23.521 ^ 0.006 23.764 ^ 0.009
150 24.233 ^ 0.011 23.922 ^ 0.009 23.735 ^ 0.007 23.664 ^ 0.007 23.714 ^ 0.015
200 24.591 ^ 0.019 24.215 ^ 0.018 23.962 ^ 0.015 23.846 ^ 0.014 23.683 ^ 0.024
Ionization equilibria of acids and bases 481
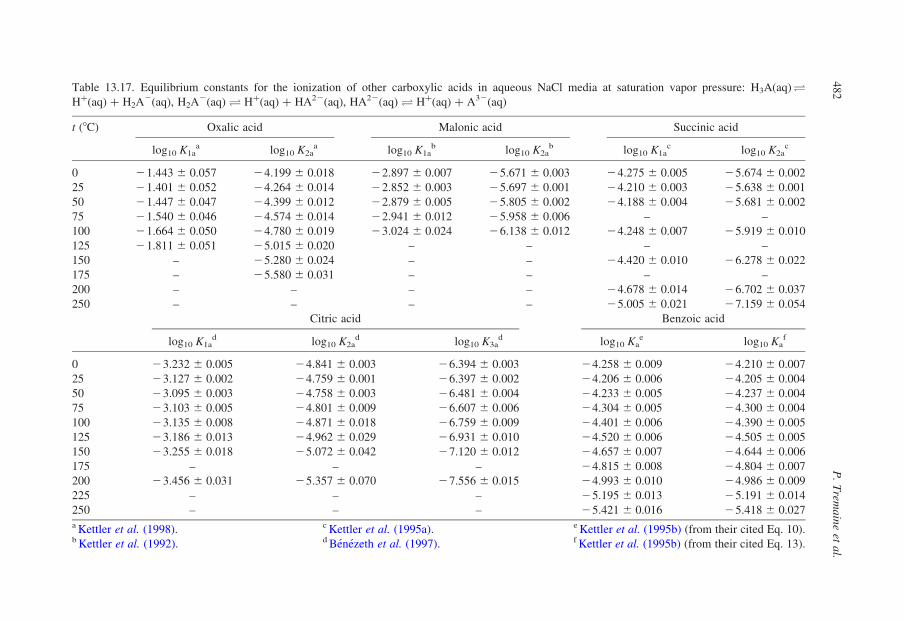
Table 13.17. Equilibrium constants for the ionization of other carboxylic acids in aqueous NaCl media at saturation vapor pressure: H3A(aq)O
Hþ(aq) þ H2A2(aq), H2A2(aq) O Hþ(aq) þ HA22(aq), HA22(aq) O Hþ(aq) þ A32(aq)
t (8C) Oxalic acid Malonic acid Succinic acid
log10 K1aa log10 K2a
a log10 K1ab log10 K2a
b log10 K1ac log10 K2a
c
0 21.443 ^ 0.057 24.199 ^ 0.018 22.897 ^ 0.007 25.671 ^ 0.003 24.275 ^ 0.005 25.674 ^ 0.002
25 21.401 ^ 0.052 24.264 ^ 0.014 22.852 ^ 0.003 25.697 ^ 0.001 24.210 ^ 0.003 25.638 ^ 0.001
50 21.447 ^ 0.047 24.399 ^ 0.012 22.879 ^ 0.005 25.805 ^ 0.002 24.188 ^ 0.004 25.681 ^ 0.002
75 21.540 ^ 0.046 24.574 ^ 0.014 22.941 ^ 0.012 25.958 ^ 0.006 – –
100 21.664 ^ 0.050 24.780 ^ 0.019 23.024 ^ 0.024 26.138 ^ 0.012 24.248 ^ 0.007 25.919 ^ 0.010
125 21.811 ^ 0.051 25.015 ^ 0.020 – – – –
150 – 25.280 ^ 0.024 – – 24.420 ^ 0.010 26.278 ^ 0.022
175 – 25.580 ^ 0.031 – – – –
200 – – – – 24.678 ^ 0.014 26.702 ^ 0.037
250 – – – – 25.005 ^ 0.021 27.159 ^ 0.054
Citric acid Benzoic acid
log10 K1ad log10 K2a
d log10 K3ad log10 Ka
e log10 Kaf
0 23.232 ^ 0.005 24.841 ^ 0.003 26.394 ^ 0.003 24.258 ^ 0.009 24.210 ^ 0.007
25 23.127 ^ 0.002 24.759 ^ 0.001 26.397 ^ 0.002 24.206 ^ 0.006 24.205 ^ 0.004
50 23.095 ^ 0.003 24.758 ^ 0.003 26.481 ^ 0.004 24.233 ^ 0.005 24.237 ^ 0.004
75 23.103 ^ 0.005 24.801 ^ 0.009 26.607 ^ 0.006 24.304 ^ 0.005 24.300 ^ 0.004
100 23.135 ^ 0.008 24.871 ^ 0.018 26.759 ^ 0.009 24.401 ^ 0.006 24.390 ^ 0.005
125 23.186 ^ 0.013 24.962 ^ 0.029 26.931 ^ 0.010 24.520 ^ 0.006 24.505 ^ 0.005
150 23.255 ^ 0.018 25.072 ^ 0.042 27.120 ^ 0.012 24.657 ^ 0.007 24.644 ^ 0.006
175 – – – 24.815 ^ 0.008 24.804 ^ 0.007
200 23.456 ^ 0.031 25.357 ^ 0.070 27.556 ^ 0.015 24.993 ^ 0.010 24.986 ^ 0.009
225 – – – 25.195 ^ 0.013 25.191 ^ 0.014
250 – – – 25.421 ^ 0.016 25.418 ^ 0.027a Kettler et al. (1998).b Kettler et al. (1992).
c Kettler et al. (1995a).d Benezeth et al. (1997).
e Kettler et al. (1995b) (from their cited Eq. 10).f Kettler et al. (1995b) (from their cited Eq. 13).
