
DM n°1 : Synthèse organique et résolution de problème
6
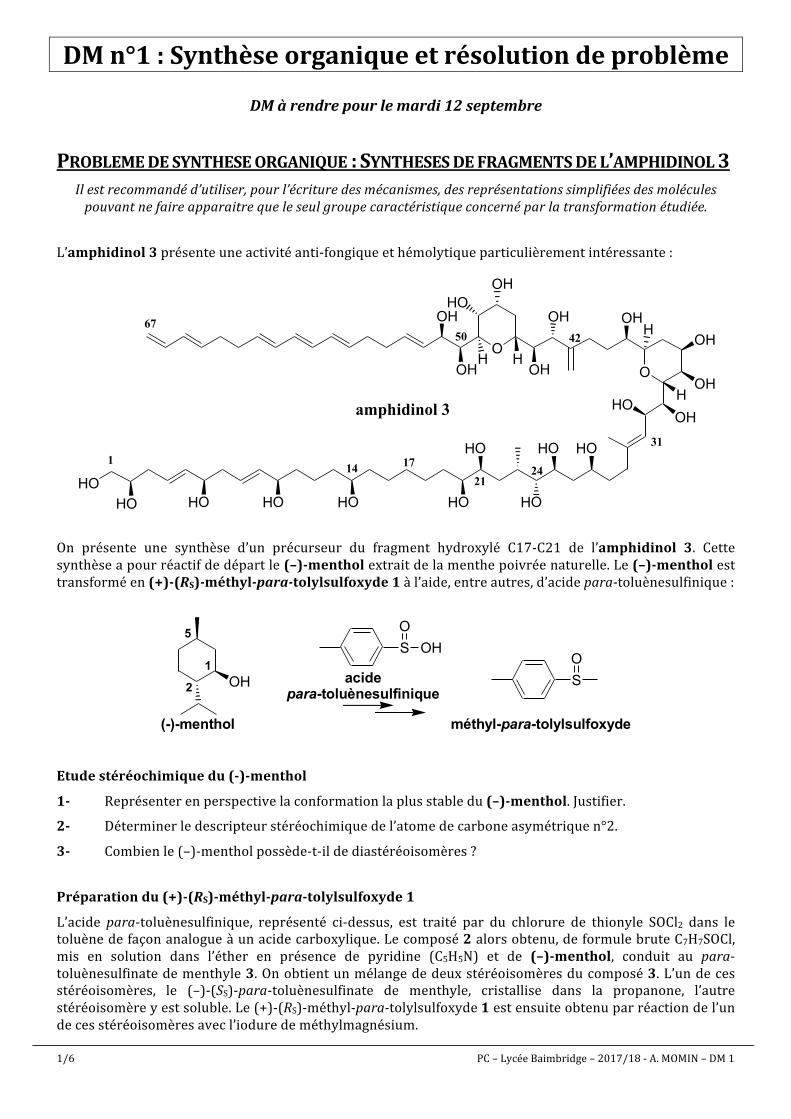
1/6 PC – Lycée Baimbridge – 2017/18 - A. MOMIN – DM 1 DM n°1 : Synthèse organique et résolution de problème DM à rendre pour le mardi 12 septembre PROBLEME DE SYNTHESE ORGANIQUE :SYNTHESES DE FRAGMENTS DE L’ AMPHIDINOL 3 Il est recommandé d’utiliser, pour l’écriture des mécanismes, des représentations simplifiées des molécules pouvant ne faire apparaitre que le seul groupe caractéristique concerné par la transformation étudiée. L’amphidinol 3 présente une activité anti-fongique et hémolytique particulièrement intéressante : O OH OH H HO OH H OH OH OH O H OH OH H OH HO HO HO HO HO HO HO HO HO amphidinol 3 1 31 42 50 67 HO HO 21 24 17 14 On présente une synthèse d’un précurseur du fragment hydroxylé C17-C21 de l’amphidinol 3. Cette synthèse a pour réactif de départ le (–)-menthol extrait de la menthe poivrée naturelle. Le (–)-menthol est transformé en (+)-(RS)-méthyl-para-tolylsulfoxyde 1 à l’aide, entre autres, d’acide para-toluènesulfinique : OH (-)-menthol 1 2 5 S O OH acide para-toluènesulfinique S O méthyl-para-tolylsulfoxyde Etude stéréochimique du (-)-menthol 1- Représenter en perspective la conformation la plus stable du (–)-menthol. Justifier. 2- Déterminer le descripteur stéréochimique de l’atome de carbone asymétrique n°2. 3- Combien le (–)-menthol possède-t-il de diastéréoisomères ? Préparation du (+)-(RS)-méthyl-para-tolylsulfoxyde 1 L’acide para-toluènesulfinique, représenté ci-dessus, est traité par du chlorure de thionyle SOCl2 dans le toluène de façon analogue à un acide carboxylique. Le composé 2 alors obtenu, de formule brute C7H7SOCl, mis en solution dans l’éther en présence de pyridine (C5H5N) et de (–)-menthol, conduit au para- toluènesulfinate de menthyle 3. On obtient un mélange de deux stéréoisomères du composé 3. L’un de ces stéréoisomères, le (–)-(SS)-para-toluènesulfinate de menthyle, cristallise dans la propanone, l’autre stéréoisomère y est soluble. Le (+)-(RS)-méthyl-para-tolylsulfoxyde 1 est ensuite obtenu par réaction de l’un de ces stéréoisomères avec l’iodure de méthylmagnésium.
