
Down-regulation of IRAK-4 is a component of LPS- and CpG DNA-induced tolerance in macrophages
7
Down-regulation of IRAK-4 is a component of LPS- and CpG DNA-induced tolerance in macrophages Dominic De Nardo, Thao Nguyen, John A. Hamilton, Glen M. Scholz ⁎ Department of Medicine and Cooperative Research Centre for Chronic Inflammatory Diseases, The University of Melbourne, Royal Melbourne Hospital, Victoria 3050, Australia abstract article info Article history: Received 23 June 2008 Received in revised form 20 October 2008 Accepted 20 October 2008 Available online 25 October 2008 Keywords: Toll-like receptor Tolerance Interleukin-1 receptor associated kinase Macrophage Protein degradation Macrophages are important mediators of the immune response to infection by virtue of, amongst other things, their ability to secrete cytokines (e.g. TNF) that trigger inflammation. However, excessive systemic release of inflammatory cytokines can cause septic shock and ultimately death. Tolerance is an adaptive mechanism that prevents macrophage activation and inflammatory cytokine production. The activation of macrophages by pathogens is largely mediated by Toll-like receptors (TLRs). IRAK-4 and IRAK-1 are proximal protein kinases in TLR signalling pathways; IRAK-1 is activated via its phosphorylation by IRAK-4. The rapid degradation of IRAK-1 following its TLR-induced activation has been proposed to represent a major mechanism for tolerance. Here, we established that IRAK-1 degradation is insufficient to cause tolerance; in the absence of IRAK-1, IRAK-4 likely activates downstream signalling proteins (e.g. NF-κB) via IRAK-2. Significantly, tolerance coincided with IRAK-4 down-regulation, which occurred at the protein level via proteolytic degradation as well as at the mRNA level. Gene silencing experiments confirmed the importance of IRAK-4 for the regulation of TNF expression. The different kinetics of IRAK-4 and IRAK-1 down-regulation may result in both quantitative and qualitative differences in TLR signalling and potentially allow macrophages to temporally modify their inflammatory responses. Furthermore, differences in the kinetics and extent of IRAK-4 down-regulation by TLR ligands may provide a mechanism whereby macrophages can tailor their inflammatory response according to the location and/or type of pathogen detected. © 2008 Elsevier Inc. All rights reserved. 1. Introduction The innate immune system represents the first line of defence against infection. Macrophages are a key component of the innate immune system as they have the capacity to secrete inflammatory cytokines (e.g. TNF and IL-6), chemokines, proteases, and reactive oxygen metabolites when they encounter pathogens [1–4]. The activation of macrophages by pathogens is primarily mediated by Toll-like receptors (TLRs) [2,5–7]. TLRs are selectively activated by specific components of pathogens referred to as pathogen-associated molecular patterns (PAMPs). For example, TLR4 is activated by bac- terial lipopolysaccharide (LPS) [8], whereas TLR9 is activated by bac- terial DNA [9]. The detection of PAMPs by TLRs triggers the activation of intracellular networks of signalling pathways that coordinate the ensuing inflammatory response of macrophages to pathogens [2,5–7]. The recruitment of MyD88 and related adaptor proteins (e.g. Mal, Tram and Trif) to the activated TLR complex is the initial event in TLR signalling. This is followed by the interaction of the protein kinases interleukin-1 receptor associated kinase (IRAK)-1 and IRAK-4 with the complex; IRAK-1 activation occurs in response to its phosphorylation by IRAK-4. The subsequent binding of TRAF6 to IRAK-1 ultimately results in the activation of additional protein kinases (e.g. IκB kinase, JNK and p38 MAP kinase) as well as transcription factors (e.g. NF-κB), which then up-regulate expression of inflammatory genes (e.g. TNF and IL-6) [2,5–7]. Excessive systemic release of inflammatory cytokines by macro- phages during the course of a bacterial infection can cause substantial tissue damage, potentially resulting in multi-organ failure and circula- tory collapse (i.e. septic shock), and even death [10]. Tolerance is an adaptive mechanism that limits the extent and duration of inflammatory cytokine production by preventing the re-activation of macrophages by bacterial pathogens and/or their products. For example, the exposure of macrophages to LPS blocks their subsequent re-activation by LPS [11]. Several other TLR ligands (e.g. CpG DNA, lipoteichoic acid and flagellin) have also been shown to induce tolerance in macrophages [12–16]. Various molecular explanations for tolerance have been suggested including down-regulation of TLR4 expression [17], expression of an inhibitory MyD88 splice variant [18], as well as changes in the com- position of NF-κB complexes [19]. Stress response proteins, such as Hsp90, may also be involved in tolerance [20]. IRAK-1 is a key molecule in TLR signalling (e.g. linking MyD88 to TRAF6) and, as such, its rapid TLR-induced degradation has been proposed to represent a major mechanism for tolerance [13,15,16,21– 24]. However, we found that LPS- and CpG DNA-induced IRAK-1 Cellular Signalling 21 (2009) 246–252 ⁎ Corresponding author. Department of Medicine, The University of Melbourne, Royal Melbourne Hospital, Parkville, VIC 3010, Australia. Tel.: +613 8344 3298; fax: +613 9347 1863. E-mail address: [email protected] (G.M. Scholz). 0898-6568/$ – see front matter © 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.cellsig.2008.10.009 Contents lists available at ScienceDirect Cellular Signalling journal homepage: www.elsevier.com/locate/cellsig
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Down-regulation of IRAK-4 is a component of LPS- and CpG DNA-induced tolerance in macrophages

Cellular Signalling 21 (2009) 246–252
Contents lists available at ScienceDirect
Cellular Signalling
j ourna l homepage: www.e lsev ie r.com/ locate /ce l l s ig
Down-regulation of IRAK-4 is a component of LPS- and CpG DNA-induced tolerancein macrophages
Dominic De Nardo, Thao Nguyen, John A. Hamilton, Glen M. Scholz ⁎Department of Medicine and Cooperative Research Centre for Chronic Inflammatory Diseases, The University of Melbourne, Royal Melbourne Hospital, Victoria 3050, Australia
⁎ Corresponding author. Department of Medicine, TheMelbourne Hospital, Parkville, VIC 3010, Australia. Tel.: +1863.