P.
Trem
ain
eet
al.
48
2
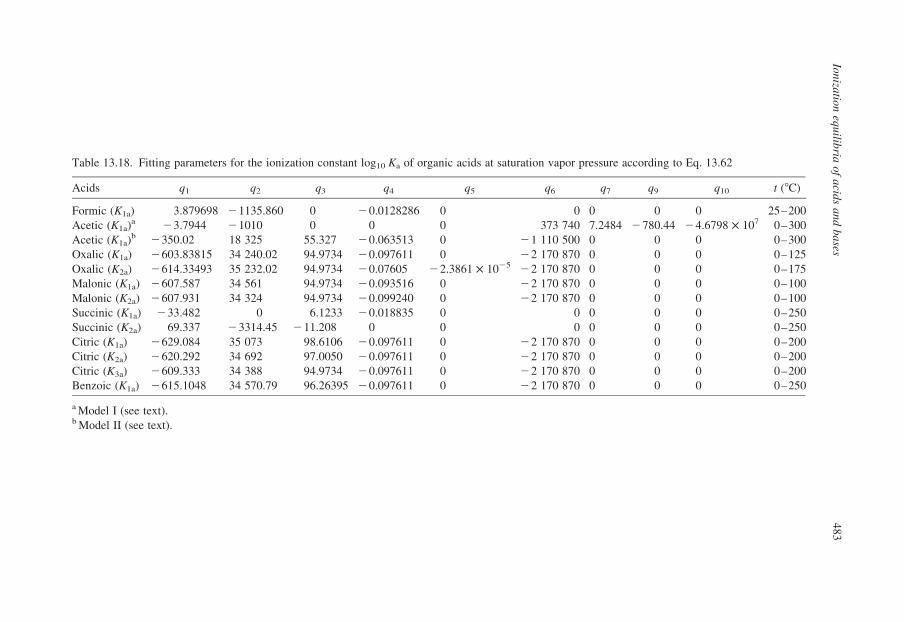
Table 13.18. Fitting parameters for the ionization constant log10 Ka of organic acids at saturation vapor pressure according to Eq. 13.62
Acids q1 q2 q3 q4 q5 q6 q7 q9 q10 t (8C)
Formic (K1a) 3.879698 21135.860 0 20.0128286 0 0 0 0 0 25–200
Acetic (K1a)a 23.7944 21010 0 0 0 373 740 7.2484 2780.44 24.6798 £ 107 0–300
Acetic (K1a)b 2350.02 18 325 55.327 20.063513 0 21 110 500 0 0 0 0–300
Oxalic (K1a) 2603.83815 34 240.02 94.9734 20.097611 0 22 170 870 0 0 0 0–125
Oxalic (K2a) 2614.33493 35 232.02 94.9734 20.07605 22.3861 £ 1025 22 170 870 0 0 0 0–175
Malonic (K1a) 2607.587 34 561 94.9734 20.093516 0 22 170 870 0 0 0 0–100
Malonic (K2a) 2607.931 34 324 94.9734 20.099240 0 22 170 870 0 0 0 0–100
Succinic (K1a) 233.482 0 6.1233 20.018835 0 0 0 0 0 0–250
Succinic (K2a) 69.337 23314.45 211.208 0 0 0 0 0 0 0–250
Citric (K1a) 2629.084 35 073 98.6106 20.097611 0 22 170 870 0 0 0 0–200
Citric (K2a) 2620.292 34 692 97.0050 20.097611 0 22 170 870 0 0 0 0–200
Citric (K3a) 2609.333 34 388 94.9734 20.097611 0 22 170 870 0 0 0 0–200
Benzoic (K1a) 2615.1048 34 570.79 96.26395 20.097611 0 22 170 870 0 0 0 0–250
a Model I (see text).b Model II (see text).
Ion
izatio
neq
uilib
riao
fa
cids
an
db
ases
48
3

‘Model I’, which is the following version of the density model:
log10 K1a ¼ q1 þ q2=T þ q6=T2 þ q10=T3 þ ðq7 þ q9=TÞ ln rw ð13:64Þ
These authors also gave a second model, Model II, based on the expression of
DCop of the form a1 þ a2T þ a3=T2 which is described in the original paper. The
values reported in Table 13.15 at infinite dilution were generated from
log10 K1a ¼ q1 þ q2=T þ q3 ln T þ q4T þ q6=T2 ð13:65Þ
Excellent fits of the results of Mesmer et al. (1989) were obtained with both
models.
The acid dissociation constants for the ionization of formic acid:
HCOOHðaqÞO HþðaqÞ þ HCOO2ðaqÞ ð13:66Þ
were measured by Bell et al. (1993) using the same approach as for acetic acid,
described above. The molal equilibrium quotients and thermodynamic properties
obtained by these authors are reported in Table 13.16 as a function of temperature
and ionic strength. The infinite dilution constants are generated from Eq. 13.62
together with the parameters reported in Table 13.18.
13.5.1.2. Polybasic Carboxylic acids and Benzoic Acid
The ionization of several polybasic carboxylic acids have been measured at
elevated temperatures. These acids are generally less stable than monobasic acids
at elevated temperatures and ionize according to the stepwise equations:
H3AðaqÞO HþðaqÞ þ H2A2ðaqÞ K1a ð13:67Þ
H2A2ðaqÞO HþðaqÞ þ HA22ðaqÞ K2a ð13:68Þ
HA22ðaqÞO HþðaqÞ þ A32ðaqÞ K3a ð13:69Þ
The ionization equilibria of oxalic, malonic, succinic and citric acid have been
studied by Kettler et al. (1992, 1995a, 1998) and Benezeth et al. (1997). Values for
log10 Ka at infinite dilution are reported in Table 13.17. The fitting parameters used
to reproduce the experimental results are listed in Table 13.18. Although the
density model was used for the fit, the density term, q9, which dominates at high
temperature, was often set to zero because the data did not extend to high enough
temperatures for this term to be significant.
