
design, synthesis and characterization of - OhioLINK ETD
449
DESIGN, SYNTHESIS AND CHARACTERIZATION OF FLUORESCENT DYES AND LIQUID CRYSTAL SEMICONDUCTORS A dissertation submitted to Kent State University in partial fulfillment of the requirements for the degree of Doctor of Philosophy by Alexander N. Semyonov August 2006
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of design, synthesis and characterization of - OhioLINK ETD

DESIGN, SYNTHESIS AND CHARACTERIZATION OF
FLUORESCENT DYES AND
LIQUID CRYSTAL SEMICONDUCTORS
A dissertation submitted to Kent State University
in partial fulfillment of the requirements for the
degree of Doctor of Philosophy
by Alexander N. Semyonov
August 2006

ii
Dissertation written by
Alexander N. Semyonov
B.S., Saratov State University, 1997
Ph.D., Kent State University, 2006
Approved by
Robert J. Twieg Dissertation Committee Chair
Carmen C. Almasan Dissertation Committee Member
Brett D. Ellman Dissertation Committee Member
Peter Palffy-Muhoray Dissertation Committee Member
John L. West Dissertation Committee Member
Accepted by
Oleg D. Lavrentovich Chair,
Chemical Physics Interdisciplinary Program
John R. D. Stalvey Dean, College of Arts and Sciences

iii
TABLE OF CONTENTS
LIST OF ABBREVIATIONS........................................................................................... vii
LIST OF FIGURES .......................................................................................................... xii
LIST OF SCHEMES..........................................................................................................xv
LIST OF TABLES.......................................................................................................... xxii
CHAPTER I. DPP FOR SINGLE-MOLECULE SPECTROSCOPY.........................1
1.1. Single-Molecule Spectroscopy and its Applications ........................................1
1.2. Dye Requirements for Single-Molecule Spectroscopy.....................................7
1.3. Review of known DPP chemistry ...................................................................17
1.3.2. Chemical Properties .....................................................................................32
1.3.3. Physical Properties.......................................................................................44
1.4. Newly prepared DPP dyes ..............................................................................60
1.4.1. Alkylation of DPPs ......................................................................................69
1.4.2. Action of bases on DPP ...............................................................................72
1.4.3. Halogenation of DPPs..................................................................................77
1.4.4. Substitution of halogen by amine in DPPs ..................................................80
1.4.5. Extension of the conjugation chain of DPP .................................................83
1.4.6. DPPs with hydrophilic solubilizing groups .................................................87
1.4.7. Hydroxy-functionalized DPPs .....................................................................88

iv
1.4.8. DPPs with a cysteine-reactive maleimide moiety........................................99
1.4.9. N-Arylated DPPs .......................................................................................102
1.4.10. Physical Properties of Newly Prepared DPPs..........................................105
1.4.11. Conclusion ...............................................................................................115
CHAPTER II. CYSTEINE-SPECIFIC FLUORESCENT TAGS: NILE RED –
MALEIMIDE AND DCDHF – MALEIMIDE............................................................116
2.1. Introduction to molecular probes and tags....................................................116
2.2. Design of the Probes .....................................................................................122
2.3. Synthesis .......................................................................................................130
2.4. Results and Discussion .................................................................................140
2.5. Conclusion ....................................................................................................149
CHAPTER III. ORGANIC LIQUID CRYSTAL SEMICONDUCTORS...............150
3.1. Introduction...................................................................................................150
3.2. Polyacenes.....................................................................................................161
3.2.1. Anthracenes................................................................................................162
3.2.1.1. 2,3,6,7-Tetraalkoxyanthracenes ..............................................................163
3.2.1.2. 2,3,6,7-tetraalkoxy-9,10-dialkyltetracenes .............................................169
3.2.1.3. 1,2,3,4,5,6,7,8-octaalkylanthracenes.......................................................171
3.2.2. Tetracenes ..................................................................................................172
3.2.3. Pentacenes..................................................................................................175
3.3. Iodoarenes .....................................................................................................195
3.3.1. Why Iodine?...............................................................................................195

v
3.3.2. Direct iodination ........................................................................................198
3.3.3. Iodo-de-diazoniation ..................................................................................200
3.3.4. Halogen exchange......................................................................................202
3.4. Liquid Crystal Semiconductors.....................................................................204
3.4.1. HAT Discotic Liquid Crystals ...................................................................204
3.4.2. Nitrated HAT5 Discotic Liquid Crystals ...................................................209
3.4.3. Conclusions................................................................................................211
CHAPTER IV. EXPERIMENTAL PART. .................................................................212
4.1. General Instrumentation and Techniques .....................................................212
Measurement of Fluorescence Quantum Yield........................................212
Measurement of Fluorophore’s Photostability.........................................215
Gas Chromatogrphy – Mass Spectroscopy (GC-MS)..............................217
HPLC-MS ................................................................................................219
Nuclear Magnetic Resonance (NMR)......................................................220
Thermal Analysis: DSC and TGA ...........................................................221
M.Braun SPS............................................................................................222
High Pressure Reactors ............................................................................222
UV-Vis.....................................................................................................223
IR..............................................................................................................224
4.2. Synthetic Procedures.....................................................................................225
CONCLUSIONS ............................................................................................................355
REFERENCES...............................................................................................................357

vi
APPENDIX A..................................................................................................................393
APPENDIX B ..................................................................................................................403
APPENDIX C ..................................................................................................................417

vii
LIST OF ABBREVIATIONS
A Absorbance (value), II0lg
Ab absorbance (subscript)
Ac acetyl, CH3CO–
Ac2O acetic anhydride
AcOH acetic acid
AFM atomic force microscopy
APCI atmospheric pressure chemical ionization
BHT 4-t-butylhydroxytoluene
Am amyl, pentyl
t-AmOH tert-amyl alcohol, 2-methyl-2-butanol
ca. circa, approximately
cf. confer, compare with
Chx cyclohexyl
Δ delta, difference between two values
Δλ Stokes’ shift
Δx refluxing solvent or at solvent’s refluxing temperature
CAS RN Chemical Abstracts Service Registry Number
CDI 1,1′-carbonyldiimidazole

viii
Cys cysteine
d doublet
DBU 1,8-diazabicyclo[5.4.0]undec-7-ene
DCC 1,3-dicyclohexylcarbodiimide
DEAD diethyl azodicarboxylate
DIB 1,4-diiodobenzene
DMAc dimethylacetamide
DMAE or deanol — N,N-dimethylaminoethanol
DMAP 4-(dimethylamino)pyridine
DMF dimethylformamide
DPP 2H,5H-dihydropyrrolo[3,4-c]pyrrole-1,4-dione
DSC differential scanning calorimetry
Em Emission
ESI electro-spray ionization
ε molar extinction coefficient, l·mol–1·cm–1
f femto, 10–15
FRET fluorescence resonance energy transfer
FCS fluorescence correlation spectroscopy
Fu furyl
HAT-5 hexapentyloxytriphenylene
HMDS 1,1,1,3,3,3-Hexamethyldisilazane
HMPA hexamethylphosphoramide

ix
ID internal diameter
i, iso prefix to denote isomeric branching at the terminus of a substituent
kF total rate of fluorescent decay
kisc intersystem crossing rate
knr total rate of non-radiative decay
kT decay rate from T1 to S0
λ wavelength
L, l liter
l, ℓ length
lg decimal logarithm
ln natural logarithm
m meter
MI maleimide residue, 1H-pyrrole-2,5-dione-1-yl
mm Hg pressure unit of millimeters of mercury
mol mole, NA number of species
μ micro, 10–6
NA Avogadro’s number, 6.0245·1023 of structural units
25Dn refractive index at 25 °C for center of sodium D-line doublet (589 nm)
n nano, 10–9
p pico, 10–12
P pressure, followed by value and units: P = 2 mm Hg
Ph phenyl

x
PPA polyphosphoric acid, 2H3PO4•P2O5
ppb parts per billion
ppm parts per million
PPTS pyridinium 4-(para)-tolenesulfonate
PTFE poly(tetrafluoroethylene), Teflon®
RET resonance energy transfer
q quartet
r.t. room temperature
s singlet
sec- secondary
sec second
σp absorption cross-section
S0 ground singlet state
S1 first excited singlet state
SMS single-molecule spectroscopy
STM scanning tunneling microscopy
Suc succinyl acyl residue, (CH2CO)2
t-, tert- tertiary
t triplet
TGA thermal gravimetric analysis
THF tetrahydrofuran
THP tetrahydropyran-2-yl

xi
TMEDA N,N,N′,N′-Tetramethylethylenediamine
TriMEDA N,N,N′-Trimethylethylenediamine
Ts tosyl, 4-toluylsulfonate (4-methylbenzenesulfonate)
TsCl tosyl chloride, 4-toluylsulfonyl chloride
UV-Vis ultra-violet and visible (spectra, properties or data)
y yocto, 10–24
τ1 fluorescence lifetime
T1 first excited triplet state
δ chemical shift
ν wavenumber
ΦF fluorescence quantum yield
φ b photobleaching quantum efficiency
[####-##-#] CAS Registry Number

xii
LIST OF FIGURES
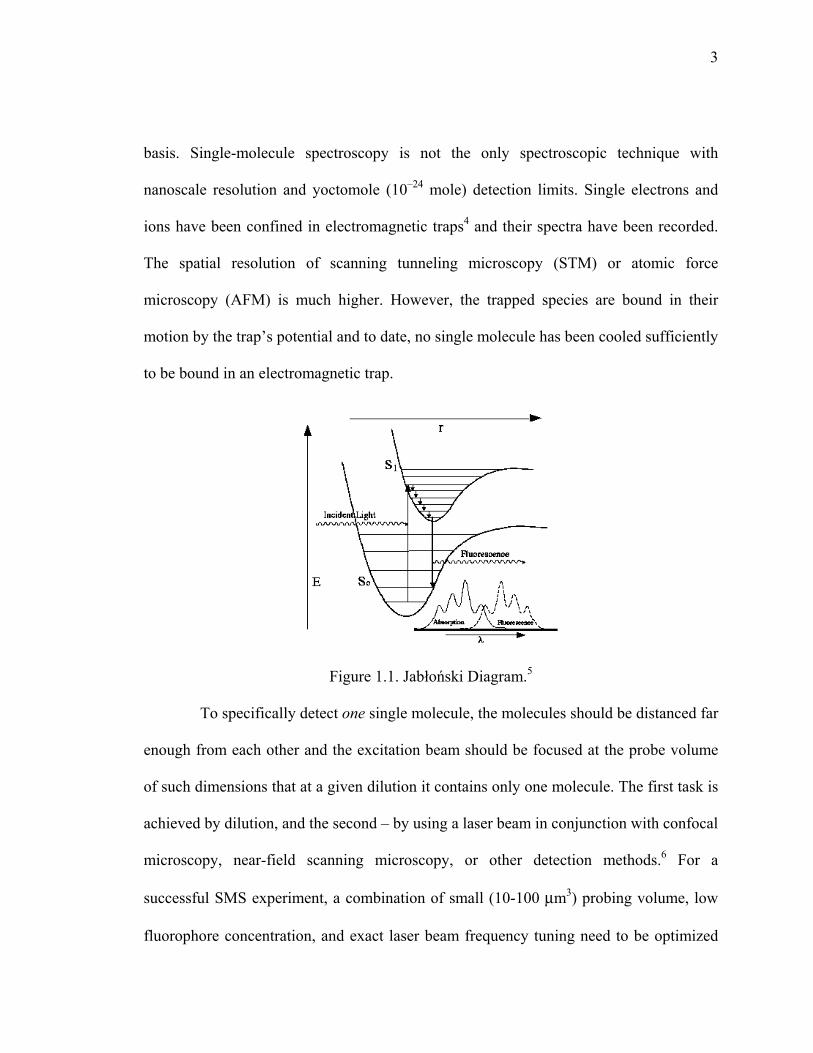
Figure 1.1. Jabłoński Diagram. ............................................................................................3
Figure 1.2. Irreversible photobleaching (at 5.6 sec) of single Cy5 molecule, immobilized
on a glass slide. .............................................................................................10
Figure 1.3. X-ray molecular structure of DPP and DPP-Me. ............................................45
Figure 1.4. Crystal structure of (a) DPP – triclinic and (b) DPP-Me – orthorombic.........46
Figure 1.5. Overlap of the two molecules along the stacking axis: (a) DPP and (b) DPP-
Me. ................................................................................................................46
Figure 1.6. DSC of (a) crude and (b) conditioned DPP. ....................................................49
Figure 1.7. TGA traces of crude DPP (red) and Br-DPP (blue). .......................................50
Figure 1.8. Visible spectrum of DPP as NMP solution (absorption) vs. solid (reflection).51
Figure 1.9. Absorption (S), fluorescence in CHCl3 solution (F), and solid state
fluorescence (SF) of DPP and 4-t-Bu-DPP...................................................55
Figure 1.10. 1H and 13C NMR spectra of 3,6-diphenyl-2,5-diallylpyrrolo[3.4-c]pyrrole-
1,4-dione. ......................................................................................................58
Figure 1.11. 1H and 13C Assigned chemical shifts for 3,6-diphenyl-2,5-diallylpyrrolo[3.4-
c]pyrrole-1,4-dione. ......................................................................................58
Figure 1.12. Effect of fast vs. slow work-up on the purity of crude DPP..........................68

xiii
Figure 1.13. Single molecules of DPP-Me 23 imaged in a PMMA film, excited at
wavelength of 488 nm with an intensity of 0.85 kW·cm–2. ........................106
Figure 1.14. Photostability of compounds 32, 33, 64, and 65. ........................................107
Figure 1.15. Photostability of compounds 32, 91, 92, and 93. ........................................108
Figure 2.1. Generalized structure of a bioconjugate fluorescent molecular probe. .........117
Figure 2.2. Common dye classes, used in molecular probes. ..........................................122
Figure 2.3. Nile Red absorption (Ab) and emission (Em) spectra in dioxane. ................125
Figure 2.4. DCDHF-6, imaged at single molecule level in PMMA film.........................127
Figure 2.5. Fluorescence intensity of 123 decreases with solvent polarity increase. ......142
Figure 2.6. Ribbon model of GroEL homotetradecamer. ................................................144
Figure 2.7. Ribbon model of GroEL protein....................................................................145
Figure 2.8. Fluorescence change after addition of (1) MDH, (2) GroES, (3) nucleotide.144
Figure 2.9. Effect of various addition orders on the fluorescence intensity. ...................145
Figure 2.10. Proposed scheme for the formation of symmetric/asymmetric complex of
GroEL/GroES with ADP/AlFx.. .................................................................147
Figure 3.1. Conductivity domains of metals, semiconductors, and insulators. ...............151
Figure 3.2. Conduction electron concentrations in different materials............................154
Figure 3.3. Temperature dependence of conductance electron concentration in Ge and Si.154
Figure 3.4. Effect of doping impurity (Sb, donor) concentrations on the resistivity of Ge
as a function of inverse temperature. ..........................................................155
Figure 3.5. Band structure of an intrinsic inorganic semiconductor................................156
Figure 3.6. Mobility of (a) ultrapurified and (b) conventionally purified perylene.........159

xiv
Figure 3.7. TGA Analysis of (a) tetracene from TCI America; (b) crude pentacene 163;
(c) pentacene from TCI America; pentacene, sublimed (d) once and (e)
twice............................................................................................................176
Figure 3.8. Crystal structure of (a) pentacene and (b) 1,2,3,4-tetrafluoro-6,13-bis(2-
diisopropulsilylethynyl)pentacene. .............................................................181
Figure 3.9. Interatomic distances in a-DIB unit cell. .......................................................196
Figure 3.10. Intermolecular iodines’ p-orbital overlap in crystalline a-DIB. ..................197 Figure 3.11. Significance of various HAT-n compounds represented as number of
publications for each member of the homologous series............................205
Figure 4.1. Photodegradation of compound 93 and its exponential fit. ...........................215

xv
LIST OF SCHEMES
Scheme 1.1. Farnum’s synthesis of DPP. ..........................................................................18
Scheme 1.2. Condensation of benzonitrile with alkyl succinate. ......................................21
Scheme 1.3. Self-condensation of alkyl succinate under basic conditions........................21
Scheme 1.4. Synthetic approaches to the intermediates 2 and 3. ......................................24
Scheme 1.5. Stobbe condensation of a Schiff base with alkyl succinate...........................26
Scheme 1.6. Preparation of DPP from benzylaniline. .......................................................26
Scheme 1.7. Mechanism of DPP formation from benzonitrile and succinate. ..................29
Scheme 1.8. Condensation of succindiamide with N,N,-dimethylbenzamide...................30
Scheme 1.10. The sole example of an unsymmetrical N,N′-diaryl DPP. ..........................31
Scheme 1.11. The sole example of aliphatic amine reaction with furo[3,4-c]furane-1,4-
dione..............................................................................................................31
Scheme 1.12. Routes to 3,6-diphenyl-furo[3,4-c]furan-1,4-dione.....................................32
Scheme 1.13. Reactive sites of DPP. .................................................................................33
Scheme 1.14. Various reactions of DPP. ...........................................................................33
Scheme 1.15. N-Alkylation of DPP...................................................................................34
Scheme 1.16. Acylation of DPP with di-tert-butyl dicarbonate. .......................................35
Scheme 1.17. Acylation of DPP with di(2-methyl-3-buten-2-yl) dicarbonate and
subsequent decomposition. ...........................................................................35

xvi
Scheme 1.18. N-Hydroxymethylation of DPP...................................................................36
Scheme 1.19. Reactions of N,N′-bis(hydroxymethyl) DPP...............................................37
Scheme 1.20. DPP bromination and chlorination products. ..............................................38
Scheme 1.21. Mechanism of DPP bromination. ................................................................38
Scheme 1.22. Sulfonation of DPP......................................................................................39
Scheme 1.23. Replacement of O in –NH–C=O by C. .......................................................39
Scheme 1.24. Replacement of O in –NH–C=O by N. .......................................................40
Scheme 1.25. Replacement of O in –NH–C=O by N–CN (cyanoimination of DPP). ......40
Scheme 1.26. Nitration and aromatic nucleophilic substitution of chlorine in DPP. ........41
Scheme 1.27. Aromatic nucleophilic substitution of bromine in DPP. .............................42
Scheme 1.28. Pd coupling of Br-DPP with CO in presence of calcium formate. .............42
Scheme 1.29. Pd coupling of Br-DPP with CO in presence of MeOH. ............................42
Scheme 1.30. Pd coupling of Br-DPP with CO in presence of butylamine.......................42
Scheme 1.31. Stille coupling polymerization of DPP........................................................43
Scheme 1.32. Suzuki coupling polymerization of DPP.....................................................43
Scheme 1.33. Design of red-shifted DPP chromophores...................................................63
Scheme 1.34. Preparation of sterically hindered di-alkyl succinates.................................64
Scheme 1.35. Preparation of 4-R-benzonitriles: R= F (13), Br (14), OMe (15)................65
Scheme 1.36. Preparation of 4-aminobenzonitriles. ..........................................................65
Scheme 1.37. Alkylation of DPP.. .....................................................................................71
Scheme 1.38. Step-wise alkylation process of DPP. .........................................................72
Scheme 1.39. High temperature basic degradation of DPP. ..............................................76

xvii
Scheme 1.40. Michael addition of OH– to DPP-Me. ........................................................77
Scheme 1.41. Bromination reactions of DPPs. ..................................................................80
Scheme 1.42. Indirect approach to iodo-DPPs. .................................................................81
Scheme 1.43. Conversion of Br-DPP-Pr to CN-DPP-Pr. ..................................................82
Scheme 1.44. Heck coupling between X-DPP-Me with 4-tert-butyl- and 4-acetoxy-
styrenes. ........................................................................................................83
Scheme 1.45. Preparation of styrenes. ...............................................................................84
Scheme 1.46. Pd-catalyzed coupling between styrene 57 and Br-DPP-Pr 44. ..................85
Scheme 1.47. Pd-catylized coupling between styrene 59 and Br-DPP-Pr 44....................85
Scheme 1.48. Preparation of thien-2-yl zinc reagents 62 and 63. Negishi coupling
between Br-DPP-Pr 44 and thien-2-yl zinc chloride 62. ..............................86
Scheme 1.49. Negishi coupling between Br-DPP-Pr 44 and 5-(4-(N,N-di-n-
hexylamino)phenyl)-thien-2-yl zinc chloride 63. .........................................87
Scheme 1.50. Introducing sulfonic acid group into DPP structure....................................88
Scheme 1.51. Proposed direct introduction of alcohol functionality into DPP by
alkylation with ω-halo-α-alcohols. ...............................................................89
Scheme 1.52. The sole successful example of DPP alkylation with ω-halo-α-alcohol in
presence of t-BuOK. .....................................................................................90
Scheme 1.53. Attempted hydroboration route to alcohol-functionalized DPPs. ...............91
Scheme 1.54. Cu/CuI-cat. coupling of 2-(ethylamino)ethanol with I-DPP-Pr 46. ............92
Scheme 1.55. Alkylation approach to alcohol-functionalized DPPs with protection-
deprotection of the hydroxy group................................................................93

xviii
Scheme 1.56. Preparation of alkylating reagents with alcohol functionality, protected
with a THP protecting group.........................................................................93
Scheme 1.57. Reaction of THP- protected alcohols 71–73 with DPP...............................95
Scheme 1.58. Preparation of alkylating reagent 75 with alcohol functionality, protected
with a benzoate ester.....................................................................................95
Scheme 1.59. Reaction of benzoate ester protected alcohol 74 with DPP and subsequent
removal of the protecting group....................................................................96
Scheme 1.60. Preparation of complimentary mono-alcohol functionalized DPPs. ...........97
Scheme 1.61. Direct alkylation of DPPs with ω-halo-α-alcohols in presence of Cs2CO3.98
Scheme 1.62. Bis(maleimide) DPP derivative.................................................................100
Scheme 1.63. An attempt towards maleimide mono-functionalized DPP 90..................101
Scheme 1.64. Direct copper-mediated N-arylation of isatins. .........................................102
Scheme 1.65. Direct copper-mediated N-arylation of DPP 1. .........................................103
Scheme 1.66. Aromatic nucleophilic substitution on DPP. .............................................104
Scheme 2.1. Fluorophores conceptually similar in structure...........................................124
Scheme 2.2. Examples of new DCDHF dyes. .................................................................126
Scheme 2.3. The target fluorescent tags with fluorophores, hook, and spacer of choice.128
Scheme 2.4. Proposed attachment of the maleimide hook via N-(2-hydroxyethyl) group.129
Scheme 2.5. Proposed synthesis of Nile Red derivative 99 and attachment of the
maleimide hook via phenol functionality. ..................................................130
Scheme 2.5. Preparation of Nile Red Phenol...................................................................131
Scheme 2.6. C6-Spacer attachment to Nile Red Phenol...................................................131

xix
Scheme 2.7. Syntheses of maleimides. ............................................................................132
Scheme 2.8. Syntheses of maleimides from maleic anhydride........................................133
Scheme 2.9. Attempted Mitsunobu reactions between Nile Reds 105 and 106 and various
hydroxy-functionalized maleimides............................................................134
Scheme 2.10. Attempted N-alkylation of maleimide according to a reported protocol. .134
Scheme 2.11. Simple alkylations and acylations of Nile Red Phenol 115. .....................135
Scheme 2.12. Diels-Alder adduct of furan and maleic anhydride Fu-MA. .....................137
Scheme 2.13. Synthesis of protected Nile Red – Maleimide 122....................................137
Scheme 2.14. Retro Diels-Alder deprotection of 122......................................................138
Scheme 2.15. Synthesis of maleimide-tagged DCDHF-2V 124......................................139
Scheme 2.16. Synthesis of maleimide-tagged DCDHF-6 125. .......................................140
Scheme 3.1. Target polysubstituted n-acenes. .................................................................162
Scheme 3.2. Various approaches to 2,3,6,7-tetraalkoxyanthracene via 2,3,6,7-tetraalkoxy-
9,10-anthraquinone. ....................................................................................164
Scheme 3.3. Attempted self-acylation of veratroyl chloride. ..........................................165
Scheme 3.4. Ortho-lithiation approach to 138. ................................................................166
Scheme 3.5. Oxidation and cyclization of 138 into anthraquinone 130. .........................167
Scheme 3.4. Attempted preparation of 133. ....................................................................167
Scheme 3.5. Preparation of 2,3,6,7-tetraalkoxy-9,10-dihydroanthracenes......................168
Scheme 3.6. Conversion of methoxy groups into hydroxyl functionalities.....................169
Scheme 3.7. Synthesis of 2,3,6,7-Tetraalkoxy-9,10-dialkylanthracenes. ........................171

xx
Scheme 3.8. Preparation of 1,2,3,4,5,6,7,8-octaalkylanthracenes by Pd-catalyzed ring
extension reaction. ......................................................................................171
Scheme 3.9. Peralkylation of 9,10-dihydroanthracene with heptyl bromide...................172
Scheme 3.10. Synthesis of tetracene 157.........................................................................173
Scheme 3.11. Synthesis of 2,3-Bis(decyloxy)tetracene 162............................................174
Scheme 3.12. Synthesis of pentacene. .............................................................................177
Scheme 3.13. Preparation of pentacenequinone 164 via 1,4-anthracenequinone 167.....182
Scheme 3.14. Synthesis of the key intermediates for 1-fluoropentacene. .......................183
Scheme 3.15. Synthesis of 2-fluoropentacene. ................................................................185
Scheme 3.16. Retrosynthetic analysis of alkoxypentacenes............................................187
Scheme 3.17. Ortho-lithiation approach to 4,5-dimethoxyphthalaldehyde 158. .............188
Scheme 3.18. Three-step synthesis of 158 from veratrole...............................................191
Scheme 3.19. Synthesis of 2,3,9,10-tetramethoxypentacene...........................................192
Scheme 3.20. Soluble and stable pentacene ethers by Anthony. .....................................193
Scheme 3.21. Attempted synthesis of 2,3,9,10-tetrahexylpentacene 183........................193
Scheme 3.22. Reversible Diels-Alder adduct of pentacene. ............................................194
Scheme 3.23. Preparation of 2,4,6,8-tetraiodoglycoluril. ................................................198
Scheme 3.24. Iodoarenes prepared by direct iodination. .................................................200
Scheme 3.25. Iodo-de-diazoniation approach to some iodotoluenes...............................201
Scheme 3.26. Routes to 1,4-diiodonaphthalene...............................................................203
Scheme 3.27. Preparation of 2,3-diiodonaphthalene .......................................................204
Scheme 3.28. Oxidative trimerization of 1,2-dialkoxybenzene to HAT-n. .....................207

xxi
Scheme 3.29. Main by-products of HAT-n synthesis by oxidative trimerization. ..........207
Scheme 3.30. Synthesis of MN-HAT-5 and TN-HAT-5.................................................210

xxii
LIST OF TABLES
Table 1.1. Typical SMS fluorophores................................................................................17
Table 1.2. DPP compounds prepared by Reformatsky reaction. .......................................20
Table 1.3. DPP compounds prepared by aromatic nitrile – succinate ester condensations.24
Table 1.4. DPP compounds prepared by step-wise condensations. ...................................26
Table 1.5. Solubilities of various DPPs in mol·liter–1........................................................48
Table 1.6. Influence of 3,6-substituents and crystal structure on absorptive properties of
DPP. .................................................................................................................52
Table 1.7. Fluorescence data for several DPPs in chloroform...........................................53
Table 1.8. Fluorescence in solution and solid state of some N-phenyl substituted DPPs. 54
Table 1.9. Fluorescence quantum yields of some N-mono-aryl and N,N′-diaryl substituted
DPPs.................................................................................................................56
Table 1.10. 1H Chemical shifts of the NH group of DPP in different solvents and at
various temperatures. .......................................................................................59
Table 1.11. 13C Chemical shifts of DPP, its mono- and di-anion. .....................................59
Table 1.12. DPP preparation reactions. .............................................................................65
Table 1.13. N-Alkylated DPPs...........................................................................................72
Table 1.14. Halogenated DPPs. .........................................................................................81
Table 1.15. Aminated DPPs...............................................................................................82
Table 1.16. Photostability of several DPPs......................................................................109

xxiii
Table 1.17. Symmetrical 4,4’–Disubstituted DPPs. ........................................................110
Table 1.18. N– and N,N′-substituted DPPs......................................................................112
Table 2.1. Amine (R2–NH2) reactive groups for molecular probes. ...............................118
Table 2.2. Thiol (R2–SH) reactive groups for molecular probes. ....................................120
Table 2.3. Optical properties of maleimide-containing fluorescent tags. ........................141
Table 3.1. Carrier mobilities m in some crystalline inorganic semiconductors at room
temperature, in cm2/V·s. ................................................................................153
Table 3.2. Key differences between metals, semimetals, intrinsic inorganic
semiconductors, and insulators at room temperature.....................................153
Table 3.3. Carrier mobilities (of electrons e– and holes p+) in some organic molecular
crystals at room temperature..........................................................................158
Table 3.4. 2,3,6,7-Tetraalkoxy-9,10-dialkylanthracenes. ................................................170

1
CHAPTER I.
DPP FOR SINGLE-MOLECULE SPECTROSCOPY
1.1. Single-Molecule Spectroscopy and its Applications
Spectroscopy (Latin spectrum – image, impression, from specĕre – to look; and
Greek σκοπια, – observation, from σκοπειν – examine, look at) is a physical method of
study of matter based on observation of radiation intensity as a function of frequency,
wavelength or other parameter (m/z, polarization, energy, etc.). The radiation may be of
electromagnetic (i.e. optical spectroscopy), sonic, or particulate (mass spectroscopy)
origin. The electromagnetic radiation may be emitted, absorbed, reflected, scattered or
transformed into another form. Luminescence (Latin lūmin, lūmen – light, genitive case
lūminis, and -ēscentem, from -ēscĕre – ‘beginning to assume a certain state’) is a physical
phenomenon, where matter, after being excited, emits light in excess of thermal radiation,
and that emission lasts significantly longer than the oscillation period of the light being
emitted. Luminescence differs from scattering, reflection, deceleration emission
(Bremsstrahlung), Vavilov-Cherenkov radiation, or parametric transformation of light. If
the excitation is electromagnetic, the phase or polarization of the emitted light does not

2
correlate with the phase of the excitation light, resulting, for example, in the
depolarization of the luminescence in isotropic solutions of otherwise randomly oriented
fluorophores. There are several types of luminescence, classified by the excitation
sources – light (photoluminescence), ionizing radiation (radioluminescence), X-ray,
electrical field (electroluminescence), beam of electrones (cathodoluminescence),
mechanical force (triboluminescence), crystallization1, or (bio)chemical reaction (chemo-
and bio-luminescence). Photoluminescence splits into phosphorescence and
fluorescence. Fluorescence is short (10–9 to 1 sec) light emission due to transitions
between excited and ground states of the same multiplicity, usually singlets (S1 S0). In
that it is different from phosphorescence, which is usually long (1 sec to days) light
emission due to forbidden (i.e. low probability) transitions between excited and ground
states of different multiplicity, usually the excited triplet state and the ground singlet
state. Fluorescence quantum yield ΦF is a ratio of number of photons emitted to the
number of photons absorbed by the same amount of the substance.
Conventional spectroscopy observes a large number of molecules at once and
the spectral signal is averaged over the statistical ensemble. Single-molecule
spectroscopy (SMS) is a relatively new (first SMS experiment was performed in 19892),
yet well-elaborated technique, which detects fluorescence from one single molecule.3
This allows the full distribution of optical values (fluorescence intensity, fluorescence
decay, emission spectrum, diffusion coefficient, fluorescence anisotropy, polarization,
and lifetime) to be recorded and molecular heterogeneity in optical properties, as well as
many molecular time-dependent state changes to be measured on a molecule by molecule
3
basis. Single-molecule spectroscopy is not the only spectroscopic technique with
nanoscale resolution and yoctomole (10–24 mole) detection limits. Single electrons and
ions have been confined in electromagnetic traps4 and their spectra have been recorded.
The spatial resolution of scanning tunneling microscopy (STM) or atomic force
microscopy (AFM) is much higher. However, the trapped species are bound in their
motion by the trap’s potential and to date, no single molecule has been cooled sufficiently
to be bound in an electromagnetic trap.
Figure 1.1. Jabłoński Diagram.5
To specifically detect one single molecule, the molecules should be distanced far
enough from each other and the excitation beam should be focused at the probe volume
of such dimensions that at a given dilution it contains only one molecule. The first task is
achieved by dilution, and the second – by using a laser beam in conjunction with confocal
microscopy, near-field scanning microscopy, or other detection methods.6 For a
successful SMS experiment, a combination of small (10-100 μm3) probing volume, low
fluorophore concentration, and exact laser beam frequency tuning need to be optimized

4
for any given fluorophore and optical set up. That assured, the signal-to-noise ratio for a
given fluorophore, generally depends (variable for different detection methods) on its
quantum yield ΦF, absorption cross-section σp, and (implicitly, via detector averaging
time) – on photostability, expressed in terms of photobleaching quantum efficiency, also
called branching ratio φb. The medium where the fluorophore is dispersed also has certain
requirements: it should be transparent in the frequency range of interest, be impurity-free
to minimize elastic (Rayleigh) or inelastic (Raman) scattering, and has minimum dark
state emission and background fluorescence.
Single-molecule spectroscopy has found numerous applications. First of all,
from a theoretical point of view, it permits the direct comparison of models drawn from
considerations of individual molecules and their properties. For example, it was SMS that
established the fact that reaction pathways for different molecules (of the same substance)
are not predetermined, but still deliver the same product. That is, reactions proceed
heterogeneously on multidimensional energy landscapes and individual molecules follow
different pathways in the phase space. Since these different pathways can occur on a wide
range of time scales, from femtoseconds to seconds, individual reaction rates for single
molecules of the same substance differ significantly.7,8
From a practical point of view, the most promising application for SMS is the
task of single strand DNA sequencing by detecting fluorophore-labelled individual bases
as they are being assembled by polymerase (direct method) or sequenced by exonuclease
(reverse method) into or from an immobilized single DNA molecule. The main idea here
is that the residence time of a freely diffusing single fluorescently labelled nucleotide

5
molecule is in the range of few microseconds. After being incorporated into the single
DNA strand, their residence time increases to several milliseconds, becoming sufficiently
long for unequivocal fluorescent identification, presuming that each nucleotide base is
labelled with its own, easily distinguishable fluorophore. By this method, the DNA
sequence is directly retrieved from the detected signal sequence. Although this goal of
DNA sequencing has not been achieved yet as a whole, each step in this scheme has been
shown to work on single-molecule level.8 In 2005 NIH gave nine grants totalling
$25,000,000 to implement this process as well as other approaches to reduce the cost of
genome sequencing below $1,000.9 Most biologically relevant SMS experiments have
been conducted in vitro. The ultimate dream, however, is to bring the SMS expertise to a
level which would allow non-invasive in-vivo analysis of living cells in their native
conditions.
An ability to precisely and specifically label several different sites in a
macromolecule allows utilization of such a technique as fluorescence resonance energy
transfer (FRET or RET), a physical phenomenon involving long-range dipole-dipole
interactions, known from 1948.10 FRET relies on nonradiative transfer of electronic
excitation from an excited donor to a ground state acceptor, which depends on the
distance between the two fluorophores (and decays as R–6), the spectral overlap of the
donor emission and the acceptor absorption (overlap integral), the refractive index of the
media, the donor quantum yield, and the relative orientation of the two fluorophores.
Thus, in combination with SMS, FRET is ideally suited for structure elucidation and
monitoring of conformational changes in biomolecules.11 Fluorescence correlation

6
spectroscopy (FCS) calculates the autocorrelation of the fluorescence intensity
fluctuations to follow time-dependent dynamics. Near-field single-molecule optical
microscopy, confocal fluorescence microscopy operates with 0.5–1.0 fL probe
volume12.
SMS was recently employed in a very elegant method, called by the authors
anti-Brownian electrophoretic trap (ABEL), to pin-point, trap, position and manipulate
nanoscale objects (down to 20 nm) in solutions at ambient temperatures.13 Two-photon
excitation (TPE) 14,15,16 is a second-order, non-linear process with extremely small cross
sections, typically on the order of 10–50 cm4. The molecular excitation rate depends
quadratically on the laser beam intensity. This method was used for two-color
colocalization of different dyes.17 Colocalization is an implementation of single-
molecule imaging to biological objects. Since the fluorophore molecules are much
smaller than the wavelength of the light they emit, they serve as point sources of light.
With the help of modern detection techniques and mathematical processing of the
resulting point-spread functions, the emitting center may be localized with high accuracy
and has been used to follow the motion of individual motor proteins and the diffusional
trajectories of labelled lipids in membranes.8 The colocalization technique gives
resolution of a few tens of nanometers and closes up the gap between far-field optical
microscopy with resolution of 200 nm and up, and FRET with resolution of ca. 2 to 8 nm.
The single biggest application of SMS nowadays is molecular imaging. The
first optical image of single pentacene molecules diluted in terphenyl was performed by
W. E. Moerner at cryogenic temperatures.2,18 Since then huge progress has been made

7
and single molecules are now imaged even in cells (living or, more often, immobilized).
Some imaging methods in biology rely on autofluorescent biomolecules,19,20,21,22 the most
famous among them being the green fluorescent protein (GFP),23 while majority of
imaging methods require labelling24 of nucleic acids, proteins, DNAs or RNAs with
fluorescent tags. The power of the molecular imaging technique may be exemplified by
the recent real-time monitoring of the infection pathway of single viruses.25 A single,
seven-nanometer (7·10–9 m) long, single-walled carbon nanotube has been imaged at a
resolution, comparable to that of scanning tunneling microscopy.26 The tags, or labels,
used for molecular imaging, have to fulfill a number of requirements.
1.2. Dye Requirements for Single-Molecule Spectroscopy
A fluorescent dye, to be successfully employed in an SMS experiment, should
possess several properties. Specifically, it should possess:
• large absorption cross section σp
• high quantum yield ΦF
• short fluorescence lifetime τ1
• high photostability = small φb
• high Stokes shift
• weak bottlenecks into dark states, i.e. small kISC

8
If the SMS experiment to be conducted is in biological media – on biomolecules,
in living or immobilized cells, in addition to the above requirements, several additional
requirements should be added (this does not imply the dye should fulfill all the
requirements above and below all the time or simultaneously):
• cell permeability
• high site binding specificity
• non-interference with the biological functions of the substrate being labeled
• emission in red region of spectrum
• high solvatochromism
Absorption cross-section σp is directly proportional to the molar extinction
coefficient ε and may be calculated from it: σp in cm2 per molecule is ( ) ( )ANNl
A ελλσ =⋅
= ,
where A is the absorbance for an optical path length l (cm), N is the number of molecules
per unit volume, and NA is Avogadro’s number. High σp at the excitation wavelength
means that the photons of the incident (exciting) light beam are efficiently absorbed and
background signals from unabsorbed photons are minimized. Since σp and ε are directly
related, everything that affects one is also true in changing the other. Typical SMS
fluorophores have ε > 20,000 L·mol–1·cm–1.
Quantum yield ΦF, defined in section 1.1, is a number, characteristic in how
efficient a fluorophore is in emission. An ideal fluorophore emits same number of
photons (counting as particles) it absorbed, producing a quantum yield of exactly unity.

9
This is the case when the radiationless decay to the ground state S0 is much slower than
radiative decay. The energy of the emitted fluorescent photons, however, is always lower
than the energy of absorbed photons because of relaxation processes (Stokes’ losses). A
good fluorophore should possess high (as close to unity as possible) fluorescence
quantum yield ΦF, but at least higher than 0.1. A product of the fluorophore’s quantum
yield and its molar extinction coefficient at the excitation wavelength is called
brightness.27
The fluorescence lifetime τ1, or lifetime of the excited state is the average time
the molecule spends in the excited state prior to decay to the ground electronic state. This
time should be fairly short (in the range of 0.5–5 nanoseconds)27 to provide as many
excitation-emission cycles per unit time, as possible. It is during this time span that the
fluorophore acquires information about its environment.
Common dye molecules employed in SMS can emit up to a million photons
before irreversible photobleaching occurs. A quantitative measure of photobleaching is
photobleaching quantum efficiency, also called branching ratio φ b, defined as
probability per optical absorption event to irreversibly generate a non-fluorescent
product. Photostability is the reciprocal of photobleaching quantum efficiency. Low
photostability of a fluorophore in practical SMS experiments results in low observation
and averaging times available to the experimenter (Fig. 1.2.).8 Photobleaching in a single,
instantaneous step is also an indirect proof that the fluorescence observed from the probe
volume is coming from only a single molecule. Photodestruction of fluorophores is one
of the most important yet least understood processes that affect the application of

10
fluorescence in biology.28 Up to date they are best described as “elusive photochemical
reactions, mainly photo-oxidation”.27,29 Depending on the time scale of the detection
method and the processes studied, photostability varies from desirable to essential. For
example, surface-immobilized fluorophore-labelled biomolecules should have long
photosurvival times, while dyes for photo histogram studies are less sensitive to
photobleaching of individual molecules as they come into and pass out of the probe
volume. Photostability may generally be increased by careful exclusion of oxygen from
the solution containing the fluorophore.30
Figure 1.2. Irreversible photobleaching (at 5.6 sec) of single Cy5 molecule, immobilized
on a glass slide.8
Generally, the higher the power of the laser pumping the fluorophore, the higher
the fluorescence response, until the corresponding transition is saturated. Saturation
intensity is at a maximum when there are no strong bottlenecks, like intersystem
crossing (ISC) from the excited singlet state S1 into the excited triplet state T1. During

11
the lifetime of the triplet state T1, no emission or absorption happens (thus, the name of
dark state), resulting in saturation of the emission rate and decrease of the absorption
cross section. Thus, another requirement for a good SMS fluorophore is small value of
kisc (rate of intersystem crossing) and large value of kT (decay rate from triplet to ground
state). One of the molecular structural classes, satisfying this requirement, are planar
aromatic compounds.31
For biologically oriented applications, it is desirable for the fluorophore to
absorb and emit in the red region of the visible spectrum (above 600 nm), since there are
only few compounds of biological origin32 that show intrinsic absorption and emission in
this region. Also, the major source of the background in SMS is scattering of the
excitation light. The intensity of the scattered light decays approximately as fourth power
of the wavelength, thus, red-shifted dyes give less background33. Non-interference with
biological functions is essential if the fluorophore’s intended use is in live cells or the
molecules, which will be tagged with such fluorophore, will undergo biochemical
transformations. For example, if the nucleotides for determining of DNA sequence are
labelled with fluorescent tags, those tags should not affect the ability of the nucleotides to
undergo synthesis under polymerase action or sequencing by exonuclease. In fact, most
natural DNA polymerases and exonucleases have been found very sensitive to such
structural changes in nucleotides as dye labelling and discriminate unlabelled nucleotides
against labelled ones. To circumvent this problem, mutant enzymes were employed. Non-
interference also means that the dye should be non-toxic.34

12
High site binding specificity of the fluorescent tag necessitates from the end-
purpose of such labelling. If a tag will attach to a wrong part of the molecule to be
imaged, the image may provide wrong information about that molecule. Binding here
means either covalent bonding to the substrate molecule, i.e. tagging, or mere preference
to position (adsorb, adhere) itself within certain parts of the substrate molecule (probing)
due to structural, steric, conformational or other peculiarities. For example, it is known
that for a dye to specifically remain in the vesicle of a cell, it is often sufficient to have
long normal alkyl chains in its structure. The unbound fluorophores create high
background fluorescence and must be washed out (usually by gel filtration) from the
labelled system as thoroughly as possible. An ideal solution to this problem would be a
tag, which becomes fluorescent only after binding to the substrate. Delivery of the dye
inside the cells (cell-loading) and then specifically to the sites to be labelled constitute a
problem in itself, current solutions of which are direct microinjections or utilizations of
liposomes.
The Stokes’ shift35 of the fluorophore is the difference between the emission
maximum and the absorption maximum, Δλ=λem–λabs. The energy decrease has many
causes, the common ones being (a) the rapid (10–12 sec) relaxation of the excited
molecule to the lowest vibrational level of S1 and (b) fluorescent decay to the higher
vibrational levels of S0. Therefore, the wavenumber of the Stokes shift is a direct measure
of the vibrational energies of the molecule. Other physical phenomena, like interactions
with solvent, excited-state reactions and energy transfers, may contribute to the Stokes’
shift as well. The high Stokes’ shift makes the fluorophore’s emission bathochromically

13
shifted, i.e. the larger it is, the more the emission maximum is shifted into the red region
of the spectrum, which allows easy discrimination of the fluorescence signal from
Rayleigh scattering and increases signal-to-noise ratio.27 Large Stokes’ shift also results
in smaller overlap integral of absorption and emission spectra, thus minimizing
concentration related self-quenching or RET homotransfer of the fluorophore.
Solvatochromism is a phenomenon involving dependence of the absorption or
emission maximum on the solvent polarity the dye or fluorophore is dissolved in.
Negative solvatochromism corresponds to a hypsochromic shift; positive
solvatochromism corresponds to a bathochromic shift with increasing solvent polarity.
The origin of the phenomenon is the same for both absorption and fluorescence and lies
in the dipole-dipole interactions (distortions) of the ground (for absorption) and excited
(for fluorescence) electronic states of the molecules with the molecules of the solvent.
Thus, solvatochromism may serve and is used indeed to assess the polarity of different
media. There is, however, an important difference between solvatochromism in
absorption and the one in emission. Absorption of light by a molecule is an instantaneous
process: it takes femtoseconds (10–15 sec) to occur. Thus absorption can provide
information only on the average ground state of dye molecules and on the solvent
molecules immediately adjacent to them. Absorption spectra are not sensitive to
molecular dynamics and the absorptional solvatochromism, regardless of number of
molecules, averages the information from the surrounding solvent shell by the very
nature of the absorption process. On the contrary, a fluorophore spends some time (10–9
to10–8 sec) in its excited state and it is during this time τ1 that it gathers information about

14
its environment. This time scale allows an excited fluorophore molecule to collide with
other species and/or reflect conformational changes in the molecule. Solvent relaxation, a
process of reorientation of polar solvent molecules to minimize dipole-dipole interactions
with the solute, occurs during a picosecond (10–12 sec) time span. Most of the
fluorophores have larger dipole moment in the excited state than in the ground state and
the solvent molecules have sufficient time to reorient themselves around the excited state
dipole, lowering its energy and resulting in a bathochromic Stokes’ shift. Thus,
solvatochromism of emission spectrum is much more sensitive to the solvent polarity
than solvatochromism of absorption spectrum of the same compound. An important
parameter for a fluorophore to sense its environment polarity is therefore its dipole
moment. Nonpolar fluorophores, like most aromatic hydrocarbons, are non- or weakly
sensitive to the solvent polarity.
When combined with single-molecule spectroscopy, solvatochromic
fluorophores can sense the local polarity of their immediate (i.e. of nanometer size)
environment – be it the closest coordination sphere in a micelle or the closest part of the
biomolecule it is confined or bound to. Since the local polarity of biomolecules often
depends on their conformation states, and the latter often dynamically change and affect
the very biological functions thereof, solvatochromic fluorescent probes and tags are
powerful tools for determination and monitoring of structural changes in biomolecules.
The possibility of successful monitoring of the conformational dynamics of individual
biomolecules with temporal resolutions comparable to those of new molecular dynamics
simulations36 raises the hope that it might be possible to relate conformational dynamics

15
directly with enzyme activity. Such studies would require site-specific labelling of the
active site of enzyme with a fluorophore, which is selectively quenched (perhaps by an
amino acid) in close proximity.
For a covalently binding fluorescent tag, an important variable to consider in its
design is the length of the spacer or linker between the fluorophore and the hook or
reactive group, which after labeling determines the proximity of the fluorophore to the
substrate. According to the opinion of Weiss and Kapanidis,27 fluorescent tags with
small, short, and rigid spacers are preferred since they tend to be less perturbative to their
local environment and ensue in fewer fluctuations of the fluorescent properties of the
single fluorophore due to unknown and uncontrolled conformational changes.27 Flexible
spacers, on the other hand, provide more sensitivity (more pronounced changes in
fluorescence parameters) to the local environment and are also more desirable in FRET
experiments. Extreme care must therefore be taken to separate out fluorophore dynamics
from the biological dynamics.28
The pool of chemical reactions that provide site-specific covalent binding of the
fluorescent labels (not necessarily detected at single-molecule level) to biosubstrates is
referred to as “bioconjugate” or “bioconjugation” chemistry. There are currently many
fluorescent dyes available with a wide range of physico-chemical parameters to choose
from.37 And yet the need for new SMS fluorophores is growing: “current dye-based
fluorescent technologies do not stand up to the challenge” (of study of individual and rare
biological processes in the living cells). The development of new fluorescent probes with
superior photophysical properties is needed.28 It is also necessary to elaborate new

16
labelling strategies and “devise chemistries that render the reagent fluorescent only after
incorporation to the site of interest, thus minimizing the background of unincorporated
reagent that will otherwise overwhelm the SMS signal.”27 Many (i.e. thousands) existing
fluorophores have never been tested for performance and applicability in SMS. There are
also no solid rules for rational design of SMS-compatible fluorophores. The
photophysical parameters of the fluorescence are media dependent as well as fluorophore
dependent. The fluorophores are not easily tunable to the desirable precision, and change
of one parameter (e.g. fluorescence wavelength) often affects the other (e.g. quantum
yield). An emerging solution to fine-tuning fluorescent probes are fluorescent
semiconductor nanocrystals, or quantum dots,38,39 which have broad excitation spectra,
narrow and tunable emission spectra, long fluorescence lifetimes, and high photostability.
They have recently been used to monitor individual eight nanometer step motions of two
molecular motors, kinesin and dynein, in vivo.40 However, their bioconjugation
chemistries are immature yet.27 Quantum mechanics and computational chemistry allow
calculations of both electronic and vibrational energy levels of a given molecule in the
gas phase with very good reproducibility and often with good accuracy. Yet the mere fact
of a molecular compound being fluorescent or not is still not reliably predicted by
modern theory.
From a molecular structure point of view, synthetic fluorophores are typically
aromatic organic compounds. With the exception of the lanthanides, individual atoms are
generally nonfluorescent in condensed phases.41 The quantum yield and lifetime can be
altered by factors which affect either of the rate constants – kF and knr. A molecule can be

17
nonfluorescent because of fast internal conversion or because of slow emission rate. It is
empirically known that the presence of a nitro-group in aromatic compounds makes knr
large and thus quenches (i.e. decreases) the fluorescence. The quenching may be caused
by intermolecular processes as well. For example, halogens such as Cl, Br, and I and
other heavy atoms quench fluorescence via spin-orbital coupling and intersystem crossing
to the triplet state. As a result, molecules containing heavy atoms, specifically bromine
and iodine, are often phosphorescent (rather than fluorescent), and the fluorescence
quantum yields of fluorophores in chlorinated and other heavy-atom containing solvents
are generally lower.
Table 1.1. Typical SMS fluorophores.27
1.3. Review of known DPP chemistry
The first representative of the dihydropyrrolo[3,4-c]pyrrole-1,4-dione (DPP)
class of dyes, 3,6-diphenyldihydropyrrolo[3,4-c]pyrrole-1,4-dione (1) was discovered in
1974 by Farnum42 in an attempt of a Reformatsky reaction on benzonitrile, ethyl

18
bromoacetate, and zinc to obtain 2-azetinone. Instead of the latter, a red pigment of
structure 1 was obtained in poor yield (Scheme 1.1).
NH NH
O
O
N
OEt
Br
O
+Xa
NH
O
1
Scheme 1.1. Farnum’s synthesis of DPP. a) Zn/Cu, toluene, 110°C, several hours.
This new class of dyes turned out to be highly insoluble materials and excellent
pigments43: highly thermally stable, photostable and fluorescent in solid state. Soluble
derivatives of DPP are reported to possess high quantum yields,44,45,46,63 large Stokes
shifts46, and high photostability47. Solubilized DPP derivatives have found applications in
“soluble (latent) pigments”48,49,50, polymers51,52,53, photorefractive,54 photoconductive,55
electrochromic,56 electronic57,58 materials, dendrimers59,60, and liquid crystals61,62. The
reported photophysical characteristics63 of the DPP family of dyes would appear to lend
itself to the field of single-molecule spectroscopy. The demands on dye stability for
applications in single-molecule spectroscopy are particularly high64 and the amazing
thermal stability of 1 up to 500°C was encouraging. The sole previous study of single-
molecule fluorescence of a DPP dye involves a diphenyl DPP core covalently imbedded
in a dendrimer in polymer films65. We decided to further examine the diphenyl DPP class
of fluorescent dyes, which might be utilized in single-molecule biological studies. Simple

19
diaryl DPP’s have solution absorptions in the range of 470-520 nm and emission in the
range of 508–540 nm. Since still longer wavelengths of fluorescence are desirable
(biological media are more transparent at longer wavelengths and with less background
autofluorescence), we also wanted to prepare DPP dyes with longer fluorescence
wavelengths by extending the conjugation of the core and by introducing donor or
acceptor groups at the termini of the conjugated system. The only systematic review of
DPP chemistry in the open literature is from Iqbal66. There are also two recent reviews of
DPPs as a class of pigments.67,68 Otherwise, virtually all DPP chemistry has been
described in patents, where over a thousand DPPs are described. Here most important
extract of that chemistry pertinent to our work follows.
The nomenclature of the DPPs is derived from the core bicyclic heterocyle DPP,
named by IUPAC 1957 system, rule B-3 or rule B-4.1(b) – Chemical Abstract system.
Accordingly, the DPP may be termed as
N
N
O
OH
H
R2
R11
2
3
4 5
6
NH
1
2
3
NH
5
4 +
a
bc
d
e
3
0
3
CAB-3
N
N
O
O
R1
R1
R
R
1
23
4 1
2
3
4 5
6 1
2 3
4
DPP 1: R1–DPP–R
► 1,4-diketo-2H,5H-dihydropyrrolo[3,4-c]pyrrole (obsolete);
► 2H,5H-dihydropyrrolo[3,4-c]pyrrole-1,4-dione;
► 3,7-diazabicyclo[3.3.0]octa-4,8-dien-2,6-dione.

20
For derivatives of 3,6-diphenyldihydropyrrolo[3,4-c]pyrrole-1,4-diones, we will
use the simplified notation of R1–DPP–R, where R1 is the substituent on the phenyl ring
of 1, and R is the substituent on nitrogen. If no position of R1 is specified, it is assumed to
be at the default 4- (para) position.
Since the Reformatsky reaction of benzonitrile gives only variable 5-20% yields
of DPP with various byproducts, Farnum’s approach to DPP preparation is only of
historical interest. Some DPP derivatives prepared by this method69,70 and their
absorption properties are presented in the Table 1.2.
Table 1.2. DPP compounds prepared by Reformatsky reaction.69
NH
NH
O
O
R
R
R λmax, nm in NMP
H 475, 504
Et2N 554, 512
Br 515, 480
CN 535, 500
F 464, 500
OMe 512, 475
Me2N 554, 512
3-COOMe 465, 505

21
The mechanistic study of the Reformatsky reaction and a retrosynthetic analysis
of 1 led Iqbal et al. in 1986 to propose another preparative approach to DPP, the
condensation of aromatic nitriles with succinate esters71 (Scheme 1.2). Since alkyl
succinates undergo self-condensation under basic conditions (Scheme 1.3), optimum
conditions for condensation with aryl nitriles must be followed to minimize unwanted
byproducts. First, the alkyl group in the succinate ester should have large steric volume.
Second, the order and rate of reagent addition is of utmost importance.
N
OAlk
O
O
OAlk+ a
NH
NH
O
O
Scheme 1.2. Condensation of benzonitrile with alkyl succinate.
a) strong base (e.g. t-BuOK), protic solvent (e.g. t-AmOH).
OAlk
O
O
OAlk
O
O
AlkO
AlkO+
O
O
COOAlk
AlkOOC
Base
Scheme 1.3. Self-condensation of alkyl succinate under basic conditions.
The rate of succinate ester self-condensation decreases in the order of alkyl
substituent bulkiness: Me > Et > i-Pr > t-Bu > t-Am. Thus, dimethyl and diethyl
succinates are least preferred for condensation with aromatic nitriles, and t-amyl
succinate is the ester of choice. Self-condensation is a bimolecular process, which rate
depends quadratically on the ester concentration. By maintaining a low concentration of

22
the ester, one decreases both the rates of ester – nitrile and ester – ester condensation, yet
the latter is decreased to a greater extent. From the practical point of view, this means
very slow addition of the t-amyl succinate by a syringe metering pump to the reaction
mixture of the base and aromatic nitrile. Some exemplary DPP representatives prepared
by this method are given in the Table 1.3. A recent patent claims that this condensation
may be conducted in an essentially “solvent-free” way.72
This new condensation of aromatic nitriles with alkyl succinates resembles the
classical Stobbe condensation of an aldehyde or Schiff base with succinic acid esters
(Scheme 1.5).73 The resulting hexahydropyrrolo[3,4-c]pyrrole-1,4-diones, obtainable also
from N-benzylaniline instead of Schiff bases, may be dehydrogenated with 2,3-dichloro-
5,6-dicyanobenzoquinone (DDQ) to the corresponding DPP compounds (Scheme
1.6).74,75 The Schiff base of aniline and 2-formylpyridine was condensed with
diethylsuccinate to give 3,6-bis(pyridin-2-yl)tetrahydropyrrolo[3,4-c]pyrrole-1,4-dione in
36% yield. N-benzylaniline was condensed with dialkylsuccinate to give 15% of
hexahydropyrrolo[3,4-c]pyrrole. The latter two compounds could be aromatized with
DDQ to yield 30% (4.5% total) of N,N′-diarylated DPP. This approach, however, gives
poor yields at both stages and requires separation of the desired compounds from various
byproducts.
The reaction in Scheme 1.2 must proceed via several steps. The corresponding
intermediates 276 and 377 were separated and the overall mechanism was suggested66,78 as
depicted in Scheme 1.6. The enamino compound 2 can be isolated at low temperature (–
78°C) condensation of diethyl succinate dianion with nitriles (both aliphatic and

23
aromatic) and later converted to lactam 3 with sodium methoxide in methanol. Both 2
and 3 condense with another equivalent of nitrile providing DPP. Asymmetric dialkyl,
diaryl, or alkyl-aryl DPPs may be obtained by this step-wise procedure (Table 1.4).
Aliphatic nitriles yield much lower yields of DPPs in the condensation with 3, compared
to their aromatic counterparts. Evidently, this is caused by the presence of α-hydrogens in
the structure of aliphatic nitriles, which, upon removal (deprotonation) by the strong base,
decreases the electrophilicity of the nitrile and engages them into side reactions. This fact
is also reflected in the number of known 3,6-diaryl DPPs relative to 3,6-dialkyl DPPs: out
of 1,190 known DPPs, 1,140 (96%) contain 3,6-diphenyl-2H,5H-dihydropyrrolo[3,4-
c]pyrrole-1,4-dione core as a substructure.79 Also noteworthy is that a Beilstein search for
the same substructures yields only 68 DPP compounds (5.7%), 52 of which have
diphenyl DPP core as a substructure.80 This, in turn, reflects the fact that over 95% of
information on DPP compounds is found in patents, most of which belong to the Ciba-
Geigy Specialty Chemicals Company, Inc., headquarters in Basel, Switzerland. The total
number of original, primary literature sources dealing with DPPs is close to a thousand
(STN: 945; Beilstein: 37; references may overlap).
The intermediates 2 (dialkyl 2-[amino(phenyl)methylene]succinate) and 3 (3-
alkoxycarbonyl-2-phenyl-pyrrolin-5-one) may be obtained as presented in the Scheme
1.4. The dianion of the succinate ester is much more stable, less prone to self-
condensation and thus is preferred to a more accessible mono-anion.

24
R1
O
COOR
COOR
OR
OR
O
O
OR
OR
O
O
–
_
OR
OR
O
O
R1
NH2 NH
R1
COOR
O
OR
OR
O
O
–
_
a
a
b
c
d e
2 3
58%
59% 60%
Scheme 1.4. Synthetic approaches to the intermediates 2 and 3.
a) LDA, THF, –70°C; b) R1COCl, Et3N; c) AcONH4, AcOH, 100°C, 16 hrs;
d) R1CN, ZnCl2, THF, –70°C, 2 hrs.

25
Table 1.3. DPP compounds prepared by aromatic nitrile – succinate
ester condensations.71 NH
NH
O
O
R1
R2
# R T, °Ca Addition time, hrsb Yield, %
1 4-Tol 97-99 3¾ 23.4
2 3-Cl-Ph 89-91 2 56.8
3 4-Cl-Ph 88-91 2 39.5
4 4-MeOOC-Ph 89-91 2¼ 6.6
5 3-CN-Ph 89-91 2¼ 77.5
6 4-CN-Ph 90-91 2½ 80.0
7 1-Np 95-97 2 4.5
8 2-Np 96-97 1¾ 24.2
9 3-CF3-C6H4- 105-110 3 56.8
10 6-AmO-3-Py 105-110 3 65.0
11 4-CF3-C6H4- 105-110 2.5 44.9
12 4-CN-C6H4-C6H4- 105-110 3 36.7
13 4-C6H4-C6H4- 105-110 2.5 10.0
14 4-t-Bu-C6H4- 105-110 2 55.2
15 3,4-Me2-C6H3- 105-110 2 52.4
16 2-Fu 90 1 17.9
17 4-Me-C6H4- 105-110 2 41.8
18 3-thienyl 85 1 42.0
19 3,5-Cl2-C6H3- 85 1 70.4
20 4-Me2N–C6H4– 120 2 3.7
a Reaction temperature; b Total addition time of diethyl succinate for a 0.2 mole reaction of aromatic nitrile.

26
+OEt
O
O
OEt N
N
Base
N
N
O
O
N
N
Scheme 1.5. Stobbe condensation of a Schiff base with alkyl succinate.
NH
Cl
Cl
N
N
O
O
Cl
Cl
Cl
Cl
a
N
N
O
O
Cl
Cl
Cl
Cl
b
Scheme 1.6. Preparation of DPP from benzylaniline.
a) t-BuOK, DMF, –10°C; b) DDQ, dichlorobenzene.

27
Table 1.4. DPP compounds prepared by step-wise condensations.76,77,81
NH
NH
O
O
R1
R2
# R1 R2 Yield, % M.p. °C UV λmax (lg ε)
1 Me Me 14 >250 380 (4.16), 392 (4.2)a
2 Me Pr 24 >250 382 (4.1), 398 (4.1)a
3 Me Ph – – 433 (4.0), 450 (4.0), 550 (2.5)b
4 Ph 4-Ph–S–C6H4– 74 – 380 (4.1), 480 (4.5), 518 (4.6)b
5 Ph 4-CN–C6H4– 80 – 271, 310, 485, 520b
6 Ph 4-Cl–C6H4– 74 – 471 (4.4), 510 (4.5)b
7 C11H23 C11H23 – 247–250 385 (4.0), 402 (4.0)b
8 Me i-Pr 11 – 383 (4.1), 397 (4.2)a
9 Me C11H23 10 – 382 (4.2), 398 (4.2)b
10 Pr 4-Cl–C6H4– 52 – 440 (4.0), 460 (4.0), 630 (2.8)c
11 4-Ph–C6H4– 4-Cl–C6H4– 70 336 (4.2), 488 (4.5), 524 (4.6)b
12 Ph 2-Cl–C6H4– – – 459 (4.2)b
13 Ph 4-Me–C6H4– 49 – 307, 312, 472, 507b
14 4-Cl–C6H4– 3-CN–C6H4– 87 – 288, 308, 450, 480, 513b
15 Ph 3-CN–C6H4– 61 – 289, 305, 445, 478, 512b
16 4-CN–C6H4– 3-CN–C6H4– 27 – 280, 310, 490, 521b

28
Table 1.4. (Continued).
# R1 R2 Yield, % M.p. °C UV λmax (lg ε)
17 4-pyridyl 4-Cl–C6H4– 76 – 268, 308, 483, 517b
18 Ph 1-Np 28 – 470 (4.2), 493 (4.2)b
19 Ph 6-MeO-1-Np 2 – 500 (4.4)b
20 Ph 2-Me–C6H4– – – 453 (4.3), 481 (4.3)b
21 Ph 2,5-Me2–C6H4– 6 – 456 (4.3), 482 (4.3)b
22 Ph PhCH2– 17 – 381 (4.1), 466 (3.1)b
23 Ph PhCH2CH2– – – 438 (4.2), 459 (4.2)b
24 Ph 9-phenanthryl 39 – 472 (4.2), 492 (4.2)b
25 Ph i-Pr 31 306–307 437 (4.1)d
26 Ph Cyclohexyl 11 subl. > 400 439 (4.2), 460 (4.2)d
27 Ph Ph2CH 29 295–296 443 (4.25), 466 (4.2)d
28 Ph 2-Norbornyl 15 subl. > 400 430 (4.2), 449 (4.2)d
29 Ph cyclohex-3-en-yl 42 358–360 –
a) in methanol; b) in NMP; c) in DMF; d) in DMSO
29
N
OAlk
O
O
OAlk+
Base
NH2
COOAlk
O
OAlk
NHCOOAlk
OAlk
O
NH
O
COOAlk
2 3
+NH
O
COOAlk
N
Base NH
O
COOAlk
NHNH
NH
O
O
NH
O
COOAlk
NH2
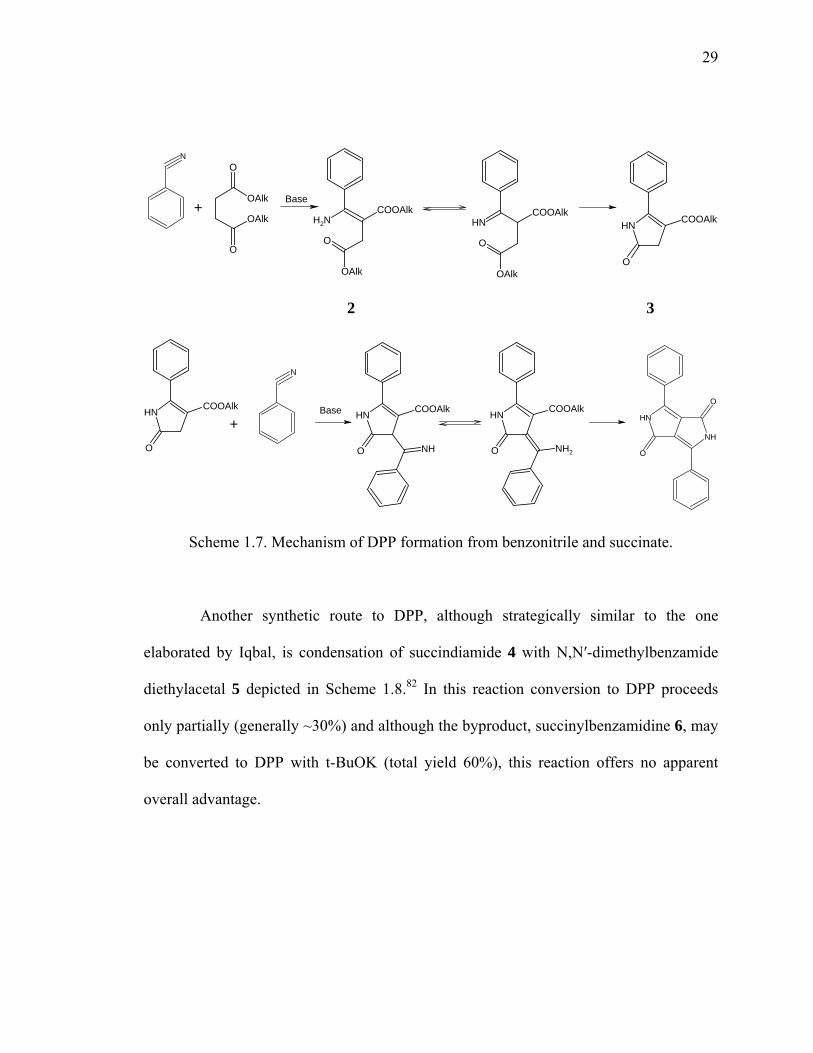
Scheme 1.7. Mechanism of DPP formation from benzonitrile and succinate.
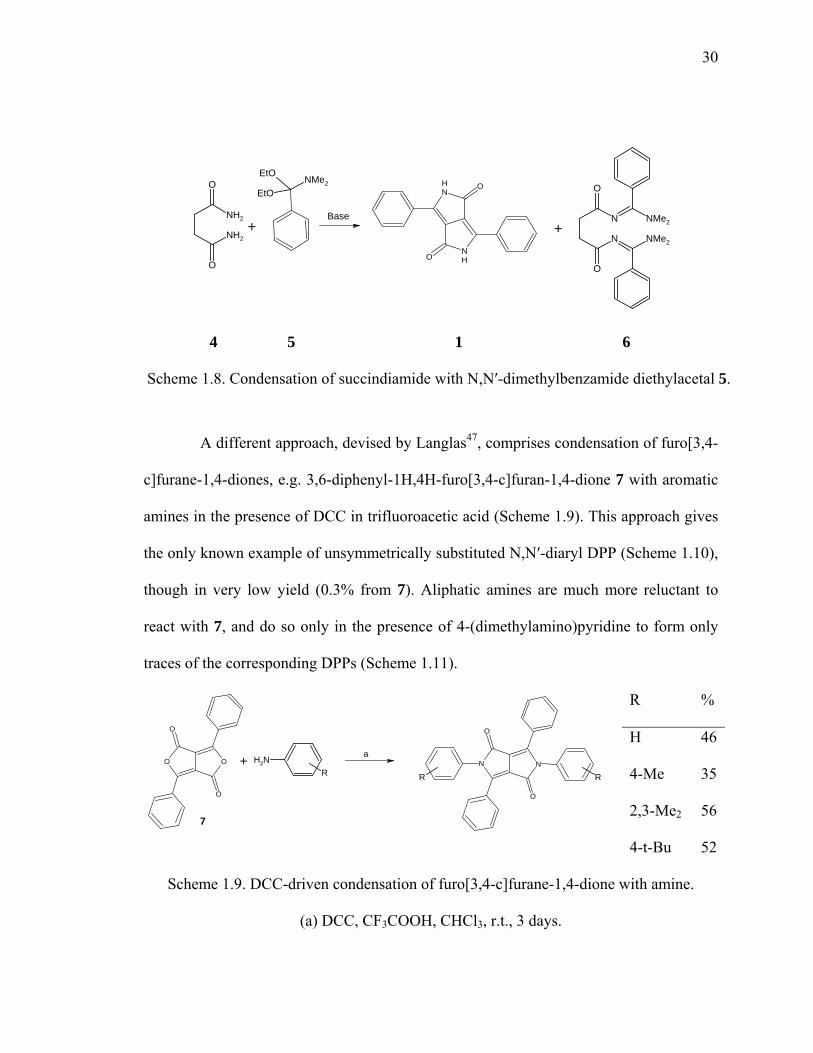
Another synthetic route to DPP, although strategically similar to the one
elaborated by Iqbal, is condensation of succindiamide 4 with N,N′-dimethylbenzamide
diethylacetal 5 depicted in Scheme 1.8.82 In this reaction conversion to DPP proceeds
only partially (generally ~30%) and although the byproduct, succinylbenzamidine 6, may
be converted to DPP with t-BuOK (total yield 60%), this reaction offers no apparent
overall advantage.
30
NMe2EtO
EtO
NH2
O
O
NH2+
Base
NH
NH
O
O
+N
O
O
N
NMe2
NMe2
4 5 1 6
Scheme 1.8. Condensation of succindiamide with N,N′-dimethylbenzamide diethylacetal 5.
A different approach, devised by Langlas47, comprises condensation of furo[3,4-
c]furane-1,4-diones, e.g. 3,6-diphenyl-1H,4H-furo[3,4-c]furan-1,4-dione 7 with aromatic
amines in the presence of DCC in trifluoroacetic acid (Scheme 1.9). This approach gives
the only known example of unsymmetrically substituted N,N′-diaryl DPP (Scheme 1.10),
though in very low yield (0.3% from 7). Aliphatic amines are much more reluctant to
react with 7, and do so only in the presence of 4-(dimethylamino)pyridine to form only
traces of the corresponding DPPs (Scheme 1.11).
R %
O O
O
O
+ NH2 N N
O
O
R RR
a
7
H
4-Me
2,3-Me2
4-t-Bu
46
35
56
52
Scheme 1.9. DCC-driven condensation of furo[3,4-c]furane-1,4-dione with amine.
(a) DCC, CF3COOH, CHCl3, r.t., 3 days.

31
O O
O
O
aN O
O
O
bN N
O
O
CH3
CH3
CH315% 2%
Scheme 1.10. The sole example of an unsymmetrical N,N′-diaryl DPP.
a) 1 eq. aniline, DCC, CF3COOH, CHCl3, r.t., 3 days; b) 4-tert-butylaniline, DCC,
CF3COOH, r.t., 3 days.
O O
O
OCH3
CH3
aCH3
NH2+ N N
O
OCH3
CH3
CH3 CH3
8
Scheme 1.11. The sole example of aliphatic amine reaction with furo[3,4-c]furane-1,4-
dione. (a) DCC, DMAP, CF3COOH (yields ‘trace’ amounts).
The 3,6-diphenyl-1H,4H-furo[3,4-c]furan-1,4-dione83 7 can be prepared
(Scheme 1.12), in turn, (a) from benzoylacetic acid ester via its oxidative dimerization84
to a mixture of meso- and racemic forms of 2,3-dibenzoylsuccinic acid diesters with
subsequent thermal cyclization85; and (b) from 1,6-diaryl-1,3,4,6-tetraoxohexane86 via its
oxidation with dinitrogen trioxide (N2O3) to 1,6-diaryl-2,5-bis(diazo)-1,3,4,6-
tetraoxohexane87 and subsequent thermal decomposition of the latter neat (25–50% yield)
or in toluene (59% yield). The laborious preparation procedures of the lactone 7 make

32
this seemingly straightforward route to N-aryl disubstituted DPPs somewhat troublesome.
Moreover, the harsh condition of the oxidative coupling in the route (a) and vigorous
thermolysis step in the route (b) exclude the presence of sensitive functional groups. In
fact, there are merely five known 3,6-diphenylfuro[3,4-c]furan-1,4-diones (and 3,7-
dioxabicyclo[3.3.0]octa-4,8-dien-2,6-diones in general) in the Beilstein database: 4,4′-
dimethyl- [76695-70-0]; 4,4′-dichloro- [76695-71-1]; 4,4′-dimethoxy- [76695-69-7]; and
the aforementioned 2,2′-dimethyl- 8.
O O
O
O
OR
RO
O
O
O
ORO
O
O
OR
O
O
a b N2
N2
O
O
O
O
O
O
O
O
cd
13% 56% 51%59%
Scheme 1.12. Routes to 3,6-diphenyl-furo[3,4-c]furan-1,4-dione.
a) Na/Et2O, I2; b) thermolysis 300°C; c) N2O3, CH2Cl2, –30°C; d) thermolysis 100°C.
1.3.2. Chemical Properties
3,6-Diphenyldihydropyrrolo[3,4-c]pyrrole-1,4-diones undergo several types of
reactions. The Scheme 1.13 shows the points of attack by nucleophiles and electrophiles.
Specific examples of each of such attack follow and are also illustrated on Scheme 1.14.

33
NHNH
O
O
X
E+
Nu–
EFG
NFG
Scheme 1.13. Reactive sites of DPP. X – halogen; EFG – electrophilic functional group;
NFG – nucleophilic functional group
NH
NH
O
O
N
N
O
O
Alk
Alk
NH
NH
S
S
N
N
O
O
ORO
RO O
NH
NH
O
O
Br
Br
NH
NH
O
O
Cl
Cl
ClCl
NH
NH
O
O
S
O
O OH
S
O
OOH
N
N
O
O
CH2OH
CH2OH
N
NH
OPOCl2
O
Cl2 Br2
RO
O
Cl
AlkOTsO
CH3
P P
S
S
S
O
CH3
S
POCl3Oleum CH2O H2SO4
Scheme 1.14. Various reactions of DPP.

34
Alkylation of one or both of the amidic nitrogens is usually performed in high-
boiling solvents — nitrobenzene (at 200…205°C) or DMF (at 140°C) — with alkyl (Me
or Et) 4-toluenesulfonate and potassium carbonate to yield 40–68% of N,N′-dialkyl DPP.
Reaction of n-butyl bromide with preformed DPP sodium salt for 20 hrs at 60°C, then 2
hrs at 100°C gave a mixture of mono- and dibutylated products (m.p. 250–252 and 123–
124°C correspondingly). Benzylation was achieved in 47% yield88,89. It is noteworthy
that only N-alkylation takes place, with no O-alkylated products corresponding to the
lactim tautomeric structure ever reported.74 The N,N′-dialkylated DPP is strikingly more
soluble compared to its dihydro precursor: at 25°C one liter of DMF dissolves a mere 110
mg of DPP vs. 3300 mg of DPP-Me.66 The solubility of mono-alkylated DPPs is in
between these two.
NH
NH
O
ON
N
OH
OH
N
N
OAlk
OAlk
N
N
O
OAlk
Alk
XAlkOTs
K2CO3
NH
N
O
O
Alk
+
Scheme 1.15. N-Alkylation of DPP.
Acylation of the amidic nitrogens is performed essentially in the same way as
alkylation, in this case using acyl chlorides. For example, benzoylation of DPP proceeded
in 38% yield. 1,4-Diketo-3,6-diaryl-2,5-bis(alkyloxycarbonyl)pyrrolo[3,4-c]pyrroles may
also be obtained in high (20–90%) yields by action of di-alkyl dicarbonates in presence of
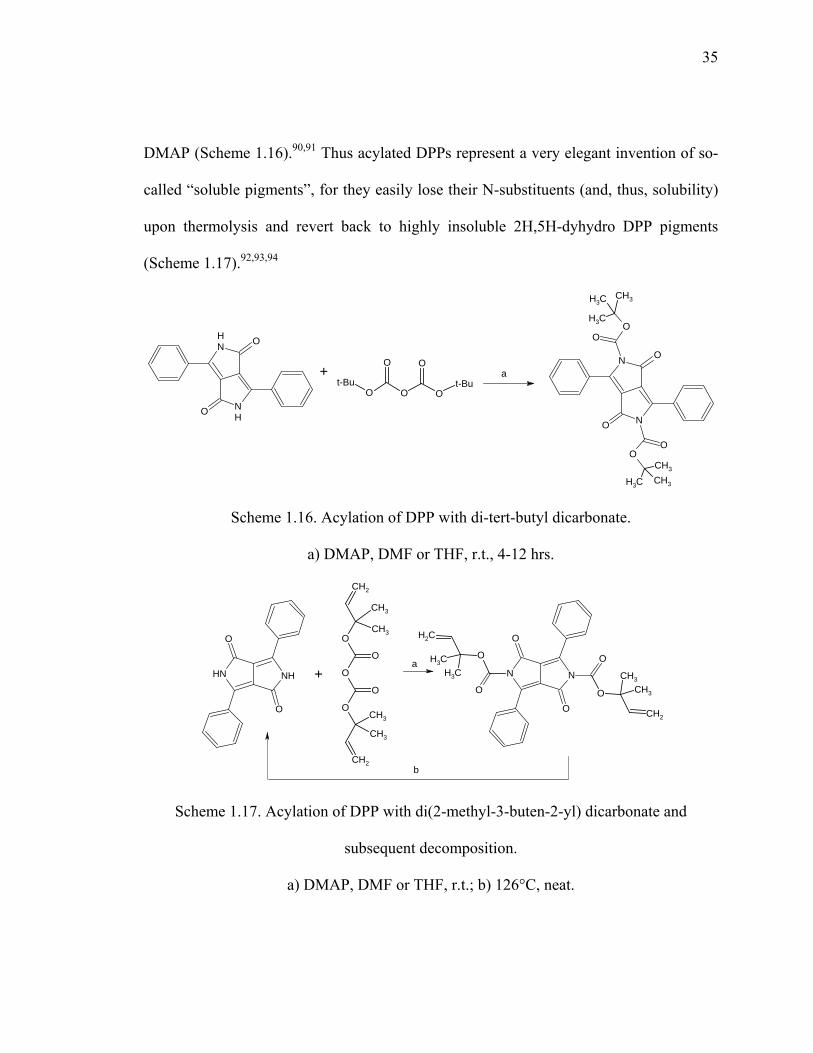
35
DMAP (Scheme 1.16).90,91 Thus acylated DPPs represent a very elegant invention of so-
called “soluble pigments”, for they easily lose their N-substituents (and, thus, solubility)
upon thermolysis and revert back to highly insoluble 2H,5H-dyhydro DPP pigments
(Scheme 1.17).92,93,94
NH
NH
O
O
a
N
N
O
O
OO
OO
CH3
CH3
CH3
CH3
CH3
CH3
t-BuO O
O
O
O
t-Bu+
Scheme 1.16. Acylation of DPP with di-tert-butyl dicarbonate.
a) DMAP, DMF or THF, r.t., 4-12 hrs.
O
O
O
O
O
CH2
CH2
CH3
CH3
CH3
CH3
NHNH
O
O
+a
b
O
O
CH2
CH3
CH3
NN
O
O
O
O
CH2
CH3
CH3
Scheme 1.17. Acylation of DPP with di(2-methyl-3-buten-2-yl) dicarbonate and
subsequent decomposition.
a) DMAP, DMF or THF, r.t.; b) 126°C, neat.

