
Diseases originate and terminate by genes: unraveling nonviral gene delivery
18
REVIEW ARTICLE Diseases originate and terminate by genes: unraveling nonviral gene delivery Rajan Swami & Indu Singh & Wahid Khan & Sistla Ramakrishna # Controlled Release Society 2013 Abstract The world is driving in to the era of transformation of chemical therapeutic molecules to biological genetic ma- terial therapeutics, and that is where the biological drugs especially “genes” come into existence. These genes worked as “magical bullets” to specifically silence faulty genes re- sponsible for progression of diseases. Viral gene delivery research is far ahead of nonviral gene delivery technique. However, with more advancement in polymer science, new ways are opening for better and efficient nonviral gene delivery. But efficient delivery method is always considered as a bottleneck for gene delivery as success of which will decide the fate of gene in cells. During the past decade, it became evident that extracellular as well as intracellular bar- riers compromise the transfection efficiency of nonviral vec- tors. The challenge for gene therapy research is to pinpoint the rate-limiting steps in this complex process and implement strategies to overcome the biological physiochemical and metabolic barriers encountered during targeting. The synergy between studies that investigate the mechanism of breaking in and breaking out of nonviral gene delivery carrier through various extracellular and intracellular barriers with desired characteristics will enable the rational design of vehicles and revolutionize the treatment of various diseases. Keywords Gene delivery . Nonviral gene delivery . Extra and intracellular barriers . Gene delivery vectors Introduction Advancements in the understanding of the molecular and genetic basis of diseases progressed humankind to an era of transformation of chemical therapeutic molecules to biological genetic material therapeutics, and this therapy with genes is called “gene therapy.” The transfer of a therapeutic or working gene copy into specific cells of an individual in order to repair a faulty gene copy is solely involved with the gene therapy. Thus, a faulty gene is generally replaced, or a new gene whose function is to cure or to favorably modify the clinical course of a condition is introduced [1, 2]. Novel opportunities to combat a large number of diseases with designed genes either in the form of therapeutic oligonucleotides (ON) or plasmids DNA (pDNA) carrying gene sequences are offered by sequenc- ing of the human genome and functional genomics [3]. Gene- based therapeutics include plasmids (containing transgenes for gene therapy), ON (for antisense and antigene applications) [4], ribosomes, DNAzymes, aptamers, small interfering RNAs (siRNAs) [5], etc. Selective recognition of molecular target and pathway which imparts tremendous recognition specificity of action as compared to available low molecular weight phar- maceuticals is offered by DNA-based drugs. These drugs can be used in mitigating disease states either prophylactically or at a very early stage, thereby preventing disease progression and its complication [6]. Gene expression can be disrupted at the transcription level (triplex DNA) [7] or translation (antisense DNA or short inference RNA) level [7–12]. Transcription is disrupted by the binding of a triplex forming ON at the pro- moter region of a target gene, and in the antisense strategy, the ON molecule corresponding to a target gene is delivered inside a cell where it binds complementarily with targeted messenger RNA (mRNA) hence, block protein synthesis [13, 14]. Vitravene, an antiretroviral drug used for local therapy of Cy- tomegalovirus retinitis, available in the market is based on similar principle of antisense ON produced and nearly 20 others in late-stage clinical trials [15]. R. Swami : I. Singh : W. Khan : S. Ramakrishna Department of Pharmaceutics, National Institute of Pharmaceutical Education & Research (NIPER), Hyderabad 500037, India S. Ramakrishna (*) Department of Pharmacology, Indian Institute of Chemical Technology, Hyderabad 500607, India e-mail: [email protected] Drug Deliv. and Transl. Res. DOI 10.1007/s13346-013-0159-6
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Diseases originate and terminate by genes: unraveling nonviral gene delivery

REVIEWARTICLE
Diseases originate and terminate by genes: unravelingnonviral gene delivery
Rajan Swami & Indu Singh & Wahid Khan &
Sistla Ramakrishna
# Controlled Release Society 2013
Abstract The world is driving in to the era of transformationof chemical therapeutic molecules to biological genetic ma-terial therapeutics, and that is where the biological drugsespecially “genes” come into existence. These genes workedas “magical bullets” to specifically silence faulty genes re-sponsible for progression of diseases. Viral gene deliveryresearch is far ahead of nonviral gene delivery technique.However, with more advancement in polymer science, newways are opening for better and efficient nonviral genedelivery. But efficient delivery method is always consideredas a bottleneck for gene delivery as success of which willdecide the fate of gene in cells. During the past decade, itbecame evident that extracellular as well as intracellular bar-riers compromise the transfection efficiency of nonviral vec-tors. The challenge for gene therapy research is to pinpoint therate-limiting steps in this complex process and implementstrategies to overcome the biological physiochemical andmetabolic barriers encountered during targeting. The synergybetween studies that investigate the mechanism of breaking inand breaking out of nonviral gene delivery carrier throughvarious extracellular and intracellular barriers with desiredcharacteristics will enable the rational design of vehicles andrevolutionize the treatment of various diseases.
Keywords Gene delivery . Nonviral gene delivery . Extraand intracellular barriers . Gene delivery vectors
Introduction
Advancements in the understanding of the molecular andgenetic basis of diseases progressed humankind to an era oftransformation of chemical therapeutic molecules to biologicalgenetic material therapeutics, and this therapy with genes iscalled “gene therapy.” The transfer of a therapeutic or workinggene copy into specific cells of an individual in order to repair afaulty gene copy is solely involved with the gene therapy. Thus,a faulty gene is generally replaced, or a new gene whosefunction is to cure or to favorably modify the clinical courseof a condition is introduced [1, 2]. Novel opportunities tocombat a large number of diseases with designed genes eitherin the form of therapeutic oligonucleotides (ON) or plasmidsDNA (pDNA) carrying gene sequences are offered by sequenc-ing of the human genome and functional genomics [3]. Gene-based therapeutics include plasmids (containing transgenes forgene therapy), ON (for antisense and antigene applications) [4],ribosomes, DNAzymes, aptamers, small interfering RNAs(siRNAs) [5], etc. Selective recognition of molecular targetand pathway which imparts tremendous recognition specificityof action as compared to available low molecular weight phar-maceuticals is offered by DNA-based drugs. These drugs canbe used in mitigating disease states either prophylactically or ata very early stage, thereby preventing disease progression andits complication [6]. Gene expression can be disrupted at thetranscription level (triplex DNA) [7] or translation (antisenseDNA or short inference RNA) level [7–12]. Transcription isdisrupted by the binding of a triplex forming ON at the pro-moter region of a target gene, and in the antisense strategy, theON molecule corresponding to a target gene is delivered insidea cell where it binds complementarily with targeted messengerRNA (mRNA) hence, block protein synthesis [13, 14].Vitravene, an antiretroviral drug used for local therapy of Cy-tomegalovirus retinitis, available in the market is based onsimilar principle of antisense ON produced and nearly 20 othersin late-stage clinical trials [15].
R. Swami : I. Singh :W. Khan : S. RamakrishnaDepartment of Pharmaceutics, National Institute of PharmaceuticalEducation & Research (NIPER), Hyderabad 500037, India
S. Ramakrishna (*)Department of Pharmacology, Indian Institute of ChemicalTechnology, Hyderabad 500607, Indiae-mail: [email protected]
Drug Deliv. and Transl. Res.DOI 10.1007/s13346-013-0159-6
However, in spite of its superior selective recognition overlow molecular weight pharmaceuticals, folding of targetRNAs and its specificity towards specific proteins still re-mains a major stumbling block in DNA-based drugs delivery.Employment of relatively high doses and low half-life havemade ON molecules less attractive for therapeutic productdevelopment [16]. Two key components in current gene ther-apy are: an effective therapeutic gene that can be expressed ata target site [17] and an efficient and safe delivery system thatdelivers the therapeutic genes to a specific target tissue ororgan [18]. But the efficient delivery method is always con-sidered a bottleneck for gene delivery as success of which willdecide the fate of gene in cells. The impetus in the develop-ment of these drugs can be attributed, in part, to their poorcellular uptake profile in vivo. The innate ability of DNA-based drugs to be internalized by target cells is minimal undernormal circumstances. In addition, poor biological stabilityand a short half-life result in unpredictable pharmacokinetics[19]. Furthermore, DNA molecules that do manage to enterthe cell are subsequently subjected to intracellular degradationalong with stringently restricted nuclear access. The resultingrandom delivery profile of DNA-based drugs is further com-plicated by a dearth of in vivo/in vitro correlation of theirpharmacological outcomes.
A comprehensive insight of various delivery systems forDNA-based drugs and the mechanism becomes anindispensible part of any gene-based drug delivery program.In current review, we intend to discuss about types of genedelivery specifically nonviral gene delivery, barriers encoun-tered during nonviral gene delivery, along with possiblemethod/techniques to overcome them.
Gene delivery systems
A major technical impediment to the gene transfer is the lackof an ideal gene delivery system. For effective in vivo delivery
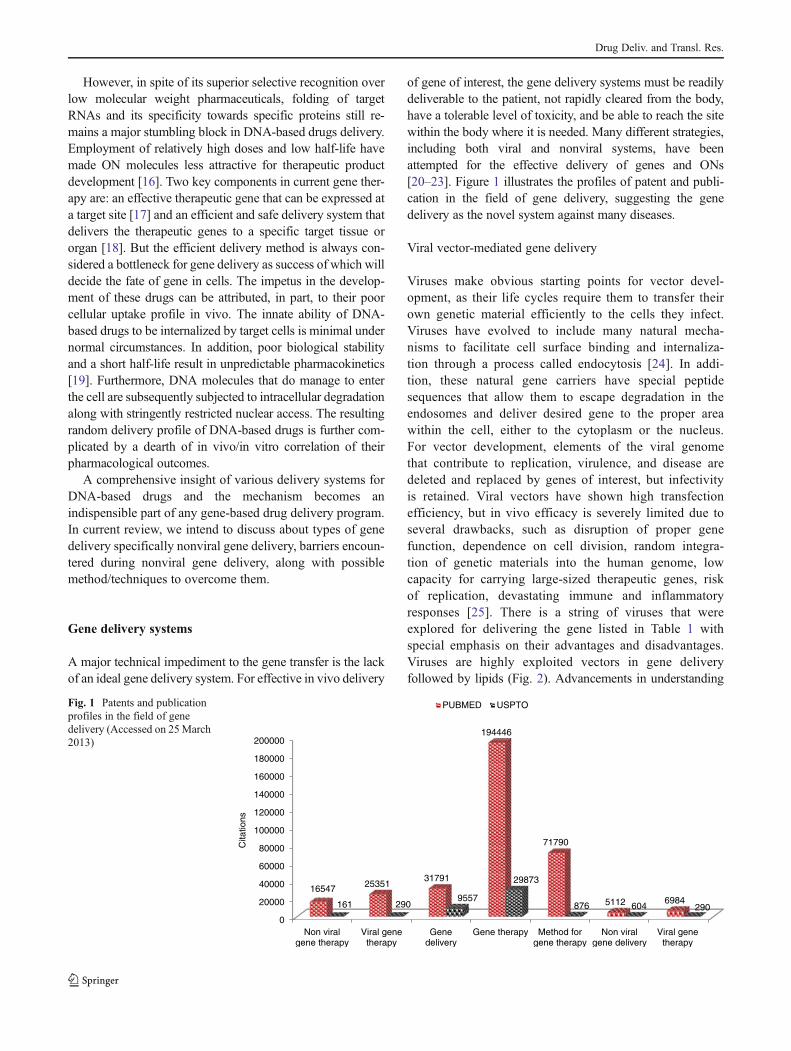
of gene of interest, the gene delivery systems must be readilydeliverable to the patient, not rapidly cleared from the body,have a tolerable level of toxicity, and be able to reach the sitewithin the body where it is needed. Many different strategies,including both viral and nonviral systems, have beenattempted for the effective delivery of genes and ONs[20–23]. Figure 1 illustrates the profiles of patent and publi-cation in the field of gene delivery, suggesting the genedelivery as the novel system against many diseases.
Viral vector-mediated gene delivery
Viruses make obvious starting points for vector devel-opment, as their life cycles require them to transfer theirown genetic material efficiently to the cells they infect.Viruses have evolved to include many natural mecha-nisms to facilitate cell surface binding and internaliza-tion through a process called endocytosis [24]. In addi-tion, these natural gene carriers have special peptidesequences that allow them to escape degradation in theendosomes and deliver desired gene to the proper areawithin the cell, either to the cytoplasm or the nucleus.For vector development, elements of the viral genomethat contribute to replication, virulence, and disease aredeleted and replaced by genes of interest, but infectivityis retained. Viral vectors have shown high transfectionefficiency, but in vivo efficacy is severely limited due toseveral drawbacks, such as disruption of proper genefunction, dependence on cell division, random integra-tion of genetic materials into the human genome, lowcapacity for carrying large-sized therapeutic genes, riskof replication, devastating immune and inflammatoryresponses [25]. There is a string of viruses that wereexplored for delivering the gene listed in Table 1 withspecial emphasis on their advantages and disadvantages.Viruses are highly exploited vectors in gene deliveryfollowed by lipids (Fig. 2). Advancements in understanding
0
20000
40000
60000
80000
100000
120000
140000
160000
180000
200000
Non viralgene therapy
Viral genetherapy
Genedelivery
Gene therapy Method forgene therapy
Non viralgene delivery
Viral genetherapy
16547 2535131791
194446
71790
5112 6984161 2909557
29873
876 604 290
Cita
tions
PUBMED USPTOFig. 1 Patents and publicationprofiles in the field of genedelivery (Accessed on 25March2013)
Drug Deliv. and Transl. Res.

