
Diagnosis and management of adrenal disorders in childhood
55
Diagnosis and management of adrenal disorders in childhood Professor Maria Craig Staff Specialist Institute of Endocrinology & Diabetes Children's Hospital at Westmead University of Sydney, University of NSW
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Diagnosis and management of adrenal disorders in childhood

Diagnosis and management of
adrenal disorders in childhood
Professor Maria CraigStaff Specialist
Institute of Endocrinology & Diabetes
Children's Hospital at Westmead
University of Sydney, University of NSW

Session Outline
• Adrenal physiology – the basics
• Case presentations
• Pheochromocytoma
• Cushing’s disease in an infant
• Hyponatraemia
• Diagnosis and management of PPGL
• Diagnosis and management of Cushing’s
• Diagnosis and management of adrenal
insufficiency and CAH

Fetal adrenal cortex
• Adrenal cortex is derived from mesodermal
gonadal ridge at 5-6wks gestation
• Gonadal ridge cells migrate, giving rise to
steroidogenic cells (adrenal and gonadal)
• At 7-8wks, sympathetic nerve cells (neural
crest/ectodermal origin) invade into primitive
adrenocortical cells – adrenal medulla
• During late gestation, placental oestrogen
promotes fetal cortisol production• Promotes maturation of lung, thyroid, liver, gut
• Fetal adrenal rapidly regresses at birth

Mineralocorticoid
Glucocorticoids
Androgens
CatecholaminesNeural crest origin
Mesothelium
origin

Adrenal cortical hormone synthesis
*
*
* CYP11B2 (in zona glomerulosa)
CYP11B1 (in zona fasciculata)

Adrenal Physiology 1
• Cortisol secretion regulated
by CRH and ACTH
• negative feedback on
both CRH and ACTH
• ACTH secretion also
regulated by cytokines
• CRH regulates ACTH
via production of POMC
• Cortisol secretion
• 7-9 mg/m2/day neonates
• 6-7 mg/m2/day children
• 100-150 mg/m2/day stress

Renin-angiotensin-aldosterone axis
Angiotensinogen Angiotensin I Angiotensin II and III
(liver)
Aldosterone secretion
Potassium ACTH
Renin (kidney) ACE (lung)
+
+ (+)
Aldosterone secretion rate
50 ug/m2/day - adults and older children
> 300 ug/m2/day neonates and young infants
Adrenal Physiology 2
Mineralocorticoid secretion regulated mainly
by RAAS, potassium and ACTH (minor)

Actions of glucocorticoids
• Glucocorticoids influence the regulation of
about 20% of the human genome !
• Intermediary metabolism • Maintenance of plasma glucose
• Lipolysis
• Cardiovascular function• Permissive effects on inotropic, chronotropic &
pressor effects of hormones
• Maintenance of muscle work capacity• muscle enzyme induction

Actions of glucocorticoids 2
• Growth and development
• Induction of fetal enzymes, surfactant
• Immunoregulatory and antiinflammatory actions• Induction of lymphocyte apoptosis
• Renal function
• GFR, sodium and water excretion
• Central nervous system / behaviour
• Bone and connective tissue
• Increase bone resorption and reduce bone formation
• Calcium balance
• Decrease intestinal ca absorption

Actions of mineralocorticoids
• Aldosterone promotes active sodium
resorption and potassium excretion in its
major target tissues
• Target tissues - kidney, colon, salivary glands
• Others: liver, pituitary, brain, mononuclear cells
• Effects via high affinity type I glucocorticoid
(mineralocorticoid) receptors
• Glucocorticoids also act weakly via these
receptors

Adrenal disorders in children
• Adrenal insufficiency
• 72% CAH, 6% other genetic, 13% Addison’s
• CAH: 1 in 15,000-20,000
• Adrenal insufficiency: 1 in 16,000
• Congenital adrenal hypoplasia
• 1 in 12,500 (Japan), less frequent elsewhere
• Phaeochromocytoma: 1 in 100,000
• Adrenal tumor: 1 in 1,000,000

