
Temperature dependence of macrobending loss in all-fiber bend loss edge filter

Does inbreeding and loss of genetic diversity decrease disease resistance??
Derek Spielman1,2, Barry W. Brook3, David A. Briscoe1 & Richard Frankham1,4,*1Key Centre for Biodiversity and Bioresources, Department of Biological Sciences, Macquarie University,NSW 2109, Australia; 2Present address: Ocean Park, Aberdeen, Hong Kong; 3Key Centre for TropicalWildlife Management, Charles Darwin University, Darwin, NT 0909, Australia; 4School of Tropical Biology,James Cook University, Townsville, QLD 4811, Australia (*Author for correspondence: Key centre forBiodiversity and Bioresources, Department of Biological Sciences, Macquarie University, NSW 2109,Australia; fax: +612-9850-8245; e-mail: [email protected])
Received 23 May 2003; accepted 1 October 2004
Key words: disease resistance, Drosophila melanogaster, genetic diversity, inbreeding, population size
Abstract
Inbreeding and loss of genetic diversity are predicted to decrease the resistance of species to disease.However, this issue is controversial and there is limited rigorous scientific evidence available. To testwhether inbreeding and loss of genetic diversity affect a host’s resistance to disease, Drosophila melano-gaster populations with different levels of inbreeding and genetic diversity were exposed separately to (a)thuringiensin, an insecticidal toxin produced by some strains of Bacillus thuringiensis, and (b) live Serratiamarcescens bacteria. Inbreeding and loss of genetic diversity significantly reduced resistance of D. mela-nogaster to both the thuringiensin toxin and live Serratia marcescens. For both, the best fitting relationshipsbetween resistance and inbreeding were curvilinear. As expected, there was wide variation among replicateinbred populations in disease resistance. Lowered resistances to both the toxin and the pathogen in inbredpopulations were due to specific resistance alleles, rather than generalized inbreeding effects, as correlationsbetween resistance and population fitness were low or negative. Wildlife managers should strive to minimiseinbreeding and loss of genetic diversity within threatened populations and to minimise exposure of inbredpopulations to disease.
Introduction
Infectious diseases are ubiquitous, and areamongst the most important and common envi-ronmental challenge encountered by species andindividuals (Haldane 1949; Weatherall 2003).Contagious diseases, whether induced by viruses,prokaryotes, or eukaryotes, are an important anduniversal selective evolutionary force (Seal 1991).Pathogens can severely affect all populations,including humans (see Dobson and May 1986;
Nicholas 1987; Meltzer 1993; Real 1996; Diamond1998).
The zoological literature is replete with exam-ples of diseases that have devastated populations,including threatened species. Pathogens intro-duced into Hawaii caused the extinction of nearlyhalf the endemic terrestrial bird species (Warner1968; van Riper et al. 1986). In the 1890’s rinder-pest from introduced cattle eliminated 90–95% ofthe wildebeest and cape buffalo herds in EastAfrica (Plowright 1982; O’Brien and Evermann1988). Pathogens that infect multiple species canseverely affect endangered species (McCallum andDobson 1995). For example, a devastating caninedistemper epizootic almost exterminated a tiny
?February–30 June 2004: Program for Evolutionary Dynamics,
Harvard University, One Brattle Square level 6, Cambridge
MA 02138, USA.
Conservation Genetics 5: 439–448, 2004.� 2004 Kluwer Academic Publishers. Printed in the Netherlands.
439

relict population of black-footed ferrets (Mustelanigripes). There are a growing number of caseswhere diseases have harmed endangered species(e.g., Warner 1968; Dobson and May 1986; vanRiper et al. 1986; O’Brien and Evermann 1988;Thorne and Williams 1988; Jacobsen et al. 1991;Lyles and Dobson 1993; Real 1996; Hedrick et al.2001; Acevedo-Whitehouse et al. 2003). Evidencefor pathogens jumping species boundaries andaffecting threatened species is growing as morespecies come into contact through human actions(Daszak et al. 2000).
Pathogens have a wide range of life cycles,enter hosts by diverse means and have differenteffects following infection (see Aiello and Mays1998). Many cause harm by infecting hosts andinvading tissues, while others produce harmfultoxins. Some toxins are ingested, including Clos-tridium botulinum (Aiello and Mays 1998), toxiccyanobacteria (Blood and Studdert 1999), red-tidealgae and ergot alkaloids from the fungus Clavi-ceps purpurea (Blood and Studdert 1999). Forexample, saxitoxin from the dinoflagellate Alex-andrium minutum has killed critically endangeredMediterranean monk seals (Monachus monachus),endangered sea otters (Enhydra lutris) and hump-back whales (Megaptera novaeanglia). Domoicacid intoxication from marine diatoms has killedpeople, pelicans, cormorants and sea lions, whileciguatoxins from dinoflagellates have killedendangered Hawaiian monk seals (Monachusschauinslandi) (Dieraut and Gulland 2001). Othertoxins are produced within a host, including thosefrom some Escherichia coli strains, tetanus (Clos-tridium tetani), gas gangrene (C. perfringens),malignant oedema (C. septicum), and bacillaryhaemoglobinuria (C. novyi) (Aiello and Mays1998).
Host responses vary according to the mode ofinfection and pathophysiology, includingbehavioural avoidance of pathogens, barriers toinfection, innate immunity (e.g. defensins) sharedby invertebrates and vertebrates, and adaptiveimmunity that is restricted to vertebrates. Severalphysiological mechanisms are used to respond totoxins. No matter what the physiological orbehavioural mechanisms of resistance to patho-gens, the genetic consequences of small populationsize are expected to be the same.
Almost every trait that has been examinedshows genetic variation in large populations
(Lewontin 1974), including disease resistance innatural animal and plant populations (reviewed byBurdon 1987; Parker 1990; Frank 1992; Hedrickand Kim 1999; Frankham et al. 2002). Threatenedpopulations are, by definition, small and ordeclining (Hilton-Taylor 2000). In small popula-tions of randomly mating species, inbreeding andloss of genetic diversity through drift areunavoidable, as indicated by the following equa-tion:
Ht=H0 ¼ ½1� 1=ð2NeÞ�t ¼ 1� F ð1Þ
where Ht is heterozygosity at generation t, H0 theinitial heterozygosity, Ne the effective populationsize and F the inbreeding coefficient (Frankhamet al. 2002). While this equation applies to neutralvariation, alleles subject to selection are also lostby drift (Frankham et al. 2002). Reduced geneticdiversity limits the ability of populations to evolvein response to new pests, climatic changes, habitatdegradation and introduced or evolving parasites(Frankham et al. 2002). Inbreeding results in re-duced reproductive fitness (inbreeding depression)in essentially all well-studied species (see Frank-ham et al. 2002). Generally, the effects ofinbreeding and loss of genetic diversity are insep-arable in naturally outbreeding species, as indi-cated by Equation (1).