of various vectors for gene delivery lead to the utilization ofmore sophisticated nonviral vectors [26]. Troubles in viralvector-assisted gene delivery lead to the exploitation of othersystems of delivery like polymeric [27, 28] lipidic [29], or moreadvanced vectors like bacterial ghosts and exosomes [30–32].
Nonviral vector-mediated gene delivery
Preliminary research focused with viral carriers, includingboth retroviruses and adenoviruses, as these vectors revealedhigh competence at transporting both DNA and RNA toseveral cell lines [6]. Though basic problems associated withviral vector systems, including toxicity, immunogenicity,and drawbacks with respect to scale-up measures, promotedthe enquiry of other prospective scaffolds exogenous DNA
into targeted tissue [7]. This leads to exploration of the vastopportunities in nonviral gene delivery carriers.
Nonviral gene delivery carriers are explored by employinggenerally of three types of following techniques [33]:
& Polyionic complexes& Encapsulated systems& Combination of polyionic and encapsulated systems
There are many types of nonviral vectors used to delivergenes by using the above mentioned techniques (Fig. 3).Beside polymeric and lipidic system of gene delivery, thereare other systems considered as novel nonviral, nonbacterialgene delivery systems including vesicles prepared as a partof cellular cycle like exosomes or bacterial ghost which aredevoid of cytoplasmic processes.
Table 1 Types of viruses used in viral gene delivery
Virus Packagingcapacity(kilobase)
Host range Advantages Disadvantages
Adeno-associatedvirus
Low <4 kb Broad, infects bothdividing andnon-dividing cells
Long-term expression, requireadenovirus co-infection,currently in clinical trials,low immunogenicity
Slow expression onset, inefficientlarge-scale virus production/manufacture, QC very difficult, safetyconcern of insertional mutagenesis,repeat dosing affected by neutralizingantibody responses
Adenovirus Medium <7.5 kb Broad, low transductionof neurons
Transient expression Strong immunogenicity, manufacture,storage, and QC are moderately difficult
Alpha virus Medium <7.5 kb Broad, neuron and glialcell-specific strains
Transient but extreme,expression level, lowimmunogenicity
–
Herpes simplexvirus
High <30 kb Broad, neurons, stemcells, muscle cells
Long-term expression, lowtoxicity , natural latency inneurons, very large packingcapacity
Latent infection, Highly infectious, earlygenerations are cytotoxic
Lentivirus Medium <8 kb Broad, dividing andnon-dividing cells
Genome integration, long-termexpression, transfecthaematopoietic stem cells
Production inefficient, moderately hightiters possible, no clinical experience
Retrovirus Medium <8 kb Restricted, dividingcells only
Genome integration, long termexpression, transfect, onlytransduces dividing cells
Transfect only proliferative cells, safetyconcern of insertional mutagenesis,manufacture, storage, and QC areextremely difficult
0.00% 5.00% 10.00% 15.00% 20.00% 25.00%
Unknown, 3.3%
Other categories, 6.3%
Herpes simplex virus, 3.1%
Lentivirus, 3.2%
Poxvirus, 4.9%
Adeno-associated virus, 5.1%
Lipofection, 5.7%
Vaccinia virus, 1.2%
Naked/Plasmid DNA, 18%
Retrovirus, 19.4%
Adenovirus, 23.20%Fig. 2 Vectors used in genetherapy clinical trials worldwide[26]
Drug Deliv. and Transl. Res.

Nonviral biological vectors
Progresses in gene delivery systems lead to the investigationof advanced biological origin vesicles having inborn activityto target particular cells in body or organelle in cells. Twomajor vesicles that have been tried are bacterial ghost andexosomes.
Bacterial ghost One disadvantage of viruses is their capacityto encapsulate foreign antigens or DNA is restricted [34].This disadvantage can be solved by using a larger biologicalvesicle system having approximately all the advantages of aviral delivery system like high transduction, better targeting,advanced level of gene expression, etc. These biologicalvessels are “bacteria.” Bacterial ghosts are a novel nonlivingbut biological vaccination technology platform, which isbased on the conditional expression of the lethal lysis geneE from bacteriophage in gram-negative bacteria [35]. Thecytoplasmic content is expelled through a cellular envelopevia a newly formed transmembrane tunnel through a bacte-rial cellular envelope, resulting in an empty bacterial cellenvelope. Bacterial ghosts retain all morphological, structur-al, and antigenic features of the cell wall and can be used asvaccine candidate per se. Thomas et al. evaluated the capac-ity ofMannheimia haemolytica ghosts as delivery system forDNA vaccines [31]. Their studies showed that bacterialghosts loaded with a plasmid carrying the green fluorescentprotein-encoding gene are efficiently taken up by APC,thereby leading to high transfection rates (52–60 %) andbetter expression. Subsequent in vivo vaccination studies inbalb/c mice demonstrated that M. haemolytica ghost-mediated DNA delivery by intradermal or intramuscularroute of a eukaryotic expression plasmid encoding forbgalactosidase under the control of a cytomegalovirus pro-moter stimulated more efficient antigen-specific humoraland cellular (CD4+ and CD8+) immune responses than
naked DNA. The results were certainly encouraging consid-ering the primary role of dendritic cells as antigen-presentingcell (APC). Bacterial ghosts not only target the DNAvaccineconstruct to APCs but also provide a strong danger signal byacting as natural adjuvants (being inactivated whole cellbacteria), thereby promoting efficient maturation and activa-tion of dendritic cells. Thus, bacterial ghosts constitute apromising technology platform for the development of moreefficient DNAvaccines. Similarly, bacterial ghosts were usedfor targeting macrophages by Paukner et al. [30]. Theyshowed that around 6,000 plasmids that can be loaded intobacterial core lead to better expression in macrophages incontrast to viruses that can load only a small piece of geneticmaterial.
Exosomes The term “exosome” was coined by Rose Johnstone in the late 1970s because the vesicles she found emerg-ing from sheep reticulocytes structurally resembledendosomes, only these particular ones were exiting materialrather than entering [36]. Exosomes evade clearance by themononuclear phagocyte system (which clears circulatingparticles >100 nm in size), maximizing their circulation timeand underlining their superiority in (systemic) intercellularcommunication. Primarily, they were regarded as waste dis-posal vesicles of cells, but later on, they were found to have arole in aiding release of antigens from B cells [37]. The mostintriguing property of exosomes, from gene delivery point ofview, is their horizontal gene transfer [38]. Exosomes arecategorized as nanoparticle based on size and can be packedwith proteins, RNA/miRNAs, and lipids [39]. Erviti et al.studied the role of exosome in siRNA delivery [40].Bioengineered exosomes expressing exosomal membraneprotein and fused to the neuron-specific RVG peptide wereloaded with exogenous siRNA by electroporation. Theseexosomes, when injected intravenously, delivered siRNAspecifically to neurons, microglia, oligodendrocytes in the
Methods for Gene delivery
• Retroviruses • Adenoviruses• Adeno-associated virus • Lentivirus
Biological vesicles•Bacterial Ghost•Exosomes
Non viral-non bacterial
Viral mediated methods
Non viral mediated methods
Polymeric Gene Delivery Lipidic Gene Delivery System
• Cationic polymers(DEAE-dextran,poly(L-lysine), poly(ethyleneimine), PAMAM, Chitosan
• Anionic polymer (PLGA, algenicacid)• Cyclodextrin
• DOTAP• DDAB• DOTMA• lipofectamine
Inorganic Nanoparticle• Calcium Phosphate• Gold• Iron •Silica etc.
Fig. 3 Methods explored forgene delivery consisting viraland nonviral-mediated method
Drug Deliv. and Transl. Res.

brain, resulting in a specific gene knockdown. Exosomes as asiRNA delivery agent have a multitude of advantages overthe typical viruses, lipid nanoparticles, and polycationic de-livery agents currently in use. Most importantly, they delivertheir cargo directly into the cytosol, bypassing the need forendosomal escape, while their inertness avoids attack andclearance in the extracellular environment [41].
Inorganic nonviral gene delivery
With the advancement in area of inorganic nanoparticlesentrapping biomolecules, they exhibited its diversity andpotential applications in many frontiers of modern materialscience. Their advantages over organic nanoparticle madethem more versatile and robust to use as nonviral genedelivery vectors. These advantages include biocompatibility,microbial attack resistance wide availability, rich functional-ity, ability to modify release, and easy preparation. Inorganicnanoparticles have an interesting potential as DNA carriersystem due to the possibility to tailor their surface reactivityand electrical surface potential that can be obtained by sur-face modification. The chemistry of inorganic nanoparticlesis highly advanced; therefore, many classes of inorganicnanoparticles have been used as carriers comprising calciumphosphate, carbon nanotubes, silica, gold, quantum dots,magnesium phosphate, double hydroxides (anionic clays),etc. Of these inorganic compounds, the most commonly usedsalt is calcium phosphate, whose complexes with DNAwerepart of research from very long. Calcium ions are known toform ionic complexes with the helical phosphates of DNA,and these complexes have easy transportability across thecell membrane via ion-channel-mediated endocytosis.Kakizawa et al. [42] reported the novel preparation route oforganic–inorganic hybrid nanocarriers entrapping siRNA,based on the self-assembly of the block aniomer, poly(eth-ylene glycol)-block-poly(methacrylic acid) with calciumphosphate crystals. These nanoconstructs showed similartransfection efficacy but increased silencing than RNAiFect.However, there in vivo use is not tolerable because of theirnontargeted behavior and particle size variations due toOstwald ripening [43].
Gold nanoparticles provide particularly attractive scaffoldsfor the creation of transfection agents. Gold colloids arebioinert, nontoxic, readily synthesized, functionalized, andhave anti-angiogenesis properties [44]. Gold nanoparticles(typical sizes, 10–20 nm) are easily taken up by cells. It wasrecently shown by that gold clusters effectively interact withDNA and can be used as anticancer agents. They also providea multifunctional platform for both therapeutic and diagnosticpurposes. The active application of gold nanoparticles centersprimarily on two key aspects that make them appropriate foruse in biological systems: (1) the intrinsic properties of thegold core and (2) the ability to tailor the functionality of the
surface. Numerous approaches exist for nanoparticle fabrica-tion, the most simplistic being the reduction of gold salts in thepresence of a reducing agent. This initiates the nucleation ofthe gold ions thus forming nanoparticles. To help preventaggregation, a stabilizing agent is often added during thesynthesis process. This method was first reported byTurkevich and later improved on by Frens [45, 46].
Gold nanoparticles were explored widely for gene delivery,and their advancements lead to usage of surface-modified goldnanoparticle for site-specific delivery. Hydrophobically mod-ified chitosan was used as a stabilizer for gold nanoparticleensuring physicochemical stability in aqueous medium atphysiological pH 7.4 [47]. It has been reported that lysinedendron-functionalized gold nanoparticles are 28-fold superi-or to polylysine in reporter gene expression [48]. Thus, byappropriate surface functionalization of gold nanoparticles, ahighly efficient and less toxic nonviral gene delivery vectormay be developed for various biomedical applications. Re-cently, Shan et al. reported a new gene delivery vector basedon dendrimer-entrapped gold nanoparticles with significantlyhigher gene transfection efficiency than that of dendrimerswithout gold nanoparticles entrapped [49]. These goldnanoparticles preserve the three-dimensional spherical mor-phology of dendrimers, allowing for more efficient interactionbetween dendrimers and DNA. Similarly, Yuhan et al. dem-onstrated the controlled synthesis of polyethyleneimine (PEI)-coated gold nanoparticles using catechol-conjugated PEI forsiRNA delivery. Since the catechol groups are reductive andmoderately hydrophobic, catechol-conjugated PEI formedspherical multi-cored micelles in aqueous solution and servedas reductive templates for the growth and synthesis of spher-ical gold nanoparticle with tunable sizes and surface charges[50]. Recently, gold nanorods were explored for better genetransfection in gene delivery. Gold nanorods have strongabsorption bands in the near-infrared region, in which lightpenetrates deeply into tissues. The absorbed light energy isconverted into heat by gold nanorods, the so-calledphotothermal effect. This photothermal effect works in asynergistic fashion with encapsulated or complexed nucleicacid. Yamashita et al. explored the use of these gold nanorodsfor the stimuli-controlled gene delivery. When the double-stranded DNA-modified gold nanorods were irradiated bynear-infrared light, the single-stranded DNA (ssDNA) wasreleased from gold nanorods due to the photothermal effect[51]. The amount of released ssDNAwas dependent upon thepower and exposure time of light irradiation.
Mesoporous silica materials are typically synthesizedthrough polymerization of silica species on surfactant tem-plate in basic or acidic conditions. Their precise shape,morphology, pore size, biocompatible nature, and the versa-tility of various chemical surface modifications extend theapplications of mesoporous silica in the field of gene deliv-ery [52–56]. Since the discovery of mesoporous M41S
Drug Deliv. and Transl. Res.

