
DNA mismatch repair deficiency in therapy related acute ...
263
DNA mismatch repair deficiency in therapy related acute myeloid leukaemia I myelodysplastic syndrome By Judith Muriel Offman A thesis submitted for the degree of Ph.D. at The University of London January 2004 Cancer Research UK London Research Institute Clare Hall Laboratories South Mimms Potters Bar Herts EN6 3EL and Department of Biochemistry University College London WC1E6BT
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of DNA mismatch repair deficiency in therapy related acute ...

DNA mismatch repair deficiency in
therapy related acute myeloid
leukaemia I myelodysplastic
syndromeBy
Judith Muriel Offman
A thesis submitted for the degree of Ph.D.
at
The University of London
January 2004
Cancer Research UK
London Research Institute
Clare Hall Laboratories
South Mimms
Potters Bar
Herts EN6 3EL
and
Department of Biochemistry
University College London
W C 1E6B T

ProQuest Number: U644338
All rights reserved
INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U644338
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.This work is protected against unauthorized copying under Title 17, United States Code.
Microform Edition © ProQuest LLC.
ProQuest LLC 789 East Eisenhower Parkway
P.O. Box 1346 Ann Arbor, Ml 48106-1346

Abstract
DNA mismatch repair (MMR) defects occur in both sporadic and familial
cancers and are associated with dramatic increases in spontaneous mutation rates.
In the majority of repair-defective sporadic tumours, the hM LH1 MMR gene is
inactivated epigenetically by promoter méthylation. In both cultured cells and in
tumours, deficient MMR is also associated with resistance to certain therapeutic
drugs - particularly methylating agents and thiopurines.
The incidence of therapy related cancer - a malignancy that follows
therapeutic treatment - is increasing. Therapy-related acute myeloid leukaemia/
myelodysplastic syndrome (t-AML/MDS) presently accounts for ^10% of all
AML/MDS cases. AML is a malignancy of myeloid progenitor cells. MDS comprises
a group of disorders characterized by abnormal myeloid stem cell differentiation and
often precedes t-AML. This thesis describes an investigation of possible
relationships between defective MMR, t-AML/MDS, and drug treatment.
DNA from a group of cancer therapy-related acute leukaemia (AL)/MDS
cases was examined for the microsatellite instability (M SI) phenotype that is
diagnostic for inactive MMR. More than 60% (16/25) were M S r compared to <4%
(0/28) of de novo cases. hMLH1 promoter méthylation was infrequent. It occurred in
less than one-third of the M S r cases although it appeared to be more common
among M S r therapy-related acute promyelocytic leukaemias.
In view of the acknowledged resistance of MMR deficient cells to 6-
thioguanine, possible connections between therapeutic thiopurine use and M S r
AML/MDS were examined. I demonstrated that chronic treatment with 6-thioguanine
could be used to select MMR defective clones from repair-proficient human cells
grown in tissue culture. The possible relationship between azathioprine - a
thiopurine prodrug, widely used as an immunosuppressant following organ
transplantation - and M S r t-AML/MDS was investigated. Together with Professor
Gerhard Opelz, I analysed the incidence of AML among > 170,000 organ transplant
patients. This revealed a significant excess of AML among transplant recipients.
MMR deficiency was found to be frequent among transplant-related AML/MDS
cases; seven of seven examined were M S I\
MMR defective (M ST) tumours are highly genetically unstable. They
accumulate frameshift mutations in coding sequences, which result in most cases in
truncated and/or inactive proteins. Caspase-5 and FancD2 were identified as targets
for addition/deletion mutations in M S f t-AML cases.

Acknowledgements
I would like to thank Dr. Peter Karran, my principal supervisor, for all his
advice, help, patience and discussion during my time in the laboratory and the
preparation of this manuscript. I am grateful to Prof. Peter Swann, my supervisor at
UCL, for his interest and support throughout my PhD.
I would also like to thank all the past and present members of the laboratory,
Steve Durant, Peter O’Donovan, Conal Perrett, Ramin Sadri, Irene Lavagi, Nathalie
Guibourt, Casey Kobayashi and Andy Massey. I am specifically grateful to Pauline
Branch for her help with the tissue culture, Peter Macpherson for carrying out the in
vitro mismatch repair assay and some of the Western blots, and Karen Gascoigne
who was a great summer student to supervise. I would like to acknowledge all my
friends and colleagues at Clare Hall for their help, advice and friendship.
I am very grateful to everyone who helped with this work: Giuseppe Leone
and Luca Mele at the Université Cattolica S. Cuore in Rome, as well as David
Cummins, Ozay Halil, Nick Banner, Margaret Burke at Harefield Hospital for
providing clinical material; Margherita Bignami and Ida Casorelli at the Institute
Superiors di Sanitâ in Rome for carrying out the MSI analysis of the chemotherapy-
related AL/MDS samples; Gerhard Opelz and his colleagues at the Collaborative
Transplant Study for the AML incidence analysis and for help with obtaining clinical
samples; Dr. Ian Tomlinson, Cancer Research UK, LRI for the colon carcinoma
samples; the oligonucleotide synthesis and cell production services at Clare Hall for
all their great work; Peter Maddox for help with the paraffin embedded tissue
sections; as well as the LIF equipment park for running sequencing and genotyping
reactions.
My studentship was generously funded by ICRF/Cancer Research UK and
the Bnai Brith Leo Baeck Lodges.
I would like to thank all my friends for their help and support. Last but not
least I would like to thank my parents and my brother Marc for their love and
support. I am also grateful to Marc for bioinformatics and computer help.

This thesis is dedicated to all my relations who think that I would be much better off
getting married, especially my late granddad Avram Hendric ‘Dedi’ Offman who I
miss very much.

Publications
Massey, A., Offman, J., Macpherson, P., Karran, P. DNA mismatch repair and
acquired cisplatin resistance in E. coli and human ovarian carcinoma cells. DNA
Repair 2003; 2: 73-89.
Casorelli, I., Offman, J., Mele, L., Pagano, L., Sica, S., D’Errico, M., Giannini, G.,
Leone, G., Bignami, M., Karran, P. Drug treatment in the development of mismatch
repair defective acute leukemia and myelodysplastic syndrome. DNA Repair 2003;
2: 547-559.
Karran, P., Offman, J., Bignami, M. Human mismatch repair, drug-induced DNA
damage, and secondary cancer. Biochimie, in press.
Offman, J., Opelz, G., Cummins, D., Halil, O., Banner, N., Burke, M., Sullivan, D.,
Macpherson, P., Karran, P. Defective DNA Mismatch Repair in Acute Myeloid
Leukemia/Myelodysplastic Syndrome in Organ Transplant Patients. Submitted to
Blood.

Table of Contents
ABSTRACT....................................................................................................................................2ACKNOW LEDGEMENTS...........................................................................................................3PUBLICATIONS........................................................................................................................... 5TABLE OF CONTENTS.............................................................................................................. 6LIST OF TABLES.......................................................................................................................10LIST OF F IG U R ES.................................................................................................................... 11LIST OF ABBREVIATIONS.....................................................................................................13
CHAPTER 1 : INTRODUCTION..............................................................................................16
1.1 DNA dam age................................................................................................................... 161.1.1 Damage caused by ionizing radiation.................................................................171.1.2 Alkylating damage....................................................................................................18
Monofunctional alkylating agents............................................................................... 18Clinically used monofunctional alkylating agents................................................... 21Bifunctional alkylating agents...................................................................................... 21Nitrogen mustards......................................................................................................... 23
1.1.3 Damage by other drugs..........................................................................................25Thiopurines..................................................................................................................... 25Cisplatin........................................................................................................................... 25
1.2 Cellular responses to DNA dam age........................................................................271.2.1 Excision pathways................................................................................................... 27
Base excision repair......................................................................................................27Nucleotide excision repair............................................................................................ 30Transcription-coupled DNA repair..............................................................................32DNA mismatch repair................................................................................................... 33
1.2.2 Reversal of DNA damage...................................................................................... 331.2.3 Double strand break repair....................................................................................35
Non-homologous end joining...................................................................................... 35Homologous recombination.........................................................................................38Fanconi anaemia............................................................................................................40Single strand annealing................................................................................................42
1.2.4 Tolerance of DNA dam age....................................................................................42Translesion synthesis................................................................................................... 42
1.3 DNA mismatch rep a ir.................................................................................................. 451.3.1 DNA mismatches......................................................................................................451.3.2 Bacterial mismatch repair...................................................................................... 461.3.3 Human mismatch repair..........................................................................................501.3.4 Biological consequences of inactive mismatch repair.....................................54
1.4 The role of DNA mismatch repair in tolerance to DNA damaging agents................................................................................................................................................... 55
1.4.1 Tolerance to methylating agents.......................................................................... 551.4.2 Tolerance to thiopurines.........................................................................................591.4.3 Tolerance to other drugs.........................................................................................61
1.5 DNA mismatch repair and cancer........................................................................... 611.5.1 Roles of MMR genes in human cancer.............................................................. 61
Familial cancer............................................................................................................... 61Sporadic cancer.............................................................................................................63Definition of M S I.............................................................................................................63Mutational target genes in MMR defective tumours...............................................63
1.5.2 Roles of MMR genes in mouse tumourigenesis...............................................65Mice with mutations is MutS homologues.................................................................66Mice with mutations MutL homologues..................................................................... 66

Mice with mutations in other MMR genes.................................................................67Response to DNA damaging agent treatment of MMR defective mice.............68
1.6 DNA mismatch repair and secondary cancer..............................................691.6.1 General haematopoiesis and leukaemia development.................................. 69
General haematopoiesis...............................................................................................70Classification of leukaemia..........................................................................................70Acute promyelocytic leukaemia...................................................................................72Myelodysplastic syndrome...........................................................................................75
1.6.2 Therapy-related acute myeloid leukaemia......................................................... 76AML after Hodgkin's disease and non-Hodgkin’s lymphoma...............................77AML after breast and ovarian cancer........................................................................77
1.6.3 Therapy-related myelodysplastic syndrome......................................................771.7 Overview of work described in this thesis...................................................78
CHAPTER 2 : MATERiALS AND METHODS.......................................................... 792.1 Materiais......................................................................................................... 79
2.1.1 Chemicals..................................................................................................................792.1.2 Cell culture................................................................................................................ 792.1.3 Cell lines and clinical samples..............................................................................792.1.4 Molecular biology reagents....................................................................................80
2.2 Celi culture techniques..................................................................................812.2.1 Maintenance of cell cultures................................................................................. 812.2.2 Cell storage............................................................................................................... 812.2.3 Generation of cell cultures from single cells......................................................812.2.4 Selection of 6-TG resistant clones.......................................................................82
HAT treatment................................................................................................................ 826-TG treatment............................................................................................................... 82Screening for cross-resistance to M NU.................................................................... 82
2.2.5 Cell survival by colony formation assay.............................................................. 846 -T G 84MNU...................................................................................................................................84
2.2.6 Cell survival by cell density....................................................................................842.2.7 Lymphoblastoid growth curves..............................................................................84
2.3 Molecular and cellular biology techniques.................................................. 852.3.1 Preparation of genomic D N A ................................................................................ 85
From animal cells...........................................................................................................85From paraffin embedded tissues................................................................................ 85
2.3.2 Preparation of total RNA.........................................................................................852.3.3 Parallel RNA and genomic DNA purification......................................................852.3.4 Determination of DNA concentrations in aqueous solutions..........................86
Spectrophotometer........................................................................................................ 86SYBR Green I stained agarose g e l........................................................................... 86
2.3.5 Determination of RNA concentrations in aqueous solutions..........................86Spectrophotometer........................................................................................................ 86Spectrofluorophotometer............................................................................................. 87
2.3.6 hMLHI promoter méthylation assay.................................................................... 872.3.7 MMR RT-PCR...........................................................................................................88
MMR RT-PCR of clinical samples..............................................................................882.3.8 Microsatellite analysis............................................................................................. 912.3.9 Target gene analysis...............................................................................................91
Genotyping.......................................................................................................................91DNA sequencing reactions..........................................................................................91
2.3.10 FancD2 RT-PCR................................................................................................... 922.3.11 Cell extracts............................................................................................................92
For MGMT assay............................................................................................................92

For Western blots...........................................................................................................92Stillman replication extract...........................................................................................93
2.3.12 Measurement of protein concentrations in aqueous solution...................... 932.3.13 In vitro mismatch repair assay........................................................................... 932.3.14 Western blotting.................................................................................................... 942.3.15 Methyltransferase assay...................................................................................... 95
Substrate and reaction conditions..............................................................................95Calculation of MGMT activity...................................................................................... 95
CHAPTER 3 : RESULTS I ..................,.....................................................................97DRUG TREATMENT AND HMLH1 PROMOTER METHYLATION IN THE DEVELOPMENT OF MMR DEFECTIVE ACUTE LEUKAEMIA AND MDS
3.1 Introduction................................................................................................. 973.2. Results.......................................................................................................... 98
3.2.1 Patients...................................................................................................................... 983.2.2 Microsatellite instability in chemotherapy-related AL/MDS.......................... 1003.2.3 Clinical sample DNA concentrations................................................................. 1033.2.4 Optimisation of the hMLHI promoter méthylation assay.............................. 1043.2.5 Sensitivity of the hMLHI promoter méthylation assay..................................1103.2.6 hMLHI promoter méthylation analysis of control samples.......................... 1103.2.7 hMLHI promoter méthylation analysis of t-AL/MDS samples.................... 1133.2.8 Optimization of MMR RT-PCR............................................................................1153.2.9 MMR gene expression in AML samples...........................................................118
3.3 Discussion................................................................................................... 1203.3.1 M S r frequency among t-AL/MDS......................................................................1203.3.2 M S r and treatment...............................................................................................1213.3.3 Cyclophosphamide and t-AML............................................................................1223.3.4 hMLHI promoter méthylation in t-AL/MDS.....................................................1233.3.5 hMLHI promoter méthylation in t-APL............................................................ 1243.3.6 Gene defects in other MMR genes in t-AL/MDS.............................................1243.3.7 t-AL/MDS and primary breast cancer................................................................125
3.4 Conclusion..................................................................... 128CHAPTER 4 : RESULTS II.....................................................................................129THE ROLE OF THIOPURINE RESISTANCE AND DEFECTIVE DNA MISMATCH REPAIR IN THE DEVELOPMENT OF ACUTE MYELOID LEUKAEMIA AFTER ORGAN TRANSPLANTS
4.1. Introduction................................................................................................. 1294.2. Results........................................................................................................ 131
4.2.1 Generation of clonal variants of A2780 resistant to 6-TG.............................1314.2.3 Mismatch repair status of thiopurine resistant cells........................................1324.2.4 HGPRT status of 6-TG resistant clones........................................................... 1334.2.5 Drug resistance of A2780-JA5-3 and JA 8-3....................................................1374.2.6 Microsatellite instability of 6-TG resistant clones........................................... 1414.2.7 AML in transplant recipients.................................................................................1444.2.8 Microsatellite instability in transplant related AML/MDS............................... 150
4.3. Discussion.................................................................................................. 1564.3.1 MMR' clonal variants of A2780 resistant to 6-TG and M N U ........................ 1564.3.2 AML after organ transplants.................................................................................1574.3.3 Microsatellite instability in transplant-related AML/MDS............................... 158
4.4 Conclusion................................................................................................... 159

CHAPTER 5 : RESULTS III....................................................................................160MUTATIONS AT MONONUCLEOTIDE REPEATS IN TARGET GENES IN MSI* HUMAN CELL LINES AND T-AML
5.1 Introduction................................................................................................. 1605.2 Results......................................................................................................... 167
5.2.1 M re ll analysis of chemotherapy-related AML samples............................... 1675.3.2 Genescan analysis of target repeats................................................................. 1675.2.3 Target gene analysis of MMR proficient and deficient tumour cell lines ..1685.2.4 Target gene analysis of t-AML samples........................................................... 1725.2.5 FancD2 repeat analysis of MMR proficient cell lines and human samples ..............................................................................................................................................1745.2.6 FancD2 analysis of MMR deficient human tumour cell lines and colon cancer samples.................................................................................................................1775.2.7 FancD2 repeat analysis of t-AML samples...................................................... 1775.2.8 FancD2 expression...............................................................................................1795.2.9 Mitomycin 0 sensitivity assay............................................................................. 1795.2.10 Functionality of FancD2..................................................................................... 183
5.3 Summary and Discussion...........................................................................1835.3.1 M r e l l ....................................................................................................................... 1835.3.2 Caspase 5 ................................................................................................................1855.3.3 F lt3 ............................................................................................................................1865.3.4 Other genes............................................................................................................ 1875.3.5 New assignment of case 8 ................................................................................... 1875.3.6 Fanconi anaemia pathway................................................................................... 187
5.4 Conclusion................................................................................................... 189CHAPTER 6 : DISCUSSION AND CONCLUSION................................................. 190REFERENCES....................................................................................................... 194APPENDIX I: FIGURES..........................................................................................208APPENDIX II: TABLES..........................................................................................214

List of Tables
Table 1.1 Human DNA glycosylases...................................................................................... 29Table 1.2 Properties of eukaryotic DNA polymerases........................................................43Table 1.3 Mammalian MutS and MutL homologues............................................................52Table 1.4 Different genes mutated at coding repeat sequences in M S r cancer 64Table 1.5 Mouse models with disrupted MMR alleles.........................................................65Table 1.6 FAB classification of AML....................................................................................... 70Table 2.1 Cell lines............................ 80Table 2.2 Primers........................................................................................................................89Table 2.3 Antibodies...................................................................................................................94Table 3.1 Characteristics of t-AL/MDS patients....................................................................99Table 3.2 Analysis of MSF in t-AL/MDS patients............................................................. 103Table 3.3 MSF and hMLHI méthylation in t-AL/MDS patients..................................... 113Table 3.4 Microsatellite instability in therapy-related and de novo AML/MDS
published in the literature................................................................................................121Table 3.5 MSF and therapy for the first tumour................................................................. 121Table 3.6 MSF and hMLHI méthylation in therapy-related and de novo APL.......... 124Table 4.1 Plating efficiency of A2780-JA clones in HAT medium..................................132Table 4.2 Comparative cytotoxicity of 6-TG resistant clones..........................................137Table 4.3 Microsatellite instability of A2780-JA subclones..............................................143Table 4.4 Consultants contacted to enquire about post-azathioprine treatment AML.
..............................................................................................................................................145Table 4.5 Details of patients................................................................................................... 149Table 4.6 Microsatellite Instability in MDS/AML cases..................................................... 151Table 5.1 Potential target genes for frameshift mutations analysed by Ida Casorelli.
160Table 5.2 Potential target genes for frameshift mutations in MMR defective cell lines.
161Table 5.3 Target gene analysis of t-AL/MDS cases by Ida Casorelli............................167Table 5.4 Target gene analysis of MMR proficient and deficient cell lines..................171Table 5.5 Target gene analysis of AML samples...............................................................173Table 5.6 FancD2 analysis of MMR^ cell lines and human samples............................ 176Table 5.7 FancD2 analysis of AML samples........................................................................178
10

List of Figures
Figure 1.1 SnI and Sn2 reactions of alkylating agents....................................................... 19Figure 1.2 Sites of méthylation on the DNA bases.............................................................. 19Figure 1.3 Chemical structures of MNU and 0®-alkylating agents in common clinical
use..........................................................................................................................................20Figure 1.4 Mechanism of action of dacarbazine and temazolamide............................... 20Figure 1.5 DNA interstrand cross-link formation after chloroethylation of guanine in
DNA....................................................................................................................................... 22Figure 1.6 Metabolism of cyclophosphamide........................................................................24Figure 1.7 Metabolism of 6-MP and 6-TG............................................................................. 26Figure 1.8 Schematic representation of the base excision repair pathways................. 28Figure 1.9 Schematic representation of the nucleotide excision repair pathway......... 31Figure 1.10 Schematic representation of the DNA non-homologous end-joining
pathway.................................................................................................................................36Figure 1.11 Double strand break repair by homologous recombination........................ 39Figure 1.12 The Fanconi anaemia / BROA pathway...........................................................41Figure 1.13 Pathways for the nonmutatgenic DNA repair of replication forks.............. 44Figure 1.14 Microsatellite instability........................................................................................47Figure 1.15 Model of methylation-dependent mismatch repair in E. coli...................... 48Figure 1.16 Initiation of mismatch repair in human cells.................................................... 51Figure 1.17 Model of the contribution of mismatch repair to the cytotoxicity of DNA
méthylation damage...........................................................................................................57Figure 1.18 Processing of 0®-meG basepairs by mismatch repair..................................58Figure 1.19 Model of the contribution of mismatch repair to the cytotoxicity of 6-TG.
60Figure 1.20 Scheme of haemopoiesis.................................................................................... 71Figure 1.21 The multiple oncogenic functions of PML-RARa........................................... 73Figure 2.1 Flowchart of the generation of 6-TG resistant A2780 clones........................83Figure 3.1 Microsatellite instability in chemotherapy-related AL/MDS..........................101Figure 3.2 DNA concentration standard curves................................................................. 105Figure 3.3 Map of the hMLHI and PCNA promoter regions........................................... 108Figure 3.4 Optimisation of the hMLHI promoter méthylation assay............................. 109Figure 3.5 hM LHI promoter méthylation assay of Ra]i/SW48 DNA titrations.............I l lFigure 3.6 Examples of the hMLHI promoter méthylation assay of clinical control
samples...............................................................................................................................112Figure 3.7 Examples of the hMLHI promoter méthylation analysis of t-AL/MDS
samples...............................................................................................................................114Figure 3.8 Optimisation of the MMR RT-PCR.................................................................... 116Figure 3.9 MMR gene expression in AML samples...........................................................119Figure 3.10 t-AL/MDS and primary breast cancer............................................................. 127Figure 4.1 Expression of MMR genes in 6-TG resistant clones.....................................134Figure 4.2 MMR protein expression in 6-TG resistant clones.........................................135Figure 4.3 In vitro MMR correction in A2780-JA5-3 and -JA8-3.................................... 136Figure 4.4 HAT and Aminopterin sensitivity assay of A2780-JA5-3 and -JA8-3........ 136Figure 4.5 MNU and 6-TG sensitivity of A2780-JA5-3 and -JA8-3................................138Figure 4.6 CsA, Pred and 6-MP sensitivity assay of A2780-JA5-3 and -JA8-3..........139Figure 4.7 Microsatellite instability analysis of A2780-JA8-3 subclones...................... 142Figure 4.8 Incidence of AML in transplant recipients........................................................ 148Figure 4.9 Microsatellite instability in transplant-related AML/MDS (cases A1-6). ...152Figure 4.10 Microsatellite instability in transplant-related AML/MDS (cases A7).......155Figure 5.1 Schematic representation of the amplified genomic region of assayed
target genes {Mre11, Chk1, Fas. Rb, Fit3 and Cbffi) and example of the multiplex PGR....................................................................................................................164
11

Figure 5.2 Schematic representation of the amplified genomic region of assayedtarget genes {FancE and FancG) and example of the multiplex PCR................. 165
Figure 5.3 Schematic representation of the amplified genomic region of assayedtarget genes {FancD2 and Casp-5) and example of the multiplex PCR..............166
Figure 5.4 Casp-5 repeat analysis of tumour cell lines................................................... 169Figure 5.5 Examples of target gene analysis of repeats in Mre11, Chk1, Fas, Rb,
Casp-5, FancE and FancG............................................................................................. 170Figure 5.6 Examples of FancD2 mutations in blood donors and cell lines....................175Figure 5.7 FancD2 RT-PCR................................................................................................... 180Figure 5.8 MMC sensitivity assay..........................................................................................182Figure 5.9 FancD2 Western blot............................................................................................184Figure 6.1 Model illustrating the development of MSF AML........................................... 192
12

List of Abbreviations
4-HC5-FU 1-meA 3-meA 3-meC 7-meG6-MP 6-TG ABVD AL ALL AML AAPAPBSCTAPLAPEATATLDATMATPATRATRAAzaBMBCNUBERBLMbpBRCA1BRCA2BSACCasp-5CbfpCGNUCisPtGibGMMLGOPPGSGsAGTXGysdHzODAMDNADNA-PKdNTPDMHDMSODSBdsDNA
4-hydroxycyclophosphamide5-fluorouracil 1-methyladenine 3-methyladenine 3-methylcytosine7-methylguanine6-mercaptopurine6-thioguanineadriamycin, bleomycin, vinblastine, dacarbazineacute leukaemiaacute lymphocytic leukaemiaacute myeloid leukaemiaadenineapurinic / apyrimidinicautologous peripheral blood stem cell transplantationacute promyelocytic leukaemiaAP endonucleaseataxia-telangiectasiaataxia-telangiectasia-like disorderataxia-telangiectasia mutatedadenosine 5-triphosphateataxia-telangiectasia relatedall-frans-retinoic acidazathioprinebone marrowN,N'-bis(2-chloroethyl)-N-nitrosourea base excision repair Bloom syndrome DNA helicase base pairbreast cancer 1 genebreast cancer 2 genebovine serum albumincytosinecaspase 5core binding factor pN-(2-chloroethyl)-N’-cyclohexyl-N-nitrosoureacisplatinchlorambucilchronic myelomonocytic leukaemiaGTX, oncovin, procarbazine, prednisoneGockayne syndromeGyclosporin Acyclophosphamidecysteinedeionised waterDNA adenine methylasedeoxyribonucleic acidDNA-dependent polymerase protein kinase deoxyribonucleotide triphosphate 1,2-dimethylhydrazine dimethyl sulfoxide double strand break double-stranded DNA
13

DTTE. coliEDTAENUeoExo1FAFABFancA-LFCSFen1FLT3GGGRGlHATHDHepesHGPRTHNPCCHRIDLIRITDLOHkbKOM62SO4
MiCMA
MGMTMLHMMCMMRMMSMNNGMNUMOPPMSHMSImRNAMBSNERNF1NHEJNHL0®-BzGG®-meGPAGEPBSPCNAPCRPMPMLpol
DIthiothreitol Escherichia colidisodium ethylenediaminetetraacetic acidethylnitrosoureaeosinophiliaExonuclease 1Fanconi anaemiaFrench-American-BritishFanconi anaemia complementation group A-Lfoetal calf serumFlap endonuclease 1FMS-like tyrosine kinase (3)guanineglobal genome repair gastrointestinalhypoxanthine, aminopterin, thymidine Hodgkin's diseaseA/-2-hydroxyethylpiparazine-A/-2-ethane sulphonic acidhypoxanthine guanine phosphoribosyl transferasehereditary nonpolyplosis colorectal cancerhomologous recombinationinsertion-deletion loopionising radiationinternal tandem duplicationloss of heterozygositykilobaseknock-outdimethylsulfatemitoxanthrone, carboplatin, methylprednisolone, cytosine arabinoside0®-methylguanine DNA methyltransferaseMutL homologuemitomycin CDNA mismatch repairmethylmethane sulphonateA/-methyl-A/-nitro-/V-nitrosoguanidineA/-methyl-/V-nitrosoureamechloroethamine, vincristine, procarbazine, prednisoneMutS homologuemicrosatellite instabilitymessenger RNANijmegen breakage syndromenucleotide excision repairneurofibromatosis type 1non-homologous end joiningnon-Hodgkin’s lymphoma0®-benzylguanine0®-methylguaninepolyacrylamide gel electrophoresisphosphate buffered salineproliferating cell nuclear antigenpolymerase chain reactionphosphoramide mustardpromyelocytic leukaemiapolymerase
14

Pred prednisolonePTLD post-transplant lymphoproliferative disordersRA refractory anaemiaRAEB refractory anaemia with excess blastsRAEB-t refractory anaemia with excess blasts ‘in transformation'RARa retinoic acid receptor aRARS refractory anaemia with ringed sideroblastsRb retinoblastoma-susceptibility proteinRNA ribonucleic acidRNAPII RNA polymerase IIRPA replication protein Arpm revolutions per minuteRT radiation therapyRT-PCR reverse transcriptase polymerase chain reactionSnI unimolecular nucieophilic substitutionSn2 bimolecular nucieophilic substitutionSAM S-adenosylmethionineSDS sodium dodecyl sulphateSSA single-strand annealingSSC squamous cell carcinomassDNA single-stranded DNAT thyminet- therapy-relatedTBE Tris Borate EDTATOR transcription coupled repairTE Tris-EDTATFIIH transcription factor IIHTGFpRII transforming growth factor p receptor type IITPMT thiopurine methyltransferasetRNA transfer RNA
Tris tris[hydroxymethyl]amino-methane(2-amino-2-hydroxy- methylpropane-1,2-diol)
U unitsUTR untranslated regionUV ultravioletVCR vincristinewt wild typexg gravitational forceXP xeroderma pigmentosum
15

Chapter 1 : Introduction
Fifty years ago James Watson and Francis Crick proposed their model for the
DNA double helix (Watson and Crick, 1953). This macromolecule was thought to be
highly stable at the time, but it has since been recognised that DNA is dynamic and
subject to continuous damage, either by environmental agents or spontaneously. To
deal with these different forms of damage cells have evolved different mechanisms
for either tolerating or repairing DNA damage (Friedberg, 2003). Failure of these
mechanisms can lead to diseases like cancer, as it is well illustrated in the human
hereditary diseases xeroderma pigmentosa (XP), hereditary non-polyposis colon
cancer (HNPCC) and some forms of breast cancer. In addition, acquired DNA repair
defects have also been implicated in the development of sporadic cancers and
chemotherapy resistance in human tumours (Peltomaki, 2003).
Paradoxically, DNA damage cannot only induce cancer, it is also involved in
treating it. Ionizing radiation (IR) and chemotherapeutic drugs are toxic to cancer
cells because they introduce DNA lesions that interfere with cell division. The
problem that this approach to treating cancer poses, is that not only cancer cells are
affected by this type of treatment. Many chemotherapeutic drugs are also toxic to
other dividing cells like bone marrow cells. This can lead to bone marrow depression
and is implicated in certain types of post-chemotherapy leukaemia.
1.1 DNA damage
Single nucleotide (point) mutations are typically generated by the replication
of DNA that has acquired some form of base damage. DNA damage can be due to
spontaneous alterations or may be environmentally induced. Normal cellular
metabolism is associated with many types of DNA lesion including hydrolytic base
loss, deamination, oxidative damage, base méthylation and base misincorporation
during replication. Environmental agents, such as chemicals in cigarette smoke,
also damage DNA. However, the main function of clinical radiation therapy and
chemotherapy is to kill rapidly dividing cells by damaging DNA.
16

1.1.1 Damage caused bv ionizing radiation
IR can induce a variety of DNA lesions. Radiation damage to DNA can be
caused by either direct or indirect effects. Direct effects result from the direct
interaction of the radiation energy with the DNA, and are thought to cause -35% of
ionizing radiation damage. Indirect effects are due to the interaction of DNA with
reactive species formed by the radiation. A major potential source of indirect
damage are reactive oxygen species formed by the radiolysis of water, specifically
•OH, O 2 and H2O2. One of the major forms of mutagenic base damage caused by
active oxygen is 8-oxoguanine, which can base-pair with adenine in replication,
leading to transversion mutations. Thymine glycol, another product of radical attack
on DNA, can block replication and also undergoes further fragmentation to products
such as methyltartronylurea and urea. One characteristic feature of ionizing
radiation damage that is not usually encountered following treatment with other
agents that generate reactive oxygen species, is the formation of more than one
damaged moiety in close proximity.
Most of the lethal effects of IR can be attributed to directly induced strand
breaks, in particular double-strand breaks. The majority of these strand breaks are
characterised by unusual or damaged termini that preclude repair by a simple DNA
ligation step. IR can induce strand breakage directly. Single-strand breaks are
initiated by radical formation at deoxyribose followed by the loss of a hydrogen
atom. The reactions involved in the formation of double strand breaks (DSB) by
radiation directly are less clear.
Radiation therapy (RT) plays an important role in the management of benign
and malignant diseases (Mundt et al., 2000). RT alone is used to treat a variety of
tumours, for example early stage head and neck and gynaecological tumours. For
certain tumours, RT administered alone produces results comparable to, if not better
than, those obtained with surgery. Examples include tumours of the oral cavity and
early stage cervical cancer. RT is more commonly used in combination with surgery
and/or chemotherapy. RT is used postoperatively for many tumours, for example
breast and lung, but may also be given before or during surgery. Chemotherapy can
be administered prior to, during, or following radiotherapy, depending on the tumour
site and stage. Early in the 20* century it became clear that RT was equally
efficacious but better tolerated when administered in divided doses. This is thought
to spare normal tissue by allowing time for repair and repopulation of normal cells.
Patients are therefore typically treated five days a week for several weeks.
17

1.1.2 Alkylating damage
Alkylating agents transfer alkyl groups to DNA. Although numerous
structurally diverse alkylating agents are used clinically, the methylating agents are
one of the most significant groups. DNA damage caused by methylating agents has
been reviewed by (Middleton and Margison, 2003; Sedgwick and Lindahl, 2002;
Singer and Grunberger, 1983). Alkylating agents produce a wide variety of base
lesions and phosphotriesters. The relative yield of each lesion depends on the
nature of the alkylating agent, its reaction mechanism, and the secondary structure
of the DNA target. One type of alkylating agent (e.g. methyl methanesulphonate
(M MS)) reacts via an Sn2 (bimolecular nucieophilic substitution) mechanism and
alkylates DNA almost exclusively at nitrogen moieties in the purine and pyrimidine
rings. The second group of alkylating agents (e.g. A/-methyl-/V-nitrosurea (MNU)),
are SnI agents (unimolecular nucieophilic substitution) and alkylate both nitrogens
and oxygens in the DNA bases as well as oxygen in the sugar-phosphate backbone.
SnI type reactions function by a stepwise mechanism, whereas the bond making
and breaking steps in a Sn2 type reaction occur at the same time via a reactive
intermediate (Fig. 1.1). The rate-limiting step in an SnI type reaction is the breaking
away of the leaving group (X ) which is followed by the reaction of the nucleophile
(Y) with the electrophilic carbon (R"). The rate-limiting step of an Sn2 reaction is
bimolecular. This step occurs when the nucleophile (Y ) begins to bind to the
electrophilic carbon (R) at the same time that the leaving group (X) begins to break
away (Morrison and Boyd, 1992).
Monofunctional alkylating agents
Alkylating agents can be further divided into mono- and bifunctional
alkylating agents. The main monofunctional methylating agents used in the
laboratory are dimethylsulfate (Me2S0 4 ), MMS, MNU and A/-Methyl-A/-nitro-A/-
nitrosoguanidine (MNNG). Me2S0 4 and MMS react mainly with the N-7 of G, N-1
and N-3 of A, and N-3 of C. The potent carcinogens MNU (Fig. 1.3) and MNNG
predominantly methylate oxygens, and the O® of G, the and of U or T and the
of C can all be alkylated. For monofunctional methylating agents, 7-
methylguanine (7-meG), 3-methyladenine (3-meA), and 0®-methylguanine (O®-
meG) are the major methylated bases generated in double-stranded DNA whereas
1-methyladenine (1-meA) and 3-methylcytosine (3-meC) comprise the majority of
modifications in single-stranded DNA (Fig. 1.2). The most abundantly formed lesion,
7-meG, is relative innocuous, as is 3-meA. It appears that these lesions are only
18
RX -► R+ + X‘ -
Sn I Reaction
-► RY + X'
Ô- Ô-RX + y -------► [Y ...R ...X ] ►RY + X'
Sn2 Reaction
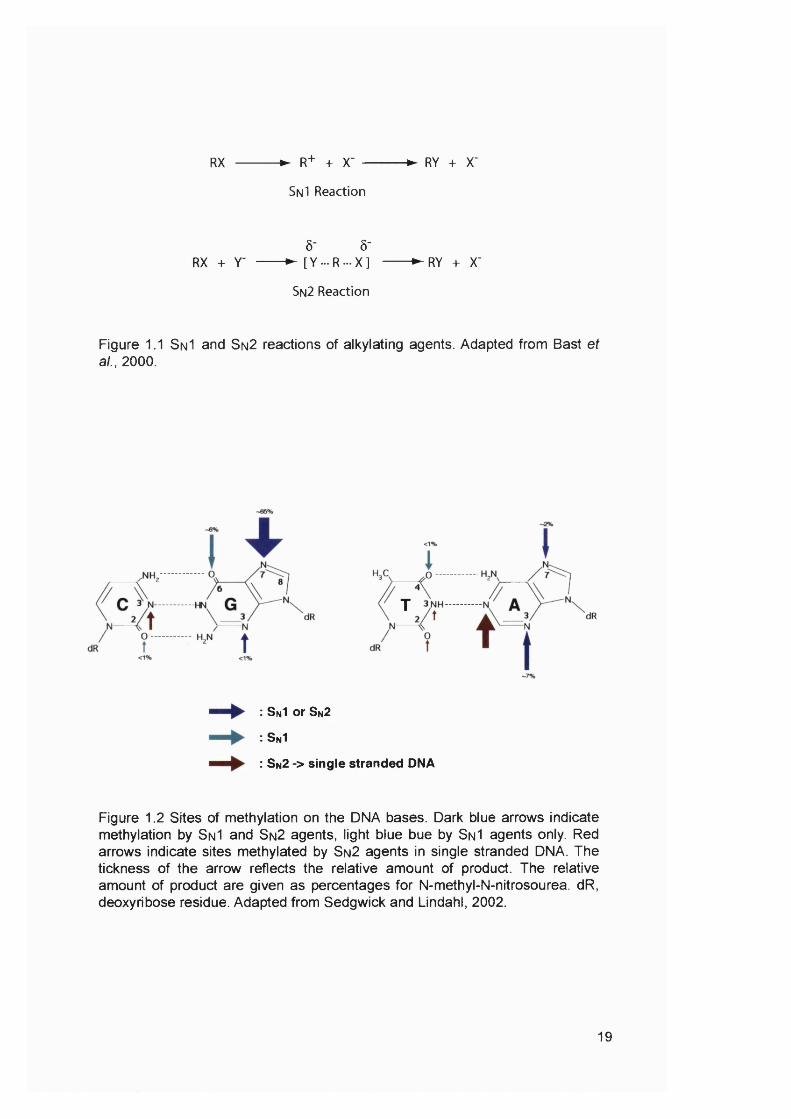
Figure 1.1 SnI and Sn2 reactions of alkylating agents. Adapted from Bast et al., 2000.
T
: S nI or S n2
: S n1
: S n2 > s ing le stranded DNA
Figure 1.2 Sites of méthylation on the DNA bases. Dark blue arrows indicate méthylation by S n I and Sn2 agents, light blue bue by Sn I agents only. Red arrows indicate sites methylated by Sn2 agents in single stranded DNA. The tickness of the arrow reflects the relative amount of product. The relative amount of product are given as percentages for N-methyl-N-nitrosourea. dR, deoxyribose residue. Adapted from Sedgwick and Lindahl, 2002.
19

0
I IH
N-methyl-N-nltrosourea
CICHjCHj.^ ^CH CH.C: CarmustlneI I H
0 f " 80 J
C H ,C H r P N N
" ""o
,C Fotemustine's'" "2 CHfHf
OH
0 — ( 0
OHCH.;— ( X . ^CH, Streptozotocin
Fig. 1.3 Chemical structures of N-methyl-N-nitrosourea and 0®-alkylating agents in common clinical use. The nitrosourea backbone is shown in bold and the alkylating moiety is shown in red. Adapted from Middleton and Margison, 2003.
Dacarbazine Temazolamide
Hepatic activation
H,N V. ^ 0
Chemical decomposition
MTIC
H . N ^ O
+ N - - CH
' N
1 Methyldiazoniumion
H,C- <X,xN NH,
<%%dR
Guanine
N NH,dR
06-methylguanine
Fig. 1.4 Mechanism of action of dacarbazine and temazolamide. The metabolic activation of dacarbazine and spontaneous degradation of temazolamide leads to the formation of O^-methylaguanine. MTIC, methyltriazenoimdazole carboxamide; AlC, aminoimidazole carboxamide; dR, deoxyribose residue. Adapted from Middleton and Margison, 2003.
20

cytotoxic when repair is incomplete and repair intermediates accumulate (Horton et
al., 2003). 0®-meG is both cytotoxic and mutagenic. 0®-meG induced mutations are
mainly guanine (G) to adenine (A) transitions that arise through mispairing by the
methylated base during replication (Karran and Bignami, 1994). The analogous
ethylating agents generally react with the same nitrogen and oxygen groups as their
methylating counterparts.
Clinically used monofunctional alkylating agents
Tetrazines are monofunctional alkylating agents that are used clinically.
They methylate DNA at the O® position of guanine via a diazonium ion intermediate.
Procarbazine and dacarbazine are both used commonly in the treatment of
Hodgkin’s disease (HD) and non-Hodgkin’s lymphoma (NHL). Temozolomide is an
imidazotetrazinone. These drugs are metabolized to reactive intermediates that
decompose to produce a methyl diazonium ion which methylates the O® position of
guanine (Fig. 1.4). Temozolomide was shown to have activity against high-grade
glioma. It was significantly more active than procarbazine and was also better
tolerated (Yung et al., 2000). This anti-tumour activity was further increased when
temozolomide treatment was combined with procarbazine (Newlands et al., 2003).
Streptozotocin, which is a naturally occurring nitrosourea, also produces DNA O®-
meG (Fig. 1.3). This drug is mainly used for pancreatic cancer.
Bifunctional alkylating agents
Bifunctional alkylating agents have two reactive groups and are able to
modify two different sites in DNA. If these sites are situated on the same strand, the
reaction product is a DNA intrastrand cross-link. If the two sites are on opposite
strands, an interstrand DNA cross-link is produced. Interstrand DNA cross-links
pose problems for the cell, as they prevent DNA strand separation and hence
constitute complete blocks to DNA replication and transcription. Exposure to
chloroethylnitrosoureas (for example BCNU, see below) leads to the formation of
interstrand cross-links between guanine and cytosine residues (Fig. 1.5).
Bifunctional agents are generally much more efficient anti-tumour agents than
monofunctional agents.
21

NH:
/3N
dR 0
Cytosine
UlI
CHjI
©CH2
/HNi V n'
= NHoN
dR
Guanine
NH:
NI
dR
0 . . .
NH-
- HgN
0 6 -chloroethylguaninc
i
NI
dR
0
dR
1 0 6 -ethanoguanine
H i3N
dR dR
l-< 3-cytosi nyl )-2 -{ 1-guanosi ntyl )-ethane
Fig. 1.5 DNA interstrand cross-link formation after chloroethylation of guanine in DNA. dR, deoxyribose residue. Adapted from Middleton and Margison, 2003.
22

Bifunctional nitrosoureas
The first nitrosourea that was found to have an anti-leukaemic activity was
MNU. When it was shown that the activity of MNU could be further increased by
substitution of a chloroethyl group, several chloroethylnitrosoureas were developed
based on the structure of MNU (Fig. 1.3). The main nitrosourea compounds used
clinically are carmustlne (or BCNU) (N,N'-bis(2-chloroethyl)-N-nitrosourea) (Fig.
1.3), lomustine (or CGNU) (N-(2-chloroethyl)-N ’-cyclohexyl-N-nitrosourea),
fotemustine (N-(2-chloreoethyl)-N'-(diethyl)ethylphosphonate-N-nitrosourea) (Fig.
1.3). Carmustlne is mainly used for HD, NHL, brain tumours and multiple myelomas,
whereas lomustine is mainly used for HD and brain tumours. Fotemustine is also
used to treat brain tumours. These drugs are all bifunctional alkylating agents. They
are highly mutagenic.
Chloroethylnitrosoureas cause crosslinks between guanine and cytosine
residues (Fig. 1.5). The chloroethyl group initially attacks the O® position of guanine,
forming an intermediate 1 -0^-ethanoguanine, which can react with the N position of
cytosine to form the crosslink. Alternatively, reaction with a protein generates a
DNA-protein cross-link. This reaction has been demonstrated for the O®-
methylguanine-DNA methyltransferase.
Nitrogen mustards
Nitrogen mustards are cyclic alkylating agents, which are also bifunctional.
Nitrogen mustards can form both monoaducts and crosslinks. Examples include
cyclophosphamide (CTX), ifosphamide, melphalan and chlorambucil (CIb). These
are the most frequently administered alkylating agents in cancer therapy and are
effective in treating a wide range of cancers. CTX and CIb have also been used as
immunosuppressive agents. The characteristic chemical constituent of the nitrogen
mustards is the bischloroethyl group, and all of these compounds react through an
aziridinium intermediate.
The nitrogen mustards undergo a complex activation to generate the
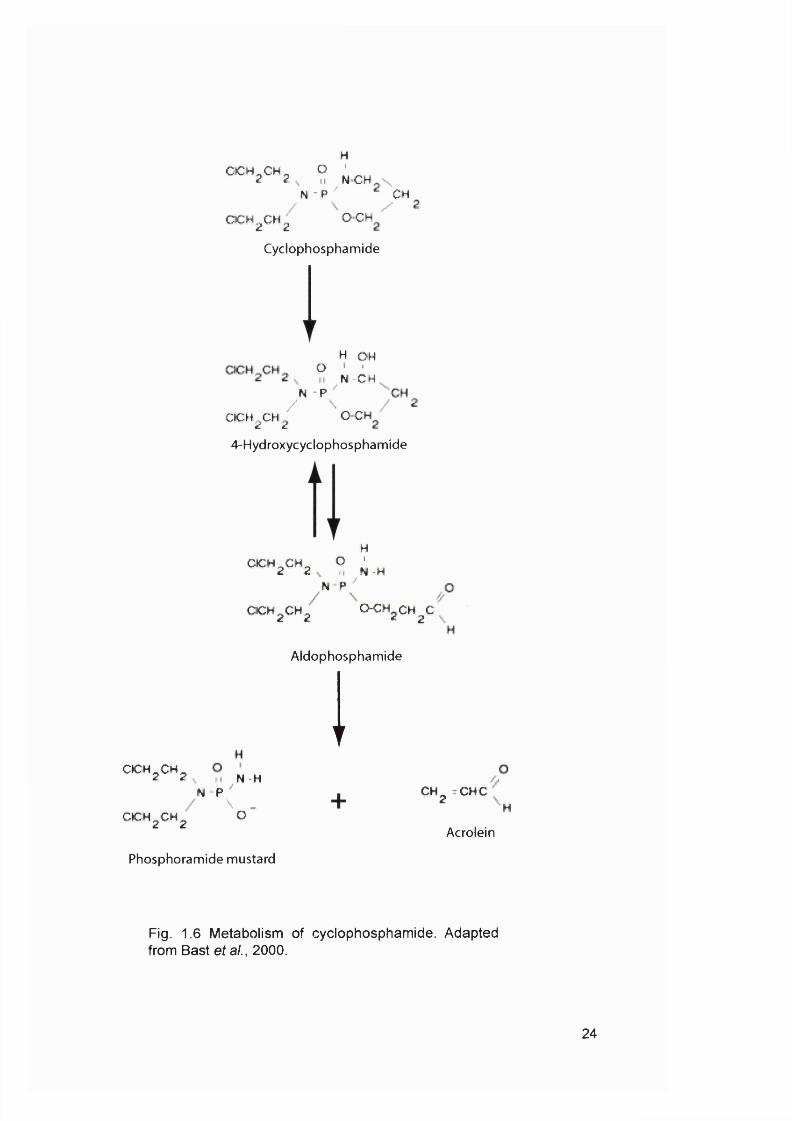
ultimate effector molecule. For example, CTX is activated by P450 in the liver to
produce 4-hydroxycyclophosphamide (4-HC), which is in spontaneous equilibrium
with the tautomer aldophosphamide (Fig. 1.6). At physiological pH, this equilibrium
is predominantly In the form of 4-HC, which readily enters target cells by diffusion.
Aldophosphamide spontaneously decomposes to produce phosphoramide mustard
23
CCHgCHg
OCH^CHg
HO 'II N CH
N -P CH
OCH.
Cyclophosphamide
H OH
° N-CH
ClCH^CHg/
N -P
OCH
4-Hydroxycyclophosphamide
CKZH^CHg
OCH^CHg
HO 'II N -H
N P "
OCH CH c « 2
Aldophosphamide
1CCHgCH^
CICHgCHg
N PN -H
" o -
CHg =CHC
Acrolein
H
Phosphoramide mustard
Fig. 1.6 Metabolism of cyclophosphamide. Adapted from Bast et a i, 2000.
24

(PM) and acrolein. PM is a reactive alkylating agent. It was suggested that one of
the chloroethyl groups cyclizes to form a chloroethyl azidinium moiety, which leads
to the formation of chloroethylazidridine. It is thought that chloroethylazidridine
contributes significantly to the alkylation and cross-linking properties of CTX.
Acrolein has been shown to inhibit MGMT and will be discussed in more detail in
Chapter 3.
1 1.3 Damage bv other drugs
Thiopurines
6-mercaptopurine (6-MP) and 6-thioguanine (6-TG), originally discovered by
Gertrude B. Elion, are cytotoxic drugs that are used in the treatment of acute
leukaemia and as immunosuppressants (Elion, 1989). Azathioprine (Aza) is a
component of a standard triple immunosuppressive therapy that is used following
solid organ transplants. It is also used, either as the sole agent or in combination
with other drugs, in treatment of autoimmune diseases. Aza is a pro-drug that is
converted in vivo to 6-MP. Both 6-MP and 6-TG are taken up in target cells,
metabolized to 2’-deoxy-6-thioguanosine triphosphate via the hypoxanthine guanine
phosphoribosyl transferase (H G PR T) pathway (Fig. 1.7) and extensively
incorporated as thioguanine into DNA (Aarbakke et al., 1997). The exact
mechanisms by which 6-TG is incorporated and functions as a cytotoxic drug will be
explained in more detail later (Chapters 1 and 4).
Cisplatin
Cisplatin is a widely used chemotherapeutic drug for a variety of tumours, for
example ovarian and bladder cancer, and it is a common component of many
chemotherapy regimes. It has been used very successfully in the treatment of
testicular carcinomas - more than 90% are now curable by this drug. Cisplatin is
cytotoxic because it forms DNA cross links (Aquilina and Bignami, 2001). 1,2-
intrastrand cross-links between N7 atoms of adjacent purines are the major
products (^80% of all adducts), with 1,2 GpG and 1,2 ApG representing 65 and 25%
respectively. 1,3 diguanyl crosslinks are less abundant.
25

SH
6-mercaptopurineH,N
6-thioguanine
SCH,
S-metfiyl-mercaptopurine
TPMTU
SH
iT \ \H G P R T V IMPD
HOP-O-CH,6-MP
SCH,
*S-methy1-thioguanine 6-thioguanine
O H -P -O -P -O -^O -C f^O OH OH OH RNA
HGPRT
OH OH6-thioguanosine5'-triphosphate
kinase
%H O -^O -C H ,
6:"')wGMPS HjN
H O -^O -C H ,
Xjc"-»kinase
XO
OH OH6-thioinosine
5 -monophosphale (TIN)
TPMT
OH OH6-thioxanthosine
5-monophosphale
OH OH6-thiogua nosine
5-monophosphate
0 n'' n H H
6-thkxjnc acid
SCH,
■ t e
OH OHS-methyl-thioinosine
5'-monophosphate (MeTIMP)
OH OH6-thioguanosine5'-diphosphate
kinase
H ^
H0-P-0-ÇH,
*H O -^O -P -O -
6 h 6 h
DNA
OH OHS-methyl-thioguanosine
5 -monophosphate
OH Hdeoxy-6-thioguanosine
5'-triphosphate
Fig. 1.7 Metabolism of 6-MP and 6-TG. Both drugs are incoorporated into DNA via the purine salvage pathway. 6-MP is catabolised via xanthine oxidase and méthylation is catalysed by the thiopurine methyltransferase (TPMT). GMPS, guanosine monphosphate synthetase; HGPRT, hypoxanthine guanine phosphoribosyl transferase; IMPD, inosine monophosphate dehydrogenase; TPMT, thiopurine methyltransferase; XO xanthine oxidase. Adapted from Aarbakke et al., 1997.
26

1.2 Cellular responses to DNA damageThis brief survey of the major DNA repair pathways is predominantly based
on three sources (Cline and Hanawalt, 2003; Fried berg et al., 1995; Hoeijmakers,
2001). The most general repair mechanism in cells is to excise the damaged or
inappropriate base and replace the normal nucleotide sequence (Friedberg et al.,
1995). Unless otherwise stated, this summary describes the different DNA repair
and damage tolerance mechanisms that operate in human cells.
1.2.1 Excision pathways
These pathways all involve recognition of DNA damage, removal of a single
stranded section of DNA that contains the damage, repair replication across the gap
and restoration of DNA continuity by ligation of the repaired strand. All three
pathways are error free, as the complementary strand is used as a template for
accurate repair.
Base excision repair
Base excision repair (BER) has been reviewed by (Krokan et al., 1997;
Lindahl, 2001; Lindahl and Barnes, 2000). The three BER pathways are
summarised in Fig. 1.8. BER is essential for the removal of endogenous DNA
lesions caused by hydrolysis, oxidation, or non-enzymatic alkylation. This repair
pathway is initiated by DNA glycosylases, which recognise and remove damaged or
modified base residues from DNA (Table 1.1). By cleaving the base-sugar bond
they produce abasic (AP) sites. Subsequently, an AP endonuclease or AP lyase
activity incises the abasic site. The terminal deoxyribose phosphate is removed
leaving a single nucleotide gap that may be filled by the insertion of a single
nucleotide. Alternatively, the gap may be slightly enlarged requiring the insertion of
up to six nucleotides for repair. Repair is completed by ligation of the newly
synthesised base or bases to the original strand.
Eleven known DNA glycosylases act on DNA lesions in human cells (Table
1.1). DNA glycosylases can be divided into two groups, monofuctional and
bifunctional. In addition to their DNA glycosylase activity the bifunctional enzymes
posses an intrinsic AP lyase that catalyses strand cleavage 3' to the abasic site after
base removal (Fig. 1.8.A). All DNA glycosylases known to remove alkylation
damage and uracil are monofuctional, whereas those participating in the repair of
27

A. OXIDATIVE LESION REPAIR
a URACILREPAIR
P T P T P I T P - r P n r p I TA DHU C A T 6T 9 9 T A c
p p _X_ p _ i_ p_ l_ P p
p T p - r p - r P - r p n - p - r A U C A T GT ? ? T A Ç_1_ p p -1— p _1_ p _ l_ p p
hNthI and XPG
’“pP^ p-rP-rP-rP-T A C A T GT G G T A Ç
p _ l_ p _ l_ p _ L _ p _ L f
I MAPI
T T T ' T ' T
I Poip
' v i ' V V T - ^I p l p l p l p A p i p
UDG
T G G T A Ç—i— p 1 p —i— p —I— p 1 p —1—p
MAPI
X.. - rA C A
P —I— P —I— P—[— P - j -
T G G T A Ç_1_ P _1_ P _1_ p _ L p p p
, Poip
i p ^ p i p i p ^ p i p
p /p
I Poip T (excision)
p - p P - f P - p P - | - P - p P A Ç C A T GÎ ? 9 T A Ç
P P p _1_ p _1_ p
I Ug IIVXRCC1
TI p i p i p l p ^ p i p
X
Poi Ô andPCNA.orPoip ^ x
^ W T V T ^T G G T A Ç
_ i_ p _L_ p _1_ p _ i_ p _ i_ p _1_ F
FEN1 and PC MA
' T T X T ' f ^ TJ_p lp lp J_p J ÎL p ip
+ L ig I
^ V T T V T JJ _ p f p f p lp A p f p
Figure 1.8 Schematic representation of the base excision repair pathways.A. The removal of oxidative lesions by a bifuctional DNA glycosylase-AP lyase. The lesion removed from DNA is dihydrouracil (DHU), a ring-saturated pyrimidine residue. 6. The main pathway is represented by the repair of a uracil residue in DNA. An alternative pathway employing several replication factors in the latter stages of the repair process is shown on the right. UDG, uracil DNA glycosylase; MAPI (or APE1), AP endonuclease 1; Pol (3, DNA polymerase p; Lig I / III, DNA ligase I / III. Adapted from Lindahl, 2000.
28

oxidised base residues are bifunctional (Seeberg et a!., 2000). After base removel,
the AP endonuclease APE1 (also called HAP1), DNA polymerase p (pol p) and DNA
ligase III/XRCC1 are required to compete repair. The XPG protein, which is also
involved in nucleotide excision repair, acts as a cofactor for glycosylase excision of
some damaged pyrimidines.
Table 1.1 Human DNA glycosylases
DNA Functionality major altered base releasedUNG mono- USMUG1 mono- UMBD4 mono- U or T opposite G at GpGTDG mono- U or T opposite GOGGI bi- 8-oxoG opposite CMYH bi- (very weak activity, not clear if
really bifunctional)A opposite 8-oxo-G
NTHL1 (NTH1) bi- ring-saturated or fragmented pyrimidines
MPG mono- 3-meA, hypoxanthineNEIL1 bi- removes thymine glycolNEIL2 bi- removes oxidative products of 0, UNEIL3 removes fragmented pyrimidines
Adapted from http://www.cgal.icnet.uk/DNA_Repair_Genes.html.
The sequential steps of BER are carefully ordered. Most DNA glycosylases
remain bound to the abasic site after cleavage of the base sugar bond. They are
displaced by APE1 which cleaves the DNA 5 ’ to the abasic site (Fig 1.8.8). Pol p
(Fig 1.8.8 left side) then removes the 5'-terminal abasic sugar-phosphate residue
and carries out the gap-filling synthetic step. DNA ligase III has been implicated in
the final step in this pathway. The role of its XRCC-1 cofactor in this process is not
entirely clear, but it is thought that XRCC1 acts as a scaffolding protein (Lindahl,
2000). Complex lesions with damage to both the base and deoxyribose residues
can be repaired by an alternative BER pathway (Fig 1 .8 .8 right side). This
subpathway leads to slightly longer repair patches of 2-6 nucleotides, and employs
replication factors such as Flap endonuclease 1 (Feni), the proliferating cell nuclear
antigen (PCNA), and DNA ligase I.
No human syndromes have been attributed to inherited dysfunctions of BER.
However, single-nucleotide polymorphism in MYH - which encodes a glycosylase
that recognises A opposite 8-oxoguanine (Table 1.1) - have been linked to an
29

increased susceptibility to colorectal cancer (Al-Tassan et al., 2002; Jones et al.,
2002). Mice with knockouts of specific DNA glycosylases have unexpectedly mild
phenotypes. Mice defective in BER functions such as APE1, pol p, or XRCC1 on the
other hand, exhibit an embryonic lethal phenotype. Embryonic fibroblasts
established from these mice are, however, viable. This apparent paradox might be
explained in two ways. DNA glycosylases might be highly redundant, or individual
BER pathways not essential. Loss of the entire system is lethal, however. This,
together with the apparent absence of inherited human syndromes associated with
non-functional BER, would suggest that BER itself is an essential function (Lindahl,
2001). Alternatively, the viability of DNA glycosylase knock-out mice indicates that
BER may not be essential. The extreme phenotype of APE1, pol p and XRCC1
knockout animals may suggest additional functions for these proteins that are
required for normal embryogenesis.
Nucleotide excision repair
The principal function of nucleotide excision repair (NER) is the removal of
DNA photoproducts caused by exposure of cells to sunlight. This excision repair
pathway was reviewed recently by (Batty and Wood, 2000; Friedberg, 2001). NER
can also act with varying efficiency on a wide variety of other bulky helix-distorting
adducts in DNA. Helix distortion plays a role in recognition by NER factors. In
general, the more a lesion distorts the normal DNA structure, the more efficiently it
is likely to be repaired by NER.
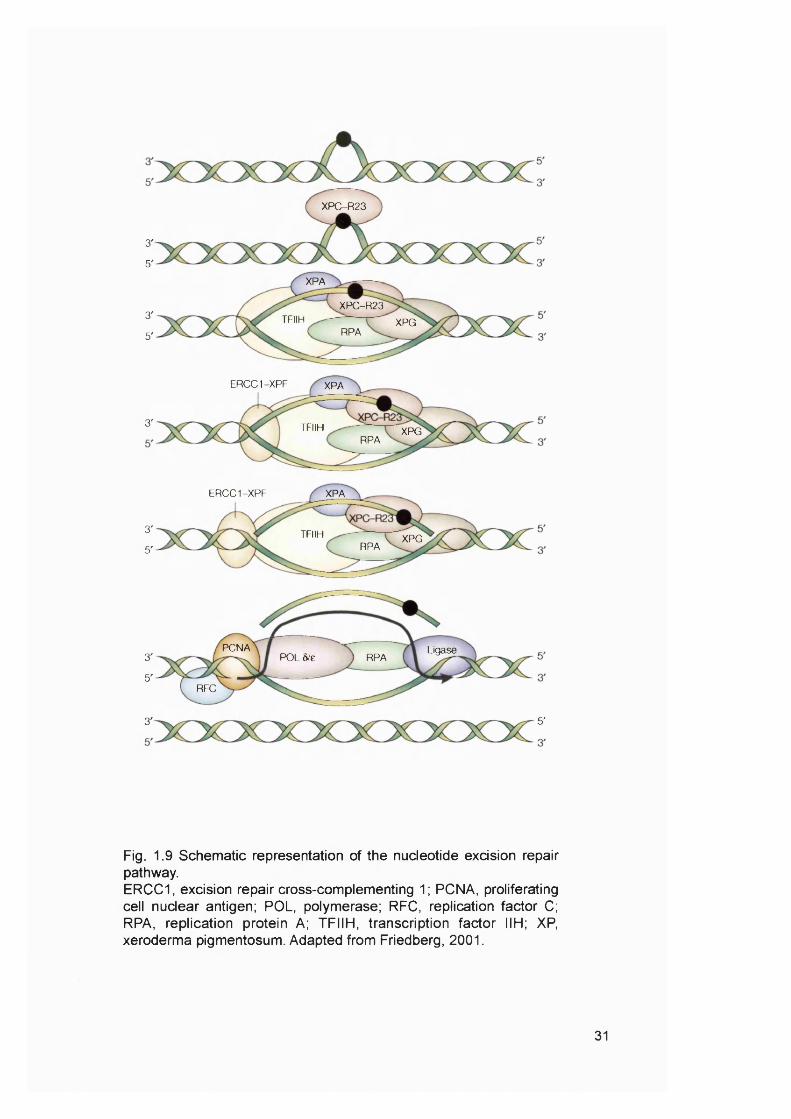
Six NER core factors, some of which have multiple subunits, are required for
the repair of most damage. These proteins are: XPA, the replication protein A (RPA)
heterotrimer, the XPC-hHR23B complex, the 6 subunit core transcription factor IIH
(TFIIH ) complex and the two structure-specific endonucleases, XPG and the
heterodimeric excision repair cross-complementing 1 (E R C C I)-X PF. These NER
factors assemble in an ordered, stepwise fashion. This generates a large
multiprotein complex (Fig. 1.9). A helix-distorting lesion is first recognised by XPC,
which is stably bound to HHRAD23B. This step is followed by binding of the XPA,
RPA, TFIIH and XPG proteins. Of these, XPA and RPA are thought to facilitate
specific recognition of the base damage. TFIIH is composed of six subunits and
contains two DNA helicase activities (XPB and XPD) that unwind the DNA duplex in
the immediate vicinity of the base damage. This generates a bubble in the DNA with
discrete junctions between double-stranded and single-stranded DNA at the edges.
30
XPC-R23
3'
5'
XPA
XPC-R233' TFIIH XPG
RPA5'
ERCC1-XPF XPA
3' TFIIH XPGRPA
ERCC1-XPF XPA
3' TFIIH XPGRPA5'
PCNA Ligase3' POL 5/e RPA
5'RFC
5'3'
Fig. 1.9 Schematic representation of the nucleotide excision repair pathway.ERCC1, excision repair cross-complementing 1; PCNA, proliferating cell nuclear antigen; POL, polymerase; RFC, replication factor C; RPA, replication protein A; TFIIH, transcription factor IIH; XP, xeroderma pigmentosum. Adapted from Friedberg, 2001.
31

These junctions define the position of incision of the DNA. Bubble formation is
followed by the binding of the ERCC1-XPF heterodimer to generate a completely
assembled NER complex. The XPG endonuclease cuts the damaged strand 3’ to
the site of damage, and the ERCC1-XPF endonuclease cuts 5’ to the damage. This
results in the release of a damage-containing oligonucleotide 24-32 residues in
length. The gap is then filled by DNA polymerase ô or e holoenzyme and sealed by
a DNA ligase.
Defects in NER result in the human autosomal recessive hereditary disease
XP. XP is characterised by a severe predisposition to skin cancers, mainly
squamous cell carcinomas (SSCs) and basal cell carcinomas. This disease
provided evidence that the inability to repair DNA damage, specifically base damage
caused by exposure to UV light, results in enhanced mutational burden and
eventually cancer.
Transcription-coupled DNA repair
Repair of DNA damage in active genes occurs much faster than in non
transcribed regions of the genome (reviewed by (Svejstrup, 2001)). Specifically it is
the transcribed DNA strand that is preferentially repaired. Lesions in the non
transcribed strand of the same gene are repaired at similar rates to those in non
transcribed regions.
Transcription coupled repair (TCR) was first thought to be a specific sub
pathway of NER, but it has become clear that several lesions removed by base
excision repair are also repaired through TCR. The proteins encoded by the
Cockayne’s Syndrome A (CSA) and CSB genes have been implicated as specific
TCR factors. Cell lines expressing defective forms of these proteins have normal
global genome repair (GGR), but are almost completely unable to effect selective
repair of lesions in the transcribed strand of active genes. An arrested RNA
polymerase II (RNAPII) is required to trigger TCR. A DNA lesion that is exposed in
the context of a DNA duplex pre-melted by RNAPII might be an excellent substrate
for the repair machinery. This would explain why the general initiator of NER, XPC,
is not required for TC R (Svejstrup, 2001). Almost all models involve the
displacement of RNAPII from the lesion by CSB and other TCR factors. The
following model was recently proposed by Svejstrup (Svejstrup, 2003). After RNAPII
stalls CSA uses DNA translocase activity to remodel the RNAPII-DNA interface,
32

possibly to push the polymerase past the obstruction or to remove it from the DNA
so that repair can take place. However, if RNAPII displacement is not possible, the
polymerase is unbiquitinated instead and eventually removed by proteolysis.
DNA mismatch repair
Will be discussed in detail in Section 1.3.
1.2.2 Reversal of DNA damage
Some alkylation damage can be repaired through direct chemical reversal by
specialised enzymes.
1-meA/3-meC DNA dioxygenases
The Escherichia coii (E. coli) AlkB protein is one of several that are induced
during the adaptive response to alkylation damage; a response that enhances the
cellular resistance to these agents (Sedgwick and Lindahl, 2002). The AlkB protein
is an a-ketogluterate-Fe(ll)-dependent dioxygenase that repairs 1-meA and 3-meC
in DNA by oxidative déméthylation (Faînes et al., 2002; Trewick et al., 2002). These
lesions are predominantly generated in single-stranded DNA by Sn2 methylating
agents such as MMS and DMS. AlkB can repair them both in single and double
stranded DNA in vitro and reverts them directly to adenine and cytosine with the
release of the oxidised methyl-group as formaldehyde. AlkB can also demethylate 1-
meA in nucleotides (Koivisto et al., 2003).
Two human AlkB homologues, hABH2 and hABH3, have been identified so
far. Both remove 1-meA and 3-meC from single- and double-stranded DNA by direct
reversal to adenine and cytosine by oxidative déméthylation (Aas et al., 2003;
Duncan et a!., 2003). Furthermore, AlkB and hABH3, but not hABH2, can repair 1-
meA and 3-meC in RNA (Aas et al., 2003). Apart from reactivation of single
stranded DNA and RNA phages inactivated by MMS méthylation (Aas et al., 2003;
Dinglay et al., 2000), no biological activity of AlkB and its human homologues has
been shown. The in vivo relevance of the déméthylation of RNA by AlkB and hABH3
has been subject of some debate, as 1-meA and 3-meC are naturally occurring
bases in tRNA molecules, which are essential for correct folding. Inappropriate
33

activity on RNA molecules could lead to cytotoxic tRNA destruction by AlkB and
hABH3 (Koivisto et al., 2003).
Repair of 0^-alkylguanlne
0^-methylguanine is a potentially mutagenic lesion because it can mispair
with thymine during semi-conservative DNA synthesis. The repair of 0®-alkylguanine
has recently been reviewed by (Margison and Santibanez-Koref, 2002; Middleton
and Margison, 2003; Pegg, 2000). The predominant pathway for the repair of O®-
meG in DNA is via the 0®-methylguanine-DNA methyltransferase (MGMT). MGMT
repairs 0®-meG in double-stranded DNA by transferring the methyl-group from the
DNA to one of its own cysteine (Cys) residues. This stochiometric reaction leads to
the inactivation of the enzyme. The most probable reaction mechanism involves the
flipping of the substrate 0®-meG out of the DNA helix into a binding pocket
containing the Cys acceptor site. Such base flipping would require the insertion of
an amino residue into the DNA to displace the base. MGMT binds to DNA via a
helix-turn-helix motif in the C-terminal domain. The second helix contains an
arginine that flips the methylated base out into the active site. The reaction
mechanism is not fully understood, but it is thought that a histidine residue and an
glutamic acid residue form a hydrogen bond network with the active site Cys residue
effecting the transfer of the methyl group to the Cys. In addition to removing methyl-
groups, longer alkyl groups including ethyl-, n-propyl-, n-butyl-, 2-chloroethyl-, 2-
hydroxyethyl-, /so-propyl-, /so-butyl- and some other adducts can be repaired. This
indicates that the active pocket must be quite flexible to recognise this wide range of
different structures. The autoinactivated protein is quickly degraded via the
ubiquitin/proteosomal system and de novo synthesis of the protein is required for
the continued repair of 0®-alkylation damage.
Since the reaction of MGMT leads to its inactivation, any substrate of the
protein acts as an irreversible inhibitor. The most commonly used inhibitor is O®-
benzylguanine (0®-BzG), which binds in the active site and the benzyl-group is
transferred to the protein. This reaction results in the formation of S-benzylcysteine
at the active site and the stoichiometric release of guanine. Even though 0®-meG in
DNA is a much better substrate for MGMT than 0®-BzG as a free base, the latter is
an effective inhibitor of the enzyme in vivo because much larger concentrations of
the free 0®-BzG base can be readily achieved.
34

Many human tumour cell lines and primary human tumours lack MGMT
expression. These cells are referred to as having a Mex' (or Mer ) phenotype. In
most cases this is due to silencing of MGMT expression rather than deletion of the
gene. However, mutations and deletions of the M G M T gene have been reported
(Wang et al., 1997).
1.2.3 Double strand break repair
Double strand break repair has been recently reviewed by (Jackson, 2002;
Karran, 2000; Lieber et al., 2003; Thompson and Schild, 2002; West, 2003). DSBs,
caused for example by radiation, can be repaired by two distinct pathways: non-
homologous end joining (NHEJ) or homologous recombination (HR). The relative
contribution of each pathway depends on the stage of the cell cycle. NHEJ
dominates in G1 and HR makes a greater contribution in the S and G2 phases
during which the presence of an additional copy facilitates recombinational
exchanges.
Non-homologous end joining
NHEJ does not require a template. It involves the simple ligation of the
broken termini that generally produces small deletions of DNA sequence. For the
two ends to be processed they must be maintained in physical proximity, known as
synapsis. The first proteins to bind to the ends at a DSB is a heterodimer of Ku70
and Ku80 (Fig. 1.10). Once loaded onto a DNA terminus the heterodimer undergoes
conformational change, which leads to the formation of a highly charged channel
through which the DNA passes. Then the DNA-dependent protein kinase catalytic
subunit (DNA-PKcs) is recruited which, together with Ku, forms the DNA-PK
holoenzyme. The protein kinase function of DNA-PKcs is activated by its interaction
with a single-stranded DNA region derived from the DSB. Ku then recruits XRCC4
along with DNA ligase IV. One likely in vivo substrate for DNA-PK is XRCC4,
phosphorylation of which may influence its activity. DNA ligase IV, which functions in
a tight complex with XRCC4, brings about the physical religation of the DNA ends.
Most DNA DSBs generated by DNA damaging agents cannot be directly
ligated, but require some processing and/or DNA polymerisation. The M r e l l-
Rad50-Nbs1 complex, which contains exo- and endo-nuclease and helicase
activities, is thought to be involved in NHEJ if the ends require processing before
35

dsi/V ' \
DSB recognition
Ku70, Ku«0
DNA-PKcs
Mrell-Rad50-NBSI
Artcnn‘t leaning up” of
DNA ends
XRCC4, I l igase IV DNA polymeri/alion.
Ligation
Inaccurate repair
Fig. 1.10 Schematic representation of the DNA Non- homologous end-joining pathway.See text for details. DSB, double strand break. Adopted from Jackson, 2002.
36

ligation. The exact role of the Mre11-Rad50-Nbs1 complex in NHEJ is not
understood yet, but recent work suggests that one of its roles may be to keep the
DNA ends in close proximity (de Jager et al., 2001). Another factor that might be
involved in processing DNA DSB ends before NHEJ is the Artemis endonuclease.
DNA-PKcs has been shown to bind to, and phosphorylate Artemis, thereby activating
its endonuclease.
Furthermore, the Mre11-Rad50-Nbs1 complex is also thought to play an
early role in a signal transduction pathway by which DSBs activate checkpoint
functions (D'Amours and Jackson, 2002). Cell cycle checkpoints pause cell cycle
progression, presumably providing time to allow DNA repair. Both M rel 1 and Nbsl
are phosphorylated in response to UV, IR, MMS and hydroxyurea. After IR
treatment this phosphorylation is dependent on the ataxia telangiectasia kinase
(ATM), whereas after UV, hydroxyurea or MMS the ATM-related kinase ATR is
probably responsible. Evidence suggests that the ATM-dependent pathway
recognises DNA DSBs, whereas ATR is triggered by bulky lesions and stalled
replication forks (O'Driscoll and Jeggo, 2003).
Deficiencies of the genes encoding proteins mentioned above involved in
NHEJ and/or DNA damage response are associated with carcinogenesis and/or
clinical radiosensitivity in people. The rejoining of V(D)J recombination intermediates
is also severely impaired in some of these patients leading to immunodeficiency.
Defects in Artemis occur in a group of radiosensitive patients with severe combined
immune-deficiency (Moshous et al., 2001). Mutations in ligase IV have been found
in a set of patients with features including immunodeficiency and developmental and
growth delay (O'Driscoll et al., 2001). Their clinical phenotype closely resembled the
DNA damage response disorder Nijmegen breakage syndrome (NBS), and the cells
from individuals with defective DNA ligase IV were radiosensitive, displayed
chromosomal instability but were proficient in cell cycle responses. NBS is a genetic
disorder involving defects in N bsl. These patients display clinical radiosensitivity,
immunodeficiency, and an elevated cancer incidence. Defects in ATM give rise to
the rare hereditary genetic disorder ataxia-telangiectasia (AT). Mutations in the
Mre11 gene (ataxia-telangiectasia-like disorder (ATLD)) have also been reported
recently in a group of patients. Both AT and ATLD patients exhibited many of the
features characteristic of NBS. It was recently shown that a splicing mutation in ATR
results in Seckel syndrome (O'Driscoll et al., 2003). This syndrome shares an
37

overlap In clinical features with the disorders described above. Seckel patients,
however, are not immunodeficient.
Homologous recombination
The different steps in HR are described in Fig. 1.11. The early steps of
recombination are promoted by proteins belonging to the RadSI family of
recombinases. The DNA at the break site is resected to expose single-stranded
(ss)DNA, which then becomes coated by the single-stranded-binding protein
replication protein A (RPA). The ssDNA-RPA complex acts as a target for the
binding of RAD52. RAD52 stimulates RadSI-mediated strand invasion through
direct interaction with RAD51 and RPA. The mammalian RadSI protein, a
homologue of the E. coli RecA protein, forms a long helical polymer that wraps
around the DNA to form a nucleoprotein filament. Invasion of a resected end of the
DSB takes place in the RADSI filament, requires ATP binding and is stimulated by
the SWI/SNF protein RADS4.
The next stages of HR are less clear, but involve either invasion of the
ssDNA tail of the second end, or simple annealing of the second ssDNA tail with the
displaced strand at the joint. One proposed model is called synthesis-dependent
strand annealing, where the first invading end could function as a primer for DNA
replication. This is followed by strand displacement of the newly synthesised DNA,
which would then be available for annealing with the complementary strand of the
second-end tail. A double Holliday junction is formed by DNA resynthesis using the
two invading ends as primers. The Holliday junction needs to be resolved to
separate both chromosomes, but this process is not yet understood in mammalian
cells. In prokaryotes this step involves the RuvA, B and C proteins.
In response to DNA damage recombination proteins that are normally found
diffused throughout the nucleus are rapidly relocalised and concentrated in nuclear
foci. After treatment with various DNA-damaging agents, nuclear RAD51 foci form.
Other proteins like RAD52, RAD54, RPA and the tumour suppressors BRCA1 and
BRCA2 (see below) colocalise with RAD51 in these foci. RAD51 foci also form in
undamaged S-phase cells, probably at sites of stalled or broken replication forks
undergoing repair.
38

1.
2.I
I
3.
4.
I
I
5.
I
6 .
Fig. 1.11 Double strand break repair by homologous recombination.1. DNA double-strand break; 2. rexection; 3. invasion of first end into a homologous duplex; 4. second-end capture; 5. continued strand exchange and DNA synthesis; 6. Holliday junction resolution. Adapted from West 2003.
39

BRCA1 and BRCA2
People who carry mutations in BRCA1 or BRCA2 are predisposed to breast
and/or ovarian cancers, and cells derived from their tumours show evidence of
genome instability. One defective copy of BRCA1 or BRCA2 is enough to confer
cancer predisposition, and the loss of the second allele is often observed in the
tumours of these patients. Both BRCA1 and BRCA2 are required for normal levels
of HR and DSB repair. BRCA2 has been shown to interact directly with RAD51, and
BRCA2 interacts either directly, or indirectly via its interaction with BRCA1, with
RAD51. Cells lines that are defective in one of these two breast cancer susceptibility
genes show gross chromosomal rearrangements indicating chromosome breakage.
Furthermore, BRCA1 and BRCA2 have been shown to interact with many different
proteins and are thought to be involved in processes like chromatin remodelling and
transcription.
Fanconi anaemia
Fanconi anaemia (FA) has been recently reviewed by (D'Andrea, 2003;
DAndrea and Grompe, 2003). FA is an autosomal recessive disease characterised
by chromosome instability and cellular hypersensitivity to DNA-crosslinking agents
like mitomycin C (MMC). FA patients have an increased predisposition to cancer,
specifically leukaemia and SSCs of the head and neck or gynaecologic systems. At
least nine separate FA complementation groups have been identified and the
FancA, 6, C, D1, D2, E, F and G, genes have been cloned. Of these genes only
F an cD 2 has homologues in other eukaryotic systems. Several FA proteins,
including A, C, E, F and G, form a constitutive multisubunit complex in the nucleus
of normal human cells (Fig. 1.12). This complex has some chromatin binding
activity, but its exact function is still unknown. It is thought to mediate translocation
of FancD2 into the nucleus and its subsequent monoubiquitination in response to
DNA damage. A new component, FancL, of the FA complex has just been identified
(Meetei et al., 2003). This protein possesses E3 ubiquitin ligase activity and has
been suggested to be the FA complex subunit required for monoubiquitination of
FancD2.
FancD2 seems to function in a downstream point in the FA pathway (Fig.
1.12). The monoubiquitinated protein is targeted to nuclear foci where it colocalises
with BRCA1 and RAD51. Indeed, BRCA1 is required for the monoubiquitylation of
FancD2. It has recently been established that FancDI and BRCA2 are the same
40
DNA Damage FANCD2
Activation and monoubiquitylation
BRCA1 ,UbFANCD2
DNA Repair
Fig. 1.12 The Fanconi anaemia / BRCA pathway.In response to DNA damage, the FA complex (FancA, C, E, F, G and L) mediates the monoubiquitylation (Ub) of FancD2. FancDI is then translocated to RAD51 (51) foci on DNA, which also contain the BRCA1 and BRCA2/FancD2 proteins. The damage is then repaired RAD51-mediated homologous recombination. Adapted from West, 2003.
41

protein (Hewlett et al., 2002). As BRCA1 and BRCA2 are required for HR and
function in the same pathway as the FA proteins, and FancD2 colocalises with
Rad51 in DNA damage induced foci, it appears that the FA pathway, HR, and DSB
repair are inextricably linked.
Single strand annealing
The single-strand annealing (SSA) pathway can be considered as a
subpathway of HR. SSA uses regions of homology to align the strands of DNA to be
rejoined, but this pathway does not involve the formation and resolution of Holliday
junctions. The Rad50-Mre11-Nbs1 complex are the main proteins involved in this
pathway. This complex probably binds to the DNA ends and holds them in close
proximity (de Jager et al., 2001). This could facilitate searches for regions of
sequence homology. Sites of limited homology may anneal to begin repair by SSA.
DNA annealing steps would create single-stranded tails, which have to be trimmed
before ligation. This is most probably performed by the endonuclease activity of the
XPF/Ercci complex. This process furthermore requires the Msh2 and Msh3 proteins
in yeast, but their exact function in SSA is not clear yet. The last step in this pathway
is ligation of the DNA ends.
1,2.4 Tolerance of DNA damage
Unrepaired DNA damage encountered during DNA replication can be
bypassed directly by translesion synthesis or indirectly by recombination.
Translesion synthesis
Translesion synthesis has been reviewed recently by (Friedberg et al., 2002;
Holmquist and Maher, 2002; Lehmann, 2002). The DNA polymerases that are part
of the replication machinery replicate DNA with high speed and extreme accuracy,
but they are unable to deal with damage in the template strands they are copying.
The first polymerase known to specialise in lesion bypass is DNA polymerase ^ ( pol
^). Since the discovery of this polymerase a number of new polymerases, mainly
belonging to the Y family of polymerase, have been characterised. Many Y
polymerases appear to be involved in replicating DNA past damaged sites. The
properties of eukaryotic DNA polymerases are summarised in Table 1.2. Bypass
polymerases are characterised by their ability to copy specific lesions or classes of
42

lesions with high genetic fidelity by incorporating the nucleotide that normally pairs
with the undamaged version of the base. Pol t], for example, seems to have evolved
to copy cyclobutane TT dimers by inserting the correct complementary bases AA.
But when copying undamaged DNA they exhibit reduced fidelity resulting in the
generation of mutations.
The following model for trans-lesion synthesis has been proposed. High-
fidelity replication is arrested at sites of DNA damage. The exact mechanism of
polymerase switching is not fully understood yet, but recent studies have shown
interactions between some of the bypass polymerases and known accessory
proteins for DNA replication like PCNA. During trans-lesion synthesis the correct or
incorrect nucleotide is incorporated depending on the polymerase. The newly
synthesised strand is extended for some distance, often by a second bypass
polymerase like pol Then high-fidelity replication is resumed. Once the site of
damage is cleared, it is subject to normal DNA replication, which can result in a
mutation if the bypass polymerase inserted the incorrect base. There are still many
unanswered questions concerning the replication of DNA damage, including how
the appropriate polymerase is selected to carry out trans-lesion synthesis.
Table 1.2 Properties of eukaryotic DNA polymerases.
DNApolymerase
Gene or synonym Proposed function Family Fidelity reading undamaged DNA
a POLA {P0L1) Replication priming B 2x10"*P POLB BER X 7x10"*Y POLG Mitochondrial replication A <1 X 10*®Ô P0LD1 {P0L3) Replication (leading
strand), repairB 2x10®
£ POLE {P0L2) Replication (lagging strand), repair, damage sensor
B <1 X 10"®
; POLZ {REV3) TLS B 10"*-10®n POLH
{RAD30A/XPV)TLS Y 10‘ -10®
0 POLO Cross-link repair A10'*-10"*1 POLI {RAD30B) TLS, BER, Ig
hypermutationY
K POLK {DINB1) TLS Y 10®-10"*X POLL {P0L4) Meiosis, BER X 9x10"*
POLM ds DNA break repair, Ig hypermuation
X
a POLS {TRF4) Sister chromatid cohesion X4) POLF {P0L5) rRNA synthesis BRev 1 REV1L TLS YTdT TDT Ig and TcR gene diversity XAdapted from (Holmquist and Maher, 2002).
43
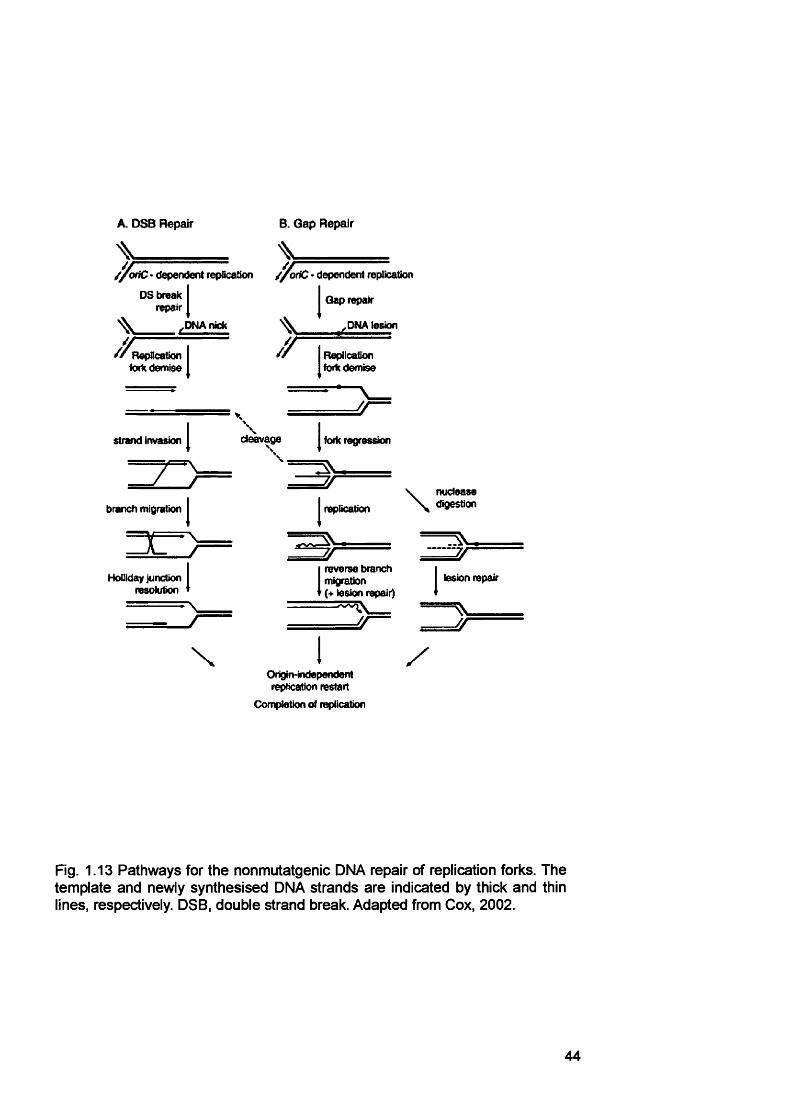
A. DSB Repair
^ ________dependent fepllcatfon //a tC • dependent repTicatlon
B. Gap Repair
DS break repair
,DNAnick
RepBcaüon kxK eternise
strand invasion
branch migration
HoQIday Junction resolution
J -
\
y
Gap repair
.DMA lesion
Replication forte derntee
JJ
d^vage fork regression
replication
■ -
\ nuclease digestion
7/---------
(reverse branch migration (+ lesion repair)
lesion repair
/ /
XOrigin-independent replication restart
Completion of replication
Fig. 1.13 Pathways for the nonmutatgenic DNA repair of replication forks. The template and newly synthesised DNA strands are indicated by thick and thin lines, respectively. DSB, double strand break. Adapted from Cox, 2002.
44

