
Dynamics of continuous-time quantum walks in restricted geometries
Upload
independentCategory
view
2download
0
1
Downregulation of Connexin40 and Increased Prevalence of Atrial
Arrhythmias in Transgenic Mice with Cardiac Restricted
Overexpression of Tumor Necrosis Factor
Sam E. Sawaya,1 Yadavendra S. Rajawat,1 Tapan G. Rami,1 Gabor Szalai,1 Robert L.
Price,2 Natarajan Sivasubramanian,1 Douglas L. Mann,1 Dirar S. Khoury1
From the 1Section of Cardiology, Department of Medicine, Baylor College of Medicine,
Houston, Texas, and 2Department of Cell and Developmental Biology and Anatomy,
School of Medicine, University of South Carolina, Columbia, South Carolina.
Inflammation and Atrial Remodeling
Address for correspondence:Dirar S. Khoury, PhDThe Methodist Hospital6565 Fannin St, F764Houston, TX 77030Tel: (713) 441-1388Fax: (713) 441-0188E-mail: [email protected]
Page 1 of 31
Copyright Information
Articles in PresS. Am J Physiol Heart Circ Physiol (November 22, 2006). doi:10.1152/ajpheart.00285.2006
Copyright © 2006 by the American Physiological Society.

2
ABSTRACT
Atrial arrhythmias, primarily atrial fibrillation, have been independently associated with
structural remodeling and with inflammation. We hypothesized that sustained
inflammatory signaling by tumor necrosis factor (TNF) would lead to alterations both in
underlying atrial myocardial structure and in atrial electrical conduction. We performed
ECG recording, intracardiac electrophysiology studies, epicardial mapping, and
connexin immunohistochemical analyses on transgenic mice with targeted
overexpression of TNF in the cardiac compartment (MHCsTNF) and on wild type (WT)
control mice (age, 8-16 wk). Atrial and ventricular conduction abnormalities were always
evident on ECG in MHCsTNF mice, including a shortened atrioventricular interval with a
wide QRS duration secondary to junctional rhythm. Supraventricular arrhythmias were
observed in 5 out of 8 MHCsTNF mice, whereas none of the mice demonstrated
ventricular arrhythmias. No arrhythmias were observed in WT mice. Left ventricular
conduction velocity during apical pacing was similar between the two mouse groups.
Connexin40 was significantly downregulated in MHCsTNF mice. In contrast,
connexin43 density was not significantly altered in MHCsTNF mice, but rather dispersed
away from the intercalated disks. In conclusion, sustained inflammatory signaling
contributed to atrial structural remodeling and downregulation of Cx40 that was
associated with an increased prevalence of atrial arrhythmias.
Keywords: Atrial fibrillation, Electrophysiology, Heart failure, Inflammation
Page 2 of 31
Copyright Information

3
INTRODUCTION
The association of inflammation with some forms of atrial fibrillation (AF) has been
demonstrated in animal models [17], in patients following cardiac surgery [5], and in
non-postoperative patients [6]. Common to these findings is the observed elevation in
C-reactive protein, which is an inflammatory marker that is driven by proinflammatory
cytokines. Statins, known to reduce C-reactive protein levels, have recently been shown
to reduce inducibility of AF in various canine models [29]. Recently, intraperitoneal
lipopolysaccharide, a potent inflammatory mediator, has been shown to attenuate cell-
to-cell communication in mouse aortic endothelial cells in part through reduction in
connexin40 (Cx40) levels, the major connexin in the atria and specialized conduction
system [31]. Persistence of atrial tachyarrhythmias has been related to structural
changes in the atria, which eventually result in AF. The relationship between altered
Cx40 expression and AF has been verified in experimental animal models [2] and in
humans [10,15].
The proinflammatory cytokine TNF is upregulated in heart failure, in which there
is an increased prevalence of AF. Previously we have generated a transgenic mouse
model with targeted overexpression of TNF in the cardiac compartment (referred to as
MHCsTNF), that progresses to a dilated cardiac phenotype by 12 weeks of age [32]. In
the present study we hypothesized that sustained inflammatory signaling in MHCsTNF
mice would lead to alterations both in underlying atrial myocardial structure and in atrial
electrical conduction.
Page 3 of 31
Copyright Information

4
MATERIALS AND METHODS
Animals
The study was performed on hemizygous MHCsTNF mice generated in an ICR x C57
background [18,32] and on age-matched wild type (WT) littermate controls. All mice
(age, 8-16 wk) were anesthetized by intraperitoneal injection (15 ml/kg) of a mixture of
sodium pentobarbital (4.1 mg/ml), sodium chloride (6.6 mg/ml) and ethanol (0.18 ml/ml).
The study protocol adhered to the PHS guidelines for the care and use of laboratory
animals.
Electrocardiographic and Intracardiac Electrophysiologic Evaluation
Intracardiac pacing and recording was performed in 8 MHCsTNF and 8 WT mice by
inserting an 8-electrode catheter (1.1 F, EPR-801, Millar Instruments; or 2 F, CIBer,
NuMED) through the jugular vein and advancing it into the right atrium and ventricle.
Electrophysiology methods were employed as previously described [1]. Electrical
stimulation was applied through the catheter electrodes using an external stimulator
(S8800, AstroMed). Inducibility of arrhythmias was investigated by delivering up to two
extra stimuli (S2 and S3) that were programmed at progressively shorter intervals
following an 8-beat conditioning drive train (S1).
Electrocardiographic limb lead signals were continuously recorded during
electrophysiology studies. In addition, to determine time-dependent effects of
MHCsTNF gene expression, ECGs were recorded in MHCsTNF and in WT mice 1 d, 1
wk, and 3 wk after birth.
Page 4 of 31
Copyright Information

5
The ECG signals were filtered between 5 and 100 Hz, and intracardiac
electrograms were filtered between 5 and 500 Hz, all sampled at 1 kHz (CardioLab, GE
Healthcare). All ECG intervals were determined from lead II.
Epicardial Activation Mapping
Anesthetized mice were ventilated with room air using a small animal respirator (tidal
volume, 0.5 cc; rate, 130 breaths/min, Columbus Instruments). The heart was then
exposed following a midline thoracotomy. An array comprised of 36 electrodes
(interelectrode spacing, 300 microns) and spanning an area of 1.5 mm × 1.5 mm was
used for electrical mapping on the epicardium of the intact beating heart (Multichannel
Systems). The array was placed on the anterior-basal aspect of the LV epicardium. A
total of 28 bipolar electrograms were filtered between 5-500 kHz, sampled at 2 kHz, and
simultaneously recorded (CardioLab).
Custom algorithms (Matlab, Mathworks) determined activation times, constructed
isochrone maps that depicted activation sequences, and computed conduction
velocities. Local activation time was determined as the time of absolute maximum value
in the bipolar electrogram [28]. The gradient of activation time as a function of distance
was computed at each data point relative to surrounding data points on a two-
dimensional grid, and conduction velocity was calculated as the inverse of the gradient.
Connexin Analysis
Western Blot analysis was performed to assess Cx40 and Cx43. The frozen heart was
pulverized and homogenized in a buffer containing (mmol/L) NaCl 50, Tris-HCl (pH 7.4)
Page 5 of 31
Copyright Information