36
Arylation of the amidic nitrogens hitherto was possible only indirectly and led
mostly to mono-arylated DPPs – see Scheme 1.10.47
Hydroxymethylation of the amidic nitrogens has been performed with para-
formaldehyde in concentrated (90–96%) aqueous sulfuric acid at 20…30°C to give 2,5-
bis(hydroxymethyl)-3,6-diarylpyrrolo[3,4-c]pyrrole-1,4-diones in unspecified yield. If
the temperature was allowed to rise above 40°C, considerable concurrent sulfonation
ensued. The diol obtained may be reacted in the same pot to condense with two additional
equivalents of the same or different DPP, resulting in DPP trimers (Scheme 1.18). The
diol also reacts with quinacridone (5,12-dihydroquino[2,3-b]acridine-7,14-dione) and
aniline – Scheme 1.19.95,96,97,98
NH
NH
O
Oa
R
R
N
N
O
O
OH OH
R
R
b N
N
O
O
N N
NH
O
ONH
O
O
R
R
R1
R1
R1
R1
Scheme 1.18. N-Hydroxymethylation of DPP. a) (CH2O)n, H2SO4, 20…30°C; b) R1–
DPP, H2SO4.

37
N
N
O
O
OH OHN
N
O
O
MeO OMea b N
N
O
O
NH NH
Scheme 1.19. Reactions of N,N′-bis(hydroxymethyl) DPP. a) MeOH, 4-TsOH, Δx; b)
aniline, DMF, Δx.
The result of the free halogen action on the parent DPP depends on the nature of
the halogen. Chlorine adds to the bicyclicDPP to give tetrachloro adduct (Scheme 1.20).
Bromine in CCl4 gives a mixture of two brominated products and unreacted starting
material. Bromination with gaseous bromine by the method of Buckles and Wheeler99,100
gives much better results, producing 3,6-di-(4-bromophenyl)-pyrrolo-[3,4-c]-pyrrole-1,4-
dione 9 in 87% yield (Scheme 1.21), but the final product still contains some mono-
bromo compound as evidenced by elemental analysis (Br found/calc 33.5/35.8)101 and
our own experience on alkylation of such a product. Bromination of DPP with N-
bromosuccinimide in sulfuric acid has been reported. Action of iodine and fluorine has
not been documented to our knowledge.

38
NH
NH
O
O
Br
Br
NH
NH
O
O
NH
NH
O
O
Cl
Cl
Cl
Cl
NH
NH
O
O
Cl
Cl
MeO
OMe
Br2 Cl2 MeOH
9 1
Scheme 1.20 DPP bromination and chlorination products.
NH
NH
O
O
NH
NH
O
O
Br
Br
Br
Br
NH
NH
O
O
BrBr
NH
NH
O
O
Br
Br
MeO
OMe
NH
NH
O
O
BrMeO
+ +Br2 MeOH
Scheme 1.21. Mechanism of DPP bromination.
DPP undergoes smooth sulfonation in fuming sulfuric acid (oleum). There is
information, scattered over claim sections of several patents, claiming control over
degree of DPP sulfonation by varying sulfuric acid concentration, temperature, and
reaction time. Thus, degree of sulfonation has been claimed to vary from zero to four
SO3H groups per DPP molecule. The disulfonic acid 10 is soluble in water and alcohols
(first three homologs) and forms salts with alkali earth metals, used as rheology-
improving (viscosity reducing) additives to other DPP pigment compositions.

39
NH
NH
O
O
NH
NH
O
O
HO3S
SO3H
fuming
H2SO4
10
Scheme 1.22. Sulfonation of DPP.
The nucleophilic attack of the Lawesson’s reagent (or P4S10 in HMPA) at the
amidic carbonyl results in an overall replacement of the carbonyl oxygen with sulphur.102
The resulting 1,4-dithio-DPP is much more susceptible to carbanion attacks and affords
overall dicyanomethylation, unattainable directly.
NH
NH
O
O
NH
NH
S
S
a
b
N
N
EtS
SEtc
NH
N
EtS
CN
NC
Scheme 1.23. Replacement of O in –NH–C=O by C. a) Lawesson’s reagent;
b) EtI, K2CO3, Me2CO; c) H2C(CN)2, THF, r.t.
Phosphorylation of the amidic carbonyl with POCl3 results in an unusually
stable (its chloride salt form has m.p. 235–237°C with decomposition) adduct (Scheme
1.24), which may subsequently be reacted with such N-nucleophiles as various aromatic
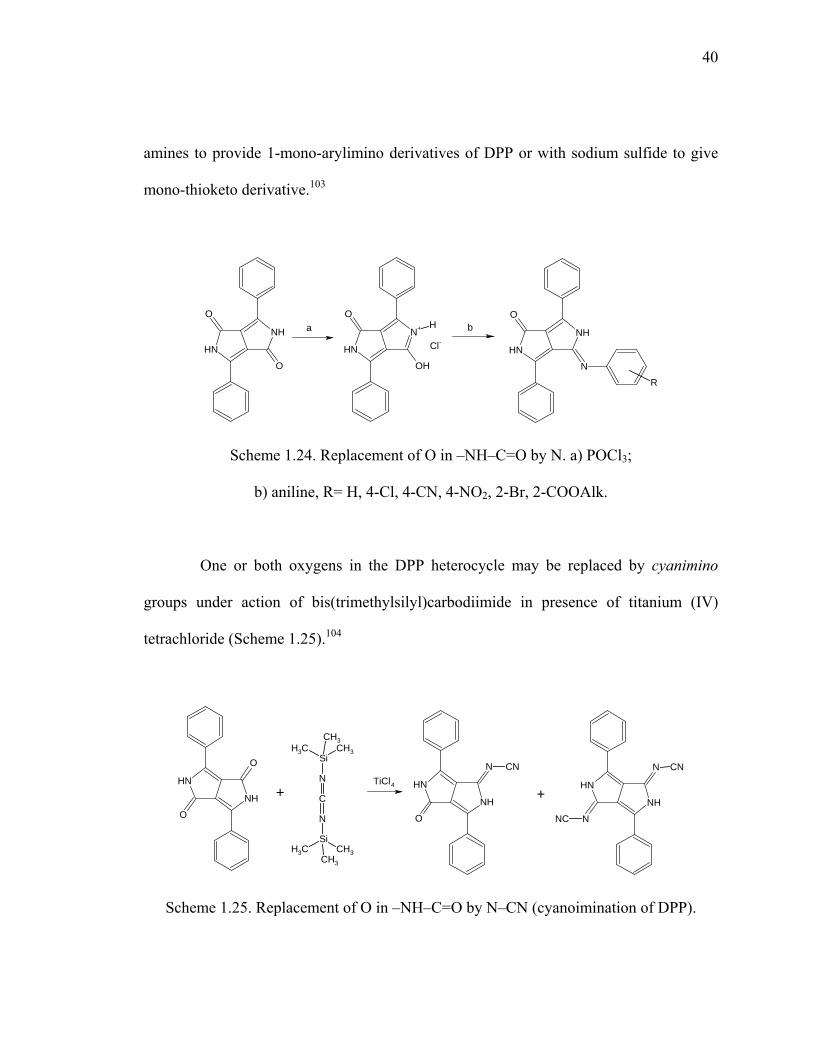
40
amines to provide 1-mono-arylimino derivatives of DPP or with sodium sulfide to give
mono-thioketo derivative.103
NH
NH
O
O
N+
NH
OH
OH
Cl-a b NH
NH
N
O
R
Scheme 1.24. Replacement of O in –NH–C=O by N. a) POCl3;
b) aniline, R= H, 4-Cl, 4-CN, 4-NO2, 2-Br, 2-COOAlk.
One or both oxygens in the DPP heterocycle may be replaced by cyanimino
groups under action of bis(trimethylsilyl)carbodiimide in presence of titanium (IV)
tetrachloride (Scheme 1.25).104
NH
NH
O
O
C
N
Si
N
SiCH3CH3
CH3
CH3 CH3
CH3
+TiCl4 NH
NH
O
N CN
NH
NH
N
N CN
NC
+
Scheme 1.25. Replacement of O in –NH–C=O by N–CN (cyanoimination of DPP).

41
Aromatic nucleophilic substitution of chlorine at para position in the phenyl
ring of several unsymmetrical N-unsubstituted 2H,5H-dihydro DPPs has been performed
with pyrrolidine and dimethylamine.105 In the same patent there is also an example of
successful nitration, followed by substitution of chlorine in the other phenyl ring (Scheme
1.26). The resulting 3-(4-nitrophenyl)-6-(4-dimethylaminophenyl)-2,5-dihydropyrrolo-
[3,4-c]pyrrole-1,4-dione is dark blue-violet pigment.
NHNH
O
O
Cl
aNHNH
O
O
Cl
N+
O-
O
bNHNH
O
O
N
N+
O-
O
CH3
CH3
Scheme 1.26. Nitration and aromatic nucleophilic substitution of chlorine in DPP.
a) KNO3, H2SO4, 0–5°C, 1 hr.; b) Me2NH, NMP, 180°C, 10 hrs in autoclave.
Aromatic nucleophilic substitution of bromine at the para position in the phenyl
ring(s) of parent 2H,5H-dihydro DPPs has been conducted with dimethylamine,
pyrrolidine, piperidine, and morpholine in 45–70% yields.106 The compounds obtained
have been claimed to be useful photoconductive substances.

42
NMP, pyrrolidine
HN
NH
O
ON
N
HN
NH
O
OBr
Br
Scheme 1.27. Aromatic nucleophilic substitution of bromine in DPP.
Palladium-mediated coupling was shown to be an efficient transformation of
bromine in the phenyl rings of DPP to carboxy (Scheme 1.28), alkoxycarboxy (Scheme
1.29) or alkylaminocarboxy (Scheme 1.30) functionality in 78–93% yields.107
PdCl2, CO, PPh3
(HCOO)2Ca, NMP
HN
NH
O
OHOOC
COOH
HN
NH
O
OBr
Br
Scheme 1.28. Pd coupling of Br-DPP with CO in presence of calcium formate.
HN
NH
O
OBr
Br
HN
NH
O
OCH3OOC
COOCH3
PdCl2, CO, PPh3
MeOH, Et3N, NMP
Scheme 1.29. Pd coupling of Br-DPP with CO in presence of MeOH.
HN
NH
O
OBr
Br
HN
NH
O
OBuNHOC
CONHBu
PdCl2, CO, PPh3
n-BuNH2, NMP
Scheme 1.30. Pd coupling of Br-DPP with CO in presence of butylamine.

43
Appropriately di-functionalized DPPs can form oligomers,60 polymers,51,52,54 and
dendrimers,59,63 as exemplified on Schemes 1.31 and 1.32.
N
N
O
O
OTf
TfO +SBu3Sn SnBu3
+OTfTfO
R
R =
N
CH2—
S OO
a
N
N
O
O
S
S
x y
n
Scheme 1.31. Stille coupling polymerization of DPP. (a) Pd(PPh3)4, LiCl, 1,4-dioxane. 51
N
N
O
O
Br
Br
C6H13
H13C6
B(OH)2(HO)2B
H13C6
C6H13
+ BrBr
H13C6
C6H13
+
a
N
N
O
OC6H13
H13C6H13C6
C6H13
H13C6
C6H13
H13C6
C6H13
x 1-x
n
Scheme 1.32. Suzuki coupling polymerization of DPP. (a) Pd(PPh3)4, K2CO3, toluene. 52

44
1.3.3. Physical Properties
The DPP and DPP-Me molecules, the crystalline molecular geometry structures
of which are shown in Fig. 1.3, both belong to point group Ci, and are not entirely planar.
The phenyl rings are twisted in the same direction, out of the plane of the planar
heterocyclic system by 7°±1° in DPP and by 31°±1° in DPP-Me. The DPP molecules
align in nearly the same molecular plane and parallel to each other due to intermolecular
hydrogen bonding. By contrast, the DPP-Me molecules are arranged in a herringbone
fashion along the stacking c axis. In DPP, the interatomic distances H(1)–H(N) and H(5)–
O(1) are 2.19 and 2.31 Å, respectively. These distances are considerably shorter than the
sum of the van der Waals radii of the corresponding atoms: 2.4 and 2.6 Å, respectively.
The C(2)–C(4) distance consequently becomes shorter than the value expected for the
carbon–carbon single bond of 1.54 Å. The observed value of 1.455 Å is much shorter, for
example, than the value of 1.496 Å found in biphenyl. The present bond shortening is
presumably caused by well-delocalized π-electrons in the pyrrolo[3,4-c]pyrrole-1,4-dione
chromophore which gives some double-bond character to the C(2)–C(4) bond. A similar
bond shortening also operates in DPP-Me. The C(2)–C(4) bond is 1.460 Å. This is
slightly longer than in DPP but still much shorter than in biphenyl.108

45
Figure 1.3. X-ray molecular structure of DPP and DPP-Me.108
In DPP there are chains of intermolecular hydrogen bonds along the ·110Ò
direction in the molecular plane between the N–H group of one molecule and the
carbonyl oxygen of the neighboring one. There are van der Waals contacts along the c
axis. In DPP-Me, by contrast, molecules face each other alternately, forming a dimeric
stacking structure. The overlap of the two molecules along the stacking axis is shown in
Fig. 1.5. There is significant overlap between the heterocyclic ring systems in DPP
causing π-π interactions (interplanar spacing 3.36 Å). On the other hand, no such overlap
is observed in DPP-Me. Instead, there are π-π contacts between the heterocyclic ring and
the phenyl ring. On the other hand, DPP-Me molecules are mainly associated together by
van der Waals forces. Because of this, the molecular arrangement is very different in DPP
compared with DPP-Me.

46
(a)
(b)
Figure 1.4. Crystal structure of (a) DPP – triclinic and (b) DPP-Me – orthorombic.108
(a) (b)
Figure 1.5. Comparison of overlap of the two molecules along the stacking axis:
(a) DPP and (b) DPP-Me.108
The astonishingly low solubility (see Table 1.5) of the parent 2H,5H-dihydro-
DPPs in most organic solvents (except, e.g. sulfuric acid and trifluoromethanesulfonic
acid) due to intermolecular hydrogen bonding and π-π interactions, results in impurity
trapping during their precipitation immediately upon their formation in the preparation
reactions. These trapped impurities are very difficult to remove by recrystallization. Even
such high boiling solvents as nitrobenzene and dimethylacetamide at their reflux

47
temperatures dissolve so little DPP (ca. several hundred milligrams per half a liter) that
recrystallization is an impractical technique for preparative scale. A mixture of diphenyl
ether and biphenyl has been used109 to change the morphology of DPP polycrystalline
powder for dyeing purposes, yet it still isn’t suitable even for gram-scale
recrystallizations. The purification method of choice is salt formation/dissolution with
strong base (NaOH, KOH) in water — polar organic solvent mixtures (DMSO, DMF),
followed by re-acidification/precipitation.110 This strategy has also been reflected in step-
wise (methanol followed by water followed by acetic acid) protolysis of DPP dianion
during work-up after the preparation reaction. In this case the formation of undesirable
by-products and degradation products can be suppressed greatly with a simultaneous
improvement in the coloristic properties.111 In our own experiments we found that, since
N-alkylation of DPP almost always forms a mixture of mono- and di-alkylated products,
the purification method of choice is chromatography, performed after the N-alkylation
step.
The thermal stability of parent 2H,5H-dihydro-DPPs is largely determined by
the lattice crystal structure and thus depends strongly on the crystal forms and
polymorphic modifications. Conditioning of crude DPP in high boiling solvents allows,
apart from partial purification, a change of the morphology of the polycrystalline
particles and an enhancement of the thermal stability. For example, boiling crude DPP in
a mixture of diphenyl ether and biphenyl at 245–260°C for 0.5…1 hr, results in the
change of the DSC thermogram as depicted on Fig. 1.6 and particle morphology change

48
to flake-like or platelet (determined by scanning electron microscopy and powder X-ray,
not shown).109
Table 1.5. Solubilities of various DPPs in mol·liter–1.46
NH
NH
O
O
[54660-00-3]
CHCl3: 8.4·10–7
PhMe: 8·10–8
MeOH: 2.3·10–6
DMSO: 3.7·10–3
NH
NH
O
O
t-Bu
t-Bu
[84632-59-7]
CHCl3: 6.4·10–6
PhMe: 1.6·10–7
MeOH: 2.6·10–6
DMSO: 1.3·10–3
NH
NH
O
Ot-Bu
t-Bu
t-Bu
t-Bu [107680-82-0]
CHCl3: 1·10–1
PhMe: 2.8·10–4
MeOH: 5.8·10–4
DMSO: 3.9·10–4
N
N
O
O
[96159-17-0]
CHCl3: 1.3·10–1
PhMe: 2.1·10–3
MeOH: 3.1·10–4
DMSO: 2.7·10–3
N
N
O
O
t-Bu
t-Bu
[107680-85-3]
CHCl3: 5.0·10–1
PhMe: 4.4·10–2
MeOH: 7.8·10–4
DMSO: 1.3·10–3
N
N
O
Ot-Bu
t-Bu
t-Bu
t-Bu [107711-05-7]
CHCl3: 5.1·10–2
PhMe: 4.2·10–3
MeOH: 3.6·10–5
DMSO: 2.4·10–5

49
Figure 1.6. DSC of (a) crude DPP and (b) conditioned DPP.
The thermal stability of 2H,5H-dihydro-DPPs in general may be characterized as
“high”, and that of several specific representatives — as not less than “amazing”. On Fig.
1.7 there are TGA traces of DPP and Br-DPP, showing that these two substances
sublime, without melting or decomposition (as determined by DSC, not shown),
continuously up to 452° and 500°C (!) correspondingly. The continuous endothermic
signal indicates the heat absorption due to sublimation. The observed residue after
sublimation is due to contaminants, trapped by DPP during its preparation reaction. Re-
sublimed samples of DPP give no residue after sublimation and their TGA traces reach
zero.
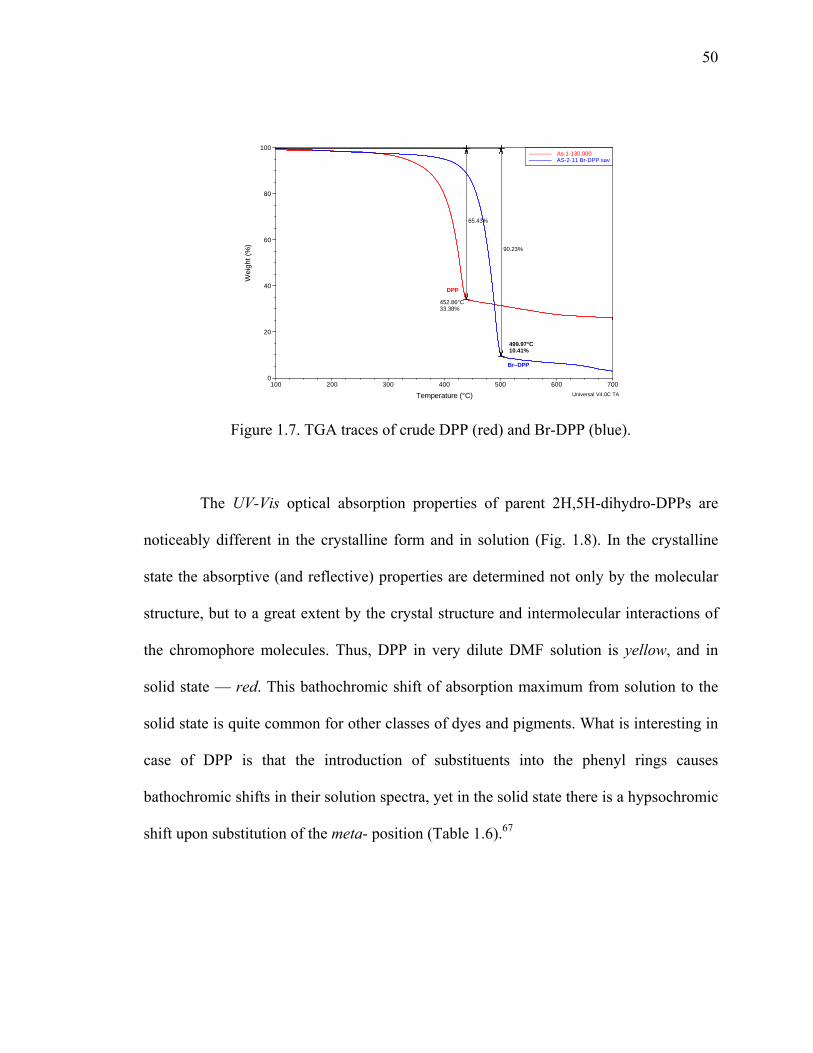
50
452.86°C33.38%
65.43%
DPP
90.23%
499.97°C10.41%
Br–DPP
0
20
40
60
80
100
Wei
ght (
%)
100 200 300 400 500 600 700
Temperature (°C)
As-1-130.000––––––– AS-2-11 Br-DPP.sav–––––––
Universal V4.0C TA
Figure 1.7. TGA traces of crude DPP (red) and Br-DPP (blue).
The UV-Vis optical absorption properties of parent 2H,5H-dihydro-DPPs are
noticeably different in the crystalline form and in solution (Fig. 1.8). In the crystalline
state the absorptive (and reflective) properties are determined not only by the molecular
structure, but to a great extent by the crystal structure and intermolecular interactions of
the chromophore molecules. Thus, DPP in very dilute DMF solution is yellow, and in
solid state — red. This bathochromic shift of absorption maximum from solution to the
solid state is quite common for other classes of dyes and pigments. What is interesting in
case of DPP is that the introduction of substituents into the phenyl rings causes
bathochromic shifts in their solution spectra, yet in the solid state there is a hypsochromic
shift upon substitution of the meta- position (Table 1.6).67
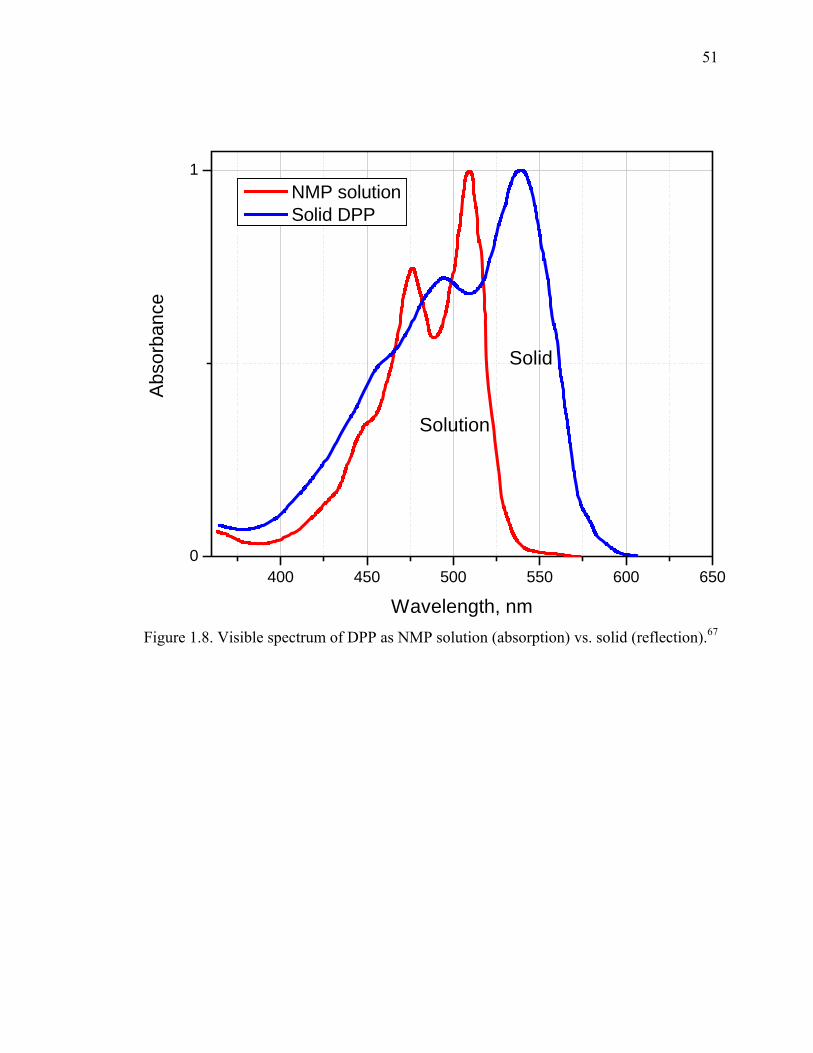
51
400 450 500 550 600 6500
1A
bsor
banc
e
Wavelength, nm
NMP solution Solid DPP
Solid
Solution
Figure 1.8. Visible spectrum of DPP as NMP solution (absorption) vs. solid (reflection).67

52
Table 1.6. Influence of 3,6-substituents and crystal structure on absorptive properties of
DPP.66
NH
NH
O
O
R
R
R Color a λmax in NMP λmax solid state b Δλmax ε
H yellow-red 504 538 34 33,000
4-Br blue-red 515 555 18 35,000
3-Cl orange 512 528 16 27,000
3-CF3 orange-
yellow
509 518 9 21,000
4-NMe2 violet-blue 554 603 51 81,500
a Determined as shade of plasticized PVC pigmented with 0.2% of corresponding DPP.
b Reflectance.
Fluorescence of parent 2H,5H-dihydro-DPPs is also very much phase-
dependent. Bathochromic shifts of fluorescence emission maxima up to 40 nm are
observed in solid samples. Vapor-deposited polycrystalline films and vapor-grown single
crystals of DPP have emission maximum shifted even further bathochromically, at 634
nm.112 This probably can be explained by more regular network of hydrogen bonds in
single crystal relative to polycrystalline powder, formed by abrupt precipitation. Intensity
of solid-state fluorescence of polycrystalline DPP increases with the size of the grains.

53
No data on solid-state fluorescence quantum yield have been found in the reviewed
literature.
Fig. 1.9. Intensity of solid state fluorescence increases with the size of the DPP grain.112
NMR properties in terms of 1H and 13C chemical shifts of some known DPPs are
presented on Fig. 1.10 and 1.11. Table 1.10 shows solvent and temperature dependence
of amidic proton chemical shift.113 The proton chemical shift increases with polarity of
solvent and appears around 11 pm, which is characteristic of a hydrogen-bonded proton.
As the temperature increases, the thermal motion disrupts hydrogen bonding and δ moves
upfield.

54
Table 1.7. Fluorescence data for several DPPs in chloroform.46
N
N
O
O
R
R
R1
R1
# R1 R λmax(lg ε) DMSO λmax λEm Δλ a ΦF
290sh, 304(4.11), 318sh,
450sh, 471(4.38), 505(4.50) 496 509 13 –
1 H H
Ref.47 496 509 13 –
3 3,5-(t-Bu)2 H 311br(4.13), 449sh,
473(4.40). 507(4.55) 500 513 13 0.63
4 2-Me H 430sh, 480sh, 453(4.29) 448 518 70 0.64
5 2-Me Me 442(4.20) 439 489 50 0.95
291(4.12), 486(4.24) 474 523 49 0.54 6 H Me
Ref.45 470 520 50 0.9±0.05
7 4-t-Bu Me 307br(4.21) 485 528 43 0.53
8 3,5-(t-Bu)2 Me 305(4.11), 475sh, 489(4.27) 484 525 41 0.56
9 4-Me Allyl Ref.45 468 521 53 0.97
a) Stokes’ shift in chloroform.
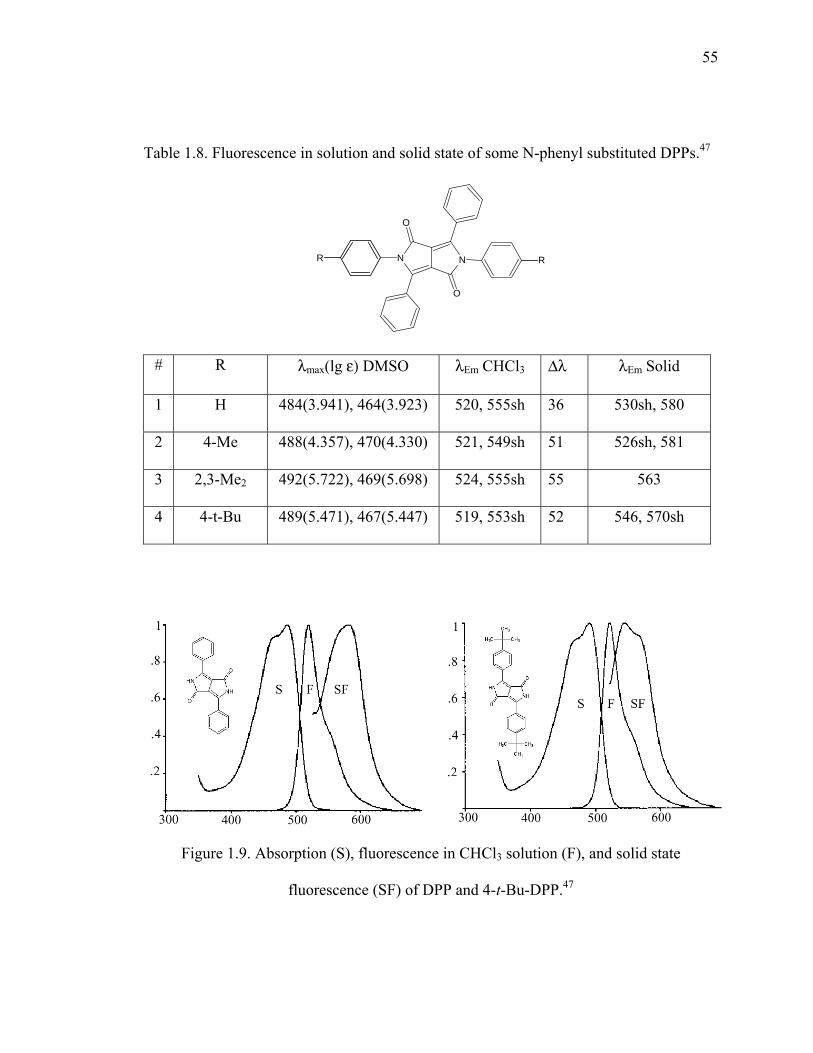
55
Table 1.8. Fluorescence in solution and solid state of some N-phenyl substituted DPPs.47
NN
O
O
RR
# R λmax(lg ε) DMSO λEm CHCl3 Δλ λEm Solid
1 H 484(3.941), 464(3.923) 520, 555sh 36 530sh, 580
2 4-Me 488(4.357), 470(4.330) 521, 549sh 51 526sh, 581
3 2,3-Me2 492(5.722), 469(5.698) 524, 555sh 55 563
4 4-t-Bu 489(5.471), 467(5.447) 519, 553sh 52 546, 570sh
Figure 1.9. Absorption (S), fluorescence in CHCl3 solution (F), and solid state
fluorescence (SF) of DPP and 4-t-Bu-DPP.47

56
Table 1.9. Fluorescence quantum yields of some N-mono-aryl and N,N′-diaryl substituted DPPs.114
N
N
O
O
R3
R4
R1
R2
# R1 R2 R3 R4 λmax(lg ε) Solvent λEm ΦF
1 H H 4-t-Bu-C6H4– H 488.5(5.42),
466.7(5.39) CHCl3 521, 561sh a Ref. 47
2 H H 4-NO2-C6H4– 4-NO2 470(4.33) D 516 0.02
3 H H H H 498(4.23) D 520 0.44
4 H H H 4-MeO 472(4.17), 500(4.20) D 521 0.03
5 H H H 4-CF3 467(4.17), 493(4.19) D 519 0.43
6 Cl Br H 4-MeO 481(4.43), 511(4.46) D 532 0.09
7 H H Me H 468(4.11) DCM 521 0.43
8 H H Me 4-MeO 471(4.19) DCM 522 0.23
9 H H Me 4-CF3 470(4.11) DCM 520 0.48
10 Cl Br Me 4-MeO 487(4.18) DCM 533 0.15
11 H H PhCH2– H 468(4.11) DCM 519 0.42
12 H H PhCH2– 4-MeO 470(4.27) DCM 522 0.27
13 H H PhCH2– 4-CF3 468(4.12) DCM 518 0.43
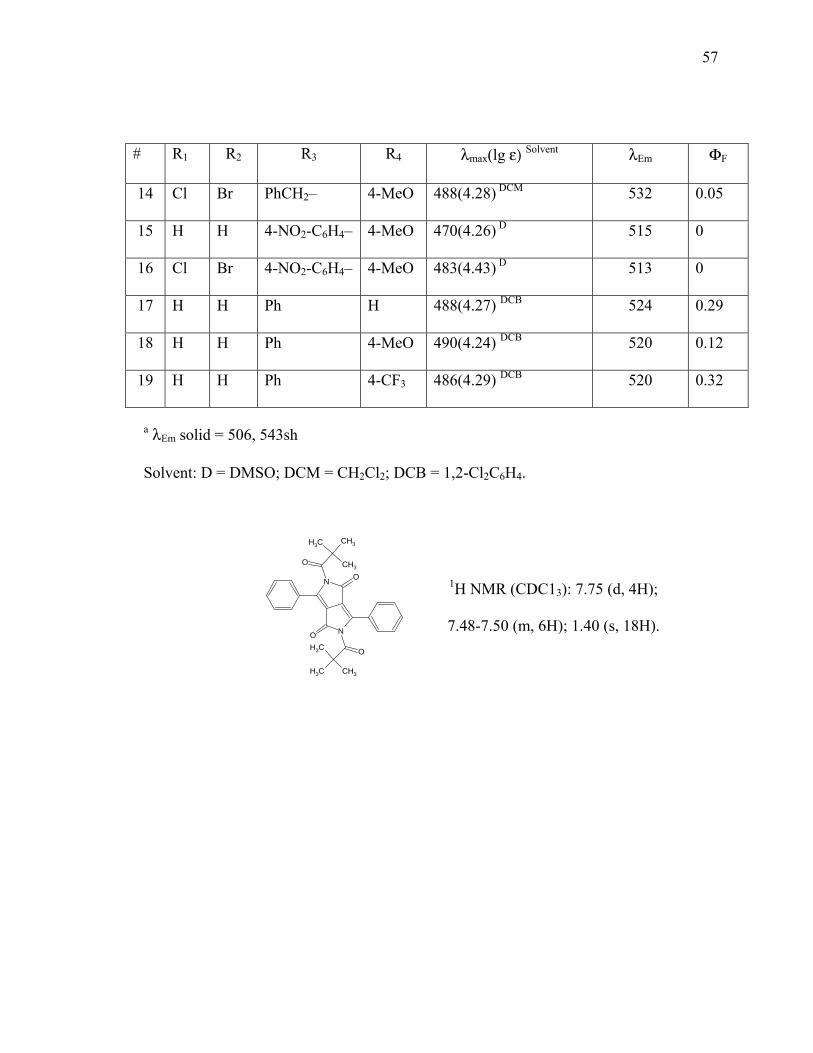
57
# R1 R2 R3 R4 λmax(lg ε) Solvent λEm ΦF
14 Cl Br PhCH2– 4-MeO 488(4.28) DCM 532 0.05
15 H H 4-NO2-C6H4– 4-MeO 470(4.26) D 515 0
16 Cl Br 4-NO2-C6H4– 4-MeO 483(4.43) D 513 0
17 H H Ph H 488(4.27) DCB 524 0.29
18 H H Ph 4-MeO 490(4.24) DCB 520 0.12
19 H H Ph 4-CF3 486(4.29) DCB 520 0.32
a λEm solid = 506, 543sh
Solvent: D = DMSO; DCM = CH2Cl2; DCB = 1,2-Cl2C6H4.
N
N
O
O
O
CH3 CH3
CH3
O
CH3CH3
CH3
1H NMR (CDC13): 7.75 (d, 4H);
7.48-7.50 (m, 6H); 1.40 (s, 18H).

58
Figure 1.10. 1H and 13C NMR spectra of 3,6-diphenyl-2,5-diallylpyrrolo[3.4-c]pyrrole-
1,4-dione.45
Figure 1.11. 1H and 13C Assigned chemical shifts for
3,6-diphenyl-2,5-diallylpyrrolo[3.4-c]pyrrole-1,4-dione. 45
59
Table 1.10. 1H Chemical shifts of the NH group of DPP in different solvents and at
various temperatures.113
Chemical shift, δ, ppm Solvent
25°C 50°C 75°C 100°C
Dioxane-d8 10.88 10.58 10.38 10.20
DMF-d7 11.08 10.81 10.57 10.43
DMSO-d6 11.35 11.14 10.96 10.76
Table 1.11. 13C Chemical shifts of DPP, its mono- and di-anion.
δ, ppm in DMSO-d6 #Ca
DPP DPP– DPP2–
1 162.4 172.0 172.5
1a 110.7 117.9 118.7
3 144.0 155.3 155.4
4 127.7 134.4 135.6
5 128.9 128.0 127.6
6 127.7 127.6 127.5
7 131.7 128.5 127.6
NH
NH
O
O
1
3
1a
45
6
7
a Numeration is shown on the right for carbon atom referencing only and not for
nomenclature purposes.

60
1.4. Newly prepared DPP dyes
The optical and fluorescent properties of DPP dyes, specifically their high
photostability, large Stokes’ shift, small size of the molecule, and high fluorescence
quantum yields prompted us to explore them in the SMS, as well as design and
preparation of a series of new fluorophores of this class. The small size (both in
molecular weight and bulkiness) of the DPP chromophore, we believe, would allow it to
permeate more easily through the cell membrane. The targeted new DPP fluorophores
should posses the following properties (separately or combined):
• red-shifted (with respect to DPP) absorption and fluorescence
• high (compared to 2,5-dihydro DPPs) solubility
• functionalization: for covalent labeling and for SMS in polymers
The need for the red shift in absorption and fluorescence transferred into the
following structural proposals. From the electronic color theory of organic dyes115,116 and
molecular orbital theory117 it follows that absorbance wavelength bathochromically shifts
as the length and degree of the conjugation chain in the dye increase (within certain
limits). Electron-donating and electron-accepting groups, introduced at certain places of
the conjugation chain and capable of participating in the delocalization of electronic
density (i.e. groups with mesomeric effect), also shift the absorbance bathochromically.1
Thus, structure-wise, bathochromically shifted absorption and emission can be achieved

61
by either extension of the length of a conjugated chain or by increase of the degree of
conjugation, or both. Application of these considerations to the DPP structure yields
structural modifications, one of which is depicted in the Scheme 1.33.
The property aim of solubility is based on the crystal structure and established
trends in solubilities of known DPP compounds, reviewed above. Rupture of the
intermolecular hydrogen bonding network by introduction of substituents at one or better
both lactam nitrogens, greatly increases the solubility of DPP compounds. Thus, we
planned to make N-alkylated DPPs and compare their solubilities. The single-molecule
spectroscopy requires relatively low solubility of the fluorophores. Simple dimethylation
increases the solubility of DPP in chloroform seven orders of magnitude. Thus, even the
solubility of mono-N-substituted DPPs might suffice for our purposes. We have prepared
and compared solubilities of N,N′-dimethyl, dipropyl, dihexyl, didecyl, didodecyl, and
dibenzyl DPPs and some of their mono- analogs. It turned out that for purification by
recrystallization the C3 chain was the most convenient one: methylated DPPs with
solubility in chloroform of order ~0.1 M required much higher solvent volumes for
recrystallization, while the didodecyl derivative (~0.8 M chloroform solubility) was too
soluble. For this reason, most of the compounds prepared here have propyl substituents
on the lactam nitrogen(s).
1 These statements are very simplified generalizations and should not be considered as all-cases rules.

62
The functionalization was first planned to be achieved via pre-functionalization
of nitriles used as starting materials in the condensation with dialkyl succinate. However,
this approach proved to be futile, at least in our hands and required exploration of post-
factum functionalization (after the formation of the DPP core), which is discussed below.
The parent DPP 1 and Br-DPP 9 are used in commercial pigment mixtures,
manufactured by Ciba-Geigy Specialty Chemicals Co., and, according to acknowledge-
ments in several papers,52,60 can be obtained from the European Research Division, in
Switzerland, as individual compounds as a favor. However, our communications with the
U.S. division of Ciba-Geigy (Ann Cardillo, CE) resulted in no fruitful answer. Thus, we
had to learn and prepare the DPP precursors ourselves. Down this road we also learned
about nitrile functionality tolerance in the DPP preparation reaction.

63
N
N
O
ON
Alk
Alk
NAlk
Alk
N
N
O
O
N
NAlk
Alk
Alk
Alk
N
N
O
ON
Alk
Alk NAlk
Alk
Con
juga
tion
Leng
th In
crea
se
Δ π−
π * E
nerg
y D
ecre
ase
Bath
ochr
omic
Shi
ft
Alk = C6H13: AS-2-69b
Alk = Et: AS-3-18
Alk = Bu: AS-3-22
Scheme 1.33. Design of red-shifted DPP chromophores.
The sterically hindered di-alkyl succinates have been prepared (Scheme 1.34):
di-iso-propyl succinate [924-88-9] 11 — by esterification of succinic acid with isopropyl
alcohol in presence of sulfuric acid; and di-tert-amyl succinate [77106-39-9] 12 — by
trans-esterification of diethyl succinate with tert-amyl alcohol (2-methyl-2-butanol) in the
presence of lithium alkoxide.118

64
OHOH
O
O
OO
O
O
11
a
OEtOEt
O
O
OO
O
O 12
b
Scheme 1.34. Preparation of sterically hindered di-alkyl succinates.
(a) i-PrOH, H2SO4 (cat.), az. H2O removal. (b) t-AmOH, Li, fractional EtOH removal.
The following aromatic nitriles have been prepared: 4-fluorobenzonitrile 13, 4-
bromobenzonitrile 14, 4-methoxybenzonitrile 15 — from corresponding aldehydes by a
corrected procedure of Wang119 (Scheme 1.35); 4-(pyrrolidin-1-yl)benzonitrile 16, 4-(N-
n-hexylamino)benzonitrile 17, and 4-(N,N-di-n-hexylamino)benzonitrile 18 — from 4-
aminobenzonitrile; 4-(N,N-di-n-butylamino)benzonitrile 19 — from 4-fluorobenzonitrile
(Scheme 1.36). The Wang procedure was corrected (and thus improved), taking two
equivalents of phthalic anhydride instead of the specified one equivalent, since two
equivalents of water are formed overall – one at the oxime formation step, and another –
at the dehydration of the latter, resulting in much better conversion and higher yield.
Also, a higher boiling point solvent, NMP, was used instead of acetonitrile, resulting in
shorter reaction times.

65
CHO
R
a
CN
RR
NH
OH
b
– H2O – H2O
Scheme 1.35. Preparation of 4-R-benzonitriles: R= F (13), Br (14), OMe (15).
(a) NH2OH, Et3N. (b) phthalic anhydride, NMP.
CN
NH2
a
CN
NHAlk
CN
N
b
CN
NAlk Alk
c
CN
F
d
16 17 R=C6H13: 18, Bu: 19
Scheme 1.36. Preparation of 4-aminobenzonitriles. (a) 1,4-diiodobutane, i-Pr2NEt, NMP.
(b) C6H13Br, HMPA. (c) C6H13Br, K2CO3, NMP. (d) Bu2N, Py, HMPA.
The results of the reactions between prepared or commercial nitriles with dialkyl
succinates are summarized in the Table 1.12. Firstly, only four out of eleven nitriles gave
any tangible amount of DPP upon condensation. Secondly, for those nitriles, from which
we did obtain corresponding DPPs, we were consistently obtaining much lower yields
than specified in the patents or literature. Because of this, we performed some
optimization of the DPP preparation reaction, based on the considerations from the
literature, patents, and our own.

66
Table 1.12. DPP preparation reactions. R # N
R
OAlk
O
O
OAlk
a
NH
NH
O
OR
R+
H
4-F
4-CN
4-Me
1
20
21
22
R Alk in
(AlkO)2Suc2 Yield, %
Lit. Yield Ref. or
[CAS RN] if N/A
H
Et
i-Pr
t-Am
23
48
56
11.5 71, 59 46
65 71
82 71
F Et
i-Pr
12
66
no prep.
[84632-57-5]69,120
Br Et 0 81.7 72, 43 121
MeO Et traces 6.6 46,51
Ph Et traces 10 71
Bu2N Me, Et, i-Pr, t-Am traces Me2N: 3.7 71
C6H13NH Et 0 –
(C6H13)2N Et 0 Me2N: 3.7 71
NO2 Et 0 [186967-03-3]122
4-C8H17–C6H4– Et 0 –
4-C11H23O–C6H4– Et 0 –
N Et 0
no prep.
from nitrile
4-CN t-Am 35 80 71
Me Et, i-Pr,
t-Am
12
38
23.4 71
48.6 71

67
Since self-condensation of dialkyl succinates slows down with the bulkiness of
the alkyl moiety, we switched in our DPP preparations from commercially available
diethyl succinate to bulkier di-iso-propyl succinate and even more bulkier di-tert-amyl
succinate. In going through this succinate ester series, the yield of the parent DPP 1
increased in our hands from 23 to 48 to 56%. The rate of the succinate ester addition to
the reaction mixture also affects the yield of DPP dramatically. For example, in our hands
4-Tol-DPP 22 wouldn’t form in tangible amounts if the 4-toluonitrile had been added to
the reaction mixture from the onset at once. However, with a syringe pump metered
addition rate of 3.4 ml/hr and use of (t-AmO)2Suc2, we obtained the latter in 38% yield.
Considering the stoichiometry of the condensation reaction, one mole of a succinate ester
reacts with two moles of nitrile. Yet if the succinate ester self-condenses in a parallel
competing reaction, the initial 1:2 ester to nitrile ratio will leave us with some unreacted
nitrile, which was indeed detected by gas chromatography. This consideration led us to
using an excess of succinate ester: the optimal ratio of di-tert-amyl succinate ester to
nitrile was determined to be 0.57 and 0.8:1.16≈0.7 for di-iso-propyl succinate.
Another important improvement, contributing to the purity of the crude DPP is
the rate of hydrolysis. Since the dianion DPP2– is quite soluble in the reaction medium
(especially compared to solubility of DPP), abrupt acidification of the latter causes, to our
presumption, occlusion, trapping, and co-precipitation of impurities, which was proved
by TGA analysis of the crude DPP (Fig. 1.12): as opposed to clean sample (recrystallized
or sublimed), crude DPP does not sublime completely at the end of TGA run, and leaves

68
a residue. Slow and step-wise increase of the acidity of the reaction mixture allows
slower crystallization of DPP, resulting in less sublimation / TGA residue.
34.52%
17.53%
10.07%
crude, fast ppt
crude, slow ppt
recr. DMAc0
20
40
60
80
100
Wei
ght (
%)
0 100 200 300 400 500 600
Temperature (°C)
As-1-130.000––––––– As-1-130.001––––––– AS-2-70.000–––––––
Universal V4.0C TA
Figure 1.12. Effect of fast vs. slow work-up on the purity of crude DPP.
Concurrently with our learning about the above DPP preparation reaction itself,
we also learned that our initial plans to prepare functionalized and derivatized DPPs by
functionalizing or derivatizing the starting nitriles were substantially compromised by our
inability to reproduce many reactions, claimed in the patents, as well as to force other
nitriles to give corresponding DPPs. The “nitrile pre-functionalization” approach became
even less attractive after our several failures to either alkylate certain DPPs or to separate
discrete products from such alkylation.

69
1.4.1. Alkylation of DPPs
The preferred method for alkylation of DPP in the patents is the action of methyl
or ethyl tosylates (4-tolylsulfonates) and potassium carbonate on DPP in DMF or
nitrobenzene at their boiling point. Alkylation with alkyl halides, especially with their
higher homologs (e.g. butyl), gives much less satisfactory yields, as well as considerable
amount of mono-alkylated DPP in addition to di-alkylated one. We tried to reproduce the
reported alkylation reactions, tried new or modified procedures, and prepared several new
mono- and di- alkylated DPPs. The methyl and ethyl tosylates are commercially
available, and iso-propyl tosylate 36 was prepared by a modified procedure of
Waldron.123
Methylation and ethylation with corresponding alkyl tosylate proceeds almost
quantitatively and, given enough alkylating reagent and time, the di-alkylated products
are formed in predominance. Separation of mono-methylated DPP, however, was
reported recently by Zambuonis.124 The alkylation reaction (Scheme 1.37) proceeds via
several steps (Scheme 1.38). Firstly, (1) DPP goes from crystalline state to solution,
where (2) it gets deprotonated to DPP– or (3) DPP2–, depending on the strength of the
base used, and either of those anions then (4) attacks alkyl halide (or tosylate), producing
mono-alkylated DPP, followed by (5) subsequent attack onto second equivalent of R–X,
producing di-alkylated DPP. This scheme suggests that the overall rate of the alkylation
may depend on the DPP concentration (low solubility may be a limiting factor) in a given
solvent, solubility of the base in the same, and the stationary concentration of the DPP
anion(s).

70
If the base used for deprotonation is weak (e.g. K2CO3), and the solvent is
nitrobenzene, the stationary concentration of DPP anions is low, and the reaction rate is
limited by the solubility of DPP and DPP– in the solvent. In this case high reaction
temperature (200°C) is necessary to complete the reaction. If the base is strong, the
equilibrium may be shifted towards almost complete (t-BuOK) or complete (NaH, KH)
deprotonation of the dissolved DPP, and limited now only by the solubility of DPP– and
DPP2– in the solvent, thus permitting lower reaction temperatures to be employed — in
the range of 60 to 120°C. The solubility of the ionized DPP species is highest in mixtures
of polar organic solvents and water (e.g. DMF:water ≈ 5:1), but our alkylation attempts in
such solvent mixture never went to completion (even with addition of phase transfer
reagents, like Aliquat 336) or gave satisfactory yields. Employment of dry DMF, DMAc,
or DMSO, however, allowed complete consumption of the starting material. The
discussion of the possible reasons for such behavior is found below, where basic
degradation of DPP is discussed. The reactivity of the alkylating agent is also an
important factor — alkylating agents with higher reactivity provide not only shorter
reaction times, but also somewhat higher yields, presumably because shorter reaction
times reduce the degradation of DPP, caused by the base. For this reason, addition of
catalytic amounts of potassium iodide to the reaction mixture whenever using alkyl
bromides as alkylating reagents, is advisable. The change in the structure of the
alkylating agent, which hinders SN2 mechanism and favors SN1 mechanism, seems to
prevent N-alkylation of DPP under the conditions studied – compare results for
compounds 26–28 in the Table 1.13 with the other entries. Some additional examples of

71
N-alkylation reactions, as well as effect of base (Cs2CO3 instead of K2CO3) are discussed
below with the functionalized DPPs.
The solubility of the prepared alkyl-DPPs follows the following trend: within the
pair, di-alkylated DPP has generally higher solubility than mono- analog. As the length of
the alkyl chain increases, however, the solubility of the mono-alkyl DPP in DMF
becomes comparable to that of di-alkyl one. The solubility in non-polar solvents (hexane,
toluene) or in solvents of low polarity (ether) also increases with the length of the alkyl
chain. For example, neither DPP-Me, nor DPP-Et is considerably soluble in ether. The
next homolog, however, DPP-Pr, dissolves in THF, and DPP-Hx is soluble in ether, while
DPP-C12 dissolves in neat hexane at room temperature.
NH NH
O
O
+ R X + BaseDMAc
or NMPN N
O
O
R R N N
O
O
R H+
Scheme 1.37. Alkylation of DPP. X = OTs for R = Me, Et, i-Pr;
X = I, Br, or Cl for R = i-Pr, n-Pr, C6H13, C10H21, C12H25, PhCH2.

72
NH NH
O
O
crystal
NH NH
O
O
solution
N NH
O
O
– N N
O
O
– –
N NH
O
O
R
Base Base
RX
N N
O
O
R RN N
O
O
RRX–
RX
Base
(1) (2) (3)
(4)
(5)
(4)
Scheme 1.38. Step-wise alkylation process of DPP.
1.4.2. Action of bases on DPP
The study of base action on DPP was prompted by two reasons: relatively low
and sometimes inconsistent yields in alkylation reactions, and contemplation of the
possibility to obtain bis-lactone 7 upon hydrolysis of DPP. The bis-lactone 7 has been
used in the (limited) preparation of N-arylated DPPs and easy access to it would be quite
handy.
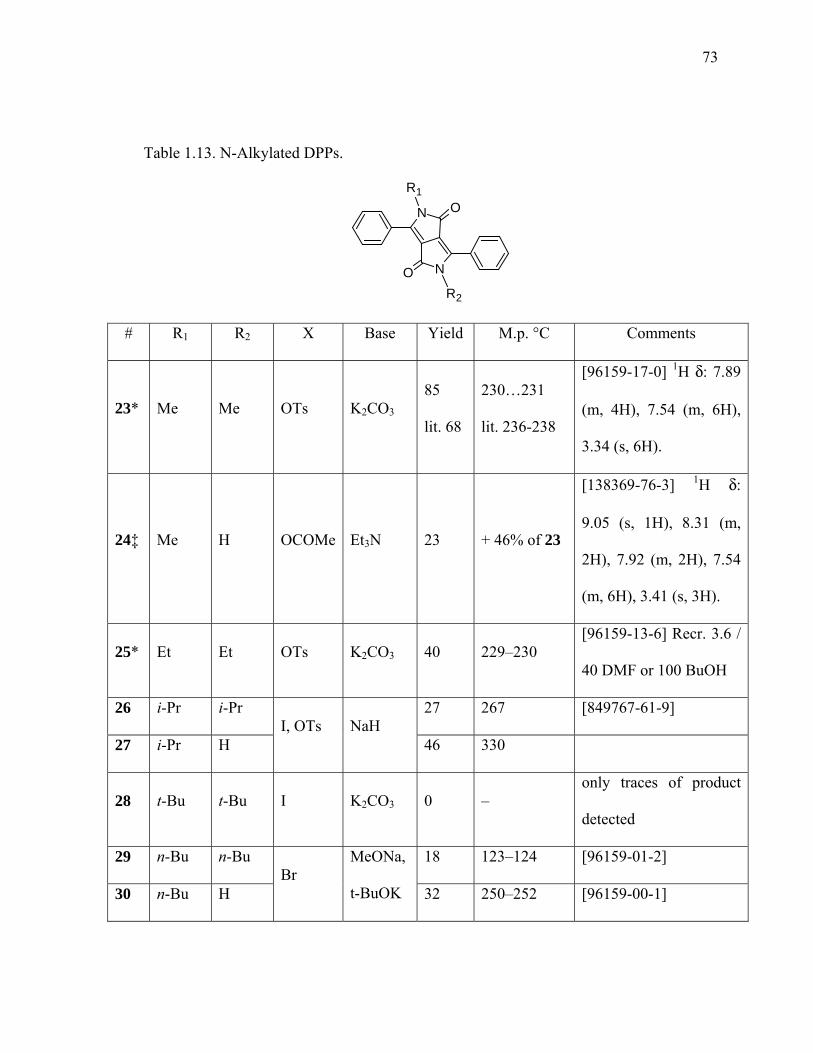
73
Table 1.13. N-Alkylated DPPs.
N
N
O
O
R1
R2
# R1 R2 X Base Yield M.p. °C Comments
23* Me Me OTs K2CO3 85
lit. 68
230…231
lit. 236-238
[96159-17-0] 1H δ: 7.89
(m, 4H), 7.54 (m, 6H),
3.34 (s, 6H).
24‡ Me H OCOMe Et3N 23 + 46% of 23
[138369-76-3] 1H δ:
9.05 (s, 1H), 8.31 (m,
2H), 7.92 (m, 2H), 7.54
(m, 6H), 3.41 (s, 3H).
25* Et Et OTs K2CO3 40 229–230 [96159-13-6] Recr. 3.6 /
40 DMF or 100 BuOH
26 i-Pr i-Pr 27 267 [849767-61-9]
27 i-Pr H I, OTs NaH
46 330
28 t-Bu t-Bu I K2CO3 0 – only traces of product
detected
29 n-Bu n-Bu 18 123–124 [96159-01-2]
30 n-Bu H Br
MeONa,
t-BuOK 32 250–252 [96159-00-1]

74
31† 2-EtHx 2-EtHx – – – – [132029-46-0]
32 Pr Pr 62 189 Sol. toluene
33 Pr H I t-BuOK
30 276 Sol. CHCl3
34 C6H13 C6H13 8 134 Sol. toluene
35 C6H13 H Br, I t-BuOK
16 252 Sol. toluene
36† C10H21 C10H21 Br t-BuOK 42 117 [132029-47-1]
37 C12H25 C12H25 47 114 Sol. Et2O, toluene,
hexane
38 C12H25 H
I t-BuOK
Sol. toluene
39* PhCH2 PhCH2 51 290–292
[96159-02-3] 1H δ: 7.75
(d, 4H), 7.49-7.43 (m,
6H), 7.30 (t, 4H), 7.24
(t, 2H), 7.19 (d, 4H),
4.99 (s, 4H).
40 PhCH2 H
Br, Cl t-BuOK
32 344
41* Allyl Allyl 42 209
lit. 216–217
[96159-07-8]
42 Allyl H
Br t-BuOK
46 309
* known compound
† These compounds have been claimed in the patent,125 but no preparation,
characterization or properties have been reported.
‡ Preparation and separation is given in the patent.124

75
A dilute (1 mg/ml) solution of DPP in a 5M solution of sodium hydroxide in
mixture of DMAc and water (5:1) has a deep crimson color, characteristic of DPP2–
anion. However, left at room temperature for five days, the color fades out completely,
indicating degradation of the chromophore. It is entirely reasonable to suggest then that in
any reaction of DPP, employing base, there will be a competing reaction of degradation.
More interestingly, even fully alkylated DPP, for example, DPP-Pr 32, subjected to the
same conditions, outlives parent DPP not for much longer.
DPP 1 was subjected to a basic aqueous hydrolysis with potassium hydroxide in
a closed high-pressure steel reactor at 120°C to give the sole aromatic degradation
product — benzoic acid (Scheme 1.39). Lower reaction temperature does not affect any
changes on DPP within 24 hrs. The mechanism of such transformation is clear up to the
sym-dibenzoylsuccinic acid decarboxylation, whereafter some oxidant needed, probably
atmospheric oxygen left in the vessel, or oxidation by means of disproportionation can be
proposed, to get the detected product.
The fact that di-alkyl DPPs undergo degradation was also noticed during the
study of the optical properties of DPP-Me 23 at Ciba-Geigy by Jin Mizuguchi. He found
that upon sodium hydroxide titration of DPP-Me solution in DMSO–H2O, the color of the
solution disappears, creating a new UV peak at 380 nm, though reversibly: titration back
with hydrochloric acid restores the absorption peak at 475 nm. Mizuguchi proposed
(based on 1H NMR evidence, not supplied though) a Michael addition of hydroxide ion to
the 1-position of the DPP ring, which could be also viewed as 4-position of α,β-
unsaturated amide. The Michael addition product is proposed to be stabilized by an

76
intramolecular hydrogen bond, as depicted in Scheme 1.40. The quantitiy of sodium
hydroxide, required to form such adduct, is “slightly more than theoretical amount, and
further addition of sodium hydroxide brings a complete discoloration of DPP-Me solution
(decomposition) due to subsequent reactions.”113 It is possible that these unspecified
subsequent reactions are lactam hydrolysis, similar to what we observed in case of basic
degradation of DPP 1. No further study in this direction has been conducted.
NH
NH
O
Oa
O OH
COO
COO
O
PhPh
O
–
–
NH
NH
OH
OPh
PhO–
O
PhPh
ONH2
OOC
NH
OPh
Ph–
HNHOOC
NH
OPh
Ph
–
NHOOC
NH
OPh
Ph–
OHOOC
NH
OPh
PhNH2
–OOOC
NH
OPh
Ph–
– 2CO2
Scheme 1.39. High temperature basic degradation of DPP. (a) KOH, EtOH, H2O, 120°C,
6 hrs.

77
N
N
O
O OH
H+
–
N
N
O
O
O
H –
Scheme 1.40. Michael addition of OH– to DPP-Me.113
1.4.3. Halogenation of DPPs
Since the “nitrile pre-functionalization” approach to the functionalized DPPs has
“kicked the bucket”, at least in our hands, we moved on to the post-functionalization, that
is modification of the DPP after the core bicyclic system has been formed. The
bromination of the parent DPP by gas-phase bromine is known. When repeated as
prescribed,101 however, it gave a material, which upon per-alkylation (with methyl
tosylate or propyl iodide, NaH, DMAc) produced a mixture of at least three compounds,
which have very close Rf on TLC. Apparently, they correspond to three precursor DPPs:
unreacted parent DPP 1, as well as mono- and di-brominated ones. Bromination,
conducted with neat liquid bromine or with a solution in CCl4 did not give cleaner
product — in full accordance with the patent’s statement. However, when we applied the
neat liquid bromine treatment after the gas-bromination, the resulting product gave upon
alkylation a good yield of Br-DPP-Pr, though still requiring purification by
chromatography. In our search for a cleaner bromination method, we investigated the
bromination of N,N′-dialkylated DPPs and found a peculiar trend (Scheme 1.41): DPP-
Me 23 undergoes bromination both in gas phase and in solution smoothly and cleanly,

78
while the higher N,N′-dialkylated homologs are destroyed by bromine even under careful
exclusion of light. For example, Pr-DPP 32 gives, upon treatment with bromine solution
in chloroform even at –30°C, a black tarry product, deficient of the DPP chromophore
system and its characteristic absorption and fluorescent properties. This degradation of
the DPP chromophore was observed for R = propyl and hexyl, while R = Me gives
brominated product (Br-DPP-Me) even in higher overall yield, than if DPP is brominated
first and methylated second. Alkylation of brominated DPP was conducted similar to that
for the parent DPP — see details in the Experimental section.
Since the intended destiny of the bromine in brominated DPPs was to use them
as reactive sites for subsequent Pd-catalyzed couplings, and since it is known that iodo-
arenes generally give better yields in such reactions, we also aimed to introduce iodine
atoms into phenyl rings of the DPP. Iodine monochloride reacts neither with parent DPP
1, nor with alkylated DPPs — neither in gaseous phase, nor in solution, nor neat and thus
the 4,4′-diiododiphenyl DPPs are not available by direct iodination with ICl.1 However,
we found that the bromine atoms at the 4-positions of the benzene rings in both parent
and alkylated DPPs can be easily replaced by iodine using an excess of KI/CuI in
DMAc126 at 180°C, thus producing the desired iodo-DPP indirectly.
The cyano group is often considered as a pseudo-halogen. For example,
dicyanogen (CN)2 has both physical and chemical properties similar to that of a diatomic

79
halogen. Likewise, reaction of the same aryl bromides 43 and 44 with KCN/CuCN gives
the nitrile derivatives 47 (X=CN, Table 1.14) in good yield (Scheme 1.43), which was
inaccessible by the attempted alkylation of the 3,6-bis(4-cyanophenyl) DPP 21. In
contrast to successful bromine-to-iodine halogen exchange, treatment of Br-DPP-Pr 44
with cesium fluoride127 did not give any substitution by fluorine. The obtained
halogenated DPPs are summarized in the Table 1.14.
1.4.4. Substitution of halogen by amine in DPPs
Aromatic nucleophilic substitution of chlorine in N-unsubstituted 2H,5H-
dihydro DPPs is known. We extended the scope of this reaction to N-substituted DPPs
with bromine atoms in the aromatic system. Reactions of Br-DPP-Me 43 and Br-DPP-Pr
44 with pyrrolidine, piperidine, dibutyl- and dihexylamines have been performed to give
the corresponding diaminated DPPs (Scheme 1.44), which are deep red in color and give
beautiful crimson fluorescence. The cyclic amines give generally higher yields than
aliphatic di-n-alkylamines. The latter also give a mixture of mono- and di-substituted
products (e.g. 51 and 52, 53 and 54), in the mono-product dominates. And if the reaction
with the cyclic amines can be pushed to a completion (no mono-product on TLC) by
1 Electrochemical iodination and iodination with other «I+»-genic species was not explored.

80
longer reaction time and higher temperature, the same strategy in the case of di-n-
alkylamines isn’t as fruitful, though addition of HMPA and use of large excess of the
amine increases the yield somewhat.
NH
NH
O
O
N
N
O
OPr
Pr
N
N
O
O
a
a
a
N
N
O
OPr
Pr
Br
Br
NH
NH
O
O
Br
Br
N
N
O
O
Br
Br
b b
c c
X
96%
94%
62% 37%
46%85%
19
8
1
7
20
Scheme 1.41. Bromination reactions of DPPs.
(a) Br2 (gas), (b) PrI, K2CO3, DMAc, (c) MeOTs, K2CO3, DMAc

81
Table 1.14. Halogenated DPPs – yield indicated is that from parent compound (R=X=H).
N
N
O
O
R
R
X
X
# X R Yield % M.p. °C
9 Br H 94 >450
43 Br Me 46 (from 9) 96 (from 23) >350
44 Br Pr 37 247
45 I Me 37 dec. 355
46 I Pr 68 260.5
47 CN Pr 83 227
N
N
O
OPr
Pr
Br
Br
N
N
O
OPr
Pr
I
I
N
N
O
O
Br
Br
N
N
O
O
I
I
a
a
46
4543
44
Scheme 1.42. Indirect approach to iodo-DPPs. (a) KI, CuI, DMAc, 180°C.
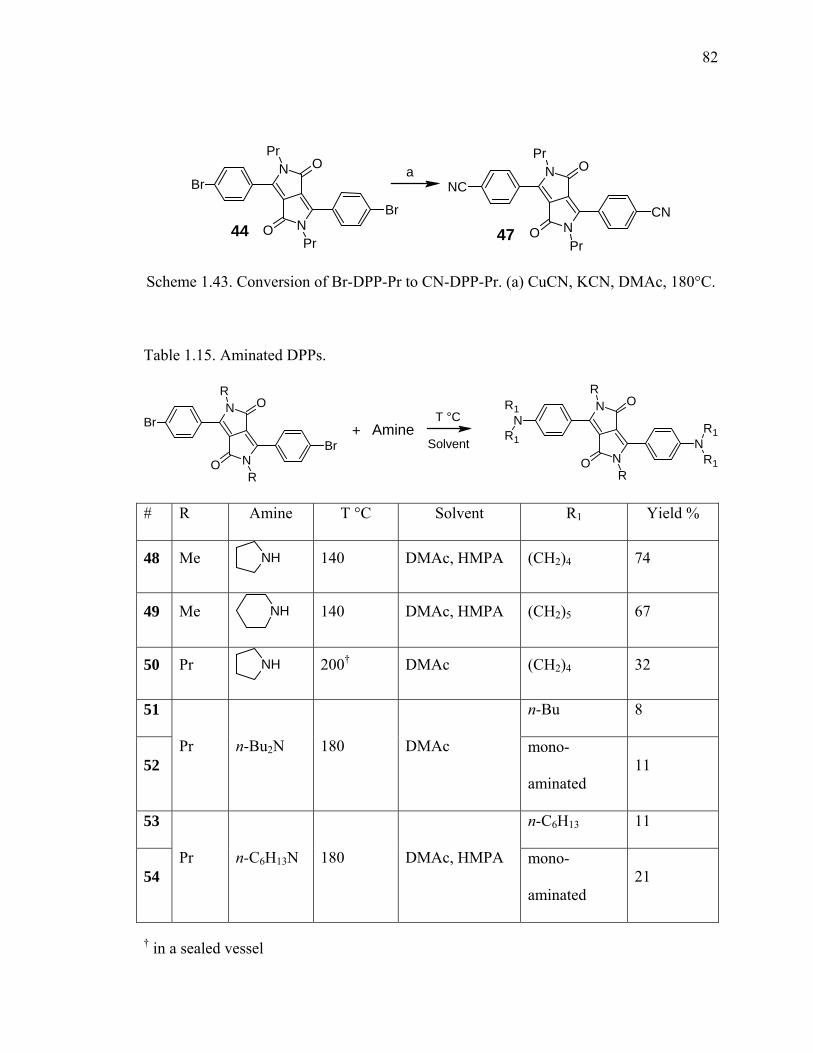
82
N
N
O
OPr
Pr
Br
Br
N
N
O
OPr
Pr
NC
CN
a
4744
Scheme 1.43. Conversion of Br-DPP-Pr to CN-DPP-Pr. (a) CuCN, KCN, DMAc, 180°C.
Table 1.15. Aminated DPPs.
N
N
O
O
R
R
Br
Br+ Amine
T °C
Solvent
N
N
O
O
R
R
N
N
R1
R1
R1
R1
# R Amine T °C Solvent R1 Yield %
48 Me NH
140 DMAc, HMPA (CH2)4 74
49 Me NH
140 DMAc, HMPA (CH2)5 67
50 Pr NH
200† DMAc (CH2)4 32
51 n-Bu 8
52 Pr n-Bu2N 180 DMAc mono-
aminated 11
53 n-C6H13 11
54 Pr n-C6H13N 180 DMAc, HMPA mono-
aminated 21
† in a sealed vessel

83
1.4.5. Extension of the conjugated chain of DPP
To make longer-wavelength absorbing and emitting DPPs, we extended the
conjugation length of the fluorophore. For this purpose we explored and applied some
Pd-catalyzed coupling reactions. Firstly, we used the Jeffery modification128 of Heck
coupling of DPP aryl halides (bromides and iodides) with various styrenes. Trial
reactions with commercially available 4-tert-butylstyrene and 4-acetoxystyrene went well
with both methylated iodo- 43 and bromo- 45 DPPs (Scheme 1.44).
N
N
O
O
R
R
N
N
O
O
X
X
R
+a
X = Br or I
R = tert-Bu:
AcO:
55
56
Scheme 1.44. Heck coupling between X-DPP-Me with 4-tert-butyl- and 4-acetoxy-
styrenes. (a) Pd(OAc)2, TDA-1, K2CO3, DMAc.
Encouraged by this fact, we prepared several other styrenes: 4′-
(diethylamino)styrene 57, 1,4-divinylbenzene 58, and 4'-(2-[(4-

84
dibutylamino)phenyl]vinyl)styrene 59 from the corresponding aldehydes by Wittig
reaction (Scheme 1.45).
N
CHO
Et EtNEt Et
CHO
CHO
NBu
Bu;
a a b
OHC CHOHOOC
COOH
c
d57
59
58
Scheme 1.45. Preparation of styrenes. (a) Ph3P+Me I–, t-BuOK, DMSO. (b) Pd(OAc)2, (o-
Tol)3Ph, Et3N, DMAc. (c) CH2(COOH)2, Py. (d) quinoline, Ph2O, Cu, hydroquinone, Δx.
The prepared styrenes 57 and 59 were coupled with Br-DPP-Pr 44 (Scheme 1.46
and 1.47 correspondingly) to give DPPs 60 and 61 with long conjugation chains and
amine donors, as well as propyl solubilizing groups. The DPP 61 was rather difficult to
purify from numerous colored by-products and it was obtained eventually in a state of
purity, which gave a good NMR and mass spectra, but eluted at least two additional
minor components on HPLC. The physical and optical properties of these compounds are
discussed below.
The Negishi coupling between Br-DPP-Pr 44 and either thien-2-yl zinc
chloride129 62 or 5-(4-(N,N-di-n-hexylamino)phenyl)-thien-2-yl zinc chloride 63 gave
another pair of DPPs with an extended conjugation chain — 64 and 65 (Schemes 1.49
and 1.50).

85
N
N
O
O
N
N
N
N
O
O
Br
Br+ N
Et
Et
a
2
44 57
60
Scheme 1.46. Pd-catalyzed coupling between styrene 57 and Br-DPP-Pr 44.
N
N
O
ON
Bu
Bu
NBu
Bu
N
N
O
O
Br
Br
a
NBu
Bu+44 59
61
Scheme 1.47. Pd-catylized coupling between styrene 59 and Br-DPP-Pr 44.
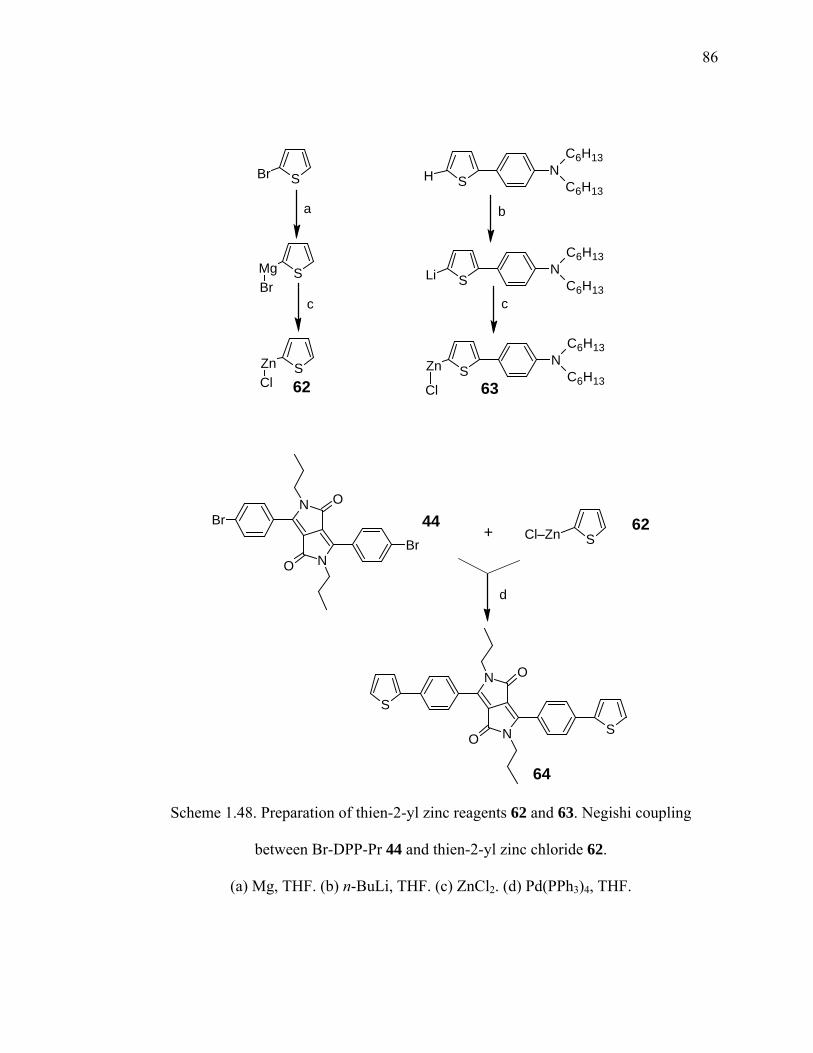
86
SZnCl
SMgBr
SBr
62 63
a b
c c
NC6H13
C6H13SH
NC6H13
C6H13SLi
NC6H13
C6H13SZn
Cl
N
N
O
O
S
S
N
N
O
O
Br
Br
d
SCl–Zn+44 62
64
Scheme 1.48. Preparation of thien-2-yl zinc reagents 62 and 63. Negishi coupling
between Br-DPP-Pr 44 and thien-2-yl zinc chloride 62.
(a) Mg, THF. (b) n-BuLi, THF. (c) ZnCl2. (d) Pd(PPh3)4, THF.

87
N
N
O
O
Br
Br
a
+44 63
65
SCl–Zn NC6H13
C6H13
N
N
O
O
SN
H13C6
H13C6S
NC6H13
C6H13
Scheme 1.49. Negishi coupling between Br-DPP-Pr 44 and 5-(4-(N,N-di-n-
hexylamino)phenyl)-thien-2-yl zinc chloride 63. (a) Pd(PPh3)4, THF.
1.4.6. DPPs with hydrophilic solubilizing groups
Apart from the known 4,4′-bis(hydroxysulfonyl) DPP derivative 10, there are no
water-soluble DPPs, to our knowledge. Even for compound 10 there was no preparation
procedure or characterization given in the original paper.68 Since biological applications
require dye solubility in polar solvents and aqueous buffers, we reproduced the
sulfonation of DPP (Scheme 1.22), worked out the separation of the product from the
reaction mixture, characterized compound 10, and prepared several other derivatives,
containing the hydroxysulfonyl group. For this purpose we alkylated the DPP’s lactam’s
nitrogen(s) with either 1,3-propanesultone or 1,4-butanesultone (Scheme 1.50). The DPP

88
with two sulfonic acid groups 66 was not isolated in crystalline form due to difficulties in
its crystallization — it is not soluble in non-polar solvents and does not crystallize from
polar ones (ethanol, water, acetone), once dissolved in them. DPPs with only one
hydroxysulfonyl group 67 and 68 did not tend to crystallize on cooling either — their
purification was achieved either by chromatography (68) or by acid re-precipitation (67).
NHNH
O
O
+ NNH
O
O
SO
O OHS
OOOH
NN
O
O
SO
O OH
N
NH
O
O
Pr
N
N
O
O
SO
OOH
Pr
33
66 67
68
SOO
O
OS
O O
a
b
b
Scheme 1.50. Introducing sulfonic acid group into DPP structure.
(a) t-BuOK, PrI, DMAc. (b) sultone, t-BuOK, DMAc.
1.4.7. Hydroxy-functionalized DPPs
For the purpose of incorporation of DPP fluorophore into a polymer chain
(performed by another researcher — Wonhee Jeong), we needed bis- and mono-alcohol

89
functionalized complimentary DPP pairs. The complementation here means similarity in
the position and reactivity of the hydroxy groups in the bis- and mono- functionalized
compounds. Based on our alkylation experience of various DPPs, we contemplated
introduction of the alcohol functionality via alkylation of DPP with ω-halo-α-alcohols
(Scheme 1.51). However, this approach, when implemented with alkali metal alkoxides
or hydrides as bases, either failed completely or worked on a sole example of already
mono-alkylated DPP only. As a possible explanation for these failures, we might
speculate that strong base t-BuOK with its conjugate acid t-BuOH correspondingly
weaker than any n-CnH2n+1OH primary aliphatic alcohol, abstracts the hydroxyl’s proton
in competition with deprotonation of lactam’s nitrogen. The formed alkoxide, although
sufficiently strong to reversibly deprotonate DPP in turn, might undergo intra- or inter-
molecular side-reactions, as well as to add to the DPP bicyclic system by Michael type
(similar to hydroxy anion, cf. Scheme 1.40), which would be impossible for the tert-
butoxide ion due to its bulkiness.
NHNH
O
O
t-BuOK, DMAc
OH(CH2)
X
n
NN
O
O
(CH2) (CH2) OHOH NNH
O
O
(CH2) OH+
X = Cl, Br, I
n n n
Scheme 1.51. Proposed direct introduction of alcohol functionality into DPP by
alkylation with ω-halo-α-alcohols.

90
NN
O
O
(CH2) OH8NHN
O
O
a
40 69
Scheme 1.52. The sole successful example of DPP alkylation with ω-halo-α-alcohol in
presence of t-BuOK. (a) t-BuOK, DMAc, 8-bromo-1-octanol.
Since the di-allylation of 3,6-bis(4-methylphenyl)pyrrolo[3,4-c]pyrrole-1,4-
dione (Tol-DPP 22) has been reported to proceed in 75% yield45, we pursued a
hydroboration approach (Scheme 1.53) to alcohol functionality via mono- and diallyl
DPPs (41 and 42), supported by two literature examples130 of terminal alkene
hydroborations in the presence of a cyclic amide. The attempted hydroboration reactions
with both BH3•THF and 9-BBN did not yield any alcohols on the DPP-containing
substrate. Moreover, those reagents destroyed the fluorophore’s core when the reaction
was forced by heating. This destructive action of boranes is definitely characteristic of the
DPP heterocycle, rather than the allyl termini, for the two above boranes similarly
destroy on warming a THF or CH2Cl2 solution of Pr-DPP 32. No analysis of the
degradation products has been conducted and the criteria for the DPP core destruction
were the loss of the DPP’s UV-Vis absorption and fluorescence.

91
NH NH
O
O
N N
O
O
NH N
O
O
+
N N
O
O OH
OH
NH N
O
O OH
a
b b
41 421
Scheme 1.53. Attempted hydroboration route to alcohol-functionalized DPPs.
(a) t-BuOK, allyl bromide. (b) BH3•THF or 9-BBN.
The first success in alcohol functionalization of DPP has been achieved with a
method, discovered and reported by Lu and Twieg:131 copper-catalyzed coupling of aryl
iodides with sec-amines. We coupled I-DPP-Pr 46 with 2-(ethylamino)ethanol in
presence of copper and copper (I) iodide in DMAc (Scheme 1.54). No DMAE, essential
as a solvent for the reaction success in other cases, can be used when this specific amine,
similar to DMAE, is used as a reactant. Although the mono-coupling product has been
detected on TLC while monitoring the progress of the reaction, the major product was 70.
Perhaps, one could use 1:1 ratio of I-DPP-Pr and 2-(ethylamino)ethanol to get
predominantly mono-coupling product, but the optical absorbtive properties of them

92
would be different due to electron-donor properties of the amino group being introduced.
Therefore, we moved back to the utilization of lactam nitrogen(s) alkylation reaction,
where the optical properties of the mono- and di- products do not differ significantly and
where they could sometimes be obtained in equal amounts, relatively easily separated and
the mono-product then can be re-alkylated with a different reagent.
N
N
O
OI
I
+ NH
OH
N
N
O
ON
N
OH
OH
a
7046
Scheme 1.54. Cu/CuI-cat. coupling of 2-(ethylamino)ethanol with I-DPP-Pr 46.
(a) Cu, CuI, DMAc, 80°C.
Revisiting the alkylation methodology, we resorted to a protection-deprotection
approach to introduce the terminal alcohol functionalized groups (Scheme 1.55). Thus,
we have prepared the THP- protected C6 alcohols 71–73 (Scheme 1.56) and benzoate
ester-protected C6 alcohol 74 (Scheme 1.57) with a halide (or tosylate) on the other
terminus of the alkyl chain.

