
Melatonin ameliorates ionizing radiation-induced oxidative organ damage in rats

2004;64:5398-5406. Cancer Res Daniel Zingg, Oliver Riesterer, Doriano Fabbro, et al. and Primary Endothelial Cells-Kinase/Akt Survival Pathway by Ionizing Radiation in Tumor
′Differential Activation of the Phosphatidylinositol 3
Updated version
http://cancerres.aacrjournals.org/content/64/15/5398
Access the most recent version of this article at:
Cited Articles
http://cancerres.aacrjournals.org/content/64/15/5398.full.html#ref-list-1
This article cites by 64 articles, 24 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/64/15/5398.full.html#related-urls
This article has been cited by 10 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

[CANCER RESEARCH 64, 5398–5406, August 1, 2004]
Differential Activation of the Phosphatidylinositol 3�-Kinase/Akt Survival Pathwayby Ionizing Radiation in Tumor and Primary Endothelial Cells
Daniel Zingg,1 Oliver Riesterer,1 Doriano Fabbro,2 Christoph Glanzmann,1 Stephan Bodis,1 and Martin Pruschy1
1Department of Radiation Oncology, University Hospital Zurich, Zurich, and 2Novartis Pharma, Inc., Department of Oncology Research, Basel, Switzerland
ABSTRACT
Ionizing radiation induces an intracellular stress response via activa-tion of the phosphatidylinositol 3�-kinase (PI3K)/Akt survival pathway. Intumor cells, the PI3K/Akt pathway is induced through activation ofmembers of ErbB receptor tyrosine kinases. Here, we investigated thereceptor dependence of radiation-induced PI3K/Akt activation in tumorcells and in endothelial cells. The integrity of both the ErbB and thevascular endothelial growth factor (VEGF) ligand-activated PI3K/Aktpathway in endothelial cells was demonstrated using specific ErbB andVEGF receptor tyrosine kinase inhibitors. Irradiation of endothelial cellsresulted in protein kinase B (PKB)/Akt activation in a similar time courseas observed in response to VEGF. More importantly, radiation-inducedPKB/Akt phosphorylation in endothelial cells was strongly down-regu-lated by the VEGF receptor tyrosine kinase inhibitor, whereas the ErbBreceptor tyrosine kinase inhibitor did not affect PKB/Akt stimulation inresponse to irradiation. An opposite receptor dependence for radiation-induced PKB/Akt phosphorylation was observed in ErbB receptor-over-expressing A431 tumor cells. Furthermore, direct VEGF receptor phos-phorylation was detected after irradiation in endothelial cells in absence ofVEGF, which was almost completely inhibited after irradiation in pres-ence of the VEGF receptor tyrosine kinase inhibitor. These data demon-strate that ionizing radiation induces VEGF ligand-independent butVEGF receptor-dependent PKB/Akt activation in endothelial cells andthat PI3K/Akt pathway activation by radiation occurs in a differential celltype and receptor-dependent pattern.
INTRODUCTION
Ionizing radiation (IR) induces a complex network of signalingprocesses at cellular sites distant from and independent of DNAdamage and many of these processes unveil new therapeutic interven-tions to combine IR with pharmacological anti-signaling agents. IR-induced activation of receptor tyrosine kinases (RTKs) is a highlyinvestigated area to understand the cellular stress response to IR(1–4). Similar to growth factor stimulation, irradiation results indefined phosphorylation of Tyr residues in the cytoplasmic domain ofRTKs, which serve as docking sites for signaling entities of additionaldownstream signal transduction pathways such as the ras/mitogen-activated protein kinase, phosphatidylinositol 3�-kinase (PI3K), andsignal transducers and activators of transcription 3 (STAT-3) path-ways, which are involved in enhanced proliferation and regulate anintrinsic survival threshold. These signaling activities might contrib-ute to radiation-induced accelerated tumor cell repopulation and en-hanced radioresistance (5, 6). These IR-induced processes representan additional rationale that RTKs are promising targets for combinedtreatment modalities using IR in combination with RTK inhibitors. Avariety of promising approaches were developed to abrogate the
signaling processes generated from mutated and overexpressed mem-bers of the epidermal growth factor (EGF) or ErbB family of type IRTKs such as EGF receptor (EGFR) (ErbB1/HER1) and HER2(ErbB2/HER2). These include strategies that target the extracellularRTK domains, e.g., with antireceptor antibodies or that inhibit theintracellular tyrosine kinase activity with low molecular weight in-hibitors (7–10). Both approaches resulted in compounds that havebeen tested in preclinical studies and that have entered clinical trialsduring recent years, alone, and in combination with chemotherapy andradiotherapy (11, 12).
The PI3K/Akt signaling pathway is activated through growth fac-tor- and radiation-induced ErbB receptor stimulation in tumor cellsand is involved in the cellular response to stress stimuli and apoptosisregulation (13–17). PI3K can directly be recruited to the specificphosphorylated docking sites of the ErbB3 isoform of the EGFRfamily through the SH2-domain of its p85 regulatory subunit (18–20).Because of the lack of an intact kinase domain ErbB3 requiresheterodimerization with other ErbB isoforms such as ErbB1 (EGFR)or ErbB2, which by themselves lack specific consensus sites for PI3Kbinding. However, PI3K can also be stimulated in an ErbB3 inde-pendent way through the interaction of RTK-activated ras with PI3K(21).
One of the downstream targets of PI3K is the serine-threonineprotein kinase PKB/Akt also referred to as protein kinase B (PKB orRAC). Activation of PKB/Akt involves the binding of PI3K-phos-phorylated phosphoinositides to the PKB/Akt-pleckstrin domain andtranslocation to the plasma membrane, where PKB/Akt is phospho-rylated by the phosphatidylinositide (PI)-dependent kinase PDK1 andan unidentified kinase referred as PDK2. Phosphorylation of the twophosphorylation sites Thr308 by PDK1 and Ser473 is required for itsactivation (22, 23). Several downstream targets of PKB/Akt have beenidentified (e.g., the bcl-2 family member Bad, caspase-9, mdm2, thetranscription factor forkhead, and glycogen synthase kinase 3), thusconferring to PKB/Akt an important role in the regulation of down-stream growth-promoting and cell survival signaling pathways(24–28).
Targeting the vascular system of tumors has been regarded as ahighly promising anticancer strategy not only because it is directedagainst the delivery of nutrients, growth factors, and oxygen but alsobecause it prevents tumor progression and the formation of metastasis(29, 30). Furthermore, targeting the vascular system is an interestingapproach because primary endothelial cells of the tumor angiogenicsystem do not undergo genetic mutations. In particular, specific tyro-sine kinase inhibitors of the vascular endothelial growth factor recep-tor (VEGFR) have been developed as inhibitors of angiogenesis(31–35). This is mainly due to the important role of the VEGF as aproangiogenic factor in pathological situations, neovascularizationand enhanced vascular permeability, and the advanced mechanisticunderstanding of the corresponding VEGFRs. The VEGFRs Flt-1(Fms-like tyrosine kinase, VEGFR-1) and KDR (kinase insertdomain-containing receptor, VEGFR-2) are predominantly located onendothelial cells (36). Both receptors have seven immunoglobulin-likedomains in their extracellular region, a single transmembrane-span-ning domain, and an intracellular split tyrosine kinase domain andbelong to the same family of receptors as EGFR, platelet-derived
Received 10/28/03; revised 4/21/04; accepted 5/24/04.Grant support: Swiss Cancer League (D. Zingg, M. Pruschy), the Radium Fonds,
Hartmann Muller-Foundation and Stiftung zur Krebsbekampfung (O. Riesterer) and anEORTC-Astra Zeneca Translational Research Fellowship (M. Pruschy).