6
20, EGTA 1, NaN3 5, b-mercaptoethanol 10, and phenylmethylsulfonyl fluoride 2 plus
protease inhibitors as described previously [3]. Protein concentration in the samples
was assessed by BCA method and 30 µg protein was loaded on each lane. The
samples were run on a 10% SDS-polyacrylamide gel and Western blotting was
performed using goat polyclonal antibody for Cx40 (0.02 µg/µL, cat #SC-20466, Santa
Cruz Biotechnology) and rabbit polyclonal antibody for Cx43 (1.6 µg/ml, cat #71-0700,
Zymed). The immunoblot was blocked in 5% non-fat milk for 2 hr at room temperature.
The blots were then scanned (OfficeJet 5510, Hewlett-Packard) and quantified (Image
J, NIH). Levels of Cx40 and Cx43 were normalized to total protein concentration in the
samples, in so far as we were unable to determine a myocardial membrane protein that
did not differ between the WT and MHCsTNF myocardium. Therefore, protein
concentration was measured multiple times before each western blot analysis, and the
average protein concentration was used for normalization.
Immunohistochemical staining was performed in the atria and ventricles in both
MHCsTNF and WT mice using standard methods described previously [24 ,26].
Samples were prepared from excised hearts that were immediately frozen in liquid
nitrogen and stored at –70°C. The hearts were sliced to a thickness of 20 microns and
stained with immunofluorescent goat anti-Connexin40 (1:100 dilution, cat #SC-20466,
Santa Cruz Biotechnology) and rabbit anti-Connexin43 (1:100 dilution, cat #C-6219,
Sigma-Aldrich), as well as a Cy-5 phalloidin stain for f-actin (1:250 dilution, Molecular
Probes) and a DAPI stain for nuclei (1:25000 dilution, Molecular Probes). The slices
were visualized using a Zeiss Confocal microscope.
Page 6 of 31
Copyright Information

7
Statistical Analysis
Continuous variables were expressed as mean ± standard error. Differences between
MHCsTNF and WT mice were compared by the Student t-test.
Page 7 of 31
Copyright Information

8
RESULTS
Electrocardiography and Electrophysiology
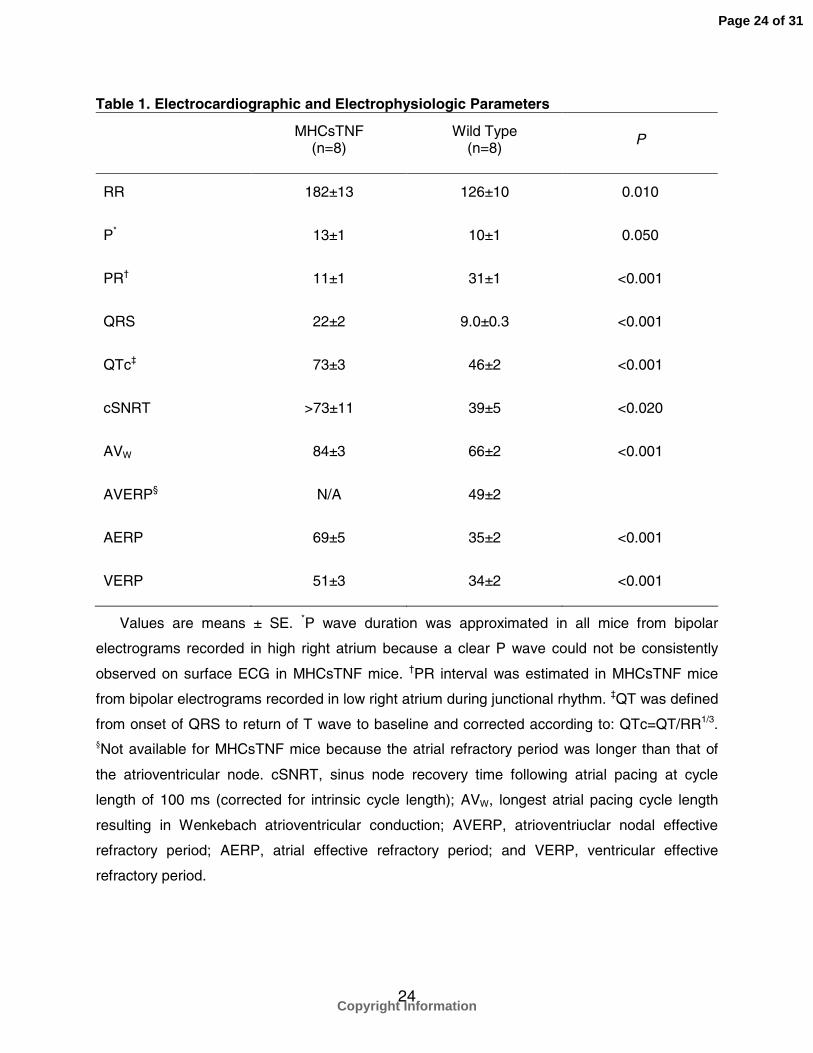
The ECG parameters in MHCsTNF and WT mice are summarized in Table 1. Unlike
normal atrioventricular (AV) activation in WT mice, the ECG in MHCsTNF mice
consistently exhibited AV junctional rhythm, with either absent or negative P wave in
Lead II, short PR interval, and wide QRS complex (Figure 1).
Representative intracardiac electrograms recorded from the right atrium of
MHCsTNF and WT mice are shown in Figure 1. The electrograms confirmed a short PR
interval in MHCsTNF mice, consistent with the presence of a junctional rhythm.
Baseline electrophysiologic parameters are summarized in Table 1. Unlike WT mice,
sinus node function was either suppressed or its impulses blocked in MHCsTNF mice,
as evidenced by (1) development of a junctional rhythm at a slow rate at baseline and
(2) regular appearance of a junctional rhythm at the conclusion of decremental atrial
pacing in MHCsTNF mice, substantiating a long sinus node recovery time. Atrial
conduction was also significantly slower in MHCsTNF compared to WT mice as
evidenced by the widened P wave duration. Decremental atrial pacing resulted in
progressively long AV nodal conduction times; however, the longest atrial pacing cycle
causing Wenckebach AV conduction was prolonged in MHCsTNF mice, suggestive of
abnormal AV nodal conduction. The ventricular refractory period was significantly longer
in MHCsTNF mice than in WT mice, consistent with long QTc intervals observed on
ECG in MHCsTNF mice. Supraventricular arrhythmias were observed in 5 out of 8
MHCsTNF mice. These arrhythmias included atrial tachycardia induced by atrial or
ventricular electrical stimulation in 4 mice and lasting more than 4 beats, spontaneous
Page 8 of 31
Copyright Information