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of DM n°1 : Synthèse organique et résolution de problème
1/6 PC–LycéeBaimbridge–2017/18-A.MOMIN–DM1
DMn°1:Synthèseorganiqueetrésolutiondeproblème
DMàrendrepourlemardi12septembre
PROBLEMEDESYNTHESEORGANIQUE:SYNTHESESDEFRAGMENTSDEL’AMPHIDINOL3Ilestrecommandéd’utiliser,pourl’écrituredesmécanismes,desreprésentationssimplifiéesdesmoléculespouvantnefaireapparaitrequeleseulgroupecaractéristiqueconcernéparlatransformationétudiée.
L’amphidinol3présenteuneactivitéanti-fongiqueethémolytiqueparticulièrementintéressante:
O
OH
OHH
HOOH
HOH
OH OH
O
HOH
OHHOH
HO
HOHO
HO
HO
HOHOHOHO
amphidinol 3
131
425067
HOHO
2124
1714
On présente une synthèse d’un précurseur du fragment hydroxylé C17-C21 de l’amphidinol 3. Cettesynthèseapourréactifdedépartle(–)-mentholextraitdelamenthepoivréenaturelle.Le(–)-mentholesttransforméen(+)-(RS)-méthyl-para-tolylsulfoxyde1àl’aide,entreautres,d’acidepara-toluènesulfinique:
OH
(-)-menthol
1
2
5SOOH
acidepara-toluènesulfinique
SO
méthyl-para-tolylsulfoxyde
Etudestéréochimiquedu(-)-menthol
1- Représenterenperspectivelaconformationlaplusstabledu(–)-menthol.Justifier.
2- Déterminerledescripteurstéréochimiquedel’atomedecarboneasymétriquen°2.
3- Combienle(–)-mentholpossède-t-ildediastéréoisomères?
Préparationdu(+)-(RS)-méthyl-para-tolylsulfoxyde1
L’acide para-toluènesulfinique, représenté ci-dessus, est traité par du chlorure de thionyle SOCl2 dans letoluènedefaçonanalogueàunacidecarboxylique.Lecomposé2alorsobtenu,deformulebruteC7H7SOCl,mis en solution dans l’éther en présence de pyridine (C5H5N) et de (–)-menthol, conduit au para-toluènesulfinatedementhyle3.Onobtientunmélangededeuxstéréoisomèresducomposé3.L’undecesstéréoisomères, le (–)-(SS)-para-toluènesulfinate de menthyle, cristallise dans la propanone, l’autrestéréoisomèreyestsoluble.Le(+)-(RS)-méthyl-para-tolylsulfoxyde1estensuiteobtenuparréactiondel’undecesstéréoisomèresavecl’ioduredeméthylmagnésium.

2/6 PC–LycéeBaimbridge–2017/18-A.MOMIN–DM1
4- A l’aide du document 1, représenter, en convention spatiale de Cram le (+)-(RS)-méthyl-para-tolylsulfoxyde1dont la formule topologique plane a été précédemment donnée. Justifier l’existence d’unmomentdipolaireimportantpourcecomposé1.
5- Par analogie avec les transformations décrites audocument 2, représenter la formule topologiqueplaneducomposé2,puiscelledupara-toluènesulfinatedementhyle3.