Repair of broken replication forks via recombination
As an alternative to translesion synthesis, which can be mutagenic, DNA
damage can also be bypassed indirectly by recombination This pathway is
nonmutagenic (reviewed by (Cox, 2002)). For example, a lesion in the template for
the leading strand synthesis causes fork arrest and results in a long single strand
gap (Fig. 1.13.8). The first step in the repair of arrested forks is probably fork
regression to generate a four-way junction that is also called a chicken foot'. The
single stranded gap that is now at the end of the short arm in the four-way junction,
can be filled in by a polymerase. A replication fork like structure is regenerated by a
reverse branch migration step. Alternatively, the ‘chicken foot’ structure can be
cleaved by a Holliday junction resolvase. This creates two double strands, one full
length and one shorter (Fig. 1.13.A), which can then be repaired by homologous
recombination. The third possible way of resolving a ‘chicken foot’ structure is to
degrade the short arm by a nuclease generating a fork like structure. As this does
not remove the lesion, DNA repair has to precede the restart of replication.
1.3 DNA mismatch repairDNA mismatch repair (MMR) is an excision repair pathway, and its primary
role is to remove replication errors from DNA. MMR has been reviewed extensively
(Aquilina and Bignami, 2001; Buermeyer et al., 1999; Harfe and Jinks-Robertson,
2000; Hsieh, 2001; Jiricny, 2000; Modrich and Lahue, 1996). This repair mechanism
is also sometimes referred to as the ‘long-patch’ MMR system to distinguish it from
short-patch’ systems that repair deaminated 5-methyl cytosine residues. MMR
proteins also recognise mismatches in heteroduplex recombination intermediates.
Here the role of MMR seems to be to suppress excessive recombination between
interspersed and diverged sequences (Datta et al., 1997; de Wind et al., 1995). In
addition, it was shown recently that MMR removes 8-oxo-dGMP incorporated during
replication (Colussi et al., 2002). This is thought to provide an additional level of
protection against DNA oxidation damage.
1.3.1 DNA mismatches
DNA mismatches can arise due to DNA biosynthetic errors that are
generated by DNA polymerases and have escaped proofreading mechanisms
(reviewed by (Kunkel, 1996; Lindahl, 1996)). The replicative DNA polymerases are
slightly error-prone enzymes, and make about one incorporation mistake for every
45

ten thousand nucleotides replicated. DNA synthetic errors can be either due to
insertion of an incorrect deoxyribonucleoside monophosphate or template-primer
slippage. Some alternative base pairs can be formed readily, for example G*T, or
between two purine nucleotides when one of them is in the syn conformation.
Frameshift mutations can arise from template-primer slippage. In this case the
terminal nucleotide of the primer relocates to pair with the same base but in the
wrong position in the template prior to its extension. This results in extrahelical loops
(Fig. 1.14).
DNA polymerases have an intrinsic editing function, which increases the
replication accuracy to 10 . This proofreading activity is provided by an associated 3 ’
5 ’ exonuclease that edits out 3’-terminal mispaired nucleotides. DNA mismatch
repair (will be discussed in detail below) further increases the accuracy by one
hundred- to one thousand-fold by excising any persistent error from the newly
synthesized daughter strand.
The human genome contains several hundred thousand extensive
repetitions of motifs of up to 6 base pairs. These sequences are called
microsatellites. As noncoding DNA regions can generally accumulate neutral
mutations, microsatellites are usually highly polymorphic (Karran, 1996). The
mononucleotide repeat Bat26, which has been shown to be quasi monomorphic, is
an exception (Hoang et al., 1997). Shorter repetitive DNA sequences (usually mono-
or dinucleotide repeats) can be found within the coding sequences of genes.
Repetitive DNA sequences are prone to frameshift mutations due to slippage of the
daughter and parental strands during replication. This misalignment generates
‘looped-out’ or 'slipped/mispaired' regions of one or more repeated element, which
are recognised and repaired by DNA mismatch repair. In the absence of correction,
further elongation of the daughter strand then fixes the mutation as a plus or minus
frameshift, depending on the configuration of the slippage (Fig. 1.14) (Karran, 1996).
1.3.2 Bacterial mismatch repair
The bacterial MMR pathway serves as a paradigm for the more complex
MMR processes in eukaryotes and will therefore be discussed first. The three-
dimensional structures of the bacterial MMR proteins have been determined by X-
ray crystallography. They provide new insights into the molecular mechanisms of
MMR.
46
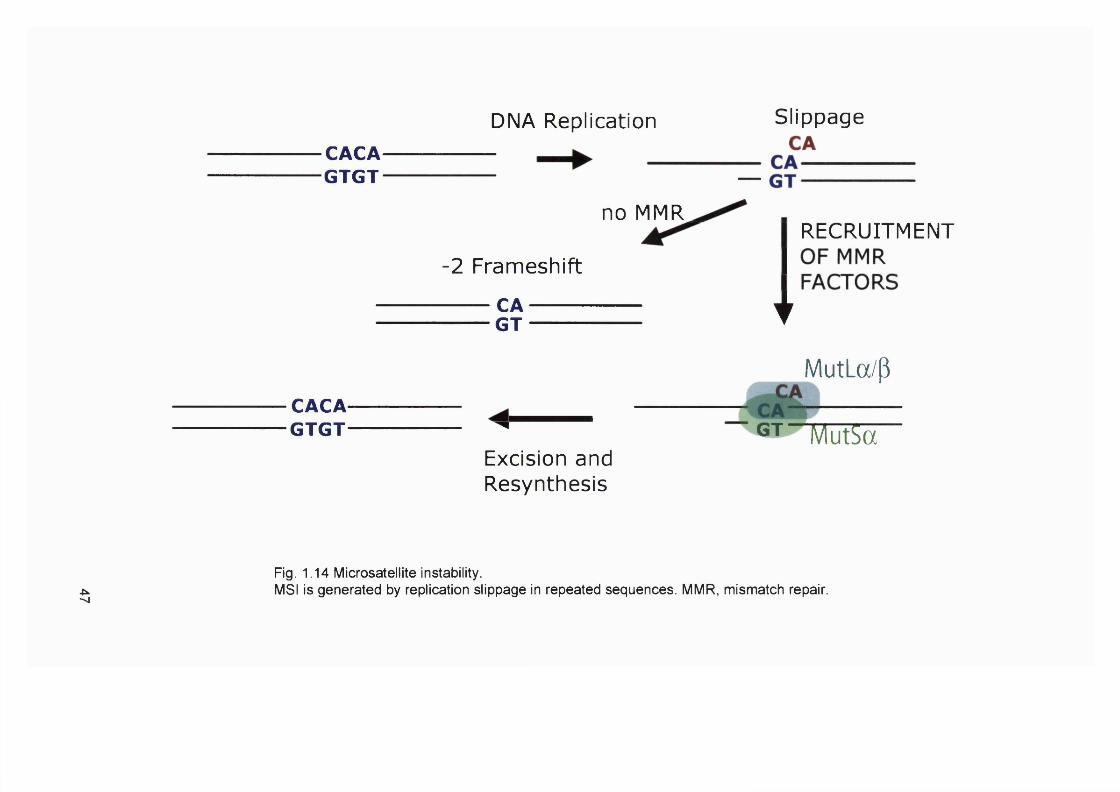
DNA ReplicationCACA------------------GTGT------------------
Slippage
no M M R _■ RECRUITMENT
-2 Fram eshift I= l î = *
CACA
MutLa/(3
GTGT------------------ - GT M utSaExcision and Resynthesis
Fig. 1.14 Microsatellite instability.^ MSI is generated by replication slippage in repeated sequences. MMR, mismatch repair.
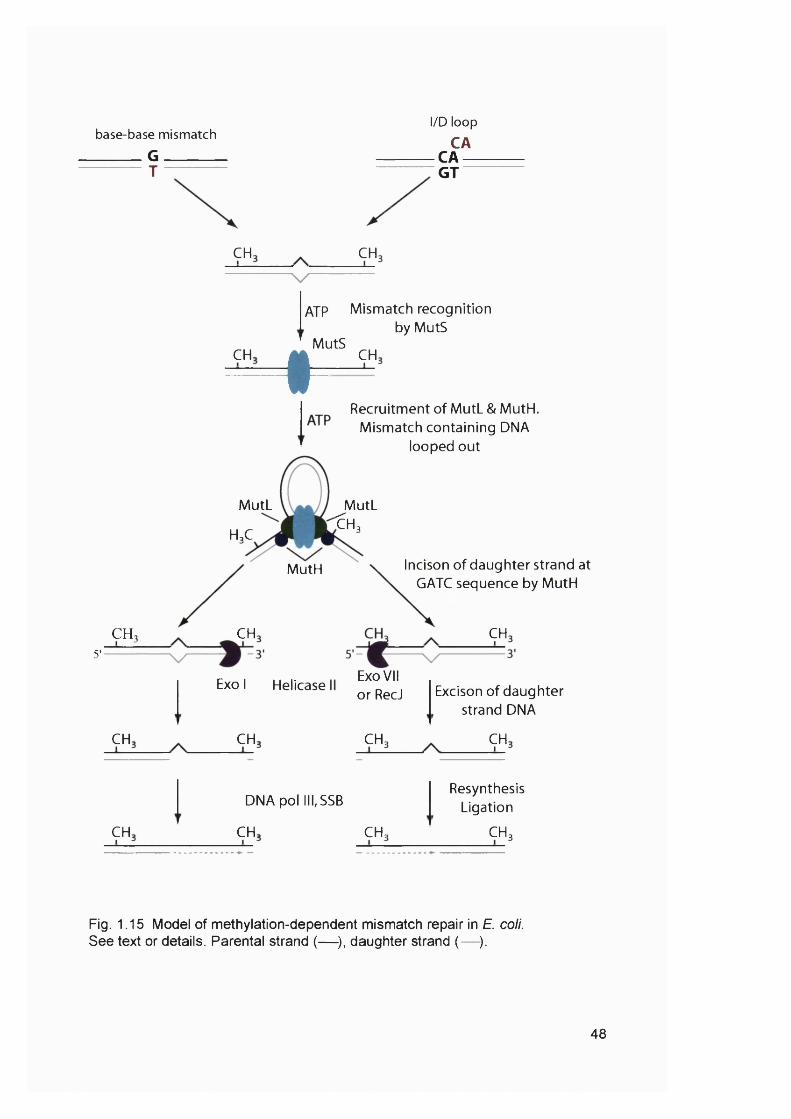
base-base mismatch
G ________ T
CH:
l/D loopCA
- C A —GT
CH CH
CH:
ATP Mismatch recognition by MutS
MutSe CH:
Recruitment of MutL & MutH. Mismatch containing DNA
looped out
MutL JVlutLCH,
H,C
Incison of daughter strand at GATC sequence by MutH
MutH
CHCH CH5'
Exo VII or RecJ
Exo I Helicase Excison of daughter strand DNA
CH: CH: CH:
CH:
DNA pollllSSB
CH: CH:
ResynthesisLigation
CH,
Fig. 1.15 Model of methylation-dependent mismatch repair in E. coli. See text or details. Parental strand (— ), daughter strand ( ).
48

The E. coli MMR system involves three dedicated proteins, MutS, MutL and MutH.
MutS recognises and binds to both base-base mismatches and insertion-deletion
loops (IDLs), MutL couples mismatch recognition by MutS to downstream
processing steps and MutH cleaves the transiently unmethylated daughter strand at
GATC sequences thereby ensuring that it, rather than the fully methylated template
strand, is corrected (Fig. 1.15).
MutS binds to mismatches as a homodimer with a substrate preference for
base-base mismatches and IDLs of up to 4 nucleotides in length. The MutS protein
possesses an intrinsic ATPase and is a member of the ABC (ATP binding cassette)
transporter superfamily of ATPases. The ATP hydrolysis activity of MutS is
essential, as mutations in the conserved ATP-binding domain are associated with a
dominant mutator phenotype in vivo. The three-dimensional structures of the MutS
proteins from E. coli and Thermus aquaticus complexed with an oligonucleotide
containing either a G*T mispair or a single unpaired thymine, respectively have been
solved (Lamers et al., 2000; Obmolova et al., 2000). MutS is present as a
homodimer. The structure has been described as resembling a pair of praying
hands in which the thumbs are folded inwards, and the DNA passes between the
fingertips and the thumbs (Lamers et al., 2000). In the case of both oligonucleotide
substrates, the DNA was bent by about 60°. These structures also showed that the
interaction between the MutS dimer and the DNA mismatch is asymmetric, as direct
interaction with the mismatch involves only one monomer.
Electron microscopy studies indicated that MutS mediates the formation of
an a-shaped loop structure in a reaction dependent on mismatch binding and ATP
hydrolysis (Allen et al., 1997). This led Modrich and colleagues to propose a model
in which MutS engages in ATP-dependent migration away from the mismatch,
resulting in the formation of an a-loop structure that contains the mismatch (Fig.
1.15). This model is contradicted by Hsieh and colleagues, who showed that
although MutS by itself diffuses rapidly from the mismatched DNA, in the presence
of MutL a stable MutS-MutL-DNA complex is formed. They proposed that this
complex activates MutH and regulates the extent of gap repair (Hsieh, 2001) in E.
coii.
MutL possesses an ATP binding site, but its precise role has not been
defined. ATP binding and hydrolysis leads to dimérisation, and it was shown that
49

mutations in the ATP binding domain can give rise to a dominant negative mutator
phenotype. MutL interacts directly with MutS, the helicase UvrD and MutH. It can
stimulate the endonuclease activity of MutH. Gel shift experiments indicate that the
presence of MutL increases the efficiency of MutS binding to the mismatch. It is
thought that one of the functions of MutL is the recruitment and assembly of a
functional repair complex. MutL can be seen as the factor that couples the initial
mismatch recognition step to downstream processing.
For MMR to selectively remove the newly incorporated mispaired base, it
has to be able to distinguish between the daughter and parental strands. The strand
discrimination signal in E. coli is provided by the transiently unmethylated state of
the newly synthesised DMA. Méthylation of DMA by the DMA adenine methylase
(Dam) protein at the sequence GATC lags slightly behind replication. This means
that, just behind the replication fork, the newly synthesised daughter strand is less
methylated than the parental strand allowing discrimination between the two
strands. The monomeric protein MutH is a méthylation sensitive endonuclease. It is
activated in vitro by a complex of MutS, MutL, and mismatched DMA and cleaves
the unmethylated strand of a hemimethylated GATC dam méthylation site in vivo.
This targets repair to the newly synthesised strand. The distance separating the
strand signal and the mismatch can be quite substantial, up to one or two kilobases
(kb). MutH can nick DNA either 5’ or 3’ to the mismatch, reflecting the bidirectionality
of this system. The MutH-directed nick serves as a point of entry for helicase II
(UvrD). Depending on where the cleavage occurred, either a single-strand specific
5' to 3' (RecJ or exonuclease VII) or 3' to 5' (exonuclease I) exonuclease degrades
the mismatch-containing nicked strand to just beyond the mismatch. The last step is
gap repair by DNA polymerase III holoenzyme and ligation of the repair product.
1.3.3 Human mismatch repair
The eukaryotic counterparts of MutS (MSH proteins for MutS homologues)
and MutL (MLH proteins for MutL homologues) are heterodimers (Table 1.3). Two
mismatch binding complexes formed by MutS homologues, hMutSa (hMSH2-
hMSH6) and hMutSp (hMSH2-hMSH3), carry out recognition of replication errors in
humans. In vitro hMutSa preferentially recognises single base mismatches and
loops of one or two bases. hMutSp on the other hand binds larger ( 2 - 8 bases) IDL.
It can also recognise loops of just one base, however (Fig. 1.16). This redundancy
50

Single base mis pair 1-2bp loop
/ \ ________
2-5bp loop
hMutSa
Primary recognition by hMutS complexes
hMSH2
$hMSH6/
hMSH3 .. hMutSP
hMLHI
Secondary recognition by hMutL complexes
hPMS2
PCNA? EX01
+ others
hMutLa
Strand discrimination
& Excision
hMSH2
hMLHI
\hPMS2
PCNA? EX01
+ others
Polymerase 6 PCNA RPA
Resynthesis & Ligation
Polymerase 5 PCNA RPA
Figure 1.16 Initiation of Mismatch Repair in Human Cells.Illustrated are the two major mismatch recognition complexes h Mut Sa (hMSH2 / hMSHS) and hMutSp (hMSH2 / hMSH3). Both have distinct yet overlapping substrate specificities. Only the major secondary recognition complex, hMutLa (hMLHI / hPMS2), is shown for simplicity. Parental strand ( — ), daughter strand ( — ).
51

for one and two base pair loops presumably ensures efficient recognition and repair
of these more common replication errors. Like the bacterial MutS proteins, the C-
termini of MSH proteins contain ATP binding sites. Several human MutL
homologues have been identified. The most important MutL homologue for DNA
repair is encoded by the hM LH I gene. The hMLHI protein can form heterodimers
with any of the remaining three MutL homologues, hPMS2, hPMS1 and hMLH3. The
hMutLa complex consists of hLMH1 and hPMS2, and seems to provide the main
MutL function, whereas the hMutLp (hMLH1/PMS1) complex provides a minor or
more specialised function. The role of the hMLH1/hMLH3 complex has not yet been
characterised. Expression of a dominant-negative hMLH3 protein in cells in culture
causes microsatellite instability, which could indicate that hMLH3 has a role in MMR
(Lipkin et al., 2000). Alternatively, hMLH3 overexpression could prevent hMutL
formation by binding all available hMLHI protein. This would also result in MSI.
Furthermore it has been shown that mlh3 has a essential role in murine meiosis
(Lipkin et al., 2002).
Table 1.3 Mammalian MutS and MutL homologues.
E. coli Mammals Mammalian Function
MutS MSH2Mismatch and loop recognitionMSH3
MSH6MSH5 MutS homologues specialised for meiosisMSH4
MutL PMS2 MutL homologue forming heterodimerMLH1PMS1 MutL homologues of unknown functionMLH3
Adapted from http://www.cgal.icnet.uk/DNA_Repair_Genes.html.
No obvious MutH homologues have been found in eukaryotes and the strand
discrimination signal in mammalian cells has not been identified either. PCNA
participates in MMR. It interacts both with MSH2 and replication proteins, and is
required at both early and late stages of MMR. A plausible role for PCNA might be
to couple MMR proteins to the replicative machinery. The termini of Okazaki
fragments could serve as strand discrimination signals. This hypothesis is supported
by the observation that yeast PCNA mutated in the binding motifs for MSH3 and
MSH6 results in a mutator phenotype (Clark et al., 2000). The involvement of
52

Okazaki fragments would be consistent with the excision of long patches of single
stranded DNA.
The degradation and resynthesis steps also remain uncharacterised.
Exonuclease I (Exo1), a 5’-3 ’ exonuclease, interacts with MutS and MutL
homologues. Exo1 was shown to participate in mismatch provoked incision directed
by a strand break either 5’ or 3’ of the mismatch in vitro (Genschel et al., 2002), and
to be required for repair of both mismatches and single-base IDL in murine ES cells
(Wei et al., 2003). A second exonuclease, Fen1, has also been implicated in MMR.
Biochemical evidence suggests that DNA polymerase Ô is used in the repair of DNA
mismatches. No helicase involved in MMR has been identified yet, but it is not clear
if this is because a helicase is simply not required or, perhaps more likely, this is
due to functional redundancy among helicases.
Two models have been proposed for the function of hMutSa. Both models
are based on the following experimental data. The ATPase activity of hMutSa is
stimulated by its binding to a DNA mismatch and ATP binding induces rapid
dissociation of the complex from the DNA. The model proposed by Modrich involves
translocation of the DNA through the hMutSa complex driven by ATP hydrolysis.
ATP hydrolysis induced the formation of a DNA loop by threading the DNA through
the hMutSa compiex (Blackwell et al., 1998). This loop is formed because the DNA
strand can diffuse freely through one of the subunits while it is being ‘held tightly’ by
the latch (L-) site of the other sub-unit. The open or closed status of the L-state is
determined by ATP or ADP occupancy of the subunit nucleotide-binding site. ATP
turnover induces a change in the occupancy of the nucleotide-binding site and
opens up the L-site. The looped DNA is then extruded through the open site and the
protein complex is translocated along the DNA by the distance of the length of the
extruded DNA loop.
Fishel has proposed an alternative model (‘hydrolysis-independent sliding
clamp model’) in which hMutSa functions like a G protein (Fishel, 2001). The
mismatch binding complexes exist in the ADP-bound form when ‘resting’ as hMutSa
binds to and hydrolyses ATP. In the absence of a mispaired base the ADP remains
associated. Oniy this hMutSa form can recognise a mispair. Mismatch binding
induces the exchange of ADP for ATP and dissociation of the heterodimer from the
mispair. This allows the dimer to slide along the DNA in the form of a sliding clamp
53

(SC). Diffusion of the 8 0 to the downstream repair machinery initiates MMR. Fishel
further proposes that the MutL heterodimers recycle/reset the MSH sliding clamps
(Fishel, 2001). The key difference between this and the ATP-hydrolysis mediated
translocation model proposed by Modrich is that SC formation and movement is
independent of ATP hydrolysis.
1.3.4 Biological consequences of inactive mismatch repair
The main role of MMR is the correction of replication errors. This is reflected
in an increased spontaneous mutation rate in MMR defective tumours. The
frequencies of transitions, transversions, and frameshift mutations are all greatly
increased in repair defective human cell lines. The mutation spectrum is somewhat
dependent on the missing MMR function, however, and there are major differences
in the mutational spectra of hM SH6- and h M L H I- or hPMS2-6e1ec{\\/e tumour cell
lines. For example, inactivation of hMutLa was associated with a greater increase in
frameshift mutations than loss of hMSH6, implying that hMutSp repairs a significant
fraction of frameshift intermediates.
As one of the major roles of MMR is to repair extrahelical loops created by
polymerase slippage, frameshift-like mutations in extensive regions of repetitive
mono- or dinucleotides called microsatellites, are used as diagnostic markers for
loss of MMR (Fig. 1.12). This mutator phenotype is known as microsatellite
instability (MSI). Microsatellite mutations in expressed genes cause frameshifts that
result in truncated, inactive proteins. Microsatellite instability is affected by the MMR
genotype. Loss of hMSH2, h M L H I or P M S 2 leads to a pronounced instability at
both mono- and dinucleotide repeats while inactivation of hM SH 6 is associated
predominantly with instability at mononucleotide runs. It has been suggested that,
due to the redundancy in the repair functions, inactivation of more than one MMR
gene is required for a strong mutator phenotype (Malkhosyan et al., 1996). This is
supported by the finding, that most MSI human tumour cell lines have mutations in
more than one MMR gene. These are often frameshift mutations in mononucleotide
repeats in the coding sequences of the hMSHS and hMSH6 genes (Malkhosyan et
al., 1996). Furthermore, the mutator phenotype is much more pronounced in tumour
tissue than in normal organs in hM SH 2 mice (Baross-Francis et al., 1998). This
could, however, reflect contribution of other metabolic changes in tumours to the
mutation rate.
54

1.4 The role of DNA mismatch repair in tolerance to DNA damaging agents
1.4.1 Tolerance to methvlatino agents
DNA damage tolerance can be defined as resistance to the cytotoxic effects
of a DNA lesion without its removal from the DNA. Méthylation tolerance was
originally shown in E. coli dam' mutants where inactivation of MMR restored wild-
type resistance to the toxicity of MNNG (Karran and Marinus, 1982).
The main cytotoxic lesion caused by methylating agents like MNU and
MNNG is 0®-meG (Section 1.1.6). Most human cells are protected against the
cytotoxicity of DNA 0®-meG by the enzyme MGMT (Section 1.2.2). Cells that do not
express this protein (designated Mex or Mer ) are extremely sensitive to killing by
these agents. Mex* cells can, however, develop tolerance to these drugs by
inactivating MMR. The following mechanism for tolerance to methylating agents was
proposed by Karran and Bignami (Fig. 1.17) (Karran and Bignami, 1994). DNA O®-
meG introduced by methylating agent treatment miscodes during replication, and
directs incorporation of a T or 0 . Neither 0®-meG*T nor 0®-meG*C fulfil the
requirements for a correct base pair and both mispairs are recognised by hMutSa,
which then recruits hMutLa. This complex initiates MMR targeted to the daughter
strand. A long DNA fragment containing the newly incorporated base is removed,
and this stretch of DNA is resynthesised. The ‘incorrect’ base 0®-meG remains in
the parental strand, however. The repair polymerase reinserts either a T or 0
opposite the methylated base. Further attempts at correction are provoked, resulting
in a futile cycle of uncomplete repair attempts. These long-lived strand interruptions
lead to cell death, probably by induction of DNA double strand breaks. These death
prone’ repair attempts can be avoided if MMR is not functional. Inactivation of MMR
creates a strong selective advantage for a cell treated with a methylating agent.
This model is supported by the following evidence.
1. Inactivation of MMR confers high levels of methylating agent resistance with
increases in the D37 (the drug dose required to kill 63% of cells) of up to 100-
fold (Aquilina et al., 1990; Branch et al., 1993; Hampson et al., 1997;
Humbert et al., 1999; Koi et al., 1994).
2. Cells defective in MSH2, MSH6, MLH1 and P M S2 all exhibit méthylation
tolerance.
55

3. If MMR is restored by transfer of the appropriate human chromosome or
oDNA in human cells, sensitivity is also restored (Branch et al., 2000; Cejka
et al., 2003; Koi et al., 1994).
4. MNU resistant clones can be isolated from human cell lines by exposing
cells to either a single cytotoxic dose or by chronic exposure to low levels of
the drug (Bignami et al., 2000; Hampson et al., 1997).
5. hMutSa can recognise 0®-meG paired with T, and to a lesser extent with C,
in an oligonucleotide (Duckett et al., 1996; Griffith et al., 1994).
6. The ATPase activity of hMutSa is stimulated by 0®-meG»T and 0®-meG»C
mispairs (Berardini et al., 2000).
7. 0®-meG»T/C mispairs are processed by MMR in vitro (Fig. 1.18) (Duckett et
al., 1999). As repair of 0®-meG«T/C mispairs where the methylated base is
in the parental strand and the T is in the daughter strand, is likely to direct
reincorporation of T a reverse system was used. A circular heteroduplex
DNA contained an 0®-meG»T lesion so that the 0®-meG is in the nicked
strand mimicking the ‘daughter strand’, and the T is in the ‘template strand".
It was shown using this system that 0®-meG*T mispairs are excised in a
reaction that depends on functional MMR (Duckett et al., 1999). Although
strictly, this is not a biological relevant experiment because the 0®-meG is
not in the ‘template strand’, it is nevertheless important because it
establishes the principle that 0®-meG base pairs provoke full processing by
MMR - not just recognition. It is easy to imagine though how, when the O®-
meG is in the parental strand and therefore not excised, complications can
arise.
An alternative model for resistance to alkylating agents was proposed by
Fishel and colleagues (Berardini et al., 2000; Fishel, 2001). In the SC model the
MMR proteins act as direct sensors capable of signalling to downstream effectors
such as the apoptotic machinery. This direct-sensor signalling mechanism would be
independent of DSBs and p53.
56
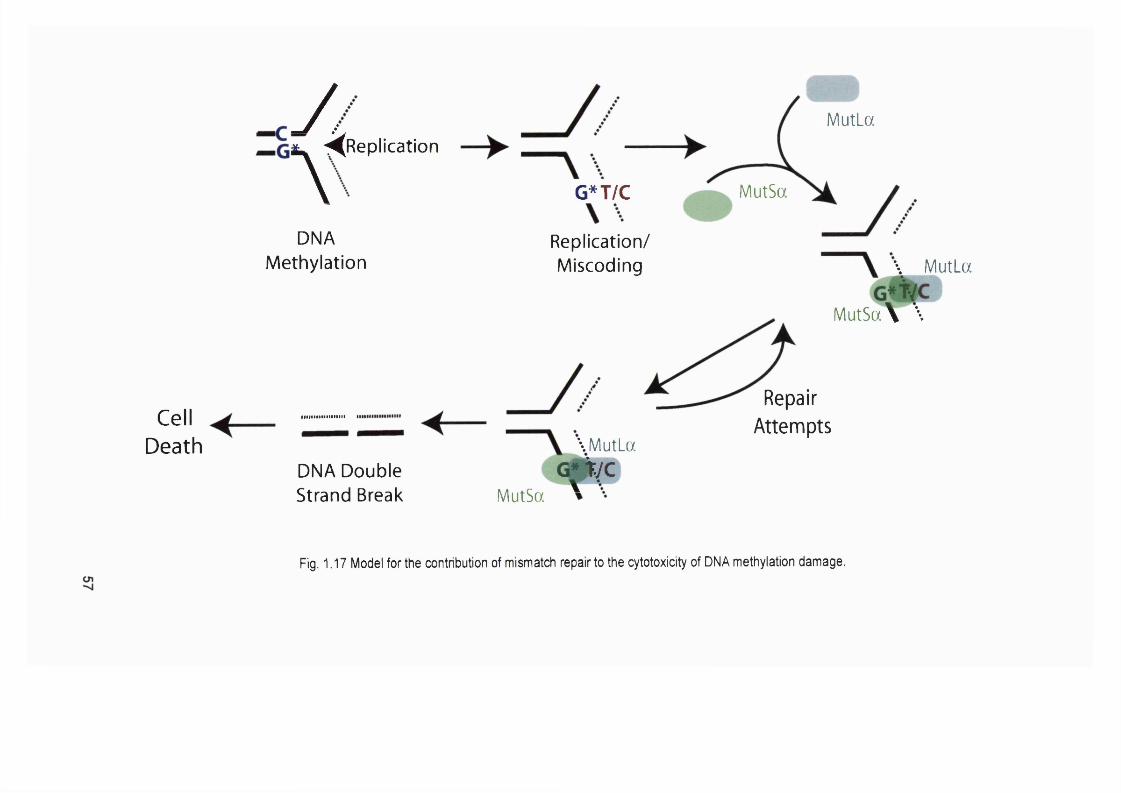
CellDeath
y MutLa^R eplication
\ MutSaG*T/C
DNAMéthylation
DNA Double Strand Break
Replication/Miscoding
\ MutLaG fty c
MutSa '
\ MutLa
MutSa\ \
RepairAttempts
Fig. 1.17 Model for the contribution of mismatch repair to the cytotoxicity of DNA méthylation damage.

'templatestrand'
'daughterstrand'
MMR
Excision
'templatestrand'
'daughterstrand'
MMR
Excision
Fig. 1.18 Processing of G^-meG basepairs by mismatch repair.G®-meG »T with G®-meG in either the 'template' (A ) or 'daughter* (B.) strand should be processed by MMR, but as G®-meG redirects incorporation of T, plasmid (B.) was used by Duckett et al., 1999. Repair here results in a A "T basepair.
58

1.4.2 Tolerance to thiopurines
MMR deficiency is also associated with resistance to the base analog 6 -TG
(Aquilina et al., 1990; Hawn et al., 1995). The 6 -TG resistance conferred by a MMR
defect is substantial (about 5 - 10-fold). It was shown by Swann and co-workers
several years ago that the molecular mechanism of this resistance involves loss of
MMR (Fig. 1.20) (Swann et al., 1996). Despite the fact that 6 -TG has been used in
the clinic for more than 50 years, its exact mechanism of action is not fully
understood. It is known that 6 -TG is incorporated into DNA via the HGPRT pathway
and that this incorporation is essential for its cytotoxicity (Section 1.1.7 and 4.1).
Once incorporated, 6 -TG can be methylated to form 6 -thiomethylguanine (6 -meTG).
This is probably by a non-enzymatic reaction most likely involving S-
adenosylmethionine (SAM). SAM is a co-factor involved in several biosynthetic
reactions, but it is also a weak methylating agent, and the thiol group of 6 -TG is a
good acceptor for the methyl-group of SAM. The cytotoxicity of 6 -TG is independent
of the Mex status of the cells treated, as 6 -meTG is a very poor substrate for
MGMT. The removal of the methyl group of 6 -meTG by MGMT is 10® times slower
than the removal of the methyl group of 0®-meG (Swann et al., 1996). During DNA
replication, thymine and cytosine are incorporated opposite 6 -meTG at similar rates,
and the resultant base pairs are recognized by hMutSa (Waters and Swann, 1997).
Just as 0®-meG"T is a better substrate for hMutSa recognition and for correction,
the S®-meG*T pair is recognised more efficiently by hMutSa than S®-meG*C. By
analogy to 0 ®-methylguanine, the methylated base will be in the template strand,
and hence not excised by MMR. This unrepairable mispair is likely to induce a futile
repair cycle that leads to cell death (Fig. 1.20). As for 0®-meG, cells could avoid the
cytotoxicity of 6 -TG by the same mechanism, inactivation of MMR.
Even though 6 -meTG has miscoding properties 6 -TG is not a strong
mutagen at sub-toxic doses, unlike MNU, which is able to induce mutations at low
toxicity. However, in a MMR-defective background in which the base analog is not
toxic, growth in high concentrations of 6 -TG induced point-mutations (Aquilina et al.,
1993). Loss of MMR therefore not only confers resistance to toxicity but also
increases the susceptibility to mutation induction by 6 -TG.
59

cOo '
o '^
TGCMéthylation
^ meTGC DNA Replication
DNA-TG
CellDeath
\ MutLa
mefG t _\ \MutSa
meTG T
RepairAttempts
Fig. 1.19 Model for the contribution of mismatch repair to the cytotoxicity of 6-TG.
g

1.4.3 Tolerance to other drugs
Evidence suggests that MMR may contribute to the cytotoxicity of the
chemotherapeutic drug cisplatin. hMutSa binds to DNA containing a 1,2 cisplatin
intrastrand cross-link (1,2 GpG), although with a lower affinity than to a G«T
mismatch. No binding is observed with a 1,3-intrastrand lesion (Duckett et al., 1996;
Yamada et al., 1997). Binding comparable to that for a G*T mispair was observed
only when the complementary strand contains T opposite the 3' G in the crosslink.
This mispair might be caused by replicative bypass of the 1,2 diguanyl intrastrand
crosslink (Yamada et al., 1997). It is generally agreed that MMR defects are
associated with a minor (^ 2 -fold) increase in cisplatin resistance compared to
méthylation or 6 -TG treatment (Aebi et al., 1997; Branch et al., 2000). A two-fold
increase in cisplatin sensitivity was observed when the MMR defect of the hM LHI-
deficient human colon carcinoma cell line HCT116 was corrected by transfer of
chromosome 3 (Lin and Howell, 1999). HCT116 was furthermore found to be more
mutable by cisplatin than HCT116+ch3.
In addition to the substrates mentioned above, purified hMutSa is able to
bind to synthetic DNA duplexes containing various single DNA lesions including 8 -
oxoG (Mazurek et al., 2002), UV photoproducts (Li et al., 1996; Mu et al., 1997),
1,2-dipurinyl intrastrand crosslinks (Duckett et al., 1996; Li et al., 1996; Yamada et
al., 1997), C8 -guanylaminofluorenes or acetylaminofluorenes (Li et al., 1996), and
benz(a)pyrene adducts (Wu et al., 1999). Binding in vitro does not necessarily
indicate that these lesions are processed in vivo, and the degree to which MMR is
involved in the cytotoxicity of these various lesions it is still a matter of debate.
1.5 DNA mismatch repair and cancer1.5.1 Roles of MMR aenes in human cancer
Familial cancer
The roles of MMR defects in the pathogenesis of human cancer has been
reviewed by several authors (Bignami et al., 2003; Duval and Hamelin, 2002; Jiricny
and Marra, 2003; Peltomaki, 2003). Germline defects in MMR genes underlie
HNPCC. This cancer syndrome predisposes individuals to colorectal cancer, other
gastrointestinal (Gl) cancer, and cancer of the female reproductive organs. MMR
mutations have been identified in 70 - 80% of HNPCC families. About 50% of the
known MMR mutations affect hM L H I, about 40% are found in hMSH2, and ^10%
affect hM SH6. hP M S I, hPMS2 and hMLHS are rare (less than 5%), and might
61

account for a small percentage (Peltomaki, 2003). The predisposing germline
mutations inactivate one allele of the MMR gene. The second allele is inactivated
somatically prior to, or during, tumour development.
h M L H I and hM S H 2 mutations generally give rise to classical' HNPCC
families, that lie within the strict clinical definition of the syndrome and give rise to
tumours with overt MSI. hPMS2 mutations are also associated with severe MSI, but
are much rarer in ‘classical’ families. hMSH6 mutations give rise to more cryptic MSI
and occur often in less typical HNPCC families. Based purely on clinical criteria, it is
thought that HNPCC accounts for 0.5 - 2% of the total colorectal cancer burden.
Exo1 and DNA pol ô may also have a role in HNPCC development, but not
enough data are available to allow any reliable assignments at the moment. These
two proteins as well as PCNA and RPA are not only involved in MMR but they also
play key roles in DNA replication. It has been suggested that inactivating mutations
in the genes encoding these polypeptides would be lethal because of this function in
replication (Jiricny and Marra, 2003). Mutations in these genes could only lead to
HNPCC if they were missense mutations affecting amino-acid residues in domains
that are only necessary for MMR and not for replication. These types of mutations
are likely to be very rare. Exo1 has been reported to be mutated in HNPCC, but
these tumours either lost the mutant allele and retained the wild-type copy, or
retained both copies (Jagmohan-Changur et al., 2003; Wu et al., 2001).
A small number of individuals homozygous for mutations in the MMR genes
h M L H I and hM SH2 have been reported. Children from consanguineous families
who had homozygous h M L H I mutations developed early onset haematological
malignancies (lymphomas and leukaemias), one glioma and clinical features of de
novo neurofibromatosis type 1 (NF1) (Ricciardone et al., 1999; Vilkki et al., 2001;
Wang et al., 1999). One patient homozygous for mutations in h M S H 2 was
diagnosed with acute lymphocytic leukaemia (ALL) and café au lait spots, a feature
of NF1 (Whiteside et al., 2002).
Furthermore, hPMS2 mutations have been described in Turcot kindreds (De
Rosa et al., 2000). Turcot’s syndrome is a genetic disease characterised by the
concurrence of primary brain tumours and colon cancer and/or multiple colorectal
adenomas. Some Turcot patients were reported to have inherited either one or two
62

mutated copies of PMS2. These tumours, and In some cases even normal tissue,
was microsatellite unstable.
Sporadic cancer
MSI occurs in approximately 15% to 25% of sporadic colorectal cancers and
tumours at other sites (for example endometrium, ovary, stomach - and others that
are common in HNPCC).
Definition of MSI
Following the discovery of MSI and its role in human cancer, different
laboratories used different micorsatellites and arbitrary definitions of instability to
determine MSI. This made it difficult to compare results and lead to confusion in the
field. In 1997 a panel of five microsatellites, two mono- (Bat25 and Bat26) and three
dinucleotide (D2S123, D5S346 and D17S250) repeats, was recommended for the
analysis of microsatellites by the National Cancer Institute Workshop on
Microsatellite Instability for Cancer Detection and Familial Predisposition' (Boland et
al., 1998). They suggested MSI be defined as instability at two or more of these five
loci or ^ 30% to 40% of all microsatellites studied. In the absence of non-tumour
tissue to which to compare the length of the altered microsatellite, the
mononucleotide marker Bat26 is often used to determine the MSI status. Bat26 is
quasimonomorphic (Hoang et al., 1997) and germ-line polymorphisms are rare in
the Caucasian population (Samowitz et al., 1999). This marker has been shown to
identify 97% of all MSI cases (Hoang et al., 1997; Loukola et al., 2001).
Mutational target genes in MMR defective tumours
In MSI tumours changes occur not only at non-coding microsatellites, but
also at repetitive sequences within the coding sequence of a gene, or at intronic
sequences adjacent to mRNA splice sites. Frameshift mutations generally result in
inactive proteins and such sequences are important targets for selective inactivation
in MMR deficient tumours. The observation that some genes with a potential tumour
suppressor function are particularly prone to mutations at repetitive coding
sequences led to the concept of target genes for instability.
63

The first repetitive sequence identified to be selectively mutated in MST
tumours was a poly (A) 10 repeat within the coding region of the transforming growth
factor p receptor type II (TGFpRII) gene (Markowitz et al., 1995). A substantial
number of potential target genes have been reported to be mutated at
polynucleotide repeats in MSI cancer (Examples in Table 1.4). These include genes
involved in apoptosis (e.g. Caspase-5, Bax), regulation of cell growth (e.g. IGFIIR),
response to DNA damage (e.g. BLM, RAD50, MRE11), MMR genes (hMSHS,
hMSH6 and hMLHS), and many others. Some of these genes will be discussed in
more detail in Chapter 5. It was shown that genes frequently found to be altered in
MMR proficient colorectal cancers, such as ARC or P5S, were rarely mutated in
M S r tumours (Olschwang et al., 1997). The number of genes found to be mutated
in M S r tumours has been increasing steadily, but it is difficult to establish which of
these alterations really contribute to carcinogenesis in MSF tumours. The frequency
of these mutations is generally taken as an indication that they were selected during
the development of the MSF tumour.
Table 1.4 Different genes mutated at coding repeat sequences in MSF cancer.
Target gene Gene functionACTRII Growth factor receptorAI M2 Interferon inducible proteinAPAF1 Pro-apoptotic factorAXIN2 Wnt signallingBax Pro-apoptotIc factorBCL10 Pro-apoptotIc factorBLM Response to cellular damageCaspase-5 Pro-apoptotic factorCDX2 Homeobox transcription factorGRB14 Growth factor receptor bound proteinshG4-1 Cell cycle proteinIGFIIR Growth factor receptorKIAA0977 Homologue of mouse cordon-blue proteinMBD4 DNA glycosylase and methyl CpG binding proteinMLH3 DNA MMRMSH3 DNA MMRMSH6 DNA MMRNADH-UOB NADH ubiquinone oxidaseOGT 0-llnked GlcNAc transferasePTEN Cell cycle proteinRAD50 Response to cellular damage and HRMRE11 Response to cellular damage and HRRHAMM Cellular motility and HA bindingRIZ Cell cycle and apoptotic proteinSEC63 ER membrane proteinSLC23AI Nucleobase transporterTCF4 Transcription factor (Wnt pathway)TGFbRII Inhibitor of cellular growthWISP-3 Growth factor (Wnt pathway)
Adopted from (Duval and Hamelin, 2002).
64

The different spectra of genes mutated in MMR proficient and deficient
tumours supports the hypothesis proposing the existence of two distinct pathways
for oncogenesis: the mutator and tumour suppressor pathways (Perucho, 1996;
Perucho et a!., 1994). Tumour suppressor gene mutations are accompanied by an
immediate capability for territorial expansion and/or a selective growth advantage. In
contrast, the mutator phenotype, caused by MSI for example, does not alter the
growth properties of the cell. This phenotype increases the likelihood of occurrence
of mutations in cancer genes and in other mutator genes, which then affect the
growth property of the cell.
1.5.2 Roles of MMR aenes in mouse tumouriaenesis
The relationship between MMR defects and tumourigenesis in mice has
been reviewed by (Buermeyer et al., 1999; Jiricny and Marra, 2003; Wei et al.,
2002). Mice heterozygous for MMR defects do not develop significant numbers of
tumours, and the tumours that eventually do develop are generally MMR proficient.
Therefore mice homozygous for MMR defects were studied. The findings from
homozygous knock-out (KO) mice are summarised in Table 1.5. Mice with
homozygous MMR defects generally succumb to lymphoma at an early age.
Table 1.5 Mouse models with disrupted MMR alleles.
Genotype Fertility(male/female)
Tumourincidence
Tumour spectrum M sr
Msh2'- yes/yes High Lymphomas, Gl, skin and yes
Msh6 '-other tumours
yes/yes Low Lymphomas, Gl and other no
Msh3*'- Msh6 '- & Msh3''
tumoursyes/yes High Gl tumours at old age yesyes/yes High Lymphomas, Gl, skin and yes
other tumoursMIhl*'- no/no High Lymphomas, Gl, skin and
other tumoursyes
Pms2'‘Pmsl* '
no/yes High Lymphomas and sarcoma yesyes/yes Low None no
Exol'- no/no High Lymphomas yes*MSI was investigated in tumour samples, or normal tissue or culture cells. Gl, gastro-intestinal; MMR, mismatch repair.Adapted from Jiricny and Marra, 2003, Current Opinion in Genetics & Development
65

Mice with mutations is MutS homologues
mshZ^' mice were developed by two independent groups (de Wind et a!.,
1995; Reitmair et a!., 1995). These mice displayed no major abnormalities, were
fertile, but a significant fraction developed lymphoid tumours at an early age. These
tumours were M S r. Older mice were also found to have intestinal and skin
neoplasms (Reitmair et al., 1996). De Wind et al. showed that immunocompromised
(reduced levels of CD 8 * T-cells) m sh2-deficient mice did not develop T-cell
lymphomas, survived relatively long, and more than half developed intestinal
tumours (de Wind et al., 1998). They furthermore showed that, even though
heterozygous animals are generally not more susceptible to death from
spontaneous cancer than wild-type mice, msh2*^' mice developed more tumours
than msh2*^* animals. These excess tumours tend to be non-GI and appear to retain
the active copy of msh2.
Animals homozygous for a null msh6 mutation had a reduced life span and
developed a variety of tumours. The most frequent malignancies were NHL followed
by tumours of the Gl tract. These malignancies were microsatellite stable
(Edelmann et al., 1997). Te Riele and co-workers reported early onset lymphomas
and epithelial cancers of the uterus and skin, but only very rare cancers of the Gl
tract in msh&^' mice (de Wind et al., 1999). msh3 deficiency on the other hand did
not predispose to cancer (de Wind et al., 1999; Edelmann et al., 2000). These mice
were viable and fertile, and developed tumours in old age with a similar frequency to
that in wild type animals. The tumour spectrum in the mutant mice was
characterised by more Gl tumours, however (Edelmann et al., 2000). Combined
m sh3/m sh6 deficiency predisposed to the development of both intestinal and
lymphoid malignancies (de Wind et al., 1999; Edelmann et al., 2000). These double
KO mice had survival rates compared to msh2^' animals. These findings are
compatible with the inactivation of both MutSa and p complexes.
Mice with mutations MutL homologues
mlh1 mutant mice were generated by two groups, and both heterozygous
and homozygous mlh1 mice were viable (Baker et al., 1996; Edelmann et al., 1996).
mlh1 homozygous mutant mice showed reduced survival compared to wild type
mice (all of the animals died within 12 months) and were susceptible to tumours at
an early age. They developed a spectrum of neoplasms similar to that in m sh2
mutant mice, including lymphomas both of T- and B-cell origin, Gl tumours and, to a
66

lesser extent, sebaceous gland tumours (Edelmann et al., 1999; Prolla et al., 1998).
As expected, m/h)-deficient tumours displayed high levels of MSI. Furthermore,
males and females were found to be sterile (Baker et al., 1996; Edelmann et
al., 1996). m//?f-deficient spermatocytes exhibited high levels of prematurely
separated chromosomes and arrested in first division meiosis. It was also shown
that mihl appears to localise to sites of crossing-over on meiotic chromosomes.
This phenotype suggests a role for mihl in the processing or resolution of
recombination intermediates, m/hf"''’ mice were also susceptible to tumour
development both outside and inside the Gl tract, but did not develop as many
tumours as homozygous mice (Edelmann et al., 1999).
Only few cases of colorectal tumours with hP M S 2 mutations have been
described. Both heterozygous and homozygous pms2-mutant mice were viable, but
the male pms2 homozygous mice were sterile due to the production of abnormal
spermatozoa. The females on the other hand were fertile (Baker et al., 1995). pm sZ
mice were susceptible to early onset cancers, but they lived longer than mlh1
animals. pms2 mice developed mainly lymphomas, but also a small number of
sarcomas, and all of these tumours displayed MSI (Prolla et al., 1998). Unlike mlh1,
msh2 and msh6 mutant mice, the pms2^' animals did not develop Gl and skin
tumours. The MSI analysis in tissues from pms2 and mlh1 KO mice revealed that
the mononucleotide repeat mutation frequency in pms2^' mice was 2- to 3-fold lower
than in mlh1' ' mice (Yao et al., 1999). These differences could influence the risk for
tumourigenesis.
pms1 homozygous mice on the other hand have no apparent phenotype.
These animals have a normal life span and do not seem to have increased cancer
susceptibility. Both sexes are fertile (Prolla et al., 1998).
Mice with mutations in other MMR genes
The majority of exo1 mutant mice developed lymphomas. These animals
also developed other tumours, for example Gl tumours, lung tumours, uterine and
ovarian cancers, and others, but these malignancies were observed at similar
frequencies in wild-type mice. Like mlh1 mice, exoT^'animals were unstable at
mononucleotide repeats in normal tissue. When dinucleotide repeats were analyzed
no MSI was found in the exo1 mice, whereas mlh1 mice showed high stability at
these markers. This supports the idea that Exol plays an important role in the repair
67

of single bp IDL and is less important in the repair of two-base I/D loop mismatches.
Furthermore, exol'^' male and female mice were infertile because of a meiotic defect
(Wei et al.. 2003).
Response to DNA damaging agent treatment of MMR defective mice
To investigate what role MMR plays in DNA damage, and specifically DNA
methylating agent, induced cancer MMR defective homozygous and heterozygous
mice were treated with different carcinogens. Treatment of mshZ^' mice with MNU
resulted in increased mutation frequencies, mainly G«C to A*T transitions, in all
tissues tested (Andrew et al., 1998). Treatment of m s h 2 KO mice with
ethyl nitrosourea (ENU) and the methylating agent 1,2-dimethylhydrazine (DMH) led
to an increase in tumour frequency. Lymphomagenesis was strongly accelerated by
ENU at a dose that did not induce tumours in wild-type mice (Claij et al., 2003).
DMH not only accelerated the development of lymphomas, but also induced
colorectal adenocarcinomas (Colussi et al., 2001). MNU treatment of mit)1 and
pm s2 KO mice at doses that did not induce lymphomas in wt mice, caused an
increase in thymic lymphoma development in the mutant animals (Kawate et al.,
1998). Thus, MMR defects are associated with an increased susceptibility to drug
induced cancer.
Treatment of heterozygous msh2 mice with MNU on the other hand did not
resulted in increased mutation frequencies (Andrew et al., 1998). Furthermore
msh2*^' mouse cell lines do not display an increased tolerance to the methylating
agent MNNG (de Wind et al., 1995). There is evidence that heterozygous MMR KO
mice may be more susceptible to drug induced cancer, however. DMH-treatment did
not affect the survival of msh2*'’ mice, but produced more tumours and a somewhat
different spectrum of tumour types in msh2*'' compared to msh2*^* animals (Colussi
et al., 2001). MNU treatment also causes significantly more Gl tumours in pms2*^'
mice than in pms2*^* animals. In this case there was no effect on overall survival,
however, and mice of both genotypes died at the same rate (Qin et al., 2000). Even
though msh2 heterozygosity does not affect the mutability by drug damage, it still
increases the susceptibility to drug induced cancer. The effect of drug treatment on
tumourigenesis is less pronounced in heterozygous than in homozygous KO though.
mgmf^' mice, similarly to human Mex' cell lines, are extremely sensitive to
killing by methylating agents like MNU. They are also susceptible to carcinogenesis
68

and even low-dose treatment with the drug induces a large number of tumours. Mice
with homozygous mutations in both mgmt and mlh1 are as resistant to MNU as wt
mice, but develop large numbers of thymic lymphomas (Kawate et al., 1998).
Evidence of haploinsufficiency for mihl was seen in this mgrnf^' background.
Survival of m g m f' mlh1*^' animals after MNU treatment was intermediate between
mgrnt^' mlh1' ‘ and wt. The drug induced thymic tumour formation in about half as
many mgrnf^' mlhl*^' animals compared to rngmf^' mlh1' ' mice (Kawate et al., 2000).
1.6 DNA mismatch repair and secondary cancerApart from sporadic HNPCC-like tumours, therapy-related acute myeloid
leukaemia is most associated with MSI. The incidence of a therapy-related or
secondary malignancy - a tumour at a different site to the first tumour that is a direct
consequence of the therapy - is increasing (Bignami et al., 2003). The risk of
developing a second malignancy has been estimated to range from 8 % to 1 2 % by
20 years after the diagnosis of a first cancer (Leone et al., 1999). Leukaemia is the
most frequent secondary neoplasm, and therapy-related acute myeloid leukaemia or
myelodysplastic syndrome (t-AMUMDS) now accounts for around 10% of all acute
myeloid leukaemia (AML) (Leone et al., 2001).
1.6.1 General haematopoiesis and leukaemia development
Acute leukaemia is described in detail in (Barrett, 1996; Schiffer and Stone,
2000; Weinstein, 1988). The acute leukaemias are defined as primary malignant
diseases of the blood forming organs characterised by a predominance of immature
myeloid and lymphoid precursors (blasts). They arise due to the unregulated
proliferation of early precursor cells that have lost their capacity to differentiate in
response to normal hormonal signals and cellular Interactions. Blast cells
progressively replace the normal bone marrow cells, which suppresses normal
maturation of erythrocytes, granulocytes, platelets and other blood cells derived
from residual precursor cells. This results in bone marrow failure and patients
become anaemic, thrombocytopenic and neutropenic. In addition blast cells can
migrate and invade other organs, for example the spleen, liver and, to a lesser
extent the lymph nodes. If untreated, acute leukaemia is fatal within a period of
months to one year.
69

General haematopoiesis
Leukaemic transformation can arise at any point during the differentiation of
the pluripotent haematopoietic stem cell. The pluripotent stem cell normally divides
infrequently to generate either more pluripotent stem cells or progenitor cells that
are irreversibly programmed to produce only a few types of blood cells (Fig. 1.20).
The earlier progenitor cells are multipotent but, as division and differentiation
proceed, later committed progenitors are formed. These progenitor cells are
stimulated to proliferate by specific growth factors, and can divide only a limited
number of times. The terminally differentiated cells have a limited lifespan and die
after several days or weeks (Alberts et al., 1994).
Classification of leukaemia
The acute leukaemias are classified morphologically by reference to the
predominant cell line involved as lymphoid (ALL) and myeloid (AML). In 1976, a
classification system was developed to quantify and standardise definitions of the
sometimes clinically and biologically distinct subtypes of AML and ALL. This is
known as FAB (French-American-British) classification (Bennett et al., 1976). ALL
was divided into three subgroups, LI to L3 and AML was into 7 subgroups. The
AML classification was later extended to 8 groups, MO to M7 (Table 1.6).
Table 1.6 FAB classification of AML.
FABClass
Common Name Subtypes Commonkaryotypes
MO Minimally differentiated AML
complex
Ml Myeloid leukaemia without maturation
Wide range
M2 Myeloid leukaemia with maturation
20-25%: t(8;21)
M3 Hypergranular promyelocytic leukaemia
Majority: t(15;17) Rare: t(11;17)
M4 Myelomonocytic leukaemia With bone marrow eosinophilia (M4E0)
Wide range M4E0: inv16 or del16q22
M5 Monocytic leukaemia Poorly differentiated (M5a); differentiated (M5b)
Del(11q23); t(-,11) M5b: t(8;16)
M6 Erythroleukaemia Del(5), del(5q), del(7), del(7q)
M7 Megakaryoblasticleukaemia
Minority: inv(3)
70

No1k.ral (NK) cell
Lymphoid piogenitw r
cell
# Neutfophil
T lymphocyte»
#B lymphocyte
Mullipotential Myeloiditerri cell progenitor
cell
Basophil
'' Eownophil
Monocyte mocrophoge
Pld'elel*
Red blood cells
Fig. 1.20 Scheme of haemopoiesis. Adapted from Bignami etal., 2003.

AML is much more common after drug treatment than ALL. Because my
work is concerned with therapy-related cancer, the remainder of this chapter
focuses on AML. The FAB morphological classification categorises the AML
subtypes according to the degree of maturation of the leukaemic cells and their
lineage differentiation (Table 1.6). AML cases are now divided into eight major
groups with subtypes for three of them (McKenna, 2000). These classification
criteria are based on morphologic and cytochemical features, but for some of the
categories immunophenotyping is necessary. In addition, some AML subtypes have
specific chromosomal translocation. Even though morphology and karyotype have
prognostic significance predicting favourable and less favourable outcomes of
treatments, they do not affect the type of treatment a patient receives. Acute
promyelocytic leukaemia (APL) is an exception. This subgroup is uniquely
responsive to all-frans-retinoic acid (ATRA).
Acute promyelocytic leukaemia
APL has been reviewed by (Pandolfi, 2001; Pitha-Rowe et al., 2003;
Ruggero et al., 2000). In almost 100% of cases, APL blasts contain reciprocal
chromosomal translocations involving chromosome 17. Nearly all these (-99% ) are
reciprocal and balanced translocations between chromosomes 15 and 17,
t(15; 17)(22;q11.2-12). The breakpoints were found to lie within the retinoic acid
receptor a {RAR a) gene on chromosome 17 and the promyelocytic leukaemia
{PM L) gene on chromosome 15. The PML-RARa fusion protein has multiple
oncogenic functions. It can block myeloid differentiation and confers a survival and
proliferative advantage on the expressing cell. It also seems to render cells
genomically unstable (Fig. 1.21.A).
PML-RARa affects both the RARa pathway and the PML function. RARs are
members of the superfamily of nuclear hormone receptors that act as retinoic acid -
inducible transcriptional activators. RARs form heterodimers with retinoid-X-
receptors (RXR), which repress transcription in the absence of retinoic acid by
recruiting nuclear receptor corepressors, which then recruit histone deacetylases
(HDACs). PML-RARa can bind to retinoic acid response elements (RARE) and
binds retinoic acid with the same affinity as RARa (Fig. 1.20.6). The fusion protein
could hence interfere with the RARa pathway at multiple levels. It was shown that at
physiological concentrations of retinoic acid, PM L-RARa acts as a potent
72
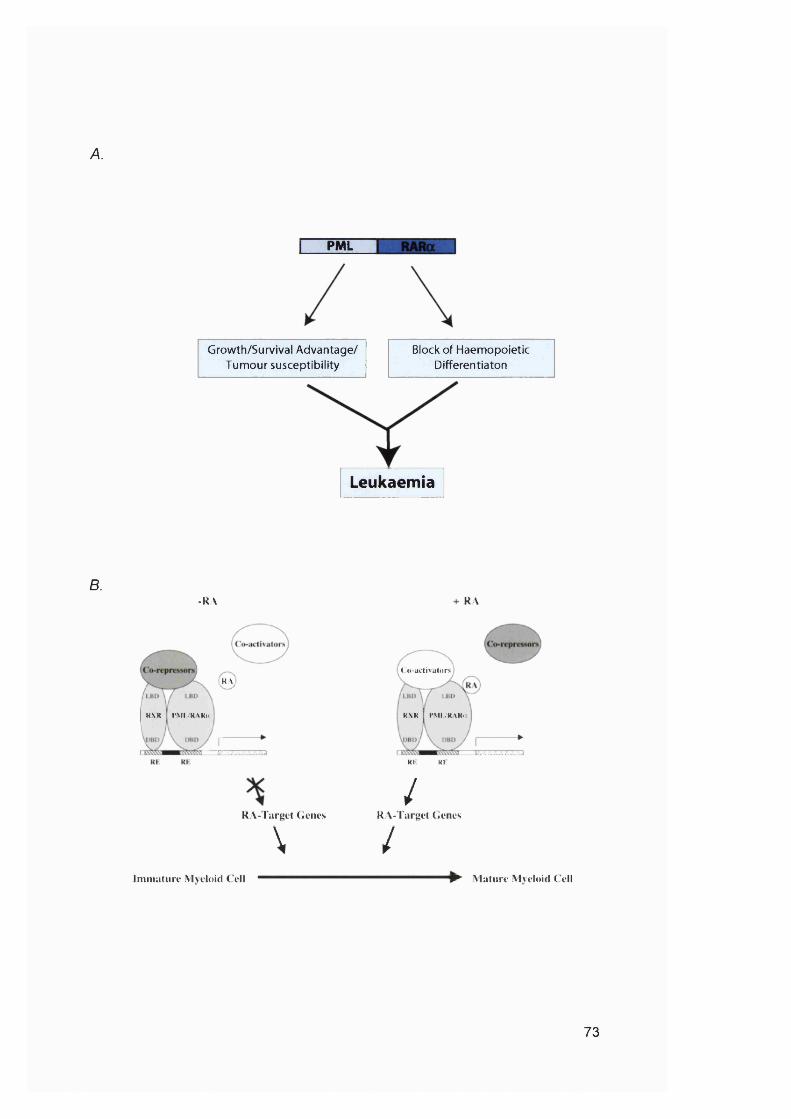
A.
PML
Leukaemia
Block of Haemopoietic Differentiaton
Growth/Survival Advantage/ Tumour susceptibility
B.-KA RA
M\
K \R l* M l iR \R ii
RA- l aryt't (ienes
\
( 'o -u c ti\a lo r!>
R \R I PMIVRXR.i
Rl RK
/R A- l urgct Genes
/Immature Mveloid ( ell Mature Mveloid C ell
73

c.
SP100CBP
RibosomalproteinsISG20
PMLPICl/SUMOl BImlnt-6
DaxxRFPp53
PML-RARa: APL
Cell cycle control
Genome stability
Apoptosis
Figure 1.21 The multiple oncogenic functions of PML-RARa.A.The fusion protein of APL can confer a proliferative and survival advantage to the myeloid haematopoietic cells. PML-RARa also has the ability to perturb myeloid differentiation. Adopted from Pandolfi, 2001.B. In the absence of pharmacologic RA dosages, PML-RARa recruits corepressors and acts as a dominant-negative inhibitor of multiple retinoid-dependent signaling pathways. This blocks cells in an immature stage of differentiation. Pharmacologic RA dosages can overcome this block by the loss of corepressor interactions and the recruitment of a coactivator complex. This results in the activation of RA target genes that activate terminal myeloid differentiation and other biological effects that lead to clinical remissions in APL. Adopted from Pitha-Rowe et al., 2003.C. PML is localised in discrete nuclear structures called PML MBs. NB components and the possible functions of these bodies are shown. Adopted from Ruggero et a/., 2000.
74

transcriptional repressor due to an increased affinity for corepressors and HDACs.
At a (high) pharmacological dose of the ligand however, the PM L-RARa-
corepressor complex is resolved and the fusion protein can transactivate RARa
target genes (Fig. 1.21.6). Treatment of APL patients with ATRA alone induces
disease remission by allowing maturation of leukaemia cells to neutrophils.
Remission is only transient however and relapse is inevitable if the patients are not
also treated with conventional chemotherapy or bone marrow transplantation post
remission.
In addition, the fusion protein can act as a dominant negative PML mutant.
PML is essential for the proper formation and stability of nuclear bodies (NB). Cells
typically contain 10 to 30 NB per nucleus. These structures contain several proteins
including transcription co-factors, oncoproteins, and ribosomal proteins (e.g. p53,
the retinoblastoma-susceptibility protein (Rb) and the Bloom syndrome DMA
helicase (BLM)) (Fig 1 .21 .0 ), some of which PML physically interacts with.
However, the actual function of this structure is still largely unknown. It has been
proposed that MBs might serve as depots for the different proteins they contain. The
PML-RARa fusion protein can form complexes with PML, which leads to disruption
of the NB. PML has also been found to have a role in growth and tumour
suppression possibly due to its interaction with proteins like tumour suppressors in
the NB. Due to this role, interference with PML function might result in a growth
advantage for leukaemic blasts. In addition, interference with PML can result in
genomic instability, probably related to the number of proteins involved in the
maintenance of genomic stability that accumulate in PML bodies. These include
BLM. Thus it appears that the PML-RARa fusion protein promotes oncogenic
growth by disrupting normal PML function.
Myelodysplastic syndrome
Secondary leukaemia associated with alkylating agent treatment is generally
preceded by a myelodysplastic phase. Myelodysplastic syndrome (M DS) is
described in detail in a number of textbooks (Catovsky, 1996; Silverman, 2000).
MDS describes a group of disorders that are characterised by hyperproliferative
bone marrow reflective of ineffective haematopoiesis. MDS is accompanied by one
or more peripheral blood cytopenias, most commonly anaemia. If not treated it
results in either bone marrow failure, which causes death from bleeding and
75

infection, or transformation to acute leukaemia. The FAB cooperative group has
proposed a classification of these heterogeneous conditions (Bennett et al., 1982).
MDS was divided into five categories on the basis of morphologic characteristics
and the percentage of blasts in the bone marrow and peripheral blood. The different
disorders are called;
(1) refractory anaemia (RA)
(2) refractory anaemia with ringed sideroblasts (RARS)
(3) refractory anaemia with excess blasts (>5 to 20% blasts) (RAEB)
(4) chronic myelomonocytic leukaemia (CMML)
(5) refractory anaemia with excess blasts ‘in transformation' (21 to <30% blasts)
(RAEB-t).
These should not be considered as rigid definitions, however, as progression
from one to another, especially from RAEB to RAEB-t occurs frequently.
Transformation to AML is the inevitable outcome in 30 to 50% of patients with RAEB
and CMML, and in more than 50% of those with RAEB-t. This course is less
common in RA and RARS (5 to 15% of cases). MDS with complex karyotypes and
more than 10% blasts have the highest risk of progression to AML. The FAB
classification has also been shown to correlate with prognosis, with RAEB and
RAEB-t generally having the worst. Other prognostic factors include the number of
bone marrow blasts, age, the severity of cytopenia and karyotypic abnormalities.
Clonal karyotypic abnormalities can be found in the marrows of 50 to 70% of
patients with MDS. These changes frequently involve chromosomes 5 (-5,5q-), 7 (-
7,7q-) and 8 (+8 ). The chromosomal abnormalities are similar to those seen in AML
patients, but none of the changes associated with specific types of AML, e.g.
t(15;17) in APL, are found in MDS. This suggests that these AML subtypes are not
preceded by a myelodysplastic phase.
1.6,2 Therapy-related acute mveloid leukaemia
t-AML was reviewed by (Bignami et al., 2003; Karp and Smith, 1997; Leone
et al., 1999). t-AML and de novo AML are clinically distinct diseases and they
respond very differently to therapy. The proportion of t-AML is increasing due to the
widespread use of chemo- and radiotherapy and the increased survival of patients.
Unfortunately t-AML, unlike do novo AML, is refractory to treatment and good
responses are rare. t-AML occurs most frequently after HD, NHL, ALL, multiple
76

myeloma, myeloproliferative diseases, breast cancer, ovarian cancer, and testicular
cancer.
t-AML has been divided into two sub-types: AML after alkylating agent
treatment and radiotherapy, and AML induced by DNA topoisomerase II (topo-ll)
inhibitors. The peak incidence of AML after alkylating agents is 4 to 6 years after
initiation of treatment for the primary malignancy, but they can occur with a latency
as short as 12 months or as long as 15 to 20 years. Complete or partial deletions of
chromosomes 5 and/or 7 are commonly found among this group of t-AML. Topo-ll
inhibitor induced AML is characterised by a shorter latency period (2 to 3 years) and
chromosomal abnormalities of the 11q23 locus.
AML after Hodgkin’s disease and non-Hodgkin’s iymphoma
Alkylating agents, but not radiotherapy or topo-ll inhibitors, are the major risk
factors for t-AML after HD and NHL. Both forms of lymphoma are often treated with
mixtures of drugs that include methylating agents such as procarbazine or
dacarbazine, for example as part of COPP (CTX, oncovin, procarbazine,
prednisone). The risk in patients treated for HD has been reported to be 10 to 80-
fold greater than among the general population.
AML after breast and ovarian cancer
Both alkylating agents and topo-ll inhibitors are risk factors for t-AML after
breast cancer. One study estimated the cumulative risk after these treatments to be
as high as 25±10% at 3 years (Andersen et al., 1990). The risk of t-AML in women
treated for ovarian cancer has also been shown to be high, specifically after
alkylating agent treatment (7% at 10 years).
1.6.3 Theraov-reiated mveiodvsolastic syndrome
t-MDS was discussed in detail in (Leone et al., 1999; Leone et al., 2001). In
classic alkylating agent related leukaemia, about two thirds of cases present with
fewer than 30% blasts in the marrow and less than 5% blasts in the blood and are
diagnosed as t-MDS. t-MDS is considered to generally have a poorer prognosis
than de novo MDS and more than 50% of t-MDS evolve into AML. Cytogenetic
abnormalities in t-MDS are similar to, but more frequent than in de novo MDS.
77

Deletion or loss of chromosomes 5 and 7 is often associated with exposure to
alkylating agents.
1.7 Overview of work described in this thesisThe work described in this thesis is concerned with the role of MMR and
drug treatment in the development of secondary leukaemia. In Chapter 3, the
involvement of MMR defects in the development of post chemotherapy AML/MDS
was investigated. hMLH1 silencing by promoter méthylation was examined as a
potential mechanism for loss of MMR function in these cases. As the cytotoxicity of
methylating agents and 6 -TG are both dependent on functional mismatch repair, the
role of MMR defects in the development of AML/MDS after thiopurine treatment was
investigated in Chapter 4. The work described in Chapter 5 was aimed at identifying
target genes for frameshift mutations in MMR defective leukaemia cases. The aim of
the projects described in the thesis was to elucidate the different steps involved in
the development of leukaemia after drug treatment.
78

Chapter 2 : Materials and Methods
2.1 Materials2.1.1 Chemicals
All chemicals were obtained from Sigma-Aldrich, Biorad Laboratories, Merck
/ BDH and Gibco Cell Culture / Invitrogen Life Technologies unless otherwise
stated. Standard solutions of 0.5M EDTA, 1M Tris-HCI, Tris-EDTA (TE), 1M MgCL,
5M NaCI, Phosphate Buffered Saline (PBS) and Tris Borate EDTA (TBE) were
produced by Cancer Research UK London Research Institute (LRI) central services.
All other stock solutions were made according to standard methods (Sambrook et
al., 1989; Sambrook and Russell, 2001).
Recrystalized MNU was a kind gift from Prof. Peter Swann. A 50mM stock of
MNU was made up in 4mM potassium acetate buffer pH4.0. 0®-BzG was a kind gift
from Dr. Juergen Thomale. A 20mM 0®-BzG stock was made up in DMSO. 6 -TG
and 6 -MP were both made up to a 5mM stock in 0.1 M sodium hydroxide. A 5mg/mL
stock of Cyclosporin A (CsA) and a lOOmM stock of prednisolone (Pred) were made
up in DMSO. A Im M stock of MMC was made up in H2O.
2.1.2 Cell culture
Dulbecco's MEM (E4), 2% RPMI 1640 and Giemsa stain were supplied by
Cancer Research UK LRI central services. Cells were grown in media
supplemented with 10% (v/v) foetal calf serum (PCS) (Gibco Cell Culture I
Invitrogen Life Technologies). Sterile disposable plastic ware was acquired from
Becton Dickinson, Corning Incorporated, Millipore Corporation, Nun and Sterile.
2.1.3 Cell lines and clinical samples
All cells used were obtained from the Cancer Research UK LRI central cell
services. The cell lines used are summarised in Table 2.1.
Chemotherapy-related AL/MDS DNA, RNA and bone marrow samples were
obtained from Dr. Margherita Bignami and Dr. Ida Casorelli, Instituto Superiore di
Sanita’, Laboratorio di Tossicologia Comparata, Rome, Italy, and Prof. Giuseppe
Leone and Dr. Luca Mele, Haematology Department, Catholic University, Rome,
Italy. Colon cancer DNA samples were contributed by Dr. Ian Tomlinson, Cancer
79

Genetics Laboratory, Cancer Research UK LRI, UK, Bone marrow and explanted
tissue samples from transplant-related AML patients were provided by Dr. David
Cummins, Dr. Ozay Halil, Dr. Nicholas R. Banner and Dr. Margaret M. Burke,
Harefield Hospital, Hill End Road, Harefield, Middlesex, UK.
Table 2.1 Cell lines
Cell Line Derived from MGMTStatus
MMRdefect
HeLa cervix carcinoma Mex+ WTRaji TK- Burkitt’s lymphoma Mex- WTRaji TK+ Burkitt’s lymphoma Mex+ WTSW480 colon carcinoma ? WTRamos Burkitt’s lymphoma ? WTBL2 Burkitt’s lymphoma ? WTA2780-SCA ovarian carcinoma Mex+ WTGM08925 peripheral blood leukocytes of an apparently normal individual ? WTGM03714 peripheral blood leukocytes of an apparently normal individual ? WTGM03469 peripheral blood leukocytes of an apparently normal individual ? WTGM01953 peripheral blood leukocytes of an apparently normal individual ? WTGM00621 peripheral blood leukocytes of an apparently normal individual ? WTGM00558 peripheral blood leukocytes of an apparently normal individual ? WTGM1056a peripheral blood leukocytes of an apparently normal individual ? WTGM00130b peripheral blood leukocytes of an apparently normal individual ? WTGM00637 peripheral blood leukocytes of an apparently normal individual ? WTRpmi 1788 peripheral blood leukocytes of an apparently normal individual ? WTJurkat T-lymphocyte leukaemia ? hMSH2SW48 colon carcinoma Mex- hMLHIHCT116 colon carcinoma Mex+ hMLHILoVo colon carcinoma Mex+ hMSH2DLD1 colon carcinoma Mex+ hMSHBHecIA endometrial carcinoma ? hMLHIREH B-lymphocyte leukaemia ? hMLHIMolt4 T-lymphocyte leukaemia ? hPMS2CCRF-GEM B-lymphocyte leukaemia ? hPMS2AN3CA endometrial carcinoma ? hMLHIDU 145 Prostate Carcinoma ? hMLHISK0V3 ovarian carcinoma ? hMLHIA278G-MNÜCI1 ovarian carcinoma Mex+ hMLHIRaji F I 2 Burkitt’s lymphoma Mex- hMSHB
2.1.4 Molecular biology reagents
All molecular biology reagents and kits were obtained from Qiagen unless
otherwise stated. dNTPs were purchased from Amersham Pharmacia. The ImProm-
II Reverse Transcriptase was obtained from Promega Corporation. DNA markers
and restriction enzymes were obtained from New England Biolabs Inc. The BigDye
Terminator v3.1 Cycle Sequencing Kit was purchased from Applied Biosystems. All
PCR amplification and sequencing reactions were performed on a Peltier Thermal
Cycler PTC-200.
80

2.2 Cell culture techniques2.2 .1 Maintenance of cell cultures
Lymphoblastoid cell lines were maintained in 2% RPMI 1640 cell culture
medium supplemented with 10% PCS. Cells were maintained at a concentration of
between 2 x 1 0 ® and 2 x 10® cells/ml at 37°C in a humidified atmosphere containing
5% CO2. Fibroblast lines, colorectal lines and the variants of A2780 were maintained
in Dulbecco's MEM (E4) supplemented with 10% PCS at 37°C and 10% CO2. Cells
were passaged approximately twice a week by detaching the cells with a thin layer
of 0.25% Trypsin in 0.1% Versene for 3 to 5 minutes at 37°C. This was neutralised
with lOmL of medium containing 10% PCS and cells replated at dilutions of between
1:5 and 1:12. Viable cell numbers were counted using an Improved Neubauer
haemocytometer. At least four large squares were counted, the number averaged
and multiplied by 10 to give the number of viable cells/mL. Long-term maintenance
of cell cultures was performed by the Cancer Research UK LRI central cell services
laboratory according to their standard methods.
2.2.2 Cell storage
Frozen cell stocks were stored at -70°C and under liquid nitrogen (LN2).
Approximately 2 to 5 x 10^ cells were pelleted, the medium removed and the pellet
resuspended in the appropriate growth medium containing 10% (v/v) DMSO. Cell
suspensions were transferred to cryovials, wrapped in tissue paper, placed at -70°C
and cooled at a rate of 1°C / minute. After 48 hours, vials were transferred to LN2
for long-term storage. Cell stocks were recovered by rapidly thawing the cryovial at
37°C and transferring the cells into an appropriate volume of fresh media.
2.2.3 Generation of cell cultures from single cells
Single cell clones were established by limiting dilution. Essentially, an
exponentially growing cell culture was diluted to 10, 20 or 30 cells per mL. 200pL of
diluted cells were seeded in each well of a 96 well plate and allowed to grow for 2-3
weeks. Mass cultures were established from wells containing only a single colony.
81

2.2.4 Selection of 6-TG resistant clones
The 6 -TG resistant clones were isolated as described in Fig. 2.1.
HAT treatment
One medium flasks containing an exponentially growing, sub-confluent
population of approximately 5 x 1 0 ® A2780-SCA cells was treated with 1 x HAT
supplement (100M hypoxanthine, 0.4M aminopterin, 16M thymidine) in growth
medium containing 10% PCS. A second flask was treated with 1 x aminopterin
(0.4M) as a control. Treatment was 10 days until the aminopterin-only treatment had
killed all cells. The medium was then replaced with drug-free medium containing
10% PCS.
6-TG treatment
The HAT treated cells were divided into five medium flasks. Once the cells
were growing exponentially each flask was treated with either 0.1 or 0.3pg/mL 6 -TG
in growth medium containing 10% PCS (Pig. 2.1). Treatment was continual until the
cells had recovered. The cells treated with 0.3pg/mL 6 -TG were maintained in 6 -TG
at this concentration for one month. The treatment of the cells grown in O.Ipg/mL 6 -
TG was increased to 0.3pg/mL after one week. These cells were then maintained in
this concentration of 6 -TG for an additional 3 weeks. After one month of continuous
treatment with 6 -TG the concentration was increased in all five flasks to either 0.7 or
I.Opg/mL. The cells were maintained in this concentration for another month and
then cloned into growth medium containing 10% PCS and 1x HAT to obtain single
HGPRT"^ clones.
Screening for cross-resistance to MNU
After 5 weeks single colonies were picked and each colony was divided into
two wells of a 24-well plate in growth medium containing 10% PCS. Cells in one of
the wells from each colony were treated with 25pM 0®-BzG and allowed to attach.
After 2 hours cells growing in medium containing 0®-BzG were treated with 200pM
MNU. Five single cell clones that were cross-resistant to MNU were picked and
expanded.
82
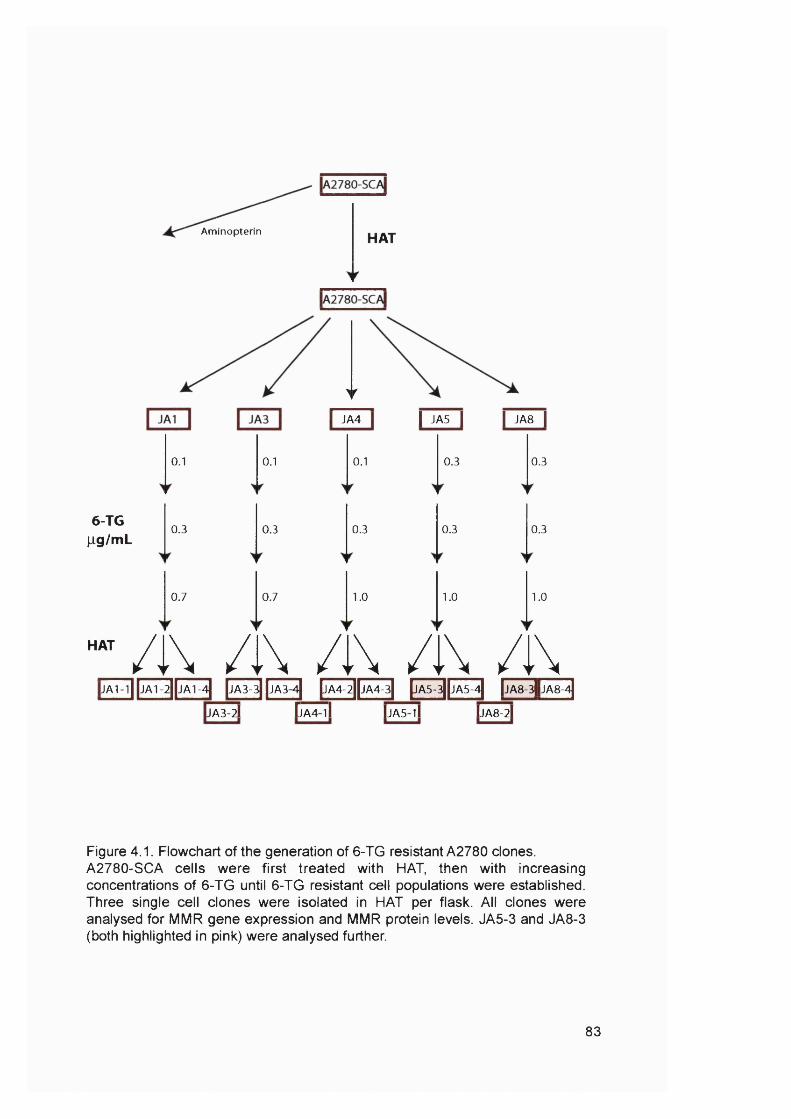
Aminopterin HAT
6-TGlig /m L
HAT
0.1
0.3
0.7
0.1
0.3
0.7
>r
I JA5 I I JA8 IJA4
0.1
0.3
1.0
/i\ /i\ /i\ /i\ /i\
0.3
0.3
1.0
0.3
0.3
1.0
[jA1-l||jA 1-2||jA1-4] |JA3-3| |jA3 4] |jA4-2||jA4-3| |jA5-3||jA5-4j |jA 8-3|jA 8-4|
A3-2I II A4 i l |JA5 i l LIA8-2I
Figure 4.1. Flowchart of the generation of 6 -TG resistant A2780 clones. A2780-SCA cells were first treated with HAT, then with increasing concentrations of 6 -TG until 6 -TG resistant cell populations were established. Three single cell clones were isolated in HAT per flask. All clones were analysed for MMR gene expression and MMR protein levels. JA5-3 and JA8-3 (both highlighted in pink) were analysed further.
83