Measurements on benzoic acid ionization have been made by emf, conductance
and UV–visible spectrophotometric methods. Benzoic acid contains both a phenyl
and aryl carboxylic acid group. The carboxylic acid group ionizes according to the
reaction:
C6H5COOHðaqÞO C6H5COO2ðaqÞ þ HþðaqÞ ð13:70Þ
P. Tremaine et al.484

Equilibrium constants have been measured by Kettler et al. (1995b) using
potentiometric titration methods. Values of log10 Ka and fitting parameters are
listed in Tables 13.17 and 13.18, respectively.
13.6. Amines and Alkanol Amines
13.6.1. Effects of Amine Structure on Ionization and the Formationof Carbamates
Amines and alkanolamines have seen wide application in the energy industry as
‘all volatile’ additives for boiler-water pH control and as ‘chemical solvents’ for
removing carbon dioxide and other acid gases from natural gas and combustion
gases. Boiler pH control applications typically involve dilute solutions of a few
parts per million at temperatures from 25 to 350 8C, while gas treatment
applications require concentrated solutions in excess of 1 mol·kg21 at tempera-
tures from 25 to 150 8C.
Factors influencing the ionization constants of amines have been reviewed by
Jones and Arnett (1974). Briefly, amines and alkanolamines consist of three alkyl,
alkanol or hydrogen substituent groups covalently bound to a central nitrogen
atom. Together with the unbonded lone pair of valence electrons, these groups give
the amine a tetrahedral structure. The basicity of the amine is controlled by the
availability of the valence band electrons for chemical bonding to Hþ(aq). Electron
donor substituent groups enhance the basicity and increase Kb, while electron-
withdrawing groups reduce the magnitude of Kb. Steric effects (i.e., crowding
effects) due to interactions among the three substituent groups may also suppress
the value of Kb. This is particularly true for tertiary amines. Steric effects are also
affected by hydration. Because of these competing factors, the relative base
strengths of primary, secondary and tertiary amines at 25 8C follow the sequence:
NH3ðaqÞ , RNH2ðaqÞ , RR0NHðaqÞ . RR0R00NðaqÞ
In addition to the ionization reaction:
RR0R00NðaqÞ þ HþðaqÞO ½RR0R00NH�þðaqÞ ð13:71Þ
ammonia, primary and secondary amines can also react with carbon dioxide to
form carbamic acid according to the reaction:
H2CO3ðaqÞ þ RR0NHðaqÞO RR0NCOO2ðaqÞ þ HþðaqÞ þ H2OðlÞ ð13:72Þ
Carbamate formation, which is a reversible reaction, does not occur in the dilute
solutions used in boiler treatment applications, and is less favored at elevated
temperatures (Roberts and Tremaine, 1985). Indeed, the decreasing stability with
increasing temperature allows carbamate formation to be used in reversible cyclic
Ionization equilibria of acids and bases 485

processes to trap CO2 (Astarita et al., 1983). The use of sterically hindered
secondary amines with bulky alkyl and alkanol substituents can prevent carbamate
formation in processes where it is detrimental.
13.6.1.1. Ammonia
Ammonia is an extremely important base in hydrothermal technology, because
of its high solubility, volatility, pH buffer properties and ability to form complexes
with metal ions. For example, it is among the most wisely used bases in
‘all-volatile’ treatments of boiler water (‘AVT’), and is also present in boiler water
as a breakdown product of hydrazine (which is added as a reducing agent).
Ammonia’s properties as a complexing agent form the basis of the Sherritt–
Gordon hydrometallurgical process, in which copper, nickel and cobalt are
extracted directly from crushed ore in large zircalloy pressure vessels, as metal–
amine complexes.
The ionization of ammonia has been studied by a number of authors, beginning
with the pioneering work of Noyes (1907). Conductance measurements by Quist
and Marshall (1968b) extended the data up to 700 8C. Hitch and Mesmer (1976)
used a flow-cell potentiometric titration technique to determine log10 Q1b vs. ionic
strength at temperatures up to 300 8C. The potentiometric values measured for the
neutralization reaction:
NH3ðaqÞ þ H2OðlÞO NHþ4 ðaqÞ þ OH2ðaqÞ ð13:73Þ
by Hitch and Mesmer (1976) are summarized in Table 13.19. Values for the
variation of log10 K1b with temperature and pressure have been used by Mesmer
et al. (1988, 1991) to derive parameters for the density model to describe ammonia
ionization at temperatures up to 800 8C.
13.6.1.2. Amines and Alkanolamines
In addition to ammonia, morpholine, cyclohexylamine, ethanolamine and
dimethylamine are the most widely used amines for ‘AVT’ boiler-water additives
by the electric power industry, particularly nuclear plants. Thermodynamic
constants for all three systems under boiler conditions have been determined by
potentiometric titrations (Mesmer and Hitch, 1977; Benezeth et al., 2001) and from
estimates based on experimental values of V o (Tremaine et al., 1997; Shvedov and
Tremaine, 1997). Values for log10 Q1b,H for the neutralization reactions:
R2NHðaqÞ þ HþðaqÞO R2NHþ2 ðaqÞ ð13:74Þ
are listed in Table 13.20.