family by the Mobil Oil Company in 1992 [57], severalnovel types of mesoporous silica materials such as SBA-15, MCM-48, and MSU have been developed by usingtemplate-assisted method [58]. The strategy of encapsulatingnucleic acid into the mesoporous enables mesoporous silicananoparticles (MSNs) to be an attractive material for genedelivery. Many researchers have demonstrated that MSN canbe efficiently endocytosed in vitro by a variety of mamma-lian cells including cancer (HeLa, CHO, lung, PANC-1),non-cancer (neural glia, liver, endothelial), macrophages,and stem cells (3TL3, mesenchymal) [59–61]. MSNs arealso being used as hybrid systems of other polymers or lipidsto exploit advantages of both the components. Li et al. de-scribed a magnetic MSNs based, polyelectrolyte (PEI) andfusogenic peptide (KALA) functionalized siRNA deliverysystem, which was highly effective for initiating target genesilencing both in vitro and in vivo and hence inhibited tumorgrowth in vivo [62]. Similarly, Xia et al. covalently attachedPEI on the surface of MSN to increase their efficiency ofintracellular delivery [63].
Nonviral nonbacterial vectors
Nonviral vector systems, combining cationic lipids and poly-mers, suggest potential routes for compacting DNA for sys-temic delivery [64]. In contrast to dissimilar viral analoguesthat have advanced means to conquer cellular barriers andimmune defense mechanisms, nonviral gene carriers con-stantly exhibit considerably reduced transfection efficiencyas they are obstructed by numerous extra and intracellularobstacles. However, biocompatibility and potential for large-scale production make these compounds increasingly attrac-tive for gene therapy [65]. As a result, a significant amount ofresearch in the past decade has focused on designing cationic
compounds that can form complexes with DNA and canavoid both in vitro and in vivo barriers for gene delivery. Inthe following sections, cationic lipids and polymers, barriersfor nonviral gene delivery will be discussed, and the currentstrategies for overcoming these obstacles will be illustratedby compound class.
Cationic lipid-based gene delivery Among the different ap-proaches to drug delivery, lipid-based colloidal carrier sys-tems have attracted much attention for gene delivery. Lipidshave always been considered as the first choice in nonviralgene delivery due to their similarity with the cell wall constit-uents. But it was in 1987 when Felgner et al. first accounted adouble chain monovalent quaternary ammonium lipid,N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammoniumchloride (DOTMA) that effectively attaches and trans-ports DNA to cultured cells [66]. Similarly, a numberof new cationic lipids have been developed [67]. These lipidsdiffer with each other by the number of charges present in theirhydrophilic head group and by the detailed structure of theirhydrophobic moiety (Fig. 4). Even though some cationic lipidsalone possess good transfection activity, they are often formu-lated as liposomes with a noncharged phospholipid or choles-terol. After combining with cationic liposomes, pDNA iscompacted into small quasi-stable particles called lipoplexesand is well shielded from nuclease degradation. Lipoplexestrigger cellular uptake and assist the release of DNA from theintracellular vesicles before reaching lysosomal compartments.
Lipid-mediated gene delivery starts from characterizationof lipoplexes prepared in low-salt solution followed by trans-fection to cells in the absence of serum. In these conditions,the transfection efficiency of lipoplexes is influenced by thechemical structure of the cationic lipid, the charge ratiobetween the cationic lipid and the DNA, the structure and
DOTMA
Dioctadecylamido-glycylspermine (DOGS)
DOPE
DC-Chol
Fig. 4 Cationic lipids for genedelivery
Drug Deliv. and Transl. Res.

proportion of the helper lipid in the complexes, the size andstructure of the liposomes, the total amount of the lipoplexesused, and the cell type. The initial four factors define thestructure, charge property, and transfection activity of thelipoplexes, whereas the remaining two describe the overalltoxicity to the treated cells, and the sensitivity of the cells to aparticular lipid-based transfection reagent. The chemicalstructure of the cationic lipids mainly affects the transfectionefficiency. Generally, multivalent lipids with long and unsat-urated hydrocarbon chains are more efficient than monovalentcationic lipids with the same hydrophobic chains. Transfec-tion normally requires that the lipoplexes have net positivecharges on the surface, and for this, the concentration ofcationic lipids should be more than the DNA. Spontaneousaddition of cationic lipids and cellular lipids in the membraneof the endocytic vesicles is vital to the endosome-releasingprocess [68]. When a nonbilayer-forming lipid such asdioleoylphosphatidylethanolamine (DOPE; a helper lipid)and bilayer-forming lipid such as dioleoylphosphatidylcholineare spontaneously mixed in endosomes, it leads to endosomelysis [69]. DOPE incorporation increases membrane fluidity,aids lipid exchange and membrane fusion between lipoplexesand the endosomal membrane. A high local concentration ofDOPE, having tough propensity to shape an inverse hexago-nal phase, may form a nonbilayer lipid structure and causemembrane perturbation and endosome destruction [70]. Thisdifference between intrinsic transfection activity of multiva-lent lipids and helper lipids indicates that multivalent lipidswork on different mechanism [71, 72]. Frequently, these cat-ionic lipopolyamines having protonable amine groups actual-ly absorb protons to retard the acidification process inside theendosomes and stopping endosome lysosome transition. It hasalso been suggested that a local destabilization effect of somemicelle-forming cationic lipids on the endosomal membrane’sintegrity is part of the underlying mechanism of lipid-basedgene delivery.
Lipoplexes are formed when cationic liposomes are mixedwith DNA. This involves electrostatic interaction betweenpolycationic liposomes and polyanionic DNA followed by aslower lipid rearrangement process [73]. The easiest method ofpreparation of lipoplexes is mixing of diluted solutions ofpDNA with preformed liposomes. Lipoplexes formed in thisway are usually heterogeneous in size and morphology. Theprime reason of this heterogeneity is the relatively large sizes ofDNA and liposomes and the multivariant nature of the interac-tion between the DNA and liposomes. Other methodsconcerning types of lipid assembly other than liposomes havebeen proposed to overcome these problems. Slow dialysismethod for lipoplex preparation has also been designed. Themethod involves DNA compaction in mixed micelles compris-ing of cationic lipid and nonionic detergent and exclusion of thedetergent by dialysis [74]. Below the critical micelle concentra-tion of single chain cationic lipids, DNA collapses into
unimolecular lipid DNA nanoparticles of smaller size (20–30 nm). Smaller particles are good for in vivo gene deliverybecause they clear slowly from blood, and therefore, they ap-proach the target cells well. Conjugating these small-sized com-plexes with polyethylene glycol (PEG) and targeting ligands ontheir surface makes it possible to make target-specific genecarriers [75]. Most of the cationic lipids are having excellenttransfection activity in cell culture, but majority of them do notact upon well in the presence of serum, and only a few are activein vivo [67]. When lipoplexes are exposed to the overwhelmingamount of negatively charged and often amphipathic proteinsand polysaccharides that are present in blood, mucus, epitheliallining fluid, or tissue matrix, their size, surface charge, and lipidcomposition change. Lipoplexes when administered in vivointeract with negatively charged blood components and formlarge aggregates that could be absorbed onto the surface ofcirculating red blood cells, trapped in a thick mucus layer, orembolized in microvasculatures, preventing them from reachingthe intended target cells in the distal location, while someundergo dissolution after introducing to the blood circulation[76, 77]. Govindarajan et al. studied a combination of magneticnanoparticles together with cationic lipid N,N-di-n-hexadecyl-N,N-dihydroxyethylammonium chloride formulated with a co-lipid cholesterol. Upon magnetofection, these nanoparticles en-hanced DNA uptake into human glioblastoma-astrocytoma,epithelial-like cell line U-87 MG, hepatocellular carcinomaHep G2, cervical cancer HeLa, and breast cancer MDA-MB-231 cells [78]. Layek et al. prepared caproic acid-grafted chito-san cationic nanocomplexes for enhanced gene delivery.Caproic acid substitution resulted in a 2-7-fold higher cellularuptake in HEK 293 cells and higher gene expression withoutaffecting biocompatibility [79].
Cationic polymer-based gene delivery Synthetic and naturalcationic polymers are another class of DNA carriers thathave been used for gene delivery. Poly-L-lysine is basicallythe first group of cationic polymers used in gene transferin vivo [80]. A large number of polymers such as cationicpolymers (linear or branched), neutral and anionic (Fig. 5)have been discovered as carriers for in vitro and in vivo genedelivery. These include polyethylenimine (PEI) [81],polyamidoamine [82, 83], polypropylamine dendrimers[84], cationic dextran [85], chitosan [86, 87], cationic pro-teins (polylysine, protamine, and histones) [88], and cationicpeptides [89]. Most of the cationic polymers condense DNAinto small particles and assist cellular uptake through endo-cytosis by charge–charge interaction with anionic sites oncell surfaces; their transfection activity and toxicity vary con-siderably. PEI is the most widely studied polymer for genedelivery. Jean Paul Behr’s group first introduced PEI as anefficient and economic synthetic polymeric gene transfer agent[72]. In PEI-mediated transfection, DNA to PEI ratios, themolecular weight and configuration of PEI, the concentration
Drug Deliv. and Transl. Res.

of DNA and polymer, and the ionic strength of the solvent forpreparation are main factors for estimating the physical prop-erties and transfection activity of the DNA/PEI complexes(polyplex). Both linear PEI and branched PEI have brillianttransfection activities in vitro and modest transfection activityin vivo. Linear PEI is apparently less toxic to cells thanbranched one. Linear PEI polyplexes are less compacted anddissociate more efficiently than branched PEI polyplexes.Studies have also revealed that linear PEI polyplex entersthe nucleus more easily than branched PEI polyplex [90].Surface modification of PEI with PEG reduces the surfacecharge and the tendency to form large aggregates in thepresence of serum. Ligand conjugation with PEI, such astransferrin, hyaluronic acid, antibody, avidin or sugar moie-ties, also provides covering to polyplexes and the possibilityof target-specific gene delivery [91].
One of the successful classes of newly designed biode-gradable cationic polymers like aminoesters have beendesigned to reduce the toxicity associated with cationicpolymer-based gene delivery systems such as PEI andpoly-L-lysine [92]. Studies also revealed that branchedpolyaminoesters are chemically more stable than linear onescontaining primary amine groups [93, 94].
Cationic polymers formed from low molecular weightpolyamines having disulfide bond linkages have shown goodtransfection efficiency and comparatively low toxicity. A“proton sponge” hypothesis explained the mechanism ofcationic polymer-based gene delivery, particularly genetransfer by PEI and its derivatives [95]. Not many lipids or
natural cationic polymers like polylysine, histone, and chi-tosan are able to trigger endosome rupture to much extent,but they can transfect cells in vitro and in vivo, yet PEI is anexception. Cationic peptides have been recently explored asa carrier for gene delivery. An arginine-rich peptide fromprotamine was able to transfect cells in vitro as effectively asPEI, whereas the protein as a whole showed minimal activity[89]. Synthetic peptides from HIV TAT protein containing adimer of arginine-rich peptides were found to be active intransfection. Lysine-rich peptides from histone H2A [96],anti-DNA antibodies [97] can also transport genes into cells.Short cationic peptides can also act as membrane penetratingmolecules bring into cells a lot of substances ranging fromsmall molecules and proteins to nanoparticles, though theexact mechanism of transport across the cell membrane is notcompletely understood. A nonviral gene delivery systemusually encounters two types of barrier namely extracellularand intracellular.
Arduous path of nonviral gene delivery vectors
Extracellular barriers to the systemic delivery of nucleicacids put nonviral gene delivery in the most quandary pathhaving many troubles. The nonviral gene delivery systemsmust show low toxicity, dodge the adaptive immune system,minimize interactions with plasma proteins, extracellularmatrices, and nontargeted cell surfaces (nonself-interactions)
Fig. 5 Polymers used in genedelivery including cationic,neutral, and anionic polymers
Drug Deliv. and Transl. Res.