There are strong theoretical grounds for sus-pecting that inbreeding and loss of genetic diver-sity will reduce the ability of populations to copewith disease (O’Brien and Evermann 1988; Sorciet al. 1997; Frankham et al. 2002). As genetic driftmay lead to increased, as well as to reducedresistance to particular diseases, replicated studiesare required to evaluate the overall impacts ofinbreeding and loss of genetic diversity. The cur-rent body of evidence is conflicting and oftenlacking in rigour and replication, largely due todifficulties in experimenting with threatened spe-cies, especially in the wild. Inbreeding and lowheterozygosity have been associated with de-creased resistance to infectious pathogens in plantsand animals (see Ferguson and Drahushchak1990; Lively et al. 1990; Black 1992; Roelke et al.1993; Vrijenhoek 1994; Clay and Kover 1996;Lively 1996; Liersch and Schmid-Hempel 1998;Baer and Schmid-Hempel 1999; Coltman et al.1999; Meagher 1999; Schmid-Hempel and Crozier1999; Hedrick et al. 2001; Acevedo-Whitehouse
440

et al. 2003). For example, the endangered Floridapanther has very low levels of genetic diversity andshows a high prevalence of infectious diseases(Roelke et al. 1993). Similarly, the cheetah has lowgenetic variability and suffered high mortality dueto an epizootic of feline infectious peritonitis(O’Brien et al. 1985). In both cases there weresingle, unreplicated populations and no controlsfrom the same species. Conversely, a controlled,replicated experimental study by Stevens et al.(1997) found most inbred flour beetle lineages wereless susceptible to parasitism than the crossbredcontrol population.
The limited and sometimes contradictoryinformation on the relationship between diseaseresistance and inbreeding and the loss of geneticdiversity has lead to scepticism about the rela-tionship (Pimm et al. 1989; Caro and Laurenson1994; Caughley 1994; Merola 1994; May 1995;Elgar and Clode 2001).
It is not practical to study the relationship be-tween inbreeding and the loss of genetic diversityand disease in endangered species as they are rareand too valuable to risk in experiments that arelikely to kill them. Ethical approvals are unlikely tobe granted for such studies in vertebrates. Further,many endangered species are slow breeders andexpensive to maintain. Consequently, such studiescan only realistically be done using laboratoryspecies with short generations where controlled,replicated experiments are possible (Frankham2000). We usedDrosophila melanogaster as a modelorganism to investigate the relationship betweengenetic diversity and resistance to a known insec-ticidal toxin and to an infectious pathogen. Dro-sophila is used extensively as a model to researchcell biology and insect immunity (Flyg et al. 1987),as well as conservation and evolutionary genetics(Frankham et al. 2002).
The choice of experimental pathogen is ex-tremely important to investigations of the effect ofinbreeding and loss of genetic diversity on diseaseresistance. We chose Bacillus thuringiensis andSerratia marcescens for our experiments. B. thur-ingiensis is a soil bacterium that can grow eitherby utilising dead organic matter or by colonisingliving insects. B. thuringiensis acts more like achemical insecticide than an infectious agent(Glazer and Nikaido 1995). It grows rapidly indead larvae and epizootics occur when foodsources become limited, forcing larvae to ingest
infected cadavers (Burges and Hurst 1977).B. thuringiensis is a model for the very wide rangeof pathogens that produce toxins (see above). Theheat stable b-exotoxin, thuringiensin is one ofseveral insecticidal toxins produced by theBerliner strain of B. thuringiensis (Benz 1966;Benz and Graf 1971). The toxin is lethal to D.melanogaster larvae when mixed into the rearingmedium at sufficient concentrations (Marec et al.1988; Paumard-Rigal and Rosenberg-Bourgin1992).
As diseases often involve infections by livingorganisms, we also sought an appropriate andconvenient living infective pathogen. Serratiamarcescens proved to be a suitable organism(Spielman 2002). It is a facultatively anaerobic,gram-negative bacterium found worldwide. It ispathogenic to over 70 species of insects, includingD. melanogaster (Flyg et al. 1980).
The aims of these investigations were todetermine whether inbreeding and loss of geneticvariability in populations decrease their resistanceto disease. Replicate populations of Drosophila,maintained for 50 generations at effective sizes of25, 50, 100, 250 and 500, were compared for theirresistance to B. thuringiensis exotoxin. In a sec-ond study, replicate highly inbred populationsand their outbred base population were comparedfor their abilities to survive infection by liveSerratia marcescens. Correlations between repro-ductive fitness in the absence of toxin or patho-gen and resistance were used to determinewhether differences in resistance among inbredpopulations were due to generalised inbreedingdepression, or to specific alleles conferring diseaseresistance. If resistance is due solely to general-ised inbreeding depression, then the correlationwill not differ significantly from one. Conversely,if resistance is due solely to specific alleles forresistance, the correlation will not differ signifi-cantly from zero. An intermediate positive cor-relation indicates that both mechanisms areinvolved.
Methods
Base populations
An outbred strain of D. melanogaster (T92) wasfounded from 272 wild-inseminated females
441

caught at Tyrrells Winery, Pokolbin, in easternNSW, Australia, in February 1992. Outbred Dro-sophila populations were expanded to 1000 adultparents per generation as rapidly as possible (bythe second generation in captivity) and maintainedat that size in 20 · 270 ml bottles per generation(about 25 pairs of parents per bottle) on potato-sugar (PS) medium with dry live yeast sprinkled onthe surface (Frankham et al. 1988). Outbred pop-ulations were maintained at 18 �C to slow thegeneration time to 4 weeks. New populations fromTyrrells’ were established in 1994 (from 79 wild-inseminated females; T94), 1995 (415 wild-insem-inated females; T95) and 1996 (273 inseminatedfemales; T96). The Tyrrells wild population hasbeen found to be stable in allozyme frequenciesfrom the 1970s to the 1990s (see Franklin 1981;Frankham and Loebel 1992), in microsatellitefrequencies from 1992 to 1996 (England 1997), andto be stable in fitness from 1992 to 1994 (Wood-worth et al. 2002). The thuringiensin experimentswere run on T92 and T94 and their derivativepopulations, the Serratia tests on T96 and itsderivatives, with T95 used only for determiningthe LC50 for thuringiensin. Inbreeding coefficientsin Tyrrells’ populations when experimental popu-lations were derived from them were all ~0, andinbreeding coefficients in experiment popula-tions were calculated relative to the foundingpopulations, as is standard (Falconer and Mackay1996).