93
NHNH
O
O
+OPG
(CH2)X
nX = Cl, Br, I
NN
O
O
(CH2) (CH2) OPGPGONNH
O
O
(CH2) OPG + n nn
Deprotection
NN
O
O
(CH2) (CH2) OHOHNNH
O
O
(CH2) OHn n n
N N
O
O
(CH2)OH Alkn
Scheme 1.55. Alkylation approach to alcohol-functionalized DPPs with protection-
deprotection of the hydroxy group. PG = Protecting Group.
OOI
OH
OH
O O
OH
O O
OTs
OH
X
O O
X
a b
c
d
e 71
72 73X = Br
= I 73
Scheme 1.56. Preparation of alkylating reagents with alcohol functionality, protected
with a THP protecting group. (a) dihydropyran (DHP), THF, Dowex 50WX8-100. (b) 4-
toluenesulfonyl chloride (TsCl), Py, Et2O. (c) HBr 48% aq. or HI 55% aq. or P2I4, CS2.
(d) DHP, THF, Dowex 50WX2-100. (e) P2I4, CS2.

94
The tosylate 71 reacted with DPP 1 in presence of t-BuOK in DMAc very
slowly up to 180°C: even after four days most of the starting material was not consumed.
When the reaction temperature was raised to 200°C, the starting material was consumed
in three hours, however, these harsh conditions seemed to destroy the THP protection and
side-reactions ensued, for very little target material was obtained. The same reaction
performed with bromide 72 went under more mild conditions and that with iodide 73 —
was complete in twelve hours at 40°C (Scheme 1.57). The THP-protected DPP diol 74
turned out to require rather strong conditions for the removal of the THP groups.
Reaction with pyridinium 4-tolenesulfonate (PPTS) in ethanol resulted in no deprotection
at all. Acetic acid removed the THP group only partially. Trifluoroacetic acid, used in
amount of 10% with respect to the substrate, did give the target compound, though as
mixture with at least three other ones with very similar retention factors. The deprotection
was confirmed by 13C NMR spectrum of the mixture, but our separation attempts did not
yield pure individual compounds. Thus, we resorted to the alkylation with a benzoate
ester-protected iodo-alcohol 75.
The iodide 75 alkylated DPP 1 at 60°C in four hours to give 76, with a little of
mono-alkylated product 77. Same reaction, repeated with mono-propyl and mono-
dodecyl DPPs 33 and 38 yielded corresponding precursors of DPP mono-alcohols 78 and
79. The removal of the benzoate ester moiety by classical methods132 also encountered
with some difficulties. Potassium hydroxide 1% solution in ethanol did perform
deprotection, but very slowly and the deprotection was never complete at room
temperature.

95
NHNH
O
O
NN
O
O
(CH2)6 (CH2)6 OHOH
NN
O
OOO
OO
O O
X+
a
171– 73
74
Scheme 1.57. Reaction of THP- protected alcohols 71–73 with DPP.
OH
OH
O
O
OH
a b O
O
I75
Scheme 1.58. Preparation of alkylating reagent 75 with alcohol functionality, protected
with a benzoate ester. (a) PhCOCl, Et3N, THF. (b) KI, H3PO4, PPA.
When pushed by heating, however, the dye underwent degradation (presumably
of the same nature as described in Chapter 1.4.2) in parallel with deprotection. A much
milder deprotecting reagent, potassium cyanide in methanol,133 successfully tested firstly
on the simplest ester — ethyl benzoate, gave a clean deprotection of 77 to 81 on a ten
milligram scale, but scaling up this reaction met with the same problem of the dye
degradation. Eventually we found that excess (over recommended catalytic amount) of

96
titanium (IV) tetra-iso-propoxide in methanol at temperatures quite a bit over those
usually employed for such a reaction (180°C vs. usual r.t. to 60°C) and for much longer
reaction time (days vs. usual hours) gave almost quantitative deprotection of 76 to 80
without any side reactions.
O
O
I
75
NHNH
O
O
+
1
a
O
O
N
NO
O
O
O
+NH
NO
O
O
O
NNH
O
O
(CH2)6 OHNN
O
O
(CH2)6 (CH2)6 OHOH
b c
76 77
80 81
Scheme 1.59. Reaction of benzoate ester protected alcohol 74 with DPP and subsequent
removal of the protecting group. (a) t-BuOK, DMAc. (b) MeOH, Ti(i-PrO)4, 180°C. (c)
KCN, MeOH.
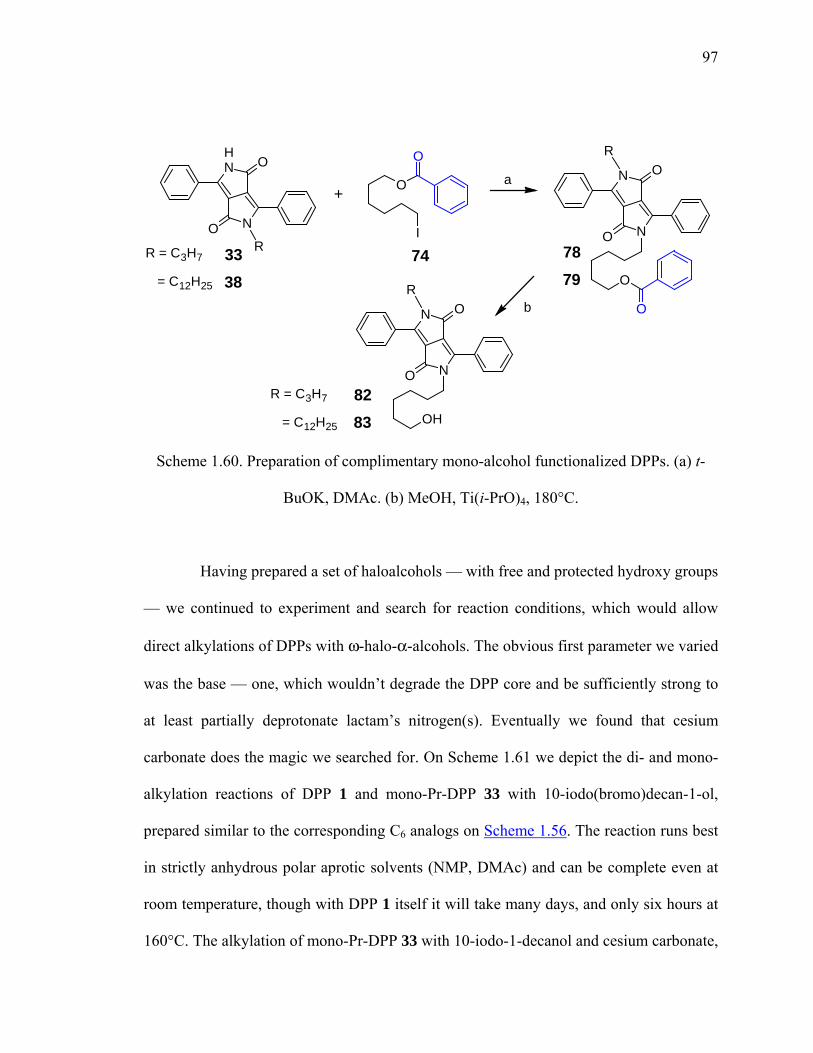
97
NH
NO
O
R
O
O
I
74
+N
NO
O
O
O
R
N
NO
O
OH
R
R = C3H7
= C12H25
3338
7879
8283
R = C3H7
= C12H25
a
b
Scheme 1.60. Preparation of complimentary mono-alcohol functionalized DPPs. (a) t-
BuOK, DMAc. (b) MeOH, Ti(i-PrO)4, 180°C.
Having prepared a set of haloalcohols — with free and protected hydroxy groups
— we continued to experiment and search for reaction conditions, which would allow
direct alkylations of DPPs with ω-halo-α-alcohols. The obvious first parameter we varied
was the base — one, which wouldn’t degrade the DPP core and be sufficiently strong to
at least partially deprotonate lactam’s nitrogen(s). Eventually we found that cesium
carbonate does the magic we searched for. On Scheme 1.61 we depict the di- and mono-
alkylation reactions of DPP 1 and mono-Pr-DPP 33 with 10-iodo(bromo)decan-1-ol,
prepared similar to the corresponding C6 analogs on Scheme 1.56. The reaction runs best
in strictly anhydrous polar aprotic solvents (NMP, DMAc) and can be complete even at
room temperature, though with DPP 1 itself it will take many days, and only six hours at
160°C. The alkylation of mono-Pr-DPP 33 with 10-iodo-1-decanol and cesium carbonate,

98
due to its higher solubility, will be complete in 6 hours at much lower temperature of
60°C.
The preparation of ω-halo-α-alcohols was deemed necessary, despite the fact
that ω-bromo-α-alkanols are commercially available, for the GC-MS and NMR analysis
of the commercial samples on our shelves proved their inferiority. For example, GC-MS
analysis of 8-bromo-1-octanol from Aldrich’s batch #09915TG revealed at least three
major components therein, with 8-bromo-1-octanol accounting to only 56% of total
integration area. 13C NMR of the same sample, in turn, gave twenty-four discrete signals,
also suggesting a mixture of three individual compounds (24/8).
NH N
O
O
Pr
NH NH
O
O
33
1
X(CH2)10OH
Cs2CO3
N N
O
O
Pr(CH2)10OH
N N
O
O
(CH2)10OH (CH2)10 OHX(CH2)10OH
Cs2CO3
84
85
Scheme 1.61. Direct alkylation of DPPs with ω-halo-α-alcohols in presence of Cs2CO3.

99
Mono-alkylation of DPP 1 is possible with 1.2 eq. of I-C10-OH. Cs2CO3 alone in
DMAc does degrade Pr-DPP on prolonged heating too.
1.4.8. DPPs with a cysteine-reactive maleimide moiety
Maleimide group has been widely utilized in the bioconjugate chemistry as a
specific binding site for the cysteine residue of proteins. In part it is due to low
abundance of the Cys unit, in part – due to high binding specificity of maleimide to the
latter. Following the successful application of maleimide-containing Nile Red dye (see
Chapter 2) for imaging of GroEL chaperonine, we were about to exploit the DPP
chemistry learned to prepare maleimide-functionalized DPPs. The furan-protected
maleimide alkylating reagent 86 (see its preparation in Chapter 2) has been used to di-
alkylate DPP 1, followed by thermal removal of furan (retro Diels-Alder reaction) to give
bis-functionalized DPP derivative 88 (Scheme 1.62).
An attempt was made to prepare mono-functionalized maleimide derivative 90,
possessing a sulfonic acid group for improved solubility in polar medium. A one-pot,
mixed, stepwise alkylation of DPP 1 was performed — firstly with 1,3-propanesultone,
followed by 2-(6-iodohexyl)-3a,4,7,7a-tetrahydro-4,7-epoxy-1H-isoindole-1,3(2H)-dione
86 to give the product 89 (Scheme 1.63). However, the subsequent retro Diels-Alder
reaction, performed on pure isolated substrate 89 in neat refluxing xylene, gave an
intractable brown tar, missing both the characteristics of DPP dye (by absorption
spectroscopy) and 13C signal of maleimide C3-carbon altogether. Apparently, the
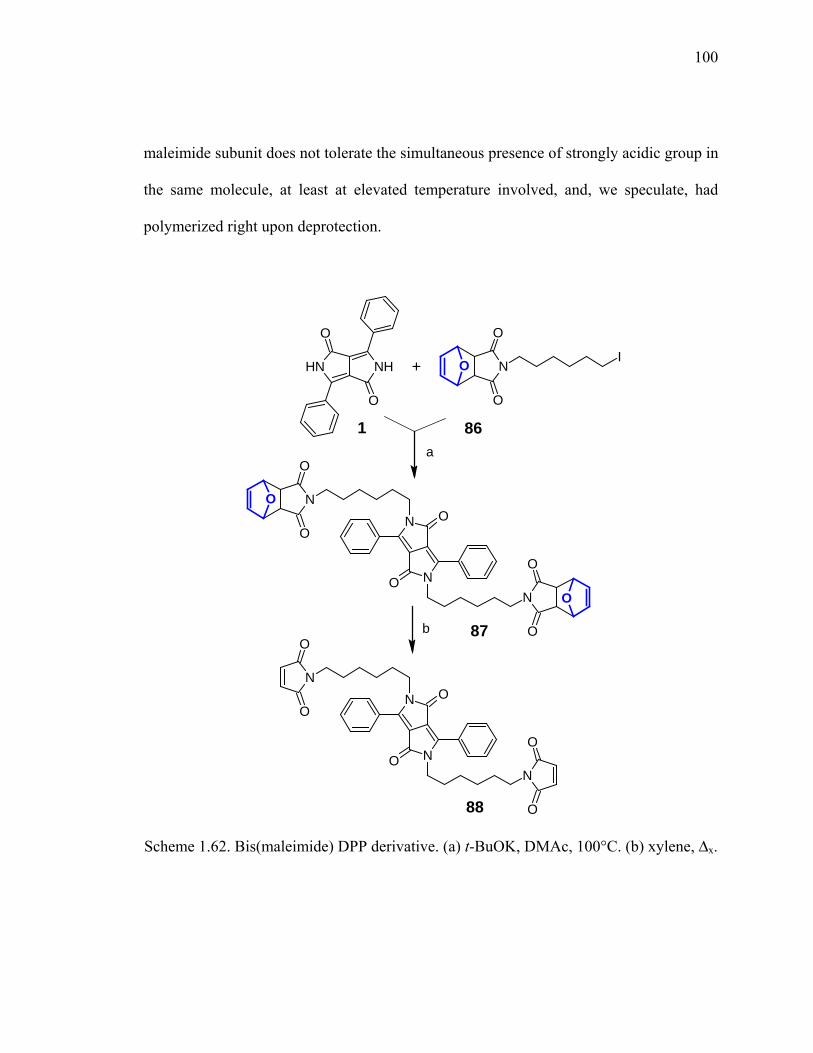
100
maleimide subunit does not tolerate the simultaneous presence of strongly acidic group in
the same molecule, at least at elevated temperature involved, and, we speculate, had
polymerized right upon deprotection.
NH NH
O
O
1
N
O
O
ON
NO
O
N
O
O
O
N
O
O
O I+
a
86
87
N
O
O
N
NO
O
N
O
O88
b
Scheme 1.62. Bis(maleimide) DPP derivative. (a) t-BuOK, DMAc, 100°C. (b) xylene, Δx.

101
N
O
O
O
N
NO
O
SO3H
NH NH
O
O
1
N
O
O
OI+
a86
SO O
O
1 2
+
89
N
O
O
N
NO
O
SO3H
90
tar
b
b
123
Scheme 1.63. An attempt towards maleimide mono-functionalized DPP 90. (a) t-BuOK,
DMAc, 100°C. (b) xylene, Δx.

102
1.4.9. N-Arylated DPPs
Langhals and Potrawa47 have suggested that absence of ‘benzylic’ hydrogens on
the lactam’s nitrogens of DPP should considerably improve the photostability of DPP
dyes. They have prepared a couple of N-arylated DPPs and characterized their
spectroscopic (NMR, UV-Vis, and fluorescence) properties, yet the “high photostability”,
claimed in the paper title, has not been discussed, measured or shown otherwise. This
argument, however, seemed reasonable to us, and since there was no general approach to
N-arylated DPPs, with only few examples, prepared indirectly, have been known, we
explored ‘direct’ approaches to N-arylated DPPs. The first approach was inspired by a
work of Copola134 on direct copper-mediated N-arylation of isatins (Scheme 1.64).
R Yield, % NH
O
O
+
Br
R
CuO
DMF NO
O
R
H
4-F
4-MeO
3,5-Me2
92
97
71
81
Scheme 1.64. Direct copper-mediated N-arylation of isatins.134
The reactions of bromo-arenes with DPP in the presence of copper (I) oxide in
DMAc were performed at 120–180°C (Scheme 1.65). In the reactions with 1-bromo-4-
(perfluorobutylsulfonyl)benzene (4′-bromophenyl perfluorobutyl sulfone) and with 1-
bromo-4-fluorobenzene in addition to the bis-adducts 91 and 94 we also separated mono-
adducts 92 and 95. When monitoring the reaction of 1-bromo-3,5-

103
bis(trifluoromethyl)benzene with DPP under the same conditions, the mono-adduct’s spot
on TLC is also detected, is major at some point of time and most surely the reaction can
be stopped at that time and the mono-adduct separated as well, but we drove the reaction
to completion and separated only the target compound 93.
NH NH
O
O
1
Br
R
+CuO
DMAcN
N O
O
R
R
N
NH
O
O
R
+
Di-arylated Yield Mono-arylated Yield
4-C4F9–SO2– 91 32% 4-C4F9–SO2– 92 18%
3,5-(CF3)2 93 37% – R =
4-F 94 12% 4-F 95 37%
Scheme 1.65. Direct copper-mediated N-arylation of DPP 1.
The second approach to direct N-arylation was based upon our hypothesis of
utilizing DPP anion(s) in nucleophilic aromatic substitution of halogen.135 The preformed
DPP2– dianion was reacted with 1-fluoro-4-nitrobenzene, and Pr-DPP– anion — with 1-
fluoro-2,4-dinitrobenzene to give the corresponding N-arylated compounds 96 and 97
(Scheme 1.66).

104
NH NH
O
O
+
F
NO2
a
N
N O
O
NO2
NO2
NH N
O
O
Pr +NO2
NO2
F
a
N
N O
O
Pr
O2N
NO2
96
97
Scheme 1.66. Aromatic nucleophilic substitution on DPP. (a) t-BuOK, DMAc.
These two direct approaches to N-arylated DPPs are, to our knowledge, new for
this class of dyes and have never been reported before. They allow easy, one-step access
to compounds, which could potentially be prepared before only via laborious multi-step
synthesis. The photostability of the prepared N,N-diarylated DPPs turned out to be
superior, compared to their N,N-dialkyl analogs and is discussed below.

105
1.4.10. Physical Properties of Newly Prepared DPPs
The physical properties of the newly prepared DPPs are summarized in the
Tables 1.17 and 1.18. There are several generalizations that can be drawn from these
data. The melting points of N,N′-di-substituted DPPs are always lower than those of their
N-mono-substituted counterparts, which is in agreement with all previous observations.
Melting points of N,N′-dialkylated DPPs decreases as the length of the alkyl chain grows.
Optical absorption spectra of N,N′-di-substituted and N-mono-substituted DPPs are quite
similar and are almost not affected by the nature of the substituent — be it aromatic or
aliphatic. The substitution of the DPP’s phenyl rings, however, brings about considerable
changes into the electronic spectra of these compounds. Extension of the conjugation
chain causes a bathochromic shift in both emission and absorption spectra, so does the
introduction of electron-donating groups at the termini of the phenyl rings. The quantum
yields vary, but generally are high to excellent and always higher than 0.5. The Stokes’
shifts span from 28 nm to a good 84 nm. Photostability of some of the prepared
compounds has been measured semi-quantitatively as the time required for a sample
solution to completely fade out under UV radiation of λ=360 nm. For most of the DPPs
this time interval falls within several days, while for DPP 1, DPP disulfonic acid and
N,N′-diarylated DPPs no complete fading even after two weeks of irradiation has been
observed. Several DPPs have been utilized (by the group of Prof. W. E. Moerner) in
single-molecule imaging and have been shown to be suitable for such applications (Fig.
1.13), though their photostability remains to be still further improved. We are looking

106
forward now to test the more photostable DPPs under SM conditions and see the
improvement over the total observation time.
Figure 1.13. Single molecules of DPP-Me 23 imaged in a PMMA film, excited by 488
nm with an intensity of 0.85 kW cm-2. A 6.2 x 6.2 μm region of the sample is shown.
Photostability of seven prepared DPPs has been measured quantitatively (see
experimental part for details) as the slope tangence of fluorescence (or absorbance) decay
vs. time under constant UV radiation flux of 1.2 mW/cm2 at λ=360 nm. The graphs
below (Fig. 1.14 and 1.15) show that under same exposure conditions, mono-alkylated
DPP 33 is ca. 5.8 times (calculated as ratio of two slope tangences: –0.176/–0.0306) more
prone to photodecomposition than its di-alkylated analog 32. Likewise, compound 65 is
ca. 2.1 times (–0.0574/–0.0272) less photostable than its counterpart 64.
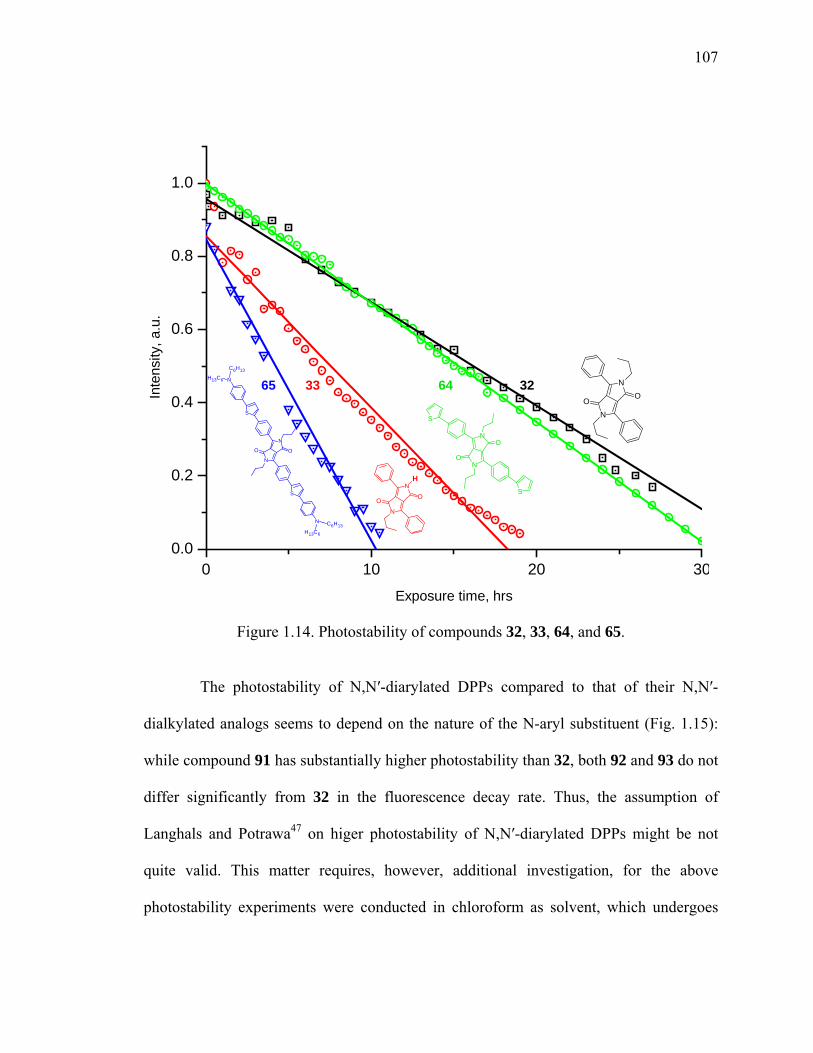
107
0 10 20 300.0
0.2
0.4
0.6
0.8
1.0
Inte
nsity
, a.u
.
Exposure time, hrs
65
N
N
O O
S
N
C6H13
H13C6
S
N C6H13H13C6
HN
NO O
N
N
O
O
S
S
64 N
NO
O3233
Figure 1.14. Photostability of compounds 32, 33, 64, and 65.
The photostability of N,N′-diarylated DPPs compared to that of their N,N′-
dialkylated analogs seems to depend on the nature of the N-aryl substituent (Fig. 1.15):
while compound 91 has substantially higher photostability than 32, both 92 and 93 do not
differ significantly from 32 in the fluorescence decay rate. Thus, the assumption of
Langhals and Potrawa47 on higer photostability of N,N′-diarylated DPPs might be not
quite valid. This matter requires, however, additional investigation, for the above
photostability experiments were conducted in chloroform as solvent, which undergoes

108
photochemical decomposition on its own, and may thus level, rather than differentiate
photostability of the fluorophores.
0 10 20 30 40 50 60 700.0
0.2
0.4
0.6
0.8
1.0
Inte
nsity
, a.u
.
Exposure time, hrs
N
NO
O
32
N
N O
O
CF3
F3C
CF3
F3C
NN
O
O
SO2(CF2)4FF(F2C)4O2S
919293
Figure 1.15. Photostability of compounds 32, 91, 92, and 93.
The quantitative data on photostability of compounds 32, 33, 64, 65, 91, 92, and
93 are summarized in the Table 1.16.
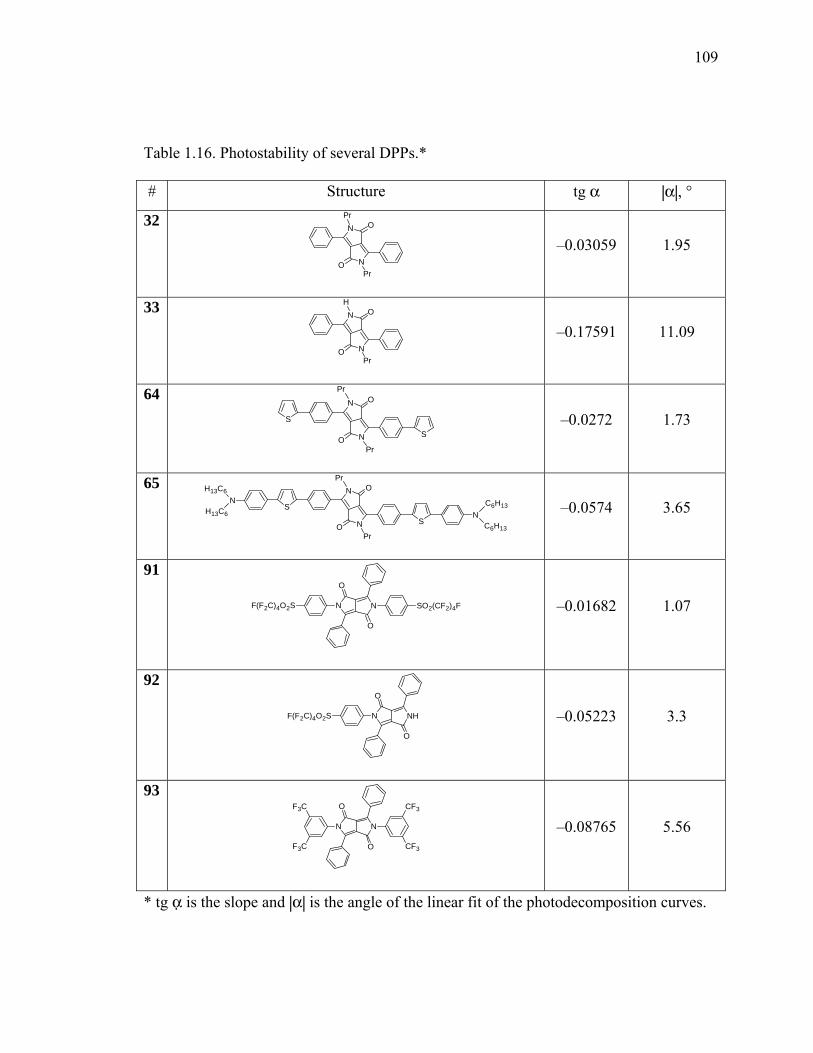
109
Table 1.16. Photostability of several DPPs.*
# Structure tg α |α|, °
32 PrN
NO
O
Pr
–0.03059 1.95
33 HN
NO
O
Pr
–0.17591 11.09
64
N
N
O
O
Pr
Pr
S
S
–0.0272 1.73
65
N
N
O
O
Pr
Pr
SN
H13C6
H13C6S
NC6H13
C6H13
–0.0574 3.65
91
NN
O
O
SO2(CF2)4FF(F2C)4O2S
–0.01682 1.07
92
NHN
O
O
F(F2C)4O2S
–0.05223 3.3
93
NN
O
O
F3C
F3C
CF3
CF3
–0.08765 5.56
* tg α is the slope and |α| is the angle of the linear fit of the photodecomposition curves.

110
Table 1.17. Symmetrical 4,4’–Disubstituted DPPs.
N
N
O
O
R1
R1
R
R
# R R1 yield, % m.p., °C λmax, absorption, nm λmax, emission, nm ΦF
142 H H 56 420 50466 508, 545b –
21 CN H 35 443 dec. 501, 538 556, 603a 0.63b
22 Me H 38 354 dec. 274, 472, 508 514, 557 0.66b
1066 HO3S– H 77 – 268, 474, 505 525, 566a 0.63c
Me Pr 60 186 306, 474 530, 575a 0.64
47 CN Pr 83 227 285, 496 568 0.65
43 Br Me 46 >350 275, 305, 491 541, 587a 0.83
44 Br Pr 37 247 275, 304, 478 541, 587a 0.86
45 I Me 37 dec. 355 276, 318, 495 544, 590a 0.89
46 I Pr 68 260.5 276, 317, 479 550, 592a 0.86

111
56 4-AcO-C6H4-CH=CH– Me 6 357.7 279, 335, 510 582 0.73
55 4-t-Bu-C6H4-CH=CH– Me 50 295 280, 337, 515 587 0.75
48 4-Pyrr– Me 74 324 dec. 282, 385, 548 575 0.95
50 4-Pyrr– Pr 32 267 273, 384, 539 577 0.94
53 (C6H13)2N– Me 11 132 282, 385, 548 575, 620a 0.92
60 4-Et2N-C6H4-CH2=CH– Pr 49 247 334, 538 620 0.72
64 S
Pr 84 281 334, 362, 532 616, 690a 0.96
65 S(H13C6)2N
Pr 32 278 241, 359, 541 615, 693a 0.82
a) Shoulder of the main peak, determined as minimum of the second derivative.
b) In dimethylformamide; c) In ethanol; d) Yield from parent DPP or Br-DPP.

112
Table 1.18. N– and N,N′-substituted DPPs. N
N
O
O
R
R1 # R R1 yield, % m.p.°C λmax, absorption, nm λmax, emission, nm ΦF
23 Me Me 85 231 265, 294, 476 522 0.9044, 0.54
32 Pr Pr 62 189 289, 466, 488 528, 568a 0.76
33 Pr H 30 276 265, 465, 488 523, 563a 0.69
68 C4H9SO3H Pr 9 oil 264, 465, 486 524, 565a 0.73
67 C3H7SO3H H 61 270 465, 486 525, 566a 0.77b
34 C6H13 C6H13 8 134 472 530, 572a 0.74
35 C6H13 H 16 252 262, 464, 481 522, 565a 0.71
36 C10H21 C10H21 42 117 268, 467 529, 573a 0.65
37 C12H25 C12H25 47 114 284, 474 529, 573a 0.66
76 C6H12OCOPh C6H12OCOPh 23 111 475 527, 563a 0.97
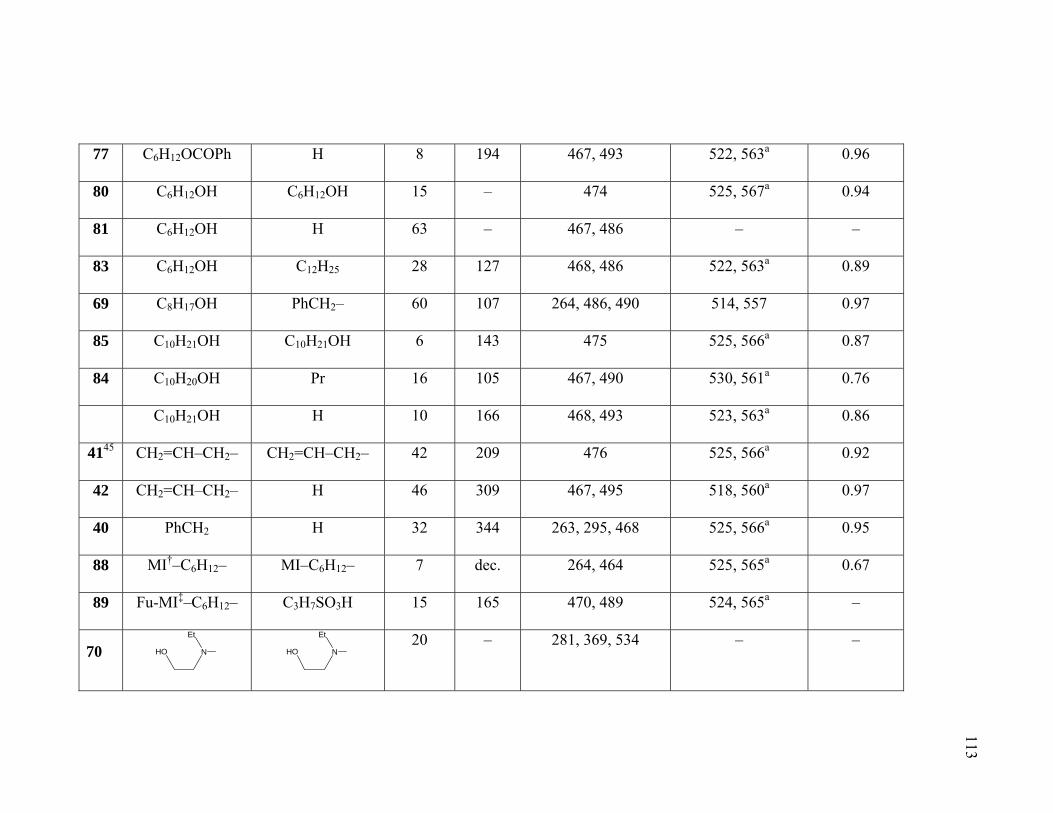
113
77 C6H12OCOPh H 8 194 467, 493 522, 563a 0.96
80 C6H12OH C6H12OH 15 – 474 525, 567a 0.94
81 C6H12OH H 63 – 467, 486 – –
83 C6H12OH C12H25 28 127 468, 486 522, 563a 0.89
69 C8H17OH PhCH2– 60 107 264, 486, 490 514, 557 0.97
85 C10H21OH C10H21OH 6 143 475 525, 566a 0.87
84 C10H20OH Pr 16 105 467, 490 530, 561a 0.76
C10H21OH H 10 166 468, 493 523, 563a 0.86
4145 CH2=CH–CH2– CH2=CH–CH2– 42 209 476 525, 566a 0.92
42 CH2=CH–CH2– H 46 309 467, 495 518, 560a 0.97
40 PhCH2 H 32 344 263, 295, 468 525, 566a 0.95
88 MI†–C6H12– MI–C6H12– 7 dec. 264, 464 525, 565a 0.67
89 Fu-MI‡–C6H12– C3H7SO3H 15 165 470, 489 524, 565a –
70 N
Et
OH
N
Et
OH
20 – 281, 369, 534 – –

114
91 4-C4F9-SO2-C6H4 4-C4F9-SO2-C6H4 32 262 271, 475 515, 555a 0.91
92 4-C4F9-SO2-C6H4 H 18 320 461, 485, 490 513, 554a 0.93
93 3,5-(CF3)C6H3– 3,5-(CF3)C6H3– 37 298 251, 315, 475 520, 552a 0.95
96 4-NO2–C6H4– 4-NO2–C6H4– 12 200 268, 470 – –
94 4-F–C6H4– 4-F–C6H4– 12 344 271, 475 520, 566a –
95 4-F–C6H4– H 37 404 271, 475 – –
97 2,4-(NO2)2-C6H4 Pr 25 dec. 263, 298, 466 non-fluor. –
†MI = maleimide residue, 1H-pyrrole-2,5-dione-1-yl.
‡ Fu-MI = 3a,4,7,7a-tetrahydro-4,7-epoxy-1,3-dioxo-1H-isoindol-2-yl.
a) Shoulder of the main peak, determined as minimum of the second derivative
b) In ethanol

115
1.4.11. Conclusion
A series of new fluorophores for single-molecule spectroscopy applications have
been designed and prepared based on derivatives of dihydropyrrolo[3,4-c]pyrrole-1,4-
dione (DPP) with substantially higher solubility, longer absorption and fluorescence
emission wavelengths, and a wide range of functionalization. A range of reliable N, N′-
alkylation, aryl halogenation and organometallic mediated C-C bond formation reactions
has been developed and conducted. A cysteine-reactive fluorescent tag and a variety of
hydroxy-functionalized and water-soluble DPP derivatives have been synthesized. Two
new methods for preparation of N,N′-diaryl-substituted DPPs have been identified and
explored. The fluorescence properties of the new DPPs were studied in detail and some
of the fluorophores have been successfully imaged at the single-molecule level. The
quantum yields were measured and vary from moderate to high and Stokes shifts as large
as 84 nm were observed. Photostability of seven new DPPs was quantified and compared
with the structures of the fluorophores.

116
CHAPTER II.
CYSTEINE-SPECIFIC FLUORESCENT TAGS:
NILE RED – MALEIMIDE AND
DCDHF – MALEIMIDE.
2.1. Introduction to molecular probes and tags
The fluorescent tags, or bioconjugate molecular probes are a subclass of
fluorescent labels, discussed briefly in the introduction to the Chapter 1. Their
characteristic feature is a specific covalent bonding of the probe’s reactive site to the
certain region(s) of a biomolecule or of a living cell due to a chemical reaction, resulting
in overall binding of the probe to the substrate. The specificity of the overall binding is
determined by that of the chemical reaction (called here conjugation, meaning binding),
which creates the new covalent bond. The structure of all bioconjugate probes may
generalized as it is depicted on Fig. 2.1: a fluorescent dye, connected to a reactive group
with a spacer of variable length.
Some of the commonly used hooks, or the reactive groups with their
corresponding bioconjugation chemistry, are summarized in Table 2.1 and Table 2.2.
Some of the dyes commonly used as fluorescent molecular probes (but not necessarily

117
N
O
Et2N
O
O
NOO
Dye
Spacer
Hook
Figure 2.1. Generalized structure of a bioconjugate fluorescent molecular probe.
generally for SMS) and their absorbtive and fluorescence ranges are summarized as a
chart in Fig. 2.2.139
As can be seen from the examples in the Tables 1.1 and 1.2 and Fig. 2.2, the
pool of the commercially available fluorescent dyes is very diverse, yet it exploits a small
number of fluorophore’s structural templates (of xanthenes — fluorescein, rhodamines;
of chromans — coumarins; of BODIPY — difluoroboradiazaindacene, of polynuclear
aromatic hydrocarbons — pyrene, perylene, coronene, prodan, of conjugated or fused
diazoles — NBD, 7-nitrobenza-2-oxa-1,3-diazoles, POPOP, 1,4-bis(5-phenyloxazol-
2yl)benzene, PPD, 2,5-diphenyl-1,3,4-oxadiazole, 2-pyridyl-2-phenyl-1,3-oxazoles;
cyanines … to name some of them). Few of them fulfill some of the requirements for the
SMS dyes, listed in Chapter 1. Specifically, the demand for dyes with high quantum yield
and photostability remains unfulfilled with many commercial dyes.

118
Table 2.1. Amine (R2–NH2) reactive groups for molecular probes.
Reactive group Structure Product Example
Isothiocyanate R1–N=C=S CS
NHR1 NH R2
benzophenone-4-isothiocyanate, and
O
HOOC
Me2N NMe2
N C S
+
Succinimidyl Ester N
O
O
OCO
R1
NHOCO
R1 R2
[243670-15-7], and
ON
OO
O
Sulfosuccinimidyl
Ester (STP) N
O
O
OCO
R1
SO3H
NHOCO
R1 R2
O
O
O
O
OO
O
O
N
OO
O
O
N
SO3H
HO3S
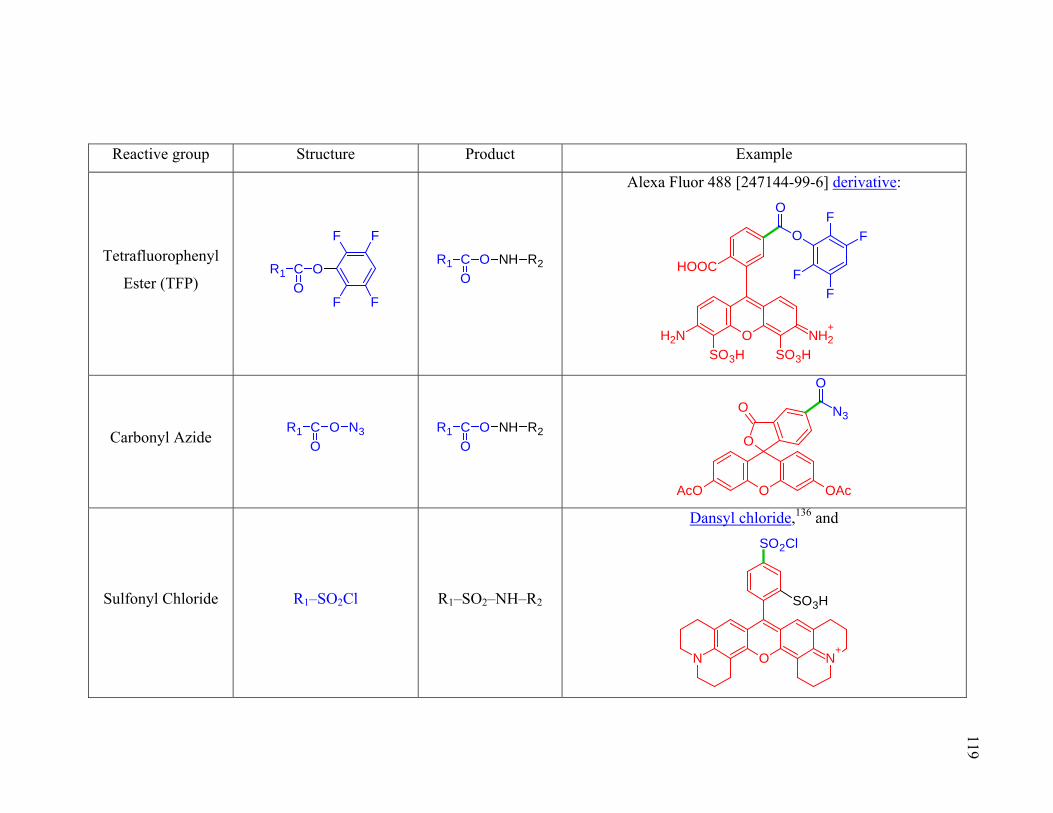
119
Reactive group Structure Product Example
Tetrafluorophenyl
Ester (TFP) OC
OR1
F F
F F
NHOCO
R1 R2
Alexa Fluor 488 [247144-99-6] derivative:
O
HOOC
NH2 NH2+
SO3HSO3H
O
OF
F
FF
Carbonyl Azide N3OC
OR1
NHOC
OR1 R2
O
O
O OAcAcO
N3
O
Sulfonyl Chloride R1–SO2Cl R1–SO2–NH–R2
Dansyl chloride,136 and
O N+
N
SO2Cl
SO3H

120
Table 2.2. Thiol (R2–SH) reactive groups for molecular probes.
Reactive group Structure Product Example
Iodoacetamide O
NH2I
O
NH2SR2
BODIPY137 [400885-92-9] derivative
NB
-N+
FF NH O
I
Maleimide N
O
O
R1
N
O
O
R1S
H
R2
O
HOOC
Me2N NMe2
NO O
+
Alkyl Halide R1–CH2–X R1–CH2–S–R2
BODIPY 138 [615574-25-9] and
NB
- N+
F F
Br

121
Figure 2.2. Common dye classes, used in molecular probes.139
To create a bioconjugate fluorescent molecular probe (fluorescent tag), suitable
for SMS detection, one needs to connect a suitable, reliable hook to an SMS-effective dye
with an appropriate spacer. Sometimes the spacer is missing (or of zero length) and the
reactive group connects directly to the fluorophore moiety. If the subsequent chemical
reaction creates a longer conjugation chain, the fluorophore’s absorption and emission
wavelengths may change. For example, dansyl chloride (5-(dimethylamino)naphthalene-
1-sulfonyl chloride, λmax Ab ≈ 350 nm) does not absorb or fluoresce in the visible region
until it reacts with an amine (λmax Em ≈ 520 and is solvent-dependent). A short spacer may

122
facilitate resonance energy transfer, while a very long one may cause steric problems and
report local polarity falsely, separating the fluorophore and the site of reactive group
attachment. The optimal length of a spacer is contigent upon specific application.
2.2. Design of the Probes
We designed and prepared several fluorescent tags with a thiol- (mercapto, or
sulfhydryl, –SH) reactive maleimide group. The synthesis of two DPP-Maleimide
fluorescent tags has been attempted, and one of them succeeded – see the details in the
Chapter 1, compounds 88 and 89, 90.
Choice of the dye moiety. Structurally new or modified dyes rarely give
improvements in several parameters, all crucial for fluorescence detection, at once. For
example, the new BODIPY dyes have excellent quantum yields, high extinction
coefficients (~80,000 M–1·cm–1), and thus — good brightness, but possess small Stokes’
shift and are not very sensitive to the local polarity (that is, their fluorescence is not
solvatochromic). The cyanine dyes give useful far-red and near-infrared absorption and
emission maxima, but also suffer from small Stokes’ shift (~30 nm). The small Stokes’
shift results in (a) large background signal due to the scattering of the excitation light
(especially important factor in the SMS experiments), and in (b) large overlap integral,
which in turn ensues significant self-quenching of the fluorophore (fluorescein is a
classically notorious example).
Such is the case with many new SMS dyes as well. This is another reason for
further research and diversification of the available pool of the dyes. In Scheme 2.1 a

123
series of fluorophores are depicted, which resemble each other structurally. Lakowicz41
comments on them (except fluorescein, acridine, and coumarin): “At present, these dyes
are less used in biochemistry owing to their lack of water solubility, their tendency to
aggregate, and the lack of conjugatable forms”.[41, p 75]. One of the depicted
fluorophores, Nile Blue140,141 [2381-85-3], has a close relative from the same oxazine
class, called Nile Red [7385-67-3]. Nile Red has an extinction coefficient142,143 of 38,000
M–1·cm–1 at λmax Ab = 519.4 nm, its fluorescence maximum (λmax Em = 580 nm in dioxane,
Fig. 2.3) is highly solvent-dependent, thus exhibiting solvatochromism144. The Stokes’
shift of Nile Red in dioxane is thus 61 nm, and its quantum yield is 0.7.145 The red
emission, large solvatochromism,146 and fair quantum yield — all these properties make
this fluorophore attractive to incorporate it into a bioconjugate form, sensitive to and
aimed to measure the local polarity in the biomolecules. We picked Nile Red Phenol147
[188712-75-6], a known phenolic derivative of Nile Red with λmax Ab = 551 nm, λmax Em =
632 nm (in MeOH), to use the existing hydroxyl group as a site for attachment of the
thiol-reactive group via an appropriate spacer.
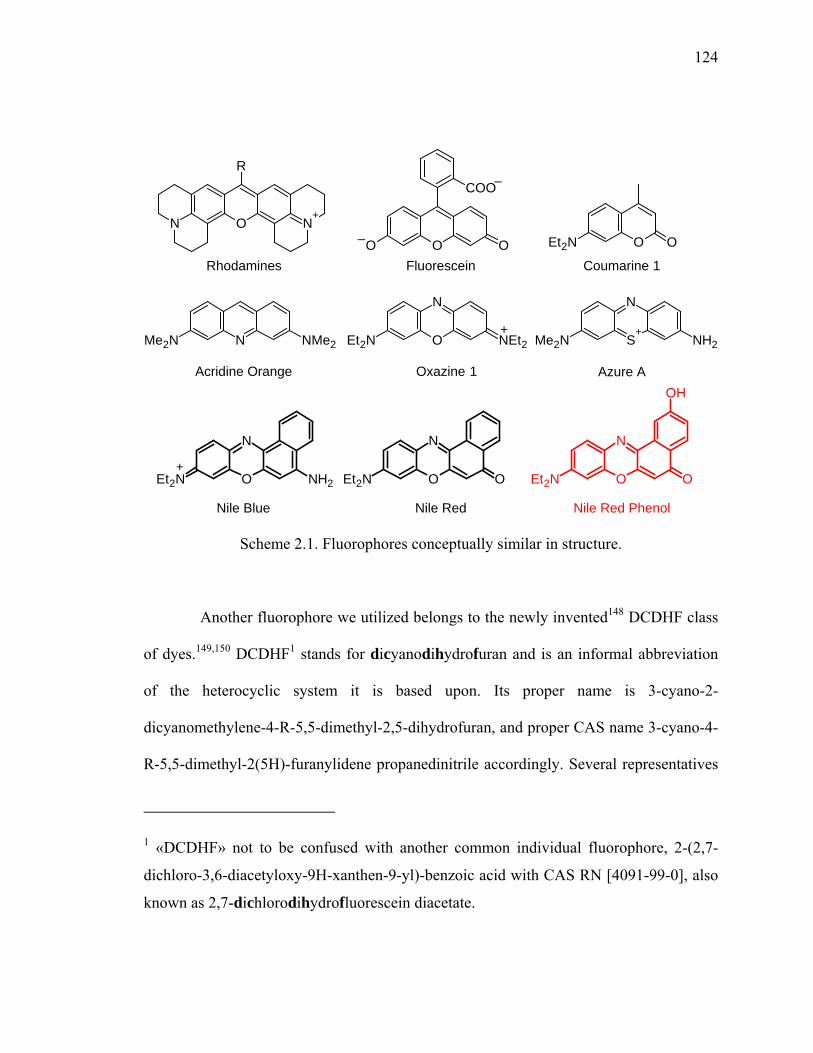
124
O N+
N
R
Rhodamines
N
S+
NH2Me2N
Azure A
N
O NEt2Et2N+
Oxazine 1
N NMe2Me2N
Acridine Orange
OEt2N O
Coumarine 1O OO
COO
–
–
Fluorescein
N
OEt2N O
N
OEt2N NH2
Nile Blue Nile Red
N
OEt2N O
OH
Nile Red Phenol
+
Scheme 2.1. Fluorophores conceptually similar in structure.
Another fluorophore we utilized belongs to the newly invented148 DCDHF class
of dyes.149,150 DCDHF1 stands for dicyanodihydrofuran and is an informal abbreviation
of the heterocyclic system it is based upon. Its proper name is 3-cyano-2-
dicyanomethylene-4-R-5,5-dimethyl-2,5-dihydrofuran, and proper CAS name 3-cyano-4-
R-5,5-dimethyl-2(5H)-furanylidene propanedinitrile accordingly. Several representatives
1 «DCDHF» not to be confused with another common individual fluorophore, 2-(2,7-
dichloro-3,6-diacetyloxy-9H-xanthen-9-yl)-benzoic acid with CAS RN [4091-99-0], also
known as 2,7-dichlorodihydrofluorescein diacetate.
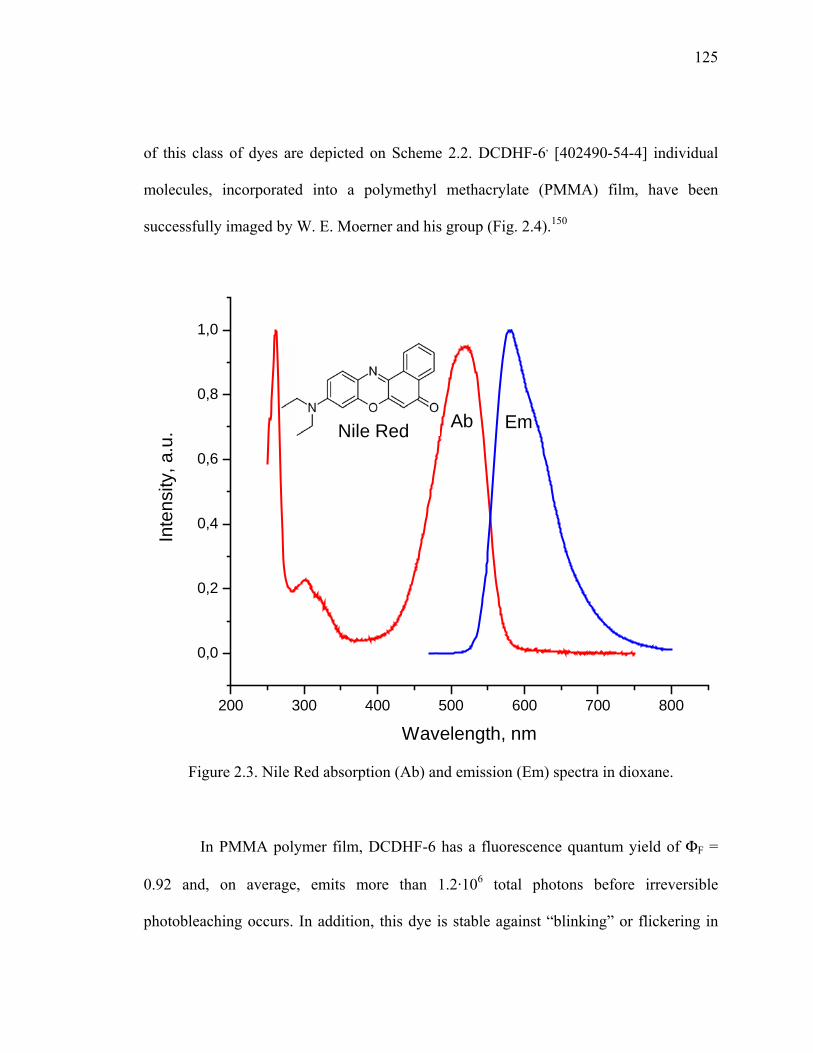
125
of this class of dyes are depicted on Scheme 2.2. DCDHF-6, [402490-54-4] individual
molecules, incorporated into a polymethyl methacrylate (PMMA) film, have been
successfully imaged by W. E. Moerner and his group (Fig. 2.4).150
200 300 400 500 600 700 800
0,0
0,2
0,4
0,6
0,8
1,0
Inte
nsity
, a.u
.
Wavelength, nm
Ab EmNile Red
Figure 2.3. Nile Red absorption (Ab) and emission (Em) spectra in dioxane.
In PMMA polymer film, DCDHF-6 has a fluorescence quantum yield of ΦF =
0.92 and, on average, emits more than 1.2·106 total photons before irreversible
photobleaching occurs. In addition, this dye is stable against “blinking” or flickering in

126
emission, with approximately 85% of the molecules showing no blinking behavior on the
100-ms time scale of the measurement.150
In toluene solutions, however, the quantum yields of the DCDHF dyes were
found to be almost an order of magnitude lower. This result suggests that there is an
environmentally sensitive path through which the molecule can return nonradiatively to
the ground state. Quantum mechanical calculations of the electronic structure of this
system suggest the presence of two excited-state minima — one radiative and the other
nonradiative — accessed through different intramolecular twists in the excited state. The
twist leading to nonradiative decay has an environmentally dependent energy barrier,
resulting in a DCDHF fluorescence quantum yield that varies with local environment.151
Therefore, these dyes are promising candidates as fluorophores in single-molecule probes
for local environment viscosity, rigidity, or polymer-free volume. 150
RO
NCCN
NCO
NCCN
NC
(H13C6)2N
DCDHF
DCDHF-6
O
NCCN
NC
(H13C6)2N
DCDHF-2v
O
NCCN
NC
S(H13C6)2N
DCDHF-Th-6
O
NCCN
NC
(H13C6)2NS
DCDHF-Th-6v
Scheme 2.2. Examples of new DCDHF dyes.

127
Figure 2.4. DCDHF-6, imaged at single molecule level in PMMA film.152
The color saturation towards red indicates increase in fluorescence intensity.
The peaks correspond to locations of single molecules.
Choice of the reactive group. The amine-reactive groups (mostly acylating
reagents) are quite indiscriminative, and most of them also react with hydroxy and
mercapto functionalities in the biomolecules. Thiol-reactive groups are much more
selective and for this reason they are used to prepare fluorescent peptides, proteins, and
oligonucleotides for probing biological structure, function, and interactions. Maleimides
are excellent reagents for the thiol groups, for they do not react with methionine,
histidine, or tyrosine, as iodoacetamides sometimes do.153 Reaction of maleimides with
amines usually requires a higher pH than reaction of maleimides with thiols. However,
hydrolysis of the maleimide to an unreactive maleamic acid can compete significantly
with thiol modification, particularly above pH 8. Furthermore, once formed, maleimide

128
adducts can hydrolyze to an isomeric mixture of maleamic acid adducts, which may
cause a significant change in the fluorescence properties of the conjugate,154 or they can
ring-open by nucleophilic reaction with an adjacent amine to yield crosslinked
products.155 Because of their overall best selectivity, the maleimide reactive group is the
hook of choice for thiols.
Choice of the spacer. Since there are many views on the spacer length role and
how it affects the fluorescence properties, it is hard to predict and assess, which spacer
length is “right”. Since lengthier and flexible spacers tend to amplify the probe’s
sensitivity to the local environment effects, we have chosen a flexible alkyl chain as our
spacer. In our opinion, C6 length of such a spacer is somewhat the optimal or at least a
good compromise to start with.
The resulting target fluorescent probes are depicted in Scheme 2.3. For the Nile
Red fluorophore, we contemplated other sites of the hook attachments, as depicted on the
Schemes 2.4 and 2.5, yet these approaches have not been implemented, and the Nile Red
Phenol with the hook attached via hydroxy functionality at the 2-position is described
below.
N
ON O
OH
NH
O
O
Nile Red Phenol Maleimide (MI)
Spacer
RO
NCCN
NC
DCDHF
NH
O
O
Spacer
Maleimide (MI)
;
Scheme 2.3. The target fluorescent tags with fluorophores, hook, and spacer of choice.
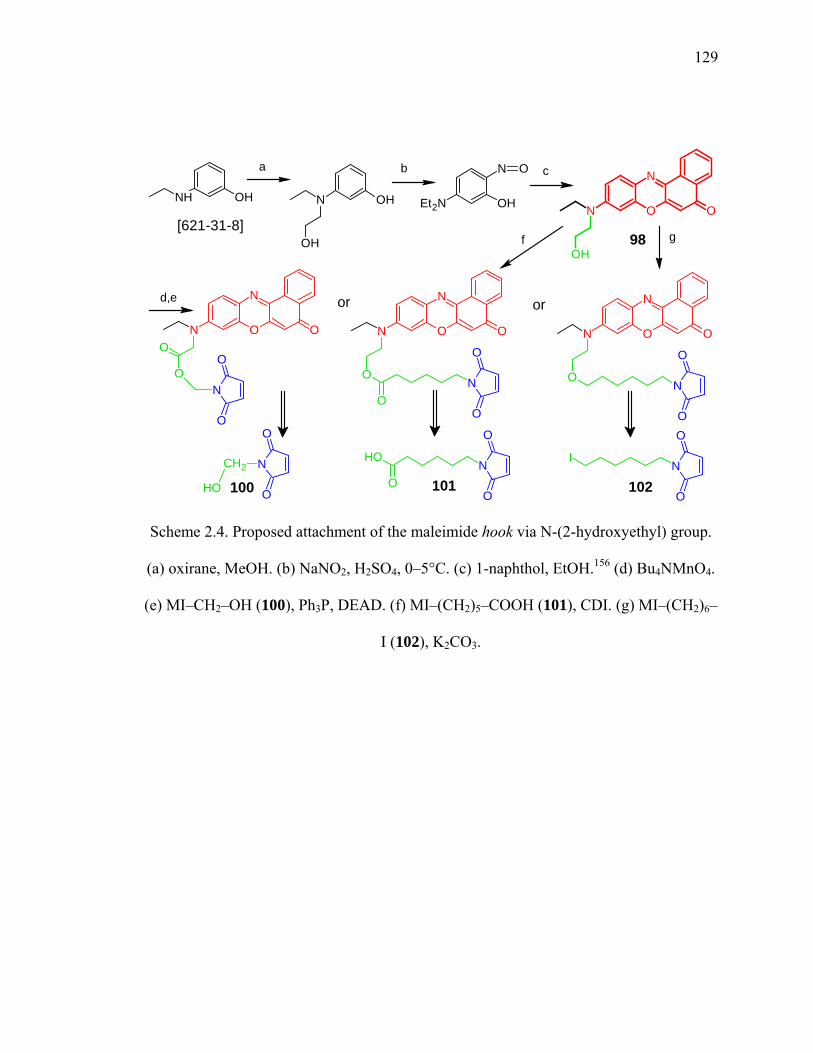
129
NH OH Et2N OH
N O
N OH
OH
N
ON O
OH
N
ON O
ON
O
O
N
ON O
O
ON
O
O
N
ON O
O
O
N
O
O
or or
a b c
d,e
f g
N
O
O
CH2
OH
OH
ON
O
O
IN
O
O
[621-31-8]98
100 101 102
Scheme 2.4. Proposed attachment of the maleimide hook via N-(2-hydroxyethyl) group.
(a) oxirane, MeOH. (b) NaNO2, H2SO4, 0–5°C. (c) 1-naphthol, EtOH.156 (d) Bu4NMnO4.
(e) MI–CH2–OH (100), Ph3P, DEAD. (f) MI–(CH2)5–COOH (101), CDI. (g) MI–(CH2)6–
I (102), K2CO3.
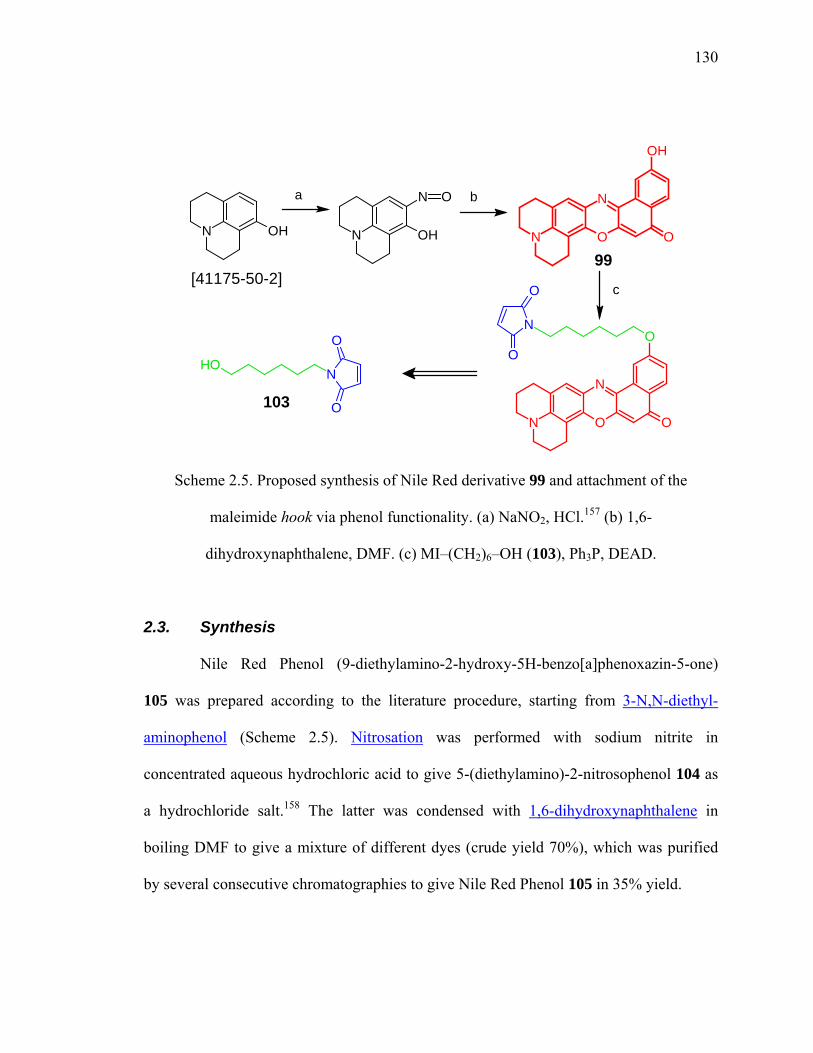
130
N OH
a
N OH
N O b
N
N
O O
OH
99
OHN
O
O103N
N
O O
ON
O
O c[41175-50-2]
Scheme 2.5. Proposed synthesis of Nile Red derivative 99 and attachment of the
maleimide hook via phenol functionality. (a) NaNO2, HCl.157 (b) 1,6-
dihydroxynaphthalene, DMF. (c) MI–(CH2)6–OH (103), Ph3P, DEAD.
2.3. Synthesis
Nile Red Phenol (9-diethylamino-2-hydroxy-5H-benzo[a]phenoxazin-5-one)
105 was prepared according to the literature procedure, starting from 3-N,N-diethyl-
aminophenol (Scheme 2.5). Nitrosation was performed with sodium nitrite in
concentrated aqueous hydrochloric acid to give 5-(diethylamino)-2-nitrosophenol 104 as
a hydrochloride salt.158 The latter was condensed with 1,6-dihydroxynaphthalene in
boiling DMF to give a mixture of different dyes (crude yield 70%), which was purified
by several consecutive chromatographies to give Nile Red Phenol 105 in 35% yield.
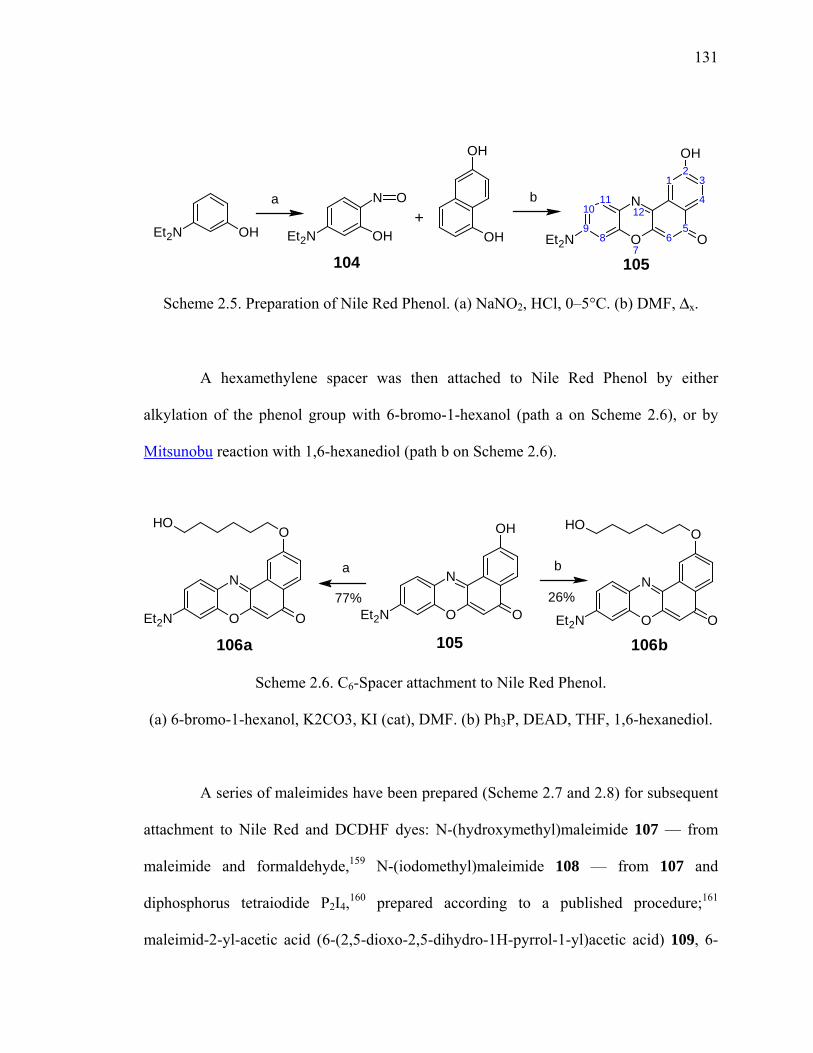
131
Et2N OH
N O
Et2N OH
a
OH
OH
+b 11
8
10
9
N12
O7
65
3
4
21
Et2N O
OH
105104
Scheme 2.5. Preparation of Nile Red Phenol. (a) NaNO2, HCl, 0–5°C. (b) DMF, Δx.
A hexamethylene spacer was then attached to Nile Red Phenol by either
alkylation of the phenol group with 6-bromo-1-hexanol (path a on Scheme 2.6), or by
Mitsunobu reaction with 1,6-hexanediol (path b on Scheme 2.6).
N
OEt2N O
OH
105 106b
N
OEt2N O
OOH
106a
a bN
OEt2N O
OOH
77% 26%
Scheme 2.6. C6-Spacer attachment to Nile Red Phenol.
(a) 6-bromo-1-hexanol, K2CO3, KI (cat), DMF. (b) Ph3P, DEAD, THF, 1,6-hexanediol.
A series of maleimides have been prepared (Scheme 2.7 and 2.8) for subsequent
attachment to Nile Red and DCDHF dyes: N-(hydroxymethyl)maleimide 107 — from
maleimide and formaldehyde,159 N-(iodomethyl)maleimide 108 — from 107 and
diphosphorus tetraiodide P2I4,160 prepared according to a published procedure;161
maleimid-2-yl-acetic acid (6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)acetic acid) 109, 6-

132
(maleimid-2-yl)caproic acid (6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanoic acid)
110 — from maleic anhydride and corresponding aminoacids, via dehydration of the
intermediate maleamic acids with acetic anhydride;162 6-(maleimid-2-yl)caproyl chloride
(6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanoyl chloride) 111 — from 110 and
thionyl chloride; N-(4-hydroxyphenyl)maleimide163 (1-(4-hydroxyphenyl)-1H-pyrrole-
2,5-dione) 112,164,165 N-(6-iodohexyl)maleimide (1-(6-iodohexyl)-1H-pyrrole-2,5-dione)
113 — from the silver salt of maleimide166 MI–Ag and 1,6-diiodohexane; N-(6-
hydroxyhexyl)maleimide 114 — from MI–Ag and 6-bromo-1-hexanol. Maleimide itself,
although commercially available, is rather expensive ($10 per gram) and was required in
plentiful amounts. Thus, it was prepared from maleic anhydride according to the
literature procedure in 60% overall yield.167
N
O
O
OHN
O
O
I
N
O
OI
NH
O
O 107 108
N
O
O
Ag 113
MI–Ag
MI
114N
O
OOH
a b
cd
e
Scheme 2.7. Syntheses of maleimides. (a) CH2O, NaOH 5% aq. pH = 9. (b) P2I4, CS2,
CHCl3, K2CO3. (c) AgNO3, EtOH, NaOH. (d) 1,6-diiodohexane. (e) 6-bromo-1-hexanol.

133
N
O
OOH
O
NO O
OHN
O
O
OHO
109
110
112 111
N
O
OCl
O
O
O
O
NH2(CH2)5OH
O+
a, d
b, d
c, d
e
Scheme 2.8. Syntheses of maleimides from maleic anhydride. (a) glycine, AcOH. (b) 6-
aminocaproic acid, AcOH. (c) 4-aminophenol, AcOH. (d) Ac2O, AcONa. (e) SOCl2.
Following a reported protocol for the preparation of N-alkylated maleimides by
the Mitsunobu reaction,168 a series of such reactions between Nile Red Phenol with (106)
a 6-hydroxyhexyl spacer, or without (105) such a spacer as one reagent, and maleimide
per se, N-(hydroxymethyl)maleimide 107, (4-hydroxyphenyl)maleimide 112, or N-(6-
hydroxyhexyl)maleimide 114 as other reagent has been performed (Scheme 2.9), but no
tangible amounts of the expected products have been obtained from any of the attempted
reactions.
Benzyl alcohol has been reported169 to react with maleimide under Mitsunobu
reaction conditions. We tried to reproduce this reaction, as well as with 1-hexanol, 1,6-
hexanediol, and 1,4-benzenedimethanol (1,4-bis(hydroxymethyl)benzene) but without
avail: although some of the expected product was separated in each case (Scheme 2.10),
most of the starting materials were recovered and the reaction was very messy overall.

134
N
OEt2N O
OH
N
OEt2N O
OOH
+
OR
X OHN
O
O
NH
O
O
OR
+Ph3P N NEtOOC
COOEt+
107112
MI
112107
114
N
OEt2N O
ON
O
O
N
OEt2N O
OX
N
O
O
105106
Scheme 2.9. Attempted Mitsunobu reactions between Nile Reds 105 and 106
and various hydroxy-functionalized maleimides.
R %
C5H11– 7
Ph– 2
OH17
OHR+Ph3P N NEtOOC
COOEt+
+ OH + NH
O
O
N
O
O
R
HO–(CH2)5– 0
Scheme 2.10. Attempted N-alkylation of maleimide according to a reported protocol.169

135
Likewise, no reaction has been detected upon treatment of Nile Red Phenol 105
with either 109 or 110 neither under Mitsunobu conditions, nor under action of 1,1-
carbonyldiimidazole and DMAP in THF or DMAc. Overall with 105, we observed only
two successful Mitsunobu reactions: with 1,6-hexanediol in 26% yield (Scheme 2.6, b)
and acylation with simple caproic acid in 17% yield. The product obtained in the latter
reaction, was identical to the one (118) obtained by acylation with caproyl chloride.
To check the actions of simple alkylating and acylating reagents on 105, the
latter was methylated to give 115 in 99% yield, alkylated with ethylene carbonate to 116,
with 1,6-diiodobenzene — to 117, and acylated with caproyl chloride to 118 (Scheme
2.11).
N
OEt2N O
OH
105 118
N
OEt2N O
O
115
a bN
OEt2N O
O
O
99% 63%
N
OEt2N O
OOH
116
N
OEt2N O
OI
117
c d42% 79%
Scheme 2.11. Simple alkylations and acylations of Nile Red Phenol 115.
(a) MeI, K2CO3, DMF. (b) AmCOCl, DBU, DMF. (c) ethylene carbonate, K2CO3, DMF.
(d) I–(CH2)6–I, K2CO3, DMF.

136
Apparently, the maleimide functionality interferes with and deteriorates the
alkylations and acylations.170 Thus, we considered a strategy of protecting the maleimide
moiety before its introduction and deprotecting it later. Protecting groups for maleimide’s
can be a diene, entering into a [2+4] cycloaddition reaction, and which can be easily
removed later by a retro Diels-Alder reaction.171 At first, we attempted to employ the
adduct of maleimide with cyclopentadiene as a protective group.172 However, it turned
out to be difficult to deprotect these adducts later — temperatures above 200°C
sometimes are required.173 Then we spotted an approach of Clevenger and Turnbull,
utilizing an adduct of furan and maleimide, which could be deprotected in 1–2 hours in
boiling anisole in most cases.174 This approach can possibly be complicated somewhat by
two diastereomeric adducts formed — endo (m.p. 126–128°C) and exo (m.p. 162°C). If
necessary, the endo-isomer (or mixture thereof, m.p. 130–132°C) can be converted into
thermodynamically more stable exo-isomer by recrystallization from a high-boiling
solvent.175
Following this strategy, the adduct of furan and maleic anhydride Fu-MA 119
(exo-cis-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic anhydride) was prepared.176 It
was then converted into Fu-MI 120 (exo-cis-7-oxabicyclo[2.2.1]hept-5-ene-2,3-
dicarboximide), best following the modified procedure of Zawadowski177 to obtain the
product in crystalline form: the internal reaction temperature must be held strictly at 125
± 5°C for 45 ± 5 min. Then, after evaporation of ca. 40% of the water on a rotovap,
boiling ethanol was added in such amount as to completely dissolve the material. This

137
modification yielded 94% yield of crystalline material, compared to the original 40%
reported.
O
O
O
+O
OO
O
O
H
H
NHO
O
O
H
H
119 120
a b
Scheme 2.12. Diels-Alder adduct of furan and maleic anhydride Fu-MA.
(a) Et2O, 90°C or r.t., then recr. (b) NH3·H2O, 125°C, 45 min.
NHO
O
O
H
H
120
aNO
O
O
H
HI
121
N
OEt2N O
OI
117
N
OEt2N O
OH
105
N
OEt2N O
ONO
O
O
H
H
122
NO
O
O
H
H
(CH2)6 I
121
+
NHO
O
O
H
H
120
+b
b
Scheme 2.13. Synthesis of protected Nile Red – Maleimide 122.
(a) I–(CH2)6–I, K2CO3, Me2CO. (b) K2CO3, DMF.

138
The Fu–MI adduct 120 was then alkylated (in DMF or, better, acetone178) with
either 1,6-diiodohexane to give 121, which was in turn used for subsequent alkylation of
Nile Red Phenol 105, or with 117. In either case, the product was the furan-protected
maleimide derivative of Nile Red Phenol 122.
The furan-protected maleimide derivative of Nile Red Phenol 122 was then
deprotected to the target Nile Red Maleimide 123 by refluxing in a mixture of xylene and
dichloromethane (added for solubility reasons) with a short Vigreaux column and no
reflux condensor — to condense and return xylene, but allowing furan to escape. Physical
and optical properties of the Nile Red Maleimide 123 and its application in protein
structure and function elucidation are described below.
N
OEt2N O
ONO
O
O
H
H
122
aN
OEt2N O
ON
O
O
123
Scheme 2.14. Retro Diels-Alder deprotection of 122. (a) xylene, CH2Cl2, Δx.
After the advantage of double bond protection of the maleimide was realized and
proved efficient on the Nile Red dye, the DCDHF Maleimide tags 124 (Scheme 2.15) and
125 (Scheme 2.16) syntheses were rather straightforward. These two tags are derived
from DCDHF-2V and DCDHF-6 correspondingly (Scheme 2.2) and also differ in the
place of the spacer and maleimide attachment — via the 2′-hydroxy group in 124 and via
the 4′-amino group in 125.
The synthesis of 124 by alkylation of the precursor with a DCDHF subunit
already in place (upper path on Scheme 2.15) was not successful, and thus the
introduction of the DCDHF heterocycle was performed after the protected maleimide

139
subunit had been introduced (lower path), followed by deprotection of the maleimide
hook.
O
NCCN
NC
Et2N
OH
CHO
NEt2
OH
O
CN
CN
CN
a
b
a O
NCCN
NC
Et2N
O
N
O
O O
H H
NO
O
O
H
HI+
c
b
124
CHO
NEt2
O
NO
O
O
H
H
Et2N 4'3' 2'
1'
O
O
CN
NCNC
NO O
121
121+
Scheme 2.15. Synthesis of maleimide-tagged DCDHF-2V 124.
(a) 3-cyano-2-dicyanomethylene-4,5,5-trimethyl-2,5-dihydrofuran, Py, AcOH, 60°C. (b)
Fu-MI-C6-I 121, K2CO3, Et2CO, Δx. (c) xylene, Δx.

140
4' 1'3' 2' O
NCCN
NC
NN
O
OZL-1-48W
O
NCCN
NC
F
NO
O
O
H
H
OH
72%
NHO
O
O
H
H
OTs
OH
57%
38%
OH
OH
a
NO
O
O
H
H
OTs
+b
c
NHNO
O
O
H
H
dNH2
H7C3
O
NCCN
NC
N
N O
O
O
H
H
+
e
f
48%
120
125
Scheme 2.16. Synthesis of maleimide-tagged DCDHF-6 125.179
(a) TsCl, Py, THF. (b) Fu-MI 120, K2CO3, DMF, 45°C. (c) TsCl, Py.
(d) 1-PrNH2, Py, THF, Δx. (e) 4′-F-DCDHF, Py. (f) xylene, Δx.
2.4. Results and Discussion
The optical properties of the prepared tags 123, 124, and 125 are summarized in
Table 2.3. The emission spectra, quantum yield measurements, and all biological studies
were performed in the group of Prof. W. E. Moerner, Prof. Arthur L. Horwich, and Prof.
Judith Frydman at Stanford University.