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
Note: D. Zingg and O. Riesterer contributed equally to this work.Requests for reprints: Martin Pruschy, Laboratory for Molecular Radiobiology,
Department of Radiation Oncology, University Hospital Zurich, Ramistr. 100, CH-8091Zurich, Switzerland. Phone: 411-255-8549; Fax: 411-255-44-35; E-mail: [email protected].
5398
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

growth factor receptor, c-Kit, c-Fms, Flt-3, and Flt-4. Flt-1 bindsVEGF-A and VEGF-B and the related placenta growth factor,whereas KDR binds VEGF-A, VEGF-C, and VEGF-D (37, 38). KDRis strongly autophosphorylated upon VEGF stimulation, and VEGFinduces signaling processes such as the PI3K/Akt pathway throughdirect recruitment of downstream targets to the phosphorylated con-sensus sites of the VEGFR (39–41)
Interestingly, IR also activates PKB/Akt in endothelial cells in aPI3K-dependent way, and PI3K inhibitors enhance radiation-inducedapoptosis and cytotoxicity in these cells (42). Thus, inhibition of theradiation-activated PI3K/Akt survival pathway might also contributeto an increased antiangiogenic effect after treatment with IR in com-bination with inhibitors of this survival pathway. However, radiation-induced activation of the PI3K/Akt pathway could be mediatedthrough different upstream RTKs, and current PI3K inhibitors (e.g.,wortmanin, LY294002) are overly toxic for clinical use (43). Wetherefore investigated radiation-induced PI3K/Akt stimulation in en-dothelial cells using clinically relevant RTK inhibitors and comparedits activation profile to radiation-induced PKB/Akt stimulation intumor cells.
MATERIALS AND METHODS
Cell Cultures, Irradiation, and Treatment with RTK Inhibitors. Thevulvar squamous carcinoma cell line A431 was grown at 37°C and 5% CO2
atmosphere in DMEM containing 10% FCS (Life Technologies Laboratories,Karlsruhe, Germany) supplemented with penicillin and streptomycin. Forserum starvation, A431 cells were washed with PBS and incubated withDMEM containing 0.5% FCS for 24 h. The primary human umbilical veinendothelial cells (HUVECs; PromoCell GmbH, Heidelberg, Germany) wereused between the second and the seventh passage number and cultured inendothelial cell growth medium kit (EGM; PromoCell GmbH) containing 5%FCS, 0.4% endothelial cell growth supplement/H, 0.1 ng/ml EGF, 1 ng/mlbasic fibroblast growth factor, 1 �g/ml hydrocortisone, 50 �g/ml gentamicin,and 50 ng/ml amphotericin B. HUVECs were plated in collagen I coated 6 cm(Becton Dickinson Labware, Bedford, MA) or 1.5% gelatin-coated 10-cm Petridishes. For growth factor starvation, HUVECs were washed with PBS andincubated with endothelial cell basal medium (EBM; PromoCell GmbH)supplemented with 0.1% BSA, amphotericin B, and penicillin/streptomycin,for 12 h. The human dermal microvascular endothelial cells (PromoCellGmbH) were used between the second and the seventh passage number andcultured in endothelial cell growth medium kit (EGM MV; PromoCell GmbH)containing 5% FCS, 0.4% endothelial cell growth supplement/H, 10 ng/mlEGF, 1 �g/ml hydrocortisone, 50 �g/ml gentamicin, and 50 ng/ml amphoter-icin B. Growth factor starvation for human dermal microvascular endothelialcells was performed as described for HUVECs. EGF and human VEGF165
(R&D Systems, Wiesbaden-Nordstadt, Germany) were added to the culturemedia for the indicated time to a final concentration of 50 and 20 ng/ml,respectively. Antihuman EGF and antihuman VEGF antibodies were pur-chased from R&D Systems and added to the culture media as indicated.Irradiation was carried out at room temperature using a Pantak Therapax300kV X-ray unit at 0.7 Gy/min. Cellular pretreatment with PKI166 (100 or500 nM; Novartis-Pharma, Basel, Switzerland) or PTK787/ZK222584 (100 nM;Novartis-Pharma) was performed for 1 and 2 h before irradiation, respectively.
Immunoblotting. Cells were harvested at different time points by scrapingoff the cells in 100 �l of SDS sample buffer. Samples were stored at �80°Cor directly resolved by SDS-PAGE followed by blotting onto polyvinylidenedifluoride membranes. Membranes were probed with rabbit polyclonal anti-PKB/Akt, anti-PKB/Akt-phosphoSer473 (New England Biolabs, Beverly MA),anti-ErbB1-phosphoTyr1086 (Calbiochem, Darmstadt Germany) or anti-VEGFR-2-phosphoTyr951 antibodies (Cell Signaling, Beverly, MA), or mousemonoclonal anti-�-actin (clone AC-15; Sigma, Buchs Switzerland), anti-ErbB1 (Oncogene, Boston, MA), and anti-VEGFR-2-antibodies (Calbiochem).Antibody detection was achieved by enhanced chemiluminescence-enhancedchemiluminescence (Amersham, Freiburg, Germany) using a horseradish per-oxidase-conjugated second antibody, according to the manufacturer’s protocol.
Immunoprecipitation. Cells were washed with ice-cold PBS and har-vested by scraping off cells in 500 �l of buffer A [20 mM Tris-HCl (pH 8.0),5% Glycerol (m/V), 1% NP40 (m/V), 2.7 mM KCl, 138 mM NaCl, 1 mM
MgCl2, 1 mM CaCl2, 50 mM sodium EDTA, and 1 mM DTT], supplementedwith protease and phosphatase inhibitors (5 mg/ml pepstatin A, 10 mg/mlleupeptin, 2 mg/ml aprotinin, 1 mM Na3VO4, 1 mM NaF, and 0.1 mM phen-ylmethylsulfonyl fluoride). After incubation on ice for 30 min, the lysate wascentrifugated at 15,000 � g for 10 min at 4°C. Protein concentration wasdetermined from the supernatant by colorimetric measurement using the Bio-Rad DC protein assay kit II according to the manufacturer’s instructions(Bio-Rad Laboratories, Hercules, CA). Supernatants (1 mg/ml protein) wereincubated with 25 �g of protein A agarose-conjugated anti-phosphoTyr mono-clonal antibody (4G10; Upstate Biotechnology, Lake Placid, NY) or 2 �g ofanti-VEGFR-2 polyclonal antibody (Calbiochem) overnight at 4°C and withprotein A-conjugated agarose beads (Upstate Biotechnology) at 4°C for 4 h.The beads were washed three times in 0.5 ml of buffer A, supplemented withall inhibitors, and immunoprecipitates were resuspended in 2� SDS samplebuffer for Western blot analysis.