E-mail address: [email protected] (G.M. Scho
0898-6568/$ – see front matter © 2008 Elsevier Inc. Aldoi:10.1016/j.cellsig.2008.10.009
a b s t r a c t
a r t i c l e i n f oArticle history:
Macrophages are importan Received 23 June 2008Received in revised form 20 October 2008Accepted 20 October 2008Available online 25 October 2008Keywords:Toll-like receptorToleranceInterleukin-1 receptor associated kinaseMacrophageProtein degradation
t mediators of the immune response to infection by virtue of, amongst otherthings, their ability to secrete cytokines (e.g. TNF) that trigger inflammation. However, excessive systemicrelease of inflammatory cytokines can cause septic shock and ultimately death. Tolerance is an adaptivemechanism that prevents macrophage activation and inflammatory cytokine production. The activation ofmacrophages by pathogens is largely mediated by Toll-like receptors (TLRs). IRAK-4 and IRAK-1 are proximalprotein kinases in TLR signalling pathways; IRAK-1 is activated via its phosphorylation by IRAK-4. The rapiddegradation of IRAK-1 following its TLR-induced activation has been proposed to represent a majormechanism for tolerance. Here, we established that IRAK-1 degradation is insufficient to cause tolerance; inthe absence of IRAK-1, IRAK-4 likely activates downstream signalling proteins (e.g. NF-κB) via IRAK-2.Significantly, tolerance coincided with IRAK-4 down-regulation, which occurred at the protein level viaproteolytic degradation as well as at the mRNA level. Gene silencing experiments confirmed the importanceof IRAK-4 for the regulation of TNF expression. The different kinetics of IRAK-4 and IRAK-1 down-regulationmay result in both quantitative and qualitative differences in TLR signalling and potentially allowmacrophages to temporally modify their inflammatory responses. Furthermore, differences in the kineticsand extent of IRAK-4 down-regulation by TLR ligands may provide a mechanism whereby macrophages cantailor their inflammatory response according to the location and/or type of pathogen detected.
© 2008 Elsevier Inc. All rights reserved.
1. Introduction
The innate immune system represents the first line of defenceagainst infection. Macrophages are a key component of the innateimmune system as they have the capacity to secrete inflammatorycytokines (e.g. TNF and IL-6), chemokines, proteases, and reactiveoxygen metabolites when they encounter pathogens [1–4]. Theactivation of macrophages by pathogens is primarily mediated byToll-like receptors (TLRs) [2,5–7]. TLRs are selectively activated byspecific components of pathogens referred to as pathogen-associatedmolecular patterns (PAMPs). For example, TLR4 is activated by bac-terial lipopolysaccharide (LPS) [8], whereas TLR9 is activated by bac-terial DNA [9].
The detection of PAMPs by TLRs triggers the activation ofintracellular networks of signalling pathways that coordinate theensuing inflammatory response of macrophages to pathogens [2,5–7].The recruitment of MyD88 and related adaptor proteins (e.g. Mal,Tram and Trif) to the activated TLR complex is the initial event in TLRsignalling. This is followed by the interaction of the protein kinasesinterleukin-1 receptor associated kinase (IRAK)-1 and IRAK-4 with the
University of Melbourne, Royal613 8344 3298; fax: +613 9347
lz).
l rights reserved.
complex; IRAK-1 activation occurs in response to its phosphorylationby IRAK-4. The subsequent binding of TRAF6 to IRAK-1 ultimatelyresults in the activation of additional protein kinases (e.g. IκB kinase,JNK and p38 MAP kinase) as well as transcription factors (e.g. NF-κB),which then up-regulate expression of inflammatory genes (e.g. TNFand IL-6) [2,5–7].
Excessive systemic release of inflammatory cytokines by macro-phages during the course of a bacterial infection can cause substantialtissue damage, potentially resulting in multi-organ failure and circula-tory collapse (i.e. septic shock), and even death [10]. Tolerance is anadaptivemechanismthat limits the extentanddurationof inflammatorycytokine production by preventing the re-activation of macrophages bybacterial pathogens and/or their products. For example, the exposure ofmacrophages to LPS blocks their subsequent re-activation by LPS [11].Several other TLR ligands (e.g. CpG DNA, lipoteichoic acid and flagellin)have also been shown to induce tolerance in macrophages [12–16].Various molecular explanations for tolerance have been suggestedincluding down-regulation of TLR4 expression [17], expression of aninhibitory MyD88 splice variant [18], as well as changes in the com-position of NF-κB complexes [19]. Stress response proteins, such asHsp90, may also be involved in tolerance [20].
IRAK-1 is a key molecule in TLR signalling (e.g. linking MyD88 toTRAF6) and, as such, its rapid TLR-induced degradation has beenproposed to represent a major mechanism for tolerance [13,15,16,21–24]. However, we found that LPS- and CpG DNA-induced IRAK-1

247D. De Nardo et al. / Cellular Signalling 21 (2009) 246–252
degradation was not sufficient to cause tolerance in primary macro-phages; instead tolerance correlated with IRAK-4 down-regulation. Inaddition, siRNA-mediated knockdown of IRAK-4 expression had agreater inhibitory effect on CpG DNA-induced TNF expression than dida similar reduction in IRAK-1 expression. Therefore, we propose thatTLR ligand-induced down-regulation of IRAK-4 is important for es-tablishing tolerance in macrophages.
2. Experimental procedures
2.1. Reagents
Cell culture medium and supplements, foetal calf serum, Super-Script III reverse transcriptase, pre-cast 10% SDS-PAGE gels and anti-V5 antibodies were from Invitrogen. Recombinant human colonystimulating factor-1 (CSF-1) was a generous gift from Chiron.Ultrapure lipopolysaccharide (E. coli 0111:B4) and the CpG DNAoligonucleotide 1668 were from InvivoGen. The mouse monoclonalanti-IRAK-1 antibody was obtained from Santa Cruz Biotechnology,Inc., while the rabbit polyclonal anti-IRAK-4 antibody was from UBI,Inc. The anti-IκBα, anti-phospho-Erk1/2, anti-phospho-JNK, anti-phospho-p38 MAP kinase, anti-p38 MAP kinase, and anti-Mycantibodies were from Cell Signaling Technology. HRP-conjugatedanti-FLAG and agarose-coupled anti-FLAG antibodies were obtainedfrom Sigma. Complete™ protease inhibitors were supplied by RocheBiochemicals and HiPerFect transfection reagent was from Qiagen.ON-TARGETplus SMARTpool siRNAs targeting mouse IRAK-1 andIRAK-4 were supplied by Dharmacon (catalogue numbers L-040116-00 and L-061264-00, respectively).