and aggregation (self-interactions). Efforts to design nonviralgene delivery systems with these characteristics are ongoing.These barriers are explained in Fig. 6.
Extracellular hurdles for nonviral gene delivery
Toxicity
A serious snag of nonviral gene delivery systems is thetoxicity of carrier and vectors untargeted delivery. Polymersused in preparing these carriers are cytotoxic. This is becauseof their charge and interaction with the cell organelles. Muchcurrent work is involved in preparing carriers that have lowertoxicity. For example, evidence shows that low molecularweight preparations of polycations such as chitosan, PEI,and grouping chitosan with PEI have been evaluated andwere found to be less toxic [98]. Different grades of PEI arealso investigated, and it is reported that linear PEI are lesscytotoxic then branched ones [99] but have less transfectionratio. However, low molecular weight fractions gave a hightransfection ratio as well as low toxicity [100]. Hwang et al.also reported that β-cyclodextrin-containing polymers aresignificantly less toxic than high molecular weightpolycations both in cultured cells and in animals, and thedistance between charge centers along the backbone of apolycation is responsible for such toxicity [101]. Recently,there is a shift of scientist’s interest towards nontoxic, bio-degradable, biocompatible polymers like poly (DL-lactide-co-glycolide) (PLGA) nano/microparticles [102, 103]. Theyare also pursued as nonviral gene delivery system with greatzeal for their obvious advantages [104]. Numerous in vivostudies have demonstrated that both single-walled carbonnanotubes (SWCNTs) and multi-walled carbon nanotubes,
when instilled into the lungs of rodents, have the potential tocause inflammation, fibrosis (scarring of the lungs), andgranuloma (small nodule) formation in the lung tissue [105,106]. Warheit et al. evaluated the acute lung toxicity ofintratracheally instilled SWCNT in rats. Pulmonary expo-sures to SWCNT in rats produced a nondose-dependentseries of multifocal granulomas, which were evidence of aforeign tissue body reaction [107]. Anna et al. demonstratedthat pharyngeal aspiration of SWCNT elicited unusual pul-monary effects in C57BL/6 mice that combined a robust butacute inflammation with early onset yet progressive fibrosisand granulomas [108]. Inorganic nanoparticles are biocom-patible, but even then, there are many findings where theyare considered toxic. Their toxicity and distribution are de-pendent upon particle size, concentration as well as thecharge on the particle [109–111]. However, some groupsreported nontoxicity of gold nanoparticles to human cells[112]. Silica nanoparticles have high affinity for phospho-lipids on the surface of nanoparticles [113], whereasmesopourous silica nanoparticle were considered consider-ably nontoxic than silica nanaoparticles because of theirmesopourous structure [113–115].
Stability
The stabilization of nonviral gene delivery particles is im-portant for extended circulation times that are required totarget particular cell types (especially in case of non-targetedparticle which depends on enhanced permeations rate effect).Highly charged particles rapidly aggregate in high ionicstrength solutions, therefore the decrease in the protectiveelectrostatic double layer. Additionally, a strong positivecharge on the particles facilitates nonspecific interactions
Fig. 6 Barriers in nonviral genedelivery A Route ofadministration of nucleic acidtherapeutic, Breticuloendothelial system(RES) uptake, C nonspecificdistribution of administerednucleic acid, D unnecessaryaggregation with oppositecharged proteins present insystemic circluation, E enzymesin systemic circulation degradenaked nucleic acid, F cellmembrane restricts the entry ofnegatively charged nucleic acid,G denaturation of nucleic acidin acidic pH of endosomes, Htransport of nucleic acid fromcytoplasm to nucleus isrestricted through high densityof cytoplasm and lower poresize of nucleus membrane
Drug Deliv. and Transl. Res.

with the extracellular matrix, cell surfaces, and plasma pro-teins (all negatively charged) and leads to cytotoxicity inother cells which eventually signals apoptosis and increasein size of particle, whereas strong negative charge can causeRES uptake by phagocytosis via the macrophage polyanionreceptor [116]. The common method to protect nanocarriersfrom the reticulo-endothelial system consists of coating thesurface of the particles with PEG, a procedure calledPEGylation. To contribute to the “stealth” characteristics ofPEGylated nanoparticles, there are three important factors,(1) the molecular weight of the PEG chain, (2) the surfacechain density, and (3) the conformation. The coating of PEGchains to the surface of nanoparticles results in an increase inthe blood circulation half-life by several orders of magni-tude. By creating a hydrophilic protective layer around thenanoparticles, steric repulsion forces repel the absorption ofopsonin proteins, thereby blocking and delaying theopsonization process [117]. This process improves the sta-bility of lipoplexes or polyplexes by decreasing self-non-selfand nonspecific interactions, for example the PEGylation ofpolyplexes by the conjugation of PEG before particle forma-tion [118], covalent grafting of PEG on the nanoparticlesurfaces [119]. Particle formation or association of PEG viainclusion complex formation with the surface of β-cyclodextrin-containing polyplexes [120] resulted in its in-creased stability against aggregation and reduced non-self-interactions, without much disruption of polyplex morphol-ogy [121, 122]. These cross-linked particles do not revealsufficient gene expression. Hence, cross-linking chemistrythat provides intracellular degradation is necessary in suchcases. Davis and colleagues [123] have prepared polyplexeswith degradable cross-linking moieties. A similar strategyhas also recently been investigated by Gosselin and co-workers [124]. Additionally, control of the surface chargeof stabilized particles will play a major role in minimizingnonspecific interactions. Davis [125] reported preparation ofstabilized polyplexes where the charge can be tuned. It isreported that PLGA and chitosan impart stability by encap-sulating DNA in matrix [126, 127] and by changing theanionic charge on gene encapsulating PLGA nanoparticleby surface modification [69].
Distribution
Targeting the distribution of gene at the site of action isanother major obstruction in nonviral gene delivery system.It is known that only minority of pDNA cure or prevent afterinjection into host, and the fate of majority remains unclear.pDNA being hydrophilic in nature tends to get partitioned inblood compartment. Zhang et al. reported similar observa-tion and showed that concentration of pDNA was high inblood, but the majority of plasmid was degraded, and itsmaximum part of it existed in circular form rather than
supercoiled form [128]. The in vivo degradation rate ofsupercoiled pDNA was 20.9 % in 10 min, 34.1 % in 1 h,86.8 % in 1 day, and 97.8 % in 1 week in sera. The secondproposed reason for resulting in much less pDNA in othertissues than in circulating blood was the excretion of theinjected plasmids in urine and feces through blood circula-tion. In case of pDNA vaccines, I.M. route is considered asthe intended route for administration, and it was confirmedby same group that the majority of DNA was detected inmuscles where DNAwas injected. pDNA vaccine long-termexistence in muscles was detectable at 1 week after I.M.injection, especially in local injected sites as an antigen pool,and it was also reported that the pDNA existence was longerin lymphatic organs than in other tissues except injectedmuscles. Hengge et al. reported that the distribution andsafety of intracutaneously injected pDNA were assessed ina relevant animal model, and mRNA expression only tookplace in the skin, regional lymph nodes, and muscular tissues[129]. However, nanotechnology can be effectively used totarget distribution of requisite gene at desired site of action.
The challenge of the targeting is multiple:
& To find the proper target for a particular disease& To find the drug that effectively treats this disease and& To find how to carry the drug
The specific targeting of nanocarriers leads to better phar-macokinetics and pharmacodynamics profiles, controlledand sustained release of drugs, an improved specificity, anincreased internalization, and more importantly lower sys-temic toxicity. The specific targeting consists of “passivetargeting” and “active targeting”; however, the activetargeting process cannot be separated from the passive be-cause it occurs only after passive accumulation in tumors[130]. Passive targeting is based on Enhanced Permeabilityand Retention (EPR) effect as discovered by Maeda et al.The EPR effect is now becoming the gold standard in cancer-targeting drug designing to supersede the barrier ofunavailability of gene at site of action [131, 132]. The EPReffect will be optimal if nanocarriers can evade immunescrutiny and circulate for a long period. Due to high EPReffect, very high local concentrations of drug-loadednanocarriers can be achieved at the tumor site, for instance10–50-fold higher than in normal tissue within 1–2 days[133]. To this end, the nanocarriers should possess threeimportant properties. First is the size of the nanoparticle.The ideal nanocarrier size should be somewhere between10 and 100 nm. Indeed, for efficient extravasation from thefenestrations in leaky vasculature, nanocarriers should bemuch less than 400 nm. On the other hand, to avoid thefiltration by the kidneys, nanocarriers need to be larger than10 nm; and to avoid a specific capture by the liver,nanocarriers need to be smaller than 100 nm. Second factoris the charge on the surface of nanoparticle. The charge of the
Drug Deliv. and Transl. Res.

particles should be neutral or anionic for efficient evasion ofthe renal elimination. The last property is stealth character-istic. The nanocarriers must be hidden from the reticulo-endothelial system, which destroys any foreign materialthrough opsonization followed by phagocytosis [134]. Con-sidering all these factors, an efficient delivery system has tobe designed. For example, carbon nanotubes having desiredsize are being exploited for EPR-assisted tumor targeting,but they are still at research level owing to their high carci-nogenicity [135].
In active targeting, targeting ligands are attached at thesurface of the nanocarrier for binding to appropriate recep-tors expressed at the target site. The ligand is chosen to bindto a receptor overexpressed by tumor cells or tumor vascu-lature and not expressed by normal cells, e.g., folate recep-tors [136], epidermal growth factor receptor [137], andplatelet-derived growth factor [138].
To explore the maximum potential of ligand-associatedactive targeting, dual targeting has been used by many re-searchers. A dual aptamer-based targeting system was devel-oped by Ban’s group for delivery of doxorubicin to bothprostate-specific membrane antigen (PSMA) (+) and PSMA(−) prostate cancers by means of an A10 RNA aptamer,which recognizes PSMA (+) prostate cancer cells, and aDUP-1 (a new prostate carcinoma binding peptide) peptideaptamer, which targets PSMA (−) prostate cancer cells, re-spectively. Streptavidin links both the targeting ligands, anddrugs were loaded onto the stem region of the A10 aptamer.As a result, the dual-targeting system showed selective de-livery and induced apoptosis of both PSMA (+) and PSMA(−) cells [139]. Nie et al. used the similar dual-targetingmechanism by attaching both RGD peptide and transferringtargeting peptide B6 with PEGylated, PEI-based polyplexes[140]. This dual-targeting system showed 60- and 20-foldincreases of the reporter gene expression on DU145 and PC3cells, respectively, when compared to control nontargetedpolyplexes. Inorganic nanoparticles have high distributionconcerns because of their nonbiodegradable nature. Terentyuket al. reported that silica gold nanoparticles having particlesize around 15 nm were observed in all organs with rathersmooth distribution over the liver, spleen, and blood, and theyalso observed size-dependent degeneration and morphologi-cal changes in accumulating organ [141].
pH and enzymes
Genes are susceptible to degradation because of alkaline pHand enzymes like nucleases DNases, etc. Hence, pH andenzymes play a key role in selection of vehicle for DNA inwhich DNA should remain stable. Cationic polyplexes arenot only smaller in size but also protect DNA from degrada-tion in serum. Many cationic polymers like PEI, poly-L-lysine, chitosan, protamine, and other cationic lipids can
compact the anionic DNA. Their size lies in the range offew nanometers depending upon n/p ratio (nitrogen in poly-mer to phosphate) in DNA. In general, it was observed thatlower n/p ratio results in more compact DNA, but also in-creases toxicity and is difficult in intracellular dissociation[142]. Dunlap and colleagues estimated that the size ofpolyplex formed between branched 25-kDa PEI or linear22-kDa PEI and DNA ranged from 20 to 40 nm when thecharge ratio was 1.6 [143]. Generally with PEI 25 kDa, N/Pratio 0.4 or above this gives good protection from enzymeand pH in vitro and in vivo [144]. A better protection for theDNA against degradation by cellular nucleases strongly de-pends on the polymer/DNA ratio, and could also account forthe higher efficiency of positively charged lipoplex [145].
The enhanced protection against nucleases has been at-tributed to a possible DNA collapse and condensation atcertain N/P ratio. Indeed, accessibility of ethidium bromideto DNA decreased with increasing N/P ratios [146]. Multi-valent lipids seem more efficient than monovalent ones inreducing this accessibility [147]. In other models such as themultilamellar model, covering of DNA strands with lipidbilayers may account for the observed protection againstnucleases [148].
Intracellular barriers
Due to the large size and lipophilic nature, the cytoplasmicmembrane is typically impermeable to nucleic acids. Manyefforts have been made to facilitate internalization of nucleicacids. One of the strategies that involve internalization of nucleicacids by physical means, i.e., electroporation, ultrasound-basedsonoporation, the use of a gene gun, and microinjection. Allphysical approaches enable nucleic acid molecules to penetrateinto cells by transiently percolating through the cellular mem-brane. However, because of their invasive nature and potentialdamage to the structure of cells, as well as poor access to deepertissues limit their use in in vivo gene delivery [149].
Nanoparticle can enter cells either by charge-mediatedinteractions with proteoglycans on cell membranes or byreceptor-mediated endocytosis (RME) by ligand receptorbinding interactions. Both methods result in uptake of vesi-cles that ultimately deliver the content to lysosomes. Intra-cellular barriers can therefore be confronted in all of theprocesses of vesicles uptake, vesicle escape, trafficking tothe nucleus, vector unpackaging, and nuclear entry that arerequired for gene delivery.
Vesicle uptake
Endocytosis is the main process by which eukaryotic cellsabsorb macromolecules, whereas, in the case of large particles,phagocytosis is the main pathway for internalization of parti-cles. There are two different mechanisms for internalization,
Drug Deliv. and Transl. Res.