Case 1 - Phaeochromocytoma
Pimonsri Hantanasiriskul
Bangkok, Thailand

PPGL: chromaffin cell tumours
• Pheochromocytoma (80%) - arises from
adrenomedullary chromaffin cells
• 1.7% of children with hypertension
• 5% of incidentally discovered adrenal masses
• Rarely biochemically silent but 1/3 of cases
normotensive and asymptomatic
• Paraganglioma (20%) - from extra-adrenal
chromaffin cells in sympathetic paravertebral
ganglia of thorax, abdomen and pelvis
• At least 1/3 of patients with PPGLs have disease-
causing germline mutations

Phaeochromocytoma: 5 Ps
• Pressure (HTN) ~90%
• Pain (Headache) ~80%
• Perspiration ~71%
• Palpitation ~64%
• Pallor ~42%
• Lack of the first 3 Ps exclude diagnosis of
phaeo

Clinical importance of PPGL
• Catecholamine hypersecretion - high
cardiovascular morbidity and mortality
• Enlargement over time – mass effect
• Earlier diagnosis and treatment in relatives if
familial disease
• Detection of malignant disease
• Metastases in nonchromaffin tissue: 10-17%
• SDHB mutations 40%

PPGL diagnosis
• Initial testing: plasma free metanephrines or
24-hr urinary fractionated metanephrines• Use liquid chromatography with mass spectrometric or
electrochemical detection methods
• Draw blood with the patient in the supine position,
having been recumbent for 30 mins
• False +ve: acetaminophen, labetalol, sotalol, stress
• Imaging if biochemical evidence of PPGL
• CT with contrast rather than MRI - excellent
spatial resolution for thorax, abdomen & pelvis

PPGL Management
• Surgery is first line therapy
• Perioperative pharmacological blockade
• α blockade: Prazosin (usually 10-30 mg TID)
• β blockade: Atenolol (β1) (12.5-25 mg BID) after
at least 1 week of alpha blockade
• Ca channel blockers (start with 5 mg OD)
• High sodium diet and fluid intake
• Duration: 7 to 14 days to allow adequate time to
normalize blood pressure and heart rate

PPGL follow up
• Post op: monitor BP, HR and BGLs
• Biochemical testing 2–4 wk after surgery to
document successful tumour removal
• Lifelong annual biochemical testing to
assess for recurrent or metastatic disease
• High likelihood of hereditary disease if
• positive family history, syndromic features,
multifocal, bilateral or metastatic disease
• even if none of these, mutation rate ~12%

Genetic testing
• Most common mutations
• SDHB 10.3%, SDHD 8.9%, VHL 7.3%,
RET 6.3%, NF1 3.3%
• Clinical feature-driven decisional algorithm
• Genes tested prioritized according to a
syndromic or metastatic presentation
• If paraganglioma – test for SDH mutation
• If metastatic disease – test for SDHB mutations

Lenders et al, JCEM 99: 1915–1942, 2014)

Summary
• Look for associated features
• Look for familial disease
• Look for location, biochemistry and
immunohistochemistry to prioritize genetic
screening in sporadic patients
• clinical feature-driven diagnostic algorithm
• Indefinite monitoring

Case 2
Cushing’s disease in an infant
Mya Sandar Thein
Myanmar

Hypercortisolism
• Physiologic States
1. Stress
2. Pregnancy
3. Chronic strenuous exercise
• Pathophysiologic States
1. Cushing syndrome
2. Psychiatric states
• Depression
• Alcoholism
• Anorexia nervosa
• Anxiety disorders
3. Malnutrition
4. Glucocorticoid resistance

Cushing’s syndrome in childhood
• 1-5 per million population per year
• 10% are paediatric cases
• Median age at diagnosis ~ 9 years*
• Median time between symptoms and
diagnosis 1 year
• F>M• * Güemes M et al, Eur J Peds 2016
• In adults
• Cushing’ s disease > adrenal Cushing’s