2.2. Plasmids
The vector pEF-V5-IRAK-1, which expresses an N-terminal V5-tagged version of mouse IRAK-1, was created by PCR using the primerpair: F1 (5′-ACG CGT GCC GGG GGG CCG GGC CCC GGG-3′) and R1 (5′-ACG CGT TCA GCT CTG GAA TTC ATC ACT TTC TTC AGG TCC-3′), and theplasmid pIC-FLAG-IRAK-1 (a generous gift from Dr S. Ghosh; YaleUniversity) as the template. The PCR product generated was digestedwith MluI and cloned in-frame into the MluI site in pEF-V5. A vectorthat expresses a kinase-dead version of FLAG-tagged IRAK-4 wascreated by overlapping PCR using the primer pairs: F2 (5′-ACG CGTAAC AAG CCG TTG ACA CCA TCG ACA TAC ATA-3′) and R2 (5′-AAC CATCGC TCC GAG CGC CGC CAC CGC CAC-3′), and F3 (5′-ACC ATC GTG GCGGTG GCG GCG CTC GGAGCG-3′) and R4 (5′-ACG CGT TTA AGC AGA CATCTC TTG TAG CAG CTG-3′), and the plasmid pTriplEx-IRAK-4(a generous gift from Dr W.C. Yeh; Ontario Cancer Institute) as thetemplate. The final PCR product generated was cut with MluI andcloned in-frame into the MluI site in pEF-FLAG. The expression vectorpcDNA3-Myc-IRAK-2 was a generous gift from Dr L. O'Neill (TrinityCollege; Dublin). The NF-κB luciferase reporter plasmid pELAM-FLwasa generous gift from Dr M. Sweet (University of Queensland), whilstthe Renilla luciferase reporter plasmid pRL-TK was from Promega.
2.3. Cell culture
The use of mice in this study was approved by the MelbourneHealth Animal Ethics Committee. Bone marrow-derived macrophageswere obtained by culturing bone marrow cells from 6 to 8-week old,female C57BL/6 mice in Dulbecco's-modified Eagle's medium (DMEM)supplementedwith 5000 U/mL recombinant CSF-1,10% FCS,100 units/mL Penicillin, 100 µg/mL Streptomycin, and 2 mM GlutaMax-1™ for3 days at 37 °C in a humidified atmosphere of 5% CO2. Non-adherentmacrophage precursors were collected and seeded in the requiredformat; they were then cultured for an additional 3–4 days until ahomogenous population of adherent macrophages was obtained.HEK293T and mouse RAW 264.7 cells were cultured in DMEM sup-
plemented with 10% FCS, 100 units/mL Penicillin, 100 µg/mL Strep-tomycin, and 2 mM GlutaMax-1™.
2.4. Induction of tolerance to TLR ligands
Tolerance was induced by treating bone marrow-derived macro-phages with 100 ng/mL LPS or 0.5 μM CpG DNA (CpG oligonucleotide1668) for 2 and 12 h. The cells were washed twice with serum-freemedium and then incubated in complete medium for 1 h before beingre-stimulated with the same TLR ligand. The tissue culture super-natants were harvested for measuring TNF and IL-6 levels, and thecells retained for the isolation of total RNA and preparation of celllysates.
2.5. Cell lysis, Western blotting, and co-immunoprecipitation assays
Following washing with ice-cold PBS, cells were scraped into NP-40 lysis buffer (20mM Tris–HCl [pH 7.4], 150mMNaCl, 1 mM EDTA, 1%Nonidet P-40, 10% glycerol, 1 mM sodium orthovanadate, 10 mM NaF,10 mM β-glycerophosphate, and Complete™ protease inhibitors) andincubated on ice for 30 min. The lysates were then clarified by cen-trifugation at 13,000 ×g for 10 min at 4 °C and the protein concen-trations of the whole cell lysates measured using a Bio-Rad proteinassay kit. Lysates were subjected to electrophoresis on 10% SDS-PAGEgels followed by Western blotting according to standard techniques.Immunoreactive bands were visualised using ECL reagents (Amer-sham Bioscience) and exposure to X-ray film (Fuji). Films werescanned using either a GS710 or GS800 Calibrated Imaging Densit-ometer (Bio-Rad) and the resulting images exported as TIFF files.FLAG-tagged kinase-dead IRAK-4 (i.e. FLAG-IRAK-4 KD) was immu-noprecipitated from cell lysates using agarose-coupled anti-FLAGantibodies. Following continual mixing for 3 h at 4 °C, the im-munoprecipitates were washed four times with lysis buffer and thensubjected to Western blotting.
2.6. Real-time PCR analysis
Total RNA was isolated from macrophages using RNeasy Mini kits(QIAGEN). The RNA was reverse-transcribed using SuperScript IIIreverse transcriptase (Invitrogen) and then converted into cDNA.Quantitative PCR was performed using an ABI PRISM 7900HT se-quence detection system and pre-developed TaqMan probe/primercombinations for TNF, IRAK-4, IRAK-M, and 18S rRNA (ABI). RelativemRNA levels were then calculated by transforming the threshold cyclenumbers using the ΔΔCt and relative value method as described by themanufacturer [20].
2.7. Measurement of cytokine levels by ELISA
TNF and IL-6 levels in cell culture supernatants were quantified byELISA using mouse OptEIA Enzyme Immunoassay kits (BD Pharmin-gen) according to the manufacturer's instructions.
2.8. NF-κB reporter assays
HEK293T cells were seeded in 12-well tissue culture plates at adensity of 4×105 cells per well and transfected the next day usingFuGENE 6™ transfection reagent. Cells were co-transfected with100 ng pELAM-FL, 10 ng pRL-TK, and 5–50 ng of pEF-V5-IRAK-1 andpcDNA3-Myc-IRAK-2 expression plasmids. The total amount of DNAtransfectedwas kept constant using empty vector where required. Thecells were lysed 6 h post-transfection using Passive lysis buffer andassayed for firefly and Renilla luciferase activity using the Dual-Glo™
luciferase assay system (Promega). The level of Renilla luciferaseactivity was used to normalise the transfection efficiencies.