141
Table 2.3. Optical properties of maleimide-containing fluorescent tags.
# Structure λmax Ab, nm (ε) λmax EmSolvent ΦF
123 N
OEt2N O
ON
O
O
536 DCM
547 MeOH
(3.62·105)
564 PhMe
635 MeOH
656 water
0.83
–
<0.1%
124 Et2N
O
O
CN
NCNC
N
O
O
589 DCM
(8.35·105) 720 DCM 0.73
125
H7C3
O
NCCN
NC
N
N
O
O
496 DCM
(9.27·105) 516 DCM 0.12
The longer conjugated path in the chromophore subunit of 124 gives
correspondingly red-shifted absorption and emission maxima, compared to that of 125.
The difference in the quantum yield between the two compounds is 5.8 times, with 124
being noticeably more efficient emitter. Maleimide-tagged Nile Red 123 showed very
strong solvatochromism, especially in its fluorescence properties: besides the
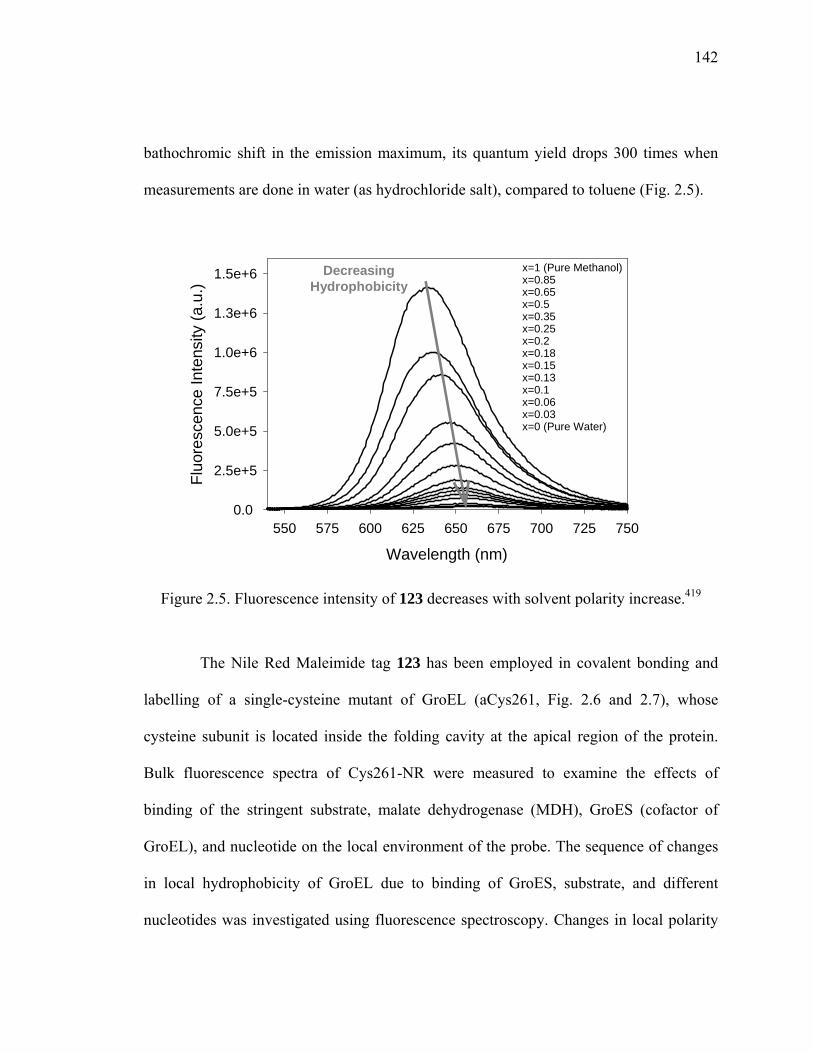
142
bathochromic shift in the emission maximum, its quantum yield drops 300 times when
measurements are done in water (as hydrochloride salt), compared to toluene (Fig. 2.5).
Wavelength (nm)
550 575 600 625 650 675 700 725 750
Fluo
resc
ence
Inte
nsity
(a.u
.)
0.0
2.5e+5
5.0e+5
7.5e+5
1.0e+6
1.3e+6
1.5e+6 x=1 (Pure Methanol)x=0.85x=0.65x=0.5x=0.35x=0.25x=0.2x=0.18x=0.15x=0.13x=0.1x=0.06x=0.03x=0 (Pure Water)
DecreasingHydrophobicity
Wavelength (nm)
550 575 600 625 650 675 700 725 750
Fluo
resc
ence
Inte
nsity
(a.u
.)
0.0
2.5e+5
5.0e+5
7.5e+5
1.0e+6
1.3e+6
1.5e+6 x=1 (Pure Methanol)x=0.85x=0.65x=0.5x=0.35x=0.25x=0.2x=0.18x=0.15x=0.13x=0.1x=0.06x=0.03x=0 (Pure Water)
Wavelength (nm)
550 575 600 625 650 675 700 725 750
Fluo
resc
ence
Inte
nsity
(a.u
.)
0.0
2.5e+5
5.0e+5
7.5e+5
1.0e+6
1.3e+6
1.5e+6 x=1 (Pure Methanol)x=0.85x=0.65x=0.5x=0.35x=0.25x=0.2x=0.18x=0.15x=0.13x=0.1x=0.06x=0.03x=0 (Pure Water)
DecreasingHydrophobicity
Figure 2.5. Fluorescence intensity of 123 decreases with solvent polarity increase.419
The Nile Red Maleimide tag 123 has been employed in covalent bonding and
labelling of a single-cysteine mutant of GroEL (аCys261, Fig. 2.6 and 2.7), whose
cysteine subunit is located inside the folding cavity at the apical region of the protein.
Bulk fluorescence spectra of Cys261-NR were measured to examine the effects of
binding of the stringent substrate, malate dehydrogenase (MDH), GroES (cofactor of
GroEL), and nucleotide on the local environment of the probe. The sequence of changes
in local hydrophobicity of GroEL due to binding of GroES, substrate, and different
nucleotides was investigated using fluorescence spectroscopy. Changes in local polarity

143
were monitored by fluorescence intensity as a function of time. In order to observe the
sequence dependence, various reagents such as GroES, substrate, and nucleotide or
nucleotide mimics were added in different orders.
A typical set of kinetic scans showing the relative peak intensity as a function of
time is shown in Fig. 2.8 for one specific sequence of adding reagents, MDH-ES-nt.
Since the preliminary study of fluorescence intensity showed linear dependence of
intensity on hydrophobicity the intensity changes on Fig. 2.8 may be quantified and
related to specific binding/conformational changes.
For example, when GroES-cofactor is added before ADP/AlFx, GroEL would
begin in the TT state, since nucleotides have not been added and GroES does not strongly
bind to GroEL without them. After adding ADP/AlFx, however, GroES can bind to
GroEL, and form either a symmetric complex or an asymmetric complex (known as the
“bullet complex”). The large fluorescence increase induced by MDH addition suggests
that the asymmetric bullet complex seems to prevail, leaving open a binding site for
unfolded MDH in the trans ring. This might be explained by a reduced binding affinity of
ADP/AlFx to the trans ring when GroES is already bound to the cis ring (ADP/AlFx bullet
complex).
We have observed a variety of local polarity changes upon the addition of
substrate MDH, nucleotide (nt), and GroES. For the most cases, a large shift to less
polarity is observed when unfolded MDH is added, while addition of nucleotide and
GroES produce shifts toward increasing polarity. For sequences in which GroES is added
last, we observe competition between GroES and substrate for the binding sites of the

144
apical domain (Fig. 2.9). For the special case in which the substrate was added last and
the nucleotide was the transition state mimic ADP/AlFx, the fluorescence changes
depended upon the order of addition of the first two components, the nucleotide and
GroES. This behavior is consistent with the formation of a doubly-capped symmetric
complex when ADP/AlFx is added first.
Intriguing results were obtained for the two sequences in which MDH was the
last reagent added and the nucleotide used was ADP/AlFx (Fig. 2.9). Specifically, if
ADP/AlFx was added after GroES (ES-ADP/AlFx-MDH), the fluorescence increase
induced by MDH (“the MDH effect”) was large (20 ± 1%, Fig. 2.9 a), while if ADP/AlFx
was added before GroES (ADP/AlFx-ES-MDH), the MDH effect was much smaller than
in all other cases (7 ± 2%, Fig 2.9 b). On the other hand, when the nucleotide added is
either ATP or ADP, the MDH effect was independent of the order of adding nt and ES
(i.e., for the sequences nt-ES-MDH and ES-nt-MDH). In these two situations, the
fluorescence increase induced by MDH was ~31% and ~28% for ATP and ADP,
respectively.

145
Figure 2.6. Ribbon model of GroEL homotetradecamer.
The monomer unit is shown in color.
These observations may be interpreted as a competition between GroES and the
substrate for the binding sites of the apical domain, and the degree of this competition,
which ultimately induces the substrate release into the cavity, is determined by the
nucleotides. For the special case in which the substrate was added last and the nucleotide
was the transition-state mimic ADP/AlFx, the fluorescence changes depended upon the
order of addition of the first two components, the nucleotide and GroES.

146
CysCys
Figure 2.7. Ribbon model of GroEL protein.
The position of the Cys subunit is shown with an arrow.
Time (sec)
0 1000 2000 3000 4000
Rel
ativ
e P
eak
Inte
nsity
0.9
1.0
1.1
1.2
1.3
1.4ATPADP/AlFx
ADP
Adding MDH
DecreasingHydro-
phobicity
DecreasingHydro-
phobicity
AddingNucleotide
Adding GroES
Figure 2.8. Fluorescence change after addition of (1) MDH, (2) GroES, (3) nucleotide.

147
0 1000 2000 3000 4000 50000.80
0.85
0.90
0.95
1.00
1.05
1.10
GroES-ADP/AlFx-MDH
Time (sec)
Rel
ativ
e P
eak
Inte
nsity
0 1000 2000 3000 4000 5000 60000.84
0.88
0.92
0.96
1.00
1.04 GroES-ADP/AlFx(45 min)-MDH
Rel
ativ
e P
eak
Inte
nsity
Time (sec)
Time (sec)0 1000 2000 3000 4000 5000
Rel
ativ
e P
eak
Inte
nsity
0.80
0.85
0.90
0.95
1.00
1.05
1.10
ADP/AlFx-GroES-MDH
Adding GroES
AddingMDH
AddingADP/AlFx MDH effect
(Decreased)
Time (sec)0 1000 2000 3000 4000 5000 6000
Rel
ativ
e P
eak
Inte
nsity
0.84
0.88
0.92
0.96
1.00
1.04ADP/AlFx - GroES (1:1 molar ratio) - MDH
AddingGroES
AddingMDH
AddingADP/AlFx
MDH Effect
(a) (b)
(d)
AddingGroES
AddingMDH
AddingADP/AlFx
MDH Effect
(c)
MDH Effect
AddingGroES
AddingADP/AlFx
AddingMDH
Figure 2.9. Effect of various addition orders on the fluorescence intensity.
On the basis of these results, we can understand the behavior as follows. First, a
substrate like MDH can bind to the GroEL apical domain in GroEL, causing increased
hydrophobicity. Although GroES exists in the solution, it cannot bind tightly to the
GroEL apical domain due to the absence of nucleotides. After addition of nucleotides,
GroES can finally bind to GroEL, and the nucleotide type determines the extent of the
conformational changes of the apical domain by GroEL-GroES complex, inducing MDH
release into the cavity. Furthermore, as noted alone, the fluorescence decay after the final
addition in Fig. 2.9 a shows a fast change followed by a slow drop. The common feature

148
of all these sequences is that the compact GroEL-GroES complex formation can only
start at the end of the sequence and, therefore, the displacement of MDH by GroES
should always occur at the end. The schematic explanation of these sequences is depicted
on Fig. 2.10.
ADP/AlFxADP/AlFx
ADP/AlFx
GroESADP/AlFx
ADP/AlFx
MDH
No MDH binding
ADP/AlFx
ADP/AlFx
RR stateTT state Symmetric complex(American Football)
ADP/AlFxADP/AlFx
ADP/AlFx
ADP/AlFx
ADP/AlFx
GroESADP/AlFx
ADP/AlFx
ADP/AlFx
ADP/AlFx
MDH
No MDH binding
ADP/AlFx
ADP/AlFx
ADP/AlFx
ADP/AlFx
RR stateTT state Symmetric complex(American Football)
No GroES bindingTT state
GroES ADP/AlFxADP/AlFx
MDHADP/AlFx
RT state(Bullet Structure)
TT state MDH bindingto the trans-ring
No GroES bindingTT state
GroES ADP/AlFxADP/AlFxADP/AlFx
MDHADP/AlFxADP/AlFx
RT state(Bullet Structure)
TT state MDH bindingto the trans-ring
(a)
(b)
Figure 2.10. Proposed scheme for the formation of symmetric/asymmetric complex of
GroEL/GroES with ADP/AlFx. (a) When GroES is added first, GroES can only bind after
adding ADP/AlFx, and the binding affinity of ADP/AlFx to the trans ring may be reduced
because of GroES binding to the cis ring, thus preventing formation of the symmetric
complex. (b). When ADP/AlFx is added first (before GroES), hindered MDH binding to
the GroEL can be explained by the formation of the symmetric football complex.

149
2.5. Conclusion
We have designed, prepared and characterized fluorescent probes with two
different fluorophores — Nile Red and DCDHF, with a hexamethylene spacer and a
maleimide reactive group. One of the probes has been utilized in local polarity probing
and conformational changes elucidation experiments after covalently binding to a single-
cysteine mutant of GroEL chaperonin of E. coli. Large solvatochromism and polarity
dependence of the prepared probe, combined with high site binding specificity, allowed
to study conformational changes of GroEL chaperonin.

150
CHAPTER III.
ORGANIC LIQUID CRYSTAL SEMICONDUCTORS.
3.1. Introduction
The electrical conductivity is a physical manifestation of charge transport
phenomenon in materials. The physical value of electrical conductivity σ is the
quantitative measure of this phenomenon — total charge, transported across a unit cross-
section area per second per unit electric field applied. Depending on their electrical
conductivity values, all materials historically and rather arbitrarily can roughly be divided
into the classes of conductors (metals), semimetals, semiconductors, and insulators.
Conductors are materials with σ between 106 and 104 Ohm–1·cm–1. Semiconductors
have σ values between 104 and 10–10 Ohm–1·cm–1. Insulators (dielectric materials) have
conductance below 10–10 Ohm–1·cm–1. Schematically these classes may be represented as
depicted on Fig. 3.1.180
Semimetals are materials, whose electrical properties lie in between of metals
and semiconductors. Semimetals have considerably lower electrical conductivity due to
lower carrier density, compared to metals and much weaker temperature dependence of
electrical conductivity on temperature compared to semiconductors. Semimetals also
possess electrical conductivity at the absolute zero of temperature (which is

151
151
characteristic of metals), while semiconductors are insulators under the same conditions.
Examples of semimetals are graphite, α-As, α-Sb, Bi, Po, At. Structurally, most
semimetals are anisotropic heterodesmic crystals.181 That is, the chemical bonds differ by
their energy (and sometimes by type) in different directions of the crystal.
Figure 3.1. Conductivity domains of metals, semiconductors, and insulators.180
The classification into the four named classes of the materials by the values of
electrical conductivity does not reflect, however, the fundamental differences between
them, which are much more complex than simply the conductance values. The sign of
temperature dependence of the conductivity and whether or not the material conducts at
absolute zero are the ultimate criteria to delineate metals from insulators. While there is
no sharp, well-defined border between semiconductors and insulators (for

152
semiconductors are insulators), the physical properties of typical representatives of each
class are markedly different.
The general phenomenological equation for conductivity is182:
he PeNe μμσ += , (3.1)
where N and P are the concentrations of negative (electrons) and positive (holes) charge
carriers of charge e. The mobility μ is the magnitude of the drift velocity v (that is,
average velocity, attained due to electric field) of a charge carrier per unit electric field E:
Ev
=μ , (3.2)
The mobility is defined to be positive for both electrons and holes, though their
drift velocities are opposite in a given field. The mobility in SI is expressed in m2/V·s and
in practical units, cm2/V·s.
Table 3.1 gives experimental values of the mobility at room temperature for
some inorganic semiconductors. The highest mobility observed in a bulk semiconductor
is 5×106 cm2/V·s in PbTe at 4 K.182
Table 3.2 summarizes the key differences between metals, semimetals.
semiconductors, and insulators. The conductivity of metals decreases with temperature,
while conductivity of semi-conductors — increases, and is strongly dependent on
temperature. For example, Fig. 3.2 shows the concentration of electrons in different
materials, and Fig 3.3 — the dependence of electron concentration (and thus,
conductivity) on temperature in Ge and Si. 182

153
Table 3.1. Carrier mobilities μ in some crystalline inorganic semiconductors at room
temperature, in cm2/V·s. 182
Crystal Electrons Holes Crystal Electrons Holes
Diamond 1800 1200 GaAs 8000 300
Si 1350 480 GaSb 5000 1000
Ge 3600 1800 PbS 550 600
InSb 800 450 PbSe 1020 930
InAs 30000 450 PbTe 2500 1000
InP 4500 100 AgCl 50 –
AlAs 280 – KBr (100 K) 100 –
AlSb 900 400 SiC 100 10–20
Table 3.2. Key differences between metals, semimetals, intrinsic inorganic
semiconductors, and insulators at room temperature.
Parameter Metal Semimetal Semiconductor Insulator
Concentration of charge carriers,
N, per structural unit
N, per cm3
~1
1023
10–2…10–5
1021…107
10–6…10–10
1013…1017
<10–10
<1013
Energy gap, eV 0 ~0 >0…6 >6
Mobility, μ, cm2/V·s ~10† 105…107 >0…105 ~0
Temperature dependence, dσ/dT – – + +
σ, Ohm–1·cm–1 106…104 104…102 104…10–10 ~10–10
† The notion of mobility has little use in metals. Calculated from Al σ by (3.1).
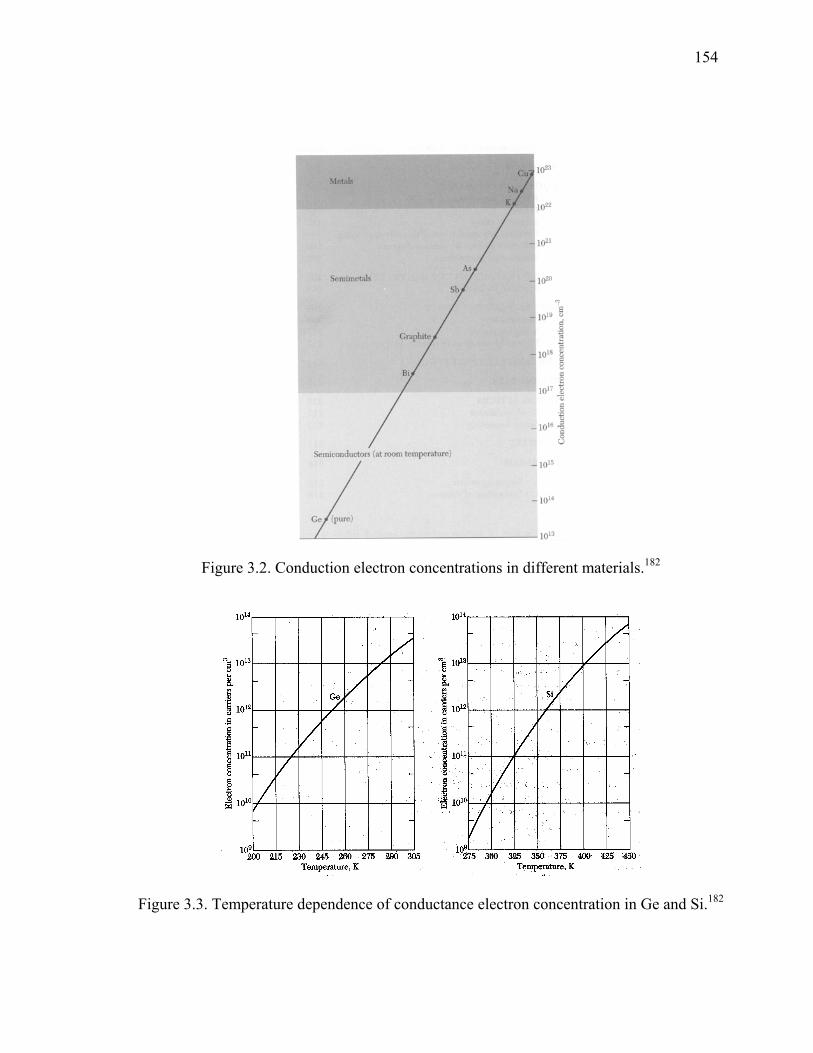
154
Figure 3.2. Conduction electron concentrations in different materials.182
Figure 3.3. Temperature dependence of conductance electron concentration in Ge and Si.182
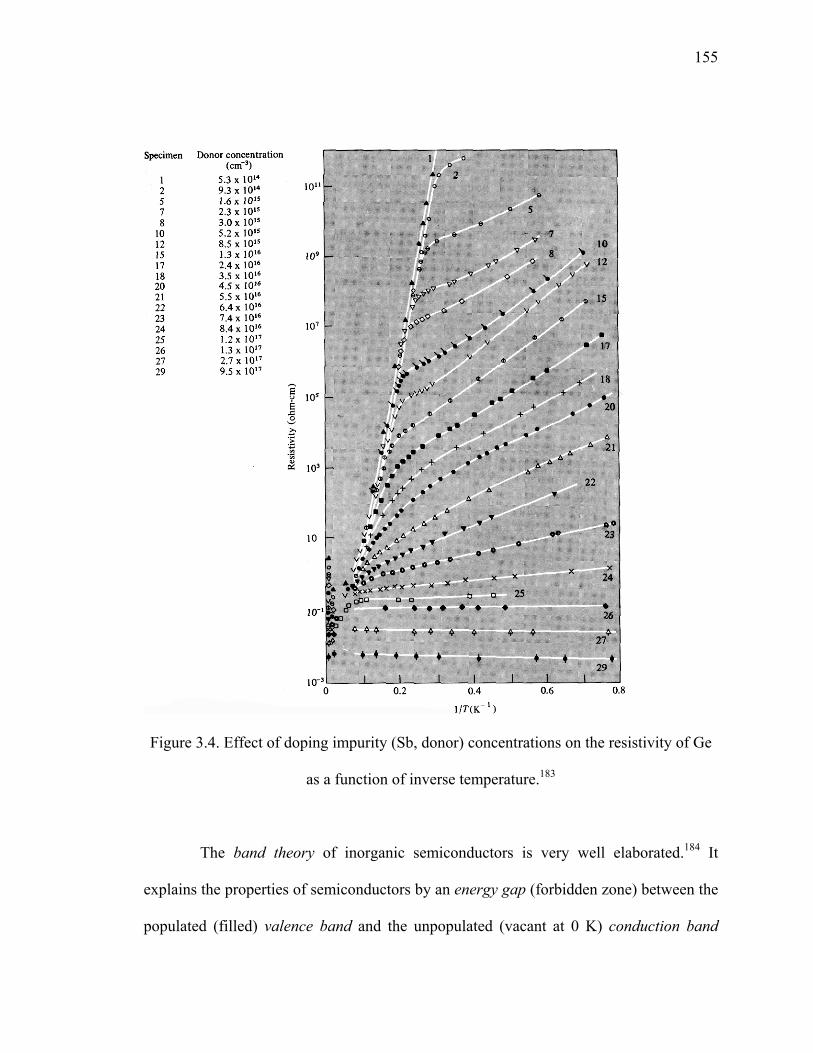
155
Figure 3.4. Effect of doping impurity (Sb, donor) concentrations on the resistivity of Ge
as a function of inverse temperature.183
The band theory of inorganic semiconductors is very well elaborated.184 It
explains the properties of semiconductors by an energy gap (forbidden zone) between the
populated (filled) valence band and the unpopulated (vacant at 0 K) conduction band

156
(Fig. 3.5). At temperatures above 0 K a fraction of the electrons may be thermally excited
from the valence band into the conductance band, creating a conducting electron and a
hole, capable of hole-type conduction. The theory gives the following expression for the
product of concentration of electrons and holes in an intrinsic semiconductor:
⎟⎟⎠
⎞⎜⎜⎝
⎛ −=
TkE
CTNPB
gexp3 , (3.3)
where C is some constant, which depends on material T is temperature, Eg is energy gap
(also depends on temperature), and kB is the Boltzmann constant. From this formula it
follows that the concentration of charge carriers rapidly increases with temperature (Fig.
3.3), in striking contrast to metals, where, though the charge carriers’ density stays about
the same as the temperature rises, the conductivity slowly decreases due to increasing
electron-phonon interactions (scattering on the vibrations of the crystal structure units).
Ener
gy
Eg
Valence Band
Conductance Band
Ener
gy
Eg
Valence Band
Conductance Band
e– p+ e–
e–
T = 0 K T > 0 K
a b
Forbidden Band Forbidden Band
k k
Figure 3.5. Band structure of an intrinsic inorganic semiconductor.

157
According to the band gap theory, a semiconductor is a material which is an
insulator at 0 K (has zero charge carriers at absolute zero of temperature), but whose
energy gap is small enough (0…6 eV, typically 0…2 eV) for thermal excitations of
electrons to the conductance zone, resulting in observable electrical conductivity at T > 0
K.184
Organic semiconductors185 are fundamentally different from inorganic ones.
All solid inorganic semiconductors are coordination compounds, while organic
semiconductors are molecular crystals or glasses and have a different mechanism of
conductivity. The origin of the conductivity in organic semiconductors lies in the
multicentered, delocalized, conjugated π-bonds of organic molecules. The conjugation
gives delocalization energy gain, resulting in a decrease of HOMO-LUMO gap for a
given molecule. In conjugated polymers this energy gap may be comparable to thermal
energy kT. The conductivity in organic semiconductors is primarily due to electron
transitions between discrete levels (not merged into bands or zones) of molecular orbitals
with different energies. The band theory, developed for crystalline inorganic
semiconductors, is often applied to the organic ones and frequently gives correct
predictions,186 but may not be quite adequate187 or not even valid for molecular
structures, where electronic levels cannot be considered as collectivized between
molecules — at least at room temperature.188
The mobility of charge carriers in many of the highest mobility organic
semiconductors at room temperature is of order of 1 cm2/V·s and strongly depends on:
purity (assay of the material and absence of impurities/dopants), state (crystal or

158
amorphous — in the latter case the mobility in them decreases to 10–3…10–5 cm2/V·s),
lattice defects (if the material is crystalline), crystallographic axis direction (if crystalline
and structurally anisotropic), or director (if liquid crystal). Table 3.3 gives several
representative values of electron and hole mobilities in organic molecular crystals.189
Table 3.3. Carrier mobilities (of electrons e– and holes p+) in some organic molecular
crystals at room temperature: μ, cm2/V·s.189
Crystal e– p+ Crystal e– p+
Benzene 1.5 – Perylene 2.0 –
Naphthalene 0.63 1.5 1,4-Diiodobenzene – 12.0
Anthracene 1.73 1.13 Terphenyl 0.34 0.8
Durene 8.0 5.0 Phenazine 0.29 –
Biphenyl – 0.42 Azulene 0.2 0.19
Pyrene 0.7 0.7 Phenothiazine 5.0 –
Tetracene – 0.85 Anthraquinone 0.02 –
1,4-Dibromonapthalene 0.017 0.66 Benzophenone 0.16 –
The purity of organic semiconductors is of no less importance than in the case
of inorganic ones. For example, on Fig. 3.6 mobility data are shown for two
monocrystalline samples of perylene: (a) ultrapurified (with zone refining of metallic
potassium treated perylene), and (b) conventionally purified.190 The two samples
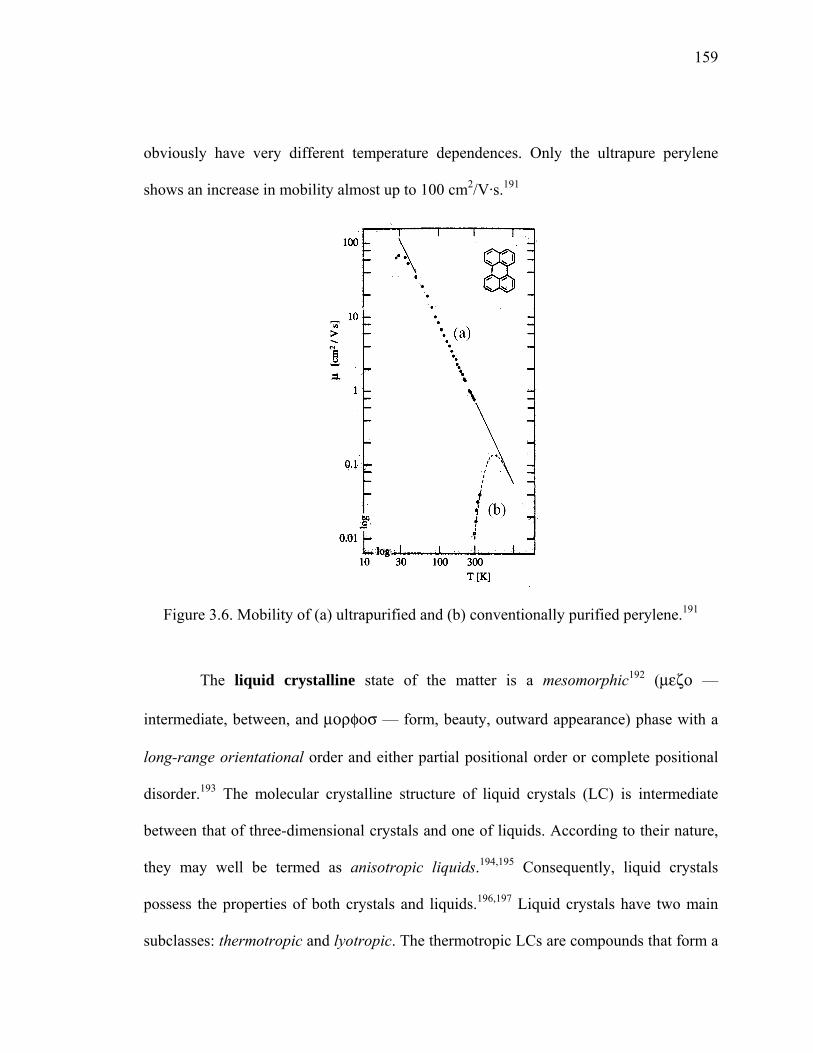
159
obviously have very different temperature dependences. Only the ultrapure perylene
shows an increase in mobility almost up to 100 cm2/V·s.191
Figure 3.6. Mobility of (a) ultrapurified and (b) conventionally purified perylene.191
The liquid crystalline state of the matter is a mesomorphic192 (μεζο —
intermediate, between, and μορφοσ — form, beauty, outward appearance) phase with a
long-range orientational order and either partial positional order or complete positional
disorder.193 The molecular crystalline structure of liquid crystals (LC) is intermediate
between that of three-dimensional crystals and one of liquids. According to their nature,
they may well be termed as anisotropic liquids.194,195 Consequently, liquid crystals
possess the properties of both crystals and liquids.196,197 Liquid crystals have two main
subclasses: thermotropic and lyotropic. The thermotropic LCs are compounds that form a

160
mesophase by heating a crystal or cooling an isotropic liquid. The lyotropic LCs form the
mesophase by dissolving an amphiphilic mesogen in suitable solvent, under appropriate
conditions of concentration and temperature. There are also amphotropic mesogens,
which can exhibit both thermotropic and lyotropic properties. We will be dealing here
only with thermotropic LCs.
Columnar liquid crystals are compounds (mesogens) that under suitable
conditions of temperature, pressure and concentration can form a mesophase, wherein
molecules are stacked in columns. Discotic liquid crystals are mesogens with relatively
flat, disc- or sheet-shaped molecules. All discotic mesogens can form columnar
mesophase(s).
Liquid crystal semiconductors (that is, organic molecular crystalline
semiconductors in a liquid crystal state) benefit from the (a) order and anisotropy and (b)
easy processability of the LC phase. The long-range orientational and (in some cases)
short-range positional order of the organic molecules in LC phase cause the columnar
LCs to generally show higher mobilities than the same compound in liquid phase.198
More ordered (compared to nematic) smectic phases of calamitic (rod-like) liquid crystals
and the columnar phases of discotic liquid crystals show electronic and hole transport199
up to 0.1 cm2/V·s 200 by either a hopping or (in the most ordered systems) a band transport
mechanism.201,202 The conductivity obtained is usually highly anisotropic — 1000:1
parallel to perpendicular with respect to the director.203
The organic semiconductors represent an attractive area of research from
theoretical and — especially — from a practical viewpoint. The mobilities measured in

161
new organic and liquid crystalline semiconductors pose a challenge to the classical band
theory and thus stimulate new models, insights, and theories on the mechanism of charge
transport in molecular crystals. Application-wise, organic semiconductors are candidates
to compete with and possibly replace amorphous silicon in industry. The obvious
advantages are the ease of processability, absence of grain boundaries, spontaneous self-
alignment between interfaces, and an ability to self-repair due to their inherent dynamic
nature.199
We studied the synthesis, purification, trace analysis, thermal properties, phase
diagrams, and mobilities of several low molecular weight semiconductors and a few
liquid crystalline semiconductors. The syntheses of tail-functionalized polyacenes are
provided below, followed by iodoarenes and triphenylene derivatives.
3.2. Polyacenes
Our targets were new n-acenes (n=3 anthracene, n=4 tetracene, and n=5
pentacene) with two, four, six, or eight substituents (Scheme 3.1). We expected that some
of them might exhibit liquid crystalline and semiconducting properties in analogy to the
behavior of other multiple tail decorated polynuclear hydrocarbons such as triphenylene
and hexa-substituted benzene esters.

162
R
RR
R
RR
RR
R
R
R
R
R
R
R
R
R
RR
R
R
R R
R R
R R
RR
R
Scheme 3.1. Target polysubstituted n-acenes. R = alkyl or alkoxy.
3.2.1. Anthracenes
For many years anthracene was one of the best known organic
semiconductors.204 It was obtained in pure enough form to observe a cyclotron resonance
at 2 K.205 We aimed to modify the anthracene core to obtain materials, that could posses a
columnar mesophase. Nematic liquid crystals containing 2-mono-substituted and 2,6-
disubstituted anthracene moieties in the core have been long known.206 Recently, their
charge transport properties in the nematic phase have been reported to be about 2·10–3
cm2/V·s, which is quite high for a disordered nematic phase and may be explained by
some π-stackings, induced at the short range by the mesophase structure.207 (In the
nematic phase there is only long-range orientational order, and no positional order.)
Nematic 9,10-bis(phenylethynyl)anthracenes were shown to posses luminescent208 and
fluorescent209 properties. Some 2,6,9,10-tetrasubstituted anthracenes for liquid crystals
have been patented.210 1,4,5,8-Tetrasubstituted anthraquinones and anthracenes showed
smectic mesophases.211,212 1,2,3,5,6,7-Hexasubstituted anthraquinones are thermotropic

163
liquid crystals, which form columnar mesophases.213,214,215 1,2,3,4,5,6,7,8-
Octa(alkanoyloxy)-substituted ester derivatives of anthraquinone also show columnar
mesophase.216 2,3,6,7-Tetra-n-alkylanthracenes are known, non-mesogenic compounds
with m.p. 88 (n-C3), 97 (n-C5), and 84 °C (n-C7). 2,3,6,7,9,10-Hexa-n-heptylanthracene
has m.p. 74 °C and does not exhibit any mesophases ether.217 2,3-Dialkoxyanthracenes
have been prepared, though with difficulties and on a small scale, as efficient
organogelators.218 We aimed to synthesize some of the 2,3,6,7-tetraalkoxyanthracenes,
2,3,6,7-tetraalkoxy-9,10-dialkylanthracenes, and 1,2,3,4,5,6,7,8-octaalkylanthracenes.
3.2.1.1. 2,3,6,7-Tetraalkoxyanthracenes
The symmetrical structure of 2,3,6,7-tetraalkoxyanthracene and the increased
reactivity of anthracene core’s meso (9- and 10-) positions naturally prompt to trace it in
the retrosynthetic analysis to the corresponding symmetrical 2,3,6,7-tetraalkoxy-9,10-
anthraquinone. 2,3,6,7-Tetramethoxy-9,10-anthraquinone (130, R=Me) is a known
compound [5629-55-0], which may be approached, in turn, via several routes (Scheme
3.2):
(1) from meta-hemipinic (4,5-dimethoxyphthalic) acid anhydride [4821-94-7]
126 by Friedel-Crafts acylation (a) of veratrole with subsequent internal dehydration (b)
of the veratroyl-veratric acid 127 in polyphosphoric or sulfuric acid; 219
(2) from veratrole and formaldehyde condensation (c) product 129,220 after the
oxidation (g) of the latter with Cr(VI) reagents; and
(3) from the bis(trimethylsilyl) ether of 2,3-butanedione dienol 132 221 by its
double [4+2] addition (d) to benzoquinone, hydrolysis/oxidation218 (e) of the adduct 133,
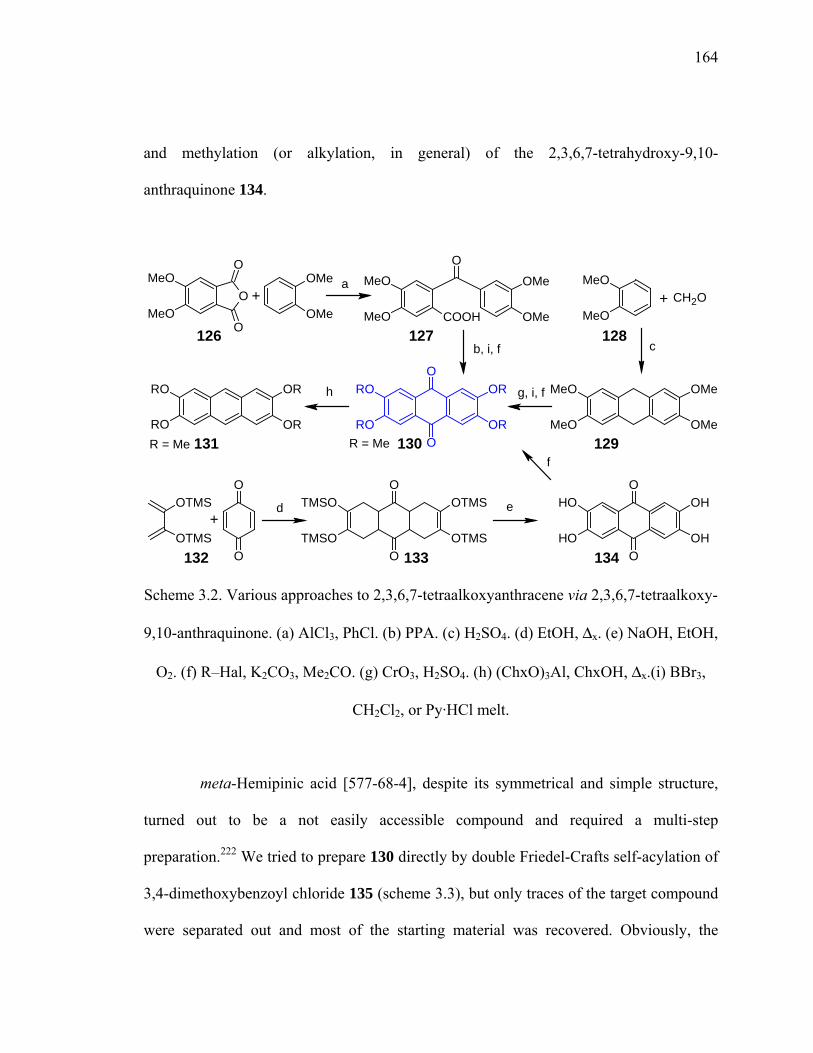
164
and methylation (or alkylation, in general) of the 2,3,6,7-tetrahydroxy-9,10-
anthraquinone 134.
OTMS
OTMS OTMS
OTMS
O
OTMSO
TMSO
RO
RO OR
OR
OR
OR
O
ORO
RO
COOH OMe
OMeMeO
MeO
O
OMe
OMeMeO
MeO
MeO
MeOCH2O
MeO
MeOO
O
OOMe
OMe+
O
O
+OH
OH
O
OOH
OH
+a
b, i, f c
d e
f
g, i, fh
126 127 128
129130131
132 133 134
R = MeR = Me
Scheme 3.2. Various approaches to 2,3,6,7-tetraalkoxyanthracene via 2,3,6,7-tetraalkoxy-
9,10-anthraquinone. (a) AlCl3, PhCl. (b) PPA. (c) H2SO4. (d) EtOH, Δx. (e) NaOH, EtOH,
O2. (f) R–Hal, K2CO3, Me2CO. (g) CrO3, H2SO4. (h) (ChxO)3Al, ChxOH, Δx.(i) BBr3,
CH2Cl2, or Py·HCl melt.
meta-Hemipinic acid [577-68-4], despite its symmetrical and simple structure,
turned out to be a not easily accessible compound and required a multi-step
preparation.222 We tried to prepare 130 directly by double Friedel-Crafts self-acylation of
3,4-dimethoxybenzoyl chloride 135 (scheme 3.3), but only traces of the target compound
were separated out and most of the starting material was recovered. Obviously, the

165
reactivity of acyl-containing benzene ring towards electrophilic substitution is not
sufficient, even despite of the two methoxy groups, present in the same ring.
MeO
MeO
O
Cl
OMe
OMe
O
Cl+
OMe
OMeMeO
MeO
O
O
a
135 130traces
Scheme 3.3. Attempted self-acylation of veratroyl chloride. (a) AlCl3, PhNO2.
Given the commercial availability of the veratric aldehyde (3,4-
dimethoxybenzaldehyde) 136 and our experiments in ortho-lithiation reactions en route to
4,5-dimethoxyphthalic dialdehyde (vide infra), we devised a route to 130 starting from
136 (scheme 3.4 and 3.5). First, we (a) brominated veratric aldehyde to 137 with bromine
in chloroform. Interestingly, this reaction never went to completion if exactly one
equivalent of bromine was used and required ca. 1.2 eq. of bromine, though the excess of
the reagent did not produce any dibromo or other byproducts. The aldehyde was (b)
protected as either a 1,3-dioxolane or a dimethyl ether (the latter is depicted in the
scheme), which was subjected to (c) halogen-lithium exchange with n-butyl lithium and
the organolithium compound was condensed with another equivalent of veratric aldehyde
to give, after acidic work-up, aldol 139. To avoid the protection of the aldehyde as an
extra step in this synthesis, we resorted to (e) an in situ protection by a lithium
diethylamide, preformed in the same flask, with almost no decrease in the overall yield.
Then we realized that the bromination step could also be dropped if the amine we used

166
for in situ protection of carbonyl, will be modified to act as ortho-director in the direct
lithiation step. Thus, (d) N,N,N′-trimethylethylenediamine (TriMEDA) was employed.
MeO
MeO
O
H
136
MeO
MeO
O
H
Br137
MeO
MeO
O
O
Br138
MeO
MeO
O Li
NN
– +
MeO
MeO
O Li
NN
Li
– +
MeO
MeO
O Li
N
– +
MeO
MeO
O Li
N
Li
– +
MeO
MeO
O
O
Li
135+
MeO
MeO
OH
CHO
OMe
OMe
139
a b
c c
c
d e
33% 34%42%
Scheme 3.4. Ortho-lithiation approach to 138. (a) Br2, CHCl3. (b) HC(OMe)3, Dowex
50W-X8-100, MeOH. (c) BuLi, TMEDA. (d) BuLi, TriMEDA. (e) BuLi, Et2NH.
The aldol 139 was oxidized by potassium dichromate in acetic acid into
veratroylveratric acid, which was then cyclized (without purification) into 130 by heating
in sulfuric acid. The overall yield of 130 from 136 was mere 5%.

167
MeO
MeO
OH
CHO
OMe
OMe
139
MeO
MeO
O
COOH
OMe
OMe OMe
OMeMeO
MeO
O
O130
a b
13%
Scheme 3.5. Oxidation and cyclization of 138 into anthraquinone 130.
(a) K2Cr2O7, AcOH. (b) H2SO4 (conc.)
The Diels-Alder condensation of 2,3-bis(trimethylsilyloxy)-1,3-butadiene 132
with benzoquinone didn’t look attractive and promising either, for the employment of the
same reagent in the similar [4+2] addition to 1,4-napthoquinone produced only 33% of
the desired product accompanied by many byproducts, and didn’t scale up well.218
Nevertheless, we prepared disilyl ether 132 223 and introduced it into reaction224 with
benzoquinone (Scheme 3.4). As determined by GC, the known mono-adduct224 was
formed predominantly, with only small amount of 133 (identified by its mass spectrum).
Close monitoring of the reaction by GC-MS revealed that the first cycloaddition proceeds
fast, and benzoquinone (the limiting reagent) disappears after 8 hrs of reflux in benzene.
Further heating results in formation of dark brown-black tar, insoluble in chloroform,
which presumably is a polymerization product of 132. This route has not been pursued
any further.
OTMS
OTMS
O
O
+
132
a
O
O
TMSO
TMSO45%
OTMS
OTMS
O
OTMSO
TMSO1336%
+
Scheme 3.4. Attempted preparation of 133. (a) PhH or EtOH, BHT (0.5 mass %), Δx.

168
Condensations of formaldehyde with veratrole, 1,2-bis(hexyloxy)benzene 140
and 1,2-bis(dodecyloxy)benzene 141 gave the corresponding 2,3,6,7-tetraalkoxy-9,10-
dihydroanthracenes 142 [26952-97-6], 143, and 144 (Scheme 3.5). The
dihydroanthracene 142 turned out to be quite resistant to oxidation and either did not
undergo any oxidation (Ag2O, KMnO4 in H2SO4) or (with CrO3 in AcOH, K2Cr2O7 in
H2SO4) gave an inseparable mixture of compounds, wherein no target anthraquinone has
been detected. After many trials, the only successful oxidant that gave any tangible, yet
not preparatively valuable, amount of 130 was KMnO4 in aq. NaOH. Later we found that
this strange oxidation resistance has been already described in the literature.225 If
formaldehyde is substituted for higher aliphatic aldehyde homologs, the condensation
with veratrole in presence of nitriles226 produces anthracenes, rather than
dihydroanthracenes, and the former can be oxidized to 130 by K2Cr2O7 in AcOH in 40%
yield.227
OR
ORRO
RO
RO
ROCH2O+
a
R = Me
C5H11
C12H25 141
140
veratrole 142
143
144
Scheme 3.5. Preparation of 2,3,6,7-tetraalkoxy-9,10-dihydroanthracenes. (a) CH3SO3H.
Both 2,3,6,7-tetramethoxy-9,10-anthraquinone 130 and its 2,3,6,7-tetramethoxy-
9,10-dihydroanthracene 142 were successfully deprotected into tetrahydroanthracenes

169
145 and 146 by Py·HCl melt and BBr3 in CH2Cl2 correspondingly (Scheme 3.6). From
these four compounds only 142 (perhaps due to its non-planar structure) has sufficient
solubility in common organic solvents to be characterized by NMR. Compound 130 was
characterized by its 1H NMR spectrum in hot DMSO-d6 and comparing its melting point
(348 °C) to literature values (340…346 °C).219
OMe
OMeMeO
MeO
O
O130
OH
OHOH
OH
O
O
a
145
OMe
OMeMeO
MeO142
OH
OHOH
OH
b
146
Scheme 3.6. Conversion of methoxy groups into hydroxyl functionalities.
(a) Py·HCl melt. (b) BBr3 CH2Cl2, 0 °C.
Aromatization of compounds 142–143 with Bu4NIO4, Pb(OAc)4,228 and SeO2
did not succeed. Aromatization used for highly alkyl-substituted dihydroanthracenes —
with n-BuLi, TMEDA, and MeI 229— was contemplated, but was not performed.
3.2.1.2. 2,3,6,7-tetraalkoxy-9,10-dialkylanthracenes
Several 2,3,6,7-tetraalkoxy-9,10-dialkylanthracenes have been prepared by
condensing230 the corresponding 1,2-bis(alkoxy)benzenes (veratrole, 140, 141) with
aliphatic aldehydes (acetaldehyde CH3CHO, caproic C5H11CHO, and capric C9H19CHO
aldehyde), as depicted in Scheme 3.7 and summarized in Table 3.4.
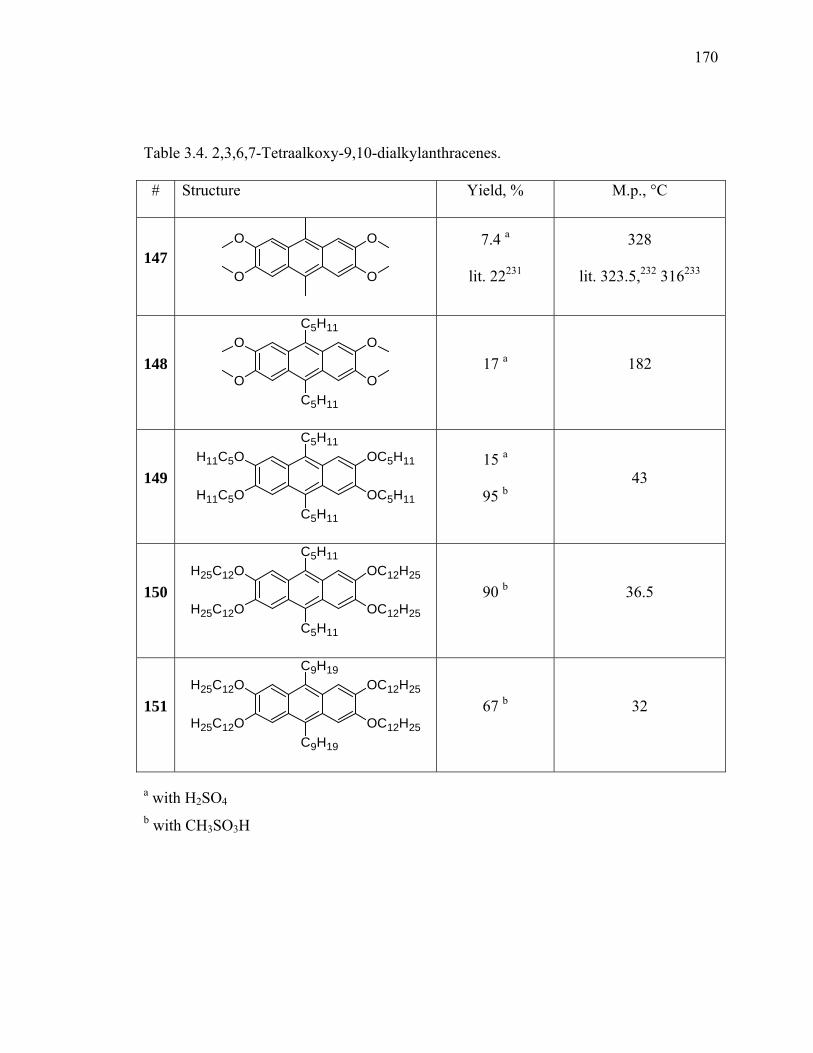
170
Table 3.4. 2,3,6,7-Tetraalkoxy-9,10-dialkylanthracenes.
# Structure Yield, % M.p., °C
147 O
O O
O
7.4 a
lit. 22231
328
lit. 323.5,232 316233
148 O
O O
O
C5H11
C5H11
17 a 182
149 H11C5O
H11C5O OC5H11
OC5H11
C5H11
C5H11
15 a
95 b 43
150 H25C12O
H25C12O OC12H25
OC12H25
C5H11
C5H11
90 b 36.5
151 H25C12O
H25C12O OC12H25
OC12H25
C9H19
C9H19
67 b 32
a with H2SO4 b with CH3SO3H
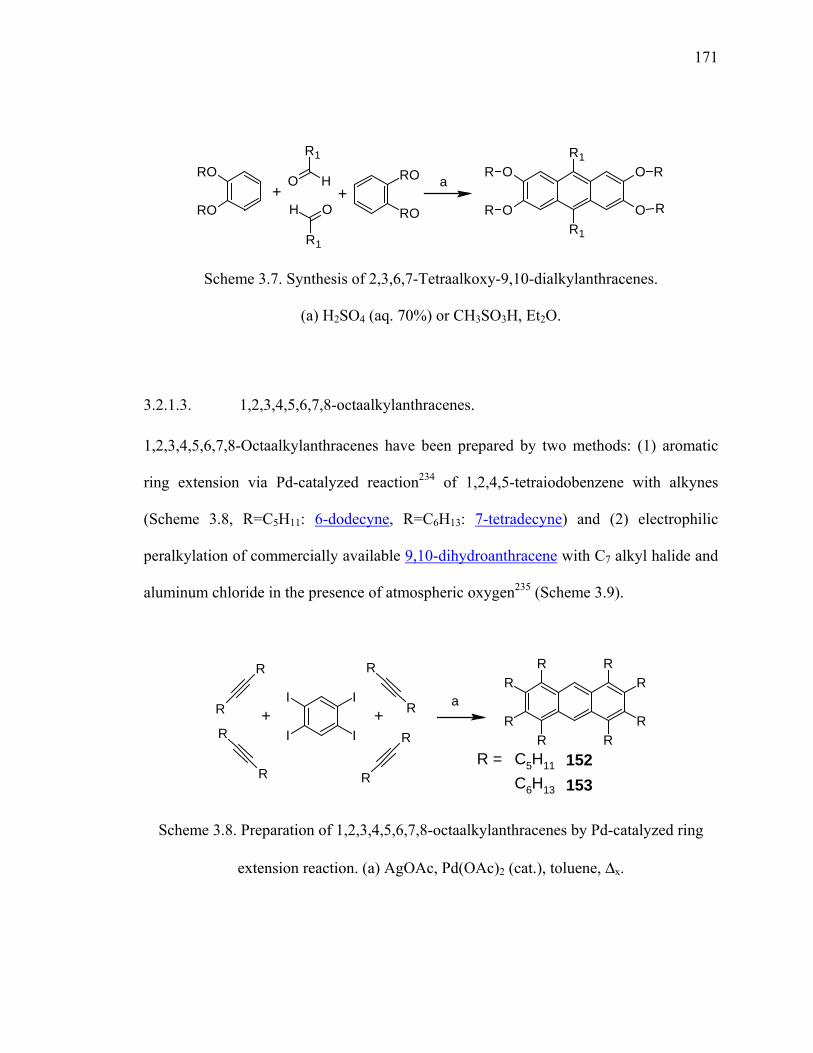
171
O
OR
R
O
O R
RR1
R1RO
RO+
R1
O H RO
RO+
R1
OH
a
Scheme 3.7. Synthesis of 2,3,6,7-Tetraalkoxy-9,10-dialkylanthracenes.
(a) H2SO4 (aq. 70%) or CH3SO3H, Et2O.
3.2.1.3. 1,2,3,4,5,6,7,8-octaalkylanthracenes.
1,2,3,4,5,6,7,8-Octaalkylanthracenes have been prepared by two methods: (1) aromatic
ring extension via Pd-catalyzed reaction234 of 1,2,4,5-tetraiodobenzene with alkynes
(Scheme 3.8, R=C5H11: 6-dodecyne, R=C6H13: 7-tetradecyne) and (2) electrophilic
peralkylation of commercially available 9,10-dihydroanthracene with C7 alkyl halide and
aluminum chloride in the presence of atmospheric oxygen235 (Scheme 3.9).
R
RR
R
RR
RR
I
I I
I
R
R
R
R
R
R
R
R+ +a
R = C5H11
C6H13
152153
Scheme 3.8. Preparation of 1,2,3,4,5,6,7,8-octaalkylanthracenes by Pd-catalyzed ring
extension reaction. (a) AgOAc, Pd(OAc)2 (cat.), toluene, Δx.

172
H15C7
H15C7C7H15
C7H15
C7H15
C7H15
C7H15
C7H15
a
154
Scheme 3.9. Peralkylation of 9,10-dihydroanthracene with heptyl bromide.
(a) C7H15Br, AlCl3, neat.
None of the anthracene compounds described here exhibited any liquid
crystalline properties.
3.2.2. Tetracenes
Although tetracene itself has not shown remarkable semiconducting properties,
it is a predecessor of pentacene (vide infra) and a catacondensed1 parent of rubrene.
Rubrene (5,6,11,12-tetraphenyltetracene) shows extraordinary mobility values of 13
cm2/V·s at room temperature 236 and 30 cm2/V·s at 200 K.237 The parent tetracene,
although commercially available as benz[b]anthracene, is expensive ($150 per gram) and
was prepared (Scheme 3.10) and thoroughly purified before single crystal growth (vide
infra). Naphthalene-1,4-quinone was reduced with either zinc dust in acetic acid or
1 Catacondensed (Greek cata: down, under, against; entirely, completely, back) aromatic hydrocarbons have all carbons on the periphery of the ring system (no interior carbons).

173
Na2S2O4 in ether/water238 to 1,4-dihydroxynaphthalene [571-60-8] 155, which was
condensed with phthalic dicarboxaldehyde in an ethanolic solution of KOH to tetracene-
5,12-dione 156. The latter quinone was reduced by Meerwein-Ponndorf-Verley
method239 with aluminum cyclohexanolate240,241 in anhydrous cyclohexanol in presence
of CCl4.242
O
O
O
O
OH
OH
a
155
O
O
OHC
OHC+
b
c
156157
Scheme 3.10. Synthesis of tetracene 157. (a) Zn, AcOH, Δx. (b) KOH, EtOH, Δx.
(c) (ChxO)3Al, ChxOH, HgCl2, CCl4, Δx.
2,3-Bis(decyloxy)tetracene was prepared likewise (Scheme 3.11): 4,5-
dimethoxyphthalic dicarboxaldehyde [43073-12-7] 158 (preparation see below) was
condensed with 1,4-dihydroxynaphthalene 155 in presence of NaOH in ethanol-THF
mixture to give 2,3-dimethyltetracene-5,12-dione 159. The latter was heated with molten
pyridinium hydrochloride at ~200 °C under an argon atmosphere to give 2,3-
dihydroxytetracene-5,12-dione 160, which was alkylated with 1-bromodecane, potassium
carbonate, and catalytic amount of potassium iodide in NMP to 2,3-bis(decyloxy)
tetracene-5,12-dione 161. This quinone was reduced with cyclohexanol and aluminum

174
cyclohexanolate into 2,3-bis(decyloxy)tetracene 162. In contrast to 2,3-
dialkoxyanthracenes, 162 is not an organogelator, is soluble in THF, CH2Cl2, CHCl3, and
can be recrystallized from ethanol. Only one crystal to isotropic phase transition and no
mesophases have been detected for 162 both on DSC trace and on the hot stage under
polarizing microscope at 130–132 °C.
OH
OH 155
CHO
CHO
O
O+
158
a
O
O
O
O159
b
O
O
OH
OH
c
O
O
O
O
H21C10
H21C10 d 160161
162
O
O
Scheme 3.11. Synthesis of 2,3-Bis(decyloxy)tetracene 162.
(a) NaOH, EtOH, THF. (b) Py·HCl melt, 200 °C. (c) C10H21Br, K2CO3, KI (cat.), NMP.
(d) (ChxO)3Al, ChxOH, HgCl2, CCl4, Δx.

175
3.2.3. Pentacenes
Pentacene 163 has attracted much attention as a semiconductor (both in single
crystalline, polycrystalline, and amorphous forms) in recent years. In fact, it has
established itself as a de-facto standard in the field of organic semiconductors, for it
consistently shows very high mobility values243, in structured films — over 1 cm2/V·s.244
163
The conductivity of linear polyacenes was predicted to grow with the number of
annealed rings.245,246 And, although the mobility of the best pentacene specimens is
already comparable with that of amorphous silicon, there are still at least two problems to
be solved: stability and solubility.
All linear polyacenes are aromatic compounds. However, their thermodynamic
and kinetic247 stability decreases, while their reactivity increases248,249,250,251 with the
number of rings.252 Naphthalene is more reactive than benzene. Anthracene is easily
oxidizable at the meso- (9,10-) position to anthraquinone, wherein two separate aromatic
rings are more thermodynamically stable than three delocalized ones. Tetracene oxidizes
above its melting point. Pentacene solutions are destroyed immediately upon contact with
atmospheric oxygen and in its absence neat pentacene can photodimerize.253 Hexacene
[258-31-1] shows similar reactivity and must be handled under an inert atmosphere.
Heptacene [258-38-8] has been long disputed by many authors to exist at all, and the
identity of the prepared samples was argued, too. Some believe it is not possible to obtain

176
this compound in pure state.254 Only derivatives of octacene [258-33-3] and nonacene
[258-36-6] are known.255
Commercial pentacene from TCI America ($325 per gram) was found by TGA
analysis (Fig. 3.7) to contain up to 34% of impurities and after two years of storage on a
shelf in a closed vial was found decomposed. Preparation of pentacene was performed
according to Scheme 3.12.
a
b
c
d
e
0
20
40
60
80
100
Wei
ght (
%)
20 120 220 320 420 520
Temperature (°C)
Tetracene TCI––––––– AS-2-14 crude pentacene––––––– Pentacene TCI––––––– AS-2-14 sublimed twice––––––– AS-2-14 sublimed once–––––––
Universal V4.0C TA Instruments
Figure 3.7. TGA Analysis of (a) tetracene from TCI America; (b) crude pentacene 163;
(c) pentacene from TCI America; pentacene, sublimed (d) once and (e) twice.
The main intermediates of the pentacene synthesis, pentacene-6,13-dione [3029-
32-1] (pentacenequinone) 164 and pentacene-5,7,12,14-tetraone [23912-79-0]
(pentacenediquinone) 166 were prepared by (a) base-catalyzed condensation of o-

177
phthalic dicarboxaldehyde with 1,4-cyclohexandione256 and (c) Friedel-Crafts acylation
of benzene with pyromellitic anhydride in an autoclave with (d) successive dehydrative
cyclization of dibenzoylterephthalic acid in boiling sulfuric acid.257 Both quinones were
then reduced with aluminum cyclohexanolate in anhydrous cyclohexanol in presence of
CCl4 and HgCl2.258
163
164
O
O
O
O
O
O166
O
O
CHO
CHO
OHC
OHC+ +
OO
O
OO
O
+ +
HOOC
OCOOH
O165
a
b
bc
d
Scheme 3.12. Synthesis of pentacene.
(a) NaOH, EtOH, Δx. (b) (ChxO)3Al, ChxOH, HgCl2, CCl4, Δx.
(c) AlCl3, autoclave, 100 °C. (d) H2SO4, 200 °C.
The quinone 164 was also prepared according to Scheme 3.13: quinizarin (1,4-
dihydroxyanthraquinone259) was reduced with sodium borohydride in methanol to 1,4-

178
anthracenequinone 167, required for other reactions (vide infra) and the latter was
reduced either under sonication with zinc dust260 in trifluoroacetic acid or with TiCl3261
into 2,3-dihydroanthracene-1,4-dione 168. Condensation262 thereof with phthalaldehyde
in methanol and pyridine afforded pentacenequinone 164 in 41% yield.
O
O
OH
OH
aO
O
O
O[81-64-1] 167 168
O
O168
OHC
OHC+
164
O
O
b
c
Scheme 3.13. Preparation of pentacenequinone 164 via 1,4-anthracenequinone 167.
(a) NaBH4, MeOH. (b) Zn, CF3COOH, sonication. (c) NaOH, EtOH, Δx.
Purification of the prepared pentacene was performed via vacuum sublimation
and single crystal vapor growth in a tube furnace with controlled temperature zones.
Sublimation of the crude product posed significant difficulties, for the crude pentacene
contains up to 40% of impurities (see Fig. 3.7, b), which in the process of sublimation
form an extremely fluffy residue, and while the sublimation per se is a very effective
method of purification (two sublimations remove ~88% of impurities), the inadvertent
transport of the fluffy residue on the bottom of a sublimator to the cooled finger may

179
devastate much of the purification effort. To increase the effectiveness of the sublimation,
we introduced the following changes into the apparatus setup and procedure:
• the crude material was loaded into the sublimer premixed with either
degreased reduced iron filings (to prevent floating of the fluffy stuff) or coarse
quartz sand (1:1 v/v);
• the layer of the above mixture was covered with a layer of degreased reduced
iron filings;
• the evacuation of the sealed sublimer was performed very slowly, with a
needle valve between the vacuum manifold and the sublimer’s outlet tube;
• the temperature was raised very slowly by means of either an oil bath or a
heating mantle;
• the outer walls of the sublimer were wrapped with a heating glass fiber ribbon
to avoid condensation of the material on parts other than the cooling finger.
The purification was followed by a TGA analysis and the sublimation was
repeated in stages until the TGA residue ceased to decrease after another purification
stage. The material thus obtained consistently gave ~1…3% residue after a TGA run,
though there was no detectable residue after sublimation (performed on neat material,
without admixtures). This observation suggested that upon prolonged heating pentacene
gradually decomposes even under inert atmosphere. Indeed, TGA analysis of the material
from the source zone of the furnace crystal grower, wherein the purified pentacene had
spent about three days, showed an increase of the non-volatile residue from 1…3% to ca.
27…30%. In addition, DSC analysis of purified pentacene in a sealed hermetic pan

180
shows an exotherm at 410 °C, immediately following by an exotherm at 411 °C, which
may be interpreted as melting with decomposition. Recently a very thorough
investigation of pentacene thermal behavior appeared, which states that neat pentacene
disproportionates at elevated temperatures.263
Pentacene crystals arrange in the crystal lattice in the order, which is often
referred to as “herringbone” packing (Fig. 3.14).264,265 This arrangement diminishes
effective face-to-face π-π interactions between the aromatic π-molecular orbitals of
pentacene molecules, thus reducing the overlap integral and decreasing the mobility of
charge carriers compared to alternative possible packing, wherein pentacene molecules
would stack upon each other more or less in columnar arrangement. To facilitate such
columnar-like arrangement in the pentacene crystal lattice, we took two approaches. First,
we prepared two pentacenes, wherein hydrogen has been replaced with fluorine: 1-
fluoropentacene 169 and 2-fluoropentacene 170. A fluorine atom is very similar to a
hydrogen atom in many respects: first of all, the size and the polarity of C–F bond. This
similarity reflects, for example, in the fact that element “hydrogen” is dot-printed in the
main subgroup of the seventh group in the Mendeleev Periodic Table of the Elements,
and there were times, when it was considered as one of the halogens only. Our intention
was to break the symmetry of the pentacene molecule by replacing one hydrogen atom by
one fluorine atom, but not to change significantly the geometrical, electronic, and other
properties of pentacene. Fluorine seemed to suit this purpose the best. In fact, after we
had completed the synthesis of the two isomeric fluoropentacenes, a paper was published
by an Anthony group, implementing similar structural modification approach: they

181
prepared halogenated pentacenes (including 1,2,3,4-tetrafluoro- and 1,2,3,4,8,9,10,11-
octafluoro-6,13-bis(2-diisopropulsilylethynyl)pentacenes) and showed that some of those
compounds switch in crystal structure from herringbone packing to face-to face
stacking.266
F
HH
H
H
H
H
H
H
H
HH
HH F
H
H
HH
H
H
H
H
H
HH
HH
169 170
The approach to 1-fluoropentacene 169 included intermediate, similar to the one
in the parent pentacene synthesis: 1-fluoropentacene-6,13-dione 173. One approach (a) to
173 was based on the strategy, depicted on Scheme 3.13, namely, we contemplated to
condense 2,3-dihydro-1,4-anthracenequinone 168 with 3-fluorophthalic aldehyde 172 and
reduce the resulting 1-fluoropentacenequinone 173. Another approach (c) to 173 was
based upon Cava’s methodology267 and reactivity of o-quinodimethanes.268,269
a b
Figure 3.8. Crystal structure of (a) pentacene270 and
(b) 1,2,3,4-tetrafluoro-6,13-bis(2-diisopropulsilylethynyl)pentacene.266

182
F169
O
O
168
OHC
OHCF
+
173
O
OF
a
172
O
O167
F174
+
bc
Scheme 3.13. Synthesis of 1-fluoropentacene 169. (a) EtOH, Py, KOH. (b) (ChxO)3Al,
ChxOH, HgCl2, CCl4, Δx. (c) DMF, Δ.
Since both 3-fluorophthalic dicarboxaldehyde 172 and 3-fluoro-o-
quinodimethane 174 retrosynthetically trace to 2,3-dimethylaniline (2,3-xylidine) 175, we
explored both approaches (Scheme 3.14). 2,3-Dimethylaniline was (a) diazotized,271 the
diazonium salt was precipitated with fluoroboric acid,272 and thermally decomposed to 1-
fluoro-2,3-dimethylbenzene 176. The latter was (b) radically dibrominated273 into the side
chains274 with N-bromosuccinimide (NBS) and benzoyl peroxide (Bz2O2) in CCl4 under
UV irradiation. The presence of two initiators (Bz2O2 and UV light) is due to the very
long induction period, which was observed (even in boiling CCl4) when only one initiator
was employed. Both 1-fluoro-2,3-dimethylbenzene 176 and 1,2-bis(bromomethyl)-3-
fluorobenzene 177 were subjected to radical tetrabromination under conditions similar to
the above, with either NBS or bromine, but these reactions has never been observed to
run to completion: an inseparable mixture of dibromo-, tribromo-, and tetrabromo- 178

183
isomers was always detected by NMR and GC. We found that this is a general outcome
for this type of reaction: “The direct bromination of two aromatic methyl groups is a very
unsatisfactory procedure. The yields are low, and in most cases a mixture of isomers of
similar properties is formed, from which the isolation of definite compounds is
difficult.”275
F
Br
BrBr
BrF
BrBr
FNH2
CHO
CHOF
172
175 176 177 178
F
OHOH
179
a b c
e d
f
F
Br
BrBr
Br F (Br)
(Br)
174
g
g
Scheme 3.14. Synthesis of the key intermediates for 1-fluoropentacene.
(a) NaNO2, HCl, 0–5 °C ; HBF4, Δ. (b) NBS, Bz2O2, hν, CCl4. (c) Br2, AIBN, hν. (d, e)
K2CO3, H2O, Aliquat 336. (f) (COCl)2, DMF, –20 °C, CH2Cl2. (g) NaI, DMF, 70 °C.
Therefore, instead of obtaining 3-fluorophthalic dicarboxaldehyde 172 by (d)
hydrolysis of 178, we resorted to (e) hydrolysis of 177 with aqueous potassium carbonate
and a phase-transfer reagent into fluorodiol 179 and oxidation thereof with a Swern
reagent276,277 into 172. Fortunately, quinodimethanes may be formed from both dibromo-
and tetrabromo-o-xylenes. The former may also be reacted with Rongalit® (sodium

184
hydroxymethanesulfinate) to form sultines, which upon heating to 80 °C are cleanly
converted into o-quinodimethanes.278,279 When 1,2-bis(bromomethyl)-3-fluorobenzene
177 was heated with 1,4-anthracenequinone 167 and sodium iodide in DMF, a dark
brown tar was formed, characteristic for the reactions involving o-quinodimethanes, from
which by a series of sublimations, 1-fluoropentacenequinone 173 was separated out in
35% yield. The same product was obtained in a near quantitative yield by a condensation
of 3-fluorophthalic dicarboxaldehyde 172 with 2,3-dihydro-1,4-anthracenequinone 168.
Reduction with cyclohexanol and aluminum cyclohexanolate afforded 1-fluoropentacene
169 in 12% yield, which after purification by sublimation dropped to 5%.
Our approach to the 2-isomer of fluoropentacene280 170 employed the
commercially available 4-fluorophthalic anhydride and did not required C–F bond
formation. The anhydride was condensed with hydroquinone in concentrated sulfuric
acid, wherein boric acid had been added to prevent side sulfonation.259 The resulting
fluoro-quinizarin 180 was reduced (cf. Scheme 3.13) with sodium borohydride in
methanol to 6-fluoroanthracene-1,4-dione 181. The latter was condensed281 with 1,3-
dihydro-2-benzofuran-1-ol282 — dihydroisobenzofuran-1-ol [81305-98-8], a tautomer of
2-hydroxymethylbenzaldeyde [55479-94-2] — in glacial acetic acid into 2-
fluoropentacenequinone 182, which was then reduced to 2-fluoropentacene 170.

185
O
OF
O
OH
O
O
O
F
OH
OH
+F
O
O
OH
OH
+O
OF
F
a b
cd
170
180 181
182
Scheme 3.15. Synthesis of 2-fluoropentacene.280 (a) H2SO4, B(OH)3.
(b) NaBH4, MeOH. (c) AcOH. (d) (ChxO)3Al, ChxOH, HgCl2, CCl4, Δx.
The crystal structure of and carrier mobility in the prepared fluoropentacenes
have yet to be determined. The compounds 169 and 170 were characterized by UV-Vis
and IR (HATR sampling) spectroscopies; TGA and DSC thermal analyses, and solid-
state, magic-angle NMR, for their solubilities in common organic solvents are very low
(enough for recording UV spectrum) and, more important, these solutions react instantly
with atmospheric oxygen, irreversibly forming peroxides. For example, a saturated
solution of 1-fluoropentacene in degassed benzene has a spectacular deep blue color,
which disappears in four minutes, when this solution, unstirred, is exposed to air.
Next, we attempted a synthesis of functionalized pentacenes with long alkoxy
chains, contemplating that such compounds could exhibit liquid crystalline properties.
Liquid crystals, containing tetracene or pentacene unit as a core, are unknown to date, to
the best of our knowledge.283 The retrosynthetic analysis of the approaches to 2,3,9,10-
tetraalkoxy- 183 and 2,3,9,10-tetraalkoxy-6,13-dialkylpentacenes 184 is shown on

186
Scheme 3.16. The grounds for the step (a) are the known aluminum cyclohexanolate
reduction of parent pentacenequinone into pentacene.258 The grounds for the step (c) are
the reported addition of phenylmagnesium bromide to pentacenequinone, which could be
reduced (b) with potassium iodide in acetic acid to 6,13-diphenylpentacene.284 Alkylation
(d) of 2,3,9,10-tetrahydroxypentacenes 184 was anticipated to go smoothly, since
alkylation of 2,3-dihydroxyanthracenequinone was reported.218 Deprotection of the
2,3,9,10-tetramethoxy-6,13-pentacenedione 188 was planned to be performed with either
BBr3 (solubility problems anticipated here, though) or pyridinium chloride in its melt.
2,3,9,10-Tetramethoxy-6,13-pentacenedione 188, the key intermediate in this approach,
was to be prepared by proven condensation262 of o-dialdehyde 158 with 1,4-
cyclohexanedione. In order to accomplish these transformations first we needed a reliable
preparation method for 4,5-dimethoxyphthalaldehyde 158.
The known approaches for 158 included: oxidation285 of 4,5-dimethoxyphthalic
alcohol,286 obtained either by reduction287 of 4,5-dimethoxyphthalide [531-88-4],
288,289,222b by oxidative cleavage of the veratrole and formaldehyde condensation trimer290;
oxidation of 4,5-dimethoxyxylylene chloride [1134-52-7]291; or from reduction/oxidation
of the esters of m-hemipinic acid.

187
O
O
O
O O
OO
O
OH
OH OH
OH
O
O O
ORR R
R
OH
OH
O
O O
O
RR R
R
R1
R1
CHO
CHO
O
O
O
O
+
O
O
O
O O
O
RR R
R
R1
R1
O
O O
O
RR R
R
183 184
185 186
187 188
158
a b
c
d
e
f
Scheme 3.16. Retrosynthetic analysis of alkoxypentacenes.
(a) (ChxO)3Al, ChxOH, HgCl2, CCl4, Δx. (b) SnCl2, HCl. (c) R1MgCl.
(d) RX, K2CO3, NMP. (e) Py·HCl, melt. (f) EtOH, NaOH.
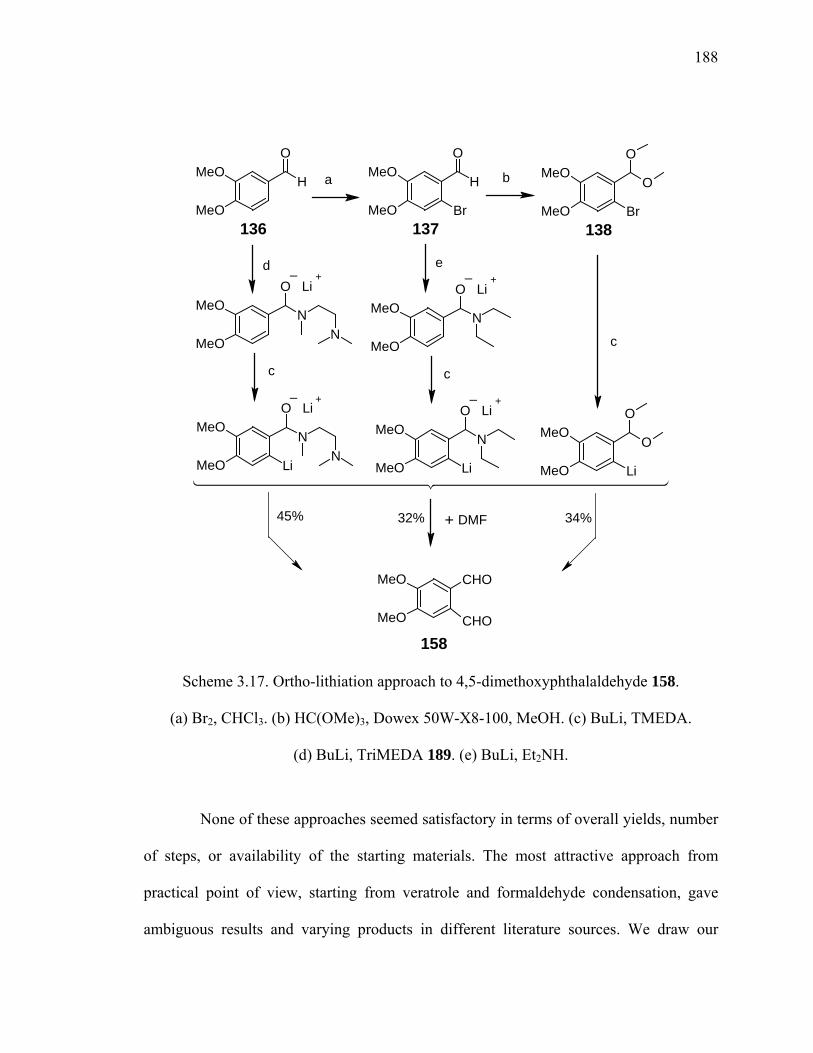
188
MeO
MeO
O
H
136
MeO
MeO
O
H
Br137
MeO
MeO
O
O
Br138
MeO
MeO
O Li
NN
– +
MeO
MeO
O Li
NN
Li
– +
MeO
MeO
O Li
N
– +
MeO
MeO
O Li
N
Li
– +
MeO
MeO
O
O
Li
+
MeO
MeO
CHO
CHO
158
a b
c c
c
d e
32% 34%45% DMF
Scheme 3.17. Ortho-lithiation approach to 4,5-dimethoxyphthalaldehyde 158.
(a) Br2, CHCl3. (b) HC(OMe)3, Dowex 50W-X8-100, MeOH. (c) BuLi, TMEDA.
(d) BuLi, TriMEDA 189. (e) BuLi, Et2NH.
None of these approaches seemed satisfactory in terms of overall yields, number
of steps, or availability of the starting materials. The most attractive approach from
practical point of view, starting from veratrole and formaldehyde condensation, gave
ambiguous results and varying products in different literature sources. We draw our

189
attention to the facts that (1) the carboxaldehyde groups in the target 158 are in ortho-
positions to each other and (2) there has been significant progress in the area of
introduction substituents into a phenyl ring specifically into an ortho-position relative to
an existing substituent. This methodology has become known as directed ortho-
metallation.292,293,294 Based on this new methodology and its application to similar
systems,295,296 we elaborated an ortho-lithiation approach to 158 (Scheme 3.17, cf.
Scheme 3.4).
Commercially available veratric aldehyde (3,4-dimethoxybenzaldehyde) 136
was (a) brominated with bromine in chloroform to 137 and then (b) protected as a
dimethyl acetal. Dioxolane protection of the aldehyde functionality with ethylene glycol
has been tried as well, but it gave much poorer results on subsequent lithiation-
formylation, since it tends to undergo fragmentation reaction (resulting in carboxylate
and ethylene) under the metallation conditions.297 The protection serves a two-fold
purpose: (1) protect the already present aldehyde functionality from the nucleophilic
attack of the organolithium reagent used for ortho-lithiation, and (2) form an ortho-
directing group. The pool of the carbonyl-derived ortho-directing groups is represented,
for example, by CONR2, CONHR, cyclohexylimines,298 α-amino alkoxides,299
CH2OAlk,300 and CH(OR)2.297,301
The protected bromoaldehyde 138 was subjected to (c) halogen-lithium
exchange with n-butyl lithium in THF and the organolithium compound was quenched
with DMF to give, after acidic work-up, 4,5-dimethoxyphthalic aldehyde 158. To avoid
the protection of the aldehyde as an extra step in this synthesis, we resorted to (e) an in

190
situ protection by a lithium diethylamide, preformed from Et2NH and n-BuLi, with
almost no decrease in the overall yield. Then it was realized that the bromination step
could also be omitted if the amine we use for in situ protection of carbonyl, will be
modified to act as an ortho-director and butyl lithium activator in the direct lithiation
step. It is known that N,N,N′,N′-tetramethylethylenediamine (TMEDA) greatly
accelerates and facilitates lithiation reactions due to intermolecular complexation with
butyl lithium via coordination of lithium with the two nitrogens, polarization of C–Li
bond, and forming a monomeric butyl lithium species.302
Thus, (d) N,N,N′-trimethylethylenediamine (TriMEDA) 189 was prepared by
alkylation303 of aqueous methylamine solution with N-(2-chloroethyl)dimethylamine
hydrochloride, prepared in turn from 2-(dimethylamino)ethanol and thionyl chloride.304
The adduct of its lithium salt to carboxaldehyde has been shown to be both ortho-director
and lithiation activator.305 By employing tetrahydropyran (THP) instead of THF as a
solvent more stable to n-BuLi action,305,306 we attained 45% yield of 158 in a one-pot
reaction from commercially available starting material.
We also tried several other approaches to aromatic o-dialdehydes, tested in the
preparation of the parent phthalaldehyde: oxidation of side chains with either CrO3 in
presence of acetic aldehyde307,308 or chromyl chloride309 (A. L. Étard reaction), and
recently reported oxidative decarboxylation of 1,2-phenylenediacetic acid with solid
KMnO4.310 All these approaches gave less than 16% yields. Later another, more
convenient route was elaborated,311 based on Blanc bis(chloromethylation) of
veratrole.312,313 Hydrolysis of 1,2-bis(chloromethyl)-4,5-dimethoxybenzene with

191
potassium carbonate and subsequent oxidation287 of the resulting diol with freshly
prepared MnO2 provided a better access to 158.
MeO
MeO
a MeO
MeO
CH2Cl
CH2Cl
MeO
MeO
CH2OH
CH2OH
MeO
MeO
CHO
CHO158
b c
Scheme 3.18. Three-step synthesis of 4,5-dimethoxyphthalic aldehyde 158 from
veratrole. (a) CH2O, HCl (conc. aq.), HCl (gas), dioxane. (b) K2CO3, H2O. (c) MnO2.
After a reliable approach to 158 was established and preparative quantities (ca.
10 g) thereof were prepared, the synthesis of 2,3,9,10-tetramethoxypentacene-6,13-
dione314 188 and 2,3,9,10-tetramethoxypentacene 189 has been performed as depicted in
Scheme 3.19. The solubility of 4,5-dimethoxyphthalic aldehyde 158 in ethanol, even at
the boiling point, was much lower compared to the parent phthalaldehyde, and use of
THF as co-solvent was necessary in the condensation step. Reduction of the thus obtained
tetramethoxypentacenequinone 188 with aluminum cyclohexanolate gave hitherto
unknown 2,3,9,10-tetramethoxypentacene 189 — crystalline compound of dark ruby-red
color, only very sparingly soluble in benzene. Its solutions are destroyed upon contact
with atmospheric oxygen within several minutes. It was purified for this reason by
sublimation in high vacuum and characterized by DSC in a sealed hermetic pan (m.p. 413
°C dec.) and by solid-state magic angle NMR.