The use of amines to control the pH of boiler water is designed to minimize
the corrosion of carbon steel components of the boiler and feed-train before and
after boiling. As a result, there is much interest in balancing the effects of
P. Tremaine et al.486
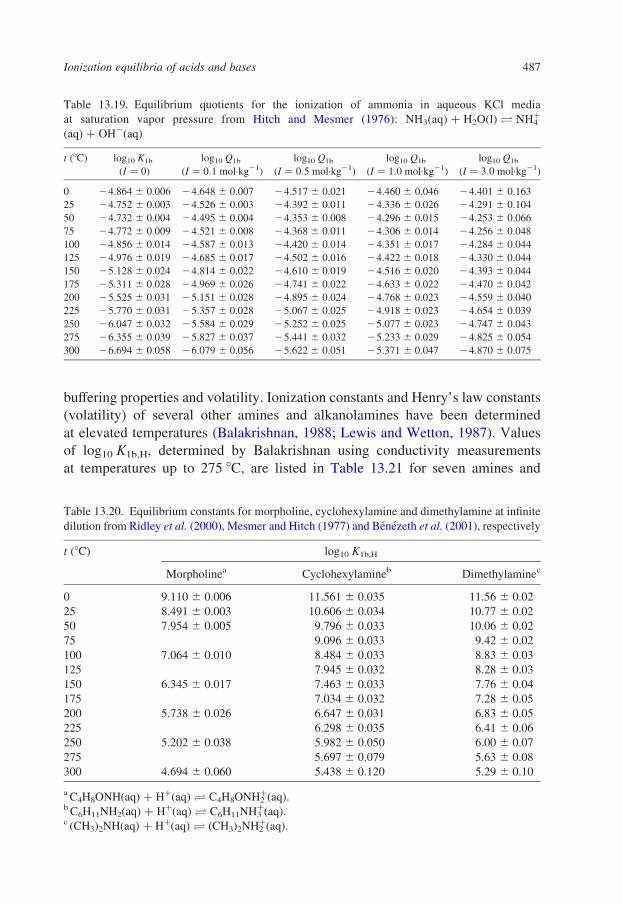
buffering properties and volatility. Ionization constants and Henry’s law constants
(volatility) of several other amines and alkanolamines have been determined
at elevated temperatures (Balakrishnan, 1988; Lewis and Wetton, 1987). Values
of log10 K1b,H, determined by Balakrishnan using conductivity measurements
at temperatures up to 275 8C, are listed in Table 13.21 for seven amines and
Table 13.19. Equilibrium quotients for the ionization of ammonia in aqueous KCl media
at saturation vapor pressure from Hitch and Mesmer (1976): NH3(aq) þ H2O(l) O NH4þ
(aq) þ OH2(aq)
t (8C) log10 K1b
(I ¼ 0)
log10 Q1b
(I ¼ 0:1 mol·kg21)
log10 Q1b
(I ¼ 0:5 mol·kg21)
log10 Q1b
(I ¼ 1:0 mol·kg21)
log10 Q1b
(I ¼ 3:0 mol·kg21)
0 24.864 ^ 0.006 24.648 ^ 0.007 24.517 ^ 0.021 24.460 ^ 0.046 24.401 ^ 0.163
25 24.752 ^ 0.003 24.526 ^ 0.003 24.392 ^ 0.011 24.336 ^ 0.026 24.291 ^ 0.104
50 24.732 ^ 0.004 24.495 ^ 0.004 24.353 ^ 0.008 24.296 ^ 0.015 24.253 ^ 0.066
75 24.772 ^ 0.009 24.521 ^ 0.008 24.368 ^ 0.011 24.306 ^ 0.014 24.256 ^ 0.048
100 24.856 ^ 0.014 24.587 ^ 0.013 24.420 ^ 0.014 24.351 ^ 0.017 24.284 ^ 0.044
125 24.976 ^ 0.019 24.685 ^ 0.017 24.502 ^ 0.016 24.422 ^ 0.018 24.330 ^ 0.044
150 25.128 ^ 0.024 24.814 ^ 0.022 24.610 ^ 0.019 24.516 ^ 0.020 24.393 ^ 0.044
175 25.311 ^ 0.028 24.969 ^ 0.026 24.741 ^ 0.022 24.633 ^ 0.022 24.470 ^ 0.042
200 25.525 ^ 0.031 25.151 ^ 0.028 24.895 ^ 0.024 24.768 ^ 0.023 24.559 ^ 0.040
225 25.770 ^ 0.031 25.357 ^ 0.028 25.067 ^ 0.025 24.918 ^ 0.023 24.654 ^ 0.039
250 26.047 ^ 0.032 25.584 ^ 0.029 25.252 ^ 0.025 25.077 ^ 0.023 24.747 ^ 0.043
275 26.355 ^ 0.039 25.827 ^ 0.037 25.441 ^ 0.032 25.233 ^ 0.029 24.825 ^ 0.054
300 26.694 ^ 0.058 26.079 ^ 0.056 25.622 ^ 0.051 25.371 ^ 0.047 24.870 ^ 0.075
Table 13.20. Equilibrium constants for morpholine, cyclohexylamine and dimethylamine at infinite
dilution from Ridley et al. (2000), Mesmer and Hitch (1977) and Benezeth et al. (2001), respectively
t (8C) log10 K1b,H
Morpholinea Cyclohexylamineb Dimethylaminec
0 9.110 ^ 0.006 11.561 ^ 0.035 11.56 ^ 0.02
25 8.491 ^ 0.003 10.606 ^ 0.034 10.77 ^ 0.02
50 7.954 ^ 0.005 9.796 ^ 0.033 10.06 ^ 0.02
75 9.096 ^ 0.033 9.42 ^ 0.02
100 7.064 ^ 0.010 8.484 ^ 0.033 8.83 ^ 0.03
125 7.945 ^ 0.032 8.28 ^ 0.03
150 6.345 ^ 0.017 7.463 ^ 0.033 7.76 ^ 0.04
175 7.034 ^ 0.032 7.28 ^ 0.05
200 5.738 ^ 0.026 6.647 ^ 0.031 6.83 ^ 0.05
225 6.298 ^ 0.035 6.41 ^ 0.06
250 5.202 ^ 0.038 5.982 ^ 0.050 6.00 ^ 0.07
275 5.697 ^ 0.079 5.63 ^ 0.08
300 4.694 ^ 0.060 5.438 ^ 0.120 5.29 ^ 0.10
a C4H8ONH(aq) þ Hþ(aq) O C4H8ONH2þ(aq).
b C6H11NH2(aq) þ Hþ(aq) O C6H11NH3þ(aq).
c (CH3)2NH(aq) þ Hþ(aq) O (CH3)2NH2þ(aq).