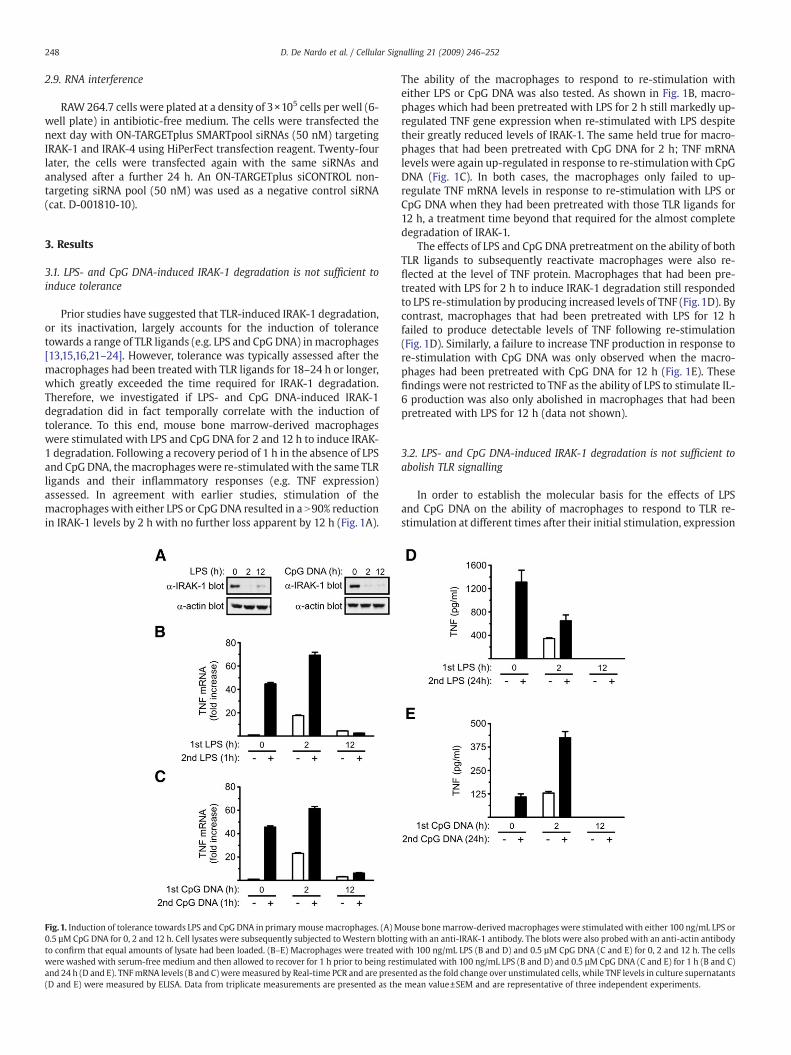
248 D. De Nardo et al. / Cellular Signalling 21 (2009) 246–252
2.9. RNA interference
RAW264.7 cells were plated at a density of 3×105 cells per well (6-well plate) in antibiotic-free medium. The cells were transfected thenext day with ON-TARGETplus SMARTpool siRNAs (50 nM) targetingIRAK-1 and IRAK-4 using HiPerFect transfection reagent. Twenty-fourlater, the cells were transfected again with the same siRNAs andanalysed after a further 24 h. An ON-TARGETplus siCONTROL non-targeting siRNA pool (50 nM) was used as a negative control siRNA(cat. D-001810-10).
3. Results
3.1. LPS- and CpG DNA-induced IRAK-1 degradation is not sufficient toinduce tolerance
Prior studies have suggested that TLR-induced IRAK-1 degradation,or its inactivation, largely accounts for the induction of tolerancetowards a range of TLR ligands (e.g. LPS and CpG DNA) in macrophages[13,15,16,21–24]. However, tolerance was typically assessed after themacrophages had been treated with TLR ligands for 18–24 h or longer,which greatly exceeded the time required for IRAK-1 degradation.Therefore, we investigated if LPS- and CpG DNA-induced IRAK-1degradation did in fact temporally correlate with the induction oftolerance. To this end, mouse bone marrow-derived macrophageswere stimulated with LPS and CpG DNA for 2 and 12 h to induce IRAK-1 degradation. Following a recovery period of 1 h in the absence of LPSand CpGDNA, themacrophages were re-stimulatedwith the same TLRligands and their inflammatory responses (e.g. TNF expression)assessed. In agreement with earlier studies, stimulation of themacrophages with either LPS or CpG DNA resulted in a N90% reductionin IRAK-1 levels by 2 h with no further loss apparent by 12 h (Fig. 1A).
Fig.1. Induction of tolerance towards LPS and CpG DNA in primarymousemacrophages. (A) M0.5 µM CpG DNA for 0, 2 and 12 h. Cell lysates were subsequently subjected toWestern blottito confirm that equal amounts of lysate had been loaded. (B–E) Macrophages were treated wwere washed with serum-free medium and then allowed to recover for 1 h prior to being resand 24 h (D and E). TNFmRNA levels (B and C) weremeasured by Real-time PCR and are prese(D and E) were measured by ELISA. Data from triplicate measurements are presented as the
The ability of the macrophages to respond to re-stimulation witheither LPS or CpG DNA was also tested. As shown in Fig. 1B, macro-phages which had been pretreated with LPS for 2 h still markedly up-regulated TNF gene expression when re-stimulated with LPS despitetheir greatly reduced levels of IRAK-1. The same held true for macro-phages that had been pretreated with CpG DNA for 2 h; TNF mRNAlevels were again up-regulated in response to re-stimulationwith CpGDNA (Fig. 1C). In both cases, the macrophages only failed to up-regulate TNF mRNA levels in response to re-stimulation with LPS orCpG DNA when they had been pretreated with those TLR ligands for12 h, a treatment time beyond that required for the almost completedegradation of IRAK-1.
The effects of LPS and CpG DNA pretreatment on the ability of bothTLR ligands to subsequently reactivate macrophages were also re-flected at the level of TNF protein. Macrophages that had been pre-treated with LPS for 2 h to induce IRAK-1 degradation still respondedto LPS re-stimulation by producing increased levels of TNF (Fig.1D). Bycontrast, macrophages that had been pretreated with LPS for 12 hfailed to produce detectable levels of TNF following re-stimulation(Fig. 1D). Similarly, a failure to increase TNF production in response tore-stimulation with CpG DNA was only observed when the macro-phages had been pretreated with CpG DNA for 12 h (Fig. 1E). Thesefindings were not restricted to TNF as the ability of LPS to stimulate IL-6 production was also only abolished in macrophages that had beenpretreated with LPS for 12 h (data not shown).
3.2. LPS- and CpG DNA-induced IRAK-1 degradation is not sufficient toabolish TLR signalling
In order to establish the molecular basis for the effects of LPSand CpG DNA on the ability of macrophages to respond to TLR re-stimulation at different times after their initial stimulation, expression
ouse bonemarrow-derivedmacrophages were stimulatedwith either 100 ng/mL LPS orng with an anti-IRAK-1 antibody. The blots were also probed with an anti-actin antibodyith 100 ng/mL LPS (B and D) and 0.5 µM CpG DNA (C and E) for 0, 2 and 12 h. The cellstimulated with 100 ng/mL LPS (B and D) and 0.5 µM CpG DNA (C and E) for 1 h (B and C)nted as the fold change over unstimulated cells, while TNF levels in culture supernatantsmean value±SEM and are representative of three independent experiments.