192
O
O
O
O O
OCHO
CHO
O
O
O
O
+a
188
O
O O
O
189
b
Scheme 3.19. Synthesis of 2,3,9,10-tetramethoxypentacene.
(a) NaOH, EtOH, THF. (b) (ChxO)3Al, ChxOH, HgCl2, CCl4, Δx.
Quite recently Anthony315 prepared a series of pentacenes, structurally similar to
2,3,9,10-tetramethoxypentacene (Scheme 3.20). Their main distinctive feature is
substitution in the meso 6,13-position, which makes them more stable in solution.
Removal of the methyl groups from 2,3,9,10-tetramethoxypentacene-6,13-dione
188 by reaction with molten pyridinium hydrochloride gave 2,3,9,10-
tetrahydroxypentacene-6,13-dione 187, which was successfully alkylated with 1-
iodohexane and potassium carbonate in NMP to provide 2,3,9,10-tetrahexylpentacene-
6,13-dione 185 (Scheme 3.21). The latter compound did not exhibit mesogenic properties
and had m.p. 190–194 °C (microscope, hot stage).

193
CHO
CHO
OZ
O
O
O
+a
O
O
OZ
O
OZ
O
bLi(i-Pr)3Si
OH
OH
OZ
O
OZ
O
Si(i-Pr)3
(i-Pr)3Si
OZ
O
OZ
O
Si(i-Pr)3
Si(i-Pr)3
c
Z = CH2,
Scheme 3.20. Soluble and stable pentacene ethers by Anthony.315
(a) KOH, EtOH. (b) (i-Pr)3Si–C≡C–Li, THF. (c) SnCl2, THF, MeCN.
O
O
O
O O
O
188
O
O
OH
OH OH
OH
187
a
bO
O
O
O O
O
RR R
R
185R = C6H13
O
O O
O
RR R
R
c
183R = C6H13
O
O O
O
RR R
RO O
183a
O2
Scheme 3.21. Attempted synthesis of 2,3,9,10-tetrahexyloxypentacene 183.
(a) Py·HCl, melt. (b) n-C6H13I, K2CO3, NMP, 180 °C. (c) (ChxO)3Al.

194
The attempted reduction of the 2,3,9,10-tetrahexyloxypentacene-6,13-dione 185
did not succeed, however. We believe that the reduction reaction per se was successful,
for (1) we observed a distinct color change of the reaction mixture from yellow,
characteristic of pentacenequinone 185 to deep cherry-red, characteristic of
tetraalkoxypentacene 183, and (2) complete consumption of the starting material by TLC.
Upon working up the reaction mixture with water, a brick-red precipitate was obtained.
Every attempt of purification thereof by chromatography was unsuccessful, however, for
even traces of oxygen caused an almost immediate change of color from cherry red to
gray-brown, presumably because of formation of peroxo-compound 183a, similar to the
case of pentacene.316,317,318 Small-scale sublimation did not yield any material on the
cooling finger either. The possible solutions for this problem would be (1) handling and
purification in a glove box, under complete exclusion of oxygen, (2) same-pot
preparation of Diels-Alder adduct, which shall be purified in solution and exposed to
deprotection under an inert atmosphere later. An example of the second approach has
been published by Afzali.319,320
+N
O
S O
NS O
O
120–200 °C
a
Scheme 3.22. Reversible Diels-Alder adduct of pentacene.
(a) MeReO3, CHCl3, Δx.319

195
3.3. Iodoarenes
3.3.1. Why Iodine?
In general, charge mobility due to a hopping mechanism321,322 depends, amongst
other factors, upon the overlap integral of the electronic wavefunctions. For example, for
a small polaron hopping model the mobility expression goes as:323
e kTE
a
a
kTEkT
Jea −
=
π
μ4
22
h
,
where e is the carrier’s charge, a is the hopping distance, h is reduced Plank’s constant, k
is Boltzmann constant, T is temperature, Ea is activation energy, and J is an overlap
integral: baba dVJ ψψψψ == ∫ * . Polynuclear acenes have high mobilities due to
overlap of their π-orbitals, which significantly increases in case of π-stacking.243,324 Other
types of orbitals may contribute to the overlap integral, too. In order for this contribution
to be significant, the orbitals should be bulky (diffuse in space), just like the π-orbitals.
The p-orbitals of iodine seem to suite this purpose well: iodine’s van-der-Waals radius is
215 pm (picometers).325,326 For example, the charge mobility in zone-refined 1,4-
diiodobenzene (DIB) single crystals has been reported as high as 12, 4, and 1.7 cm2/V·sec
in the a, b, and c directions of the orthorombic unit cell,327 which is an order of magnitude
higher than the highest mobility values for pentacene reported so far.243,244

196
The computer-aided density functional theory calculations for a series of iodine-
containing low molecular weight aromatic organic compounds, made by Prof. Ellman,328
revealed, for the case of 1,4-diiodobenzene, that (1) the shortest iodine-iodine and
carbon-iodine distances (1.47 and 1.9 Å) between two nearest DIB molecules is less than
the sum of two iodines’ (2×1.4 Å) and iodine–carbon (1.4 + 0.7 Å) Slater radii. This
suggests the possible principle role bulky electronic orbitals of iodine may play in the
electronic properties of the DIB crystal. Fig. 3.10 shows the intermolecular overlap of the
iodine’s p-orbitals in red color. (2) Iodine’s contribution to the HOMO molecular orbital
of DIB is the largest.
Figure 3.9. Interatomic distances in α-DIB unit cell.

197
Figure 3.10. Intermolecular iodines’ p-orbital overlap in crystalline α-DIB.329
Therefore we purified some of commercially available iodoaromatic
compounds, prepared several of them, and elaborated reliable purification protocols for
their use as organic single-crystal semiconductors.
The pool of preparative methods for introduction of iodine atoms into aromatic
molecules includes274 iodo-de-diazoniation1 (Sandmeyer reaction of aryldiazonium salt
with iodide anion), halogen exchange,330,331,332 iodo-de-metallation333 (of electrophilic
metalloorganic species: e.g. reaction of Grignard, aryl lithium, aryl thallium,334 aryl
nickel,335,336 or aryl mercury,337 reagents with free iodine), and various direct iodination
1 The organic reaction naming convention, elaborated by Jerry March takes the group
being introduced (iodine), and separates it by reaction type (overall formal replacement,
thus ‘-de-’) from the group being replaced (diazo group in this case).

198
methods338 (electrophilic attack of «I+» species339 onto the aromatic system), including
electrochemical iodination.340,341
3.3.2. Direct iodination
Direct iodination of aromatic compounds is electrophilic aromatic substitution of
hydrogen by «I+» species, usually prepared in situ from I–/I –3O , I–/I –
4O , I2/HNO3/H2SO4,
or other iodine-containing reagents. We used the above combinations of inorganic iodine
salts, as well as tetraiodoglycoluril, benzyltrimethylammonium dichloroiodate, and
bis(pyridine)iodonium (I) tetrafluoroborate, prepared according to the literature
procedures as described below.
Tetrahydroimidazo[4,5-d]imidazole-2,5-dione 190 was prepared from glyoxal
and urea,342,343 brominated to 191,344 and the resulting tetrabromide 191 was converted345
into 2,4,6,8-tetraiodoglycoluril346 (I4Glu) 192 and used for iodination in 90% sulfuric347
or trifluoroacetic346 acid media.
NH NH
NH NH
O
O
NH2 NH2
O
CHO
CHO+
a b N N
N N
O
O
BrBr
Br Br
c N N
N N
O
O
II
I I
190 191 192
Scheme 3.23. Preparation of 2,4,6,8-tetraiodoglycoluril.345
(a) H2O, HCl, pH=1.5…2.0. (b) H2O, Br2, pH=9…10. (c) I2, Ac2O.

199
Benzyltrimethylammonium dichloroiodate (BnMe3NICl2) was prepared from
benzyltrimethylammonium chloride and iodine monochloride.348 Bis(pyridine)iodonium
(I) tetrafluoroborate synthesis started from preparation of mercury (II) oxide –
tetrafluoroboric acid impregnation on silica gel.349 This impregnation HgO–HBF4/SiO2
was treated with iodine in dichloromethane to yield Py2I+B –4F ,350 which was used for
iodination with triflic acid.351 Both reagents are yellow crystalline solids and may be
stored, after recrystallization, in pure form on the shelf for at least several months without
decomposition.
The following iodoarenes (Scheme 3.24) have been prepared by the direct
iodination methods (the reagents used are specified in parentheses): diiododurene352,353
193 (I4Glu/H2SO4/dioxane; Py2I+B –4F , KIO4/I2/H2SO4; HIO4·2H2O/I2/H2SO4
354),
iodopentamethylbenzene355,356 [64349-91-3] 194 (KIO4/I2/H2SO4; HIO4·2H2O/I2/H2SO4),
1,2,4,5-tetraiodo-3,6-dimethylbenzene357 [27059-93-4] 195 (HIO4·2H2O/I2/H2SO4),
1,2,4,5-tetraiodobenzene358,359 196 (HIO4·2H2O/I2/H2SO4), hexaiodobenzene358,360 197,
2,6-diiodo-4-methylaniline 198 (I2/NaHCO3; ICl; BnMe3NICl2), 2-iodo-4-
methylacetanilide 199 (ICl; HIO4·2H2O/I2/AcOH/H2O; BnMe3NICl2).

200
INH2
I INH
O
I
II
II
I
I
II
I
I I
II
I
II
II
I
I
193 194 195 196
197 198 199
Scheme 3.24. Iodoarenes prepared by direct iodination.
Noteworthy, hexaiodobenzene has been extensively studied in the last few years
and has been shown to change its crystal361 and molecular362 structure under high
pressure, shows insulator-to metal transition and becomes a superconductor at 35GPa and
2 K. Some authors363 present evidence that the increase in conductivity is mainly due to
enhanced charge transfer interaction generated by the intermolecular overlap of a 5pz
orbital of I and a p-orbital of C, while others364 argue the significance of the overlap
integral increase in superconductivity of C6I6, though support this mechanism for iodanil
(that is, iodanil's conductivity does improve from the overlap integral increase, while
cause of C6I6 metallization is different).
3.3.3. Iodo-de-diazoniation
The Sandmeyer reaction has been the most widely used method for preparation
of iodoarenes, for, in fact, it was the only main general method known to organic

201
chemists up until the 1950s.338 According to this method, still widely employed, an
aromatic amine is diazotized into a diazonium salt, which is then subjected to solution of
iodide anion, resulting in nitrogen evolution and overall displacement of diazo-group
with an iodine atom.274 Modern modifications of the Sandmeyer reaction have been
elaborated as well.365 We employed the classical version of Sandmeyer reaction for the
synthesis of 3,5-diiodotoluene366,367 [49617-79-0] 200, 3,4,5-triiodotoluene366,367,368
[89677-87-2] 201, and 3,4-diiodotoluene366,369 202 (Scheme 3.25).
INH2
I
198
a
IN2
+I
b
II
I
201c
II
200
NH2
II
INH
O 199
d, e
BnMe3NICl2
a, b
202
Scheme 3.25. Iodo-de-diazoniation approach to some iodotoluenes.
(a) NaNO2, H2SO4, 0…5 °C. (b) KI, H2O. (c) H2O, r.t. (d) Ac2O, AcOH, Δx.
(e) HIO4·2H2O/I2/AcOH/H2O.

202
3.3.4. Halogen exchange
Direct iodination of many substrates is either difficult to accomplish, or it gives
mixtures or undesired isomer(s). For example, we needed an easy access to highly pure
1,4-diiodonaphthalene370 [36316-83-3] 203 and tried to prepare it by direct
electrochemical iodination of naphthalene,340,341 but only incomplete reaction and a
mixture of various isomers of different degree of iodination has been obtained. The only
reported attempt on direct iodination of 1,5-dialkoxynaphthalene was also negative.371 On
the contrary, highly selective bromination of naphthalene has been recently reported.372
Therefore, 1,4-diiodonaphthalene has been prepared by halogen-exchange reactions.
There are several variants of such reaction, involving formation of various organometallic
intermediate species of Li, Cu, Ni, Tl and Hg. We explored the lithium, copper, and
nickel-mediated reactions (Scheme 3.26). Nickel-mediated halogen exchange of 1,4-
dibromonapthalene gave unsatisfactory results to make 1,4-diiodonaphthalene, both in
yield and purity. Copper-mediated variant worked well in terms of yield both with and
without HMPA, originally reported as solvent. However, the conversion of 93% (after
one run) left us with a mixture of dibromo and diiodonaphthalenes, which we failed to
separate efficiently. Use of excess of KI and repeated re-subjection of the separated
products to the same reaction conditions improved the purity of the product via increase
of the conversion rate, but 1-bromo-4-iodonaphthalene was always still detectable by
GC-MS as a major (3% after first run) impurity even after two subsequent runs. Thus, we
resorted to lithium-bromine exchange reaction with subsequent quenching of the dilithio-
derivative with iodine.371 The use of tert-butyl lithium373 was advantageous over n-BuLi,

203
for the metal-halogen exchange proceeds irreversibly (with iso-butylene escaping as gas)
and no similar by-products (like 1-bromo-4-iodonaphthalene) are formed. The main by-
product detected in tiny amount by GC-MS was 1-iodonapthalene, which was easily
separable by repeated recrystallization.
Br
Br
a
I
I
Li
Li
I
I
Br
I
I
+
+
b
f
or c
d or e
202
Scheme 3.26. Routes to 1,4-diiodonaphthalene.
(a) Br2, CH2Cl2, –30 °C. (b) KI, Ni powder, DMF, Δx. (c) KI, CuI, DMAc (HMPA),
160…180 °C. (d) n-BuLi, Et2O, –20 °C. (e) tert-BuLi, Et2O, –20 °C. (f) I2.
A positional isomer of 202, 2,3-diiodonaphthalene374,375 [27715-43-1] 203 has
been prepared from 2,3-dihydroxynaphthalene (Scheme 3.27) via a Bucherer reaction,376
followed by diazotation of 2,3-diaminonaphthalene and Sandmeyer reaction. The low
yield (18%) of the Sandmeyer reaction may be excused perhaps due to the ortho-
relationship of the substituents, which make the intermediate bis(diazo)cation presumably
less stable and prone to decomposition.

204
OH
OH
a NH2
NH2
c I
I203
N2+
N2+
b
Scheme 3.27. Preparation of 2,3-diiodonaphthalene 203.
(a) NH3·H2O aq. 28%, NaHSO3, 140…160 °C, pressure vessel.
(b) NaNO2, H2SO4, 0…5 °C. (c) KI, H2O.
The prepared iodoarenes have been thoroughly purified by repeated preparative
column chromatography, series of careful recrystallizations and zone refinings, and single
crystals of some of them were grown either by vapor furnace or Bridgeman methods. The
single crystals obtained are currently being studied for charge mobility in the laboratory
of Professor Brett Ellman.
3.4. Liquid Crystal Semiconductors
3.4.1. HAT Discotic Liquid Crystals
2,3,6,7,10,11-Hexaalkyloxytriphenylenes (HAT-n) constitute the single most
important subclass in the realm of discotic liquid crystals, largely because of their
applications as quasi-one-dimensional conductors377 and photoconductors.378 In fact, one
compound from this family, 2,3,6,7,10,11-hexapentyloxytriphenylene (HAT-5) [69079-
52-3] 205 has established itself as a de-facto media standard for study of charge mobility
in columnar liquid crystals. The chart on Fig. 3.18 shows total number of publications

205
(registered in CAPLUS database as per March of 2006) vs. alkyl chain length of HAT-n
compounds. Note that the first three members of this homolog series are not liquid
crystals, and the first member has been known for years (from 1965379) and deserved its
attention as the series’ progenitor.
2 4 6 8 10 12 14 16 18 20
0
20
40
60
80
100
120
140
Num
ber o
f Pub
licat
ions
Alkyl chain length
O
O
O
O
O
O
CnH2n+1
CnH2n+1
CnH2n+1
CnH2n+1
H2n+1Cn
H2n+1Cn
Figure 3.11. Significance of various HAT-n compounds represented as number of
publications for each member of the homologous series.
Despite being the de-facto research standard, HAT5 (and HAT6) compounds are
still not commercially available, one of the possible reasons being the very high degree of
purity required for them to be useful, at least as semiconductor applications are

206
concerned. Generally, symmetrical HAT-n compounds are prepared (Scheme 3.28) by
oxidative trimerization of 1,2-dialkoxybenzene with chloranil in sulfuric acid,380 FeCl3,381
FeCl3/Al2O3,382 MoCl5,383 VOCl3,384,385 and electrochemically.386
O
O
O
O
O
O
a
OO
OO
O
O
+6e– + 6H+
E0
205204
Scheme 3.28. Oxidative trimerization of 1,2-dialkoxybenzene to HAT-n.
(a) VOCl3, CH2Cl2.
Unfortunately, use of any reagents listed above and any conditions in this
trimerization reaction leads to formation of at least two by-products, in amounts
minimum 3%: mono-hydroxy-pentaalkoxytriphenylene 206 and α-chloro-hexaalkoxytri-
phenylene 207 (Scheme 3.29).385 The amounts of these undesired by-products might be
kept at minimum if strictly anhydrous conditions are employed, none or only catalytic
(0.3%)381 amount of sulfuric acid used (necessary only when FeCl3 is used as oxidant),
dichloromethane used as solvent, for no reaction happens in THF, MeCN or AcOH,385

207
and the reaction is quenched with anhydrous methanol before addition of water to
perform the work-up.
RO
OR
OR
OR
RO
OH
RO
OR
OR
OR
RO
OR
Cl
206 207
Scheme 3.29. Main by-products of HAT-n synthesis by oxidative trimerization.385
After reviewing the available literature on the mechanism of this trimerization,
we came to a conclusion that formation of these by-products cannot be avoided by the
virtue of the reaction and the nature of the compounds formed. The standard redox
potential E0 for 1,2-dipentyloxybenzene (M) oxidation to triphenylene (T) is 1.05 V,387
while E0 of the next step, oxidation of triphenylene (T) to radical-cation (T+•) is 1.0 V.388
The increase in the anodic potential has been shown to give higher charged species,
dications-diradicals T++•• (ΔE01-2 = 300 mV) and trication-radicals T+++• (ΔE0
2-3 = 450
mV)389 and even tetracations (E01-4 = 2.27 V), if electrolytic reaction is conducted in
CH2Cl2/CF3COOH, which stabilizes these species.388 All these cations are highly
susceptible to attack by nucleophiles and at temperatures above –70 °C polymerize very
quickly. The formation of dication-diradical T++•• is irreversible above –40 °C.388 A
couple of other side reactions have been observed as well.390 The electrochemical
potentials of all three oxidants traditionally utilized in HAT-n preparation (FeCl3, MoCl5,

208
VOCl3) are high enough to cause the trimerization of dialkoxybenzene, therefore they
will inevitably cause side reactions and form by-products. This is also why all attempts to
conduct the preparative trimerization of dialkoxybenzene electrochemically have been
inferior to chemical oxidants-employing reactions.391 Therefore, we aimed to elaborate a
reliable protocol for effective purification of HAT5 prepared with any of the known
methods.
We tried all three reported oxidants and found that FeCl3 gives crude product of
the poorest quality, while MoCl5, if employed at ambient temperature, gives crude
product of the best quality. The high cost of anhydrous molybdenum (V) chloride,
however, made vanadyl chloride our reagent of choice.
After having tried many standard purification techniques and combinations
thereof, including those developed by other researchers,392 we arrived at the following
purification protocol. Firstly, neutral alumina (as opposed to usually employed silica gel)
is activated at 450–500 °C under argon atmosphere for 12 hrs, cooled under argon and
transferred into a tightly closed container. Second, commercial activated charcoal is
subjected to a two-stage activation procedure: (a) under ambient atmosphere at 550 °C
for 4 hrs, followed by (b) twelve hours at 700 °C under argon atmosphere, then cooled
under argon stream to room temperature and sieved. The fraction 16–35 mesh is
collected. Thirdly, the crude HAT-5 is impregnated onto (five times its weight) activated
neutral alumina and left in a thinly distributed layer open to air, but in the dark392 for 12
hrs. This process can be shortened by air suction through the bed of HAT-5 impregnated
alumina. The impregnation changes color from white to yellow to brown.

209
A flash suction column with a sintered coarse glass at the bottom is then packed
in four layers (from the bottom): (1) a 3…4 mm layer of Celite® 545 (to retain fine
charcoal particles and avoid quick clogging); (2) 150…170 mm of activated alumina; (3)
30…35 mm of activated carbon; (4) aged HAT-5 alumina impregnation. The solvents
(hexanes and dichloromethane) must be distilled or of residue-free grade. Gradient
elution from neat hexane to 35% dichloromethane in hexane yields, after evaporation of
solvents, snow-white crystalline HAT-5, which remains snow-white and does not change
color (to pink or purple) upon shelf storage.
Samples of HAT-5 solutions in dichloromethane, chloroform, 1-propanol, iso-
octane and mixtures thereof intentionally left exposed to open air and especially to direct
sunlight turned yellowish in several weeks. Therefore, all recrystallizations of HAT-5
after the chromatographic purifications (a series of three chromatographies usually
suffices if conducted as described above) in (distilled and micron-filtered) 1-propanol and
chloroform mixtures were conducted under inert atmosphere and in the dark.
3.4.2. Nitrated HAT-5 Discotic Liquid Crystals
The chemical structure of HAT-n liquid crystals has received quite a few
modification attempts in the search for wider mesophase, increase of dipole moment,
change of molecular symmetry, etc. Apart from unsymmetrical HAT-n,382 several post-
derivatizations of trimerized HAT-5 have been reported: nitration to mono-nitro-HAT-n

210
(MN-HAT-n),393 selective mono-dealkoxylation to mono-hydroxy-penta-
alkoxytriphenylene, and bromination.394 We attempted synthesis of di- and tri-nitro
substituted HAT-5. Only 1,5,9-trinitro-2,3,6,7,10,11-hexapentyloxytriphenylene (TN-
HAT-5) was isolated in pure form and characterized, while two isomeric dinitro-HAT-5
compounds have been detected by HPLC-MS only and have not been individually
isolated due to difficulties in their separation and purification.
RO
OR
OR
OR
RO
OR
RO
OR
OR
OR
RO
ORNO2
RO
OR
OR
OR
RO
ORNO2
NO2
O2N
a b
Scheme 3.30. Synthesis of MN-HAT-5 and TN-HAT-5. (a) 1 eq. HNO3, CH2Cl2,
CH3NO2, room temperature. (b) 3.2 eq. HNO3/Al2O3, CH3NO2, –20 to room temperature.
Both mono- and trinitro-HAT-5 isomers exhibit columnar liquid crystalline
properties: MN-HAT-5: Cr–Colh –37°C, Colh–I 139°C; TN-HAT-5: Cr–Colh 34°C, Colh–
I 142°C. Both compounds are being measured for charge transport properties in their
liquid crystal phases in the laboratory of Prof. Brett Ellman.

211
3.4.3. Conclusions
We have attempted preparation of polyacene compounds, aimed to exhibit liquid
crystalline properties. None of the synthesized tail-equipped anthracenes, anthraquinones,
and pentacenequinones showed expected mesogenic behavior.
Iodine-containing arenes we prepared and purified are to be employed in further
study, pertaining to measurements of their mobilities.
A reliable purification protocol for HAT-5 discotic semiconductor has been
elaborated.

212
Chapter IV.
Experimental Part
4.1. General Instrumentation and Techniques
Measurement of Fluorescence Quantum Yield
There are several approaches to measure fluorescence quantum yield.41,395 The
most simple and widely employed method is a secondary (or relative) method, elaborated
originally by Parker and Rees.396 This method is based on comparing the quantum yield
of a sample to that of a standard at the same conditions.397 Ideally, the standard and
sample should have absorption and emission spectra matched (overlapped) as close as
possible and quantum yield of standard should be of approximately same value as that of
sample.398
The standard’s emission spectrum and quantum yield ideally should be
independent of excitation wavelength, temperature, and concentration. The standard
should be stable in solution, be easily purified or commercially available in pure form,
and ideally be quenched with oxygen as little as possible. There are very few compounds
that fulfill all these requirements. For example, fluorescein’s fluorescence is easily
quenched by oxygen and photo bleached, and its emission spectrum depends on pH of the

213
solution. Rhodamine 6G’s quantum yield is temperature dependent.399,400 Thus, the
choice of standard is the most crucial step in the whole technique of relative quantum
yield measurement. Some good standards are Rhodamine 101,401 perylene, perylene
tetracarboxylates – esters and salts,402 pyrene. Good sources of information on common
fluorophore’s properties are http://fluorophores.org and Photochem CAD Database. After
a standard has been chosen, the following is the sequence:
• The solvents should be of spectrophotometric grade with no inhibitors and as low
cut-off as possible. Chlorinated, nitrated and other heavy-elements containing solvents
will generally quench fluorescence.
• Deoxygenate the solvent by passing a stream of argon or helium into it and/or
sonicate it (in a special degas mode of the sonicator) at the same time.
• Weigh out precisely 0.1–0.5 mg of sample (i.e. both sample and standard) on
microbalances (weighing is optional and can be used for simultaneous extinction
coefficient determination). Dissolve the sample in 10.00 ml of deoxygenated solvent of
choice and perform a series of dilutions to bring the solution’s absorbance below 0.05.
Generally (with compounds having ε of order 105 l·mol–1·cm–1), it means to dilute to
some micromolar concentration range ~10–5 M = 1-10 μM.
• Take the UV-Vis spectrum and note the optical density A at the wavelength, which
will be used for excitation (λEx). Note: the samples at micromolar concentrations might
have very low absorbances at wavelengths other than λmax. When taking absorbances of
dilute samples at such wavelengths, big errors are possible. Thus, it is better to measure,
for example, 10× times more concentrated solution (A of penultimate solution, i.e. of that

214
in the dilution series, from which the solution for fluorescence measurement is prepared),
taking the optical density (which is taken most accurately within 0.1–0.45 range) at the
necessary wavelength and calculating the optical density of the final solution by dividing
the obtained number by the dilution factor.1
• Ideally, the dilutions should give you such solutions that the absorbances of the
standard and the sample solutions should be exactly the same at the excitation
wavelength. At any rate, they should be very close to each other and less that 0.05.
• Record fluorescence spectra of the standard and of the sample solutions as soon as
possible after each other and after the absorbance measurements and at EXACTLY the
same conditions. Note the conditions – temperature, λEx, PMT voltage/gain, excitation
monochromator and detector slits. Recording fluorescence of blank solvent(s) to ensure
clear fluorescence background is recommended.
• Chose an excitation wavelength ~20 nm hypsochromic (blue-shifted) off the λmax
to get most of the emission spectrum. Start collection of emission at least 10 nm (depends
on detector slit) bathochromic (red-shifted) from λEx.
• Keep in mind scattering signal at 2λEx, which should be excluded from integration
or be the same for standard and sample (that is, not absorbed by both). Keep fluorescence
1 “…and there is difficulty in determining the low absorbances. This latter problem is
generally overcome by accurately diluting more concentrated solutions of measured
absorbances and/or using long-path-length cells.”397

215
intensity within specification range of the fluorimeter by adjusting concentration and/or
PMT voltage/gain. Integrate both sample and standard fluorescence spectra.
• Plug the values obtained into the equation (4.1):
( )( )
( )( )Ex
2
2
F λλ
λλ
λλA
A
dF
dF
nn Exref
FFref
FF
refrefF ××Φ=Φ
∫∫ , (4.1)
which is an approximation of more precise equations:
( )( )
( )
( )
( )( )
( )[ ]( )[ ] ,
10lnexp110lnexp1
101101
Ex
Ex2
2
2
2
Ex
λλ
λλ
λλ
λλ
λλλ
λ
AA
dF
dF
nn
ordF
dF
nn
ref
FFref
FF
refrefFF
A
A
FFref
FF
refrefFF
Exref
−−−−
××Φ=Φ
−−××Φ=Φ
∫∫∫∫
−
−
(4.2)
where ΦF is quantum yield, n – refractive index of solvent, A(λEx) – absorbance at the
excitation wavelength, and F(λF) – fluorescence spectrum trace to be integrated. Note that
optical density of the standard goes into nominator, while all other values of the standard
— into denominator.
Measurement of Fluorophore’s Photostability
The photobleaching quantum yield φ b is the slope of the least square fit line
relating the photobleaching rate of a sample to the excitation rate of the laser.403 The
bleaching curves should be recorded at several excitation intensities (that is, five or more
fluxes of the excitation light) and fit to an exponential decay functions (see Fig. 4.1).
Often, two exponents and an offset are required to obtain a good fit for all samples and

216
intensities. Using the exponential fit, the bleaching rate is calculated from the initial (t=0)
slope, i.e. from the fastest exponential.404
-2 0 2 4 6 8 10 12 14 16 18 20 22 240.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
y = A + B*xR^2 = 0.99409A = 1.33525 ± 0.0089B = -0.08765 ± 0.00138
y = a*exp(b*x)R^2 = 0.98043a = 1.40277 ± 0.02219b = -0.1116 ± 0.00303
RT-3-188
Exposure, hrs
Abs
orba
nce
Figure 4.1. Photodegradation of compound 93 and its exponential fit.
Although it is possible to control the light intensity by varying the distance from
a conventional light source (I ~ d–3), it is a suitable mean for only decreasing the flux.
Moreover, in order to see an exponential photobleaching rate, a source of very strong
light intensity is required, i.e. a laser.404 Since we did not have a laser source with a
tunable intensity, we were able to perform measurements of photobleaching rates for our
compounds at a single and low excitation intensity from a conventional UV lamp.

217
The measurements have been performed on chloroform solutions in a standard
1 cm quartz cuvette, illuminated at λ = 360 nm by a UVP 8W UV lamp model 3UV-38,
distanced from the cuvette by 4 cm. The intensity of UV radiation was measured with an
International Light photometer model IL1400 to be 1.2 mW/cm2. For most of the samples
we observed a linear decay in fluorescence (or absorbance, since the photobleached dyes
are colorless — see Fig. 1.15, 1.16). The tangence of the slope of the least square fit gave
the following photobleaching rates at the specified flux: 32 (–0.03059), 33 (–0.17591), 64
(–0.0272), 65 (–0.0574), 91 (–0.01682), 92 (–0.05223), 93 (–0.08765).
Gas Chromatography – Mass Spectroscopy (GC-MS)
All GC-MS runs have been performed on a Thermo Electron Trace GC 2000,
coupled to a Polaris Q ion trap MS. The injection of a series of samples has been
performed with autosampler AI 3000. The compound being injected into Trace GC must
be proven (by usual GC or TGA) to be volatile and contain no non-volatile or thermally
decomposable matters. That is, no reaction mixtures or crude products with non-volatile
impurities may be injected. The specification sensitivity of Polaris Q MS is 10 picogram
of decafluorobenzophenone. Both volume and amount/concentration of sample being
injected matter, for the injector liner cannot accept more vapor volume than equivalent to
3 μl of liquid. The injector was usually used in split mode (with split liner installed) with
split ratios 10…500. Inject no more than 1–100 ng (nanogram) = 10–9 g = 10–6 mg of

218
sample, dissolved in no more than 1 μl (microliter) = 10–3 ml of solvent. To prepare such
solutions, use one of the three methods below. Useful to notice is that 1 ng/μl = 1 μg/ml =
1 mg/liter, and 100 ng/μl = 0.1 mg/ml = 1 mg/10 ml.
Method 1. Prepare Solution#1 by dissolving 1 mg in 10 ml. Take an aliquot of 1
ml and dilute to 10 ml of Solution#2. Take an aliquot of 1 ml of Solution#2 and dilute to
10 ml of Solution#3. Inject 1 μl of Solution#3. If no signal appears on GC-gram, inject 1
μl of Solution#2.
Method 2. Prepare Solution A by dissolving 1 mg in 10 ml. Take an aliquot of 1
μl and dilute to 1 ml of Solution B. Inject 1 μl of Solution B. If no signal appears on GC-
gram, take an aliquot of 10 μl of Solution A, dilute to 1 ml of Solution C and inject 1 μl
thereof.
Method 3. Prepare Solution Z by dissolving 1 μg (0.001 mg) in 1 ml. Inject 1 μl
of Solution Z.
The GC column used was Restek #12623 RTX-5MS 30 m × 0.25 mm × 0.25 μm
film, crosslinked 5% diphenyl – 95% dimethylpolysiloxane. Column maximum
temperature is 350 °C. The carrier gas used was ultra-high purity helium (grade 5.0) at 1
ml/min with vacuum compensation. Injector nominal temperature was 225 °C. Transfer
line nominal temperature was always 300 °C. MS ion source nominal temperature was
200 °C. The complete, illustrated, step-by-step instructions for running a GC-MS
experiment can be found in Appendix A.

219
HPLC-MS
High-performance liquid chromatography (HPLC) was performed with an
Agilent 1100 HPLC, equipped with an Agilent «Zorbax NH2» column, which has ID 4.6
mm; ℓ= 250 mm; average particle size 5 μm; inverse phase is medium-polar 3-amino-
propyldiethoxysilane. Column volume was calculated to be 4.15 ml; void volume was
calculated from hexane void time as ~3ml. Typical flow rates were 1…1.5 ml/min. The
mobile phase was a combination of usually two or three solvents from the following: iso-
octane (2,2,4-trimethylpentane), chloroform, THF, acetonitrile, methanol. All solvents
were either of HPLC grade or were distilled twice on a dedicated rotovap and filtered
through 0.3 μm PTFE micron filter. When used as standalone instrument, a diode array
detector (DAD) was used to register and identify components in the effluent. Normally,
UV-Vis spectrum of the effluent in the range 190–900 nm (in 2 nm resolution) was
recorded at the sampling rate of 2 sec. The HPLC-gram’s ordinate axis was set in
correspondence with 254 (350), 360 (500), or 400 (500) nm absorption of the effluent
(reference wavelength in parentheses).
Bruker Daltonics Esquire 3000+ ion trap mass spectrometer was used both
standalone for identification of single-component analytes by direct infusion and coupled
with the Agilent 1100 HPLC for analysis of multi-component mixtures. Atmospheric
pressure chemical ionization (APCI) ion source was generally used. In some cases of
low-volatile analytes electro-spray ionization (ESI) ion source was employed. Drying gas
temperature was usually set to 300 °C, APCI heater’s temperature varied in the range of
300…450 °C. When used as HPLC detector, the ion trap parameter ICC was set to

220
20,000, the signal was averaged over 7 measurements and “rolling average” option was
checked on.
Nuclear Magnetic Resonance (NMR)
NMR spectra were recorded for 1H, 13C, and 19F nuclei. Instruments used were:
Bruker AMX 300 and Bruker Biospin Avance 400 spectrometers with 1H base
frequencies 300 (7.05 T) and 400 MHz (9.4 T superconducting magnet) correspondingly.
Both instruments were equipped with 5 mm probes. The 5-mm NMR glass tube should
be filled with a deuterated solvent solution to a minimum depth of 5.0…5.5 cm (about
0.60…0.75 ml). The amount (concentration) of sample required for a proton spectrum
ranges from less than 1 mg/ml to about 20mg/ml (for a compound with Mw ~ 400). Too
much sample can result in a loss of resolution or a distorted 1H spectrum. This includes
not just the sample of interest, but any proton source such as protonated buffers, residual
protonated solvents, and water. About 5mg/ml is sufficient maximum for 1H. For 13C
spectra the higher the concentration used the better. The solution should be free from any
solid, such as undissolved solute, or dust. Cloudy solutions were routinely filtered
through either a Pasture pipette with a tiny cotton swab, or a syringe filter. The following
deuterated solvents were used: chloroform-d (CDCl3, used by default, if another solvent
not specified explicitly), acetone-d6, dimethylsulfoxide-d6, and trifluoroacetic acid-d
(CF3COOD), prepared from trifluoroacetic anhydride and D2O. Step-by-step instructions
to run a 1D 1H and 13C experiments on Bruker AMX 300 can be found in Appendix B.

221
Thermal Analysis: DSC and TGA
Thermal Analysis (TA) Instruments differential scanning calorimeter (DSC)
models 2920 and Q10 were used to routinely measure melting points of the samples.
Both instruments were calibrated in temperature and heat capacity against a certified
indium standard. Non-hermetic 5 μl aluminum pan was charged with 1…10 mg of
sample, topped with corresponding lid and crimped in a press. A blank pan charged with
no sample, also crimped with a lid, served as a reference. Liquid samples were loaded
into hermetic pans and crimped with corresponding lid. The sample and reference pans
were placed onto their corresponding thermocouples inside DSC cell, and the cell was set
to constant purge with nitrogen gas at 5 ml/min. Standard heating rate (ramp) was 10
°C/min, if not specified otherwise. On DSC traces endothermal peaks show up,
exothermal peaks — down. Melting point, followed by decomposition is reported as
melting point onset1 temperature, followed by “dec.”, while decomposition without
concurrent melting is reported as “dec.” followed by onset decomposition temperature.
Thermal stability, decomposition temperature and sublimation range were
measured on thermal gravimetrical analyzer (TGA) model 2950 by the same
manufacturer (TA Instruments). The instrument was calibrated in temperature with nickel
standard by determining Curie point transition, and with calcium oxalate standard in
1 Onset m.p. is independent of the sample amount charged into the pan, while peak
maximum shifts to bigger temperature values proportionally to the sample mass.

222
mass. Platinum 100 μl sample pan was tared to zero and charged with no more than 1…5
mg of sample. Standard heating rate (ramp) was 10 °C/min, if not specified otherwise.
M.Braun SPS
M.Braun solvent purification system (SPS) is a safer alternative to solvent
stills.405 The solvent is purified therein by passage through a bed of activated alumina,
contained in a hermetic steel reservoir. Hydrocarbons can also be deoxygenated by a pass
through another column with activated copper. Inert gas is used to push the solvent
through the columns. Complete, step-by-step illustrated instructions on how to operate
SPS can be found in Appendix C.
High Pressure Reactors
Reactions, required high pressure (e.g. amination with pyrrolidine or Bucherer
reaction) were performed either in glass pressure vessel CG-1880 by ChemGlass, or in
Parr Instrument High Pressure Reactor, model 4563, depending on the load and
maximum expected pressure.
Glass pressure vessel CG-1880 can be used for reactions (see, for example, 83),
where maximum possible pressure does not exceed ~10…15 atm. It should never be
filled more than 1/3 of its volume and always be used behind a safety shield. When
aggressive media (like concentrated nitric acid) are employed, the Viton® O-ring needs to
replaced with a lead (Pb) gasket, custom-cut from a sheet. This glass pressure vessel has

223
also been employed in combination with Parr Instrument High Pressure Reactor 4563, to
run reactions in mineral acids, which would severely attack the stainless steel body of
4563 reactor. Such reaction mixtures were placed inside the glass pressure vessel (sealed
with its original Teflon® stopper and Viton® O-ring or with a custom-made Pb gasket),
which was placed inside the 4563 reactor, filled with sufficient amount of appropriate
solvent. The solvent was chosen such that at the reaction temperature the pressure from
the solvent onto the glass reactor would approximately match the pressure from the
reaction mixture (e.g. water and benzene mixture for reactions in nitric acid).
Parr Instrument High Pressure Reactor 4563 was used with either a Teflon®or
glass insert (liner). The Teflon® liner can be used up to 250°C. The glass liner has the
same maximum operating temperature as the reactor itself — 350°C, which limit is
imposed by PTFE self-sealing gasket of the reactor. Whenever the reactor is heated above
100 °C, cooling water must be set running through the cooling jacket around the stirrer
coupling. The thumbscrew on the drop band (encircling the split ring closure) should fit
into the slot in the stand’s bracket to prevent slipping of the whole reactor when the
stirrer is turned on. Never operate the heating mantle without reactor lowered into it.
UV-Vis
Ultra-violet and visible (UV-Vis) spectra were recorded on Agilent / HP 8453
diode array spectrophotometer. Samples were dissolved in a suitable solvent of
photometric grade and diluted down so that their maximum absorbance would fall within

224
0.1…0.45 range (units of optical density). Standard quartz cuvettes of 1.0 cm path-length
were generally employed. Blank solvent was measured before the sample spectrum was
recorded. According to Bouguer-Lambert-Beer law, lCA ε= , thus for 1 cm path we
get:
][]/[•][•
][•][]/[•][•
mgmmolgMmlVA
cmlgmmolgMlVA
ClA ===ε
For a solution of m mg per 10 ml of solution and 1 cm path:
10][
]/[•]••[ 1–1– ×=mgm
molgMAcmmollε .
This formula has been used to calculate the molar extinction coefficients from
the UV-Vis spectra of solutions with a known concentration of solute.
IR
Bruker Optics Vector 33 Fourier-transform infra-red spectrometer (FT-IR) was
used to record infra-red (IR) spectra. Sampling was performed in either of three ways: (1)
neat on the MIRacle™ Horizontal Attenuated Total Reflectance (HATR) stage by Pike
Technologies; (2) solution on the MIRacle™ HATR; (3) between KBr plates. If not
specified otherwise, IR spectra are reported as neat (both solids and liquids) on MIRacle™
HATR.

225
4.2. Synthetic Procedures
3,6-Diphenyldihydropyrrolo[3,4-c]pyrrole-1,4-dione, DPP 1.
NH
NH
O
O
N O
O
O
O+
12 1
t-BuOK
t-AmOH
DPP was prepared by modification of the published procedure.71 A five-liter
reactor was topped with a three-neck lid. The central neck was fitted with a mechanical
stirrer, and a thermometer/nitrogen inlet adaptor into another neck. The reactor was
flushed with nitrogen and charged with tert-amyl alcohol (3 liters). The third neck was
fitted with a reflux condenser and the reactor was heated with stirring on a heating mantle
until reflux of the solvent (102°C) began. At that temperature potassium tert-butoxide
(560 g, 5.0 mol) was added in portions and heating and stirring continued until all of the
t-BuOK dissolved (the temperature rose to 120°C). After the temperature was lowered to
110°C, benzonitrile (206.24 g, 2.0 mol) was added at once. Di-tert-amyl succinate
(258.35 g, 1.0 mol) was placed into a syringe and added by means of a syringe pump
during 4 hrs. In the middle of the addition and near the end of the addition, additional
portions of tert-amyl alcohol (500 ml) were added to facilitate stirring. After the addition
of succinate was complete, the reaction mixture was stirred at 110°C for an additional
four hours, and then cooled to 60°C and methanol (1 liter) was added in 100 ml portions,
followed by acetic acid (350 ml). The resulting slurry was filtered on two 2-liter Buchner
funnels, and then gradually suspended (with help of sonication) in methanol and filtered

226
from methanol (2×2 liters), hot water (2×1 liter), and boiling ethanol (4 liters). The filter
cake was dried overnight to give 161.5 g (56%) of 1 as fine red powder. This material
was characterized as 2,5-dimethyl-3,6-diphenyldihydropyrrolo[3,4-c]pyrrole-1,4-dione.
M.p. 231°C (lit.74 m.p. 233-234°C); 1H δ: 7.87 (d, 2H, J=8.7 Hz), 7.61 (d, 2H, J=8.7 Hz),
3.31 (s, 3H). 13C δ: 138.35, 130.50, 29.60. ΦF=0.79.
3,6-bis(4-bromophenyl)-2,5-dimethyl-pyrrolo[3,4-c]pyrrole-1,4-dione, Br-DPP 9.
NH
NH
O
O
NH
NH
O
OBr
Br
Br2 gas
A Petri dish (14 cm internal diameter) was filled with finely ground 3,6-
diphenyl-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (22.3 g, 0.077 mol). Bromine (124
g, 0.775 mol) was placed into a 50 ml evaporation dish at the bottom of a desiccator (15
cm internal diameter) and topped with a porcelain insert, over which the Petri dish was
placed. The desiccator was left with closed lid and slightly open vent in the dark for ten
days. After the reaction was complete, the Petri dish was removed, left open to the air for
2 hours, then placed into a vacuum oven and dried at 75°C: for 2 hours at 40 mm Hg, and
for 4 hours at 1 mm Hg to remove absorbed bromine and hydrogen bromide and then was
weighed out to check the mass gain (19.8 g, 162% of theory). The crude product was
suspended in water (300 ml) and ethanol (200 ml). To that vigirously stirred suspension,
sodium hydrocarbonate (sat. aq., 150 ml) was added until CO2 evolution ceased. The

227
neutralized suspension was filtered, and gradually washed with water (200 ml), methanol
(200 ml), ether (100 ml) and vacuum-dried to give 32.5 (94%) of 9 as fine red powder.
3,6-bis(4-hydroxysulfonylphenyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione 10.
NH
NH
O
O
SO
OOH
SO
OOH
NH
NH
O
O
H2SO4·SO3
A 200 ml round bottom flask with a magnetic stir bar was charged with DPP
(6.0 g, 20.8 mmol), and oleum (45 g). The reaction mixture was heated in an oil bath at
40°C for 4 hours, cooled in an acetone – dry ice bath, and a mixture of ice (200 g) and
dry ice (40 g) was added slowly. The dark red precipitate formed was filtered through
fritted glass. The thick paste on the filter was re-suspended (3×) in acetone (50 ml) aided
by sonication, and filtered again to allow, after vacuum drying, 7.2 g (77%) of dark-red
powder. M.p. > 400°. 1H (NaOD, DMSO-d6/D2O) δ: 8.2 (br., 2H), 7.75 (br., 2H), 4.55
(br.). 13C (NaOD, DMSO-d6/D2O) δ: 162.8, 147.8, 144.3, 128.7, 128.2, 126.5, 111.5.
UV-Vis (H2O) λmax: 268, 477, 505. Fluorescence (EtOH) λmax: 525, 566sh. ΦF=0.63.
Di-iso-propyl succinate 11.
OHOH
O
O
OO
O
O
OHOH2+ 2 + 2
H2SO4

228
A two-liter, two-neck round bottom flask with a stir-bar was charged with
succinic acid (236.1 g, 2 mol), iso-propyl alcohol (480.8 g, 8 mol), benzene (500 g, Note
1), and sulfuric acid (20 ml). One neck was fitted with a nitrogen inlet adapter, and
another – with a short Vigreaux column, topped with a Dean-Stark trap and a condensor.
The reaction mixture was heated with a heating mantle (465 W, 50% of 120 V, 195°
mantle temperature) at reflux with a slow stream of nitrogen, introduced at the nitrogen
adapter inlet, until no more water was separated in the Dean-Stark trap. The trap should
be emptied periodically from separated water, total amount of which was ca. 36 ml, 2 mol
(50% of this amount separates during first four hours, the remaining amount – during
additional 20 hrs). After azeotropic removal of water ceased, the solvents were removed
on a rotovap at reduced pressure in two stages (first at P=20 mm Hg with water aspirator,
then at P=1 mm Hg with oil pump). The residue was vacuum-distilled on a rotovap at
P=0.1 mm Hg, yielding 336 g (83%) of colorless liquid, 25Dn = 1.4151 (lit. 406 25
Dn =
1.4177). The residue after vacuum distillation was extracted with hexane (200 ml, Note
2) in a separatory funnel, hexane was removed from the extract on a rotovap and the
residue of the extract was vacuum-distilled as above to give additional 90 g of colorless
liquid, 25Dn = 1.4165. The total yield was 381 g (94%). 1H NMR δ: 5.02 (quintet, 2H, J =
6.3 Hz), 2.56 (s, 4H), 1.24 (d, 12H, J = 6.3 Hz) — in accordance with lit. data.407
Note 1. The amount of benzene was calculated, based on the azeotropic data
from408: PhH:H2O = 91:9 (mass), az. b.p. 69°C. To distill out 2 moles of water, 365 g of
benzene are required. However, iso-propanol forms a ternary azeotrope with benzene and
water, requiring more benzene. Note 2. At the end of the first vacuum distillation,

229
considerable gas evolution and foaming occurs, indicating possible decarboxylation.
Extraction with hexane helps to remove the component susceptible to decarboxylation,
accumulated at the end of the distillation.
tert-Amyl succinate 12.
OO
O
O
OEtOEt
O
O
OH+Li
+ EtOH
tert-Amyl succinate was prepared by a modified procedure of a U.S. Patent409. A
two-liter, two-neck round bottom flask with a stir-bar was charged with diethyl succinate
(174.2 g, 1 mol), tert-amyl alcohol (970 g, 11 mol), and lithium (wire, 0.7 g, 0.1 mol).
One neck of the flask was fitted with a nitrogen inlet adapter, and another – with a long
(80 cm), efficient, double-section fractionating column, wrapped with asbestos tape for
thermal insulation. The column was topped with a Würtz adapter, thermometer, and a
condenser. The reaction mixture was heated with a heating mantle to reflux with a slow
stream of nitrogen, introduced at the nitrogen adapter inlet, continuously distilling out
ethanol with admixture of tert-amyl alcohol. The amount of alcohol distilled off the flask
was supplemented by equal amount of tert-amyl alcohol (350 ml of t-AmOH added in
total). The progress of the reaction was monitored by nD of the distillate ( 25Dn of EtOH is
1.3595, 25Dn of t-AmOH is 1.41021, and 25
Dn of (EtO)2Suc – 1.4178). After 38 hrs the
reaction mixture was cooled down to ~60°C and the alcohols were distilled off on a
rotovap (Note 1), leaving 340 ml of brown-green residue. To that residue, hexane (80 ml)

230
was added and the mixture was consecutively washed with water (200 ml), HCl (3M, 80
ml in 200 ml of water), water (250 ml × 5 times), and brine (50 ml), leaving a solution of
light yellow color and strong blue fluorescence under λ=365 nm UV lamp, but noticeable
under normal light too. The solution was dried with MgSO4, hexane was removed on a
rotovap, and the residue was vacuum-distilled twice on a rotovap with oil pump, followed
by traditional vacuum distillation, to give 238 g (92%, cf. to 77% in the patent) of
colorless liquid, b.p. 89–100°C at 0.2 mm Hg. 25Dn = 1.4273. 1H NMR (CDCl3) δ: 2.5 (s,
2H), 1.8 (quartet, 2H), 1.4 (s, 6H), 0.9 (t, 3H).
Note 1. The distilled alcohol mixture (~670 ml) was washed three times with
water (200 ml), then water-brine (1:1, 200 ml), and brine (200 ml). The water-immiscible
layer after all washings was dried over CaCl2, then over Na, and vacuum-distilled twice
on a rotovap to give 300 ml of recovered t-AmOH.
4-Fluorobenzonitrile 13.
4-Fluorobenzonitrile was prepared similar to 15, starting from 4-fluorobenzaldehyde.
M.p. 35–37 °C (lit.410 m.p. 32–34 °C). B.p. 70°C at 10 mm Hg. Mixture with a
commercial sample did not give depression in m.p. 13C NMR δ: 162.9, 131.3, 113.7,
112.7, 104.5.
4-Bromobenzonitrile 14.
4-Bromobenzonitrile was prepared similar to 15, starting from 4-bromobenzaldehyde.
M.p. 112–115 °C (lit.411 m.p. 110–115 °C). Mixture with a commercial sample did not

231
give depression in m.p. IR ν, cm–1: 3100 – 2850, 2240, 1590, 1450, 1260, 1040, 830, 670.
EI-MS: m/z (%): 182 (M+). 13C NMR δ: 162.3, 132.5, 127.8, 117.8, 111.2.
4-Methoxybenzonitrile 15.
CHO
OMe
+ NH2OH
OMe
NOH
— H2O
CN
OMe
4-Methoxybenzonitrile was prepared by a modified procedure of Wang.119 A two-liter
round bottom flask with a heavy stir-bar was charged with anisaldehyde (4-
methoxybenzaldehyde, 272 g, 2.0 mol), hydroxylamine hydrochloride (166 g, 2.4 mol),
triethylamine1 (137 g, 190 ml, 1.35 mol), and NMP (anhydrous, 500 ml). The mixture
was stirred for 30 min, and phthalic anhydride (352 g, 2.3 mol) was added at once. The
flask was topped with reflux condensor and nitrogen bubbler, and heated to reflux on a
heating mantle (mantle temperature 150–170°C) for five hours. The reaction mixture was
cooled down, poured into cold water (4 liters), and chilled in a fridge, resulting in
crystallization of gray crystals. After 12 hrs in the fridge, the solids were filtered off on a
Büchner funnel, suspended in sodium hydrocarbonate solution (sat. aq.), stirred at 40°C
for 30 min to remove phthalic acid, and then recrystallized once from ethanol with
activated charcoal (2% wt/v) and four times from ethanol–water mixtures, increasing the

232
water content each time from 10 to 40% in 10% increments, yielding 91.7 g (69%) of 15
as off-white needles. M.p. 57°C, lit.412 m.p. 55–60°C. 13C NMR δ: 162.9, 104.0, 134.0,
114.7, 55.5, 119.2. The mother liquors from all recrystallizations may be diluted with
water, chilled in a fridge, and the precipitated oil, after recrystallization from ethanol with
activated charcoal, would give additional amount of 15.
4-(Pyrrolidin-1-yl)benzonitrile 16.
CN
NH2
II
+i-Pr2NEt
CN
N
A 250 ml round bottom flask with a stirbar was charged with 4-
aminobenzonitrile (10.0 g, 0.085 mol), 1,4-diiodobutane (26.25 g, 0.085 mol), Hunig’s
base (N,N-diisopropylethylamine, 21.88 g, 0.17 mol), HMPA (10 g), and NMP (20 g).
The reaction mixture was stirred at 100°C for two days, cooled down, poured into water
(200 ml), and filtered on Büchner funnel. The residue on filter was recrystallized twice
from ethanol (30 and 50 ml correspondingly) to give 8.1 g (55%) of off-white crystals.
M.p. 89°C, lit. m.p. 81413 and 89°C.414 1H NMR (CDCl3) δ: 7.4 (d, 2H), 6.5 (d, 2H), 3.3
(t, 4H), 2.0 (m, 4H).
1 The amount of Et3N has been reduced due to the basicity of NMP used as solvent.

233
4-(N-n-hexylamino)benzonitrile 17.
CN
NH2
CN
NHC6H13
+ I
4-(N-n-hexylamino)benzonitrile was prepared according to published procedure
for a similar (hexadecylamino-) compound.415 A 250 ml round bottom flask with a stir-
bar was charged with 4-aminobenzonitrile (23.6 g, 0.2 mol), 1-bromohexane (16.5 g, 0.1
mol), and HMPA (200 ml). The reaction mixture was stirred at 120°C under argon for 22
hrs, cooled down, poured into water (300 ml), and extracted with ether (2×100 ml).1 The
extract was evaporated from the solvent and the residue was vacuum-distilled to give
18.9 g (93.6%) of colorless liquid, b.p. 167–170°C at 2 mm Hg, crystallizing on standing.
Since TLC of this product (eluent hexane:EtOAc = 2:1) showed presence of some
starting material, the distilled material was chromatographed with the same eluent to give
17.7 g (88%) of 17 as white crystals. M.p. 48–49°C. Lit.416 m.p. 38–39°C. 1H NMR
(CDCl3) δ: 7.4 (d, 2H), 6.5 (d, 2H), 4.3 (s, br, 1H), 3.1 (m, 2H), 1.6 (m, 2H), 1.4 (m, 6H),
0.9 (t, 3H). 13C NMR (CDCl3) δ: 151.7, 133.9, 120.9, 112.2, 98.3, 43.4, 31.8, 29.3, 26.9,
22.8, 14.3.

234
4-(N,N-di-n-hexylamino)benzonitrile 18.
4-(N,N-di-n-hexylamino)benzonitrile was prepared similar to 19, starting from 4-
fluorobenzonitrile and di-n-hexylamine. Yield 23%. 1H NMR (CDCl3) δ: 7.4 (d, 2H), 6.6
(d, 2H), 3.3 (t, 4H), 1.6 (pentet, 4H), 1.4 (m, 8H), 0.9 (t, 6H). 13C NMR (CDCl3) δ: 150.8,
133.7, 121.2, 111.3, 96.3, 50.9, 31.8, 29.3, 26.9, 22.8, 14.3..
4-(N,N-di-n-butylamino)benzonitrile 19.
CN
F
+ NHPy
HMPA
CN
NBu Bu
A 250 ml round bottom flask with a stir-bar was charged with 4-
fluorobenzonitrile (12.1 g, 0.1 mol), di-n-butylamine (30 g, 0.23 mol), and pyridine (10
ml). The reaction mixture was heated at 60°C for 24 hrs and monitored by TLC (neat
hexane or hexane:ether = 8:2) – no new spots detected. HMPA (15 g) was added and
temperature was raised to 100°C. After 48 hrs the reaction mixture was cooled down,
poured into water (200 ml), diluted with HCl (3M, aq., 40 ml), and extracted with ether
(2×100 ml). Ether was evaporated on a rotovap and the residue was vacuum-distilled.
The fraction, boiling 150–160°C at 0.4 mm Hg was collected and amounted to 12.0 g
1 To check that diethyl ether does not extract considerable amount of HMPA from water solution, 1.00 g of HMPA was dissolved in 40.0 g of water and extracted with 15.0 g of ether. The ethereal layer was separated, and evaporated to give 11.5 mg of residue.

235
(52%). Lit.417 b.p. 176–179 at 2 mm Hg, which corresponds to 150°C at 0.4 mm Hg. 1H
NMR (CDCl3) δ: 7.4 (d, 2H), 6.6 (d, 2H), 3.3 (t, 4H), 1.6 (pentet, 4H), 1.4 (pentet, 4H),
0.9 (t, 6H). 13C NMR (CDCl3) δ: 150.8, 133.7, 121.2, 111.3, 96.3, 50.9, 29.3, 20.5, 14.2.
3,6-Bis(4-fluorophenyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione, F-DPP 20.
NH
NH
O
O
F
F
N
F
+t-BuOK
t-AmOHOO
O
O13 11 20
A five liter reaction vessel was charged with tert-amyl alcohol (2000 ml),
potassium tert-butoxide (561 g, 5 mol), topped with a three-neck lid, and flushed with
nitrogen. The central neck was fitted with a mechanical stirrer, and a
thermometer/nitrogen inlet adaptor into another neck. The third neck was fitted with a
reflux condenser and the reactor was heated with stirring on a heating mantle (590 W,
60% of 120 V) to reflux (102°C) until all t-BuOK dissolved. After that a solution of 4-
fluorobenzonitrile (140 g, 1.156 mol) in tert-amyl alcohol (warm, 200 ml) was added
slowly from an addition funnel, placed on top of the reflux condensor – the temperature
of the reaction mixture rose to 113°C. Then a solution of di-iso-propyl succinate (161.8 g,
0.8 mol) in tert-amyl alcohol (150 ml) was added from a syringe, using a syringe pump,
at a rate of 40 ml/hr. After all di-iso-propyl succinate had been added, the reflux
condensor was replaced with a 15-cm Vigreaux column and a Liebig condensor and the
alcohols were distilled out (ca. 800–850 ml of distillate) until nD of the distillate reached

236
that of tert-amyl alcohol ( 20Dn = 1.4050). When the distillation had been finished, the
heating was continued for 6 additional hours, and then the reaction mixture was cooled
down and transferred to a 4000 ml Erlenmeyer flask, equipped with a mechanical stirrer.
To that mixture, methanol (1000 ml) was added in portions of 100 ml so that the
temperature did not rise above 60°C. After the methanol addition was complete, acetic
acid (350 ml) was added from a syringe pump at a rate of 40 ml/hr, with vigorous stirring
(the viscosity of the mixture increases considerably), followed by water (200 ml), and
added in one portion. The suspension obtained was filtered on two 2-liter Buchner
funnels, and the thick red mud on the filter was then gradually suspended (with help of
sonication) in and filtered from: methanol (2×2 liters), hot water (2×1 liter), and boiling
ethanol (4 liters). The filter cake was dried overnight to give 123.85 g (66%) of 20 as fine
red powder. Direct characterization was not performed due to low solubility. Alkylation
gave an inseparable mixture of products.
3,6-Bis(4-cyanophenyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione 21.
NH
NH
O
OCN
NC
CN
CN
O
O
O
O+
12 21
A one liter three-neck round bottom flask was fitted with a mechanical stirrer, a
thermometer/septum inlet adaptor, and a reflux condenser. The flask was charged with
tert-amyl alcohol (250 ml) and heated with stirring on a heating mantle until reflux of the

237
solvent (102°C) began. At that temperature sodium tert-pentoxide (33 g, 0.3 mol) was
added in portions and heating and stirring continued until all of the t-AmONa dissolved
(the temperature rose to 110°C). After the temperature was lowered to 110°C, 1,4-
dicyanobenzene (25.6 g, 0.2 mol) was added at once. Di-tert-amyl succinate (33.6 g, 0.13
mol) was added fro a syringe pump during 2 hrs period. After the addition was complete,
the reaction mixture was stirred at 110°C for four additional hours, cooled to 60°C and
methanol (60 ml) was added, followed by acetic acid (30 ml). The resulting slurry was
filtered, and then re-suspended in and filtered from ethanol (2×200 ml), hot water (2×100
ml), and boiling ethanol (200 ml). The filter cake was air-dried to allow 23.75 g (35%) of
dark red crystals. 1H NMR (NaOD, DMSO-d9/D2O) δ: 8.64 (m, 2H), 7.75 (m, 2H).
3,6-Bis(4-methylphenyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione 22.
NH
NH
O
O
CNO
O
O
O+
A one liter three neck round bottom flask was flushed with argon and charged
with tertiary amyl alcohol (2-methyl-2-butanol, 320 g). The central neck of the flask was
fitted with a mechanical stirrer, the side neck – with a thermometer/nitrogen inlet adaptor,
and the third neck – with a reflux condenser. The reaction mixture was heated with
stirring on a heating mantle until reflux of the solvent (102°C) began. At that temperature
sodium tert-amylate (110 g, 1 mol) was added in portions and heating and stirring
continued until all of the t-AmONa dissolved. 4-Toluonitrile (47 g, 0.4 mol) was added at

238
once. While maintainig reaction temperature 105-110°C, di-iso-propyl succinate (AS-2-
09, 54 g, 0.267 mol) was added at vigirous stirring from a syringe via syringe pump at a
rate of 20 ml/hr. After the addition was complete, the reaction mixture was stirred at
110°C for four additional hours, and then cooled to 60°C and methanol (50 ml) was
added dropwise, followed by acetic acid (50 ml). The resulting slurry was filtered on a
Buchner funnel, and then gradually suspended (with help of sonication) in and filtered
from: hot water (400 ml), hot ethanol (2×400 ml) and hot acetone (2×400 ml). The filter
cake was dried overnight: 24 g (37.8%) – compare to 56% for parent Ph-DPP-H. UV-Vis
(DMAc) λmax: 269, 444, 473, 508; (sodium salt, DMAc): 277, 400, 417, 531, 565. 1H
NMR (DMSO-d6) δ: 8.4 (d, 2H, J=8.5 Hz), 7.4 (d, 2H, J=8.5 Hz), 2.4 (s, 3H).
2,5-dimethyl-3,6-diphenyldihydropyrrolo[3,4-c]pyrrole-1,4-dione74 23.
NH
NH
O
O
N
N
O
O
A 500 ml round bottom flask with a stir bar was charged with DPP 1 (14.42 g,
0.05 mol) and DMAc (300 ml). To the stirred reaction mixture sodium hydride (2.5 g,
0.105 mol) was added in portions. When the addition was complete, the reaction mixture
was heated at 80°C for 30 min and then methyl p-toluenesulfonate (30.0 g, 0.161 mol)
and additional dimethylacetamide (70 ml) were added at once. The reaction mixture was
stirred at 150°C for 24 hrs, cooled down, poured into water (200 ml), and filtered. The
residue on the filter was re-dissolved in boiling chloroform (200 ml), filtered from

239
insolubles and after evaporation of solvent and recrystallization from DMAc gave 13.45
g (85%) of orange-red crystals. M.p. 231°C (lit.46 m.p. 233-234°C); 1H δ: 7.87 (d, 2H,
J=8.7 Hz), 7.61 (d, 2H, J=8.7 Hz), 3.31 (s, 3H). 13C δ: 138.35, 130.50, 29.60. ΦF=0.79.
2,5-diethyl-3,6-diphenyldihydropyrrolo[3,4-c]pyrrole-1,4-dione 25 was prepared
similar to 23, starting from DPP 1 and ethyl p-toluenesulfonate. Yield 78%. M.p. 229 °C.
2,5-diisopropyl-3,6-diphenyldihydropyrrolo[3,4-c]pyrrole-1,4-dione 26 was prepared
similar to 23, starting from DPP 1 and iso-propyl tosylate. Yield 66%. M.p. 267 °C
(DSC, 20 °C/min).
3,6-Diphenyl-2,5-dipropylpyrrolo[3,4-c]pyrrole-1,4-dione 32.
NH NH
O
O
N N
O
O
N NH
O
O
+
1 32 33
A one liter round bottom flask with a stir bar was charged with 3,6-Diphenyl-
2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (20.0 g, 0.07 mol), potassium tert-butoxide
(24.0 g, 0.24 mol), dimethylacetamide (200 ml), fitted with an air condenser and heated
at 140°C for 4 hours, distilling out tert-butanol. After distillation ceased, the reaction
mixture was cooled down to room temperature and propyl iodide (47.0 g, 0.27 mol) was

240
added dropwise with stirring. When the addition was completed, the reaction mixture was
heated at 140°C for 12 hours and monitored by TLC. After 12 hours reaction mixture was
cooled down and poured into water (500 ml). The precipitate was collected under suction
filtration, air-dried, dissolved in chloroform (200 ml), applied onto silica gel, and
chromatographed with dichloromethane.
32. M.p. 189°C (DMAc). 1H δ: 7.82-7.85 (m, 2H), 7.56-7.50 (m, 3H), 3.72 (t,
2H), 1.59 (sextet, 2H), 0.83 (t, 3H). 13C δ: 162.2, 148.7, 131.3, 129.3, 129.0, 128.8,
128.4, 43.6, 23.0, 11.3. UV-Vis λmax, nm: 289, 466, 488. Fluorescence λmax: 528, 568sh.
ΦF=0.76.
33. (3,6-diphenyl-2-propyl-5H-pyrrolo[3,4-c]pyrrole-1,4-dione) M.p. 276°C
(DMAc). 1H (CDCl3/DMSO-d9) δ: 11.00 (s, 1H), 8.53-8.48 (m, 2H), 7.80-7.77 (m, 2H),
7.59-7.50 (m, 6H), 3.75 (t, 2H, J=7.8 Hz), 1.61 (m, 2H), 0.83 (t, 3H, J=7.8Hz). 13C
(CDCl3/DMSO-d9) δ: 163.1, 162.9, 146.7, 131.7, 130.6, 128.8, 128.7, 128.4, 128.2,
109.4, 42.8, 23.1, 11.9. UV-Vis λmax, nm: 265, 465, 488. Fluorescence λmax: 523, 563sh.
ΦF=0.69.
3,6-Diphenyl-2,5-dihexylpyrrolo[3,4-c]pyrrole-1,4-dione 34.
NH NH
O
O
N N
O
O
H13C6 C6H13 N NH
O
O
H13C6+
1 34 35

241
3,6-Diphenyl-2,5-dihexylpyrrolo[3,4-c]pyrrole-1,4-dione 34 was prepared
similar to 32 starting from DPP 1 (2.88 g, 10 mmol) and 1-iodohexane (2.12 g, 10 mmol).
Chromatography yielded 0.4 g (8%) of 34 and 0.6 g (16%) of 35.
34. M.p. 134°C (DMAc). 1H δ: 7.82-7.78 (m, 1H), 7.56-7.49 (m, 4H), 3.74 (t,
2H, J=7.8 Hz), 1.58 (pentet, 2H, J=7.8 Hz), 1.18 (m, 6H), 0.82 (t, 3H, J=7.8Hz). 13C δ:
162.9, 148.7, 131.2, 129.0, 128.8, 128.4, 109.9, 42.0, 31.4, 29.5, 26.5, 22.6, 14.1. UV-Vis
λmax, nm: 472. Fluorescence λmax: 530, 572sh. ΦF=0.74.
35. M.p. 252°C (DMAc). 1H (CDCl3/DMSO-d9) δ: 11.00 (s, 1H), 8.53-8.48 (m,
2H), 7.80-7.77 (m, 2H), 7.59-7.50 (m, 6H), 3.80 (t, 2H, J=7.8 Hz), 1.58 (pentet, 2H,
J=7.8 Hz), 1.21 (m, 6H), 0.83 (t, 3H, J=7.8Hz). 13C (CDCl3/DMSO-d9) δ: 163.1, 162.9,
146.7, 131.7, 130.6, 128.8, 128.7, 128.4, 128.2, 109.4, 41.6, 30.9, 29.1, 26.1, 22.1, 13.9.
UV-Vis λmax, nm: 262, 464, 481. Fluorescence λmax: 522, 565sh. ΦF=0.71.
3,6-diphenyl-2,5-didecyl-dihydropyrrolo[3,4-c]pyrrole-1,4-dione 36 was
prepared similar to 32 starting from DPP 1 (1.44 g, 5 mmol) and iododecane (2.68 g, 10
mmol). Chromatography with dichloromethane and recrystallization from 1-propanol
gave 1.2 g (42%) of orange crystals. M.p. 117°C. 1H (300 MHz, CDCl3) δ: 7.78-7.82 (m,
2H), 7.51-7.55 (m, 3H), 3.74 (t, 2H), 1.19-1.25 (m, 16H), 0.86 (t, 3H). 13C δ: 162.9,
148.7, 131.3, 129.1, 128.9, 128.5, 109.9, 42.1, 33.0, 32.0, 29.6, 29.5, 29.2, 26.9, 25.9,
22.8, 14.3. UV-Vis (CH2Cl2, λmax, nm): 268, 467. Fluorescence λmax: 529, 573sh.
ΦF=0.65.

242
3,6-Diphenyl-2,5-didodecylpyrrolo[3,4-c]pyrrole-1,4-dione 37 was prepared
similar to 36, substituting 1-iododecane for 1-bromododecane. Yield 44%. M.p. 114°C.
1H δ: 7.8 (m, 1H), 7.6-7.5 (m, 4H), 3.7 (t, 2H, J=7.8 Hz), 1.6 (pentet, 2H, J=7.8 Hz), 1.3–
1.15 (m, 18H), 0.82 (t, 3H, J=7.8Hz). 13C δ: 162.9, 148.7, 131.3, 129.1, 128.9, 128.5,
110.0, 42.1, 32.0, 29.6 (double intensity), 29.5, 29.2, 26.9, 22.9, 14.3. UV-Vis λmax: 284,
474. Fluorescence λmax: 527, 563sh. ΦF=0.97.
iso-Propyl tosylate.
SO
OCl
OH
Na
ONa ONa+ S
O
OO;
– NaCl
A one-liter round bottom flask with a large stir-bar was charged with iso-propyl
alcohol (300 g, 5 mol), THF (200 ml), and sodium (48 g, 2 mol), topped with a reflux
condensor, and heated at 80°C for two hours. After all sodium had been dissolved, the
heating mantle was replaced with an ice bath and the reaction mixture was cooled to
below 5°C. At that temperature tosyl chloride (360 g, 1.89 mol) was added in portions —
smaller at the beginning of the addition, larger towards the end, at such a rate that the
temperature did not rise above 15-20°C. After the TsCl addition was complete, the
reaction mixture was heated at 70°C for an hour, cooled down, and poured into cold
water (1000 ml). The layers were separated and the organic layer was gradually washed
with water (2×300 ml), HCl (1M, 200 ml), ammonium chloride (sat. aq., 200 ml), dried
with MgSO4, and in the vacuum oven. The crude product thus obtained (308 g, 76%) is a
dark brown oil and may be used for the alkylation reactions without further purifications.

243
If colorless material is desired, the crude product may be purified either by careful
vacuum distillation – on large scale (decomposition occurs if overheated!), or by column
chromatography – on small scale.
3,6-Bis(4-methylphenyl)-2,5-dipropylpyrrolo[3,4-c]pyrrole-1,4-dione.
N
N
O
O
NH
NH
O
O
A 500 ml round bottom flask with a stir bar was charged with 3,6-bis(4-tolyl)-
2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione 22 (20.7 g, 65.4 mmol), potassium tert-
butoxide (18.4 g, 164 mmol), dimethylacetamide (150 ml), fitted with air condenser and
heated at 60°C for 4 hours, distilling out tert-butanol. After distillation ceased, the
reaction mixture was cooled down to ~65°C and propyl iodide (25 g, 147 mmol) was
added dropwise with stirring. When the addition was completed, the reaction mixture was
heated at 80°C for 12 hours. The reaction mixture was cooled down and poured into
water (500 ml). The precipitate was collected by suction filtration, air-dried (49.7 g),
dissolved in chloroform (250 ml), filtered from insoluble matter, applied (37 g) onto
silica gel, and chromatographed with hexane:dichloromethane from 8:2 to 1:1 ratio
yielding two fractions.
Fraction #1 is Tol-DPP-Pr (3,6-bis(4-methylphenyl)-2,5-dipropylpyrrolo[3,4-
c]pyrrole-1,4-dione), recrystallized from DMAc (12 ml) to give 15.9 g (60%) of brown-
orange crystals. M.p. 186°C. 1H δ: 7.75 (d, 2H), 7.34 (d, 2H), 3.76 (t, 2H), 2.46 (s, 3H),

244
1.65 (sextet, 2H), 0.87 (t, 3H). 13C δ: 162.7, 148.2, 141.4, 129.5, 128.7, 125.7, 109.6,
43.5, 22.8, 21.5, 11.0. UV-Vis λmax, nm (lg ε): 270 (4.35), 306 (4.16), 474 (4.27).
Fluorescence λmax: 530, 575sh. ΦF=0.64.
Fraction #2 is 3,6-bis(4-methylphenyl)-2-propyl-5-hydropyrrolo[3,4-c]pyrrole-
1,4-dione, recrystallized from DMAc (20 ml) to give 0.68 g (3%) of red-orange crystals.
M.p. 294°C. 1H δ: 8.16 (d, 2H), 7.72 (d, 2H), 7.33 (AB, 4H), 3.80 (t, 2H), 2.43 (s, 3H),
2.44 (s, 3H), 1.65 (sextet, 2H), 0.87 (t, 3H). 13C δ: 162.9, 142.7, 141.5, 138.9, 129.9(CH),
129.6(CH), 128.7(CH), 127.7(CH), 125.6, 125.0, 43.7, 22.7, 21.7, 21.5, 11.1 (other peaks
do not show up due to low solubility). UV-Vis λmax, nm (lg ε): 268 (4.47), 304 (4.17),
469 (4.36), 495 (4.36). Fluorescence λmax, nm: 524, 565; ΦF=0.90.
2-Benzyl-3,6-diphenyldihydropyrrolo[3,4-c]pyrrole-1,4-dione 40.
NH
N
O
O
NH
NH
O
O
2-Benzyl-3,6-diphenyldihydropyrrolo[3,4-c]pyrrole-1,4-dione was prepared
similar to 32 starting from DPP 1 (722mg, 2.5 mmol) and benzyl bromide (428mg, 2.5
mmol). Chromatography with dichloromethane and recrystallization from 1-propanol
gave 303 mg (32 %) of 40 as orange crystals. M.p. 344°C. 1H (300 MHz, DMSO) δ:
11.36 (s, 1H), 8.5 (m, 2H), 7.7 (m, 2H), 7.6 (m, 4H), 7.5 (m, 2H), 7.3 (m, 4H), 7.1 (m,

245
1H), 5.0 (s, 2H). UV-Vis (CH2Cl2, λmax, nm): 263, 295, 468. Fluorescence λmax: 525,
566sh. ΦF=0.95.
3,6-diphenyl-2,5-diallylpyrrolo[3,4-c]pyrrole-1,4-dione 41.
NH NH
O
O
N N
O
O
N NH
O
O
+
1 41 42
3,6-diphenyl-2,5-diallylpyrrolo[3,4-c]pyrrole-1,4-dione 41 was prepared similar
to 32 starting from DPP 1 (5.76 g, 20 mmol), sodium tert-amylate (6.6 g, 60 mmol), NMP
(150 ml), and allyl bromide (8.0 g, 66 mol). Chromatography with neat dichloromethane,
followed by dichloromethane : ethyl acetate = 1:1 gave 3.13 g (42%) of 41 and 3.0 g
(46%) of 42.
41. M.p. 209°C. 1H δ: 7.95-7.90 (m, 2H), 7.53-7.51 (m, 3H), 6.0-5.9 (m, 1H),
5.24-5.18 (m, 2H), 4.4 (d, 2H). 13C δ: 162.3, 148.6, 133.4, 131.2, 129.0, 128.7, 128.1,
116.9, 109.9, 44.5. UV-Vis λmax: 476. Fluorescence λmax: 525, 566sh. ΦF=0.92.
42. (3,6-diphenyl-2-allyl-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione) M.p. 289°
(DMAc). 1H (DMSO) δ: 8.5 (dd, 2H), 7.8 (dd, 2H), 7.6 (m, 6H), 5.9 (m, 1H), 5.1 (dd,
1H), 5.0 (dd, 1H), 4.4 (m, 2H). 13C (DMSO) δ: 163.0, 161.8, 146.7, 146.5, 134.3, 132.7,
131.5, 129.5, 129.2, 129.0, 128.4, 128.2, 128.0, 116.7, 111.8, 108.9, 44.1. UV-Vis λmax:
467, 495. Fluorescence λmax: 518, 560sh. ΦF=0.97.