9
atrial fibrillation in one mouse, and spontaneous atrial bigeminy in 3 mice (Figure 2).
None of the MHCsTNF mice had spontaneous or inducible ventricular arrhythmias. No
arrhythmias were observed in WT mice.
Figure 3 shows ECGs in MHCsTNF and WT mice. At 1 day of age, none of the
MHCsTNF mice (0/4) had AV junctional rhythm phenotype. Further, at 1 wk of age only
3 out of 5 MHCsTNF mice had the phenotype. At 3 wk of age and older, all MHCsTNF
mice (14/14) exhibited the junctional phenotype.
Left Ventricular Conduction Pattern
Epicardial electrical mapping revealed that the sequence of LV activation in MHCsTNF
mice was reversed during intrinsic rhythm when compared to the sequence of LV
activation in WT mice. That is, whereas early activation originated from LV apex in WT
mice (Figure 4A), early LV activation originated from anterior basal region in MHCsTNF
mice (Figure 4B). Furthermore, during intrinsic rhythm, apparent conduction velocity in
MHCsTNF mice (0.64±0.03 m/s, n=4) was significantly slower than the apparent
conduction velocity in WT mice (1.13±0.08 m/s, n=4); P<0.01. However, as summarized
in Figure 4C, the conduction velocities in both mouse genotypes were similar during
pacing at LV apex, and increased appropriately with increasing pacing cycle length.
Connexin Expression
We performed Western blot analysis for Cx40 and Cx43 to explore the potential reasons
for the abnormalities in atrial and ventricular conduction in the MHCsTNF mice. The
analysis (Figure 5) showed that the relative density of Cx40 in the atria was significantly
Page 9 of 31
Copyright Information

10
less in MHCsTNF compared to WT mice (33±12% vs. 100%, n=4, P=0.02). Meanwhile,
Western blot analysis showed that Cx43 existed in multiple phosphorylated isoforms.
However, no significant differences were observed in Cx43 levels between WT and
MHCsTNF mice (Figure 5). Immunohistochemical analysis showed that the distribution
of Cx43 was dispersed throughout the sarcolemma and intercalated disks of cardiac
myocytes in the MHCsTNF, whereas Cx43 was primarily located in the intercalated
disks in the WT mice (Figure 6A and Figure 6B), respectively.
Representative Cx40 immunostaining in the atria of WT and MHCsTNF mice are
shown in Figure 7A and Figure 7B, respectively. As depicted, the amount of Cx40 was
decreased and less uniformly distributed in the atria of MHCsTNF when compared to
WT mice. Figure 7C and Figure 7D show connexin expression in the bundle branch and
adjacent ventricular myocardium in WT and MHCsTNF mice, respectively. As shown,
Cx40 in the bundle branch and Cx43 in the ventricular myocardium were decreased in
MHCsTNF mice relative to WT mice.
Page 10 of 31
Copyright Information

11
DISCUSSION
Atrial arrhythmias, primarily AF, have been independently associated with inflammation
and with structural remodeling caused by downregulation of Cx40. To determine
whether sustained inflammatory signaling through targeted overexpression of TNF was
sufficient to lead to alterations in underlying atrial myocardial structure, alterations in
atrial electrical conduction, and atrial arrhythmias, we performed ECG screening,
intracardiac electrophysiology studies, epicardial mapping, and connexin
immunohistochemical analyses on MHCsTNF and WT control mice. The major new
finding of this study was that sustained inflammatory signaling contributed to atrial
structural remodeling and downregulation of Cx40 that was associated with an
increased prevalence of atrial arrhythmias.
The study showed that MHCsTNF mice develop abnormalities of both atrial and
ventricular conduction, including a shortened AV interval with a wide QRS duration
secondary to junctional rhythm. Atrial arrhythmias were observed in the majority of
MHCsTNF mice, whereas none of the mice demonstrated ventricular arrhythmias. The
LV epicardial activation pattern was reversed and apparent conduction velocity slowed
during intrinsic rhythm in MHCsTNF mice compared with WT mice, consistent with a
bundle branch block pattern. However, both the activation pattern and conduction
velocity during LV pacing (independent of the specialized conduction system) were
similar between the two mouse groups suggesting that ventricular conduction velocity
was not altered in the MHCsTNF mice. Connexin40, which is the major connexin in
atrial myocytes and the specialized conduction system, was significantly downregulated
Page 11 of 31
Copyright Information

12
in MHCsTNF mice. In contrast, the major ventricular myocyte connexin, Cx43, was
dispersed away from the intercalated disks in MHCsTNF mice, which did not appear to
be functionally significant in our model.
Atrioventricular junctional rhythm, presenting within the first week after birth in
some mice, is liable to be caused by TNF expression rather than heart failure.
Specifically, the atrial electrical abnormalities observed in the MHCsTNF mice are most
likely due to the observed abnormalities of Cx40. Indeed, studies in Cx40 deficient mice
are consistent with the electrical phenotype observed in our MHCsTNF mice, including
depressed sinus node activity, increased AV Wenckebach periodicity, increased
inducibility of atrial tachyarrythmias, and reduced conduction in the His-purkinje system
with preserved conduction velocity in the ventricular myocardium [4,12,30,35,36]. Our
results of atrial arrhythmias in a mouse model with overexpression of TNF confirm
previous observations by others [27]. However, a novel finding of our study was the role
of Cx40 in the setting of inflammation. Furthermore, while myocardial tissue fibrosis was
evident in TNF mice aged 2 – 9 mo [27], it is unlikely that fibrosis played a major role in
the pathogenesis of atrial arrhythmias in our model, insofar as fibrosis does not develop
significantly in this model until 12 wk of age [32].
Ion channel currents are altered in heart failure; specifically it is thought that L-
type calcium currents are upregulated and transient outward potassium currents are
downregulated [14]. A recent study showed that action potential prolongation and
abnormal calcium handling could contribute to the initiation of atrial and ventricular
arrhythmias observed with TNF overexpression in mice aged 3 to 9 months [19]. The
younger ages of the MHCsTNF mice that we studied (2 to 4 months) and absence of
Page 12 of 31
Copyright Information