6- Quelestl’intérêtdelapyridinedanscettesynthèse?Onrappellequel’ordredegrandeurdupKaducouplepyridinium/pyridine C"H"NH%/C"H"N estde5,ceuxdescouplesalcool/alcoolategénéralementde16à18.
7- Pourquoil’undesstéréoisomèresde3cristallise-t-ildanslapropanonealorsquel’autreyestsoluble?
8- Quel réactif, de l’iodométhane ou de l’iodure de méthylmagnésium, pourrait être retenu pourtransformerlecomposé3enméthyl-para-tolylsulfoxyde1?Justifiervotreréponse.
Transformationdu(+)-(RS)-méthyl-para-tolylsulfoxyde1
La g-butyrolactone, ester cyclique de formule brute C4H6O2, subit une réaction de saponification (voirdocument3).Onisoleunsolideionique4,deformulebruteC4H7O3Na,dontlespectreRMN1H,réalisédansl’eaudeutéréeD2O,présentelessignauxregroupésdansletableauci-dessous:
Proton Déplacementchimiqueenppm Multiplicité ConstantedecouplageenHz Intégration
Ha 1,8 multiplet 2
Hb 2,5 triplet 7,5 2
Hc 3,8 triplet 6,1 2
Le spectre infrarouge de 4 présente, entre autres, deux bandes larges centrées vers 𝟑𝟑𝟐𝟎𝐜𝐦-𝟏 et𝟐𝟗𝟓𝟎𝐜𝐦-𝟏ainsiqu’unebandevers𝟏𝟓𝟔𝟎𝐜𝐦-𝟏.
9- Représenterlaformuletopologiquedusolideionique4.
10- Quelestleprotonde4dontlesignaln’estpasobservéenRMN1HdansunsolvantprotiquetelqueD2O?Enexpliquerlaraison.
11- Attribuer les bandes IR aux liaisons concernées. Expliquer pourquoi la bande à 1560cm-8 estobservéeàunsifaiblenombred’onde.
12- Attribuer l’ensembledes signauxobservésenRMN 1HauxdifférentsprotonsnotésHa,HbetHcducomposé4.JustifierlamultiplicitédessignauxobservéspourlesprotonsHbetHc.
Le solide ionique 4, mis en solution dans le diméthylformamide (DMF, (CH3)2NCHO, solvant polaireaprotique), est traitépar l’iodométhane.Onobtient le composé5, non isolé, qui après ajoutd’hydruredesodium(NaHou(Na%, H-))etobservationd’undégagementgazeux,esttransforméen6parlechlorurede4-méthoxybenzyle.Cesdeuxdernierscomposéssontreprésentésci-après:
13- Représenter la formule topologique du composé 5. Proposer un mécanisme pour la réaction detransformationde4en5etlenommer.Justifierlechoixdumécanisme.
14- Indiquerlanaturedugazformélorsdelatransformationde5en6.Quelestlerôledel’hydruredesodium?Quelautreréactifaurait-onpuemployerpourcettetransformation?
15- Proposer une suite de transformations chimiques pour préparer le chlorure de4-méthoxybenzyleà partir de 4-bromophénol. Préciser les réactifs et solvants pour chacune de ces
7/14
Le solide ionique 4, mis en solution dans le diméthylformamide [DMF, (CH3)2NCHO], est traité par l’iodométhane. On obtient le composé 5, non isolé, qui après ajout d’hydrure de sodium (NaH) et observation d’un dégagement gazeux, est transformé en 6 par le chlorure de 4-méthoxybenzyle. Ces deux derniers composés sont représentés ci-après :
chlorure de 4-méthoxybenzyle
OO O
O
6
O
B1.12 Représenter la formule topologique du composé 5. Proposer un mécanisme pour la réaction de transformation de 4 en 5 et le nommer.
B1.13 Indiquer la nature du gaz formé lors de la transformation de 5 en 6. Quel est le rôle de l’hydrure de sodium ? Aurait-on pu utiliser à sa place de l’hydroxyde de sodium ?