Experimental populations
The thuringiensin experiments used populationsmaintained with effective sizes (Ne) of 500 (tworeplicates), 250 (3), 100 (4), 50 (6) and 25 (8) for 50generations. These were derived from the T92 basepopulation two generations after it was foundedand maintained as separate pedigreed populationsin vials at 25 �C (for details see Montgomery et al.2000). Expected inbreeding coefficients for the fiveNe treatments were 0.049, 0.095, 0.222, 0.395 and0.636 respectively. Analyses of pedigrees indicatedthat effective population sizes were close to thoseintended (correlation of intended and actualNe ¼ 0.997: Woodworth et al. 2002). Loss of al-lozymic variation in these populations does notdiffer from that predicted by Equation (1)(Montgomery et al. 2000). After 50 generations,each experimental population was maintained
using 750 parents per generation in 15 · 270 mlbottles at 18 �C.
Two highly inbred populations were also usedin the thuringiensin experiments. They were de-rived from the T94 base population by subjectinglines to 35 generations of full-sib mating (Frank-ham et al. 1999), and thus had inbreeding coeffi-cients of 0.999 (Falconer and Mackay 1996).
The Serratia experiment involved comparing11 highly inbred populations with their T96 basepopulation. After 10 generations in captivity, 50inbred populations were begun and full-sibs inbredcontinuously for 20 generations. They had pedi-gree inbreeding coefficient of 0.986 (Falconer andMackay 1996), and markedly reduced allozymicvariation (Gilligan, Briscoe and Frankham, sub-mitted). Subsequently, they were maintained bymass mating in bottles. Eleven highly inbredpopulations survived until the experimental expo-sures.
Thuringiensin experiments
Two experiments were carried out, one for totalfitness and the other for larval-adult survival. Inthe former, exposure to the LC50 concentration ofthuringiensin occurred in 100 · 25 mm2 glass vialswith 10 ml of PS medium. For each population,five pairs of young flies were allowed to lay for sixhours in each of 12 vials per population, six vialswith thuringiensin at the calculated LC50, and sixcontrol vials without the toxin. Emerged male andfemale flies were counted and removed daily fromday 8 to 16. Resistance to thuringiensin for eachpopulation was measured as the ratio of fliesemerging in vials with the toxin to that in vialswithout the toxin.
Effects on larval survival were determined bytransferring 25 first instar larvae to each of 6–12vials per population, split between those withthuringiensin at the calculated LC50, and controlvials without the toxin. Emerging male and femaleflies were counted and removed daily from day 8 to16.
All experiments were done at 25 �C on PSmedium (Frankham et al. 1988). Thuringiensinwas added to the water at volumes to provide thefinal LC50 concentrations of the medium. B. thur-ingiensis serotype HD 1, supplied by Prof D. Pin-nock of the University of Adelaide, SouthAustralia was fermented in a heart and brain broth
442

(Oxoid) at 32 �C and thuringiensin extracted bystandard methods and the solution autoclaved, asdescribed in Spielman (2002). Thuringiensin con-centrations were assayed using HPLC assays fol-lowing the method of Campbell et al. (1987) bycomparing the results with those from a thurin-giensin standard of 0.1 mg/ml (Spielman 2002).The LC50 for thuringiensin was determined byallowing five pairs of young T95 flies to lay for sixhours in vials containing medium with thurin-giensin supernatant concentrations of 0%, 0.25%,0.50%, 1.00%, 1.25%, 1.50%, 1.75% or 2.00% (sixvials per concentration), counting emerging fliesfrom day 8 to 16 and regressing the total numberof emerged flies against thuringiensin concentra-tion. The LC50 concentration was determined asthe concentration where half as many flies emergedas in the control vials.
Serratia marcescens experiment
The impact of Serratia marcescens on 11 highlyinbred populations was compared with that fortheir T96 base population.
Experimental vials for this experiment con-tained PS medium without dry yeast, but withnystatin added to the water to provide a finalconcentration of 1% nystatin in the medium.Nystatin increased the susceptibility of the flies toSerratia marcescens and improved the repeatabil-ity of results, presumably by making the peri-trophic membrane leak (Spielman 2002). Liveyeast was not used as it prevented the pathogenfrom killing D. melanogaster. Instead, two dropsof a banana solution were added to the surface ofthe medium in each vial to stimulate female flies tolay.
A culture of S. marcescens, supplied by Prof.Duncan Veal, Macquarie University, Australia,was used to produce a pure, actively growingculture to add to the experimental vials at the re-quired time. The purity of the cultures waschecked microscopically under oil immersion andthe activity of the broth was determined visuallyby the deepness of the red colour due to prodigi-osin.
One to two day-old inseminated femaleD. melanogaster (10–20 females with 8–15 malesper vial) were allowed to lay consecutively in twosets of test vials until ~25–70 eggs were laid in eachof eight vials per population. The numbers of eggs
laid were counted and the pathogen added toalternate vials, and sterile broth to the other vials,forming exposed and unexposed pairs. The num-ber of emerged female and male D. melanogaster ineach vial were counted. For logistical reasons, egglaying by the 11 inbred populations was carriedout in four sets of two to four populations, eachwith contemporary vials of the T96 outbred con-trol.
D. melanogaster populations were exposed toS. marcescens by adding 16 drops of S. marcescensculture evenly to the surface of the medium inalternate vials 18–24 h after the eggs were laid.Sterile broth (16 drops) was similarly added to thecontrol vials from the first lay. Paired vials fromthe second lay received the opposite (i.e., sterilebroth or culture). Six more drops of the appro-priate liquid were also added 24 and 48 h later.
Resistance to the Serratia pathogen wasdetermined by calculating the ratio of emergencesto eggs laid in an exposed vial over the ratio in thepaired unexposed vial, i.e. ([exposed flies/eggs]/[unexposed flies/eggs]). This measure for eachinbred population was compared with that for theT96 control.
To determine whether resistance in the inbredpopulations was correlated with general inbreed-ing depression, several measures of fitness in theabsence of the pathogen were determined andcorrelated with resistance.