Immunofluorescence Labeling and Confocal Laser-Scanning Micros-copy. HUVECs were treated in 2-well collagen-coated plates (Biocoat; Bec-ton Dickinson) as indicated above, followed by fixation with 3% paraformal-dehyde for 15 min and cell permeabilization with 0.5% Triton X-100 in PBSfor 2.5 min. The primary rabbit polyclonal anti-VEGFR-2 phosphoTyr996
antibody (Calbiochem) was used at a dilution of 1:50. The secondary TexasRed X-conjugated affinity-purified goat antirabbit IgG (H � L) antibody(Molecular Probes, Eugene, OR) was used at a dilution of 1:100. Bothantibodies were incubated for 1 h with the cells. Cells were costained with4�,6-diamidino-2-phenylindole (10 �g/ml) for detection of nuclei. The cover-slips were mounted on glass slides and embedded in Mowiol (Calbiochem). Allsteps were performed at room temperature. A Zeiss Axioplan fluorescencemicroscope (Zeiss, Jena, Germany) equipped with a confocal scanning unitMRC-600 (Bio-Rad Laboratories, Herts, United Kingdom) and an argon-krypton laser was used to acquire images, which were subsequently processedusing Adobe PhotoShop, system 6.0 (San Jose, CA) and reconstructed usingsimple PC software.
RESULTS
IR Activates PKB/Akt in Human Endothelial Cell. The PI3K/Akt survival pathway is activated in different cell lines in response tovarious stress factors. PKB/Akt activation is a multistep process andincludes phosphorylation of PKB/Akt at the specific residue Ser473
also in response to irradiation with IR (44). To determine PI3K/Aktpathway activation in endothelial cells, we investigated the PKB/Aktphosphorylation status in HUVECs after treatment with low doses ofIR using the Ser473 site-specific anti-phospho-Akt antibody. HUVECswere serum starved for 12 h, and the PKB/Akt phosphorylation statuswas then analyzed by Western blotting of whole cellular lysates afterirradiation with 2 Gy (Fig. 1A). A clear increase in the phosphoryla-tion level was observed at 20, 30, 60, and 120 min after irradiation,which is in agreement with recent data reported by Edwards et al.(42). A dose-dependent effect of PKB/Akt phosphorylation was in-vestigated after treatment with increasing doses of IR. Interestingly,irradiation with doses as high as 10 Gy did not additionally increasethe Ser473-PKB/Akt phosphorylation level (Fig. 1B).
The PI3K/Akt pathway is activated in proliferating cells via growthfactor receptor stimulation, and interestingly, IR-induced PKB/Aktphosphorylation has been demonstrated in epithelial tumor cells to bemediated through activation of ErbB receptors (13). Thus, IR mightalso induce PKB/Akt phosphorylation in HUVECs via upstreamRTKs. To investigate the mechanism of IR-dependent PKB/Akt ac-tivation in greater detail, we analyzed at first the effect of PKB/Aktphosphorylation in endothelial cells in response to the two intrinsicligands of the ErbB and VEGFR systems.
HUVECs were serum deprived for 12 h before treatment with thetwo growth factors EGF (50 ng/ml) and VEGF (20 ng/ml), respec-
5399
VEGF RECEPTOR-DEPENDENT PKB/Akt ACTIVATION
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

tively, followed by analysis of the Ser473-Akt phosphorylation statusby Western blotting of whole cellular protein extracts. PKB/Aktphosphorylation could be observed 5 and 10 min after EGF stimula-tion, whereas growth factor-specific up-regulation of the PKB/Aktphosphorylation status was only observed as early as 10 min afterstimulation with VEGF (Fig. 2). The delayed kinetics of PKB/Aktphosphorylation in response to VEGF mirrored PKB/Akt stimulationafter irradiation. Furthermore, these results show that the PI3K/Aktpathway can be activated both via EGF or VEGF in endothelial cells.
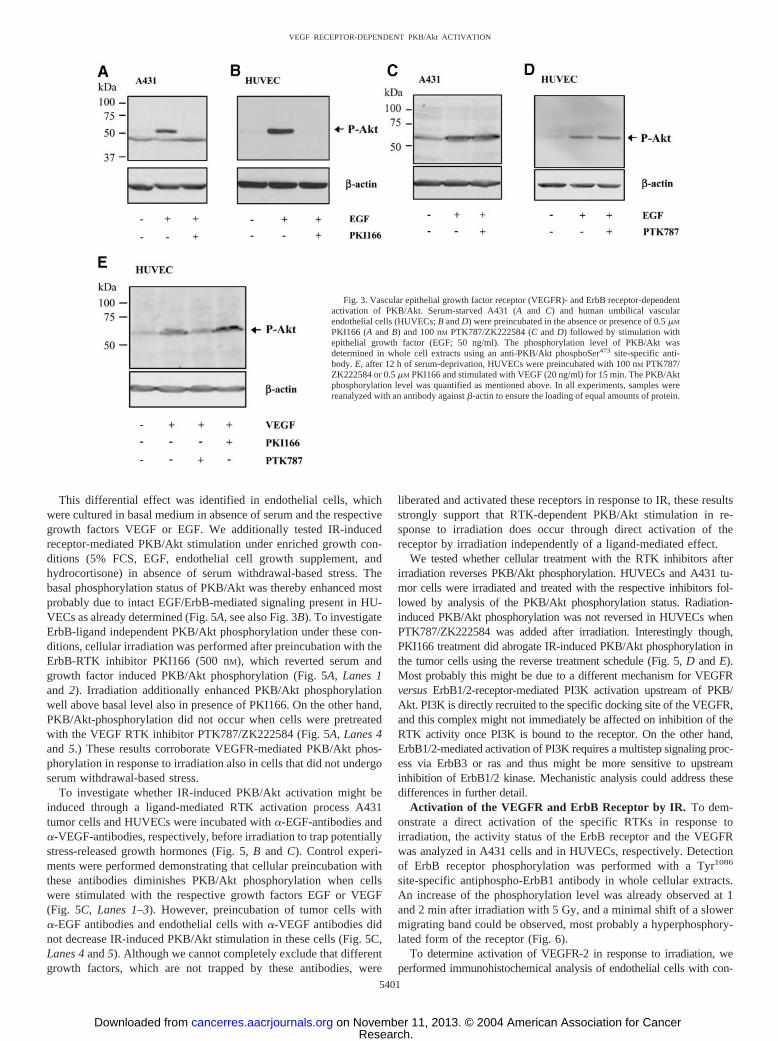
VEGF and ErbB Receptor-Mediated Activation of PKB/Akt.Clinically relevant inhibitors of the VEGF receptor system (the phta-lazine derivative PTK787/ZK222584) and the ErbB1/2 receptors (thepyrrolopyrimidine derivative PKI166) were used to dissect whetheractivation of specific RTKs is required for IR-induced PKB/Aktphosphorylation. On the basis of the high selectivity of these rationaldesigned pharmacological compounds, they are suitable as smallmolecular probes to dissect signal transduction cascades. Both theErbB1/2 inhibitor PKI166 and the VEGFR inhibitor PTK787/ZK222584 inhibit the kinase activity of their respective receptors inthe submicromolar range, with an overlapping inhibitory effect only athigher doses levels (7). To determine the required concentration forselective receptor inhibition, growth factor-dependent PKB/Akt acti-vation was first tested in HUVECs and the ErbB1-overexpressinghuman tumor cell line A431. Both cell lines were serum starved for12 h and stimulated with EGF (50 ng/ml) with or without pretreatmentwith PKI166 (500 nM) or PTK787/ZK222584 (100 nM) for 1 h.EGF-induced PKB/Akt phosphorylation was completely abrogated inA431 and HUVECs, respectively, when cells were pretreated withPKI166 (Fig. 3, A and B; 500 nM). On the other hand, cellularpretreatment with PTK787/ZK222584 (100 nM) did not affect EGF-
induced PKB/Akt phosphorylation in either cell line (Fig. 3, C and D).Furthermore, PKB/Akt phosphorylation was also tested in HUVECsin response to VEGF. Although the ErbB inhibitor PKI166 did notdown-regulate VEGF-dependent PKB/Akt phosphorylation, the phos-phorylation status was strongly decreased on pretreatment with theVEGF receptor inhibitor PTK787/ZK222584 (Fig. 3E). These resultsconfirm that EGF- and VEGF-induced PKB/Akt activation are medi-ated through their corresponding RTKs and demonstrate the growthfactor receptor-selective inhibitory potential of the two RTK inhibi-tors to dissect the potential link between RTKs and IR-inducedPI3K/Akt stimulation in tumor and endothelial cells.