B1.14 Proposer une suite de transformations chimiques pour préparer le chlorure de 4-méthoxybenzyle à partir de 4-bromophénol. Préciser les réactifs et solvants pour chacune de ces transformations chimiques :
chlorure de 4-méthoxybenzyle
O HO
4-bromophénol
Le (+)-(5S)-méthyl-SDUD�tolylsulfoxyde 1, en solution dans le THF, est traité à basse température par du diisopropylamidure de lithium (LDA). A cette solution est ajouté le composé 6. Après hydrolyse et traitement usuel, on isole le composé solide 7 dont une représentation plane est donnée ci-dessous :
O SO O
7
O
B1.15 En raisonnant par analogie avec la réactivité des composés carbonylés, indiquer les protons à caractère acide du (+)-(5S)-méthyl-SDUD�tolylsulfoxyde 1. Justifier leur acidité.
B1.16 Proposer un mécanisme pour la formation de 7 à partir des composés 6 et 1. Dessiner une représentation spatiale de 7 en convention de Cram.
Le composé 7, traité dans l’éthanol par l’hydrure de diisobutylaluminium [(DIBAL-H), ((CH3)2CHCH2)2Al-H], conduit à un mélange de deux stéréoisomères 8. La comparaison des spectres IR des composés 7 et 8 montre la disparition, lors de cette transformation, d’une bande à 1 711 cm-1 au profit d’une bande large vers 3 400 cm-1 alors qu’une bande intense persiste à 1 030 cm-1.
B1.17 Représenter la formule topologique plane du composé 8 en analysant les données IR.
Le sulfoxyde 8 est transformé en aldéhyde 9, précurseur du fragment C17-C21 de l’amphidinol 3,selon un réarrangement de Pummerer présenté dans le document 4, page 12.
B1.18 Représenter la formule topologique plane de l’aldéhyde 9.

3/6 PC–LycéeBaimbridge–2017/18-A.MOMIN–DM1
transformationschimiques:
NB:Onprécisequel’onpeuttransformerunalcoolenchloroalcaneparactiondechloruredethionyleSOCl>(classiquemaisthéoriquementhors-programme).Cen’estpaslaréponseattendueici.
La finde la synthèse fait intervenir le (+)-(RS)-méthyl-para-tolylsulfoxyde1,puis la chimiedessulfoxydes(document1).
Document1–Lessulfoxydes
LessulfoxydessontdesmoléculesorganiquescontenantungroupefonctionnelsulfinyleSO.Lessulfoxydesprésententunfortmomentdipolaire.L’énergienécessaireà l’inversiondeconfigurationde l’atomedesoufreest trèsélevée(del’ordrede150à180kJmol-8).Àtempératureambiante,l’énergierequisepourinverserlecentredechiralitéestdoncsuffisammentélevéepourqu’unsulfoxydeoptiquementactifneseracémisepas:
R1
O
SR2
R1
O
S R2
150-180 kJ.mol-1
Onnotera,respectivementRSetSS, lestéréodescripteurRouSdel’atomedesoufre.Ainsi,si l’oxygèneestprioritaireselonlesrèglesdeCahnIngoldetPrelog(règlesCIP)surungroupealkyleR1,cedernierprioritairesurungroupealkyleR2, lui-mêmeprioritairesurledoubletd’électronsnonliant,représentépar«••»etaffectéd’unnuméroatomiqueetd’unnombredemassenuls,ona:
Parailleurs,lessulfoxydesontlaparticularitédestabiliserleschargesnégativessurunatomedecarbonesituéen𝛼deSO.Lessulfoxydessontainsiutiliséspourgénérerdesnucléophilescarbonéstrèsutilesensynthèseorganique.
Enfin,lasingularitédessulfoxydesrésideenleurstransformationsaiséesend’autresfonctionsorganiquesprésentéesci-dessous:
O
SR2R1
R1
R1OH
R1O
SR2R1
SR2R1
O O
R1
réarrangementde Pummerer
réduction
oxydation
coupure réductrice
pyrolyse
NuR1
substitution
nucléophile
Le réarrangement de Pummerer permet notamment la transformation des sulfoxydes en aldéhydes. Toutefois, ceréarrangementselimiteauxsulfoxydespossédantunhydrogèneacideen𝛼deSOetauxsubstratsnonsensiblesàdesmilieuxacides.Eneffet,laréactions’effectuegénéralementaurefluxdel’acideéthanoïque.
7/14
Le solide ionique 4, mis en solution dans le diméthylformamide [DMF, (CH3)2NCHO], est traité par l’iodométhane. On obtient le composé 5, non isolé, qui après ajout d’hydrure de sodium (NaH) et observation d’un dégagement gazeux, est transformé en 6 par le chlorure de 4-méthoxybenzyle. Ces deux derniers composés sont représentés ci-après :
chlorure de 4-méthoxybenzyle
OO O
O
6
O
B1.12 Représenter la formule topologique du composé 5. Proposer un mécanisme pour la réaction de transformation de 4 en 5 et le nommer.