246
3,6-bis(4-bromophenyl)-2,5-dimethyl-pyrrolo[3,4-c]pyrrole-1,4-dione 43.
N
N
O
O23
N
N
O
O
Br
Br
43
(a) A Petri dish (14 cm internal diameter) was filled with finely ground 3,6-
diphenyl-2,5-dimethylpyrrolo[3,4-c]pyrrole-1,4-dione 23 (8.0 g, 0.025 mol). Bromine
(40.0 g, 0.25 mol) was placed into a 50 ml evaporation dish at the bottom of a desiccator
(15 cm internal diameter) and topped with a porcelain insert, over which the Petri dish
was placed. The desiccator was left with lid closed and a slightly open vent in the dark
for ten days. After the reaction was complete, the Petri dish was removed, left open to the
air for 2 hours, then placed into a vacuum oven and dried at 75°C: for 2 hours at 40 mm
Hg, and for 4 hours at 1 mm Hg to remove absorbed bromine and hydrogen bromide and
then was weighed out to check the mass gain (5.5 g, 137% of theory). The crude product
was recrystallized from DMAc (45 ml) to yield 11.7 g (96%) of ruby red crystals. M.p.
343°C. 1H (CDCl3) δ: 7.75 (AB, 2H), 7.65 (AB, 2H), 3.31 (s, 3H). 13C(CDCl3) δ: 205.3,
132.4, 130.6, 126.8, 126.2, 29.6. UV-Vis λmax, nm: 275, 305, 491. Fluorescence λmax:
541, 587sh. ΦF=0.83.
NH
NH
O
O
Br
Br
N
N
O
O
Br
Br
9 43

247
(b) 3,6-bis(4-bromophenyl)-2,5-dimethyl-pyrrolo[3,4-c]pyrrole-1,4-dione 43
was also prepared from 9: 3,6-bis(4-bromophenyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-
dione 9 (9.7 g, 0.0217 mmol) was placed into a 250 ml round bottom flask together with
potassium carbonate (30.0 g, 0.217 mol), methyl p-toluenesulfonate (16.0 g, 0.086 mol),
and dimethylacetamide (70 ml). The reaction mixture was stirred at 150°C for 24 hrs,
cooled down, poured into water (200 ml), and filtered. The residue on the filter was re-
dissolved in boiling chloroform (200 ml), filtered from insolubles and after evaporation
of solvent gave 4.30 g (43%) of orange-red crystals with the same characteristics as
above.
3,6-bis(4-bromophenyl)-2,5-dipropylpyrrolo[3,4-c]pyrrole-1,4-dione 44 was
prepared similar to 43 starting from Br-DPP 9. Yield 37% of Br-DPP-Pr 44 as yellow
crystals. M.p. 247°C (DMAc). 1H δ: 7.68 (AA′, 4H), 3.72 (t, 2H), 1.60 (m, 2H), 0.86 (t,
3H). 13C δ: 162.5, 147.4, 132.3, 130.0, 127.0, 125.8, 109.9, 43.4, 22.8, 11.2. ES-MS:
530.8. UV-Vis λmax, nm: 275, 304, 478. Fluorescence λmax: 541, 587sh. ΦF=0.86.
3,6-Bis(4-iodophenyl)-2,5-dimethylpyrrolo[3,4-c]pyrrole-1,4-dione 45.
N
N
O
O
I
I
43 45
N
N
O
O
Br
Br

248
A 200 ml pear-shaped flask with a magnetic stirbar was charged with 3,6-bis(4-
bromophenyl)-2,5-dimethylpyrrolo[3,4-c]pyrrole-1,4-dione (2.37 g, 5 mmol), potassium
iodide (12.50 g, 75 mmol), copper (I) iodide (4.75 g, 25 mmol), and dimethylacetamide
(50 ml). The reaction mixture was stirred under argon at 165°C for 115 hours, cooled to
~80°C, and mixed with water (200 ml). The resulting slurry was filtered, washed with
water (2×100 ml) and ethanol (30 ml), and air-dried, resulting in 7.7 g of pink powder,
which was boiled with chloroform (3×300 ml), and filtered through a #50 filter. The
filtrate, after evaporation, gave 1.1 g (37%) of red powder, m.p. 355°C dec. 1H δ: 7.87 (d,
2H, J=8.7 Hz), 7.61 (d, 2H, J=8.7 Hz), 3.31 (s, 3H). 13C δ: 138.35, 130.50, 29.60. UV-Vis
λmax, nm: 276, 318, 495. Fluorescence λmax: 544, 590sh. ΦF=0.89.
3,6-Bis(4-iodophenyl)-2,5-dipropylpyrrolo[3,4-c]pyrrole-1,4-dione 46 was
prepared similar to 45 starting from Br-DPP-Pr 44 to give 5.74 g (92%) of red powder.
Recrystallization from dimethylacetamide (5 ml/g) gave 4.25 g (68%) of ruby red
crystals. M.p. 260.5°C dec. 1H δ: 7.86 (d, 2H, J=8.7 Hz), 7.53 (d, 2H, J=8.7 Hz), 3.70 (s,
2H), 1.58 (m, 2H), 0.83 (t, 3H). 13C δ: 162.5, 147.6, 138.4, 130.1, 127.9, 110.34, 97.9,
43.8, 23.2, 11.4. ES-MS: 624.7 (M+). UV-Vis λmax, nm (ε): 277 (21520), 317 (18923),
479 (21350). Fluorescence λmax: 550, 592sh. ΦF=0.86.
3,6-Bis(4-cyanophenyl)dihydropyrrolo[3,4-c]pyrrole-1,4-dione 47 was
prepared similar to 1, using 1,4-dicyanobenzene instead of benzonitrile. Yield 35% of

249
dark red crystals. 1H (NaOD, DMSO-d9/D2O) δ: 8.64 (m, 2H), 7.75 (m, 2H). UV-Vis
(DMAc) λmax: 556, 603sh. ΦF=0.63.
3,6-bis(4-[pyrrolydin-1-yl]phenyl)-2,5-dimethylpyrrolo[3,4-c]pyrrole-1,4-dione 48.
N
N
O
O
N
N
43 48
N
N
O
O
Br
Br
A 50 ml pear shaped flask with a magnetic stirbar was charged with Br-DPP-Me
43 (300 mg, 0.63 mmol), dimethylacetamide (3 g), and pyrrolidine (3.0 g, 42 mmol). The
reaction mixture was stirred at 140°C for 12 hours (the starting material spot is gone on
TLC after 2 hrs, the di-substituted, lower Rf spot becomes major on TLC after 6 hrs),
cooled to room temperature and water (30 ml) was added dropwise. The precipitate was
filtered, and air-dried, leaving 213 mg (74%) of dark red crystals. M.p. 324°C dec. 1H δ:
7.94 (d, 2H, J=9 Hz), 6.64 (d, 2H, J=9 Hz), 3.40 (m, 7H), 2.04 (m, 4H). 13C δ: 163.2,
149.4, 147.4, 131.1, 115.3, 111.5, 107.0, 47.4, 30.1, 25.6. UV-Vis (λmax, CHCl3): 240,
282, 385, 548. Fluorescence λmax: 575. ΦF=0.95.
3,6-bis(4-[piperidin-1-yl]phenyl)-2,5-dimethylpyrrolo[3,4-c]pyrrole-1,4-
dione 49 was prepared similar to 48, starting from Br-DPP-Me 43 and piperidine. Yield
45%.
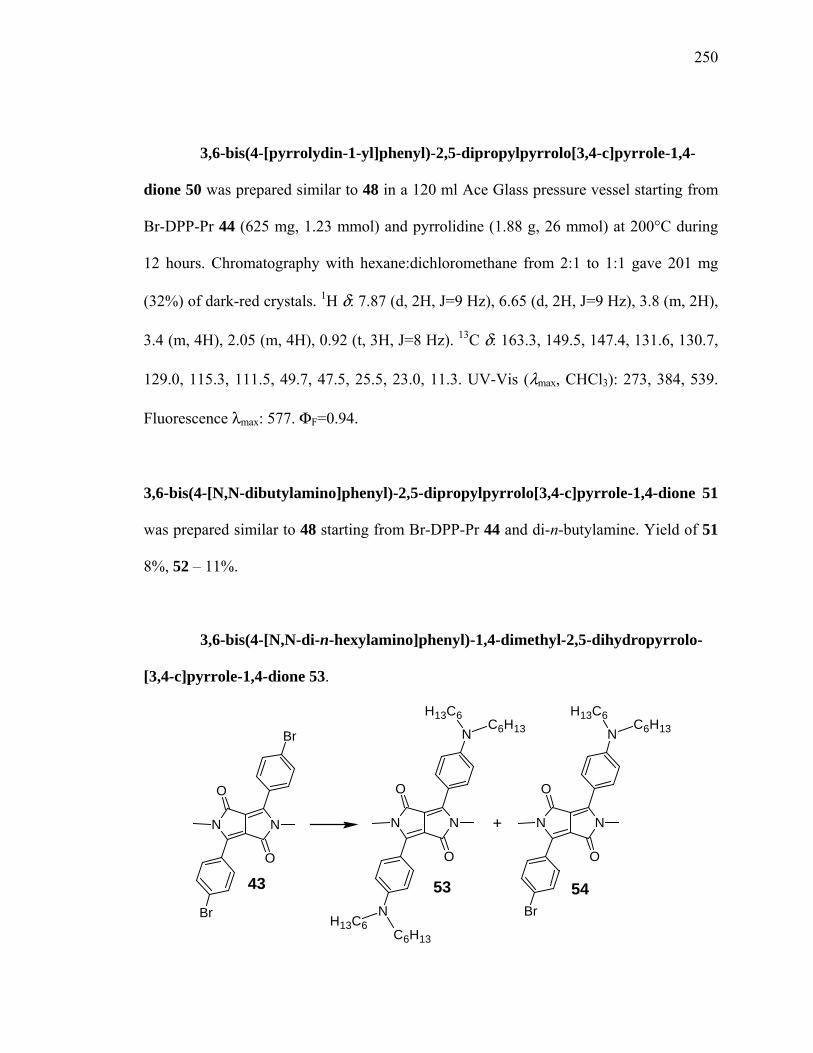
250
3,6-bis(4-[pyrrolydin-1-yl]phenyl)-2,5-dipropylpyrrolo[3,4-c]pyrrole-1,4-
dione 50 was prepared similar to 48 in a 120 ml Ace Glass pressure vessel starting from
Br-DPP-Pr 44 (625 mg, 1.23 mmol) and pyrrolidine (1.88 g, 26 mmol) at 200°C during
12 hours. Chromatography with hexane:dichloromethane from 2:1 to 1:1 gave 201 mg
(32%) of dark-red crystals. 1H δ: 7.87 (d, 2H, J=9 Hz), 6.65 (d, 2H, J=9 Hz), 3.8 (m, 2H),
3.4 (m, 4H), 2.05 (m, 4H), 0.92 (t, 3H, J=8 Hz). 13C δ: 163.3, 149.5, 147.4, 131.6, 130.7,
129.0, 115.3, 111.5, 49.7, 47.5, 25.5, 23.0, 11.3. UV-Vis (λmax, CHCl3): 273, 384, 539.
Fluorescence λmax: 577. ΦF=0.94.
3,6-bis(4-[N,N-dibutylamino]phenyl)-2,5-dipropylpyrrolo[3,4-c]pyrrole-1,4-dione 51
was prepared similar to 48 starting from Br-DPP-Pr 44 and di-n-butylamine. Yield of 51
8%, 52 – 11%.
3,6-bis(4-[N,N-di-n-hexylamino]phenyl)-1,4-dimethyl-2,5-dihydropyrrolo-
[3,4-c]pyrrole-1,4-dione 53.
N N
O
O
N
N
H13C6C6H13
C6H13H13C6
43 53
N N
O
O
Br
Br
N N
O
O
Br
N
H13C6C6H13
54
+

251
A 50 ml pear shaped flask with a magnetic stir bar was charged with Br-DPP- 43
(1.0 g, 2.11 mmol), dimethylacetamide (5.0 g), hexamethylphosphoramide (7.8 g) and
dihexylamine (1.5 g, 8.1 mmol). The reaction mixture was stirred at 180°C for 96 hours
(the starting material spot was still present on TLC after 36 hrs, the di-substituted, lower
Rf spot becomes major on TLC after 50 hrs, six spots on TLC total), cooled to room
temperature and water (60 ml) was added dropwise. The organic layer was extracted with
chloroform (150 ml), applied on silica (15 g) and flash-chromatographed: 430 mg of
crude material (six spots on TLC). A second chromatography yielded 158 mg (11%) of
diaminated product 53 and 257 mg (21%) of monoaminated product 54.
53. 1H (CDCl3) δ: 7.93 (d, 2H), 6.70 (d, 2H), 3.42 (s, 3H), 3.33 (t, 4H), 1.62 (m,
4H), 1.33 (m, 12H), 0.907 (t, 6H). 13C δ: 163.2, 149.9, 148.1, 132.1, 131.8, 131.2, 111.1,
51.2, 31.8, 30.127.4, 26.9, 22.8, 14.2. UV-Vis (λmax): 282, 385, 548. Fluorescence λmax:
575. ΦF=0.92.
54. 1H (CDCl3) δ: 7.93 (d, 2H), 7.66 (d, 2H), 7.53 (d, 2H), 6.64 (d, 2H), 3.36 (s,
3H), 3.33 (t, 4H), 3.30 (s, 3H), 1.62 (m, 4H), 1.33 (m, 12H), 0.91 (t, 6H). 13C δ: 163.3,
162.2, 151.0, 150.6, 131.9, 131.8, 130.6, 127.6, 124.6, 114.2, 111.0, 106.2, 51.3, 31.8,
30.1, 29.4, 27.4, 26.9, 22.8, 14.2.

252
2,5-dimethyl-3,6-bis(4-[2-{4-t-butylphenyl}vinyl]phenyl)-2,5-dihydropyrrolo[3,4-
c]pyrrole-1,4-dione (55).
N
N
O
O
N N
O
O
I
I
+
a) In a 50 ml pear-shaped flask with a magnetic stirring bar was placed 2,5-dimethyl-3,6-
bis(4-iodophenyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (4, 109.4 mg, 0.193
mmol), 4-tert-butylstyrene (67.2 mg, 0.42 mmol), potassium carbonate (82.8 mg, 0.6
mmol), TDA-1 (tris(3,6-dioxaheptyl)amine418, 6 mg, 0.02 mmol), and dimethylacetamide
(5 g). The flask was fitted with an oil bubbler and warmed to 100°C in a heating mantle
while degassing with argon. After 10 min palladium (II) acetate (1 mg, 0.004 mmol) was
added. The reaction mixture was stirred at 100°C for 10 hr, cooled to room temperature,
diluted with water (40 ml) and extracted with chloroform (4×60 ml). Montmorillonite
clay and magnesium sulfate were added to the combined extracts, filtered, solvent
evaporated to small volume and the residue was impregnated onto silica (13 g). Flash-
chromatography on 50 g of silica gel with hexane to hexane-dichloromethane (1:1)
yielded, after removal of solvent, 54 mg of dark solid, which was washed with pentane to
give 51 mg (43%) of dark crystals, m.p.295°C. 1H NMR δ: 7.93 (d, J=9 Hz), 7.65 (d, J=9
Hz), 7.50 (d, J=9 Hz), 7.41 (d, J=9 Hz), 7.24 (d, J=16.2 Hz), 7.12 (d, J=16.2 Hz). ES-MS

253
(APCI, CHCl3) m/z: 633.3 (M+1). 13C δ: 162.7, 151.6, 148.1, 140.5, 134.2, 131.0, 129.7,
127.1, 126.8, 126.7, 125.9, 109.4, 34.9, 31.4, 29.7.
b) 2,5-Dimethyl-3,6-bis(4-bromophenyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (8)
reacted as above for 10 hrs at 150°C gave 50% yield of the same compound.
2,5-dimethyl-3,6-bis(4-[2-{4-acetoxyphenyl}vinyl]phenyl)-2,5-dihydropyrrolo[3,4-
c]pyrrole-1,4-dione (56).
N
N
O
O
O
O
O
O
N N
O
O
I
I
O
+
A 200 ml pear-shaped flask with a magnetic stir bar was charged with 2,5-dimethyl-3,6-
bis(4-iodophenyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (4, 607 mg, 1.07 mmol), 4-
acetoxystyrene (357 mg, 2.2 mmol), TDA-1 (tris(dioxa-3,6-heptyl)amine, 78 mg, 0.24
mmol), potassium carbonate (504 mg, 3.6 mmol), and dimethylacetamide (30 g). The
flask was fitted with an oil bubbler and warmed to 80°C in an oil bath while degassing
with nitrogen. After 30 min palladium (II) acetate (5 mg, 0.02 mmol) was added. The
reaction was stirred at 100°C for 4 hr, cooled down to room temperature, diluted with
water (150 ml), filtered, washed with water, and air-dried. The crude product, dark red
crystalline material, 535 mg (83%), was dissolved in chloroform (350 ml), impregnated
on silica gel (48 g) and flash-chromatographed with dichloromethane yielding, after

254
solvent removal, 40.5 mg (6%) of dark red crystals. M.p. 357.7°C (DSC, 10°C/min). 1H
(CDCl3, DMSO-d9) δ: 7.96 (d, 2H, J=8.7 Hz), 7.74 (d, 2H, J=8.7 Hz), 7.64 (d, 2H, J=8.7
Hz), 7.31 (AB, 2H, J=14.9), 7.1 (d, 2H, J=8.7 Hz), 3.4 (s, 3H), 2.26 (s, 3H). UV-Vis
(λmax): 279, 335, 510.
4-Diethylaminostyrene 57.
NO
N
A 500 ml round bottom flask with a stir bar was charged with
methyltriphenylphosphonium bromide (11.0 g, 30.8 mmol) and DMSO (70 ml). To that
stirred mixture, potassium tert-butoxide (3.5 g, 31.2 mmol) was added at once and
stirring continued for 30 min while yellow ylide color was developing. To the stirred
reaction mixture, 4-diethylaminobenzaldehyde (5.4 g, 30.5 mmol) in DMSO (30 ml) was
added dropwise during 40 min period and stirring continued for 4 additional hours. After
the reaction was complete (TLC, hexane:ethyl acetate = 9:1), the reaction mixture was
diluted with water (300 ml), extracted with ether (2×100 ml), and chromatographed on
silica gel with hexane:ethyl acetate = 9:1 to allow, after evaporation of solvents, 1.82 g
(34%) of pale yellow oil. 24Dn =1.540 (lit. 1.5904). 1H NMR (CDCl3) δ: 7.57 (d, 2H, J=9
Hz), 6.95 (m, 1H), 6.90 (d, 2H, J=9 Hz), 5.83 (dd, 1H, J1=17 Hz, J2=1.2 Hz), 5.30 (dd,
1H, J1=17 Hz, J2=1.2 Hz), 3.59 (q, 4H, J=7 Hz), 1.42 (t, 6H, J=7 Hz). 13C NMR (CDCl3)
δ: 147.7, 137.1, 127.8, 125.5, 111.8, 108.8, 44.6, 12.9. ES-MS: 176.2 (M+H).

255
1,4-divinylbenzene 58.
O
O
An oven-dried 500 ml three-neck round bottom flask with a stir bar was fitted
with a gas inlet, cooled under argon flow and charged with methyltriphenylphosphonium
bromide (17.9 g, 0.05 mol) and anhydrous ether (150 ml). The flask was topped with a
reflux condenser and a septum. To the above stirred mixture, n-butyl lithium solution (2.5
M in hexanes, 20 ml, 0.05 mol) was added from a syringe during 30 min period. The
reaction mixture turned yellow and was stirred at room temperature for 4 hrs. After that
time, terephthalic aldehyde (3.35 g, 0.05 mol) in ether (100 ml) was added dropwise from
an additional funnel and reaction continued for 24 additional hours. The white precipitate
of Ph3PO was filtered off, re-suspended in ether (300 ml) and filtered again. The
combined ether filtrates were evaporated on rotovap, applied onto silica gel and
chromatographed with neat distilled hexane to yield, after evaporation of hexane, 469 mg
(14%) of clear oil, which crystallizes upon standing to clear plates. M.p. 30°C (lit. m.p.
31°C from AcOH-H2O). 1H NMR (CDCl3) δ: 7.54 (m, 2H, J=1.5 Hz), 6.88 (ddt, 1H,
J1=17 Hz, J2=11 Hz, J3=1.5 Hz), 5.93 (d, 1H, J=17 Hz), 5.42 (d, 1H, J=11 Hz). 13C NMR
(CDCl3) δ: 137.3, 136.7, 126.6, 113.9. IR (HATR, neat solid, ν, cm–1): 3088, 3005, 1626,
1508, 1398, 985, 902, 840.

256
N,N-di-n-butyl-4-[2-(4-vinylphenyl)vinyl]aniline 59.
NBu
Bu
I NBu
Bu+
A 250 ml round bottom flask with a stir bar was charged with N,N-dibutyl-4-
iodoaniline (12.4 g, 37.4 mmol), 1,4-divinylbenzene (#3-8c, 4.9 g, 37.6 mmol),
diisopropylethylamine (6 g, 46.5 mmol), tri-o-tolylphosphine (35 mg, 0.11 mmol), and
dimethylacetamide (50 ml). This mixture was stirred under argon for 30 min and then
palladium acetate (8 mg, 0.03 mmol) was added. The reaction mixture was stirred at
120°C for 5 hrs, cooled down, poured into water, and extracted with chloroform (3 × 100
ml). The combined extracts were washed with water (3 × 100 ml), saturated aq.
ammonium chloride (2 × 100 ml), dried with MgSO4, and evaporated on rotavap to give
12.0 g (96%) of pale yellow oil, which crystallized on standing to yellow crystals. M.p.
65°C. 1H NMR (CDCl3) δ: 7.47-7.39 (m, 6H), 7.06 (dd, 1H, J1=16 Hz, J2=3 Hz), 6.90
(dd, 1H, J1=16 Hz, J2=3 Hz), 6.66 (d, 2H, J=8 Hz), 5.77 (d, 1H, J=16 Hz), 5.24 (d, 1H,
J=16 Hz), 3.32 (t, 4H, J=7 Hz), 1.62 (pentet, 4H, J=7 Hz), 1.45-1.35 (m, 4H), 1.00 (t, 6H,
J=7 Hz). 13C NMR (CDCl3) δ: 147.7, 136.7, 129.2, 128.7, 128.2, 127.7, 126.2, 125.6,
124.7, 123.6, 111.7, 111.0, 50.9, 29.6, 20.5, 14.1.
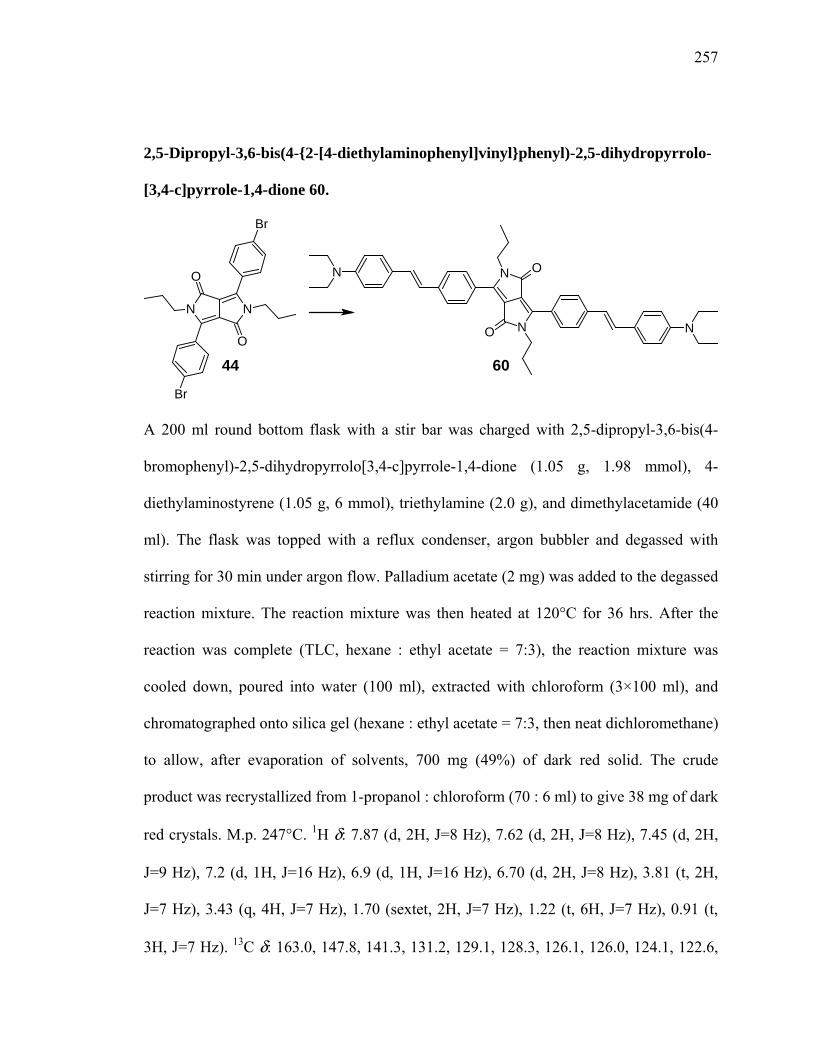
257
2,5-Dipropyl-3,6-bis(4-{2-[4-diethylaminophenyl]vinyl}phenyl)-2,5-dihydropyrrolo-
[3,4-c]pyrrole-1,4-dione 60.
NN
O
O
Br
Br
N
N
O
O
N
N
44 60
A 200 ml round bottom flask with a stir bar was charged with 2,5-dipropyl-3,6-bis(4-
bromophenyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (1.05 g, 1.98 mmol), 4-
diethylaminostyrene (1.05 g, 6 mmol), triethylamine (2.0 g), and dimethylacetamide (40
ml). The flask was topped with a reflux condenser, argon bubbler and degassed with
stirring for 30 min under argon flow. Palladium acetate (2 mg) was added to the degassed
reaction mixture. The reaction mixture was then heated at 120°C for 36 hrs. After the
reaction was complete (TLC, hexane : ethyl acetate = 7:3), the reaction mixture was
cooled down, poured into water (100 ml), extracted with chloroform (3×100 ml), and
chromatographed onto silica gel (hexane : ethyl acetate = 7:3, then neat dichloromethane)
to allow, after evaporation of solvents, 700 mg (49%) of dark red solid. The crude
product was recrystallized from 1-propanol : chloroform (70 : 6 ml) to give 38 mg of dark
red crystals. M.p. 247°C. 1H δ: 7.87 (d, 2H, J=8 Hz), 7.62 (d, 2H, J=8 Hz), 7.45 (d, 2H,
J=9 Hz), 7.2 (d, 1H, J=16 Hz), 6.9 (d, 1H, J=16 Hz), 6.70 (d, 2H, J=8 Hz), 3.81 (t, 2H,
J=7 Hz), 3.43 (q, 4H, J=7 Hz), 1.70 (sextet, 2H, J=7 Hz), 1.22 (t, 6H, J=7 Hz), 0.91 (t,
3H, J=7 Hz). 13C δ: 163.0, 147.8, 141.3, 131.2, 129.1, 128.3, 126.1, 126.0, 124.1, 122.6,

258
44.4, 43.7, 22.9, 12.7, 11.2. ES-MS: 719.2 (M+H). IR (HATR, neat solid, ν, cm–1): 2951,
1671, 1586, 1528, 1401, 1359, 1185, 1153, 1084, 1010, 960, 825. UV-Vis λmax, nm (lgε):
333 (4.99), 538 (5.08). Fluorescence λmax: 620. ΦF=0.72.
2,5-Dipropyl-3,6-bis(4-thien-2-yl-phenyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-
dione 64.
N
N
O
O
SS
64
N
N
O
O
Br
Br
44
A 200 ml round bottom flask with a stir bar was charged with magnesium
turnings (0.20 g, 8.3 mmol), 2-bromothiophene (1.0 g, 6.1 mmol), anhydrous THF (20
ml), and refluxed for 4 hrs. After the Grignard reagent had been formed, its THF solution
was decanted from excess of magnesium, and to that solution anhydrous zinc chloride
(2.0 g, 14 mmol) in anhydrous THF (30 ml) was added at once and the reaction mixture
was stirred for 1 hr at room temperature while precipitating magnesium chloride. Then a
solution of Br-DPP-Pr 44 (0.68 g, 1.28 mmol) in warm THF (120 ml), followed by
tetrakis(phenylphospine)palladium (0) (37 mg, 0.03 mmol) were added and the resulting
mixture was refluxed for 12 hrs. Chromatographic separation on silica gel with neat
dichloromethane gave 1.25 g of dark red solid, which after recrystallization from
chloroform-ethanol yielded 576 mg (84%) of ruby red crystals. M.p. 281°C. 1H δ: 7.90

259
(d, 2H, J=8Hz), 7.80 (d, 2H, J=8Hz), 7.45 (dd, 1H, J1=3.5 Hz, J2=1.3 Hz), 7.4 (dd, 1H,
J1=5 Hz, J2=1.3 Hz), 7.14 (m, 1H), 3.81 (t, 2H, J=7.5Hz), 1.70 (sextet, 2H, J=7.5 Hz),
0.91 (t, 3H, J=7.5 Hz). 13C (CDCl3, 50°C) δ: 162.9, 147.8, 143.5, 137.1, 129.5, 128.4,
127.3, 126.2, 124.4, 110.2, 43.8, 23.0, 11.3. UV-Vis λmax, nm: 315, 497. Fluorescence
λmax: 616, 690sh. ΦF=0.96.
3,6-Bis{4-[5-(4-dihexylaminophenyl)thien-2-yl]phenyl}-2,5-dipropylpyrrolo[3,4-c]-
pyrrole-1,4-dione 65.
N
N
O
O
Br
BrN
N
O
O
S
S
N(C6H13)2
(H13C6)2N
44 65
A 500 ml round bottom flask with a stir bar was charged with 2-(4-
dihexylaminophenyl)thiophene (5.5 g, 16 mmol), anhydrous ether (100 ml), fitted with an
argon bubbler and cooled to –20°C. To that stirred mixture, butyl lithium (1.6 M in
hexanes, 10 ml, 16 mmol) was added dropwise via syringe, the cooling bath was
removed, the mixture was stirred for 30 min and cooled to 0°C. A solution of anhydrous
zinc chloride (3.5 g, 25 mmol) in anhydrous THF (20 ml) was added at once, the reaction
mixture was allowed to warm up to room temperature and stirred for 1 hr. A suspension
of Br-DPP-Pr 44 (3.9 g, 7.3 mmol) in anhydrous THF (200 ml) was added at once
folowed by tetrakis(triphenylphosphine)palladium (35 mg) and the resulting mixture was

260
refluxed for 24 hrs. After the reaction was complete, the reaction mixture was cooled
down, solvents evaporated on a rotovap, the residue was dissolved in chloroform (200
ml) and washed with HCl (2M aq., 2×100 ml), water (200 ml) and applied onto silica gel.
Chromatography with gradient elution from dichloromethane to chloroform-methanol
allowed, after solvent evaporation, 2.5 g (32%) of dark purple crystals. M.p. 278°C. 1H δ:
7.91 (d, 2H), 7.76 (d, 2H), 7.51 (d, 2H), 7.37 (d, 1H), 7.15 (d, 1H), 6.69 (d, 2H), 3.84 (t,
2H), 3.33 (t, 4H), 1.65 (m, 6H), 1.38 (m, 12H), 0.97-0.87 (m, 9H). 13C δ: 162.9, 148.4,
147.8, 146.9, 140.0, 137.6, 129.5, 127.1, 126.7, 125.5, 125.4, 121.8, 121.6, 112.3, 110.2,
51.3, 43.9, 31.8, 27.5, 27.0, 23.0, 22.7, 14.0, 11.2. UV-Vis λmax, nm (ε): 354 (37652), 539
(47585). Fluorescence λmax: 615, 693 sh. ΦF=0.82.
3,6-Diphenyl-2-(3-hydroxysulfonylpropyl)-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione 67.
NH
NH
O
O
NH
N
O
O
SO
O
OH
1 67
A 250 ml round bottom flask with a stirbar was charged with DPP 1 (1.44 g, 5
mmol), potassium tert-butoxide (1.2 g, 10.5 mmol), dimethylacetamide (50 ml), and
heated at 80°C for 30 min while the tert-butanol formed was removed under reduced
pressure. To the cooled reaction mixture, 1,3-propane sultone (1.2 g, 10.0 mmol) was
added dropwise resulting in an exothermic reaction. After the addition was complete (10
min), the reaction mixture was heated at 80°C for an additional hour and cooled down.

261
Water (50 ml) was added and stirred for 2 hrs, the resulting solution was filtered, cooled
in a refrigerator and acidified with cold hydrochloric acid (12M, 30 ml). The acidified
solution upon refrigeration deposited a precipitate, which was filtered off and redissolved
in a mixture of 1-propanol (30 ml), water (2 ml), and hydrochloric acid (12M, 1 ml).
Evaporation of the solvents allowed 1.3 g (61%) of dark brown crystals with blue luster.
M.p. 270°C. 1H (300 MHz, DMSO-d6) δ: 11.0 (s, 1H), 8.5 (m, 2H), 7.82 (m, 2H), 7.57-
7.50 (m, 3H), 3.96 (t, 2H), 2.85 (t, 2H), 2.11 (m, 2H). 13C (75 MHz, DMSO-d6) δ: 162.4,
161.6, 146.0, 145.8, 132.2, 131.0, 129.1, 128.9, 128.7, 127.9, 127.7, 127.5, 110.8, 108.4,
49.0, 40.6, 24.9. UV-Vis (EtOH) λmax, nm: 465, 486. Fluorescence (EtOH) λmax: 525,
566sh. ΦF=0.77.
3,6-Diphenyl-2-(4-hydroxysulfonylbutyl)-5-propylpyrrolo[3,4-c]pyrrole-1,4-dione 68.
N
NH
O
O
PrN
N
O
O
SO
OOH
Pr
33 68
OS
O O
+
A 200 ml round bottom flask with a stir bar was charged with 3,6-diphenyl-2-
propyl-5H-pyrrolo[3,4-c]pyrrole-1,4-dione 21 (1.0 g, 3.3 mmol), dimethylacetamide (20
ml), and sodium hydride (0.42 g, 17.5 mmol). The reaction mixture was stirred at 40°C
for an hour, 1,4-butanesultone (5.0 g, 36.7 mmol) was added dropwise and stirred at
room temperature for 12 hrs. After the reaction was completed, the solvent was rotary
evaporated, the residue dissolved in chloroform (150 ml), applied onto silica gel and

262
chromatographed with CH2Cl2:MeOH = 9:1 to give 145 mg of dark brown oil. 1H δ: 8.05
(d, 2H), 7.33-7.52 (m, 8H), 4.52 (t, 4H), 3.13 (m, 2H), 2.23 (m, 2H), 1.82 (m, 2H), 0.83
(t, 3H). UV-Vis λmax, nm: 275, 465, 486. Fluorescence (EtOH) λmax: 524, 565sh.
ΦF=0.73.
3,6-bis(4-[ethyl(2-hydroxyethyl)amino]phenyl)-2,5-dipropylpyrrolo[3,4-c]pyrrole-
1,4-dione 70.
N
N
O
O
N
NOH
OHN
N
O
O
I
I
46 70
A 100 ml round bottom flask with a stirbar was charged with I-DPP-Pr 46 (286
mg, 0.458 mmol), 2-(ethylamino)ethanol (10 g), copper powder (5 mg, 0.08 mmol),
copper (I) iodide (10 mg, 0.05 mmol), dimethylacetamide (5 ml), fitted with air
condenser and heated under argon at 80°C. After 12 hours the reaction mixture was
cooled down to room temperature, diluted with chloroform (70 ml) and filtered. The
filtrate was washed with water (3×150 ml) in a separatory funnel, dried with MgSO4 and
applied onto silica gel. Chromatography with 20 to 80% of ethyl acetate in
dichloromethane gave 50 mg (20%) of the product as a dark red oil. 1H δ: 7.8 (d, 4H), 6.8
(d, 4H), 3.8-3.3 (m, 12H), 1.74 (m, 4H), 1.12 (t, 6H), 0.9 (t, 6H). 13C δ: 163.4, 150.0,

263
145.0, 130.8, 129.2, 115.8, 111.6, 107.7, 60.1, 52.4, 45.6, 44.0, 23.1, 12.0, 11.4. UV-Vis
λmax: 281, 369, 534.
6-(tetrahydro-2H-pyran-2-yloxy)hexan-1-ol.
OH
OH
O O
OH
A 100 ml round bottom flask with a stir bar was charged with 1,6-hexanediol
(8.2 g, 0.07 mol), Dowex 50WX2-100 resin (7 g), and tetrahydrofuran (50 ml). To that
mixture, dihydropyran (10.0 g, 0.118 mol) was added dropwise (during 30 min) at room
temperature with vigouros stirring. After the addition was complete, stiring was
continued for three additional hours. The resin was filtered off and the solvent was
evaporated. The colorless oil was impregnated onto silica gel and chromatographed with
hexane : ethyl acetate = 4:1 to elute 1,6-bis(tetrahydropyran-2-yloxy)hexane (3.65 g,
18%), followed by hexane : ethyl acetate = 1:1 to elute 6-(tetrahydropyran-2-
yl)oxyhexan-1-ol (6.32 g, 45%) as colorless oil. 25Dn =1.4777 (lit. 21
Dn = 1.457). EI-MS: 203
(M++1). IR (neat) ν, cm–1: 3406, 2938, 2863, 1454, 1441, 1353, 1201, 1138, 1121, 1077,
1026, 982, 868, 813. 1H NMR δ: 4.52 (m, 1H), 3.81 (m, 1H), 3.7-3.6 (m, 2H), 3.57-3.54
(m, 1H), 3.47-3.43 (m, 1H), 3.38-3.32 (m, 1H), 1.83-1.75 (m, 1H), 1.68-1.62 (m, 1H),
1.58-1.44 (m, 8H), 1.35 (m, 4H). 13C NMR (CDCl3) δ: 98.8, 67.4, 62.5, 62.3, 32.6, 30.8,
29.7, 26.1, 25.5, 25.4, 19.5.
6-(tetrahydro-2H-pyran-2-yloxy)hexyl 4-methylbenzenesulfonate 71.

264
O O
OH
O O
OTs71
A 200 ml round bottom flask with a stir bar was charged with 6-
(tetrahydropyran-2-yl)oxyhexan-1-ol (10.0 g, 0.05 mol), pyridine (8 g), and ether (100
ml). The reaction mixture was cooled in an ice-acetone bath to 0°C and powdered 4-
toluenesulfonyl chloride (12.0 g, 0.063 mol) was added at once. The reaction was
allowed to warm up in the bath during 12 hrs and then was filtered off. The precipitate on
filter was washed with ether (2×100 ml). The combined ethereal filtrates were
chromatographed with hexane : ethyl acetate = 4:1 to elute elute 6-(tetrahydropyran-2-
yl)oxyhex-1-yl 4-methylbenzenesulfonate (6.4 g, 36%) as colorless oil. 1H NMR δ: 7.76
(m, 2H), 7.33 (m, 2H), 4.52 (m, 1H), 4.0 (m, 2H), 3.82 (m, 1H), 3.67 (m, 1H), 3.46 (m,
1H), 3.32 (m, 1H), 2.43 (s, 3H), 1.78 (m, 1H), 1.63 (m, 1H), 1.50 (m, 8H), 1.30 (m, 4H).
13C NMR (CDCl3) δ: 144.7, 133.1, 129.8, 127.8, 98.9, 70.6, 67.3, 62.4, 30.7, 29.5, 28.7,
25.6, 25.4, 25.2, 21.6, 19.7 (16 C). IR: the band 3406 cm–1 is gone.
2-(6-iodohexyloxy)tetrahydro-2H-pyran 73.
O O
I
O O
OH73
A 250 ml round bottom flask with a stir bar was charged with diphosphorus
tetraiodide (2.4 g, 4.2 mmol) and carbon disulfide (100 ml). The mixture was sonicated
until P2I4 dissolved. To that stirred mixture, 6-(tetrahydro-2H-pyran-2-yloxy)hexan-1-ol
(3.3 g, 16.3 mmol) was added at once to form brown precipitate. After one hour, solid

265
potassium carbonate (1.5 g, 0.01 mol) was added and stirring continued for additional 12
hrs. Saturated aqueous potassium carbonate (15 ml) was added, the layers separated. The
CS2 layer was dried with MgSO4 and applied onto silica gel. Chromatography with 10 to
20% of dichloromethane in hexane gave, after removal of solvents, 3.4 g (66%) of
transluscent oil. 22Dn =1.5265. (Compare with the refractive index of the s.m. = 1.4589
and that of 1,6-diiodohexane = 1.5834). 1H NMR (CDCl3) δ: 4.51 (m, 1H), 3.81 (m, 2H),
3.68 (m, 2H), 3.48 (m, 2H), 3.34 (m, 2H), 3.16 (t, 2H), 1.8 (m, 2H), 1.5 (m, 4H), 1.35 (m,
4H). 13C (CDCl3) δ: 98.9, 67.5, 62.5, 33.3, 30.5, 29.7, 29.5, 25.7, 25.4, 19.8, 7.05.
3,6-diphenyl-2,5-bis(6-(tetrahydropyran-2-yl)oxyhex-1-yl)pyrrolo[3,4-c]pyrrole-1,4-
dione 74.
NHNH
O
O
NN
O
OOO
OO
O O
I+
173
74
A 100 ml round bottom flask with a stir bar was charged with 3,6-diphenyl-2,5-
dihydropyrrolo[3,4-c]pyrrole-1,4-dione (1.44 g, 5 mmol), potassium tert-butoxide (2.0 g,
17.8 mmol), and dimethylacetamide (35 ml). The reaction mixture was sonicated for five
minutes, placed onto rotovap and tert-butanol was evaporated under 1 mm Hg vacuum.
To that mixture, 1-(6-iodohexyloxy)tetrahydropyran (3.12 g, 10 mmol) was added
dropwise with vigorous stirring at room temperature (~20 min). After the addition was

266
complete, the flask was fitted with air condenser and heated under argon at 40°C. After
12 hours the reaction mixture was cooled down to room temperature, diluted with
chlorofrom (150 ml) and filtered. The filtrate was washed with water (3×150 ml) in a
separatory funnel, dried with MgSO4 and appled onto silica gel. Chromatography with 40
to 80% of ethyl acetate in dichloromethane gave, after removal of solvents, 1.1 g (34%)
of dark red oil. 13C (CDCl3) δ: 162.5, 131.0, 128.8, 128.6, 128.2, 109.6, 98.7, 67.5, 62.2,
41.7, 30.7, 29.7, 29.5, 26.1, 25.6, 25.4.
6-(benzoyloxy)-1-hexanol.
OH
OH
O
O
OH
A 250 ml round bottom flask with a stir bar was charged with 1,6-hexanediol
(5.0 g, 42.3 mmol), triethylamine (4.3 g, 42.2 mmol), and tetrahydrofuran (30 ml). The
mixture was cooled in an ice bath to ~5°C and cold benzoyl chloride (5.95 g, 42.3 mmol)
was added dropwise during a 30 min period, with vigorous stirring so that the
temperature did not rise above 10°C. After the addition was complete, the reaction
mixture was stirred for six additional hours. The precipitate of triethylammonium
chloride was filtered off and the filtrate evaporated on a rotovap. The residue was
chromatographed first with 20% ether in hexane to elute diester (3.1 g, 22%) and then
with neat ether to elute 6-(benzoyloxy)hexan-1-ol (5.6 g, 60%). 1H (CDCl3) δ: 8.0 (dd,
2H), 7.5 (t, 1H), 7.41 (t, 2H), 4.3 (t, 2H), 3.6 (t, 2H), 2.6 (s, br, 1H), 1.8 (pentet, 2H), 1.6

267
(pentet, 2H), 1.4 (m, 4H). 13C (CDCl3) δ: 167.0, 132.9, 130.4, 129.5, 128.3, 65.0, 62.5,
32.5, 28.7, 25.9, 25.5.
6-(benzoyloxy)-1-iodohexane 75.
O
O
OH
O
O
I75
A one liter two neck round bottom flask with a stir bar was charged with
phosphoric acid (85% aq., 32g, 0.277 mol), polyphosphoric acid (84%, 2H3PO4×P2O5, 32
g, 0.094 mol), the mixture was swirled until a homogeneous solution was formed, and
was allowed to cool to room temperature. To that mixture, potassium iodide (36.5 g, 0.22
mol) and 6-(benzoyloxy)-1-hexanol (48 g, 0.216 mol) were added at once, the flask was
fitted with a nitrogen/thermometer adapter, and a nitrogen bubbler. The reaction mixture
was heated to 110±10°C on a heating mantle for four hours (monitored by TLC with neat
hexane or 10% ether in hexane), cooled down and ether (300 ml) added. The layers were
separated and the organic layer was washed with sodium sulfite (10% aq., 2×100 ml),
water (2×200 ml), dried with MgSO4, and the solvent was removed on a rotovap. The
residue was chromatographed with gradual elution from neat hexane to ether.
Fraction 1 was eluted with neat hexane: 15.4 g, identified as a mixture of 1,6-
diiodohexane (traces) and 6-(benzoyloxy)-1-iodohexane. Fraction 2 was eluted with 6%
ether in hexane: 43 g (60%) of (benzoyloxy)-1-iodohexane as a colorless liquid.
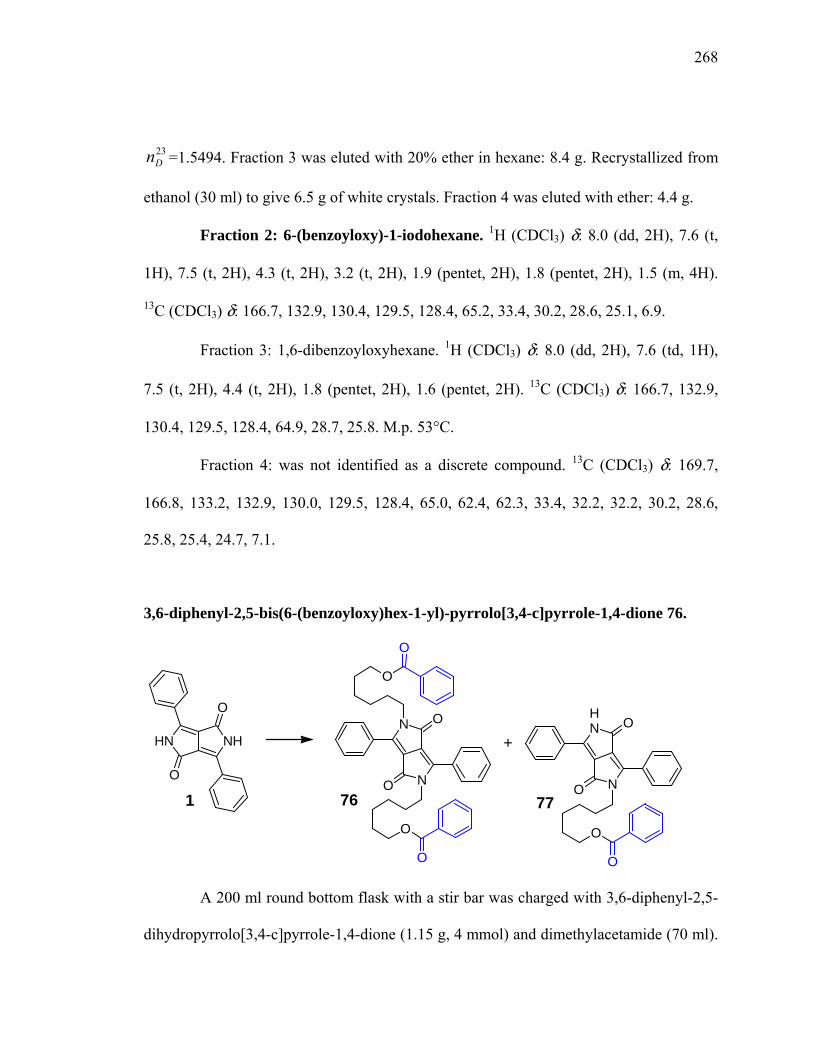
268
23Dn =1.5494. Fraction 3 was eluted with 20% ether in hexane: 8.4 g. Recrystallized from
ethanol (30 ml) to give 6.5 g of white crystals. Fraction 4 was eluted with ether: 4.4 g.
Fraction 2: 6-(benzoyloxy)-1-iodohexane. 1H (CDCl3) δ: 8.0 (dd, 2H), 7.6 (t,
1H), 7.5 (t, 2H), 4.3 (t, 2H), 3.2 (t, 2H), 1.9 (pentet, 2H), 1.8 (pentet, 2H), 1.5 (m, 4H).
13C (CDCl3) δ: 166.7, 132.9, 130.4, 129.5, 128.4, 65.2, 33.4, 30.2, 28.6, 25.1, 6.9.
Fraction 3: 1,6-dibenzoyloxyhexane. 1H (CDCl3) δ: 8.0 (dd, 2H), 7.6 (td, 1H),
7.5 (t, 2H), 4.4 (t, 2H), 1.8 (pentet, 2H), 1.6 (pentet, 2H). 13C (CDCl3) δ: 166.7, 132.9,
130.4, 129.5, 128.4, 64.9, 28.7, 25.8. M.p. 53°C.
Fraction 4: was not identified as a discrete compound. 13C (CDCl3) δ: 169.7,
166.8, 133.2, 132.9, 130.0, 129.5, 128.4, 65.0, 62.4, 62.3, 33.4, 32.2, 32.2, 30.2, 28.6,
25.8, 25.4, 24.7, 7.1.
3,6-diphenyl-2,5-bis(6-(benzoyloxy)hex-1-yl)-pyrrolo[3,4-c]pyrrole-1,4-dione 76.
NHNH
O
O
1
O
O
N
NO
O
O
O
+NH
NO
O
O
O
76 77
A 200 ml round bottom flask with a stir bar was charged with 3,6-diphenyl-2,5-
dihydropyrrolo[3,4-c]pyrrole-1,4-dione (1.15 g, 4 mmol) and dimethylacetamide (70 ml).

269
To this reaction mixture, a solution of lithium diisopropylamide (prepared from n-butyl
lithium 1.7M, 2.3 ml, and diisopropylamine, 4 ml) in ether (30 ml) was added at once.
The reaction mixture was allowed to stir at room temperature for one hour and 6-
(benzoyloxy)-1-iodohexane (3.12 g, 9.4 mmol) was added dropwise with vigorous
stirring at room temperature. After the addition was complete, the flask was fitted with air
condenser and heated under argon, gradually increasing the temperature from 80 to
160°C. After 26 hours the reaction mixture was cooled down to –10°C, and ice-cold
hydrochloric acid (12M, 15 ml) was added dropwise. The precipitate formed was filtered
off, washed with water (3×100 ml), dissolved in chloroform (50 ml), dried with MgSO4
and appled onto silica gel. Chromatography with 10% ethyl acetate in dichloromethane to
neat ethyl acetate gave, after removal of solvents, two fractions. Fraction 1 was 76: 0.64
g (23%) of brown-orange crystals. M.p. 111°C (1-propanol). 1H δ: 8.1–8.0 (m, 2H), 7.8
(dd, 2H), 7.6-7.5 (m, 3H), 7.47–7.43 (m, 3H), 4.3 (t, 2H), 3.7 (t, 2H), 1.7 (m, 4H), 1.4 (m,
4H). 13C δ: 166.6, 162.7, 148.5, 132.8, 131.2, 130.4, 129.5, 128.9, 128.7, 128.3, 128.2,
109.7, 64.8, 41.7, 29.3, 28.6, 26.4, 25.5. UV-Vis λmax: 475. Fluorescence λmax: 527,
563sh. ΦF=0.97. Fraction 2 was 77: 0.155 g (8%) of bright orange crystals, m.p. 194°C.

270
3,6-diphenyl-2-(6-(benzoyloxy)hex-1-yl)-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione 77.
NHNH
O
O
1
NH
NO
O
O
O77
A 200 ml round bottom flask with a stir bar was charged with 3,6-diphenyl-2,5-
dihydropyrrolo[3,4-c]pyrrole-1,4-dione (0.288 g, 1 mmol), potassium tert-butoxide (0.33
g, 2.9 mmol), and dimethylacetamide (70 ml). The reaction mixture was sonicated for
five minutes, placed onto rotovap and tert-butanol was evaporated under 1 mm Hg
vacuum. To that mixture, 6-(benzoyloxy)-1-iodohexane (0.71 g, 2.1 mmol) was added
dropwise with vigorous stirring at room temperature. After the addition was complete, the
flask was fitted with air condenser and heated under argon at 60°C. After 6 hours the
reaction mixture was cooled down to room temperature, diluted with chlorofrom (100 ml)
and filtered from unreacted starting material. The filtrate was washed with water (3×100
ml) in a separatory funnel, dried with MgSO4 and appled onto silica gel. Chromatography
with 20% ether in hexane to neat ether gave, after removal of solvents, 80 mg (11%) of
red crystals. M.p. 194°C (1-propanol). 1H δ: 10.1 (s, br., 1H), 8.4 (dd, 2H), 8.0 (dd, 2H),
7.8 (m, 1H), 7.6 (m, 4H), 7.5 (m, 1H), 4.3 (t, 2H), 3.9 (t, 2H), 1.7 (m, 4H), 1.4 (m, 4H).
13C (CDCl3 or DMSO-d6) δ: 166.3, 162.8, 162.1, 146.7, 133.4, 132.5, 131.2, 129.4,
129.3, 129.2, 129.0, 128.9, 128.4, 111.5, 109.1, 64.9, 41.5, 28.9, 28.4, 26.0, 25.3. UV-Vis
λmax: 467, 493. Fluorescence λmax: 522, 563sh. ΦF=0.96.

271
3,6-diphenyl-2-(6-(benzoyloxy)hex-1-yl)-5-propylpyrrolo[3,4-c]pyrrole-1,4-dione 78
was prepared similar to 79, starting from 3,6-diphenyl-2-propyl-5-hydropyrrolo[3,4-
c]pyrrole-1,4-dione 33 and using LDA (prepared in situ from n-BuLi and i-Pr2NH) as a
base.
3,6-diphenyl-2-(6-(benzoyloxy)hex-1-yl)-5-dodecylpyrrolo[3,4-c]pyrrole-1,4-dione 79.
NH
NO
O
C12H25
O
O
I74
+N
NO
O
O
O
H25C12
38 79
A 200 ml round bottom flask with a stir bar was charged with 3,6-diphenyl-2-
dodecyl-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione (0.205 g, 0.449 mmol) and
dimethylformamide (70 ml, to insure there is no precipitation on cooling). The reaction
mixture was cooled in an ice bath and tert-butyl lithium (1.7 M solution in hexanes, 1.0
ml) was added via syringe under argon. (t-BuLi probably reacts with DMAc, but the
enolate formed should be a good base as well.) The ice bath was removed and the
mixture was stirred at room temperature for 1 hr. 6-(Benzoyloxy)-1-iodohexane (0.250 g,
0.75 mmol) was added at once and the reaction mixture was heated at 80°C for 52 hrs.
After the reaction was complete by TLC (starting material is gone, two new fluorescent
spots), it was cooled down to room temperature, diluted with chlorofrom (100 ml),
washed with water (3×100 ml) in a separatory funnel, dried with MgSO4 and appled onto

272
silica gel. Chromatography with 10 to 35% ether in hexane to neat ether gave, after
removal of solvents, 0.1 g (34%) of orange crystals, m.p. 74°C. 13C (CDCl3) δ: 166.6,
162.7, 148.7, 148.4, 132.9, 131.1, 130.4, 129.6, 129.0, 128.9, 128.7, 128.6, 128.4, 128.2,
109.8, 109.6, 64.8, 42.1, 32.6, 31.8, 29.5, 29.3, 29.2, 29.0, 28.7, 28.6, 26.7, 26.4, 25.9,
25.6, 22.7, 14.1.
3,6-diphenyl-2,5-bis(6-hydroxyhex-1-yl)-pyrrolo[3,4-c]pyrrole-1,4-dione 80.
O
O
N
NO
O
O
O
76
NN
O
O
(CH2)6 (CH2)6 OHOH
80
A 350 ml glass pressure vessel with a stir-bar was charged with 28 (38 mg, 0.05
mmol), anhydrous methanol (50 ml), and titanium triisopropoxide (0.1 ml, 0.34 mmol).
The vessel was closed with a Teflon plug and heated, with stirring at 180°C (mantle
temperature) for 72 hrs. Each 24 hr the vessel was cooled and an aliquot was taken to
monitor reaction progress by TLC (neat EtOAc as eluent). After the reaction was
complete, the reaction mixture was cooled down to room temperature and methanol was
evaporated under reduced pressure. The residue was impregnated onto silica gel.
Chromatography with neat ethyl acetate gave 17 mg (64%) of orange crystals. 1H δ: 7.8
(dd, 2H), 7.6-7.5 (m, 3H), 3.8 (t, 2H), 3.6 (t, 2H), 1.65-1.57 (pentet, 2H), 1.50 (pentet,
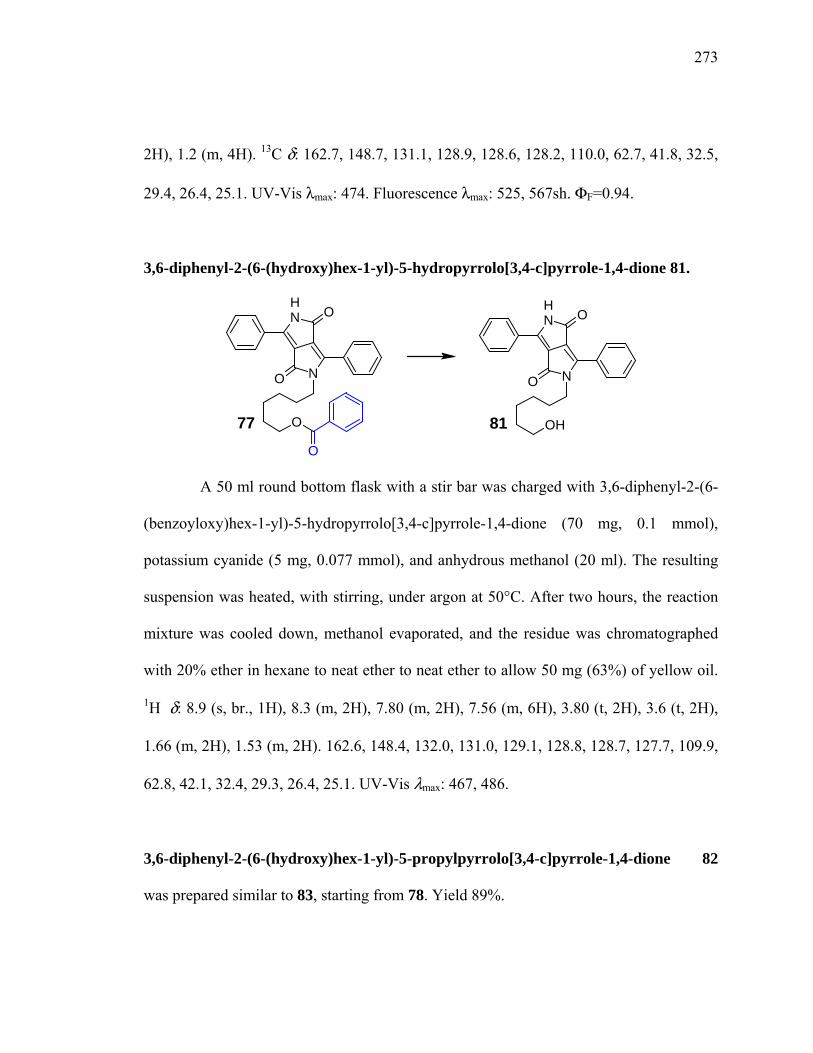
273
2H), 1.2 (m, 4H). 13C δ: 162.7, 148.7, 131.1, 128.9, 128.6, 128.2, 110.0, 62.7, 41.8, 32.5,
29.4, 26.4, 25.1. UV-Vis λmax: 474. Fluorescence λmax: 525, 567sh. ΦF=0.94.
3,6-diphenyl-2-(6-(hydroxy)hex-1-yl)-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione 81.
NH
NO
O
O
O
77 81
NH
NO
O
OH
A 50 ml round bottom flask with a stir bar was charged with 3,6-diphenyl-2-(6-
(benzoyloxy)hex-1-yl)-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione (70 mg, 0.1 mmol),
potassium cyanide (5 mg, 0.077 mmol), and anhydrous methanol (20 ml). The resulting
suspension was heated, with stirring, under argon at 50°C. After two hours, the reaction
mixture was cooled down, methanol evaporated, and the residue was chromatographed
with 20% ether in hexane to neat ether to neat ether to allow 50 mg (63%) of yellow oil.
1H δ: 8.9 (s, br., 1H), 8.3 (m, 2H), 7.80 (m, 2H), 7.56 (m, 6H), 3.80 (t, 2H), 3.6 (t, 2H),
1.66 (m, 2H), 1.53 (m, 2H). 162.6, 148.4, 132.0, 131.0, 129.1, 128.8, 128.7, 127.7, 109.9,
62.8, 42.1, 32.4, 29.3, 26.4, 25.1. UV-Vis λmax: 467, 486.
3,6-diphenyl-2-(6-(hydroxy)hex-1-yl)-5-propylpyrrolo[3,4-c]pyrrole-1,4-dione 82
was prepared similar to 83, starting from 78. Yield 89%.

274
3,6-diphenyl-2-(6-(hydroxy)hex-1-yl)-5-dodecylpyrrolo[3,4-c]pyrrole-1,4-dione 83.
N
NO
O
OH
H25C12
83
N
NO
O
O
O
H25C12
79
A 350 ml glass pressure vessel with a stir-bar was charged with 3,6-diphenyl-2-
(6-(benzoyloxy)hex-1-yl)-5-dodecylpyrrolo[3,4-c]pyrrole-1,4-dione (100 mg, 0.15
mmol), anhydrous methanol (70 ml), and titanium triisopropoxide (0.5 ml, 1.7 mmol).
The vessel was closed with a Teflon screw-in plug and heated, with stirring at 180°C
(mantle temperature) for 72 hrs. Each 24 hr the vessel was cooled and an aliquote taken
to monitor reaction progress by TLC (neat EtOAc as eluent). After the reaction was
complete, the reaction mixtu.re was cooled down to room temperature and methanol
evaporated under reduced pressure. The residue was impregnated onto silica gel.
Chromatography with neat ethyl acetate gave, after removal of solvents, 68 mg (81%) of
83 as orange crystals. 1H δ: 7.8 (dd, 4H), 7.5 (m, 6H), 3.8 (m, 4H), 3.6 (t, 2H), 1.6 (m,
2H), 1.5 (m, 2H), 1.3–1.2 (m, 26H), 0.9 (t, 3H). 13C δ: 162.73, 162.70, 148.7, 148.3,
131.1, 128.9, 128.7, 128.2, 109.8, 109.6, 62.6, 41.9, 41.6, 32.4, 31.8, 29.7, 29.5, 29.4,
29.3, 29.2, 29.0, 26.7, 26.4, 25.0, 22.7, 14.1. UV-Vis λmax: 468, 486. Fluorescence λmax:
522, 563sh. ΦF=0.89.

275
3,6-diphenyl-2-(10-hydroxy-1-decyl)-5-propylpyrrolo[3,4-c]pyrrole-1,4-dione 84.
NH N
O
O
Pr
33
N N
O
O
Pr(CH2)10OH
84
A 100 ml round bottom flask with a stir-bar was charged with 3,6-diphenyl-2-
propyl-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione (0.5 g, 1.51 mmol), cesium carbonate (1.0
g, 3.0 mmol), 10-bromo-1-decanol (0.5 g, 2.1 mmol) and dimethylacetamide (40 ml). The
reaction mixture was stirred at 60°C (mantle temperature) under argon and monitored by
TLC (DCM as eluent). After 6 hours the reaction mixture was cooled down to –10°C, and
ice-cold hydrochloric acid (12M, 5 ml) was added dropwise. The precipitate formed was
extracted with chloroform (80 ml) and appled onto silica gel. Chromatography with 50%
ethyl ether in hexane to neat ether gave, after removal of solvents, 490 mg of brown oil.
The oil crystallized upon sonication with cold ether, was recrystallized from ether-
chloroform to give 370 mg (49%) of 35 as bright yellow crystals. M.p. 105°C. 1H δ: 7.8
(m, 4H), 7.5 (m, 6H), 3.75 (m, 4H), 3.6 (t, 2H), 1.6-1.5 (m, 6H), 1.3-1.2 (m, 12H), 0.9 (t,
3H). 13C δ: 162.7, 162.63, 148.5, 131.1, 128.9, 128.7, 128.6, 128.2, 109.75, 109.71, 63.0,
43.3, 41.8, 32.7, 29.39, 29.32, 29.25, 28.91, 26.6, 25.7, 22.7, 11.1. UV-Vis λmax: 467,
490. Fluorescence λmax: 530, 561sh. ΦF=0.76.

276
3,6-diphenyl-2,5-bis(10-hydroxy-1-decyl)pyrrolo[3,4-c]pyrrole-1,4-dione 85.
NH NH
O
O
85
N N
O
O
(CH2)10OH (CH2)10 OH
1
A 200 ml round bottom flask with a stirbar was charged with DPP 1 (1.0 g, 3.47
mmol), cesium carbonate (3.4 g, 10.4 mmol), 10-bromo-1-decanol (1.8 g, 7.6 mmol) and
dimethylacetamide (70 ml). The reaction mixture was stirred at 120°C (mantle
temperature) under argon and monitored by TLC (DCM:EtOAc = 1:1). After 6 hours the
reaction mixture was cooled down to –10°C, and ice-cold hydrochloric acid (12M, 7 ml)
was added dropwise. The precipitate formed was extracted with chloroform (80 ml),
washed with water (2×50 ml), dried with MgSO4 and applied onto silica gel.
Chromatography with 50% ethyl acetate in dichloromethane gave 700 mg (34%) of dark
brown oil, which was crystallized from 1-propanol to give 125 mg (6%) of orange
crystals. The mother liquors after recrystallization contain only the target (by TLC) but
fail to deposit second crop of crystals. M.p. 143°C. 1H δ: 7.8 (dd, 2H), 7.5 (m, 3H), 3.8 (t,
2H), 3.6 (t, 2H), 1.6-1.5 (m, 4H), 1.3-1.2 (m, 12H). 13C δ: 162.6, 148.4, 130.9, 128.8,
128.7, 127.6, 109.9, 63.0, 41.8, 32.8, 29.3, 29.2, 28.9, 26.6, 25.6. UV-Vis λmax: 475.
Fluorescence λmax: 525, 566sh. ΦF=0.87.

277
3,6-diphenyl-2-(10-hydroxy-1-decyl)-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione.
NH NH
O
O
N NH
O
O
(CH2)10OH
3,6-diphenyl-2-(10-hydroxy-1-decyl)-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione
was prepared similar to 85 starting from DPP (1.0 g, 3.47 mmol) and 10-bromo-1-
decanol (1.8 g, 7.5 mmol). Chromatography with a gradient of 0-100% ethyl acetate :
dichloromethane, gave 0.87 g of brown oil. The oil was crystallized from 1-propanol to
give 0.7 g (34%) of mono-(HOC10H21)DPP as orange crystals. M.p. 166°C. 1H δ: 7.8 (dd,
2H), 7.5 (m, 3H), 3.8 (t, 2H), 3.8 (t, 1H), 3.6 (t, 2H), 1.6–1.5 (m, 2H), 1.4–1.2 (m, 14H).
13C δ: 162.6, 148.4, 130.9, 128.8, 128.7, 128.0, 109.9, 63.0, 41.8, 32.8, 29.3, 29.2, 29.1,
28.9, 28.7, 26.6, 25.6. UV-Vis λmax: 468, 493. Fluorescence λmax: 523, 563sh. ΦF=0.86.
2-(6-iodohexyl)-3a,4,7,7a-tetrahydro-4,7-epoxy-1H-isoindole-1,3(2H)-dione 86.
CAS RN [874998-65-9]
NHO
O
O120
NO
O
O
I
86
A 100 ml round-bottom flask with a stirring bar was charged with the adduct of
furan and maleimide (1.65 g, 0.01 mol), 1,6-diiodohexane (3.7 g, 0.011 mol), potassium

278
carbonate (1.4 g, 0.11 mol), and 35 ml of acetone. The flask was topped with a reflux
condenser and the reaction mixture was stirred at 40°C for 12 hours. The course of the
reaction was monitored by TLC with 1:1 eluent of hexane and ethyl acetate. The solvent
was evaporated; the residue dissolved in a minimum amount of chloroform and
chromatographed with 3:1 to 1:1 mixture of hexane and ethyl acetate. Recrystallization
from ethyl acetate yields 1.0 g (26.6%) of white crystalline material, m.p. 71.5°C (DSC,
10°/min). 1H NMR (CDCl3), δ: 6.52 (s, 1H); 5.25 (s, 1H); 3.46 (t, 2H, J=6.6Hz); 3.17 (t,
2H, J=6.6Hz); 2.84 (s, 2H); 1.8 (m, 2H); 1.56 (m, 2H); 1.45-1.26 (m, 4H). 13C NMR
(CDCl3), δ: 176.4, 136.7, 81.1, 47.6, 38.9, 33.4, 30.1, 27.5, 25.7, 7.1.
3,6-Diphenyl-2,5-bis(6-[3a,4,7,7a-tetrahydro-4,7-epoxy-1,3-dioxo-1H-isoindol-2-yl]-
hexyl)pyrrolo[3,4-c]pyrrole-1,4-dione 87.
N
O
O
ON
NO
O
N
O
O
O
NH NH
O
O
1
N
O
O
O
I
+
86 87
A 200 ml round bottom flask fitted with a stirbar, air condenser and nitrogen
bubbler was charged with DPP 1 (2.88 g, 10 mmol), potassium tert-butoxide (2.5 g, 22
mmol), dimethylacetamide (80 ml), and heated at 80°C for 2 hrs. The resulting mixture
was cooled to room temperature and a solution of 2-(6-iodohexyl)-3a,4,7,7a-tetrahydro-
4,7-epoxy-1H-isoindole-1,3(2H)-dione419 (7.54 g, 20 mmol) in dimethylacetamide (10

279
ml) was added at once and the reaction mixture was stirred at room temperature for 12
hrs, then at 100°C for 12 hrs, and cooled down. The solvent was evaporated to 50 ml,
hydrochloric acid (70 ml 2M) was added, the resulting precipitate was filtered off, air-
dried (5.11 g), dissolved in chloroform (150 ml), filtered, applied onto silica gel and
chromatographed with gradual elution from hexane – ethyl acetate (1:1) to neat ethyl
acetate. Evaporation of solvents afforded 0.59 g (7%) of Fu-MI-(CH2)6-DPP as red
crystals. 1H δ: 8.32 (m, 1H), 7.81 (m, 2H), 7.51-7.58 (m, 4 H), 6.49 (m, 2H), 5.23 (m,
1H), 3.81 (t, 2H), 3.41 (t, 2H), 2.81 (s, 1H), 1.64-1.47 (m, 8H). 13C δ: 176.4, 136.7,
132.3, 131.4, 129.4, 129.1, 128.9, 128.0, 81.0, 47.5, 42.1, 38.9, 29.5, 27.5, 26.4, 26.2.
3,6-Diphenyl-2,5-bis(6-[2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl]hexyl)pyrrolo[3,4-
c]pyrrole-1,4-dione 88.
N
O
O
ON
NO
O
N
O
O
O87
N
NO
O
N
O
O
N
O
O
88
A 100 ml round bottom flask fitted with a stirbar, air condenser and nitrogen
bubbler was charged with Fu-MI-(CH2)6-DPP (265 mg, 0.345 mmol), xylenes (mixture
of isomers, 70 ml) and refluxed for 24 hrs. The solvent was evaporated and the residue
was chromatographed with gradual elution from neat chloroform to 5% ethyl acetate in
chloroform. Evaporation of solvents gave orange glass (194 mg, 87%). Recrystallization

280
from dimethylacetamide (4 ml) gave 33 mg (14%) of orange crystals. 1H δ: 7.79 (dd,
2H), 7.54 (dd, 2H), 7.18 (m, 1 H), 6.66 (s, 2H), 3.74 (t, 2H), 3.44 (t, 2H), 1.53-1.39 (m,
8H). 13C δ: 171.0, 163.2, 148.1, 134.2, 131.3, 129.1, 128.8, 128.6, 128.2, 41.7, 37.7, 29.5,
27.5, 26.4, 26.2. ES-MS: 647.2 (M+1), 527.1. UV-Vis (CH2Cl2) λmax, nm (lg ε): 231
(3.98), 264 (3.79), 464 (3.31). Fluorescence λmax: 525, 565sh. ΦF=0.67.
3,6-Diphenyl-2-(6-[3a,4,7,7a-tetrahydro-4,7-epoxy-1,3-dioxo-1H-isoindol-2-yl]hexyl)-
5-(3-hydroxysulfonylpropyl)-pyrrolo[3,4-c]pyrrole-1,4-dione 89.
N
O
O
ON
NO
O
SO3H
NH NH
O
O
1N
O
O
O I
+
86
SO O
O
1
2
+89
A 100 ml round bottom flask fitted with a stirbar, air condenser and nitrogen
bubbler was charged with DPP 1 (1.44 g, 5 mmol), potassium tert-butoxide (1.2 g, 10.5
mmol), dimethylacetamide (50 ml), and heated at 80°C for 12 hrs. The resulting mixture
was cooled to room temperature and a solution of 1,3-propanesultone (0.61 g, 5 mmol) in
dimethylacetamide (20 ml) was added dropwise. After the addition was complete, the
reaction mixture was stirred at room temperature for 1 hr, and then at 140°C for 4 hrs,
and cooled back to room temperature. A solution of 2-(6-iodohexyl)-3a,4,7,7a-
tetrahydro-4,7-epoxy-1H-isoindole-1,3(2H)-dione (1.88 g, 5 mmol) in dimethylacetamide
(20 ml) was added at once and the reaction mixture was stirred at room temperature for

281
12 hrs, then at 120°C for 4 hrs, and cooled down. The solvent was evaporated to 50 ml,
hydrochloric acid (70 ml 2M) was added, the resulting precipitate was filtered off, air-
dried, dissolved in a mixture of chloroform and methanol (100 ml), applied onto silica gel
and chromatographed with gradual elution from hexane – ethyl acetate (1:1) to neat ethyl
acetate. Evaporation of solvents afforded 500 mg (15%) of 41 as red crystals. M.p.
165°C. 1H δ: 10.27 (s, 1H), 8.3 (m, 1H), 8.13 (m, 3H), 7.8 (m, 1H), 7.4-7.6 (m, 8 H), 6.49
(m, 1H), 5.24 (m, 1H), 3.81 (t, 2H), 3.41 (t, 2H), 2.82 (s, 1H), 1.65-1.14 (m, 12H). 13C δ:
176.5, 171.3, 164.2, 149.2, 144.8, 140.5, 136.7, 133.8, 132.3, 131.6, 130.3, 129.4, 129.2,
128.9, 128.6, 128.0, 81.0, 47.5, 42.1, 38.9, 32.1, 29.8, 27.5, 26.2, 26.1, 22.9, 14.3. UV-
Vis λmax: 470, 489. Fluorescence λmax: 524, 565sh.
3,6-Diphenyl-2,5-bis(4-perfluorobutylsulfonylphenyl)-pyrrolo[3,4-c]pyrrole-
1,4-dione 91.
N N
O
O
C4F9–SO2 SO2–C4F9NH NH
O
O
1 91
A 100 ml round bottom flask with a stirbar was charged with DPP 1 (720 mg,
2.5 mmol), copper (II) oxide (795 mg, 10 mmol), 1-bromo-4-(perfluorobutylsulfonyl)-
benzene (2.81 g, 7.0 mmol), and dimethylformamide (15 ml). The reaction mixture was
stirred and refluxed for three days, cooled down, poured into water (100 ml). The

282
precipitate formed was filtered off and treated with boiling ethyl acetate (5×70 ml).
Combined hot ethyl acetate solutions were filtered from insoluble matter, solvent
evaporated, and the residue was recrystallized from 1-propanol to give 786 mg (32%) of
43 as orange crystals. M.p. 262°C. 1H δ: 8.05 (d, 2H), 7.57-7.55 (m, 2H), 7.52-7.47 (m,
3H), 7.41-7.36 (m, 2H). 13C δ: 142.8, 132.6, 132.1, 130.8, 129.8, 129.1, 128.3, 126.4.
UV-Vis λmax: 271, 475. Fluorescence λmax: 515, 555sh. ΦF=0.91.
3,6-Diphenyl-2-(4-perfluorobutylsulfonylphenyl)-5(H)pyrrolo[3,4-c]pyrrole-
1,4-dione 92. The residue, insoluble in ethyl acetate was dissolved in hot
dimethylformamide, filtered off while hot and after cooling gave 297 mg (18%) of 92 as
orange crystals. M.p. 320°C. 1H (DMSO) δ: 11.48 (s, 1H), 8.46 (dd, 2H), 8.02 (d, 2H),
7.55-7.45 (m, 7H), 7.37-7.29 (m, 3H). 13C δ: 162.7, 160.0, 148.5, 143.5, 142.1, 132.3,
131.4, 130.6, 128.9, 128.7, 128.6, 128.3, 128.1, 127.2, 126.8. UV-Vis λmax: 461, 485,
490. Fluorescence λmax: 513, 554sh. ΦF=0.93.
3,6-Diphenyl-2,5-bis(3,5-bis[trifluoromethyl]phenyl)-pyrrolo[3,4-c]pyrrole-
1,4-dione 93 was prepared similar to 91, starting from DPP 1 (2.88 g, 10 mmol) and 1-
bromo-3,5-bis(trifluoromethyl)benzene (14.6 g, 50 mmol). Yield: 2.66 g (37%). M.p.
298°C. 1H δ: 7.85 (m, 2H), 7.66 (m, 4H), 7.57 (m, 2H), 7.55 (m, 3H), 7.50 (m, 2H), 7.47
(m, 1H), 7.43 (m, 2H), 7.41 (m, 2H), 7.38 (t, 1H). UV-Vis λmax: 251, 315, 475.
Fluorescence λmax: 520, 552sh. ΦF=0.95.

283
3,6-Diphenyl-2,5-bis(4-fluorophenyl)-pyrrolo[3,4-c]pyrrole-1,4-dione 94.
N N
O
O
F FNH NH
O
O
1 94
A 50 ml pear-shaped flask with a stir bar and a reflux condenser was charged
with DPP 1 (576 mg, 2 mmol), 1-bromo-4-fluorobenzene (1.05 g, 6 mmol), copper (I)
oxide (1.15 g, 8 mmol), and dimethylformamide (30 ml). The reaction mixture was
stirred and refluxed for 8 days. After the reaction was complete by TLC (CHCl3), it was
cooled, diluted with chloroform (70 ml), and applied onto silica gel. Flash-
chromatography on silica gel with CH2Cl2 as eluent afforded 115 mg (12%) of bright
yellow crystals. M.p. 344°C. 1H δ: 7.56-7.53 (m, 1H), 7.46-7.37 (m, 2H), 7.32-7.28 (m,
2H). 13C not available due to low solubility.
3,6-Diphenyl-2-(4-fluorophenyl)-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione 95.
N NH
O
O
FNH NH
O
O
1 95
A 100 ml pear-shaped flask with a stir bar and a reflux condenser was charged
with 3,6-diphenyl-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (2.88 g, 10 mmol), 1-iodo-

284
4-fluorobenzene (9.0 g, 40 mmol), copper (I) oxide (6.0 g, 42 mmol), and
dimethylformamide (60 ml). The reaction mixture was stirred and refluxed for 24 hours,
cooled down, diluted with chloroform (70 ml), filtered through #50 filter and applied
onto silica gel. Flash-chromatography with hot chloroform as eluent afforded, after
evaporation of solvent, 1.8 g (37%) of 48 as bright yellow crystals. M.p. 404°C. 1H δ:
10.4 (s, 1H), 7.64-7.56 (m, 4H), 7.36-7.31 (m, 4H), 7.23-7.07 (m, 6H).
2,5-Bis-(4-nitrophenyl)-3,6-diphenyl-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione 96.
N N
O
O
O2N NO2NH NH
O
O
1 96
A 100 ml round bottom flask with a stirbar was charged with DPP 1 (1.44 g, 5
mmol), potassium tert-butoxide, and dimethylacetamide (70 ml). The reaction mixture
was heated at 80°C for 30 min and the tert-butanol formed was removed under reduced
pressure. To the cooled reaction mixture, 4-fluoro-1-nitrobenzene (3.0 g, 21 mmol) was
added and the reaction mixture was stirred for 48 hrs at 80°C and then 96 hrs at 140°C.
The reaction mixture was cooled down, poured into water (150 ml) and filtered off. The
solid on the filter was air dried and recrystallized from dimethylacetamide (15 ml) to give
320 mg (12%) of 96 as orange crystals. M.p. 200°C. 1H (300 MHz) δ: 8.27 (d, 4H), 8.06

285
(d, 1H), 7.16 (d, 4H), 6.57 (d, 1H). 13C (75 MHz) δ: 160.8, 154.4, 144.3, 132.2, 129.0,
128.5, 127.9, 124.8, 124.4, 119.5, 110.3. UV-Vis λmax: 268, 470.
3,6-diphenyl-2-(2,4-dinitrophenyl)-5-propylpyrrolo[3,4-c]pyrrole-1,4-dione 97.
N N
O
O
Pr NO2N NH
O
O
Pr
33 97
A 100 ml round bottom flask with a stir bar was charged with 3,6-diphenyl-2-
propyl-5-hydropyrrolo[3,4-c]pyrrole-1,4-dione 21 (0.3 g, 0.91 mmol), cesium carbonate
(0.75 g, 2.3 mmol), 1-fluoro-2,4-dinitrobenzene (0.37 g, 2 mmol), and dimethylacetamide
(45 ml). The reaction mixture was stirred at 80°C and monitored by TLC (eluent neat
chloroform). After 8 hrs the mixture was cooled down, diluted with cold hydrochloric
acid (3 M, 50 ml), extracted with chloroform (3×50 ml) and applied onto silica gel.
Chromatography with 60 to 100% dichloromethane in hexane gave, after evaporation of
solvents and recrystallization from 1-propanol : chloroform (5 ml) 113 mg (25%) of
orange crystals. M.p. 234°C dec. ES-MS: 497 (M++1). 1H δ: 8.9 (d, 1H, J=2.5 Hz), 8.4
(dd, 1H, J1=8.8 Hz, J2=2.5 Hz), 7.8 (m, 2H), 7.6 (m, 2H), 7.5 (m, 3H), 7.4 (m, 3H), 7.3
(M, 1H), 3.8 (t, 2H), 1.7 (m, 2H), 0.9 (t, 3H). 13C δ: 162.7, 159.5, 151.9, 146.0, 145.6,
143.8, 135.0, 132.1, 131.8, 131.2, 129.1, 129.0, 128.9, 127.3, 126.7, 121.1, 113.9, 107.5,
43.9, 22.7, 11.2. UV-Vis λmax (lg ε): 263 (4.45), 298 (4.31), 466 (4.26).