13
spontaneous or inducible ventricular arrhythmias favor structural problems and
downregulation of Cx40 as the likely mechanism for the observed atrial arrhythmias in
the MHCsTNF mice, although we cannot exclude other ion channel mediated events.
As noted above, apparent ventricular conduction in MHCsTNF mice was
significantly slowed during intrinsic rhythm, as demonstrated by wide QRS on ECG and
by epicardial mapping. Meanwhile, the epicardial conduction velocity during LV pacing
(which bypassed the specialized conduction system) was similar between MHCsTNF
and WT mice. Interestingly, Cx40 knockout mice have been shown to exhibit slowing or
block in the bundle branches with a LV sequence of activation propagating from base to
apex [34], similar to what has been observed in the present study.
Conduction between ventricular myocytes depends on the excitability of
cardiomyocytes, the geometrical relationship between adjacent cardiomyocytes, and the
distribution of Cx43, which is primarily located in the intercalated disks of cardiac
myocytes, and has not been localized in the fibers of the specialized conduction system.
Previous studies have shown that reducing levels of Cx43 by 50% (a fraction similar to
that observed in human heart failure [16]) leads to a reduction in cardiomyocyte
conduction velocity [8,11], whereas other studies have not found this to be the case
[21,33]. Disruption of Cx43 has also been correlated with increased propensity for
ventricular tachyarrhythmias [25]. We did not find Cx43 levels to be downregulated;
however, by immunohistochemisty, we found Cx43 to be redistributed away from the
sarcolemma [23]. As noted above, the alterations in Cx43 expression in MHCsTNF mice
were probably not sufficient to cause a decrease in ventricular myocyte conduction
velocity or to provoke ventricular arrhythmias. However, these observations neither
Page 13 of 31
Copyright Information

14
exclude the formal possibility that greater disruptions of Cx43 might be functionally
significant, nor preclude a potentially important role for Cx43 in transgenic models that
do develop ventricular conduction disturbances.
Since many proteins were up- and down-regulated in MHCsTNF hearts [9,32],
we were unable to find a good normalization control (calsequestrin levels - data not
shown) for the membrane fractions of atrium and left ventricle that did not change
between littermate controls and MHCsTNF animals. Hence, the total protein
concentration was used to normalize the levels of Cx40 and Cx43. Therefore, the
observed abnormalities in both atrial and ventricular conduction in MHCsTNF
myocardium may be additionally due to the other proteins involved in this function
whose levels might have changed in MHCsTNF hearts.
Previously we found that MHCsTNF mice die prematurely when compared to age
matched littermate controls [13]. Progression of the observed atrioventricular conduction
block in MHCsTNF mice may eventually advance into complete heart block and
ultimately result in bradyarrhythmic death, although we cannot exclude other reasons
for death such as metabolic and respiratory causes. Continuous telemetry in our
conscious MHCsTNF mice remains to be performed; however, the mode of death in a
different line of transgenic mice with targeted overexpression of TNF was previously
noted to be secondary to bradyarrhythmias [19]. Interestingly, bradyarrhythmias were
also the cause of death in mice with downregulated Cx40 [22]. Additionally, the major
mode of cardiac arrest in patients with nonischemic heart failure has been found to be
due to bradyarrhythmias [20], raising the interesting possibility that inflammation and
Cx40 downregulation may play a contributory role. Finally, it is worth mentioning that
Page 14 of 31
Copyright Information

15
other cardiomyopathies that are associated with inflammation, including Lyme disease,
viral myocarditis, Sjogren’s syndrome, and Chagas disease myocarditis all result in a
higher proportion of patients with conduction system block.
We have generated several lines of mice that overexpress TNF in the cardiac
compartment. The line of mice (MHCsTNF) generated in the present study has TNF
protein and mRNA levels that are in excess of what one would expect to see in human
heart failure. However, we have generated additional lines of mice (MHCsTNF2) that
have levels of TNF proteins and mRNA that are similar to human heart failure [7]. The
major difference between these two lines is that the time course for development of
cardiac remodeling is greatly accelerated in the line of mice with higher TNF levels.
Conclusions
Sustained inflammatory signaling, provoked by overexpression of TNF in MHCsTNF
mice, induced both atrial structural remodeling by downregulation of Cx40 and
increased prevalence of atrial arrhythmias
Grants
This research was supported by research funds from the Howard Hughes Medical
Institute, Veterans Administration, and National Institutes of Health (P50 HL-06H, R01
HL58081-01, R01 HL61543-01, and HL-42250-10/10).
Disclosures
The authors have no conflicts to disclose.
Page 15 of 31
Copyright Information

16
REFERENCES
1. Appleton GO, Li Y, Taffet GE, Hartley CJ, Michael LH, Entman ML, Roberts R,
and Khoury DS. Determinants of cardiac electrophysiological properties in mice. J
Interv Card Electrophysiol 11: 5-14, 2004.
2. Ausma J, van der Velden HM, Lenders MH, van Ankeren EP, Jongsma HJ,
Ramaekers FC, Borgers M, and Allessie MA. Reverse structural and gap-
junctional remodeling after prolonged atrial fibrillation in the goat. Circulation 107:
2051-2058, 2003.
3. Beardslee MA, Laing JG, Beyer EC, and Saffitz JE. Rapid turnover of connexin43
in the adult rat heart. Circ Res 83: 629-635, 1998.
4. Bevilacqua LM, Simon AM, Maguire CT, Gehrmann J, Wakimoto H, Paul DL,
and Berul CI. A targeted disruption in connexin40 leads to distinct atrioventricular
conduction defects. J Interv Card Electrophysiol 4: 459-467, 2000.
5. Bruins P, te Velthuis H, Yazdanbakhsh AP, Jansen PG, van Hardevelt FW, de
Beaumont EM, Wildevuur CR, Eijsman L, Trouwborst A, and Hack CE.
Activation of the complement system during and after cardiopulmonary bypass
surgery: postsurgery activation involves C-reactive protein and is associated with
postoperative arrhythmia. Circulation 96: 3542-3548, 1997.
6. Chung MK, Martin DO, Sprecher D, Wazni O, Kanderian A, Carnes CA, Bauer
JA, Tchou PJ, Niebauer MJ, Natale A, and Van Wagoner DR. C-reactive protein
elevation in patients with atrial arrhythmias: inflammatory mechanisms and
persistence of atrial fibrillation. Circulation 104: 2886-2891, 2001.
Page 16 of 31
Copyright Information