Receptor-Dependent PKB/Akt Stimulation by IR in Tumor andEndothelial Cells. Using these pharmacological inhibitors we testedRTK-dependent PKB/Akt phosphorylation after irradiation in endo-thelial cells and in the tumor cell line A431. A431 cells were serumstarved, pretreated with PKI166 (500 nM) or PTK787/ZK222584 (100nM) for 1 h and irradiated with 5 Gy of IR. The ErbBR inhibitorcompletely abrogated IR-induced PKB/Akt phosphorylation whereasthe VEGFR inhibitor did not affect PKB/Akt phosphorylation in thesecells (Fig. 4, A and B). These results are in accordance with previousdata demonstrating that IR activates PKB/Akt in tumor cells via anErbB receptor-mediated process (13, 45, 46).
In parallel, PKB/Akt stimulation was investigated in two endothe-lial cell lines. Interestingly, PKI166 did not decrease the IR-inducedPKB/Akt phosphorylation status in HUVECs, whereas pretreatmentwith PTK787/ZK222584 down-regulated IR-dependent PKB/Aktphosphorylation (Fig. 4, C and D). Likewise, IR-induced PKB/Aktphosphorylation in microvascular endothelial cells, which show adifferent proliferative activity than umbilical vein endothelial cells,was also sensitive to PTK787/ZK222584 but not to PKI166 (Fig. 4, Eand F). These data strongly suggest a differential RTK-dependentstimulation of the PI3K/Akt pathway by IR in tumor and endothelialcells, respectively.
Fig. 2. Epithelial growth factor (EGF) and vascular epithelial growth factor (VEGF)activate PKB/Akt with a different kinetic in human endothelial cells. After 12 h of serumdeprivation, human umbilical vascular endothelial cells (HUVECs) were stimulated withEGF (50 ng/ml; A) or with VEGF (20 ng/ml; B) for the indicated times. The phospho-rylation level of PKB/Akt was determined in whole cell extracts using an anti-PKB/AktphosphoSer473 site-specific antibody. Samples were reanalyzed with an anti-�-actinantibody to ensure the loading of equal amounts of protein.
Fig. 1. Ionizing radiation induces PKB/Akt phosphorylation in human endothelialcells. A, after 12 h of serum deprivation, human umbilical vascular endothelial cells(HUVECs) were irradiated with 2 Gy radiation and analyzed at the indicated time points.The phosphorylation level of PKB/Akt was determined in whole cell extracts using ananti-PKB/Akt phosphoSer473 site-specific antibody. B, cells were irradiated with increas-ing doses of ionizing radiation (IR) and PKB/Akt phosphorylation level was determinedat the 30-min time point as in A. Samples were reanalyzed using anti-�-actin or anti-Aktantibodies to ensure the loading of equal amounts of protein.
5400
VEGF RECEPTOR-DEPENDENT PKB/Akt ACTIVATION
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from
This differential effect was identified in endothelial cells, whichwere cultured in basal medium in absence of serum and the respectivegrowth factors VEGF or EGF. We additionally tested IR-inducedreceptor-mediated PKB/Akt stimulation under enriched growth con-ditions (5% FCS, EGF, endothelial cell growth supplement, andhydrocortisone) in absence of serum withdrawal-based stress. Thebasal phosphorylation status of PKB/Akt was thereby enhanced mostprobably due to intact EGF/ErbB-mediated signaling present in HU-VECs as already determined (Fig. 5A, see also Fig. 3B). To investigateErbB-ligand independent PKB/Akt phosphorylation under these con-ditions, cellular irradiation was performed after preincubation with theErbB-RTK inhibitor PKI166 (500 nM), which reverted serum andgrowth factor induced PKB/Akt phosphorylation (Fig. 5A, Lanes 1and 2). Irradiation additionally enhanced PKB/Akt phosphorylationwell above basal level also in presence of PKI166. On the other hand,PKB/Akt-phosphorylation did not occur when cells were pretreatedwith the VEGF RTK inhibitor PTK787/ZK222584 (Fig. 5A, Lanes 4and 5.) These results corroborate VEGFR-mediated PKB/Akt phos-phorylation in response to irradiation also in cells that did not undergoserum withdrawal-based stress.
To investigate whether IR-induced PKB/Akt activation might beinduced through a ligand-mediated RTK activation process A431tumor cells and HUVECs were incubated with �-EGF-antibodies and�-VEGF-antibodies, respectively, before irradiation to trap potentiallystress-released growth hormones (Fig. 5, B and C). Control experi-ments were performed demonstrating that cellular preincubation withthese antibodies diminishes PKB/Akt phosphorylation when cellswere stimulated with the respective growth factors EGF or VEGF(Fig. 5C, Lanes 1–3). However, preincubation of tumor cells with�-EGF antibodies and endothelial cells with �-VEGF antibodies didnot decrease IR-induced PKB/Akt stimulation in these cells (Fig. 5C,Lanes 4 and 5). Although we cannot completely exclude that differentgrowth factors, which are not trapped by these antibodies, were
liberated and activated these receptors in response to IR, these resultsstrongly support that RTK-dependent PKB/Akt stimulation in re-sponse to irradiation does occur through direct activation of thereceptor by irradiation independently of a ligand-mediated effect.