249D. De Nardo et al. / Cellular Signalling 21 (2009) 246–252
levels and activation states of proteins involved in TLR4 and TLR9signalling were examined. Stimulation of naïvemacrophages with LPSresulted in the rapid degradation of IRAK-1 and IκBα; it also resultedin the activation of Erk1/2, JNK and p38 MAP kinase (Fig. 2A). Similarobservations were made when macrophages were stimulated withCpG DNA (Fig. 2B). Despite the fact that IRAK-1 levels in macrophagesthat had been pretreated with LPS or CpG DNA for 2 h were greatlyreduced (N90%), TLR4 and TLR9 signalling was still intact, albeit themagnitude and kinetics of signallingwere altered, when the cells werere-stimulated (Fig. 2A and B). By contrast, TLR4 and TLR9 signallingwas abolished in macrophages that had been pretreated with LPS orCpG DNA for 12 h (Fig. 2A and B). Thus, TLR-induced IRAK-1 degra-dation may only be sufficient to partially suppress TLR signalling.
3.3. Binding to IRAK-4 and activation of NF-κB by IRAK-2
It has recently been suggested that IRAK-1 and IRAK-2 functionredundantly to mediate TLR signalling [25]. Therefore, the ability ofmacrophages that had been pretreated with LPS or CpG DNA for 2 h torespond toTLR re-stimulationmay have been due to IRAK-4mediatingTLR signalling via IRAK-2 in the absence of IRAK-1. This possibilitywas tested by transiently co-expressing FLAG-tagged IRAK-4 with V5-
Fig. 2. TLR signalling in LPS- and CpG DNA-pretreated macrophages. Mouse bonemarrow-derivedmacrophages were stimulated with 100 ng/mL LPS (A) and 0.5 µM CpGDNA (B) for 0, 2 or 12 h. The cells were washed with serum-free medium and thenallowed to recover for 1 h prior to being restimulated with 100 ng/mL LPS (A) and0.5 µM CpG DNA (B) for 0, 30, 60 and 90min. Cell lysates were subsequently subjected toWestern blotting with the indicated antibodies. The blots were also probed with ananti-p38 antibody to confirm that equal amounts of cell protein had been loaded. Thedata are representative of three independent experiments.
Fig. 3. Comparative interaction of IRAK-1 and IRAK-2with IRAK-4 and activation of NF-κB.(A) HEK293T cells were transiently transfected with plasmids expressing V5-taggedIRAK-1, Myc-tagged IRAK-2 and FLAG-tagged kinase-dead IRAK-4. Twenty-four h laterIRAK-4 was immunoprecipitated from lysates of the transfected cells using anti-FLAGantibodies. The immunoprecipitateswere then subjected toWestern blottingwith theindicated antibodies to detect the co-immunoprecipitation of V5-IRAK-1 and Myc-IRAK-2. The whole cell lysates (WCL) were also subjected to Western blotting.(B) HEK293T cells were transfected with a NF-κB luciferase reporter plasmid alongwith the indicated amounts of V5-IRAK-1 and Myc-IRAK-2 expression plasmids.Luciferase activity was subsequently measured using the Dual-Glo™ luciferase assaysystem. NF-κB reporter activity is given as fold increase over levels in cells notexpressing V5-IRAK-1 orMyc-IRAK-2. Data from duplicate transfections are presentedas the mean value±SEM and are representative of three independent experiments.
IRAK-1 and Myc-IRAK-2 in HEK293T cells; complex formation wasthen assessed via co-immunoprecipitation assays. As shown in Fig. 3A,both IRAK-1 and IRAK-2 were capable of forming complexes withIRAK-4. The relative abilities of IRAK-1 and IRAK-2 to activate NF-κBwere also tested using a luciferase-based reporter assay. The activity ofthe NF-κB reporter plasmid was markedly increased upon its co-transfection with plasmids expressing IRAK-1 and IRAK-2 (Fig. 3B).Interestingly, IRAK-2 appeared to be more effective than IRAK-1 atinducing NF-κB reporter activity when the cells were transfected withrelatively low levels of the IRAK-1 and IRAK-2 expression plasmids; athigher plasmid concentrations, however, IRAK-1 was the better acti-vator of NF-κB (Fig. 3B).
3.4. Temporal correlation of IRAK-4 down-regulation with the inductionof tolerance
Because of the importance of IRAK-4 in TLR signalling [26,27] weinvestigated the effects of LPS and CpG DNA on IRAK-4 expression. Asshown in Fig. 4A, IRAK-4 levels were reduced ~25% inmacrophages thathad been stimulatedwith LPS for 2 h (Fig. 4A). A comparable decrease inIRAK-4 levels was also apparent in macrophages that had beenstimulated with CpG DNA for the same time (Fig. 4B). However, IRAK-4 levels had declined by around 60% and 80% in response to LPS and CpGDNA, respectively, after 12 h (Fig. 4A and B). Given these findings, weexamined more closely the kinetics of IRAK-4 down-regulation by LPSand CpG DNA. A ~50% decrease in IRAK-4 levels occurred within 8 h of

Fig. 4. Down-regulation of IRAK-4 levels in LPS- and CpG DNA-stimulated macrophages.Mouse bone marrow-derived macrophages were stimulated with 100 ng/mL LPS (A) and0.5 µM CpG DNA (B) for 0, 2 or 12 h. The cells were washed with serum-free medium andthen allowed to recover for 1 h prior to being restimulated with 100 ng/mL LPS (A) and0.5 µM CpG DNA (B) for 0, 30, 60 and 90 min. Cell lysates were subsequently subjected toWestern blotting with an anti-IRAK-4 antibody. The blots were also probed with an anti-actin antibody to confirm that equal amounts of cell protein had been loaded.
250 D. De Nardo et al. / Cellular Signalling 21 (2009) 246–252
LPS stimulation,whichwas reduced furtherby24h (Fig. 5AandB). In thecase of CpG DNA stimulation, the rate and extent of the reduction wasgreater than that achieved with LPS (Fig. 5A–D). While IRAK-1 levelsdeclined rapidly initially (N90% decrease after 2 h), they began to berestored around 16 h post-stimulation (Fig. 5A and C). Together, thesefindings suggest that down-regulation of IRAK-4 may contribute to theinduction of tolerance towards LPS and CpG DNA.
3.5. Down-regulation of IRAK-4 mRNA and protein by LPS and CpG DNA
The mechanism underlying the down-regulation of IRAK-4 wasinvestigated by first testing the effects of LPS and CpG DNA on IRAK-4
Fig. 5. Kinetics of LPS- and CpG DNA-induced down-regulation of IRAK-4 protein. Mousebone marrow-derived macrophages were stimulated with 100 ng/mL LPS (A and B) and0.5 µM CpG DNA (C and D) for the indicated time. Cell lysates were subsequentlysubjected to Western blotting with anti-IRAK-4 and anti-IRAK-1 antibodies. The blotswere also probed with an anti-p38 antibody to confirm that equal amounts of cellprotein had been loaded. (B and D) Quantified data presented as the expression level ofIRAK-4 relative to that in untreated macrophages. Data from at least three independentexperiments are presented as the mean value±SEM.
mRNA. Both LPS and CpG DNA induced a relatively rapid decrease(~50–60%) in IRAK-4 mRNA levels, which then remained at thesereduced levels for at least a further 18 h (Fig. 6A). It was not possible todirectly test if the reduction in IRAK-4 mRNA levels was due to areduction in IRAK-4 gene expression and/or a decrease in the half-lifeof IRAK-4 transcripts as simultaneous treatment of the macrophageswith actinomycin D and LPS or CpG DNA resulted in rapid cell death(data not shown). Concomitant with the reduction in IRAK-4 mRNAwas an increase in the levels of IRAK-M transcripts (Fig. 6B).