Ionization equilibria of acids and bases 487
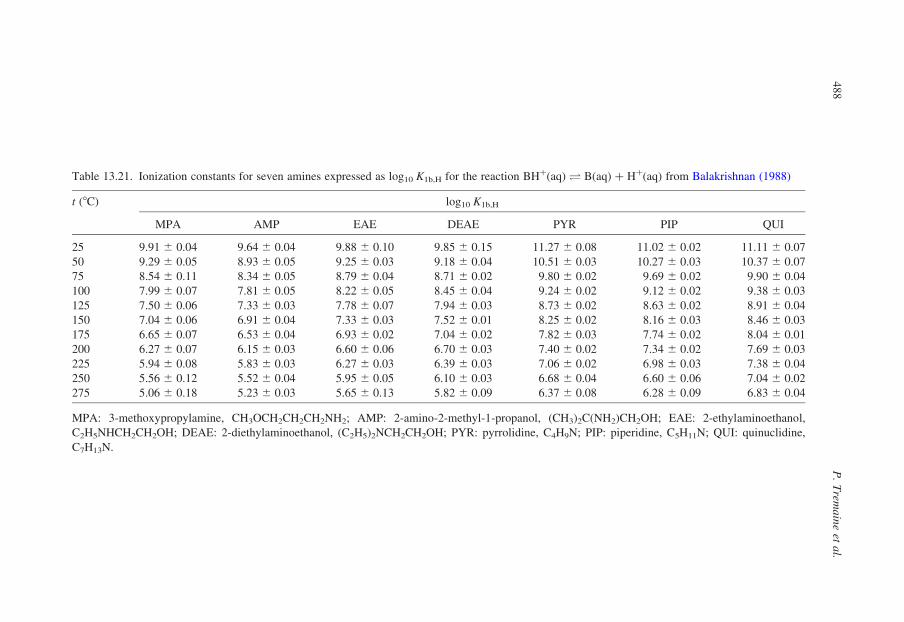
Table 13.21. Ionization constants for seven amines expressed as log10 K1b,H for the reaction BHþ(aq) O B(aq) þ Hþ(aq) from Balakrishnan (1988)
t (8C) log10 K1b,H
MPA AMP EAE DEAE PYR PIP QUI
25 9.91 ^ 0.04 9.64 ^ 0.04 9.88 ^ 0.10 9.85 ^ 0.15 11.27 ^ 0.08 11.02 ^ 0.02 11.11 ^ 0.07
50 9.29 ^ 0.05 8.93 ^ 0.05 9.25 ^ 0.03 9.18 ^ 0.04 10.51 ^ 0.03 10.27 ^ 0.03 10.37 ^ 0.07
75 8.54 ^ 0.11 8.34 ^ 0.05 8.79 ^ 0.04 8.71 ^ 0.02 9.80 ^ 0.02 9.69 ^ 0.02 9.90 ^ 0.04
100 7.99 ^ 0.07 7.81 ^ 0.05 8.22 ^ 0.05 8.45 ^ 0.04 9.24 ^ 0.02 9.12 ^ 0.02 9.38 ^ 0.03
125 7.50 ^ 0.06 7.33 ^ 0.03 7.78 ^ 0.07 7.94 ^ 0.03 8.73 ^ 0.02 8.63 ^ 0.02 8.91 ^ 0.04
150 7.04 ^ 0.06 6.91 ^ 0.04 7.33 ^ 0.03 7.52 ^ 0.01 8.25 ^ 0.02 8.16 ^ 0.03 8.46 ^ 0.03
175 6.65 ^ 0.07 6.53 ^ 0.04 6.93 ^ 0.02 7.04 ^ 0.02 7.82 ^ 0.03 7.74 ^ 0.02 8.04 ^ 0.01
200 6.27 ^ 0.07 6.15 ^ 0.03 6.60 ^ 0.06 6.70 ^ 0.03 7.40 ^ 0.02 7.34 ^ 0.02 7.69 ^ 0.03
225 5.94 ^ 0.08 5.83 ^ 0.03 6.27 ^ 0.03 6.39 ^ 0.03 7.06 ^ 0.02 6.98 ^ 0.03 7.38 ^ 0.04
250 5.56 ^ 0.12 5.52 ^ 0.04 5.95 ^ 0.05 6.10 ^ 0.03 6.68 ^ 0.04 6.60 ^ 0.06 7.04 ^ 0.02
275 5.06 ^ 0.18 5.23 ^ 0.03 5.65 ^ 0.13 5.82 ^ 0.09 6.37 ^ 0.08 6.28 ^ 0.09 6.83 ^ 0.04
MPA: 3-methoxypropylamine, CH3OCH2CH2CH2NH2; AMP: 2-amino-2-methyl-1-propanol, (CH3)2C(NH2)CH2OH; EAE: 2-ethylaminoethanol,
C2H5NHCH2CH2OH; DEAE: 2-diethylaminoethanol, (C2H5)2NCH2CH2OH; PYR: pyrrolidine, C4H9N; PIP: piperidine, C5H11N; QUI: quinuclidine,
C7H13N.
P.
Trem
ain
eet
al.
48
8

alkanolamines: 3-methoxypropylamine (MPA), 2-amino-2-methyl-1-propanol
(AMP), 2-ethylaminoethanol (EAE), 2-diethylaminoethanol (DEAE), pyrrolidine,
piperidine and quinuclidine.
13.7. Other Sources of Data
Values for the ionization constants of many acids and bases at 25 8C are listed in
compilations by Pettit and Powell (1997), Smith and Martell (1997), Christensen
et al. (1976) and Oscarson et al. (1992). Some high-temperature values are
included. Proprietary databases of high-temperature ionization constants are
maintained and developed for electric power industry applications by EPRI
(Alexander and Liu, 1989) and for more general chemical industry applications by
OLI Systems Inc. (Anderko, 1995).