We tested whether cellular treatment with the RTK inhibitors afterirradiation reverses PKB/Akt phosphorylation. HUVECs and A431 tu-mor cells were irradiated and treated with the respective inhibitors fol-lowed by analysis of the PKB/Akt phosphorylation status. Radiation-induced PKB/Akt phosphorylation was not reversed in HUVECs whenPTK787/ZK222584 was added after irradiation. Interestingly though,PKI166 treatment did abrogate IR-induced PKB/Akt phosphorylation inthe tumor cells using the reverse treatment schedule (Fig. 5, D and E).Most probably this might be due to a different mechanism for VEGFRversus ErbB1/2-receptor-mediated PI3K activation upstream of PKB/Akt. PI3K is directly recruited to the specific docking site of the VEGFR,and this complex might not immediately be affected on inhibition of theRTK activity once PI3K is bound to the receptor. On the other hand,ErbB1/2-mediated activation of PI3K requires a multistep signaling proc-ess via ErbB3 or ras and thus might be more sensitive to upstreaminhibition of ErbB1/2 kinase. Mechanistic analysis could address thesedifferences in further detail.
Activation of the VEGFR and ErbB Receptor by IR. To dem-onstrate a direct activation of the specific RTKs in response toirradiation, the activity status of the ErbB receptor and the VEGFRwas analyzed in A431 cells and in HUVECs, respectively. Detectionof ErbB receptor phosphorylation was performed with a Tyr1086
site-specific antiphospho-ErbB1 antibody in whole cellular extracts.An increase of the phosphorylation level was already observed at 1and 2 min after irradiation with 5 Gy, and a minimal shift of a slowermigrating band could be observed, most probably a hyperphosphory-lated form of the receptor (Fig. 6).
To determine activation of VEGFR-2 in response to irradiation, weperformed immunohistochemical analysis of endothelial cells with con-
Fig. 3. Vascular epithelial growth factor receptor (VEGFR)- and ErbB receptor-dependentactivation of PKB/Akt. Serum-starved A431 (A and C) and human umbilical vascularendothelial cells (HUVECs; B and D) were preincubated in the absence or presence of 0.5 �M
PKI166 (A and B) and 100 nM PTK787/ZK222584 (C and D) followed by stimulation withepithelial growth factor (EGF; 50 ng/ml). The phosphorylation level of PKB/Akt wasdetermined in whole cell extracts using an anti-PKB/Akt phosphoSer473 site-specific anti-body. E, after 12 h of serum-deprivation, HUVECs were preincubated with 100 nM PTK787/ZK222584 or 0.5 �M PKI166 and stimulated with VEGF (20 ng/ml) for 15 min. The PKB/Aktphosphorylation level was quantified as mentioned above. In all experiments, samples werereanalyzed with an antibody against �-actin to ensure the loading of equal amounts of protein.
5401
VEGF RECEPTOR-DEPENDENT PKB/Akt ACTIVATION
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

focal laser-scanning microscopy. Unfortunately and due to low detectionquality, no Western blotting experiments with specific antibodies againstthe activated, phosphorylated form of the VEGFR-2 could be used foradditional analysis of immunoprecipitates or whole cellular extracts fromprimary endothelial cells. HUVECs were serum starved for 3 h andirradiated with 2 Gy in the presence or absence of PTK787/ZK222584.Immunofluorescent labeling was performed with the Tyr996 site-specificanti-phospho-VEGFR-2 polyclonal antibody. In parallel the nuclei werestained with 4�,6-diamidino-2-phenylindole to facilitate cellular localiza-tion. In comparison to control cells, irradiated cells displayed a markedlyenhanced, almost continuous rim staining at the site of the plasma cellmembrane. On the other hand, pretreatment of cells with PTK787/ZK222584 before irradiation resulted in a less pronounced rim stainingwith only a few dotted structures, similar to the staining of control cells(Fig. 7). Together with the data showing RTK inhibitor-dependent con-trol of IR-induced PKB/Akt phosphorylation, these results demonstrateVEGFR-mediated activation of the PI3K/Akt pathway in endothelial
cells and ErbB receptor-dependent activation of this survival pathway inthe A431 tumor cells (Fig. 8).
DISCUSSION
The relevance of the PKB/Akt protein kinase for cellular survivalhas been investigated in different cell types and in response to manystress factors (47–50). Radiation-induced PKB/Akt activation in en-dothelial cells has previously been demonstrated to contribute to anincrease in radioresistance both in vitro and in vivo; however, theupstream molecular entities leading to an activated PI3K/Akt pathwayhave not been addressed thus far in endothelial cells (42). In thisarticle we have compared RTK dependence of radiation-inducedPI3K/Akt activation in tumor cells with the activation pattern inendothelial cells using clinically relevant inhibitors of the ErbB1/2class receptors and the VEGFR. Both growth factor ligands EGF andVEGF stimulated PKB/Akt phosphorylation through their corre-
Fig. 4. Differential receptor-dependent PKB/Akt stimulation by ion-izing radiation in tumor and endothelial cells. A431 (A and B), humanumbilical vascular endothelial cells (HUVECs; C and D), and HDMEcells (E and F) were serum deprived for 24 or 12 h, respectively,followed by preincubation in the absence or presence of 0.5 �M PKI166(A, C, and E) or 100 nM PTK787/ZK222584 (B, D, and F) and irradiatedwith 5 Gy of IR. The phosphorylation level of PKB/Akt was determinedin whole cell extracts using an anti-PKB/Akt phosphoSer473 site specificantibody 30 min after irradiation. Samples were reanalyzed with anantibody against �-actin to ensure the loading of equal amounts ofprotein. All experiments were repeated at least twice.
5402
VEGF RECEPTOR-DEPENDENT PKB/Akt ACTIVATION
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

sponding growth factor receptors, which could be abrogated by theRTK inhibitors PKI166 and PTK787/ZK222584, respectively, indi-cating an intact growth factor-induced signaling network from theRTKs to PKB/Akt in endothelial cells. Interestingly, IR dose andtime-dependent activation of PKB/Akt in these cells closely followedthe VEGF- but not the EGF-mediated activation pattern of this sur-vival pathway. Radiation-induced PKB/Akt phosphorylation wasstrongly down-regulated by the VEGF-RTK inhibitor in the endothe-lial cells, whereas the ErbB receptor inhibitor did not decrease IR-induced PKB/Akt-stimulation in these cells. An opposite receptordependence for radiation-induced PKB/Akt-phosphorylation was ob-served in the tumor cell line A431, similar to previously observedErbB-receptor mediated activation of PKB/Akt in response to IR intumor cells. Specific VEGF-independent but VEGFR-mediated PKB/Akt-activation was additionally corroborated through the direct acti-vation of the PTK787/ZK222584-sensitive VEGFR-2 by IR.
Using PKI166 and PTK787/ZK222584, we could selectively dis-criminate between ErbB1/2- and VEGFR protein tyrosine kinase-
mediated PKB/Akt phosphorylation in response to IR. Both inhibitorswere intensively characterized with regard to their in vitro and in vivospecificity (35, 51), and especially PTK787/ZK222584 displays highVEGFR-specific inhibitory activity at the concentration used in thisarticle (Ref. 35 and J. Wood, personal communication). Nevertheless,we cannot exclude that other kinases are (co-)activated by IR, coin-hibited by PTK787/ZK222584, and at the same time contribute toIR-induced PKB/Akt-phosphorylation. On the other hand, the signal-ing pathway linking an activated VEGF-receptor with PI3K/Akt iswell established in endothelial cells. More importantly, we demon-strated a differential mechanism for PKB/Akt activation in endothelialand tumor cells in response to irradiation and pinpoint to a responsibleRTK activity and a relevant inhibitor. This is important with regard tothe establishment of novel combined treatment modalities becausegrowth factor-independent but IR-induced RTK-dependent PKB/Aktactivation will also contribute to the resistance of irradiated vascularendothelium (52).