B1.13 Indiquer la nature du gaz formé lors de la transformation de 5 en 6. Quel est le rôle de l’hydrure de sodium ? Aurait-on pu utiliser à sa place de l’hydroxyde de sodium ?
B1.14 Proposer une suite de transformations chimiques pour préparer le chlorure de 4-méthoxybenzyle à partir de 4-bromophénol. Préciser les réactifs et solvants pour chacune de ces transformations chimiques :
chlorure de 4-méthoxybenzyle
O HO
4-bromophénol
Le (+)-(5S)-méthyl-SDUD�tolylsulfoxyde 1, en solution dans le THF, est traité à basse température par du diisopropylamidure de lithium (LDA). A cette solution est ajouté le composé 6. Après hydrolyse et traitement usuel, on isole le composé solide 7 dont une représentation plane est donnée ci-dessous :
O SO O
7
O
B1.15 En raisonnant par analogie avec la réactivité des composés carbonylés, indiquer les protons à caractère acide du (+)-(5S)-méthyl-SDUD�tolylsulfoxyde 1. Justifier leur acidité.
B1.16 Proposer un mécanisme pour la formation de 7 à partir des composés 6 et 1. Dessiner une représentation spatiale de 7 en convention de Cram.
Le composé 7, traité dans l’éthanol par l’hydrure de diisobutylaluminium [(DIBAL-H), ((CH3)2CHCH2)2Al-H], conduit à un mélange de deux stéréoisomères 8. La comparaison des spectres IR des composés 7 et 8 montre la disparition, lors de cette transformation, d’une bande à 1 711 cm-1 au profit d’une bande large vers 3 400 cm-1 alors qu’une bande intense persiste à 1 030 cm-1.
B1.17 Représenter la formule topologique plane du composé 8 en analysant les données IR.
Le sulfoxyde 8 est transformé en aldéhyde 9, précurseur du fragment C17-C21 de l’amphidinol 3,selon un réarrangement de Pummerer présenté dans le document 4, page 12.
B1.18 Représenter la formule topologique plane de l’aldéhyde 9.
12/14
Document 4 - Les sulfoxydes
Les sulfoxydes sont des molécules organiques contenant un groupe fonctionnel sulfinyle SO. Les
sulfoxydes présentent un fort moment dipolaire ainsi qu’une stabilité optique. En effet, l’énergie
nécessaire à l’inversion de configuration de l’atome de soufre est très élevée (de l’ordre de 150 à
180 kJ.mol-1
). A température ambiante, l’énergie requise pour inverser le centre de chiralité est
donc suffisamment élevée pour qu’un sulfoxyde optiquement actif ne se racémise pas :
O
S
O
S150-180 kJ.mol-1
Figure 4.1 - Barrière d’inversion de la configuration d’un sulfoxyde
bloquant la racémisation
On notera, respectivement 5S et 6S, le stéréodescripteur 5 ou 6 de l’atome de soufre S. Ainsi, si
l’oxygène est prioritaire selon les règles de Cahn Ingold et Prelog (règles CIP) sur un groupe alkyle
R1, ce dernier prioritaire sur un groupe alkyle R2, lui-même prioritaire sur le doublet d’électrons non
liant, représenté par « » et affecté d’un numéro atomique et d’un nombre de masse nuls, on a :
O
S
O
SS S O
ar
Figure 4.2 - Détermination du descripteur stéréochimique 5 ou 6d’un atome de soufre stéréogène
Par ailleurs, les sulfoxydes ont la particularité de stabiliser les charges négatives sur un atome de
carbone situé en de SO. Les sulfoxydes sont ainsi utilisés pour générer des nucléophiles carbonés
très utiles en synthèse organique.
Enfin, la singularité des sulfoxydes réside en leurs transformations aisées en d’autres fonctions
organiques présentées ci-dessous :
O
S
OH
O
S
SO O
r arrar r
r
a
p r r r
p r
p
Figure 4.3 - Transformations possibles d’un sulfoxyde
Le réarrangement de Pummerer permet notamment la transformation des sulfoxydes en aldéhydes.
Toutefois, ce réarrangement se limite aux sulfoxydes possédant un hydrogène acide en de SO et
aux substrats non sensibles à des milieux acides. En effet, la réaction s’effectue généralement au
reflux de l’acide éthanoïque.