Statistical analyses
For the data from the thuringiensin total fitnessexperiment, resistance for populations (ratio offlies in vials with the toxin to that in vials lackingit) did not differ from normality and the signifi-cance of the relationship between resistance andinbreeding coefficient was the same for the rankcorrelation and the regression analyses describedbelow. Population resistance was regressed on theinbreeding coefficient, as well as on F2 and astepwise multiple regression done with both F andF2 as predictors. The significance of the variationin resistance among populations within treatmentswith the same F was assessed using an F-test of theamong populations within inbreeding treatmentsby thuringiensin treatment interactions against thevariation among vials within populations (onsquare root transformed data). The correlationbetween resistance and fitness of populations in
443
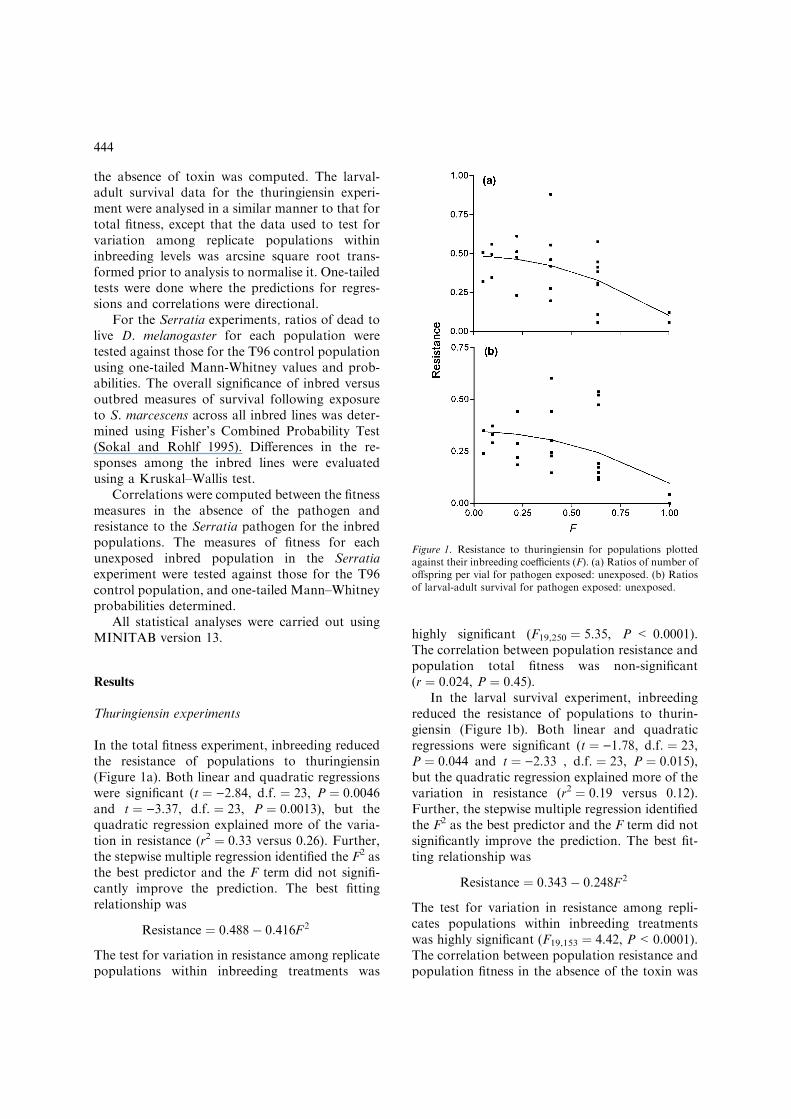
the absence of toxin was computed. The larval-adult survival data for the thuringiensin experi-ment were analysed in a similar manner to that fortotal fitness, except that the data used to test forvariation among replicate populations withininbreeding levels was arcsine square root trans-formed prior to analysis to normalise it. One-tailedtests were done where the predictions for regres-sions and correlations were directional.
For the Serratia experiments, ratios of dead tolive D. melanogaster for each population weretested against those for the T96 control populationusing one-tailed Mann-Whitney values and prob-abilities. The overall significance of inbred versusoutbred measures of survival following exposureto S. marcescens across all inbred lines was deter-mined using Fisher’s Combined Probability Test(Sokal and Rohlf 1995). Differences in the re-sponses among the inbred lines were evaluatedusing a Kruskal–Wallis test.
Correlations were computed between the fitnessmeasures in the absence of the pathogen andresistance to the Serratia pathogen for the inbredpopulations. The measures of fitness for eachunexposed inbred population in the Serratiaexperiment were tested against those for the T96control population, and one-tailed Mann–Whitneyprobabilities determined.
All statistical analyses were carried out usingMINITAB version 13.
Results
Thuringiensin experiments
In the total fitness experiment, inbreeding reducedthe resistance of populations to thuringiensin(Figure 1a). Both linear and quadratic regressionswere significant (t ¼ )2.84, d.f. ¼ 23, P ¼ 0.0046and t ¼ )3.37, d.f. ¼ 23, P ¼ 0.0013), but thequadratic regression explained more of the varia-tion in resistance (r2 ¼ 0.33 versus 0.26). Further,the stepwise multiple regression identified the F2 asthe best predictor and the F term did not signifi-cantly improve the prediction. The best fittingrelationship was
Resistance ¼ 0:488� 0:416F 2
The test for variation in resistance among replicatepopulations within inbreeding treatments was
highly significant (F19,250 ¼ 5.35, P < 0.0001).The correlation between population resistance andpopulation total fitness was non-significant(r ¼ 0.024, P ¼ 0.45).
In the larval survival experiment, inbreedingreduced the resistance of populations to thurin-giensin (Figure 1b). Both linear and quadraticregressions were significant (t ¼ )1.78, d.f. ¼ 23,P ¼ 0.044 and t ¼ )2.33 , d.f. ¼ 23, P ¼ 0.015),but the quadratic regression explained more of thevariation in resistance (r2 ¼ 0.19 versus 0.12).Further, the stepwise multiple regression identifiedthe F2 as the best predictor and the F term did notsignificantly improve the prediction. The best fit-ting relationship was
Resistance ¼ 0:343� 0:248F 2
The test for variation in resistance among repli-cates populations within inbreeding treatmentswas highly significant (F19,153 ¼ 4.42, P<0.0001).The correlation between population resistance andpopulation fitness in the absence of the toxin was
Figure 1. Resistance to thuringiensin for populations plottedagainst their inbreeding coefficients (F). (a) Ratios of number ofoffspring per vial for pathogen exposed: unexposed. (b) Ratiosof larval-adult survival for pathogen exposed: unexposed.