17
7. Diwan A, Dibbs Z, Nemoto S, DeFreitas G, Carabello BA, Sivasubramanian N,
Wilson EM, Spinale FG, and Mann DL. Targeted overexpression of noncleavable
and secreted forms of tumor necrosis factor provokes disparate cardiac phenotypes.
Circulation 109: 262-268, 2004.
8. Eloff BC, Lerner DL, Yamada KA, Schuessler RB, Saffitz JE, and Rosenbaum
DS. High resolution optical mapping reveals conduction slowing in connexin43
deficient mice. Cardiovasc Res 51: 681-690, 2001.
9. Flesch M, Hoper A, Dell'Italia L, Evans K, Bond R, Peshock R, Diwan A, Brinsa
TA, Wei CC, Sivasubramanian N, Spinale FG, and Mann DL. Activation and
functional significance of the renin-angiotensin system in mice with cardiac restricted
overexpression of tumor necrosis factor. Circulation 108: 598-604, 2003.
10.Gollob MH, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP,
Tang ASL, Stewart AFR, Tesson F, Klein GJ, Yee R, Skanes AC, Guiraudon
GM, Ebihara L, and Bai D. Somatic mutations in the connexin 40 gene (GJA5) in
atrial fibrillation. N Engl Med 354: 2677-2688, 2006.
11.Guerrero PA, Schuessler RB, Davis LM, Beyer EC, Johnson CM, Yamada KA,
and Saffitz JE. Slow ventricular conduction in mice heterozygous for a connexin43
null mutation. J Clin Inves 99: 1991-1998, 1997.
12.Hagendorff A, Schumacher B, Kirchhoff S, Lüderitz B, and Willecke K.
Conduction disturbances and increased atrial vulnerability in Connexin40-deficient
mice analyzed by transesophageal stimulation. Circulation 99: 1508-1515, 1999.
13.Hodgson DM, Zingman LV, Kane GC, Perez-Terzic C, Bienengraeber M, Ozcan
C, Gumina RJ, Pucar D, O'coclain F, Mann DL, Alekseev AE, and Terzic A.
Page 17 of 31
Copyright Information

18
Cellular remodeling in heart failure disrupts K(ATP) channel-dependent stress
tolerance. Embo Journal 22: 1732-1742, 2003.
14.Janse MJ. Electrophysiological changes in heart failure and their relationship to
arrhythmogenesis. Cardiovasc Res 61: 208-217, 2004.
15.Kanagaratnam P, Cherian A, Stanbridge RD, Glenville B, Severs NJ, and Peters
NS. Relationship between connexins and atrial activation during human atrial
fibrillation. J Cardiovasc Electrophysiol 15: 206-2016, 2004.
16.Kostin S, Rieger M, Dammer S, Hein S, Richter M, Klövekorn WP, Bauer EP,
and Schaper J. Gap junction remodeling and altered connexin43 expression in the
failing human heart. Mol Cell Biochem 242: 135-144, 2003.
17.Kumagai K, Khrestian C, and Waldo AL. Simultaneous multisite mapping studies
during induced atrial fibrillation in the sterile pericarditis model. Insights into the
mechanism of its maintenance. Circulation 95: 511-521, 1997.
18.Li X, Moody MR, Engel D, Walker S, Clubb FJ, Sivasubramanian N, Mann DL,
Reid MB. Cardiac-specific overexpression of tumor necrosis factor-alpha causes
oxidative stress and contractile dysfunction in mouse diaphragm. Circulation 102:
1690-1696, 2000.
19.London B, Baker LC, Lee JoonS, Shusterman V, Choi BR, Kubota T, Mctiernan
CF, Feldman AM, and Salama G. Calcium-dependent arrhythmias in transgenic
mice with heart failure. Am J Physiol Heart Circ Physiol 284: H431-H441, 2003.
20.Luu M, Stevenson WG, Stevenson LW, Baron K, and Walden J. Diverse
mechanisms of unexpected cardiac arrest in advanced heart failure. Circulation 80:
1675-1680, 1989.
Page 18 of 31
Copyright Information

19
21.Morley GE, Vaidya D, Samie FH, Lo C, Delmar M, and Jalife J. Characterization
of conduction in the ventricles of normal and heterozygous Cx43 knockout mice
using optical mapping. J Cardiovasc Electrophysiol 10: 1361-1375, 1999.
22.Mungrue IN, Gros R, You X, Pirani A, Azad A, Csont T, Schulz R, Butany J,
Stewart DJ, and Husain M. Cardiomyocyte overexpression of iNOS in mice results
in peroxynitrite generation, heart block, and sudden death. J Clin Invest 109: 735-
743, 2002.
23.Musil LS, Cunningham BA, Edelman GM, and Goodenough DA. Differential
phosphorylation of the gap junction protein connexin43 in junctional communication-
competent and -deficient cell lines. J Cell Biol 111: 2077-2088, 1990.
24.Nakayama M, Yan X, Price RL, Borg TK, Ito K, Sanbe A, Robbins J, and Lorell
BH. Chronic ventricular myocyte-specific overexpression of angiotensin II type 2
receptor results in intrinsic myocyte contractile dysfunction. Am J Physiol Heart Circ
Physiol 288: H317-H327, 2005.
25.Peters NS, Coromilas J, Severs NJ, and Wit AL. Disturbed connexin43 gap
junction distribution correlates with the location of reentrant circuits in the epicardial
border zone of healing canine infarcts that cause ventricular tachycardia. Circulation
95: 988-996, 1997.
26.Price RL, Chintanowonges C, Shiraishi I, Borg TK, and Terracio L. Local and
regional variations in myofibrillar patterns in looping rat hearts. Anat Rec 245: 83-93,
1996.
27.Saba S, Janczewski AM, Baker LC, Shusterman V, Gursoy EC, Feldman AM,
Salama G, McTierman CF, and London B. Atrial contractile dysfunction, fibrosis,
Page 19 of 31
Copyright Information