Aetiology of Cushing’s
• >7 years old: 85% ACTH dependent • Of these, 80% pituitary dependent, 20% ectopic ACTH
• 15% ACTH independent
• Adenoma 30%, carcinoma 70%
• <7 years old: adrenal tumors (esp infants)
• UK series (all ages)
• 50% pituitary dependent CS
• 36% adrenal dependent CS
• 7% (2 cases) – ectopic ACTH
• Güemes M et al, Eur J Peds 2016
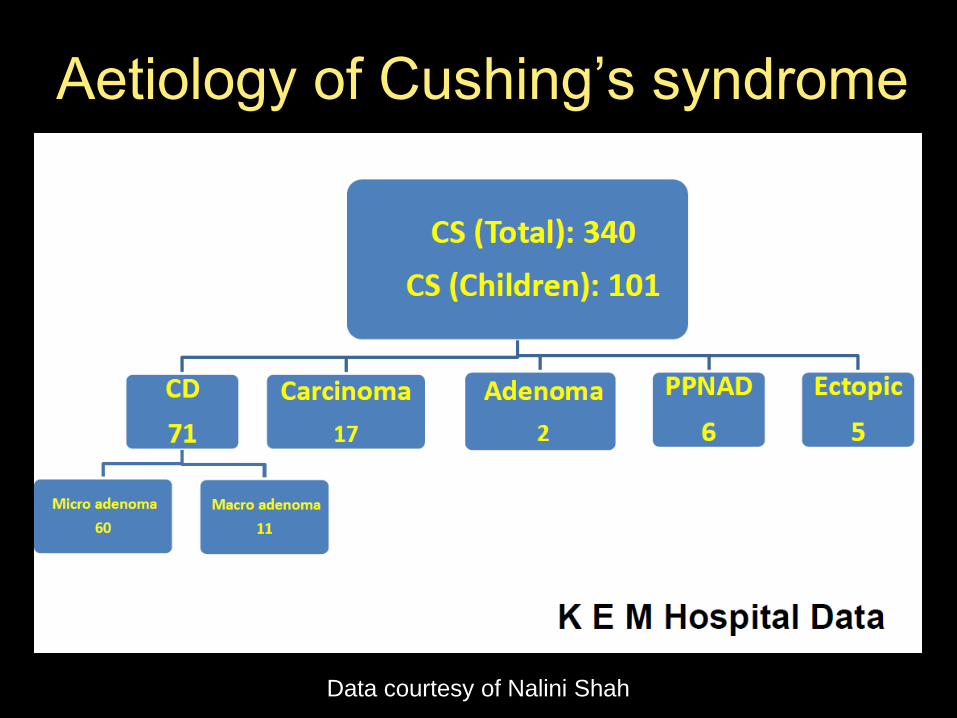
Aetiology of Cushing’s syndrome
Data courtesy of Nalini Shah

Mechanisms of clinical signs
• Excess glucocorticoids
• Excess mineralocorticoids
• Excess adrenal androgens

Clinical symptoms & signs
• Weight gain 77%,
• Hirsutism 57%
• Acne 50%
• Hypertension 50%
• Poor school performance 43%
• Fatigue or weakness 40%
• Growth retardation 37%
• Striae 25%
• Median BMI +2.1 SDS (-6.5 to +4.6)
• Güemes M et al, Eur J Peds 2016

Evaluation of Cushing’s syndrome
1. Establish diagnosis
2. Differentiate causes

Diagnosis of Cushing’s syndrome
• Drug history to exclude iatrogenic CS
• Testing recommended for specific groups
• Patients with unusual features for age (eg
osteoporosis, hypertension)
• Patients with multiple and progressive features
• Children with decreasing height percentile and
increasing weight
• Patients with adrenal incidentaloma compatible
with adenoma

Diagnostic
work up
in Cushing’s
Syndrome

Localisation of endogenous
hypercortisolism
• Clinical assessment
• ACTH : basal, midnight
• Imaging : MRI

Pituitary Adenoma vs.
Ectopic ACTH Secreting Tumour
• False positive (1-3%)
• Normal corticotrophs not fully suppressed in
adrenal disorder
• False negative (6 – 9%)
• Technical factors
• Anatomical variation
• Anomalous venous drainage
• Hypo plastic IPS
• Ectopic tumour in CD

Treatment of Cushing’s syndrome
• Principles:
• Normalize cortisol levels or action at its receptors
to eliminate symptoms and signs of cortisol
• do not treat based on biochemistry alone
• Treat comorbidities
• Multidisciplinary team approach
• Provide educational materials
• Surgery for CD, ectopic and adrenal causes
• 1-2 weeks post op assess for pit hormone def
• 1-3 months post op – MRI