444
negative, rather than positive and close to one(r ¼ )0.57, P ¼ 0.999).
Serratia marcescens experiment
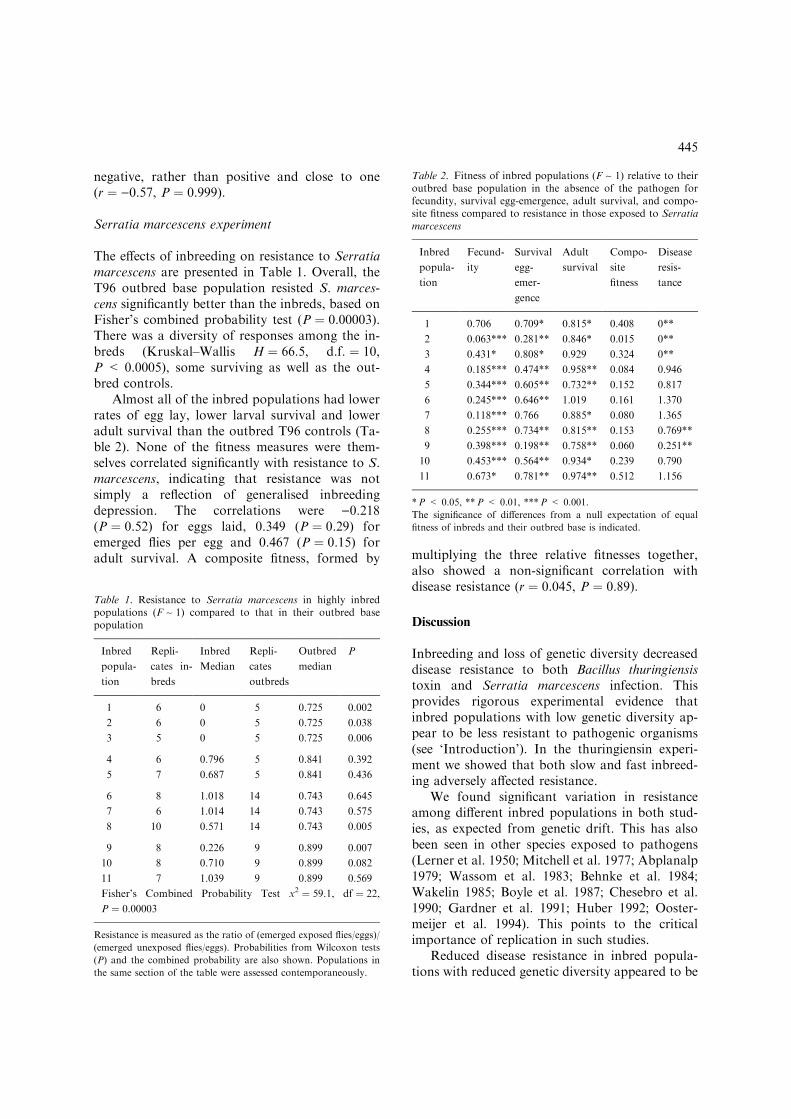
The effects of inbreeding on resistance to Serratiamarcescens are presented in Table 1. Overall, theT96 outbred base population resisted S. marces-cens significantly better than the inbreds, based onFisher’s combined probability test (P ¼ 0.00003).There was a diversity of responses among the in-breds (Kruskal–Wallis H ¼ 66.5, d.f. ¼ 10,P < 0.0005), some surviving as well as the out-bred controls.
Almost all of the inbred populations had lowerrates of egg lay, lower larval survival and loweradult survival than the outbred T96 controls (Ta-ble 2). None of the fitness measures were them-selves correlated significantly with resistance to S.marcescens, indicating that resistance was notsimply a reflection of generalised inbreedingdepression. The correlations were )0.218(P ¼ 0.52) for eggs laid, 0.349 (P ¼ 0.29) foremerged flies per egg and 0.467 (P ¼ 0.15) foradult survival. A composite fitness, formed by multiplying the three relative fitnesses together,
also showed a non-significant correlation withdisease resistance (r ¼ 0.045, P ¼ 0.89).
Discussion
Inbreeding and loss of genetic diversity decreaseddisease resistance to both Bacillus thuringiensistoxin and Serratia marcescens infection. Thisprovides rigorous experimental evidence thatinbred populations with low genetic diversity ap-pear to be less resistant to pathogenic organisms(see ‘Introduction’). In the thuringiensin experi-ment we showed that both slow and fast inbreed-ing adversely affected resistance.
We found significant variation in resistanceamong different inbred populations in both stud-ies, as expected from genetic drift. This has alsobeen seen in other species exposed to pathogens(Lerner et al. 1950; Mitchell et al. 1977; Abplanalp1979; Wassom et al. 1983; Behnke et al. 1984;Wakelin 1985; Boyle et al. 1987; Chesebro et al.1990; Gardner et al. 1991; Huber 1992; Ooster-meijer et al. 1994). This points to the criticalimportance of replication in such studies.
Reduced disease resistance in inbred popula-tions with reduced genetic diversity appeared to be
Table 2. Fitness of inbred populations (F ~ 1) relative to theiroutbred base population in the absence of the pathogen forfecundity, survival egg-emergence, adult survival, and compo-site fitness compared to resistance in those exposed to Serratiamarcescens
Inbred
popula-
tion
Fecund-
ity
Survival
egg-
emer-
gence
Adult
survival
Compo-
site
fitness
Disease
resis-
tance
1 0.706 0.709* 0.815* 0.408 0**
2 0.063*** 0.281** 0.846* 0.015 0**
3 0.431* 0.808* 0.929 0.324 0**
4 0.185*** 0.474** 0.958** 0.084 0.946
5 0.344*** 0.605** 0.732** 0.152 0.817
6 0.245*** 0.646** 1.019 0.161 1.370
7 0.118*** 0.766 0.885* 0.080 1.365
8 0.255*** 0.734** 0.815** 0.153 0.769**
9 0.398*** 0.198** 0.758** 0.060 0.251**
10 0.453*** 0.564** 0.934* 0.239 0.790
11 0.673* 0.781** 0.974** 0.512 1.156
*P < 0.05, **P < 0.01, ***P < 0.001.