2.2.5 Cell survival bv colony formation assay
6-TG
Cells from an exponentially growing culture were seeded at between 200
and 5000 cells per well of a 6-well plate and allowed to attach for about 2-4 hours.
An appropriate concentration of 6-TG was added. The cells were incubated for 10
days and then colonies counted after staining with Giemsa. Every treatment was
performed in duplicate.
MNU
Cells from an exponentially growing culture were seeded at between 200
and 5000 cells per well of a 6-well plate into growth medium containing 10% PCS
and 25pM 0®-BzG and allowed to attach for about 2-4 hours. An appropriate
concentration of MNU was added. The cells were incubated for 10 days and then
colonies counted after staining with Giemsa. Every treatment was performed in
duplicate.
2.2.6 Cell survival bv cell density
Cells from an exponentially growing culture were seeded at 2 x 10 cells per
well of a 6-well plate into growth medium containing 10% PCS and allowed to attach
for about 2-4 hours. An appropriate concentration of 6-MP, CsA and Pred was
added. The cells were incubated for 10 days. The colonies were then stained with
Giemsa. The relative resistance of the treated cultures was reflected by the intensity
of the staining.
2.2.7 Lymphoblastoid growth curves
Cells from exponentially growing cell stocks were seeded at a concentration
of 5 X 10® cells per mL in a 24 well plate and grown in the continuous presence of
MMC. Cell numbers were determined by daily cell counts using an improved
Neubauer haemocytometer. Exponential growth of cells was maintained by dilution.
These experiments were carried out by Karen Gascoigne.
84

2.3 Molecular and cellular biology techniques2.3.1 Preparation of genomic DNA
From animal cells
2 X 10^ cells were pelleted, washed twice in PBS and either used
immediately or stored at -70®C. Genomic DNA was prepared from these cells using
a DNeasy Tissue Kit according to the manufacturer's instructions. An RNase step
was included.
From paraffin embedded tissues
10pm thick sections were cut from the paraffin blocks by Dr. Peter Maddox.
The DNA was extracted following the manufacturer’s instructions. The paraffin was
removed by extraction with xylene as described. An RNase step was included. Due
to the small amount of starting material 5pg of carrier DNA (poly(dl)»poly(dC) double
strand (Amersham Pharmacia Biotech)) was added to the buffer AL (before the
precipitation step). The DNA was precipitated with 230pL instead of 200pL ethanol
and in the last step eluted with 60pL buffer AE.
2.3.2 Preparation of total RNA
5 X 10^ cells were pelleted, washed twice in PBS and either used
immediately or stored at -70°C. Total RNA was prepared using an RNeasy Mini Kit
according to the manufacturers instructions. Qiashredders were used to
homogenise the samples. A DNase digestion step was included using the RNase-
Free DNase Set according to manufacturer’s instructions.
2.3.3 Parallel RNA and genomic DNA purification
Bone marrow samples were obtained suspended in TRIzol reagent
(Invitrogen Life Technologies). RNA and DNA were isolated following the
manufacture’s instructions.
85

2.3.4 Determination of DNA concentrations in aqueous solutions
Spectrophotometer
Concentrations of DNA in solution were determined according to the method
of (Sambrook et a!., 1989; Sambrook and Russell, 2001). The absorbance of
appropriately diluted solutions was determined at 260 and 280nm (A260 and A280) in
quartz cuvettes (Jencons) using a GeneQuant (Amersham Pharmacia Biotech,
APB) spectrophotometer. An A260 equal to 1.0 corresponds to 50pg/mL dsDNA or
33pg/mL ssDNA or oligonucleotide. The ratio of A260/A280 is an estimate of purity and
should be between 1.8 and 2.0 for pure dsDNA.
SYBR Green I stained agarose gel
The method described by (Kiltie and Ryan, 1997) was used to determine the
concentration of DNA samples that were too low to be determined using the
spectrophotometer. DNA samples were separated on a 0.7 to 1% agarose gel using
0.5 X TBE buffer. The gel was dried at room temperature for 30 and then at 60®C for
60 minutes using a gel drier (Biorad model 583). The gel was stained with 1 x SYBR
green (Molecular Probes, Invitrogen Life Technologies) in 0.5 x TBE pH 8.0 buffer
for 30 minutes to 24 hours protected from light. The DNA was visualised using the
blue fluorescent channel on the Storm 860 Scanner. A set of Lambda DNA
standards of known amounts were run at the same time as the DNA samples of
unknown concentration. The intensity of each band was calculated using the IQ Mac
version 1.2 software and plotted against the known amounts of DNA. The DNA
concentration of the unknown samples was calculated using the slope of the
standard curve.
2.3.5 Determination of RNA concentrations in aqueous solutions
Spectrophotometer
Concentrations of RNA in solution were determined according to the method
of (Sambrook et al., 1989; Sambrook and Russell, 2001). The absorbance of
appropriately diluted solutions was determined at 260 and 280nm (A260 and A280) in
quartz cuvettes (Jencons) using a GeneQuant (Amersham Pharmacia Biotech,
APB) spectrophotometer. An A260 equal to 1.0 corresponds to 40pg/mL RNA. The
ratio of A260/A280 is an estimate of purity and should be between 1.9 and 2.1 for pure
RNA.
86

Spectrofluorophotometer
Concentrations of RNA in solution were determined using the RiboGreen
RNA Quantitation Reagent (Molecular Probes, Invitrogen Life Technologies)
following the manufacture’s instructions. The excitation maximum for RiboGreen
bound to RNA is ~500nm and the emission is ~525nm. The sample fluorescence
was m easured in disposable fluorim eter cuvettes (K arte ll) using a
spectrofluorophotometer (Model RF-1501, Shimadzu) and standard fluorescein
wavelengths (excitation ~480nm, emission ~520nm). An RNA standard curve was
prepared using RNA purified from Raji cells. The concentration of the Raji RNA had
previously been determined using the spectrophotometer. The RNA concentration of
the unknown sample was determined from the standard curve.
2.3.6 hM LHI promoter méthylation assay
The promoter méthylation assay was adapted from Kane et al. (Kane et al.,
1997). The PCNA promoter sequence from -982 to +45 used as a control contains
the ATG start codon and five Hpall/MspI restriction sites. Genomic DNA (10pg to
2ng) was digested in 10pL buffer (NEB) with no enzyme, SOU Hpall, or 100U Mspl.
Incubation was for 8 hours at 37"C followed by nuclease inactivation for 10 minutes
at 6 5 X .
Digested DNA was analyzed by multiplex PCR in SOpL 1 x Qiagen PCR
buffer containing 2.5mM MgCb, 0.2mM each dNTP, 2.5U HotStarTaq DNA
Polymerase and lOpmol of each primer. The primers are described in Table 2.2.
Amplification was performed after initial dénaturation at 95®C for 15 minutes. Each
cycle was: 1 minute at 9 5 X , 1 minute at 68“C, 1 minute at 72®C. Thirty to 35 cycles
were followed by a final extension at 72®C for 7 minutes.
After 35 cycles, unamplified samples were cycled further with nested
primers. For the hM LH I promoter, IpL of the multiplex PCR product was analyzed
in 50pL buffer containing I.SmM MgCI2, 0.2mM each dNTP, 2.5U HotStarTaq DNA
Polymerase and lOpmol of each primer (Table 2.2). hM LH I amplifications were
performed as described above with an annealing temperature of 59”C after an initial
dénaturation at 95‘*C for 15 minutes. Twenty cycles of 1 minute at 9 5 X , 1 minute at
59®C, 1 minute at 7 2 X were performed followed by a final extension of 72°C for 7
minutes. Conditions for PCNA were identical except that the MgCb concentration
was 2.5mM, the annealing temperature was 69®C and each cycle consisted of 1
87

minute at 95°C, 1 minute at 69®C, and 1 minute at 72®C followed by 7 minutes at
72®C. The primers are described in Table 2.2. Products were separated on an
agarose (1.5% ) gel and visualised with ethidium bromide or SYBR Green I
(Molecular Probes, Invitrogen Life Technologies).
2.3.7 MMR RT-PCR
The MMR RT-PCR was adapted from (Wei et al., 1997). Ipg of total cellular
RNA was reverse transcribed to cDNA in 20pL 1 x ImProm-ll Reaction Buffer
containing 20U of RNasin Ribonuclease Inhibitor (Ambion), lOmM of each dNTP,
3mM MgCl2, 0.5pg pd(N)e random hexamer primers (Amersham Pharmacia Biotech,
APB) and Ip L ImProm-ll Reverse Transcriptase following the manufacturer’s
instructions. 2pL of the RT product was amplified in a multiplex PCR reaction
containing 2pmol p-actin, 25pmol hMSH2, 12.5pmol hPMS2, Spmol hMSH6, 25pmol
hMLHI and 6.25pmol hPMSI primers as described in Table 2.2. p-actin was used
as a positive control. Reactions (50pL) contained 2.5mM MgCb, 200pM of each
dNTP and 2.5U HotStarTaq. Amplification was performed for 15 minutes at 95®C, 30
cycles of 95®C for 30 seconds, 58®C for 30 seconds and 72®C for 45 seconds
followed by a final extension of 72“C for 10 minutes. PCR products were separated
on a 2% agarose gel and detected by ethidium bromide staining.
MMR RT-PCR of clinical samples
To increase the sensitivity of this assay for the use with clinical samples, the
number of cycles was increased to 50 and the amount of HotStarTaq was doubled.
Alternatively, nested primers were designed for amplification of samples of
low RNA concentration and quality (Table 2.2). The primers from the first PCR
reaction were removed using the QIAquick PCR Purification Kit following the
manufacturer’s instructions. 0.1 to 2pL of the multiplex PCR product were amplified
in a multiplex PCR reaction containing lOpmol of each primer (Table 2.2). Reactions
(50pL) contained 4mM MgCb, 200pM of each dNTP and 2.5U HotStarTaq.
Amplification was performed for 15 minutes at 95®C, 30 cycles of 95®C for 30
seconds, 58®C for 30 seconds and 72°C for 45 seconds followed by a final
extension of 72®C for 10 minutes. PCR products were separated on a 2% agarose
gel and detected by ethidium bromide staining.
88

Table 2.2 Primers
00CO
Gene / Locus Primer sequence (S' to 3’) PCR annealing temperature, °C
MgCb concentration, mM
fragment size (bp)
Reference
hMLHI promoter méthylation assayhMLHI promoter CCA CAT ACC GCT CGT AGT ATT CGT GC
CCT CAG TGC CTC GTG CTC ACG TTC55 / 68 multiplex 2.5 618 Kane et al., 1997
hMLHI promoter nested GGG TTG CTG GGT CTC TTC GTC CCT CC CGC GTT CGC GGG TAG CTA CGA TGA GG
59 1.5 449
hPCNA promoter GGC AGG AGA CTC ACT TGA ACC TGG CCT AGA AAG ACA ACG ACC ACT CTG C
67/ 68 multiplex 2.5 1010
hPCNA promoter nested GAA TGT TAA GAG GAT GAT AGG GAG C GCG GGG AAG ACT TTA GGG CCA ATC G
69 2.5 440
MMR RT-PCRP-actin ACA CTG TGC CCA TCT ACG AGG
AGG GGC CGG ACT CGT CAT ACT58 2.5 621 Wei et al., 1997
p-actin nested TCG TGC GTG ACA TTA AGG AG TGA TCC ACA TCT GCT GGA AG
57 4 450
hMSH2 GTC GGC TTC GTG CGC TTC TTT TCT CTG GCC ATC AAC TGC GGA
58 2.5 429 Wei et al., 1997
hMSH2 nested CTT CGT GCG CTT CTT TCA G CCT TGG ATG CCT TAT TTC CA
57 4 287
hPMS2 TGC ATG CAG CGG ATT TGG AAA GAA CCC CTC AGA ATC CAC GGA
58 2.5 358 Wei et al., 1997
hPMS2 nested TTT GGA AAA GCC CAT GGT AG GAG ATC TCA GGA CGC CTT TG
57 4 239
hMSH6 CCC TCA GCC ACC AAA GAA GCA CTG CCA CCA CTT CCT CAT CCC
58 2.5 288 Wei et al., 1997
hMSHB nested CAG CCA CCA AAC AAG CAA CT CCT CCT CCT GTG CTC ATC TC
57 4 194
hMLHI GTG CTG GCA ATC AAG GGA CCC CAC GGT TGA GGC ATT GGG TAG
58 2.5 215 Wei et al., 1997
hMLHI nested CAA CAT AGC CAC GAG GAG AA GGT TGA GGC ATT GGG TAG TG
57 4 166
hPMS1 GCG GCA ACA GTT CGA CTC CTT AGC CTT GAT ACC CTC CCC GTT
58 2.5 174 W eieta l., 1997
MSI analysis Dietmaier et al., 1997Bat25 TCG CCT CCA AGA ATG TAA GT 55 2 -9 0

TCT GCA TTT TAA CTA TGG CTCBat26 TGA CTA CTT TTG ACT TCA GCC
AAC CAT TCA ACA TTT TTA ACC C55 1.5 -80-100
Bat40 ATT AAC TTC CTA CAC CAC AAC GTA GAG CAA GAC CAC CTT G
55 1.5 -80-100
D2S123 AAA CAG GAT GCC TGC CTT TA GGA CTT TCC ACC TAT GGG AC
55 2 197-227
D5S346 ACT CAC TCT AGT GAT AA TCG AGC AGA TAA GAC AGT ATT ACT AGT T
55 2 96-122
D17S250 GGA AGA ATC AAA TAG ACA AT GCT GGC CAT ATA TAT ATT TAA ACC
55 2 -150
Target gene analysisCaspase-5 CAG AGT TAT GTC TTA GGT GAA GG
AAC ATG AAG AAC ATC TTT GCC CAG55 3 141 Schwartz et al., 1999
Mre11 AAT ATT TTG GAG GAG AAT CTT AGG G AAT TGA AAT GTT GAG GTT GCC
57 3 122 Giannini et al., 2002
Chk1 CTG CCA TGC CTA TCC TGA TT TCA CAC AAT GAT GAA ACC ACC T
57 3 175
Fas ATC ACC TGG CCA TTT TCT TG GGT TGG GGG AAA GGA GAA TA
57 3 205
Rb AAG AAA GAA AAT CTT TAC CAT GCT G CAG CCT TAG AAC CAT GTT TGG
57 3 336
Fits GAA CTC GCT GCA GAA ATC CT GGG ATT CTT GCC TGA CTC AA
57 3 402
CBFp CAT CAC AAC TGC CAT GGT TT ACA GAA GCC AGC CTG TGA CT
57 3 480
FancD2 CTT GCA AAG AGC CAT CTG CT ATC CTG TGT TCC CGC TAT TT
55 3 300
FancE GGG AGC TTC TCC ACT GTC TG TGA GTC CTT TCT GCG TTT CC
63 3 160
FancG AGG AAA GCC AGA GTG TGT GG ACT GCA GCT GGA GAG AAA GG
63 3 358
FancD2 RT-PCRFancD2 exon 5-9 TTT GTC TTG TGA GCG TCT GC
GGA TCT CAG GTA GGC TGG TG57 3 314
COo

2.3.8 Microsatellite analysis
The standard microsatellites BAT26, BAT25, D2S123, D17S250, D5S346
and BAT40 were amplified by PCR using fluorescent primers (Table 2.2). The sense
primer of each primer set was FAM-labelled. Every marker was analysed
separately. Genomic DNA (10pg to 20ng) were amplified in 1 x Qiagen PCR buffer
containing 1.5 or 2mM MgC^ (Table 2.2), 0.2mM each dNTP, 2.5U HotStarTaq DNA
Polymerase and lOpmol of each primer. Amplification was performed after initial
dénaturation at 95®C for 15 minutes. Each cycle was: 1 minute at 95®C, 1 minute at
55®C, 1 minute at 72®C. Thirty to 35 cycles were followed by a final extension at
72®C for 10 minutes. Products were analyzed by the Cancer Research UK LRI
Equipment Park either on an ABI Prism 377 Sequencer or an ABI Prism 3100
capillary system. The results obtained were analysed using the Genotyper 1.1
software.
2.3.9 Target gene analysis
Genotyping
The repeat sequences in Caspase-5, Mre11, Chk1, Fas, Rb, Flt3, Cbf^,
FancD2, FancE and FancG were amplified by PCR using fluorescent primers (Table
2.2). The sense primer of each primer set was FAM-labelled. These genes were
amplified as three multiplex PCRs: Caspase-5 and FancD2; Mre11, Chk1, Fas, Rb,
Flt3 and Cbf^; FancE and FancG. Genomic DNA (lOpg to 20ng) was amplified in 1
X Multiplex PCR Master Mix containing lOpmol of each primer. Amplification was
performed after initial dénaturation at 95®C for 15 minutes. Each cycle was: 30
seconds at 94®C, 90 seconds at 55-63°C (Table 2.2), 60 seconds at 72®C. To
increase the sensitivity of the assay for use with clinical samples the annealing time
was increased from 90 seconds to 3 minutes. Thirty to 40 cycles were followed by a
final extension at 72®C for 10 minutes. Products were analyzed by the Cancer
Research UK LRI Equipment Park either on an ABI Prism 377 sequencer or an ABI
Prism 3100 capillary system. The results obtained were analysed using the
Genotyper 1.1 software.
DNA sequencing reactions
First multiplex PCR reactions were performed as described above with the
exception that primers were not fluorescently labelled. The PCR products were
separated by agarose gel electrophoresis (2% agarose). The bands were excised
91

from the gel and the PCR products were purified with the Gel Purification Kit. One to
5pL of purified products were sequenced using the BigDye Terminator v3.1 Cycle
Sequencing Kit according to the manufacturer's instructions. Essentially, reactions
contained 8pL of BigDye mix, and 3.2pmol primer in a total volume of 20pL. The
DNA was then sequenced for 25 cycles of 96°C for 30 seconds, 50°C for 15
seconds and 60°C for 4 minutes (at a ramp rate of 1°C/second). At the end of the
reaction, excess dye terminators were removed using the DyeEx 2.0 Spin Kit.
Electrophoresis was carried out by the Cancer Research UK LRI Equipment Park
either on an ABI Prism 377 sequencer or an ABI Prism 3100 capillary system.
Sequences were analysed on the Sequencher 4.1 software.
2.3.10 FancD2 RT-PCR
The reverse transcriptase reaction was performed as described in Section
2.3.7. The FancD2 cDNA was amplified with p-actin as a positive control as a
multiplex PCR. 4pl of the RT product was amplified in 1 x Multiplex PCR Master Mix
containing lOpmol of each primer. Amplification was performed after initial
dénaturation at 95*C for 15 minutes. Each cycle was: 30 seconds at 94°C, 90
seconds at 57®C, 60 seconds at 72"C. Thirty-five cycles were followed by a final
extension at 72"C for 10 minutes. The FancD2 product was purified and sequenced
as described in Section 2.3.9.
2.3.11 Cell extracts
For M GM T assay
Approximately 10 cells were lysed in Triton extraction buffer (50mM Tris-
HCI pH 7.5, lOmM DTT, Im M EDTA, 0.2% (v/v) Triton X-100) for 5 minutes on ice.
Lysates were clarified by centrifugation at 15 000 x g at 4°C for 15 minutes. The
protein concentration in the supernatant was determined.
For Western blots
One medium flask (or around 2x10^ cells) were pelleted, washed and
resuspended in 100-200pL of extraction buffer (1% NP40, lOmM NaF, Im M
NaV3Ü 4, Im M NaPP0 4 , Mini EDTA free protease inhibitor cocktail tablet (Roche) in
PBS) and incubated on ice for 1 hour. Samples were centrifuged at 15 000 x g for
30 minutes at 4°C and the protein concentration determined.
92

Stillman replication extract
Cell extracts were prepared from either 20 triple flasks or 5 litres of cell
culture (Cell Production Unit, Cancer Research UK LRI Clare Hall). The cells were
washed first in hypotonic buffer (20mM Hepes-KOH pH 7.5, 5mM KCI, 1.5mM MgCl2
and 0.5mM DTT) containing 250mM sucrose then in hypotonic buffer alone and
allowed to swell on ice for 5 minutes. Cells were then dounce homogenised with a B
pestle and centrifuged for 20 minutes at 10 000 x g at 4°C. The supernatant was
centrifuged again at 50,000 x rpm in a Beckman TL100.2 rotor for 1 hour at 4°C.
Aliquots of the supernatant were snap frozen and stored at -70°C. All solutions were
kept at less than 4°C and all steps carried out on ice. These cells extracts were
prepared by Peter Macpherson.
2.3.12 Measurement of protein concentrations in aqueous solution
Protein concentrations were determined according to the method of
Bradford. An appropriately diluted IpL aliquot of extract was added to iOOpL of
dH20 and to this, 900pL of Bradford reagent. After thorough mixing and incubation
at room temperature for 5 minutes, the absorbance at 595nm (A595) was determined
and the background (900pL + lOOpI dH20) subtracted. The protein concentration
was determined from a standard curve of the A595 from known amounts of BSA.
Bradford Reagent: 100mg Coomassie Brilliant Blue
50mL 95% (v/v) Ethanol
lOOmL Orthophosphoric Acid
dH20 to 1L
The solution was stored at +4°C, protected from light but was warmed to room
temperature prior to use.
2.3.13 In vitro mismatch repair assay
The substrate was prepared and the assay carried out by Mr. P. Macpherson
in the laboratory according to the previously described method (Oda et al., 2000).
Mismatch correction was assayed in 25pL 30mM Hepes-KOH pH 8.0, 7mM
MgCb, 0.5mM DTT, 100pM each dNTP, 4mM ATP, 40mM phosphocreatine, Ipg
creatine phosphokinase (rabbit muscle type I), 70mM KCI, 90ng DNA substrate (T*C
mispair) and up to 200pg cell extract. Extracts were prepared according to the
method of Stillman described earlier. Mixtures were incubated at 37°C for 15
93

minutes and then terminated by the addition of 10mM EDTA, 0.5% SDS. Proteins
were removed by proteinase K digestion and phenol extraction and the DNA
precipitated with 1/10*'’ volume 3M NaAc and 2 volumes ethanol. After extensive
washing with 70% ethanol and drying, the DNA was dissolved in restriction enzyme
buffer (NEB) and digested with Mlul which is diagnostic for the removal of the
mismatch. Digestion products were separated on a 0.8% agarose gel and detected
by ethidium bromide staining.
2.3.14 Western blotting
SOpg of whole cell extract (total volume of lOpL) per well was loaded onto a
8% SDS-PAGE gels along with lOpL of prestained Perfect Protein markers (lane 1)
and lOpL Kaleidoscope markers (lane 10, both Biorad). Separated proteins were
transferred to a PVDF membrane (Immobilon-P, Millipore) for 1 hour at 18V using a
Transblot semi-dry transfer cell (Biorad) and 25mM Tris, 192mM Glycine, 0.037%
SDS and 20% MeOH as the transfer buffer. Membranes were blocked overnight in
5% (w/v) non-fat powdered milk in PBS-Tween (0.1% Tween20) at 4°C with gentle
rocking. Membranes were washed three times with PBS-Tween before being
incubated with the primary antibody diluted in 5% BSA / 0.1% sodium azide / PBS-
Tween for 1 hour at room temperature. Membranes were again washed three times
with PBS-Tween and immunoreactive proteins detected using a HRP-conjugated
secondary antibody diluted according to the manufacturers instructions in 5% BSA /
PBS-Tween; anti-mouse (hMSH2, hM LHI, hPMS2, Biorad) or anti-goat (hMSH6,
Chemicon). Incubation was for 1 hour at room temperature. Membranes were
washed a further four times before antibody complexes were detected using ECL
substrate and quantitated on Hyperfilm ECL film (APB). Transfer efficiency was
ascertained by staining the membranes with Ponceau after immunogen detection.
Table 2.3 Antibodies.
Immunogen Manufacturer Clone Concentration (pg/ml)hMSH2 Pharmingen G219-1129 1hMSH6 Santa Cruz Q-20 2hPMS2 Pharmingen A16-4 1hMLHI Pharmingen G168-15 0.5
94

2.3.15 Methvltransferase assay
Substrate and reaction conditions
M. leutus DNA was methylated with A/-[methyl - ^H]-A/-nitrosourea
(Amersham Biosciences) as enzyme substrate; heat treatment of the DNA removes
labile /V-methylated bases thus increasing the proportion of 0®-meG in the total
amount of méthylation damage. The substrates were prepared by Peter
Macpherson.
Enzyme reactions were performed in 100pL assay buffer (70mM Hepes
KOH pH7.8; 10mM DTT; 1mM EDTA; substrate so that approximately 1.5 x 10
counts per lOOmL; stored at -20®C) by addition of 0, 25 and 50mg. Reactions were
incubated 20 minutes at 37°C, protein and DNA precipitated by addition of 125pL
ice cold 0.8M trichloroacetic acid and lOpL carrier DNA (2mg/mL heat denatured
herring sperm DNA) and incubated on ice for 5 minutes. The precipitate was
pelleted by centrifugation at 13,000 x g for 10 minutes at 4°C and the supernatant
removed and discarded. 0®-pH]meG in DNA was hydrolysed by incubation with
0.1 M HCI. This treatment did not hydrolyse pHJmethyl groups associated with the
methyltransferase. Thus transferred and non-transferred methyl groups could be
distinguished. After incubation on ice for 5 minutes and centrifugation (1 minutes,
1000 X g at room temperature) 80mL of the hydrolysate were added to 5mL
EcoscintA (National Diagnostics) and activity measured in a liquid scintillation
counter (Tricarb 1500 Liquid Scintillation Counter, Packard).
Calculation of MGMT activity
In order to estimate the amount of radioactivity removed from the DNA by
incubation with cell extracts, the count of radioactive disintegrations per minutes
(dpm) in the sample containing extract was subtracted from the number of
disintegrations detected in the control reaction without protein. The specific activity
of the substrate is approximately 5 x 10^ dpm per pmole DNA, the counting
efficiency is assumed to be 20% thus approximately 10 counts per minute (cpm)
per pmole substrate will be recorded. If the data are plotted as amount of
radioactivity removed versus the amount of protein used in the incubation, the slope
of the graph represents the cpm removed per mg of protein incubated with.
95

The MGMT activity of the extract can thus be calculated as:
slope _ cpm!mg
10"* cpm/pmole
with 1 unit of MGMT being able to remove 1 pmole of 0®meGua.
96

Chapter 3 : Results I
Drug treatment and hM LHI promoter méthylation in the development of MMR defective acute leukaemia and MDS
3.1 Introductiont-AML is particularly associated with treatment for HD and NHL (Section
1.6.2). Both diseases are often treated with mixtures of drugs that include
methylating agents such as procarbazine or dacarbazine. These drugs are cytotoxic
and cells can acquire resistance by inactivating their MMR (Section 1.4.1) (Karran
and Bignami, 1994). Thus, the question arises whether methylating agents exert the
same selective pressure in a clinical situation - in particular, on the cells of patients
who are treated with these drugs - as they do on cells in the laboratory. Bone
marrow cells are particularly sensitive to methylating agents and treatment dosage
for these drugs is usually limited by this myelotoxicity. The tissue in which the
highest selective pressure to inactivate MMR occurs is therefore the bone marrow.
This led us to propose that t-AML after methylating agent treatment could be the
clinical counterpart to selection for méthylation tolerance and loss of MMR in the
laboratory. This hypothesis would be supported by the detection of MSI among
methylation-treatment related myeloid leukaemia.
When I started working on this project the estimates of the frequency of MSI
in t-AML/MDS varied widely. Three studies had investigated MSI among t-AML and
reported frequencies of 94% (Ben-Yehuda et al., 1996), 0% (Boyer et al., 1998) and
20% (Horiike et al., 1999), so the role of MMR in these malignancies was far from
clear. Guided by the hypothesis that selection for loss of MMR might play a role in
the development of therapy related leukaemia, a collaboration with Professor G.
Leone (Université Cattolica, Rome) and Dr. M. Bignami (Instituto Superiore di
Sanità, Rome) was initiated. Ideally we wanted to selectively investigate the
possible association between MSI and methylating agent treatment compared to
other drugs. Unfortunately we were not able to obtain a sufficiently large number of
post-methylating agent AML cases to address this question directly. We therefore
decided to examine all available t-AML samples from Prof. Leone and his
colleagues. Firstly, we analysed these samples for MSI to identify MMR defective
cases. Secondly, we addressed the mechanism by which the MMR defect arose in
MSF cases.
97

hM LH I promoter méthylation is the most common mechanism of loss of
MMR in sporadic M S r tumours. It occurs in most (80% ) M S r sporadic colorectal
(Kane et al., 1997), (Herman et al., 1998), endometrial (Simpkins et al., 1999), and
gastric (Fleisher et al., 1999) tumours. DNA méthylation is a post-replication
modification that is predominantly found in cytosines of the CpG sequence
(reviewed by (Herman and Baylin, 2003; Jaenisch and Bird, 2003)). Most CpG sites
in the genome are methylated. The CpG islands, in which the frequency of CpG
dinucleotides is significantly higher than in the genome overall, are the exception.
CpG islands are found in the promoters of some genes and méthylation of CpG
dinucleotides in these regions generally correlates with silencing of the gene.
Many types of cancers show both reduced CpG méthylation in non-promoter
regions and increased méthylation in gene promoter sequences. This promoter CpG
méthylation is associated with transcriptional silencing and is, for example, one
mechanism for inactivation of tumour suppressor genes. Somatic inactivation of
both alleles of a gene by point mutations is improbable. Loss of gene expression
more commonly involves more high frequency events such as promoter silencing.
Numerous genes involved in critical growth regulatory mechanisms like cell cycle
control, DNA repair, apoptosis and others are known to be silenced in human
cancers.
The h M L H I promoter contains a CpG island. It was first shown by Baylin
and co-workers that the h M L H I gene was silenced by hypermethylation of this 5’
CpG island in the overwhelming majority of MSF sporadic primary colorectal
cancers (Herman et al., 1998). Silencing of the h M L H I gene confers an identical
phenotype to gene mutation and/or loss of heterozygosity (LOH). I therefore decided
to investigate the frequency of hM LH I promoter méthylation among the therapy-
related AML cases.
3.2. Results3.2.1 Patients
We examined 25 Italian cases from the Division of Haematology, Université
Cattolica of Rome. Patient details are summarized in Table 3.1. This group of
patients consisted of 19 AML and 2 MDS cases, but also included 4 cases of ALL.
The median ages at diagnosis of the primary and secondary malignancy were 52
years (range 22-73) and 58 years (range 39-75), respectively.
98
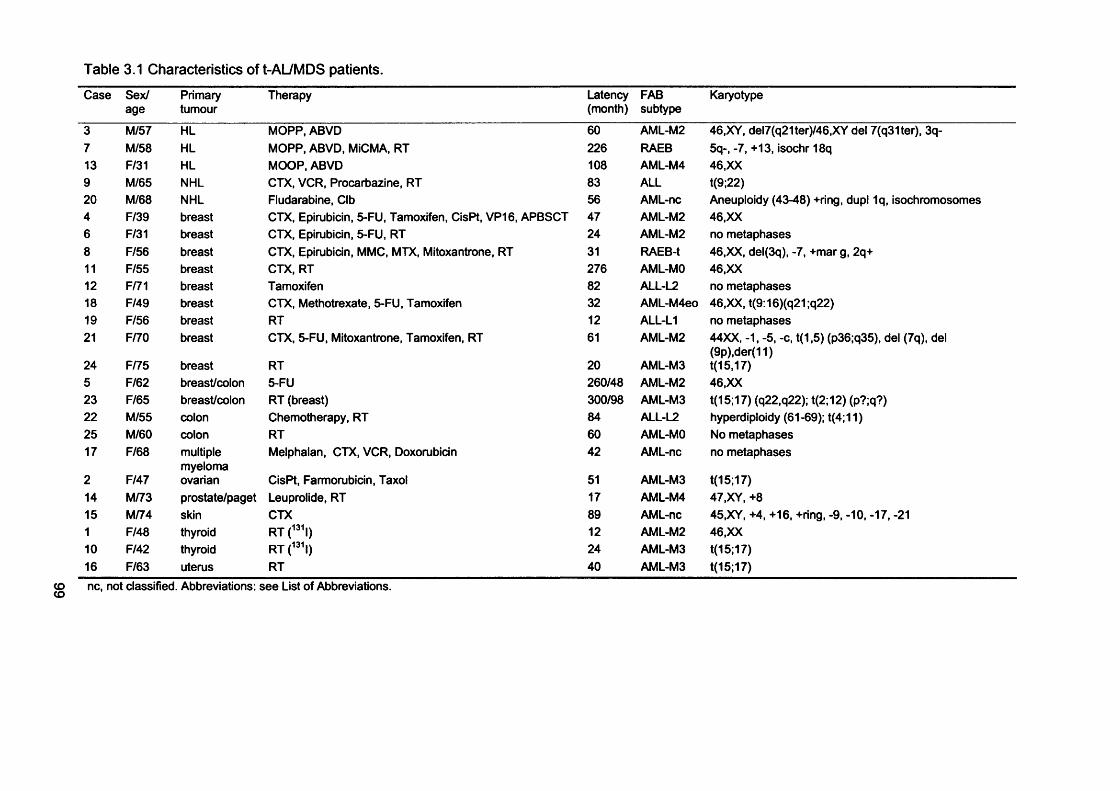
Table 3.1 Characteristics of t-AL/MDS patients.
Case Sex/age
Primarytumour
Therapy Latency(month)
FABsubtype
Karyotype
3 M/57 HL MOPP, ABVD 60 AML-M2 46,XY, del7(q21ter)/46,XY del 7(q31ter), 3q-7 M/58 HL MOPP, ABVD, MiCMA, RT 226 RAEB 5q-, -7, +13, isochr 18q13 F/31 HL MOOP, ABVD 108 AML-M4 46,XX9 M/65 NHL CTX, VCR, Procarbazine, RT 83 ALL t(9;22)20 M/68 NHL Fludarabine, CIb 56 AML-nc Aneuploidy (43-48) +ring, dupl 1q, isochromosomes4 F/39 breast CTX, Epirubicin, 5-FU, Tamoxifen, CisPt, VP16, APBSCT 47 /UVIL-M2 46,XX6 F/31 breast CTX, Epirubicin, 5-FU, RT 24 AML-M2 no metaphases8 F/56 breast CTX, Epirubicin, MMC, MTX, Mitoxantrone, RT 31 RAEB-t 46,XX, del(3q), -7, +mar g, 2q+11 F/55 breast CTX, RT 276 AML-MO 46,XX12 F/71 breast Tamoxifen 82 ALL-L2 no metaphases18 F/49 breast CTX, Methotrexate, 5-FU, Tamoxifen 32 AML-M4eo 46,XX,t(9:16)(q21;q22)19 F/56 breast RT 12 ALL-LI no metaphases21 F/70 breast CTX, 5-FU, Mitoxantrone, Tamoxifen, RT 61 AML-M2 44XX, -1, -5, -c, t(1,5) (p36;q35), del (7q), del
(9p),der(11)24 F/75 breast RT 20 AML-M3 t(15,17)5 F/62 breast/colon 5-FU 260/48 AML-M2 46,XX23 F/65 breast/colon RT (breast) 300/98 AML-M3 t(15;17) (q22,q22); t(2;12) (p?;q?)22 M/55 colon Chemotherapy, RT 84 ALL-L2 hyperdiploidy (61-69); t(4;11)25 M/60 colon RT 60 AML-MO No metaphases17 F/68 multiple
myelomaMelphalan, CTX, VCR, Doxorubicin 42 AML-nc no metaphases
2 F/47 ovarian CisPt, Farmorubicin, Taxol 51 AML-M3 t(15;17)14 M/73 prostate/paget Leuprolide, RT 17 AML-M4 47,XY, +815 M/74 skin CTX 89 AML-nc 45,XY, +4, +16, +ring, -9, -10, -17, -211 F/48 thyroid RT(^^'l) 12 AML-M2 46,XX10 F/42 thyroid RT(^^\) 24 AML-M3 t(15;17)16 F/63 uterus RT 40 AML-M3 t(15;17)
g ne, not classified. Abbreviations: see List of Abbreviations.

Cytogenetic analysis was performed at the time of diagnosis. Deletion of the
long arm or loss of an entire chromosome 7 was detected in only four cases (3, 7, 8,
and 21) and cases 7 and 21 also had a deletion of 5q or loss of the whole
chromosome 5. This is consistent with a previous suggestion that these events are
infrequent in t-AML in Italian patients (Pagano et al., 2001). The 5 t-ARLs (cases 2,
10,16, and 24) each had the classical t(15:17) translocation.
We also examined 28 de novo acute leukaemias (ALs) with a median age at
diagnosis of 43.5 years (range 16-75) and a similar distribution between females
and males (13 and 15, respectively). These cases included 8 do novo APL.
3.2.2 Microsatellite instabilitv in chemotherapv-related AUM DS
The microsatellite analysis was performed by Ida Casorelli and Margherita
Bignami at the Institute Superiore di Sanita’ of Rome. Where the patient’s normal
DNA was available, the standard panel of five microsatellites recommended by
Boland et al. (Boland et al., 1998) was analyzed. Examples of MSI are shown in Fig.
3.1. Evidence of instability at two or more loci was found in 10 of 15 (67%) cases.
These were designated MSF. Non-tumour DNA was not available for the remaining
10 patients. Tumour DNA was analyzed at the Bat26 locus in these cases. This
locus is considered to be essentially monomorphic among the general population
and alterations at Bat26 are considered to be a good indication of MSF (Loukola et
al., 2001). As expected, the frequency of MSF was similar in this group and six
cases (60%) were altered at Bat26. These were also designated MSF. We conclude
that the overall frequency of MSF among our patients is around 60% (16/25, 64%)
(Table 3.2). The frequency of MSF was similar in t-AL/MDS (11/16, 68%), t-APL
(3/5, 60%), and t-ALL (2/4, 50%) considered separately. In contrast to the therapy-
related cases, none (0/28) of the de novo leukaemias were MSF. This low (<3.5%)
frequency of MSF in de novo disease is in good agreement with published estimates
(Ohyashiki et al., 1996; Sill et al., 1996).
100
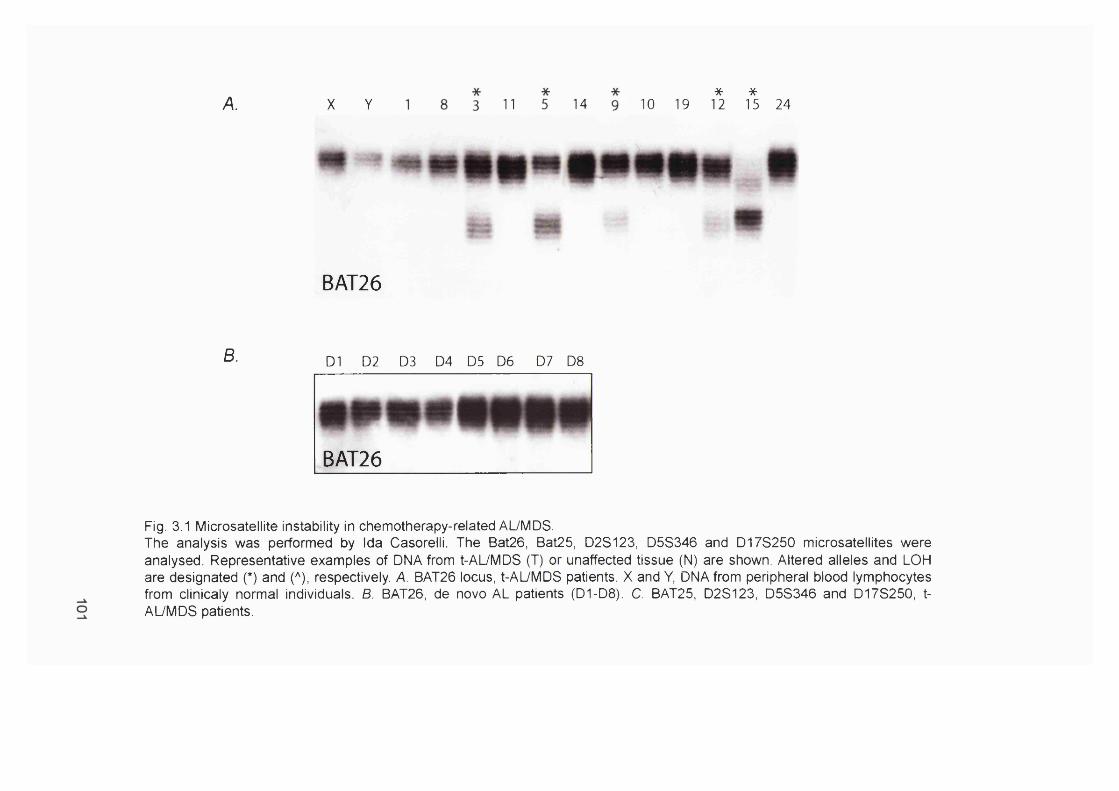
A. X Y 11 14 10 19*12
*15 24
B.
BAT26
D1 D2 D3 D4 D5 D6 D7 D8
BAT26
Fig. 3.1 Microsatellite instability in chemotherapy-related AUMDS.The analysis was performed by Ida Casorelli. The Bat26, Bat25, D2S123, D5S346 and D178250 microsatellites were analysed. Representative examples of DNA from t-AUMDS (T) or unaffected tissue (N) are shown. Altered alleles and LOH are designated (*) and (*), respectively. A. BAT26 locus, t-AUMDS patients. X and Y, DNA from peripheral blood lymphocytes from clinicaly normal individuals. B. BAT26, de novo AL patients (D1-D8). C. BAT25, D2S123, D5S346 and D17S250, t- AUMDS patients.

c.
BAT25
t ’ ° n N T T^ '* N
# * '
D5S3462? #
T N
*T N T
« # 1
m # # mm S 3i
mm»I
D2S123
*
T N T N T ^ N
*N
D175250
Oro

Table 3.2 Analysis of M S r in t-AL/MDS patients.
Case Primary tumour Bat26 Bat25 D2S123 D178250 D5S346 diagnosisHD/NHL
3 + + + - No DNA U7 + U9 + U13 + + + . No DNA U20 + + + - - U
Breast4 + U5 + + - - - U6 na + + - - U8 - 811 - 812 + U18 + + - - - U19 - - - - - S21 + + - LOH U23 + - + + No DNA U24 - - - - - S
Others22 - - - - - 825 - 81 - - - - - S10 + 82 + + - + + U14 - 815 + U16 + U17 + + + + + ULOH, loss of heterozygosity; U, unstable; 8, stable; na, not amplifiable.
3.2.3 Clinical sample DNA concentrations
DNA samples from the t-AL/MD8, de novo AML and blood donor control
patients were obtained from Ida Casorelli. The DNA concentration and quality of the
samples varied somewhat. As t-AML is still a relatively rare disease most hospitals
see only a small number of cases every year. Most of the t-AL/MD8 DNA samples
had therefore been purified from archival material, in some cases from sections
scraped off slides. These samples were often of very poor quality and low DNA
concentration. By contrast, DNA from cases 2, 10, and 21 - 25, which had been
purified from bone marrow samples recently taken from patients, were of higher
concentration and better quality. This emphasizes the desirability of working with
material freshly prepared from clinical samples without intervening periods of
storage. The blood donor and do novo ALL DNA samples had also been purified
from fresh material, and were of good quality. To be able to obtain consistent results
from these samples the DNA concentration had to be determined accurately and the
103

PCR assays optimised to achieve the highest level of amplification possible. The
conventional method of measuring DNA concentrations by A260 did not result in a
reading that was above background for most of the t-AL/MDS samples. I therefore
investigated the use of SYBR Green I stain, following the method described by Kiltie
and Ryan (Kiltie and Ryan, 1997) to determine the DNA concentration (Figure
3.2.AIB). 1pL of the t-AML samples (cases 3 - 1 2 shown as examples) was run on a
0.7% agarose gel together with a titration of Lambda DNA (5 - 80ng). To determine
the sensitivity of the dye, a second titration (0.1 - 20ng) was performed. After
electrophoresis, the gel was dried and stained with SYBR Green I. Stained DNA
bands were visualised using the Storm 860 Scanner and IQMac v1.2 software. The
intensities of the Lambda DNA bands were used to construct a standard curve from
which the DNA concentrations of the t-AML samples were determined. Figure 3.2.C
shows that as little as O.Sng Lambda DNA can be detected using this method. The
correlation between DNA concentration and the intensity of the bands is not linear
below 5ng (Figure 3.2.D). Nevertheless, the method will usefully detect < 1 ng DNA
with reasonable precision. Despite this high sensitivity, I was not able to detect any
DNA in seven out of ten samples. This indicates that the concentration of these
samples is less that 0.5ng/pL. The concentration of most of the other 15 samples
was equally low.
In addition to low concentrations, some the t-AL/MDS DNA samples ran as
heterogeneous smears on electrophoresis (data not shown). This indicates that
these DNA samples are severely degraded and contain many breaks.
3.2.4 Optimisation of the hMLHI promoter méthylation assay
hMLH1 promoter méthylation was examined by Hpall/M spI digestion. Hpall
and MspI are isoschizomers. The recognition sequence of both enzymes is CCGG
and hence contains a CpG site. Hpall is méthylation sensitive and only digests non-
methylated DNA. M spI on the other hand, cleaves both methylated and non-
methylated DNA. In the basic approach, DNA is first digested with either MspI or
Hpall. The region of interest can only be amplified by PCR if the CpG sites within
the recognition sequence are methylated and the DNA is resistant to cleavage by
Hpall. MspI digestion serves as an internal control for digestion, because the DNA is
always cleaved it can never give rise to a detectable amplification product.
104

A. tAL/MDS Samples
o o o o o«— fN ^ o o o o ' ^ L o v o r ^ o o O N f — fN
8 ,
D N A S ta n d a r d s
700000
600000 4
500000
§ 300000 4
200000 -
100000
0 ^
y = 7908.6X
0 20 40 60
ng Lam bda DNA
80
Fig. 3.2 DNA concentration standard curve.A. Increasing amounts of genomic Lambda DNA (5 - 80 ng) were, together with 1 pi of each tAML sample (samples 3-12), separated on a 0.7% agarose gel. After electrophoresis the gel was dried and stained with SYBR Green I. The stained DNA was visualised using the Storm 860 Scanner. 8. The intensity of each band was calculated using the IQ Mac version 1.2 software and plotted against the known amounts of DNA. The DNA concentration of the tAML samples was calculated using the slope of the standard curve. The SYBR Green I sensitivity was determined repeating the experiment (C.) electrophoresing smaller amounts of Lambda DNA (0.1 - 20 ng) and (D.) from these plotting the DNA standard curve.
105

XDNA
D.
r - und d
o o(N r - fN
D N A S ta n d a rd s
80000
70000= 6822.6X
60000
50000
40000
30000
20000
10000
ng Lam bda DNA
106

The hM LH I promoter méthylation assay was adapted from Kane et al. (Kane
et ai., 1997). The region between nucleotides -6 7 0 to -6 7 of the promoter amplified
by PCR analysis consists of 58.4% G+C (Fig. 3.3.A). Hpall I MspI recognition sites
are located at nucleotide positions -567 , -527 , -347 , and -341 . Kane et al. showed
that lack of hMLHI expression correlated with cytosine méthylation at these four
sites in the h M L H I promoter region in a group of sporadic colon tumours, the
sporadic colon tumour cell line SW48 and the sporadic endometrial tumour cell line
AN3CA.
The promoter region of the PCNA gene was included as an internal standard
for enzyme digestion. This gene is ubiquitously expressed in dividing cells and its
promoter is méthylation free. The sequence amplified (-982 to +45) contains the
start codon and 5 Hpall/MspI restriction sites. The sequence amplified comprises
part of the CpG island (-3 0 9 to 190) identified by Marc Offman (personal
communication) using the EMBOSS tool. The SW48 cell line that has a methylated
h M LH I promoter (Kane et al., 1997) and does not express hM LHI protein
detectable by Western blotting, was used as a positive control. The Raji Burkitt's
lymphoma cell line, in which hM LHI gene expression and protein levels are normal
served as a negative control.
When the assay was first set up it worked very well with DNA isolated from the
SW48 and Raji cell lines. I was unable to amplify DNA from the clinical samples,
however. It seemed likely that this was due to their low DNA concentration and poor
quality. To increase the sensitivity of this assay, I designed a set of nested primers
for PCR of both the h M L H I and PCNA promoters (Fig. 3.3). Each of the PCR
reactions was optimised to obtain the highest possible level of amplification (Fig.
3.4). A MgCb titration was carried out to determine the optimal Mg^ concentration
(Fig. 3.4.A, B, D, F). This was 2.5mM for both PCNA and hM LH I. For the PCR
reactions with the nested primers the same PCR reaction using the determined
MgCb concentration was performed over a range of different temperatures to
identify the optimal temperature and to minimize non-specific amplification (Fig.
3.4.E, G). The hM LH I and PCNA promoters were then amplified in a multiplex PCR
using 2.5mM MgCb This PCR was performed over a temperature gradient to identify
the optimal temperature which was found to be 68®C. The nested PCRs were then
set up using a small amount of the h M L H I and PCNA multiplex PCR. Fig. 3 .4 .0
shows that the hM LH I PCR with the set of nested primers is sufficiently sensitive to
detect 0.1 to Ing of Raji DNA. When a nested PCR using 1 pL of a hM LH I PCR of
107
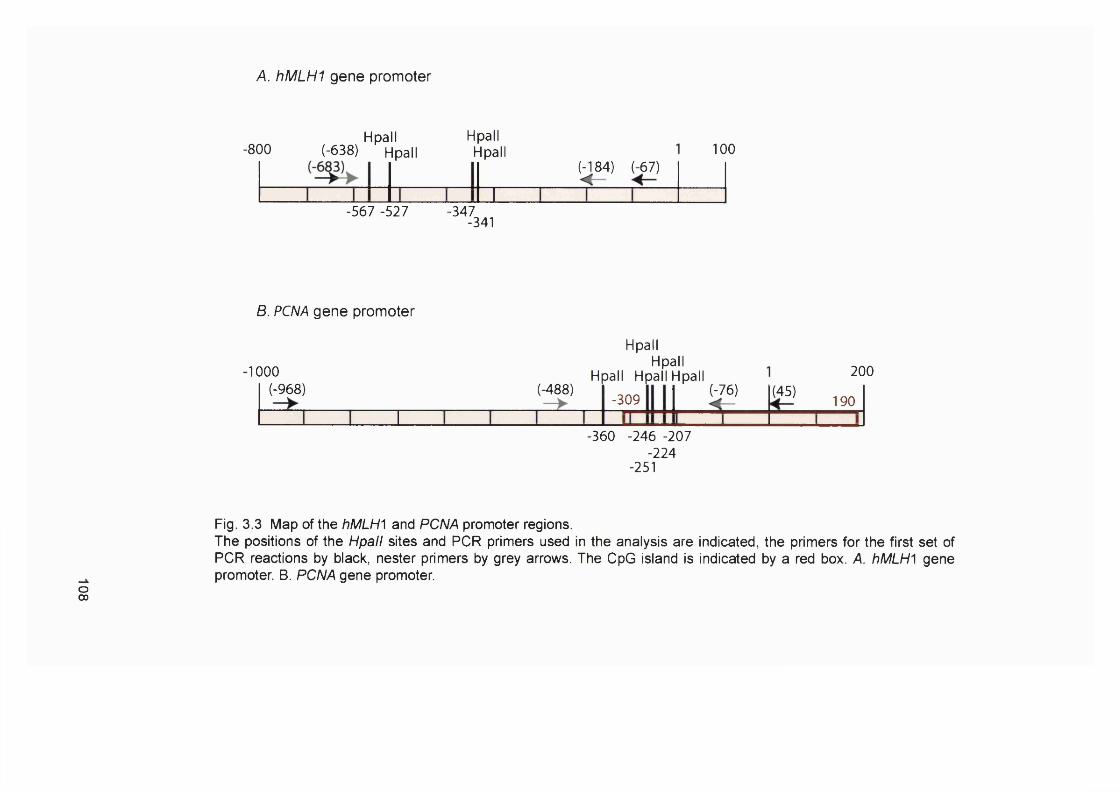
A. hM LHI gene promoter
-800Hpall Hpall
(-638) Hpall Hpall 100(-184) (-67)
-567 -527- '% 4 1
B. PCNA gene promoter
1000 (-968) (-488)
H
Hpall Hpall
Hpallpall Hpall
-309(-76) (45)
200
190
-360 -246 -207 -224
-251
Fig. 3.3 Map of the hMLH^ and PCNA promoter regions.The positions of the Hpall sites and PCR primers used in the analysis are indicated, the primers for the first set of PCR reactions by black, nester primers by grey arrows. The CpG island is indicated by a red box. A. hMLH^ gene promoter. B. PCNA gene promoter.
o00
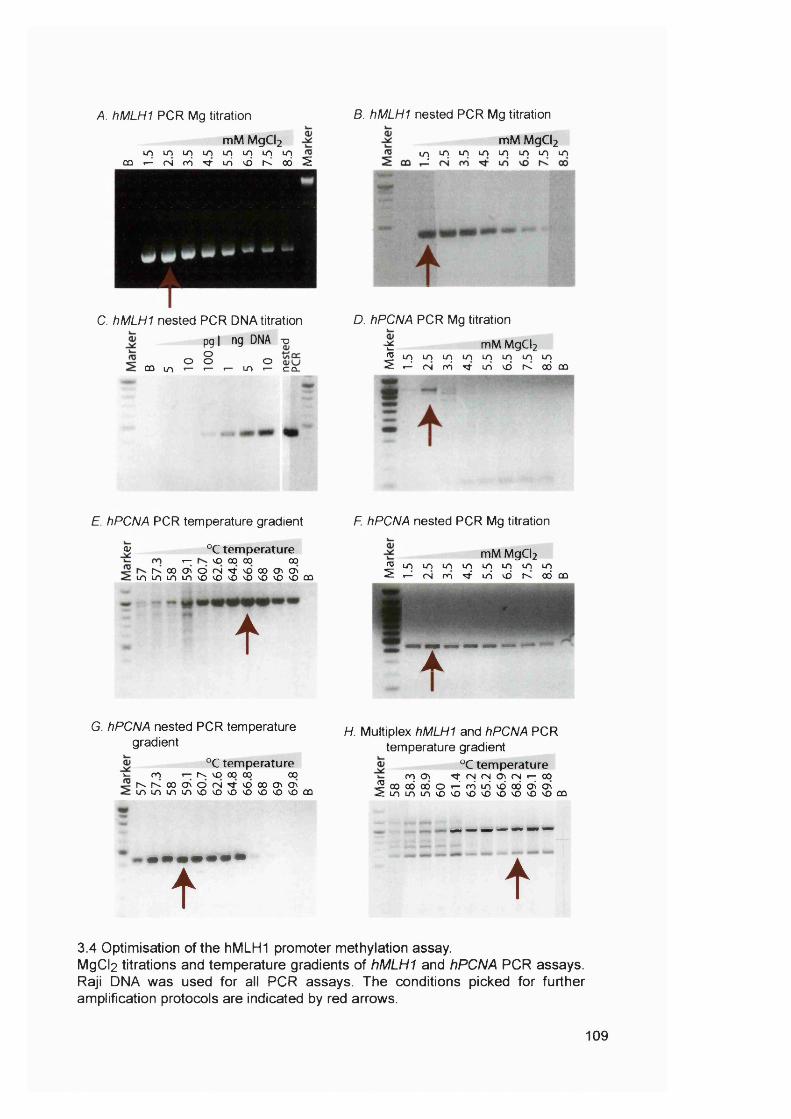
A. hMLHI PCR Mg titration
mM MgCl2L nLnLALnLn i nLnLn
c û r - ^ ( N r Ô T t u S v d r < o ô I E
C. hMLHI nested PCR DNA titration
pgl ng DNA -o
0 8 oÛÛ LO 1— «— «— U1 «— C O -
B. hMLHI nested PCR Mg titration
m M MgCl2ur j u^u^i nuoLOLOLn
D. hPCNA PCR Mg titration
^ mM MgCl2^ u n L O L o u n u T j u o i r j L n^ « - ^ ( N r Ô T t L O v d t ^ o d o û
E. hPCNA PCR temperature gradient
<u °C tem perature •— vo 00 00 00
r>. 00 <T> o <N Tt m U-) Lo Lo vo vo vo vo 00 <Ji CTv
vO "vO vO vO ÛÛ
t
F. hPCNA nested PCR Mg titration
mM MgCl2W i n L O L O L r j L n L T j L O L n^ « - ^ ( N r o ^ L r i v d r v o d û û
- r —
G. hPCNA nested PCR temperature gradient
k. ro°C tem perature
rv vo 00 00 00r ^r^ooo\ Of N^voooo\ o\L O L n i n t o v o v o v o v o v o v o v o c û
H. Multiplex hMLHI and hPCNA PCR temperature gradient
°C tem perature^ <N fS O i fN «— COk . m G \
2coodoc>Oi -^rôuSvdo6c3^c^ ^ L O L n i n v o v o v o v o v O v o v o v o c û
t t3.4 Optimisation of the hMLHI promoter méthylation assay.MgCl2 titrations and temperature gradients of hMLHI and hPCNA PCR assays. Raji DNA was used for all PCR assays. The conditions picked for further amplification protocols are indicated by red arrows.
109

10pg Raji DNA, was performed with these primers under the same conditions, it
resulted in clear amplification products (Fig. 3.4.C right panel).
3.2.5 Sensitivity of the hMLHI promoter méthylation assay
To determine the limits of detection of this assay different amounts of Raji
and SW48 genomic DNA were mixed and assayed. Figure 3.5.A shows that, after a
single PCR of 2ng of total DNA input, the limit of detection is 10% (0.2ng)
methylated DNA. An Increase in sensitivity was obtained using nested PCR (Figure
3.5 .6 ), where the input into the PCR reaction was only around lOOpg DNA and as
little as 2% (2pg) methylated DNA resulted in a positive signal. As one human cell
contains between 5 to lOpg of DNA, lOOpg is equivalent to DNA from only 10 to 20
cells. Since as little as 2% (2pg) of methylated DNA can be detected, this method is
clearly very sensitive. It appears that as little as a single methylated sequence is
detectable.
This high sensitivity of the assay can be a source of problems. As enzymatic
digestion of DNA is unlikely to be complete, even very small amounts of undigested
DNA can produce false positives. To minimise this risk of false positives the digest
was carried out with a large amount of restriction enzyme for more than 16 hours. In
addition, every sample was assayed at least three times to ensure reproducibility.
3.2.6 hMLHI promoter methvlation analysis of control samples
To ensure that this assay does not give rise to false positives, twenty MMR
proficient control samples were analysed. I assayed 8 de novo APL, 10 blood donor
and 12 de novo ALL DNA samples. These samples were all shown to be MSI' by
Ida Casorelli. Examples are shown in Figure 3.6. Raji and SW48 were used as
controls in all assays. These samples were of better quality and higher DNA
concentration than the majority of the t-AL/MDS samples. The do novo APL
samples were analysed both by nested hM LH I PCR (Fig. 3 .6 .A ) and single
multiplex PCR (Fig. 3 .6 .6). No promoter méthylation was detected using either
method. The high sensitivity of the nested PCR indicates that less than 2% of each
DNA sample and therefore also of the leukaemic cell population was methylated. As
the same result was obtained by both assays, the remaining control samples (blood
donor and do novo ALLs) were analysed by single PCR only. This assay is not as
sensitive as the nested PCR. None of the blood donor (Fig. 3 .6 .C) or the de novo
110

A.
Percentage SW48 DNA Controls
^ 0% 1% 2% 5% 10% 20%
S H M U H M U H M U H M U H M U H M U H M U H M U H M H M S
hPCNA
hMLHI
B. Percentage SW48 DNA Controls
1 c 0% 2% 4% 6% 8% 10% 15% 20% 100% Buffer Enz cÎ55
fO00 H M U H M U H M U H M U H M U H M U H M U H M U H M U H M H M
ro00
— ^ w * . V . ^ hMLHI
Fig.3.5 hMLHI promoter méthylation assay of Raji/SW48 DNA titrations.Different amounts of SW48 DNA (methylated hMLH^ promoter) were mixed with Raji DNA. A. 2ng total DNA, single PCR. B. 100pg total DNA, nested PCR. The DNA mixture was digested as discribed in Chapter 2 and amplified by PCR. H, Hpall digested. M, MspI digested. U, undigested. Controls: Blank, no DNA added to PCR; Buffer, digest without enzyme used as template; Enz, digest withour buffer used as template for PCR.

APL 3 APL 4 APL 8 Raji*
SW48
H M U H M U H M U H M U H M U
— hPCNA
mm hMLHI
B.APL 3 APL 4 APL 8 Raji
*SW48
H M U H M U H M U H M U H M U
hMLHI
BDl BD2 BD3 BD4 BD5 Raji*
SW48
H M U H M U H M U H M U H M U H M U H M U
♦ hMLHI
D. ALL 10 ALL 11 Raji*
SW48
H M U H M U H M U H M U
hMLHI
Fig. 3.6 Examples of the hMLHI promoter méthylation assay of clinical control samples.DNA aliquots were digested with either Hpall (H) or MspI (M) or left undigested (U). The cell lines SW48 (methylated hM LHI promoter) and Raji (non- methylated) were used as controls. The hMLHI fragments described in Figure3.3.A were amplified as described in Chapter 2. An hM LHI PCR product following digestion with Hpall, but not with MspI, is diagnostic for promoter méthylation provided both Hpall and MspI digestion abolish the PCNA PCR product. Samples containing a methylated hMLHI promoter are marked (*). A. de novo APL multiplex PCR; 8. da novo APL samples hMLHI nested PCR; C. blood donor samples single hMLHI PCR; D. da novo ALL single hMLHI PCR
112

ALL (Fig. 3.6.D) samples was methylated at the sites analysed, indicating that less
than 10% of the DNA was methylated. More than 90% of the DNA from these
control samples was therefore negative ior hM LHI promoter méthylation.
3.2.7 hMLHI promoter methvlation analysis of t-AL/MDS samples
The t-AL/MDS samples described earlier were examined for hMLH^
promoter méthylation. Each sample was analysed by the sensitive nested PCR
assay. The more recently collected samples (2, 10, 21-25), which were of better
quality and quantity, were also analysed by single multiplex PCR (not shown). In
cases where both types of analysis were used congruent results were obtained.
Examples of the analysis of the t-AL/MDS samples are shown in Fig. 3.7, the
remaining examples can be found in the Appendix. Evidence of hM LH I promoter
méthylation was seen in six t-AL/MDS cases (2, 4, 5, 14, 16, 23) (Table 3.3).
Table 3.3 MSF and hMLHI méthylation in t-AL/MDS patients.
Case Primarytumour
Therapy M sr hMLHIméthylation
HUNHL MOPP, ABVD + -
37 MOPP, ABVD, MiCMA, RT +9 COPP, RT + -
13 MOPP, ABVD + -
20 Fludarabine, CIb + -
4 Breast CTX, Epirubicin, 5-FU, Tamoxifen, Pt, VP16, APBSCT
+ M
5 5-FU + M6 CTX, Epirubicin, 5-FU, RT + -
8 CTX, Epirubicin, MTX, Mitoxantrone, MMC, RT - -
11 CTX, RT - -
12 Tamoxifen + -
18 CTX, 5-FU, Methotrexate, Tamoxifen + -
19 RT - -
21 CTX, 5-FU, Mitoxantron, Tamoxifen, RT (50Gy) + -
23 RT (breast) + M24 RT - -
22 Others 5-FU, RT - -
25 RT - -
1 RT(;^;i) - -
10 RT(^^ l) - -
2 CisPt, Epirubicin, Paclitaxel + M14 CTX - M15 CTX + -
16 RT + M17 VCR, Doxorubicin, CTX, Melphalan + -
M, methylated:
113
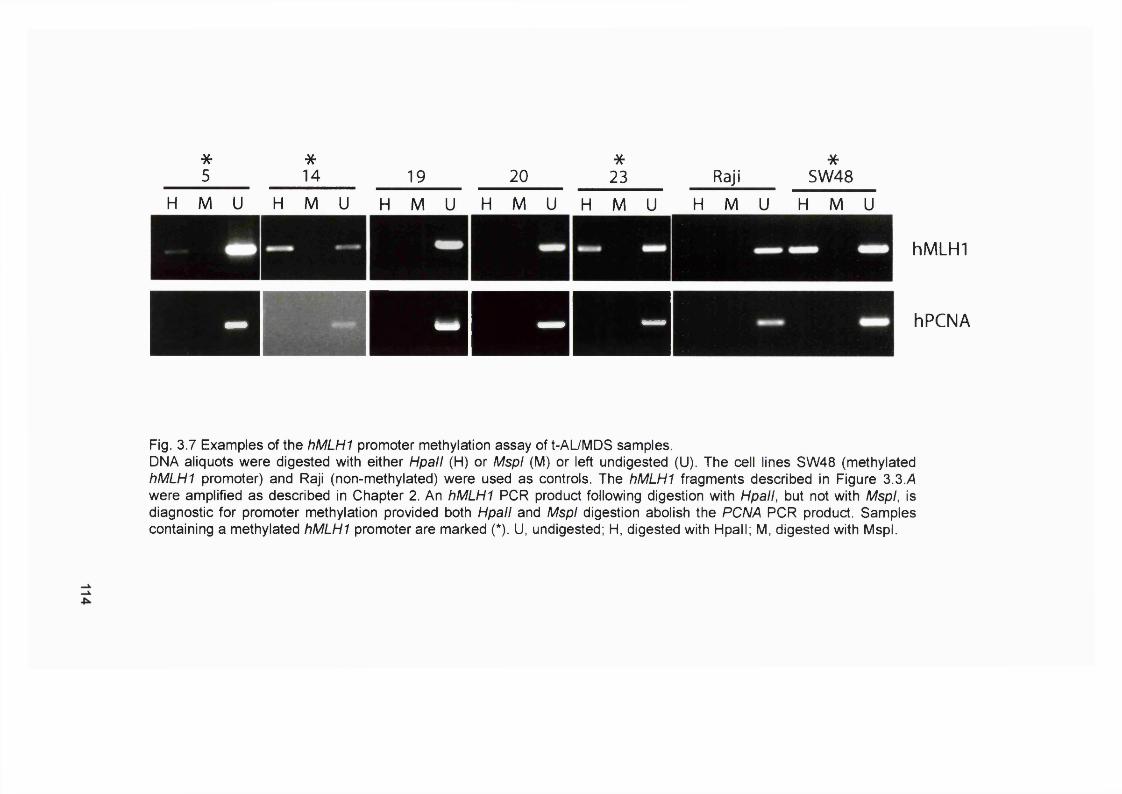
*5
*14 19 20
*23 Raji
*SW48
H M U H M U H M U H M U H M U H M U H M U
hMLHI
hPCNA
Fig. 3.7 Examples of the hMLHI promoter méthylation assay of t-Al_/MDS samples.DNA aliquots were digested with either Hpall (H) or MspI (M) or left undigested (U). The cell lines SW48 (methylated hMLHI promoter) and Raji (non-methylated) were used as controls. The hMLHI fragments described in Figure 3.3.A were amplified as described in Chapter 2. An hMLHI PCR product following digestion with Hpall, but not with MspI, is diagnostic for promoter méthylation provided both Hpall and MspI digestion abolish the PCNA PCR product. Samples containing a methylated hMLHI promoter are marked (*). U, undigested; H, digested with Hpall; M, digested with MspI.

Raji and SW48 DNA were always included as negative and positive controls
for hM LH I méthylation. Case 5, 14 and 23 were positive for promoter méthylation at
the sites examined, and amplification was observed after Hpall digestion (H-lane)
(Fig. 3.7). Case 19 and 20 are shown as examples of samples negative for hM LH I
promoter méthylation. Five of the méthylation positive samples were MSF. The
proportion of t-AL/MDS cases that are MMR defective due to silencing of hM LH I by
promoter méthylation is about 30%, much lower than the frequency in other
malignancies.
3.2.8 Optimization of MMR RT-PCR
RT-PCR offers an alternative way to investigate gene expression. mRNA
levels of hM LH I, hPMS2, hP M S I, hMSH2 and hMSH6 were examined by this
method. The assay used was adopted from Wei et al. (Wei et al., 1997). RNA was
purified and transcribed into cDNA using random hexamers. Fragments of easily
distinguishable length of each target gene were amplified from the cDNA using the
primers described in Chapter 2 (Fig. 3.8.A-F). p-actin was used as an internal
control and to monitor possible DNA contamination (Fig. 3.8.A). The 621 bp
fragment of p-actin spans the exon4/intron 4/5 (95 bp)/exon 5/intron 5/6 (112
bp)/exon 6 boundaries. If the cDNA amplified was contaminated with genomic DNA,
the PCR product would also contain an additional 828 bp product containing introns
4/5 and 5/6, which would be visible on an agarose gel.
A series of control experiments was carried out to optimize conditions.
Firstly, each fragment was amplified independently from Raji cDNA (Fig. 3 .8 .G).
Secondly, all fragments were amplified as a multiplex PCR using cDNA from
different control cell lines (Fig 3.8./-/). Raji was used as a positive control. Three
MMR defective colorectal carcinoma cell lines served as negative controls. To
obtain similar levels of amplification from each gene, the concentration of each
primer pair in the PCR mix was adjusted to the optimal concentration previously
determined in several primer titration assays (not shown). All five MMR fragments
and p-actin were amplified from Raji cDNA. The hMLHI product was absent from
SW48 - consistent with the known silencing of the hMLHI promoter in these cells
(Kane et al., 1997). Similarly, no hMSH2 product was recovered from LoVo cDNA.
LoVo cells contain a homozygous hMSH2 deletion (Umar et al., 1994). The DLD-1
cell line contains two mutations in the hMSH6 gene (Papadopolous et al., 1995).
115

PCR primers
A p-actin 621 bp
1 exon 4 I--..... - 1 exon 5 1- .....- | exon 6 :95 bp 1 1 2 bp
RhMSH2 429 bp 4-
1 ATG exon 1 1 exon 2 1 exon 3 I
C. hPMS2 389 bp ^
1 exon 1 1 1
D. hMSH6 288 bp ^
1 exon 4
E hMLHI 215 bp ^
I exon 5 I exon 6 I exon T
EhPMSI ^ 1 74 bp^
I ATG exon 1| exon 2 1
116

G. Primers H.Cell Line
(U
m
c. rN r \i vOV-> X to X _ c V IuTO1
un V I — IQ_ o_
CO. _ c _ c _ c _ c
00
I
p-actin
hMSH2hPMS2hMSH6hMLhlhPMSl
fOGC
00
p-actin
hMSH2hPMS2hMSH6hM Lhl
Fig 3.8 Optimisation of the MMR RT-PCR.RNA was transcribed into cDNA and amplified by PCR as described in Chapter 2. Products were separated by agarose gel electrophoresis, stained with ethidium bromide and visualized under UV. A-F. Diagrams of gene regions amplified. G. Separate PCRs. Raji cDNA was amplified with each of the five sets of primers separately. H. Multiplex PCR of control samples. cDNA from control cell lines Raji (MMR proficient). SW48 (hMLHI deficient). LoVo (hMSH2 deficient). DLD-1 (hMSH6 deficient) was amplified as a multiplex PCR of all five target genes. /. Nested RT-PCR. I pi of the Raji and SW48 multiplex PCR each were amplified using nested sets of primers.
117

Although RT-PCR under these conditions is not quantitative, I consistently observed
a reduced level of hMSH6 amplification from this cell line (Fig. 3.8 .H). This is
consistent with a reduced level of hMSH6 mRNA in DLD-1. This approach
confirmed normal levels of hMLHI transcription in a series of cisplatin-resistant
variants of A2780 ovarian carcinoma cells (Massey et al., 2003).
In an attempt to increase the sensitivity of the assay, a set of nested primers
(Table 2.2) was designed and I pL of the SW48 and Raji multiplex MMR RT-PCR
each were amplified using these primers. Fig 3.8./ shows that nested PCR revealed
low levels of hMLHI transcription even in the ‘fully methylated’ SW48 cells. This can
be explained in two ways. Méthylation silencing of hMLHI may be leaky and a very
small amount of the transcript is detected by this highly sensitive nested RT-PCR
approach. Alternatively nested RT-PCR may detect a small sub-population of
‘revertant’ SW48 cells that express normal levels of hM LHI. These two possibilities
are not mutually exclusive. I decided that this extreme sensitivity and the
concomitant risk of false positive signals was not an advantage in analysing the
clinical samples.
3.2.9 MMR gene expression in AML samples
To examine the feasibility of RT-PCR analysis with the clinical samples, RNA
prepared from two de novo AML cases was analysed. The RNA from these cases
was of good quality and I considered that it was likely to be superior to that from the
therapy-related cases. Figure 3.9.A shows that even though the assay worked well
with RNA purified from cell lines, it does not amplify the different MMR gene
transcripts consistently using mRNA from clinical samples. For example, I could
amplify p-actin, hPMS2 and hMSH6 from AML-1 RNA, but I could not amplify any of
the cDNA fragments from AML-2 RNA. The reasons for this are not known, but may,
for example, reflect degradation of some of the RNA molecules due to inadequate
storage before or after extraction.
I obtained cDNA from patients 5, 8, 11, 17, 18, 21, 24 and 25. I wanted to
assay these samples with the highest sensitivity possible, but without increasing the
risk for false positives by using nested PCR. These cDNAs were therefore amplified
by a single PCR, but for 50 cycles. Unfortunately this protocol did not improve the
conditions compared to the nested PCR with regards to false positives, nor did it
118
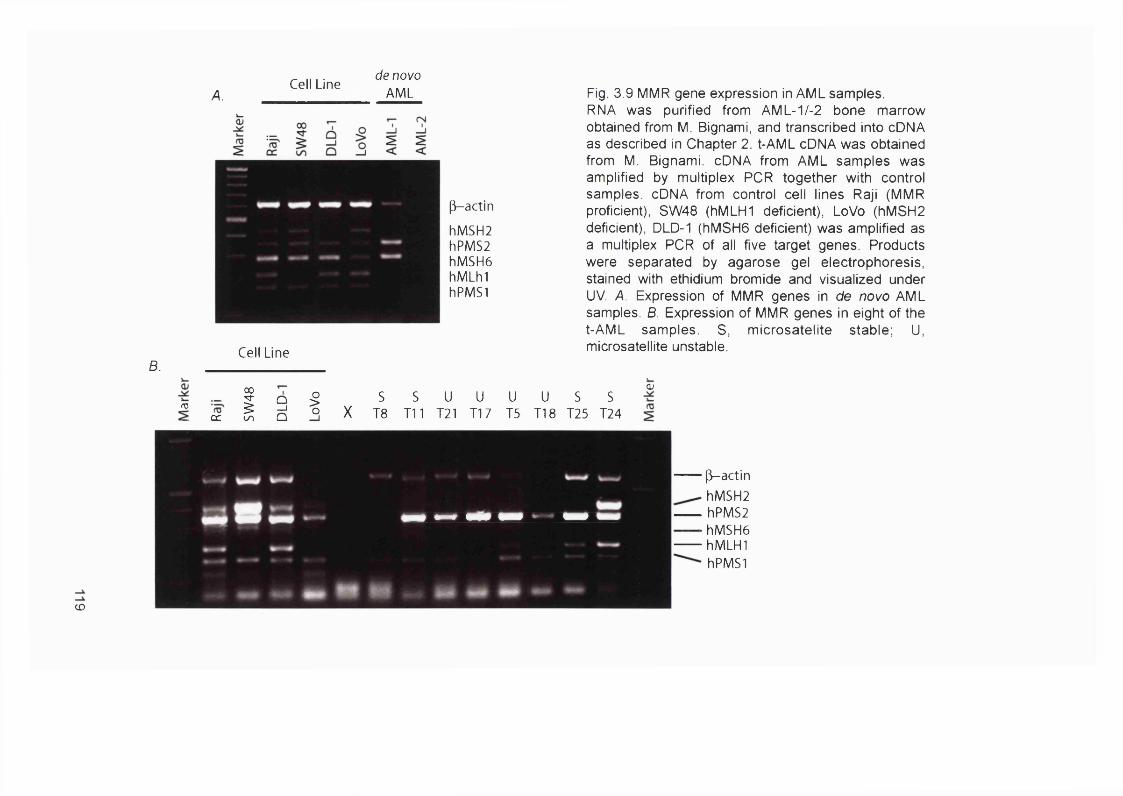
A.Cell Line
de novo AML
p-actin
hMSH2hPMS2hMSH6hMLhlhPMSI
B.Cell Line
Fig. 3.9 MMR gene expression in AML samples.RNA was purified from AML-1/-2 bone marrow obtained from M. Bignami, and transcribed into cDNA as described in Chapter 2. t-AML cDNA was obtained from M. Bignami. cDNA from AML samples was amplified by multiplex PCR together with control samples. cDNA from control cell lines Raji (MMR proficient), SW48 (hMLHI deficient), LoVo (hMSH2 deficient), DLD-1 (hMSH6 deficient) was amplified as a multiplex PCR of all five target genes. Products were separated by agarose gel electrophoresis, stained with ethidium bromide and visualized under UV. A. Expression of MMR genes in de novo AML samples. B. Expression of MMR genes in eight of the t-AML samples. S, microsatelite stable; U, microsatellite unstable.
(T3ÛC
00
sunQ_ jQ I X
sT8
S U T il 121
UT17
U15
U S S TIB T25 T24
(U
CO
p-actinhMSH2
hPMS2■ hMSH6
hMLHIhPMSI

ensure reproducible amplification of the p-actin positive control. The results obtained
can therefore only be used as an indication.
The problems with this assay are illustrated in Fig. 3.9.6. Cases 8, 11, 25
and 24 are microsatellite stable, whereas 5, 17, 18 and 21 are unstable. Case 5 was
furthermore shown to have a methylated hM LH I promoter. The p-actin cDNA, used
as a positive control, could not be amplified from any of the samples.
hM S H 6 cDNA could not be amplified in any of the clinical samples, and
hM SH2 could only be amplified from sample 24. In contrast, cDNA fragments from
both genes were amplified from controls.
hMLHI cDNA was amplified from all of the control samples, including
hMLHV S\N4S, and from cases 24 and 25 cDNA, which are both MSI'. No hMLHI
signal could be detected from the other AML samples apart from case 5, a patient
that was both shown to be MSF and have a methylated hM LHI promoter.
hPMS2 cDNA was amplified from all samples, including the controls, apart
from case 8. The ease and reproducibility of hPMS2 amplification in the other
samples suggest that this finding for case 8 may be valid.
3.3 Discussion3.3.1 M S r frequency among t-AUMDS
Cancer therapy clearly plays an important role in the development of
secondary leukaemia with defective MMR. Since we started this project a number of
studies of MSI among t-AML cases have appeared (Table 3.4). The reported
frequencies for MSI lie between 0 and 94% with an average of about 40%. The
variation is probably influenced by factors such as primary cancer and type of
treatment. In the absence of a generally accepted methodology for MSI
determination and an agreed definition of MSI in leukaemia, a contribution of
different methods of analysis cannot be ruled out, however. Notwithstanding this
reservation, there is a clear consensus that the M SF phenotype is much more
common in t-AL/MDS than in de novo AML. Our data fully support this conclusion.
Almost two-thirds (64%) of our therapy-related cases were MSF compared to none
of 28 de novo cases.
120
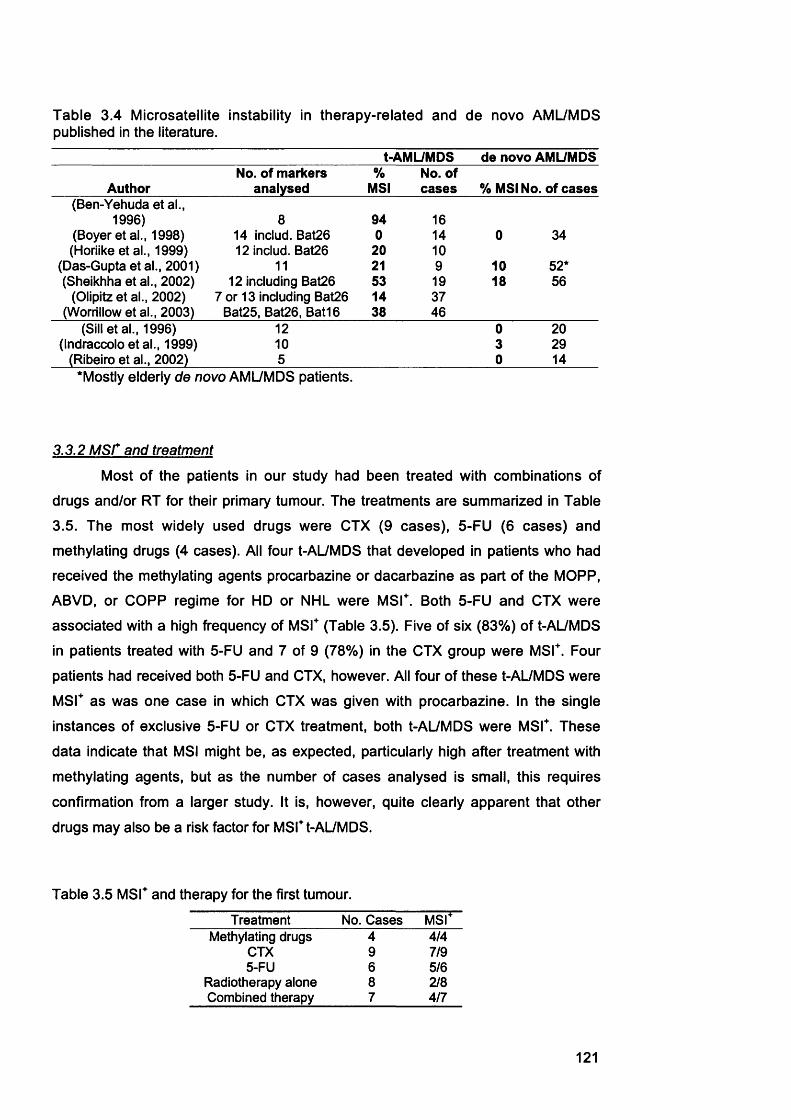
Table 3.4 Microsatellite instability in therapy-related and de novo AML/MDS published in the literature.
t-AMUMDS de novo AML/MDSNo. of markers % No. of
Author analysed MSI cases % MSI No. of cases(Ben-Yehuda et al.,
1996) 8 94 16(Boyer et al., 1998) 14 includ. Bat26 0 14 0 34(Horiike et al., 1999) 12 includ. Bat26 2 0 1 0
(Das-Gupta et al., 2001) 1 1 2 1 9 1 0 52*(Sheikhha et al., 2002) 12 including Bat26 53 19 18 56
(Olipitz et al., 2002) 7 or 13 including Bat26 14 37(Worrillow et al., 2003) Bat25, Bat26, Bat16 38 46
(Sill et al., 1996) 1 2 0 2 0
(Indraccolo et al., 1999) 1 0 3 29(Ribeiro et al., 2002) 5 0 14
'Mostly elderly de novo AML/MDS patients.
3.3.2 M S r and treatment
Most of the patients in our study had been treated with combinations of
drugs and/or RT for their primary tumour. The treatments are summarized in Table
3.5. The most widely used drugs were CTX (9 cases), 5-FU (6 cases) and
methylating drugs (4 cases). All four t-AL/MDS that developed in patients who had
received the methylating agents procarbazine or dacarbazine as part of the MOPP,
ABVD, or COPP regime for HD or NHL were MSF. Both 5-FU and CTX were
associated with a high frequency of MSF (Table 3.5). Five of six (83%) of t-AL/MDS
in patients treated with 5-FU and 7 of 9 (78%) in the CTX group were MSF. Four
patients had received both 5-FU and CTX, however. All four of these t-AL/MDS were
M S r as was one case in which CTX was given with procarbazine. In the single
instances of exclusive 5-FU or CTX treatment, both t-AL/MDS were MSF. These
data indicate that MSI might be, as expected, particularly high after treatment with
methylating agents, but as the number of cases analysed is small, this requires
confirmation from a larger study. It is, however, quite clearly apparent that other
drugs may also be a risk factor for MSF t-AL/MDS.
Table 3.5 MSF and therapy for the first tumour.
Treatment No. Cases MsrMethylating drugs 4 4/4
CTX 9 7/95-FU 6 5/6
Radiotherapy alone 8 2 / 8
Combined therapy 7 4/7
121