To establish if a decrease in IRAK-4 mRNA could account for thedecrease in IRAK-4 protein levels in response to LPS and CpG DNAstimulationwe determined the half-life of IRAK-4 protein.Macrophageswere treated with the protein synthesis inhibitor cycloheximide over atime course of 8 h and IRAK-4 levels then measured. No significantdecrease in IRAK-4was observed over the time-course of cycloheximidetreatment (Fig. 6C); thus indicating that its half-life in unstimulatedmacrophages is greater than8h. Thehalf-life of IRAK-1was also found tobe greater than 8 h (Fig. 6C), which was consistent with an earlier study[20]. The fact that CSF-1 receptor levels quickly declined under the sameconditions (Fig. 6C) confirmed the concentration of cycloheximide usedwas sufficient to inhibit protein synthesis as the CSF-1 receptor isinternalised and degraded following its ligand-induced activation [28].Taken together, our findings indicate that LPS- and CpG DNA-induced
Fig. 6. Regulation of IRAK-4 mRNA and protein levels by LPS and CpG DNA. (A and B)Mouse bone marrow-derived macrophages were stimulated with 100 ng/mL LPS (■)and 0.5 µM CpG DNA (○) for the indicated time. (A) IRAK-4 and (B) IRAK-MmRNA levelswere subsequently measured by Real-time PCR. IRAK-4 and IRAK-M mRNA levels inunstimulated macrophages was given an arbitrary value of 1.0. Data from fourindependent experiments (triplicate measurements) are presented as the mean value±SEM. (C) Macrophages were treated with 10 μg/mL cycloheximide for the indicatedtime. Cell lysates were subsequently subjected to Western blotting with anti-IRAK-4,anti-IRAK-1 and anti-CSF-1 receptor (CSF-1R) antibodies. The blots were also probedwith an anti-p38MAP kinase antibody to confirm that equal amounts of cell protein hadbeen loaded.
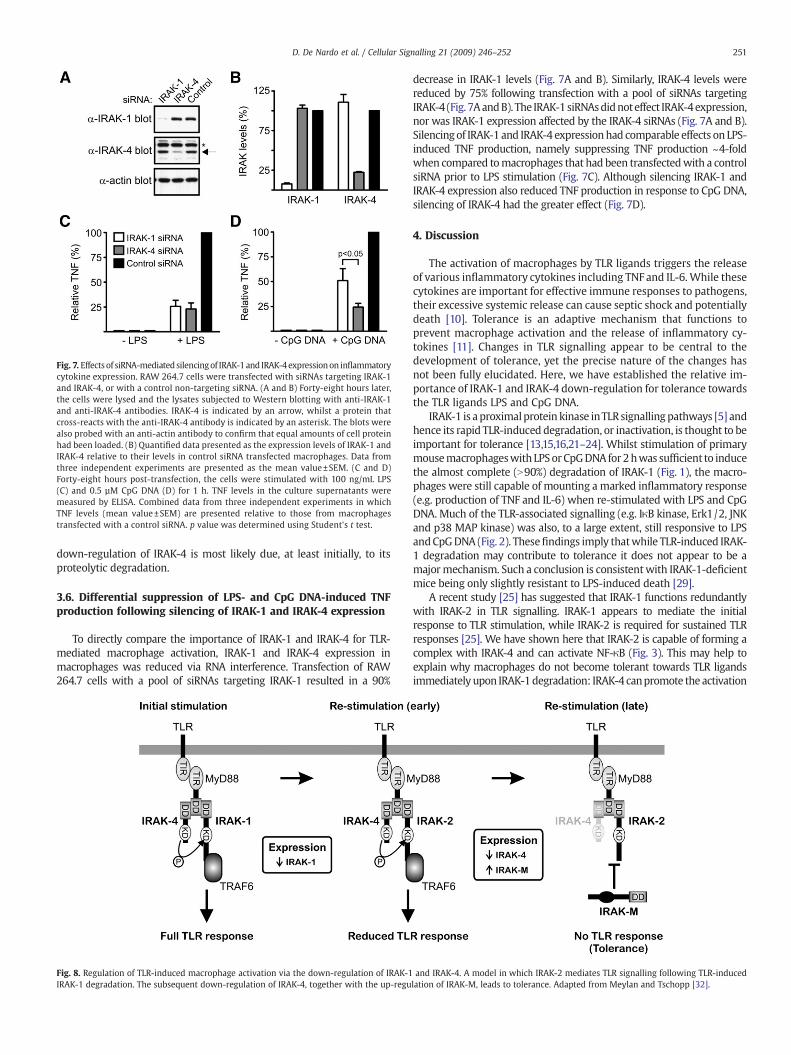
Fig. 7. Effects of siRNA-mediated silencingof IRAK-1and IRAK-4expressionon inflammatorycytokine expression. RAW 264.7 cells were transfected with siRNAs targeting IRAK-1and IRAK-4, or with a control non-targeting siRNA. (A and B) Forty-eight hours later,the cells were lysed and the lysates subjected to Western blotting with anti-IRAK-1and anti-IRAK-4 antibodies. IRAK-4 is indicated by an arrow, whilst a protein thatcross-reacts with the anti-IRAK-4 antibody is indicated by an asterisk. The blots werealso probed with an anti-actin antibody to confirm that equal amounts of cell proteinhad been loaded. (B) Quantified data presented as the expression levels of IRAK-1 andIRAK-4 relative to their levels in control siRNA transfected macrophages. Data fromthree independent experiments are presented as the mean value±SEM. (C and D)Forty-eight hours post-transfection, the cells were stimulated with 100 ng/mL LPS(C) and 0.5 µM CpG DNA (D) for 1 h. TNF levels in the culture supernatants weremeasured by ELISA. Combined data from three independent experiments in whichTNF levels (mean value±SEM) are presented relative to those from macrophagestransfected with a control siRNA. p value was determined using Student's t test.
251D. De Nardo et al. / Cellular Signalling 21 (2009) 246–252
down-regulation of IRAK-4 is most likely due, at least initially, to itsproteolytic degradation.