20
and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific
overexpression of tumor necrosis factor-α. Am J Physiol Heart Circ Physiol 289:
H1456-H1467, 2005.
28.Shenasa M, Borggrefe M, and Breithardt G. Cardiac Mapping. New York: Futura
Publishing Company, 1993, p. 17-18.
29.Shiroshita-Takeshita A, Schram G, Lavoie J, and Nattel S. Effect of simvastatin
and antioxidant vitamins on atrial fibrillation promotion by atrial-tachycardia
remodeling in dogs. Circulation 110: 2313-2319, 2004.
30.Simon AM, Goodenough DA, and Paul DL. Mice lacking connexin40 have cardiac
conduction abnormalities characteristic of atrioventricular block and bundle branch
block. Curr Biol 8: 295-298, 1998.
31.Simon AM, McWhorter AR, Chen H, Jackson CL, and Ouellette Y. Decreased
intercellular communication and connexin expression in mouse aortic endothelium
during lipopolysaccharide-induced inflammation. J Vasc Res 41: 323-333, 2004.
32.Sivasubramanian N, Coker ML, Kurrelmeyer KM, Maclellan WR, Demayo FJ,
Spinale FG, and Mann DL. Left ventricular remodeling in transgenic mice with
cardiac restricted overexpression of tumor necrosis factor. Circulation 104: 826-831,
2001.
33.van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, Jongsma HJ,
Opthof T, and de Bakker JM. Slow conduction and enhanced anisotropy increase
the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of
connexin43. Circulation 109: 1048-1055, 2004.
Page 20 of 31
Copyright Information

21
34.van Rijen HV, van Veen TA, van Kempen MJ, Wilms-Schopman FJ, Potse M,
Krueger O, Willecke K, Opthof T, Jongsma HJ, and de Bakker JM. Impaired
conduction in the bundle branches of mouse hearts lacking the gap junction protein
connexin40. Circulation 103: 1591-1598, 2001.
35.Vanderbrink BA, Sellitto C, Saba S, Link MS, Zhu W, Homoud MK, Estes NA,
Paul DL, and Wang PJ. Connexin40-deficient mice exhibit atrioventricular nodal
and infra-Hisian conduction abnormalities. J Cardiovasc Electrophysiol 11: 1270-
1276, 2000.
36.Verheule S, van Batenburg CA, Coenjaerts FE, Kirchhoff S, Willecke K, and
Jongsma HJ. Cardiac conduction abnormalities in mice lacking the gap junction
protein connexin40. J Cardiovasc Electrophysiol 10: 1380-1389, 1999.
Page 21 of 31
Copyright Information

22
FIGURE LEGENDS
Fig. 1. Intrinsic ECG (leads I, II, and III) and intracardiac electrograms in a WT mouse
(left panel) and in a MHCsTNF mouse (right panel). RA, right atrial electrogram; and
RV, right ventricular electrogram.
Fig. 2. Examples of supraventricular arrhythmias observed in MHCsTNF mice. The
arrhythmias include spontaneous bigeminy (A), atrial tachycardia induced by ventricular
extrastimulation (B and C), atrial tachycardia induced by atrial extrastimulation (D), and
spontaneous atrial fibrillation (E). I, ECG lead I; LRA, intracardiac low right atrial
electrogram; and HRA, intracardiac high right atrial electrogram.
Fig. 3. Electrocardiograms and PCRs of WT and MHCsTNF mice 1 d (left), 1 wk
(middle), and 3 wk (right) after birth. A different group of mice was studied at each time
interval. Mice demonstrating AV junctional rhythm are marked by an asterisk.
Fig. 4. Representative maps of LV epicardial activation sequences during intrinsic
rhythm in WT mice (A) and in MHCsTNF mice (B). Arrows reflect conduction velocities.
The maps are displayed such that the top aspect corresponds with the base and the left
aspect with the left anterior descending coronary artery. Panel C summarizes LV
epicardial conduction velocities in WT and MHCsTNF mice while pacing at LV apex.
Page 22 of 31
Copyright Information

23
Fig. 5. Western blots for Cx40 and Cx43 in WT and MHCsTNF mice. NP, non-
phosphorylated isoform; P1, least phosphorylated isoform; P2, most phosphorylated
isoform.
Fig. 6. Sample immunofluorescence data from ventricular myocardium in WT (A) and
MHCsTNF (B) mice stained for Cx43 (red) both at 40X magnification. Nuclei are stained
blue. Scale bar = 100 microns.
Fig. 7. Sample immunofluorescence data from atrial (A and B) and bundle branch (C
and D) regions in WT and MHCsTNF mice stained for Cx40 (green) and Cx43 (red) all
at 40X magnification. Nuclei are stained blue. Scale bar = 100 microns.
Page 23 of 31
Copyright Information
24
Table 1. Electrocardiographic and Electrophysiologic Parameters
MHCsTNF(n=8)
Wild Type(n=8)
P
RR 182±13 126±10 0.010
P* 13±1 10±1 0.050
PR† 11±1 31±1 <0.001
QRS 22±2 9.0±0.3 <0.001
QTc‡ 73±3 46±2 <0.001
cSNRT >73±11 39±5 <0.020
AVW 84±3 66±2 <0.001
AVERP§ N/A 49±2
AERP 69±5 35±2 <0.001
VERP 51±3 34±2 <0.001
Values are means ± SE. *P wave duration was approximated in all mice from bipolar
electrograms recorded in high right atrium because a clear P wave could not be consistently
observed on surface ECG in MHCsTNF mice. †PR interval was estimated in MHCsTNF mice
from bipolar electrograms recorded in low right atrium during junctional rhythm. ‡QT was defined
from onset of QRS to return of T wave to baseline and corrected according to: QTc=QT/RR1/3.§Not available for MHCsTNF mice because the atrial refractory period was longer than that of
the atrioventricular node. cSNRT, sinus node recovery time following atrial pacing at cycle
length of 100 ms (corrected for intrinsic cycle length); AVW, longest atrial pacing cycle length
resulting in Wenkebach atrioventricular conduction; AVERP, atrioventriuclar nodal effective
refractory period; AERP, atrial effective refractory period; and VERP, ventricular effective
refractory period.
Page 24 of 31
Copyright Information

Fig. 1. Intrinsic ECG (leads I, II, and III) and intracardiac electrograms in a WT mouse (left panel) and in a MHCsTNF mouse (right panel). RA, right atrial electrogram; and RV,
right ventricular electrogram.
Page 25 of 31
Copyright Information

Fig. 2. Examples of supraventricular arrhythmias observed in MHCsTNF mice. The arrhythmias include spontaneous bigeminy (A), atrial tachycardia induced by ventricular extrastimulation (B and C), atrial tachycardia induced by atrial extrastimulation (D), and
spontaneous atrial fibrillation (E). I, ECG lead I; LRA, intracardiac low right atrial electrogram; and HRA, intracardiac high right atrial electrogram.
Page 26 of 31
Copyright Information