D’après la thèse de doctorat de N. Rival (2012) 9HUV�OD�V\QWKqVH�WRWDOH�GH�O¶DPSKLGLQRO���

4/6 PC–LycéeBaimbridge–2017/18-A.MOMIN–DM1
D’aprèslathèsededoctoratdeN.Rival(2012)Verslasynthèsetotaledel’amphidinol3
Document2–Transformationdesacidescarboxyliquesenesters
Document3–Réactiondesaponificationd’unesternoncyclique
Donnéesspectrales
DonnéesRMN1H:gammededéplacementschimiquesenppm
ProtonH -CH-C- -CH-C=C- -CH-C=O -CH-OR -CH=C- -CH=O𝛿(ppm) 0,9-1,3 1,6-2,5 2,0-3,0 3,3-3,7 4,5-6,0 9,5-10,0
DonnéesINFRAROUGE:nombresd’ondeassociéàlavibrationd’élongationdequelquesliaisons
Liaison O–H C–H C=C C=O S=O
𝜎(cm-8) 3300-3600 2910-2970 1580-1620 1710-1750 1030-1050
R C
O
OH
SOCl2 R C
O
Cl– SO2(g)– HCl
R C
O
Cl
R'OHR C
O
OR'– HCl
R C
O
OR'
+ HO– R C
O
O–
+ R'OH

5/6 PC–LycéeBaimbridge–2017/18-A.MOMIN–DM1
RESOLUTIONDEPROBLEME:UNEETAPEDESYNTHESEVERSLANEPETALACTONE
Lanépétalactoneestlamoléculeactivedel’«herbeàchat».Ils’agitenréalitéd’unpsychotropeagissantsur l’espèceféline(voirci-contre).
Une étape de synthèse de lanépétalactoneparWOLINSKIetcoll.estretranscrite ci-dessous, à partir d’undérivédu(+)-menthol:
Expliquezlatransformationréaliséed’unpointdevuemécanistique.Vousvousappuierezsurlesdocumentsprésentésci-dessous.Vousnevousintéresserezpas,dansunpremiertemps,àlastéréochimie.
[5/2seulement]Enbonusultime,vouspourrezvouspenchersur lastéréochimieducarboneasymétriquenouvellementcréédansleproduitfinalmajoritaire.
Document4–Additiond’halogènessurlesalcènes(d’après«Chimieorganiqueavancée»,F.A.CAREY&R.J.SUNDBERG,volume2,éd.DeBoeckSupérieur,1997)
L’additiondechloreetdebromesurlesalcènesestuneréactiontrèsgénérale.Lacompréhensiondumécanismedecesadditionsd’halogènesestengrandepartiedueauxétudesportantsurlastéréochimiedecesréactions.Laplupartdestypesd’alcènessontconnuspourréaliser l’additiondubromed’unemanièrestéréospécifiqueaboutissantauproduitd’addition anti. Les ions bromonium intermédiaires cycliques chargés positivement, fournissent une explicationséduisantedelastéréospécificitéobservée,puisqu’ilssubissentuneouverturenucléophileselonuneréactiondeSN2delapartdel’ionbromure:
Beilstein J. Org. Chem. 2012, 8, 1246–1255.
1248
Scheme 1: Route from (S)-pulegone to the mixture of dihydronepetalactones a and b, consequently following Wolinsky's approach [23]. Reactionconditions: a) Br2, HOAc, 0 °C; b) KOH, H2O, reflux (15%, 2 steps); c) MeOH/HCl, rt, 96 h (82%); d) 2,6-lutidine, reflux, 72 h (75% of 12 + 13);e) BH3·SMe2, 0 °C, THF, NaOH, H2O2 (86% mixture of diastereomers); f) p-TsOH, toluene, reflux (17% of a + b).
Figure 3: Structures and gas chromatographic retention times of trans-fused dihydronepetalactones on a conventional FFAP column (FFAP)and on an enantioselective cyclodextrin column (cyclo). For experi-mental details see Supporting Information File 1. The racemates b/b'and d/d', which coeluted on FFAP, could be separated from each otheron DB5 (data not shown).
tions of diastereomeric mixtures. Therefore, as an alternative,we present a novel stereoselective route to trans-fused dihy-dronepetalactones starting from pure, cheaply available enan-tiomers of limonene.