Both enhanced cell proliferation and DNA repair as well as reducedapoptosis are part of a cytoprotective response and are induced inresponse to growth factor ligand- and IR-activated RTKs. PrimaryIR-induced RTK activation has been intensively investigated for ErbBreceptor tyrosine kinases in tumor cells; however, the exact mecha-nism for their activation is still unclear (45, 53). The generation ofreactive oxygen and nitrogen species may shift the steady-state tyro-sine phosphorylation status of RTKs to its phosphorylated active formdue to the deactivation of critical cysteine residues in the catalyticcenter of corresponding protein phosphatases (54–59). On the otherhand, reactive oxygen species also affect lipid bilayer compositionand rigidity, which might ultimately lead to a proximity effect andtransphosphorylation of plasma membrane integrated RTKs (60).Interestingly, although ligand-independent, IR-induced RTK (auto-)transphosphorylation is induced to a lower level in comparison toligand-mediated RTK activation, downstream PKB/Akt phosphoryl-ation is induced to a similar extent by IR and the correspondinggrowth factor (46, 61, 62). We also observed a lower level of IR-induced VEGFR activation in endothelial cells, but again, the level ofPKB/Akt phosphorylation was enhanced to a similar extent by IR and
Fig. 5. Ligand-independent receptor-mediated PKB/Akt stimulation by ioniz-ing radiation. In A, human umbilical vascular endothelial cells (HUVECs) werecultured under serum and growth factor-enriched growth conditions and prein-cubated in presence or absence of PKI166 (0.5 �M, 3 h) and PTK787/ZK222584(100 nM, 2 h) before irradiation with 5 Gy. For growth factor trapping A431 cells(B) or HUVECs (C) were pretreated with an anti-epithelial growth factor (EGF;0.5 �g/ml) or an anti-vascular epithelial growth factor (VEGF) antibody (0.5�g/ml), respectively, added twice (5 and 30 min) before stimulation with EGF (1ng/ml), VEGF (10 ng/ml), or irradiation (5 Gy). In D and E, A431 cells (D) andHUVECs (E) were serum deprived for 24 or 12 h, respectively, followed byirradiation with 5 Gy. PKI166 (0.5 �M; D) or PTK787/ZK222584 (0.1 �M; E)was added to the respective culture medium 20 min after irradiation. Thephosphorylation level of PKB/Akt was determined in all whole cell extracts usingan anti-Akt phosphoSer473 site-specific antibody 30 min after irradiation. Sam-ples were reanalyzed with an antibody against �-actin to ensure the loading ofequal amounts of protein. All experiments were repeated at least twice.
Fig. 6. Ionizing radiation activates the ErbB1 receptor in tumor cells. After 24 h ofserum deprivation, A431 cells were irradiated with 5 Gy and analyzed at the indicatedtime points or after treatment with epithelial growth factor (50 ng/ml) as positive control.The phosphorylation level of ErbB1 receptors was determined in whole cell extracts usingan anti-ErbB1 receptor phosphoTyr1086 site-specific antibody. Samples were reanalyzedwith an antibody against ErbB1 receptor to ensure the loading of equal amounts of protein.
5403
VEGF RECEPTOR-DEPENDENT PKB/Akt ACTIVATION
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

VEGF. Activation of multiple ErbB receptor isoforms in parallelmight explain that low level of RTK phosphorylation on the individ-ual isoform level is still sufficient to induce strong downstreamsignaling in a cumulative way or to overcome a signaling threshold.This might also apply to the low level of receptor phosphorylation ofthe different VEGFR isoforms, which could be determined by West-ern blotting in response to IR (data not shown). More importantly,IR-induced site-specific VEGFR-2 phosphorylation could be detectedby confocal microscopy on the individual cellular level and could beabrogated by pretreatment with the VEGFR inhibitor PTK787/ZK222584.
Ligand-dependent activation of RTKs are mainly determined by theaffinity of the ligand to the corresponding receptor-isoform. In-depthanalysis by Dent et al. on the level of ErbB receptors revealed thatIR-induced RTK activation does not discriminate between the differ-ent ErbB receptor isoforms and their different ligand affinities (61).Rather, it is the expression level of the isoforms that predetermines
IR-mediated RTK isoform activation. These studies have mainly beenperformed in tumor cells in which ErbB-mediated signaling dominateproliferation and survival processes not least because of a high ex-pression level of these RTKs. We used primary, not established,proliferating endothelial cells for these studies, which most probablyresemble in vivo endothelial cell structures with regard to RTKexpression. Thus, the differential RTK dependence of IR-inducedPKB/Akt phosphorylation might be due to a shifted RTK expressionprofile toward elevated VEGFRs in endothelial cells in comparison toErbB receptor overexpression in ErbB receptor-dominated tumor cellsor due to a change in the relative importance of a specific pathway.However, we cannot exclude a distinct activation of VEGFRs by IR,which might be linked to inter- and intramolecular cross-talks be-tween the different VEGFR isoforms or the release of trapped signal-ing agonists such as VEGF or PIGF (63). However, our irradiationexperiments performed in presence of trapping anti-VEGF antibodiesdo not support such a scenario. A detailed quantification and analysisof PI3K recruitment to the different VEGFR isoforms will furtherclarify the mechanism of RTK-dependence for PI3K/Akt pathwayactivation in response to irradiation.
IR is a very powerful therapy against cancer because of its cyto-toxicity mainly induced by DNA damage. However, IR also inducesstress responses promoting cell survival and repopulation eventuallyleading to treatment resistance. Activation of the ErbB receptorsfollowed by downstream activation of the PI3K/Akt pathway upon IRexposure was proposed to be a central step favoring tumor cellsurvival and proliferation. Moreover, this mechanism was shown tostimulate ErbB-receptor mediated VEGF expression from tumor cellsindirectly promoting an angiogenic response or at least an antiapop-totic stimulus, which further contributes to enhanced radioresistance.
However, irradiation targets both tumor and endothelial cells, andinterestingly, the radiosensitivity of the tumor microvasculature andmicrovascular damage strongly contributes to the tumor response toradiation (64). Thus, targeting an intrinsic treatment threshold inendothelial cells of the tumor vasculature sensitizes the tumor for IR.Besides the cytotoxic effect of IR on the endothelial cell level, a directcytoprotective stress response is induced by the activation of thePI3K/Akt pathway additionally promoting endothelial cell survival
Fig. 7. Ionizing radiation activates the vascularepithelial growth factor receptor (VEGFR) in hu-man endothelial cells. Human umbilical vascularendothelial cells were serum deprived for 3 h in theabsence or presence of 500 nM PTK787/ZK222584and then irradiated with 2 Gy. The cells were fixed5 min after treatment, and immunofluorescencelabeling was performed with the rabbit polyclonalantibody anti-VEGFR-2-phosphoTyr996 site spe-cific antibody. Nuclei were stained with 4�,6-diamidino-2-phenylindole (DAPI). The cells weresubsequently examined by confocal laser-scanningmicroscopy. The experiments were repeated atleast three times and representative images aredisplayed.