286
Basic hydrolysis of DPP 1.
HOOC
COOHO
O
NH
NH
O
O
O OH
An autoclave was charged with DPP (4.35 g), sodium dodecylsulfate (0.1 g, as a
dispersinf agent), water (200 ml), potassium hydroxide (10 g) and ethanol (3 ml). The
reaction mixture was stirred in an autoclave at 150°C for 12 hrs and cooled down. The
colorless homogeneous solution formed was acidified with hydrochloric acid and the
precipitate formed was filtered off, washed with water and air-dried to give 1.25 g of gray
solid. The crude product was chromatographed with ethyl acetate and recrystallized from
1-propanol to give 1.2 g of white crystals, which were identified as benzoic acid. M.p.
121.5°C. 1H NMR (acetone-d6) δ: 8.1 (dd, 2H), 7.6 (td, 1H), 7.5 (t, 2H). 13C NMR
(acetone-d6) δ: 168.7, 134.0, 131.2, 130.6, 129.4.
6-[(3-carboxypropanoyl)amino]caproic acid.
OHNH
O
O
OH
OO
O
O
NH2 OH
O+
A 500 ml round bottom flask with a stir bar was charged with ω-aminocaproic
acid (13.1 g, 0.1 mol), maleic anhydride (9.8 g, 0.1 mol), and acetic acid (400 ml). The
reaction mixture was stirred at room temperature for 24 hrs, the precipitate formed was

287
filtered off on a Büchner funnel, washed with water (200 ml), and air-dried to give 22.3
(97%) of maleamic acid 101. M.p. 170 °C. Lit.420 m.p. 171–173 °C.
6-(maleimid-2-yl)caproic (6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)caproic) acid 101.
N
O
O
OH
OOHNH
O
O
OH
O
A 500 ml round bottom flask with a stir bar was charged with . maleamic acid
101 (12.3 g, 0.05 mol), acetic anhydride (50 ml), and sodium acetate (2.5 g, 0.03 mol).
The reaction mixture was stirred at 90 °C for 2 hrs, cooled, and poured onto ice (100 g).
The aqueous solution was extracted with ether (3×50 ml) and the extracts were
impregnated onto silica gel. Chromatography with hexane – ethyl acetate (1:2), followed
by recrystallization from isopropanol–water, gave (49%) of ω-maleimidylcaproic acid as
white crystals. M.p. 88–89.5 °C. Lit.420 m.p. 88–91 °C. 1H NMR (CDCl3) δ: 11.5 (s, br,
1H), 6.7 (s, 2H), 3.5 (t, 2H), 2.3 (t, 2H), 1.6 (m, 6H), 1.3 (m, 2H). 13C NMR (CDCl3) δ:
180.0, 170.2, 135.1, 38.9, 34.7, 28.0, 26.0, 24.0. IR (neat solid, HATR) ν, cm–1: 3429,
3313, 3051, 2946, 2869, 1708, 1624, 1570, 1512. ES-MS: 210.15 (M+). Analysis
(found/calc): C 56.67/56.86, H 6.10/6.20, N 7.50/6.63.
1,6-Diiodohexane
OH OH I I

288
A one liter three neck round bottom flask was charged with polyphosphoric acid
(H3PO4·½P2O5, 150 g), o-phosphoric acid (85% aq., 200 g), potassium iodide (342 g,
2.06 mol), and 1,6-hexanediol (118 g, 1 mol). The flask was fitted with a mechanical
stirrer, reflux condenser, and a nitrogen/thermometer adapter and placed into a heating
mantle. The reaction mixture was stirred at 120-130°C for five hours under nitrogen
while dense oil separates at the bottom of the flask. The reaction mixture was cooled to
room temperature, water (400 ml) and ether (300 ml) were added and the contents
transferred to extraction funnel. The separated ether layer was washed with water (200
ml), dried with MgSO4 and ether evaporated on rotavap. The dark oily residue (297 g,
88%) was mixed with copper powder (2 g) and vacuum distilled (100-110°C at 0.5-0.3
mm) to give 271 g (80%) of clear product. 20Dn 1.583. Notes: (1) Mixture of o-phosphoric
and polyphosphoric acids must be allowed to cool down to room temperature before KI
addition to avoid excessive loss of HI and I2 evolution, if exposed to air. (2) Vaccum
distillation w/o copper powder produces a dark brown distillate.
1-(6-Iodohexyl)maleimide 102
N
O
O
I
A 250 ml round bottom flask with a stir bar was charged with 1-(6-
hydroxyhexyl)maleimide 103 (1.9 g, 0.01 mol), chloroform (20 ml), and carbon disulfide
(20 ml). To that stirred mixture, a solution of diphosphorus tetraiodide (1.42 g, 2.5 mmol)

289
in carbon disulfide (100 ml) was added at once. After 30 min of stirring, solid potassium
carbonate (5.0 g), followed by sat. aq. solution of potassium carbonate (30 ml) were
added. The layers were separated and the aqueous layer was extracted with ether (3×50
ml). The combined organic layers were dried with MgSO4, solvents evaporated, and the
oily residue was filtered through a short silica pad, washing it with hexane – ethyl acetate
(1:2). The filtrate, after removal of the solvents, gave 2.1 g (68 %) of 1-(6-
iodohexyl)maleimide as colorless oil. The product is unstable and should be used
immediately or may be stored under argon in freezer for a few days. 1H NMR (CDCl3) δ:
6.6 (s, 2H), 3.4 (t, 2H), 3.2 (t, 2H), 1.5 (m, 8H). 13C NMR (CDCl3) δ: 175.6, 136.4, 47.6,
33.3, 30.0, 27.5, 25.5, 7.0. The same compound was also prepared by an alternative
procedure from maleimide silver salt MI-Ag and 1,6-diiodohexane.
1-(6-Hydroxyhexyl)maleimide 103.
N
O
O
OH
1-(6-hydroxyhexyl)maleimide was prepared similar to 1-(6-iodohexyl)male-
imide, starting from maleimide silver salt MI-Ag and 6-bromo-1-hexanol. 1H NMR
(CDCl3) δ: 6.7 (s, 2H), 3.5 (t, 2H), 3.4 (t, 2H), 1.5 (pentet, 2H), 1.4 (pentet, 2H), 1.3 (m,
4H). 13C NMR (CDCl3) δ: 175.4, 135.6, 61.8, 46.5, 38.6, 33.3, 28.0, 27.4, 26.5.

290
5-(diethylamino)-2-nitrosophenol 104.
Et2N OH
N O
Et2N OH104
A one-liter beaker, equipped with a mechanical stirrer, was charged with 3-
(N,N-diethylamino)phenol (50.0 g, 0.3 mol), ice (100 g), and hydrochloric acid (aq.,
12M, 130 ml). The beaker was immersed into an ice bath and the mixture was stirred
until a homogeneous solution was formed. To that stirred solution, a solution of sodium
nitrite (22.0 g, 0.32 mol) in water (200 ml), precooled to 5 °C, was added with vigorous
stirring at such a rate that temperature of the reaction mixture did not rise above 5 °C.
After the addition was complete, the reaction mixture was stirred for additional 6 hrs, and
filtered off on a Büchner funnel. The solid on filter was washed with cold water (2×100
ml), ethanol (2×100 ml, precooled to –20 °C) and vacuum dried at room temperature to
give 68.2 g (97%) of 5-(diethylamino)-2-nitrosophenol as hydrochloride salt. The product
decomposes above 50 °C and its recrystallization is thus not recommended. The product
had the same Rf = 0.67 (EtOAc) as the authentic commercial sample from TCI America.
9-Diethylamino-2-hydroxy-5H-benzo[a]phenoxazin-5-one (Nile Red Phenol) 105.
Et2N OH
N O
OH
OH
+N
OEt2N O
OH
105104

291
A 500 ml round bottom flask with a stir bar was charged with 5-(diethylamino)-
2-nitrosophenol hydrochloride (9.2 g, 0.04 mol), 1,6-dihydroxynaphthalene (6.0 g, 0.375
mol), and DMF (200 ml). The flask was topped with a reflux condensor and the reaction
mixture was stirred at reflux for 6 hrs. The reaction progress was monitored by TLC
(hexane : ethyl acetate = 1:2, target’s Rf = 0.48) and at least eight colored products can be
distinguished. After the reaction was complete, DMF was removed on a rotovap with an
oil pump, the residue was dissolved in methanol and impregnated onto silica gel.
Repeated chromatographies, performed on a dry-packed columns (Note 1) with hexane :
ethyl acetate = 1:1 (target’s Rf = 0.25) gave crude Nile Red Phenol, which was further
purified by several recrystallizations from 1-propanol to give 5.4 g (43%) of pure
material. IR (neat solid, HATR) ν, cm–1: 3375, 2964, 1645, 1590, 1561. UV-Vis (MeOH)
λmax (lg ε): 210 (4.37), 265 (4.44), 326 (4.76), 547 (4.60). Fluorescence λmax = 632 nm.
1H NMR (DMSO-d6) δ: 10.4 (s, br, 1H), 7.96 (d, 1H, J = 8.7 Hz), 7.87 (d, 1H, J = 2.4
Hz), 7.56 (d, 1H, J = 9.0 Hz), 7.08 (dd, 1H, J1 = 8.6 Hz, J2 = 2.5 Hz), 6.78 (dd, 1H, J1 =
9.0 Hz, J2 = 2.5 Hz), 6.61 (d, 1H, J = 2.5 Hz), 6.14 (s, 1H), 3.48 (q, 4H, J = 7.0 Hz), 1.15
(t, 6H, J = 7 Hz). 13C NMR (DMSO-d6) δ: 181.6, 160.6, 151.5, 150.6, 146.3, 138.6,
133.7, 130.8, 127.5, 123.8, 118.3, 109.9, 108.2, 107.9, 104.1, 96.0, 44.4, 12.4.

292
9-Diethylamino-2-(6-hydroxyhexyloxy)-5H-benzo[a]phenoxazin-5-one 106.
N
OEt2N O
OH
105 106
N
OEt2N O
OOH
A 125 ml round bottom flask with a stir bar was charged with Nile Red Phenol
105 (0.5 g, 1.5 mmol), 6-bromo-1-hexanol (0.8 g, 4.4 mmol), potassium carbonate (0.45
g, 3.3 mmol), potassium iodide (50 mg), and DMF (50 ml). The reaction mixture was
heated at 80 °C for 14 hrs and monitored by TLC (hexane : ethyl acetate = 1:2). After the
reaction was complete, DMF was evaporated on a rotovap with an oil pump, the residue
was dissolved in ethyl acetate (50 ml), filtered from insolubles, and impregnated onto
silica gel. Chromatography in hexane : ethyl acetate with gradual elution from = 1:1 to
1:2, followed by recrystallization from iso-octane/1-propanol, gave 0.5 g (71%) of 9-
diethylamino-2-(6-hydroxyhexyloxy)-5H-benzo[a]phenoxazin-5-one as dark red crystals.
Analysis (found/calc): C 68.21/71.87, H 6.93/6.96, N 6.17/6.45. 1H NMR (CDCl3) δ:
8.21 (d, 1H, J = 8.7 Hz), 8.04 (d, 1H, J = 2.4 Hz), 7.60 (d, 1H, J = 9.0 Hz), 7.16 (dd, 1H,
J1 = 8.6 Hz, J2 = 2.5 Hz), 6.65 (dd, 1H, J1 = 9.0 Hz, J2 = 2.5 Hz), 6.46 (d, 1H, J = 2.5 Hz),
6.30 (s, 1H), 4.17 (t, 2H, J = 6.3 Hz), 4.1 (t, 2H, J = 6.6 Hz), 3.47 (q, 4H, J = 7.2 Hz),
1.26 (t, 6H, J = 7.2 Hz). 13C NMR (DMSO-d6) δ: 183.4, 161.9, 152.2, 150.9, 147.0,
140.1, 134.2, 131.2, 127.9, 125.8, 124.9, 118.4, 109.7, 106.7, 105.4, 96.4, 68.4, 64.7,
45.3, 29.3, 28.8, 26.0, 21.2, 12.8.

293
N-(Hydroxymethyl)maleimide 107.
NH
O
O
+ N
O
OOH
CH2O
To a suspension of maleimide (9.8 g, 0.1 mol) in formaldehyde (37% aq., 8.1
ml) at 30°C was added NaOH (5% aq., 0.3 ml). Within 10 min all maleimide had
dissolved and a mildly exothermic reaction ensued, rising the temperature to 35 °C.
Separation of the product began promptly. After 2.5 hrs at room temperature the reaction
mixture was filtered to yield 9.6 g (75%) of product m.p. 100–102°C. Once recrystallized
from EtOAc gave m.p. 103-104°C. Lit.421 m.p. 104–106°C. 1H NMR (DMSO-d6) δ: 7.0
(s, 2H), 6.23 (t, 1H, J = 7.1 Hz), 4.79 (d, 2H, J = 7.1 Hz).
N-(2-hydroxyethyl)maleimide.
NH
O
O
N
O
O
OHNH2 OH+
A half a liter round bottom flask with a stir bar was charged with maleic
anhydride (20 g, 0.20 mol), benzene (240 ml), and 2-aminoethanol (13 g, 0.21 mol). The
flask was fitted with a Dean-Stark trap and the reaction mixture was refluxed for 2 hrs
while separating water in the trap. After the reaction has been finished (no cloudy
condensate in the trap), benzene was evaporated on rotavap and the yellow oil (28 g) was
recrystallized from methanol (200 ml) to give 3.2 g (11%) of clear oil. 1H NMR (DMSO-

294
d6) δ: 7.01 (s, 2H), 6.23 (t, 1H), 4.79 (d, 2H), 3.47 (m, 2H). 13C NMR (DMSO-d6) δ:
171.01, 134.40, 57.87, 39.88. The yield is comparable to that in the literature.422
Diphosphorus tetraiodide P2I4. P4(α) + 4 I2 = 2 P2I4.
Commercial carbon tetrachloride was purified (Note 1) from traces of sulfur by
shaking it with metallic mercury, drying with CaH2 (no alkali metals can be used!), and
then distilling on a rotovap. The water-cooled condensor was replaced with a finger
condensor, filled with dry ice-acetone mixture and distillation was done under argon.
White phosphorus (20.0 g, 0.161 mol of P4) was washed with acetone, ether,
dried under argon, weighed into an argon-flushed flask with purified carbon disulfide
(100 ml), and filtered through an Acrodisk micron filter under argon pressure (do not use
syringe!); some additional CS2 was used to wash the filter. Iodine (164.0 g, 0.645 mol)
was dissolved in purified carbon disulfide (800 ml) with help of sonication. Solubility of
I2 in CS2 at 25 °C is 197 g I2 per 1000 g CS2, but it safer to use excess of CS2 to avoid
crystallization.
A two-liter, three-neck ChemGlass reaction vessel, equipped with a mechanical
stirrer, an efficient reflux condensor, and a dropping funnel, was flushed with argon and
charged with white phosphorus (20.0 g, 0.161 mol of P4) solution in carbon disulfide
(100 ml). Iodine (164.0 g, 0.645 mol) solution in purified carbon disulfide (800 ml) was
added from the addition funnel during 30 min period of time, with stirring. An
exothermic reaction starts almost immediately. After the addition was complete, the
reaction mixture was stirred for 24 hrs, and cannula transferred under argon into a two-

295
liter pear-shaped flask. The solvent was evaporated under argon on a rotovap (Note 2)
until signs of P2I4 crystallization started to appear. The solution was then removed from
the rotovap and left for crystallyzation – first, at room temperature and then in a fridge.
The precipitate was filtered off on a Büchner funnel and recrystallized once more from
clean CS2 to give 79.8 g of crop 1. The combined mother liquors were evaporated, left to
crystallize, and the precipitate was recrystallized from fresh CS2 (Note 3) to give 62.9 g
of crop 2. The combined yield was 142.7 g (78%) of P2I4 as red-orange crystals. M.p.
126.8 °C. Lit.423 m.p. 124.5 °C. The material should be stored in a fridge under argon
(sensitive to moisture, but not air).
Note 1. If unpurified CS2 has been used, the m.p. of resulting P2I4 was 110 °C.
Note 2. The recovered CS2 was mixed with Ph3P to remove iodine, dried with CaH2, and
redistilled. Note 3. Heat the solution very gently, for P2I4 decomposes very easily to PI3
and β-P4. For the same reason, m.p. depends on the heating rate: the higher the rate, the
higher recorded m.p. value.
N-(Iodomethyl)maleimide 108.
N
O
O
OHN
O
O
I
107 108
N-(Iodomethyl)maleimide was prepared similar to 1-(6-iodohexyl)maleimide
102 from 107 and P2I4 in 37% yield as yellowish liquid. 1H NMR (CDCl3) δ: 6.8 (s, 1H),

296
5.3 (s, 1H). 13C NMR (CDCl3) δ: 175.5, 136.5, 10.5. The compound may be stored under
argon in freezer for a week. Readily decomposes on open air and at room temperature.
Maleimid-2-yl-acetic acid (6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)acetic acid) 109.
N
O
O
CH2—COOHO
O
O
NH2 OH
O
+OHNH
O
O
OH
O
109
Maleimid-2-yl-acetic acid was prepared similar to 6-(maleimid-2-yl)caproic acid
101, starting from glycine and maleic anhydride, in 44% overall yield. M.p. 110 °C (from
water, lit.424 m.p. 111–113.5 °C). 1H NMR (D2O) δ: 6.7 (s, 2H), 4.2 (s, 2H). 13C NMR
(D2O) δ: 172.5, 171.7, 130.5, 39.4,
Silver salt of maleimide MI–Ag.
NH
O
O
N
O
O
Ag
MI-AgMI
A one liter Erlenmeyer flask with a stir bar was charged with a solution of
maleimide (7.1 g, 0.073 mol) in anhydrous ethanol (300 ml) and a solution of silver
nitrate (12.4 g, 0.073 mol) in DMSO (50 ml). To that mixture, a solution of sodium
hydroxide (aq., 0.4 M, 182.5 ml, 2.92 g NaOH) was added dropwise during 2 hrs at
vigorous stirring. The precipitate formed was filtered off on a Büchner funnel, washed

297
with ethanol (100 ml), water (250 ml), methanol (200 ml), acetone (200 ml). The product
was air-dried, followed by vacuum drying in a desiccator over P2O5 at 20 mm Hg to give
14.0 g (94%) of MI-Ag.
6-(Maleimid-2-yl)caproyl chloride (6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanoyl
chloride) 111.
N
O
O
OH
ON
O
O
Cl
O
A 125 ml round bottom flask with a stir bar was charged with thionyl chloride
(45 ml) and the flask was cooled in a dry ice – acetone bath to –10 °C. To the cooled
thionyl chloride, 6-(maleimid-2-yl)caproic acid 101 (2.0 g, 9.5 mmol) was added at
stirring, followed by DMF (1 drop). The reaction mixture was allowed to warm up and
then was heated for 3 hrs at 80 °C. Thionyl chloride was evaporated on a rotovap and its
traces were removed by azeotropic distillation with benzene (2×80 ml). The residue was
dissolved in dichloromethane (25 ml) and filtered through a thin pad of silica gel on a
Büchner funnel to give, after solvent removal, 2.0 g (92%) of colorless liquid, which was
directly utilized in acylation.
N-(4-hydroxyphenyl)maleimide (1-(4-hydroxyphenyl)-1H-pyrrole-2,5-dione) 112
was prepared similar to 6-(maleimid-2-yl)caproic acid 101, starting from 4-aminophenol
and maleic anhydride and substituting acetic anhydride for acetyl chloride in the

298
dehydration step, in overall yield of 68%. M.p. 185 °C (lit. m.p. 154425…195426). 1H
NMR (DMSO-d6) δ: 9.9 (br, 1H), 7.10 (d, 2H), 7.05 (d, 2H), 6.8 (s, 2H). 13C NMR
(DMSO-d6) δ: 171.2, 157.9, 135.4, 129.3, 123.3, 116.3.
N-(6-iodohexyl)maleimide (1-(6-iodohexyl)-1H-pyrrole-2,5-dione) 113.
N-(6-iodohexyl)maleimide was prepared similar to 1-(6-iodohexyl)maleimide
102, starting from maleimide silver salt MI-Ag and 1,6-diiodohexane, in 96% yield. 1H
NMR (CDCl3) δ: 6.6 (s, 2H), 3.4 (t, 2H), 3.2 (t, 2H), 1.5 (m, 8H). 13C NMR (CDCl3) δ:
175.6, 136.4, 47.6, 33.3, 30.0, 27.5, 25.5, 7.0.
9-Diethylamino-2-methoxy-5H-benzo[a]phenoxazin-5-one 115.
N
OEt2N O
OH
105
N
OEt2N O
OMe
115
A 250 ml round bottom flask with a stir bar was charged with Nile Red Phenol
(0.5 g, 1.5 mmol), iodomethane (0.25 g, 1.76 mmol), potassium carbonate (0.96 g, 7
mmol), and DMF (70 ml). The reaction mixture was stirred at 80 °C for 3 hrs and
monitored by TLC (ethyl acetate – hexane 2:1, product Rf = 0.57). After the reaction was
complete, the reaction mixture was cooled down and applied directly to a silica gel
column. Chromatography with ethyl acetate – hexane (1:1) gave, after evaporation of
solvents and recrystallization from 1-propanol, 0.43 g (77%) of dark red crystals. IR (neat
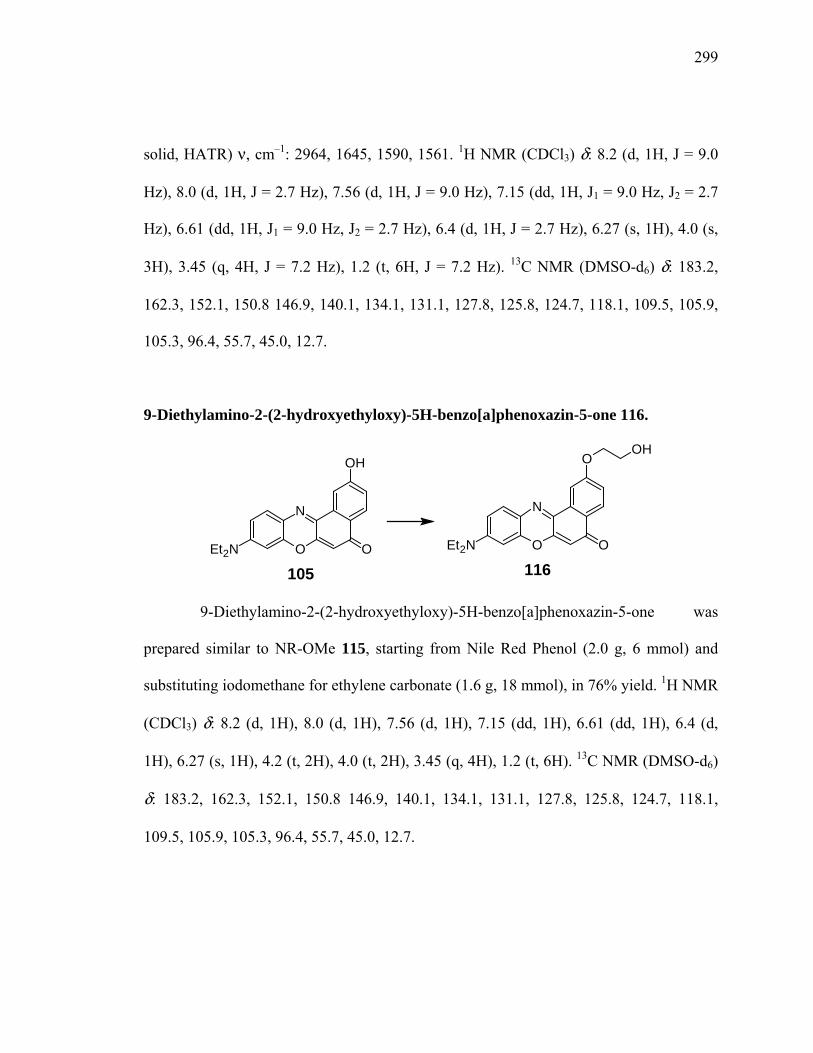
299
solid, HATR) ν, cm–1: 2964, 1645, 1590, 1561. 1H NMR (CDCl3) δ: 8.2 (d, 1H, J = 9.0
Hz), 8.0 (d, 1H, J = 2.7 Hz), 7.56 (d, 1H, J = 9.0 Hz), 7.15 (dd, 1H, J1 = 9.0 Hz, J2 = 2.7
Hz), 6.61 (dd, 1H, J1 = 9.0 Hz, J2 = 2.7 Hz), 6.4 (d, 1H, J = 2.7 Hz), 6.27 (s, 1H), 4.0 (s,
3H), 3.45 (q, 4H, J = 7.2 Hz), 1.2 (t, 6H, J = 7.2 Hz). 13C NMR (DMSO-d6) δ: 183.2,
162.3, 152.1, 150.8 146.9, 140.1, 134.1, 131.1, 127.8, 125.8, 124.7, 118.1, 109.5, 105.9,
105.3, 96.4, 55.7, 45.0, 12.7.
9-Diethylamino-2-(2-hydroxyethyloxy)-5H-benzo[a]phenoxazin-5-one 116.
N
OEt2N O
OH
105
N
OEt2N O
OOH
116
9-Diethylamino-2-(2-hydroxyethyloxy)-5H-benzo[a]phenoxazin-5-one was
prepared similar to NR-OMe 115, starting from Nile Red Phenol (2.0 g, 6 mmol) and
substituting iodomethane for ethylene carbonate (1.6 g, 18 mmol), in 76% yield. 1H NMR
(CDCl3) δ: 8.2 (d, 1H), 8.0 (d, 1H), 7.56 (d, 1H), 7.15 (dd, 1H), 6.61 (dd, 1H), 6.4 (d,
1H), 6.27 (s, 1H), 4.2 (t, 2H), 4.0 (t, 2H), 3.45 (q, 4H), 1.2 (t, 6H). 13C NMR (DMSO-d6)
δ: 183.2, 162.3, 152.1, 150.8 146.9, 140.1, 134.1, 131.1, 127.8, 125.8, 124.7, 118.1,
109.5, 105.9, 105.3, 96.4, 55.7, 45.0, 12.7.

300
9-Diethylamino-2-(6-iodohexyloxy)-5H-benzo[a]phenoxazin-5-one 117.
N
OEt2N O
OH
105
N
OEt2N O
OI
117
A 50 ml pear-shaped flask was charged with a stirring bar, Nile Red Phenol (9-
diethylamino-2-hydroxy-5H-benzo[a]phenoxazin-5-one 105, 66 mg, 0.2 mmol), 1,6-
diiodohexane (134 mg, 0.4 mmol), potassium hydroxide (55 mg, 0.4 mmol), and
dimethylacetamide (10 ml). The reaction mixture was stirred at 90°C for one hour (most
of the starting material was consumed during first several minutes, as monitored by TLC
with 1:1 eluent of hexane and ethyl acetate). The solvent was evaporated and the residue
was chromatographed on silica gel with 1:1 mixture of hexanes and ethyl acetate. Yield
74 mg (79%) of dark ruby red material, which is used directly in next step. 1H NMR
(CDCl3), δ: 8.16 (d, 1H, J=9Hz); 7.97 (d, 1H, J=2.4Hz); 7.56 (d, 1H, J=9Hz); 7.13 (dd,
1H, J1=9Hz, J2=2.4Hz); 6.68 (dd, 1H, J1=9Hz, J2=2.4Hz); 6.43 (d, 1H, J=2.4Hz); 6.37 (s,
1H); 4.15 (t, 2H, J=6.6Hz); 3.47 (q, 4H, J=7.2Hz); 3.19 (t, 2H, J=6.6Hz); 1.84 (m, 2H);
1.54 (m, 2H); 1.42 (m, 2H); 1.25 (t, 6H, J=7.2Hz). 13C NMR (CDCl3), δ: 182.2, 162.0,
152.1, 151.3, 147.2, 139.5, 139.4, 134.2, 131.5, 127.8, 125.8, 125.2, 118.5, 110.7, 106.7,
104.8, 96.7, 68.4, 45.6, 33.6, 29.9 (double intensity), 29.6, 12.8, 7.0.
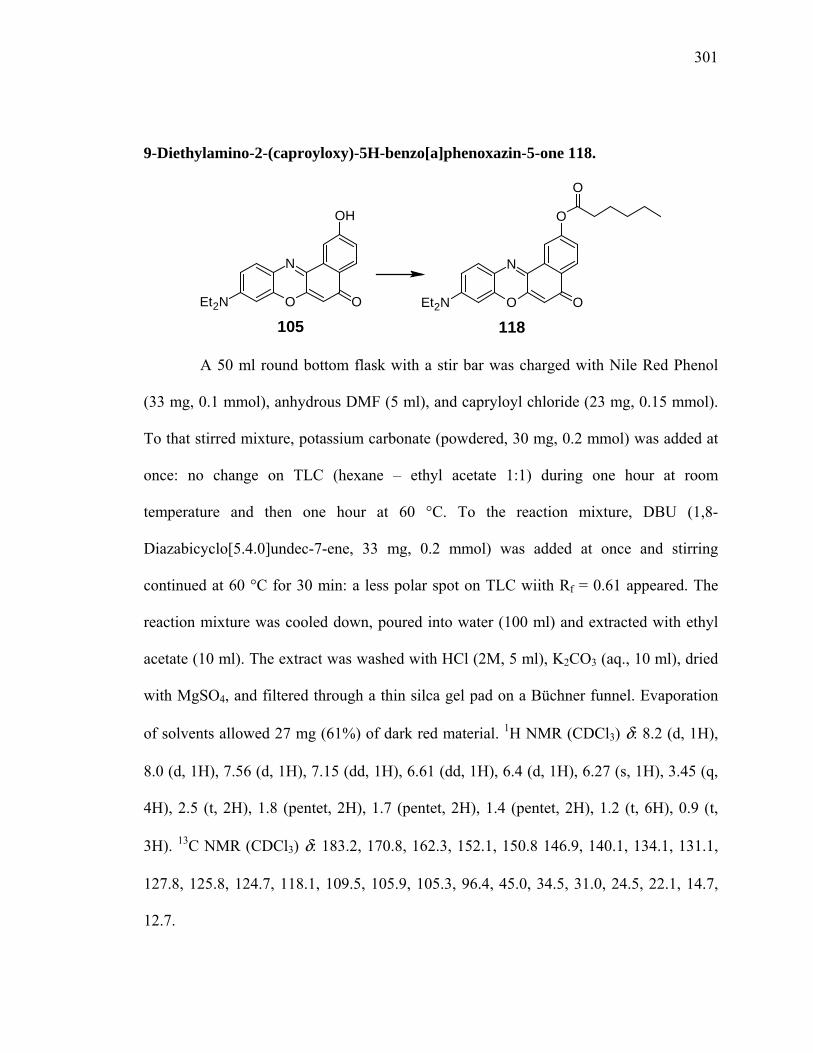
301
9-Diethylamino-2-(caproyloxy)-5H-benzo[a]phenoxazin-5-one 118.
N
OEt2N O
OH
105
N
OEt2N O
O
O
118
A 50 ml round bottom flask with a stir bar was charged with Nile Red Phenol
(33 mg, 0.1 mmol), anhydrous DMF (5 ml), and capryloyl chloride (23 mg, 0.15 mmol).
To that stirred mixture, potassium carbonate (powdered, 30 mg, 0.2 mmol) was added at
once: no change on TLC (hexane – ethyl acetate 1:1) during one hour at room
temperature and then one hour at 60 °C. To the reaction mixture, DBU (1,8-
Diazabicyclo[5.4.0]undec-7-ene, 33 mg, 0.2 mmol) was added at once and stirring
continued at 60 °C for 30 min: a less polar spot on TLC wiith Rf = 0.61 appeared. The
reaction mixture was cooled down, poured into water (100 ml) and extracted with ethyl
acetate (10 ml). The extract was washed with HCl (2M, 5 ml), K2CO3 (aq., 10 ml), dried
with MgSO4, and filtered through a thin silca gel pad on a Büchner funnel. Evaporation
of solvents allowed 27 mg (61%) of dark red material. 1H NMR (CDCl3) δ: 8.2 (d, 1H),
8.0 (d, 1H), 7.56 (d, 1H), 7.15 (dd, 1H), 6.61 (dd, 1H), 6.4 (d, 1H), 6.27 (s, 1H), 3.45 (q,
4H), 2.5 (t, 2H), 1.8 (pentet, 2H), 1.7 (pentet, 2H), 1.4 (pentet, 2H), 1.2 (t, 6H), 0.9 (t,
3H). 13C NMR (CDCl3) δ: 183.2, 170.8, 162.3, 152.1, 150.8 146.9, 140.1, 134.1, 131.1,
127.8, 125.8, 124.7, 118.1, 109.5, 105.9, 105.3, 96.4, 45.0, 34.5, 31.0, 24.5, 22.1, 14.7,
12.7.

302
Exo-cis-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic anhydride Fu-MA 119.
O
O
O
O + OO
O
O
H
H
Maleic anhydride (98 g, 1 mol) was placed in a 2 liter Erlenmeyer flask,
dissolved in 900 ml of ether, furan (100 ml, 1.34 mol) added, the flask was sealed with
rubber stopper and aluminum foil and left undisturbed for a week. Crystals formed were
filtered and air-dried giving 152.5 g (91.8%) of the exo-cis Diels-Alder adduct.
Recrystallization of the direct product is not recommended since fine crystals give too
violent reaction with ammonia. M.p. 116.5°C with decomposition. Analysis
(Found/Calc): C 57.59/57.84; H 3.71/3.64.
Exo-cis-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboximide Fu-MI 120.
(10-oxa-4-azatricyclo[5.2.1.02,6]dec-8-ene-3,5-dione)
OO
O
O
H
H
NHO
O
O
H
H
(a) A one liter pear-shaped round bottom flask was charged with exo-cis-7-
oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic anhydride (150 g, 0.9 mol), 150 ml of ice-
cold water and a stirring bar. Aqueous ammonia (450 ml of 29% solution, 3.7 mol) was
added dropwise to the stirred suspension, then the flask was equipped with a reflux
condenser, immersed in an oil bath with temperature 130-140°C and refluxed for 45 min
(at the end of this time period the temperature of the reaction mixture rises to

303
101…103°C). Longer reaction time and higher bath temperature cause decomposition of
the product and reduce the yield. After cooling most of the solvent was rotoevaporated
and 95% ethanol was added in such amount that all solid dissolved at boiling. The
solution was cooled slowly, precipitated crystals filtered off, washed with 85% ethanol,
then absolute ethanol and vacuum dried. Concentration of the mother liquors allowed
additional crop of the crystals, combined yield is 140.6 g (94%). M.p. 157.5°C. Analysis
(Found/Calc): 57.50/58.18; H 4.33/4.27; N 8.43/8.48. Rf = 0.34 (hexane : ethyl acetate =
1:1, visualized with KMnO4).
Fu-MI [42074-03-3] 120.
NH
O
O
O + NHO
O
O
H
H
120
Maleimide (7.4 g, 76 mmol) was placed in a 250 ml round bottom flask,
dissolved in 150 ml of ether, furan (8.5 ml, 116 mmol) was added, the flask was covered
with aluminum foil and left undisturbed for 24 hrs. Crystals formed were filtered and air-
dried to give 11.8 g (94%) of the Diels-Alder adduct. M.p. 168.3°C (lit.427 m.p. 162 °C
for exo isomer, 126–128°C for endo isomer, and 126–128°C for mixture thereof.) IR
(cm–1): 3028, 3012, 2970, 1738, 1726, 1365, 1353, 1287, 1229, 1216, 1205, 1142, 1090,
1018, 927, 896, 854, 821, 791, 733, 688, 633, 582. Analysis (Found/Calc): 58.22/58.18;
H 4.29/4.27; N 8.64/8.48. Rf = 0.34 (hexane : ethyl acetate = 1:1, visualized with
KMnO4).

304
N-(6-iodohexyl)-exo-cis-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboximide 121.
(4-(6-iodohexyl)-10-oxa-4-azatricyclo[5.2.1.02,6]dec-8-ene-3,5-dione)
CAS RN [874998-65-9]
NHO
O
O
H
H
120
NO
O
O
I
H
H
121
Exo-cis-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboximide (10.0 g, 0.06 mol)
was dissolved in 220 ml of acetone, then 1,6-diiodohexane (23.7 g, 0.07 mol) and
potassium carbonate (16.6 g, 0.12 mol) were added and the mixture was refluxed for 12
hours upon which time the color gradually changes to deep red. Acetone was evaporated
and 50 ml of chloroform added, the solution was filtered off of insoluble inorganic salts,
applied to a dry-packed silica gel column (ID 40 mm, l=30 cm) and eluted with hexane :
ethyl acetate mixture = 3:1 , then 1:1 composition. The former elutes out 1,6-
diiodohexane, and the latter elutes the product. One may avoid chromatography and
increase the yield if the solid obtained after evaporation of the chloroform filtrate is
thoroughly washed with cold (–20°C) hexane and then recrystallized from hexane –
chloroform mixture. Yield 6.2 g (27%). M.p. 71.5°C (DSC, 5°C/min). Analysis
(Found/Calc): C 44.84/44.82; H 4.83/4.84; N 3.78/3.73. Rf = 0.47 (hexane : ethyl acetate
= 1:1). 1H NMR, δ: 6.52 (s, 2H); 5.25 (s, 2H); 3.46 (t, 2H, 3J=6 Hz); 3.17 (t, 2H, 3J=6
Hz); 2.84 (s, 2H); 1.8 (pentet, 2H, 3J=6 Hz); 1.54 (pentet, 2H, 3J=6 Hz); 1.38, 1.27 (m,
4H, 3J=6 Hz). 13C NMR, δ: 176.4; 136.7; 81.1; 47.6; 38.9; 33.4; 30.1; 27.5; 25.7; 7.1.

305
4-[6-(9-Diethylamino-5-oxo-5H-benzo[a]phenoxazin-2-yloxy)-hexyl]-10-oxa-4-aza-
tricyclo[5.2.1.02,6]dec-8-ene-3,5-dione [874998-66-0] 122.
N
OEt2N O
ONO
O
O
H
H
122
Diels-Alder protected Nile Red maleimide 122 was prepared by two methods.
(a) A 125 ml round-bottom flask with a stirring bar was charged with Nile Red phenol (9-
diethylamino-2-hydroxy-5H-benzo[a]phenoxazin-5-one, 370 mg, 1.1 mmol), 2-(6-
iodohexyl)-3a,4,7,7a-tetrahydro-4,7-epoxy-1H-isoindole-1,3(2H)-dione (500 mg, 1.33
mmol), potassium carbonate (400 mg, 2.9 mmol), and dimethylformamide (3 ml). The
reaction mixture was stirred at 65°C for 12 hours and then chromatographed on silica gel
with 1:1 mixture of hexane and ethyl acetate. After evaporation of the solvent, the residue
was washed with distilled hexane (10 ml × 2), and dried in vacuum, Yield 418 mg (65%).
1H NMR (CDCl3), δ: 8.18 (d, 1H, J=9Hz); 8.01 (d, 1H, J=2.4Hz); 7.57 (d, 1H, J=9Hz);
7.14 (dd, 1H, J1=9Hz, J2=2.4Hz); 6.63 (dd, 1H, J1=9Hz, J2=2.4Hz); 6.50 (d, 1H,
J=0.9Hz); 6.42 (s, 1H), 6.27 (d, 1H, J=0.9Hz); 5.26 (s, 2H), 4.14 (t, 2H, J=6.6Hz); 3.53-
3.41 (m, 6H, overlap of N1–H (q) and N2–H (t)); 2.88 (s, 2H); 1.85 (m, 2H); 1.75-1.52
(m, 4H); 1.25 (t, 6H, J=6.6Hz). 13C NMR (CDCl3), δ: 183.3, 176.3, 161.8, 152.1, 150.7,
146.6, 140.1, 136.5 (double intensity), 134.1, 131.1, 127.7, 125.6, 124.7, 118.3, 109.5,
106.7, 105.3, 96.3, 80.9, 68.2, 49.9, 47.4, 38.9, 25.6, 26.4, 27.5, 29.0, 12.7.

306
(b) Same as (a) except that 9-Diethylamino-2-(6-iodohexyloxy)-5H-benzo[a]phenoxazin-
5-one (74 mg, 0.156 mmol) and furan-maleimide adduct (33 mg, 0.2 mmol) were used as
reactants. Yield 55 mg (61%).
1-[6-(9-Diethylamino-5-oxo-5H-benzo[a]phenoxazin-2-yloxy)-hexyl]-pyrrole-2,5-dione.
[874998-67-1] 123 (Nile Red maleimide).
N
OEt2N O
ONO
O
O
H
H
122
N
OEt2N O
ON
O
O
123
A one liter pear-shaped flask with a stirring bar was charged with Diels-Alder
protected Nile Red maleimide (4-[6-(9-Diethylamino-5-oxo-5H-benzo[a]phenoxazin-2-
yloxy)-hexyl]-10-oxa-4-aza-tricyclo[5.2.1.02,6]dec-8-ene-3,5-dione, 4.0 g, 6.87 mmol),
dichloromethane (100 ml), toluene (250 ml), and topped with a 40 cm Vigreaux column.
The flask was heated on a 140°C heating mantle with gentle reflux for 12 hours, allowing
of gradual evaporation of dichloromethane and evolved furan. The solvent was
evaporated, the residue dissolved in dichloromethane and chromatographed on silica gel
with a 1:1 mixture of hexane and ethyl acetate. Yield 2.26 g (64%) of dark red material,
which could be converted to a crystalline form by very slow evaporation of
dichloromethane solution thereof. Analysis (Found/Calc): C 69.26/70.16, H 6.20/6.08, N
8.10/8.18. 1H NMR (CDCl3), δ: 8.20 (d, 1H, J=9Hz); 8.01 (d, 1H, J=2.4Hz); 7.58 (d, 1H,
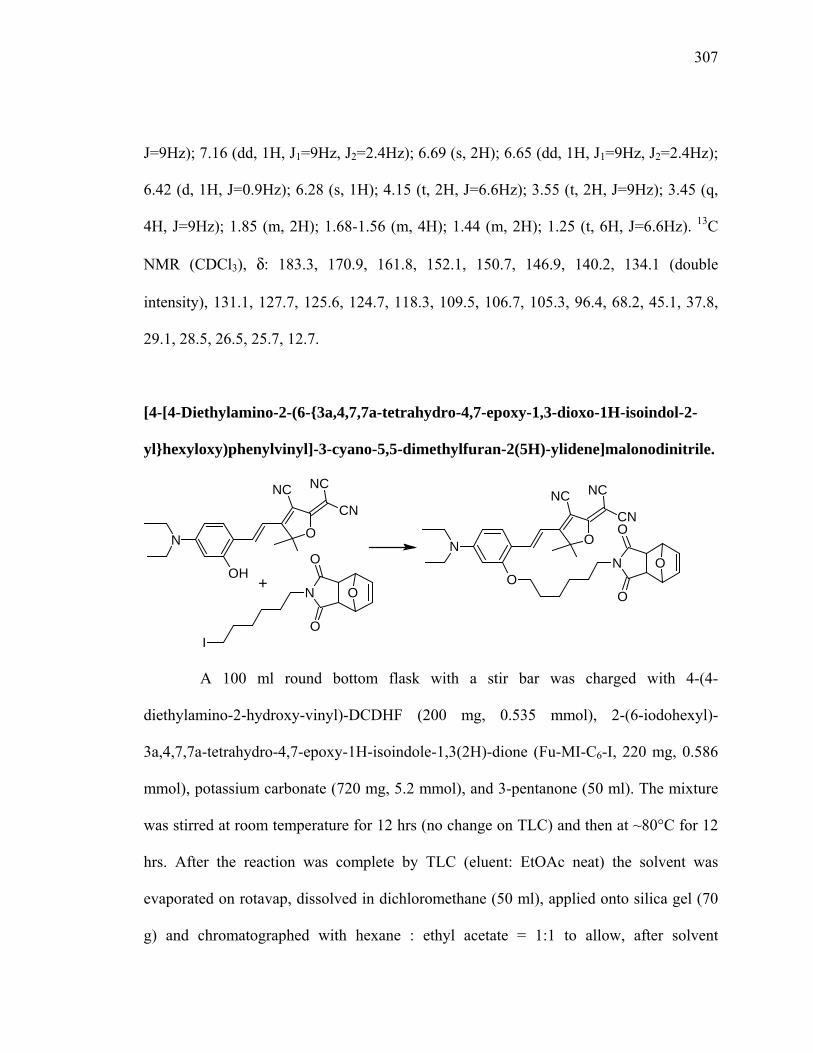
307
J=9Hz); 7.16 (dd, 1H, J1=9Hz, J2=2.4Hz); 6.69 (s, 2H); 6.65 (dd, 1H, J1=9Hz, J2=2.4Hz);
6.42 (d, 1H, J=0.9Hz); 6.28 (s, 1H); 4.15 (t, 2H, J=6.6Hz); 3.55 (t, 2H, J=9Hz); 3.45 (q,
4H, J=9Hz); 1.85 (m, 2H); 1.68-1.56 (m, 4H); 1.44 (m, 2H); 1.25 (t, 6H, J=6.6Hz). 13C
NMR (CDCl3), δ: 183.3, 170.9, 161.8, 152.1, 150.7, 146.9, 140.2, 134.1 (double
intensity), 131.1, 127.7, 125.6, 124.7, 118.3, 109.5, 106.7, 105.3, 96.4, 68.2, 45.1, 37.8,
29.1, 28.5, 26.5, 25.7, 12.7.
[4-[4-Diethylamino-2-(6-{3a,4,7,7a-tetrahydro-4,7-epoxy-1,3-dioxo-1H-isoindol-2-
yl}hexyloxy)phenylvinyl]-3-cyano-5,5-dimethylfuran-2(5H)-ylidene]malonodinitrile.
N
O
O
CN
NC
N O
O
O
NC
+
I
N O
O
O
N
OH
O
CN
NCNC
A 100 ml round bottom flask with a stir bar was charged with 4-(4-
diethylamino-2-hydroxy-vinyl)-DCDHF (200 mg, 0.535 mmol), 2-(6-iodohexyl)-
3a,4,7,7a-tetrahydro-4,7-epoxy-1H-isoindole-1,3(2H)-dione (Fu-MI-C6-I, 220 mg, 0.586
mmol), potassium carbonate (720 mg, 5.2 mmol), and 3-pentanone (50 ml). The mixture
was stirred at room temperature for 12 hrs (no change on TLC) and then at ~80°C for 12
hrs. After the reaction was complete by TLC (eluent: EtOAc neat) the solvent was
evaporated on rotavap, dissolved in dichloromethane (50 ml), applied onto silica gel (70
g) and chromatographed with hexane : ethyl acetate = 1:1 to allow, after solvent

308
evaporation, 43 mg (13%) of dark blue solid. Rf=0.42 (neat EtOAc). 1H NMR (CDCl3) δ:
7.90 (d, 1H, J=16.2 Hz), 7.46 (d, 1H, J=9 Hz), 6.85 (d, 1H, J = 16 Hz), 6.50 (s, 2H), 6.34
(d, 1H, J = 9 Hz), 6.00 (s, 1H), 5.25 (s, 2H), 4.0 (t, 2H), 3.60 (m, 4H), 3.46 (m, 2H), 3.17
(m, 2H), 2.82 (s, 6H), 1.71 (s, 6H), 1.56-1.32 (m, 8H), 1.06 (t, 6H). 13C NMR (CDCl3) δ:
180.8, 179.2, 176.7, 176.6, 136.7, 106.2, 93.8, 68.3, 62.76, 62.73, 47.5, 46.0, 45.3, 38.9,
32.6, 27.6, 26.3, 25.3, 12.94.
4-Diethylamino-2-(6-[3a,4,7,7a-tetrahydro-4,7-epoxy-1,3-dioxo-1H-isoindol-2-yl]-
hexyl)benzaldehyde.
N
O
O N O
O
ON
O
OH
A 200 ml round bottom flask with a stir bar was charged with 4-
diethylaminosalicylaldehyde (10.3 g, 53.3 mmol), 1-iodo-6-[3a,4,7,7a-tetrahydro-4,7-
epoxy-1,3-dioxo-1H-isoindol-2-yl]hexane (Fu-MI-C6-I, 20 g, 53.3 mmol), potassium
carbonate (10 g), and acetone (120 ml). The reaction mixture was stirred at reflux for 3
days, filtered, the solid on filter washed with acetone (50 ml), and the combined acetone
filtrates were impregnated onto silica gel. Chromatography with hexane:ethyl acetate
from 3:1 to neat ethyl acetate allowed, after solvent evaporation, 18 g (77%) of greenish
yellow oil. ES-MS: 441.1 (M+1) and 373.2 (M+1–Fu). 1H NMR (CDCl3) δ: 10.11 (s,
1H), 7.65 (d, 1H), 6.50 (s, 2H), 6.23 (dd, 1H), 5.97 (d, 1H), 5.22 (s, 2H), 3.97 (t, 2H),
3.45 (t, 2H), 3.38 (q, 4H), 2.80 (s, 2H), 1.78 (pentet, 2H), 1.56 (m, 2H), 1.47 (m, 2H),

309
1.33 (m, 2H), 1.18 (t, 6H). 13C NMR δ: 187.0, 176.4, 163.9, 153.8, 136.5, 130.0, 114.2,
104.2, 93.1, 80.9, 67.8, 47.3, 44.7, 38.7, 28.9, 27.4, 26.2, 25.6, 12.6.
[4-[4-Diethylamino-2-(6-{3a,4,7,7a-tetrahydro-4,7-epoxy-1,3-dioxo-1H-isoindol-2-
yl}hexyloxy)phenylvinyl]-3-cyano-5,5-dimethylfuran-2(5H)-ylidene]malonodinitrile.
N
O
O
CN
NC
N O
O
O
NC
N
O
O N O
O
O
O
CN
NCNC+
A 200 ml round bottom flask with a stir bar was charged with 4-diethylamino-2-
(6-[3a,4,7,7a-tetrahydro-4,7-epoxy-1,3-dioxo-1H-isoindol-2-yl]hexyl)benzaldehyde (AS-
3-04, 3.7 g, 8.4 mmol), 2-dicyanomethylene-3-cyano-4,5,5-trimethylfuran (1.6 g, 8.0
mmol), pyridine (21 g), and acetic acid (50 mg, 6 drops). The reaction mixture was
stirred at 60°C for 12 hrs, cooled and the solvent was removed by rotary evaporation. The
residue chromatographed on silica gel with gradient of hexane : ethyl acetate = 1:1
through neat ethyl acetate. Fractions containing pure product were combined and
concentrated to give 4.7 g (90%) of dark blue crystals. M.p. 110°C. 1H NMR (CDCl3) δ:
7.96 (d, 1H, J=16 Hz), 7.50 (d, 1H, J=9 Hz), 6.87 (d, 1H, J=16 Hz), 6.53 (s, 2H), 6.37
(dd, 1H, J1=9 Hz, J2=2 Hz), 6.05 (d, 1H, J=2 Hz), 5.26 (s, 2H), 4.04 (t, 2H, J=6 Hz), 3.50
(m, 6H), 1.89 (m, 2H), 1.74 (s, 6H), 1.66-1.36 (m, 6H), 1.27 (t, 6H, J=7 Hz). 13C NMR
(CDCl3) δ: 176.7, 176.4, 175.1, 162.3, 153.9, 144.1, 136.5, 113.3, 112.5, 112.2, 111.9,

310
108.2, 106.2, 96.3, 93.7, 80.97, 68.2, 47.4, 45.2, 38.7, 28.9, 27.50, 27.0, 26.2, 25.8, 12.8.
ES-MS: 554.2 (M+H–C4H4O). IR (HATR, neat, ν, cm–1): 2984, 2940, 2871, 2220 (m),
1710 (s), 1615, 1583, 1550, 1513 (s, br), 1251 (s), 1186 (s, br), 1073, 1011, 960, 873,
811, 694, 651, 596. IR (HATR, CHCl3 solution, ν, cm–1): 1710, 1517, 1419, 1361, 1259,
1088, 909, 643, 527.
3-cyano-2-dicyanomethylene-4-(2-[2-{6-<maleimid-2-yl>hexyloxy}-4-{N,N-diethyl-
amino}phenyl]vinyl)-5,5-dimethyl-2,5-dihydrofuran 124.
N
O
O
CN
NC
N O
O
O
NC
N
O
O
CN
NC
N
O
O
NC
A 250 ml round bottom flask with a stir bar was charged with [4-[4-
Diethylamino-2-(6-{3a,4,7,7a-tetrahydro-4,7-epoxy-1,3-dioxo-1H-isoindol-2-yl}hexyl-
oxy)phenylvinyl]-3-cyano-5,5-dimethylfuran-2(5H)-ylidene]malonodinitrile (4.7 g, 7.56
mmol) and xylene (mixture of isomers, 70 ml). The reaction mixture was refluxed for 2
hrs, at which time the starting material was absent by TLC analysis. The reaction was
cooled and the solvent was removed by rotary evaporation. The residue was
chromatographed on silica gel with a gradient of hexane : ethyl acetate = 1:1 through neat
ethyl acetate to give 2.0 g (47%) of dark blue crystals, which was recrystallized from 1-
propanol to give 1.76 g (41 %) of pure product. 1H NMR (CDCl3) δ: 8.00 (d, 1H, J=16
Hz), 7.50 (d, 1H, J=9 Hz), 6.84 (d, 1H, J=16 Hz), 6.7 (s, 2H), 6.4 (dd, 1H, J1=9 Hz, J2=2

311
Hz), 6.05 (d, 1H, J=2 Hz), 4.05 (t, 2H, J=6 Hz), 3.50 (m, 6H), 1.89 (m, 2H), 1.74 (s, 6H),
1.66-1.36 (m, 6H), 1.27 (t, 6H, J=7 Hz). 13C NMR (CDCl3) δ: 176.8, 175.0, 170.9, 162.2,
154.0, 144.1, 134.1, 113.4, 112.6, 112.2, 112.0, 108.0, 106.3, 96.3, 93.7, 68.2, 45.2, 37.6,
28.9, 28.5, 27.0, 26.4, 25.8, 12.8. ES-MS: 554.2 (M+H). UV-Vis (CHCl3) λmax: 590 nm.
M.p. 174.8°C. IR (HATR, neat solid, ν, cm–1): 2972, 2934, 2870, 2220 (s), 1705 (s),
1617, 1581, 1553, 1511 (s, br), 1377, 1348, 1313, 1235 (s, br), 1182 (s, br), 1119, 1077,
1006, 964, 872, 822 (s), 709, 692, 653.
4,5-Dimethoxyphthalaldehyde.
O
O
ClCl
O
O
CHO
CHO
A 200 ml round bottom flask with a stir bar was charged with 4,5-dimethoxy-
1,2-bis(chloromethyl)benzene (2.35 g, 0.01 mol), tetra-n-butylammonium periodate (9.16
g, 0.02 mol), dioxane (30 ml), fitted with a reflux condenser and refluxed for 29 hrs. The
mixture was cooled down, poured into water (200 ml), filtered, air-dried (7.43 g), and
flash-chromatographed on silica (50 g) with gradual elution from hexane-ethyl acetate
(4:1) to ethyl acetate. Solvent evaporation and recrystallization of the second
chromatographic fraction from ethanol allowed 0.8 g (41%) of 4,5-
dimethoxyphthalaldehyde, identical to an authentic sample by TLC (ethyl acetate) and
NMR.

312
2,3,6,7-tetramethoxy-9,10-dihydroanthracene 129.
O
O O
H
H
O
O
O
O+
A one liter Erlenmeyer flask equipped with a mechanical stirrer was charged
with sulfuric acid (70%, 850 g) and veratrole (50 g, 0.36 mol). To that vigorously stirred
mixture formaldehyde (37% aq., 70 g) was added dropwise during 30 min and stirring
was continued for 2 additional hrs. The reaction mixture was poured onto crashed ice
(500 g), filtered, washed with water (500 ml), air-dried, and recrystallized from a mixture
of 1-propanol (450 g) and chloroform (350 g). The crystals were washed with cold 1-
propanol (3×50 ml) and air-dried, 42.2 g (78%). M.p. 232°C (lit.428 mp. 230°C). 1H NMR
(CDCl3) δ: 6.83 (s, 2H), 4.76 (d, 1H, J=13.5 Hz), 3.84 (s, 6H), 3.54 (d, 1H, J=13.5 Hz).
13C NMR (CDCl3) δ: 147.8, 131.9, 113.2, 56.2, 36.6.
1,1,1-Trichloro-2,2-bis(3,4-dimethoxyphenyl)ethane.
MeO
MeO+
CCl3
OH OH
CCl3MeO
MeO
OMe
OMe
A 200 ml round bottom flask with a stirring bar was charged with chloral
hydrate (50.0 g, 0.3 mol), acetic acid (100 ml), sulfuric acid (50 ml), and heated to ~40
°C. To that stirred mixture, veratrole (37.6 g, 0.2 mol) was added from a syringe pump at
3.4 ml/hr. After the addition was complete (which takes ca. 11 hrs), the reaction mixture
was stirred at 40 °C for additional 24 hrs, and poured onto crushed ice (150 g). The

313
precipitate formed was filtered off on a Büchner funnel, air-dried (51.0 g, 83%) and
recrystallized from 1-propanol to give 31.0 g (51%) of white crystals. M.p. 117°C (lit.429
m.p. 115–116 °C). 1H NMR δ: 7.2 (dd, 2H), 7.1 (d, 2H), 6.9 (d, 2H), 4.9 (s, 1H), 3.9
(2×s, 12H). 13C NMR δ: 149.8, 149.5, 131.6, 123.2, 115.4, 112.4, 102.7, 70.7, 56.6, 56.3.
2,3,6,7-tetramethoxy-9,10-anthraquinone 130.
CCl3MeO
MeO
OMe
OMe
CHCl2
CCl3
OMeO
MeO
OMe
OMeO
A 200 ml round bottom flask with a stirring bar was charged with 1,1,1-
Trichloro-2,2-bis(3,4-dimethoxyphenyl)ethane (20.0 g, 5 mmol), chloral hydrate (18.0 g,
0.11 mol), and acetic acid (62 ml). To that stirred mixture, sulfuric acid (120 ml) was
added from an addition funnel during 4 hrs. After the addition was complete, the reaction
mixture was stirred for additional 3 hrs, and poured onto crushed ice (400 g). The
precipitate formed was filtered off on a Büchner funnel, air-dried (41 g) and dissolved in
potassium hydroxide solution (aq., 10%, 400 ml). To that stirred mixture, a solution of
potassium permanganate (28 g, 0.177 mmol) in water (120 ml) was added dropwise from
an addition funnel. After the addition was complete, the reaction mixture was heated to
80 °C and stirred at that temperature for 30 min. The solution was cooled and filtered on
a Büchner funnel. The solid on filter was recrystallized from DMAc to give 1.1 g (67%)
of 2,3,6,7-tetramethoxy-9,10-anthraquinone as yellow crystals. M.p. 348 °C (lit.430 345–

314
346 °C). 1H NMR (CF3COOD) δ: 7.8 (s, 4H), 4.1 (s, 12H). 13C NMR (CF3COOD) δ:
187.0, 156.1, 130.8, 111.6 (108.5*), 57.8 (56.6*). *Value in CDCl3.
2-Bromo-4,5-dimethoxybenzaldehyde 137.
CHOO
O
CHO
Br
O
O137
A one-liter two-neck round bottom flask equipped with a mechanical stirrer and
an addition funnel, was charged with 4,5-dimethoxybenzaldehyde (83.2 g, 0.5 mol) and
chloroform (500 ml). To the stirred reaction mixture bromine (160.0 g, 1 mol) was added
dropwise from the addition funnel during ca. 2 hrs. After ~¼ of all bromine had been
added, the reaction mixture became warm and a precipitate began to deposit and an
increase in power input to the mechanical stirrer was necessary. After all bromine had
been added, the reaction mixture was stirred for additional two hours, cooled in frige to –
20 °C and filtered off on a Büchner funnel. The solid on filter was suspended in water (1
liter) and potassium carbonate (sat. aq., ~70 ml) was carefully added until basic reaction
to pH paper, followed by sodium sulfite solution (sat. aq., ~120 ml) until bromine color
disappeared. The decolorized suspension was filtered off on a Büchner funnel, washed
with water (3×200 ml), air-dried, and recrystallized from acetic acid to give 192.0 g
(52%) of 2-bromo-4,5-dimethoxybenzaldehyde as white crystals. M.p. 150 °C (lit.431 m.p.
148–150 °C). 1H NMR (CDCl3) δ: 10.15 (s, 1H), 7.4 (s, 1H), 7.0 (s, 1H), 3.93 (s, 3H),
3.90 (s, 3H). 13C NMR (CDCl3) δ: 190.8, 154.5, 148.9, 126.5, 120.4, 115.4, 110.4, 56.5,
56.2.

315
2-Bromo-4,5-dimethoxybenzaldehyde dimethyl acetal 138.
Br
O
O
O
OCHO
Br
O
O137 138
A 250 ml round bottom flask with a stir bar was charged with 2-Bromo-4,5-
dimethoxybenzaldehyde (24.5 g, 0.1 mol), trimethoxymetane (31.2 g, 0.3 mol),
anhydrous methanol (150 ml), and Dowex 50W-X8 ion exchange resin (10.0 g). The
flask was topped with a Vigreaux column (Note 1) and the reaction mixture was refluxed
for 20 hrs and monitored by TLC (CH2Cl2 neat). After the reaction was complete, the
reaction mixture was filtered off on a Büchner funnel, the filtrate was evaporated from
solvents and the oily residue was vacuum distilled, collecting fraction b.p. 105–120 °C at
0.05 mm Hg: 16.51 g (58%) of colorless oil. 1H NMR (CDCl3) δ: 7.1 (s, 1H), 7.0 (s, 1H),
5.5 (s, 1H), 3.91 (s, 3H), 3.86 (s, 3H), 3.4 (s, 6H). 13C NMR (CDCl3) δ: 149.8, 148.5,
129.3, 115.4, 113.2, 110.7, 103.3, 56.27, 56.17, 54.1.
2,3,6,7-tetrakis(pentyloxy)-9,10-dihydroanthracene 143.
O
OH11C5
H11C5O
OH11C5
H11C5
O
OC5H11C5H11
A 250 ml Erlenmeyer flask with a stirring bar was charged with sulfuric acid
(70%, 50 ml) and 1,2-dipentyloxybenzene (5 g, 20 mmol). To that vigorously stirred
mixture formaldehyde (37% aq., 4 g) was added dropwise during 10 min and stirring was

316
continued for 2 additional hrs, while a viscous oil separated out. The oily product was
extracted with dichloromethane (2×50 ml), applied onto silica gel and chromatographed
with hexane – ethyl acetate (gradual elution from 8:1 to 4:1). Evaporation of solvent
afforded 2.4 g (45%) of colorless opalescent oil, which solidified in two days to slightly
yellow crystals. M.p. 51°C.
2,3,6,7-tetrahydroxy-9,10-dihydroanthracene 146.
O
O O
O OH
OH OH
OH
A 500 ml round bottom flask with a stir bar was charged with 2,3,6,7-
tetramethoxy-9,10-dihydroxyanthracene (1.0 g, 3.33 mmol), chloroform (19 g) and
cooled in an ice bath for 20 min. To the cooled stirred solution, boron tribromide (6.6 g,
26 mmol) was added dropwise from a syringe during 30 min. The reaction mixture was
allowed to warm up to room temperature during 12 hrs and then methanol (20 ml) was
added dropwise followed by water (50 ml) all at once. The white precipitate formed was
dissolved in ethanol (150 ml), filtered off, evaporated, air- and vacuum dried: 0.6 g
(74%). M.p. 161.8°C. 1H (DMSO) δ: 11.36 (s, 4H), 9.17 (d, 2H), 7.90 (d, 2H). 13C
(DMSO) δ: 143.3, 130.8, 116.7, 34.9.
9,10-Dimethy1-2,3,6,7-tetramethoxyanthracene 147.

317
O
OOH
O
O
O
O+
A half liter round bottom flask with a stir bar was charged with veratrole (13.8 g,
0.1 mol), acetic acid (30 ml), and acetaldehyde (12 ml, 0.21 mol). To this mixture, cooled
in an ice bath, was added sulfuric acid (20 ml) dropwise during 20 min. The ice bath was
removed and the reaction mixture was stirred for 6 hrs at room temperature. The deep-red
mixture was poured onto crushed ice (300 g), extracted with chloroform (2×100 ml) and
chromatographed on silica (150 g) with dichloromethane. Solvent evaporation afforded
1.2 g (7.4%) of off-white crystals. M.p. 328°C (DSC, 10°C/min). Lit. m.p. >340°C,432
323.5°C.433 1H NMR (CDCl3) δ: 7.40 (s, 2H), 4.08 (s, 6H), 2.94 (s, 3H). 13C NMR
(CDCl3) δ: 148.8, 125.9, 124.01, 102.7, 55.8, 14.9.
2,3,6,7-Tetramethoxy-9,10-dipentylanthracene 148.
O
O
O
HH11C5
O
O O
OC5H11
C5H11
+
A 200 ml round bottom flask with a stirring bar was charged with veratrole (5.0
g, 36 mmol) and hexanal (5.0 g, 50 mmol). To the stirred reaction mixture
methanesulfonic acid (15 g, 0.15 mol) was added dropwise during 20 min period. After
the addition was complete, the reaction mixture was stirred at room temperature for 2 hrs,
poured onto crushed ice (150 g), and extracted with chloroform. Chromatography with

318
gradual elution from neat hexane to 5% EtOAc in hexane, followed by solvent
evaporation and recrystallization from 1-propanol, allowed 1.3 g (16%) of light yellow
crystals. M.p. 182°C. 1H NMR δ: 7.4 (s, 2H), 4.1 (s, 6H), 3.35 (t, 2H), 1.8 (pentet, 2H),
1.6-1.5 (m, 4H), 0.9 (t, 3H). 13C NMR δ: 149.0, 129.7, 125.5, 102.7, 55.8, 32.7, 30.1,
28.9, 22.8, 14.4. UV-Vis (CHCl3) λmax, nm: 277, 357, 373, 392. (Cf. anthracene: 256,
342, 360, 379).
2,3,6-Tripentyloxy-7-hydroxyanthracene.
O
OH11C5
H11C5 O
HH11C5
O
O
H11C5
O
OH
C5H11
C5H11
C5H11
H11C5+
A 100 ml round bottom flask with a stirring bar was charged with 1,2-
dipentyloxybenzene (2.0 g, 8 mmol) and hexanal (1.8 g, 18 mmol). To the stirred reaction
mixture methanesulfonic acid (10 g, 104 mmol) was added dropwise over 30 min. After
the addition was complete, the reaction mixture was stirred for 2 hrs at room temperature,
poured onto crushed iced (100 g) and extracted with chloroform (100 ml).
Chromatography with gradual elution from neat hexane to 3% EtOAc in hexane allowed
0.8 g (20%) of the title compound as light yellow oil. 1H NMR (CDCl3) δ: 6.75 (m, 2H),
3.98 (t, 2H), 1.90-1.80 (m, 6H), 1.5 (m, 6H), 0.90 (m, 6H). 13C NMR (CDCl3) δ:149.1,
147.7, 138.7, 120.2, 114.4, 114.0, 69.51, 69.48, 36.4, 32.2, 30.0, 29.7, 28.5, 28.0, 22.8,
22.7, 14.3.

319
2,3,6,7-Tetrapentyloxy-9,10-dipentylanthracene 149.
O
OH11C5
H11C5O
HH11C5
O
O
H11C5
O
O
C5H11
C5H11
C5H11
H11C5
C5H11+
A 100 ml round bottom flask with a stirring bar was charged with 1,2-
dipentyloxybenzene (5.0 g, 20 mmol), methanesulfonic acid (20 g, 208 mmol), and
cooled in an ice bath. To the stirred reaction mixture hexanal (2.1 g, 21 mmol) was added
dropwise over 10 min. After the addition was complete, the reaction mixture was stirred
for 2 hrs at room temperature, chloroform (20 ml) was added, stirring continued for 12
hrs and then water (100 ml) was added. The mixture was extracted with chloroform
(3×50 ml) and chromatographed with gradual elution from neat hexane to 1-2% EtOAc in
hexane to allow 6.3 g (95%) of light yellow oil. Recrystallized from pentane in dry ice:
white crystals, m.p. 43°C. 1H NMR (CDCl3) δ: 6.8 (s, 2H), 3.98 (t, 4H), 3.76 (t, 2H),
1.90-1.80 (m, 9H), 1.5 (m, 12H), 0.90 (m, 9H). 13C NMR (CDCl3) δ: 149.1, 147.6, 138.7,
120.2, 114.3, 113.9, 69.5, 69.48, 50.5, 36.3, 32.1, 29.3, 28.5, 22.7, 14.2.
1,2,3,4-Tetrapentylnaphthalene.
I
IH11C5 C5H11
C5H11C5H11
C5H11C5H11
+
Ten ml round bottom flask with a stir bar was charged with 1,2-diiodobenzene
(131 μl, 1 mmol), 6-dodecyne (1.06 g, 6.3 mmol, 6×), silver acetate (347 mg, 2.07 mmol),

320
and anhydrous toluene (6.25 g). The mixture was refluxed under argon for 20 min, cooled
below b.p. and palladium acetate (11 mg, 49 μmol, 5 mol%) was added at once. The
reflux was continued for 12 more hours, until the peak of diiodobenzene almost
disappeared on GC-MS. Conversion (by GC-MS) >90%. EI-MS: 409.36 (37), 408.41
(100), 295.24 (39), 282.24 (57), 281.21 (44), 239.28 (34), 225.26 (38), 211.19 (26),
183.20 (29), 169.19 (29). The solvent was evaporated on rotovap, and the residue was
chromatographed on argenated silica gel (Note 1) to allow 392 mg (93%) of yellow oil.
1H NMR: 7.98-7.95 (m, 2H), 7.41-7.37 (m, 2H), 2.50 (t, 4H), 2.36 (t, 4H), 1.46-1.15 (m,
24H), 0.93 (t, 12H).
Note 1. Argentation of silica gel. Silver nitrate (14 g) was dissolved with
sonication in methanol (550 ml). To this solution silica gel (300 g) was added and solvent
evaporated on rotovap. The free-flowing solid was dried at 110°C in the dark for three
hrs.
1,2,3,4,5,6,7,8-Octapentylanthracene 152.
I
I
I
IH11C5 C5H11
C5H11
H11C5
H11C5
C5H11 C5H11C5H11
C5H11C5H11
+
A 200 ml round bottom flaskwith a stir-bar was charged with charged with
1,2,4,5-tetraiodobenzene (5.82 g, 0.01 mmol), 6-dodecyne (10 g, 0.06 mol, 6×), silver
acetate (7.0 g, 0.042 mol), and anhydrous, degassed toluene (60 ml). The mixture was
refluxed under argon for 20 min, cooled below b.p. and palladium acetate (112 mg, 0.5

321
mmol, 5 mol%) was added at once and reflux was continued for 24 more hours. The
reaction mixture was cooled down, filtered, washed with ether (2×50 ml), the solvent
from combined washings was evaporated and the residue was chromatographed with neat
hexane to allow 3.7 g (50%) of yellow oil, part of which crystallizes on standing to
yellow crystals. M.p. 87°C (no LC phases). 1H NMR: 8.68 (s, 1H), 3.17 (t, 4H), 2.79 (t,
4H), 1.66-1.45 (m, 28H), 0.99 (t, 12H). 13C NMR: 135.9, 133.7, 129.1, 119.4, 33.2, 33.0,
31.5, 31.3, 30.6, 29.7, 23.0, 22.7, 14.4, 14.3. ES-MS: 739.9 (M+), 661.3, 590, 488.4,
424.4.
1,2,3,4,5,6,7,8-Octaheptylanthracene 154.
C7H15
H15C7
H15C7
C7H15 C7H15C7H15
C7H15C7H15
A 250 ml round bottom flask with a stir-bar was charged with 9,10-
dihydroanthracene (1.8 g, 0.01 mol) and 1-bromoheptane (20 ml). To that stirred solution
aluminum chloride (8 g, 0.07 mol) was added in small portions over 30 min period and
stirring continued under air exposure at room temperature for 24 hrs. The reaction
mixture was poured onto crushed ice (300 g) and extracted with ether (3×150 ml). The
combined extracts were evaporated and the residue was chromatographed on argenated
silica with neat hexane to afford 7.7 g (80%) of 154 as opalescent oil. 1H NMR: 8.68 (s,
1H), 3.17 (t, 4H), 2.79 (t, 4H), 1.66-1.25 (m, 36H), 0.89 (t, 12H). 13C NMR: 135.9, 133.7,
129.1, 119.4, 33.2, 33.0, 31.5, 31.3, 30.6, 29.7, 23.0, 22.7, 15.6, 15.3, 14.4, 14.3

322
1,2,3,4,5,6,7,8-Octa(decyl)anthracene.
C10H21C10H21
C10H21C10H21C10H21
H21C10
H21C10
C10H21
A 500 ml round bottom flask with a stir-bar was charged with 9,10-
dihydroanthracene (5.4 g, 0.03 mol), aluminum chloride (32 g, 0.24 mol), and heptane
(70 ml). To that stirred mixture, 1-bromodecane (55 ml, 0.26 mol) was added dropwise
from an addition funnel over 2 hr period and stirring continued under air exposure at
room temperature for 24 hrs. The reaction mixture was poured onto crushed ice (300 g)
and extracted with ether (3×150 ml). The combined extracts were evaporated and the
residue was chromatographed on silica gel with neat distilled hexane to afford, after
solvent evaporation, 21.2 g (54%) of pale yellow oil. Tg –15°C. 1H NMR (CDCl3) δ:
7.18 (s, 1H), 2.5-2.4 (m, 8H), 1.9-0.9 (m, 77H). UV-Vis (CHCl3) λmax: 263 nm.
2,3-Anthracenedicarbaldehyde.434
CHO
CHOO
OCH3
OCH3
CHO
CHO+
A 100 ml round bottom flask was charged with phthalic aldehyde (10 g, 0.074
mol), 2,5-dimethoxytetrahydrofuran (20 g, 0.15 mol), acetic acid (7.5 ml), water (7.5 ml),
piperidine (10 drops), and was heated under reflux for 24 hrs. The reaction mixture was

323
cooled down, the dark red-brown precipitate was filtered and washed with water and
methanol: 3.48 g (20%). The crude material was sublimed at 180°C and 0.1 mm Hg to
allow 2.84 g of lemon yellow crystals, 2.84 g (16%). 1H NMR (CDCl3) δ: 10.65 (s, 1H),
8.64 (s, 1H), 8.62 (s, 1H), 8.03 (dd, 1H), 7.64 (dd, 1H). 13C NMR (CDCl3) δ: 192.6,
136.6, 132.2, 131.1, 130.2, 129.7, 128.8, 127.9
2,3-Diazatetracene.
CHO
CHO
NN
A 200 ml round bottom flask was charged with 2,3-anthracenedicarbaldehyde
(1.0 g, 4.2 mmol), ethanol (120 ml), hydrazine (3 ml), and refluxed for an hour. The
reaction mixture was cooled down, filtered, and the precipitate was air-dried, 0.6 g
(70%). Dec. 287°C. 1H NMR (CDCl3) δ: 9.59 (s, 1H), 8.84 (s, 1H), 8.77 (s, 1H), 8.11
(dd, 1H), 7.58 (dd, 1H). 13C NMR (CDCl3) δ: 152.2, 133.2, 132.4, 128.5, 128.2, 127.8,
127.2, 122.3.
4,5-Dimethoxyphthalic aldehyde 158.
CHO
Br
O
O137
CHO
CHO
O
O158
An oven-dried one-liter two-neck round bottom flask with a stir bar was charged
TriMEDA (17.3 g, 0.169 mol) and THP (150 ml). The necks were fitted with a

324
thermomter/argon adapter and septum. The mixture was cooled under argon in a dry ice –
acetone bath to –40 °C and a solution of n-butyl lithium (5M in hexanes, 30 ml) was
added dropwise via a syringe with vigorous stirring, while the temperature rose to –
10 °C. After the addition of n-BuLi was complete, the reaction mixture was stirred at –20
°C for 20 min and cooled down to –40 °C, at which temperature a solution of 2-bromo-
4,5-dimethoxybenzaldehyde (30.0 g, 0.122 mol) in THP (450 ml) was added via a
syringe pump. Additional THP (100 ml) used to wash the syringe, was added to the
reaction mixture at once. After the addition of bromoaldehyde was complete, the reaction
mixture was stirred at –20 °C for 20 min and cooled down to –40 °C. A solution of n-
butyl lithium (5M in hexanes, 29 ml) was added dropwise via a syringe with vigorous
stirring, while the temperature rose to –20 °C. After the addition of n-BuLi was complete,
the reaction mixture was stirred at –20 °C for 20 min and anhydrous DMF (20 ml) was
added at once. The cooling bath was removed and the reaction mixture was allowed to
warm up to room temperature.
The solvents were removed to ~1/4 of initial volume on a rotovap, the solution
was cooled to 0 °C and hydrochloric acid (aq., 3M, ~350 ml) was added slowly at
vigorous stirring. The brown precipitate formed was filtered off on a Büchner funnel,
washed with water (3×200 ml), ice-cold ethanol (3×100 ml), and air-dried to give 19.0 g
(80%) of 4,5-dimethoxyphthalic aldehyde as brownish-gray crystals. The crude material
was recrystallized from ethanol (aq. az., 30 ml/g) to give 10.0 g (42%) of white crystals.
M.p. 170 °C (lit.435 m.p. 168–169 °C). 1H NMR (CDCl3) δ: 10.6 (s, 1H); 7.5 (s, 1H); 4.00
(s, 3H); 13C NMR (CDCl3) δ: 190.1, 153.2, 131.0, 111.6, 56.5.

325
2,3-Dimethoxytetracene-6,11-dione 159.
OH
OH 155
CHO
CHO
O
O+
158
O
O
O
O159
A 500 ml round bottom flask with a stir bar was charged with a solution of 4,5-
dimethoxyphthalic aldehyde (3.88 g, 0.02 mol) in a hot mixture of ethanol (150 ml) and
THF (150 ml) and a solution of 1,4-dihydroxynaphthalene (3.20 g, 0.02 mol) in hot
ethanol (50 ml). To that stirred mixture, sodium hydroxide solution (aq., 10%, 5 ml) was
added at once and the reaction mixture was stirred for 30 min. The yellow precipitate
formed was filtered off on a Büchner funnel, washed with hot ethanol (200 ml), acetone
(100 ml), and air-dried to give 5.1 g (80%) of 2,3-dimethoxytetracene-6,11-dione as
bright orange crystals. M.p. 323 °C (lit.436 m.p. 328–329 °C). 1H NMR (CDCl3) δ: 8.7 (s,
2H), 8.4 (m, 2H), 7,8 (m, 2H), 7,3 (s, 2H), 4.1 (s, 6H). 13C NMR (CDCl3) δ: 183.1, 152.3,
134.5, 133.9, 131.8, 128.6, 127.4, 127.3, 107.9, 56.2.
2,3-Dihydroxytetracene-6,11-dione 160.
O
O
OH
OH160
O
O
O
O159
A 500 ml round bottom flask was charged with pyridine hydrochloride (60.0 g,
0.17 mol) and lithium iodide (10 g, 0.075 mol), and the flask was heated under nitrogen

326
at 150 °C for 12 hrs. To thus dried molten salts, 2,3-dimethoxytetracene-6,11-dione (8.3
g, 0.026 mol) was added at once (the color of the starting material solution in the melt is
deep orange) and the heating was continued at 200 °C for 30 min (color gradually
changes to brown-green). After the reaction was complete (TLC, CHCl3 neat), the
reaction mixture was cooled down, water (150 ml) was added and the flask was shaken
until the bulb of solid salts has dissolved. Hydrochloric acid (aq., 12 M, 5 ml) was added
and the formed suspension was filtered off on a Büchner funnel. The solid on filter was
washed with water (50 ml) and air-dried to give 7.3 g (96%) of 2,3-dihydroxytetracene-
6,11-dione as green-brown powder. 1H NMR (acetone-d6) δ: 8.6 (s, 2H), 8.3 (dd, 2H), 7.9
(dd, 2H), 7.2 (s, 2H). 13C NMR (CDCl3) δ: 185.2, 152.4, 138.6, 134.7, 131.8, 128.4,
127.4 (double intensity), 108.9.
2,3-Didecyloxytetracene-6,11-dione 161.
O
O
OH
OH
O
O
O
O
H21C10
H21C10160 161
A 250 ml round bottom flask with a stir bar was charged with 2,3-
dihydroxytetracene-6,11-dione (4.0 g, 13.8 mmol), 1-bromodecane (12.2 g, 55 mmol),
potassium carbonate (7.5 g, 55 mmol), potassium iodide (75 mg), and NMP (120 ml).
The reaction mixture was stirred at 80 °C for 12 hrs and monitored by TLC (neat CHCl3).
After the reaction was complete, it was cooled down and poured into an ice-water
(250 ml) with vigorous stirring. The precipitate was filtered off on a Büchner funnel,

327
washed with water (200 ml), ethanol (20 ml), hot ethanol (30 ml) and air-dried. The crude
product was dissolved in boiling chloroform (2×100 ml) and filtered from insoluble
matters. The filtrate was reduced in volume to ~75 ml and impregnated onto silica gel.
Chromatography with neat chloroform allowed, after evaporation of solvent, 1.32 g
(17%) of 2,3-didecyloxytetracene-6,11-dione as bright orange gel. Recrystallization
proved to be difficult, for the material is a strong gelator in most solvents (CHCl3, PrOH,
AcOH), and for this reason a m.p. was not measured. 1H NMR (CDCl3) δ: 8.6 (s, 2H), 8.3
(dd, 2H), 7.8 (dd, 2H), 7.2 (s, 2H), 4.1 (t, 4H), 1.9 (pentet, 4H), 1.5–1.3 (m, 28H), 0.9 (t,
6H). 13C NMR (CDCl3) δ: 185.1, 152.4, 138.6, 134.7, 131.9, 128.4, 127.4, 108.9, 69.2,
32.2, 29.9, 29.8, 29.7, 29.6, 29.2, 26.3, 22.9, 14.4.
2,3-Didecyloxytetracene 162.
O
O
O
O
H21C10
H21C10161
O
O
H21C10
H21C10162
A 250 ml round bottom flask with a stir bar was charged with anhydrous
cyclohexanol (distilled from sodium, 20 ml), aluminum (1.0 g, 37 mmol), and mercry (II)
chloride (22 mg, 0.08 mmol). The flask was topped with a reflux condensor, flushed with
nitrogen, and carefully heated over flame of a Bunsen burner until hydrogen evolution
started. At that point the flask was removed from the flame and reaction went
spontaneously. After the hydrogen evolution ceased, carbon tetrachloride (0.5 ml) was
added, followed by 2,3-didecyloxytetracene-6,11-dione (1.32 g, 2.3 mmol). The reaction

328
mixture was heated at 120 °C and monitored by TLC (dichloromethane : hexanes = 1:1).
After the reaction was complete (~3 hrs), the reaction mixture was cooled to ~70 °C and
the following reagents were added dropwise in this order: ethanol (95% aq., 40 ml),
acetic acid (4 ml), water (15 ml), hydrochloric acid (aq., 2M, 5 ml). The precipitate
formed was filtered off on a Büchner funnel, washed with water (70 ml), ethanol (2×20
ml), and air-dried to give 1.1 g (88%) of 2,3-didecyloxytetracene as off-orange crystals.
The crude product was chromatographed on a 150 mm Biotage column with hexanes :
dichloromethane (gradient elution from 2:1 to 1:1) to give, after evaporation of solvents,
140 mg (11%) of pure 2,3-didecyloxytetracene as orange crystals. M.p. (EtOH) 130–
132 °C (microscope, hot stage). UV-Vis (CHCl3) λmax: 240, 304, 393. 1H NMR (CDCl3)
δ: 8.8 (s, 2H), 8.1 (dd, 2H), 7.7 (s, 2H), 7.6 (dd, 2H), 7.3 (s, 2H), 4.2 (t, 4H), 1.9 (pentet,
4H), 1.5 (pentet, 4H), 1.3 (m, 24H), 0.9 (t, 6H). 13C NMR (CDCl3) δ: 154.2, 135.2, 130.3,
130.2, 129.5, 129.4, 129.3, 109.7, 69.6, 32.1, 29.8 (two peaks), 29.6 (double intensity),
29.2, 26.2, 22.9, 14.3.
Pentacene 163.
O
O164 163
Preparation of aluminum cyclohexanolate stock solution. Commercial
cyclohexanol (Acros, 98%, 20Dn = 1.4625) was found to contain 1…2% of water and was
purified by distillation from sodium under normal pressure, employing a 15 cm Vigreaux

329
column, thermally insulated with an asbestos tape, wrapped around. The forerun boiling
below 160 °C (50 ml from a liter of commercial cyclohexanol) was discarded and the
fraction boiling 160…161 °C was collected and stored over powdered437 3Å molecular
sieves. A one-liter three-neck round bottom flask with a stirbar was charged with
cyclohexanol (500 ml), aluminum turnings (25 g, 0.925 mol), mercury (II) chloride
(0.625 g, 2.3 mmol), and carbon tetrachloride (12.5 ml). The flask was topped with a
reflux condensor, thermometer, and nitrogen inlet and the reaction mixture was heated
under nitrogen on an IR hot plate to ~160 °C until hydrogen evolution started. The
temperature was lowered to ~120 °C or as necessary to keep the hydrogen evolution
vigorous, but not out of control (intermittent cooling of the flask by immersing it into an
ice-water bath was used to control the reaction rate). After the hydrogen evolution had
ceased (~3 hrs) and all aluminum had dissolved, the solution of aluminum
cyclohexanolate was cooled down, and another portion of carbon tetrachloride (7 ml) was
added.
To the warm (60…80 °C) solution of aluminum cyclohexanolate in
cyclohexanol obtained above, pentacene-6,13-dione (25.0 g, 0.08 mol) was added at once
under nitrogen and the reaction mixture was refluxed for 4 hrs. The precipitate of
pentacene (insoluble in cyclohexane) started to appear after 2 hrs of reflux. The reaction
mixture was cooled down, acetic acid (500 ml), followed by methanol (1250 ml) were
added and stirred for an hour. The suspension was then filtered off on a Büchner funnel,
washed with methanol (300 ml), and air-dried to give 12.8 g (57%) of crude pentacene.
Triple sublimation in vacuum (p = 0.1 mm Hg) gave 7.4 (33%) of purified pentacene.