Treatment of Cushing’s disease
• Successful management
• Localisation is essential
• Treatment of choice
• Trans-sphenoid surgery
• For failed surgery or persistent/recurrent CD
• radiotherapy
• bilateral adrenalectomy
• medical management
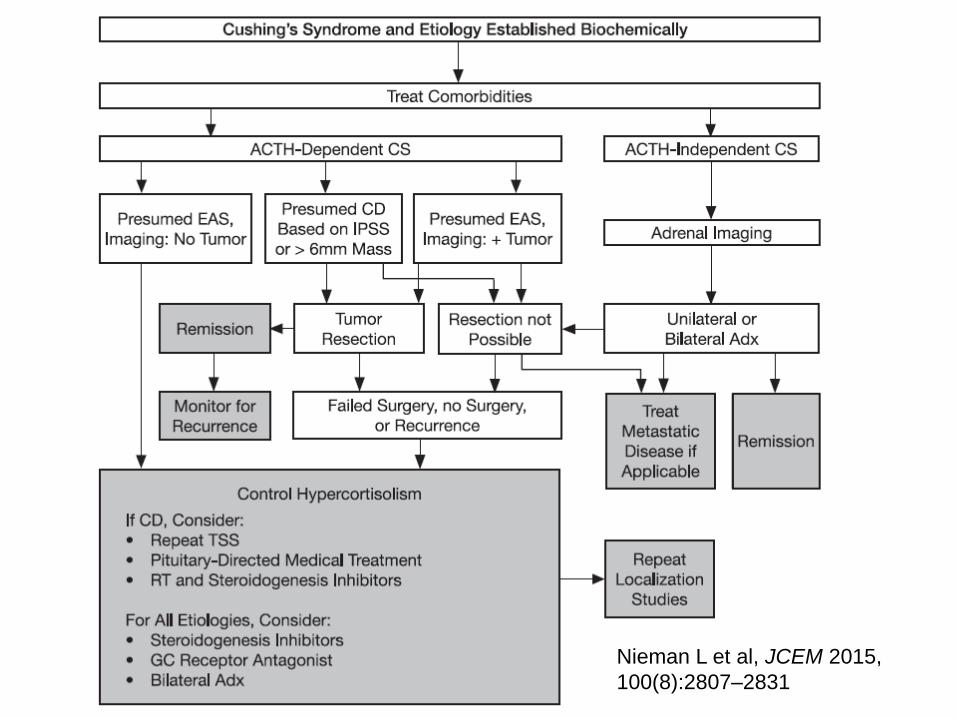
Nieman L et al, JCEM 2015,
100(8):2807–2831

Is there a role for biopsy in
adrenal disease?
• NO, because needle biopsy
• Cannot distinguish a functional cortical
adenoma from a non-functional adenoma
• Cannot reliably distinguish adrenocortical
carcinoma from benign adenoma
• Unfavourable to violate an adrenal cortical
carcinoma with needle biopsy
• Biopsy of an unknown and/or untreated
phaeochromocytoma may result in
haemorrhage and hypertensive crisis
= potentially fatal

Case 3 - Hyponatraemia
Lisa Power
Auckland, New Zealand

Primary Adrenal Insufficiency
ADRENAL PATHOLOGY
1. Autoimmune (Addison’s disease, most common ~80%)
2. Congenital
• Congenital adrenal hyperplasia
• Congenital adrenal hypoplasia (DAX-1, SF-1, IMAGe syn)
• Adrenoleukodystrophy, adrenomyeloneuropathy
• ACTH resistance (FGD: MC2R, MRAP, NNT, TXNRD2, NNT)
• Resistance: glucocorticoid/mineralocorticoid receptor
3. Infection / destruction / haemorrhage / infiltration
• TB, fungal, CMV, meningococcus, anticoagulants, Trauma, emboli, tumour, Sarcoid, Amyloid, Hemochromatosis
4. Surgical – adrenalectomy
5. Adrenal suppressive or antagonist drugs
• Ketoconazole, cyproterone, steroid withdrawal, T4

AAA syndrome
• Alacrima-Achalasia-Adrenal Insufficiency
• AAAA syndrome + autonomic disturbance
• Mutations in ADRACALIN (AAAS) gene (12q13)
• Encodes ALADIN protein of nuclear pore complexes
• Progressive loss of cholinergic function, or melanocortin
receptor signalling
• Alacrimia earliest & most consistent feature
• Crying without tears
• Recurrent vomiting, dysphagia, failure to thrive
• Neurological manifestations at later age