The significance of differences from a null expectation of equal
fitness of inbreds and their outbred base is indicated.
Table 1. Resistance to Serratia marcescens in highly inbredpopulations (F ~ 1) compared to that in their outbred basepopulation
Inbred
popula-
tion
Repli-
cates in-
breds
Inbred
Median
Repli-
cates
outbreds
Outbred
median
P
1 6 0 5 0.725 0.002
2 6 0 5 0.725 0.038
3 5 0 5 0.725 0.006
4 6 0.796 5 0.841 0.392
5 7 0.687 5 0.841 0.436
6 8 1.018 14 0.743 0.645
7 6 1.014 14 0.743 0.575
8 10 0.571 14 0.743 0.005
9 8 0.226 9 0.899 0.007
10 8 0.710 9 0.899 0.082
11 7 1.039 9 0.899 0.569
Fisher’s Combined Probability Test x2 ¼ 59.1, df ¼ 22,
P ¼ 0.00003
Resistance is measured as the ratio of (emerged exposed flies/eggs)/
(emerged unexposed flies/eggs). Probabilities from Wilcoxon tests
(P) and the combined probability are also shown. Populations in
the same section of the table were assessed contemporaneously.
445

due to specific polymorphic loci affecting diseaseresistance, rather than a consequence of general-ised inbreeding depression, as correlations be-tween resistance and fitness in the absence of thetoxin or pathogen were low or negative. Our re-sults are consistent with the work of Lerner et al.(1950) who found that the resistance of inbredchicken lines to coryza (Hemophilus gallinarum)was not correlated with the general vigour of theflocks.
Our results contrast with those of Stevens et al.(1997), who found no overall decrease in resistanceto a tapewormamong 31 inbred flour beetle lineagescomparedwith their large ancestral population.Ourstudies differ from theirs in taxon of model, patho-gen, type of base population and exposuremethods.We doubt that the former three are the reason forthe difference in results. As their exposure method(beetles had to consume paper impregnated withtapeworm oocysts) meant that inbred beetles mayhave consumed fewer tapeworms than ‘outbred’beetles (Yan and Norman 1995), there are groundsfor scepticism about their conclusions. In ourexperiments, both inbred and outbred D. melano-gaster larvae could not have avoided ingesting thethuringiensin or S. marcescens in the medium.Consequently, our results should be amore realisticmodel for disease responses in natural populations.
The effects of inbreeding and loss of geneticdiversity in increasing the susceptibility to infec-tious disease are expected to be greater for naturalpopulations subject to many different infectiousagents than we found for single pathogens. Inbredpopulations that are relatively resistant to onepathogen are likely to be susceptible to most otherunrelated pathogens (Frankham et al. 2002).
What are the conservation implications of ourfindings? Managers should strive to minimiseinbreeding and the loss of genetic diversity withinpopulations of wild species, not only because ofdepressed reproductive fitness and loss of evolu-tionary potential, but especially where populationsare at increased risks of exposure to virulentpathogens (e.g., in zoological and botanical col-lections, in proximity to expanding human settle-ments and agriculture, popular sites forinternational tourists, etc.). Exposure of inbredand threatened populations to pathogens shouldbe avoided or at least minimised. This may oftenmean minimising exposure to potential carriers orvectors of pathogens such as tourists, pets, live-
stock and their products and agricultural andhorticultural plants. Great care about diseaseneeds to be taken when moving animals betweenzoos or animals or plants among fragmentedpopulations to mitigate inbreeding and loss ofgenetic diversity.
Governments should factor disease risks intothe cost-benefit analyses of increasing free tradeand loosening quarantine requirements. The im-pacts of the recent international outbreaks of footand mouth (10 million animals slaughtered in theUK; Uhlig 2002) and ‘mad cow’ diseases stronglyunderscore these recommendations.
In conclusion, inbreeding and loss of geneticdiversity reduced the ability of Drosophila popu-lations to cope with disease caused by twopathogens.
Acknowledgements
We thank M. Montgomery, L. Woodworth andE. Lowe for developing the populations with dif-ferent effective sizes, D. Veal, K. Fomiatti andJ. McIntosh for providing microbiological advice,Serratia marcescens and laboratory facilities,D. Pinnock for supplying Bacillus thuringiensis, foruse of his laboratory facilities, to his staff fortraining DS in methods for producing and assay-ing thuringiensin, D. Cooper for assistance inproducing thuringiensin, K. Formiatti, L. Melvilleand A. Janmat for providing access to the Berri-mah Veterinary Laboratory, Northern Territory,R. Hinde for suggesting the use of nystatin,K. Belov, A. Shuetrim and two anonymousreviewers for comments on the manuscript andD. Gilligan, E. Lowe, J. Lewis, M. Montgomery,K. Lees, E.Mardones and J.Walmsley for technicalassistance. This work was supported by AustralianResearch Council and Macquarie University Re-search grants and by an Australian PostgraduateAward to DS. This is publication 380 of the KeyCentre for Biodiversity and Bioresources.
References
Abplanalp H (1979) The role of genetics in the immune re-sponse. Avian Dis., 23, 299–308.
Acevedo-Whitehouse K, Gulland F, Greig D, Amos W (2003)Inbreeding: Disease susceptibility in California sea lions.Nature, 422, p. 35.
446

Aiello SE, Mays A (eds.) (1998) The Merck Veterinary Manual,8th edn. Merck & Co. Inc. Whitehouse Station, NJ, USA.
Baer B, Schmid-Hempel P (1999) Experimental variation inpolyandry affects parasite loads and fitness in a bumble-bee.Nature, 397, 151–154.
Behnke JM, Ali NMH, Jenkins SN (1984) Survival to patencyof low-level infections with Trichuris muris in mice concur-rently infected with Nematospiroides dubius. Ann. Trop. Med.Hyg., 78, 509–517.
Benz G (1966) On the chemical nature of the heat stable exo-toxin of Bacillus thuringiensis. Experientia, 22, 81–82.
Benz G, Graf E (1971) Antagonism of terramycin on action ofBacillus thuringiensis ‘exotoxin’ in Drosophila melanogaster.Experientia, 27, 73–75.
Black FL (1992) Why did they die? Science, 258, 1739–1740.Blood DC, Studdert VP (1999) Saunders Comprehensive Vet-
erinary Dictionary, 2nd edn. WB Saunders, London.Boyle JF, Weismiller DG, Holmes KV (1987) Genetic resistance
to mouse hepatitis virus correlates with absence of virus-binding activity on target tissues. J. Virol., 61, 185–189.