In this regard, it is of note that combination therapy (RT + chemotherapy)
was associated with a higher frequency of M S r than RT alone. Of seven t-AL/MDS
in patients treated with combination therapy, four (57%) were M S r. In contrast,
there were only two M S r cases in the RT group (25%). This suggests that for M S r
t-AL/MDS, RT is relatively low-risk. The probability of M S r t-AL/MDS is notably
increased by drug treatment.
3.3.3 CvcloDhosDhamide and t-AML
CTX is commonly used to treat a wide range of malignancies. CTX
exposure was also highly represented among our group of t-AL/MDS patients (Table
3.1). CTX is activated to 4-HC by P450 in the liver. 4-HC can then tautomerise to
aldophosphamide, which fragments to acrolein and PM (Lind and Ardiet, 1993)
(Section 1.1.6 and Fig. 1.6).
Lee et al. showed that concentrations of acrolein similar to serum levels after
high dose CTX treatment, but not of CTX inhibit recombinant MGMT in vitro. PM
also inhibited the enzyme but only at much higher concentrations than are used to
treat patients (Lee et al., 1992). If acrolein inhibits MGMT in vivo, it would function
like 0®-BzG when given in combination with méthylation agents. Acrolein would
deplete MGMT activity and thereby increases the effective alkylating agent dose.
This effect would be most pronounced if CTX was given in combination with a
methylating agent such as procarbazine as it is, for example, in the COPP protocol.
Under these conditions, acrolein might serve to enhance the leukaemogenicity of
the methylating drug.
I carried out a preliminary investigation of the effect of acrolein on MGMT
activity. The MGMT activity of cultured cells was reduced by a 4 to 8 hour treatment
with 30 - 60 pM acrolein (data not shown). The decrease in MGMT activity was both
time and drug dose dependent: 60pM acrolein for 8h provided the maximum
inactivation. It is not clear whether this inhibitory effect is due to a direct inactivation
of MGMT by acrolein or requires the production of a DNA lesion that is repaired by
MGMT by an autoinactivating mechanism. Whatever the mechanism, it would be
interesting to investigate if methylating agent treatment in combination with acrolein
can potentiate the effect of the methylating agent.
122

3.3.4 hMLHI promoter methvlation in t-AUMDS
hM LH I promoter méthylation was infrequent (6/25 or 24%) in t-AL/MDS and
accounts for at most one-third (between 31% and 35%, 5/16 or 6/17) of M S r cases.
Only five of the six cases in which a methylated h M LH I promoter was identified
were M S r. Patient 14, who had been designated MSI* based on Bat26 only, also
appeared to have a methylated promoter. This apparently paradoxical promoter
méthylation may result from intra-tumour heterogeneity or from méthylation of a
single h M L H I promoter, as the assay is not quantitative and a positive signal is
produced if only a minor fraction of the DNA is methylated. In case 14, Bat26 may
simply have been uninformative and it may be MSI". I note in this connection that
one other case (case 8) was subsequently reassigned unstable based on a more
detailed molecular analysis (Chapter 5). Bat26 is known to be occasionally
uninformative in colorectal carcinomas.
Deng et al. showed that the region of the h M L H I promoter analysed using
this assay can be partially methylated without silencing of the hM LH I gene. They
nevertheless found a correlation between full méthylation and gene silencing (Deng
et a!., 1999). The region from -2 4 8 to -1 7 8 (8 CpG) sites seemed to be the most
indicative of silencing, but does not contain any Hpall sites. This could be a further
explanation for the MSI and hM LH I promoter méthylation results. It is not possible
to distinguish between these possibilities using my analysis.
The frequency of about 30% obtained for h M L H I promoter méthylation
represents an upper limit as this sensitive method can detect even a very minor
population of méthylation positive leukaemic cells. Even though this value is
considerably lower than the frequency (>80% ) among sporadic MSI" colorectal
tumours (Herman et al., 1998), it is higher than the frequency among t-AML cases
reported by others. Sheikhha et al. identified one methylated case among eight MSI"
t-AMLs. Seedhouse et al. did not identify any méthylation among 23 t-AML cases.
This apparent discrepancy may simply reflect the higher sensitivity of the assay I
used and its ability to detect very low levels of methylated sequences.
123

3.3.5 hMLHI promoter methvlation in t-APL
The M S r frequency among the t-APL cases was 3/5 (60%), similar to the
overall frequency of 64%. The incidence of h M L H I promoter méthylation was,
however, strikingly higher among the MSrt-APLs (Table 3.6).
Table 3.6 M S r and hM LHI méthylation in therapy-related and de novo APL.
t-APL Primarytumour Therapy MSI" hMLHI
méthylation
T2 ovary CisPt, Epirubicin, Paclitaxel
+ M
T10T16
thyroiduterus
RT(^^ I)RT + M
T23 breast/colon RT (breast) + MT24 breast RT - -
APL 1-8 de novo - - -
Méthylation was observed in all (3/3) M S r t-APLs (cases 2, 16, and 23).
This contrasts with only one fifth (15% (2/13) or 21% (3/14)) of the remaining MSI"
cases. None (0/8) of the de novo APL cases were MSI" and none contained a
methylated h M L H I promoter (Figure 3.6.A /8). Although this observation requires
confirmation from a larger number of patients, the possibility that t-APL is associated
with a high frequency of hM LHI promoter méthylation is consistent with the known
derangement of epigenetic gene regulation in APL. Pelicci and colleagues
demonstrated that the PM L-R A R a fusion (Section 1 .6 .1) induces gene
hypermethylation and silencing by recruiting DNA methyltransferases to target
promoters. This hypermethylation contributed to the leukaemic potential of the PML-
R AR a fusion (Di Croce et al., 2002). A possible scenario for the development of
MSI" t-APL could be as follows: The initial event is the translocation between
chromosomes 15 and 17. This would result in a general ‘méthylation phenotype' as
a result of which a large number of genes is switched off. Silencing of the hMLHI
promoter would then result in a ‘mutator phenotype’ which, in addition to the
‘méthylation phenotype’, would further accelerate leukaemogenesis.
3.3.6 Gene defects in other MMR aenes in t-AL/MDS
The RT-PCR assay was developed to provide a direct assessment of the
expression of the MMR genes. Unfortunately unresolved problems remain with this
analysis. Low RNA/cDNA quality and quantity did not allow reliable amplification of
t-AL/MDS samples. The results provided only an indication of gene expression.
124

Eight of the t-AL/MDS cases together with the control cell lines were
analysed by RT-PCR. hMLH1 cDNA was amplified from all of the control samples,
including SW48, which is hMLHV due to a methylated hMLH1 promoter. The only t-
AL/MDS samples that resulted in visible hMLH1 amplification was case 5. This
patient though had been shown to be both M S r and have a methylated hMLH1
promoter. The fact that both SW48, the negative control for hMLH1, and case 5 are
hMLH1 positive indicates that this RT-PCR assay is probably not informative for loss
of expression by hMLH1 méthylation at this high sensitivity.
hPMS2 cDNA was amplified from all samples including the controls apart
from case 8 which had previously been assigned stable, but by using Bat26 only.
More detailed molecular analysis (Chapter 5) of this case at a later time led to a
reassignment of case 8 as unstable. As hPMS2 was amplified from all samples, and
p-actin and hPMS1 could be amplified from case 8, it seems possible that case 8 is
hPMS2 deficient. Inactivation of hPMS2 is encountered infrequently among HNPCC
families and sporadic M S r tumours. If these data are confirmed, case 8 would be to
my knowledge, the first example of loss of hPMS2 in M S r t-AL/MDS.
A recent study reported an increase in incidence of an intron splice acceptor
polymorphism in hMSH2 among t-AML cases (Worrillow et al., 2003). They
identified two of thirteen MST t-AML cases that were homozygous for this
polymorphism compared to an incidence of homozygosity of 1.6% among the
general population. They found one M S r case that was heterozygous for this
polymorphism compared to the frequency of heterozygotes of 12% among the
general population Together with Karen Gascoigne, I identified this polymorphism in
two of the M S r t-AL/MDS cases (cases 5 and 6; 2/16,12.5% ). Both cases were
heterozygous (data not shown). As there was no evidence for an overrepresentation
of this polymorphism among our t-AL/MDS samples it is not clear to what degree
this hMSH2 variant is involved in leukaemogenesis after drug treatment.
3.3.7 t-AUMDS and primary breast cancer
The largest subgroup - 11 patients - had been treated for a primary breast
cancer. M S r in this group (7/11, 64%) and the frequency of hMLH1 promoter
méthylation among the M S r cases (3/7, 42%) were similar to the overall incidences
(64% and 35%, respectively). There appeared to be a trend towards early onset
breast cancer among these cases.
125

The Gruppo Italiano Malattie Ematologiche dell'Adulto (GIMEMA) Archive of
Adult Acute Leukaemia yielded information on 37 more primary breast cancer
patients who had been excluded from the MSI / promoter méthylation study because
no DNA was available. The study population of the GIMEMA Archive comprised
patients with newly diagnosed acute leukaemia registered in 62 Italian Haematology
Divisions during the period July 1992 to June 1996 (Pagano et al., 2001). Inclusion
of the age of onset data for these additional patients supported the apparent bias
towards early primary disease. Fig. 3.10 compares the age at diagnosis of breast
cancer for the patients in the GIMEMA Archive (48 cases total) with similar data for
all breast cancer patients from the Cancer Registries of six Italian provinces (Zanetti
et al., 1992). Only 6% of registry cases were diagnosed between 20 years and 39
years. For t-AL/MDS patients, the corresponding figure was 24%. Overall, more
than half (54%) of the breast cancer cases in the GIMEMA group occurred at <50
years. The corresponding value for patients in the registries was 23.5%. The data
from the GIMEMA Archive and the cancer registries were supplied by Ida Casorelli
and Luca Mele.
Interestingly, a number of patients who developed acute leukaemia either
simultaneously or several years after surgical removal of breast cancer were
described by Rosner et al. and Carey et al. (Carey et al., 1967; Rosner et al., 1978).
Several surgery only' AML cases after breast cancer also appear in the GIMEMA
Archive. These studies indicate a thirty-fold higher risk for secondary AML in breast
cancer patients than in the general population. Taken together, these reports point
towards a possible involvement of one of the breast cancer susceptibility genes
{BRCA1 and 2) in the development of secondary AML. This possibility is further
supported by recent data showing that BRCA1 and BRCA2 function in the same
pathway as the FA proteins (discussed in Chapter 5).
126

20-29 30-39 40-49 50-59 60-69Age at diagnosis (years)
>70
Fig. 3.10 tAUMDS and primary breast cancer.Age at diagnosis of a primary breast tumour in t-AL7MDS patients and in the general population. Light blue bars: breast cancer cases (6664 total) retrieved from the cancer registries of six Italian provinces (Torino, Genova, Parma, Firenze, Latina and Ragusa) (1983 - 1987) (Zanetti, et al., 1992). Dark blue bars: breast cancer patients in the GIMEMA Archive, 48 cases total.
127

3.4 ConclusionOur study shows that MSI is high among t-AL/MDS cases (>60%). Loss of
hMLH1 expression due to promoter silencing was found to be relatively infrequent
and to accounts for at most 30% of the MSI cases. Many questions remain,
however. Foremost among these is the mechanism by which MMR is inactivated in
the other MSF cases. With improved clinical sample collection and handling, it
should be possible to address these issues using more informative approaches
such as real-time RT-PCR and bisulfite analysis of promoter méthylation. With these
more incisive methods - which are crucially dependent on good quality DNA and
RNA - it will be possible to investigate the mechanism of loss of MMR and to further
clarify the connections between specific drugs and possible genetic predisposition
and the development of t-AML/MDS.
128

Chapter 4: Results II
The role of thiopurine resistance and defective DNA mismatch repair in the development of acute myeloid leukaemia after organ transplants
4.1. IntroductionAza Is part of the triple immunosuppressive therapy following solid organ
transplants, and it is also used, either as the sole agent or in combination with other
drugs, for autoimmune diseases (Appendix Table 4.7). Aza Is a pro-drug that is
converted in vivo to 6-MP. Both 6-MP and 6-TG are taken up by target cells,
metabolized to 2’-deoxy-6-thioguanosine triphosphate via the hypoxanthine guanine
phosphoribosyl transferase (HGPRT) and incorporated as thioguanine into DNA
(Fig. 1.7) (Aarbakke et al., 1997). Both drugs are metabolised by a slightly different
route. 6-TG is directly converted to 6-thioguanosine-5’-monophosphate (6-TGMP).
6-MP, on the other hand, is first converted to 6-thioinosine-5’-monophosphate (6-
TIMP), which is then converted to 6-TGMP in two steps by inosine monophosphate
dehydrogenase (IMPD) and guanosine monophosphate synthetase (GMPS) (Fig.
1.7).
The delayed cytotoxicity and chromosome damage that are characteristic of
these drugs are associated with their incorporation, and these properties involve
MMR (Swann et al., 1996) (discussed in more detail in Section 1.4.2). Cells avoid
the cytotoxicity of 6-TG by the same mechanism as for 0®-meG, inactivation of
MMR. Thus, MMR-deficient cells can tolerate 6-meTG in their DNA and are resistant
to killing by TG. As functional MMR is also required for the cytotoxicity of
methylating agents, 6-TG resistant cells exhibit cross-resistance to methylating
agents like MNU (Aquilina et al., 1990; Green et al., 1989).
Patients with low thiopurine methyltransferase (TPMT) (Fig. 1.7) activity
cannot catabolize thiopurines efficiently. When they are treated with thiopurines,
they accumulate abnormally high levels of the thioguanine nucleotides. These
patients have a significantly increased risk of t-AML/MDS following thiopurine
treatment (Thomsen et al., 1999) suggesting that thiopurine-induced DNA damage
may contribute to cancer development.
Interested in identifying a potential link between Aza treatment and
leukaemia, I started searching the literature for a possible connection. More than 30
publications reported the occurrence of AML after thiopurine treatment, either for
129

autoimmune diseases (Appendix Table 4.7.A) or after organ transplants (Appendix
Table 4.7 .8). All of these patients had been treated with Aza, with the exception of
two cases that had been treated with 6-Mercaptopurine. In most cases Aza was
administered in combination with other drugs. Interestingly, in more than half of the
AML cases for which the karyotype was described, there was an involvement of loss
of all or part of chromosomes 5 and/or 7, which is also very common among
alkylating agent-related AML. The large number of these reports and the
involvement of loss of chromosomes 5 and/or 7 was taken as an indication that
there might be a connection between thiopurine treatment and AML.
This led us to suggest the following hypothesis. If a patient is treated for a
prolonged period of time with the myelotoxic drug Aza, the selective pressure for
loss of MMR would be high in the bone marrow. This MMR defective cell would
have a growth advantage and would undergo clonal expansion. It would have a
mutator phenotype and rapidly accumulate the mutations necessary to give rise to
M S r myeloid leukaemia. The hypothesis was tested by several approaches.
1. Cells were treated with 6-TG in vitro to examine whether this selected for
MMR defective clones.
2. The incidence of AML after Aza treatment was investigated.
3. AML samples from patients treated with Aza for organ transplants were
assayed for MSI.
MMR defective clones can be isolated by either chronic or acute methylating
agent treatment. The A2780-MNUcl1 subclone of the human ovarian carcinoma cell
line A2780 was isolated by MNU treatment (see below) (Aquilina et al., 2000).
A2780 cells are MMR proficient and p53 positive. A2780-MNUcl1 exhibited a 100-
fold increased resistance to MNU and a high degree of cross-resistance to 6-TG. It
was additionally shown that the p53-dependent DNA damage response was
defective in this subclone due to a dominant negative p53 mutation. It seems likely
that this resistant variant was a member of a pre-existing drug-resistant
subpopulation {hMLH1' and p5S) within A2780.
To mimic the selective pressure experienced by bone marrow cells of organ
transplant recipients tissue culture cells were treated with increasing concentrations
of 6-TG for a prolonged period (Fig. 2.1), using a protocol similar to that used by
Aquilina et al. to select for 6-TG resistant CHO cells (Aquilina et al., 1990). Cells can
become resistant to 6-TG by losing HGPRT. This enzyme is central to the purine
130

salvage pathway that incorporates 6-TG into DNA (Stout and Caskey, 1985). To
avoid selecting a pre-existing H G PR T clone, A2780-SCA cells were first treated
with HAT (hypoxanthine, aminopterin and thymidine) for 10 days. Aminopterin
blocks de novo nucleotide synthesis. It kills all cells that do not have purine or
pyrimidine salvage pathways.
4.2. Results4.2.1 Generation of clonal variants of A2780 resistant to 6-TG
The A2780 cell line was used to investigate the development of thiopurine
resistance in vitro. They are widely-used as a model for drug resistance. As this cell
line harbours a sub-population of cells defective both in hMLHI and p53, a single
cell clone (A2780-SCA) previously isolated by Andrew Massey in this laboratory,
was used. A2780-SCA cells are proficient in both hMLHI and p53 (Massey et el.,
2003).
Preliminary experiments showed that A 2780-SC A are sensitive to
aminopterin. Cultures were then treated with HAT. After 10 days the HAT treated
cells were divided into five flasks, the HAT medium was removed and each flask
was treated with escalating concentrations of 6-TG for 2 months (Fig. 2.1). The cells
were exposed continuously to the drug, and were allowed to recover and repopulate
the flask after each increase in dose. After 2 months of continuous 6-TG treatment
the cell populations as a whole grew normally in to 1 pg/mL 6-TG.
To isolate single cell clones 2, 4 or 6 cells per well were inoculated into 96-
well plates in medium containing HAT. The plating efficiency was between 50 and
83% (Table 4.1), which is similar to A2780-SCA. This indicates that the 6-TG
resistance of the five cell populations was not mainly due to loss of HGPRT. The
HAT selection at the end of the cloning procedure ensured that any 6-TG resistant
clones picked for further analysis had functional HGPRT.
Approximately 30 single cell clones from each of the five 6-TG resistant
cultures were screened for resistance to MNU. Resistance to methylating agents
like MNU is a common characteristic of MMR defective cells (Bignami et al., 2000;
Hampson et al., 1997). As an indicator of a possible MMR defect, each clone was
assayed for cross-resistance to MNU. Because A2780 cells express normal levels
of MGMT, all MNU treatment was performed in the presence of the MGMT inhibitor
131

0®-BzG. Each HAT-resistant clone was split into two wells in a 24-well plate. 0®-bzG
was added to one well to inactivate MGMT followed by MNU after 2h. The second
well was left untreated, so that these cells could be used for further analysis. Most
clones (140/154) did grow after MNU treatment. Clones (not treated with MNU) were
picked from the independent resistant cultures each and expanded for further
analysis.
Table 4.1 Plating efficiency of A2780-JA clones in HAT medium.
PE% Average PE %JA1 80.2
2/well 864/well 96.86/well 57.8JA3 83.4
2/well 53.44/well 96.86/well 100JA4 59.6
2/well 51.94/well 73.96/well 53JA5 45.8
2/well 43.84/well 56/well 43.6JA8 47
2/well 46.34/well 36.86/well 57.8
PE, Plating efficiency.
The clones were named A2780 after the parental cell line. JA indicates the
selection protocol described above. The first digit specifies the flask the clone
originated from and the second digit describes the number of the clone isolated from
each flask. A2780-MNUcl1 (see above) was used as a MMR defective control
subclone in the following experiments performed to characterise the isolated clones.
4.2.3 Mismatch repair status of thiopurine resistant cells
MMR gene expression was analyzed in all fifteen 6-TG resistant clones
using the RT-PCR method described and established in Chapter 3. Most expressed
detectable hMSH2, hM LHI, hPMS2, hMSHS, and hPMSI mRNAs. hMLHI mRNA
was absent in clones 5-1, 5-3, 5-4 and 8-3 and diminished in some clones, however
132

(Fig. 4.1). As clones 5-1, 5-3 and 5-4 originated from the same culture they are
possibly siblings originating from the same hM LHI' clone.
Independent clones that did not express measurable hMLHI mRNA were
analyzed further. The hMLHI protein was either not detectable or was present at
severely reduced levels in extracts of all of the clones analysed (Fig. 4.2). It was
present in extracts of parental A2780-SCA cells. hPMS2, the heterodimer partner of
hM LH I, was also undetectable, or barely detectable, in all extracts including JA5-3
and JA8-3. The hPMS2 protein is unstable in the absence of hM LHI (Li and
Modrich, 1995) and this finding is consistent with a primary defect in hM LH I gene
expression. The hMSH2 and hMSH6 proteins were present at similar levels in
extracts from the parental and most of the resistant clones (Fig. 4.2). The absence
of detectable MMR protein in the JA3-5 extract was probably due to general low
protein concentrations in this extract. Similarly, the faint hMSH2 and hMSH6 bands
from JA5-1 and JA5-4 extracts might also be a consequence of low protein levels,
protein degradation or reduced protein transfer during the blotting procedure. An
hMSH6 defect in clone JA5-1 and JA5-4 cannot be excluded without further assays,
however.
Biochemical assays confirmed the MMR deficiency of JA5-3 and JA8-3. The
mismatch repair assay was carried out by Peter Macpherson. Unlike parental
A2780-SCA cell extracts, JA5-3 and JA8-3 cell extracts did not correct a C/T mispair
in a standard substrate (Figure 4.3).
4.2.4 HGPRT status of 6-TG resistant clones
To confirm that the 6-TG resistant subclones retained active HGPRT their
sensitivity to aminopterin was tested. A2780-SCA, -JA5-3, -JA8-3 and -M NUcll
were treated with both HAT and aminopterin at the same concentrations used
during the selection procedure. After drug treatment for 10 days the colonies formed
were stained with GIEMSA (as described in Chapter 2). All four clones grew
normally in HAT, but did not in the presence of aminopterin (Fig. 4.4). This indicates
that this dose of aminopterin efficiently blocks de novo nucleotide synthesis and all
four clones retained an active purine salvage pathways.
133

6-TG-resistant clones" j i ? I g T ,CN m fN ro ro ro
P-actin
hMSH2hPMS2hMSH6hMLHIhPMSI
* *
6 TG resistant clones
P-actin
hMSH2 hPMS2 hMSH6hMLHI hPMSI
^ *
Figure 4.1 Expression of MMR genes in 6-TG resistant clones.RNA from representative 6-TG and MNU-resistant A2780-SCA subclones was amplified by RT-PCR. RNA from control cell lines Raji (MMR proficient), SW48 (hMLHI deficient). LoVo (hMSH2 deficient), DLD-1 (hMSH6 deficient) were included. Products were separated by agarose gel electrophoresis, stained with ethidium bromide and visualized under UV. Clones A2780-JA5-1, -JA5-3 and -JA5-4 (*) arose in the same flask. A2780-JA8-3 (*) was isolated from a different flasks.
134

VO A2780 clones
hMLHI
hPMS2
hMSH2
hMSH6
r— rnLO 00U TO u < <X OC VA
• 4
A2780 clones^ r\l LA
< 1 I If— m m
^ < < <
A2780 clones
hMLHI
hPMS2
hMSH2
hMSH6
lA «—< 4 lÀ lA 00U < < < <lA
A2780 clonesf— rN m (N m
<UlA < <
hMLHI
hPMS2
hMSH2
hMSH6
< i
Figure 4.2 MMR protein expression in 6-TG resistant clones.Extracts from parental A2780-SCA, the control MMR defective cells, and A2780-JA clones were separated by SDS polyacrylamide gel electrophoresis, blotted and probed sequentially with antibody against hMLHI, hMSH2, hMSHB, and hPMS2.
135

um
Figure 4.3 In vitro mismatch correction in A2780-JA5-3 and -JA8-3.Extracts (lOOpg) of HeLa, A2780-SCA, -JA5-3, or -JA8-3 were incubated with a standard nicked circular DNA substrate containing a T:C mispair. DNA was recovered, digested with Mlul and products separated by agarose gel electrophoresis. The band shown arrowed is diagnostic for correction. This assay was carried out by P. Macpherson.
JA5-3 JA8-3 SCA5
IxHAT
MNUcll
#IxA
Figure 4.4 HAT and Aminopterin sensitivity assay of A2780-JA5-3 and -JA8-3. 1x10^ cells were plated per well into medium containing either 1x HAT or 1x A. Cells were stained after 5 - 1 0 days with Giemsa stain. Cell lines: A2780- SCA, -M NUcll, -JA5-3 and JA8-3. A. aminopterin, HAT. hypoxanthine, aminopterin, thymidine.
136

4.2.5 Drug resistance of A2780-JA5-3 and JA8-3
As the MNU screen as well as the MMR analysis suggested that all of the
clones tested were MMR deficient due to loss of hM LH I, two independent clones,
A2780-JA5-3 and -JA8-3 were picked for further analysis. The MNU and 6 -TG
sensitivities of these two clones were determined by clonogenic survival. The MNU
treatment was performed in the presence of 0®-bzG. JA5-3 and JA8-3 were both
highly resistant to MNU and 6 -TG (Fig. 4.5). Their D37 values were estimated 770
pM and 680 pM for MNU in the presence of 0®-bzG and 3.00 and 2.45 for 6 -TG
respectively (Table 4.2). JA5-3 is therefore 39-fold more resistant to MNU and 6.7-
fold more resistant to 6 -TG compared to the parental cell line A2780-SCA. The
resistance is similar for JA8-3, 34-fold for MNU and 5.4-fold for 6 -TG. The small
differences in resistance between the two clones is probably not significant.
Table 4.2 Comparative cytotoxicity of 6 -TG resistant clones.
MNU (pM) 6 -TG (pg/mL)Clone D3 7 RF D3 7 RFSCA 2 0 0.45
JA5-3 770 39 3.00 6.7JA8-3 680 34 2.45 5.4
MNUcll 1600 80 2.35 5.2D 3 7 values (the dose of MNU / 6 -TG required to kill 63% of the cells) were estimated from clonal survival curves. RF, Fold resistance compared to the wild type.
Thiopurines in organ transplant patients are usually administered in
combination with CsA and Pred. To see if the development of AML could also be
due to selection for resistance to CsA and/or Pred the sensitivity of the MMR'
subclones A2780-JA5-3 and -JA8-3 to these two drugs was compared to the MMR
proficient parental cell line. 2 x1 0 cells were plated per well, treated with increasing
concentrations of the drug and stained after 5 - 1 0 days. These assays show that
the difference in sensitivity to CsA or Pred was less than 2-fold (Fig. 4.6.A/B) which
indicates that loss of MMR does not significantly affect the cytotoxicity of these
drugs.
Aza is converted to 6 -MP in vivo. As 6 -MP and 6 -TG are metabolised slightly
differently, the cross-resistance of the 6 -TG resistant clones to 6 -M P was
investigated. Figure 4.6C shows that the MMR defective clones are resistant to 6 -
137
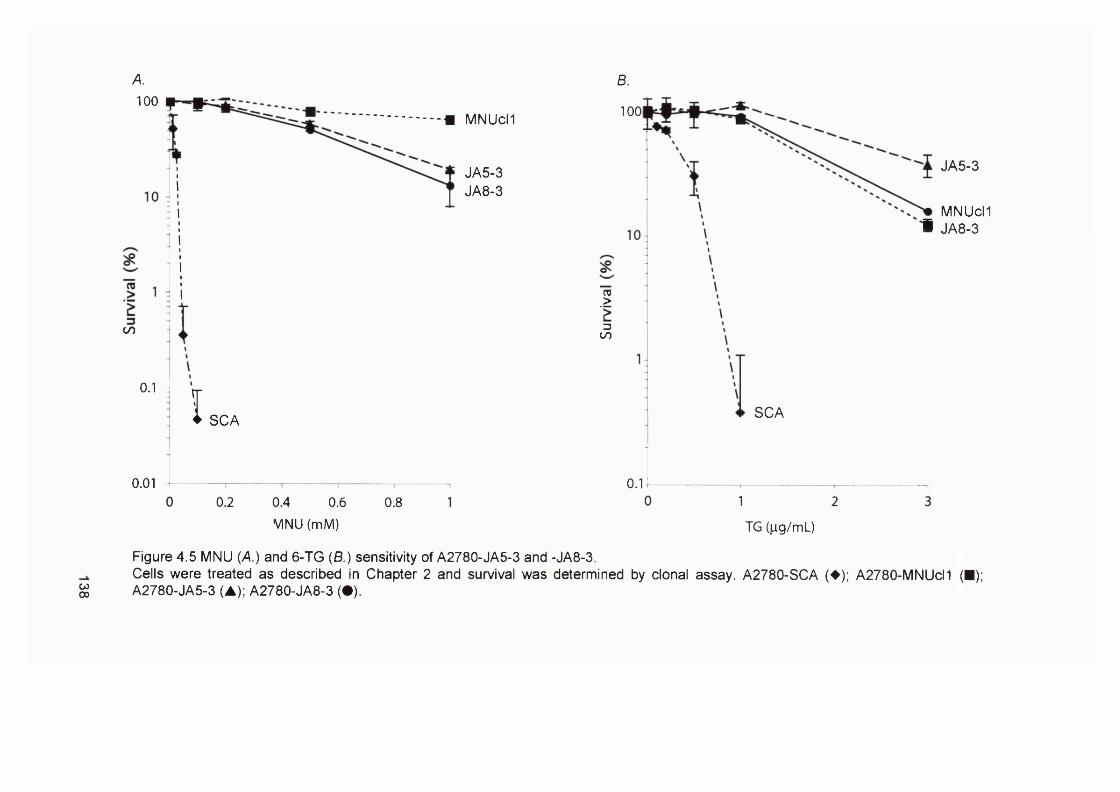
A. B.
w00
0.1 4 ISCA
0.010.2 0.4 0.6
MNU (mM)0.8
100m MNUch
f JA5-3 JA8-3
00I
TO.>
300
1001
JA5-3
MNUcM # JA8-3
0.1
SCA
1 2
TG (M-g/mL)
Figure 4.5 MNU (A.) and 6-TG (B.) sensitivity of A2780-JA5-3 and -JA8-3.Cells were treated as described in Chapter 2 and survival was determined by clonal assay. A2780-SCA (♦); A2780-MNUcl1 (■); A2780-JA5-3 (A); A2780-JA8-3 ( • ) .

A. Prednisolone - |iM100 250 500 1000
MNUcll
A2780
JA5-3
JA8-3
B. Cyclosporin A - ng/mL
A2780
SCA
T I
MNUcll
TI
139
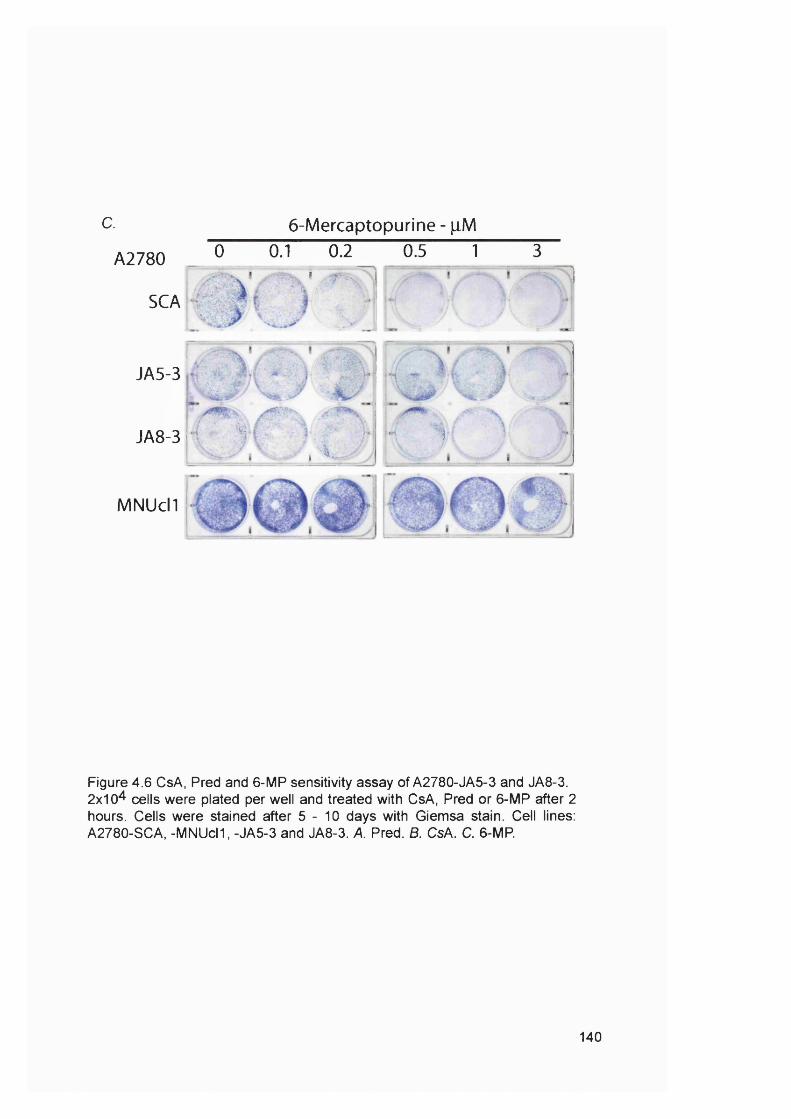
c. 6-Mercaptopurine - pM
A2780 0 0.1 0.2I " ÿ -
SCA
JA5-3
0.5 1 3
JA8-3 i
MNUcll
'Vi
i L .'i
;■;. ■*«
Figure 4.6 CsA, Pred and 6-MP sensitivity assay of A2780-JA5-3 and JA8-3. 2x10^ cells were plated per well and treated with CsA, Pred or 6-MP after 2 hours. Cells were stained after 5 - 1 0 days with Giemsa stain. Cell lines: A2780-SCA, -MNUcll, -JA5-3 and JA8-3. A. Pred. B. CsA. C. 6-MP.
140

MP. Most of the A2780-SCA cells were killed by 0.2|jM 6-TG, whereas the A2780-
JA5-3 and -JA8-3 variants were resistant to more than 1|jM of the drug. The
difference in resistance between the parental A2780-SCA cells and the A2780-JA5-
3 and -JA8-3 clones lies between five and ten-fold, similar to 6-TG.
4.2.6 Microsatellite instability of 6-TG resistant clones
Analysis of the standard panel of five mono- and dinucleotide repeat
microsatellites (Boland et al., 1998) in 28 subclones of A2780-JA5-3 and -JA8-3
cells, compared to 20 subclones of A2780-SCA indicated an M S r phenotype.
Examples are shown in Fig. 4.7. MSF was defined by instability at two or more
markers compared to the originally isolated A2780-JA5-3 and -JA8-3 clones. The
subclones were isolated after continuous growth for approximately 50 generations
from the originally isolated A2780-JA5-3 and -JA8-3 subclones. Alterations at two or
more loci were detected in 14 of 28 (50%) A2780-JA5-3 subclones (Table 4.3.A)
and in 20 of 28 (71%) A2780-JA8-3 subclones (Table 4 .3 .8 ) compared to 0 of 20
(0%) A2780-SCA subclones.
Thus, cells that survived chronic treatment with 6-TG had impaired hMLHI
gene expression that resulted in loss of the hMutLa repair complex. Representative
clones were MSF, tolerant to DNA damaging drugs, and deficient in MMR in
biochemical assays. All these observations are consistent with MMR defects.
Inactivation of MMR appears to be a frequent response to 6-TG and all (5/5) treated
cultures yielded MNU/6-TG resistant clones. Thiopurine treatment hence provides
an effective selection for MMR defective clones from an initially repair competent
population of cells. None of the clones tested were resistant to the
immunosuppressants CsA and Pred, indicating that these two drugs are not
involved in the selection. Thus A2780 cells provide a model for the ability of chronic
thiopurine treatment to select for rare MMR defective variants.
141
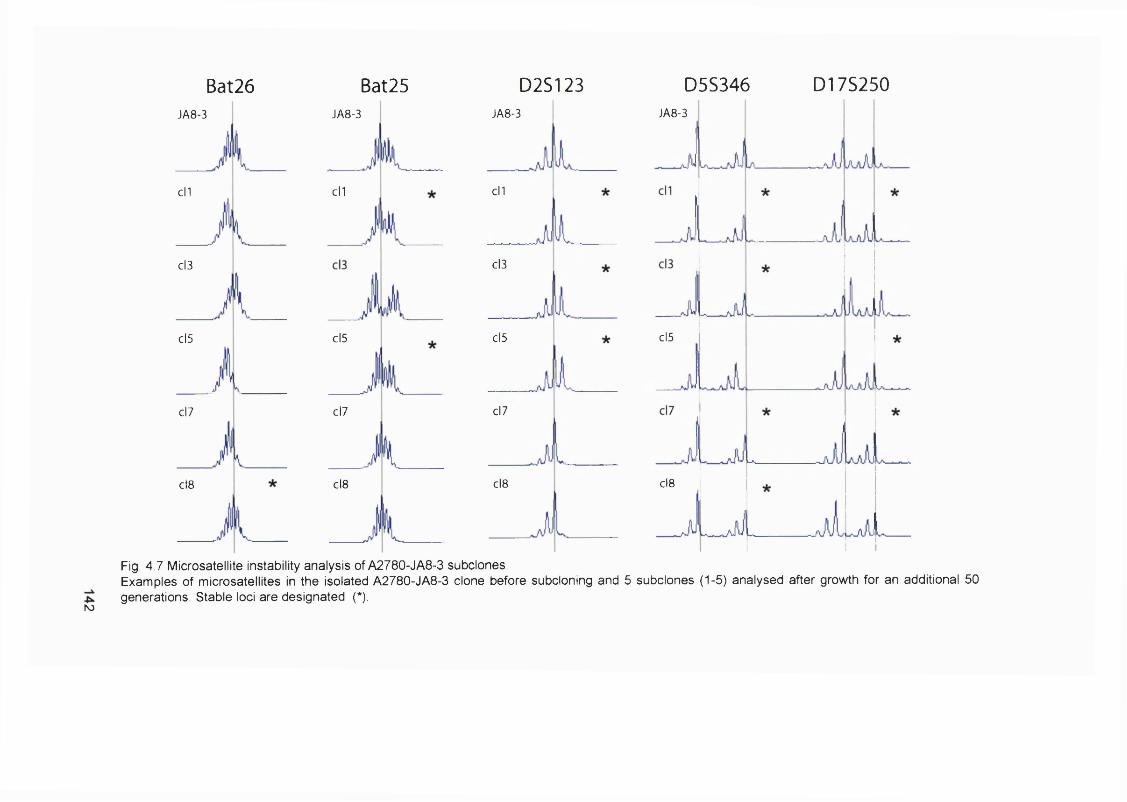
Bat26 Bat25JA8-3 JA8-3
cll
Cl3
cl5
cl7
cl8
cll
cl5
cl7
cl8
D2S123JA8-3
cll
cl3
cl5
cl7
cl8
D5S346 D17S250JA8-3
cl5
cl8
N)
Fig. 4.7 Microsatellite instability analysis of A2780-JA8-3 subclones.Examples of microsatellites in the isolated A2780-JA8-3 clone before subcloning and 5 subclones (1-5) analysed after growth for an additional 50 generations. Stable loci are designated (*).
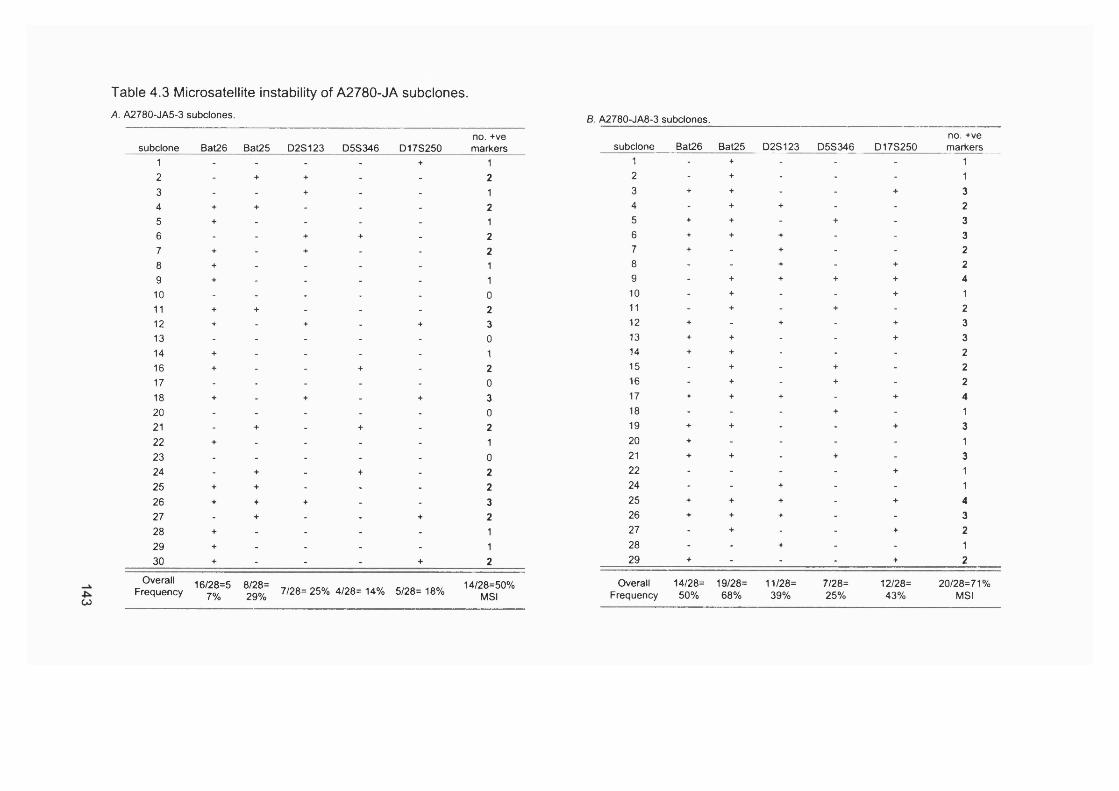
Table 4.3 Microsatellite instability of A2780-JA subclones.A. A2780-JA5-3 subclones. B. A2780-JA8-3 subclones.
CO
no.+vesubclone Bat26 Bat25 D2S123 D5S346 D17S250 markers
1 - - - - + 12 - + + - - 23 - - + - - 14 + + - - - 25 + - - - - 16 - - + + - 27 + - + - - 28 + - - - - 19 + - - - - 110 - - - - - 011 + + - - - 212 + - + - + 313 - - - - - 014 + - - - - 116 + - - + - 217 - - - - - 018 + - + - + 320 - - - - - 021 - + - + - 222 + - - - - 123 - - - - - 024 - + - + - 225 + + - - - 226 + + + - - 327 - + - - + 228 + - - - - 129 + - - - - 130 + - - - + 2
OverallFrequency
16/28=57%
8/28=29% 7/28= 25% 4/28= 14% 5/28= 18%
14/28=50%MSI
no. +vesubclone Bat26 Bat25 D2S123 D 5 S ^ 6 _ D17S250 m arkers___
1 - + - - - 12 - + - - - 13 + + - - + 34 - + + - - 25 + + - + - 36 + + + - - 37 + - + - - 28 - - + - + 29 - + + + + 410 - + - - + 111 - + - + - 212 + - + - + 313 + + - - + 314 + + - - - 215 - + - + - 216 - + - + - 217 + + + - + 418 - - - + - 119 + + - - + 320 + - - - - 121 + + - + - 322 - - - - + 124 - - + - - 125 + + + - + 426 + + + - - 327 - + - - + 228 - - + - - 129 + - - - + 2
Overall 14/28= 19/28= 11/28= 7/28= 12/28= 20/28=71%Frequency 50% 68% 39% 25% 43% MSI

4.2.7 AML in transplant recipients
Because my data from experiments with A2780 suggested a connection
between MSI, drug treatment and thiopurine resistance, I examined the hypothesis
that chronic thiopurine use might be associated with an increased risk of M S r AML.
This was approached in two ways. I searched the literature for case reports
describing this AML associated with thiopurine treatment for autoimmune diseases
or following organ transplants (Appendix Table 4.7). This provided evidence that
immunosuppressive therapy related AML occurs both after organ transplants and
immunosuppressive treatments for other diseases like rheumatoid arthritis. I then
contacted 42 specialists mainly in the fields of haematology, rheumatology and
nephrology (specialised in transplantation) mostly in the UK (Table 4.4). Twenty-
eight consultants replied, but they mainly reported lymphomas and SSCs. Dr. Sue
Snowden, a specialist in kidney transplantations at St. George’s Hospital in London
reported one case of AML after 21 years of Aza treatment (Table 4.4).
The incidence of AML after organ transplantations reported in the literature
appears to be higher after heart/lung transplants than after kidney transplants
(Appendix Table 4 .7 .8). Huebner et al. reported 5 AML cases among 631 cardiac
transplants (Huebner et al., 2000) and Krikorian et al. one among 124 cases
(Krikorian et al., 1978). This compares to one case among 620 renal transplants
(Ondrus et al., 1999) and one case among 316 renal transplants (Barbanel et al.,
1975). Furthermore, it has been suggested that the risk of malignancy is higher after
cardiac than after kidney transplants (Penn, 1993). This applies specifically for
lymphoma which is more common in heart than in kidney recipients (Opelz et al.,
1993). This led me to contact specialists at the eight cardiac transplant centres in
the UK. Dr. Jayan Prameshwar at Papworth Hospital and Dr. Nick Banner at
Harefield hospital reported two and six cases of AML/MDS, respectively (Table 4.4
and 4.5).
144

Table 4.4 Consultants contacted to enquire about post-azathioprine treatment AML.
Name Speciality Hospital AMUMDS after azathioprine AML/MDS after immunosuppressants Other malignancies after
immunosuppression
Paul Sweny Nephrology Royal Free Hospital none none SSC and occasionally EBV- induced lymphomas
A E S Gimson Hepatology Addenbrooke's Hospital - - -
Neil Turner Nephrology Royal Infirmary of Edinburgh, Transplantation Unit none none N/l
John Forsythe Transplant Surgery Royal Infirmary of Edinburgh, Transplantation Unit none none N/l
Peter Amlot Pathology Royal Free Hospital, London - - -R M Bernstein Rheumatology Manchester Royal Infirmary - - -Terence Gibson Rheumatology Guy’s Hospital, London none none N/lIan Chikanza Rheumatology Royal London Hospital - - -Chandra Chattopadhyay Rheumatology Royal Preston Hospital - - -J Isaacs Rheumatology Leeds General Infirmary none none rarely lymphomas and myelomasPatrick Kiely Rheumatology St. George’s Hospital, London none none N/lMark Peakman Immunology King’s College Hospital N/l N/l N/lJohn D Tayler General Surgery Guy’s Hospital - - -
A N Warrens Nephrology and Immunology Hammersmith Hospital none none lymphomas
mainly skin but occasional solid
Stephen Smith General Medicine Victoria Hospital, Lichfield none none organ and haematological malignancies, mainly EBV
related lymphomaColin D Short Nephrology Manchester Royal Infirmary none none N/l
D Throssell Renal Medicine Northern General Hospital, Sheffield none none
a number of cases of lympho- proliferative disease in renal
transplant recipientsAntonio Pagliuca Haematology King’s College Hospital - -Ghulam Mufti Haematology King’s College Hospital none none
t-MDS/AML after CTC and CIb forpost immunosuppression PTLDs
Jamie Cavenagh Haematology St Bartholomew's Hospital none immunosuppression include. 4 Wegener's syndrome patients who had only been treated
with CTX
N/l
David D'Cruz one case of MDS after extensive treatment withRheumatology St. Thomas' Hospital none methotrexate and CTX (but debatable whether N/l
4OlMDS disease or treatment related)
Brian Bourke Rheumatology St. George’s Hospital, London - - -
John Tapson Nephrology Freeman Hospital, Newcastle none none N/lHisashi Yamada Haematology Jikei Hospital, Japan none none noneLeo Kinlen Epidemiology University of Oxford - - -
Brendan Madden CardiothoracicMedicine St. George’s Hospital, London none none N/l
Hannah Short Information Analyst UK Transplant none none noneJudith Marsh Haematology St. George’s Hospital, London none none noneSteve Nelson Haematology St. George’s Hospital, London - - -
J. Treleaven Leukaemia Royal Marsden, Sutton - - -
R. Powles Leukaemia Royal Marsden, Sutton - - -
Jayan Prameshwar
Jackson Wong
Andrew Murday
Nick Banner
Tony H Walker
Gareth Parry
J E. Deanfield
Judith Chessels
Alison Leiper
Sue Snowden
Gerhard Opelz
Cardiology
Cardiology
CardiothoracicSurgery
Cardiology
Cardiology
Cardiology
Cardiology
Haematology and Oncology
Haematology
Nephrology
Collaborative Transplant Study
Cardiac Transplant Unit, Papworth Hospital
Cardiac Transplant Unit, Papworth Hospital
Heart Transplant Unit, Royal Infirmary, Glasgow
Heart Transplant Unit, Harefield Hospital
Heart Transplant Unit, Wythenshawe Hospital
Transplant Unit, Freeman Hospital, Newcastle
Cardiothoracic Unit, Great Ormond Street Hospital
Great Ormond Street Hospital
Great Ormond Street Hospital
St. George's Hospital London
Institute of Immunology, University of Heidelberg
2 AML following either a heart or heart and lung transplant; tx)th on long term immunosuppression with Aza and Pred, one patient also
on CsA
none none
5 cases of AML/MDS (details in Table 4.6)
N/l
N/l
none
none
none
N/l
N/l
none
N/l
N/l
N/l
1 case of AML after 21 years of Aza + possibly other drugs Same patient also had lung® cancer
Identified about 80 centres that had reported cases of post-transplant AML and contacted these to_________________________ ask for samples for MSI analysis for us._________________________
N/l, no information; -, no reply; CTX, cyclophosmamide; Aza, azathioprine.

As a result of these enquiries I contacted Prof. Gerhard Opelz, the head of
the Collaborative Transplant Study, who had previously reported a higher incidence
of lymphoma in heart transplant recipients compared with kidney transplants (Opelz
et al., 1993). By coincidence he had analysed the frequency of AML among organ
transplant recipients using information from more that 300 transplant centres
worldwide. His analysis provides the first unequivocal evidence that organ
transplantation is associated with an increased risk of AML. These data are
reproduced in Figure 4.8.A. The relative risk of AML among a total of 29,077 heart
and/or iung transplant patients compared to control individuals matched for age, sex
and geographical origin was 5.3 (p < 0.0001). The frequency of AML among
141,749 cadaver kidney recipients was lower but was also significantly greater than
in the controls (relative risk = 2.1; p < 0.0001) (Figure 4.8 .8). It is notable that the
excess of AML was less marked in the first three to four years post-transplant and
diverged sharply from the expected incidence thereafter. These findings establish
organ transplantation/immunosuppression as a risk factor for AML/MDS. They also
indicate that the relative risk is higher for cardiac than kidney transplants. This
accords with my experiences and anecdotal evidence from the kidney transplant
centres.
Gerhard Opelz contacted about 80 transplant centres that had reported at
least one AML case. He sent a letter explaining this project, and asking for samples
for MSI analysis. We received one sample from Prof. Dr. G. Kirste (Germany) (case
A7) (Table 4.5). I also received material from 6 post transplant AML/MDS cases
from Nick Banner and colleagues. Sue Snowden contacted the pathologist in charge
of the samples from her patient, but the paraffin embedded tissue blocks had
unfortunately been lost. The details on Dr. Banner’s patients can be found in Table
4.5 (cases A1-6). For all 6 cases I received one or two blocks of a paraffin
embedded bone marrow trephine, and one block of paraffin embedded explanted
tissue (heart or lung). In cases A2 the two trephines had been taken at different
times. From Prof. Dr. Kirste I received one block of paraffin embedded trephine, but
no malignant tissue to be used as a control. The histopathologist Peter Maddox cut
a number of 10pm sections of each block. These sections were used to extract
genomic DNA as described in Chapter 2.
147

A.
I8
_fODE3U
450
400
350
5 300
c<u■D "u c
i
250
200
150
100
50
0
n = 29077
Expected
0 4 6
Years
10
B.
120 18
n = 141749
80-
60-
Expected40-
2 0 -
Years
Fig. 4.8 Incidence of AML in transplant recipients.A Heart and/or lung. Expected incidence in control subjects matched for age, sex, and geographical location is included for comparison. B. Cadaver kidney, n, patient numbers.
148

Table 4.5 Details of patients.
CaseNumber
Age at Diagnosis of AML/MDS
Type and year of
transplantExplant Histology Clinical details Alive/
dead BM aspirate findings CytogeneticsAza
treatment(months)
TotalAza
(mg/kg)Other treatment
A1 68 OCT 1995 Coronary heart disease
Pancytopenia Jan 2002 A 01/02 Trilineage MDS
(18% blasts)Deletion of 6q
&7q 14.7 275 Steroids FX506 ATG
A2 62 SLT 1993 Fibrosingalveolitis
Pancytopenia1999 D
07/99 Trilineage MDS 08/99 Trilineage MDS
(7% blasts)
Deletion of 9q Absence of 21 74 554 CsA Steroids
A3
A4
A5
60
72
50
OCT 1995
OCT 1986
HLT 1991
Dilatedcardiomyopathy
Coronary heart disease
Bronchiectasis
2/52 h/o gum hypertrophy 1999
Pancytopenia1994
Unexplainedanaemia
D
D
A
06/99 AMML (M4Eo)
l)8 /94lÿpopi^ticM D S (10% blasts)
04/95JfipS (26% blastsi 11/85 Acquired
sideroblastic anaemia (35% ring sideroblasts)
Deletion of 16q distal to q22
5q-; monosomy 7 and trisomy 8
No details
43.4
91.6
59.8
1696
3080
1325
CsA Steroids ATG
CsA Steroids TLI
CsA Steroids ATG
A6 59 SLT 1989Alpha 1
Antitrypsin deficiency
Pancytopenia DHypoplastic
Trilineage dysplasia 18% blasts
Complex karyotype
Monosomy 5 &7
132 2571
CsAFK506
SteroidsTLI
A7 48 CK 1987/91 Glomerularnephritis Leukocytosis D AML M4 No details 55 960 CsA Steroids
HLT, Heart/lung transplant; SLT, Single lung transplant; OCT, Cardiac transplant; CK, Cadaver kidney transplant; ATG, Anti-thymocyte globulin; TLI, total lymphoid irradiation.
è

4.2.8 Microsatellite instability in transplant related AMUMDS
MSI was analyzed in the seven cases of AML or MDS from individuals
who had undergone organ transplantation. Available treatment histories are
presented in Table 4.5. There were three heart transplant recipients, one
heart/lung, and two lung only. One patient had received a cadaver kidney. All had
been treated with Aza, usually combined with CsA and steroids. Cytogenetic
analysis at diagnosis revealed three examples of loss of all or part of
chromosome 7 and two examples of loss of all or part of chromosome 5 -
alterations that are common in alkylating agent-related AML/MDS in cancer
patients (Leone et al., 1999). For the six patients (case A1 to 6) for whom a
biopsy of the explanted tissue was available as a source of matched non-tumour
DNA, the standard panel of microsatellites - two mononucleotides Bat26 and
Bat25, and three dinucleotides D2S123, D5S346 and D17S250 - (Boland et al.,
1998) was analyzed. Examples are shown in Figure 4.9 and the Appendix. The
results are summarized in Table 4.6. Two or more microsatellites were altered in
each of the five bone marrow samples that were compared at all loci. These meet
the criteria for MSI.
1. Case A1 (Fig. 4.9.A) is unstable at three of the five markers. The length
of the Bat25 marker amplified from the AML sample appeared to have
increased by 3 bps. A second, smaller peak appeared besides the Bat26
peak in the tumour compared to the normal sample. Both D2S123 and
D5S346 are stable, which can be seen by the unchanged length of these
markers. D 178250 is unstable.
2. Case AS (Fig. 4 .9 .8) was unstable at four of the five markers analysed. A
second smaller peak appeared next to the main Bat26 peak of the AML
sample. The Bat25 peak in the tumour sample has broadened by what
appears to be a second peak. D2S123 is the only locus that is stable from
this sample. Both D5S346 and the D173250 have broadened.
3. Case AS (Fig. 4 .9 .0 ) was unstable at two of the 5 loci analysed. Even
though the Bat26 and Bat25 peaks obtained from AML and normal
material look marginally different the width of the peaks did not change.
They were hence scored as stable. D5S346 was also stable. Both
D2S123 and D178250 were unstable. A smaller second peak appeared in
150

the tumour D2S123 product. The tumour D17S250 peak seemed to have
broadened due to two duplication events.
4. Cases A2, A3 and A4 (Appendix to Fig. 4.9) were unstable at all five
markers analysed.
Bat26 is essentially monomorphic, with 96-98% of the population carrying
the same allele. It has therefore been proposed for the detection of MSI in the
absence of normal tissue (Perucho, 1999). Hartmann et al. showed that the use
of the three mononucleotide markers Bat25, Bat26 and Bat40 resulted in 100%
detection of MSF tumours in urinary tract carcinomas (Hartmann et al., 2002).
The mononucleotide length in the AML samples was compared to blood donor
samples as normal controls. Bat26 has the same length in nine blood donor
samples tested confirming it being monomorphic (Fig. 4.10). The length of Bat25
on the other hand seems to vary by one base (Appendix). Comparing it to the
blood donor samples case A7 appeared unstable. The Bat26 marker has
obtained a second smaller peak and the main peak has shifted slightly to the
right, indicating a shortening of the microsatellite (Fig. 4.10). The Bat25
microsatellite of case A7 has both broadened and shifted to the left by about four
bases compared to the blood donors (Appendix to Fig. 4.10). As this difference is
larger than the variation observed among the blood donors, this marker was also
scored unstable. Case A7 was therefore designated MSF (Table 4.6).
Table 4.6 Microsatellite Instability in MDS/AML cases.
Case Number BAT26 BAT25 D2S123 D5S346 D17S250Al + + - - +A2 + + + +/LOH +A3 + + + + +A4 + + + + +AS + + - + +A6 - - + - +A7 + +
+, Unstable; -, Stable; LOH, Loss of heterozygosity; N/D, not determined.
Thus, all seven transplant-related AML/MDS cases analysed were MSF.
Although this is a small number of patients, the findings suggest that a large
proportion of organ transplant related AML/MDS cases may be defective in MMR.
151
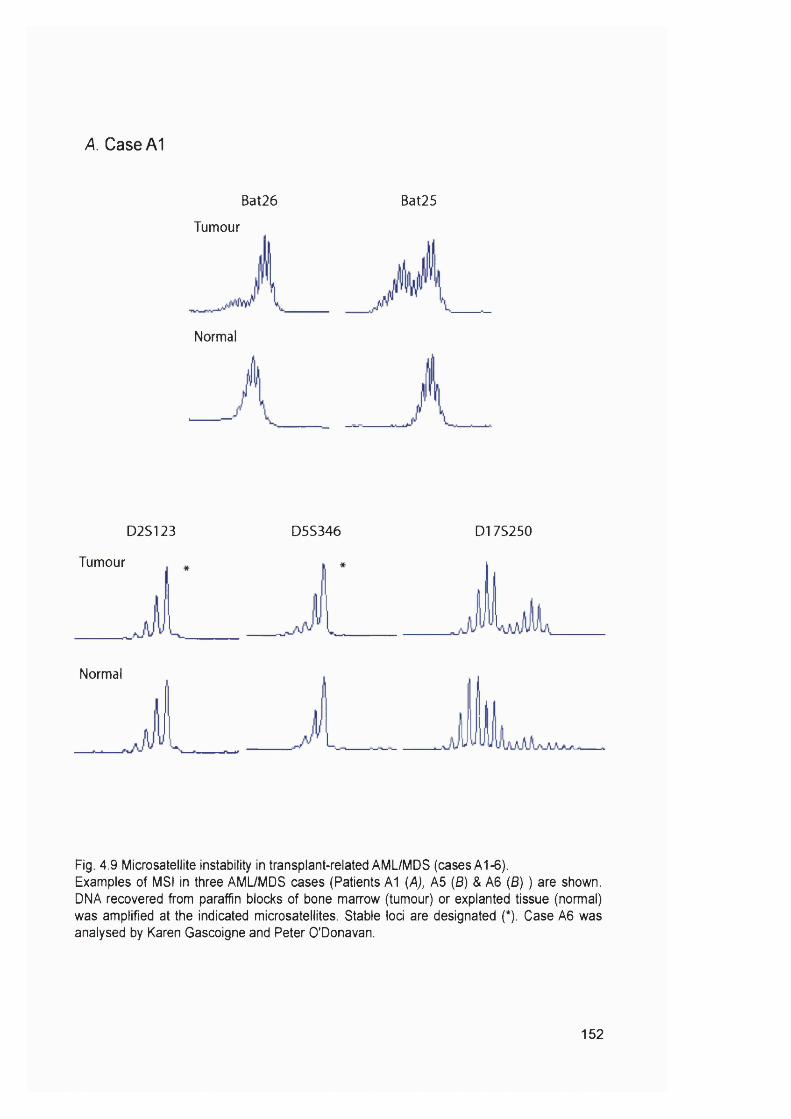
A. Case A1
Bat26 Bat25
Tumour
Normal
D2S123 D5S346 D17S250
Tumour
Normal
Fig. 4.9 Microsatellite instability in transplant-related AML/MDS (cases A1-6).Examples of MSI in three AML/MDS cases (Patients A1 {A}, A5 (B) & A6 (6) ) are shown. DNA recovered from paraffin blocks of bone marrow (tumour) or explanted tissue (normal) was amplified at the indicated microsatellites. Stable loci are designated (*). Case A6 was analysed by Karen Gascoigne and Peter O'Donavan.
152

6. Case A5
Tumour Tumour
Normal Normal
Bat26 Bat25
Tumour * Tumour Tumour
Normal Normal Normal
D2S123 D5S346 D17S250
153

C. Case A6
Bat26 Bat25
Tumour
Normal
D2S123 D5S346 D17S250
Tumour
... UIAiUUaJLLNormal
154

Bat26 Bat26
BD1 BD7
BD2
BD3
BD4
BD8
BD9
case A 7
BD5
BD6
Fig. 4.10 Microsatellite instability in transplant-related AML/MDS (case A7).Examples of MSI in nine blood donors (BD1-9) and one AML bone marrow samples (Patients A7) are shown. The blood donor DNA had been obtained from Ida Casorelli (Chapter 3). DNA recovered from paraffin blocks of bone marrow (case A7) and blood donor DNA was amplified at Bat26. As Bat26 is monomorphic blood donor DNA was used as a normal control.
155

4.3. Discussion4.3 .1 M M R clonal variants ofA2780 resistant to 6-TG and MNU
This study showed that chronic treatment with 6-TG selects for MMR
defective variants from a population of repair proficient human cells. The
carcinoma A2780 cell line contains a preexisting hMLH1 deficient cell population
that has a mutated p53 gene (Aquilina et al., 2000). To avoid isolating subclones
from this preexisting subpopulation, A2780-SCA (p53^, hMLH1*), was isolated by
single cell cloning. All 6-TG resistant A2780 subclones obtained were cross-
resistant to MNU and h M L H f. This probably reflects the tendency of A2780 cells
to silence hMLH1 (Strathdee et al., 1999) and need not implicate hM LH 1
silencing in transplant-related AML/MDS. Indeed, hMLH1 promoter méthylation is
infrequent in chemotherapy-related MSI" AML/MDS (Chapter 3 and (Sheikhha et
al., 2002)).
A2780-MNÜCI1 (Fig. 4.3 and 4.4) was used as a positive control in the
drug sensitivity assays. This subclone, which is probably a representative of the
subpopulation described above, is hM LH V and is also deficient in the p53-
dependent response to DNA damaging agents. This loss of p53 function could
explain the 2-fold higher MNU resistance of A2780-MNUcl1 than A2780-JA5-3
and -JA8-3. The p53-dependent DNA damage response was not tested in
A2780-JA5-3 and -JA8-3 due time limitations, but it seems unlikely that they
would have acquired a p53 as well as an MMR defect.
Transplant patients are treated with a combination of drugs. I tested the
sensitivity of the isolated clones to CsA and Pred. Loss of MMR did not
detectably alter the toxicity of either drug and it seems unlikely that CsA or Pred
play an important role in the development of MSI" post-transplant AML/MDS.
Although immunosuppression might contribute to AML/MDS simply by providing
a permissive environment for the clonal expansion of altered cells, this does not
provide a ready explanation for the high frequency of MSI.
Most immunosuppressed patients are treated with Aza, which is
converted to 6-MP. My MMR defective clones, however, were isolated with 6-TG.
These two drugs are metabolized slightly differently. Méthylation of 6-MP and 6-
TG bases by TPM T is inactivating, producing S-m ethyl-thioinosine-5’-
monophosphate (M eTIM P) and S-methyl-thioguanosine-5’-monophosphate
(M eTGMP), respectively (Fig. 1.7). TPMT thereby removes 6-MP and 6-TG
156

nucleotides from the pool of bases to be incorporated into DNA. It has been
suggested however, that the production of MeTIMP nucleotides may contribute to
the antiproliferative properties of the 6-MP through inhibition of de novo purine
synthesis, whereas 6-TG is inactivated by TPMT (Dervieux et al., 2001). As these
experiments were carried out in MMR' cell lines (for example CCRF-CEM) (Gu et
al., 2002) the results obtained by the authors are not likely to reflect treatment of
a normal, MMR proficient individual. To ensure that the difference in sensitivity
between MMR deficient and proficient cells is the same for 6-TG and 6-MP, the
effect of 6-MP on A2780-SCA, -JA5-3, -JA6-3, and -M NUcll was determined.
The MMR' clones were found to be almost equally resistant to 6-MP and to 6-TG.
Aza and 6-MP treatment is therefore likely to exert the same selective pressure
on cells as 6-TG.
4.3.2 AML after organ transplants
To investigate the possible link between thiopurine treatment, MMR and
AML I contacted British clinicians to enquire if they had previously treated a
patient with post-immunosuppression AML. In the case of a positive reply I asked
if I could obtain some material for my study. These enquiries gave me the
impression that the incidence of AML was much higher after heart/lung
transplants than after kidney transplants or treatment for autoimmune diseases.
This was confirmed by Gerhard Opelz (Collaborative Transplant Study). He
provides evidence that the relative risk of AML after heart/kidney transplants is
more than 5 and after kidney transplants more than 2. The reason for the
difference in incidence of AML after heart/lung and kidney transplants is not
clear. However, diagnostic and therapeutic radiation is also a potential risk factor
for t-AML/MDS and thoracic organ transplant patients generally receive more
radiation than kidney recipients.
The incidence of AML after both types of transplantation increases quite
dramatically after three to four years post-transplant. This apparent lag period
distinguishes AML from the more common transplant-related non-Hodgkin
lymphomas, the majority of which occur within the first two years (Opelz et al.,
1993). This suggests that these secondary malignancies develop by different
mechanisms. However, a second type of post-transplant lymphoproliferative
disorders (PTLDs) that arise several years after the transplant, exist. It has been
suggested that late onset PTLDs may be pathologically different from PTLDs
157

arising earlier (Randhawa et al., 1989). This is supported by Larson et al. who
found MSI only among cases of late onset PTLD (two cases). This lag-period and
loss of MMR indicates that the development of these two cases late onset PTLD
might be similar to the development of post-transplant AML.
4.3.3 Microsatellite instability in transDlant-related AMUMDS
I obtained material from 7 post-transplant AML/MDS cases. Six of these
cases were obtained from Harefield hospital, whereas case A7 was obtained
through the help of Gerhard Opelz. The clinical information on these patients
provided two things of interest. Three of the six thoracic transplant AML/MDS
cases for which cytogenetic data were available had chromosome 5/7
involvement (Table 4.5). These changes are common in alkylating agent induced
AML. This high incidence of chromosome 5/7 involvement reflects the frequency
among the case reports summarised in Table 4.7 (Appendix). I also noticed that
the majority of the cases (5 of 6 cases) obtained from Harefield hospital were not
actually AML but MDS. As the letter that Gerhard Opelz wrote to all transplant
centres only asked about AML and not MDS cases, we might have been able to
obtain more material if we had requested AML and MDS. We are considering this
option.
Microsatellite analysis of the transplant-related AML/MDS cases showed
that all seven cases were unstable. Until now, apart from de novo AML in elderly
patients (Das-Gupta et al., 2001), MSI AML/MDS was largely confined to cases
secondary to alkylating agent treatment (Chapter 3). Thiopurines and alkylating
agents are linked by a shared ability to produce DNA damage that interacts with
MMR, and the cross-resistance of MMR deficient cells to both types of drug is
well established. In addition to the high frequency of MSI, alkylation- and
immunosuppression-related AML/MDS share other features that are consistent
with a common etiology. For example, loss of all or part of chromosome 5 and/or
7 is both common in AML/MDS secondary to immunosuppression (Tables 4.5
and 4 .7 ) and alkylating agent (Leone et al., 1999). Alkylating agents and
thiopurines also share a dose-limiting myelotoxicity (Cummins et al., 1996;
Lennard et al., 1989). The data obtained here and the similarity between
alkylation- and immunosuppression-related AML/MDS are all consistent with
common steps in the developments of both malignancies.
158

4.4 ConclusionMy data show that thiopurines may contribute significantly to
leukaemogenesis in organ transplant recipients. There are preliminary data to
indicate that the incidence of AML after transplantation may be Aza dose
dependent (Gerhard Opelz, personal communication). It therefore seems likely
that low TPMT activity due to a polymorphism could increase the risk of AML. A
less efficient metabolic inactivation would result in a higher effective dose and
therefore more 6-TG being incorporated into DNA. Children diagnosed with t-
AML/MDS after treatment with 6-MP had lower TPMT activities compared to the
remaining patients on the treatment (Thomsen et al., 1999). Pre-treatment testing
for T P M T polymorphisms is being debated at the moment because of life-
threatening toxicity in high-risk patients with polymorphisms in both copies of the
gene (Marshall, 2003). These tests, however, could also benefit patients that
have a lower risk of life-threatening complications but might have a higher risk of
developing t-AML/MDS.
159

Chapter 5 : Results III
Mutations at mononucleotide repeats in target genes in M S r human cell lines and t-AML
5.1 IntroductionMMR defective (MST) tumours are highly genetically unstable. They
accumulate frameshift mutations in coding sequences, which result in most cases
in truncated and/or inactive proteins. For example, Caspase 5 (Casp-5)
(discussed in more details below) was shown to accumulate frameshift mutations
in M S r tumours (Schwartz et al., 1999).
Frameshift-like mutations in intronic polynucleotide repeats close to splice
sites can affect RNA splicing. These mutations could lead to exon skipping which
can result in premature stop-codons or inactive proteins. For example,
addition/deletion mutations in one of the introns of Mre11 (discussed in more
detail below) was shown to cause exon skipping. The list of genes containing
coding repeat sequences that are found mutated in MSF tumours is increasing
constantly (also discussed in Section 1.5.1).
To identify potential target genes in the development of MSF t-AML genes
known to be mutated in tumours at other sites, and newly identified potential
target genes were analysed. A number of known target genes was analysed by
Ida Casorelli in the chemotherapy related AML samples (Table 5.1).
Table 5.1 Potential target genes for frameshift mutations analysed by Ida Casorelli.
Gene function type of repeat locationBax promotes apoptosis (G)8 _ exon 3
TGFpRII tumour suppressor (A)10 exon 3BRCA1 (A)8 exon 9
BLM ....... (A)^ _ exon 6ATM genome integrity (T)7 exon 5
RAD50 (A)9 exon 13M rell (T)11 intron 4/5
I examined the sequences of genes known to be involved in
leukaemogenesis or haematopoiesis for the presence of small mono- or
dinucleotide repeats. Table 5.2 lists the genes I identified as potential targets in t-
160

AML development. Mre11 (discussed below) was included in this analysis as a
control.
Table 5.2 Potential target genes for frameshift mutations in MMR defective cell lines.
gene function type of repeat locationCaspase 5 apoptosis/inflammation (A)10_ exon 2
Fas apoptosis (T)9 intron 4/5Fits haematopoiesis (T)10 intron 10/11Rb
FancD2jynipur suppressor i m _____
(T)10__intron 6/7
intron 5/6FancEFancG
genome integrity (0)7(TG)5
exon 4 intron 3/4
Chkl (A)9 exon 7The repeats are also described in Figures 5.1, 5.2 and 5.3.
These genes can be divided into two categories: genes that have been found to
be involved in tumourigenesis in general, and genes which are important for
haematological development or thought to be involved in leukaemogenesis.
Genes involved in general tumourigenesis:
1. The Mre11 protein is part of the Rad50-Mre11-NBS1 complex which is
essential for a normal cellular response to DSBs (reviewed in (Thompson
and Schild, 2002)). M rel 1 is thought to be required for DSB repair and
cell cycle checkpoint signalling (discussed in Chapter 1). Recently
mutations in the Mre11 gene were identified in four patients described as
having the cancer predisposition syndrome "ataxia telangiectasia-like
disorder' (ATLD). Giannini and co-workers identified mutations of an
intronic poly(T)11 repeat in the human M RE11 gene in MMR defective
cell lines and colon tumours. These mutations resulted in aberrant
splicing, giving rise to a truncated, unstable transcript (Giannini et al.,
2002). This is the first and, as far as I am aware, the only example of an
intronic mutational target in MSF cells leading to a splice defect that
affects protein function.
2. Casp-5 was first identified as a target for frameshift mutations in MSF
tumours of the endometrium, colon and stomach by Schwartz et al.
(Schwartz et al., 1999). Members of the caspase family of proteases are
161

involved in apoptosis, and in immune and inflammatory responses. Casp-
5 is thought to act in an inflammatory context, but its exact function has
not been determined (Creagh et al., 2003). Exon 2 contains a (A) 10
repeat (Fig. 5 .3 .8 ) that has been shown to be mutated in M S r colon,
stomach and endometrial tumours, and cell lines (Schwartz et al., 1999).
3. C h k l is required for multiple DNA damage checkpoints (reviewed by
(Melo and Toczyski, 2002; Shiloh, 2001). Chkl is, for example,
phosphorylated by ATR in response to UV and IR damage. This leads to
activation of the G2/M checkpoint, which inhibits cell cycle progression.
Exon 7 of the Chkl gene contains two poly(A) repeats, one nine and one
seven nucleotides long (Fig 5.1.D).
4. The Fas protein (CD95) is a member of the tumour necrosis factor (TNF)
superfamily. It is involved in initiating the apoptotic cell signalling cascade
by activating members of the caspase family. Fas expression is up-
regulated by cytokines such as interferon^ and TNF, and Fas-mediated
apoptosis is triggered by its natural ligand, FasL. Loss of Fas function
occurs frequently during human tumour progression (Reichmann, 2002).
Intron 4/5 of the Fas gene contains a (T)9 that is separated from the
splice acceptor site by two bases (Fig. 5.1.0).
5. The retinoblastoma-susceptibility gene {Rb) is a tumour suppressor gene.
The Rb protein interacts with the E2F family of transcription factors
causing the repression of genes that are required for DNA synthesis. This
leads to an inhibition of cell growth. Hence, the inactivation of Rb is a
prerequisite for cell proliferation. Loss of Rb function occurs in many
tumour types, and can be due to either promoter méthylation or mutation
(Chau and Wang, 2003). Mutations in genes that are part of the ‘Rb
pathway' occur so frequently that it has been proposed that disabling this
pathway may be essential for cancer development (Sherr and
McCormick, 2002). Intron 6/7 of the Rb gene contains a (T)9 repeat
separated from the splice acceptor site by one base (Fig. 5.1.D).
162

Genes involved in haematopoiesis:
6. Mutations in the FMS-like tyrosine kinase 3 {Flt3) gene have been found
to be the most common genetic event in AML, occurring in ~25% of cases
(reviewed in (Levis and Small, 2003; Stirewalt and Radich, 2003)). Flt3
encodes a membrane-bound receptor tyrosine kinase that has a crucial
role in normal haematopoiesis. This kinase is involved in the proliferation,
differentiation and apoptosis of haematopoietic cells. Flt3 is normally
expressed mainly in early myeloid and lymphoid progenitor cells. Binding
of Flt3 ligand (Flt3L), produced by many haematopoietic cells, promotes
dimerization and activation of Flt3. The most common form of Flt3
mutations is an internal tandem duplication (ITD) in exons 14 and 15.
Point mutations in heavily conserved areas of the intracellular tyrosine
kinase domain are also common. Intron 10/11 of the Flt3 gene contains a
(T)11 repeat that precedes the splice acceptor site (Fig 5.1.E).
7. The Fanconi anaemia pathway is discussed in more detail in Chapter 1.
Disruption of this pathway leads to the characteristic cellular and clinical
abnormalities observed in FA. One feature of FA is a high incidence of
AML (Alter, 2003; D'Andrea and Grompe, 2003). Interestingly, cytogenetic
abnormalities similar to those found in patients with t-AML occur
frequently among FA associated AML. These abnormalities include 5q-,
monosomy 7, 7q-. Abnormalities of chromosome 7 are particularly
common (Lensch et al., 1999). Intron 5/6 of the FancD2 gene contains a
(T)10 repeat two bases 5' of the splice site (Fig. 5.3.A). Exon 4 of FancF
contains a (0 )7 repeat (Fig. 5.2.A). Intron 10/11 of F an cG contains a
(TG)5 repeat 10 bases upstream of the splice site (Fig. 5.2.6).
Part of the work in this chapter (as indicated) was carried out by Karen
Gascoigne, a summer student in the laboratory, under my supervision.
163

MS-PCR primers
A M re ll 124 bo ^
I exon 4........|- ....(T )nA A G --| exon 5 I
6. Chkl 173 bp ^
I exon 6 I — —I GG(A)hG(A)qCA GGÇAlyTC |— —| exon 8 I
205 bp ^
I exon 4 |- ....(T)qCTAG--| exon 5 I
336 bp ^
I exon 6 |- ..... (T)qC A G -| exon? I
£Flt33 402 bp ^
I exon 10 |- ....(T)ioCAG--1 exon 11 I
F. Cbfp ^ 484 bp ^ _
- |st5p:;:...AG(T)« CA(T)^aA(T)„GT. .nA(T)^G(T)^.....TTTCj[T),^.:...:i
6.
«- O Ü)
<Ü(/) CD o ^ ^
CM h -
m x Q : ( O Q i t n < ^ a D X _ i Q x a : ^ o < Q w < Q : m ^
FasChk1Mre11
Fig. 5.1 Schematic representation of the amplified genomic region of assayed target genes and example of multiplex PCR.AG splicing signals are highlighted in bold. Primers used to amplify the region containing the poly-T stretch are indicated by blue arrows. The nucleotide repeat of interest is highlighted in red and bold. A. M re ll. B. Chkl. C. Fas. D. Rb. E. Flt3. F. Cbfp (was not analysed further). G. Multiplex PCR of all 5 gene target sequences from tumour cell line DNA. The PCR was performed as described in Chapter 2, the products separated on a 2% agarose gel and visualised under UV light.
164

MS-PCR primers
A FancE
I exon 3 I- . ^ 160 bp ^ __
I atg(C)7AGT..... f ~ --1 exon 5
B. FancG
exon 10358 bp ^
(TG) JATACTCTACAG-I exon 11
CMultiplex PCR example
c<0
2o
lO< 2Ü 111to ÜÔ LL
X CO 01LU CN ÜX < Ü
o m CN
ICD
FancGControlFancE
Fig. 5.2 Schematic representation of the amplified genomic region of assayed target genes and example of multiplex PCR.AG splicing signals are highlighted in bold. Primers used to amplify the region containing the poly-T stretch are indicated by blue arrows. The nucleotide repeat of interest is highlighted in red and bold. A. Fanconi anaemia complementation group E (FancE). B. Fanconi anaemia complementation group G (FancG). C. Example of multiplex PCR of both Fanconi gene target sequences. A region downstream of the FancF served as a control. The PCR was performed as described in Chapter 2, the products separated on a 2% agarose gel and visualised under UV light.
165
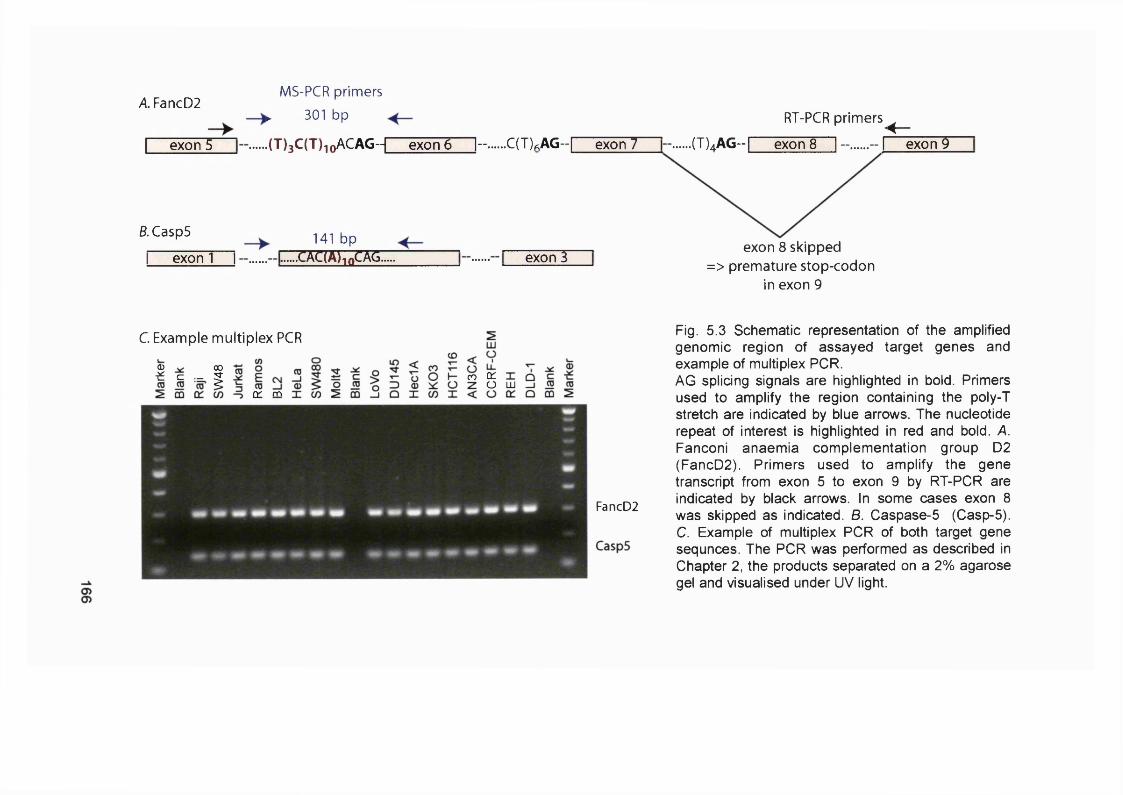
A FancD2
exon 5
MS-PCR primers
► 301 bp
.(T)3C(T)ioACAG--[ exon 6 |- .....C(T)fiAG-r exon 7
RT-PCR primers ^
.(T)^AG-r" exon 8 I - ..... - I exon 9
B. CaspS
I exon 1 I141 bp
.CAC(A),nCAG. exon 3exon 8 skipped
=> premature stop-codon in exon 9
C Example multiplex PCR
I c - I CO 0:O LU
O ' m
FancD2
Fig. 5.3 Schematic representation of the amplified genomic region of assayed target genes and example of multiplex PCR.AG splicing signals are highlighted in bold. Primers used to amplify the region containing the poly-T stretch are indicated by blue arrows. The nucleotide repeat of interest is highlighted in red and bold. A. Fanconi anaemia complementation group D2 (FancD2). Primers used to amplify the gene transcript from exon 5 to exon 9 by RT-PCR are indicated by black arrows. In some cases exon 8 was skipped as indicated. B. Caspase-5 (Casp-5). C. Example of multiplex PCR of both target gene sequnces. The PCR was performed as described in Chapter 2, the products separated on a 2% agarose gel and visualised under UV light.
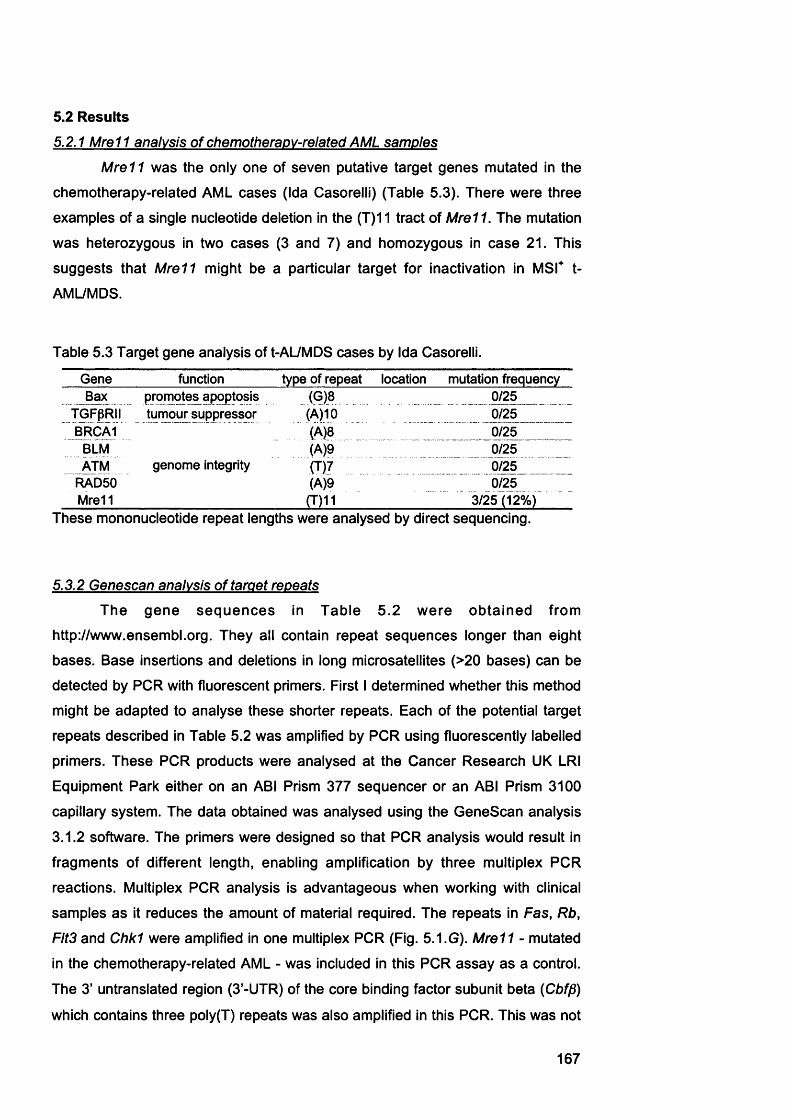
5.2 Results5.2.1 Mre11 analysis of chemotherapy-related AML samples
M r e l l was the only one of seven putative target genes mutated in the
chemotherapy-related AML cases (Ida Casorelli) (Table 5.3). There were three
examples of a single nucleotide deletion in the (T)11 tract of M re ll. The mutation
was heterozygous in two cases (3 and 7) and homozygous in case 21. This
suggests that M r e ll might be a particular target for inactivation in M S r t-
AML/MDS.
Table 5.3 Target gene analysis of t-AL/MDS cases by Ida Casorelli.
Gene function type of repeat location mutation frequencyBax promotes apoptosis (G)8 0/25
TGFpRII tumour suppressor (A)10 0/25BRCA1 (A)8 _ 0/25
BLM (A)9 0/25ATM genome integrity (TJ7 0/25
RAD50 (A)9 0/25Mrell (T)11 3/25 (12%)
These mononucleotide repeat lengths were analysed by direct sequencing.
5.3.2 Genescan analysis of target repeats
The gene sequences in Table 5 .2 w ere obtained from
http://www.ensembl.org. They all contain repeat sequences longer than eight
bases. Base insertions and deletions in long microsatellites (>20 bases) can be
detected by PCR with fluorescent primers. First I determined whether this method
might be adapted to analyse these shorter repeats. Each of the potential target
repeats described in Table 5.2 was amplified by PCR using fluorescently labelled
primers. These PCR products were analysed at the Cancer Research UK LRI
Equipment Park either on an ABI Prism 377 sequencer or an ABI Prism 3100
capillary system. The data obtained was analysed using the GeneScan analysis
3.1.2 software. The primers were designed so that PCR analysis would result in
fragments of different length, enabling amplification by three multiplex PCR
reactions. Multiplex PCR analysis is advantageous when working with clinical
samples as it reduces the amount of material required. The repeats in Fas, Rb,
Fits and C hkl were amplified in one multiplex PCR (Fig. 5 .1.G). M r e l l - mutated
in the chemotherapy-related AML - was included in this PCR assay as a control.
The 3' untranslated region (3'-UTR) of the core binding factor subunit beta {Cbfp>)
which contains three poly(T) repeats was also amplified in this PCR. This was not
167

analysed further because the PCR product was too long to be analysed on the
system as it was set up for the other, shorter PCR products. Adjusting the system
would have been prohibitively expensive. Casp-S and FancD2 (Fig. 5.3.C), and
F a n c E and FancG (Fig. 5 .2 .C) were amplified as separate multiplex PCR
reactions. A 207 bp long DNA fragment situated downstream of the FancF gene
was amplified as an internal control.
5.2.3 Target aene analysis of MMR proficient and deficient tumour cell lines
To investigate whether the putative targets could be mutated in a MMR
defective genetic background I first analysed a panel of MMR proficient and
deficient tumour cell lines (Table 5.4). The series included six MMR proficient and
fourteen deficient lines. They are described in more detail in Table 2.1. Examples
of these PCR assays are shown in Figures 5.1.G, 5 .3 .0 and 5 .2 .0 . To ensure
that this method of analysis is accurate the C asp-5/FancD 2 genotypes were
confirmed by direct sequencing. The sequencing data was analysed using
Sequencer 4.1 software. Examples of both types of analysis of the Casp-5 repeat
are shown in Fig. 5.4. This comparison shows that both genotyping and direct
sequencing leads to the same results. Genotyping of both Raji and Jurkat
resulted in one peak only (Fig.5.4.A), which is reflected by one single sequencing
trace (Fig. 5.4.8). In the cases where more than one peak was obtained from
genotyping, sequencing resulted in two overlaying traces. Genotyping of SW48
DNA, for example, gave rise to two peaks, the shorter one for (A)9 and the
second one for (A)10 (Fig.5.4.A). These two genotypes resulted in two
sequencing traces, which overlapped until the ninth A (Fig. 5.4.8). After the ninth
A the (A)9 trace reads CAGTT. The (A) 10 trace carries on with one more T and
then CAGTT. These two overlaying traces cannot be read accurately by the
Sequencer 4.1 software and result in the sequence CNGNTA. The (A)9 peak is
slightly higher than the (A)10 peak, which is reflected by the fact that the peaks of
(A)9 trace are also slightly higher than the peaks of the (A) 10 trace. Since these
and other data confirm that the two methods produce identical results,
genotyping was used to analyse the other putative target genes.
Examples of the repeat length analysis of the other genes are shown in
Fig. 5.5. The results of this analysis are summarised in Table 5.4.
168

A. Casp-5 (A) 10
8.
Raji
SW48
C C A C A A A A A A A A A N C A G T T A AC CflCflflflflflfinflflNCflGTTflfl
C C A C: f l A A A A A A A A C M G M T A
CCflCflflflflflflnflfiCNGNTfln, n n n, n n n ,n, n n n
Fig. 5.4 Casp-5 repeat analysis of tumour cell lines.All samples were assayed both by genotyping (A.) and genomic sequencing (8 .). Examples of Raji, SW 48 and Jurkat analysis are shown. The results obtained are summarised in Table 5.4.
Jurkat
C C f l C f l f l f l f l f l R f l A f l C f l G T T A A
c cncnnRRRf l RBRCRGTTnn
/W '
O)CD
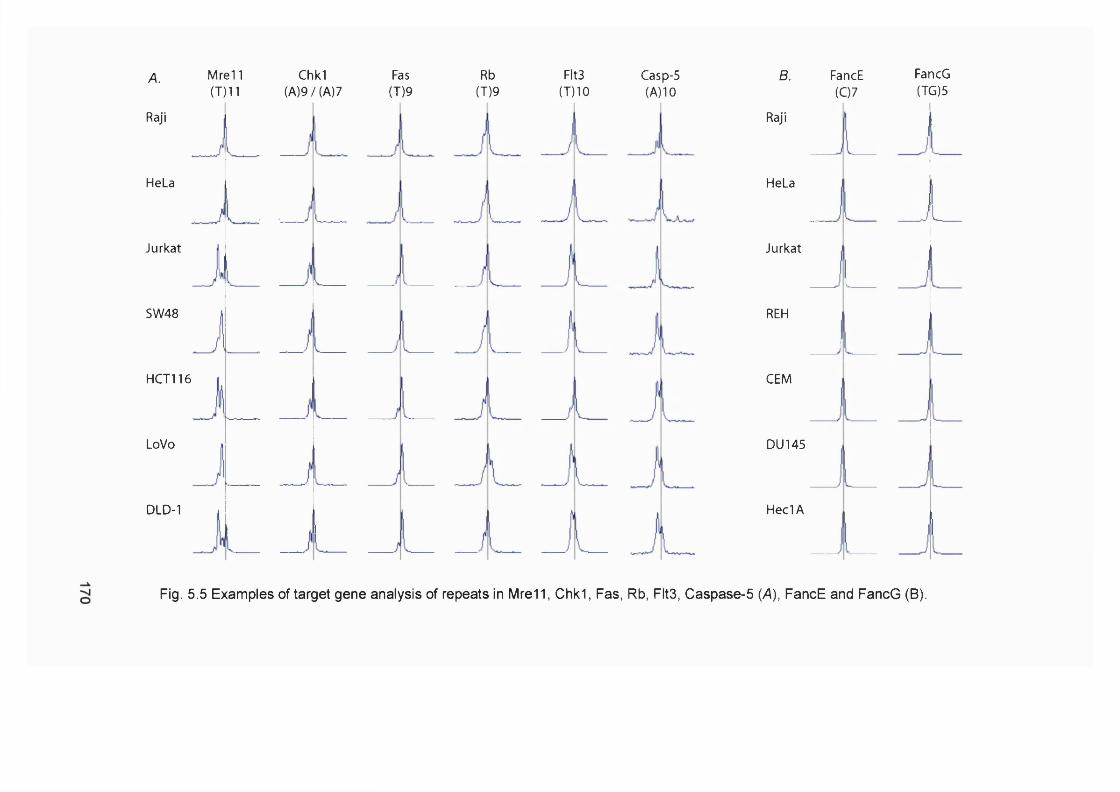
A. Mrel 1 (T)11
Raji
HeLa
Jurkat
SW48
HCT116
LoVo
DLD-1
Chkl(A)9/(A)7
Fas(T)9
Rb(T)9
Fits(T)10
Casp-5(A)10
B. FancE (07
Raji
REH
CEM
DU 145
Heel A
HeLa
Jurkat
FancG(TG)5
Fig. 5.5 Examples of target gene analysis of repeats in Mre11, Chk1, Fas, Rb, Flt3, Caspase-5 (A), FancE and FancG (B).