namely fluid phase endocytosis and RME occurring in areceptor-independent and receptor-dependent manner, respec-tively. Unlike fluid phase endocytosis, RME involves complexintracellular signaling following binding of a high affinityligand. Charge-dependent or receptor-mediated endocytosisoccurs through clathrin-dependent endocytosis [150]. Chargeon the particle is another important criterion of internalization.The cell membrane is negatively charged, and hence, it repelsnegative charged particles, but positive charged particles getinternalized due to strong electrostatic attractions. Hence, cat-ionic particles are highly internalized in cell than neutral andanionic particles. Endocytosis is strongly a size-dependent pro-cess, where internalization of nanospheres with a diameter<200 nm involved clathrin-coated pits, and with particles of∼500 nm in size, the caveolae-mediated internalization becomesthe predominant pathway of entry for particles [151, 152].
Delivery of a wide range of cargos including nucleic acidshas been demonstrated using cell-penetrating peptides (CPP)[153]. CPPs including TAT peptide have been successfullyused for intracellular delivery of a broad variety of carriersincluding various nano-particulate pharmaceutical carriers (li-posomes, micelles, nanoparticles). TAT peptide, the most fre-quently used CPP, is derived from the transcriptional activatorprotein encoded by human immunodeficiency virus type 1[154]. TAT can be covalently coupled with nanocarrier ornoncovalently with DNA for better uptake [155]. Recently,Arthnari et al. showed increase in transfection and gene silenc-ing by sh RNA noncovalently attached fused HIV-TAT peptideand cationic membrane active peptide LK15 (Tat–LK15) [156].
Champion et al. came up with an interesting finding. Theystudied the role of target geometry in phagocytosis andreported a surprising finding that particle shape, not size,plays a dominant role in phagocytosis [157]. Particlesrepresenting six distinct geometric shapes, spheres (radius1.0–12.5 μ), oblate ellipsoids (major axis 4 μ, aspect ratio 4),prolate ellipsoids (major axis 2–6 μ, aspect ratio 1.3–3),elliptical disks (major axis 3–14 μ, aspect ratio 2–4, thick-ness 400–1,000 nm), rectangular disks (major axis 4–8 μ,aspect ratio 1.5–4.5), and UFOs (sphere radius 1.5 μ, ringradius 4 μ) were fabricated. They found that all shapes werecapable of initiating phagocytosis.
Endosomal escape
Acidity and enzyme load of endosome are increased afterfusing of endosome with lysosomes (pH∼5 to 2). This in-crease in acidity renders enclosed DNA to degrade. Manyattempts have been made to increase the DNA escape fromearly endosome state. One approach involves usingfusogenic lipids or peptides, such as GALA and hyaluronicacid (HA-2) from influenza haemagglutinin, to disrupt theendosome membrane. The neutral lipid DOPE or DOTMAare generally employed as a fusogenic helper lipid in
lipoplex. A cationic counterpart of GALA is the peptideKALA which is formed by substitution of the alanine ofGALA with lysine and a decrease in content of glutamicacid. But membrane lytic activity was retained by theKALA/DNA complex, leading to gene transfection in dif-ferent cell lines [158, 159]. Later, it was observed by Wanget al. that the MCF-7-specific phage fusion protein bears pH-sensitive, fusogenic property required for endosomal escape.Modifying liposomes with the phage fusion protein enablescytoplasmic delivery of a liposome carried drug [160].
There is a second approach which includes the use of pH-sensitive liposomes and PEI as gene transfer agents, havinghigh buffering capacity and the flexibility to swell; whenprotonated, these systems lead to rupturing of endosomalmembrane [27]. Because of the great number of secondaryamines, PEI behaves as a “proton sponge,” able to buffer thelow pH in the apical endosomes, resulting in the inhibition ofthe low pH-activated nucleases. This leads to a large increasein the ionic concentration inside the endosome, finallyresulting in osmotic swelling due to water intake and ruptureof the organelles [95]. Recently, Agarwal et al. demonstratethat siRNA-carrying dendriworms can be readily internal-ized by cells and enable endosomal escape across a widerange of loading doses, whereas dendrimers or nanowormsalone are inefficient [161]. Panyam et al. demonstrated an-other principle of endosomal lysis in PLGA. They showedthat PLGA surface undergoes charge reversal in endosomewhich becomes responsible for endosomal lysis [162]. Wanget al. reported an important endosome escape property ofMSNs; later, they found a significant correlation betweenRNA interference silencing and MSN’s endosome escapecapability [163]. After endosomal escape and cytoplasmicrelease of nonviral vectors, nucleic acids should be deliveredto their intracellular targets to obtain desired biological ef-fects [149].
Cytoplasmic barriers
The cytoplasm is highly dense because of high concentrationof proteins (100 mg/ml), and cytoskeleton imposes signifi-cant molecular crowding of the cytoplasm. The cytoskeletonis responsible for the mechanical resistance of the cell, aswell as the cytoplasmic transport of organelles and largecomplexes as ribonucleotide particles [164]. Because of highdense nature of cytoplasm, it becomes difficult for macro-molecule to diffuse in cytoplasm. Dowty et al. were the firstto discover that the diffusional mobility of pDNA was neg-ligible in the cytoplasm of microinjected myotubes [165].pDNA remained predominantly at the site of microinjectionduring incubation at 37 °C, suggesting that the cytoplasmsieving could represent an impediment to gene transfer. Inanother study by Lukacs et al., they quantitate and determinethe diffusion mobility of FITC-conjugated pDNA (3–6 kb)
Drug Deliv. and Transl. Res.

and DNA fragments (20 bp–2 kb) in living cells, by usingspot photobleaching technique [166]. Following the micro-injection of FITC-labeled DNA into the cytoplasm or thenucleus, a 0.4-Å diameter spot was bleached with a high-intensity laser beam, and the time course of fluorescencerecovery was recorded. Nucleic acids larger than 2 kb havea very limited mobility and were virtually immobile duringthe course of the measurements (few minutes) in the cyto-plasm of HeLa cells. Diffusion of larger DNA fragmentsbecame remarkably slower in the cytoplasm. Diffusion of250 and 2,000 base pair fragments in cytoplasm was 17 and100 times slower, respectively, than diffusion in water. Therestricted mobility of pDNA relative to that of dextran withsimilar molecular mass could be explained by molecularcrowding, immobile cytoplasmic obstacles, or associationof the nucleic acids with cytosolic DNA-binding proteins.
Dissociation of DNA complex
A gene can only express itself if it can get separated from itscargo. Most of the separation happens at the time of endo-some lysis, where both DNA and polymer separately comeout. Sometime generally in the case of scaffolds or hybridDNA particles where the polyplexes or lipoplexes are en-capsulated in another polymer’s matrix. In such case, thepolyplexes do not come in direct contact with endosome, andtheir release occurs in cytoplasm [167]. Hence, there shouldbe some cell machinery which separates the cargo intracel-lularly. Okuda et al. demonstrated the presence of DNA-releasing factor in cytosol fraction having ability to dissoci-ate DNA and polymer complex [168]. Furthermore, thisDNA-releasing ability of the cytosolic fraction was proteasesensitive. In another experiment, Iida et al. have reportedmany proteins responsible for segregation of polyplex due toformation of protein complex with DNA [169]. Proteins thatare bound to polyplex have various molecular weights andisoelectric points. Actin and beta-tubulin are among thesecytoskeletal proteins. Other proteins were very specific, met-abolic enzymes which are very abundant in cytoplasm, butthey didn’t show quantitative correlation with bounded pro-tein with polyplex. However, they could not clarify roles ofthese proteins for intracellular gene transfer and whetherproteins positively or negatively affected gene expression.In addition, Itaka et al. have observed dissociation of linearPEI polyplex, branched PEI polyplex, poly-L-lysine polyplexin the cytoplasm by single cell observation using fluorescenceresonance energy transfer of double-labeled pDNA [170].pDNA complexed with LPEI underwent a rapid escape fromthe endosomes, spreading uniformly into the cytoplasm with asubstantial decrease in fluorescence resonance energy transfer(FRET) efficiency due to the disintegration of linear PEIpolyplex structure. pDNA complexed with branched PEI alsoachieved a rapid escape from the endosomes. Nevertheless, the
pDNA retained high FRETefficiency even after 24 h, indicatingan appreciable stability of the branched PEI polyplex to keeppDNA in a condensed state. In the PLL polyplexes, neitherendosome escape nor pDNA decondensation was observed.
Nuclear import
The nuclear envelope separates the genetic material of thecell from the surrounding cytoplasm. It also represents aphysical barrier for nuclear import of macromolecules suchas pDNA. Whenever DNA is used as a macromoleculardrug, it is utmost important for DNA to enter inside nucleusfor expression, thus making nuclear import a rate-limitingstep. During nonviral gene transfer, exogenous DNA entersinto the nucleus at the time of cell division, i.e., when thenuclear envelope breaks down. Generally, the transport ofproteins of size 40–45 kDa easily occurs through nuclearpore complexes (NPCs) having diameter of 9 nm. A fullyrelaxed, circular plasmid has a size of around 10 nm or more;thus, this suggests that the passive transport of plasmid innuclear is unlikely through NPC [171]. Several attempts toimprove entry of pDNA into the nucleus have been pub-lished. These include use of electrostatic binding of DNA tocationic NLS containing proteins [172, 173] or lipids [174],as well as sequence-specific binding of DNA to karyophilicproteins [175]. Yet such “piggyback” nuclear transport relieson the unpredictable stability of the complexes within thecytoplasm. Recent work by the group of Wolff [176] withdigitonin-permeabilized cells demonstrated nuclear accumu-lation of fluorescently labeled DNA that was randomlytagged with hundreds of NLS peptides, but nuclei of intactcells did not take up the modified DNA. Recently, Yang at el.[177] reported the role of PEI in activation and translocationof NF-КB from cytoplasm to nucleus, which is critical forNF-КB-dependent gene expression. However, in anotherresearch by a different group, the NF-КB inhibition by PEI-based/p65 shRNA complex in the inhibition of metastasis andgrowth of breast cancer was reported [178]. Kim et al. [179]used gold nanoparticle and reported gold nanoparticles asuniversal nanoparticle in nuclear delivery of genetic material.
Conclusion
Gene therapy is a relatively new paradigm in medicine withenormous therapeutic potential. However, a number of wide-ly reported adverse events have focused attention on associ-ated risks ahead of the exciting therapeutic progress beingmade. Nonviral vector-mediated gene delivery, compared toviral vector-mediated gene delivery, is a promising tool forsafe and effective delivery of therapeutic DNA in genetic aswell as acquired diseases. A viable nonviral gene vector forsystemic delivery depends on its capacity to bypass a series
Drug Deliv. and Transl. Res.