APECED / APS-1: spectrum of illness
• Uncommon – approx 1 per 100,000• 1/9,000 Iranian Jews, 1/10,000,000 Japanese
• 21q22.3 (AIRE – autoimmune regulator)
• High phenotypic variability, even within families• Chronic mucocutaneous candidiasis (~97%)
• Hypoparathyroidism (~96%)
• Addison’s disease (78%)
• Hypothyroidism
• Type 1 diabetes
• Primary gonadal failure
• Chronic active hepatitis
• Pernicious anaemia

Secondary Adrenal Insufficiency
HYPOTHALAMIC - PITUITARY PATHOLOGY
1. HPA axis suppression
• Exogenous or endogenous glucocorticoids
2. Pituitary or hypothalamic disorders
• aplasia, hypoplasia
• Note: 44% of adults with childhood GHD - ACTH deficient
• tumours, surgery, radiation
• autoimmune
• infection
• haemorrhage
• infiltration

AI – metabolic consequences
PRIMARY (ie adrenal pathology)
• Glucocorticoid AND mineralocorticoid deficiency
• Hypotension Acidosis Hyperpigmentation
• Hyponatraemia Renal salt loss Failure to thrive
• Hypoglycaemia Hyperkalaemia (Hypocalcaemia)
SECONDARY OR TERTIARY (ie pituitary or hypothalamic)
• Glucocorticoid deficiency only
• Signs of AI may be non-specific in neonates: • respiratory distress, prolonged cholestatic jaundice, lethargy, FTT,
recurrent vomiting, poor feeding, sepsis
• + hypoglycaemia, seizures
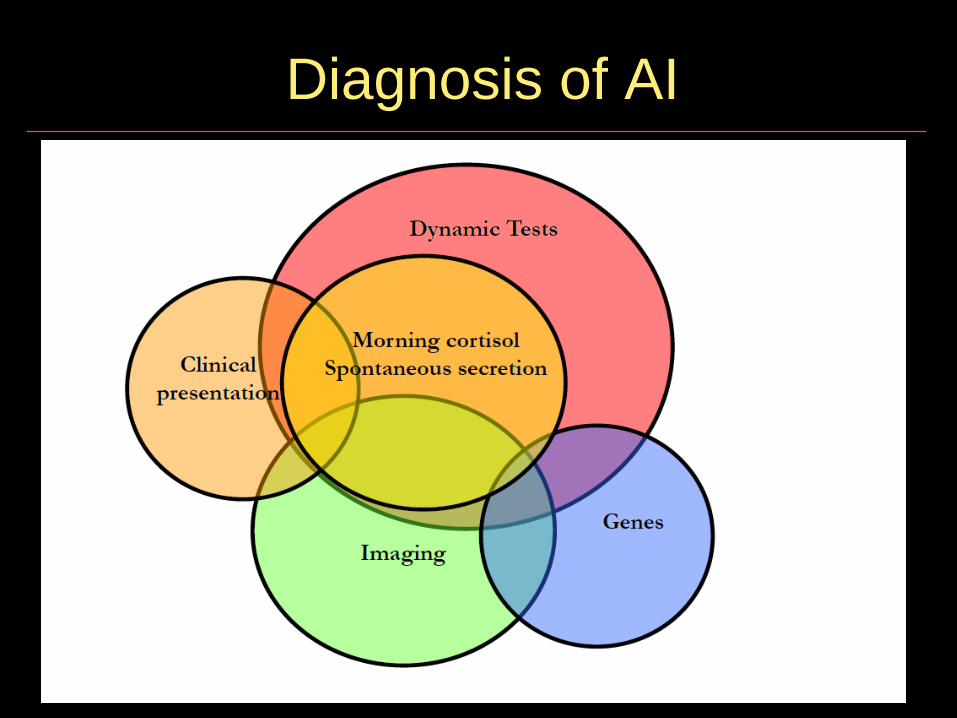
Diagnosis of AI

Diagnostic approach to PAI
Bornstein et al, JCEM 2015, 10(2) 364-389

Confirming the diagnosis of AI
• Site of pathology often evident from clinical presentation
• Serum cortisol during stress • > 500 nmol/l (but may be higher)
• Normal am cortisol 140 – 500nmol/L (afternoon ~ 1/3 am value)
• Short synacthen test • Synacthen dose 250 ug > 2 yr, 125ug <2 yr, 15ug/kg infants
• At 60 minutes peak cortisol > 500 nmol/l ; increment > 280
• Reliable in primary adrenal disease
• Useful in secondary adrenal insufficiency; but note limitations:• Not within 2 weeks of trauma, surgery, change in steroid dose
• Risk of false “normal” results
• If synacthen not possible, morning cortisol + ACTH level
• Renin + aldosterone levels