Table 5.4 Target gene analysis of MMR proficient and deficient cell lines.
Cell Line MMR Mre11 (T)11 alleles
Chk1 (A)9 + (A)7 alleles
Fas (T)9 alleles
FLT3 (T)10 alleles
Caspase 5 (A) alleles
FancD2 (T)10 alleles
FancE(C)7alleles
FancG(TG)5allele
Rb (T)9 alleles
HeLa + (T)11 (A)9 + (A)7 (T)9 (T)10 (A)10 (T)8/(T)10 (C)7 (TG)5 (T)9Raji + (T)11 (A)9 + (A)7 (T)9 (T)10 (A)10 (T)8/(T)10 (C)7 (TG)5 (T)9SW480 + (T)11 (A)9 + (A)7 (T)9 (T)10 (A)10 (T)10 (C)7 (TG)5 (T)9Ramos + (T)11 (A)9 + (A)7 (T)9 (T)10 (A)10 (T)10 (C)7 (TG)5 (T)9BL2 + (T)11 (A)9 + (A)7 (T)9 (T)10 (A)10 (T)10 (C)7 (TG)5 (T)9/V2780-SCA + (T)11 (A)9 + (A)7 (T)9 m io N/D (T)8/(T)10 (C)7 (TG)5 (T)9Jurkat - (T)9/(T)11 (A)9 + (A)7 (T)9 (T)9/(T)10 (A)9 (T)9/(T)10 (C)7 (TG)5 (T)9SW48 - (T)10 (A)9 + (A)7 (T)9 (T)9/(T)10 (A)9/(A)10 (T)10/(T)11 (C)7 (TG)5 (T)9HCT116 - (T)9/(T)10 (A)9 + (A)7 (T)9 (T)10 (A)9/(A)10 (T)10/(T)11 (C)7 (TG)5 (T)9LoVo - (T)10 (A)9 + (A)7 (T)9 (T)9/(T)10 (A)9/(A)10 (T)10 (C)7 (TG)5 (T)9DLD1 - (T)9/(T)11 (A)9 + (A)7 (T)9 (T)9/(T)10 (A)9/(A)10 (T)8/(T)10 (C)7 (TG)5 (T)9Hec1A - (T)10/(T)11 (A)9 + (A)7 (T)9 (T)10 A)10 (T)10 (C)7 (TG)5 (T)9REH - (T)11 (A)9 + (A)7 (T)9 (T)10 A)10 (T)10/(T)11 (C)7 (TG)5 (T)9Molt4 - (T)10/(T)11 (A)9 + (A)7 (T)9 (T)10 (A)9/(A)10 (T)8 (C)7 (TG)5 (T)9CCRF-CEM - (T)10/(T)11 (A)9 + (A)7 (T)9 (T)10 (A)9 (T)10 (C)7 (TG)5 (T)9AN3CA - (T)9 (A)9 + (A)7 (T)9 (T)9/(T)10 (A)10 (T)8 (C)7 (TG)5 (T)9DU145 - (T)9/(T)10/(T)11 (A)9 + (A)7 (T)9 (T)10 (A)9/(A)10 (T)10 (C)7 (TG)5 (T)9SK0V3 - (T)11 (A)9 + (A)7 (T)9 (T)10 A)10 (T)8/(T)10 (C)7 (TG)5 (T)9A2780-MNUcl1 - (T)10/(T)12 (A)9 + (A)7 (T)9 (T)10 N/D (T)8/(T)10 (C)7 (TG)5 (T)9RajiF12 - (T)11 (A)9 + (A)7 (T)9 (T)10 N/D (T)8/(T)10 (C)7 (TG)5 (T)9The lengths of the different mono- and dinucleotide repeat were analysed as described in chapter 2. N/D, not determined.

1. C a s p - 5 and M r e l l were confirmed as targets for insertion/deletion
mutations in a MMR defective background. The Mre11 (T)11 repeat was
mutated in nearly all MMR defective cell lines apart from REH, SK 0V3 and
RajiF12. Jurkat, DLD1, Hec1A, Molt4, CCRF-CEM and DU145 retained one
wt copy of the gene. No repeat length changes were observed in the MMR
proficient cell lines. Caspase-5 was not mutated in the MMR defective cell
lines N e d A, REH, AN3CA and SK0V3, but the (A)10 repeat was shortened
by one base in one copy of the gene in SW48, HCT116, LoVo, DLD1, Molt4
and DU 145. The second copy remained at wt length. Both copies were
mutated to (A)9 in Jurkat and CCRF-CEM.
2. C h k l , Fas, Rb, F an cE and FancG were not targets for insertion/deletion
mutations in the MMR defective cell lines analysed.
3. Fits was identified as a new target for frameshift-like mutations in a MMR
defective background. Changes in the length of the repeat were observed in
Jurkat, SW48, LoVo, DLD1 and AN3CA as a second peak in the analysis.
These changes were all heterozygous for a -1 deletion suggesting that a
functional copy of the gene might be required for cell viability.
5.2.4 Target aene analysis of t-AML samples
To examine whether any of these putative target sequences were altered in
M S r t-AML both chemotherapy- and organ transplant-related samples were
analysed. Table 5.5 summarises the data.
Casp-5 was identified as a target in t-AML. Three chemotherapy-related
(cases 2, 8 and 23) and two transplant-related (A2 and A3) AMLs were
heterozygous for a one bp deletion (Tables 5.5.A and B). Thus the frequency of
Caspase-5 frameshift mutations is approximately 30% (6/20) (Table 5.5.0).
Only nine chemotherapy related AML samples were analysed for mutations
in Fas, Flt3, Rb and Chk1. Mre11 was reanalyzed as a control (Table 5.5.A). As
Fas, Rb and Chk1 were not mutated in the nine t-AML cases and the MMR deficient
cell lines the analysis was not extended further. The transplant related AML samples
were not analysed due to lack of time. It seems unlikely, however, that Fas, Rb and
Chk1 are targets MSF t-AML (Table 5 .5 .0 ). Flt3 was not mutated in the AML
172

samples assayed, but due to time constraints the analysis was not extended to all
samples. This gene was identified as a potential target in M S r cell lines, however. It
would seem appropriate to investigate more M S r AMLs for changes in Flt3.
Table 5.5 Target gene analysis of AML samples.
A. Target gene analysis of chemotherapy related AML samples.
AML sample MSI Casp-5 Fas Fits Rb Cheki M rell1 - (A)10 N/D N/D N/D N/D N/D2 + (A)9/(A)10 N/D N/D N/D N/D N/D3 + (A)10 N/D N/D N/D N/D N/D4 + N/D N/D N/D N/D N/D N/D5 + (A)10 N/D N/D N/D N/D N/D6 + (A)10 N/D N/D N/D N/D N/D7 + (A)10 N/D N/D N/D N/D N/D8 - (A)9/(A)10 N/D N/D N/D N/D N/D9 + (A)10 N/D N/D N/D N/D N/D10 - (A)10 N/D N/D N/D N/D N/D11 - (A)10 N/D N/D N/D N/D N/D12 + (A)10 N/D N/D N/D N/D N/D13 + (A)10 N/D N/D N/D N/D N/D14 - (A)10 N/D N/D N/D N/D N/D15 + N/D N/D N/D N/D N/D N/D16 + N/D N/D N/D N/D N/D N/D17 + (A)10 (T)9 (T)10 (T)9 (A)9 (T)1118 + (A)10 (T)9 (T)10 (T)9 (A)9 N/D19 - n/d (T)9 (T)10 (T)9 (A)9 (T)1120 + (A)10 (T)9 (T)10 (T)9 (A)9 (T)1121 + (A)10 (T)9 (T)10 (T)9 (A)9 N/D22 - N/D (T)9 N/D N/D (A)9 N/D23 + (A)9/(A)10 (T)9 (T)10 (T)9 (A)9 (T)1124 - (A)10 (T)9 (T)10 (T)9 (A)9 (T)1125 - (A)10 (T)9 N/D (T)9 N/D (T)11
N/D, not determined.
B. Target gene analysis of transplant related AML samples.
AML sample Tissue Casp-5Ala BM (A)10Alb Heart (A)10A2a BM1 (A)9/(A)10A2b BM2 (A)9/(A)10A2c Lung (A)10A3a BM (A)9/(A)10A3b Heart (A)10A4a BM (A)10A4b Heart (A)10A5a BM (A)10A5b Heart/Lung (A)10A6a BM N/DA6b Lung N/DA7 BM (A)10
BM, bone marrow.
173
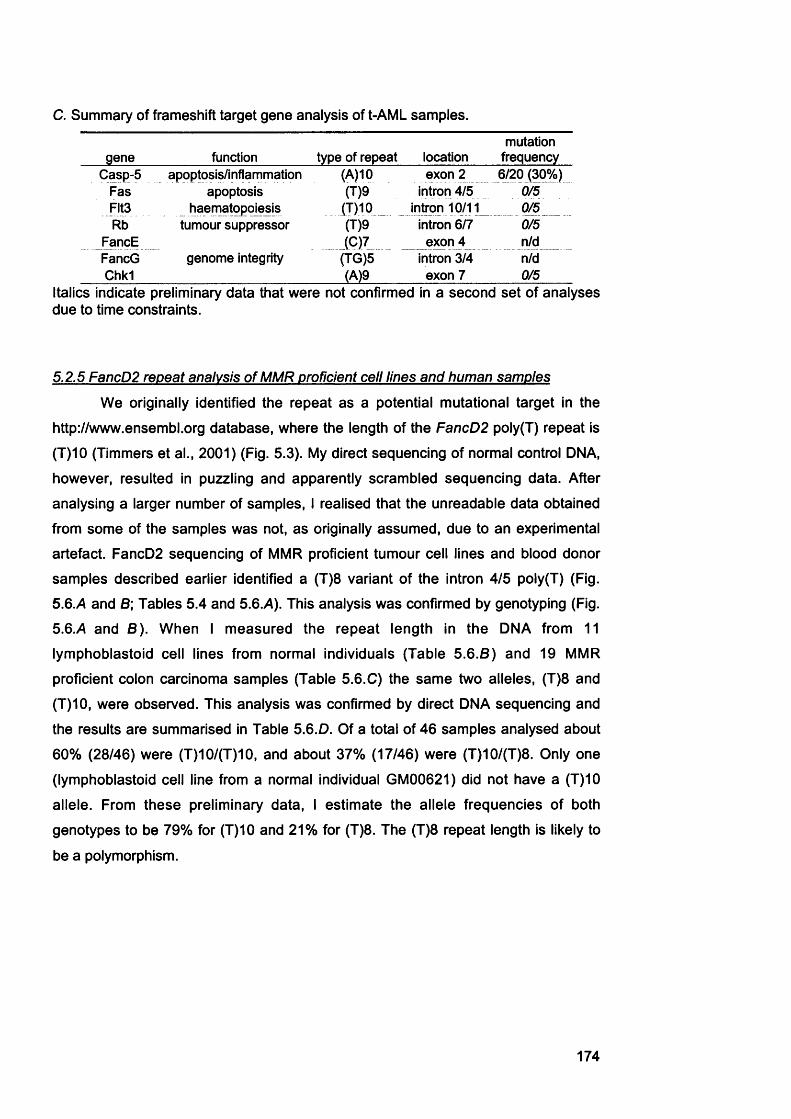
c. Summary of frameshift target gene analysis of t-AML samples.
gene function type of repeat locationmutation
frequencyCasp-5 apoptosis/inflammation (A)10 exon 2 6/20 (30%)
Fas apoptosis (T)9 intron 4/5 0/5Flt3 haematopoiesis (T)10 _ _ intron 10/11 0/5Rb tumour suppressor (T)9 intron 6/7 0/5
FancE (C)7 exon 4 n/dFancG genome integrity (TG)5 intron 3/4 n/dChkl (A)9 exon 7 0/5
Italics indicate preliminary data that were not confirmed in a second set of analyses due to time constraints.
5.2.5 FancD2 repeat analysis of MMR proficient cell lines and human samples
We originally identified the repeat as a potential mutational target in the
http://www.ensembl.org database, where the length of the FancD2 poly(T) repeat is
(T)10 (Timmers et al., 2001) (Fig. 5.3). My direct sequencing of normal control DNA,
however, resulted in puzzling and apparently scrambled sequencing data. After
analysing a larger number of samples, I realised that the unreadable data obtained
from some of the samples was not, as originally assumed, due to an experimental
artefact. FancD2 sequencing of MMR proficient tumour cell lines and blood donor
samples described earlier identified a (T)8 variant of the intron 4/5 poly(T) (Fig.
5.6.A and 8; Tables 5.4 and 5.6.A). This analysis was confirmed by genotyping (Fig.
5.6.A and 8 ) . When I measured the repeat length in the DNA from 11
lymphoblastoid cell lines from normal individuals (Table 5 .6 .8 ) and 19 MMR
proficient colon carcinoma samples (Table 5.6.C) the same two alleles, (T)8 and
(T)10, were observed. This analysis was confirmed by direct DNA sequencing and
the results are summarised in Table 5.6.D. Of a total of 46 samples analysed about
60% (28/46) were (T)10/(T)10, and about 37% (17/46) were (T)10/(T)8. Only one
(lymphoblastoid cell line from a normal individual GM00621) did not have a (T)10
allele. From these preliminary data, I estimate the allele frequencies of both
genotypes to be 79% for (T)10 and 21% for (T)8. The (T)8 repeat length is likely to
be a polymorphism.
174
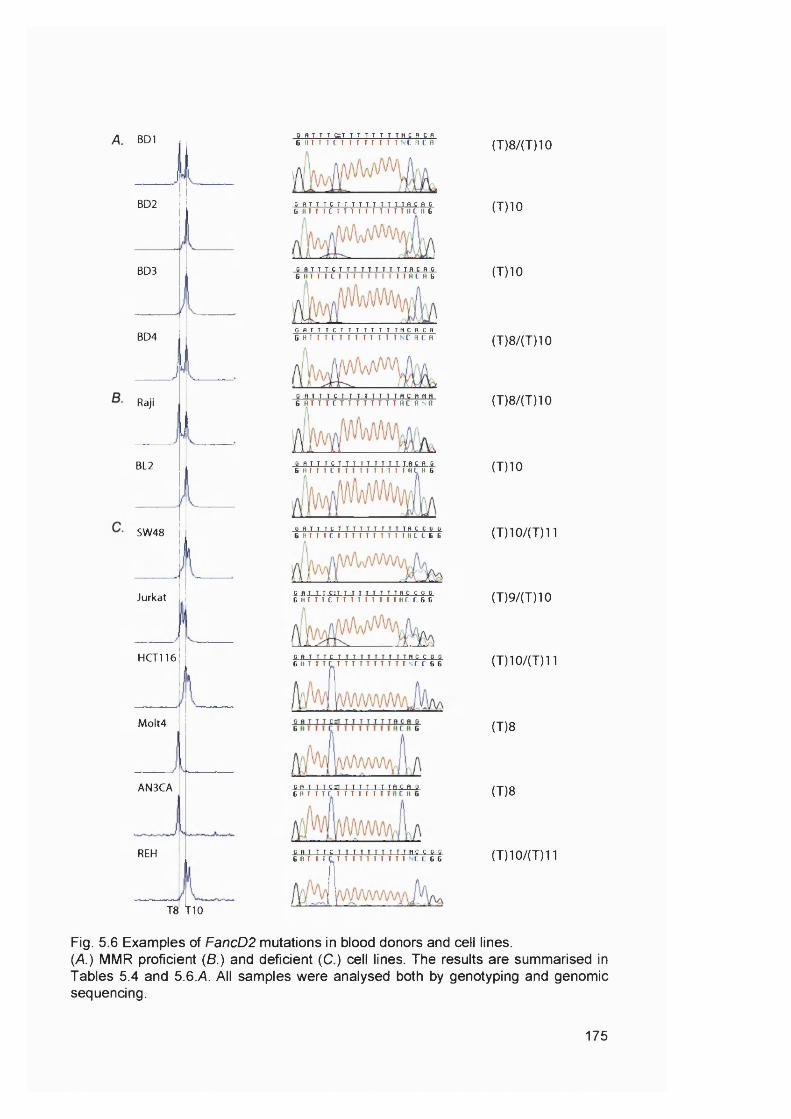
A. BD1
BD2
BD3
BD4
Raji
BL2
SW48
Jurkat
H CT116
M o lt4
AN3CA
REH
T8 t lO
g n T T T S T T T T T T T T H C n C Pe n T T T C T T T T T T T T N C R C n
C R T T T C T T T T T T T T T T f l C R CG B T T T C T T T T T T T I T T H C f l 6
C R T T T C T T T T T T T T T T R C n C6 B T I T C T T T T T T T T T T H C H G
G R T T T C T T T T T T T T M C R C RG B T T T C T T T T T T T T N C n C H
O R T T T C T T T T T T T T R C R H HS B T T T C T T T T T T T T B C B n H
B R T T T C T T T T T T T T T T f l C R G6 H T T T C T T T T I T T T T T B C H 6
l H l l l l U U I l l H S U S i
C R T T T C I T T T T T T T T T R C C G BG B T T T C T T T T T I T T T B C C G G
G R T T T C T T T T T T T T T T H C C G G G B -------------------------------------------- ----------------C C G G
G R T T T C a r i T T T T T T R C R G G B T T T C T T T T T T .................... .....
T R C RH
G R T T T C T T T T T T T T T T H C C G G G B T T T C T T T T T T T T T T C C G G
(T)8/(T)10
(T)10
(T)10
(T)8/(T)10
(T)8/(T)10
(T)10
(T)10/(T)11
(T)9/(T)10
(T)10/(T)11
(T)8
(T)8
(T) 10/(7)11
Fig. 5.6 Examples of FancD2 mutations in blood donors and cell lines.(A ) MMR proficient (8.) and deficient (C.) cell lines. The results are summarised in Tables 5.4 and 5.6.A. All samples were analysed both by genotyping and genomic sequencing.
175
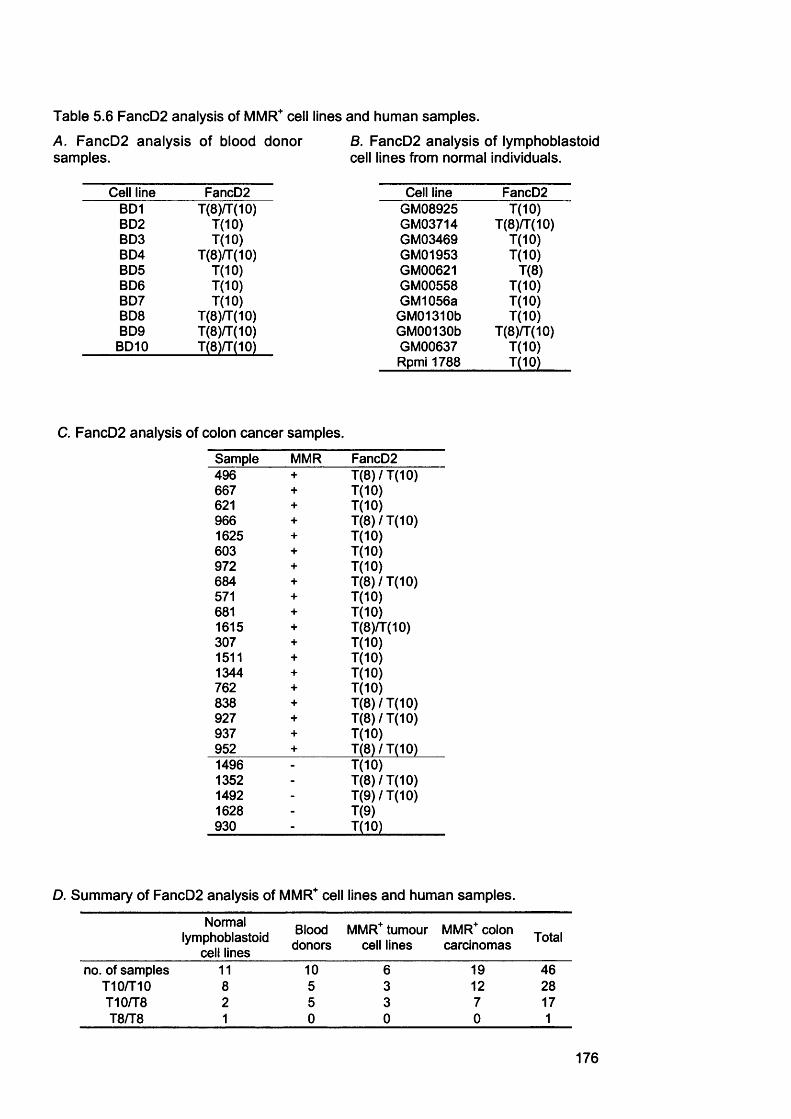
Table 5.6 FancD2 analysis of MMR" cell lines and human samples.
A. FancD2 analysis of blood donor samples.
8. FancD2 analysis of lymphoblastoid cell lines from normal individuals.
Cell line FancD2 Cell line FancD2BD1 T(8)/T(10) GM08925 7(10)BD2 1(10) GM03714 T(8)/T(10)BD3 1(10) GM03469 7(10)BD4 T(8)7T(10) GM01953 7(10)BD5 T(10) GM00621 7(8)BD6 1(10) GM00558 7(10)BD7 T(10) GM1056a 7(10)BD8 T(8)/T(10) GM01310b 7(10)BD9 T(8)/T(10) GM00130b 7(8)/7(10)
BD10 T(8KT(10) GM00637 7(10)Rpmi 1788 T(10)
C. FancD2 analysis of colon cancer samples.
Sample MMR FancD2496 + 7(8)/7(10)667 + 7(10)621 + 7(10)966 + 7(8)/7(10)1625 + 7(10)603 + 7(10)972 + 7(10)684 + 7(8)/7(10)571 + 7(10)681 + 7(10)1615 + 7(8)fr(10)307 + 7(10)1511 + 7(10)1344 + 7(10)762 + 7(10)838 + 7(8)/7(10)927 + 7(8)/7(10)937 + 7(10)952 + 7(8)/7(10)1496 - 7(10)1352 - 7(8)/7(10)1492 - 7(9)/7(10)1628 - 7(9)930 - 7(10)
D. Summary of FancD2 analysis of MMR'’ cell lines and human samples.
Normal lymphoblastoid
cell lines
Blooddonors
MMR* tumour cell lines
MMR* colon carcinomas Total
no. of samples 11 10 6 19 46710/710 8 5 3 12 28710/78 2 5 3 7 1778/78 1 0 0 0 1
176

5.2.6 FancD2 analysis of MMR deficient human tumour cell lines and colon cancer
samples
To Investigate if this repeat is mutated in a MMR deficient background, the
panel of MMR' human tumour cell lines (Table 5.4) and five MMR' human colorectal
carcinoma samples (Table 5.6.C) were analysed. The FancD2 repeat length varied
between (T)8 and (T)11 among MMR deficient samples (Examples in Fig 5 .6 .C).
Four of the cell lines are homozygous for (T)10, whereas two did not have a
detectable (T)10 allele. Four cell lines were heterozygous (T)8/(T)10. In addition to
the (T)10 and (T)8 alleles seen in MMR proficient cells and tissues there were also
examples of (T)9 and (T)11, for example (T)9/(T)10 (Jurkat) and (T)10/(T)11
(HCT116). Two of the MMR defective colon carcinoma samples had at least one
(T)9 allele, whereas (T)11 was not observed (Table 5 .6 .C). The (T)11 and (T)9
alleles were only observed among MSF samples, but in most cases one (T)10 allele
was retained. In addition, there seems to be an overrepresentation of (T)8 only (two
cases) among the MSF cell lines, indicating that this genotype arose either due to
mutation or LOH.
5.2.7 FancD2 repeat analysis oft-AML samples
Tables 5.7.7\ and B show that FancD2 is mutated in MMR defective t-AML.
Among the chemotherapy-related AML cases, FancD2 mutations resulting in nine
Ts occurred twice (Table 5.7A ). One appeared to be mixed and retained both (T)8
and (T)10 wt genotypes. The second case was either homozygous for (T)9 or had
lost the second allele. For the transplant-related AML cases samples of normal
tissue DNA were available. The analysis indicates that in case A2 the FancD2 (T)10
allele was either mutated or lost in the AML. In A3 the presence of (T)8 and (T)10
alleles suggests a mutational event.
177

Table 5.7 FancD2 analysis of AML samples.
A. FancD2 target repeat length of chemotherapy related AML samples.
New sample no. MSI FancD21 - (1)102 + (1)93 + (T)104 + N/D5 + (T)106 + (1)107 + (T)108 - (T)8/T(9)/(T)109 + (T)1010 - (T)1011 - (T)8/(T)1012 + (T)1013 + (1)814 - (T)1015 + N/D16 + N/D17 + (T)8/(T)1018 + (T)1019 - (T)1020 + (T)1021 + (T)8/(T)1022 - (T)1023 + (T)8/(T)1024 - (T)1025 - (T)10
B. FancD2 target repeat length of chemotherapy related AML samples.
AML sample Tissue FancD2Ala BM (T)8/(T)10Alb Heart (T)8/(T)10A2a BM1 (T)8A2b BM2 (T)8/(T)10A2c Lung (T)8/(T)10A3a BM (T)8/(T)10A3b Heart (T)10A4a BM (T)8/(T)10A4b Heart (T)8/(T)10A5a BM (T)8/(T)10A5b Heart/Lung (T)8/(T)10A6a BM N/DA6b Lung N/DA7 BM (T)8/(T)10
BM, bone marrow.
178

5.2.8 FancD2 expression
To see if an altered repeat length affects RNA splicing, RT-PCR was
performed. In this assay a FancD2 fragment spanning exon 5 to 9 was amplified
{FancD2 gene diagram Fig. 5.3; RT-PCR assay Fig. 5.7A ). The p-actin fragment,
(Chapter 3), was again used a control. The size of the major RT-PCR amplification
product was not affected by alterations in the length of the poly(T) repeat. An
additional, faint band appeared (~ 250bp in length) from amplification of LoVo,
HCT116, AN3CA and REH DNA. Sensitive staining with SYBR Green I (data not
shown) indicated that this band was produced from all cell lines, independent of the
poly(T) tract lengths. The short product from all fifteen cell lines was excised,
purified and sequenced. The sequences obtained were identical and were used to
perform a ‘human genome BLAST 2' search, in which the 250bp band was aligned
with part of the FancD2 cDNA (Fig. 5 .7 .8). This alignment indicated that the
reduction in fragment size was due to skipping of exon 8, and not exon 6, which is
the closest to the repeat sequence. This identifies an exon 8 deleted mRNA as a
minor splice variant of FancD2.
5.2.9 Mitomycin C sensitivity assay
FA cells are hypersensitive to DNA cross-linking agents like MMC. This
property is used a diagnostic test for the FA phenotype (Grompe and D'Andrea,
2001). To see if different FancD2 genotypes affect MMC sensitivity Karen
Gascoigne assayed the effect of the drug on growth of three representative cell
lines, Raji TK' ((T)10/(T)8), Molt4 ((T)8/(T)8) and REH ((T)10/(T)11). She treated
these cells with varying concentrations of the drug, ranging from 10 to lOOOnM, and
monitored growth by cell counts at 24 hour intervals. The growth curves (Fig. 5.8) do
not indicate significant differences in sensitivity among the three cell lines. The
growth rates of both Raji TK' and Molt4 are reduced at 300nM and fully inhibited at
lOOOnM, whereas the growth rate of REH cells seems to be reduced at lOOnM. This
apparent slight sensitivity is most likely due to a generally slower growth rate of REH
cells. The length of the poly(T) repeat in FancD2 does not appear to affect the MMC
sensitivity of these cells. In particular, Molt4 which has only the (T)8 allele, was not
notably sensitized to MMC. This is consistent with the lack of an exon 6 splicing
defect and with (T)8 being a polymorphism.
179

A.
<z §
i I % 3
£Q Xa d I & Iuoc u
inmem § t
p-actin
FancD2
Fig. 5.7 FancD2 RT-PCR.7\. A fragment of the FancD2 RNA transcript spanning exon 5 to 9 and a p-actin fragment were amplified by mulitplex RT-PCR. (Fig. 5.3 and experimental details in Chapter 2) The p-actin fragment served as a control as described previously (Chapter 3). The PCR products were separated on a 2% agarose gel and visualised with ethidium bromide. The arrow indicates the band that was amplified from DNA from all cell lines assayed, even though the band is not visible in this image of the gel. B. This band was excised from the gel, purified and sequenced. A homology search was performed with these sequences using the human genome BLAST 2. The sequence aligment is shown. The sequence of the RT-PCR product is highlighted is blue.
180
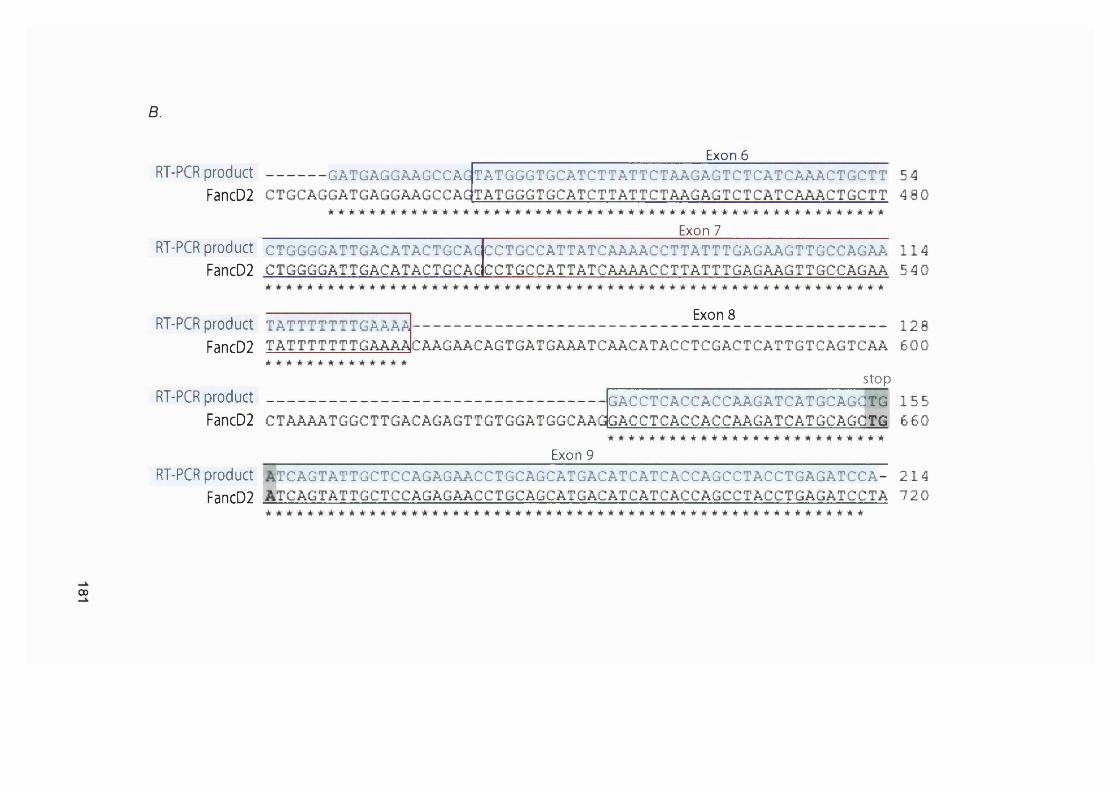
B.
RT-PCR p r o d u c t -------------- GATGAGGAAGCCAGFancD2 c t g c a g g a t g a g g a a g c c a g
Exon 6____________________TATGGGTGCATCTTATTCTAAGAGTCTCATCAAACTGCTT 54 TATGGGTGCATCTTATTCTAAGAGTCTCATCAAACTGCTT 4 8 0
Exon 7RT-PCR product CTGGGGATTGACATACTGCAG CCTGCCATTATCAAAACCTTATTTGAGAAGTTGCCAGAA 1 1 4
FancD2 CTGGGGATTGACATACTGCAGCCTGCCATTATCAAAACCTTATTTGAGAAGTTGCCAGAA 54 0
RT-PCR product t a t t t t t t t g a a à a
FancD2 T A T T T T T T T G A A A A
Exon 8 1 2 8
CAAGAACAGTGATGAAATCAACATACCTCGACTCATTGTCAGTCAA 6 0 0
RT-PCR productstop
GACCTCACCACCAAGATCATGCAGGTG 1 5 5 FancD2 CTAAAATGGCTTGACAGAGTTGTGGATGGCAAdGACCTCACCACCAAGATCATGCAGCTG 6 6 0
Exon 9
RT-PCR product STCAGTATTGCTCCAGAGAACCTGCAGCATGACATCATCACCAGCCTACCTGAGATCCA- 2 1 4 FancD2 ATCAGTATTGCTCCAGAGAACCTGCAGCATGACATCATCACCAGCCTACCTGAGATCCTA 7 2 0
00

A. Raji(T)8/(T)10 B.l.E+09
1.E+08
.X
X
z
'#-----
1.E+06 ,jr'
1.E+050 102 4 6 8
Molt4 (T)8l.E+09
l.E+08
..-X
= l.E+07
. - r '
l.E+06
l.E+050 2 6 8 104
Days Days
C. REH (T)10/(T)11l.E+09
l.E+08
l.E+06
l.E+050 2 4 6 8 10
Fig. 5.8 Mitomycin C sensitivity assay.Raji TK- [A), Moit4 (B.) and REH (C.) were treated with the following concentrations of Mitomycin C:
— - 0 nM — ■— 1 0 nM — - A - - - 30 nM — X— 100 nM — * — 300 nM — #— 1000 nM
Cell growth was monitored by cell counts at 24 hour intervals. This experiment was carried out by Karen Gascoigne under my supervision.
Days
182

5.2.10 Functionality of FancD2
In normal cells, the FancD2 protein is activated by monoubiquitination in
response to DNA damage (Garcia-Higuera et al., 2001). The FancD2 protein is
present in two isoforms, designated FancD2-S and FancD2-L. FancD2-S, normally
the predominate form is not modified, but MMC or IR treatment activates its
monoubiquitination. This activation can be observed on Western blots as an
apparent increase in size (Fig. 5.9.A). Peter Macpherson assayed the FancD2
response of four cell lines, Raji ((T)10/(T)8), GM00621 ((T)8), Molt4 ((T)8) and
AN3CA ((T)8). The FancD2-S to L shift after treatment with gamma irradiation was
observed in all four cell lines (Fig. 5.9.8). Thus the repeat (T)8 genotype does not
appear to affect FancD2 monoubiquitination, at least as seen by this simple
approach. In particular, S to L conversion was efficient in GM00621, the only (T)8
example among normals. Again these date are consistent with a polymorphism that
does not overtly affect FancD2 function.
5.3 Summary and DiscussionThe aim of the experiments described in this chapter was to examine genes
which might be targets for insertion/deletion mutations in M S r t-AML. Ultimately, the
identification of mutational targets is a prerequisite for understanding how M S r t-
AML develops.
5.3.1 M re ll
My data confirms that M r e l l is a target in MMR defective tumour cell lines.
M r e l l was first described as a target by Giannini et al. who identified frequent
M r e l l frameshift-like mutations in human colorectal cancer cell lines and primary
tumours (Giannini et al., 2002). Their SW48 ((T)10), HCT116 ((T)9/(T)10) and LoVo
((T)9) have the same mutations that I identified in the Clare Hall stocks of these cell
lines. This suggests that the mutations originally occurred in the tumour. The
convergence of data also serves to validate my method of analysis. Mutations in
M r e l l were identified in three of the chemotherapy-related AML cases. It would
have been very interesting to analyse the transplant AML samples for M r e l l
mutations, but this was not possible due to time constraints.
The functional implications of a -1 deletion in the intronic (T)11 tract of
MRE11 remain unclear. M re ll is a key participant in recombinational DNA repair
183

A.
FancD2-LFancD2-S IR FancD2-L
FancD2-S
e.
GM00621 (T8)
noIR +15 GY
_-'FancD2-L — FancD2-S
Raji (T10/8)
noIR +15 GY
mm fancD2-LFancD2-S
M0LT4 (T8)
noIR +15 GY
AN3CA (T8)
-FancD2-L■FancD2-S
noIR +15 GY
-FancD2-L■FancD2-S
Fig. 5.9 FancD2 Western Blot.A. Diagram of FancD2 activation by monoubiquitination. FancD2-L (long), high molecular weight form of FancD2. FancD2-S (short), low molecular weight isoform of FancD2. 8. Whole molecular weight extracts were prepared from the indicated cell lines with or without exposure to gamma irradiation (15 Gy) and immunoblotted with an anti-FancD2 antibody. IR treatment and immunoblotting was carried out by Peter Macpherson.
184

and cell cycle checkpoints (discussed in Chapter 1). This suggests that HR and
checkpoint functions are compromised in M S r tumours. The Mre11-Nbs1-Rad50
protein complex is important in imposing the intra-S phase checkpoint and in the
early steps of recombinational repair (Thompson and Schild, 2002). Inactivation of
the complex facilitates the development of cancer. Individuals with inherited
deficiencies in Nbs1 or Mre11 (AT-LD) are prone to lymphoid malignancies (Shiloh,
1997). NBS1 has been reported to be mutated in 15% of childhood ALL (Varon et
al., 2001), and hMre11 mutations have been noted in lymphoma and breast
carcinoma (Fukuda et al., 2001). Although null mutations in RadSO are lethal in
mice, hypomorphic mutations are associated with chromosome instability and have
a particularly severe effect on development of haematopoietic cells (Bender et al.,
2002). These preliminary observations reinforce the idea that a reduced ability or
efficiency of recombinational repair of DNA breaks and/or cell cycle check point
signalling may be important in the development of AML. In addition to MSF, it
appears that a significant proportion of t-AMLs has compromised HR and
checkpoint functions.
5.3.2 Caspase 5
C asp-5 was first identified as a target for frameshift mutations in MSF
endometrial and gastrointestinal tumours (Schwartz et al., 1999). In agreement with
this I identified heterozygous one base deletions in six and homozygous one base
deletions in two MSF tumour cell lines. We also identified Casp-5 mutations in about
30% of our M S r t-AML cases. Recently three groups analysed leukaemia cell lines
or clinical samples for C asp-5 mutations. Takeuchi et al. reported frameshift
mutations in three of 12 MSF lymphoma and leukaemia cell lines (Olipitz et al.,
2002; Scott et al., 2003; Takeuchi et al., 2003). They observed one base deletions
in CCRF-CEM, Jurkat (both T-lineage ALL cell lines) and REH (B-lineage ALL cell
line). These findings are consistent with my data. I also found our CCRF-CEM ((A)9)
and Jurkat ((A)9) to be mutated, but I did not detect any mutations in REH. These
authors did not detect C asp-5 mutations in the T-lineage ALL cell line Molt4,
whereas I found a heterozygous -1 frameshift in the same line. These discrepancies
may reflect long-term propagation in culture with a MMR' phenotype. They suggest
that the mutations may have arisen during growth in the laboratory rather than in the
tumours from which the lines were established.
185

Olipitz et al. did not identify any Casp-5 mutations in 5 therapy-related
leukaemia patients (Olipitz et al., 2002). Scott et al. identified -1 or +1 frameshift
mutations in four of 16 human t-ALL cases (Scott et al., 2003). We identified Casp-5
mutations in 6 of 20, MSI+ t-AML cases, two of which were transplant-related. Our
results are consistent with those obtained by Scott et al. and taken together, the
data suggest that Casp-5 mutations occur in about 25% of t-AMLs.
It is not known if the Casp-5 mutations found in tumours have a functional
role in tumourigenesis. Their effects are difficult to predict due to the lack of
knowledge of the precise function of the Casp-5 protein. Furthermore, the caspase
family of proteins contains more than ten members, and little is known about their
exact function and the degree of redundancy among them. Proteases of the
caspase family are though to play a role in both apoptosis, and immune and
inflammatory responses (Creagh et al., 2003). Inhibition of either or both might play
a role in tumour progression.
5.3.3 Flt3
Flt3 mutations were identified in the MMR defective cell lines. Jurkat, SW48,
LoVo, DLD-1 and AN3CA were heterozygous for a one base deletion in the intronic
(T)10 tract in Flt3. Only five of the 20 MSF t-AML samples were analysed. None
were mutated. As the frequency of these mutations among cell lines is lower than
the frequencies obtained from Mre11 and Casp-5, mutations among MSI* t-AML
samples could have been missed by examining only five cases. It would be
interesting to carry out a more extensive analysis of this gene in t-AML samples in
future.
Even though mutations in Flt3 were identified in MSI* tumour cell lines, it is
not clear if this mutation would affect Flt3 function. The (T)10 repeat analysed is
located close to a splice site in intron 10/11 of the gene and frameshift mutations
would only have an effect if they affect splicing of e x o n ll. Moreover, the Flt3
mutations found in human tumours (either ITDs or point mutations) are activating
mutations (Stirewalt and Radich, 2003). Skipping of exon 11 due to a splice defect
would lead to a premature stop codon in exon 13. This would result in a truncated
protein and likely loss of function. Inactivation of one copy might contribute to
tumour development by increasing the risk that the remaining copy might succumb
to activating mutations.
186

5.3.4 Other genes
The other genes assayed for frameshift mutations in repeat sequences were
Fas, Rb and Chk1. These genes were not mutated in any of the M S r cell lines or
leukaemia samples tested, and hence do not appear to be targets. It is possible that
mutations in these genes would be highly detrimental for the cell and would
therefore be selected against.
5.3.5 New assignment of case 8
One chemotherapy-related AML case (case 8) was designated microsatellite
stable (Chapter 3). As no non-tumour material was available from this patient, this
assignment was based on the analysis of Bat26 alone. Even though Bat26 is
thought to identify more than 90% of all M S r cases, it can lead to false negatives.
Casp-5 and FancD2 analysis of this case shows that both genes are mutated, which
indicates that case 8 was misassigned. This increases the frequency of M S r cases
in this series to 17/25 (68%), a marginal change.
5.3.6 Fanconi anaemia pathway
Two genes part of the FA pathway, FancG and FancE were analysed for
frameshift mutations in MSF cells. Neither the exonic (0 )7 tract in FancE, not the
intronic (TG)5 repeat in FancG were found to be mutated in the panel of MMR
deficient cell lines tested.
My analysis of the intronic poly(T) repeat of FancD2 - designated (T)10 in
the EMBL database - revealed what appears to be a previously unidentified
polymorphic variant, (T)8. Analysis of DNA from a number of MMR proficient tumour
cell lines, lymphoblastoid cell lines from normal individuals, MSI' colon cancers and
blood donors (46 samples in total) suggested that the allele frequency of (T)10 is
about 80% and the frequency of the (T)8 allele is about 20%. W e found only a single
example of apparent homozygosity for (T)8 among non-MSF cells.
Analysis of DNA from MSF tumours or cell lines identified (T)9 or (T)11
alleles, and an apparent overrepresentation of (T)8 which is probably also due to
mutation. As the (T)9 and (T)11 alleles appear only in a MMR defective background
they are most likely due base addition/deletion mutations. The overrepresentation of
(T)8 could be either due to mutations or LOH. Analysis of MSF t-AML samples
187

identified two cases which were mutated. Of these cases one was heterozygous
and another apparently homozygous for (T)9. Analysis of AML/MDS and control
tissue samples of the transplant-related leukaemia cases showed that in one case
the (T)8 genotype originated from the original (T)8/(T)10 genotype (case A2), either
by mutation or LOH. In a second case (case A3) the (T)8/(T)10 genotype arose due
to mutation of (T)10 in the normal control sample. It hence seems that even though
the (1)8 allele does occur in normal healthy individuals it can also arise from (T)10
due to frameshift mutations.
I did not find evidence that the length of the intronic poly(T) tract affects
splicing of the FancD2 mRNA, FancD2 protein function, or activity of the FA
pathway. RT-PCR of a FancD2 fragment spanning exon 5 to 9 showed that the
length of this repeat does not detectably affect splicing of exon 6. Skipping of exon 6
would result in a premature stop codon in exon 8. These aberrantly spliced
transcripts would most likely be degraded by nonsense-mediated decay (NMD)
(Hentze and Kulozik, 1999). RT-PCR of FancD2 revealed a second, shorter
(~250bp) band due to skipping of exon 8 which appears to represent a minor splice
variant. Skipping of this exon also results in a premature stop codon, in this case in
exon 9. As this aberrant transcript was identified by the RT-PCR assay, it serves as
a positive control for the apparent absence of a transcript lacking exon 6 (would also
result in a ~250bp fragment). Furthermore, no evidence for a truncated FancD2
protein was found by Western blotting.
Western blots of the FancD2 protein showed that it is monoubiquitinated
after DNA damage independently of the length of the poly(T) tract. In cell lines of the
(T)8 and (T)8/(T)10 genotype IR treatment activated the conversion of FancD2-S to
FancD2-L with apparently similar efficiency. This suggests that the product of the
(T)8 isoform is likely to be functional.
Sensitivity to MMC, one of the factors used for FA diagnosis (D'Andrea and
Grompe, 2003), was also apparently independent of the FancD2 genotype. The
growth curves after MMC treatment for Raji TK" ((T)10/(T)8), Molt4 ((T)8/(T)8) and
REH ((T)10/(T)11) were similar. The slight difference in sensitivity of REH is most
likely due to their generally slower growth rate and I conclude that the (T)8 FancD2
allele is functionally adequate.
188

In summary, even though we identified FancD2 as a target for frameshift-like
mutations, these mutations did not seem to have an effect on the functioning of the
FA pathway.
Circumstantial evidence implicates a defective FA pathway in the
development of t-AML. FA patients are particularly susceptible to AML. Cytogenetic
abnormalities found in FA AML patients are often strikingly similar to those found in
t-AML rather than de novo cases (mainly 5q-, monosomy 7, 7q-) (Alter, 2003;
D'Andrea and Grompe, 2003). The FA pathway furthermore links BRCA1 and
BRCA2 to leukaemia. The role of the five FA proteins FANCA, C, E, F and G is to
facilitate the interaction of FANCD2 with BRCA1 (Garcia-Higuera et al., 2001). In
addition, BRCA2 has recently been identified as the FA gene FANCD1 (Hewlett et
al., 2002). A link between AML and breast cancer susceptibility is consistent with the
observed overrepresentation of early onset breast cancer in our chemotherapy-
related AML cases (Chapter 3). Lack of BRCA2/FancD2 expression was identified in
3 of 15 AML cell lines (Shimamura et al., 2003), but this awaits confirmation in
clinical samples. All these pieces of circumstantial evidence suggest that the
possible involvement of the FA pathway in t-AML is worth pursuing.
5.4 Conclusion
The data presented in this chapter identify Flt3 and FancD2 as targets for
frameshift mutations in MMR defective cell lines. C asp -5 and F a n c D 2 were
identified as targets for addition/deletion mutations in MSF t-AML cases. Although
this adds to the list of potential targets for selective inactivation in MSI* AML, the
detailed description of events during leukaemogenesis awaits a more thorough
analysis of genetic changes in this disease.
189

Chapter 6 : Discussion and Conclusion
The work described here shows that MSI Is common (>60% ) among
chemotherapy-related AML/MDS cases. Instability was associated with methylating
agent treatment, but also occurred after treatment with other drugs. Due to the small
number of patients in each treatment group, statistically significant correlations
between specific treatments and development of MSI t-AML could not be identified.
In some cases loss of MMR was due to hMLH1 promoter méthylation. This occurred
less frequently than was expected from the frequency of hMLH1 promoter
méthylation among M S r HNPGC-type cancers and I conclude that it is a relatively
rare event in M S r AML. A possible connection between t-APL and hMLH1 promoter
méthylation was identified, although again there were only a few cases. We also
noted an overrepresentation of early onset breast cancer cases among our group of
t-AML patients. This might indicate a connection between one of the breast cancer
susceptibility genes and AML development, and identifies an interesting area for
future investigation.
I also showed that treatment with 6-TG can, like MNU, select for MMR
defective, drug resistant cells. Analysis of the data of Gerhard Opiez at the
‘Collaborative Transplant Study’ indicates a greater than five-fold risk of AML
following heart/lung transplantation and immunosuppression. Clinical material from
seven transplant-related AML/MDS cases was obtained and analysed for MSI. All
seven of these cases were unstable. It seems probable that selection occurs in
patients’ bone marrow and that azathioprine is acting as a leukaemogenic agent.
Casp-5 and FancD2 were identified as targets for frameshift mutations in
M S r t-AML. As Flt3 was a mutational target in MMR defective human tumour cell
lines, this gene could also be a target in M S r t-AML. There was no time to analyse
this observation further, but it suggests a topic for further investigation. We also
identified a novel form of the FancD2 gene. This has an alteration in an intronic
ploy(T) tract. The length of this repeat did not appear to affect FancD2 function or
the general function of the FA pathway and appears to be a polymorphism. This
analysis requires further validation, however.
This study throws some light on the steps involved in the development of
chemotherapy- and organ transplantation-related AML/MDS. The data show that
loss of MMR is involved in more than half of t-AML cases. Similar frequencies for
190

MSI in chemotherapy-related t-AML have been reported by others (discussed in
Chapter 3). The findings support the original hypothesis that development of MSI" t-
AML could be the clinical counterpart to the laboratory-based selection for loss of
MMR by drug treatment. This is outlined in Fig. 6.1. A pluripotent haemopoietic stem
cell gives rise to myeloid precursors. An occasional cell might contain a MMR defect
following a rare random event. Under normal conditions this MMR' cell would
probably disappear owing to a selective disadvantage. If the patient's bone marrow
is exposed to either a methylating agent or a thiopurine, the MMR defective cell now
has a significant growth advantage. Its clonal expansion will give rise to a population
of genetically unstable precursors capable of accumulating mutations at an
increased rate. These mutations promote leukaemogenesis and lead to the
development of MSI" t-AML.
This model is supported by the following study. When mice with
reconstituted bone marrow containing a mixture of wild-type and msh2^' cells were
treated with the methylating agent temozolomide the proportion of MMR deficient
marrow cells was significantly increased (Reese et al., 2003). The same
experiments also showed that, without any selective pressure, the mshZ^' cells
disappeared from the bone marrow consistent with their reduced fitness in the
absence of selective pressure.
How does MSI" t-AML arise in patients that were not treated with methylating
agents or thiopurines? A possible explanation lies in the observation that MMR
defective cells are often hypermutable by DNA damage that may not differentially
affect their survival. MMR defective hamster cells are, for example, considerably
more susceptible to mutation by persistent UV-induced DNA damage (Nara et al.,
2001). msh2'^'mouse cells are hypermutable by the ethylating agent ENU even
though MMR defects are associated with only a slight increased resistance to the
drug (Claij et al., 2003). During drug treatment this susceptibility to DNA damage-
induced mutations would be superimposed on the already high spontaneous
mutation rate of MMR defective cells and accelerate leukaemogenesis. This is
supported by the observation that msh2^' mice are more susceptible to ENU
induced tumours than their MMR" counterparts (Claij et al., 2003).
191
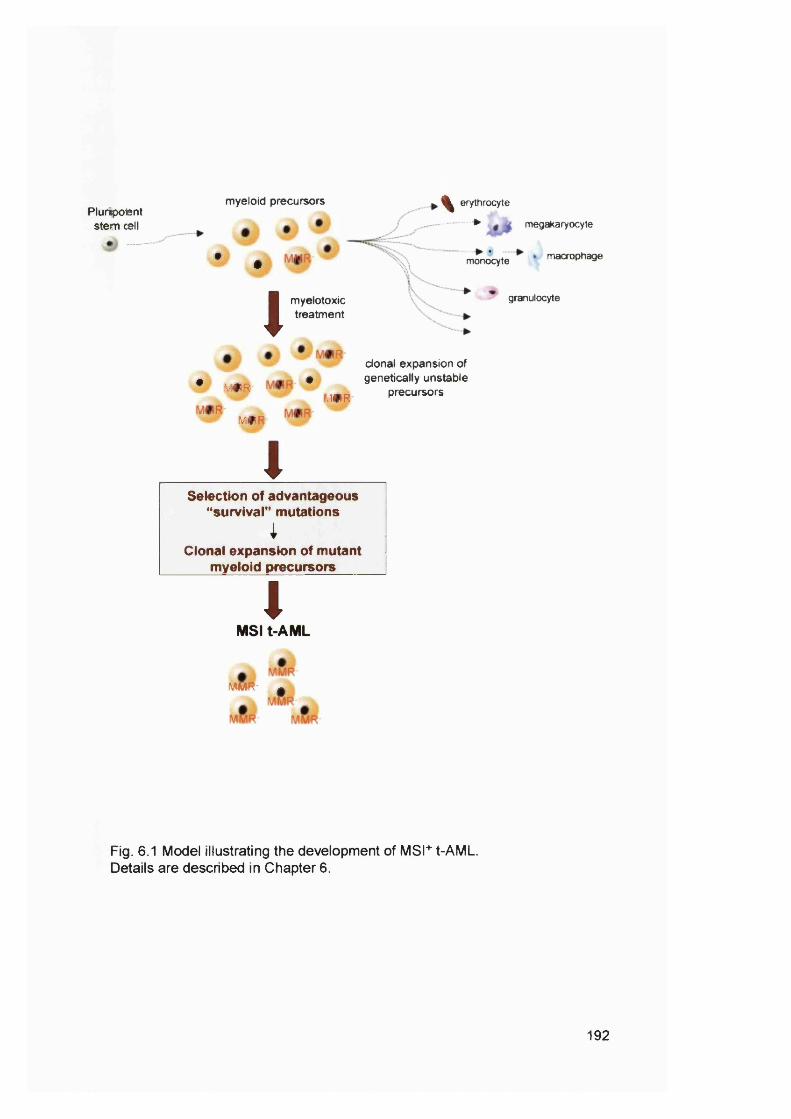
Plurmpoient stem cell
myeloid precursors
• #
, ^ erythrocyte
> megakaryocyte
\ \mooocyte macrophage
imyelotoxictreatment
granulocyte
• ,V#R *
l..*R'MR
clonal expansion of genetically unstable
precursors
ISelection of advantageous
"survival" mutationsi
Clonal expansion of mutant myeloid precursors
IMSI t-AML
mm- ^
Fig. 6.1 Model illustrating the development of MSI+ t-AML. Details are described in Chapter 6.
192

We are slowly starting to refine our ideas about how t-AML arises after
chemotherapy and organ transplants. Unfortunately, our knowledge is not yet at the
point where it can help clinicians prevent this, mostly incurable, disease. Today
cancer treatment is based to a large degree on the oncologist’s intuition, and “not
giving your patient AML is simply the hallmark of being a good doctor”.
193

References
Aarbakke, J., Janka-Schaub, G. and Elion, G.B. (1997) Thiopurine biology and pharmacology. Trends Pharmacol. Sc/., 18, 3-8.
Aas, P.A., Otterlei, M., Falnes, P.O., Vagbo, C.B., Skorpen, F., Akbari, M.,Sundheim, O., Bjoras, M., Slupphaug, G., Seeberg, E. and Krokan, H.E. (2003) Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature, 421, 859-863.
Aebi, S., Fink, D., Gordon, R., Kim, H.K., Zheng, H., Fink, J.L. and Howell, S.B.(1997) Resistance to cytotoxic drugs in DNA mismatch repair-deficient cells. Clin. Cancer Res., 3, 1763-1767.
Al-Tassan, N., Chmiel, N.H., Maynard, J., Fleming, N., Livingston, A.L., Williams, G.T., Hodges, A.K., Davies, D.R., David, S., Sampson, J R. and Cheadle, J.P. (2002) Inherited variants of MYH associated with somatic G:C-> T:A mutations in colorectal tumors. Nat. Genet., 30, 227-232.
Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K. and Watson, J.D. (1994) Molecular biology of the cell. Garland Publishing, Inc., New York.
Allen, D.J., Makhov, A., Grilley, M., Taylor, J., Thresher, R., Modrich, P. and Griffith, J.D. (1997) MutS mediates heteroduplex loop formation by a translocation mechanism. EMBO J., 16, 4467-4476.
Alter, B. (2003) Cancer in Fanconi anemia, 1927-2001. Cancer, 97, 425-440.Andersen, M., Philip, P. and Pedersen-Bjergaard, J. (1990) High risk of therapy
related leukemia and preleukemia after therapy with prednimustine, methotrexate, 5-fluorouracil, mitoxantrone and tamoxifen for advanced breast cancer. Cancer, 65, 2460-2464.
Andrew, S.E., McKinnon, M., Cheng, B.S., Francis, A., Penney, J., Reitmair, A.H., Mak, T.W. and Jirik, F.R. (1998) Tissues of MSH2-deficient mice demonstrate hypermutability on exposure to a DNA methylating agent. Proc. Natl. Acad. Sci. U. S. A., 95, 1126-1130.
Aquilina, G. and Bignami, M. (2001) Mismatch repair in correction of replication errors and processing of DNA damage. J. Cell. Physiol., 187,145-154.
Aquilina, G., Biondo, R., Dogliotti, E. and Bignami, M. (1993) Genetic consequences of tolerance to méthylation DNA damage in mammalian cells. Carcinogenesis, 14, 2097-2103.
Aquilina, G., Ceccotti, S., Martinelli, S., Soddu, S., Crescenzi, M., Branch, P.,Karran, P. and Bignami, M. (2000) Mismatch repair and p53 independently affect sensitivity to /V-(2-chloroethyl)-/V-cyclohexyl-/V-nitrosourea. Clin. Cancer Res., 6, 671-680.
Aquilina, G., Giammarioli, A.M., Zijno, A., Di Muccio, A., Dogliotti, E. and Bignami,M. (1990) Tolerance to 0®-methylguanine and 6-thioguanine cytotoxic effects: a cross-resistant phenotype in A/-methyl-/V-nitrosourea-resistant Chinese hamster ovary cells. Cancer Res., 50,4248-4253.
Baker, S.M., Bronner, C.E., Zhang, L., Plug, A.W., Robatzek, M., Warren, G., Elliott,E.A., Yu, J., Ashley, T., Arnheim, N., Flavell, R.A. and Liskay, R.M. (1995) Male mice defective in the DNA mismatch repair gene pms2 exhibit abnormal chromosome synapsis in meiosis. Cell, 82, 309-319.
Baker, S.M., Plug, A.W., Prolla, T.A., Bronner, C.E., Harris, A.C., Yao, X., Christie,D.-M., Monnell, C., Arnheim, N., Bradley, A., Ashley, T. and Liskay, R.M.(1996) Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat. Genet, 13, 336-342.
Barbanel, C., Kreis, H. and Crosnier, J. (1975) Utilisation au long cours desimmunosuppresseurs chez les malades porteurs d'un rein transplante. Ann. Med. Interne (Paris). 2, 69-74.
194

Baross-Francis, A., Andrew, S.E., Penney, J.E. and Jirik, F.R. (1998) Tumors of DNA mismatch repair-deficient hosts exhibit dramatic increases in genomic instability. Proc. Natl. Acad. Sci. U. S. A., 95, 8739-8743.
Barrett, A.J. (1996) Acute myeloblastic leukaemia. In Weatherall, D.J., Ledingham, J.G.G. and Warrell, D.A. (eds.), Oxford textbook of medicine. Oxford University Press, Oxford, Vol. 3, pp. 3404-3410.
Bast, R.C.J., Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Holland, J.F., Frei III,E. and Gansler, T.S. (2000) Cancer Medicine. BC Decker Inc., Hamilton, Ontario.
Batty, D.P. and Wood, R.D. (2000) Damage recognition in nucleotide excision repair of DNA. Gene, 241, 193-204.
Ben-Yehuda, D., Krichevsky, S., Caspi, O., Rund, D., Polliack, A., Abeliovich, D., Zelig, O., Yahalom, V., Paltiel, O., Peretz, T., Ben-Neriah, S., Yehuda, O. and Rachmilewitz, E.A. (1996) Microsatellite instability and p53 mutations in therapy-related leukemia suggest mutator phenotype. Biood, 88,4296-4303.
Bender, C.F., Sikes, M.L., Sullivan, R., Huye, I.E ., Le Beau, M., Roth, D.B.,Mirzoeva, O.K., Oltz, E.M. and Petrini, J.H.J. (2002) Cancer predisposition and hematopoietic failure in RadSO^^ mice. Genes & Deveipment, 16.
Bennett, J.M., Catovsky, D. and Daniel, M.T. (1982) The French-American-British cooperative group. Proposals for the classification of the myelodysplastic syndromes. Br. J. Haematol., 51, 189-199.
Bennett, J.M., Catovsky, D., Daniel, M.T., Flandrin, G., Galton, D.A., Gralnick, H.R. and Sultan, C. (1976) Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br. J. Haematol., 33, 451-458.
Berardini, M., Mazurek, A. and Fishel, R. (2000) The effect of 06-methylguanine DNA adducts on the adenosine nucleotide switch functions of hMSH2- hMSH6 and hMSH2-hMSH3. J. Biol. Chem., 275, 27851-27857.
Bignami, M., Casorelli, I. and Karran, P. (2003) Mismatch repair and response to DNA-damaging antitumour therapies.
Bignami, M., O'Driscoll, M., Aquilina, G. and Karran, P. (2000) Unmasking a killer: DNA 0®-methylguanine and the cytotoxicity of méthylation agents. Mutat. Res., 462, 71-82.
Blackwell, L.J., Martik, D., Bjornson, K.P., Bjornson, E.S. and Modrich, P. (1998) Nucleotide-promoted release of hMutSalpha from heteroduplex DNA is consistent with an ATP-dependent translocation mechanism. J. Biol. Chem., 273, 32055-32062.
Boland, C.R., Thibodeau, S.N., Hamilton, S.N., Sidransky, D., Eshleman, J.R., Burt, R.W., Meltzer, S.J., Rodriguez-Bigas, M.A., Fodde, R., Ranzani, G.N. and Srivastava, S. (1998) A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res., 58, 5248-5257.
Boyer, J.C., Risinger, J.l. and Faber, R.A. (1998) Stability of microsatellites in myeloid neoplasias. Cancer Genet. Cytogenet, 106, 54-61.
Branch, P., Aquilina, G., Bignami, M. and Karran, P. (1993) Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature, 362, 652-654.
Branch, P., Masson, M., Aquilina, G., Bignami, M. and Karran, P. (2000)Spontaneous development of drug resistance: mismatch repair and p53 defects in resistance to cisplatin in human tumor cells. Oncogene, 19, 3138- 3145.
Buermeyer, A.B., Deschenes, S.M., Baker, S.M. and Liskay, R.M. (1999)Mammalian DNA mismatch repair. Anna. Rev. Genet., 33, 533-564.
195

Carey, R.W., Holland, J.F., Sheghe, P.R. and Grahams, S. (1967) Association ofcancer of the breast and acute myelocytic leukemia. Cancer, 20,1988-1993.
Catovsky, D. (1996) Myelodysplastic syndromes. In Weatherall, D.J., Ledingham, J.G.G. and Warrell, D.A. (eds.), Oxford textbook of medicine. Oxford University Press, Oxford, Vol. 3, pp. 3425-3431.
Cejka, P., Stojic, L., Mojas, N., Russell, A.M., Helnlmann, K., Cannavo, E., dl Pietro, M., Marra, G. and Jlrlcny, J. (2003) Methylatlon-lnduced G2/M arrest requires a full complement of the mismatch repair protein hM LHI. EMBO J., 22, 2245-2254.
Chau, B.N. and Wang, J.Y. (2003) Coordinated regulation of life and death by RB. Nat Rev Cancer, 3,130-138.
Claij, N., van der Wal, A., Dekker, M., Jansen, L. and te RIele, H. (2003) DNA mismatch repair deficiency stimulates A/-ethyl-A/-nltrosourea-lnduced mutagenesis and lymphomagenesls. Cancer Res., 63, 2062-2066.
Clark, A.B., Valle, F., Drotschmann, K., Gary, R.K. and Kunkel, T.A. (2000)Functional Interaction of proliferating cell nuclear antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J. Biol. Chem., 275, 36498-36501.
Cline, S.D. and Hanawalt, P.C. (2003) Who's on first In the cellular response to DNA damage? Nat. Rev. Mol. Cell Bio., 4, 361.
ColussI, C., Flumlcino, S., Giuliani, A., Roslnl, S., MuslanI, P., Maori, C., Potten,C.S., Crescenzi, M. and Bignami, M. (2001) 1,2-dlmethylhydrazlne-lnduced colon carcinoma and lymphoma In msh2" ' mice. J. Natl. Cancer Inst, 93, 1534-1940.
ColussI, C., ParlantI, E., Degan, P., Aquilina, G., Barnes, D., Macpherson, P., Karran, P., Crescenzi, M., Dogliotti, E. and Bignami, M. (2002) The mammalian mismatch repair pathway removes DNA 8-oxodGMP Incorporated from the oxidized dNTP pool. Curr. Biol., 12, 912-918.
Cox, M.M. (2002) The nonmutatgenic repair of broken replication forks via recombination. Mutat. Res., 510,107-120.
Creagh, E.M., Conroy, H. and Martin, S.J. (2003) Caspase-actlvatlon pathways In apoptosis and Immunity. Immunol. Rev., 193,10-21.
Cummins, D., Sekar, M., Halil, O. and Banner, N. (1996) Myelosuppresslonassociated with azathloprlne-allopurlnol Interaction after heart and lung transplantation. Transplantation, 61,1661-1662.
D'Amours, D. and Jackson, S.P. (2002) The Mrel 1 complex; at the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Bio., 3, 317-327.
D'Andrea, A.D. (2003) The Fanconi road to cancer. Genes Dev., 17,1933-1936.D'Andrea, A.D. and Grompe, M. (2003) The Fanconi anaemla/BRCA pathway. Nat.
Rev. Cancer, 3, 23.Das-Gupta, E.P., Seedhouse, C.H. and Russell, N.H. (2001) MIcrosatelllte Instability
occurs In defined subsets of patients with acute myeloblastic leukaemia. Br. J. Haematol., 114, 307-312.
Datta, A., Hendrix, M., LIpsltch, M. and JInks-Robertson, S. (1997) Dual roles for DNA sequence Identity and the mismatch repair system In the regulation of mitotic crossing-over In yeast. Proc. Natl. Acad. Sci. U. S. A., 94, 9757-9762.
de Jager, M., van Noort, J., van Gent, D.C., Dekker, C., Kanaar, R. and Wyman, C. (2001) Human Rad50/Mre11 Is a flexible complex that can tether DNA ends. Mol. Ce//, 8,1129-1135.
De Rosa, M., Fasano, C., Panarlello, L., Scarano, M.I., Belli, G., lannelll. A., CIclllano, F. and Izzo, P. (2000) Evldene for a recessive Inherltence of Turcot's syndrome caused by compound heterozygous mutations with the PMS2gene. Oncogene, 19,1719-1723.
de Wind, N., Dekker, M., Berns, A., Radman, M. and te RIele, H. (1995) Inactivation of the mouse msh2 gene results In mismatch repair deficiency, méthylation
196

tolerance, hyperrecombination, and predisposition to cancer. Cell, 82, 321- 330.
de Wind, N., Dekker, M., Claij, N., Jansen, L., van Klink, Y., Radman, M., Riggins,G., van der Valk, M., van't Wout, K. and te Riele, H. (1999) HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. N at Genet, 23, 359-362.
de Wind, N., Dekker, M., van Rossum, A., van der Valk, M. and te Riele, H. (1998) Mouse models for hereditary nonpolyposis colorectal cancer. Cancer Res,58, 248-255.
Deng, G., Chen, A., Hong, J., Chae, M.S. and Kim, Y.S. (1999) Méthylation of CpG in a small region of the hMLhH promoter invariably correlates with the absence of gene expression. Cancer Res., 59, 2029-2033.
Dervieux, T., Blanco, J.G., Krynetski, E.Y., Vanin, E.F., Roussel, M.F. and Railing, M.V. (2001) Differing contribution of thiopurine methyltransferase to mercaptopurine versus thioguanine effects in human leukemic cells. Cancer Res, 61, 5810-5816.
Di Croce, L , Raker, V.A., Corsaro, M., Fazi, F., Fanelli, M., Faretta, M., Fuks, F., Lo Coco, F., Kouzarides, T., Nervi, C., Minucci, S. and Pelicci, P.G. (2002) Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science, 295,1079-1082.
Dinglay, S., Trewick, S.C., Lindahl, T. and Sedgwick, B. (2000) Defective processing of methylated single-stranded DNA by E. coll alkB mutants. Genes Dev., 14, 2097-2105.
Duckett, D.R., Bronstein, S.M., Taya, Y. and Modrich, P. (1999) hMutSalpha - and hMutLalpha -dependent phosphorylation of p53 in response to DNA methylator damage. Proc. Natl. Acad. Sci. U. S. A., 96,12384-12388.
Duckett, D.R., Drummond, J.T., Murchie, A.I.H., Reardon, J.T., Sancar, A., Lilley,D.M.J. and Modrich, P. (1996) Human MutSalpha recognizes damaged DNA base pairs containing 06-methylguanine, 04-methylthymine, or the cisplatin- d(GpG) adduct. Proc. Natl. Acad. Sci. U. S. A., 93, 6443-6447.
Duncan, T., Trewick, S.C., Koivisto, P., Bates, P.A., Lindahl, T. and Sedgwick, B. (2003) Reversal of DNA alkylation damage by two human dioxygenases.Proc. Natl. Acad. Sci. U. S. A., 99, 16660-16665.
Duval, A. and Hamelin, R. (2002) Genetic instability in human mismatch repair deficient cancers. Ann. Genet, 45, 71-75.
Edelmann, W., Cohen, P.E., Kane, M.F., Lau, K., Morrow, B., Bennett, S., Umar, A., Kunkel, T.A., Cattoretti, G., Chaganti, R., Pollard, J.W., Kolodner, R.D. and Kucherlapati, R. (1996) Meiotic pachytene arrest in MLH1-deficient mice.Ce//, 85, 1125-1134.
Edelmann, W., Umar, A., Yang, K., Heyer, J., Kucherlapati, M., Lia, M., Kneitz, B., Avdievich, E., Fan, K., Wong, E., Crouse, G., Kunkel, T., Lipkin, M.,Kolodner, R.D. and Kucherlapati, R. (2000) The DNA mismatch repair genes Msh3 and Msh6 cooperate in intestinal tumor suppression. Cancer Res, 60, 803-807.
Edelmann, W., Yang, K., Kuraguchi, M., Heyer, J., Lia, M., Kneitz, B., Fan, K.,Brown, A.M.C., Lipkin, M. and Kucherlapati, R. (1999) Tumorigenesis in Mlh1 and Mlh1/Apc1638N mutant mice. Cancer Res, 59,1301-1307.
Edelmann, W., Yang, K., Umar, A., Heyer, J., Lau, K., Fan, K., Liedtke, W., Cohen, P.E., Kane, M.F., Lipford, J.R., Yu, N., Crouse, G.F., Pollard, J.W., Kunkel, T.A., Lipkin, M., Kolodner, R.D. and Kucherlapati, R. (1997) Mutation in the mismatch repair gene Msh6 causes cancer susceptibility. Cell, 91,467-477.
Elion, G.B. (1989) The purine path to chemotherapy. Science, 244,41-47.Falnes, P.O., Johansen, R.F. and Seeberg, E. (2002) AlkB-mediated oxidative
déméthylation reverses DNA damage in Escherichia coll. Nature, 419 ,178- 182.
197