3.6. Differential suppression of LPS- and CpG DNA-induced TNFproduction following silencing of IRAK-1 and IRAK-4 expression
To directly compare the importance of IRAK-1 and IRAK-4 for TLR-mediated macrophage activation, IRAK-1 and IRAK-4 expression inmacrophages was reduced via RNA interference. Transfection of RAW264.7 cells with a pool of siRNAs targeting IRAK-1 resulted in a 90%
Fig. 8. Regulation of TLR-induced macrophage activation via the down-regulation of IRAK-1IRAK-1 degradation. The subsequent down-regulation of IRAK-4, together with the up-regu
decrease in IRAK-1 levels (Fig. 7A and B). Similarly, IRAK-4 levels werereduced by 75% following transfection with a pool of siRNAs targetingIRAK-4 (Fig. 7AandB). The IRAK-1 siRNAsdidnoteffect IRAK-4expression,nor was IRAK-1 expression affected by the IRAK-4 siRNAs (Fig. 7A and B).Silencingof IRAK-1 and IRAK-4 expressionhad comparable effects on LPS-induced TNF production, namely suppressing TNF production ~4-foldwhen compared tomacrophages that had been transfectedwith a controlsiRNA prior to LPS stimulation (Fig. 7C). Although silencing IRAK-1 andIRAK-4 expression also reduced TNF production in response to CpG DNA,silencing of IRAK-4 had the greater effect (Fig. 7D).
4. Discussion
The activation of macrophages by TLR ligands triggers the releaseof various inflammatory cytokines including TNF and IL-6.While thesecytokines are important for effective immune responses to pathogens,their excessive systemic release can cause septic shock and potentiallydeath [10]. Tolerance is an adaptive mechanism that functions toprevent macrophage activation and the release of inflammatory cy-tokines [11]. Changes in TLR signalling appear to be central to thedevelopment of tolerance, yet the precise nature of the changes hasnot been fully elucidated. Here, we have established the relative im-portance of IRAK-1 and IRAK-4 down-regulation for tolerance towardsthe TLR ligands LPS and CpG DNA.
IRAK-1 is aproximalprotein kinase inTLRsignallingpathways [5] andhence its rapid TLR-induced degradation, or inactivation, is thought to beimportant for tolerance [13,15,16,21–24]. Whilst stimulation of primarymousemacrophageswith LPSor CpGDNA for 2hwas sufficient to inducethe almost complete (N90%) degradation of IRAK-1 (Fig. 1), the macro-phages were still capable of mounting a marked inflammatory response(e.g. production of TNF and IL-6) when re-stimulated with LPS and CpGDNA. Much of the TLR-associated signalling (e.g. IκB kinase, Erk1/2, JNKand p38 MAP kinase) was also, to a large extent, still responsive to LPSandCpGDNA (Fig. 2). Thesefindings imply thatwhile TLR-induced IRAK-1 degradation may contribute to tolerance it does not appear to be amajormechanism. Such a conclusion is consistentwith IRAK-1-deficientmice being only slightly resistant to LPS-induced death [29].
A recent study [25] has suggested that IRAK-1 functions redundantlywith IRAK-2 in TLR signalling. IRAK-1 appears to mediate the initialresponse to TLR stimulation, while IRAK-2 is required for sustained TLRresponses [25]. We have shown here that IRAK-2 is capable of forming acomplex with IRAK-4 and can activate NF-κB (Fig. 3). This may help toexplain why macrophages do not become tolerant towards TLR ligandsimmediately upon IRAK-1degradation: IRAK-4 canpromote the activation
and IRAK-4. A model in which IRAK-2 mediates TLR signalling following TLR-inducedlation of IRAK-M, leads to tolerance. Adapted from Meylan and Tschopp [32].

252 D. De Nardo et al. / Cellular Signalling 21 (2009) 246–252
of TRAF6, and ultimately that of NF-κB and MAP kinases, via IRAK-2(Fig. 8). Our finding that IRAK-2 is not as effective as IRAK-1 in activatingNF-κB may also help to explain why TLR signalling is intact for severalhours after the initial exposure of macrophages to LPS and CpG DNA butthe magnitude of signalling is partially reduced, and the kinetics delayed.
We, aswell asothers [30], havenowshown that someTLR ligands canalso induce the down-regulation of IRAK-4 (Figs. 4 and 5). Proteolyticdegradation of IRAK-4 appears to be themajormechanismunderlying itsLPS- and CpG DNA-induced down-regulation. However, we cannotexclude the possibility that its down-regulation may also be due todecreases in IRAK-4mRNA levels (Fig. 6). This contrasts with Hatao et al.[30], who reported that IRAK-4 mRNA levels were unaffected by TLRligands. The reason for this discrepancy is unclear, but itmay relate to theuse of primary macrophages (in this report) versus a macrophage cellline (Hatao et al.). Whether IRAK-M, a TLR-induced and catalytically-inactive protein kinase that inhibits TLR signalling by preventing thedissociation of IRAK-1/IRAK-4 from MyD88 [31], plays a role in IRAK-4degradation has not been explored. While the kinetics of IRAK-4 down-regulation by LPS and CpG DNA were different, the magnitude andkinetics of their induction of IRAK-M were similar. Thus, IRAK-4 degra-dation most likely occurs independently of its interaction with IRAK-M.
A marked reduction (e.g. N50% decrease) in IRAK-4 levels onlyoccurred after the macrophages had been stimulated for around 8 h orlonger, by which time they were also no longer responsive to TLR re-stimulation. This temporal correlation suggests IRAK-4 down-regulationmaybe a keyevent in thedevelopmentof tolerance towards LPS andCpGDNA. Indeed, siRNA-mediated down-regulation of IRAK-4, to levelscomparablewith those inmacrophages that hadbeen treatedwith LPSorCpG DNA for 12 h to induce tolerance, was sufficient to suppress TNFproduction in response to TLR stimulation. However, overexpression ofIRAK-4 in RAW 264.7 cells, at levels ~5-fold higher than those of endo-genous IRAK-4, did not appear to be sufficient to prevent tolerance (datanot shown). This would suggest that down-regulation of IRAK-4 is un-likely to completely explain tolerance; instead a number of mechanismsare likely to be involved. Specifically, the coordinated and overlappingkinetics of IRAK-4 down-regulationwith the up-regulation of IRAK-M byTLR ligands may in fact provide a mechanismwhereby TLR signalling isgradually dampened prior to the establishment of tolerance (Fig. 8).