Route to trans-fused dihydronepetalactonesa and b starting from (S)-pulegoneFor comparison, the synthesis of a and b was carried outfollowing Wolinsky’s approach: (S)-Pulegone (9) was trans-formed to trans-pulegenic acid 10 via bromination, Favorskiirearrangement, and subsequent elimination (Scheme 1). Stereo-
selective addition of hydrochloric acid afforded the chloride 11,and subsequent elimination of hydrochloric acid gave a mixtureof the methyl esters 12 and 13 (methyl trans-pulegenate) [20-22] which could be separated by chromatography on silica gel.Hydroboration and lactonization of 12 furnished a mixture ofthe C4-epimers a and b that once again needed to be separatedby chromatography on silica gel [23].
Analytical data of the first eluting component a were in accor-dance with those reported in the literature [24]. The samesequence starting from (R)-pulegone yielded a mixture ofdiastereomers a' and b'. The relative configuration of a at C4was assigned according to NOESY experiments. DecisiveNO-effects were found between the protons 4-H and 7a-H aswell as between 7a-H and 7-CH3 (Figure 4).
Figure 4: Configuration of the dihydronepetalactone a.
Basically, the sequence developed by Wolinsky could alsoprovide access to the diastereomers c and d (and their enan-tiomers) if trans-pulegenic acid (10) would be replaced by cis-
Beilstein J. Org. Chem. 2012, 8, 1246–1255.
1248
Scheme 1: Route from (S)-pulegone to the mixture of dihydronepetalactones a and b, consequently following Wolinsky's approach [23]. Reactionconditions: a) Br2, HOAc, 0 °C; b) KOH, H2O, reflux (15%, 2 steps); c) MeOH/HCl, rt, 96 h (82%); d) 2,6-lutidine, reflux, 72 h (75% of 12 + 13);e) BH3·SMe2, 0 °C, THF, NaOH, H2O2 (86% mixture of diastereomers); f) p-TsOH, toluene, reflux (17% of a + b).
Figure 3: Structures and gas chromatographic retention times of trans-fused dihydronepetalactones on a conventional FFAP column (FFAP)and on an enantioselective cyclodextrin column (cyclo). For experi-mental details see Supporting Information File 1. The racemates b/b'and d/d', which coeluted on FFAP, could be separated from each otheron DB5 (data not shown).
tions of diastereomeric mixtures. Therefore, as an alternative,we present a novel stereoselective route to trans-fused dihy-dronepetalactones starting from pure, cheaply available enan-tiomers of limonene.
Route to trans-fused dihydronepetalactonesa and b starting from (S)-pulegoneFor comparison, the synthesis of a and b was carried outfollowing Wolinsky’s approach: (S)-Pulegone (9) was trans-formed to trans-pulegenic acid 10 via bromination, Favorskiirearrangement, and subsequent elimination (Scheme 1). Stereo-
selective addition of hydrochloric acid afforded the chloride 11,and subsequent elimination of hydrochloric acid gave a mixtureof the methyl esters 12 and 13 (methyl trans-pulegenate) [20-22] which could be separated by chromatography on silica gel.Hydroboration and lactonization of 12 furnished a mixture ofthe C4-epimers a and b that once again needed to be separatedby chromatography on silica gel [23].
Analytical data of the first eluting component a were in accor-dance with those reported in the literature [24]. The samesequence starting from (R)-pulegone yielded a mixture ofdiastereomers a' and b'. The relative configuration of a at C4was assigned according to NOESY experiments. DecisiveNO-effects were found between the protons 4-H and 7a-H aswell as between 7a-H and 7-CH3 (Figure 4).
Figure 4: Configuration of the dihydronepetalactone a.
Basically, the sequence developed by Wolinsky could alsoprovide access to the diastereomers c and d (and their enan-tiomers) if trans-pulegenic acid (10) would be replaced by cis-
O
OH
H

6/6 PC–LycéeBaimbridge–2017/18-A.MOMIN–DM1
Laformationdel’ionbromoniumestsoumiseàdescontraintesstériquesrelativesàl’approchedudibromesurl’uneetl’autredesfacesdel’alcène:
Document5–Aciditéenad’ungroupementcarbonyle
Dans un dérivé carbonylé (aldéhyde ou cétone), les éventuels atomes d’hydrogène portés par un atome de carbonevoisindugroupementcarbonyleC=O,dit«enalphadeC=O»,présententuncaractèreacide:
Enmilieubasique,cesdérivéscarbonylésdit“énolisables”sontdoncenéquilibreavecunionénolate,quiestunbonnucléophilecarboné.