Fishel, R. (2001) The selection for mismatch repair defects in hereditarynonpolyposis colorectal cancer; revising the mutator hypothesis. Cancer Res., 61, 7369-7374.
Fleisher, A.S., Esteller, M., Wang, S., Tamura, G., Suzuki, H., Yin, J., Zou, T.-T., Abraham, J.M., Kong, D., Smolinski, K.N., Shi, Y.-Q., Rhyu, M.-G., Powell, S.M., James, S.P., Wilson, K.T., Herman, J.G. and Meltzer, S.J. (1999) hypermethylation of the hMLHI gene promoter in human gastric cancers with microsatellite instability. Cancer Res, 59, 1090-1095.
Friedberg, E.C. (2001) How nucleotide excision repair protects against cancer. Nat. Rev. Cancer, 1, 22-33.
Friedberg, E.C. (2003) DNA damage and repair. Nature, 421,436-440.Friedberg, E.G., Wagner, R. and Radman, M. (2002) Specialized DNA polymerases,
cellular survival, and the genesis of mutations. Science, 296,1627-1630.Friedberg, E.G., Walker, G.G. and Siede, W. (1995) DNA Repair and Mutagenesis.
ASM Press, Washington DG.Fukuda, T., Sumiyoshi, T., Takashi, M., Kataoka, T., Asahara, T., Inui, H., Watatani,
M., Yasutomi, M., Kamada, N. and Miyagawa, K. (2001) Alterations of the double-strand break repair gene MRE11 in cancer. Cancer Res., 61, 23-26.
Garcia-Higuera, I., Taniguchi, T., Ganesan, S., Meyn, M.S., Timmers, G., Hejna, J., Grompe, M. and D'Andrea, A.D. (2001) Interaction of the Fanconi anemia proteins and BRGA1 in a common pathway. Mol. Cell, 7, 249-262.
Genschel, J., Bazemore, L.R. and Modrich, P. (2002) Human exonuclease I is required for 5' and 3' mismatch repair. J. Biol. Chem., 277,13302-13311.
Giannini, G., Ristori, E., Gerignoli, F., Rinaldi, G., Zani, M., Viel, A., Ottini, L.,Crescenzi, M., Martinotti, S., Bignami, M., Frati, L , Screpanti, I. and Gulino, A. (2002) Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO reports, 3, 248-254.
Green, M.H., Lowe, J.E., Petit-Frere, G., Karran, P., Hall, J. and Kataoka, H. (1989) Properties of N-methyl-N-nitrosourea-resistant, Mex-derivaties of an SV40- immortalized human fibroblast cell line. Carcinogenesis, 10, 893-898.
Griffith, S., Branch, P., Xu, Y.-Z. and Karran, P. (1994) DNA mismatch binding and incision at modified guanine bases by extracts of mammalian cells: implications for tolerance to DNA méthylation damage. Biochemistry, 33, 4787-4793.
Grompe, M. and D'Andrea, A. (2001) Fanconi anemia and DNA repair. Human Molecular Genetics, 10, 2253-2259.
Gu, L., Gline-Brown, B., Zhang, F., Qiu, L. and Li, G.-M. (2002) Mismatch repair deficiency in hematological malignancies with microsatellite instability. Oncogene, 21, 5758-5764.
Hampson, R., Humbert, O., Macpherson, P., Aquilina, G. and Karran, P. (1997) Mismatch repair defects and 0^-methylguanine-DNA methyltransferase expression in acquired resistance to methylating agents in human cells. J. Biol. Chem., 272, 28596-28606.
Harfe, B.D. and Jinks-Robertson, S. (2000) DNA mismatch repair and genetic instability. Annu. Rev. Genet, 34, 359-399.
Hartmann, A., Zanardo, L., Bocker-Edmonston, T., Blaszyk, H., Dietmaier, W.,Stoehr, R., Gheville, J.G., Junker, K., Wieland, W., Knuechel, R., Rueschoff, J., Hofstaedter, F. and Fishel, R. (2002) Frequent microsatellite instability in sporadic tumors of the upper urinary tract. Cancer Res., 62, 6796-6802.
Hawn, M.T., Umar, A., Garethers, J.M., Mara, G., Kunkel, T.A., Boland, G.R. and Koi, M. (1995) Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res., 55, 3721-3725.
Hentze, M.W. and Kulozik, A.E. (1999) A perfect message: RNA surveillance and nonsense-mediated decay. Cell, 96, 307-310.
198

Herman, J.G. and Baylin, S.B. (2003) Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med., 349, 2042-2054.
Herman, J.G., Umar, A., Polyak, K., Graff, J.R., Ahuja, N., Issa, J.-P.J., Markowitz, S., Willson, J.K.V., Hamilton, S.R., Kinzler, K.W., Kane, M.F., Kolodner,R.D., Vogelstein, B., Kunkel, T.A. and Baylin, S.B. (1998) Incidence and functional consequences of hM LHI promotor hypermethylation in colorectal carcinoma. Proc. Natl. Acad. Sci. U. S. A., 95, 6870-6875.
Hoang, J.-M., Cottu, P.H., Thuille, B., Salmon, R.J., Thomas, G. and Hamelin, R.(1997) BAT-26, an indicator of the replication error phenotype in colorectal cancers and cell lines. Cancer Res., 57, 300-303.
Hoeijmakers, J.H.J. (2001) Genome maintenance mechanisms for preventing cancer. Nature, 411, 366-374.
Holmquist, G.P. and Maher, V. (2002) The bypass of DNA lesions by DNA and RNA polymerases. Mutat. Res., 510,1-7.
Horiike, S., Misawa, S., Kaneko, H., Sasai, Y., Kobayashi, M., Fujii, H., Tanaka, S., Yagita, M., Abe, T., Kashima, K. and Taniwaki, M. (1999) Distinct genetic involvement of the TP53 gene in therapy-related leukemia and myelodysplasia with chromosomal losses of Nos 5 and/or 7 and its possible relationship to replication error phenotype. Leukemia, 13,1235-1242.
Horton, J.K., Joyce-Gray, D.F., Pachkowski, B.F., Swenber, J.A. and Wilson, S.H. (2003) Hypersensitivity of DNA polymerase p null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair, 2, 27-48.
Howlett, N.G., Taniguchi, T., Olson, S., Cox, B., Waisfisz, Q., de Die-Smulders, 0 ., Persky, N., Grompe, M., Joenje, H., Pals, G., Ikeda, H., Fox, E.A. and D'Andrea, A.D. (2002) Biallelic inactivation ot BRCA2 in Fanconi anemia. Science, 297, 606-609.
Hsieh, P. (2001) Molecular mechanisms of DNA mismatch repair. Mutat. Res., 486, 71-87.
Huebner, G., Karthaus, M., Pethig, K., Freud, M. and Ganser, A. (2000)Myelodysplastic syndrome and acute myelogenous leukemia secondary to heart transplantation. Transplantation, 70, 688-690.
Humbert, O., Fiumicino, S., Aquilina, G., P., B., Oda, S., Zijno, A., Karran andBignami, M. (1999) Mismatch repair and differential sensitivity of mouse and human cells to methylating agents. Carcinogenesis, 20, 205-214.
Indraccolo, S., Minuzzo, S., Nicoletti, L , Cretella, E., Simon, M., Papakonstantinou,G., Hehlmann, R., Mion, M., Bertorelle, R., Roganovic, J. and Chieco- Bianchi, L. (1999) Mutator phenotype in human hematopoietic neoplasms and its association with deletions disabling DNA repair genes and bcl-2 rearrangements. Biood, 94, 2424-2432.
Jackson, S.P. (2002) Sensing and repairing DNA double-strand breaks. Carcinogenesis, 23, 687-696.
Jaenisch, R. and Bird, A. (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet, 33, 245- 254.
Jagmohan-Changur, S., Poikonen, T., Vilkki, S., Launonen, V., Wikman, F., Orntoft, T.F., Moller, P., Vasen, H., Tops, 0 ., Kolodner, R.D., Mecklin, J.-P.,Jarvinen, H., Bevan, S., Houlston, R.S., Aaltonen, LA., Fodde, R., Wijnen, J. and Karhu, A. (2003) EX01 variants occur commonly in normal population: evidence against a role in hereditary nonpolyposis colorectal cancer. Cancer Res, 63,154-158.
Jiricny, J. (2000) Mismatch repair: The praying hands of fidelity. Curr. Bioi., 10, R788-R790.
Jiricny, J. and Marra, G. (2003) DNA repair defects in colon cancer. Curr. Opin. Genet. Dev., 13, 61-69.
199

Jones, S., Emmerson, P., Maynard, J., Best, J.M., Jordan, S., Williams, G.T.,Sampson, J.R. and Cheadie, J.P. (2002) Biallelic germiine mutations in MYH predispose to multiple colorectal adenoma and somatic G:C->T:A mutations. Hum. Mol. Genet, 11, 2961-2967.
Kane, M.F., Loda, M., Gaida, G.M., Lipman, J., Mishra, R., Goldman, H., Jessup, J.M. and Kolodner, R. (1997) Méthylation of the hMLH1 Promotor correlates with lack of expression of hMLHI in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res., 57, 808-811.
Karp, J.E. and Smith, M.A. (1997) The molecular pathogenesis of treatment-induced (secondary) leukemias: foundations for treatment and prevention. Semin. Oncol., 24, 103-113.
Karran, P. (1996) Microsatellite instability and DNA mismatch repair in human cancer. Semin. Cancer Biol., 7 ,15-24.
Karran, P. (2000) DNA double strand break repair in mammalian cells. Curr. Opin. Genet. Dev., 10,144-150.
Karran, P. and Bignami, M. (1994) DNA damage tolerance, mismatch repair and genome instability. BioEssays, 16, 833-839.
Karran, P. and Marinus, M.G. (1982) Mismatch correction at 0®-methylguanine residues in E. coll DNA. Nature, 296, 868-869.
Kawate, H., Itoh, R., Sakumi, K., Nakabeppu, Y., Tsuzuki, T., Ide, F., Ishikawa, T., Noda, T., Nawata, H. and Sekiguchi, M. (2000) A defect in a single allele of the Mlh1 gene causes dissociation of the killing and tumorigenic actions of an alkylating carcinogen in methyltransferase-deficient mice.Carcinogenesis, 21, 301-305.
Kawate, H., Sakumi, K., Tsuzuki, T., Nakatsuru, Y., Ishikawa, T., Takahashi, S., Takano, H., Noda, T. and Sekiguchi, M. (1998) Separation of killing and tumorigenic effects of an alkylating agent in mice defective in two of the DNA repair genes. Proc. Natl. Acad. Sci. U. S. A., 95, 5116-5120.
Kiltie, A.E. and Ryan, A.J. (1997) SYBR Green I staining of pulsed field agarose gels is a sensitive and inexpensive way of quantitating DNA double-strand breaks in mammalian cells. Nucleic Acids Res., 25, 2945-2946.
Koi, M., Umar, A., Chauhan, D.P., Cherian, S.P., Carethers, J.M., Kunkel, T.A. and Boland, C.R. (1994) Human chromosome 3 correts mismatch repair deficiency and microsatellite instability and reduces /V-methyl-A/- nitrosoguanidine tolerance in colon tumor cells with homozygous hMHL1 mutation. Cancer Res., 54, 4308-4312.
Koivisto, P., Duncan, T., Lindahl, T. and Sedgwick, B. (2003) Minimal methylatedsubstrate and extended substrate range of Escherichia co//AlkB protein, a 1- methyladenine-DNA dioxygenase. J. Biol. Chem.
Krikorian, J.G., Anderson, J.L., Bieber, C.P., Penn, I. and Stinson, E.B. (1978)Malignant neoplasms following cardiac transplantation. The Joumai of the American Medical Association, 240, 639-643.
Krokan, H.E., Standal, R. and Slupphaug, G. (1997) DNA glycosylases in the base excision repair of DNA. Biochem. J., 325,1-16.
Kunkel, T.A. (1996) DNA synthesis errors, mutators and cancer. In Lindahl, T. (ed.). Genetic instability in Cancer. Cold Spring Harbor Laboratory Press, Vol. 28, pp. 3-20.
Lamers, M.H., Perrakis, A., Enzlin, J.H., Winterwerp, H.H.K., de Wind, N. and Sixma, T.K. (2000) The crystal structure of DNA mismatch binding repair protein MutS binding to a G:T mismatch. Nature, 407, 711-716.
Lee, S.M., Crowther, D., Scarffe, J.H., Dougal, M., Rafferty, J.A. and Margison, G.P. (1992) Cyclophospahamide decreases 0®-alkylguanine-DNA alkyltransferase activity in peripheral lymphocytes of patients undergoing bone marrow transplantation. Br. J. Cancer, 66, 331-336.
200

Lehmann, A. (2002) Replication of damaged DNA in mammalian cells: new solutions to an old problem. Mutat. Res., 509, 23-34.
Lennard, L., Van Loon, J.A. and Weinshilboum, R.M. (1989) Pharmacogenetics of acute azathioprine toxicity: relationship to thiopurine methyltransferase genetic polymorphism. Clin. Pharmacol. Thar., 46,149-154.
Lensch, M.W., Rathbun, R.K., Olon, S.B., Jones, G.R. and Bagby, G.C.J. (1999) Selective pressure as an essential force in molecular evolution of myeloid leukemic clones: a view from the window of Fanconi anemia. Leukemia, 13, 1784-1789.
Leone, G., Mele, L., Pulsoni, A., Equitani, F. and Pagano, L. (1999) The incidence of secondary leukemias. Haematologica, 84.
Leone, G., Voso, M.T., Sica, S., Morosetti, R. and Pagano, G. (2001) Therapy related leukaemias: susceptibility, prevention and treatment. Leak.Lymphoma, 41,255-276.
Levis, M. and Small, D. (2003) FLT3: ITDoes matter in leukemia. Leukemia, 17, 1738-1752.
Li, G.-M. and Modrich, P. (1995) Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologues. Proc. Natl. Acad. Sci. U. S. A., 92,1950-1954.
Li, G.-M., Wang, H. and Romano, L.J. (1996) Human MutSalpha specifically binds to DNA containing aminofluorene and acetylaminofluorene adducts. J. Biol. Chem., 271, 24084-24088.
Lieber, M.R., Ma, Y., Pannicke, U. and Schwarz, K. (2003) Mechanism andregulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Bio., 4, 712-720.
Lin, X. and Howell, S.B. (1999) Effect of loss of DNA mismatch repair ondevelopment of topotecan-, gemcitabine-, and paclitaxel-resistant variants after exposure to cisplatin. Mol Pharmacol, 56, 390-395.
Lind, M. and Ardiet, C. (1993) Pharmacokinetics of alkylating agents. In Cancer Surveys 17: Pharmacokinetics and Cancer Chemotherapy, pp. 169-176.
Lindahl, T. (1996) The Croonian Lecture, 1996: Endogenous damage to DNA.Philos. Trans. R. Soc. Lond. B. Biol. Sci., 351,1529-1538.
Lindahl, T. (2000) Suppression of spontaneous mutatgenesis in human cells by DNA base excision-repair. Mutat. Res., 462,129-135.
Lindahl, T. (2001) Past, present and future aspects of base excision-repair. Prog. Nucleic Acid Res. Mol. Biol., 68, xvii-xxx.
Lindahl, T. and Barnes, D.E. (2000) Repair of endogenous damage. Cold Spring Harb. Symp. Quant. Biol., LXV, 127-133.
Lipkin, S.M., Moens, P.B., Wang, V., Lenzi, M., Shanmugarajah, D., Gileous, A., Thomas, J., Cheng, J., Touchman, J.W., Green, E.D., Schwartzberg, P., Collins, F.S. and Cohen, P.E. (2002) Meiotic arrest and aneuploidy in MLH3- deficient mice. Nat. Genet, 31, 385-390.
Lipkin, S.M., Wang, V., Jacoby, R., Banerjee-Basu, S., Baxevanis, A.D., Lynch,H.T., Elliott, R.M. and Collins, F.S. (2000) MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. N at Genet, 24, 27-35.
Loukola, A., Ekiin, K., Laiho, P., Salovaara, R., Kristo, P., Jarvinen, H., Mecklin, J.- P., Launonen, V. and Aaltonen, L.A. (2001) Microsatellite marker analysis in screening for hereditary nonpolyposis colorectal cancer (HNPCC). Cancer Res., 61, 4545-4549.
Malkhosyan, S., Rampino, N., Yamamoto, H. and Perucho, M. (1996) Frameshift mutator mutations. Nature, 382,499-500.
Margison, G.P. and Santibanez-Koref, M.F. (2002) 0®-alkylguanine-DNAalkyltransferases: Role in carcinogenesis and chemotherapy. BioEssays, 24, 255-266.
201

Markowitz, S., Wang, J., Myeroff, L , Parsons, R., Sun, L.Z., Lutterbaugh, J., Fan, R.S., Zborowska, E., KInzler, K.W., Vogelstein, B., Brattain, M. and Willson, J.K.V. (1995) Inactivation of the type II TGF-p receptor in colon cancer cells with microsatellite instability. Science, 268,1336-1338.
Marshall, E. (2003) Preventing toxicity with a gene test. Science, 302, 588-590.Massey, A., Offman, J., Macpherson, P. and Karran, P. (2003) DNA mismatch
repair and acquired cisplatin resistance in E. coli and human ovarian carcinoma cells. DNA Repair, 2, 73-89.
Mazurek, A., Berardini, M. and Fishel, R. (2002) Activation of human MutShomologs by 8-oxo-guanine DNA damage. J. Biol. Chem., 277, 8260-8266.
McKenna, R.W. (2000) Multifaceted approach to the diagnosis and classification of acute leukemias. din. Chem., 46,1252-1259.
Meetei, A.R., de Winter, J.P., Medhurst, A.L., Wallisch, M., Waisfisz, Q., van de Vrugt, H.J., Oostra, A.b., Yan, Z., Ling, C., Bishop, C.E., Hoatlin, M.E., Joenje, H. and Wang, W. (2003) A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet, 35,165-170.
Melo, J. and Toczyski, D. (2002) A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Bid., 14, 237-245.
Middleton, M.R. and Margison, G.P. (2003) Improvement of chemotherapy efficacy by inactivation of a DNA-repair pathway. Lancet Oncol., 4, 37-44.
Modrich, P. and Lahue, R. (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Anna. Rev. Biochem., 65,101-133.
Morrison, R.T. and Boyd, R.N. (1992) Organic Chemistry. Prentice-Hall, Inc., Englewood Cliffs, New Jersey.
Moshous, D., Callebaut, I., de Chasseval, R., Corneo, B., Cavazzana-Calvo, M., Le Deist, F., Tezcan, I., Sanal, O., Bertrand, Y., Philippe, N., Fischer, A. and de Villartay, J.P. (2001) Artemis, a novel DNA double-strand break repairA/(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell, 105,177-186.
Mu, D., Tursun, M., Duckett, D., Drummond, J., Modrich, P. and Sancar, A. (1997) Recognition and repair of compound DNA lesions (base damage and mismatch) by human mismatch repair and excision repair systems. Mol. Cell. Biol., 17, 760-769.
Mundt, A.J., Roeske, J.C. and Weichselbaum, R.R. (2000) Physical and biological basis of radiation oncology. In Bast, R.C.J., Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Holland, J.F. and Frei III, E. (eds.). Cancer Medicine. BC Decker Inc.
Nana, K.-i., Nagashima, F. and Yasui, A. (2001) Highly elevated ultraviolet-induced mutation frequency in isolated Chinese hamster cell lines defective in nucleotide excision repair and mismatch repair proteins. Cancer Res, 61, 50- 52.
Newlands, E.S., Foster, T. and Zaknoen, S. (2003) Phase I study of temozolamide (TMZ) combined with procarbazine (PCB) in patients with gliomas. Br. J. Cancer, 89, 248-251.
O'Driscoll, M., Cerosaletti, K.M., Girard, P.M., Dai, Y., Stumm, M., Kysela, B.,Hirsch, B., Gennery, A., Palmer, S.E., Seidel, J., Gatti, R.A., Varon, R., Oettinger, M.A., Neitzel, H., Jeggo, P.A. and Concannon, P. (2001) DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell, 8,1175-1185.
O'Driscoll, M. and Jeggo, P. (2003) Clinical impact of ATR checkpoint signalling failure in humans. Ceil Cycle, 2 ,194-195.
O'Driscoll, M., Ruiz-Perez, V.L., Woods, C.G., Jeggo, P. and Goodship, J.A. (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3- related protein (ATR) results in Seckel syndrome. Nat. Genet, 33,497-501.
202

Obmolova, G., Ban, C., Hsieh, P. and Yang, W. (2000) Crystal structure ofmismatch repair protein MutS and its complex with a substrate DNA. Nature, 407, 703-710.
Oda, S., Humbert, O., Fiumicino, S., Bignami, M. and Karran, P. (2000) Efficient repair of A/C mismatches in mouse cells deficient in long-patch mismatch repair. EMBOJ., 19,1711-1718.
Ohyashiki, J.H., Ohyashiki, K., Aizawa, S., Kawakubo, K., Shimamoto, T., Iwama,H., Hayashi, S. and Toyama, K. (1996) Replication errors in hematological neoplasias: genomic instability in progression of disease is different among different types of leukemia. Clin. Cancer Res., 2 , 1583-1589.
Olipitz, W., Hopfinger, G., Aguiar, R.C.T., Gunsilius, E., Girschikofsky, M., Bodner,C., Hiden, K., Linkesch, W., Haefler, G. and Sill, H. (2002) Defective DNA- mismatch repair: A potential mediator of leukemogenic susceptibility in therapy-related myelodysplasia and leukemia. Gene Chromosomes Cancer, 34, 243-248.
Olschwang, S., Hamelin, R., Laurent-Puig, P., Thuille, B., De Rycke, Y., Li, Y.-J., Muzeau, P., Girodet, J., Salmon, R.-J. and Thomas, G. (1997) Alternative genetic pathways in colorectal carcinogenesis. Proc. Natl. Acad. Sci. U. S. A., 94, 12122-12127.
Ondrus, D., Pribylincova, V., Breza, J., Bujdak, P., Miklosi, M., Reznicek, J. andZvara, V. (1999) The incidence of tumours in renal transplant recipients with long-term immunosuppressive therapy. Int. Urol. Nephrol., 31,417-422.
Opelz, G., Henderson, R. and Study, f.t.C.T. (1993) Incidence of non-Hodgkinlymphoma in kidney and heart transplant recipients. The Lancet, 342,1514- 1516.
Pagano, L., Pulsoni, A., Mele, L., Tosti, M.E., Cerri, R., Visani, G., Melillo, L.,Candoni, A., Clavio, M., Nosari, A., Petti, M.C., Martino, B., Mele, A., Levis,A., Allione, B., Almici, C., Equitani, F., Leone, G. and Mandelli,F.f.t.G.I.M.E.d.A.G. (2001) Acute myeloid leukemia in patients previuosly diagnosed with breast cancer: experience of the GIMEMA group. Ann. Oncol., 12, 203-207.
Pandolfi, P.P. (2001) Oncogenes and tumor suppressors in the molecularpathogenesis of acute promyelocytic leukemia. Hum. Mol. Genet, 10, 769- 775.
Papadopolous, N., Nicolaides, N.C., Liu, B., Parsons, R., Lengauer, C., Palombo,F., D'Arrigo, A., Markowitz, S., Willson, J.K.V., Kinzler, K.W., Jiricny, J. and Vogelstein, B. (1995) Mutations of GTBP in genetically unstable cells. Science, 268,1915-1917.
Pegg, A.E. (2000) Repair of 0®-alkylguanine by alkyltransferases. Mutat. Res., 462, 83-100.
Peltomaki, P. (2003) Role of DNA mismatch repair defects in the pathogenesis of human cancer. J. d in. Oncoi., 21,1174-1179.
Penn, I. (1993) Incidence and treatment of neoplasia after transplantation. The Joumai of Heart and Lung Transplantation, 12, S328-S336.
Perucho, M. (1996) Microsatellite instability: the mutator that mutates the other mutator. Nat. Med., 2, 630-631.
Perucho, M. (1999) Correspondence re: C. R. Boland et a t. National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsetellite instability in colorectal cancer. Cancer Research., 58: 5248-5257, 1998. Cancer Res., 59, 249-256.
Perucho, M., Peinado, M.A., Ionov, Y., Casares, S., Malkhosyan, S. and Stanbridge,E. (1994) Defects in replication fidelity of simple repeated sequences reveal a new mutator mechanism for oncogenesis. Coid Spring Harb. Symp. Quant. 6/0/., 59, 339-348.
203

PItha-Rowe, I., Petty, W.J., Kitareewan, S. and Dmitrovsky, E. (2003) Retinoid target genes in acute promyelocytic leukemia. Leukemia, 17,1723-1730.
Prolla, T.A., Baker, S.M., Harris, A.C., Tsao, J.-L., Yao, X., Bronner, C.E., Zheng,B., Gordon, M., Reneker, J., Arnheim, N., Shibata, D., Bradley, A. and Liskay, R.M. (1998) Tumour susceptibility and spontaneous mutation in mice deficient in MIhl, Pmsi and Pms2 DNA mismatch repair. N at Genet, 18, 276-279.
Qin, X., Shibata, D. and Gerson, S.L. (2000) Heterozygous DNA mismatch repair gene PMS2-knockout mice are susceptible to intestinal tumor induction with A/-methyl-A/-mitrosourea. Carcinogenesis, 21, 833-838.
Randhawa, P.S., Yousem, S.A., Paradis, I.L., Dauber, J.A., Griffith, B.P. andLocker, J. (1989) The clinical spectrum, pathology, and clonal analysis of Epstein-Barr virus-associated lymphoproliferative disorders in heart-lung transplant recipients. Am. J. din. Pathol., 92,177-185.
Reese, J.S., Liu, L. and Gerson, S.L. (2003) Repopulating defect of mismatch repair-deficient hematopoietic stem cells. Blood.
Reichmann, E. (2002) The biological role of the Fas/FasL system during tumor formation and progression. Semin. Cancer Biol., 12, 309-315.
Reitmair, A., Redston, M., Cai, J., Chuang, T., Bjerknes, M., Cheng, H., Hay, K., Gallinger, S., Bapat, B. and Mak, T. (1996) Spontaneous intestinal carcinomas and skin neoplasms in Msh2-deficient mice. Cancer Res, 56, 3842-3849.
Reitmair, A.H., Schmits, R., Ewel, A., Bapat, B., Redston, M., Mitri, A., Waterhouse, P., Mittruecker, W.G., Wakeham, A., Liu, B., Griesser, H., Gallinger, S., Ballhausen, W.G., Fishel, R. and Mak, T.W. (1995) MSH2 deficient mice are viable and susceptible to lymphoid tumours. N at Genet, 11, 64-70.
Ribeiro, E.M.S.F., Rodriguez, J.M., Coser, V.M., Sotero, M.G., Fonseca Neto, J.M., Pasquini, R. and Cavalli, I.J. (2002) Microsatellite instability and cytognetic survey in myeloid leukemias. Braz. J. Med. Biol. Res., 35,153-159.
Ricciardone, M.D., Oezcelik, T., Cevher, B., Oezdag, H., Tuncer, M., Guergey, A., Uzunalimoglu, O., Cetinkaya, H., Tanyeli, A., Erken, E. and Oeztuerk, M. (1999) Human MLH1 deficiency predisposes to hematological malignancy and Neurofibromatosis type 1. Cancer Res., 59, 290-293.
Rosner, F., Carey, R.W. and Zarrabi, M.H. (1978) Breast cancer and acute myeloid leukemia: report of 24 cases and review of the literature. American Joumai of Medicine, 4,151-172.
Ruggero, D., Wang, Z.-G. and Pandolfi, P.P. (2000) The puzzling multiple lives of PML and its role in the genesis of cancer. BioEssays, 22, 827-835.
Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Molecular Cloning - A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor.
Sambrook, J. and Russell, D.W. (2001) Molecular Cloning - A laboratory manual. Cold Spring Harbor Laboratory press. Cold Spring Harbor.
Samowitz, W.S., Slattery, M.L., Potter, J.D. and Leppert, M.F. (1999) BAT-26 and BAT-40 instability in colorectal adenomas and carcinomas and germiine polymorphisms. Am J Pathol, 154,1637-1641.
Schiffer, C.A. and Stone, R.M. (2000) Acute myeloid leukemia in adults. In Bast, R.C.J., Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Holland, J.F., Frei,E.l. and Gansler, T.S. (eds.). Cancer Medicine. BC Decker, Hamilton.
Schwartz, S., Yamamoto, H., Navarro, M., Maestro, M., Reventos, J. and Perucho, M. (1999) Frameshift mutations at mononucleotide repeats in caspase-5 and other target genes in endometrial and gastrointestinal cancer of the microsatellite mutator phenotype. Cancer Res., 59, 2995-3002.
Scott, S., Kimura, T., Ichinohasama, R., Bergen, S., Magliocco, A., Reimer, C., Kerviche, A., Sheridan, D. and DeCoteau, J.F. (2003) Microsatellite mutations of transforming growth factor-p receptor type II and caspase-5
204

occur in human precursor T-cell lymphoblastic lymphomas/leukemias in vivo but are not associated with hMSH2 or hMHL1 promoter méthylation. Leuk. Res., 27, 23-34.
Sedgwick, B. and Lindahl, T. (2002) Recent progress on the Ada response for inducible repair of DNA alkylation damage. Oncogene, 21, 8886-8894.
Seeberg, E., Luna, L., Morland, I., Bide, L., Johnsen, B., Larsen, E., Alseth, I., Dantzer, F., Baynton, K., Aamodt, R., Kristiansen, K.J., Rognes, T.,Klugland, A. and Bjoras, M. (2000) Base removers and strand scissors; different strategies employed in base excision and strand incision at modified base residues in DNA. Cold Spring Harb. Symp. Quant. Biol., LXV, 135-142.
Sheikhha, M.H., Tobal, K. and Yin, J.A.L. (2002) High level of microsatelliteinstability but not hypermethylation of mismatch repair genes in therapy- related and secondary acute myeloid leukaemia and myelodysplastic syndrome. Br. J. Haematol., 117, 359-365.
Sherr, C.J. and McCormick, F. (2002) The RB and p53 pathways in cancer. Cancer Cell, 2 , 103-112.
Shiloh, Y. (1997) Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Anna. Rev. Genet, 31, 635-662.
Shiloh, Y. (2001) ATM and ATR: networking cellular responses to DNA damage. Current Opinion in Genetics & Development, 11, 71-77.
Shimamura, A., Macari, T., Moreau, L.A., Svenson, J.L. and D'Andrea, A.D. (2003) The Fanconi anemia DNA repair pathway is deficient in acute myeloid leukaemia. Pediatr. Res., 53, 268A.
Sill, H., Goldman, J.M. and Cross, N.C. (1996) Rarity of microsatellite alterations in acute myeloid leukaemia. Br. J. Cancer, 74, 255-257.
Silverman, L.R. (2000) Myelodysplastic syndrome. In Bast, R.C.J., Kufe, D.W.,Pollock, R.E., Weichselbaum, R.R., Holland, J.F., Frei, E.l. and Gansler, T.S. (eds.). Cancer Medicine. BC Decker, Hamilton.
Simpkins, S., Bocker, T., Swisher, E., Mutch, D., Gersell, D., Kovatich, A., Palazzo, J., Fishel, R. and Goodfellow, P. (1999) MLH1 promoter méthylation and gene silencing is the primary cause of microsatellite instability in sporadic endometrial cancers. Hum. Mol. Genet, 8, 661-666.
Singer, B. and Grunberger, D. (1983) Molecular biology of mutatgens and carcinogens. Plenum Press, New York.
Stirewalt, D.L. and Radich, J.P. (2003) The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer, 3, 650-665.
Stout, J.T. and Caskey, C.T. (1985) HPRT: gene structure, expression, and mutation. Ann. Rev. Genet, 19,127-148.
Strathdee, G., MacKean, M.J., llland, M. and Brown, R. (1999) A role for méthylation of the hM LHI promotor in loss of hMLHI expression and drug resistance in ovarian cancer. Oncogene, 18, 2335-2341.
Svejstrup, J.Q. (2001) Mechanisms of transcription-coupled DNA repair. Nat. Rev. Mol. Ce//B/o., 3, 21-29.
Svejstrup, J.Q. (2003) Rescue of arrested RNA polymerase II complexes. J. Cell Sc/., 116, 447-451.
Swann, P.F., Waters, T.R., Moulton, D.C., Xu, Y.-Z., Zheng, Q., Edwards, M. andMace, R. (1996) Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science, 273,1109-1 111.
Takeuchi, S., Takeuchi, N., Fermin, A.C., Taguck, H. and Koeffler, H.P. (2003)Frameshift mutations in caspase-5 and other target genes in leukaemia and lymphoma cell lines having microsatellite instability. Leuk. Res., 27, 359-361.
Thompson, L.H. and Schild, D. (2002) Recombinational DNA repair and human disease. Mutat. Res., 509,49-78.
205

Thomsen, J.B., Schroder, H., Krlstinsson, J., Madsen, B., Szumlanski, C., Weinshilboum, R., Brandt Andersen, J. and Schmiegelow, K. (1999)Possible carcinogeneic effect of 6-mercaptopurine on bone marrow stem cells. Cancer, 86,1080-1086.
Timmers, C., Taniguchi, T., Hejna, J., Reifsteck, C., Lucas, L., Bruun, D., Thayer,M., Cox, B., Olson, S., D'Andrea, A.D., Moses, R. and Grompe, M. (2001) Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol. Cell, 7, 241-248.
Trewick, S.C., Henshaw, T.F., Hausinger, R.P., Lindahl, T. and Sedgwick, B. (2002) Oxidative déméthylation by Escherichia coll AlkB directly reverts DNA base damage. Nature, 419,174-178.
Umar, A., Boyer, J.C., Thomas, D.C., Nguyen, D.C., Risinger, J.I., Boyd, J., Ionov, Y., Perucho, M. and Kunkel, T.A. (1994) Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability. J. Biol. Chem., 269,14367-14370.
Varon, R., Reis, A., Henze, G., Einsiedel, H.G.v., Sperling, K. and Seeger, K. (2001) Mutations in the nijmegen breakage syndrome gene {NBS1) in childhood acute lymphoblastic leukemia (ALL). Cancer Res, 61, 3570-3572.
Vilkki, S., Tsao, J.-L., Loukola, A., Poyhonen, M., Vierimaa, O., Herva, R., Aaltonen, L.A. and Shibata, D. (2001) Extensive somatic microsatellite mutations in normal human tissue. Cancer Res, 61,4541-4544.
Wang, L., Zhu, D., Zhang, C., Mao, X., Wang, G., Mitra, G., Li, B.F.L., Wang, X. and Wu, M. (1997) Mutations of 0®-methylguanine-DNA methyltransferase gene in esophageal cancer tissues from northern China. Int. J. Cancer, 71, 719- 723.
Wang, Q., Lasset, C., Desseigne, F., Frappaz, D., Bergeron, C., Navarro, C.,Ruano, E. and Puisieux, A. (1999) Neurofibromatosis and early onset of cancers in W LHf-deficient children. Cancer Res, 59, 294-297.
Waters, T.R. and Swann, P.P. (1997) Cytotoxic mechanism of 6-thioguaninehMutSa, the human mismatch binding heterodimer, binds to DNA containing S®-methylthioguanine. Biochemistry, 36, 2501-2506.
Watson, J.D. and Crick, F.H. (1953) Molecular structure of nucleic acids. Nature, 4356, 737-738.
Wei, K., Clark, A.B., Wong, E., Kane, M.F., Mazur, D.J., Parris, T., Kolas, N.K.,Russell, R., Hou, H., Jr., Kneitz, B., Yang, G., Kunkel, T.A., Kolodner, R.D., Cohen, P.E. and Edelmann, W. (2003) Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev., 17, 603-614.
Wei, K., Kucherlapati, R. and Edelmann, W. (2002) Mouse models for human DNA mismatch-repair gene defects. Trends Mol. Med., 8, 346-353.
Wei, Q., Guan, Y., Cheng, L., Radinsky, R., Bar-Eli, M., Tsan, R., Li, L. andLegerski, R.J. (1997) Expression of five selected human mismatch repair genes simultaneously detected in normal and cancer cell lines by a nonradioactive multiplex reverse transcription-polymerase chain reaction. Pathoblology, 65, 293-300.
Weinstein, H.J. (1988) The acute leukemias. In Wyngaarden, J.B. and Smith, L.H. (eds.), Cecil Textbook of Medicine. W. B. Saunders Company, pp. 1001- 1007.
West, S.C. (2003) Molecular views of recombination proteins and their control. Nat. Rev. Mol. Cell Bio., 4, 435-445.
Whiteside, D., McLeod, R., Graham, G., Steckley, J.L., Booth, K., Somerville, M.J. and Andrew, S.E. (2002) A homozygous germ-line mutation in the human MSH2 gene predisposes to hematological malignancy and multiple Cafe-au- Lait spots. Cancer Res, 62, 359-362.
206

Worrlllow, L.J., Travis, L.B., Smith, A.G., Rollinson, S., Smith, A.J., Wild, C.P.,Holowaty, E.J., Kohler, B A , Wiklund, T., Pukkala, E., Roman, E., Morgan, G J. and Allan, J.M. (2003) An intron splice accepyor polymorphism in hMSH2 and risk of leukemia after treatment with chemotherapeutic alkylating agents. Clin. Cancer Res., 9, 3012-3020.
Wu, J., Berends, M.J.W., Post, J.G., Mensink, R.G.J., Verlind, E., van der Sluis, T., Kempinga, C., Sijmons, R.H., van der Zee, A.G., Hollema, H., Kleibeuker, J.H., Buys, C.H.C.M. and Hofstra, R.M.W. (2001) Germiine mutations of Exo1 gene in patients with hereditary nonpolyposis colorectal cancer (HNPCC) and atypical HNPCC forms. Gastroenterology, 120,1580-1587.
Wu, J., Gu, L., Wang, H., Geacintov, N.E. and Li, G.-M. (1999) Mismatch repair processing of carcinogen-DNA adducts triggers apoptosis. Mol. Cell. Biol.,19, 8292-8301.
Yamada, M., O'Regan, E., Brown, R. and Karran, P. (1997) Selective recognition of a cisplatin-DNA adduct by human mismatch repair proteins. Nucl. Acids. Res., 25, 491-496.
Yao, X., Buermeyer, A.B., Narayanan, L., Tran, D., Baker, S.M., Prolla, T.A., Glazer, P.M., Liskay, R.M. and Arnheim, N. (1999) Different mutator phenotypes in Mlh1~ versus PmsZ-deficient mice. Proc. Nati. Acad. Sci. U. S. A., 96, 6850- 6855.
Yung, W.K.A., Albright, R.E., Olson, J., Fredericks, R., Fink, K., Prados, M.D.,Brada, M., Spence, A., Hohl, R.J., Shapiro, W., Glantz, M., Greenberg, H., Selker, R.G., Vick, N.A., Rampling, R., Friedman, H., Phillips, P., Bruner, J., Yue, N., Osoba, □., Zaknoen, S. and Levin, V.A. (2000) A phase II study of temozolomide vs. procarbazine in patients with glioblastoma muliforme at first relapse. Br. J. Cancer, 83, 599-593.
Zanetti, R., Crosignani, P. and Rosso, S. (1992) Cancer in Italy: Incidence Data From Cancer Registries, 1988-1992 (Lega Italiana per ia lotta contro i tumori: II pensiero scientifico editore), vol. 2.
207

Appendix I: Figures
208

hMLHI promoter méthylation data for Fig. 3.7.
3 6 8 9 13 17 18 24 25H M U H M D H M U H M U H M U H M U H M U H M U H M D
hMLHI
hPCNA
1 2 4 7 10 11 15 21H M U H M U H M U H M U H M U H M U H M U H M U
hMLHI

Microsatellite Data for Fig. 4.9.
D. Case A2
Tumour 1 Tumour 1 Tumour 1
Tumour 2 Tumour 2
Normal Normal Normal
Tumour 1 Tumour 1
Tumour 2 Tumour 2 Tumour 2
Normal N o rm a l
Bat26 Bat25 D2S123 D5S346 D17S250

Microsatellite Data for Fig. 4.9.E. Case A3
Tum our Tum our
Normal Normal
Bat26 Bat25
Tum our Tum our Tum our
Normal Normal Normal
ro D2S123 D5S346 D17S250
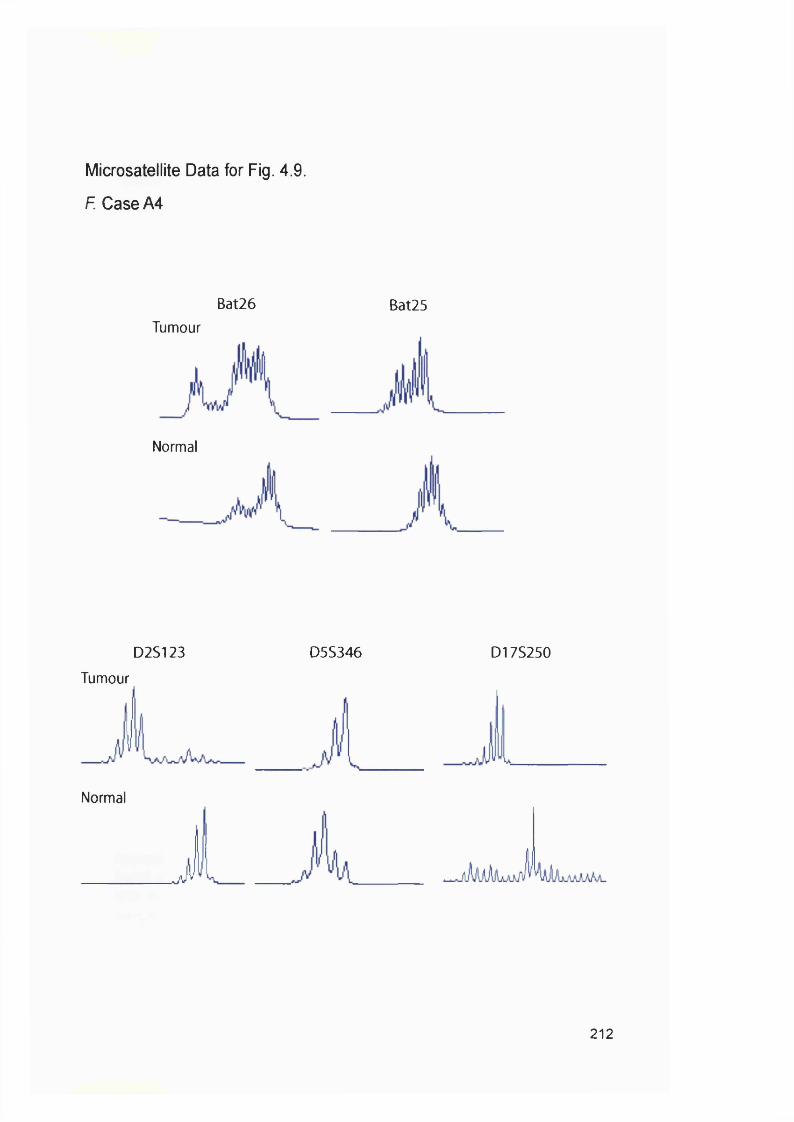
Microsatellite Data for Fig. 4.9.
F. Case A4
Bat26 Bat25
Tumeur
Normal
D2S123
Tumeur
Normal
J
D5S346 D17S250
212

Microsatellite Data for Figure 4.11
Bat25BD3
BD2
BD10
Icase A7
Appendix to Fig. 4.10.Bat25 microsatellite instability in transplant-related AML/MDS. Examples of MSI in three blood doners (BD2, 3 & 10) and one AML bone marrow samples (Patients A7) are shown.
213

Appendix II: Tables
214
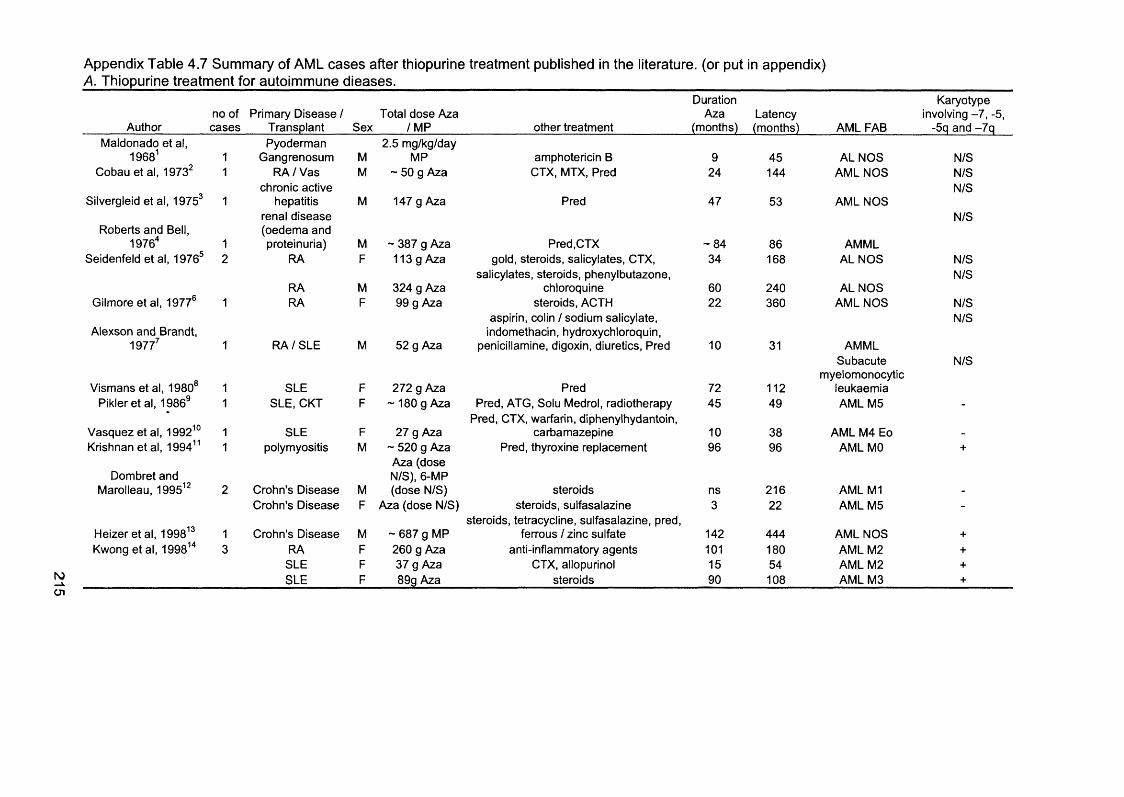
Appendix Table 4.7 Summary of AML cases after thiopurine treatment published in the literature, (or put in appendix)A. Thiopurine treatment for autoimmune dieases.
N)Ü1
Duration Karyotypeno of Prim ary D isease / Total dose A za A za Latency involving - 7 , -5,
Author cases Transplant Sex /M P other treatm ent (months) (months) A M L FAB -5q and -7 qMaldonado et al,
1968^Pyoderm an 2 .5 m g/kg/day
1 Gangrenosum M M P amphotericin B 9 45 A L N O S N/SCobau et al, 1973^ 1 RA / Vas
chronic activeM - 50 g A za C TX, M TX, Pred 24 144 A M L N O S N/S
N/SSilvergleid et al, 1975^ 1 hepatitis
renal d iseaseM 147 g A za Pred 47 53 A M L N O S
N/SRoberts and Bell, (oedem a and
1976'' 1 proteinuria) M - 387 g A za Pred,C TX - 8 4 86 A M M LSeidenfeld et al, 1976® 2 RA F 113 g A za gold, steroids, salicylates, CTX,
salicylates, steroids, phenylbutazone.34 168 A L N O S N/S
N/SRA M 324 g A za chloroquine 60 240 A L N O S
Gilmore et al, 1977® 1 RA F 99 g Aza steroids, A C TH aspirin, colin / sodium salicylate.
22 360 A M L N O S N/SN/S
Alexson and Brandt, indomethacin, hydroxychloroquin.1977^ 1 R A /S L E M 52 g Aza penicillamine, digoxin, diuretics, Pred 10 31 A M M L
Subacute N /S
Vism ans et al, 1980®myelomonocytic
1 SLE F 2 72 g A za Pred 72 112 leukaem iaPikier et al, 1986® 1 SLE, C K T F ~ 180 g A za Pred, A TG , Solu Medrol, radiotherapy 45 49 A M L M 5 -
Pred, C TX, warfarin, diphenylhydantoin.V asquez et al, 1992^° 1 SLE F 27 g Aza carbam azepine 10 38 A M L M 4 Eo -
Krishnan et al, 1994” 1 polymyositis M ~ 520 g A za A za (dose
Pred, thyroxine replacem ent 96 96 A M L MO +
Dom bret and N/S), 6 -M PMarolleau, 1995^^ 2 Crohn’s Disease M (dose N /S ) steroids ns 216 AM L M l -
Crohn's Disease F A za (dose N /S ) steroids, sulfasalazine 3 22 A M L M 5 -
H eizer et al, 1998^®steroids, tetracycline, sulfasalazine, pred.
1 Crohn's Disease M ~ 687 g M P ferrous / zinc sulfate 142 4 44 AM L NO S +
Kwong et al, 1 998” 3 RA F 260 g A za anti-inflam matory agents 101 180 A M L M 2 +SLE F 37 g Aza C TX, allopurinol 15 54 A M L M2 +
SLE F 89g A za steroids 90 108 A M L M 3 +

Arnold et al, 1999^^ 1 Derm atomyositls F 109 g A za Pred, enalapril, bendroluazide 24 27 A M L M2 +
Asten et al, 1999^®6/ N/S
1289 RA F A za (dose N /S ) 31 32 A M L N O SRA F A za (dose N /S ) 25 40 A M L N O S N/S
SLE F A za (dose N /S ) 8 5.4 A M L NO S Myelomonocytic
N/SN/S
P M R F A za (dose N /S ) 13 83 leukaem iaVA S F A za (dose N /S ) CIb 28 95 A M L NO S N/SRA F A za (dose N /S ) 76 109 N/S
Mauritzson et al. +2002^^ 7 Arth F 140 g A za M TX , CIb 70 70 A M L M 5
Gn F 185 g A za C TX 111 111 A M L M 6 +
Gn M 2.4 g Aza C TX 12 24 refractory anaem ia +
Arth F 21 g Aza 20 20 refractory anaem ia +
C TD M 9.5 g A za 26 26 RARS -
Arth M 170 g A za CIb 116 144 RAEB +
W egener M 120 g A za 100 mg/day for
CIb, C TX 124 131 RAEB +
Heesen et al, 2003^®several months several
1 M S F A za Interfe ro n-b eta ,, M TX months 192 A M L M 4 Eo -
B. Thiopurine treatment after organ transplants.Duration Karyotype
AuthorNo. of cases
Prim ary D isease / Transplant Sex
Total dose Aza /M P other treatm ent
A za(months)
Latency(months) A M L FAB
involving - 7 , -5, -5q and -7 q
Silvergleid et al, 1975® 1 LRKT M 114 g A za Solu Medrol, AcD, Pred 46 54 A M L N O S N/SSw eeney et al, 1976^® 1 LRKT M 650 g Aza Radiotherapy, Pred 95 95 A M L M 6 N/S
60 .3 g A za N/SBlanc et al, 1977^° 1 KT M 6.55 g M P Pred, PredI 30 36 A M M L
3 mg/kg/day N/Sfor 3 years +
Ellerton et ai, 1979^^ 1 G n /K T M 18 g Aza Pred, CTX, radiotherapy 39 77 A M L M 6Leithner et al, 1981®® 1 C K T M 262 g A za A TG , Pred 84 104 A M L N O S N/S
Hoy et al, 1982®® 1 LRKT M 390 g A za A TG , Pred 89 89 AM L M 6 N/StsjO)

Pikier et al, 1986® 1 SLE, C K T F ~ 180 g A za Pred, A TG , Solu Medrol, radiotherapy 45 4 9 A M L M 5 -
Butler et al 1990^^^ 1 C K T F 81 g A za CsA, PredI, A TG 42 60 A M L N O SSubar et al. 1996^® 2 C K T F A za (dose N /S ) Pred 156 156 A M L M 3 -
LRKT F A za (dose N /S ) 192 204 A M L NO SGorschlueter et al.
1997^® 1 C K T M > 50 g Aza C TX, Pred 50 50 A M L M 4 Eo -
Thalham m er-Scherreret al, 1999^^ 3 SLT M A za (dose N /S ) A TG , CsA, Pred 10 10 A M L M 5a N/S
KT M A za (dose N /S ) A TG , CsA, Pred > 6 2 78 A M L M 4 -
Ondrus et al, 1999^®CK T M A za (dose N /S ) A TG , CsA, Pred N/S 75 A M L M l -
1/620 CK T A za (dose N /S ) Pred, CsA M D S N/S
Huebner et al, 2000^®75 - 150
5/631 O C T M m g/day Aza Pred, CsA 48 48 A M L M 7 +
O C T M A za (dose N /S ) Pred, CsA 92 92 A M L M lO C T M 132 g A za Pred, CsA 58 69 M D S RAEB +
O C T F 96 g A za 5 0 -75 mg/day
Pred, CsA 67 67 A M L M 6 M D S RAEB (1996)
O C T M A za Pred, CsA 65 7 7 / 8 0 /A M L M 2 (1997) +
Mauritzson et al,2002^^ KT M 2 00 g A za CIb, C TX 182 182 RAEB +
Barbanel et al, 1975®°3-5 m g/kg/day N/S
1/316 KT A za + possibly others A L N O S
Krikorian et al, 1978®^1 0 0 -2 0 0 N/S
1/124 O C T m g/day Aza Pred, ActD, A TG , C T X n/s 46 A M L N O S
N)N
F, female; M; maie; N /S, not specified; CsA, Cyclosporin A; MP, 6-mercaptopurine; M TX, methotraxate; C TX, Cyclophosphamide; A CTH, ; Pred, prednisone;
PredI, prednisolone; Aza, azathioprine; A TG , antithymocyte globulin; ActD, actinomycin D; CIb, chlorambucil; Gn, glomeruoonephritis; VAS, vasculitis; CTD,
connective tissue disease; SLE, Systemic Lupus Erythematosus ;RA, rheumatoid arthritis; Arth, arthrosis/arthritis; VAS, vasculitis; LT, liver transplant; CKT,
Cadaver kidney transplant; LRKT, living related kidney transplant; KT, kidney transplant; HLT, heart/lung transplant; SLT, single lung transplant; OCT, cardiac
transplant; PT, pancreas transplant; SCT, stem cell transplantation; RARS, refractory anemia with ringed sideroblasts; RAEB, refractory anem ia with excess
o f b l a s t s ; A M L N O S , a c u t e m y e l o i d l e u k a e m i a n o t o t h e r w i s e s p e c i f i e d .

1. M aldonado, N., Torres, V . M ., M endez-C ashion, D., Perez-S antiago, E. & Caceres de Costas, M. Pyoderm a gangrenosum treated with 6-m ercaptopurine and followed by acute leukem ia. The Journal of Pediatrics 72, 4 0 9 -414 (1968).
2. C obau, C. D., Sheon, R. P. & Kirsner, A. B. Im m unosuppressive Drugs and Acute Leukem ia. Ann Intern Med 79. 131-132 (1973).
3. Silvergleid, A. J. & Schrier, S. L. Acute M yelogenous Leukem ia in Tw o Patients Treated with Azath ioprine for N onm alignant D iseases. The American Journal of Medicine 57 , 8 8 5 -8 8 8 (1974).
4 . Roberts, M . M . & Bell, R. Acute Leukaem ia after Im m unosuppressive Therapy. The Lancet, 7 6 8 -7 7 0 (1976).
5. S e idenfe ld , A. M ., Sm ythe, H. A ., O gyzio , M . A ., Urow itz, M . B. & Dotten, D. A. A cute L e u k e m ia in R h eu m ato id Arthritis T re a te d with C yto tox ic A g en ts . The Journal of Rheumatology Z, 2 9 5 -3 0 4 (1976).
6. G ilm ore, I. T ., Holden, G . & Rodan, K. S. A cute leukaem ia during azath ioprine therapy. Postgraduate Medical Journal 53, 173-174 (1977).
7. A lexon, E. & Brandt, K. D. Acute leukem ia after azathioprine treaatm ent of connective tissue disease. The American Journal of the Medical Sciences 273, 35 5 -3 4 0 (1977).
8. V ism ans, J. J., Briet, E ., M eijer, K. & den O tto lander, G . J. A zath ioprine and Subacute M yelomonocytic Leukem ia. Acta Med Scand 207, 315 -3 1 9 (1980).
9. Pikier, G. M ., Say, B. & Stam per, S. Cytogenetic Finding in Acute M onocytic Leukem ia in a Renal Allograft Recipient. CAncer Genet Cytogenet20, 101-107 (1986).
10. V asq u ez, S ., K avanaugh, A. P., Schneider, N. R., W acho ltz, M . C. & Lipsky, P. E. A cute N on lym p ho cytic L eu kem ia A fte r T re a tm e n t o f S ystem ic Lupus E ry th em atosu s with Imm unosuppressive Agents. J Rheumatol ^9, 1625-7 (1992).
11. K rishnan, K., A dam s, P ., S ilveira , S ., S heldon, S. & D abich , L. Therapy-te lated acute m eyeloid leukaem ia following imm unosuppression with azathioprine for polymyositis. Clin. lab. Haematol. 16, 2 8 5 -2 8 9 (1994).
12. Dom bret, H. & M aroleau. De novo acute myeloid leukem ia in patients with Crohn's disease. Nouvelle Revue Française Hématologie 37, 193-196 (1995).
13. H eizer, W . D. & Peterson, J. L. Acute M yeloblastic l_eukemia Following Prolonged Treatm ent of Crohn's D isease with 6-M ercaptopurine. Digestive Diseases and Sciences A3, 1 791-1793 (1998).
14. K w ong, Y . L., Au, W . Y . & Liang, R. H. S. A cute M yeloid Leukem ia a fter Azathioprine treatm ent for Autoim m une Diseases: association with -7 /7q -. Cancer gent Cytogenet 103, 94- 9 7 (1 9 9 8 ).
15. A rno ld , J. A ., R anson, S . A. & A b da lla , S . H. A zath ioprine-associated acute m yeloid leukaem ia with trilineage dysplasia and com plex karyotype: a case report and review of the literature. Clin. Lab. Haem. 21, 289 -292 (1999).
16. Asten, P., Barrett, J. & Sym m ons, D. Risk of Developing Certain M alignacies Is Related to Duration of Im m unosuppressive Drug Exposure in PAtients with R heum atic D iseases. The Journal of Rheumatology 26, 1705-1714 (1999).
17. M auritzson, N. et al. Pooled analysis of clinical and cytogenetic in tretam ent-related and de novo adult acute myeloid leukem ia and m yelodysplastic syndrom es based on a consecutive series of 761 patiets analyzed 1 9 7 6 -1 9 9 3 and on 5 0 9 8 unselected cases reported in the literature 1974-2001. Leukemia 16, 2 3 6 6 -2 3 7 8 (2002).
18. H eesen , C. et al. Therapy-re la ted acute m yelogenous leukaem ia (t-A M L) in a patient with multiple sclerosis treated with mitoxantrone. Multiple Sclerosis 9, 21 3 -2 1 4 (2003).
19. S w een ey , W . T ., M adge, G. E. & P ierce, J. C. Acute Erythrém ie Myelosis Occurring Eight Y ears After Renal Allograft. South Med J 69, 9 4 9 -950 (1976).
20. B lanc, A. P., G astaut, J .-A ., Dalivoust, P. & C arcassonne, Y . M alignant blood dyscrasias occuring during imm unosuppressive treatm ent. 4 new cases. Nouv. Presse med., 2 5 0 3 -2 5 0 9 (1977).
21. E llerton, J. A ., D e V e b e r, G . A. & Baker, M . A. Erythro leukem ia in a Renal Transplant Recipient. C ancer 43, 1924-1926 (1979).
22. Leithner, C ., Kuehboeck, J., W interleitner, H., P avelka , M . & Krisch, K. Akute myeloische Leukaem ie nach Nierentransplantation. Acta Medica Austriaca 8, 2 9 -3 2 (1981).
23. Hoy, W . E., Packm an, C. H. & Freem an , R. B. Evolution of A cute Leukem ia in a Renal Transplant Patient-? Relationship to Azathioprine. Transplantation 33, 3 3 1 -3 3 3 (1982).
24. Butler, J., Korb, S. & Light, J. Acute m yelogenous leukem ia in a renal allograft recipient receiving cyclosporin therapy. Transplantation A9, 81 3 -8 1 5 (1990).
25. Subar, M ., Gucalp, R., Bernstein, J., W illiam s, G . & W iernik, P. H. Acute leukaem ia following renal transplantation. Medical Oncology ^3, 9-13 (1996).
26. G orschlueter, M . et al. Successful induction and consolidation therapy of acute myeloid leukaem ia in a renal allograft recipient. Nephrol Dial Transplant 12, 593 -5 9 5 (1997).
218

27. Thalham m er-Scherrer, R. et al. Post-transplant acute myeloid leukem ia (P T -A M L). Leukemia 13, 3 2 1 -326 (1999).
28. O ndrus, D. et al. T he incidence of tum ours in renal transplant recipients with long-term immunosuppressive therapy. International Urology and Nephrology , 4 1 7 -4 2 2 (1999).
29. Huebner, G ., Karthaus, M ., Pethig, K., Freud, M. & G anser, A. Myelodysplastic syndrome and acute myelogenous leukem ia secondary to heart transplantation. Transplantation 70, 6 88 -690 (2000).
30. Barbanel, C., Kreis, H. & Crosnier, J. Utilisation au long cours des im m unosuppresseurs chez les m alades porteurs d'un rein transplante. Annales de Medecine Interne 2, 6 9 -74 (1975).
31. Krikorian, J. G., Anderson, J. L., Bieber, C. P., Penn, I. & Stinson, E. B. M alignant neoplasm s following cardiac transplantation. The Journal of the American Medical Association 2 4 0 , 639- 6 4 3 (1 9 7 8 ).
219

ELSEVIER
Available online at www.sciencedirect.com
S C IENCE r/nri D IR E CT *I N C E ^ I
D N A Repair 2 (2003) 73 -89
%
REPAIRw ww .elsevier.com /locate/dnarepair
DNA mismatch repair and acquired cisplatin resistance in E. coli and human ovarian carcinoma cells
Andrew Massey*, Judith Offman, Peter Macpherson, Peter Karran*Clare H a ll Laboratories, London Research Institute, Cancer Research UK, South M imms, Herts E N 6 3LD , U K
Received 11 Ju ly 2002; received in revised fo rm 23 September 2002; accepted 24 September 2002
Abstract
The contribution of defective DNA mismatch repair (MMR) to acquired resistance to c/5-diamminedich]oroplatinum(II) (cisplatin) has been investigated in two model systems: E coli dam mutants and the A2780 ovarian carcinoma cell line. Inactivation of MMR—as indicated by the acquisition of an elevated spontaneous mutator phenotype—was observed frequently among survivors of cisplatin-treated dam mutants. These survivors exhibited a stable resistance to further cisplatin treatment. In contrast, none of twelve independent clones of A2780 that had survived cisplatin exposure and acquired stable drug resistance were repair defective. None exhibited the hallmark méthylation tolerant phenotype associated with a MMR defect, mRNAs encoding five MMR proteins were easily detectable in all twelve variants, and the levels of four key MMR proteins were similar to those in the repair proficient parental cells. Further analysis indicated two different mechanisms of acquired resistance in A2780. The first was a protective effect that reduced the level of DNA platination. The second was observed as a reduced sensitivity to cell cycle aiiest after cisplatin treatment and a consequent reduced apoptosis. The data suggest that although loss of MMR is a significant mechanism of acquired drug resistance in dam bacteria, alterations related to DNA protection or cell cycle progression after drug damage appear to be more probable than abrogation of MMR as resistance modulators in human cells. © 2002 Elsevier Science B.V. All rights reserved.
Keywords: C isp la tin ; E. c o li ’, M M R
1. Introduction
c/.y-DiamminedichIoropIatinum(II) (cisplatin) is widely used in the treatment of cancer and the emergence of drug-resistant disease is a significant clinical problem. The use of in vitro models of human cancer has helped define numerous factors that influence cisplatin toxicity. These include variations in drug uptake, different capacities for drug detoxification and alterations in genes controlling cell proliferation [I]. DNA repair— nucleotide excision
* Corresponding author.
' Present address; RiboTargets L td ., Granta Park, Cam bridge
C B l 6 G B , U K .
repair (NER) and DNA mismatch repair (MMR)— is also regarded as a significant modulator of cisplatin toxicity. It has been suggested that the good clinical response of testicular carcinomas to cisplatin reflects their intrinsically low capacity for removal of potentially cytotoxic cisplatin-DNA adducts by NER [2]. The contribution of the M MR pathway is less easy to understand. Nevertheless, numerous independent studies have indicated a correlation between M MR deficiency and cisplatin resistance [3-6]. Although there are speculative models for this involvement, the exact contribution of M MR inactivation to acquired drug resistance remains undefined.
Mismatch repair corrects replication errors. Its inactivation is associated with a significant increase in
1568-7864/02/$ - see fron t m atter © 2002 E lsev ie r Science B .V . A ll righ ts reserved.
PIT: 8 1 5 6 8 - 7 8 6 4 ( 0 2 ) 0 0 1 8 7 - 8

74 A. Massey et a l /D N A R epa ir 2 (2003) 73-89
spontaneous mutation rates owing to the persistence of uncorrected errors (for recent review, see [7]). In M MR deficient tumours, this is seen as the microsatellite instability (MSI+) phenotype. Germ-line defects in genes encoding key MMR factors— in particular, hM LHI and HMSH2— confer a predisposition to early colorectal, endometrial and other cancer in the HNPCC syndrome. In addition, all MMR-defective cells are highly resistant to methylating agents— including clinical drugs like dacarbazine and temozolomide. The connection between M M R deficiency and methylating agent resistance was originally made in E. coli dam mutants [8]. These strains are deficient in biological méthylation of DNA adenine and are hypersensitive to the lethal effects of non-biological DNA méthylation damage by methylnitrosourea (MNU) and methyl- nitrosoguanidine (MNNG)— direct acting analogs of the clinical methylating drugs. Introduction of a second mutation in a M M R gene— either mutS" (the functional homologue of the hMSH2/hMSH6 dimer) or mutL" (homologous to hMLHl/hPMS2)— reverses the drug sensitivity and dammutS or dammutL strains exhibit wild-type resistance to MNNG. E. coli dam mutants are also significantly more cisplatin sensitive than their dam+ counterparts. Introducing a second M MR mutation again reverses drug sensitivity [9]. The cisplatin resistance of E. coli dammutS or dammutL strains and the parallels with methylating agent resistance has provided a pragmatic foundation for the investigations of the connection between drug resistance and M MR defects in human tumours.
Model human cell lines resemble E. coli dam mutants in regard to methylating agent resistance. The level of a DNA repair factor, the DNA 0^-methyl- guanine-DNA methyltransferase (MGMT), together with mismatch repair capacity provide an excellent predictor of sensitivity. The extreme sensitivity of M GMT negative cells to methylating drugs is completely reversed by introduction of an hMLHI, hMSH2, or another M MR defect [10]. The difference in resistance is sufficient to confer a remarkable selective survival advantage. This has been exploited to select MMR-defective variants of numerous human cell lines [11-13]. For cisplatin, the relationship between resistance and M MR is more uncertain. The effects of defective M MR can sometimes be obscured by other factors so that although there is often an association between M MR deficiency and drug
resistance, there are notable exceptions (for example, in [14,15]). In particular, although circumstantial evidence links MMR defects to cisplatin resistance, the connection between M M R inactivation and exposure to the drug— a situation analogous to the development of a drug-resistant tumour— is indirect. We therefore examined the ability of cisplatin to provide a selective pressure for the inactivation of M MR in a human tumour model.
We chose A2780 ovarian carcinoma cells for this investigation because they are acknowledged to be cisplatin sensitive and provide a reasonable model for a drug-naive tumour. A2780 is also one of several cell lines that have provided evidence for a connection between MMR deficiency and drug resistance [5,16]. Their behaviour was compared to that of E. coli dam mutants that provided the initial example of the connection. We report here that inactivation of MMR is a frequent occurrence in E. coli dam mutants treated with cisplatin. In contrast, among 12 independent cisplatin-resistant A2780 variants, none were detectably deficient in MMR. Our findings suggest that— at least, in these well-studied model human cells— inactivation of M M R is not a frequent route of escape from cisplatin toxicity. The emergence of resistant cells was more frequently associated with drug detoxification or changes affecting the level of DNA damage. Altered cell cycle progression after DNA damage was also revealed as a common property among resistant A2780 variants.
2. Materials and methods
2.1. E. coli
The E. coli strains used in this study were kind gifts from Dr s. J. Essigmann and M. Marinus. Their genotypes are listed as follows.
Strain Genotype
GM112 F~ thr-1 ara-14 leuB6 à(gpt-proA)62 lacYI tsx-33 supE44 galK2 hisG4 metBl rfbD l mgl-51 rpsL260 kdgK51 m tl-I thi-1 thyA12 deoB16
GM113 G U m dam-3GM150 GM112 dam-3 mutL45I

A. Massey et a l /D N A R epa ir 2 (2003) 73 -89 75
Strain Genotype
AB1157 F~ thr-1 ara-14 leuB6 A.{gpt-proA)62 lacYl tsx-33 glnV44(AS) galK2{0c) hisG4{0c) rfbD l mgl-51 rpoS396{Am) rpsL31{StrR) JcdgKSl xylAS mtl-1
.argE3{0c) thi-1 I GM3819 ABU 51 dam-16::Kan . GM5556 AB1157 dam-16::Kan mutS::TnlO ÆS1582 AB1157 m«rL25!
2.2. E. coli survival
I Cisplatin was dissolved in water at 37 °C for 4-6 h. j The drug concentration was determined by A301 (e = I 131). Log phase bacteria were pelleted, resuspended in M9 salts at a concentration of 1 x 10 to 2 x 10 ml“ ', and treated for 2h at 37 °C. Treated cells were washed twice by resuspension in M9 salts and plated on L-agar. Surviving colonies were scored after overnight incubation at 37 °C.
Cisplatin-resistant variants were generated by treating approximately 5 x 10 A2780-SCA cells with cisplatin (3.33 mM stock in 1% NaCl, David Bull Laboratories, Vic., Australia) in media containing 10% PCS for 1 h at 37 °C. After treatment, cells were allowed to recover in cisplatin-free medium for 2-3 weeks before the next treatment. Single cell clones were isolated from the resistant population by limiting dilution.
2.6. Cell survival
Cell survival was determined by clonal assay. 10 to 2 X 10 cells from an exponentially growing culture were seeded in six well plates, allowed to attach for 4 h and then treated in duplicate with 0-20 p,M cisplatin (David Bull Laboratories) for 1 h in medium containing 10% PCS at 37 °C. Cells were supplemented with drug-free medium and colonies of greater than 50 cells were scored under a microscope after 7 days growth at 37 T .
2.3. Selection o f cisplatin-resistant E. coli
Log phase cells were treated with 100 jxM aquated cisplatin as above. After each treatment, cells were washed twice with M9 salts. Pollowing overnight growth in L-broth, survivors were either diluted in L-broth and grown to log phase for a subsequent round of cisplatin treatment or plated on L-agar. Single colonies were picked and expanded for phenotype analysis. Resistant clones were generated from a total of four cisplatin treatments.
2.4. Cell lines
The A2780 cell line was kindly provided by Prof. R. Brown, University of Glasgow. The méthylation tolerant variants A2780-MNU1A and A2780-clone 1 have been described previously [17,18]. All cells were grown and treated in DMEM supplemented with 10% fetal calf serum in humidified incubators containing 10% CO2.
2.5. Selection o f cisplatin-resistant A2780 clones
A single cell clone, A2780-SCA, was isolated from A2780 by limiting dilution in 96-well plates.
2.7. Western blotting
2.7.1. p53andp21 inductionPor the determination of p53 and p21 levels, about
10 cells were treated with 30|xM cisplatin for 1 h at 37 °C. The medium was replaced and the cells harvested 24 h later, lysed in 1% NP40, 10 mM NaP, 1 mM NaV3 0 4 , mini EDTA-free protease inhibitor tablet (Roche) at 0°C for 1 h. Extracts were clarified by centrifugation at 15000 x g for 30 min. 50|xg cell extract was loaded onto a 10% (p53) or 12% (p21) SDS-PAGE gel. Separated proteins were transferred to a PVDP membrane (Immobilon-P, Millipore) using a Transblot semi-dry transfer cell (BioRad). Membranes were blocked with 5% non-fat milk and p53 detected with PL393 rabbit polyclonal (1 |JLg/ml, Santa Cruz) or p21 with EAIO mouse monoclonal (0.1|jLg/ml, Oncogene Research) antibodies. Antigen-antibody complexes were detected with HRP-conjugated anti-mouse or anti-rabbit antibodies (Biorad) and ECL substrate on Hyperfilm ECL (Amersham Pharmacia Biotech).
2.7.2. MMR proteins50 |jLg cell extract was separated on 8 % SDS-poly-
acrylamide gels, and proteins transferred to PVDP.

76 A. Massey et a l. /D N A R epa ir 2 (2003) 73-89
Antibodies to hMSH2 (G219-1129, Pharmingen, 1 fJLg/ml), hMSH6 (Q-20, Santa Cruz, 2 [xg/ml), hMLHI (G168-15, Pharmingen, 0.5 jjig/ml) or hPMS2 (A 16-4, Pharmingen, 1 |ig/ml) and ECL were used to detect MMR proteins.
2.8. p53 Amplification and sequencing
The primer sequences used to amplify the exons of p53 have been described previously [17,19]. PCR reactions contained lOOng genomic DNA (prepared using QIAGEN DNeasy tissue kit), 2.5 mM MgCl2, 200 |iM each dNTP, 0.5 |jlM primers and 2.5 U Hot- Star Taq (QIAGEN) in manufacturer’s buffer to a final volume of 50 |il. Exons 2-4 were amplified by: 15 min at 95 °C, 35 cycles of Im in at 95 °C, 1 min at 55 °C and 1 min at 72 °C followed by a final extension of 10 min at 72 °C. Exons 5-8 were amplified using the same conditions except the annealing temperature was increased to 65 °C. Exons 6 and 7 required the addition of Q-solution (QIAGEN) to the reaction. PCR products were purified on QIAquick PCR purification spin columns (QIAGEN) and sequenced on an ABI Prism 377 Sequencer.
The presence of the exon 4 Arg polymorphism was detected by digesting 200 ng of purified PCR product with 5U BstUI. Products were separated on a 1.8% agarose gel. PCR products containing the pro codon remain full length (298 bp) following BstUI digestion; those containing the Arg sequence are digested to products of 100 and 198 bp.
2.9. DNA platination
DNA platination levels were determined by atomic absorption spectroscopy as previously described [20]. Approximately 4 x 10 cells were treated with 0-100 |iM cisplatin for 2h at 37 °C. Cells were washed, harvested and the DNA extracted, ethanol precipitated and extensively washed. DNA was dried and hydrolysed in 0.2% nitric acid overnight at 37 °C.
2.10. In vitro mismatch repair assay
Mismatch repair assays were carried out as previously described [21].
2.11. RT-PCR
RNA was prepared from about 5 x 10 cells using the RNeasy minikit (QIAGEN) with a DNase step. 1 fxg of RNA in 20 |jl1 was converted to cDNA using ImProm-II Reverse Transcriptase (Promega) in the presence of 20 U of RNasin Ribonuclease Inhibitor (Ambion). The 2 jjlI RT product was amplified in a multiplex PCR reaction containing 0.04 jjlM (3-actin, 0.5 |jlM hMSH2, 0.25 p.M hPMS2, 0.1 fJiM hMSH6, 0.5 fiM hMLHI and 0.125 jjlM hPMSl primers as described in [22]. Reactions (5 0 |ul1) contained 2.5 mM MgCL, 200 jJuM each dNTP and 2.5 U HotStar Taq. Amplification was performed for 15 min at 95 °C, 30 cycles of 95 °C for 30 s, 58 °C for 30 s and 72 °C for 45 s followed by a final extension of 72 °C for 10 min. PCR products were separated on a 2% agarose gel and detected by ethidium bromide staining.
2.12. BAT26 microsatellite analysis
Single cell clones generated by limiting dilution were expanded to around 10 cells. DNA was extracted and BAT26 amplified (95 °C for 15 min followed by 35 cycles of 95 °C for 1 min, 55 °C for 1 min and 72 °C for 1 min with a final extension of 72 °C for 10 min) with fluorescently labelled primers [23]. Products were sized using an ABI Prism 377 Sequencer.
2.13. Méthylation tolerance screen
A total of 200 cells were plated in duplicate in 6-well plates, allowed to attach for 2 h. Following a 2 h pre-treatment with 25 jiM 0^-benzylguanine (Sigma), 200 (jlM recrystallised MNU (kindly provided by Prof. Peter Swann, University College London) was added. Cultures were grown for a further 7 days and colonies scored under a microscope.
2.14. Cell cycle analysis
2.14.1. Propidium iodide staining Exponentially growing cell cultures were treated
with 3 0 |jlM cisplatin for Ih . After incubation in drug-free medium, aliquots of cells were harvested and approximately 10 cells fixed in 70% ethanol for 1 h at 4 °C. Prior to analysis by flow cytometry, ethanol was removed by washing with PB SA and.

A. Massey et a l. /D N A R epa ir 2 (2003) 73-89 77
after treatment with RNaseA, cells were incubated with propidium iodide (50 fjtg/ml) for 15 min at room temperature.
2.14.2. BrdU labelling Exponentially growing cells were pulse labelled
with 10p,M BrdU for 10 min immediately prior to a 1 h treatment with 30 |jlM cisplatin. Cells were fixed and washed as above before being treated with 2 M HCl for 30 min at room temperature. Following extensive washing to remove acid, cells were incubated with anti-BrdU antibody (Becton Dickson) for 20 min at room temperature in the dark. Antibody-antigen complexes were detected with FITC-conjugated rabbit anti-mouse P(ab')2 fragments (Dako) for 15 min at room temperature in the dark. DNA was then stained with propidium iodide (50 p-g/ml) for 30 min before analysis by flow cytometry.
I 2.14.3. Sub-Gl DNA contentThe sub-Gl apoptotic cell population was measured
by propidium iodide staining as described above except fixed cells were washed with phosphate-citrate buffer (0.192M Na2HP0 4 , 4m M citric acid pH 7.8) instead of PBSA.
3. Results
3.1. Cisplatin-resistant E. coli dam strains
We investigated whether exposure of dam mutants to cisplatin provided a strong selective pressure for inactivation of M MR and whether dam mutants with a mutator phenotype were the major class of cisplatin-resistant dam isolates.
3.1.1. Sensitivity o f dam and dam MMR~ strains to cisplatin
Both GM113 (dam-3) and GM3819 (dam-16:.-Kan) were around 4-fold more sensitive to cisplatin than their respective dam~ parents (Fig. la and b). Introduction of either a mutS or mutL mutation reduced their sensitivity without fully restoring WT cisplatin resistance. For example, GM150 (dam-3 mutL), GM169 (dam-3 mutS), and GM5556 (dam-16::Kan mutS::TnlO) were all about two-fold more sensitive than GM112 (dam~^) and AB1157(Am"^). As
expected, inactivation of M M R did not alter cisplatin resistance in a dam'^ background and the responses of dam~ FS1582 (mutL) and A B I 157 to cisplatin treatment were indistinguishable. These observations confirm that the E. coli mismatch repair pathway plays an important role in cisplatin sensitivity that is only apparent in a Dam-deficient background [9]. They also indicate that inactivation of M M R in dam strains restores cisplatin resistance to a level that approaches, but does not reach, that of wild-type, dam'^ cells.
3.1.2. Selection fo r cisplatin resistant dam strainsWe examined the development of cisplatin resis
tance in GM3819 (dam-16::Kan) following repeated exposure to toxic concentrations of the drug. Exponentially growing cultures were treated up to four times with 100 p.M cisplatin, a dose that reduced survival to approximately 10“ . After each treatment cycle, the phenotype of 50 surviving clones from several independent cultures was investigated by examining resistance to 2-AP, the presence of a mutator phenotype, and the sensitivity of DNA to digestion with Dpnl.
After a single round of cisplatin treatment, about 6% of GM3819 survivors were 2-AP'' consistent with either a dam'^ or dammutS/L genotype. All of these resistant variants exhibited a strong mutator phenotype indicative of a mismatch repair defect. Further, since the dam gene in this strain is inactivated by a transposon insertion, reversion to dam'^ is unlikely. In agreement with this, DNA from all cisplatin-resistant GM3819 colonies remained sensitive to Dpnl digestion. After a second round of selection, the mutator population accounted for 66% of the total and after a further two treatments, all cells examined had this phenotype. Following the fourth round of selection, the cisplatin resistance of three independent isolates was examined quantitatively. Two clones were more cisplatin-resistant than dam mutS or dam mutL strains. The sensitivity of the remaining clone was intermediate between that of dam and W T strains and was compatible with inactivation of M M R in a dam background (Fig. la). The observations that all surviving cisplatin-resistant colonies exhibited a strong mutator phenotype, were resistant to 2-aminopurine and their DNA was insensitive to Dpnl digestion (for example, see Fig. Ic and d), suggest that inactive M M R is the predominant route by which E. coli dam mutants escape killing by cisplatin. In addition, the recovery of
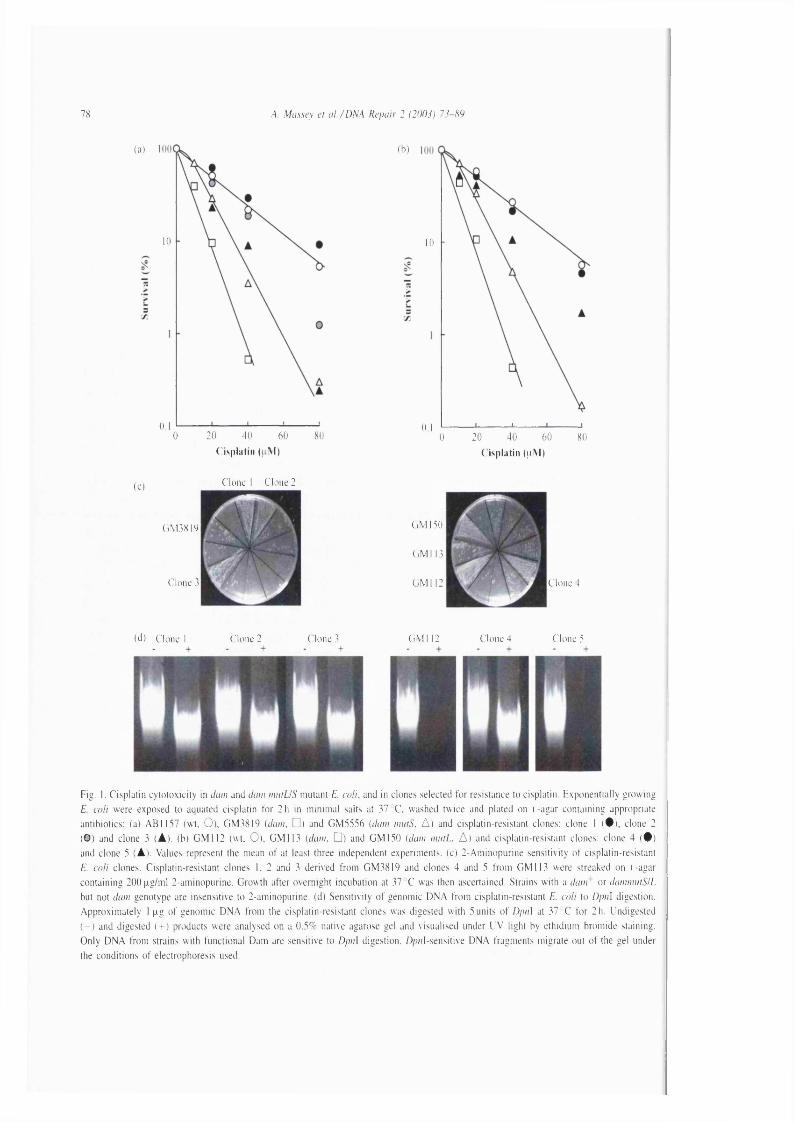
78 ,4. Massey et a i /D M A Repair 2 (2003) 73-89
(a)
10
0.120 40 60 800
( b )
10
0,120 40 600 80
C'isplatin (u M ) ( isplatin (iiM )
(0 Clone I Clone 2
(iM 3819
Clone 3
C M 130
C M 113
CM 112 Clone 4
(d) Clone 1 Clone 2 Clone 3 C M 112 Clone 4 Clone 5
Fig. 1. C isplatin cyto tox ic ity in dam and dam m iitU S mutant E. co ll, and in clones selected fo r resistance to cisplatin. Exponentially grow ing
E. co ll were exposed to aquated cisplatin fo r 2h in m inim al salts at 37 C, washed tw ice and plated on i.-agar containing appropriate
antibiotics: (a) AB1157 (w t, O), GM 3819 (dam. □ ) and GM 5556 (dam mutS. A) and cisplatin-resistant clones: clone 1 ( # ) , clone 2
(O ) and clone 3 (À ) . (b) GM 112 (w t. 0), GM 113 (dam, □ ) and G M 150 [dam m iit i. A) and cisplatin-resistant clones: clone 4 ( • )
and clone 5 (A). Values represent the mean o f at least three independent experiments, (c) 2-Am inopurine sensitivity o f cisplatin-resistant
E. co ll clones. Cisplatin-resistant clones 1, 2 and 3 derived from GM3819 and clones 4 and 5 from GM 113 were streaked on i.-agar
containing 200|xg/m l 2-aminopurine. Growth after overnight incubation at 37 C was then ascertained. Strains w ith a dam ' or damimitSIL
but not dam genotype are insensitive to 2-aminopurine. (d) Sensitivity o f genomic D N A from cisplatin-resistant E. co li to Dpiû digestion.
Approxim ate ly 1 p.g o f genomic D N A from the cisplatin-resistant clones was digested w ith 5 units o f Dpn] at 37 C fo r 2h. Undigested
( - ) and digested (-h) products were analysed on a 0.3% native agarose gel and visualised under U V light by ethidium bromide staining.
O nly D N A from strains w ith functional Dam are sensitive to Dpn\ digestion. Dpnl-sensitive D N A fragments migrate out o f the gel under
the conditions o f electrophoresis used.