Along similar lines, the different kinetics of TLR-induced IRAK-4 andIRAK-1 down-regulationmay be important in determining the nature ofthe inflammatory response. For example, both quantitative and qual-itative differences in TLR signalling are likely to occur as a consequenceof the different rates at which IRAK-4 and IRAK-1 are down-regulated.Such modulation of the repertoires of signalling molecules activated, aswell as themagnitude and duration of their activation,may enablemac-rophages to temporally tailor their inflammatory response topathogens.In the same way, differences in the kinetics and extent of IRAK-4 down-regulation by TLR ligands (e.g. LPS versus CpG DNA) may provide amechanism whereby macrophages can modify their inflammatory re-sponse depending on the location (e.g. extracellular versus endosomal)and/or type of pathogen detected.
5. Conclusions
We have established that TLR-induced IRAK-1 degradation isinsufficient to cause tolerance, most likely because IRAK-2 can mediate
TLR signalling in its absence. By contrast, down-regulation of IRAK-4appears to be important for LPS- and CpG DNA-induced tolerance.Differences in the kinetics and degree of IRAK-4 down-regulation by TLRligands may allow macrophages to tailor their inflammatory responseaccording to the location and/or type of pathogen detected, while thediffering kinetics of IRAK-4 and IRAK-1 down-regulation may enablethem to temporally modify their inflammatory response.
Acknowledgements
The authorswish to thankDrs S. Ghosh, L. O'Neill,M. Sweet, andW.C.Yeh for generously providing plasmids. This work was supported in partby the Cooperative Research Centre for Chronic Inflammatory Diseasesand the National Health and Medical Research Council.
References
[1] R. Medzhitov, C.A. Janeway Jr., Semin. Immunol. 10 (1998) 351–353.[2] C.A. Janeway Jr., R. Medzhitov, Annu. Rev. Immunol. 20 (2002) 197–216.[3] D.T. Fearon, R.M. Locksley, Science 272 (1996) 50–53.[4] S. Gordon, Res. Immunol. 149 (1998) 685–688.[5] S. Akira, S. Uematsu, O. Takeuchi, Cell 124 (2006) 783–801.[6] B. Beutler, K. Hoebe, X. Du, R.J. Ulevitch, J. Leukoc. Biol. 74 (2003) 479–485.[7] S.M. Miggin, L.A. O'Neill, J. Leukoc. Biol. 80 (2006) 220–226.[8] K. Hoshino, O. Takeuchi, T. Kawai, H. Sanjo, T. Ogawa, Y. Takeda, K. Takeda, S. Akira,
J. Immunol. 162 (1999) 3749–3752.[9] T.H. Chuang, J. Lee, L. Kline, J.C. Mathison, R.J. Ulevitch, J. Leukoc. Biol. 71 (2002)
538–544.[10] E.S. van Amersfoort, T.J. van Berkel, J. Kuiper, Clin. Microbiol. Rev. 16 (2003)
379–414.[11] E. Zeisberger, J. Roth, Ann. N.Y. Acad. Sci. 856 (1998) 116–131.[12] M.A. Dobrovolskaia, A.E. Medvedev, K.E. Thomas, N. Cuesta, V. Toshchakov, T. Ren,
M.J. Cody, S.M. Michalek, N.R. Rice, S.N. Vogel, J. Immunol. 170 (2003) 508–519.[13] R. Jacinto, T. Hartung, C. McCall, L. Li, J. Immunol. 168 (2002) 6136–6141.[14] S.B. Mizel, J.A. Snipes, J. Biol. Chem. 277 (2002) 22414–22420.[15] M. Siedlar, M. Frankenberger, E. Benkhart, T. Espevik, M. Quirling, K. Brand, M.
Zembala, L. Ziegler-Heitbrock, J. Immunol. 173 (2004) 2736–2745.[16] S.J. Yeo, J.G. Yoon, S.C. Hong, A.K. Yi, J. Immunol. 170 (2003) 1052–1061.[17] F. Nomura, S. Akashi, Y. Sakao, S. Sato, T. Kawai, M. Matsumoto, K. Nakanishi, M.
Kimoto, K. Miyake, K. Takeda, S. Akira, J. Immunol. 164 (2000) 3476–3479.[18] K. Burns, S. Janssens, B. Brissoni, N. Olivos, R. Beyaert, J. Tschopp, J. Exp. Med. 197
(2003) 263–268.[19] H.W. Ziegler-Heitbrock, A.Wedel,W. Schraut,M. Strobel, P.Wendelgass, T. Sternsdorf,
P.A. Bauerle, J.G. Haas, G. Riethmuller, J. Biol. Chem. 269 (1994) 17001–17004.[20] D. De Nardo, P. Masendycz, S. Ho, M. Cross, A.J. Fleetwood, E.C. Reynolds, J.A.
Hamilton, G.M. Scholz, J. Biol. Chem. 280 (2005) 9813–9822.[21] M. Adib-Conquy, J.M. Cavaillon, J. Biol. Chem. 277 (2002) 27927–27934.[22] C.H. Li, J.H. Wang, H.P. Redmond, J. Leukoc. Biol. 79 (2006) 867–875.[23] L. Li, S. Cousart, J. Hu, C.E. McCall, J. Biol. Chem. 275 (2000) 23340–23345.[24] A.E. Medvedev, A. Lentschat, L.M. Wahl, D.T. Golenbock, S.N. Vogel, J. Immunol. 169
(2002) 5209–5216.[25] T. Kawagoe, S. Sato, K. Matsushita, H. Kato, K. Matsui, Y. Kumagai, T. Saitoh, T.
Kawai, O. Takeuchi, S. Akira, Nat. Immunol. 9 (2008) 684–691.[26] T. Kawagoe, S. Sato, A. Jung, M. Yamamoto, K. Matsui, H. Kato, S. Uematsu, O.
Takeuchi, S. Akira, J. Exp. Med. 204 (2007) 1013–1024.[27] N. Suzuki, S. Suzuki, G.S. Duncan, D.G. Millar, T. Wada, C. Mirtsos, H. Takada, A.
Wakeham, A. Itie, S. Li, J.M. Penninger, H. Wesche, P.S. Ohashi, T.W. Mak, W.C. Yeh,Nature 416 (2002) 750–756.
[28] J.R. Downing, C.W. Rettenmier, C.J. Sherr, Mol. Cell. Biol. 8 (1988) 1795–1799.[29] J.L. Swantek, M.F. Tsen, M.H. Cobb, J.A. Thomas, J. Immunol. 164 (2000) 4301–4306.[30] F. Hatao, M. Muroi, N. Hiki, T. Ogawa, Y. Mimura, M. Kaminishi, K. Tanamoto,
J. Leukoc. Biol. 76 (2004) 904–908.[31] K. Kobayashi, L.D. Hernandez, J.E. Galan, C.A. Janeway Jr., R. Medzhitov, R.A. Flavell,
Cell 110 (2002) 191–202.[32] E. Meylan, J. Tschopp, Nat. Immunol. 9 (2008) 581–582.




