Burdon JJ (1987) Diseases and Plant Population Biology.Cambridge University Press, Cambridge, UK.
Burges HD, Hurst JA (1977) Ecology of Bacillus thuringiensis instorage moths. J. Invert. Pathol., 30, 131–139.
Campbell DP, Dieball DE, Brackett JM (1987) Rapid HPLCassay for the b-exotoxin of Bacillus thuringiensis. J. Agr.Food Chem., 35, 156–158.
Caro TM, Laurenson MK (1994) Ecological and genetic factorsin conservation: A cautionary tale. Science, 263, 485–486.
Caughley G (1994) Directions in conservation biology. J. Anim.Ecol., 63, 215–244.
Chesebro B, Miyazawa M, Britt WJ (1990) Host genetic controlof spontaneous and induced immunity to Friend murineretrovirus infection. Annu. Rev. Immunol., 8, 477–499.
Clay K, Kover PX (1996) The red queen hypothesis and plant/pathogen interactions. Annu. Rev. Phytopathol., 34, 29–50.
Coltman DW, Pilkington JG, Smith JM, Pemberton JM (1999)Parasite-mediated selection against inbred Soay sheep in afree-living island population. Evolution, 53, 1259–1267.
Daszak P, Cunningham AA, Hyatt AD (2000) Emerginginfectious diseases of wildlife: threats to biodiversity andhuman health. Science, 287, 443–449.
Dieraut LA, Gulland MD (2001) CRC Handbook of MarineMammal Medicine, 2nd edn. CRC Press, Boca Paton.
Diamond JM (1998) Guns, Germs and Steel: A Short History ofEverybody for the Last 13,000 Years. Vintage, London, UK.
Dobson AP, May RM (1986) Disease and conservation. In:Conservation Biology: The Science of Scarcity and Diversity(ed. Soule ME), pp. 345–365. Sinauer, Sunderland, Massa-chusetts.
Elgar MA, Clode D (2001) Inbreeding and extinctions in islandpopulations: A cautionary tale. Conserv. Biol., 15, 284–286.
England PR (1997) Conservation Genetics of Population Bot-tlenecks. Ph.D. thesis, Macquarie University, Sydney.
Falconer DS, Mackay TFC (1996) Introduction to QuantitativeGenetics, 4th edn. Longman, Harlow, UK.
Ferguson MM, Drahuschchak LR (1990) Disease resistanceand enzyme heterozygosity in rainbow trout. Heredity, 64,413–417.
Flyg C, Dalhammar G, Rasmuson B, Boman HG (1987) Insectimmunity – inducible antibacterial activity in Drosophila.Insect Biochem., 17, 153–160.
Flyg C, Kenne K, Boman HG (1980) Insect pathogenic prop-erties of Serratia marcescens: Phage-resistant mutants with a
decreased resistance to Cercropia immunity and a decreasedvirulence to Drosophila. J. Gen. Microbiol., 120, 173–181.
Frank SA (1992) Models of plant-pathogen coevolution. TrendsGenet., 8, 213–219.
Frankham R (2000) Modeling problems in conservationgenetics using laboratory animals. In: Quantitative Methodsfor Conservation Biology (eds. Ferson S, Burgman MA), pp.259–273. Springer-Verlag, New York.
Frankham R, Briscoe DA, Ballou JD (2002) Introduction toConservation Genetics. Cambridge University Press, Cam-bridge, UK.
Frankham R, Lees K, Montgomery ME, England PR, Lowe E,Briscoe DA (1999) Do population size bottlenecks reduceevolutionary potential? Anim. Conserv., 2, 255–260.
Frankham R, Loebel DA (1992) Modeling problems in con-servation genetics using captive Drosophila populations:Rapid adaptation to captivity. Zoo Biol., 11, 333–342.
Frankham R, Yoo BH, Sheldon BL (1988) Reproductive fitnessand artificial selection in animal breeding: culling on fitnessprevents a decline in reproductive fitness in lines of Dro-sophila melanogaster selected for increased inebriation time.Theor. Appl. Genet. 76, 909–914.
Franklin IR (1981) An analysis of temporal variation inisozyme loci in Drosophila melanogaster. In: Genetic Studiesof Drosophila Populations (eds. Gibson JB, Oakeshott JG),pp. 217–236. Australian National University Press,Canberra.
Gardner MB, Kozak CA, O’Brien SJ (1991) The Lake Casitaswild mouse: Evolving genetic resistance to retroviral disease.Trends Genet., 7, 22–27.
Glazer AN, Nikaido H (1995) Microbial Biotechnology: Fun-damentals of Applied Microbiology. WH Freeman andCompany, New York.
Haldane JBS (1949) Disease and evolution. Ricerca Sci.,19(Suppl.), 3–10.
Hedrick PW, Kim TJ (1999) Genetics of complex polymor-phisms: Parasites and maintenance of MHC variation. In:Evolutionary Genetics: From Molecules to Man (eds. SinghRS, Krimbas CB), pp. 204–234. Cambridge University Press,Cambridge, UK.
Hedrick PW, Kim TJ, Parker KM (2001) Parasite resistanceand genetic variation in the endangered Gila topminnow.Anim. Conserv., 4, 103–109.
Hilton-Taylor C (2000) 2000 IUCN Red List of ThreatenedSpecies. IUCN, Gland, Switzerland.
Huber BT (1992) Mls genes and self-superantigens. TrendsGenet., 8, 399–402.
Jacobsen ER, Gaskin JM, Brown MB, Harris RK, GardinerCH, La Pointe JL, Adams HP, Reggiardo C (1991) Chronicupper respiratory tract disease of free-ranging desert tor-toises (Xerobates agassizii). J. Wildl. Dis., 27, 296–316.
Lewontin RC (1974) The Genetic Basis of Evolutionary Change.Columbia University Press, New York.
Lerner IM, Taylor LW, Beach JR (1950) Evidence for geneticvariation in resistance to a respiratory infection in chickens.Poultry Sci., 29, 862–869.
Liersch S, Schmid-Hempel P (1998) Genetic variability withinsocial insect colonies reduces parasite load. Proc. Royal Soc.Lond. B, 265, 221–225.
Lively CM (1996) Host-parasite coevolution and sex. BioScience, 46, 107–114.
Lively CM, Craddock C, Vrijenhoek RC (1990) Red queenhypothesis supported by parasitism in sexual clonal fish.Nature, 344, 864–866.