A. Massey et a l. /D N A R epa ir 2 (2003) 73 -89 79
MMR-defective dam clones with W T or greater cisplatin resistance suggests that they may have acquired additional mutations that further reduce their cisplatin sensitivity.
Essentially similar results were obtained following cisplatin treatment of GM113 {dam) in which the dam gene is inactivated by a point mutation. In this case, resistant clones fell into two major classes. The majority exhibited the intermediate cisplatin sensitivity associated with dammutS or dammutL (clone 4). Most of the remainder had the properties consistent with reversion to dam~ (DNA sensitive to Dpnl and full
I cisplatin resistance— Fig. Ib-d, clone 5). i These observations indicate that inactive MMR is
frequent among cisplatin-resistant E. coli dam mutants. Some cisplatin-resistant MMR-defective E. coli dam survivors may harbour additional mutations that increase their cisplatin resistance to the wild-type
I level. The mutator effect that accompanies inactiva- I tion of M M R may facilitate the acquisition of these
extra mutations.
3.2. Cisplatin resistant human cells
3.2.1. SelectionThe A2780 human ovarian carcinoma cell line is
widely used as a model for drug resistance. Cultures of A2780 are known to harbour a small sub-population of cells that are defective in mismatch repair because of a methylated HMLHI promoter [18]. These cells also contain a dominant negative p53 mutation that affects their cisplatin resistance [17,18]. A single cell clone of this original population A2780-SC1, was previously shown to have normal M M R and a wild-type p53 response [17]. The present study was initiated with a freshly-isolated sub-clone of A2780-SC1 designated A2780-SCA which also retained both functional M M R and p53 responses (see in the following sections).
Cisplatin-resistant variants of A2780-SCA were isolated by two slightly different procedures. Resistant clones designated CPI A to CP6A were isolated after a single exposure to 30 jxM and two subsequent treatments of surviving cells at 50 p.M cisplatin. For clones CP7A to CP 12A, after an initial dose of 10|jlM cisplatin, survivors were treated with 20, 30 p,M and two rounds of 50 |iM . All clones isolated were of independent origin.
All 12 clones (C P IA -12A) displayed a stable resistance to further cisplatin exposure. The extent of resistance varied from around 1.5-2.5-fold. Examples are shown in Fig. 2.
3.2.2. MMR status
3.2.2.1. Méthylation tolerance. A high level resistance to MNU is a hallmark of defective MMR. All the cisplatin-resistant variants were screened for M NU resistance. Each clone was plated in duplicate wells of a 6-well plate at 200 cells per well. After 2-h incubation with 0^-benzylguanine to deplete MGMT, cells were treated with 200 p,M MNU, a dose that kills >95% of M MR proficient A2780 cells but does not detectably reduce survival of MMR-defective A2780 variants [24]. Surviving colonies were scored after 7 days growth in the continuous presence of 0^-benzylguanine. As expected, no colonies were observed in wells containing parental A2780-SCA cells. None of the cisplatin-resistant clones CP1A-12A was as resistant as a MMR-defective variant and no survivors (<1% of untreated values) were observed in wells containing CPI A or CP4A-12A. A low level of survival was observed for CP2A (6%) and CP3A (17%). In neither case was the resistance equivalent to the hMLHl-defective A2780 derivative A2780-MNU1A (100% survival). Thus, none of the cisplatin-resistant clones is méthylation tolerant.
3.2.2.2. MMR gene expression. Expression of the hM LH l, hPMS2, hMSH2 and hMSH6, M MR genes was examined by RT-PCR. Reverse transcribed mRNA was amplified by multiplex PGR using primers designed to generate M MR cDNA fragments of easily distinguishable length. A (3-actin fragment served as an internal control. Fig. 3a shows that all five M MR cDNA fragments were amplified from the two control cell lines Raji and A2780-SCA, and from all 12 cisplatin-resistant A2780 lines. Three MMR-defective colorectal carcinoma cell lines served as negative controls. The hM LHl product was absent from SW48— consistent with the known silencing of the hMLHl promoter in these cells [25]. Similarly, no hMSH2 product was recovered from LoVo cells that contain an hMSH2 deletion [26]. hMSH6 is mutated in DLD-1 [27]. The ]evel of hMSH6

80 A. Massey et a l. /D N A Repair 2 {2 0 0 i) 73-89
(a) 10 -
8 -
6 -
4 -
7 -
SCA CP3A CP5A CP7A CP8A I 200 10
( isplatin (jiiM)
Fig. 2. C isplatin cy to tox ic ity in A2780 variants. Parental cells A278()-SCA or cisplatin-resistant clonal variants were treated w ith cisplatin
fo r 6 0m in at the concentrations indicated, the drug removed and cells grown fo r a further 7 days in drug-free media. Colonies o f greater
than 50 cells were scored under a microscope, (a) values were calculated from straight line semi-log sur\iva l plots, (b) C isplatin
cyto tox ic ity in A2780-SCA; ( □ ) and clonal variants CP3A (O) and CP5A (A). Values represent the mean o f at least two independent
experitnents ± the standard error.
cDNA amplification product from these cells was consistently reduced, suggesting that the mutated mRNA might be unstable. We conclude that MMR gene, and in particular hMLHl, silencing does not underlie the cisplatin resistance of the CP1A-12A clones.
3.2.2.3. MMR protein levels. The levels of hMLHl, hPMS2, hMSH2 and hMSH6, proteins in the 12 resistant variants were investigated by western blotting. The amounts of hMSH2, hMLHl, and hPMS2 were reproducibly indistinguishable from those of A2780-SCA. Some variability in levels of hMSH6 was encountered. Differences were not reproducible, however, and we conclude that all four MMR proteins
are present at levels similar to those of the parental A2780-SCA cells (Fig. 3b).
3.2.2.4. Mismatch repair. The MMR capacity of four representative clones was examined in a standard MMR assay. Two, CP3A and CP5A, were from the first treatment schedule, and two, CP7A and CP8A, from the second. Extracts of all four clones coiTected a single C:T mispair in a nicked circular heteroduplex substrate with an efficiency similar to that of the parental A2780-SCA cells (Fig. 3c). As expected, extracts of two cell lines that are hMLH I -defective, the HCTl 16 colorectal carcinoma and the MNU-resistant A2780-MNU1 [17] were unable to carry out repair. This direct biochemical assay confirms that

/\. Massey et a l. /D N A Repair 2 (2003) 73-89
p -A c iiii
MSI 12
M S H 6
M i l
(b) SC'A C P IA CP2A CP3A C'P4AC'P5A CP6 A C’P7A CPKA ('P 9A ('P lO A C PU A C P I 2A Jiirkal
hMSH2
I1M S II6
SCA CP IA CP2A CP3A CP4A CP5A CP6 A CP7A CPKA CP9A CPiO A C PU A C P I2 A 1 IC T II6
liM l,
hPMS2
( c ) SCA CP3A CP7A CPKA M N U I
No
cxiract Hcl.a IIC T I1 6SCA
CP5A
No
extract CPKA
Fig. 3. M ismatch repair status o f A2780 variants, (a) M u ltip lex RT-PCR co-am plification o f [3-actin, hM SH2. hPMS2, I1M S H 6 , h M L H l
and hPM Sl gene products from A2780 clonal variants. Control lines Raji T K - (w t), SW48 { l iM L H I - } , LoVo (hM S H 2~) and D L D l
{I1M SH 6 - ) are shown in the left panel. The PCR products were electrophoresed through a 2% agarose gel, stained w ith ethidium bromide
and visualised w ith U V light, (b) M M R protein expression by Western blot. Cell extracts were separated on an 8 % agarose gel, transferred
to PVD F membrane and probed w ith anti-hM SH2, an ti-liM S H 6 , a n ti-h M L H l or anti-hPMS2 antibodies. Antibody-antigen complexes were
detected using ECU on H yperfilm ECU. (c) In v itro mismatch repair activity. Extracts from A2780-SCA, CP3A, CP.3A, CP7A or CP8 A
were assayed fo r the a b ility to correct a T/C mismatch and restore an M lu \ restriction endonuclease site in a standard nicked circular
duplex substrate. Fo llow ing the incubation o f the D N A substrate w ith extract, D N A was digested w ith M lii\ and the products analysed on
a 0.8% agarose gel in the presence o f ethidium bromide. The arrowed band corresponds to D N A molecules in which the mismatch has
been corrected.

82 A. Massey et a !./D N A R epa ir 2 (2003) 73-89
these four representative clones are MMR proficient.
3.2.2.5. Microsatellite instability. The BAT26 mononucleotide microsatellite is monoraorphic in the general population [28]. Changes at this locus provide a sensitive indicator of an underlying MST^ phenotype. Analysis of BAT26 in 20 subclones of CP5A provided no indication of alteration in length (data not shown). These data are consistent with the absence of an MSI"^ phenotype and with normal levels of MMR in CP5A.
In summary, there was no evidence of a MMR defect in any of the twelve cisplatin-resistant A2780-SCA clones. None had a méthylation tolerant phenotype. All resistant clones had normal levels of MMR mRNA. Key M MR proteins were present at wild-type levels in each. In an in vitro assay extracts of four representative clones showed an unimpaired ability to correct a single base mismatch. Examination of one of these four provided no evidence of an MSl"^ phenotype. We conclude that the changes underlying the cisplatin resistance of the CP1A-12A variants are unrelated to MMR.
3.2.3. Other potential resistance factorsOther possible factors that might underlie the cis
platin resistance of the A2780 variants were examined.
3.2.3.1. p53 status. p53 is a significant— albeit unpredictable— modifier of cisplatin resistance in A2780 cells. We examined the p53 response of the 12 cisplatin-resistant clones. Western blotting indicated that none of the clones had high constitutive levels of p53 protein and most responded to cisplatin treatment with increased p53 levels (Fig. 4a). Twenty-four hours after treatment with 30 fxM cisplatin, there was a clear (> 10-fold) induction of p53 in the parental A2780-SCA cells. In CP3A and CP7A (also possibly CPIOA), this response was apparently defective and the increase in p53 levels was reproducibly lower (< 3-fold) than in the parental cells (Fig. 4a). Consistent with this, p21 induction was also attenuated in CP3A and CP7A. p53 response of these clones was examined in more detail. In contrast to the behaviour after cisplatin exposure, p53 stabilisation and p21 induction after treatment with other DNA damaging
agents was normal. Fig. 4b shows that the response of CP7A to 5 Gy ionising radiation or 20J/m- UVC was not attenuated compared to parental A2780-SCA cells. Similar results were obtained with CP3A (data not shown). In a separate series of experiments, a more quantitative analysis of the p53 response after cisplatin exposure indicated that the p53 responses of CP3A and CP7A were suboptimal. Both clones retained the ability to respond to cisplatin but 2^-fold higher cisplatin concentrations were required to elicit this p53 response (examples are shown in Fig. 4c).
The p53 genes of CP3A, CP5A, CP7A, and CP8A were sequenced directly. No mutations were found in p53 exons 2-8 in any of the four variants (data not shown). The common sequence polymorphism (pro) at codon 72 in exon 4 was found in the parental cells and in all four variants. Homozygosity for Pro at codon 72 was confirmed by resistance to BstUl digestion (data not shown). Thus, the altered p53 response of the resistant A2780 variants does not reflect a mutated p53 gene.
3.2.3.2. DNA platination. The levels of DNA platinum were determined by atomic absorption spectroscopy. The extent of DNA platination was significantly lower in CP7A and CP3A than in A2780-SCA over a range of cisplatin concentrations. DNA platinum levels in CP7A were around half of those in the parental cells (0.8units/p.M drug versus 1.6units/p,M drug). For CP3A, platination was reduced to around two thirds (1.2 units per p,g drug). DNA platinum levels of CP5A and CP8A were slightly higher than those of the parental cells (Fig. 4d). Increased levels of GSH did not account for the reduced levels of DNA platinum in CP7A and CP3A. Steady-state GSH levels were closely similar in the parental cells and in CP3A, CP7A and CP8A. The extent of GSH induction by cisplatin treatment varied somewhat among the resistant clones but there was again no correlation between susceptibility to DNA platination and GSH (data not shown).
We conclude that the increased survival and the selective attenuation of the cisplatin-induced p53 response in CP3 A and CP7A most likely reflects a lower level of cisplatin-DNA damage. The precise nature of the protective process in these variants remains unclear but it seems unlikely to be due to altered steady-state or induced levels of GSH.
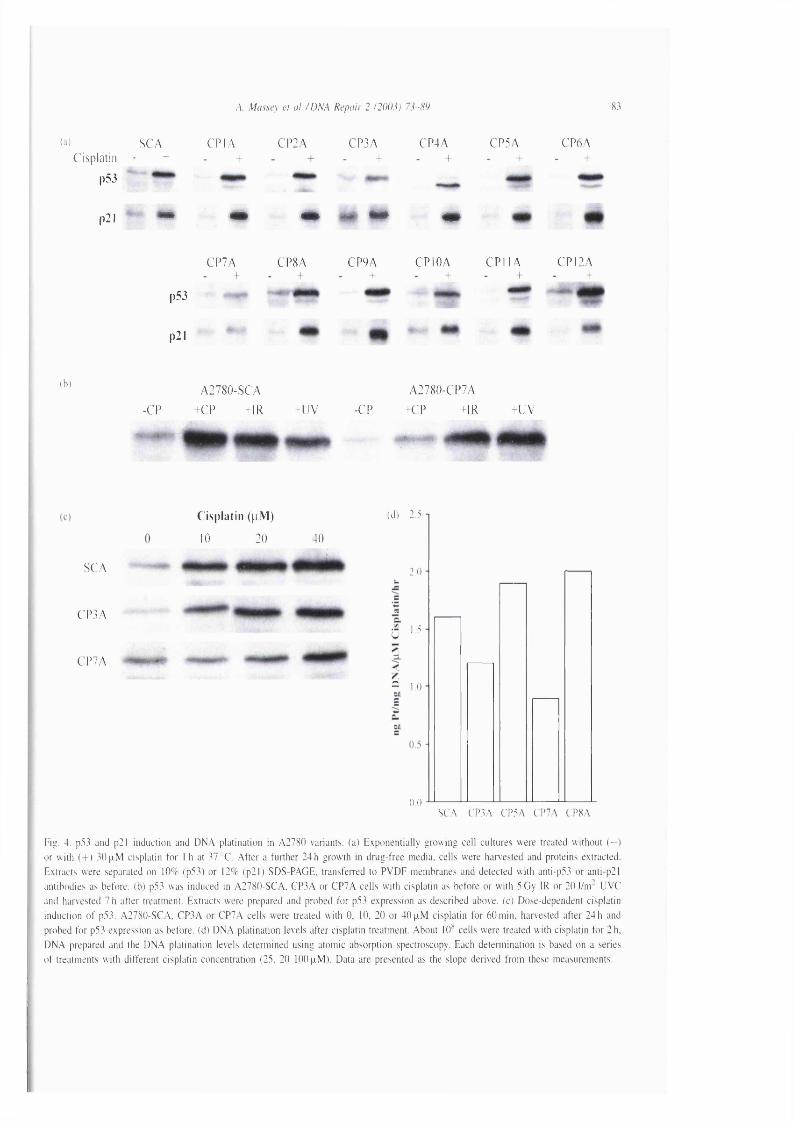
A. Massey et a l. /D N A Repair 2 (2003) 73-89 83
W SCACisplatin - +
p53
CP IA CP2A CP3A CP4A CP5A CP6A
p2l
CP7A CP8A CP9AI
p53
p21
CPIOA C P I IA CPI2A
(b)
-CPA2780-SCA
+CP +IR +UV -CPA2780-CP7A
-C P M R - U V
Cisplatin ( p M )
10 20 40
SCA
CP3A
CP7A
(d) 2.5
2.0
1.5
= 1.0
0.0.SCA CP3A CP5A CP7A CP8A
Fig. 4. p53 and p2l induction and D N A platination in A2780 variants, (a) Exponentially grow ing cell cultures were treated w ithout ( - )
or w ith ( + ) 3 0 plM cisplatin fo r I h at 37 C. A fte r a further 24 h growth in drug-free media, cells were harvested and proteins extracted.
Extracts were separated on 10% (p53) or 12% (p21) SDS-PAGE, transfeiTed to PVD F membranes and detected w ith anti-p53 or anti-p21
antibodies as before, (b) p53 was induced in A2780-SCA, CP3A or CP7A cells w ith cisplatin as before or w ith 5 Gy IR or 20 j / n r U VC
and harvested 7 h after treatment. Extracts were prepared and probed fo r p53 expression as described above, (c) Dose-dependent cisplatin
induction o f p53. A2780-SCA, CF3A or CP7A cells were treated w ith 0, 10, 20 or 40 p.M cisplatin fo r 60 m in, harvested after 24 h and
probed fo r p53 expression as before, (d) D N A platination levels after cisplatin treatment. About 10^ cells were treated w ith cisplatin fo r 2 h.
D N A prepared and the D N A platination levels determined using atomic absorption spectroscopy. Each detem iination is based on a series
o f treatments w ith d ifferent cisplatin concentration (25. 20 100 p.M). Data are presented as the slope derived from these measurements.

84
(a)
A. Massey et a l. /D N A Repair 2 (2003) 73-89
A2780-SCA A2780-CP5A
Oh
G2/M
DNA DNA
( b )
c
30
10
048 720 24 96
T im e (h is )
Fig. 5. C ell cycle response and apoptosis in resistant A2780 variants, (a) D N A content o f exponentially growing cell cultures o f SCA
or CP5A cells after 30p.M cisplatin fo r 1 h was determined by propidium iodide staining (le ft panels) at various time points as shown.
A lternatively, cells were pulsed w ith l()p .M BrdU fo r 10m in immediately prio r to cisplatin treatment and the fate o f S-phase cells fo llow ed
using anti-B rdU antibodies (righ t panels), (b) Apoptosis induction in SCA (0) or CP5A ( □ ) after treatment w ith 3()|xM cisplatin fo r 1 h.
The fraction o f cells w ith a sub-G l D N A content was detennined by modified propidium iodide staining at the times indicated.

A. Massey et a l. /D N A R epa ir 2 (2003) 73-89 85
3.2.3.3. Cell cycle. Cisplatin is known to induce cell-cycle arrest in human cells. We examined the effect of cisplatin treatment on cell cycle progression in the four representative cisplatin-resistant clones CP3A, CP5A, CP7A, and CP8A. The DNA content of cisplatin-treated cells was determined by propidium iodide staining and FACS analysis. In addition, FACS was also used to examine the fate of BrdU-labelled cells that were in S-phase at the time of cisplatin treatment.
The predominant effect of treatment of A2780-SCA cells with 30 |xM cisplatin was accumulation in late S-phase. The S -f G2/M fraction increased from 38 to 76% at 18-24h and reached a maximum of around 90% by 42-48 h post-treatment (Fig. 5a and data not shown). Analysis of BrdU-labelled cells that had been in S-phase at the time of cisplatin treatment confirmed the S-phase arrest. The block to progression appeared to be long-term and the distribution of cells remained essentially unchanged for up to 96 h post-treatment (data not shown).
The response of A2780-CP5A cells to 30 [jlM cisplatin was quite different. At 24 h, there was a modest increase in the proportion of cells in S-phase (from29 to 36%) accompanied by a more dramatic accumulation in G2/M. This is clearly demonstrated by the analysis of the fate of the BrdU-labelled S-phase cells. By 48 h, the G2/M block was partially relieved and a significant fraction of cells had re-entered the cycle. This distribution more closely resembled that of untreated cells (Fig. 5a, right panel). A fraction of the cells remained arrested, however, and the distribution had not returned to normal even at 96 h post treatment (data not shown).
The behaviour of CP8A cells after treatment with30 |xM cisplatin was similar to that of CP5A. The response of CP3 A— that sustain less cisplatin damage— was intermediate between the parental A2780-SCA and CP5A cells. In particular, the reduced S-phase progression and substantial accumulation in late S resembled the parental cells. These events occurred somewhat later (at 48 h) than in A2780-SCA, however. Unlike the parental cells, the block did not persist and cells progressed into G2/M where, like CP5A, they arrested (data not shown). It seems likely that these somewhat attenuated responses reflect the lower level of cisplatin induced DNA damage in CP3A cells.
3.2.3.4. Apoptosis. Sub-Gl DNA content is regarded as a measure of apoptotic cell death. At 24 and 48 h after cisplatin treatment, the fraction of cells with a sub G1 DNA content was not significantly different among A2780-SCA, CP5A and CP8A. As expected from their relative cisplatin sensitivity, analysis at 72h revealed a significantly greater fraction of sub-Gl A2780-SCA cells than CP5A or CP8A cells (an example is shown in Fig. 5b). Thus, alterations in cell cycle progression and reduced induction of apoptosis are two characteristic features of cisplatin resistance in CP5A and CP8A cells.
4. Discussion
Our data confirm the key role played by M M R in cisplatin toxicity in dam strains and the absence of a detectable contribution of M MR in a dam'^ background. They also indicate for the first time, that loss of M MR provides a substantial selective survival advantage for cisplatin-treated E. coli dam mutants. Clones with inactive MMR are widely represented among dam survivors of cisplatin exposure. The observation that further cisplatin treatment results in selection of clones with resistance greater than that of dammutS or dammutL strains suggests the acquisition of additional secondary mutations. The frequency of these secondary mutations may be increased by the loss of MMR.
Inactivation of M M R is now widely regarded as one of the major routes by which human tumours can acquire significant cisplatin resistance. The twelve independent A2780 variants we isolated displayed degrees of cisplatin resistance (2-3-fold) similar to those generally associated with MMR inactivation. The extreme levels of resistance such as the 60- or 39-fold exhibited by the cisplatin-selected A2008/A or A2780CP70 lines [16,29] are atypical and are most likely not simply related to MMR. We found no evidence of M MR defects at either the mRNA or protein level in any of the twelve, however. None was tolerant to DNA méthylation damage— an invariant hallmark of defective MMR. In agreement with these general findings, extracts prepared from four representative cisplatin-resistant clones were fully MMR proficient in vitro. p53 status is a known modifier of drug sensitivity. Analysis of the DNA damage response and

A. Massey et a l . / D NA R epa ir 2 (2003) 73-89
direct sequencing of the p53 exons in which most of the inactivating mutations lie, revealed that the cisplatin resistance of our clones was not a consequence of an abrogated p53 response to DNA damage. Thus, two acknowledged contributors to cisplatin sensitivity, M MR and p53, apparently functioned normally in all our resistant variants.
The finding that all 12 cisplatin-resistant variants were proficient in M M R was, perhaps, unexpected in view of the widely-held notion that MMR contributes significantly to cisplatin sensitivity. Most of the evidence that links defective M MR and cisplatin resistance is indirect, however. Previous cisplatin-resistant variants of A2780 [5,16] contain multiple alterations, many of which contribute to overall drug resistance. Altered intracellular drug accumulation or DNA modification, and dominant p53 mutations are among those known to coexist with defective MMR. In these cases, it is clearly inappropriate to ascribe the cisplatin resistance to MMR inactivation. When the relative contributions of MMR and other factors to the cisplatin resistance of méthylation tolerant A2780 cells was assessed, MMR inactivation was found to exert only a minor influence [18]. The findings reported here, based on a different approach designed to examine acquired cisplatin resistance, confirm this and indicate further that the emergence of MMR-defective variants is an infrequent consequence of cisplatin treatment.
A connection between cisplatin resistance and M MR inactivation has been inferred from clinical studies on tumours as well as from in vitro models. It is noteworthy in this regard that cisplatin resistance is frequently associated with hM LH l promoter méthylation. There are numerous examples of this MMR gene silencing among cisplatin-resistant A2780 variants [30]. In addition, many of the clinical immunohisto- chemical data are consistent with reduced, or absent, expression of this gene in cisplatin-resistant tumours. HMLHl promoter méthylation is fairly common and it underlies the MMR defects of many non-familial MSI"^ colorectal, endometrial and gastric tumours [31-33]. We suggest that HMLHl promoter méthylation in cisplatin-resistant cell lines or tumours may simply be a marker for altered epigenetic gene regulation. There is increasing evidence that promoter cytosine méthylation affects many genes in tumours with this phenotype. One, or several, of these silencing events may contribute to cisplatin resistance. This
provides a possible explanation for the paradoxical simultaneous reduced expression of the presumably epistatic HMLHl and HMSH2 genes in cisplatin- treated tumours [34] and the absence of correlation between HMLHl reactivation and cisplatin sensitivity in A2780 derivatives [30]. The absence of association between M MR status and cisplatin resistance of the 12 A2780-SCA variants may simply indicate that A2780-SCA does not have the necessary deregulated epigenetic control. It is implicit in this argument that inactivation of MMR may not, in itself, confer a sufficient survival advantage on cells treated with cisplatin. Any small contribution from inactive repair must be combined with additional changes to provide the cell with significant resistance.
The relation between p53 and cisplatin resistance is complex. In general, p53 mutations are associated with cisplatin resistance and replacement of W T p53 has been suggested as a means of clinical chemosensi- tisation. A dominant negative p53 increases cisplatin resistance in A2780 cells and in combination with a silent HMLHl promoter is responsible for the high level cisplatin resistance in many variants of A2780. The absence of p53 alterations in our cisplatin- resistant clones may simply indicate that, without a mutator phenotype, the probability of the necessary type of p53 mutation is low. A non-exclusive explanation is that most spontaneous p53 mutations do not confer sufficient resistance in a M M R proficient background for selection under our experimental conditions. It may be significant in this respect that M MR defects and p53 mutations coexist in several established cisplatin-resistant cell lines.
We identified two contributors to cisplatin resistance in the A2780 clones. The first was a protective function that significantly reduced the level of DNA platination. Although the precise mechanism of this effect remains undefined, the extent of protection is most likely sufficient to account for the increased resistance of those clones. Protective effects of this nature are widespread among cisplatin variants. They confer cross-resistance to numerous drugs and they are known to contribute to the cisplatin resistance of the much studied A2780CP series of Ozols et al [16,35].
We found examples in which drug resistance was associated with changes in the way replicating cells responded to cisplatin treatment. Although we did not examine all 12 variants by FACS, the finding that

A. Massey et a l. /D N A R epa ir 2 (2003) 73-89 87
changes in S-phase progression were apparent in two (CP5A and CP8A) of four clones suggests that this is likely to be a common phenotype. After treatment with cisplatin, the predominant effect in the parental A2780 cells was a significant block to progression through S-phase. In CP5A and CP8A, the effects of an equivalent level of DNA cisplatin damage were less pronounced. The behaviour of CP5A and CP8A was more consistent with a reversible G2 arrest associated with a reduction in cisplatin-induced apoptosis. Possible explanations for the altered S-phase response include abrogation of inter-S-phase checkpoints and an enhanced ability to process cisplatin DNA lesions by recombinational or lesion bypass mechanisms. It has been suggested that A2780 variants— and other tumour cell lines with M MR deficiencies— perform more ‘replicative bypass of cisplatin DNA adducts’ [4] and that ability is a direct consequence of the M M R defect. Increased’ cisplatin lesion bypass has also been inferred in resistant cells that are M M R proficient, however [36]. At present, we are unable to distinguish whether the impaired S-phase progression of CP5A and CP8A reflects increased elongation (lesion bypass), decreased responsiveness to signals preventing replicon firing (S-phase checkpoint), or a combination of both. It is possible that, because they are tumour-derived, the parental A2780 cells may already have a dysfunctional S-phase checkpoint. Whatever the underlying mechanism, it is clear that the S-phase effects in CP5A and CP8A are not dependent on defective MMR.
In regard to lesion bypass, it may be significant that cisplatin is a potent inducer of the E. coli SOS response. Two key components of this response, are encoded by the umuDC operon. The UmuD2UmuC protein complex is considered to function as a DNA replication checkpoint [37]. The related UmuD^ UmuC trimer, in which the UmuD protein is truncated, is DNA polymerase V. Both DNA polymerase V and the related DNA polymerase IV, encoded by the SOS regulated dinB'^ gene, perform translesion synthesis on damaged DNA. The human DNA polymerases eta and kappa can carry out bypass of cisplatin lesions [38,39]. Factors related to replication of cisplatin-damaged DNA are attractive alternatives to mismatch repair deficiency in acquired cisplatin resistance. Although the properties of the cisplatin-resistant A2780 clones do not exclude a role for defective MMR
in cisplatin resistance in some circumstances, they suggest that effects related to S-phase checkpoints and/or DNA lesion bypass might be more common. These may be fruitful areas for future investigation of acquired cisplatin resistance in human tumour cells.
Acknowledgements
We thank Drs. John Essigmann and Martin Mari- nus for kindly providing E. coli lines and advice on E. coli cisplatin toxicity. We are grateful to Dr. Ciaran O’Neill for carrying out the Atomic Absorption Spectroscopy analysis. The skilful assistance of members of the Cancer Research UK Cell Services Laboratory, Flow Cytometry Laboratory, and Equipment Park is greatly appreciated.
References
[1] R.P. Perez, Cellular and molecular determinants of cisplatin resistance, Eur. J. Cancer 34 (1998) 1535-1542.
[2] B. Koberle, J.R.W. Masters, J.A, Hartley, R.D. Wood, Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours, Curr. Biol. 9 (1999) 273-276.
[3] D. Fink, S. Nebel, S. Aebi, H. Zheng, B. Cenni, A. Nehme, R.D. Christen, S B. Howell, The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 56 (1996) 4881- 4886.
[4] A. Vaisman, M. Varchenko, A. Umar, T.A. Kunkel, J.I. Risinger, J.C. Barrett, T.C. Hamilton, S.G. Chaney, The role of hMLHl, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 58 (1998) 3579-3585.
[5] D.A. Anthoney, A.J. Mcllwrath, W.M. Gallagher, A.R.M. Edlin, R. Brown, Microsatellite instability, apoptosis, and loss of p53 function in drug-resistant tumour cells. Cancer Res. 56 (1996) 1374-1381.
[6] J.G. Gong, A. Costanzo, H.-Q. Yang, G. Melino, W.G. Kaelin Jr, M. Levrero, J.Y.J. Wang, The tyrosine kinase c-Abl regulates p73 in apoptoic response to cisplatin-induced DNA damage. Nature 399 (1999) 806-809.
[7] B.D. Harfe, S. Jinks-Robertson, DNA mismatch repair and genetic instability, Annu. Rev. Genet. 34 (2000) 359-399.
[8] P. Karran, M.G. Marinus, Mismatch correction at O -methyl- guanine residues in E. co li DNA, Nature 296 (1982) 868-869.
[9] R.J. Fram, P.S. Cusick, J.M. Wilson, M.G. Marinus, Mismatch repair of c«-diamminedichloroplatinum(II)-induced DNA damage. Mol. Pharmacol. 28 (1985) 51-55.
[10] M. Bignami, M. O’Driscoll, G. Aquilina, P. Karran, Unmasking a killer: DNA 0 -methylguanine and the cytotoxicity of methylating agents, Mutat. Res. 462 (2000) 71-82.

A. Massey et al. /D N A R epa ir 2 (2003) 73-89
[11] G. Aquilina, P. Hess, S. Fiumicino, S. Ceccotti, M. Bignami, A mutator phenotype characterises one of two complementation groups in human cells tolerant to méthylation damage. Cancer Res. 55 (1995) 2569-2575.
[12] R. Hampson, 0. Humbert, P. Macpherson, G. Aquilina, P. Karran, Mismatch repair defects and 0 -methylguanine-DNA methyltransferase expression in acquired resistance to methylating agents in human cells, J. Biol. Chem. 272 (1997) 28596-28606.
[13] M. O’Driscoll, S. Martinelli, C. Ciotta, P. Karran, Combined mismatch and nucleotide excision repair defects in a human cell line: mismatch repair processes méthylation but not UV- or ionising-induced DNA damage. Carcinogenesis 20 (1999) 799-804.
[14] S.W. Johnson, P.B. Laub, J.S. Beesley, R.F. Ozols, T.C. Hamilton, Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Cancer Res. 57 (1997) 850-856.
[15] A. Johnsson, I. Zeelenberg, Y. Min, J. Hilinski, C. Berry, S B. Howell, G. Los, Identification of genes differentially expressed in association with acquired cisplatin resistance, Brit. J. Cancer 83 (2000) 1047-1054.
[16] B.C. Behrens, T.C. Hamilton, H. Masuda, K.R. Grotzinger, J. Whang-Peng, K.G. Louie, T. Knutsen, W.M. McKoy, R.C. Young, R.F. Ozols, Characteristaion of a cir-diammine- dichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res. 47 (1987) 414-418.
[17] G. Aquilina, S. Ceccotti, S. Martinelli, S. Soddu, M. Crescenzi, P. Branch, P. Karran, M. Bignami, Mismatch repair and p53 independently affect sensitivity to A-(2-chloroethyl)- N'-cyclohexyl-#-nitrosourea, Clin. Cancer Res. 6 (2000) 671- 680.
[18] P. Branch, M. Masson, G. Aquilina, M. Bignami, P. Karran, Spontaneous development of drug resistance: mismatch repair and p53 defects in resistance to cisplatin in human tumour cells. Oncogene 19 (2000) 3138-3145.
[19] J. Toguchida, T. Yamaguchi, B. Ritchie, R.L. Beauchamp, S.H. Dayton, G.E. Herrera, T. Yamarauro, Y. Kotoura, M.S. Sasaki, J.B. Little, R.R. Weichselbaum, K. Ishizaki, D.W. Yandell, Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res. 52 (1992) 6194-6199.
[20] B. Koberle, K.A. Grimaldi, A. Sunters, J.A. Hartley, L.R. Kell and, J.R.W. Masters, DNA repair capacity and cisplatin sensitivity of human testis tumour cells. Int. J. Cancer 70(1997) 551-555.
[21] S. Oda, 0. Humbert, S. Fiumicino, M. Bignami, P. Karran, Efficient repair of A/C mismatches in mouse cells deficient in long-patch mismatch repair, EMBO J. 19 (2000) 1711-1718.
[22] Q. Wei, M.L. Bondy, L. Mao, Y. Guan, L. Cheng, J. Cunningham, Y. Fan, J.M. Bruner, W.K.A. Yung, V.A. Levin, A.P. Kyritsis, Reduced expression of mismatch repair genes measured by multiplex reverse transcription-polymerase chain reaction in human gliomas. Cancer Res. 57 (1997) 1673- 16777.
[23] W. Dietmaier, S. Wallinger, T. Bocker, F. Kullmann, R. Fishel, J. Ruschoff, Diagnostic microsatellite instability: definition
and correlation with mismatch repair protein expression. Cancer Res. 57 (1997) 4749^756.
[24] 0. Humbert, S. Fiumicino, G. Aquilina, P. Branch, S. Oda, A. Zijno, P. Karran, M. Bignami, Mismatch repair and differential sensitivity of mouse and human cells to methylating agents. Carcinogenesis 20 (1999) 205-214.
[25] M.F. Kane, M. Loda, G.M. Gaida, J. Lipman, R. Mishra, H. Goldman, J.M. Jessup, R. Koldner, Méthylation of the hMLHl promoter correlates with lack of expression of hMLHl in sporadic colon tumours and mismatch repair-defective human tumour cell lines. Cancer Res. 57 (1997) 808-811.
[26] A. Umar, J.C. Boyer, D.C. Thomas, D.C. Nguyen, J.I. Risinger, J. Boyd, Y. Ionov, M. Perucho, T.A. Kunkel, Defective mismatch repair in extracts of colorectal and endometrial cancer cell lines exhibiting microsatellite instability, J. Biol. Chem. 269 (1994) 14367-14370.
[27] N. Papadopoulos, N.C. Nicolaides, B. Liu, R. Parsons, C. Lengauer, F. Palombo, A. D’Arrigo, S. Markowitz, J.K.V. Willson, K.W. Kinzler, J. Jiricny, B. Vogelstein, Mutations of GTBP in genetically unstable cells. Science 268 (1995) 1915-1917.
[28] J.-M, Hoang, PH. Cottu, B. Thuille, R.J. Salmon, G. Thomas, R. Hamelin, BAT-26, an indicator of the replication error phenotype in colorectal cancers and cell lines. Cancer Res. 57 (1997) 300-303.
[29] S. Aebi, B. Kurdi-Haidar, R. Gordon, B. Cenni, H. Zheng, D. Fink, R.D. Christen, C.R. Boland, M. Koi, R. Fishel, S B. Howell, Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 56 (1996) 3087-3090.
[30] G. Strathdee, M.J. MacKean, M. Illand, R. Brown, A role for méthylation of the hMLHl promoter in loss of hMLHl expression and drug resistance in ovarian cancer. Oncogene 18 (1999) 2335-2341.
[31] A.S. Fleisher, M. Esteller, S. Wang, G. Tamura, H. Suzuki, J. Yin, T.-T. Zou, J.M. Abraham, D. Kong, K.N. Smolinski, Y- Q. Shi, M.-G. Rhyu, S.M. Powell, S.P. James, K.T. Wilson, J.G. Herman, S.J. Meltzer, Hypermethylation of the hMLHl gene promoter in human gastric cancers with microsatellite instability. Cancer Res. 59 (1999) 1090-1095.
[32] M. Esteller, R. Levine, S B. Baylin, L.H. Ellenson, J.G. Herman, MLHl Promoter hypermethylation is associated with the microsatellite instablility phenotype in sporadic endometrial carcinomas. Oncogene 16 (1998) 2413-2417.
[33] J.G. Herman, A. Umar, K. Polyak, J.R. Graff, N. Ahuja, J.-P.J. Issa, S. Markowitz, J.K.V. Willson, S.R. Hamilton, K.W. Kinzler, M.F. Kane, R.D. Kolodner, B. Vogelstein, T.A. Kunkel, S B. Baylin, Incidence and functional consequences of hmlhl promoter hypermethylation in colorectal cancer, Proc. Natl. Acad. Sci. U.S.A. 95 (1998) 6870-6875.
[34] G. Samimi, D. FinS, N.M. Varki, A. Husain, W.J. Hoskins, D.S. Alberts,' S.B. Howell, Analysis of MLHl and MSH2 expression in ovarian cancer before and after platinum drug-based chemotherapy, Clin. Cancer Res. 6 (2000) 1415- 1421,
[35] K. Hamaguchi, A.Ki Godwin, M. Yakushiji, P.J. O’Dwyer, R.F. Ozols, T.C. Hamilton, Cross-resistance to diverse drugs is associated with primary cisplatin resistance in ovarian carcinoma cell lines. Cancer Res. 53 (1993) 5225-5232.

A. Massey et a l. /D N A R epair 2 (2003) 73-89
[36] E.L. Mamenta, E.E. Poma, W.K. Kaufmann, D.A. Delmastro, H.L. Grady, S.G. Chaney, Enhanced replicative bypass of platinum-DNA adducts in cisplatin-resistant human ovarian carcinoma cell lines. Cancer Res. 54 (1994) 3500-3505.
[37] A.E. Ferentz, G.C. Walker, G. Wagner, Converting a DNA damage checkpoint effector (UmuD2 C) into a lesion bypass polymerase (UmuD^C), EMBO J. 20 (2001) 4287- 4298.
[38] A. Vaisman, C. Masutani, F. Hanaoka, S.G. Chaney, Efficient translesion replication past oxaliplatin and cisplatin GpG adducts by human DNA polymerase t\. Biochemistry 39 (2000) 4575^580.
[39] E. Ohashi, T. Ogi, R. Kusumoto, S. Iwai, C. Masutani, F. Hanaoka, H. Ohmori, Error-prone bypass of certain DNA lesions by the human DNA polymerase k , Genes Dev. 14 (2000) 1589-1594.

Reprinted from
DNA Repair
Available online at www.sciencedirect.com
S C I E N C E D I R E C T *
DNA Repair 2 (2003) 547-559
Drug treatment in the development of mismatch repair defective acute leukemia and myelodysplastic syndrome
Ida Casorelli^'^, Judith Offman^, Luca Mele^, Livio Pagano^, Simona Sica‘S, Mariarosaria D ’Errico^, Giuseppe Giannini^, Giuseppe Leone
Margherita Bignami^’*, Peter Karran^® Istitulo Superiore di Sanita’, Laboratorio d i Tossicologia Comparata, Rome, Italy
** Cancer Research UK, Clare H a ll Laboratories, South Mimms, UK
Hematology Department, Catholic University, Rome, Ita ly Department o f Experimental Medicine and Pathology, “La Sapienza ” University, Rome, Ita ly
Accepted 13 January 2003
E L S E V IE R

Aims and scopeDNA REPAIR provides a forum for the comprehensive coverage of cellular responses to DNA damage in living cells. The journal publishes original observations on genetic, cellular, biochemical and molecular aspects of DNA repair, mutagenesis, cell cycle regulation, apoptosis and other biological responses to cells exposed to genomic insult, as well as their relationship to human diseases. Original full-length papers are published monthly. In addition, the journal will publish a smaller number of peer-reviewed Brief Reports on original research findings of special interest, as well as invited Mini-reviews on selected topics that provide ‘state-of-the-art’ synopses of cellular responses to DNA damage. Book reviews and meeting reports will be regularly featured and the Journal welcomes Correspondence from the scientific community, especially as they relate to papers previously published in the journal. These are handled directly by the Editor-in-Chief and may be accompanied by responses solicited from relevant individuals.
Editors:
Editor-in-Chief:E C. Friedberg , Department of Pathology, University of Texas Southwestern Medical Centre, 5323 Harry Hines Blvd, Dallas, TX, 75390-9072, USA. Fax: (I) 214 648 4067; e-mail: [email protected]
European Receiving Editor:A.A. van Zeeland, Department of Toxicogenetics, Leiden University Medical Center, Sylvius Laboratory, Wassenaarseweg 72, 2333 AL Leiden, The Netherlands. Fax: (31) 71 527 6173; e-mail: [email protected]
Associate Editors:Bryn Bridges, Genome Damage and Stability Centre, University of Sussex, Falmer, Brighton BN I 9RR, United Kingdom, e-mail: [email protected] Hoeijmakers, Department of Cell Biology & Genetics, Erasmus University, P.O. 1738, 3000 DR Rotterdam, The Netherlands, e-mail: [email protected] Shiloh, Dept of Human Genetics, Tel Aviv University Medical School, Tel Aviv 69978, Israel, e-mail: [email protected] Tanaka, Osaka University, Institute of Molec. Cell. Biol. 1-3 Yamada-oka, Suita Osaka 565-0871, Japan, e-mail: [email protected] H. Wilson, Laboratory of Structural Biology, National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH), P.O. Box 12233, I I I T.W. Alexander Drive, Research Triangle Park, NC 27709-2233, USA. e-mail: [email protected] D. Wood, University of Pittsburgh Cancer Institute, Hillman Cancer Centre, Research Pavillion, Suite 2-3 5117 Centre Avenue, Pittsburgh, PA 15213-1863, USA. e-mail: [email protected]
Editorial Board
Daniel F. Bogenhagen, Stony Brook, NY, USAVilhelm A. Bohr, Baltimore MD, USASerge Boiteux, Fontenay Aux Roses, FranceJaap Brouwer, Leiden, The NetherlandsKeith W. Caldecott, Brighton, UKAntony M. Carr, Brighton, UKPriscilla K. Cooper, Berkeley, CA, USAGilbert de Murcia, Strasbourg, FranceBruce Demple, Boston, MA, USAGrigory L. Dianov, Oxfordshire, UKEugenia Dogliotti, Rome, Ita lyJean-Marc Egly, Illk irch , FranceJohn M. Essigmann, Cambridge, MA, USARichard Fishel, Philadelphia, PA, USAAlbert J. Fomace, Jr., Bethesda, MD, USARobert P.P. Fuchs, Strasbourg, FranceAnn Ganesan, Stanford, CA, USAJean Gautier, New York, NY, USAMyron F. Goodman, Los Angeles, CA, USANora Goosen, Leiden, The NetherlandsArthur P. Grollman, Stony Brook, NY, USAThanos D. Halazonetis, Philadelphia, PA, USAFumio Hanaoka, Osaka, JapanPhilip C. Hanawalt, Stanford, CA, USAIan D. Hickson, Oxford, UKPeggy Hsieh, Bethesda, MD, USASteve P. Jackson, Cand)ridge, UKPenny A. Jeggo, Brighton, UKS. Jinks-Robertson, Atlanta, GA, USABemd Kaina, Mainz, GermanyRoland Kanaar, Rotterdam, The NetherlandsPeter B. Karran, South Mimms, UKCaroline I. Kisker, Stony Brook, NY, USAHannah Klein, New York, NY, USARichard Kolodner, La Jolla, CA, USAHans E. Krokan, Trondheim, NorwayThomas A. Kunkel, Research Triangle park, NC, USAJacques Laval, Cedex, FranceChristopher W. Lawrence, Rochester, NY, USARandy Legerski, Houston, TX, USAAlan R. Lehmann, Brighton, UKMichael R. Lieber, Los Angeles, CA, USATomas Lindahl, South Mimms, UKStuart Linn, Berkeley, CA, USAMichael Liskay, Portland, OR, USAZvi Livneh, Rehovot, Israel
Lawrence A. Loeb, Seattle, WA, USAVeronica M. Maher, East Lansing, M l, USAIsabel Mellon, Lexington, KY, USAJeffrey H. Miller, Los Angeles, CA, USAPaul L. Modrich, Durham, NC, USADale W. Mosbaugh, Corvallis, OR, USALeon H.F. Mullenders, Leiden, The NetherlandsHanspeter Naegeli, Zurich, SwitzerlandYusaku Nakabeppu, Fukuoka, JapanVesna Rapic Ortin, Pittsburgh, PA, USAThomas D. Petes, Chapel H ill, NC, USAJohn H.J. Petrini, Madison, Wl, USAMiroslav Radman, Paris, FranceMichael A. Resnick, Research Triangle Park, NC, USASusan M. Rosenberg, One Baylor PlazaRodney Rothstein, New York, NY, USAL. Samson, Cambridge, MA, USAAlain R. Sarasin, Villejuif, FranceRoger A. Schultz, Dallas, TX, USAErling Seeberg, Oslo, NorwayMutsuo Sekiguchi, Fukuoka, JapanHideo Shinagawa, Osaka, JapanMiria Stefanini, Pavia, Ita lyBruce Stillman, C old Spring Harbor, NY, USAPatrick Sung, San Antonio, TX, USAJesper Svejstrup, South Mimms, UKJohn Tainer, La Jolla, CA, USAJohn Thacker, Harwell, UKFritz Thoma, Zurich, SwitzerlandLarry H. Thompson, Livermore, CA, USAAlan Tomkinson, San Antonio, TX, USABennett Van Houten, Research Triangle Park, NC, USAHarry Van Steeg, Bilthoven, The NetherlandsAshok R. Venkitaraman, Cambridge, UKWim Vermeulen, Rotterdam, The NetherlandsHarry Vrieling, Leiden, The NetherlandsGeoffrey M. Wahl, La Jolla, CA, USAGraham C. Walker, Cambridge, MA, USASusan S. Wallace, Burlington, VT, USAZhigang Wang, Lexington, KY, USARay Waters, Swansea, UKSteve West, South Mimms, UKRoger Woodgate, Bethesda, MD, USAAkira Yasui, M iyag i JapanMalgorzala Z. ^zienicka, Leiden, The Netherlands
Instructions to Authors and addresses for submission of articles are provided at the back of each issue and at the website http://www.elsevier.com/locate/dnarepair
PH: 5 1 5 6 8 - 7 8 6 4 ( 0 3 ) 0 0 0 5 0 - 8

Available online at www.sciencedirect.com
S C I E Ii N C E ^ ^ D I R E C T * D N A
REPAIRE LS E V IE R DNA Repair 2 (2003) 547-559 ....... ........................
www.elsevier.comAocate/dnarepair
Drug treatment in the development of mismatch repair defective acute leukemia and myelodysplastic syndrome
Ida Casorelli^'^, Judith Offman^, Luca Mele^, Livio Pagano^, Simona Sica‘S, Mariarosaria D ’Errico^, Giuseppe GianniniGiuseppe Leone‘S,
Margherita Bignami^’*, Peter Karran^® Istituto Superiore di Sanita’, Laboratorio di Tossicologia Comparata, Rome, Italy
Cancer Research UK, Clare H all Laboratories, South Mimms, UK Hematology Department, Catholic University, Rome, Italy
Department o f Experimental Medicine and Pathology, “La Sapienza" University, Rome, Italy
Accepted 13 January 2003
Abstract
DNA from therapy-related acute leukemia/myelodysplastic syndrome cases (tAL/MDS) from the G IM E M A [Gruppo Ital- iano Malattie Ematologiche Maligne dell’Adulto] A rch ive was examined for the microsatellite instability (MSI+) phenotype that is diagnostic for defective DNA mismatch repair. More than 60% (16/25) of tAL/MDS cases were MSP in contrast to <4% (0/28) of de novo cases. H M L H I gene silencing was rare and evidence of promoter méthylation was found in less than one-third of the MSP cases. Among the G IM E M A patients who had been treated for breast cancer there was an apparent trend towards early onset primary breast disease. This suggests that there might be common predisposing factors for breast cancer and tAL/MDS. There were also three examples of mutations in the M R E I I gene among the 25 tAL/MDS cases suggesting that defective recombinational DNA repair may promote the development of secondary mahgnancy. M SP tAL/MDS was significantly associated with previous chemotherapy and the frequency of M SP among radiotherapy patients was considerably lower. In view of the estabhshed relationship between drug resistance and mismatch repair defects, we suggest that selection for therapeutic drug resistance may contribute to the incidence of MSP tAL/MDS.© 2003 Elsevier Science B.V. All rights reserved.
Keywords: Mismatch repair; Acute myeloid leukemia; Chemotherapy
Abbreviations: tAL, therapy-related acute leukemia; tMDS, therapy-related myelodysplastic syndrome; M M R, mismatch repair; M S P , microsatellite instability; tAPL, therapy-related acute promyelocytic leukemia; tA M L, therapy-related acute myeloid leukemia; tA LL,
therapy-related acute lymphocytic leukemia; HNPCC, Hereditary Non-Polyposis Colorectal Cancer; FAB, French-American-British; 5-FU, 5-fluorouracil; CTX, cyclophosphamide; RT, radiotherapy; MGMT, 0^-methylguanine-DNA methyltransferase; MOPP, mechloroethamine, vincristine, procarbazine, prednisone; COPP, cyclophosphamide, vincristine, procarbazine, prednisone; ABVD, adriamycin, bleomycin,
vinblastine, dacarbazine* Corresponding author. Tel.: -1-39-06-49902355; fax: -1-39-06-49902355.
E-mail address: [email protected] (M. Bignami).
1568-7864/03/$ - see front matter © 2003 Elsevier Science B.V. A ll rights reserved.
doi:10.1016/S1568-7864(03)00020-X

548 I. C asore lli et a l. /D N A R epa ir 2 (2003) 547 -559
1. Introduction
As a consequence of improved therapeutic regimes, more cancer patients are surviving for longer. The frequency of therapy-related cancer— an unrelated secondary malignancy following therapy for a first tumor— has increased in parallel. Acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) are the most prevalent forms of secondary cancer and therapy-related AML (tAML) now accounts for 10% of all AML cases [1]. tAML and tMDS are distinguished from de novo disease by characteristic cytogenetic alterations [2]. tAML is generally more refractory to treatment than de novo AML and tAML patients have a significantly poorer prognosis [3-5]. Therapy-related acute lymphocytic leukemia (tALL) is less common [6].
Recent years have seen an improved understanding of how therapeutic drugs kill tumor cells and of the cellular changes that can accompany acquired drug resistance. The cytotoxicity of some chemotherapeutic agents has been linked to DNA mismatch repair (MMR) and inactivation of MMR is associated with drug resistance. MMR is an important pathway for correcting DNA replication errors. Two important het- erodimeric protein complexes are required for most MMR: hMutSa (hMSH2 and hMSH6) and hMutLa (hMLHl and hPMS2). M MR is defective in some cancers. In the Hereditary Non-Polyposis Colorectal Cancer (HNPCC) syndrome, a high frequency of early onset colorectal, endometrial and other tumors [7] is associated with a germ line mutation in either the hMSH2 or the hM LHl MMR gene. Mutations in hMSH6 and hPMS2, are less frequent [8,9]. In HNPCC tumors, the remaining functional MMR allele is inactivated by a somatic event and the tumor cells are repair deficient. In addition to this familial tumor predisposition, a considerable fraction of non-HNPCC tumors are also MMR defective. In these sporadic cases, the repair deficiency is frequently a consequence of epigenetic inactivation of the hM LHl gene involving cytosine méthylation within the promoter region [10]. A susceptibility to epigenetic silencing has also been noted for the hMSH6 gene [11]. A MMR defect confers a spontaneous mutator phenotype and repair-deficient cells exhibit a dramatic increase in the rate of frameshift-like mutations in repetitive DNA sequences. This is observed experimentally as mi
crosatellite instability (MSI” ) and the MSI^ phenotype is considered to be diagnostic for defective MMR.
Resistance to DNA damaging drugs, particularly to methylating agents and to thiopurines, is an acknowledged property of cells with defective MMR. In the case the methylating agents with which this resistance phenomenon has been most widely studied, resistance does not reflect increased removal of DNA lesions and for this reason it is known as tolerance to DNA méthylation damage [12]. In the laboratory, treatment with methylating agents is a straightforward way of selecting for MMR defective cells [13,14]. In addition to its effect on susceptibility to methylating drugs, impaired M MR has been associated with an increased resistance to a number of other therapeutic agents, including 6-thioguanine [15], cisplatin [16,17], doxorubicin, [18], and 5-fluorouracil (5-FU) [19,20],
The MSI^ phenotype is rare in de novo AML [21,22]— with the possible exception of AML in elderly individuals [23,24]. Estimates of the frequency of MSI^ in tAML/MDS vary widely. There is increasing evidence that it is more widespread than among de novo cases, however [25,26]. Here, we examine the hypothesis that MSI^ AL/MDS is the clinical counterpart of selection for MMR-deficient drug tolerant cells in the laboratory. We confirm a high frequency of MSI"^ in tAL/MDS but not in de novo AL. The MSI"*" phenotype of tAL/MDS cases was significantly associated with chemotherapy rather than radiation treatment. Silencing of the hM LHl gene by promoter méthylation is infrequent in MSI"^ tAL/MDS, although it may be particularly associated with therapy-related acute promyelocytic leukemia (tAPL). The findings are consistent with the hypothesis that tAL/MDS may sometimes reflect the selection of clones with tolerance to drug-induced chemical DNA damage.
2. Patients and methods
2.1. Patients
The study involved 25 Italian tAL/MDS patients who were admitted, treated, and followed-up at the Division of Hematology of the Catholic University of Rome from March 1992 to September 2001. Patient details were recorded in the GIMEMA [Gruppo

I. C asore lli et a l. /D N A R epa ir 2 (2003) 547 -559 549
Italiano Malattie Ematologiche Maligne dell’Adulto] Archive. Written informed consent was obtained from each subject. There were 19 tAML— including 5 tAPL-4 tALL, and 2 tMDS cases (details summarized in Table 1).
The median ages at diagnosis of the primary and secondary malignancy were 52 years (range 22-73 years) and 58 years (range 31-75 years), respectively. Breast carcinoma was the first malignancy in 11 cases. This accounted for the prevalence of females (17) over males (8).
Cytogenetic analysis of bone marrow cells was performed at the time of diagnosis. Deletion of the long arm or loss of an entire chromosome 7 was detected in four tAML/MDS cases (T3, T7, T8, T21) and cases T7 and T21 also had a deletion of 5q or loss of the whole chromosome 5.
Twenty-eight cases of de novo AL (10 AML, 8 APL and 10 ALL) were also analyzed. Their median age at diagnosis was 43.5 years (range 16-75 years) with a similar distribution between females and males (13 and 15, respectively). All APLs in both therapy and control groups had the characteristic t(15;17) translocation and diagnosis was confirmed by RT-PCR for the PML-RARa gene fusion.
2.2. Cell separation and DNA isolation
Bone marrow cells collected at diagnosis were separated on Ficoll gradients. DNA was extracted from cryopreserved mononuclear cells using TRI- zol (GIBCO BRL, Gaithersburg, MD). In two cases, paraffin blocks of skin were available from which normal DNA was isolated (DNA extraction kit— Takara shuzo Co., Japan). In 13 cases, normal DNA was extracted from the roots of 3 or 4 freshly plucked hairs [27].
2.3. Microsatellite analysis
To measure microsatellite instability, five mono- or dinucleotide microsatellites (BAT25, BAT26, D2S123, D17S250, D5S346) were examined [28]. PCR reactions were carried out in 25 p-1 1 x buffer (20 mM Tris-HCl, pH 8.4, 50 mM KCl), 1.5 mM MgCl], 0.1 mM each dNTP, 1U AmpliTaq DNA polymerase (PE Applied Biosystems), 15 ng genomic DNA and 0.5 p,M primers. Amplifications were performed on
a Perkin-Elmer Thermal Cycler after an initial dénaturation at 95 °C for 5 min. Thirty cycles of 1 min at 90 °C, 30 s at 55 °C, 30 s at 70 °C were followed by a final extension of 72 °C for 10 min. Products were separated on 6% denaturing polyacrylamide gels and transferred to Hybond N+ membrane (Amer- sham Italia, Milan, Italy). Membranes were probed overnight at 42 °C with a radiolabeled PCR primer in 130 mM sodium phosphate, pH 7.0, 250 mM NaCl, 10% polyethyleneglycol (Mr 4000; Sigma, Milan, Italy), and 7% sodium dodecylsulfate. Hybridization products were detected by autoradiography.
2.4. HMLHI promoter méthylation assay
The promoter méthylation assay was adapted from Kane et al. [10]. The PCNA promoter sequence from -98 2 to -1-45 used as a control contains the ATG start codon and five HpaU/Mspl restriction sites. Genomic DNA (lOpg to 2 ng) 10 p.1 buffer (NEB) was digested with no enzyme, 50 U H pa ll (NEB), or lOOU MspI (NEB). Incubation was for 8 h at 37 °C followed by nuclease inactivation for 10 min at 65 °C.
Digested DNA was analyzed by multiplex PCR in 50 p.1 1X Qiagen PCR buffer containing 2.5 mM MgCl2, 0.2 mM each dNTP, 2.5 U HotStarTaq DNA Polymerase (Qiagen) and lOpmol each primer. For hM LHl, the forward primer was:
5'-CCACATACCGCTCGTAGTATTCGTGC-3'
the reverse primer:
5'-CCTCAGTGCCT CGTGCTCACGTTC-3'.
For PCNA, the forward primer was:
5-CCTAGAAAGACAACGACCACTCTGC-3'
the reverse primer:
5'-GGCAGGAGACTCACTTGAACCTGG-3'.
Amplification was performed on a Peltier Thermal Cycler PTC-200 after initial dénaturation at 95 °C for 15 min. Each cycle was: 1 min at 95°, 1 min at 68°, 1 min at 72 °C. Thirty to 35 cycles were followed by a final extension at 72 °C for 7 min. After 35 cycles, unamplified samples were cycled further with nested primers. For the HMLHI promoter, 1 |xl of the multiplex PCR product was analyzed in 50 p-1 buffer
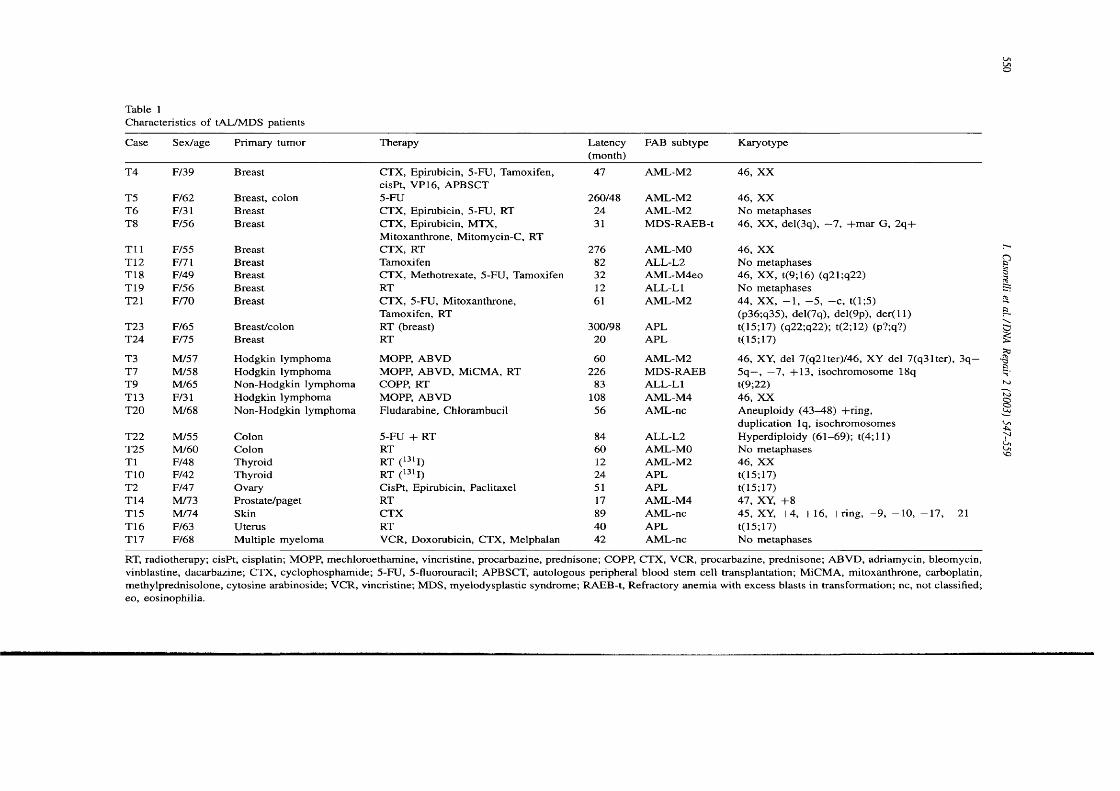
Table ICharacteristics o f tAL/MDS patients
Case Sex/age Primary tumor Therapy Latency(month)
FAB subtype Karyotype
T4 F/39 Breast CTX, Epirubicin, 5-FU, Tamoxifen, cisPt, VP16, APBSCT
47 AM L-M 2 46, X X
T5 F/62 Breast, colon 5-FU 260/48 AM L-M2 46, XXT6 F/31 Breast CTX, Epirubicin, 5-FU, RT 24 AM L-M2 No metaphasesT8 F/56 Breast CTX, Epirubicin, MTX,
Mitoxanthrone, Mitomycin-C, RT31 M DS-RAEB-t 46, XX, del(3q), —7, -t-mar G, 2q-|-
T i l F/55 Breast CTX, RT 276 AML-MO 46, XXT12 F/71 Breast Tamoxifen 82 A L L-L2 No metaphasesT18 F/49 Breast CTX, Methotrexate, 5-FU, Tamoxifen 32 AM L-M 4eo 46, XX, t(9;l6) (q21;q22)T19 F/56 Breast RT 12 ALL-Ll No metaphasesT21 F/70 Breast CTX, 5-FU, Mitoxanthrone,
Tamoxifen, RT61 AM L-M 2 44, XX, —1, —5, —c, t(l;5)
(p36;q35), del(7q), del(9p), d e r ( ll)T23 F/65 Breast/colon RT (breast) 300/98 APL t(15;17) (q22;q22); t(2;12) (p?;q?)T24 F/75 Breast RT 20 APL t(l5;17)
T3 M/57 Hodgkin lymphoma MOPP, ABVD 60 AM L-M2 46, XY, del 7(q21ter)/46, XY del 7(q31ter), 3q—T7 M/58 Hodgkin lymphoma MOPP, ABV D, MiCMA, RT 226 M DS-RAEB 5q—, —7, -1-13, isochromosome 18qT9 M/65 Non-Hodgkin lymphoma COPP, RT 83 A LL-Ll t(9;22)T13 F/31 Hodgkin lymphoma MOPP, ABVD 108 AM L-M 4 46, XXT20 M/68 Non-Hodgkin lymphoma Fludarabine, Chlorambucil 56 AML-nc Aneuploidy (4 3 ^ 8 ) -bring,
duplication Iq, isochromosomesT22 M/55 Colon 5-FU -t- RT 84 ALL-L2 Hyperdiploidy (61-69); t(4;l 1)T25 M /60 Colon RT 60 AML-MO No metaphasesT1 F/48 Thyroid RT (^311) 12 AM L-M2 46, XXTIO F/42 Thyroid RT (13'I) 24 APL t(15;17)T2 F/47 Ovary CisPt, Epirubicin, Paclitaxel 51 APL t(15;17)T14 M/73 Prostate/paget RT 17 AM L-M4 47, XY, -F8T15 M/74 Skin CTX 89 AML-nc 45, XY, -|-4, -|-16, -bring, —9, —10, —17, —21T16 F/63 Uterus RT 40 APL t(15;17)T17 F/68 Multiple myeloma VCR, Doxorubicin, CTX, Melphalan 42 AML-nc N o metaphases
RT, radiotherapy; cisPt, cisplatin; MOPP, mechloroethamine, vincristine, procarbazine, prednisone; COPP, CTX, VCR, procarbazine, prednisone; ABV D, adriamycin, bleomycin, vinblastine, dacarbazine; CTX, cyclophosphamide; 5-FU, 5-fluorouracil; APBSCT, autologous peripheral blood stem cell transplantation; M iCMA, mitoxanthrone, carboplatin, methylprednisolone, cytosine arabinoside; VCR, vincristine; M DS, myelodysplastic syndrome; RAEB-t, Refractory anemia with excess blasts in transformation; nc, not classified; eo, eosinophilia.
II

I. C asore lli et a l. /D N A R epa ir 2 (2003) 547 -559 551
containing 1.5 mM MgC^, 0.2 mM each dNTP, 2.5 U HotStarTaq DNA Polymerase (Qiagen) and lOpmol each primer.
hM LHl:
5'-GGGTTGCTGGGTCTCTTCGTCCCTCC-3'
5'-CGCGTTCGCGGGTAGCTACGATGAGG-3'.
PCNA:
5'-GCGGGGAAGACTTTAGGGCCAATCG-3'
5'-GAATGTTAAGAGGAT GATAGGGAGC-3'.
hM LHl amplifications were performed as described above with an annealing temperature of 58.5 °C after an initial dénaturation at 95 °C for 15 min. Twenty cycles of 1 min at 95 °C, 1 min at 58.5 °C, 1 min at 72 °C were performed followed by a final extension of 72 °C for 7 min. Conditions for PCNA were identical except that the MgCl2 concentration was 2.5 mM, the annealing temperature was 59 °C and each cycle consisted of 1 min at 95 °C, 1 min at 59 °C, and 1 min at 72 °C followed by 7 min at 72 °C. Products were analyzed by agarose gel electrophoresis.
2.5. Target genes
Mononucleotide repeats inside BAX (19ql3.3- ql3.4), TGF^~RII (3p22), BRCA] (17q21), BLM (15q26.1), A IM (Uq22-q23), RAD50 (5q31), M R E ll (llq21) were amplified using previously described primers [29-33]. The PCR mixture contained 25 ng DNA, 1.5 mM MgCh, 0.25 mM each dNTP, 1 x buffer (20 mM Tris-HCl, pH 8.4, 50 mM KCl), 1U AmpliTaq DNA polymerase (PE, Applied Biosystems) and 0.5 p,M primers. Amplifications were performed on a Perkin-Elmer Thermal Cycler after an initial dénaturation at 95 °C for 5 min. Thirty cycles of 1 min at 90°C, 30 s at specific 30 s at 70 °C were followedby a final extension of 72 °C for 10 min. PCR products were purified with the QIAquick PCR purification kit (Qiagen, Hilden, Germany) and sequenced in both sense and antisense directions on an ABI Prism 310 Genetic Analyzer (PE, Applied Biosystems).
Exons 5-10 of the p53 gene were amplified by 30 cycles at 95 °C for 2 min, 55 °C for 2 min, and 72 °C for 3 min using previously described primers [34]. PCR reactions (50 p,l) contained 25 ng DNA,
1.5 mM MgCl], 0.25 mM each dNTP, Ix buffer (20mM Tris-HCl, pH 8.4, 50 mM KCl), 0.5 U AmpliTaq DNA polymerase (PE, Applied Biosystems) and 25 pmol specific primers. PCR products were purified and sequenced as described above.
3. Results
3.1. Microsatellite instability and hM LHl promoter méthylation
Where the patient’s normal DNA was available, the standard panel of five microsatellites recommended by Boland et al. [35] was analyzed. Examples of MSI are shown in Fig. 1. Evidence of instability at 2 or more loci was found in 10 of 15 (67%) cases. These were designated MSI"^. Non-tumor DNA was not available for the remaining 10 patients. Tumor DNA was analyzed at the BAT26 locus in these cases. This locus is considered to be essentially monomorphic among the general population and alterations at BAT26 have been shown to be predictive of MSI^ with >93% accuracy [36]. As expected, the frequency of MSI"^ was similar in this group and six cases (60%) were altered at BAT26. These were also designated MSI^. We conclude that the overall frequency of MSI"*" among our patients is around 16/25 (64%) (Table 2). The frequency of MSI"^ was similar in tAL/MDS (11/16, 68%), tAPL (3/5, 60%), and tALL (2/4, 50%) considered separately. In contrast to the therapy-related cases, none (0/28) of the de novo ALs were MSl"^. This low (<3.5%) frequency of MSl"^ in de novo AL is in good agreement with published estimates [21,22].
HMLHI promoter méthylation was examined by Hpall/Mspl digestion. The colorectal carcinoma cell line SW48 that has a methylated HMLHI promoter [10] and does not express hMLHl protein detectable by Western blotting was used as a positive control. The Raji Burkitt’s lymphoma cell line in which HMLHI gene expression and protein levels are normal served as a negative control. The promoter of the PCNA gene that is ubiquitously expressed in dividing cells was included as an internal standard for enzyme digestion. Examples of the analysis are shown in Fig. 2. Evidence of HMLHI promoter méthylation was seen in six tAMLs (T2, T4, T5, T14, T16, T23) (Table 3). Five of these were MS1+. The exception

552
(a)
I. Casorelli el a i /D N A Repair 2 (2003) 547-559
(C)* * * * *
A B T1 T8 T 3T 11 T5 T14 T9 T10 T19 T12 T 1 5 I2 4 D I D2 D3 D4 D5 06 0 7 08
BAT26
BAT26
* *(b) 15 T19 T18 T20
I N I N T N I N
* *
12 13 T5 T23T N T N T N T N
# # # * #BAT25 D17S250
* oT10 T21 T22 T24
T N T N T N T NT2 T17 T5 T19
T N T N T N T N
# #D5S346
#mm
D2S123
Fig. I. M icrosate iiile instability. The BAT26, BAT25, D2SI23, D I7S250 and D5S346 niicrosateilites were am plified as described in Section
2. Products were separated and located by hybrid ization. Representative examples o f D NA from tA L /M D S ( I ) or unaffected tissue (N )
are shown. Altered alleles and LO H are designated ( * ) and (O ), respectively, (a) BAT26 locus, tA L /M D S patients. A and B, D N A
from peripheral blood lymphocytes from c lin ica lly normal individuals, (b) BAT25, D I7S250, D 2S I23 and D5S346. tA L /M D S patients, (c)
BAT26, de novo A L patients (D I-D 8 ) .
was patient T14 from whom no normal D N A was available and only BAT26 was analyzed. The apparent promoter méthylation in T14 may result from intra-tumor heterogeneity. Alternatively, T14 may indeed be M S I+ and BAT26 is simply uninformative in this case, as it is occasionally in colorectal carcinomas [36]. We cannot distinguish between these possibilities. Notwithstanding the status o f T14, we conclude that hM LH I promoter méthylation is infrequent (6/25 or 24% ) in tA L /M D S and accounts for at most one-third (between 31% and 35%, 5/16 or 6/17) of MSI"*" cases. This value is considerably lower than the frequency (>80% ) among sporadic M S I+ colorec
tal tumors 137]. We note, however, that although the M S l^ frequency among the tA PL cases (3/5, 60% ) was similar to the overall frequency o f 64%, evidence of a methylated hM LH I promoter was found in (3/3) M S I+ tAPLs (cases T2, T16, and T23, Table 3). None (0/8) of the de novo A PL cases was M S I^ and none contained a methylated h M L H l promoter.
3.2. Mutations in po tentia l target genes
No mutations were found in exons 5 -1 0 of the p53
gene o f the tA L /M D S cases. This region contains most of the known mutations in tumors (http://p53.curie.fr).
I. C asore lli et a l. /D N A R epa ir 2 (2003) 547-559 553
Table 2Analysis of MST*" in tAL/MDS patients
Primary tumor Case BAT26 BAT25 D2S123 DI7S250 D5S346 Diagnosis
Breast 14 + UT5 + + - - - uT6 na + + - - uT8 - sT il - sT12 + uT18 + + - - - uT19 - - - - - sT21 + + - - LOH uT23 + - + + No DNA uT24 - - - - - s
Others T3 + + + - No DNA uT7 + uT9 + uT13 + + + - No DNA uT20 + + + - - uT22 - - - - - sT25 - sT1 - - - - - sTIO - - - - + sT2 + + - + + uT14 - sT15 + uT16 + uT17 + + + + + u
LOH, loss of heterozygousity; U, unstable; S, stable; na, not amplifiable.
No frameshifts were found in genes containing nucleotide repeats including BAX (G§ repeat), TGF^-R ll (Aio), BRCA] (Ag), BLM (Ag), ATM (T?), or RAD50 (Aq) genes.
There were three examples of a single nucleotide deletion in the T i l tract of M R E ll. This suggests that M R E ll might be a particular target for inactivation in MSI^ tAML/MDS. The mutation was heterozygous in two cases (T3 and T7) and homozygous in T21 (Fig. 3, Table 3).
3.3. tAL/MDS and primary breast cancer
The largest subgroup— 11 patients— had been treated for a primary breast cancer. MST^ in this group (7/11, 64%) and the frequency of hM LHl promoter méthylation among the MSI^ cases (3/7, 42%) were similar to the overall incidences (64% and 35%, respectively). There appeared to be a trend towards early
onset breast cancer among these cases. The GIMEMA Archive yielded information on 37 more primary breast cancer patients who had been excluded from the MSI study because no DNA was available. Inclusion of age of onset data for these additional patients supported the apparent bias towards early primary disease. Fig. 4a compares the age at diagnosis of breast cancer for the patients in the GIMEMA Archive (48 cases total) with similar data for all breast cancer patients from the Cancer Registries of six Italian provinces [38]. Only 6% of registry cases were diagnosed between 20 years and 39 years. For tAL/MDS patients, the corresponding figure was 24%. Overall, more than half (54%) of the breast cancer cases in the GIMEMA group occurred at <50 years. The corresponding value for patients in the registries was 23.5%. The possibility that there is simply a general bias toward younger patients in the GIMEMA group was investigated by comparing age of onset data for all non-breast cancer in the archive. (The age distribution for Hodgkin lymphoma

5M /. Casorelli el a l. /D N A Repair 2 (2003} 547-550
* * * *
T2 T19 T5 T14 T23
H M U H M U H M U H- r i l l
hMLH1
PCNA
T20 Raji SW48
H M U H M U H M U
hMLH1
PCNA
Fig. 2. ItM L H I promoter méthylation. D N A was extracted I'rom tA L /M D S and from the control cell lines SW48 (methylated h M L H I
promoter) and Raji (non-methylated). A liquots were digested w ith either H pa ll (H ) or MspI (M ) or left undigested (U ). The h M L H I
promoter fragment and a fragment o f the PCNA promoter containing m ultip le C'pG sites ( -9 8 2 to +48 ) were am pliiied as described in
Section 2. Products were identilied fo llo w in g agarose gel electrophoresis. An l iM I .H I PCR product fo llow ing digestion w ith H pa ll, hut not
w ith M spI, is diagnostic fo r promoter méthylation provided that both H pa ll and MspI digestion abolish the PCNA PCR product. Tumors
contain ing a methylated h M I.H I promoter are marked ( * ).
is known to be atypically skewed and data from these patients were therefore excluded from the analysis,) Fig. 4b compares the age distribution of all remaining primary cancers in the archive (107 eases total) with the 48 breast cancer cases. The two distributions differ significantly {P = 0.03; Kolmogorov-Smirnov comparison). Although this analysis is not sufficiently rigorous to exclude all potential sources o f bias, the data suggest an association between early onset breast cancer and tA L /M D S that might warrant further investigation.
3.4. M S I^ and treatment
Most patients in the group (10/25) had received chemotherapy for their primary tumor. In most cases this involved treatment with more than one type of drug. Eight had received radiotherapy (RT) and the remaining seven had been treated with combination drug/RT (Table 3).
There was a significant association between chemotherapy and M S l^ . A ll 10 cases in the
chemotherapy group were M SI ^. This compares to only two o f eight patients in the RT group ( P =
0.0015, Fisher's exact test). The association o f M S I^ with combined therapy was not significantly d ifferent from that o f M SI with chemotherapy alone (P > 0.05). These observations indicate that drug treatment carries a significantly a higher risk than RT for the development o f M S I * tA L /M D S .
4. Discussion
Cancer therapy is clearly implicated in the development o f secondary leukemia with defective M M R . Our findings based on a study o f 53 patients support a growing consensus that the M S I+ phenotype is common in tA L /M D S but rare in de novo A L. Almost two-thirds (64% ) o f our therapy-related cases were M S I^ but none o f 28 de novo cases. The latter value o f < 3 .5% , taken together with estimates from the published literature indicates that the frequency of M S I^ in de novo A M L is low.
/. C asore lli et a l. /D N A R epa ir 2 (2003) 547 -559 555
Table 3MSI"*", h M L H I méthylation and M R E ll mutation in tAL/MDS patients
Primary tumor Case Therapy MSI+ H M L H I méthylation M R E ll mutation
Breast T4 Chemotherapy + M na15 Chemotherapy + M -16 Combined + - -T8 Combined - - na111 Combined - - -112 Chemotherapy + - -T18 Chemotherapy + - -TI9 Radiotherapy - - -121 Combined + -T23 Radiotherapy + M -T24 Radiotherapy - - -
Others T3 Chemotherapy + - +T7 Combined + - +T9 Combined + - -T13 Chemotherapy + - -T20 Chemotherapy + - -T22 Combined - - -T25 Radiotherapy - - -T1 Radiotherapy - - -TIO Radiotherapy - - -T2 Chemotherapy + M -T14 Radiotherapy - M -T15 Chemotherapy + - -T16 Radiotherapy + M -T17 Chemotherapy + - -
M, methylated; na, not amplifiable.® Both alleles showed -1 deletion.
Epigenetic silencing of the hM LHI M M R gene does not account for the high frequency of MSI'*' tAL. HMLHI promoter méthylation occurs in most (80%) MSI+ sporadic colorectal [10,37], endometrial [39], and gastric [40] tumors. This event is clearly uncommon in tAL. We found evidence of méthylation in less than one-third of our cases. This frequency represents an upper limit as this sensitive method can detect even a very minor population of méthylation positive leukemic cells. We did observe HMLHI promoter méthylation in all (3/3) MST^ tAPLs, however. Both MSI^ and promoter méthylation appear to be therapy-related— we found no examples of either among the eight de novo APLs. Although this observation requires confirmation from a larger number of patients, the possibility that tAPL is associated with a high frequency of HMLHI promoter méthylation is consistent with the known derangement of epigenetic gene regulation in APL [41],
The distribution of ages at diagnosis for primary breast cancer cases in the GIMEMA Archive differed from that for breast cancer in the general population. Early ages were significantly over-represented among the tAL patients. Attention has previously been drawn to a similar bias among Israeli tAML cases [25]. Although the numbers involved are small and many sources of potential bias have not been rigorously excluded, the differences appear significant. The possibility that this altered distribution reflects a common genetic susceptibility to both early onset breast cancer and tAL/MDS merits further consideration. The two best characterized breast cancer susceptibility genes BRCAI and BRCA2, both encode proteins that are implicated in recombinational repair of broken DNA [42]. There is increasing evidence to connect the recombinational DNA repair pathway to leukemia. The genetic disorder Fanconi Anemia (FA) highlights this connection. The incidence of A M L among FA patients

556 I. Casorelli el a l. /D N A Repair 2 (2003) 547-559
T A A C T T T T T T T T T T T A A G 80 90
Wild-type
a c t t t t t t t t t t n a n g g t t n0 70
A A C T T T T T T T T T T A A G G G
homozygous-T heterozygous-T
Fig. 3. M RE I I mutations in tAL/MDS. The M R E ll poly(T)l I tract was sequenced as described in Section 2. Top: wild-type Ti I sequence; bottom: homozygous TIO allele identified in patient T21 (left); heterozygous Tl 1/TlO alleles representative of -1 deletion mutations identified in patients T3 and T7 (right).
is extremely high. Five of the FA complementation group proteins (FANCA, C, E, F and G) facilitate the interaction of a sixth (FANCD2) with BRCAI |43|. BRCA2 has recently been identified as a seventh FA gene, FANCDI |44|. Thus, both breast cancer susceptibility genes are inextricably linked to FA and to AML.
Our finding of three examples of a - I deletion in the intronic Tn tract of M R E ll that is frequently mutated in MSI'*' colorectal tumors [33] may also be pertinent to recombinational DNA repair. In this case, a key function in recombinational DNA repair is at increased risk of inactivation because of MSI^. The MREl 1/NBS1/RAD50 protein complex is important in imposing the intra-S phase checkpoint and in the early steps of recombinational repair [45]. Inactivation of the complex facilitates the development of cancer. As with FA, individuals with inherited deficiencies
in NBSI (Nijmegen Breakage Syndrome) or MREl I (Ataxia-Telangiectasia-Like Disorder) are prone to lymphoid malignancies |46|. NBSI has been reported to be mutated in 15% of childhood ALL |47|, and hMREII mutations have been noted in lymphoma and breast carcinoma |48|. Although null mutations in Rad5() are lethal in mice, hypomorphic mutations are associated with chromosome instability and have a particularly severe effect on development of hematopoietic cells [49]. In two of three of our cases, the liMREI I mutation was accompanied by loss of the 5q chromosome arm. This is a common chromosomal alteration in tAL. We note that the RAÜ5Ü gene is located on chromosome 5q. These preliminary observations reinforce the idea that a reduced ability or efficiency of recombinational repair of DNA breaks may be important in the development of AML.
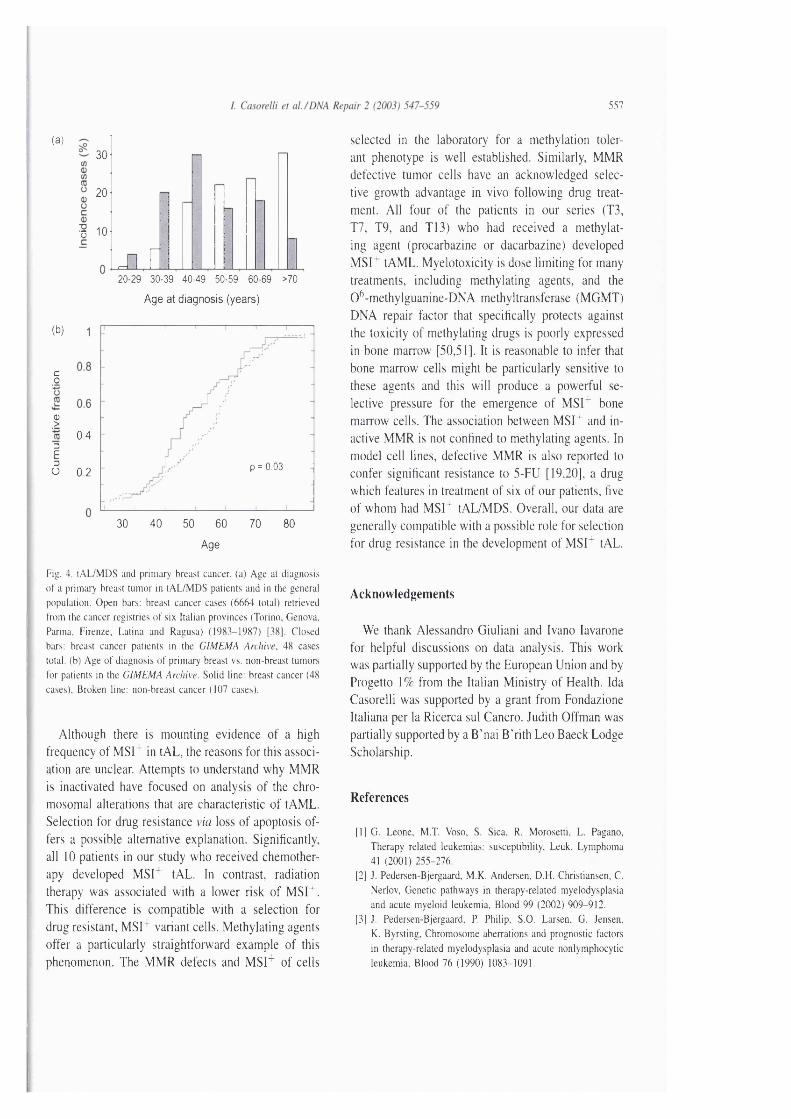
557
(a)30
20
n 10
JiX20-29 30-39 40-49 50-59 60-69 >70
Age at diagnosis (years)
(b)
co
CD>
3E3U
0.4
p = 0.030.2
30 40 50 60 70 80
Age
Fig. 4, lAL/MDS and primary breast cancer, (a) Age at diagnosis of a primary breast tumor in tAL/MDS patients and in the general population. Open bars: breast cancer cases (6664 total) retrieved from the cancer registries of six Italian provinces (Torino, Genova, Parma, Firenze, Latina and Ragusa) (1983-1987) [38]. Closed bars: breast cancer patients in the G IM EM A Archive, 48 cases total, (b) Age of diagnosis of primary breast vs. non-breast tumors for patients in the G IM EM A Archive. Solid line: breast cancer (48 cases). Broken line: non-breast cancer (107 cases).
Although there is mounting evidence of a high frequency of MSI^ in tAL, the reasons for this association are unclear. Attempts to understand why MMR is inactivated have focused on analysis of the chromosomal alterations that are characteristic of tAML. Selection for drug resistance via loss of apoptosis offers a possible alternative explanation. Significantly, all 10 patients in our study who received chemotherapy developed MSI'^ tAL. In contrast, radiation therapy was associated with a lower risk of MSI+. This difference is compatible with a selection for drug resistant, MSI^ variant cells. Methylating agents offer a particularly straightforward example of this phenomenon. The MMR defects and M S r of cells
selected in the laboratory for a méthylation tolerant phenotype is well established. Similarly, MMR defective tumor cells have an acknowledged selective growth advantage in vivo following drug treatment. All four of the patients in our series (T3, TV, T9, and T13) who had received a methylating agent (procarbazine or dacarbazine) developed MSL tAML. Myelotoxicity is dose limiting for many treatments, including methylating agents, and the 0^-methylguanine-DNA methyltransferase (MGMT) DNA repair factor that specifically protects against the toxicity of methylating drugs is poorly expressed in bone marrow [50,51]. It is reasonable to infer that bone marrow cells might be particularly sensitive to these agents and this will produce a powerful selective pressure for the emergence of M SF bone marrow cells. The association between MSI^ and inactive MMR is not confined to methylating agents. In model cell lines, defective MMR is also reported to confer significant resistance to 5-FU [19,20], a drug which features in treatment of six of our patients, five of whom had MSl^ tAL/MDS. Overall, our data are generally compatible with a possible role for selection for drug resistance in the development of M S F tAL.
Acknowledgements
We thank Alessandro Giuliani and Ivano lavarone for helpful discussions on data analysis. This work was partially supported by the European Union and by Progetto 1% from the Italian Ministry of Health. Ida Casorelli was supported by a grant from Fondazione Italiana per la Ricerca sul Cancro. Judith Offman was partially supported by a B’nai B’rith Leo Baeck Lodge Scholarship.
References
[1] G. Leone, M.T. Voso, S. Sica. R. Morosetti, L. Pagano, Therapy related leukemias: susceptibility, Leuk. Lymphoma 41 (2001) 255-276.
[2] J. Federsen-Bjergaard, M.K. Andersen, D.H. Christiansen. C. Nerlov, Genetic pathways in therapy-related myelodysplasia and acute myeloid leukemia. Blood 99 (2002) 909-912.
[3] J. Federsen-Bjergaard, F. Philip, S.O. Larsen, G. Jensen, K. Byrsting, Chromosome aberrations and prognostic factors in therapy-related myelodysplasia and acute nonlymphocytic leukemia. Blood 76 (1990) 1083-1091.