Laboratory evaluation of adrenal insufficiency
Test Criteria Utility Limitations
Random cortisol
Normal cortisol >600 nmol/l.
Definite AI <35
Acutely ill or hypoglycemic patients.
Useful if high.
Often indeterminate
Morning cortisol
Normal >600 Abnormal<140
Measure of HPA function in stable patients
Often indeterminate, not a good predictor alone
Urinary free cortisol
Not well established
? Normal in 20% with AI
ACTH level Primary AI : ACTH > 100 pg/ml (22 pmol/l)
Localizing. Best test to separate primary from central AI
Not useful as a diagnostic test for AI. Difficult assay.

Acute adrenal crisis
• Fluids - volume resuscitation, glucose
• 0.9% NaCl 20 ml/kg (up to 60ml/kg for shock)
• For hypoglycaemia dextrose 0.5-1.0 g/kg)
• Hydrocortisone bolus 100 mg/m2 BSA
• repeat if poor response, then 100 mg/m 2 QID
• Cardiovascular monitoring and support if needed
• Collect blood for EUC, glucose, ACTH, cortisol,
plasma renin
• Measures to lower K – usually only if very elevated
• Consider and investigate possible aetiology

Long term management
• Hydrocortisone replacement
• 8-12 mg/m2/day in 3-4 doses (start with 8)
• Higher doses for primary vs secondary AI
• Fludrocortisone 0.1 to 0.2 mg daily (start with 0.1)
• Sodium supplements for neonates & infants < 12 mths
• Education of parents, carers, school
• Written instructions for stress cover
• Review instructions at least annually
• Teach family to use IM hydrocortisone
• Medical alert precautions (bracelet, card)
• On call endocrine team contact details
• Screen for other autoimmune diseases periodically

Monitoring replacement
• Monitoring replacement
• Clinical judgement• Symptoms (wellness / energy), height, weight, BP, pigmentation
• CAH• 17OHP, androstenedione, testosterone, PRA, (electrolytes)
• Addison’s disease • PRA, ACTH, (electrolytes)
• Secondary adrenal insufficiency • clinical
• cortisol levels not usually helpful, 24 hour profiles in some cases

Treatment principles for CAH
1. Physiological steroid replacement
2. Suppression of excess ACTH
• To prevent pigmentation
3. Suppression of excess androgens
• To avoid virilisation in females
• To avoid precocious puberty males & females
4. Achieve normal growth
5. Avoid obesity
6. Fertility
7. Psychological well being

Initial treatment
• Hydrocortisone• Starting dose 20 mg/m²/day in 3 divided doses
• Can use higher doses in infancy, but reduce asap
• Do not use suspension
• Fludrocortisone
• 0.15-0.2 mg/day
• Neonates are relatively insensitive to
mineralocorticoids and need larger doses
• + Salt supplementation 3-4 mmol/kg/day
• 1 mmol/kg.day

Long term treatment
• Hydrocortisone 10-15 mg/m²/day
• Over treatment• Growth delay
• Cushingoid features
• PCOS
• Under treatment• Hyperandrogenism
• Advanced bone age
• Precocious puberty
• Short adult height (early epiphyseal closure) − Note pubertal growth is diminished
• Fludrocortisone 0.05-0.2 mg/day

Additional considerations
• Stress dosing for febrile illness (>38.5 C),
gastroenteritis with dehydration, surgery with
general anaesthesia, major trauma
• Not for mental and emotional stress, minor illness or
before physical exercise
• Medical alert precautions
• Monitoring:
• Consistently timed hormone measurement – avoid
complete suppression of androgen levels
• Clinical: height, weight, and physical examination
• Annual bone age after age 2 years

Summary and key points
• Wide spectrum of adrenal disease in
childhood – understanding adrenal gland
development and physiology is essential to
aid diagnosis and management
• Tailor therapy to aetiology and age
• Careful clinical & assay monitoring to avoid
consequences of over or under-treatment
• Education of patients and carers is critical,
with regular review of stress management




