of physiological barriers and on its efficiency in carryingnucleic acids to a targeted site within a cell. Optimization ofnonviral gene delivery has so far mostly focused on design ofparticulate carriers that are endowed with desirable mem-brane targeting, internalization, and endosomal escape prop-erties. Comparatively less attention has been paid to under-stand and exploit the factors driving nonviral vectors intoone of the variably attractive endocytic pathways. The con-cept of a multifunctional delivery system helps to bring asolution to the problems associated with successive barriers.This review highlighted the barrier involved in gene deliveryand stimulates interest in a number of strategies that haveemerged and provide tactics for synergistic integration ofmultiple functionalities of different gene delivery vectors.
Acknowledgments RS and IS are thankful to the Department ofPharmaceuticals, Ministry of Chemicals and Fertilizers for the awardof PhD fellowship. The authors are also thankful to the Project Director,NIPER for encouragement. The work was supported by the financialassistance from CSIR, under the project ADD (CSC 0302).
Conflict of interest All authors declare no conflict of interest.
References
1. Ali M, Lemoine N, Ring CJ. The use of DNA viruses as vectorsfor gene therapy. Gene Ther. 1994;1(6):367.
2. Culver KW, Michael Blaese R. Gene therapy for cancer. TrendsGenet. 1994;10(5):174–8.
3. Cantor CR. How will the Human Genome Project improve ourquality of life? Nat Biotechnol. 1998;16(3):212–3.
4. SioudM. Therapeutic siRNAs. Trends Pharmacol Sci. 2004;25(1):22–8.
5. Stull RA, Szoka J, Francis C.Antigene, ribozyme and aptamer nucleicacid drugs: progress and prospects. Pharm Res. 1995;12(4):465–83.
6. Kohn DB, Parkman R. Gene therapy for newborns. FASEB J.1997;11(8):635–9.
7. Seidman MM, Glazer PM. The potential for gene repair via triplehelix formation. J Clin Invest. 2003;112(4):487–94.
8. Braasch DA, Corey DR. Novel antisense and peptide nucleicacid strategies for controlling gene expression. Biochemistry.2002;41(14):4503–10.
9. De Fougerolles A, Vornlocher H-P, Maraganore J, Lieberman J.Interfering with disease: a progress report on siRNA-based thera-peutics. Nat Rev Drug Discov. 2007;6(6):443–53.
10. Dykxhoorn DM, Lieberman J. The silent revolution: RNA inter-ference as basic biology, research tool, and therapeutic. Annu RevMed. 2005;56:401–23.
11. Lysik MA, Wu-Pong S. Innovations in oligonucleotide drug de-livery. J Pharm Sci. 2003;92(8):1559–73.
12. Uprichard SL. The therapeutic potential of RNA interference.FEBS Lett. 2005;579(26):5996–6007.
13. Baker BF, Monia BP. Novel mechanisms for antisense-mediated reg-ulation of gene expression. Biochim Biophys Acta. 1999;1489(1):3.
14. Crooke ST. Molecular mechanisms of action of antisense drugs.Biochim Biophys Acta. 1999;1489(1):31–43.
15. Patil SD, Rhodes DG, Burgess DJ. DNA-based therapeutics andDNA delivery systems: a comprehensive review. AAPS J.2005;7(1):61–77.
16. Bertrand JR, Pottier M, Vekris A, Opolon P, Maksimenko A,Malvy C. Comparison of antisense oligonucleotides and siRNAsin cell culture and in vivo. Biochem Biophys Res Commun.2002;296(4):1000–4.
17. Vacik J, Dean B, Zimmer W, Dean D. Cell-specific nuclear importof plasmid DNA. Gene Ther. 1999;6(6):1006.
18. Schaffer DV, Lauffenburger DA. Optimization of cell surfacebinding enhances efficiency and specificity of molecular conju-gate gene delivery. J Biol Chem. 1998;273(43):28004–9.
19. Nishikawa M, Takakura Y, Hashida M. Theoretical considerationsinvolving the pharmacokinetics of plasmid DNA. Adv Drug DelivRev. 2005;57(5):675–88.
20. Domb AJ, Khan W. Biodegradable polymers as drug carriersystems. Polymeric Biomaterials. Boca Raton: CRC Press; 2013.p. 135–76.
21. Khan W, Hosseinkhani H, Ickowicz D, Hong PD, Yu DS, DombAJ. Polysaccharide gene transfection agents. Acta Biomater.2012;8(12):4224–32.
22. Khan W, Muthupandian S, Domb AJ. Cationic polymers for thedelivery of therapeutic nucleotides. In: Peer D, editor.Nanotechnology for the delivery of therapeutic nucleic acids.8 Temasek Boulevard: Pan Stanford Publishing; 2013. p. 27–56.
23. Khan W, Muthupandian S, Farah S, Kumar N, Domb AJ.Biodegradable polymers derived from amino acids. MacromolBiosci. 2011;11(12):1625–36.
24. Robbins PD, Ghivizzani SC. Viral vectors for gene therapy.Pharmacol Ther. 1998;80(1):35–47.
25. Naldini L. Viral vectors for gene therapy: the art of turning infec-tious agents into vehicles of therapeutics. Nat Med. 2001;7(1):33.
26. Ginn SL, Alexander IE, Edelstein ML, Abedi MR, Wixon J. Genetherapy clinical trials worldwide to 2012—an update. J GeneMed.2013;15(2):65–77.
27. Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D,Demeneix B, et al. A versatile vector for gene and oligonucleotidetransfer into cells in culture and in vivo: polyethylenimine. ProcNatl Acad Sci U S A. 1995;92(16):7297–301.
28. Koping-Hoggard M, Varum K, Issa M, Danielsen S, ChristensenB, Stokke B, et al. Improved chitosan-mediated gene deliverybased on easily dissociated chitosan polyplexes of highly definedchitosan oligomers. Gene Ther. 2004;11(19):1441–52.
29. Martin B, Sainlos M, Aissaoui A, Oudrhiri N, Hauchecorne M,Vigneron J-P, et al. The design of cationic lipids for gene delivery.Curr Pharm Des. 2005;11(3):375–94.
30. Paukner S, Kudela P, Kohl G, Schlapp T, Friedrichs S, Lubitz W.DNA-loaded bacterial ghosts efficiently mediate reporter genetransfer and expression in macrophages. Mol Ther. 2005;11(2):215–23.
31. Ebensen T, Paukner S, Link C, Kudela P, de Domenico C, LubitzW, et al. Bacterial ghosts are an efficient delivery system for DNAvaccines. J Immunol. 2004;172(11):6858–65.
32. Tan A, Rajadas J, Seifalian AM. Exosomes as nano-theranosticdelivery platforms for gene therapy. Adv Drug Deliv Rev. 2013;65:357–67.
33. Wong SY, Pelet JM, Putnam D. Polymer systems for gene delivery—past, present, and future. Prog Polym Sci. 2007;32(8):799–837.
34. Lundstrom K. Latest development in viral vectors for gene ther-apy. Trends Biotechnol. 2003;21(3):117–22.
35. Eko FO, Witte A, Huter V, Kuen B, Fürst-Ladani S, Haslberger A,et al. New strategies for combination vaccines based on the extendedrecombinant bacterial ghost system. Vaccine. 1999;17(13):1643–9.
36. Pan BT, Johnstone RM. Fate of the transferrin receptor duringmaturation of sheep reticulocytes in vitro: selective externalizationof the receptor. Cell. 1983;33(3):967–78.
37. Raposo G, Nijman HW, Stoorvogel W, Liejendekker R, HardingCV, Melief C, et al. B lymphocytes secrete antigen-presentingvesicles. J Exp Med. 1996;183(3):1161–72.
Drug Deliv. and Transl. Res.

38. Zomer A, Vendrig T, Hopmans ES, van Eijndhoven M,Middeldorp JM, Pegtel DM. Exosomes: fit to deliver smallRNA. Commun Integr Biol. 2010;3(5):447–50.
39. Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyorsof immune responses. Nat Rev Immunol. 2009;9(8):581–93.
40. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ.Delivery of siRNA to the mouse brain by systemic injection oftargeted exosomes. Nat Biotechnol. 2011;29(4):341–5.
41. van den Boorn JG, Schlee M, Coch C, Hartmann G. SiRNA deliverywith exosome nanoparticles. Nat Biotechnol. 2011;29(4):325.
42. Kakizawa Y, Furukawa S, Ishii A, Kataoka K. Organic–inorganichybrid-nanocarrier of siRNA constructing through the self-assembly of calcium phosphate and PEG-based block aniomer. JControl Release. 2006;111(3):368–70.
43. Uskokovic V, Uskokovic DP. Nanosized hydroxyapatite and othercalcium phosphates: chemistry of formation and application asdrug and gene delivery agents. J Biomed Mater Res B ApplBiomater. 2011;96(1):152–91.
44. Mukherjee P, Bhattacharya R, Wang P, Wang L, Basu S, Nagy JA,et al. Antiangiogenic properties of gold nanoparticles. Clin CancerRes. 2005;11(9):3530–4.
45. Turkevich J, Stevenson PC, Hillier J. A study of the nucleationand growth processes in the synthesis of colloidal gold. DiscussFaraday Soc. 1951;11:55–75.
46. Frens G. Controlled nucleation for the regulation of the particle sizein monodisperse gold suspensions. Nature. 1973;241(105):20–2.
47. Bhattarai SR, Remant Bahadur KC, Aryal S, Bhattarai N, Kim SY,Yi HK. Hydrophobically modified chitosan/gold nanoparticles forDNA delivery. J Nanoparticle Res. 2008;10(1):151–62.
48. Ghosh PS, Kim C-K, Han G, Forbes NS, Rotello VM. Efficientgene delivery vectors by tuning the surface charge density ofamino acid-functionalized gold nanoparticles. ACS Nano.2008;2(11):2213–8.
49. Shan Y, Luo T, PengC, ShengR, Cao A, CaoX, et al. Gene deliveryusing dendrimer-entrapped gold nanoparticles as nonviral vectors.Biomaterials. 2012;33(10):3025.
50. Lee Y, Lee SH, Kim JS, Maruyama A, Chen X, Park TG.Controlled synthesis of PEI-coated gold nanoparticles using re-ductive catechol chemistry for siRNA delivery. J Control Release.2011;155(1):3–10.
51. Yamashita S, Fukushima H, Akiyama Y, Niidome Y, Mori T,Katayama Y, et al. Controlled-release system of single-strandedDNA triggered by the photothermal effect of gold nanorods and itsin vivo application. Bioorg Med Chem. 2011;19(7):2130–5.
52. Du L, Song H, Liao S. Tuning the morphology of mesoporoussilica by using various template combinations. Appl Surf Sci.2009;255(23):9365–70.
53. Kim TW, Chung PW, Lin VSY. Facile synthesis of monodispersespherical MCM-48 mesoporous silica nanoparticles with con-trolled particle size. Chem Mater. 2010;22(17):5093–104.
54. ChenQ,HanL,GaoC,CheS. Synthesis ofmonodispersedmesoporoussilica spheres (MMSSs) with controlled particle size using geminisurfactant. Microporous Mesoporous Mater. 2010;128(1):203–12.
55. Chiang YD, Lian HY, Leo SY, Wang SG, Yamauchi Y, Wu KCW.Controlling particle size and structural properties of mesoporoussilica nanoparticles using the Taguchi method. J Phys Chem C.2011;115(27):13158–65.
56. Kortesuo P, Ahola M, Karlsson S, Kangasniemi I, Yli-Urpo A,Kiesvaara J. Silica xerogel as an implantable carrier for controlleddrug delivery—evaluation of drug distribution and tissue effectsafter implantation. Biomaterials. 2000;21(2):193–8.
57. Kresge C, Leonowicz M, Roth W, Vartuli J, Beck J. Orderedmesoporous molecular sieves synthesized by a liquid-crystal tem-plate mechanism. Nature. 1992;359(6397):710–2.
58. Wan Y, Zhao D. On the controllable soft-templating approach tomesoporous silicates. Chem Rev. 2007;107(7):2821.
59. Slowing I, Trewyn BG, Lin VSY. Effect of surface functionalizationof MCM-41-type mesoporous silica nanoparticles on the endocyto-sis by human cancer cells. J AmChem Soc. 2006;128(46):14792–3.
60. Lu J, LiongM, Zink JI, Tamanoi F. Mesoporous silica nanoparticlesas a delivery system for hydrophobic anticancer drugs. Small.2007;3(8):1341–6.
61. Slowing II, Trewyn BG, Lin VSY. Mesoporous silica nanoparticlesfor intracellular delivery of membrane-impermeable proteins. J AmChem Soc. 2007;129(28):8845–9.
62. Li X, Chen Y, Wang M, Ma Y, Xia W, Gu H. A mesoporous silicananoparticle–PEI–Fusogenic peptide system for siRNA deliveryin cancer therapy. Biomaterials. 2013;34(4):1391–401.
63. Xia T, Kovochich M, Liong M, Meng H, Kabehie S, George S,et al. Polyethyleneimine coating enhances the cellular uptake ofmesoporous silica nanoparticles and allows safe delivery ofsiRNA and DNA constructs. ACS Nano. 2009;3(10):3273–86.
64. El-Aneed A. An overview of current delivery systems in cancergene therapy. J Control Release. 2004;94(1):1–14.
65. Behr JP. Synthetic gene-transfer vectors. Acc Chem Res.1993;26(5):274–8.
66. Felgner PL, Gadek TR, Holm M, Roman R, Chan HW, Wenz M,et al. Lipofection: a highly efficient, lipid-mediated DNA-transfectionprocedure. Proc Natl Acad Sci U S A. 1987;84(21):7413–7.
67. Liu D, Ren T, Gao X. Cationic transfection lipids. Curr MedChem. 2003;10(14):1307–15.
68. Xu Y, Szoka Jr FC. Mechanism of DNA release from cationicliposome/DNA complexes used in cell transfection. Biochemistry.1996;35(18):5616–23.
69. Farhood H, Serbina N, Huang L. The role of dioleoyl phosphati-dylethanolamine in cationic liposome mediated gene transfer.Biochim Biophys Acta. 1995;1235(2):289–95.
70. El Ouahabi A, Thiry M, Pector V, Fuks R, Ruysschaert J,Vandenbranden M. The role of endosome destabilizing activityin the gene transfer process mediated by cationic lipids. FEBSLett. 1997;414(2):187–92.
71. Litzinger DC, Huang L. Phosphatidylethanolamine liposomes:drug delivery, gene transfer and immunodiagnostic applications.Biochim Biophys Acta. 1992;1113(2):201–27.
72. Behr JP, Demeneix B, Loeffler JP, Perez-Mutul J. Efficient genetransfer into mammalian primary endocrine cells with lipopolyamine-coated DNA. Proc Natl Acad Sci U S A. 1989;86(18):6982–6.
73. de Pedroso de Lima MC, Simoes S, Pires P, Faneca H, DuzgünesN. Cationic lipid–DNA complexes in gene delivery: from biophysicsto biological applications. Adv Drug Deliv Rev. 2001;47(2):277–94.
74. Hofland H, Shephard L, Sullivan SM. Formation of stable cationiclipid/DNA complexes for gene transfer. Proc Natl Acad Sci U S A.1996;93(14):7305–9.
75. Dauty E, Remy J-S, Zuber G, Behr J-P. Intracellular delivery ofnanometric DNA particles via the folate receptor. BioconjugChem. 2002;13(4):831–9.
76. Li S, Tseng W, Stolz DB, Wu S, Watkins S, Huang L. Dynamicchanges in the characteristics of cationic lipidic vectors afterexposure to mouse serum: implications for intravenouslipofection. Gene Ther. 1999;6(4):585.
77. Simberg D, Weisman S, Talmon Y, Faerman A, Shoshani T,Barenholz Y. The role of organ vascularization and lipoplex-serum initial contact in intravenous murine lipofection. J BiolChem. 2003;278(41):39858–65.
78. Govindarajan S, Kitaura K, Takafuji M, Ihara H, Varadarajan K,Patel AB et al. Gene delivery into human cancer cells by cationiclipid-mediated magnetofection. Int J Pharm. 2013; 446(1-2): 87–99
79. Layek B, Singh J. Caproic acid grafted chitosan cationicnanocomplexes for enhanced gene delivery: effect of degree ofsubstitution. Int J Pharm. 2013; 447(1-2):182–91.
80. Wu GY,Wu CH. Receptor-mediated gene delivery and expressionin vivo. J Biol Chem. 1988;263(29):14621–4.
Drug Deliv. and Transl. Res.