Document6–Mécanismed’additionnucléophilesuivied’uneélimination(AdN+E)
Cemécanismepermetlasubstitutionsurunatomedecarbonetrigonald’ungroupementXpartantparungroupementYentrant,issud’unréactifnucléophile:
Document7–TensionsdecyclepargroupementCH2danslescycloalcanes
On peut calculer à partir de données thermodynamiques la «tension de cycle», qui désigne la déstabilisation ducycloalcaneparrapportàl’alcaneacycliquelinéairecorrespondant,dueauxcontraintesd’anglesdeliaisonsetd’anglesdetorsionissuesdelagéométrieducycle.Onlacomptabiliseicien(kJmol-8(CH>)-8) Pluselleestélevée,moinslecycloalcaneeststable,etplusilauratendanceàêtrefacilementouvertenconditionsnucléophiles.
Cyclopropane 9,2 Cyclobutane 6,6Cyclopentane 1,3 Cyclohexane 0,0Cycloheptane 0,9 Cyclooctane 1,3Cyclononane 1,4 Cyclodécane 1,4Cycloundécane 1,1 Cyclododécane 0,2
520 C h a p t e r 1 2 R e a c t i o n s o f A l k e n e s
Stereospecifi c 2-Butene Bromination
Br
BrH
C
(
cis-2-Butene
C C CH
G
GC(
G
@&H
H3C
C CG
G PG
H
GCH3
Br2, CCl4
G
GBr
GBr
CH3
R R
(2R,3R)-2,3-Dibromobutane
!H3C
H
CH3
S S
(2S,3S)-2,3-Dibromobutane
Br
BrH
C
(trans-2-Butene
C
H
C C
H
G
GC(
G
@&HH3C
C CG
G PG
HG
CH3
Br2, CCl4CH3C
G
GBr
GBr
H3C S R
meso-2,3-Dibromobutane
!
CH3H
CH3
R S
Identical
Racemic mixture of two enantiomers
@&
HCH3C
@&ð"
ð""
ð""
"
ð"
ð""
ð""
"
ð""
ð""
Cyclic bromonium ions explain the stereochemistryHow does bromine attack the electron-rich double bond even though it does not appear to contain an electrophilic center? The answer lies in the polarizability of the Br – Br bond, which is prone to heterolytic cleavage upon reaction with a nucleophile. The p-electron cloud of the alkene is nucleophilic and attacks one end of the bromine molecule, with simultaneous displacement of the second bromine atom as bromide ion in an SN2-like process. What is the product of this process? We might expect a carbocation, in analogy with the proton additions discussed in Sections 12-3 and 12-4. However, if the fi rst step of addition of bromine to cyclohexene were to give a carbocation, the bromide ion released in this process could attack the positively charged carbon atom from either the same or the opposite side of the ring, resulting in a mixture of cis- and trans-1,2-dibromocyclohexanes. However, as shown previously, only the trans product is obtained. How can we explain this result?
The stereochemistry of bromination is explained if we propose that initial attack of bromine on the double bond gives a cyclic bromonium ion, in which the bromine bridges both carbon atoms of the original double bond to form a three-membered ring (Figure 12-3). The structure of this ion is rigid, and it may be attacked by bromide ion only on the side opposite the bridging bromine atom. The three-membered ring is thus opened stereo-specifi cally (compare nucleophilic ring opening of oxacyclopropanes in Section 9-9). The leaving group is the bridging bromine. In symmetric bromonium ions, attack is equally probable at either carbon atom, thereby giving the racemic (or meso) products observed.
REACTION
MODEL BUILDING
BA
C C C C
Br −
Br −Br Br +
Br +
δ+
Br δ−
Br δ+
Br δ−
C C C C
Figure 12-3 (A) Electron-pushing picture of cyclic bromonium ion formation. The alkene (red) acts as a nucleophile to displace bromide ion (green) from bromine. The molecular bromine behaves as if it were strongly polarized, one atom as a bromine cation, the other as an anion. (B) Orbital picture of bromonium ion formation.
CC
O
H
αBase
CC
O
CC
O
ion+énolate
YR C X
O
Y
+'''XC
O
YRC
O
XR




