558 I. C asore lli et a l. /D N A R epair 2 (2003) 547 -559
[4] G. Leone, L. Mele, A. Pulsoni, F. Equitani, L. Pagano, The incidence of secondary leukemias, Haematologica 84 (1999) 937-945.
[5] L. Pagano, A. Pulsoni, M E. Tosti, G. Avvisati, L. Mele, M. Mele, et al.. Clinical and biological features of acute myeloid leukaemia occurring as second malignancy: GIMEMA archive of adult acute leukaemia, Br. J. Haematol. 112 (2001) 109- 117.
[6] M.K. Andersen, D.H. Christiansen, B.A. Jensen, P. Emst,G. Hauge, J. Pedersen-Bjergaard, Therapy-related acute lymphoblastic leukaemia with MLL rearrangements following DNA topoisomerase II inhibitors, Br. J. Haematol. 114 (2001) 539-543.
[7] H.T. Lynch, T.C. Smyrk, P. Watson, S.J. Lan spa, I.E. Lynch, P.M. Lynch, et al.. Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: an updated review, Gastroenterology 104 (1993) 1535-1549.
[8] P. Peltomaki, H.F.A. Vasen, HNPCC ICG. Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. Gastroenterology 113 (1997) 1146-1158.
[9] J. Jiricny, M. Nystrom-Lahti, Mismatch repair defects in cancer, Curr. Opin. Genet Dev. 10 (2000) 157-161.
[10] M.F. Kane, M. Loda, G. Gaida, J. Lipman, R. Mishra,H. Goldman, et al.. Méthylation of the hMLHl promoter correlates with lack of expression of hMLHl in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 57 (1997) 808-811.
[11] A. Bearzatto, M. Szadkowski, P. Macpherson, J. Jiricny, P. Karran, Epigenetic regulation of the MGMT and hMSH6 DNA repair genes in cells resistant to methylating agents. Cancer Res. 60 (2000) 3262-3270.
[12] P. Karran, M. Bignami, DNA damage tolerance, mismatch repair and genome instability, BioEssays 16 (1994) 833-839.
[13] P. Branch, G. Aquilina, M. Bignami, P. Karran, Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature 362 (1993) 652-654.
[14] A. Kat, W.G. Thilly, W.-H. Fang, M.J. Longley, G.-M. Li, P. Modrich, An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair, Proc. Natl. Acad. Sci. U.S.A. 90 (1993) 6424-6428.
[15] P.F. Swann, T.R. Waters, D.C. Moulton, Y.-Z. Xu, Q. Zheng, M. Edwards, et al.. Role of post-replicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 273 (1996) 1109-1111.
[16] S. Aebi, H.-B. Kurdi, R. Gordon, B. Cenni, H. Zheng, D. Fink, et al.. Loss of DNA mismatch repair in acquired resistance to cisplatin. Cancer Res. 56 (1996) 3087-3090.
[17] P. Branch, M. Masson, G. Aquilina, M. Bignami, P. Karran, Spontaneous development of drug resistance: mismatch repair and p53 defects in resistance to cisplatin in human tumor cells. Oncogene 19 (2000) 3138-3145.
[18] A. Fedier, V.A. Schwarz, H. Walt, R.D. Carpini, U. Haller, D. Fink, Resistance to topoisomerase poisons due to loss of DNA mismatch repair. Int. J. Cancer 93 (2001) 571-576.
[19] E. Tokunaga, S. Oda, M. Fukushima, Y. Maehara, K. Sugimachi, Differential growth inhibition by 5-fluorouracil
in human colorectal carcinoma cell lines, Eur. J. Cancer 36(2000) 1998-2006.
[20] J.M. Carethers, D P. Chauhan, D. Fink, S. Nebel, R.S. Bresalier, S B. Howell, et al., Mismatch repair proficiency and in vitro response to 5-fluorouracil, Gastroenterology 117 (1999) 123-131.
[21] H. Sill, J.M. Goldman, N.C. Cross, Rarity of microsatellite alterations in acute myeloid leukaemia, Br. J. Cancer 74 (1996) 255-257.
[22] J.H. Ohyashiki, K. Ohyashiki, S. Aizawa, K. Kawakubo, T. Shimamoto, H. Iwama, et al.. Replication errors in hematological neoplasias: genomic instability in progression of disease is different among different types of leukemia, Clin. Cancer Res. 2 (1996) 1583-1589.
[23] Y.M. Zhu, E.P. Das-Gupta, N.H. Russell, Microsatellite instability and p53 mutations are associated with abnormal expression of the MSH2 gene in adult acute leukemia. Blood 94 (1999) 733-740.
[24] E.P. Das-Gupta, C.H. Seedhouse, N.H. Russell, Microsatellite instability occurs in defined subsets of patients with acute myeloblastic leukaemia, Br. J. Haematol. 114 (2001) 307- 312.
[25] D. Ben-Yehuda, S. Krichevsky, 0. Caspi, D. Rund, A. Polliack, D. Abeliovich, et al.. Microsatellite instability and p53 mutations in therapy-related leukemia suggest mutator phenotype. Blood 88 (1996) 4296-4303.
[26] M.H. Sheikhha, K. Tobal, J.A.L. Yin, High level of microsatellite instability but not hypermethylation of mismatch repair genes in therapy-related and secondary acute myeloid leukaemia and myelodysplastic syndrome, Br. J. Haematol. 117 (2002) 359-365.
[27] R. Higuchi, C.H. von Beroldingen, G.E. Sensabaugh, H.A. Erlich, DNA typing from single hairs. Nature 332 (1988) 543-546.
[28] W. Dietmaier, S. Wallinger, T. Bocker, F. Kullmann, R. Fishel, J. Ruschoff, Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res. 57 (1997) 4749-4756.
[29] N. Rampino, H. Yamamoto, Y. Ionov, H. Li Sawai, J.C. Reed, M. Perucho, Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 275 (1997) 967-969.
[30] R. Parsons, L.L. Myeroff, B. Liu, K.V. Willson, S.D. Markowitz, K.W. Kinzler, et al.. Microsatellite instability and mutations of the transforming growth factor |3 type II receptor gene in colorectal cancer. Cancer Res. 55 (1995) 5548-5550.
[31] N.G. Kim, Y.R. Choi, M.J. Back, Y.H. Kira, H. Kang, N.K. Kim, et al., Frameshift mutations at coding mononucleotide repeats of the hRAD50 gene in gastrointestinal carcinomas with microsatellite instability. Cancer Res. 61 (2001) 36-38.
[32] G. Calin, G.N. Ranzani, D. Amadori, V. Herlea, I, Matei, G. Barbanti-Brodano, et al.. Somatic frameshift mutations in the Bloom syndrome BLM gene are frequent in sporadic gastric carcinomas with microsatellite mutator phenotype, BMC Genet. 2 (2001) 14.
[33] G. Giannini, E. Ristori, F. Cerignoli, C. Rinaldi, M. Zani, A. Viel, et al.. Human MREll is inactivated in mismatch repair-deficient cancers, EMBO Rep. 3 (2002) 248-254.

I. C asore lli et a l. /D N A R epa ir 2 (2003) 547-559 559
[34]
[35]
[36]
[37]
[38]
i [39]
[40]
[41]
[42]
S. Takeuchi, C. Bartram, R. Ludwig, B. Royer-Pokora, S. Schneider, J. Imamura, et al., Mutations of p53 in Wilms’ tumors. Mod. Pathol. 8 (1995) 483^87.C.R. Boland, S.N. Thibodeau, S.R. Hamilton, D. Sidransky, J R. Eshelman, R.W. Burt, et al., A National Cancer Institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 58 (1998) 5248-5257.A. Loukola, K. Eklin, P. Laiho, R. Salovaara, P. Kristo, H. Jarvinen, et al., Microsatellite marker analysis in screening for hereditary nonpolyposis colorectal cancer (HNPCC), Cancer Res. 61 (2001) 4545-4549.J.G. Herman, A. Umar, K. Polyak, J. Graff, N. Ahuja, J. Issa, et al.. Incidence and functional consequences of hMLHl promoter hypermethylation in colorectal carcinoma, Proc. Natl. Acad. Sci. U.S.A. 95 (1998) 6870-6875.R. Zanetti, P. Crosignani, S. Rosso, Cancer in Italy: Incidence Data From Cancer Registries, I988-I992 (Lega Italiana per la lotta contro i tumori: II pensiero scientifico editore), vol. 2, 1992.S B. Simpkins, I . Bocker, E.M. Swisher, D.G. Mutch, D.J. Gersell, A.J. Kovatich, et al., MLHl promoter méthylation and gene silencing is the primary cause of microsatellite instability in sporadic endometrial cancers. Hum. Mol. Genet. 8 (1999) 661-666.A.S. Fleisher, M. Esteller, S. Wang, G. Tamura, H. Suzuki, J. Yin, et al., Hypermethylation of the hMLHl gene promoter in human gastric cancers with microsatellite instability. Cancer Res. 59 (1999) 1090-1095.L. Di Crook, V.A. Raker, M. Corsaro, F. Fazi, M. Fanelli, M. Faretta, et al., Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295 (2002) 1079-1082.A.R. Venkitaraman, Cancer susceptibility and the functions of BRCAl and BRCA2, Cell 108 (2002) 171-182.
[43] I. Garcia-Higuera, T. Taniguchi, S. Ganesan, M.S. Meyn, C. Timmers, J. Hejna, et al.. Interaction of the Fanconi anemia proteins and BRCAI in a common pathway. Mol. Cell 7(2001) 249-262.
[44] N.G. Hewlett, T. Taniguchi, S. Olson, B. Cox, Q. Waisfisz, C. De Die-Smulders, et al., Biallelic inactivation of BRCA2 in Fanconi anemia, Science 297 (2002) 606-609.
[45] J.P. Carney, R.S. Maser, H. Olivares, E.M. Davis, M. Le Beau, J.R. Yates IH, et al.. The hMrelI/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response, Cell 93 (1998) 477-486.
[46] Y. Shiloh, Ataxia-Telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart, Annu. Rev. Genet. 31 (1997) 635-662.
[47] R. Varon, A. Reis, G. Henze, H.G. von Einsiedel, K. Sperbng, K. Seeger, Mutations in the Nijmegen Breakage Syndrome gene (NBSI) in childhood acute lymphoblastic leukemia (ALL), Cancer Res. 61 (2001) 3570-3572.
[48] T. Fukuda, T. Sumiyoshi, M- Takahashi, T. Kataoka, T. Asahara, H. Inui, et al., Alterations of the double-strand break repair gene MREll in cancer. Cancer Res. 61 (2001) 23-26.
[49] C.F. Bender, M.L. Sikes, R. Sullivan, L.E. Huye, M.M. Le Beau, D.B. Roth, et al,, Cancer predisposition and hematopoietic failure in Rad50(S/S) mice, Genes Dev. 16(2002) 2237-2251.
[50] T. Moritz, W. Mackay, B.J. Glassner, D.A. Williams, L. Samson, Retrovirus-mediated expression of a DNA repair protein in bone marrow protects hematopoietic cells from nitrosourea-induced toxicity in vitro and in vivo. Cancer Res. 55 (1995) 2608-2614.
[51] J. Jelinek, L.J. Fairbaim, T.M. Dexter, J.A. Rafferty, C. Stocking, et al.. Long-term protection of hematopoiesis against the cytotoxic effects of multiple doses of nitrosourea by retrovirus-mediated expression of human 0 -alkylguanine- DNA-alkyltransferase, Blood 87 (1996) 1957-1961.

Publication information: DNA Repair (ISSN 1568-7864). For 2003, volume 2 (12 issues) are scheduled for publication. Subscription prices are available upon request from the Publisher or from the Regional Sales Office nearest you or from this journaPs website (http://www.elsevier. com/locate/dnarepair). Further information is available on this journal and other Elsevier Science products through Elsevier's website (http;// www.elsevier.com). Subscriptions are accepted on a prepaid basis only and are entered on a calendar year basis. Issues are sent by standard mail (surface within Europe, air delivery outside Europe). Priority rates are available upon request. Claims for missing issues should be made within six months of the date of dispatch.
Orders, claims, and product enquiries: please contact the Customer Support Department at the Regional Sales Office nearest you:
New Yorlc: Elsevier Science, PC Box 945, New York, NY 10159-0945, USA; phone: (4-1) (212) 633 3730 [toll free number for North American customers: 1-888-4ES-INFO (437-4636)]; fax: (4-1) (212) 633 3680; e-mail: [email protected]: Elsevier Science, PC Box 211,1000 AE Amsterdam,The Netherlands; phone: (4-31) 20 4853757; fax: (4-31) 20 4853432; e-mail: nlinfo-f@ elsevier.comTokyo: Elsevier Science, 9-15 Higashi-Azabu 1-chome, Minato-ku, Tokyo 106-0044, Japan; phone: (+81) (3) 5561 5033; fax: (+81) (3) 5561 5047; e-mail: [email protected]: Elsevier Science, 3 Killiney Road, #08-01 Winsland House I, Singapore 239519; phone: (+65) 6349 0200; fax: (+65) 67331510; e-mail: asiai nfo @ elsevier. com. sgRk) de Janeiro: Elsevier Science, Rua Sete de Setembro 111/16 Andar, 20050-002 Centro, Rio de Janeiro-RJ, Brazil; phone: (+55) (21) 509 5340; fax: (+55) (21) 507 1991; e-mail: [email protected] [Note (Latin America): for orders, claims and help desk information, please contact the Regional Sales Office in New York as listed above]
Author enquiries: For enquiries relating to the submission of articles (including electronic sutjmission), ttie status of accepted articles, the preparation of electronic artwork and any other enquiries relating to Elsevier Science, please consult http://authors.elsevier.com
Contact details for questions arising after acceptance of an article, especially tfK>se relating to proofs, are provided after registration of an article for publication.
Electronic manuscripts: Electronic manuscripts have the advantage that there is no need for the rekeying of text, thereby avoiding ttie possibility of introducing errors and resulting in reliable and fast delivery of proofs.
For the initial submission of manuscripts for consideration, hardcopies are sufficient. For ttie processing of accepted papers, electronic versions are preferred. After final acceptance, your disk plus one, final and exactly matching printed version should tie submitted together. Double density (DD) or high density (HD) diskettes are acceptable. It. is important that the file saved is in the native format of the wordprocessor program used Label the disk with the name of the computer and wordprocessing package used your name, and the name of the file on the disk. Furtfier information may be obtained from the Publisher.
USA mailing notice: DNA Repair (ISSN 1568-7864) is published monthly by Elsevier Science B.V. (P.O. Box 211,1000 AE Amsterdam, The Netfier- lands). Annual subscription price in the U.S.A. US$ 1,308 (valid in North, Central and South America), including air speed delivery. Periodical postage rate paid at Jamaica, NY 11431.USA POSTMASTER: Send address changes to DNA Repair, Publications Expediting Inc., 200 Meacfiam Avenue, Almont, NY 11003.AIRFREIGHT AND MAILING in the USA by Publications Expediting Inc., 200 Meacham Avenue, Elmont, NY 11003.
Authors in Japan please note: Upon request, Elsevier Science K.K. will provide authors with a list of people who can check and improve the English of their paper {before submission). Please contact our Tokyo office: Elsevier Science K.K., 1-9-15 Higashi-Azabu, Minato-ku, Tokyo 106-0044; Tel.: (03)-5561-5032; Fax: (03) 5561-5045.
Manuscripts can be submitted to a Managing Editor of the appropriate section (for addresses see tfie Instructions to Authors; last pages of this issue.
DNA Repair has no page charges
Printed in The Netherlands
© The paper used in this publication meets the requirements of ANSI/NISO Z39.48-1992 (Permanence of Paper)
ax: +4| E-n\
D 2003 oi:10.1

IARTICLE IN PRESS
Available online at www.sciencedirect.com
S C I E N C E D I R E C T *
ELSEVIER Biochimie 00 (2003) 000-000
BIOCHIMIE
www.elsevier.com/locate/biochi
Human mismatch repair, drug-induced DNA damage, and secondary cancer
Peter Karran Judith Offman Margherita Bignami
' Cancer Research UK, Imperial Cancer Research Fund, London Research Institute, Clare H all Laboratories, Blanche Lane,
South Mimms, Herts EN6 3LD, UK ’’ Istituto Superiore di Sanitd, Yiale Regina Elena 299, 0 0 I6 I Rome, Italy
Received 29 September 2003; accepted 8 October 2003
) Abstract
'# DNA mismatch repair (MMR) is an important replication error avoidance mechanism that prevents mutation. The association of defective jl MMR with familial and sporadic gastrointestinal and endometrial cancer has been acknowledged for some years. More recently, it has become ! apparent that MMR defects are common in acute myeloid leukaemia/myelodysplastic syndrome (AML/MDS) that follows successful ) chemotherapy for a primary malignancy. Therapy-related haematological malignancies are often associated with treatment with alkylating I agents. Their frequency is increasing and they now account for at least 10% of all AML cases. There is also evidence for an association between ) MMR deficient AML/MDS and immunosuppressive treatment with thiopurine drugs. Here we review how MMR interacts with alkylating
agent and thiopurine-induced DNA damage and suggest possible ways in which MMR defects may arise in therapy-related AML/MDS.
|l © 2003 Published by Éditions scientifiques et médicales Elsevier SAS.
I Keywords: MMR; DNA damage; Cancer
L Introduction
k The contribution of defective mismatch repair (MMR) to I the development of human cancer has been acknowledged # for more than a decade (for recent review see [1 ]). Tumours
with the microsatellite instability (MSI) that is characteristic of inactive M M R occur in both familial and sporadic settings. In the former, genetic inactivation of M M R by combined germline and somatic mutations is represented by the widely
|5 studied HNPCC syndrome. In sporadic cancers, the MMR !1 defect is generally the result of epigenetic gene silencing (see \ for example [2,3]). More recently, attention has been focused j! on the interaction between MMR and DNA damage. Active
MMR is implicated in the lethal effects of some DNA damage and abrogated M M R is clearly linked to a type of drug resistance or DNA damage tolerance [4,5]. Because an MMR
* Corresponding author. Tel.: +44-207-269-3870; fax: +44-207-269-3812.
E-mail address: [email protected] (P. Karran).
© 2003 Published by Éditions scientifiques et médicales Elsevier SAS. doi:10.1016/j.biochi.2003.10.007
defect does not confer any obvious selective growth advan- 36 tage whereas tolerance to DNA damage might, it has been 37 suggested that selection for resistance to endogenous or envi- 38 ronmentally produced DNA damage may contribute to the 39 development of MMR defective tumours [4,6]. Chemo- 40 therapy provides one of the most obvious exampies of delib- 41 erately induced DNA damage and most anticancer therapeu- 42 tic agents inflict it, either directly or indirectly. There is a 43 growing awareness that M M R defects are widespread among 44 secondary cancers that arise after successful chemotherapy. 45 Acute myeloid leukaemia/myelodysplastic syndrome 46 (AML/MDS) is one of the most common forms of secondary 47 cancer and now comprises around 10% of AML cases [7]. We 48 have suggested [8] that this AML/MDS may sometimes 49 reflect the clonal expansion of cells with significant resis- 50 tance to therapy-inflicted potentially lethal DNA damage. In 51 this review, we examine the circumstantial and experimental 52 evidence for this proposal. We also review recent findings 53 relating cellular MMR capacity and responses to drug- 54 induced DNA damage that may shed new light on the asso- 55 ciation of MMR defects with therapy-related cancer. 56
BIOCHI-00000154

ARTICLE IN PRESSp. Karran et al. /B iochim ie 00 (2003) 000-000
17 2. The role of M M R in spontaneous mutation avoidance
9 MMR— along with DNA replication proof-0 reading— ensures an acceptably low level of spontaneous1 mutation. Both MMR and proof-reading correct the misin-2 sertion errors committed by the replicative DNA poly-3 merases and restore normal Watson-Crick base pairs. The4 MMR pathway also prevents recombination between slightly5 divergent DNA (homologous) sequences with a degree of6 non-identity that exceeds a defined threshold. MMR is a7 DNA excision repair pathway. In common with nucleotide excision (NER) and base excision repair (HER), recognition
9 and removal of the targeted sequence— in this case a mispair0 or structural anomaly produced by DNA strand1 misalignment— precedes its replacement by non-2 semiconservative DNA synthesis. The MMR repair tract is3 long— one or two orders of magnitude longer than those4 associated with NER or HER, respectively. MMR is particu-5 larly adept at correcting structural anomalies that occur dur- J ing the replication of repetitive mono- or dinucleotide re- 7 gions of DNA. If left uncorrected, these aberrant I structures— generally envisioned as single or tandem bases 9 that become extrahelical because of misalignment of reitera- Î tive template and daughter DNA strands— cause heritable1 increases or decreases in the length of the repetitive tract. In2 expressed genes, this results in frameshifts and in truncated,3 inactive proteins. In non-expressed microsatellite regions, it4 causes MSI. Frameshifts in repetitive regions of expressed5 genes and MSI are the defining features of M M R deficiency.
I 3. How M M R processes DNA mismatches?
il Mismatch correction is initiated by the binding of one of I two mismatch recognition heterodimers, which have par- 9 tially overlapping specificities. hMutSa comprising the i hMSH2 and hMSH6 proteins, has a particular affinity for 1 single base mispairs and structural anomalies involvingI mono- or dinucleotide repeats. hMSH2 forms a second het-
i erodimer with the hMSH3 protein. This hMutSp complex, *4 which accounts for around 10% of the cell’s hMSH2, has a preference for rather more complex structures. A degree of
^.functional redundancy between the hMutSa and hMutSP *! heterodimers ensures that correction of all common replica-II tion errors is initiated efficiently but since most of the major t replication errors are recognised by hMutSa, hMSH2 and
( hMSH6 are more important than hMSH3 in this process. Mismatch recognition precedes the recruitment of hMutLa, a
C heterodimer of hMLHl and hPMS2. This rather enigmatic (3 factor appears to provide a molecular coupling between the It recognition factors and the downstream proteins involved in K the excision and replacement steps of repair. Although there K are similar factors, notably hMutLp, comprising hMLHl and H the related hMLH3, there appears to be less redundancy in I this step of repair. For most MMR events, the important R proteins are hMSH2, hM LHl, hMSH6, and hPMS2.
4. M M R processes some damaged DNA bases into 110lethal lesions 111
hMutSa recognises, and MMR processes, some types of 112DNA damage. DNA 0^-methylguanine (0^-meGua) is se- 113lectively lethal in M M R proficient cells. Cells that do not 114express 0^-meGua-DNA methyltransferase (MGMT) that 115directly reverses DNA 0^-meGua by in situ déméthylation 116are extremely sensitive to methylating agents like A-methyl- 117A-nitrosourea (MNU) or A-methyl-A'-nitro-A- 118 nitrosoguanidine (MNNG). Full resistance to— but not repair 119of— DNA 0^-meGua is restored by inactivating one of the 120genes encoding a component of hMutSa or hMutLa [4]. This 121phenomenon is known as méthylation tolerance. MMR pro- 122cesses DNA that contains 0^-meGua base pairs. In an in vitro 123MMR system, 0^-meGua:T pairs in which 0^-meGua in the 124nicked strand of a circular DNA heteroduplex (analogous to 125the daughter strand) is paired to T in an uninterrupted strand 126(analogous to the template), are efficiently corrected to A:T 127pairs. G^-meGua:C pairs in the same configuration are re- 128stored to G:C with a lower efficiency that is consistent with 129their poorer recognition by hMutSa [91. Although strictly not 130a biologically relevant experiment because the 0^-meGua is 131not in the template DNA strand, it is nevertheless important 132because it establishes the principle that 0^-meGua base pairs 133provoke full processing by MMR— not just recognition. 134When the methylated base is in the daughter DNA stand, 135processing proceeds to a successful conclusion. When the 136miscoding 0^-meGua is in the template strand and not, 137therefore, designated for removal, it is easy to envisage how 138complications might arise to prevent completion of process- 139ing and generate unresolved intermediates. We have sug- 140gested that the inability of 0^-meGua to form a structurally 141acceptable base pair with incoming nucleotides during the 142repair process underlies the cytotoxicity of the 0^- 143meGua/MMR interaction. The interaction of MMR with 0^- 144meGua is quite selective. MMR is not implicated in lethal 145processing of other common DNA méthylation products, 146such as 7-meGua or 3-meAde, that are produced along with 1470^-meGua. Whatever the mechanism of DNA lesion recog- 148nition by hMutSa, M M R clearly does not provide a general 149DNA damage recognition system. 150
In addition to extreme tolerance of DNA méthylation 151damage, MMR defective cells are resistant to the base ana- 152logue 6-thioguanine (6-TG) [10,11]. The 6-TG resistance 153conferred by an MMR defect is substantial (about 5-10- 154fold). The toxicity of 6-TG requires its conversion into 6-TG 155nucleotides and incorporation into nucleic acids, particularly 156into DNA. Surprisingly, it is a methylated form of DNA 1576-TG, 6-thiomethylguanine (S^-meGua), produced by rare in 158situ chemical méthylation, that interacts with MMR [12]. 159S^-meGua-containing pairs, like 0^-meGua-containing base 160pairs, are recognised by hMutSa [13]. Furthermore, just as 161G^-meGua:T is a better substrate for hMutSa recognition and 162for correction, the S^-meGua:T pair is recognised more effi- 163ciently by hMutSa than S^-meGua:C. Both S^-meGua:T and 164

iM ARTICLE IN PRESSp. Karran et al. /B iochim ie 00 (2003) 000-000
)5 0^-meGua:T base pairs can only be formed by replication of 6 the methylated bases. This requirement for replication to17 produce the ‘DNA damage’ that provokes DNA damage18 responses— including M M R— is consistent with the delayed19 biological effects of methylating agent [14] or 6-TG [15]0 treatment. It seems likely that the interaction of MMR with1 DNA méthylation damage simply reflects the recognition of '2 replication ‘errors’ . In order to interact with MMR, the initial3 DNA lesions must be capable of replication and the products4 must bear sufficient resemblance to a normal replication5 error to trigger hMutSa recognition and subsequent process-6 ing. Of the numerous DNA lesions produced by MNU and 1 MNNG, only 0^-meGua, meets these criteria. Other methy-8 lated bases are either not efficiently replicated (3-meAde) or9 are not perceived as mismatches when they are (7-meGua,0 O'^-meThy). Significantly, the ethyl counterpart of 0^-1 meGua, 0^-ethylGua, which is not efficiently bypassed dur-2 ing DNA replication, is not processed by M M R into a cyto- 5 toxic lesion and repair defective cells are only slightly4 tolerant to N-ethyl-N-nitrosourea (ENU) and not signifi-5 cantly to chloroethylating agents [16].
5. Some M M R processing of damaged DNA bases is il not associated with cell death
8 In addition to substrates containing 0^-meGua and S -9 meGua, purified hMutSa is able to bind to synthetic DNA *0 duplexes containing various other single DNA lesions in- !t eluding 8-oxoguanine (8-oxoGua) [17], UV photoproducts ’! [18], 1,2-dipurinyl cisplatin intrastrand crosslinks [9,19], 9 C8-guanylaminofluorenes or acetylaminofluorenes [20], and 'i benz(a)pyrene adducts [21 ]. Binding in vitro does not neces- sarily indicate that these lesions are processed in vivo, how-
I ever, and the extent to which MMR is involved in converting diverse DNA lesions into lethal damage has been the subject of debate. As indicated in the preceding section, we believe
fi that a significant degree of lethal processing only occurs with I a limited range of damaged bases of which 0^-meGua and i the closely related S^-meGua, are the only unequivocally ( documented representatives. Lethal processing of other DNA ( lesions may differ mechanistically and is generally of minor # significance since the survival advantages conferred by inac- i tive MMR are slight.t On the other hand, there is increasing evidence that MMR f does process a variety of DNA lesions. This processing has a I significant biological impact but is not associated with nota-I bly increased resistance in repair defective cells. For ex-II ample, DNA 8-oxoGua is an extremely important lesion I produced by numerous carcinogens. It is one of several È oxidised bases that accumulate to a greater extent in msh2~'~
6 mouse fibroblasts and h M L H l defective human tumour cells I exposed to ionising radiation or oxidants [22,23]. There is no f consistent evidence that persistent 8-oxoGua in MMR defec- ( tive cells is associated with a significant degree of resistance t to killing [22,24,25]. DNA 8-oxoGua is, however, highly I mutagenic. Importantly, there is evidence [22] that MMR
defective cells are particularly mutable by treatments that 219 produce DNA 8-oxoGua. 220
There are other examples of the connection between 221 MMR defects and increased mutability by DNA damage 222 with no, or only a minor, concomitant effect on survival. 223 Thus, hMutSa binds to UV DNA photoproducts in the form 224 of compound lesions [18] and MMR defective cells are 225 considerably more susceptible to mutation by persistent UV- 226 induced DNA damage [26]. Further, although their MMR 227 defects are associated with only a slight increased resistance 228 to killing, msh2~'~ mouse cells are hypermutable by ENU 229[27]. hMutSa binds to DNA containing a 1,2 cisplatin intras- 230 trand lesion, a major product of the therapeutic agent cispl- 231 atin [9,19]. It is generally agreed that M M R defects are 232 associated with a minor (twofold) increase in cisplatin re- 233 sistance [28,29]. HCTl 16— an hMLHl-defective colorectal 234 carcinoma cell line— is, however, considerably more mu- 235 table by cisplatin than HCT116+ch3, in which the repair 236 defect is corrected by chromosome transfer [30]. Much atten- 237 tion has been focused on the therapeutic implications of the 238 slightly higher cisplatin resistance of MMR defective cells. 239 Perhaps it might be more appropriate to consider their in- 240 creased mutability by this, and other, DNA damaging drugs. 241 A final example of DNA damage that is more mutagenic in an 242 MMR defective background is provided by the food-related 243 colon carcinogen 2-amino-l-methyl-6-phenylimidazo[4,5- 244 b]pyridine (PhIP). Despite an extremely small differential 245 cytotoxicity, PhIP is a significantly better mutagen in MMR 246 defective M T 1 cells than in the related repair proficient TK6 247 cells [31]. This hypermutability is apparent in the intact 248 animal and msH2~'~ mice are more susceptible than m sh2'^ '^ 249 animals to PhlP-induced mutations in colonic cells [32]. 250
All these findings establish the important principle that 251 although MMR may process a variety of DNA lesions, with 252 the notable exceptions of 0^-meGua produced by treatment 253 with methylating agents and S^-meGua introduced via 6-TG, 254 this need not result in significant lethality. It can, however, 255 have a major impact on mutation and MMR deficient cells 256 are at increased risk of being mutated by DNA damaging 257 treatments to which they are not significantly tolerant. We 258 will call this enhanced susceptibility to DNA damage- 259 induced mutation that is superimposed on the already high 260 spontaneous mutation rate of MMR defective cells, a super- 261 mutator phenotype. We suggest that it might be more appro- 262 priate to direct attention to the drug-related supermutator 263 phenotype of MMR defective cells rather than the minor 264 effects on cell survival that characterise the majority of DNA 265 damage/MMR interactions. 266
6. M M R defects, DNA damage and cancer 267
It has been proposed that some form of DNA damage 268 might be involved in the development of cancer in HNPCC 269 individuals [4,6]. The suggestion was advanced to confront 270 the tricky problem that loss of MMR appears incompatible 271 with the clonal evolution of tumours in which successive 272
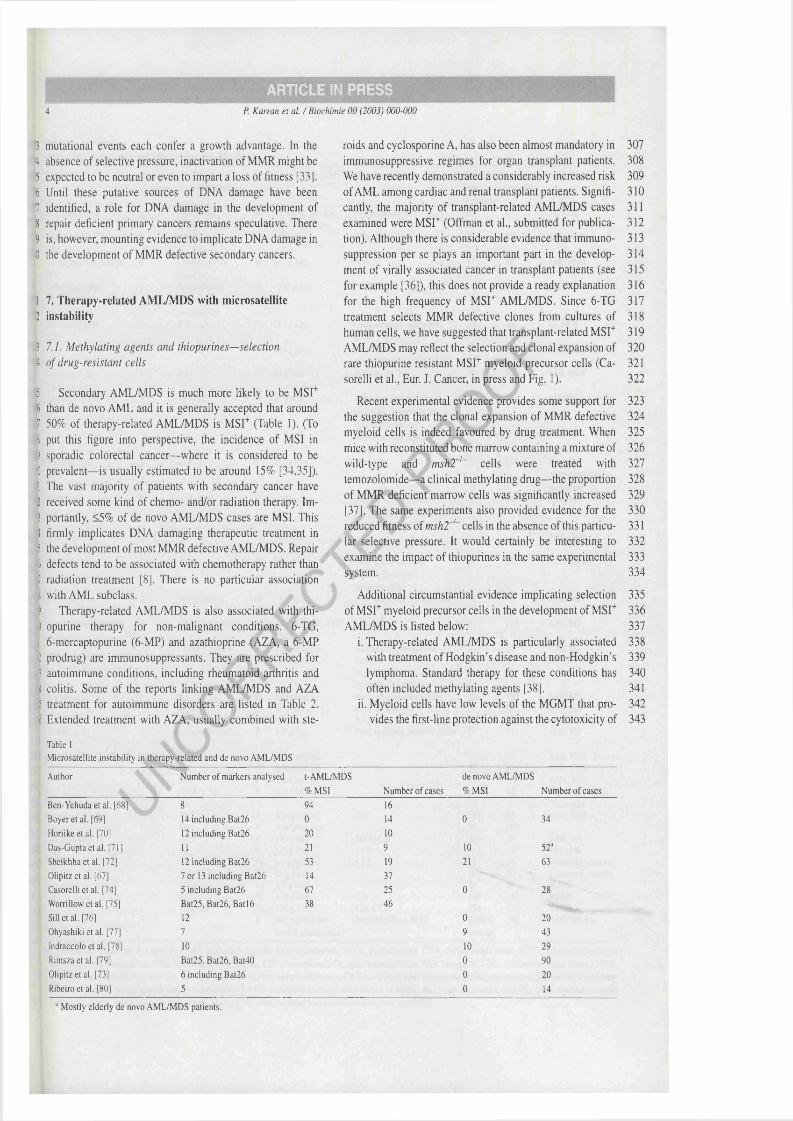
ARTICLE IN PRESSp. Karran et al. /B iochim ie 00 (2003) 000-000
mutational events each confer a growth advantage. In the H absence of selective pressure, inactivation of MMR might be '5 expected to be neutral or even to impart a loss of fitness [33]. '6 Until these putative sources of DNA damage have been 1 identified, a role for DNA damage in the development of '8 repair deficient primary cancers remains speculative. There '9 is, however, mounting evidence to implicate DNA damage in 0 the development of MMR defective secondary cancers.
1 7. Therapy-related AM L/M DS with microsatellite2 instability
3 7.1. M e th y la t in g agen ts a n d th io p u r in e s — se le c tio n
4 o f d ru g -re s is ta n t c e lls
5 Secondary AML/MDS is much more likely to be MSU '5 than de novo AML and it is generally accepted that around7 50% of therapy-related AML/MDS is M ST (Table 1 ). (To8 put this figure into perspective, the incidence of MSI in9 sporadic colorectal cancer— where it is considered to be0 prevalent— is usually estimated to be around 15% [34,35]). ij The vast majority of patients with secondary cancer have2 received some kind of chemo- and/or radiation therapy. Im-3 portantly, <5% of de novo AML/MDS cases are MSI. This1 firmly implicates DNA damaging therapeutic treatment in 5 the development of most MMR defective AML/MDS. Repair8 defects tend to be associated with chemotherapy rather than ] radiation treatment [8]. There is no particular association I with AML subclass.9 Therapy-related AML/MDS is also associated with thill opurine therapy for non-malignant conditions. 6-TG, il 6-mercaptopurine (6-MP) and azathioprine (AZA, a 6-MP S prodrug) are immunosuppressants. They are prescribed for 8 autoimmune conditions, including rheumatoid arthritis and 8 colitis. Some of the reports linking AML/MDS and AZA 8 treatment for autoimmune disorders are listed in Table 2. 8 Extended treatment with AZA, usually combined with ste-
Table 1Microsatellite instability in therapy-related and de novo AML/MDS
roids and cyclosporine A, has also been almost mandatory in 307 immunosuppressive regimes for organ transplant patients. 308 We have recently demonstrated a considerably increased risk 309 of AML among cardiac and renal transplant patients. Signifi- 310 cantly, the majority of transplant-related AML/MDS cases 311 examined were MSU (Offman et al., submitted for publica- 312 tion). Although there is considerable evidence that immuno- 313 suppression per se plays an important part in the develop- 314 ment of virally associated cancer in transplant patients (see 315 for example [36]), this does not provide a ready explanation 316 for the high frequency of MSU AML/MDS. Since 6-TG 317 treatment selects MMR defective clones from cultures of 318 human cells, we have suggested that transplant-related MSU 319 AML/MDS may reflect the selection and clonal expansion of 320 rare thiopurine resistant MSU myeloid precursor cells (Ca- 321 sorelli et al., Eur. J. Cancer, in press and Fig. 1 ). 322
Recent experimental evidence provides some support for 323 the suggestion that the clonal expansion of MMR defective 324 myeloid cells is indeed favoured by drug treatment. When 325 mice with reconstituted bone marrow containing a mixture of 326 wild-type and m sh2"'~ cells were treated with 327 temozolomide— a clinical methylating drug— the proportion 328 of MMR deficient marrow cells was significantly increased 329[37]. The same experiments also provided evidence for the 330 reduced fitness of m sh2~'~ cells in the absence of this particu- 331 lar selective pressure. It would certainly be interesting to 332 examine the impact of thiopurines in the same experimental 333 system. 334
Additional circumstantial evidence implicating selection 335 of M S r myeloid precursor cells in the development of MSU 336 AML/MDS is listed below; 337
i. Therapy-related AML/MDS is particularly associated 338 with treatment of Hodgkin’s disease and non-Hodgkin’s 339 lymphoma. Standard therapy for these conditions has 340 often included methylating agents [38]. 341
ii. Myeloid cells have low levels of the MGMT that pro- 342 vides the first-line protection against the cytotoxicity of 343
Author Number o f markers analysed t-AML/MDS
%MS1 Number o f cases
de novo AML/MDS
%MS1 Number o f cases
Ben-Yehuda etal. [68] 8 94 16
Boyer et al. [69] 14 including Bat26 0 14 0 34
Horiikeetal. [70] 12 including Bat26 20 10
Das-Gupta et al. [71] 11 21 9 10 52'
Sheikhha et al. [72] 12 including Bat26 53 19 21 63
Olipitz et al. [67] 7 or 13 including Bat26 14 37
Casorelli et al. [74] 5 including Bat26 67 25 0 28
Worrillow et al. [75] Bat25,Bat26, Batl6 38 46
Sill et al. [76] 12 0 20
Ohyashiki et al. [77] 7 9 43
Indraccolo et al. [78] 10 10 29
Rimsza et al. [79] Bat25,Bat26, Bat40 0 90
Olipitz et al. [73] 6 including Bat26 0 20
Ribeiro et al. [80] 5 0 14
“ Mostly elderly de novo AML/MDS patients.
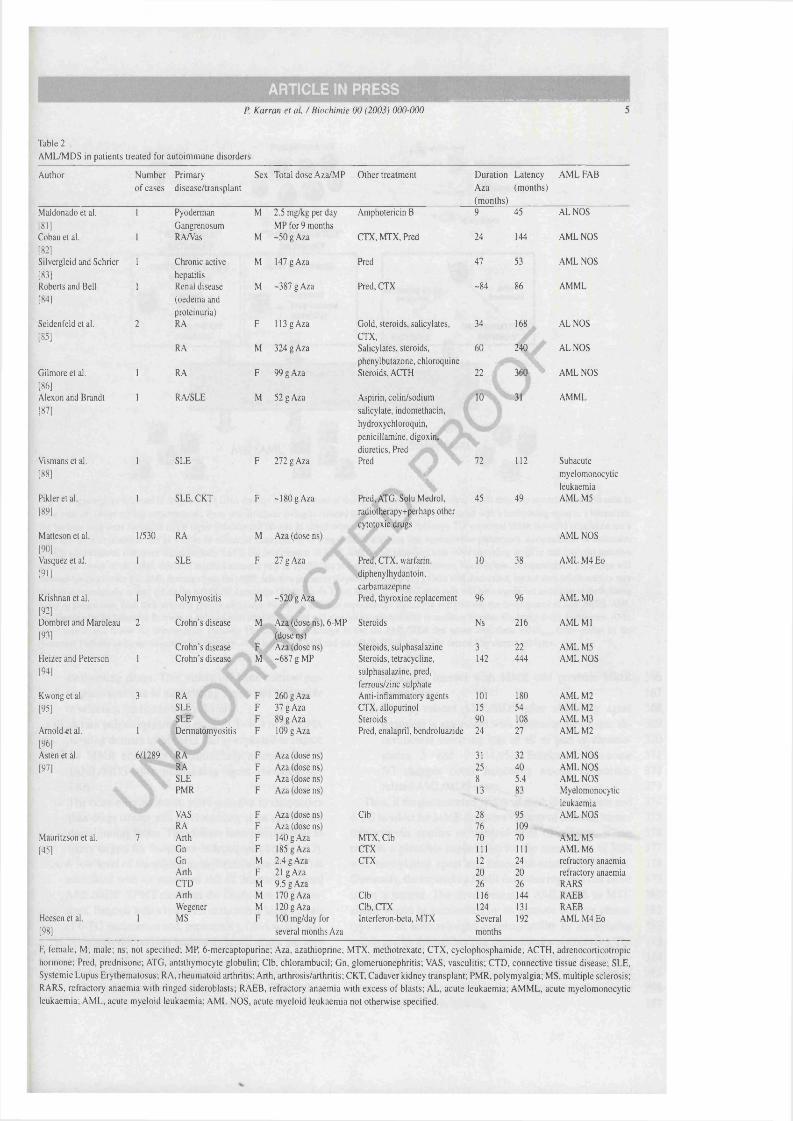
ARTICLE IN PRESSp. Karran et al. /B iochim ie 00 (2003) 000-000
Table 2AM L/M DS in patients treated for autoimmune disorders
Author Number Primary Sex Total dose Aza/MP Other treatment Duration Latency A M L FAB
o f cases disease/transplant Aza(months)
(months)
Maldonado et al. 1 Pyoderman M 2.5 mg/kg per day Amphotericin B 9 45 ALNOS
[81] Gangrenosum MP for 9 monthsCobau et al. 1 RA/Vas M -50 g Aza CTX, MTX, Pred 24 144 AM L NOS
[82]Silvergleid and Schrier 1 Chronic active M 147 g Aza Pred 47 53 AM L NOS
[83] hepatitisRoberts and Bell 1 Renal disease M -387 g Aza Pred, CTX -84 86 AM M L
[84] (oedema and proteinuria)
Seidenfeld et al. 2 RA F 113 g Aza Gold, steroids, salicylates. 34 168 ALNOS
[85] CTX,RA M 324 g Aza Salicylates, steroids. 60 240 ALNOS
phenylbutazone, chloroquineI k NGilmore et al. 1 RA F 99 g Aza Steroids, ACTH 22 f AM L NOS
[86]Alexon and Brandt 1 RA/SLE M 52 g Aza Aspirin, colin/sodium 10 ‘ : 31 AM M L
[87] salicylate, indomethacin, hydroxychloroquin,
penicillamine, digoxin, diuretics, Pred
Vismans et al. 1 SLE F 272 g Aza Pred , 72 112 Subacute
[88] i myelomonocyticleukaemia
Pikleretal. 1 SLE, CKT F -180 g Aza Pred, ATG, Solu Medrol, 45 49 AM LM 5[89] radiotherapy+perhaps other
cytotoxic drugsMatteson et al. 1/530 RA M Aza (dose ns) AM L NOS[90]Vasquez et al. 1 SLE F 27 g Aza Pred, CTX, warfarin. 10 38 A M LM 4E 0
[91] diphenylhydantoin,carbamazepine
Krishnan et al. 1 Polymyositis M -520 g Aza Pred, thyroxine replacement 96 96 AM L MO[92]Dombret and Maroleau 2 Crohn’s disease M Aza (dose ns), 6-MP Steroids Ns 216 AM L M l
[93] (dose ns)Crohn’s disease F Aza (dose ns) Steroids, sulphasalazine 3 22 AM LM 5
Heizer and Peterson 1 Crohn’s disease M -687 g MP Steroids, tetracycline. 142 444 AM L NOS[94] sulphasalazine, pred,
ferrous/zinc sulphateKwong et al. 3 RA F 260 g Aza Anti-inflammatory agents 101 180 AM L M2[95] SLE F 37 g Aza CTX, allopurinol 15 54 AM L M2
SLE F 89 g Aza Steroids 90 108 AM L M3Arnold et al. 1 Dermatomyositis F 109 g Aza Pred, enalapril, bendroluazide 24 27 AM L M2[96]Asten et al. 6/1289 RA F Aza (dose ns) 31 32 AM L NOS[97] RA F Aza (dose ns) 25 40 AM L NOS
SLE F Aza (dose ns) 8 5.4 AM L NOSPMR F Aza (dose ns) 13 83 Myelomonocytic
leukaemiaVAS F Aza (dose ns) Clb 28 95 AM L NOSRA F Aza (dose ns) 76 109
Mauritzson et al. 7 Arth F 140 g Aza MTX, Clb 70 70 AM LM 5[45] Gn F 185 g Aza CTX 111 111 A M LM 6
Gn M 2.4 g Aza CTX 12 24 refractory anaemiaArth F 21 g Aza 20 20 refractory anaemiaCTD M 9.5 g Aza 26 26 RARSArth M 170 g Aza Clb 116 144 RAEBWegener M 120 g Aza Clb, CTX 124 131 RAEB
Heesen et al. 1 MS F 100 mg/day for Interferon-beta, MTX Several 192 AM L M4Eo[98] several months Aza months
F, female; M ; male; ns; not specified; MP, 6-mercaptopurine; Aza, azathioprine; M IX , methotrexate; CTX, cyclophosphamide; ACTH, adrenocorticotropic hormone; Fred, prednisone; ATG, anti thymocyte globulin; Clb, chlorambucil; On, glomeruonephritis; VAS, vasculitis; CTD, connective tissue disease; SLE, Systemic Lupus Erythematosus; RA, rheumatoid arthritis; Arth, arthrosis/arthritis; CKT, Cadaver kidney transplant; PMR, polymyalgia; MS, multiple sclerosis; RARS, refractory anaemia w ith ringed sideroblasts; RAEB, refractory anaemia with excess o f blasts; A L , acute leukaemia; A M M L, acute myelomonocytic leukaemia; A M L, acute myeloid leukaemia; A M L NOS, acute myeloid leukaemia not otherwise specified.

ARTICLE IN PRESSp. Karran et al. /B iochim ie 00 (2003) 000-000
Plufipotent stem cell
•TOLERANCE'PATHWAY
myeloid stem cell . ^ erythrocyte
■— megekeryocyte
monocyte
grenulocyte
•SUPERMUTATOR'PATHWAY
methylating drugs/ thiopurine
• Survival • 1 advantage
• increased drug induced mutability
other treatments
•Increased drug induced mutability
a
methylating d ru g ^ thiopurin^
TOLERANCE*PATHWAY
other^treatments
•SUPERMUTATOR* PATHWAY
Selection of advantageous 'survival' mutations
Clonal expansion of mutant myeloid stem cells
Sà- ^MSltAM L
mmI-
Stable tAML
Fig. 1. Suggested involvement o f drug-induced DNA damage in development o f therapy-related AML. Occasional MMR defective myeloid stem cells arise in
bone marrow. Under normal circumstances, these w ill disappear owing to reduced fitness. I f the individual is treated with a methylating agent or a thiopurine, the intrinsic high-level resistance o f the repair deficient cell favours its clonal expansion (Tolerance Pathway). The expanded MMR defective population has a high spontaneous mutation rate as well as an enhanced susceptibility to drug-induced mutation (the supermutator phenotype). Accumulation o f successive growth advantageous mutations w ill eventually lead to the development o f MSU AM L. After treatment with other alkylating drugs or radiation, the selective growth advantage of the MMR defective myeloid stem cell may be insufficient to promote its clonal expansion. Nevertheless, its supermutator phenotype w ill increase the probability that AM L develops from the MMR defective precursor (Supermutator Pathway). Cells with diminished, but not zero, MMR activity may
also arise occasionally or may reflect the individual’s genotype (Right panel). These MMR,g^ myeloid stem cells w ill have significant resistance to methylating agent or thiopurines. Both their selective growth advantage (tolerance) and their supermutator phenotype w ill promote the development o f AML/MDS. A M L w ill develop by the Tolerance Pathway. A reduced level o f MMR activity w ill also confer susceptibility to mutation by other alkylating drugs. In this case, AM L development w ill follow the Supermutator Pathway. The important difference is that the AML/MDS that arises from these MMR,o^, cells— either by the Tolerance Pathway or by the Supermutator Pathway— w ill not exhibit MSI and the MMR defect w ill not be detected by current analyses.
methylating drugs. This makes the bone marrow particularly sensitive to methylating agents and vulnerable to selection for inactive MMR (39).
iii. A rare polymorphism (-6 exon 13 T->C) in the DNA binding domain of hMSH2 that is expected to reduce its M M R efficiency is particularly associated with tAML/MDS after methylating agent chemotherapy[40].
iv. The bone marrow is also more sensitive to thiopurines than other tissues and myelotoxicity is an established dose-limiting factor. This defines bone marrow cells as likely targets for thiopurine-induced selection [41,42].
V. A low level of thiopurine methyltransferase (TPMT) is associated with an increased risk of thiopurine-related AML/MDS. TPMT catalyses the catabolism of thiopurines. Patients with a low level accumulate higher levels of 6-TG nucleotides and, presumably, DNA 6-TG [43].
vi. Although thiopurines and methylating drugs are very different— the former react directly with DNA whereas the latter are scavenged by the purine salvage pathway for metabolic incorporation into DNA— they are united by a shared ability to form methylated DNA
bases that interact with MMR and promote MMR 366inactivation. 367
vii. Therapy-related AML/MDS after alkylating agent 368treatment is associated with particular karyotypic ab- 369normalities involving loss of all or part of chromo- 370somes 5 and 7 [44,45]. Similar chromosome 3715/7 changes occur frequently among thiopurine- 372related AML/MDS cases. 373
Thus, if the documented ability of methylating agents and 3746-TG to select for M M R defective variants of cultured human 375tumour cells applies in myeloid cells of patients, it can 376provide a plausible explanation for the association of MSI 377with methylating agent and thiopurine-related AML/MDS. 378Obviously, the expanding MMR defective myeloid cell clone 379is not a tumour. The development of AML/MDS in M S r 380clones would be accelerated by its intrinsic mutator pheno- 381type and its acknowledged hypermutability by methylating 382agents and by thiopurines (the supermutator phenotype). The 3 83contribution from drug-induced mutation might be particu- 384larly significant: cancer patients undergo numerous cycles of 385treatment and thiopurine immunosuppression for organ 386transplant patients is lifelong. 387

ÂBTICLE IN PRESSp. Karran et al. /B iochim ie 00 (2003) 000-000
18 7.2. Other alkylating agents—MMR defects and enhanced19 drug-induced mutation and carcinogenesis
Direct selection of drug-resistant M S r myeloid cells provides a coherent explanation for M S r AM L/M DS after treatment with methylating agents or thiopurines. This is clearly not the full story because M S r therapy-related AM L/M DS is not confined to patients treated with these drugs. It occurs following treatment with other alkylating agents to which a M M R defect may not furnish a significant selective survival advantage. Therapy frequently involves simultaneous treatment with a combination of different agents. Although it is possible that a significant selective growth advantage might accrue from their additive effects, recent findings from experiments with M M R deficient KO mice suggest a more plausible alternative hypothesis. They indicate that, even in the absence of a selective growth advantage, the supermutator phenotype of M M R defective cells may have a significant impact on cancer.
M M R KG mice are highly susceptible to tumour induction by methylating agents. M N U treatment increases the already high incidence of lymphoma in msh2~'~ mice [46]. Similarly, treatment with dimethylhydrazine, an acknowledged colon carcinogen, produces more lymphoma and colorectal tumours in mshT'~ mice than in their M M R proficient counterparts [47]. There is increasing evidence that this increased susceptibility to drug-induced cancer is not confined to agents to which M M R defects confer tolerance, however. For example, the supermutator phenotype of ENU treated msh2~'~ mouse cells is paralleled by a heightened susceptibility of msh2~'~ mice to tumour induction by the same drug[27].
The apparently paradoxical interaction between M M R and NER defects in UVB-induced skin cancer in mice may provide a second example of the contribution of the DNA damage related supermutator phenotype to carcinogenesis. Although M M R defects are not associated with significant cellular U V resistance, mshT'~ mice nevertheless display a predisposition to U V light induced skin tumours that is additive with xpa~'~ otxpc~'~ [48,49]. An increased susceptibility of msh2~'~ skin cells to mutation by U V provides a simple explanation for this enhanced cancer susceptibility. Finally, chronic inflammation-related colitis is associated with a higher incidence of colorectal cancer and high-grade dysplasia in msh2~'~ mice than in wild-type animals [50]. Although the involvement of DNA lesions in this chemically induced inflammation has not been formally demonstrated, it seems likely that oxidised DNA bases are an important factor. This may be another example of the impact of a DNA damage related supermutator phenotype on the early events in carcinogenesis.
Thus, it seems feasible that the supermutator phenotype that accompanies DNA damage in a M M R defective cell will contribute to the development of MSU therapy-related cancer (Fig. 1). For most types of DNA damage, the contribution of enhanced survival to selection of the M SU myeloid clone
will be minimal. The hypermutability of drug-treated MSU 443 myeloid cells significantly increases the probability that they, 444 rather than their M M R proficient counterparts, will sustain 445 the advantageous mutations/genetic rearrangements that 446 confer loss of growth control. The resulting AM L/M DS will 447 be M S r. 448
8. The impact of reduced levels of M M R on various 449M M R related functions 450
Recent findings indicate that changes in M M R protein 451 levels do not have a uniform influence on the different pro- 452 cesses in which M M R is involved. The differential impact of 453 reduced M M R capacity has particular implications for M M R 454 defective cells exposed to DNA damaging drugs, and possi- 455 bly for the development of treatment related malignancy. 456
There is no evidence of haploinsufficiency for mismatch 457 correction in humans [51,52]: normal tissue from both 458 hMSH2 and hM LH l heterozygous individuals is fully M M R 459 competent and does not exhibit MSI. An early study did not 460 find any evidence for increased sensitivity to drug-induced 461 chromosome aberrations in lymphoid cell lines established 462 from HNPCC family members bearing either an hMSH2 or 463 hM LH l mutation [53]. More recently, it has been shown that 464 hMSH2 protein levels are around 50% of normal in lympho- 465 blastoid cell lines established from hMSH2 heterozygotes. 466 This does not dramatically compromise their M M R and there 467 is a very modest reduction in mismatch correction activity 468 and mismatch binding by cell extracts. These cells are, how- 469 ever, significantly tolerant to the methylating agent temozo- 470 lomide [54]. Thus, for lethal processing of DNA 0^-meGua, 471 there is evidence of haploinsufficiency for hMSH2. (Curi- 472 ously, this does not apply to hM LHl. hM LH l protein levels 473 in heterozygous cells are not significantly lower than normal, 474 M M R is unaffected, and they are not méthylation tolerant). 475 Experiments in which M SU cell lines have been engineered 476 to express a low level of the missing M M R activity provide 477 additional evidence that reduced M M R capacity is compat- 478 ible with efficient DNA mismatch correction but not with 479 DNA damage processing. Thus, 20% of the normal level of 480 hMSH6 expressed in defective HOT 15 cells significantly 481 ameliorated their mutator phenotype and MSI. This low level 482 of expression also largely restored normal levels of in vitro 483 mismatch binding and repair capability. Despite the restora- 484 tion of reasonably effective M M R, the extreme méthylation 485 tolerance of the HOT 15 cells was unaffected [55]. Methyla- 486 tion tolerance is also associated with reduced levels of 487 hMutLa. A low level of hM LH l expression in M M R- 488 defective 293 human embryonic kidney cells was sufficient 489 to correct their M SU and fully complemented the mismatch 490 correction defect of cell extracts. The same low hM LH l 491 levels did not restore M NNG sensitivity and the cells retained 492 full méthylation tolerance [56]. The observation that M NNG 493 treatment does not activate the G^/M checkpoint in the 494 h M L H l c e l l s suggests that MMR-dependent lethal pro- 495

p. Karran et al. /Biochimie 00 (2003) 000-000
cessing of DNA 0^-meGua and Gj/M checkpoint activation are connected.
A reduced level of the mouse msh2 protein also has a different impact on DNA damage processing and correction of replication errors. The effect of heterozygosity on drug resistance is less apparent in mouse cells. m sh2^'~ c & \h do not have an overt méthylation tolerant phenotype [57] but they are, however, more resistant than m sh 2^ '^ cells to the induction of sister chromatid exchanges by MNU treatment— a very sensitive indicator of MMR-mediated processing of DNA 0^-meGua [58]. A more profound reduction in msh2 protein is associated with méthylation tolerance. msh2~'~
cells that express 10% of the W T level of msh2 are fully MNNG tolerant indicating that the corresponding level of mMutSa is insufficient to initiate lethal processing of 0^-meGua. The minimal msh2 expression in cellsnevertheless reverses their mutator phenotype, suppresses homologous recombination, and prevents MSI. Significantly, in addition to méthylation tolerance, cells retain thesupermutator phenotype of msh2~'~ cells and are particularly susceptible to mutation induction by MNNG treatment [59].
Although data are scarce, there is some evidence that heterozygous KO MMR mice may be more susceptible to drug-induced cancer. Heterozygous animals are not generally regarded as more susceptible than wild-type mice to death from spontaneous cancer although m sh2^'~ mice develop more tumours than m sh2'^''^ animals. The excess tumours tend to be non-gastrointestinal and appear to retain the active copy of m sh2 [60]. Heterozygosity for m sh2 affects carcinogen susceptibility. DM H treatment produced more tumours and a somewhat different spectrum of tumour types in msh2'^'~ compared to animals [47]. On the otherhand, examination of mutation frequencies in colonic crypts of drug-treated mhs2'^'~ mice did not provide evidence of increased sensitivity to induced mutagenesis. MNU treatment induces more thymic lymphoma in mice and these animals are less sensitive than the
controls to the lethal effects of the drug [61]. MNU treatment also causes significantly more gastrointestinal tumours in p m s2^ '~ mice than in pm s2'^''^ animals. In this case there was no effect on overall survival, however, and mice of both genotypes died at the same rate [62]. There are two significant caveats, however. Mouse cells are often intrinsically more méthylation tolerant than human cells and inactivation of MMR increases their méthylation resistance only marginally [63]. This is consistent with the less dramatic impact of heterozygosity for MMR functions on méthylation tolerance in mouse cells. In the absence of carcinogen treatment, MMR heterozygous mice are not notably prone to gastrointestinal cancer. In this regard, they are not a particularly good analogy for human heterozygotes in HNPCC. In contrast, MMR null mice more faithfully recapitulate the cancer predisposition of MMR null humans [64,65]. Thus, although homozygous MMR deficient mouse and human cells share many properties, it may be inappropriate to extrapolate from the heterozygous mouse to humans.
It is clear that more studies are needed to define the 552properties of human cells with heterozygous or significantly 553reduced MMR levels. If the preliminary indications that a 554low MMR capacity does not impair normal repair functions 555but does incapacitate lethal processing of drug-induced DNA 556damage are substantiated, these findings have significant 557implications for therapy-related cancer. If human and mouse 558cells do maintain considerable reserve MMR capability, the 559limits of MMR capacity are unlikely to be breached by the 560‘normal’ substrates of MMR— replication errors or mis- 561paired recombination intermediates. Exposure to cytotoxic 562drugs is not a ‘normal’ situation. Cells with a reduced MMR 563capacity resemble those in which repair is completely abro- 564gated, they are resistant to and/or hypermutable by drug- 565induced DNA damage. Thus, although fully competent at 566repair of replication errors, individuals with reduced MMR 567will be at increased risk of cancer following drug treatment 568(Fig. 1). It is easy to envisage that MMR capacity might be 569reduced by low penetrance mutations or by certain combina- 570tions of MMR gene polymorphisms. Importantly, because 571normal MMR functions would not be overtly impaired, the 572cancer cells would not have a spontaneous mutator pheno- 573type and the tumour would not be MSU. Thus, subtle MMR 574defects— from reduced MMR protein expression— that 575might not necessarily contribute sufficiently to primary ma- 576lignancy to be classified as HNPCC, may nevertheless have a 577significant impact on therapy-related cancer. These subtle 578MMR deficiencies might be invisible to current analytical 579approaches. 580
9. Concluding remarks 581
In this brief review, we have addressed the increasingly 582apparent association of MMR defects and secondary cancer. 583Because most therapeutic treatments produce DNA damage, 584we have summarised recent studies of the interaction of 585MMR with DNA damage. It is clear that normal and MMR 586repair defective cells respond differently to DNA damage. 587This has important implications for therapy-related cancer. 588In extreme cases— represented by methylating drugs or 589thiopurines— a combination of high level resistance and mu- 590tability associated with a repair deficiency provides a plau- 591sible explanation for the association of MSI with 592tAML/MDS. For other drugs, the findings that MMR defec- 593tive cells are hypermutable by DNA damaging agents to 594which they are not significantly tolerant should shift the 595emphasis away from the, often small, increases in DNA 596damage sensitivity that accompany inactivation of MMR and 597focus attention on the increased susceptibility of repair de- 598fective cells to DNA damage-induced mutation. It remains to 599be seen whether this supermutator phenotype may be suffi- 600cient in itself to contribute significantly to the high frequency 601of MSI among therapy-related AML/MDS cases. 602
The second factor that has implications for therapy-related 603cancer is the differential impact of reduced— but not 604zero— repair capacity on the response to ‘normal’ DNA mis- 605

m n ARTICLE IN PRESSp. Karran et al. / Biochimie 00 (2003) 000-000
)6 matches and to DNA damage. A significantly reduced MMR17 capability is probably not detrimental under normal circum-18 stances but it may be following exposure to DNA damaging19 drugs. Since the frequency of hMSH2 heterozygotes in the 0 general population is quite high [35], and there is also evi- ,1 dence that some hMSH2 polymorphisms may be associated2 with less than full activity, it is obviously important to extend3 the interesting preliminary studies on methylating agents and4 to look at the responses of cells from heterozygous individu-5 als or those with MMR gene polymorphisms to the toxic and6 mutagenic effects of other therapeutic drugs. From a clinical7 perspective, the high frequency of M S r among tAML/MDS8 cases is now well established. As a next step, retrospective9 studies to examine the frequency of MMR gene (particularly !0 hMSH2) heterozygotes and the incidence of MMR gene !1 polymorphisms among individuals who developed12 tAML/MDS would be worth considering. Already there are13 some intriguing observations. Olipitz et al. [67] reported that14 the primary tumour of three of four patients who developed15 MSI tAML, was also MSI. If these findings are confirmed on16 a wider scale, they have important implications for chemo-17 therapy in individuals heterozygous for a MMR gene. There18 is also a recent report that a particular hMSH2 polymorphism19 is significant overrepresented in tAML/MDS following me- 0 thylating agent treatment [40].
II AML/MDS arises in myeloid precursor/stem cells. We 2 have summarised the evidence that suggests that myeloid ■3 cells are likely to be particularly vulnerable to methylating4 agents and thiopurines. What is needed now is a thorough5 examination of the DNA repair capabilities of myeloid pre- ■6 cursor cells. Ideally, this would address the question of the17 variation of DNA repair, and particularly MMR, capacity in 8 individual members of the precursor population. For ex- '9 ample, is there evidence of a significant frequency of cells •0 with reduced or null MMR activity? Are the NER and BER •1 pathways fully active in removing the potentially mutagenic •2 DNA lesions that, if left unrepaired, would have particularly *3 severe effects in cells with compromised MMR? Do signifi-4 cant changes in repair capacity accompany differentiation5 into the various myeloid cell types? In order to follow up <6 these intriguing preliminary findings, careful quantitation of >1 DNA repair levels in the potential target myeloid cell popu-18 lations is a clear priority.
19 Uncited Reference
10 [66].
11 References
12 [1] P. Peltomaki, Role o f DNA mismatch repair defects in the pathogen-13 esis o f human cancer, J. Clin. Oncol. 21 (2003) 1174-1179.
[2] J.G. Herman, A. Umar, K. Polyak, J.R. Graff, N. Ahuja, J.-PJ. Issa, 654S. Markowitz, J.K.V. Willson, S.R. Hamilton, K.W. Kinzler, 655M.F. Kane, R.D. Kolodner, B. Vogelstein, T.A. Kunkel, S B. Baylin, 656Incidence and functional consequences o f hMLH 1 promoter hyperm- 657éthylation in colorectal carcinoma, Proc. Natl. Acad. Sci. USA 95 658(1998) 6870- 6875. 659
[3] J. Young, L A . Simms, K.G. Biden, C. Wynter, V. Whitehall, R. Kara- 660matic, J. George, J. Goldblatt, I. Walpole, S.A. Robin, M.M. Borten, 661R. Stitz, J. Searle, D. McKeone, L. Fraser, D R. Purdie, K. Podger, 662R. Price, R. Buttenshaw, M.D. Walsh, M. Barker, B.A. Leggett, 663J.R. Jass, Features o f colorectal cancers with high-level microsatellite 664instability occurring in familial and sporadic settings; parallel path- 665ways o f tumorigenesis. Am. J. Pathol. 159 (2001) 2107-2116. 666
[4] P. Karran, M. Bignami, DNA damage tolerance, mismatch repair and 667genome instability, BioEssays 16 (1994) 833-839. 668
[5] G. Aquilina, M. Bignami, Mismatch repair in correction o f replication 669errors and processing o f DNA damage, J. Cell. Physiol. 187 (2001) 670145-154. 671
[6] R. Fishel, The selection for mismatch repair defects in hereditary 672nonpolyposis colorectal cancer: revising the mutator hypothesis. Can- 673cer Res. 61 (2001) 7369-7374. 674
[7] G. Leone, L. Mele, A. Pulsoni, F. Equitani, L. Pagano, The incidence 675of secondary leukemias, Haematologica 84 (1999) 937-945. 676
[8] I. Casorelli, J. Offman, L. Mele, L. Pagano, M. D ’Errico, S. Sica, 677G. Giannini, G. Leone, M. Bignami, P. Karran, Drug treatment in the 678development o f mismatch repair defective acute leukemia and myelo- 679dysplastic syndrome, DNA Repair 2 (2003) 547-559. 680
[9] D.R. Duckett, J.T. Drummond, A.l.H. Murchie, J.T. Reardon, A. San- 681car, D M. Lilley, P. Modrich, Human MutSa recognizes damaged 682DNA base pairs containing 0^-methylguanine, 0 ‘*-methylthymine, or 683the cisplatin-d(GpG) adduct, Proc. Natl. Acad. Sci. USA 93 (1996) 6846443-6447. 685
[10] G. Aquilina, A.M. Giammarioli, A. Zijno, A. DiMuccio, E. Dogliotti, 686M. Bignami, Tolerance to 0^-methylguanine and 6-thioguanine cyto- 687toxic effects: a cross-resistant phenotype in A-methylnitrosourea- 688resistant Chinese hamster ovary cells. Cancer Res. 50 (1990) 4248- 6894253. 690
[11] M.T. Hawn, A. Umar, J.M. Carethers, G. Marra, T.A. Kunkel, 691C.R. Boland, M. Koi, Evidence for a connection between the mis- 692match repair system and the G^ cell cycle checkpoint. Cancer Res. 55 693(1995) 3721-3725. 694
[12] P.F. Swann, T.R. Waters, D.C. Moulton, Y.-Z. Xu, M. Edwards, 695R. Mace, Role o f postreplicative DNA mismatch repair in the cyto- 696toxic action o f thioguanine. Science 273 (1996) 1109-1 111. 697
[13] T.R. Waters, P.F. Swann, Cytotoxic mechanism o f 6-thioguanine: 698hMutSalpha, the human mismatch binding heterodimer, binds to 699DNA containing S^-methylthioguanine, Biochemistry 36 (1997) 7002501- 2506. 701
[14] N. Zhukovskaya, P. Branch, G. Aquilina, P. Karran, DNA replication 702arrest and tolerance to DNA méthylation damage. Carcinogenesis 15 703(1994)2189-2194. 704
[15] J. Maybaum, H.G. Mandel, Unilateral chromatid damage: a new basis 705for 6-thioguanine cytotoxicity. Cancer Res. 43 ( 1983) 3852-3856. 706
[16] J. Dosch, M. Christmann, B. Kaina, Mismatch G-T binding activity 707and MSH2 expression is quantitatively related to sensitivity o f cells to 708methylating agents. Carcinogenesis 19 (1998) 567-573. 709
[17] A. Mazurek, M. Berardini, R. Fishel, Activation of human MutS 710homologs by 8-oxo-guanine DNA damage, J. Biol. Chem. 277 (2002) 7118260-8266. 712
[18] D. Mu, M. Tursun, D.R. Duckett, J.T. Drummond, P. Modrich, A. San- 713car. Recognition and repair o f compound DNA lesions (base damage 714and mismatch) by human mismatch repair and excision repair sys- 715tems. Mol. Cell. Biol. 17 (1997) 760-769. 7 1 6
[19] M. Yamada, E. O'Regan, R. Brown, P. Karran, Selective recognition 717o f a cisplatin-DNA adduct by human mismatch repair proteins, 718Nucleic Acids Res. 25(1997) 4 9W 95. 719

ARTICLE IN PRESSp. Karran et al. / Biochimie 00 (2003) 000-000
51 [53] N.M. Lindor, S.M. Jalad, T.J. VanDeWalker, J.M. Cunningham,52 R.J. Dabi, S.N. Thibodeau. Search for chromosome instability in53 lymphocytes with germ-line mutations in DNA mismatch repair54 genes, Cancer Genet. Cytogenet. 104 (1998) 48-51,55 [54] G. Marra, S. D ’Atri, C. Corti, L. Bonmassar, M.S. Cataruzza, P. Sch-56 weizer, K. Heinmann, Z. Bardosova, M. Nystrom-Lahti, J. Jiricny,57 Tolerance of human MSH2*'' ly mphoblastoid cells to the methylating58 agent temozolomide, Proc. Natl. Acad. Sci. USA 98 (2001) 7164-
59 7169.50 [55] T. Lettieri, G. Marra, G. Aquilina, M. Bignami, F. Palombo, J. Jiricny,51 Effect o f HMSH6 cDNA expression on the phenotype o f mismatch52 repair-deficient colon cancer cell line H C T l5, Carcinogenesis 2053 (1999)373-382.54 [56] p. Cejka, L. Stojic, N. Mojas, A.M. Russell, K. Heinimann, E. Can-55 navo, M. di Pietro, G. Marra, J. Jiricny, Methylation-induced G2/M )6 arrest requires a full complement o f the mismatch repair protein )7 hMLH 1, EMBO J. 22 (2003) 2245-2254.58 [57] N. de Wind, M. Dekker, A. Bems, M. Radman, H.T. Riele, Inactiva-59 tion o f the mouse MsH2 gene results in postreplicational mismatch '0 repair deficiency, méthylation tolerance, hyperrecombination, and '1 predisposition to tumorigenesis. Cell 82 (1995) 321-330.?2 [58] S.D. Bouffler, N. Holland, R. Cox, R. Fodde, Evidence for Msh2 13 haploinsufficiency in mice revealed by MNU-induced sister chroma- 4 tid exchange analysis, Br. J. Cancer 83 (2000) 1291-1294.
'5 [59] N. Claij, H.T. Riele, Méthylation tolerance in mismatch repair profi- '6 cient cells with low MSH2 protein level. Oncogene 21 (2002) 2873- n 2879.8 [60] N. de Wind, M. Dekker, A. van Rossum, M. van der Valk, H.T. Riele,
?9 Mouse models for hereditary nonpolyposis colorectal cancer. Cancer1 0 Res. 58 (1998) 248-255.11 [61] H. Kawate, R. Itoh, K. Sakumi, Y. Nakabeppu, T. Tsuzuki, F. Ide,12 T. Ishikawa, T. Noda, H. Nawata, M. Sekiguchi, A defect in a single13 allele of the M lh l gene causes dissociation of the killing and tumori- 4 genic actions of a alkylating carcinogen in methyltransferase- 15 deficient mice. Carcinogenesis 21 (2000) 301-305.'6 [62] X. Qin, D. Shibata, S.L. Gerson, Heterozygous DNA mismatch repair n gene PMS2-knockout mice are susceptible to intestinal tumour induc-18 tion with N-methyl-A-nitrosourea, Carcinogenesis 21 (2000) 833-19 838.
•0 [63] 0 . Humbert, S. Fiumicino, G. Aquilina, P. Branch, S. Oda, A. Zijno, 4 P. Karran, M. Bignami, Mismatch repair and differential sensitivity of •2 mouse and human cells to methylating agents. Carcinogenesis 20 '3 (1999) 205-214.4 [64] Q. Wang, C. Lasset, F. Desseigne, D. Frappaz, C. Bergeron, 15 C. Navarro, E. Ruano, A. Puisieux, Neurofibromatosis and early onset '6 o f cancers in hMLH 1 -deficient children. Cancer Res. 59 ( 1999) 294-7 297.8 [65] M.D. Ricciardone, T. Ozcelik, B. Cevher, H. Ozdag, M. Tuncer,9 A. Gurgey, 0 . Uzunallimoglu, H. Cetinkaya, A. Tanyeli, E. Erken,10 M. Ozturk, Human M L H l deficiency predisposes to hematological 1] malignancy and neurofibromatosis type 1, Cancer Res. 59 (1999) '2 290-293.
[66]
[67]
[68][69]
[70]
[71]
[72]
[73][74]
[75][76]
[78][79][80]
[81][82]
[83][84][85]
[87][88][89]
[90][91][92][93]
[94]
[95]
[96]
[97]
[98]
D. Whiteside, R. McLeod, G. Graham, J.L. Steckly, K. Booth, M.J. Somerville, S.E. Andrew, A homozygous germ-line mutation in the human MSH2 gene predisposes to hematological malignancy and multiple cafe-au-lait spots. Cancer Res. 62 (2002) 359-362.W. Olipitz, G. Hopfinger, R.C.T. Aguiar, E. Gunsilius, M. Girschikof-
sky, C. Bodner, K. Hiden, W. Linkesch, G. Hoefler, H. Sill, Defective DNA mismatch repair: a potential mediator o f leukemogenic susceptib ility in therapy-related myelodysplasia and leukemia. Genes Chromosomes Cancer 34 (2002) 243-248.D. Ben-Yehuda, et al., Blood 88 (1996) 4296-4303.J.C. Boyer, J.l. Risinger, R.A. Faber, Cancer Genet. Cytogenet. 106(1998) 54-61.S. Horiike, Misawa, et al.. Leukaemia 13 (1999) 1235-1242.E.P. Das-Gupta, C.H. Seedhouse, N.H. Russell, Br. J. Haematol. 114 (2001)307-312.M.H. Sheikhha, K. Tobal, J.A.L. Yin, Br. J. Haematol. 117 (2002)
359-365.W. Olipitz, et al., Br. J. Haematol. 112 (2001) 248-253.1. Casorelli, et al., DNA Repair 142 (2003) 1-13.L.J. Worrillow, et al., Clin. Cancer Res. 9 (2003) 3012-3020.H. Sill, J.M. Goldman, N.C. Cross, Br. J. Cancer 74 (1996) 255-257.
[77] Ohyashiki, et al.. Clin. Cancer Res. 2 (1996) 1583-1589.S. Indraccolo, et al.. Blood 94 (1999) 2424-2432.L.M. Rimsza, et al., Leukaemia 14 (2000) 1044-1051.E.M.S.F. Ribeiro, et al., Brazilian J. Med. Biol. Res. 35 (2002) 153-
159.N. Maldonado, et al., J. Pediatr. 72 (1968) 409-414.C D. Cobau, R.P. Sheon, A.B. Kirsner, Ann. Intern. Med. 79 (1973) 131-132.A.J. Silvergleid, S.L. Schrier, Am. J. Med. 57 (1974) 885-888.M.M. Roberts, R. Bell, Lancet (1976) 768-770.A.M. Seidenfeld, et al., J. Rheumatol. 3 (1976) 295-304.I T. Gilmore, G. Holden, K.S. Rodan, Postgrad. Med. J. 53 (1977) 173-174.E. Alexon, K.D. Brandt, Am. J. Med. Sci. 273 (1977) 355-440.
J.J. Vismans, et al., Acta Med. Scand. 207 (1980) 315-319.G.M. Pikler, B. Say, S. Stamper, Cancer Genet. Cytogenet. 20 (1986) 101-107.E.L. Matteson, et al., J. Rheumatol. 18 (1991) 809-814.S. Vasquez, et al., J. Rheumatol. 19 (1992) 1625-1627.K. Krishnan, et al.. Clin. Lab. Haematol. 16 (1994) 285-289.H. Dombret, Maroleau, Nouvelle Revue Française Hematol. 37(1995) 193-196.W.D. Heizer, J.L. Peterson, Digest. Dis. Sci. 43 (1998) 1791-1793. Y.L. Kwong, W.Y. Au, R.H.S. Liang, Cancer Genet. Cytogenet. 103(1998)94-97.J.A. Arnold, S.A. Ranson, S.H. Abdalla, Clin. Lab. Haematol. 21(1999) 289-292.P. Asten, J. Barrett, D. Symmons, J. Rheumatol. 26 (1999) 1705- 1714.C. Heesen, et al.. Multiple Sclerosis, Multiple Sclerosis, 9, 2003, pp. 213-214213-214.
903904905906907908909910911912913914915916917918919920921922923924925926927928929930931932933934935936937938939940941942943944945946947948949950951952953954




