Fig. 3. Electrocardiograms and PCRs of WT and MHCsTNF mice 1 d (left), 1 wk (middle), and 3 wk (right) after birth. A different group of mice was studied at each time interval.
Mice demonstrating AV junctional rhythm are marked by an asterisk.
Page 27 of 31
Copyright Information
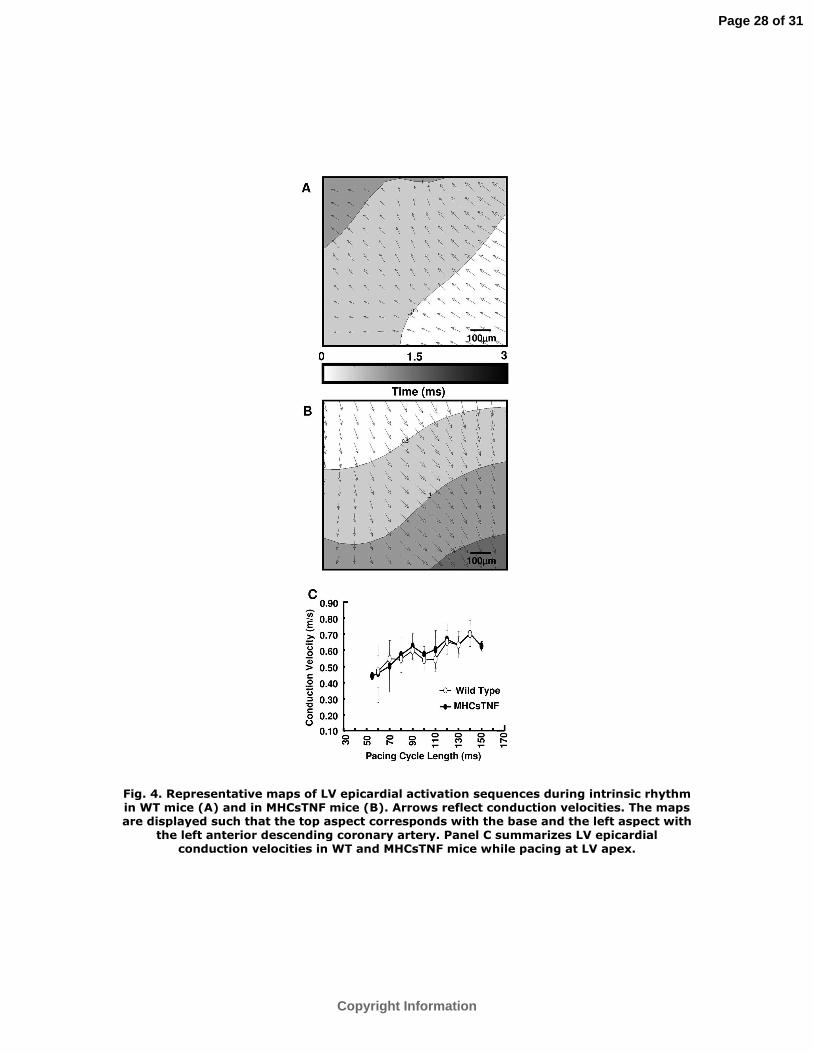
Fig. 4. Representative maps of LV epicardial activation sequences during intrinsic rhythm in WT mice (A) and in MHCsTNF mice (B). Arrows reflect conduction velocities. The maps are displayed such that the top aspect corresponds with the base and the left aspect with
the left anterior descending coronary artery. Panel C summarizes LV epicardial conduction velocities in WT and MHCsTNF mice while pacing at LV apex.
Page 28 of 31
Copyright Information
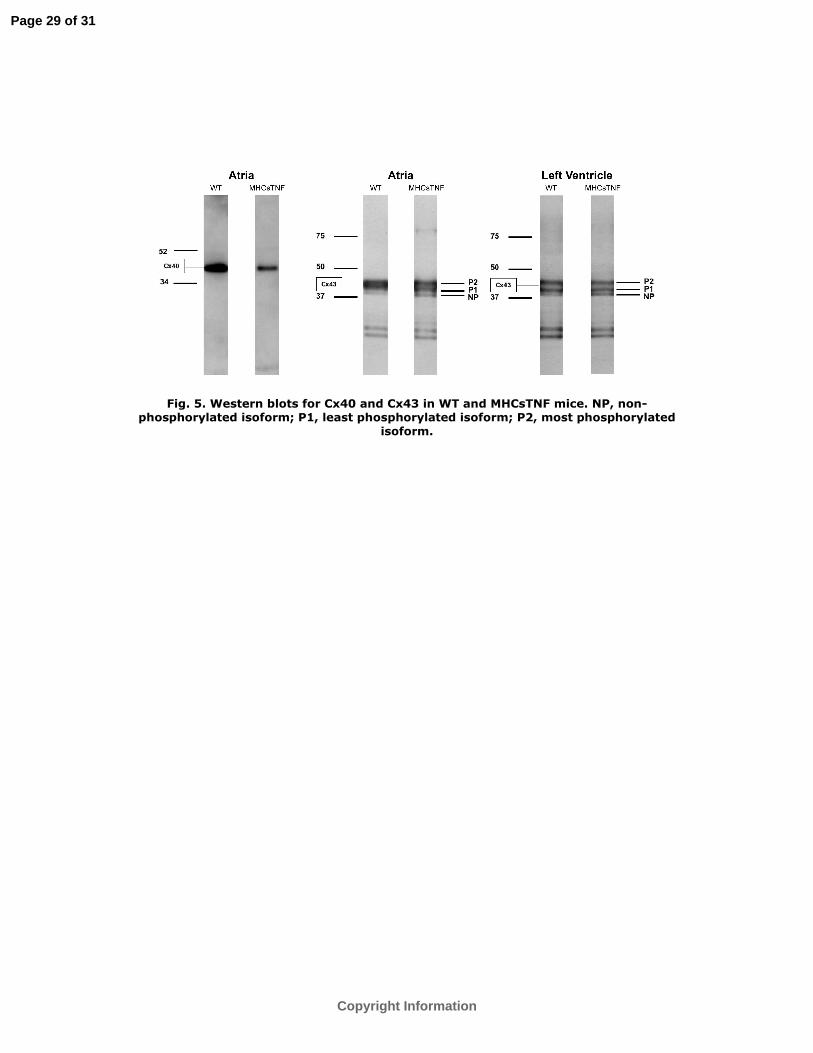