A large database and computer code for calculating the thermodynamic
properties of inorganic ions and their reactions at elevated temperature and pressure
is incorporated in the public database SUPCRT92 (Johnson et al., 1992). This is
based on the HKF equation of state, and primarily on experimental data available
before the late 1980s. Many of the standard partial molar heat capacity and volume
data at that time were limited to temperatures below 50 8C, and the HKF
extrapolations are not always in agreement with later experiments. The papers on
which the program is based (Shock and Helgeson, 1988) are a good source of
literature related to experimental studies. The HKF equation of state has also been
used to predict the ionization and complexation constants of many organic species
under hydrothermal conditions (Shock and Helgeson, 1990; Shock et al., 1992;
Shock and Koretsky, 1993, 1995; Amend and Helgeson, 1997). Because the HKF
model has no theoretical basis for neutral species, high-temperature values of
log10 K should be treated with caution unless the results have been obtained by
direct fitting of high-temperature experimental data for the species in question.
These papers are exhaustively referenced and are a valuable guide to the literature.
Finally, a number of authors have reported functional-group additivity models
for organic species from which heat capacity and volume functions can be
calculated. We particularly note the treatments by Sedlbauer et al. (2000) and
Plyasunov et al. (2000a,b), which appear to provide reliable extrapolations of
log10 K for many reactions up to about 250 8C.
References
Alexander, J.H. and Liu, L., Users Manual, Vol. I. EPRI Report NP-5561-CCM, 1989.
Amend, J.P. and Helgeson, H.C., J. Chem. Soc., Faraday Trans., 93, 1927–1941 (1997).
Anderko, A., 18th OLI Users Conference, Morristown, NJ, October 10, 1995.
Anderson, G.M., Castet, S., Schott, J. and Mesmer, R.E., Geochim. Cosmochim. Acta, 55,
1769–1779 (1991).
Ionization equilibria of acids and bases 489

Angell, C.A., Annu. Rev. Phys. Chem., 34, 593–630 (1983).
Archer, D.G., J. Phys. Chem. Ref. Data, 21, 793–823 (1992).
Arnett, E.M. and McKelvey, D.R. In: Coetzee, J.F. and Ritchie, C.D. (Eds.), Solute–Solvent
Interactions. Marcel Dekker, New York, 1969.
Astarita, G., Savage, D.W. and Bisio, A., Gas Treating with Chemical Solvents. Wiley, New York,
1983.
Baes, C.F. Jr. and Mesmer, R.E., The Hydrolysis of Cations. Robert E. Krieger Publishing, Malibar,
FL, 1986, Reprint with corrections of the 1976 edition by Wiley.
Balakrishnan, P.V., J. Solution Chem., 17, 825–840 (1988).
Bassett, W.A., Shen, A.H. and Bucknum, M., Rev. Sci. Instrum., 64, 2340–2345 (1993).
Bell, J.L.S., Wesolowski, D.J. and Palmer, D.A., J. Solution Chem., 22, 125–136 (1993).
Benezeth, P., Palmer, D.A. and Wesolowski, D.J., J. Solution Chem., 26, 63–84 (1997).
Benezeth, P., Palmer, D.A. and Wesolowski, D.J., J. Chem. Eng. Data, 46, 202–207 (2001).
Busey, R.H. and Mesmer, R.E., Inorg. Chem., 16, 2444–2450 (1977).
Busey, R.H. and Mesmer, R.E., J. Chem. Eng. Data, 23, 175–176 (1978).
Chlistunoff, J., Ziegler, K.J., Lasdon, L. and Johnston, K.P., J. Phys. Chem. A, 103, 1678–1688
(1999).
Christensen, J.J., Hansen, L.D. and Izatt, R.M., Handbook of Proton Ionization Heats and Related
Thermodynamic Quantities. Wiley, New York, 1976.
Clarke, R.G., Hnedkovsky, L., Tremaine, P.R. and Majer, V., J. Phys. Chem. B, 104, 11781–11793
(2000).
Cotton, F.A. and Wilkinson, G., Advanced Inorganic Chemistry, 5th edn. Wiley-Interscience,
New York, 1988.
Diakonov, I., Pokrovski, G., Schott, J., Castet, S. and Gout, R., Geochim. Cosmochim. Acta, 60,
197–211 (1996).
Dickson, A.G., Wesolowski, D.J., Palmer, D.A. and Mesmer, R.E., J. Phys. Chem., 94, 7978–7985
(1990).
Ding, K. and Seyfried, W.E. Jr., Geochim. Cosmochim. Acta, 59, 4769–4773 (1995).
Ding, K. and Seyfried, W.E. Jr., J. Solution Chem., 25, 421–433 (1996).
Ellis, A.J., J. Chem. Soc., 2299–2310 (1963).
Fernandez-Prini, R., Trans. Faraday Soc., 65, 3311–3313 (1969).
Fernandez-Prini, R., Corti, H.R. and Japas, M.L., High-Temperature Aqueous Solutions:
Thermodynamic Properties. CRC Press, Boca Raton, FL, 1992.
Franck, E.U., Z. Phys. Chem., 8, 107–126 (1956).
Franck, E.U., Angew. Chem., 73, 309–322 (1961).
Franck, E.U., Savolainen, J.E. and Marshall, W.L., Rev. Sci. Instrum., 33, 115–117 (1962).
Giggenbach, W., Inorg. Chem., 10, 1333–1338 (1971).
Gruszkiewicz, M.S. and Wood, R.H., J. Phys. Chem. B, 101, 6549–6559 (1997).
Helgeson, H.C., Kirkham, D.H. and Flowers, G.C., Am. J. Sci., 281, 1249–1516 (1981).
Hepler, L.G. and Hopkins, H.P. Jr., Rev. Inorg. Chem., 1, 303–332 (1979).
Hettiarachchi, S., Makela, K., Song, H. and MacDonald, D.D., J. Electrochem. Soc., 139, L3–L4
(1992).