Fig. 8. Scheme of the differential phosphatidylinositol 3�-kinase (PI3K)/Akt pathwayactivation in tumor and endothelial cells by ionizing radiation. Ionizing radiation depend-ent PI3K/Akt pathway activation is mediated by ErbB receptors in tumor cells andvascular epithelial growth factor receptors (VEGFRs) in endothelial cells, subsequentlypromoting cell survival. Ligand-specific activation is represented by dotted arrows.Ionizing radiation (IR)-specific activation is represented by solid arrows.
5404
VEGF RECEPTOR-DEPENDENT PKB/Akt ACTIVATION
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

(41, 51). Consequently, PI3K inhibitors such as LY294002 or wort-manin increase IR-induced cytotoxicity in endothelial cells and alsoenhance radiation-induced tumor growth delay in animal tumor mod-els. Unfortunately, these inhibitors are overly toxic for clinical studies.
Other preclinical studies demonstrated that specific inhibitors ofangiogenesis such as neutralizing antihuman VEGF165 antibodies andanti-VEGFR-2 monoclonal antibodies enhance radiation-induced tu-mor growth control. In addition, many studies are performed in vitroand in vivo using small molecular pharmacological inhibitors of theVEGFR-2 tyrosine kinase, such as PTK787/ZK222548, in combina-tion with IR and demonstrate a strongly potentiated effect of thiscombined treatment modality compared with each treatment modalityalone (65–68). A combined treatment modality using anti-VEGFantibodies in combination with IR does induce an enhanced antian-giogenic effect by decreasing the growth and survival promotingeffect mediated by the growth factor ligand VEGF. On the other hand,a specific clinically relevant VEGFR tyrosine kinase inhibitor willadditionally abrogate direct IR-induced and receptor-mediated signal-ing in the endothelial tumor compartment, further contributing to acooperative antitumoral effect of IR in combination with inhibitors ofangiogenesis.
ACKNOWLEDGMENTS
We thank Dr. M. Hoechli from the service center for microscopical structureanalysis (University of Zurich) for help in running the confocal microscopyexperiments. We also thank Peter Traxler and Jeanette Wood (Novartis-Pharma) for the kind gift of PKI166 and PTK787/ZK222584.
REFERENCES
1. Tenzer A, Zingg D, Riesterer O, Vuong V, Bodis S, Pruschy M. Signal transductioninhibitors as radiosensitizers. Curr Med Chem Anticancer Agents 2002;2:727–42.
2. Ethier SP. Signal transduction pathways: the molecular basis for targeted therapies.Semin Radiat Oncol 2002;12:3–10.
3. Schmidt-Ullrich RK, Mikkelsen RB, Dent P, et al. Radiation-induced proliferation ofthe human A431 squamous carcinoma cells is dependent on EGFR tyrosine phos-phorylation. Oncogene 1997;15:1191–7.
4. Goldkorn T, Balaban N, Shannon M, Matsukuma K. EGF receptor phosphorylationis affected by ionizing radiation. Biochim Biophys Acta 1997;1358:289–99.
5. Schmidt-Ullrich RK, Contessa JN, Dent P, et al. Molecular mechanisms of radiation-induced accelerated repopulation. Radiat Oncol Investig 1999;7:321–30.
6. Dent P, Yacoub A, Contessa J, et al. Stress and radiation-induced activation ofmultiple intracellular signaling pathways. Radiat Res 2003;159:283–300.
7. Traxler P, Bold G, Buchdunger E, et al. Tyrosine kinase inhibitors: from rationaldesign to clinical trials. Med Res Rev 2001;21:499–512.
8. Harari PM, Huang SM. Epidermal growth factor receptor modulation of radiationresponse: preclinical and clinical development. Semin Radiat Oncol 2002;12:21–6.
9. Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in thebiology and treatment of cancer. J Clin Oncol 2003;21:2787–99.
10. Arteaga CL, Chinratanalab W, Carter MB. Inhibitors of HER2/neu (erbB-2) signaltransduction. Semin Oncol 2001;28:30–5.
11. Ciardiello F. An update of new targets for cancer treatment: receptor-mediatedsignals. Ann Oncol 2002;13 (Suppl 4):29–38.
12. Sartor CI. Epidermal growth factor family receptors and inhibitors: radiation responsemodulators. Semin Radiat Oncol 2003;13:22–30.
13. Contessa JN, Hampton J, Lammering G, et al. Ionizing radiation activates Erb-Breceptor dependent Akt and p70 S6 kinase signaling in carcinoma cells. Oncogene2002;21:4032–41.
14. Vanhaesebroeck B, Alessi DR. The PI3K-PDK1 connection: more than just a road toPKB. Biochem J 2000;346 (Pt 3):561–76.
15. Datta K, Bellacosa A, Chan TO, Tsichlis PN. Akt is a direct target of the phosphati-dylinositol 3-kinase. Activation by growth factors, v-src and v-Ha-ras, in Sf9 andmammalian cells. J Biol Chem 1996;271:30835–9.
16. Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science (Wash. DC)1997;275:665–8.
17. Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha.Curr Biol 1997;7:261–9.
18. Stern DF. Tyrosine kinase signalling in breast cancer: ErbB family receptor tyrosinekinases. Breast Cancer Res 2000;2:176–83.
19. Neve RM, Sutterluty H, Pullen N, et al. Effects of oncogenic ErbB2 on G1 cell cycleregulators in breast tumour cells. Oncogene 2000;19:1647–56.
20. Altiok N, Altiok S, Changeux JP. Heregulin-stimulated acetylcholine receptor geneexpression in muscle: requirement for MAP kinase and evidence for a parallelinhibitory pathway independent of electrical activity. EMBO J 1997;16:717–25.
21. Downward J. Lipid-regulated kinases: some common themes at last. Science (Wash.DC) 1998;279:673–4.
22. Wymann MP, Pirola L. Structure and function of phosphoinositide 3-kinases. Bio-chim Biophys Acta 1998;1436:127–50.
23. Alessi DR, Andjelkovic M, Caudwell B, et al. Mechanism of activation of proteinkinase B by insulin and IGF-1. EMBO J 1996;15:6541–51.
24. Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survivalsignals to the cell-intrinsic death machinery. Cell 1997;91:231–41.
25. Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9by phosphorylation. Science (Wash. DC) 1998;282:1318–21.
26. Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes trans-location of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA2001;98:11598–603.
27. Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylatingand inhibiting a Forkhead transcription factor. Cell 1999;96:857–68.
28. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition ofglycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature (Lond.)1995;378:785–9.
29. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol2002;29:15–8.
30. Carmeliet P. Angiogenesis in health and disease. Nat Med 2003;9:653–60.31. Matter A. Tumor angiogenesis as a therapeutic target. Drug Discov Today 2001;6:
1005–24.32. Fong TA, Shawver LK, Sun L, et al. SU5416 is a potent and selective inhibitor of the
vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinasecatalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res1999;59:99–106.
33. Laird AD, Vajkoczy P, Shawver LK, et al. SU6668 is a potent antiangiogenic andantitumor agent that induces regression of established tumors. Cancer Res 2000;60:4152–60.