Fig. 5. Western blots for Cx40 and Cx43 in WT and MHCsTNF mice. NP, non-phosphorylated isoform; P1, least phosphorylated isoform; P2, most phosphorylated
isoform.
Page 29 of 31
Copyright Information
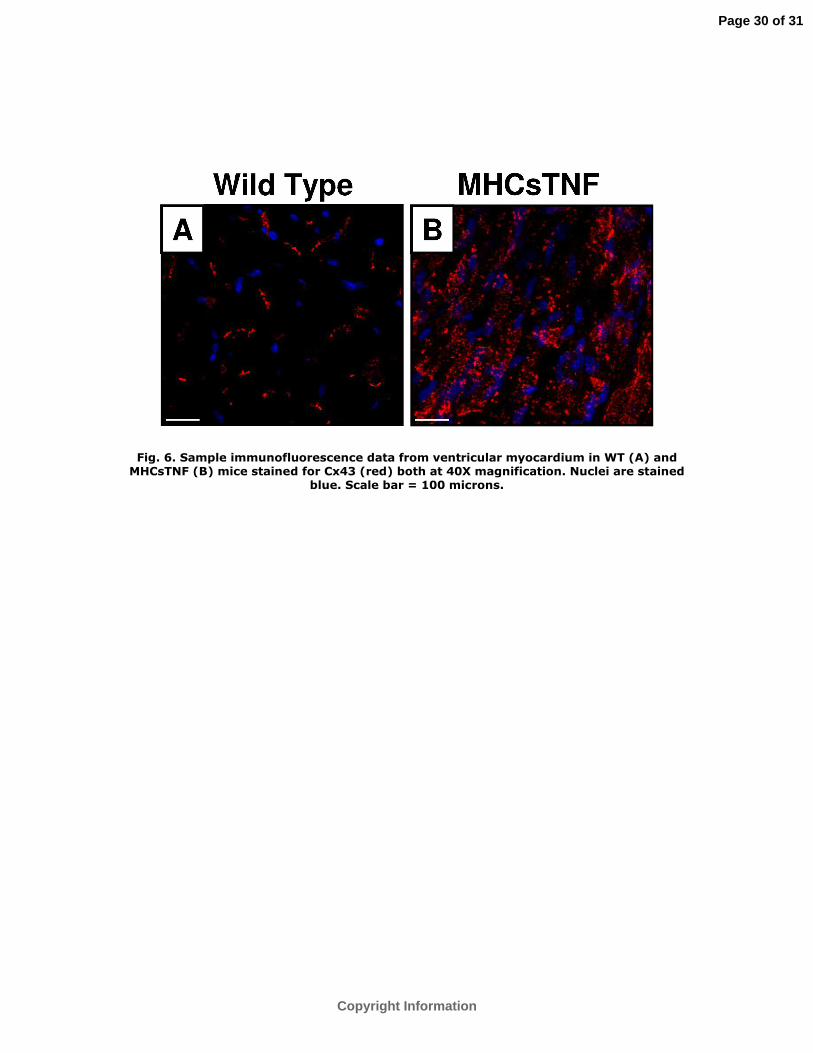
Fig. 6. Sample immunofluorescence data from ventricular myocardium in WT (A) and MHCsTNF (B) mice stained for Cx43 (red) both at 40X magnification. Nuclei are stained
blue. Scale bar = 100 microns.
Page 30 of 31
Copyright Information
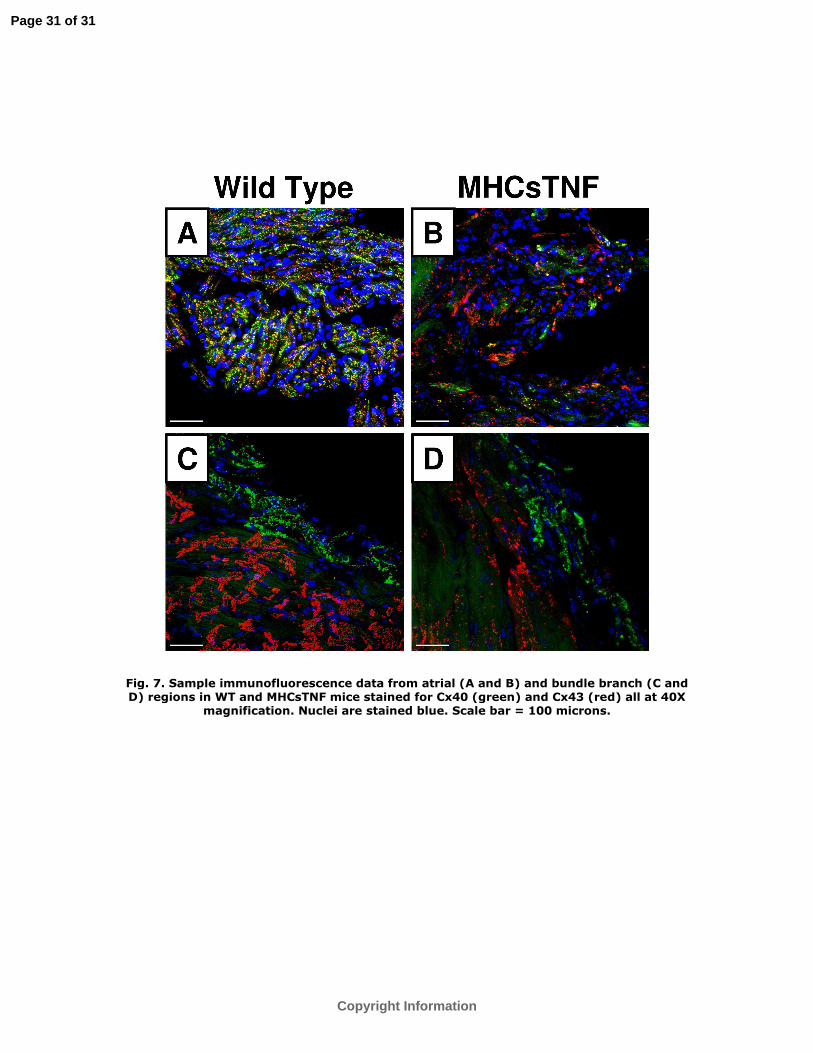
Fig. 7. Sample immunofluorescence data from atrial (A and B) and bundle branch (C and D) regions in WT and MHCsTNF mice stained for Cx40 (green) and Cx43 (red) all at 40X
magnification. Nuclei are stained blue. Scale bar = 100 microns.
Page 31 of 31
Copyright Information
Copyright © 2022 FDOKUMEN




