Hitch, B.F. and Mesmer, R.E., J. Solution Chem., 5, 667–680 (1976).
Ho, P.C. and Palmer, D.A., J. Solution Chem., 24, 753–769 (1995).
Ho, P.C. and Palmer, D.A., J. Solution Chem., 25, 711–729 (1996).
Ho, P.C. and Palmer, D.A., Geochim. Cosmochim. Acta, 61, 3027–3040 (1997).
Ho, P.C. and Palmer, D.A., J. Chem. Eng. Data, 43, 162–170 (1998).
Ho, P.C., Palmer, D.A. and Mesmer, R.E., J. Solution Chem., 23, 997–1017 (1994).
Ho, P.C., Bianchi, H., Palmer, D.A. and Wood, R.H., J. Solution Chem., 29, 217–235 (2000a).
Ho, P.C., Palmer, D.A. and Wood, R.H., J. Phys. Chem. B, 104, 12084–12089 (2000b).
P. Tremaine et al.490

Ho, P.C., Palmer, D.A. and Gruszkiewicz, M.S., J. Phys. Chem. B, 105, 1260–1266 (2001).
Holmes, H.F., Busey, R.H., Simonson, J.M., Mesmer, R.E., Archer, D.G. and Wood, R.H., J. Chem.
Thermodyn., 19, 863–890 (1987).
Johnson, J.W., Oelkers, E.H. and Helgeson, H.C., Comput. Geosci., 18, 899–947 (1992).
Johnston, K.P., Balbuena, P., Xiang, T., Wofford, W. and Flanagin, L. In: Palmer, D.A. and
Wesolowski, D.J. (Eds.), Proceedings of the 5th International Symposium on Hydrothermal
Reactions, Gatlinburg, TN, 1997, pp. 15–17.
Jones, F.M. and Arnett, E.M., Progr. Phys. Org. Chem., 11, 263–322 (1974).
Kettler, R.M., Palmer, D.A. and Wesolowski, D.J., J. Solution Chem., 20, 905–927 (1991).
Kettler, R.M., Wesolowski, D.J. and Palmer, D.A., J. Solution Chem., 21, 883–900 (1992).
Kettler, R.M., Palmer, D.A. and Wesolowski, D.J., J. Solution Chem., 24, 65–87 (1995a).
Kettler, R.M., Wesolowski, D.J. and Palmer, D.A., J. Solution Chem., 24, 385–407 (1995b).
Kettler, R.M., Wesolowski, D.J. and Palmer, D.A., J. Chem. Eng. Data, 43, 337–350 (1998).
Kriksunov, L., MacDonald, D.D. and Millett, P.J., J. Electrochem. Soc., 141, 3002–3005 (1994).
Larson, J.W., Zeeb, K.G. and Hepler, L.G., Can. J. Chem., 60, 2141–2150 (1982).
Laughton, P.M. and Robertson, R.E. In: Coetzee, J.F. and Ritchie, C.D. (Eds.), Solute–Solvent
Interactions. Marcel Dekker, New York, 1969.
Levelt Sengers, J.M.H. In: Bruno, J.J. and Ely, J.F. (Eds.), Supercritical Fluid Technology. CRC
Press, Boca Raton, FL, 1991, pp. 1–56.
Lewis, G.G. and Wetton, E.A.M., Central Electricity Generating Board Report, Report No. OED/
STN/87/20083/M, 1987.
Lindsay, W.T. Jr. In: Pichal, M. and Sifner, O. (Eds.), Properties of Water and Steam. Hemisphere,
New York, 1989, pp. 29–38.
Lindsay, W.T. Jr. In: Cohen, P. (Ed.), The ASME Handbook on Water Technology for Thermal Power
Systems. American Society of Mechanical Engineers, New York, 1990, pp. 341–544, chapter 7.
Lvov, S.N., Zhou, X.Y. and Macdonald, D.D., J. Electroanal. Chem., 463, 146–156 (1999).
MacDonald, D.D., Hettiarachchi, S. and Lenhart, S.J., J. Solution Chem., 17, 719–732 (1988).
Marshall, W.L. and Franck, E.U., J. Phys. Chem. Ref. Data, 10, 295–304 (1981).
Marshall, W.L. and Slusher, R., J. Inorg. Nucl. Chem., 37, 1191–1202 (1975a).
Marshall, W.L. and Slusher, R., J. Inorg. Nucl. Chem., 37, 2165–2170 (1975b).
Mesmer, R.E. and Baes, C.F. Jr., J. Solution Chem., 3, 307–322 (1974).
Mesmer, R.E. and Herting, D.L., J. Solution Chem., 7, 901–913 (1978).
Mesmer, R.E. and Hitch, B.F., J. Solution Chem., 6, 251–261 (1977).
Mesmer, R.E., Baes, C.F. Jr. and Sweeton, F.H., J. Phys. Chem., 74, 1937–1942 (1970).
Mesmer, R.E., Baes, C.F. Jr. and Sweeton, F.H., Inorg. Chem., 11, 537–543 (1972).
Mesmer, R.E., Marshall, W.L., Palmer, D.A., Simonson, J.M. and Holmes, H.F., J. Solution Chem.,
17, 699–718 (1988).
Mesmer, R.E., Patterson, C.S., Busey, R.H. and Holmes, H.F., J. Phys. Chem., 93, 7483–7490 (1989).
Mesmer, R.E., Palmer, D.A. and Simonson, J.M. In: Pitzer, K.S. (Ed.), Activity Coefficients in
Aqueous Solutions, 2nd edn. CRC Press, Boca Raton, FL, 1991, chapter 8.
Mesmer, R.E., Palmer, D.A., Simonson, J.M., Holmes, H.F., Ho, P.C., Wesolowski, D.J. and
Gruszkiewicz, M.S., Pure Appl. Chem., 69, 905–914 (1997).
Millero, F.J. In: Pytkowicz, M. (Ed.), Activity Coefficients in Electrolyte Solutions. Vol. 2, CRC
Press, Boca Raton, FL, 1979.