81. Goula D, Remy J, Erbacher P, Wasowicz M, Levi G, Abdallah B,et al. Size, diffusibility and transfection performance of linear PEI/DNA complexes in the mouse central nervous system. Gene Ther.1998;5(5):712.
82. Tang MX, Redemann CT, Szoka Jr FC. In vitro gene delivery bydegraded polyamidoamine dendrimers. Bioconjug Chem.1996;7(6):703–14.
83. Rudolph C, Lausier J, Naundorf S, Muller RH, Rosenecker J. Invivo gene delivery to the lung using polyethylenimine and frac-tured polyamidoamine dendrimers. J Gene Med. 2000;2(4):269–78.
84. Schatzlein AG, Zinselmeyer BH, Elouzi A, Dufes C, ChimYTA, Roberts CJ, et al. Preferential liver gene expression withpolypropylenimine dendrimers. J Control Release. 2005;101(1):247–58.
85. Hosseinkhani H, Azzam T, Tabata Y, Domb A. Dextran–sperminepolycation: an efficient nonviral vector for in vitro and in vivogene transfection. Gene Ther. 2004;11(2):194–203.
86. Leong K, Mao H, Roy K, Walsh S, August J. DNA-polycationnanospheres as non-viral gene delivery vehicles. J ControlRelease. 1998;53(1–3):183.
87. Erbacher P, Zou S, Bettinger T, Steffan AM, Remy JS. Chitosan-based vector/DNA complexes for gene delivery: biophysical char-acteristics and transfection ability. Pharm Res. 1998;15(9):1332–9.
88. Balicki D, Beutler E. Histone H2A significantly enhances in vitroDNA transfection. Mol Med. 1997;3(11):782.
89. Park YJ, Liang JF, Ko KS, Kim SW, Yang VC. Low molecularweight protamine as an efficient and nontoxic gene carrier: in vitrostudy. J Gene Med. 2003;5(8):700–11.
90. Wightman L, Kircheis R, Rössler V, Carotta S, Ruzicka R, KursaM,et al. Different behavior of branched and linear polyethyleniminefor gene delivery in vitro and in vivo. J Gene Med. 2001;3(4):362–72.
91. Kichler A. Gene transfer with modified polyethylenimines. J GeneMed. 2004;6(S1):S3–S10.
92. Akinc A, Anderson DG, Lynn DM, Langer R. Synthesis ofpoly(beta-amino ester) optimized for highly effective gene deliv-ery. Bioconjug Chem. 2003;14(5):979–88.
93. Lim YB, Han SO, Kong HU, Lee Y, Park JS, Jeong B, et al.Biodegradable polyester, poly[alpha-(4-aminobutyl)-L-glycolicacid], as a non-toxic gene carrier. Pharm Res. 2000;17(7):811–6.
94. Yb L, Sm K, Suh H, Park JS. Biodegradable, endosome disruptive,and cationic network-type polymer as a highly efficient and nontoxicgene delivery carrier. Bioconjug Chem. 2002;13(5):952–7.
95. Sonawane ND, Szoka FC, Verkman A. Chloride accumulation andswelling in endosomes enhances DNA transfer by polyamine-DNApolyplexes. J Biol Chem. 2003;278(45):44826–31.
96. Balicki D, Putnam CD, Scaria PV, Beutler E. Structure and func-tion correlation in histone H2A peptide-mediated gene transfer.Proc Natl Acad Sci U S A. 2002;99(11):7467–71.
97. Avrameas A, Ternynck T, Nato F, Buttin G, Avrameas S.Polyreactive anti-DNA monoclonal antibodies and a derived pep-tide as vectors for the intracytoplasmic and intranuclear translocationof macromolecules. Proc Natl Acad Sci U S A. 1998;95(10):5601–6.
98. Ouji Y, Yoshida-Terakura A, Hayashi Y, Maeda I, Kawase M,Yamato E, et al. Polyethyleneimine/chitosan hexamer-mediatedgene transfection into intestinal epithelial cell cultured in serum-containing medium. J Biosci Bioeng. 2002;94(1):81–3.
99. Lungwitz U, Breunig M, Blunk T, Göpferich A. Polyethylenimine-based non-viral gene delivery systems. Eur J Pharm Biopharm.2005;60(2):247–66.
100. Werth S, Urban-Klein B, Dai L, Hobel S, Grzelinski M,Bakowsky U, et al. A low molecular weight fraction ofpolyethylenimine (PEI) displays increased transfection efficiencyof DNA and siRNA in fresh or lyophilized complexes. J ControlRelease. 2006;112(2):257–70.
101. Hwang SJ, Bellocq NC, Davis ME. Effects of structure of β-cyclodextrin-containing polymers on gene delivery. BioconjugChem. 2001;12(2):280–90.
102. Jensen DMK, Cun D, Maltesen MJ, Frokjaer S, Nielsen HM,Foged C. Spray drying of siRNA-containing PLGA nanoparticlesintended for inhalation. J Control Release. 2010;142(1):138–45.
103. Wang H, Zhao P, Su W, Wang S, Liao Z, Niu R, et al. PLGA/polymeric liposome for targeted drug and gene co-delivery.Biomaterials. 2010;31(33):8741–8.
104. Raghavachari N, Fahl WE. Targeted gene delivery to skin cellsin vivo: a comparative study of liposomes and polymers as deliv-ery vehicles. J Pharm Sci. 2002;91(3):615–22.
105. Chou CC, Hsiao HY, Hong QS, Chen CH, Peng YW, Chen HW,et al. Single-walled carbon nanotubes can induce pulmonary in-jury in mouse model. Nano Lett. 2008;8(2):437–45.
106. Lam CW, James JT, McCluskey R, Hunter RL. Pulmonary toxic-ity of single-wall carbon nanotubes in mice 7 and 90 days afterintratracheal instillation. Toxicol Sci. 2004;77(1):126–34.
107. Warheit DB, Laurence B, Reed KL, Roach D, Reynolds G, WebbT. Comparative pulmonary toxicity assessment of single-wallcarbon nanotubes in rats. Toxicol Sci. 2004;77(1):117–25.
108. Shvedova AA, Kisin ER, Mercer R, Murray AR, Johnson VJ,Potapovich AI, et al. Unusual inflammatory and fibrogenic pul-monary responses to single-walled carbon nanotubes in mice. AmJ Physiol Lung Cell Mol Physiol. 2005;289(5):L698–708.
109. Goodman CM, McCusker CD, Yilmaz T, Rotello VM. Toxicity ofgold nanoparticles functionalized with cationic and anionic sidechains. Bioconjug Chem. 2004;15(4):897–900.
110. Chen Y-S, Hung Y-C, Liau I, Huang GS. Assessment of the in vivotoxicity of gold nanoparticles. Nanoscale Res Lett. 2009;4(8):858–64.
111. Lin Y-S, Haynes CL. Impacts of mesoporous silica nanoparticlesize, pore ordering, and pore integrity on hemolytic activity. J AmChem Soc. 2010;132(13):4834–42.
112. Connor EE, Mwamuka J, Gole A, Murphy CJ, Wyatt MD. Goldnanoparticles are taken up by human cells but do not cause acutecytotoxicity. Small. 2005;1(3):325–7.
113. Pera H, Kleijn JM, Leermakers FA. Interaction of silica nanoparticleswith phospholipid membranes. Chem Lett. 2012;41(10):1322–4.
114. Slowing II, Vivero-Escoto JL, Wu C-W, Lin VS-Y. Mesoporoussilica nanoparticles as controlled release drug delivery and genetransfection carriers. Adv Drug Deliv Rev. 2008;60(11):1278–88.
115. Slowing II, Wu CW, Vivero–Escoto JL, Lin VSY. Mesoporoussilica nanoparticles for reducing hemolytic activity towards mam-malian red blood cells. Small. 2009;5(1):57–62.
116. Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G,Szewczyk A. A two-stage poly (ethylenimine)-mediated cytotoxicity:implications for gene transfer/therapy. Mol Ther. 2005;11(6):990–5.
117. Owens DE, Peppas NA. Opsonization, biodistribution, and pharma-cokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102.
118. Merkel OM, Librizzi D, Pfestroff A, Schurrat T, Buyens K,Sanders NN, et al. Stability of siRNA polyplexes from poly(ethylenimine) and poly (ethylenimine)-g-poly (ethylene gly-col) under in vivo conditions: effects on pharmacokinetics andbiodistribution measured by Fluorescence Fluctuation Spectroscopyand Single Photon Emission Computed Tomography (SPECT) im-aging. J Control Release. 2009;138(2):148–59.
119. Kircheis R, Blessing T, Brunner S, Wightman L, Wagner E.Tumor targeting with surface-shielded ligand–polycation DNAcomplexes. J Control Release. 2001;72(1):165–70.
120. Liu F, Huang L. Development of non-viral vectors for systemicgene delivery. J Control Release. 2002;78(1):259–66.
121. Dash PR, Read ML, Fisher KD, Howard KA, Wolfert M, OupickyD, et al. Decreased binding to proteins and cells of polymeric genedelivery vectors surface modified with a multivalent hydrophilic
Drug Deliv. and Transl. Res.