447

Lyles AM, Dobson AP (1993) Infectious disease and intensivemanagement: population dynamics, threatened hosts, andtheir parasites. J. Zoo Wildife Med., 24, 315–326.
Marec F, Matha V, Weiser J (1988) Analysis of the genotoxicactivity of Bacillus thuringiensis beta-exotoxin by means ofthe Drosophila wing spot test. J. Invert. Pathol., 53, 347–353.
May RM (1995) The cheetah controversy.Nature, 574, 309–310.McCallum H, Dobson A (1995) Detecting disease and parasite
threats to endangered species and ecosystems. Trends Ecol.Evol., 10, 190–194.
Meagher S (1999) Genetic diversity and Capillaria hepatica(Nematoda) prevalence in Michigan deer mouse popula-tions. Evolution, 53, 1318–1324.
Meltzer DGA (1993) Historical survey of disease problems inwildlife populations: Southern Africa mammals. J. ZooWildl. Med., 24, 237–244.
Merola M (1994) A reassessment of homozygosity and thecase for inbreeding depression in the cheetah, Acinonyx juba-tus: Implications for conservation. Conserv. Biol., 8, 961–971.
Mitchell GF, Goding JW, Rickard MD (1977) Studies on im-mune responses to larval cestodes in mice: Increased sus-ceptibility of certain mouse strains and hypothymic mice toTaenia taeniaeformis and analysis of passive transfer withserum. Aust. J. Exp. Biol. Med., 55, 165–186.
Montgomery ME, Woodworth LM, Nurthen RK, GilliganDM, Briscoe DA, Frankham R (2000) Relationship betweenpopulation size and loss of genetic diversity: Comparisons ofexperimental results with theoretical predictions. Conserv.Genet., 1, 33–43.
Nicholas FW (1987) Veterinary Genetics. Clarendon Press,Oxford.
O’Brien SJ, Evermann JF (1988) Interactive influence ofinfectious disease and genetic diversity in natural popula-tions. Trends Ecol. Evol., 3, 254–259.
O’Brien SJ, Roelke ME, Marker L, Newman A, Winkler CA,Meltzer D, Colly L, Evermann JF, Bush M, Wildt DE (1985)Genetic basis for species vulnerability in the cheetah. Sci-ence, 227, 1428–1434.
Oostermeijer JGB, Van Eijck MW, den Nijs JMC (1994) Off-spring fitness in relation to population size and genetic var-iation in the rare perennial plant species Gentianapneumonanthe (Gentianaceae). Oecologia, 97, 289–296.
Parker MA (1990) The pleiotropy theory for polymorphism ofdisease resistance genes in plants. Evolution, 44, 1872–1875.
Paumard-Rigal S, Rosenberg-Bourgin M (1992) Increase in theresistance of the Bacillus thuringiensis supernatant effect in aDrosophila melanogaster wild type Oregon R line. Heredity,69, 539–546.
Pimm SL, Gittleman JL, McCracken GF, Gilpin M (1989)Plausible alternatives to bottlenecks to explain reduced ge-netic diversity. Trends Ecol. Evol., 4, 176–178.
Plowright W (1982) The effects of rinderpest and rinderpestcontrol on wildlife in Africa. Sym. Zool. Soc. Lond. 50, 1–28.
Real LA (1996) Sustainability and the ecology of infectiousdisease. Bioscience, 46, 88–97.
Roelke ME, Martenson JS, O’Brien SJ (1993) The conse-quences of demographic reduction and genetic depletion inthe endangered Florida panther. Curr. Biol., 3, 340–350.
Schmid-Hempel P, Crozier R (1999) Polyandry versus polygynyversus parasites. Proc. Roy Soc. Lond. B, 354, 507–515.
Seal US (1991) Disease and captive conservation of threatenedspecies. SSC Working Group meeting 28–29 May 1991,National Zoo, Washington, DC.
Sokal RR, Rohlf FJ (1995) Biometry: The Principles andPractice of Statistics in Biological Research, 3rd edn. WHFreeman and Company, New York.
Sorci G, Moller AP, Boulinier T (1997) Genetics of host–par-asite interaction. Trends Ecol. Evol., 12, 196–199.
Spielman D (2002) Does Inbreeding and Loss of Genetic Diver-sity Decrease Resistance to Disease? Ph.D. thesis, MacquarieUniversity, Sydney, Australia.
Stevens L, Yan G, Pray LA (1997) Consequences of inbreedingon invertebrate host susceptibility to parasitic infection.Evolution, 51, 2032–2039.
Thorne ET, Williams ES (1988) Disease and endangered spe-cies: The black-footed ferret as a recent example. Conserv.Biol., 2, 66–74.
Uhlig R (2002) 10 million animals were slaughtered in foot andmouth cull. The Telegraph 23 Jan 2002 (//www.telegraph.co.uk/news/main.jhtml?xml=%2Fnews%2F2002%2F01%2F).
van Riper C, van Riper SG, Goff ML, Laird M (1986) Theepizootiology and ecological significance of malaria inHawaiian land birds. Ecol. Monogr., 56, 327–344.
Vrijenhoek RC (1994) Genetic diversity and fitness in smallpopulations. In: Conservation Genetics (eds. Loeschcke V,Tomiuk J, Jain SK), pp. 37–53. Birkhauser Verlag, Basel,Switzerland.
Wakelin D (1985) Genetics, immunity and parasite survival. In:Ecology and Genetics of Host–Parasite Interactions (eds.Rollison D, Anderson RM), pp. 39–54. Academic Press,London.
Warner RE (1968) The role of introduced diseases in theextinction of the endemic Hawaiian avifauna. Condor, 70,101–120.
Wassom DL, Brooks BO, Babisch JG, David CS (1983) A genemapping between the S and D regions of the H-2 complexinfluences resistance to Trichinella spiralis infections of mice.J. Immunogenet., 10, 371–378.
Weatherall D (2003) Evolving with the enemy. New Scient.,180(2422), 44–47.
Woodworth LM, Montgomery ME, Briscoe DA, Frankham R(2002) Rapid genetic deterioration in captivity: Causesand conservation implications. Conserv. Genet., 3, 277–288.
Yan G, Norman S (1995) Infection of Tribolium beetles with atapeworm: Variation in susceptibility within and betweenbeetle species and among genetic strains. J. Parasitol., 81,37–42.
448
Copyright © 2022 FDOKUMEN




