Noyes, A.A., The Electrical Conductivity of Aqueous Solutions, Publication No. 63, 1907, Carnegie
Institution of Washington.
O’Connell, J.P., Sharygin, A.V. and Wood, R.H., Ind. Eng. Chem. Res., 35, 2808–2812 (1996).
Olofsson, G. and Hepler, L.G., J. Solution Chem., 4, 127–143 (1975).
Oscarson, J.L., Gillespie, S.E., Izatt, R.M., Chen, X. and Pando, C., J. Solution Chem., 21, 789–801
(1992).
Ionization equilibria of acids and bases 491

Palmer, D.A. and Drummond, S.E., J. Solution Chem., 17, 153–164 (1988).
Palmer, D.A. and van Eldik, R., Chem. Rev., 83, 651–731 (1983).
Palmer, D.A., Benezeth, P. and Wesolowski, D.J., PowerPlant Chem., 2, 261–264 (2000).
Patterson, C.S., Slocum, G.H., Busey, R.H. and Mesmer, R.E., Geochim. Cosmochim. Acta, 46,
1653–1663 (1982).
Patterson, C.S., Busey, R.H. and Mesmer, R.E., J. Solution Chem., 13, 647–661 (1984).
Pettit, L.D. and Powell, K.J., IUPAC Stability Constants Database SC-Database for Windows.
IUPAC, 1997.
Pitzer, K.S. (Ed.), Activity Coefficients in Electrolyte Solutions, 2nd edn. CRC Press, Boca Raton,
FL, 1991.
Pitzer, K.S., Thermodynamics, McGraw-Hill Series in Advanced Chemistry, 3rd edn. McGraw-Hill,
New York, 1995.
Plyasunov, A.V., O’Connell, J.P. and Wood, R.H., Geochim. Cosmochim. Acta, 64, 495–512 (2000a).
Plyasunov, A.V., O’Connell, J.P., Wood, R.H. and Shock, E.L., Geochim. Cosmochim. Acta, 64,
2779–2795 (2000b).
Quist, A.S. and Marshall, W.L., J. Phys. Chem., 72, 684–703 (1968a).
Quist, A.S. and Marshall, W.L., J. Phys. Chem., 72, 3122–3128 (1968b).
Rao, R.S. and Hepler, L.G., Hydrometallurgy, 2, 293–299 (1977).
Read, A.J., J. Solution Chem., 4, 53–72 (1975).
Ridley, M.K., Xiao, C., Palmer, D.A. and Wesolowski, D.J., J. Chem. Eng. Data, 45, 502–507
(2000).
Roberts, B. and Tremaine, P.R., Can. J. Chem. Eng., 63, 294–300 (1985).
Rudolph, W., Brooker, M.H. and Tremaine, P.R., J. Solution Chem., 26, 757–777 (1997).
Sedlbauer, J., O’Connell, J.P. and Wood, R.H., Chem. Geol., 163, 43–63 (2000).
Sharygin, A.V., Mokbel, I., Xiao, C. and Wood, R.H., J. Phys. Chem. B, 105, 229–237 (2001).
Shoesmith, D.W. and Lee, W., Can. J. Chem., 54, 3553–3558 (1976).
Shock, E.L. and Helgeson, H.C., Geochim. Cosmochim. Acta, 52, 2009–2036 (1988).
Shock, E.L. and Helgeson, H.C., Geochim. Cosmochim. Acta, 54, 915–945 (1990).
Shock, E.L. and Koretsky, C.M., Geochim. Cosmochim. Acta, 57, 4899–4922 (1993).
Shock, E.L. and Koretsky, C.M., Geochim. Cosmochim. Acta, 59, 1497–1532 (1995).
Shock, E.L., Oelkers, E.H., Johnson, J.W., Sverjensky, D.A. and Helgeson, H.C., J. Chem. Soc.,
Faraday Trans., 88, 803–826 (1992), and references cited therein.
Shvedov, D. and Tremaine, P.R., J. Solution Chem., 26, 1113–1143 (1997).
Smith, R.M. and Martell, A.E., NIST Critically Selected Stability Constants of Metal Complexes
Database Version 4.0. National Institute of Standards and Technology, Gaithersburg, MD, 1997.
Suleimenov, M. and Seward, T.M., Geochim. Cosmochim. Acta, 61, 5187–5198 (1997).
Svishchev, I.M. and Kusalik, P.G. In: White, H.J. Jr., Sengers, J.V., Neumann, D.B., and Bellows,
J.C. (Eds.), Physical Chemistry of Aqueous Systems: Meeting the Needs of Industry, Proceedings
of the 12th International Conference on the Properties of Water and Steam. Begell House,
New York, 1995, pp. 222–228.
Sweeton, F.H., Mesmer, R.E. and Baes, C.F. Jr., J. Solution Chem., 3, 191–214 (1974).
Tanger, J.C. and Helgeson, H.C., Am. J. Sci., 288, 19–98 (1988).
Tremaine, P.R., Shvedov, D. and Xiao, C., J. Phys. Chem. B, 101, 409–419 (1997).
Turq, P., Blum, L., Bernard, O. and Kunz, W., J. Phys. Chem., 99, 822–827 (1995).
Ulmer, G.C. and Barnes, H.L., Hydrothermal Experimental Techniques. Wiley-Interscience,
New York, 1983.
Wofford, W.T., Gloyna, E.F. and Johnston, K.P., Ind. Eng. Chem. Res., 37, 2045–2051 (1998).
Xiang, T., Johnston, K.P., Wofford, W. and Gloyna, E.F., Ind. Eng. Chem. Res., 35, 4788–4795
(1996).
Zimmerman, G.H., Gruszkiewicz, M.S. and Wood, R.H., J. Phys. Chem., 99, 11612–11624 (1995).
P. Tremaine et al.492




