34. Wedge SR, Ogilvie DJ. Inhibition of VEGF signal transduction. Identification ofZD4190. Adv Exp Med Biol 2000;476:307–10.
35. Wood JM, Bold G, Buchdunger E, et al. PTK787/ZK 222584, a novel and potentinhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairsvascular endothelial growth factor-induced responses and tumor growth after oraladministration. Cancer Res 2000;60:2178–89.
36. Kaipainen A, Korhonen J, Pajusola K, et al. The related FLT4, FLT1, and KDRreceptor tyrosine kinases show distinct expression patterns in human fetal endothelialcells. J Exp Med 1993;178:2077–88.
37. Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE2001;2001:RE21.
38. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med2003;9:669–76.
39. Gerber HP, McMurtrey A, Kowalski J, et al. Vascular endothelial growth factorregulates endothelial cell survival through the phosphatidylinositol 3�-kinase/Aktsignal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem1998;273:30336–43.
40. Dayanir V, Meyer RD, Lashkari K, Rahimi N. Identification of tyrosine residues invascular endothelial growth factor receptor-2/FLK-1 involved in activation of phos-phatidylinositol 3-kinase and cell proliferation. J Biol Chem 2001;276:17686–92.
41. Wu LW, Mayo LD, Dunbar JD, et al. Utilization of distinct signaling pathways byreceptors for vascular endothelial cell growth factor and other mitogens in theinduction of endothelial cell proliferation. J Biol Chem 2000;275:5096–103.
42. Edwards E, Geng L, Tan J, Onishko H, Donnelly E, Hallahan DE. Phosphatidyli-nositol 3-kinase/Akt signaling in the response of vascular endothelium to ionizingradiation. Cancer Res 2002;62:4671–7.
43. Berrie CP. Phosphoinositide 3-kinase inhibition in cancer treatment. Expert OpinInvestig Drugs 2001;10:1085–98.
44. Tenzer A, Zingg D, Rocha S, et al. The phosphatidylinositide 3�-kinase/Akt survivalpathway is a target for the anticancer and radiosensitizing agent PKC412, an inhibitorof protein kinase C. Cancer Res 2001;61:8203–10.
45. Schmidt-Ullrich RK, Contessa JN, Lammering G, Amorino G, Lin PS. ERBBreceptor tyrosine kinases and cellular radiation responses. Oncogene 2003;22:5855–65.
46. Contessa JN, Reardon DB, Todd D, et al. The inducible expression of dominant-negative epidermal growth factor receptor-CD533 results in radiosensitization ofhuman mammary carcinoma cells. Clin Cancer Res 1999;5:405–11.
47. Downward J. Mechanisms and consequences of activation of protein kinase B/Akt.Curr Opin Cell Biol 1998;10:262–7.
48. Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: a hard Akt tofollow. Trends Biochem Sci 2001;26:657–64.
49. Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinaseB/Akt. FEBS Lett 2003;546:108–12.
50. Alessi DR, Cohen P. Mechanism of activation and function of protein kinase B. CurrOpin Genet Dev 1998;8:55–62.
51. Bruns CJ, Solorzano CC, Harbison MT, et al. Blockade of the epidermal growthfactor receptor signaling by a novel tyrosine kinase inhibitor leads to apoptosis ofendothelial cells and therapy of human pancreatic carcinoma. Cancer Res 2000;60:2926–35.
52. Tan J, Hallahan DE. Growth factor-independent activation of protein kinase Bcontributes to the inherent resistance of vascular endothelium to radiation-inducedapoptotic response. Cancer Res 2003;63:7663–7.
5405
VEGF RECEPTOR-DEPENDENT PKB/Akt ACTIVATION
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from

53. Bowers G, Reardon D, Hewitt T, et al. The relative role of ErbB1–4 receptor tyrosinekinases in radiation signal transduction responses of human carcinoma cells. Onco-gene 2001;20:1388–97.
54. Tuttle S, Horan AM, Koch CJ, Held K, Manevich Y, Biaglow J. Radiation-sensitivetyrosine phosphorylation of cellular proteins: sensitive to changes in GSH contentinduced by pretreatment with N-acetyl-L-cysteine or L-buthionine-S,R-sulfoximine.Int J Radiat Oncol Biol Phys 1998;42:833–8.
55. Schmidt-Ullrich RK, Dent P, Grant S, Mikkelsen RB, Valerie K. Signal transductionand cellular radiation responses. Radiat Res 2000;153:245–57.
56. Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB. Ionizingradiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen.Cancer Res 2001;61:3894–901.
57. Leach JK, Black SM, Schmidt-Ullrich RK, Mikkelsen RB. Activation of constitutivenitric-oxide synthase activity is an early signaling event induced by ionizing radiation.J Biol Chem 2002;277:15400–6.
58. Mikkelsen RB, Wardman P. Biological chemistry of reactive oxygen and nitrogen andradiation-induced signal transduction mechanisms. Oncogene 2003;22:5734–54.
59. Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK)complex. Oncogene 1999;18:6867–74.
60. Bhosle SM, Pandey BN, Huilgol NG, Mishra KP. Membrane oxidative damage andapoptosis in cervical carcinoma cells of patients after radiation therapy. Methods CellSci 2002;24:65–8.
61. Dent P, Reardon DB, Park JS, et al. Radiation-induced release of transforming growthfactor alpha activates the epidermal growth factor receptor and mitogen-activatedprotein kinase pathway in carcinoma cells, leading to increased proliferation andprotection from radiation-induced cell death. Mol Biol Cell 1999;10:2493–506.
62. Reardon DB, Contessa JN, Mikkelsen RB, et al. Dominant negative EGFR-CD533and inhibition of MAPK modify JNK1 activation and enhance radiation toxicity ofhuman mammary carcinoma cells. Oncogene 1999;18:4756–66.
63. Autiero M, Waltenberger J, Communi D, et al. Role of PlGF in the intra- andintermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med2003;9:936–43.
64. Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapyregulated by endothelial cell apoptosis. Science (Wash. DC) 2003;300:1155–9.
65. Gorski DH, Beckett MA, Jaskowiak NT, et al. Blockage of the vascular endothelialgrowth factor stress response increases the antitumor effects of ionizing radiation.Cancer Res 1999;59:3374–8.
66. Mauceri HJ, Hanna NN, Beckett MA, et al. Combined effects of angiostatin andionizing radiation in antitumour therapy. Nature (Lond.) 1998;394:287–91.
67. Abdollahi A, Lipson KE, Han X, et al. SU5416 and SU6668 attenuate the angiogeniceffects of radiation-induced tumor cell growth factor production and amplify thedirect anti-endothelial action of radiation in vitro. Cancer Res 2003;63:3755–63.
68. Hess C, Vuong V, Hegyi I, et al. Effect of VEGF receptor inhibitor PTK787/ZK222584 [correction of ZK222548] combined with ionizing radiation on endothelialcells and tumour growth. Br J Cancer 2001;85:2010–6.
5406
VEGF RECEPTOR-DEPENDENT PKB/Akt ACTIVATION
Research. on November 11, 2013. © 2004 American Association for Cancercancerres.aacrjournals.org Downloaded from
Copyright © 2022 FDOKUMEN




