polymer and retargeting through attachment of transferrin. J BiolChem. 2000;275(6):3793–802.
122. Trubetskoy VS, Loomis A, Slattum PM, Hagstrom JE, BudkerVG, Wolff JA. Caged DNA does not aggregate in high ionicstrength solutions. Bioconjug Chem. 1999;10(4):624–8.
123. Supramolecular complexes containing therapeutic agents. WOPatent WO/2000/033,885; 2000.
124. Gosselin MA, Guo W, Lee RJ. Efficient gene transfer usingreversibly cross-linked low molecular weight polyethylenimine.Bioconjug Chem. 2001;12(6):989–94.
125. Davis ME. Non-viral gene delivery systems. Curr OpinBiotechnol. 2002;13(2):128–31.
126. Csaba N, Sanchez A, Alonso M. PLGA: poloxamer and PLGA:poloxamine blend nanostructures as carriers for nasal gene deliv-ery. J Control Release. 2006;113(2):164–72.
127. Nafee N, Taetz S, Schneider M, Schaefer UF, Lehr C-M.Chitosan-coated PLGA nanoparticles for DNA/RNA delivery:effect of the formulation parameters on complexation and transfec-tion of antisense oligonucleotides. Nanomedicine. 2007;3(3):173–83.
128. Zhang HY, Sun SH, Guo YJ, Chen ZH, Huang L, Gao YJ, et al.Tissue distribution of a plasmid DNA containing epitopes of foot-and-mouth disease virus in mice. Vaccine. 2005;23(48):5632–40.
129. Hengge UR, Dexling B, Mirmohammadsadegh A. Safety and phar-macokinetics of naked plasmid DNA in the skin: studies on dissem-ination and ectopic expression. J Invest Dermatol. 2001;116(6):979–82.
130. Bae YH. Drug targeting and tumor heterogeneity. J ControlRelease. 2009;133(1):2.
131. Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted deliv-ery of macromolecular drugs, including the EPR effect in solidtumor and clinical overview of the prototype polymeric drugSMANCS. J Control Release. 2001;74(1):47–61.
132. Maeda H, Bharate G, Daruwalla J. Polymeric drugs for efficienttumor-targeted drug delivery based on EPR-effect. Eur J PharmBiopharm. 2009;71(3):409.
133. Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhancedpermeability and retention effect for tumor targeting. Drug DiscovToday. 2006;11(17):812–8.
134. Gullotti E, Yeo Y. Extracellularly activated nanocarriers: a new par-adigm of tumor targeted drug delivery. Mol Pharm. 2009;6(4):1041–51.
135. Cheung W, Pontoriero F, Taratula O, Chen AM, He H. DNA andcarbon nanotubes asmedicine. AdvDrugDeliv Rev. 2010;62(6):633–49.
136. Low PS, Kularatne SA. Folate-targeted therapeutic and imagingagents for cancer. Curr Opin Chem Biol. 2009;13(3):256–62.
137. Acharya S, Dilnawaz F, Sahoo SK. Targeted epidermal growthfactor receptor nanoparticle bioconjugates for breast cancer ther-apy. Biomaterials. 2009;30(29):5737–50.
138. Prakash J, de Jong E, Post E, Gouw AS, Beljaars L, Poelstra K. Anovel approach to deliver anticancer drugs to key cell types intumors using a PDGF receptor-binding cyclic peptide containingcarrier. J Control Release. 2010;145(2):91–101.
139. Min K, Jo H, SongK, ChoM, ChunY-S, Jon S, et al. Dual-aptamer-based delivery vehicle of doxorubicin to both PSMA (+) and PSMA(−) prostate cancers. Biomaterials. 2011;32(8):2124–32.
140. Nie Y, Schaffert D, Rodl W, Ogris M, Wagner E, Gunther M.Dual-targeted polyplexes: one step towards a synthetic virus forcancer gene therapy. J Control Release. 2011;152(1):127–34.
141. Terentyuk GS, Maslyakova GN, Suleymanova LV, Khlebtsov BN,Kogan BY, Akchurin GG, et al. Circulation and distribution ofgold nanoparticles and induced alterations of tissue morphology atintravenous particle delivery. J Biophotonics. 2009;2(5):292–302.
142. Biri S, Stock F, Adib A, Erbacher P. Delivery of biomolecules withnon-viral vectors. Cells and Culture. 2010; 4:115–9.
143. Dunlap DD, Maggi A, Soria MR, Monaco L. Nanoscopic struc-ture of DNA condensed for gene delivery. Nucleic Acids Res.1997;25(15):3095–101.
144. Mullen PM, Lollo CP, Phan QC, Amini A, Banaszczyk MG,Fabrycki JM, et al. Strength of conjugate binding to plasmidDNA affects degradation rate and expression level in vivo.Biochim Biophys Acta. 2000;1523(1):103–10.
145. Xu Y, Hui S-W, Frederik P, Szoka Jr FC. Physicochemical char-acterization and purification of cationic lipoplexes. Biophys J.1999;77(1):341–53.
146. Miyata K, Kakizawa Y, Nishiyama N, Harada A, Yamasaki Y,Koyama H, et al. Block catiomer polyplexes with regulated den-sities of charge and disulfide cross-linking directed to enhancegene expression. J Am Chem Soc. 2004;126(8):2355–61.
147. Elouahabi A, Ruysschaert JM. Formation and intracellular traf-ficking of lipoplexes and polyplexes. Mol Ther. 2005;11(3):336–47.
148. Gershon H, Ghirlando R, Guttman SB, Minsky A. Mode offormation and structural features of DNA–cationic liposome com-plexes used for transfection. Biochemistry. 1993;32(28):7143–51.
149. Akita H, Ito R, Khalil I, Futaki S, Harashima H. Quantitativethree-dimensional analysis of the intracellular trafficking of plas-mid DNA transfected by a nonviral gene delivery system usingconfocal laser scanning microscopy. Mol Ther. 2004;9(3):443–51.
150. Rejman J, Bragonzi A, Conese M. Role of clathrin- and caveolae-mediated endocytosis in gene transfer mediated by lipo- andpolyplexes. Mol Ther. 2005;12(3):468–74.
151. Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependentinternalization of particles via the pathways of clathrin- andcaveolae-mediated endocytosis. Biochem J. 2004;377(Pt 1):159.
152. Prabha S, Zhou WZ, Panyam J, Labhasetwar V. Size-dependencyof nanoparticle-mediated gene transfection: studies with fraction-ated nanoparticles. Int J Pharm. 2002;244(1):105–15.
153. Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery oflarge molecules and small particles by cell-penetrating proteinsand peptides. Adv Drug Deliv Rev. 2005;57(4):637–51.
154. Vijayanathan V, Thomas T, Thomas T. DNA nanoparticles anddevelopment of DNA delivery vehicles for gene therapy.Biochemistry. 2002;41(48):14085–94.
155. Ko Y, HartnerW, Kale A, Torchilin V. Gene delivery into ischemicmyocardium by double-targeted lipoplexes with anti-myosin an-tibody and TAT peptide. Gene Ther. 2008;16(1):52–9.
156. Arthanari Y, Pluen A, Rajendran R, Aojula H, Demonacos C.Delivery of therapeutic shRNA and siRNA by Tat fusion peptidetargeting bcr–abl fusion gene in chronic myeloid leukemia cells. JControl Release. 2010;145(3):272–80.
157. Champion JA, Mitragotri S. Role of target geometry in phagocy-tosis. Proc Natl Acad Sci U S A. 2006;103(13):4930–4.
158. Min SH, Kim DM, Kim MN, Ge J, Lee DC, Park IY, et al. Genedelivery using a derivative of the protein transduction domainpeptide, K-Antp. Biomaterials. 2010;31(7):1858–64.
159. Mok H, Park TG. Self-crosslinked and reducible fusogenic peptidesfor intracellular delivery of siRNA. Biopolymers. 2008;89(10):881–8.
160. Wang T, Yang S, Petrenko VA, Torchilin VP. Cytoplasmic deliveryof liposomes into MCF-7 breast cancer cells mediated by cell-specific phage fusion coat protein. Mol Pharm. 2010;7(4):1149–58.
161. Agrawal A, Min DH, Singh N, Zhu H, Birjiniuk A, Von MaltzahnG, et al. Functional delivery of siRNA in mice using dendriworms.ACS Nano. 2009;3(9):2495–504.
162. Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V. Rapidendo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles:implications for drug and gene delivery. FASEB J. 2002;16(10):1217–26.
163. Wang M, Li X, Ma Y, Gu H. Endosomal escape kinetics ofmesoporous silica-based system for efficient siRNA delivery. IntJ Pharm. 2013; 448(1):51–7.
Drug Deliv. and Transl. Res.

164. Luby Phelps K. Cytoarchitecture and physical properties of cyto-plasm: volume, viscosity, diffusion, intracellular surface area. IntRev Cytol. 1999;192:189–221.
165. Dowty ME, Williams P, Zhang G, Hagstrom JE, Wolff JA.Plasmid DNA entry into postmitotic nuclei of primary ratmyotubes. Proc Natl Acad Sci U S A. 1995;92(10):4572–6.
166. Lukacs GL, Haggie P, Seksek O, Lechardeur D, Freedman N,Verkman A. Size-dependent DNA mobility in cytoplasm andnucleus. J Biol Chem. 2000;275(3):1625–9.
167. Zhang C, Yadava P, Hughes J. Polyethylenimine strategies forplasmid delivery to brain-derived cells. Methods. 2004;33(2):144–50.
168. Okuda T, Niidome T, Aoyagi H. Cytosolic soluble proteins induceDNA release from DNA–gene carrier complexes. J Control Release.2004;98(2):325–32.
169. Iida T, Mori T, Katayama Y, Niidome T. Overall interaction ofcytosolic proteins with the PEI/DNA complex. J Control Release.2007;118(3):364–9.
170. Itaka K, Harada A, Yamasaki Y, Nakamura K, Kawaguchi H,KataokaK. In situ single cell observation by fluorescence resonanceenergy transfer reveals fast intra-cytoplasmic delivery and easyrelease of plasmid DNA complexed with linear polyethylenimine.J Gene Med. 2004;6(1):76–84.
171. Tachibana R, Harashima H, Shinohara Y, Kiwada H. Quantitativestudies on the nuclear transport of plasmid DNA and gene expressionemploying nonviral vectors. AdvDrugDelivRev. 2001;52(3):219–26.
172. Dean DA, Dean BS, Muller S, Smith LC. Sequence requirementsfor plasmid nuclear import. Exp Cell Res. 1999;253(2):713–22.
173. Collas P, Alestrom P. Nuclear localization signals: a driving forcefor nuclear transport of plasmid DNA in zebrafish. Biochem CellBiol. 1997;75(5):633–40.
174. Remy JS, Kichler A, Mordvinov V, Schuber F, Behr JP. Targetedgene transfer into hepatoma cells with lipopolyamine-condensedDNA particles presenting galactose ligands: a stage toward artifi-cial viruses. Proc Natl Acad Sci U S A. 1995;92(5):1744–8.
175. Fominaya J, Wels W. Target cell-specific DNA transfer mediatedby a chimeric multidomain protein. Novel non-viral gene deliverysystem. J Biol Chem. 1996;271(18):10560–8.
176. Sebestyen MG, Ludtke JJ, Bassik MC, Zhang G, Budker V,Lukhtanov EA, et al. DNA vector chemistry: the covalentattachment of signal peptides to plasmid DNA. Nat Biotechnol.1998;16(1):80–5.
177. Ma YF, Yang YW. Delivery of DNA-based cancer vaccine withpolyethylenimine. Eur J Pharm Biopharm. 2010;40(2):75–83.
178. Xiao J, Duan X, Yin Q, Miao Z, Yu H, Chen C et al. The inhibitionof metastasis and growth of breast cancer by blocking the NF-κBsignaling pathway using bioreducible PEI-based/p65 shRNAcomplex nanoparticles. Biomaterials. 2013; 34(21):5381–90.
179. Kim DW, Kim JH, Park M, Yeom JH, Go H, Kim S, et al.Modulation of biological processes in the nucleus by delivery ofDNA oligonucleotides conjugated with gold nanoparticles.Biomaterials. 2011;32(10):2593–604.
Drug Deliv. and Transl. Res.




