330
Subsequent sublimations did not increase the purity of pentacene (determined as residue
after sublimation by TGA). Analysis (found/calculated): C 94.34/94.93, H 5.66/5.07.
Pentacene-6,13-dione 164.
O
O164
CHO
CHO
O
O
+
o-Phthalic aldehyde (10.0 g, 0.075 mol) was dissolved in ethanol (95.6%, aq.,
250 ml). 1,4-Hexanedione was dissolved in ethanol (95.6%, aq., 150 ml). The solutions
were mixed into a one-liter Erlenmeyer flask and stirred with a large stir bar. To that
mixture, potassium hydroxide (aq., 5%, 7.5 ml) was added at once. Marsh-green
suspension was formed immediately. The reaction mixture was heated at 50 °C for an
hour, during which time the color changed to yellow-brown. The hot reaction mixture
was filtered off on a Büchner funnel, washed with hot ethanol (300 ml), methanol (200
ml), and air-dried to give 10.1 g (87%) of pentacene-6,13-dione as yellow crystals. The
crude material may be recrystallized from DMAc (80 ml per gram) to give orange-yellow
crystals, m.p. 390 °C or sublimed to give yellow needles, m.p. 389 °C. Lit.256 m.p. 377–
394 °C.

331
2,5-Dibenzoylterephthalic acid and 2,4-Dibenzoylisophthalic acid [165].438
OO
O
O O
O
O
O
COOH
HOOC
O COOH
O COOH
+ +
A 600 ml autoclave fitted with a mechanical stirrer and a thermocouple was
charged with pyromellitic dianhydride (25 g, 0.115 mol), benzene (200 ml, 2.25 mol),
and anhydrous aluminum chloride (60 g, 0.45 mol). The autoclave was closed and heated
with stirring for 4 hours at 100°C (internal temperature). After cooling, the contents were
poured into a 2 l beaker with ice (500 g), and hydrochloric acid (37% aq., 50 ml), and
stirred for 15 min until the dark color disappeared. The precipitate formed was filtered,
washed with hydrochloric acid (2M, 200 ml), water (300 ml), and air-dried resulting in
brown-gray powder (48 g). The crude product was dissolved in hot solution of potassium
hydroxide (22 g, 0.4 mol) in water (200 ml), filtered, and the residue on filter was treated
with additional solution of potassium hydroxide (7 g, 0.125 mol) in water (70 ml), and
filtered. The combined filtrates were cooled to ~5°C and slowly acidified with
hydrochloric acid (2M, 350 ml). The white precipitate formed was filtered, washed with
water (300 ml), and air-dried to allow 17 g (40%) of white powder, which was used in
subsequent reaction.

332
Pentacene-5,7,12,14-tetraone 166.
O
OHOOC
COOH
O
HOOC
O
COOH
O
O O
O
+
A 250 ml Erlenmeyer flask with a stir bar was charged with a mixture of two
isomeric dibenzoylphthalic acids (AS-3-25a, 16.8 g, 0.045 mol), sulfuric acid (98%, 135
ml), the neck of the flask was sealed with Parafilm, and the reaction mixture was heated
at 100-120°C for four hours, being occasionally stirred with a teflon spatula. After
cooling, the reaction mixture was poured onto ice (400 g), filtered on fiber glass filter,
washed with sodium bicarbonate (sat.aq., 100 ml), hot water (300 ml), and hot ethanol
(200 ml). The brown-gray residue on the filter was then suspended in boiling ethanol
(2×100 ml), filtered, and air-dried to give 14.0 g (92%) of the pentacene-5,7,12,14-
tetraone as a greenish-yellow powder. M.p. 407°C (lit.438 m.p. 408°C).
1,4-Anthracenequinone 167.
O
O
OH
OH
O
O 167
A two-liter two-neck round bottom flask equipped with a mechanical stirrer and
a reflux condensor, was charged with quinizarine (Note 1, 57.0 g, 0.2375 mol),

333
anhydrous methanol (1000 ml). While stirring, the flask was cooled in an acetone — dry
ice bath to ca. –30 °C and sodium borohydride (37.8 g, 1 mol) was added during half an
hour from a powder-addition funnel, placed atop of the reflux condensor. When the
addition has been complete, the cooling bath was removed, the reaction mixture was
warmed up to room temperature and heated on a heating mantle to reflux for 24 hrs. The
reaction mixture was cooled and most of methanol was removed on a rotovap. To the
residue after evaporation water (700 ml) was added and the resulting solution was filtered
(Note 2). To the filtrate hydrochloric acid (aq., 3 M, 400 ml) was added dropwise at
stirring, causing at pH~7 an abrupt precipitation of thick yellow mass. The yellow
precipitate was filtered on a Büchner funnel, washed with water (300 ml), air- and
vacuum-dried to give 46.4 g (94%) of 1,4-anthracenequinone as yellow crystals. The
crude material is of sufficient purity, but may be recrystallized from BuOAc (800 ml per
30 g) with 72% recovery. M.p. 224 °C (lit. m.p. 204–206,439 225 °C440). 1H NMR δ: 8.6
(s, 2H), 8.0 (dd, 2H, J = 3.3 Hz), 7.7 (dd, 2H, J = 3.3 Hz), 7.1 (s, 2H). 13C NMR δ: 184.7,
140.0, 134.8, 130.2, 129.6, 128.9, 128.4.
Note 1. Commercial quinizarine from Acros was recrystallized from acetic acid
(14 g AcOH per g). Note 2. The filtration of the water solution before acidification is
essential to get pure product.

334
2,3-Dihydro-1,4-anthracenequinone 168.
O
O167
O
O168
A 250 ml Erlenmeyer flask was charged with 1,4-anthracenequinone (3.0 g, 14.4
mmol), trifluoroacetic acid (85 g), and zinc dust (3.78 g, 5.8 mmol). The reaction mixture
was sonicated for 5 min, and filtered. The filtrate was poured into water (300 ml) and the
precipitate formed was filtered off on a Büchner funnel. The crude product (2.8 g, 92%)
was washed with water (200 ml), cold ethanol (30 ml), dichloromethane (100 ml), and
recrystallized from ethanol (aq. az., 90 ml) — chloroform (10 ml) mixture to give 2.6 g
(86%) of 2,3-dihydro-1,4-anthracenequinone as crystals of marsh-green color. M.p.
227 °C (lit.262c m.p. 175 °C). 1H NMR δ: 8.6 (s, 2H), 8.0 (dd, 2H, J = 3.3 Hz), 7.7 (dd,
2H, J = 3.3 Hz), 7.1 (s, 2H), 2.16* (s, 4H). 13C NMR δ: 199.7*, 184.6, 140.0, 134.8,
130.2, 129.6, 128.8, 128.3, 21.7*.
*quickly disappears due to isomerization into 1,4-dihydroxyanthracene
1-Fluoropentacene 169.
O
OF 173 F 169
1-Fluoropentacene was prepared similar to pentacene 163, starting from 1-
fluoropentacene-6,13-dione 173 (2.78 g, 8.5 mmol). Crude material (1.1 g, 43%) was

335
sublimed: two times covered with degreased iron filings and two times neat to give 110
mg (4%) of dark blue crystals. M.p. 289 °C. UV-Vis (benzene, degased) λmax: 279, 291,
303, 322, 349. IR (neat solid, HATR) ν, cm–1: 3014, 1443, 1371, 1306, 1230, 1220, 814,
734. Analysis (found/calc.): C 88.19/89.17, H 4.85/4.42.
2-Fluoropentacene 170.
2-Fluoropentacene was prepared by Kihong Park similar to 1-fluoropentacene. The crude
material received was sublimed in vacuum (p = 0.15 mm Hg) at 180–210 °C. Sublimes in
inert atmosphere without decomposition, TGA peak (2nd derivative maximum) at 385 °C.
Analysis (found/calc.): C 87.89/89.17, H 4.94/4.42.
3-Fluorophthalaldehyde 172.
CHO
CHOF 172F
OHOH
179
A 250 ml round bottom flask with a stir bar was charged with oxalyl chloride (5
ml, 55 mmol) and dichloromethane (120 ml). The flask was topped with an addition
funnel and cooled in a dry ice – acetone bath to –40 °C under argon. From the addition
funnel, a solution of DMSO (7 ml, 90.5 mmol) in dichloromethane (20 ml) was added
dropwise during 10 min. The reaction mixture was stirred for 15 min and 1,2-
bis(hydroxymethyl)-3-fluorobenzene (3.0 g, 19.2 mmol) in dichloromethane (10 ml) was
added dropwise from the same addition funnel during 5 min. The reaction mixture was

336
stirred for additional 30 min and triethylamine (30 ml, 0.2 mol) was added at once. The
reaction mixture was allowed to warm up to room temperature, poured onto ice (300 g)
and extracted with ether (3×150 ml). The combined extracts were dried with MgSO4,
solvents evaporated on rotovap, and the oily residue was chromatographed with hexane –
ethyl acetate (7:3) to give, after removal of solvents, 2.1 g (72%) of 3-
fluorophthalaldehyde as colorless oil. 1H NMR (CDCl3) δ: 10.6 (s, 1H), 10.5 (s, 1H), 8.0
(m, 1H), 7.8 (m, 1H), 7.7 (m, 1H).
1-Fluoropentacene-6,13-dione 173.
F
O
O
O
O167
CH2Br
CH2BrF
+
177 173
A 250 ml round bottom flask with a stir bar was charged with 1,4-anthraquinone
167 (4.16 g, 0.02 mol), 1,2-bis(bromomethyl)-3-fluorobenzene 177 (10.0 g, 0.036 mol),
sodium iodide (20 g.0.13 mol), and DMF (150 ml). The reaction mixture was heated at
70 °C for 24 hrs (dark brown tar forms), cooled down, poured into water (450 ml). The
precipitate formed was filtered on a Büchner funnel, washed with water (300 ml), and air-
driedto give 7.3 g of dark-brown solid. The crude product was sublimed in vacuum twice
ti give 2.6 g (35%) of 1-fluoropentacene-6,13-dione as brownish crystals. The NMR
characterization was not performed due to low solubility.

337
1-Fluoro-2,3-dimethylbenzene 176.
F
176
NH2
A four-liter beaker, equipped with a mechanical stirrer, was charged with water
(500 ml), and o-xylidine (2,3-dimthylaniline, 200.0 g, 1.65 mol). The beaker was
immersed into a dry ice – acetone cooling bath (bath temperature –10 °C) and with
stirring, ice water mixture (1500 g) was added. When the temperature in the beaker had
reached 10 °C, a solution of hydrochloric acid (aq., 12M, 176 ml) in water (200 ml) was
added at once. When the temperature had lowered to 0–5 °C after HCl addition, a
precooled to 5 °C solution of sodium nitrite (125.3 g, 1.8 mol) in water (300 ml) was
added with vigorous stirring at such a rate that temperature did not rise above 5 °C. After
the addition was complete, the reaction mixture was stirred for additional 15 min and
fluoroboric acid solution (aq., 39%, 380 g, 1.7 mol), precooled to 5 °C was added in 20-
30 ml portions, simultaneously increasing the power input for the mechanical stirrer.
After the addition of HBF4 was complete, the suspension was stirred for 10 additional
min, and filtered off on a Büchner funnel. The solid on filter was washed with cold water
(200 ml), methanol (200 ml, precooled to –30 °C), and ether (300 ml, precooled to –30
°C). After suction drying on the filter, the solid was transferred into a one-liter pear-
shaped flask and additionally dried on the rotovap at 30–35 °C and 20 mm Hg, followed
by 40–45 °C at 0.1 mm Hg. After the drying was complete, the flask was removed from
the rotovap, topped with a long reflux condensor, and heated with a heating gun on one
side until local decomposition of the diazonium salt started. Heating was continued for 20

338
min until all solid had decomposed and nitrogen evolution had ceased. The liquid
resulting from decomposition was distilled twice at normal pressure, collecting fraction
with b.p. 144–147 °C. Lit.441 b.p. 146–148 °C. 1H NMR (CDCl3) δ: 6.9–7.0 (m, 3H), 2.2
(s, br, 3H), 2.1 (d, 3H, JH–F = 2 Hz). 13C NMR (CDCl3) δ: 161.9 (d, JC–F = 240 Hz), 139.0
(d, JC–F = 5 Hz), 126.7 (d, JC–F = 10 Hz), 125.0 (d, JC–F = 3 Hz), 123.6 (d, JC–F = 17 Hz),
112.9 (d, JC–F = 22), 19.5 (d, JC–F = 3 Hz), 10.8 (d, JC–F = 7 Hz). EI-MS: 124 (M+).
1,2-bis(bromomethyl)-3-fluorobenzene 177
CH2Br
CH2BrF 177F 176
A two-liter round bottom flask with a stir bar was charged with N-
bromosuccinimide (127.2 g, 0.707 mol, Note 1), anhydrous carbon tetrachloride (500
ml), 1-fluoro-2,3-dimethylbenzene (39.9 g, 0.321 mol), and benzoyl peroxide (1.1 g, 4.5
mmol). The flask was topped with a reflux condensor, and the reaction mixture was
heated to reflux on a heating mantle and irradiated with a 450 W medium pressure Hg
Hanovia lamp. After approximately 15 min of irradiation, a vigorous reaction was
initiated. Reflux and irradiation was continued for an additional hour. The reaction
mixture was cooled down, filtered from succinimide on a Büchner funnel and the filtrate
was rotovaped. The oily residue after evaporation was vacuum distilled, collecting
fraction b.p. 100–130 °C at 0.5 mm Hg: 81.5 g (91%), which partially solidified on
standing: m.p. 38–39 °C (lit.442 m.p. 41–42 °C). 1H NMR (CDCl3) δ: 7.5–6.9 (m, 3H), 4.8

339
(d, 2H, JH–F = 2 Hz), 4.6 (s, 2H). 13C NMR (CDCl3) δ: 161.9 (d, JC–F = 240 Hz), 139.0 (d,
JC–F = 5 Hz), 131.0 (d, JC–F = 10 Hz), 127.0 (d, JC–F = 3 Hz), 125.0 (d, JC–F = 17 Hz),
116.0 (d, JC–F = 22 Hz), 29.0 (d, JC–F = 3 Hz), 21.0 (d, JC–F = 7 Hz). EI-MS: 282.9 (55),
281.0 (100), 279,3 (55), 202.1 (22), 200.1(21), 122.3 (28), 121.3 (51), 102.2 (14), 101.2
(18), 75.4 (20), 74.3 (8).
Note 1. When exactly two equivalents of NBS have been used, a mixture of
mono- and dibromo- products in 2:1 ratio was separated (determined by GC-MS).
Increasing the NBS amount to four equivalents, however, gave an 85:5 mixture of
dibromo- and tribromo- isomers only (determined by GC-MS). Only traces (on GC-MS)
of o-fluoro-tetrabromo-o-xylene 178 have been detected when dibromo- compound 176
was subjected to radical bromination with 4 equivalents of bromine.
1,2-Bis(hydroxymethyl)-3-fluorobenzene 179.
F
OHOH
179
CH2Br
CH2BrF 177
A 2000 ml round bottom flask with a stir bar was charged with 1,2-
bis(bromomethyl)-3-fluorobenzene (87.2 g, 0.31 mol), potassium carbonate (87.0 g, 0.63
mol), water (1000 ml), and tetrabutylammonium tetrafluoroborate (5.0 g). The reaction
mixture was stirred at 80 °C for 8 hrs, cooled down, and extracted with ether (5×200 ml).
The combined extracts were dried with MgSO4, and ether evaporated to give 38.0 g
(79%) of 1,2-bis(hydroxymethyl)-3-fluorobenzene as colorless liquid. 1H NMR (CDCl3)

340
δ: 7.8 (d, 1H, JH–F = 8 Hz), 7.4 (m, 1H), 7.1 (s, 2H), 7.0 (m, 1H), 4.7 (m, 2H). 13C NMR
(CDCl3) δ: 160 (d), 143.8, 131.0, 126.3, 119.8, 116.5 (d), 61.3, 37.0. The diol becomes
pink if left to open air for more than a day.
2,3,9,10-Tetrahexyloxypentacene-6,13-dione 185.
O
O
OH
OH
OH
OH187
O
O
H13C6O
H13C6O
OC6H13
OC6H13185
2,3,9,10-Tetrahexyloxypentacene-6,13-dione was prepared similar to 2,3-
didecyloxytetracene-6,11-dione 161, starting from 2,3,9,10-tetrahydroxypentacene-6,13-
dione in 4% yield. M.p. 189.5–194.5 °C (microscope, hot stage). 1H NMR (CDCl3) δ: 8.6
(s, 1H), 7.3 (s, 1H), 4.2 (t, 2H), 1.9 (pentet, 2H), 1.6 (pentet, 2H), 1.4 (m, 4H), 0.9 (t, 3H).
13C NMR (CDCl3) δ: 183.4, 152.4, 131.9, 129.5, 127.4, 108.9, 69.2, 31.8, 29.1, 25.9,
22.8, 14.2.
2,3,9,10-Tetrahydroxypentacene-6,13-dione 187.
O
O
O
O
O
O188
O
O
OH
OH
OH
OH187
2,3,9,10-Tetrahydroxypentacene-6,13-dione was prepared similar to 2,3-
dihydroxytetracene-6,11-dione 160, starting from 2,3,9,10-tetramethoxypentacene-6,13-
dione in 90% yield. M.p. 409 °C (dec.). TGA decomposition maximum at 440 °C. IR

341
(solid, HATR) ν, cm–1: 3220, 3150, 3050, 1670, 1640, 1630, 1567, 1270, 1256, 1190,
1150, 1138, 1100, 951, 770, 711.
2,3,9,10-Tetramethoxypentacene-6,13-dione 188.
O
O
O
O
O
O188
CHO
CHO
O
O
O
O
+
158
4,5-Dimethoxyphthalic aldehyde (6.6 g, 0.034 mol) was dissolved in boiling
ethanol (95.6%, aq., 400 ml). 1,4-Hexanedione (2.0 g, 0.018 mol) was dissolved in warm
ethanol (95.6%, aq., 20 ml). The solutions were mixed into a 500 ml Erlenmeyer flask
and stirred with a large stir bar. To that mixture, potassium hydroxide (aq., 10%, 0.8 ml)
was added at once. Marsh-green suspension was formed immediately. The reaction
mixture was heated at 70 °C for an hour, during which time the color changed to yellow-
brown. The hot reaction mixture was filtered off on a Büchner funnel, washed with hot
ethanol (300 ml) and air-dried to give 6.7 g (93%) of 2,3,9,10-tetramethoxy pentacene-
6,13-dione as yellow crystals. M.p. 400 °C (lit.314 m.p. >320 °C). 1H NMR (CDCl3) δ: 8.7
(s, 4H), 7.3 (s, 4H), 4.1 (s, 12 H). 13C NMR (solid-state) δ: 183.0, 150.5, 128.2, 107.7,
55.2. IR (neat, HATR) ν, cm–1: 1670, 1620, 1590, 1510, 1480, 1435, 1390, 1250, 1210,
1160.

342
2,3,9,10-Tetramethoxypentacene 189.
O
O
O
O
O
O188
O
O
O
O189
2,3,9,10-Tetramethoxypentacene was prepared similar to pentacene from
2,3,9,10-Tetramethoxypentacene-6,13-dione 188 (4.0 g, 9.3 mmol) and aluminum
cyclohexanolate solution in cyclohexanol (100 ml) to give 2.4 g (65%) of 189 as dark
ruby red crystals. M.p. 413 °C (subl.). 13C NMR (solid-state) δ: 154.6, 132.6, 128.1,
108.7, 59.8, 58.2 (two peaks for methoxy groups are probably due to their magnetic
nonequivalence in crystalline lattice).
HgO/SiO2 Mercury tetrafluoroborate on silica gel.349
2 HBF4 + HgO + SiO2 = Hg(BF4)2–SiO2 + H2O
A one liter pear-shaped flask was charged with hydrofluoroboric acid (50% aq.
solution, 175.6 g, 1 mol) and yellow mercury (II) oxide (107 g, 0.495 mol) was added at
once. The flask was swirled until clear solution was formed and silica gel (107 g) was
added. The slurry was evaporated on rotavap (80°C, 0.2 mm Hg) till constant mass.
Bis(pyridine) iodonium tetrafluoroborate Py2I+B –4F .
2 Py + I2 + Hg(BF4)2 / SiO2 + CH2Cl2 = Py2I+ BF4–
A one liter Erlenmeyer flask was charged with mercury tetrafluoroborate
impregnated on silica gel (50 wt%, 215 g, 0.5 mol), dichloromethane (900 ml), pyridine

343
(96 ml), and large stir bar. To vigorously stirred reaction mixture, iodine (127 g, 0.5 mol)
was added in portions (10 g) and stirring was continued for two hours. Reaction mixture
warmed up (~40°C) and changed color to yellow as the reaction progressed. The solvent
was evaporated to 200 ml and ether (300 ml) was added to precipitate yellow
bis(pyridine) iodonium tetrafluoroborate. The precipitate was filtered, washed with ether
(200 ml) and air dried to yield 146.8 g (79%). M.p. 153°C (lit.350 149-151°C).
Iodopentamethylbenzene 194.
I
A one liter two neck round bottom flask with a stir bar was charged with
pentamethylbenzene (44.35 g, 0.3 mol), acetic acid (225 g), and iodine (34.3 g, 0.135
mol). The flask was fitted with a reflux condensor, thermometer, and an addition funnel
containing periodic acid dihydrate (44.1 g, 0.193 mol) in water (60 ml). This solution was
added dropwise to the stirred reaction mixture during 20 min. After the addition was
complete, the reaction mixture was brought to 110°C and refluxed for 4 hrs. When the
reaction was complete, the reaction mixture was cooled down to room temperature,
diluted with water (500 ml), filtered off and the precipitate was washed with water (200
ml), cold ethanol (2 × 50 ml), and air-dried to give 63 g (76%) of crude product. The
crude product was recrystallized twice from 1-propanol (250 ml) to give 38 g (46%) of
product. M.p. 132°C (lit. m.p. 127-135°C). EI-MS: 274.03 (100), 147.13 (80), 128.07

344
(10), 119.11 (38), 91.05 (39), 77.11 (19), 63.03 (10). 1H NMR (CDCl3) δ:2.51 (s, 6H),
2.27 (s, 6H), 2.18 (s, 3H). 13C NMR: 137.0, 135.1, 133.4, 109.3, 28.4, 18.7, 17.1.
1,2,4,5-Tetraiodo-p-xylene 195.
I
I
I
I
A one liter three neck round bottom flask was charged with sulfuric (500 ml)
and periodic (26 g, 0.114 mol) acids, fitted with a mechanical stirrer and a thermometer
and stirred until clear solution was formed (~30 min). The flask was cooled then in dry
ice – acetone bath (–15°C) to –5°C and finely ground iodine (87 g, 0.343 mol) was added
at once. After a short induction period (~10 min) the temperature raised to +10°C and
more dry ice was added to the bath to keep the temperature of the reaction mixture below
0°C. To the reaction mixture cooled to –5°C, p-xylene (21.5 g, 0.2 mol) was added
dropwise from an addition funnel during 20 min. After the addition was complete, the
cooling bath was replaced with a heating mantle and reaction was stirred at 80°C for
three days, while monitored by GC-MS. Upon completion (GC peak of triiodo-p-xylene
disappeared), the reaction mixture was cooled down, poured onto crushed ice (2 kg),
filtered, the precipitate on filter washed with water (500 ml), re-suspended in water (1000
ml), filtered, re-suspended in sodium bicarbonate (10% aq., 500 ml), filtered, washed
with sodium bisulfite (10% aq., 200 ml), water (500 ml), re-suspended and sonicated in
ethanol (500 ml), filtered, and air-dried to yield crude dark-brown product 108.2 g (88%).

345
The crude material was sublimed (3×, 230°C, 0.015 mm Hg) to yield 45.4 g (37%) of
white crystals. M.p. 245.5°C. (Lit.443 m.p. 245-248°C). EI-MS: 609.69 (87), 482.70 (39),
483.78 (20.5), 355.83 (74), 229.01 (60.5), 102.06 (100), 103.05 (17), 91.08 (2), 74.02 (9),
75.04 (6).
1,2,4,5-Tetraiodobenzene 196.
I
I
I
I
II
II
I
I+
A one liter three neck round bottom flask was charged with sulfuric (500 ml)
and periodic (26 g, 0.114 mol) acids, fitted with a mechanical stirrer and a thermometer
and stirred until clear solution was formed (~30 min). The flask was cooled then in dry
ice – acetone bath (–15°C) to –5°C and finely ground iodine (87 g, 0.343 mol) was added
at once. After a short induction period (~10 min) the temperature raised to +10°C and
more dry ice was added to the bath to keep the temperature of the reaction mixture below
0°C. To the reaction mixture cooled to –5°C, benzene (15.6 g, 0.2 mol) was added
dropwise from an addition funnel during 20 min. After the addition was complete, the
cooling bath was removed and reaction was stirred at room temperature for three days,
while monitored by GC-MS. Upon completion (GC peak of triiodobenzene disappeared),
the reaction mixture was poured onto crushed ice (1 kg), filtered, the precipitate on filter
washed with water (500 ml), sodium bisulfite (10% aq., 200 ml), and air-dried. The crude
brown-gray product was washed with hot BuOAc (500 ml) to remove most of the dark

346
color and recrystallized from di(ethylene glycol) monoethyl ether (Note 1) to give 67.8 g
(58%) of tetraiodobenzene 196 as yellowish crystals. The residue insoluble in di(ethylene
glycol) monoethyl ether was recrystallized from nitrobenzene to give 9.9 g of yellow
crystals of tetraiodobenzene and 2.7 g of orange powder of hexaiodobenzene443 (m.p.
423°C dec 430°C), insoluble in hot nitrobenzene, but recrystallizable from boiling
nitrobenzene. Total yield of tetraiodobenzene was 77.7 g (73%). M.p. 252°C. (Lit.444 m.p.
249-254°C). EI-MS: 581.75 (M+, 81), 455.79 (100), 329.90 (15), 74.00 (30).
Note 1. Di(ethylene glycol) monoethyl ether (one liter) was brought to boiling
and added to the crude tetraiodobenzene, boiled with stirring for ten minutes and rapidly
filtered. The filtrate was allowed to cool down to room temperature, filtered from the
formed crystals of tetraiodobenzene, and returned for recrystallization. This operation
was repeated three times. The recrystallized material may be further purified by
sublimation (two times, 150-210°C, 0.05 mm Hg) to leave behind 4.7 g of
hexaiodobenzene and then repeatedly recrystallized from dimethoxyethane (50 g of
solvent per gram of material) until material with a pure white color was obtained.
2,6-Diiodo-6-methylaniline 198.
NH2 NH2
II
A 300 ml round bottom flask with a stir bar was charged with p-toluidine (2.56
g, 0.02 mol), iodine (13 g, 0.051 mol), ethanol (150 ml), and water (70 ml). The mixture

347
was stirred for three hours at room temperature and then for 24 hrs at 80°C. After this
time the reaction mixture was cooled, diluted with water (100 ml) and sodium bisulfite
solution (10% aq., 50 ml), filtered off and the filtrate washed with water (200 ml) and
cold ethanol (50 ml) to give the crude product 5.3 g (62%) as a black powder. The crude
product was recrystallized from 1-propanol to give 2.22 g (25%) of brown crystals. M.p.
124°C.
N-Acetyl-4-methylaniline.
NH2 NH
O
A 500 ml round bottom flask with a stir bar was charged with p-toluidine (35.5
g, 0.331 mol) and acetic acid (100 ml). The flask was topped with an addition funnel,
filled with acetic anhydride (40 g, 0.392 mol), which was added to the reaction mixture
dropwise during 30 min. After the addition was complete, ethanol (100 ml) and water (50
ml) were added and the resulting mixture was recrystallized to afford 31.4 g (63.5%) of
N-Acetyl-4-methylaniline. M.p. 152.2°C (lit. m.p. 148-155°C). EI-MS: 149.08 (23),
107.07 (70), 106.10 (100), 79.09 (10), 77.1 (24).

348
N-Acetyl-2-iodo-4-methylaniline 199.
NH
O
NH
O
I
A 200 ml round bottom flask with a stir bar was charged with N-acetyl-4-
methylaniline (5.3 g, 0.035 mol), sodium hydrocarbonate (6.5 g, 0.077 mol),
dichloromethane (200 ml), methanol (10 ml), water (3 ml), and iodine monochloride
(11.9 g, 0.079 mol). The reaction mixture was monitored on TLC (hexane : ethyl acetate
= 1:1) and by GC-MS as it was stirred at room temperature for 12 hrs, then refluxed for
12 hrs (s.m. still present on TLC), then dichloromethane was removed on rotavap and
replaced with 1,2-dichloroethane (200 g) and pyridine (20 g) and refluxed for another 12
hrs. The reaction mixture was cooled down, filtered, washed with NaHSO3 (10% aq.,
2×100 ml), HCl (10% aq., 2×200 ml) and an aliquot injected into GC-MS: 63%
conversion to N-acetyl-2-iodo-4-methylaniline, EI-MS: 274.96 (14), 233.01 (32), 148.11
(100), 106.1 (60), 77.07 (39).
3,4,5-Triiodotoluene 201.
NH2
III
II
A 125 ml Erlenmeyer flask with a stir bar was charged with 2,6-diiodo-6-
methylaniline (1.38 g, 3.84 mmol), sulfuric acid (40 g) and cooled down in an ice-acetone

349
bath until the amine completely dissolves. To the cooled reaction mixture an ice-cold
solution of sodium nitrite (0.3 g, 4.3 mmol) in water (2 ml) was added dropwise at
vigorous stirring and left for diazotization for 30 min. Then an ice-cold solution of
potassium iodide (0.76 g, 4.57 mmol) in water (10 ml) was poured into the reaction
mixture at once, resulting in gas evolution. When the gas evolution ceased, the flask was
removed from the ice-acetone bath, allowed to warm up to room temperature, stirred for
2 hrs, and poured onto crushed ice (50 g). The black precipitate was filtered off, washed
with water (100 ml) and cold ethanol (50 ml) and sublimed twice (110°C, 1 mm Hg) to
give 1.6 g (88%). The sublimed product was recrystallized from 1-propanol (50 ml) to
give 1.2 g (66%). M.p. 119.5°C. EI-MS: 469.81 (100), 342.92 (22), 216.02 (18), 89.11
(28), 63.01 (35). 1H NMR: 7.69 (s, 2H), 2.17 (s, 3H). 13C NMR: 141.5, 139.8, 116.9,
106.9, 20.0.
1,4-Diiodonaphthalene 202.
A two-liter three-neck round bottom flask with a stir bar was charged with 1,4-
dibromonaphthalene (50.0 g, 0.175 mol) and anhydrous ether (1000 ml). The flask was
fitted with a thermometer, nitrogen inlet, and a septum. The reaction mixture was cooled
to –20 °C in a dry ice – acetone bath and tert-BuLi (393 g, 595 ml of 1.7 M solution in
hexane, 0.88 mol, 5 eq.) was added dropwise during a course of 40 min via a cannula
from a bottle, tared on top-load balances. After addition was complete, the reaction
mixture was allowed to warm up to room temperature, stirred at that temperature for 20
min, and cooled back to –20 °C. To the cooled reaction mixture, iodine (152.0 g, 0.6 mol,

350
3.3 eq.) was added in 5-7 g portions via a powder funnel under a nitrogen counterflow at
such a rate that the temperature did not rise above –15 °C. After addition was complete,
the reaction mixture was allowed to warm up to room temperature, stirred at that
temperature for 1 hour and quenched with Na2S2O3 (aq., 10%, 200 ml).
The solution was transferred into a separatory funnel and the organic layer was
washed in this order with: Na2S2O3 (aq., 10%, 200 ml), water (3×200 ml), hydrochloric
acid (aq., 2M, 2×200 ml), water (3×200 ml), treated with freshly activated carbon (2×70
g), and dried with MgSO4. The solvents were evaporated on a rotovap and the residue
crystallized to give 28.0 g (40%) of 1,4-diiodonaphthalene as canary yellow crystals.
M.p. 108.5 °C (lit.373 m.p. 110–111 °C). 1H NMR δ: 8.05 (dd, 2H, J = 3.3 Hz), 7.8 (s,
2H), 7.6 (dd, 2H, J = 3.3 Hz). 13C NMR δ: 138.3, 134.9, 133.2, 128.8, 100.9.
The ultimate purification was performed via a series of column
chromatographies with doubly distilled solvents (hexane – ether 8:2 and hexane –
dichloromethane 8:2), treatment of cyclohexane solution with freshly activated charcoal
and neutral alumina, and recrystallization from residue-free 1-propanol. Before the final
recrystallization, a saturated solution in chloroform was micron-filtered, chloroform
evaporated, and the material was recrystallized from micron-filtered 1-propanol.
2,3-Diaminonaphthalene.
OH
OH
NH2
NH2

351
A 100 ml autoclave was charged with a stir bar, 2,3-dihydroxynaphthalene (7.7
g, 48 mmol), ammonium sulfite monohydrate (9.2 g, 68 mmol), and ammonium
hydroxide (29% aq. sln, 80 ml). The reaction mixture was heated in a mantle filled with
iron filings to 250°C (mantle temperature) until pressure reached 520 psi (35 bar, takes ~
1.5 hr) and then heating continued for 12 hours. The cooled reaction mixture was poured
into water (200 ml), filtered, washed with water (2×200 ml), and air dried to allow 6.4 g
(84%) of yellow crystals of 2,3-diaminonaphthalene. M.p. 193°C (lit. 190-199°C). The
crude material was recrystallized from 1-propanol (170 ml) to yield 4.8 g (63%) of tan-
yellow crystals. EI-MS: 159.18 (M+1, 12), 158.14 (M+, 100), 141.11 (5), 140.14 (6),
131.12 (17), 130.12 (62), 114.11 (7), 103.10 (16), 77.05 (12).
2,3,6,7,10,11-Hexakis(pentyloxy)triphenylene 205.
H11C5O
H11C5O
OC5H11OC5H11
OC5H11OC5H11
OC5H11
OC5H11
Two-neck 250 ml round bottom flask with a stir bar was charged with 1,2-
bis(pentyloxy)benzene (F.W. 250.38, 17.0 g, 67.9 mmol) and anhydrous dichloromethane
(150 ml). The flask was fitted with a thermometer/nitrogen inlet and vanadium
oxychloride (27.1 g, 156 mmol) was added dropwise from an addition funnel at such a
rate that the temperature of the reaction mixture does not rise above 30-34°C (addition
takes approximately 20 min, the color changes to bluish-green). The reaction mixture was

352
stirred for additional 20 min and anhydrous methanol (100 ml) followed by water (250
ml) was added at once. The mixture was extracted with dichloromethane (4×200 ml) and
subjected to two consecutive chromatographic separations with hexane:dichloromethane
= 1:1 (first) and 9:1 (second) to afford, after solvent evaporation, 14.4 g (85.4%) of
grayish crystals.
The crude product (14.4 g, ~19.3 mmol) was re-alkylated with 1-bromopentane
(F.W. 151.05, 5.8 g, 38 mmol), potassium carbonate (6.0 g, 43 mmol), potassium iodide
(0.1 g, 0.6 mmol), and 2-butanone (150 ml) at 90°C for 12 hrs. The reaction mixture was
cooled to room temperature, filtered, solvent evaporated on a rotavap and the residue
dissolved in boiling cyclohexane. The warm solution was treated with charcoal (2×15 g),
filtered, and applied onto silica gel (50 g). The impregnated material was placed on top of
a 1500 ml flash chromatography column, filled with silica gel (25 cm of column height),
was eluted with neat hexane (3×500 ml) to remove excess of 1-bromopentane and left
under suction for 12 hrs. Then elution was resumed with dichloromethane (1 to 10%) in
hexane. First ~10% (pink) of the HAT-5 fraction was discarded, the rest of the fraction
was collected, solvents evaporated on a rotavap to allow 8.3 g (49%) of white crystals.
The purified material was recrystallized from 1-propanol/chloroform (residue-free),
dissolved in dichloromethane (residue-free), the solution was filtered through 0.45μ
syringe filter, solvent evaporated and the residue was recrystallized from cyclohexane (10
ml/g, with charcoal and alumina) to provide 6.04 g (35%) of snow-white crystals, which
do not change color when exposed to air and ambient light for two weeks.
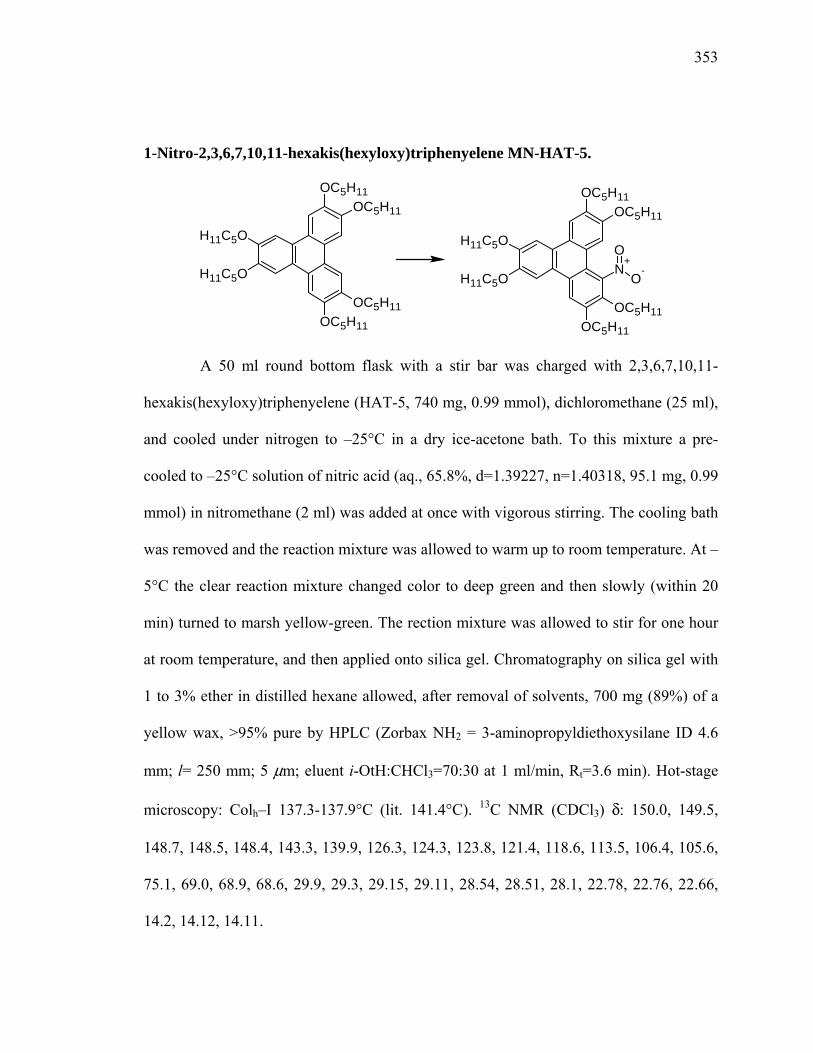
353
1-Nitro-2,3,6,7,10,11-hexakis(hexyloxy)triphenyelene MN-HAT-5.
H11C5O
H11C5O
OC5H11OC5H11
OC5H11OC5H11
H11C5O
H11C5O
OC5H11OC5H11
OC5H11OC5H11
N+
O-
O
A 50 ml round bottom flask with a stir bar was charged with 2,3,6,7,10,11-
hexakis(hexyloxy)triphenyelene (HAT-5, 740 mg, 0.99 mmol), dichloromethane (25 ml),
and cooled under nitrogen to –25°C in a dry ice-acetone bath. To this mixture a pre-
cooled to –25°C solution of nitric acid (aq., 65.8%, d=1.39227, n=1.40318, 95.1 mg, 0.99
mmol) in nitromethane (2 ml) was added at once with vigorous stirring. The cooling bath
was removed and the reaction mixture was allowed to warm up to room temperature. At –
5°C the clear reaction mixture changed color to deep green and then slowly (within 20
min) turned to marsh yellow-green. The rection mixture was allowed to stir for one hour
at room temperature, and then applied onto silica gel. Chromatography on silica gel with
1 to 3% ether in distilled hexane allowed, after removal of solvents, 700 mg (89%) of a
yellow wax, >95% pure by HPLC (Zorbax NH2 = 3-aminopropyldiethoxysilane ID 4.6
mm; l= 250 mm; 5 μm; eluent i-OtH:CHCl3=70:30 at 1 ml/min, Rt=3.6 min). Hot-stage
microscopy: Colh–I 137.3-137.9°C (lit. 141.4°C). 13C NMR (CDCl3) δ: 150.0, 149.5,
148.7, 148.5, 148.4, 143.3, 139.9, 126.3, 124.3, 123.8, 121.4, 118.6, 113.5, 106.4, 105.6,
75.1, 69.0, 68.9, 68.6, 29.9, 29.3, 29.15, 29.11, 28.54, 28.51, 28.1, 22.78, 22.76, 22.66,
14.2, 14.12, 14.11.

354
1,5,9-Trinitro-2,3,6,7,10,11-hexakis(hexyloxy)triphenyelene TN-HAT-5.
H11C5O
H11C5O
OC5H11OC5H11
OC5H11OC5H11
N+
O-
O
N+
O-
O
N+
O-
O
H11C5O
H11C5O
OC5H11OC5H11
OC5H11OC5H11
A 100 ml round bottom flask with a stir bar was charged with 2,3,6,7,10,11-
hexakis(hexyloxy)triphenyelene (HAT-5, 4.13 g, 5.5 mmol), dichloromethane (50 ml),
and cooled under nitrogen to –10°C in a dry ice-acetone bath. To this mixture an
impregnation of nitric acid (aq., 65.8%, d=1.39227, n=1.40318, 2.66 g, 27.7 mmol) on
silica gel (15 g) was added at once with vigorous stirring. The cooling bath was removed
and the reaction mixture was allowed to warm up to room temperature and stir for 48
hours at room temperature. The solvent was evaporated and the resulting brown
impregnation was was placed at the top of a silica gel column and chromatographed with
1 to 7% dichloromethane in distilled hexane. The first bright yellow fraction gave, after
removal of solvents, 0.6 g (12%) of a yellow oil, >80% pure by HPLC (Zorbax NH2 = 3-
aminopropyldiethoxysilane ID 4.6 mm; l= 250 mm; 5 μm; eluent i-octane:CHCl3=70:30
at 1 ml/min, Rt=3.0 min). APCI-MS: 879 (M+). UV-Vis (CH2Cl2) λmax (lg ε): 283 (4.87).
1H NMR (CDCl3) δ: 7.55 (s, 1H), 4.25 (t, 2H), 4.11 (t, 2H), 1.92 (pentet, 2H), 1.80
(pentet, 2H), 1.6-1.4 (m, 8H), 1.02-0.96 (m, 6H). 13C NMR (CDCl3) δ: 151.9, 143.9,
141.6, 123.0, 114.4, 108.3, 75.4, 69.3, 29.7, 28.7, 28.3, 27.9, 22.4, 14.0, 13.97. Cr–Colh
33.9-34.3°C (microscope hot stage, 0.5°C/min). Colh–I 142°C (DSC, 10°C/min).

355
CONCLUSIONS
1. We have prepared a series of fluorescent 3,6-diphenyl-2,5-dihydropyrrolo[3,4-
c]pyrrole-1,4-dione dyes, useful for the field of single-molecule spectroscopy (SMS) and
have applied several of them to the detection and characterization of single biological
molecules. The dyes have been shown to posses high quantum yields and useful (up to 84
nm) Stoke’s shifts. Photobleaching measurements have been conducted for several of the
synthesized dyes and revealed that the anticipated structure–photostability does not hold
true in all instances. Maleimide, hydroxy, halogen and some other functional groups have
been incorporated into the DPP bicyclic structure. N,N′-Diarylated DPPs have been
prepared by two new methods.
2. A cysteine-specific fluorescent probe has been synthesized and applied to testing of the
local polarity and conformational changes in a single-cysteine mutant of GroEL
chaperonin of E. coli. The methodology to introduce a maleimide moiety, learned during
the synthesis, has been utilized to synthesize other maleimide-containing fluorophores.
3. A series of iodinated aromatic compounds has been prepared and purified to study
charge transport in organic molecular crystals. Several iodination approaches have been
explored and compared to attain the highest possible purity of the final compounds.

356
4. Several polyalkyl- and polyalkoxy- acenes (anthracenes, tetracenes) and some
important key intermediates en route to polyalkoxypentacenes have been prepared. None
of the synthesized compounds exhibited mesogenic properties.
5. A reliable, highly effective purification protocol has been elaborated for purification of
hexapentyloxytriphenylene and its analogs. New trinitro-HAT-5 has been synthesized
and characterized as a discotic liquid crystal in the temperature range 34…140 °C.

357
REFERENCES
1 Chandra, B. P.; Kalia, V; Datt, S. C. Crystalloluminescence: a new tool to determine the
critical size of a crystal nucleus. J. Phys. D: Appl. Phys. 1985, 18, L189–L193. 2 Moerner, W. E.; Kador, L. Phys. Rev. Lett. 1989, 62, 2535–2538. 3 Moerner, W. E. A Dozen Years of Single-Molecule Spectroscopy in Physics,
Chemistry, and Biophysics. J. Phys. Chem. B 2002, 106, 910–927. 4 a) Itano, W. M.; Bergquist, J. C.; Wineland, D. J. Science 1987, 237, 612; b) Dehmelt,
H; Paul W., Ramsey, N. F. Rev. Mod. Phys. 1990, 62, 525. 5 Jabłoński, A. Über den Mechanisms des Photolumineszenz von Farbstoffphosphoren. Z.
Phys. 1935, 94, 38–46. 6 Moerner, W. E.; Fromm, D. P. Methods of Single-Molecule Fluorescence Spectroscopy
and Microscopy. Rev. Sci. Instrum. 2003, 74, 3597–3619. 7 Haw Y.; Guobin L.; Pallop K.; Tai-Man L.; Ivan R.; Sergio C.; Luying X.; X. Sunney,
X. Protein Conformational Dynamics Probed by Single-Molecule Electron Transfer.
Science 2003, 302, 262–266. 8 Tinnefeld, P.; Sauer, M. Branching Out of Single-Molecule Fluorescence Spectroscopy:
Challenges for Chemistry and Influence on Biology Angew. Chem. Int. Ed. (Eng.) 2005,
44 (18), 2642–2671. 9 Arnaud, C. H. The $1,000 Genome Chem. Eng. News 2005, 83 (51), 60–61. 10 Förster, T. Ann. Phys. 1948, 2, 55–75. 11 Tuschl. T.; Gohlke, C.; Jovin, T. M.; Westhof, E.; Eckstein, F. Science 1994, 266, 785–
789.

358
12 Rigler, R.; Mets, Ü.; Widengren, J.; Kask, P. Eur. Biophys. J. Biophys. Lett. 1993, 22,
169–175. 13 Cohen, A. E.; Moerner, W. E. Method for trapping and manipulating nanoscale objects
in solution. Appl. Phys. Lett. 2005, 86 (9), 0931091–0931093. 14 Denk, W.; Strickler, J. H.; Webb, W. W. Science 1990, 248, 73–76. 15 Willets, K. A.; Nishimura, S. Y.; Schuck, P. J.; Twieg, R. J.; Moerner, W. E. Nonlinear
Optical Chromophores as Nanoscale Emitters for Single-Molecule Spectroscopy.
Accounts of Chemical Research 2005, 38 (7), 549–556. 16 Schuck, P. J.; Willets, K. A.; Fromm, D. P.; Twieg, R. J.; Moerner, W. E.. A novel
fluorophore for two-photon-excited single-molecule fluorescence. Chemical Physics
2005, 318 (1–2), 7–11. 17 Heinze, K. G.; Rarbach, M.; Jahnz, M.; Schwille, P. Biophys. J. 2002, 83, 1671–1681. 18 Moerner, W. E.; Ambrose, W. P. Phys. Rev. Lett. 1991, 66, 1376. 19 Ormo, M., et al., Science 1996, 273, 1392-5. 20 Xue, Q.; Yeung, E. S. Nature 1995, 373, 681. 21 Craig, D. B.; Arriaga, E. A.; Wong, J. C. Y.; Lu, H.; Dovichi, N. J. J. Am. Chem. Soc.
1996, 118, 5245. 22 Lu, H. P.; Xun, L.; Xie, X. S. Science 1998, 282, 1877. 23 Tsien, R. Y. Rev. Biochem. 1998, 67, 509. 24 Single Molecule Optical Detection, Imaging, and Spectroscopy, Ed. T. Basche, W. E.
Moerner, M. Orrit, and U. P. Wild, Verlag Chemie, Munich, 1997. 25 Seisenberger, G.; Ried, M. U.; Endreß, T.; Büning, H.; Hallek, M.; Bräuchle, C.
Science 2001, 294, 1929–1932. 26 Nano Lett. 2006, 6, 45. 27 Kapanidis, A. N.; Weiss, S. Fluorescent probes and bioconjugation chemistries for
single-molecule fluorescence analysis of biomolecules. J. Chem. Phys. 2002, 117, 10953-
10964.

359
28 Weiss, S. Fluorescence Spectroscopy of Single Biomolecules. Science 1999, 283,
1676-1683. 29 Wilkinson, F.; McGarvey, D. J.; Olea, O.F. J.Phys.Chem. 1994, 98, 3762. 30 Hubner, C. G.; Renn, A.; Renge, I.; Wild, U.P. J. Chem. Phys. 2001, 115, 9619. 31 Single-Molecule Optical Detection, Imaging and Spectroscopy Basché, T.; Moerner,
W. E.; Orrit, M; Wild, U. P. Eds.; VCH Verlaggesellschaft: 1996; p 10. 32 Zarrin , F.; Dovichi, N. J. Anal. Chem. 1985, 57, 2690–2692. 33 Enderlein, J. Recent advances in single molecule fluorescence spectroscopy. Reviews
in Fluorescence. Kluwer Academic Publishers/Plenum Publishing: New York, NY, 2004,
pp 121–163. 34 Miller, L. W.; Cornish, V. W. Selective Chemical Labeling of Proteins in Living Cells.
Current Opinion in Chemical Biology 2005, 9, 56–61. 35 Stokes, G. G. On the Change of Refrangibility of Light. Phil. Tans. R. Soc. London
1852, 142, 463–562. 36 Bruches, M.; Moronne, M.; Gin, P.; Weiss, S.; Alivisatos, A. P. Science 1998, 281,
2013–2016. 37 Handbook of Fluorescent Probes and Research Products, Ninth Edition –
http://probes.invitrogen.com/handbook/ 38 Bruchez, M. P.; Moronne, M.; Gin, P.; Weiss, S.; Alivisatos, A.P. Science, 1998, 281,
2013. 39 Michalet, X.; Pinaud, F.; Lacoste, T. D.; Dahan, M., Bruchez, M.P.; Alivisatos, A.P.;
Weiss, S. Single Molecules 2001, 2, 261. 40 J. Phys. Chem. B 2005, 109, 24220. 41 Lakowicz, J. R. Principles of Fluorescence Spectroscopy 2nd ed. Kluwer Academic
Publishers/Plenum Publishing: New York, NY, 1999 42 Farnum, D. G.; Mehta, G.; Moore, G. G. I.; Siegal, F. P. Attempted Reformatskii
reaction of benzonitrile. 1,4-Dioxo-3,6-diphenylpyrrolo[3,4-c]pyrrole, a lactam analog of
pentalene. Tetrahedron Lett. 1974, 29, 2549–52.

360
43 a) Jost M.; Iqbal A.; Rochat, A. C. 1,4-Diketopyrrolopyrrole Pigments. U.S. Patent
4,791,204, 1988; b) Wooden, G.; Guy de Weck; Wallquist O. Diketopyrrolopyrroles and
Pigments Thereof. U.S. Patent 5,424,452, 1995; c) Lamatsch B.; Wallquist O.; Scloeder
I. Yellow Diketopyrrolopyrrole Pigments. U.S. Patent 5,672,716, 1997; d) Hendi, S. B.
1,4- Diketo-3,6-diarylpyrrolo[3,4]cpyrrole Pigment Derivatives. U.S. Patent 5,786,487,
1998; e) Wallquist O.; Lamatsch B.; Ruch T. Blue Diketopyrrolopyrrole Pigments. U.S.
Patent 5,817,832, 1998. 44 Srivatsavoy, V. J. P.; Eschle, M.; Moser, J.-E.; Grätzel, M. Excited State Electron and
Energy Transfer of a Highly Fluorescent Heterocyclic Dye: a Laser Flash Photolysis
Study of 2,5-Dimethyl-3,6-diphenylpyrrolo[3,4-c]pyrrole-1,4-dione. J. Chem. Soc. Chem.
Commun. 1995, 3, 303–304. 45 Fukuda, M.; Kodama, K.; Yamamoto, H.; Mito, K. Solid State Laser with Newly
Synthesized Pigment. Dyes and Pigments 2002, 53, 67–72. 46 Potrawa, T.; Langhals, H. Fluorescent dyes with large Stokes shifts – soluble
dihydropyrrolopyrrolediones. Chem. Ber. 1987, 120 (7), 1075–8. 47 Langhals, H.; Grundei, T.; Potrawa, T.; Polborn, K. Highly photostable organic
fluorescent pigments – a simple synthesis of N-arylpyrrolopyrrolediones (DPP). Liebigs
Ann. Chem. 1996, 5, 679–682. 48 Zambounis, J. S.; Hao, Z.; Iqbal, A. Latent pigments activated by heat. Nature
(London) 1997, 388 (6638), 131–132. 49 Hall-Goulle, V. Soluble Chromophores Containing Solubilizing Groups Which Can Be
Easily Removed. U.S. Patent 6,063,924, 2000. 50 Hall-Goulle, V., Bize, A. Soluble Chromophores Having Improved Solubilizing
Groups U.S. Patent 6,274,728, 2001. 51 Chan, W. K.; Chen, Y.; Peng, Z.; Yu, L. Rational Designs of Multifunctional
Polymers. J. Am. Chem. Soc. 1993, 115, 11735–11743.

361
52 Beyerlein, T.; Tieke, B. New photoluminescent conjugated polymers with 1,4-dioxo-
3,6-diphenylpyrrolo[3,4-c]pyrrole (DPP) and 1,4-phenylene units in the main chain.
Macromol. Rapid Commun. 2000, 21 (4), 182–189. 53 Heim, I.; Tieke, B.; Lenz, R. et al. Novel Diketopyrrolopyrrole Polymers. Int. Patent
WO 2005/049695 A1, 2005. 54 Yu, L.; Chan, W. K.; Peng, Z.; Gharavi, A. Multifunctional Polymers Exhibiting
Photorefractive Effects. Acc. Chem. Res. 1996, 29, 13–21. 55 Rochat, A. C.; Wallquist, O.; Iqbal, A.; Mizuguchi, J. Process for the Preparation of
Aminated Diketobis (aryl or heteroaryl)pyrrolopyrroles and the Use Thereof as
Photoconductive Substances. U.S. Patent 5,973,146, 1999. 56 Mizuguchi, J.; Iqbal, A.; Giller, G. Preparation of Electrochromic Diketopyrroles for
Electrochromic Display Devices. Ger. Offen. DE 4435211 A1, 1995-04-27, 1995. 57 Mizuguchi, J. Pyrrolopyrrole pigments and their electronic applications. Shikizai
Kyokaishi 1997, 70 (6), 393–403. 58 Heim, I.; Tieke, B.; Lenz, R.; Schmidhalter, B.; Rabindranath, A. R.; Dueggeli, M.
Novel diketopyrrolopyrrole polymers for electronic devices. Int. Patent WO
2005/049695, 2005. 59 Hofkens, J.; Verheijen, W.; Shukla, R.; Dehaen, W.; De Schryver, F. C. Detection of a
Single Dendrimer Macromolecule with a Fluorescent Dihydropyrrolopyrroledione (DPP)
Core Embedded in a Thin Polystyrene Polymer Film. Macromolecules 1998, 31 (14),
4493–4497. 60 Smet, M.; Metten, B.; Dehaen, W. Construction of rod-like diketopyrrolopyrrole
oligomers with well-defined length. Tetrahedron Lett. 2001, 42 (37), 6527–6530. 61 Hao, Z.; Iqbal, A.; Tebaldi, N.; Praefke, K. Liquid crystalline diketopyrrolopyrroles.
U.S. Patent 5,969,154, 1999. 62 Praefcke, K.; Jachmann, M.; Blunk, D.; Horn, M. Novel family of liquid crystals based
on a known biheterocyclic pigment material: mesomorphic derivatives of 2,5-
dihydropyrrolo[3,4-c]pyrrole-1,4-dione. Liq. Cryst. 1998, 24 (1), 153–156.

362
63 Wendy V.; Johan H.; Bert M.; Jo V.; Ramesh S.; Mario S.; Wim D.; Yves E.; Frans De
Schryver The Photo Physical Properties of Dendrimers Containing 1,4-Dioxo-3,6-
Diphenylpyrrolo[3,4-c]pyrrole (DPP) as a Core. Macromol. Chem. Phys. 2005, 206, 25–
32. 64 Eggeling, C.; Widengren, J.; Rigler, R.; Seidel, C. A. M. Photostability of fluorescent
dyes for single-molecule spectroscopy: Mechanisms and experimental methods for
estimating photobleaching in aqueous solution. In Applied Fluorescence in Chemistry,
Biology and Medicine; Rettig, W., Strehmel, B., Schrader, S., Seifert, H., Eds.; Springer-
Verlag: New York, 1999; pp 193–240. 65 Hofkens, J.; Verheijen, W.; Shukla, R.; Dehaen, W.; De Schryver, F. C. Detection of a
Single Dendrimer Macromolecule with a Fluorescent Dihydropyrrolopyrroledione (DPP)
Core Embedded in a Thin Polystyrene Polymer Film. Macromolecules 1998, 31 (14),
4493–4497. 66 Iqbal, A.; Jost, M.; Kirchmayr, R.; Pfenninger, J.; Rochat, A.; Wallquist, O. Synthesis
and properties of 1,4-diketopyrrolo[3,4-c]pyrroles. Bull. Soc. Chim. Belg. 1988, 97 (8–9),
615–43. 67 Lenz, R.; Wallquist, O. DPP Chemistry – Continuous Innovation. Surface Coatings
International, Part B: Coatings Transactions, 2002, 85 (B1), 19–26. 68 Hao, Z.; Iqbal, A. Some Aspects of Organic Pigments Chem. Soc. Rev. 1997, 26, 203–
213. 69 Iqbal A.; Cassar, L. Process for Dyeing High-Molecular Organic Material, and Novel
Polycyclic Pigments U.S. Patent 4,415,685, 1983. 70 Iqbal A.; Cassar, L. 1,4-Diketopyrrolo[3,4-c]pyrroles. U.S. Patent 4,490,542, 1984. 71 Rochat, A.; Cassar, L.; Iqbal, A. Preparation of Pyrrolo[3,4-c]pyrroles. U.S. Patent
4,579,949, 1986. 72 Kaul, B. Solvent-free prodn. and use of pyrrolo[3,4-c]pyrroledione pigments. Int.
Patent WO 2004076457, 2004. 73 Sparke, J. M.; Watson, K.D. J. Chem. Soc. Perkin Trans. 1 1976, 5.

363
74 Jost, M.; Iqbal, A.; Rochat, A. C. N-Substituted 1,4-Diketopyrrolo[3,4-c]pyrroles. U.S.
Patent 4,585,878, 1986. 75 Jost, M.; Iqbal, A.; Rochat, A. C. Compositions Pigmented with N-Substituted 1,4-
Diketopyrrolo[3,4-c]pyrroles. U.S. Patent 4,666,455 , 1987 76 Pfenninger, J.; Iqbal, A.; Rochat, A. C. Pyrrolo[3,4-c]-pyrroles and process for their
preparation. Eur. Patent EP0184982, 1986. 77 Pfenninger, J.; Iqbal, A.; Rochat, A. C. Process for the Preparation of Pyrrolo[3,4-c]-
pyrroles. U.S. Patent 4,778,899, 1988. 78 Pfenninger, J.; Iqbal, A.; Rochat, A. C.; Wallquist, O. Process for the Preparation of
Pyrrolo[3,4-c]-pyrroles and Novel Pyrrolo[3,4-c]-pyrroles. U.S. Patent 4,659,775, 1987. 79 STN substructure search in CA/CAplus/CASREACT databases, performed on
February, 7, 2006. 80 MDL Crossfire Commander substructure search in Beilstein database (ver. 2005/03). 81 Morton, Colin J. H.; Gilmour, Ryan; Smith, David M.; Lightfoot, Philip; Slawin,
Alexandra M. Z.; MacLean, Elizabeth J. Synthetic studies related to diketopyrrolopyrrole
(DPP) pigments. Part 1: The search for alkenyl-DPPs. Unsaturated nitriles in standard
DPP syntheses: a novel cyclopenta[c]pyrrolone chromophore. Tetrahedron 2002, 58 (27),
5547–5565. 82 Closs, F.; Gompper, R. 2,5-Diazapentalene. Angew. Chem. 1987, 99 (6), 564–567;
Angew. Chem., Int. Ed. Engl. 1987, 26 (6), 552–3. 83 Knorr, L.; Scheidt, M. Ber. Dtsch. Chem. Ges. 1894, 27, 1167–1168. 84 a) Pfleger, R.; Reinhardt, F. Chem. Ber. 1957, 90, 2404–2411; b) Wu, A.; Zhao, Y.;
Chen, N.; Pan, X. A Modification of the Knorr Oxidative Coupling Method for
Preparation of 1,4-Diketones. Synth. Commun. 1997, 331; c) J. Chem. Soc. Perkin 1,
1980, 2670; d) Pelter, A.; Ward, R. S.; Watson, D. J.; Jack, I. R. Synthesis and NMR
Spectra of 2,6- and 2,4-Diaryl-3,7-dioxabicyclo[3.3.0]octanes J. Chem. Soc. Perkin 1,
1982, 183–190; e) Synth. Commun. 1997, 2087; f) Tetr. Lett. 1978, 1509; g) J. Am.

364
Chem. Soc., 1964, 2392; h) Thorp, L.; Brunskill, E. R. J. Am. Chem. Soc., 1915, 37,
1258. 85 a) Knorr, L.; Scmidt, J. Liebigs Ann. Chem. 1896, 293, 111; b) Lohaus, H. Liebigs Ann.
Chem. 1934, 509, 130. 86 a) Bromme, E.; Claisen, L. Chem. Ber. 1888, 21, 1134; b) Scmidt, P. F. Chem. Ber.
1895, 28, 1206; c) Keglević, D.; Malnar, M.; Tomljenović, T. Arkiv Chemi, 1954, 26, 67. 87 Rubin, M. B.; Bargurie, M.; Kaftory, M.; Kosti, S. Synthesis and reactions of 1,6-
diaryl-2,5-bis(diazo)-1,3,4,6-tetraoxohexanes J. Chem. Soc., Perkin Trans. 1 1980 (12),
2670–2677. 88 Jost, M.; Iqbal, A.; Rochat, A. C. N-Substituted 1,4-Diketo-3,6-diarylpyrolo[3,4-
c]pyrroles. U.S. Patent 4,585,878, 1986. 89 Jost, M.; Iqbal, A.; Rochat, A. C. Compositions Pigmented with N-Substituted 1,4-
Diketo-3,6-diarylpyrolo[3,4-c]pyrroles. U.S. Patent 4,666,455, 1987. 90 Zambounis, J. S.; Hao, Z.; Iqbal, A. Pyrrolo[3,4-c]pyrroles. U.S. Patent 5,484,943,
1996 91 Zambounis, J. S.; Hao, Z.; Iqbal, A. Pyrrolo[3,4-c]pyrrole synthesis. U.S. Patent
5,616,725, 1997. 92 Hall-Goulle, V. Soluble chromophores containing solubilising groups which can be
easily removed U.S. Patent 6,063,924, 2000. 93 Hall-Goulle, V.; Bize, A. Soluble chromophores having improved solubilizing groups.
U.S. Patent 6,274,728, 2001. 94 Mizuguchi, J. A pigment precursor based on 1,4-diketo-3,6-diphenyl-pyrrolo-[3,4-c]-
pyrrole and its regeneration into the pigment. Journal of Imaging Science and
Technology 2005, 49 (1), 35–40. 95 Hendi, S. B. 1,4-Diketo-3,6-diarylpyrolo[3,4-c]pyrroles. U.S. Patent 5,785,750, 1998. 96 Hendi, S. B. Process for Preparing Diketopyrrolopyrrole Derivatives. U.S. Patent
5,840,907, 1998.

365
97 Hendi, S. B. Process for Preparing Diketopyrrolopyrrole Derivatives. U.S. Patent
5,786,487, 1998. 98 Hendi, S. B. U.S. Process for Preparing Diketopyrrolopyrrole Derivatives. Patent
5,919,945, 1999. 99 Buckles, R. E.; Hausman, E. A.; Wheeler, N. G. The Action of Bromine Vapor on
Solid Aromatic Compounds J. Am. Chem. Soc. 1950, 72, 2494. 100 Buckles, R. E.; Wheeler, N. G. 4,4′-Dibromobiphenyl. Org. Synth. Coll. Vol. 4, 256. 101 Wallquist, O.; Iqbal, A.; Pfenninger, J.; Rochat, A. Process for the Preparation of
Brominated Pyrrolo[3,4-c]pyrroles and Mixtures Thereof. U.S. Patent 4,810,802, 1989. 102 Rochat, A. C.; Iqbal, A.; Ettingen; Jeanneret, R.; Mizuguchi, J. Dithioketo
Pyrrolopyrroles U.S. Patent 4,760,151, 1988. 103 Rochat, A. C.; Iqbal, A.; Wallquist, O. Monoketopyrrolopyrrole. U.S. Patent
5,017,706, 1991. 104 Zambounis, J. S.; Hao, Z.; Iqbal, A. Pyrrolo[3,4-cipyrroles containing cyanimino
groups. U.S. Patent 5,527,922, 1996. 105 Wallquist, O.; Lamatsch, B.; Ruch, T. Blue diketopyrrolopyrrole pigments. U.S.
Patent 5,817,832, 1998. 106 Rochat, A. C.; Wallquist, O.; Iqbal, A.; Mizuguchi, J. Process for the preparation of
aminated diketobis (aryl or heteroaryl)pyrrolopyrroles and the use thereof as
photoconductive substances U.S. Patent 5,973,146, 1999. 107 Lamatsch, B.; Wallquist, O. Process for the preparation of
diketopyrrolopyrrolecarboxylic acids and their esters and amides. U.S. Patent 5,874,588,
1999. 108 Mizuguchi, J.; Grubenmann, A.; Gary W.; Rihs, J. Structures of 3,6-
Diphenylpyrrolo[3,4-clpyrrole-l,4-dione and 2,5-Dimethyl-3,6-diphenylpyrrolo[3,4-
c]pyrrole-l,4-dione. Acta Cryst. 1992, B48, 696–700. 109 Bäbler, F. Process for Preparing 1,4-Diketo-3,6-diphenylpyrrolo-[3,4-c]-pyrrole. U.S.
Patent 5,347,014, 1994.

366
110 Bäbler, F. Process for the Preparation of Diaryldiketopyrrolopyrrole Pigments. U.S.
Patent 5,565,578, 1996. 111 Surber, W.; Iqbal, A.; Stern, C. Process for the preparation of pyrrolo[3,4-c]pyrroles.
U.S. Patent 4,931,566, 1990. 112 Mizuguchi, J.; Rihs, G. Electronic spectra of 1,4-diketo-3,6-diphenyl-pyrrolo-[3,4-c]-
pyrrole in the solid state. Ber. Bunsen-Gess. Chem. 1992, 96 (4), 597–606. 113 Mizuguchi, J.; Wooden, G. A large bathochromic shift from the solution to the solid
state in 1,4-diketo-3,6-diphenyl-pyrrolo-[3,4-c]-pyrrole. Ber. Bunsen-Gess. Chem. 1991,
95 (10), 1264–1274. 114 Riggs, R. L.; Morton, C. J. H.; Slawin, A. M. Z.; Smith, D. M.; Westwood, N. J.;
Austen, W. S. D.; Stuart, K. E. Synthetic studies related to diketopyrrolopyrrole (DPP)
pigments. Part 3: Synthesis of tri- and tetra-aryl DPPs. Tetrahedron, 2005, 61, 11230–
11243. 115 Brooker, L. G. S.; Van Lare, E. J. Color and constitution of organic dyes. Kirk-Othmer
Encyclopedia of Chemical Technology, 2-nd Ed., 1964, 5, 763–88. 116 Dewar, M. J. S. The electronic theory of organic chemistry. Clarendon Press: Oxford,
1949. 117 Dewar, M. J. S.; Dougherty, R. C. The Molecular Orbital Theory of Organic
Chemistry. McGraw-Hill: New York, 1969. 118 Frei, U.; Kirchmayr, R. Process for the preparation of tert-alkyl esters. U.S. Patent
4,904,814, 1990. 119 Wang, E. C. Tetrahedron Lett. 1998, 39, 4047–4050. 120 Wallquist, O. Cosmetic formulations comprising diketodiphenyl pyrrolopyrrole
pigments. Int. Patent WO 2005039513, 2005. 121 Fukuda, M. Kodama, K.; Yamamoto, H.; Mito, K. Evaluation of new organic
pigments as laser-active media for a solid-state dye laser. Dyes and Pigments 2004, 63
(2), 115–125

367
122 Enokida, T.; Tamano, M. Organic electroluminescent device and pyrrolo[3,4-c]pyrrol-
based electron-transporting material for it. Patent JP 09003448, 1997. 123 Waldron, W. R.; Franklin, R. C. Process for preparing aromatic sulfonic esters of
branched chain aliphatic alcohols. U.S. Patent 2,728,788, 1953. 124 Zambounis, J.; Bize, A. Process for producing N-methylated organic pigments. Int.
Patent WO 9608537, 1996. 125 Kitao, T.; Yoshida, O.; Kaieda, O.; Shimoyama, F.. Pyrrolopyrrole derivatives as
petroleum product identifying agents and method of adding the agents. Patent JP
02216457, 1990. 126 Goldfinger, M. B.; Crawford, K. B.; Swager, T. M. Directed Electrophilic
Cyclizations: Efficient Methodology for the Synthesis of Fused Polycyclic Aromatics J.
Am. Chem. Soc. 1997, 119, 4578. 127 Mallory, F. B.; Mallory, C. W.; Butler, K. E.; Lewis, M. B.; Xia, A. Q.; Luzik, E. D.,
Jr.; Fredenburgh, L. E.; Ramanjulu, M. M.; Van, Q. N.; Francl, M. M.; Freed, D. A.;
Wray, C. C.; Hann, C.; Nerz-Stormes, M.; Carroll, P. J.; Chirlian, L. E. Nuclear Spin-
Spin Coupling via Nonbonded Interactions. 8.1 The Distance Dependence of Through-
Space Fluorine-Fluorine Coupling J. Am. Chem. Soc. 2000, 122, 4108. 128 Jeffery, T. Palladium-catalysed Vinylation of Organic Halides under Solid-Liquid
Phase Transfer Conditions. J. Chem. Soc. Chem. Commun. 1984, 1287. 129 (a) Mignani J. Tetrahedron Lett. 1990, 31 (33), 4743. (b) Littke A. F. J. Am. Chem.
Soc. 2000, 122 (17), 4020. 130 a) Momose, T.; Uchida, S.; Imanishi, T. Octahydro-7(1H)-quinolones. VII. A
stereoselective synthesis of cis,trans-decahydro-3H,8H-benzo[i,j]quinolizine-3,8-dione.
Chem. Pharm. Bull. 1979, 27 (1), 215–21. b) Scammells, P. J.; Baker, S. P.; Belardinelli,
L.; Olsson, R. A. Substituted 1,3-Dipropylxanthines as Irreversible Antagonists of A1
Adenosine Receptors. J. Med. Chem. 1994, 37 (17), 2704–12. 131 Lu, Z.; Twieg, R. J. Tet. Lett. 2005, 46, 2997–3001.

368
132 Greene T.; Wuts, P. G. M. Protective Groups in Organic Synthesis. 3-d Ed. Wiley and
Sons: New York, 1999; p 173–178. 133 Herzig, J.; Nudelman, A.; Gottlieb, H. E.; Fischer, B. Studies in Sugar Chemistry. 2. A
Simple Method for O-Deacylation of Polyacylated Sugars. J. Org. Chem. 1986, 51, 727–
730. 134 Copola, G. M. N-Arylation of Isatins. A Direct Route to N-Arylisatioc Anhydrides. J.
Heterocycl. Chem. 1987, 24, 1249–1251. 135 Smith, M. B.; March, J. Aromatic Nucluophilic Substitution. (Chapter 13) In: March’s
Advanced Organic Chemistry. Reactions, Mechanisms, and Structure. Wiley and Sons:
New York, 2001; pp 850–893. 136 Weber, G. Polarization of the Fluorescence of Macromolecules. Biochem J. 1952, 51,
155, 13136. 137 Bergstroem, F.; Mikhalyov, I.; Haeggloef, P.; Wortmann, R.; Ny, T.; Johansson, L. B.
A. Dimers of dipyrrometheneboron difluoride (BODIPY) with light spectroscopic
applications in chemistry and biology. J. Am. Chem. Soc. 2002, 124 (2), 196–204. 138 Amat-Guerri, F.; Liras, M.; Carrascoso, M. L.; Sastre, R. Methacrylate-tethered
analogs of the laser dye PM567-synthesis, copolymerization with methyl methacrylate
and photostability of the copolymers. Photochem. Photobiol. 2003, 77 (6), 577–584 139 Haugland, R. P. Handbook of Fluorescent Probes and Research Products. 9-th Ed. p
3. http://probes.invitrogen.com/handbook/figures/0667.html 140 Nietzki, R.; Becker, V. Oxazine Dyes. Ber. Dtsch. Chem. Ges. 1908, 40, 3397–3400. 141 Kehrmann, F.; Grillet, E.; Borgeaud, P. New syntheses of oxazine dyes. Helvetica
Chimica Acta 1926, 9, 866–880. 142 Du, H.; Fuh, R.-C. A.; Li, J.; Corkan, L. A.; Lindsey, J. S. Photochem CAD: a
computer-aided design and research tool in photochemistry. Photochemistry and
Photobiology 1998, 68 (2), 141–142. 143 Davis, M. M.; Hetzer, H. B. Titrimetric and equilibrium studies using indicators
related to Nile Blue A. Anal. Chem. 1966, 38, 451–461.

369
144 Sahyun, M. R. V. Total luminescence spectroscopy in a reverse micellar system. J.
Phys. Chem. 1998, 92, 6028–6032. 145 Sackett, D. L.; Wolff, J. Nile red as a polarity-sensitive fluorescent probe of
hydrophobic protein surfaces. Anal. Biochem. 1987, 167, 228–234, 146 Golini, C. M.; Williams, B. W.; Foresman, J. B. Further Solvatochromic,
Thermochromic, and Theoretical Studies on Nile Red. J. Fluoresc. 1998, 8 (4), 395–403. 147 Briggs, M. S. J.; Bruce, I.; Miller, J. N.; Moody, C. J.; Simmonds, A. C.; Swann, E.
Synthesis of functionalized fluorescent dyes and their coupling to amines and amino
acids. J. Chem. Soc., Perkin Trans. 1. 1997, (7), 1051–1058. 148 Moerner, W. E.; Twieg, R. J.; Kline, D. W.; He, M. Fluorophore Compounds and
Their Use in Biological Systems. U.S. Patent Appl. 20050009109, 2005. 149 Wright, D.; Gubler, U.; Roh, Y.; Moerner, W. E.; He, M.; Twieg, R. J. High-
performance photorefractive polymer composite with 2-dicyanomethylen-3-cyano-2,5-
dihydrofuran chromophore. Applied Physics Letters 2001, 79 (26), 4274–4276. 150 Willets, K. A.; Ostroverkhova, O.; He, M.; Twieg, R. J.; Moerner, W. E. Novel
fluorophores for single-molecule imaging. J. Am. Chem. Soc. 2003, 125 (5), 1174–1175. 151 Willets, K. A.; Callis, P. R.; Moerner, W. E. Experimental and Theoretical
Investigations of Environmentally Sensitive Single-Molecule Fluorophores. J Phys.
Chem. B 2004, 108 (29), 10465–10473. 152 Willets, K. A.; Twieg, R. J.; Moerner, W. E. Single-Molecule Magic. SPIE’s OE
Magazine of Photonics Technologies and Applications 2004, 4 (6), 13–15. 153 (a) Chem. Pharm. Bull. 1977, 25, 1289. (b) Anal. Biochem. 1977, 79, 83. (c) Chem.
Pharm. Bull. 1977, 25, 1678. 154 J. Hystochem. Cytochem. 1993, 41, 1413. 155 Haugland, R. P. Handbook of Fluorescent Probes and Research Products. 9-th Ed. p
82. http://probes.invitrogen.com/handbook/sections/0201.html 156 Long, J.; Wang, Y.-M.; Matsuura, T.; Meng, J.-B. J. Heterocycl. Chem. 1999, 36 (4),
895–900. (Condensation of 1,3-dihydroxy-2-carbethoxy-naphthalene [BRN 2217406]

370
with N,N-bis-(2-hydroxyethyl)-4-nitrosoaniline · HCl [BRN 3737456] in ethanol to give
6-carbethoxy-9-bis(2-hydroxyethyl)amino-5H-benzo[a]phenoxazin-5-one [BRN
8364710] in 60% yield.) 157 Bris, M.-T. L. J. Heterocycl. Chem. 1989, 26, 429–433. (Preparation of 9-nitroso-
2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-8-ol [BRN 4457827] in 60% yield.) 158 (a) J. Heterocycl. Chem. 1985, 1275. (b) Org, Synth. 1943, Coll. Vol. 2, 223.
http://www.orgsyn.org/orgsyn/prep.asp?rxntypeid=182&prep=CV2P0223 159 (a) J. Org. Chem. 1961, 26, 19. (b) J. Org. Chem. 1972, 37, 392–393. 160 Lauwers Tetrahedron Lett. 1979, 1801–1804. 161 (a) Germann, F. J. Am. Chem. Soc. 1927, 49, 307–312. (from α-P4 and I2) (b)
Tetrahedron Lett. 1979, 1801–1804. (c) Newkome, G. J. Chem. Soc. Chem. Commun.
1975, 885. (from PCl3 and KI in Et2O) 162 J. Med. Chem. 1996, 39, 1700. 163 J. Chem. Res. Synopsis 1998, 272–273. 164 Choi, D. H.; Song, S. Jahng, W. S. Optimal synthetic design of second-order nonlinear
optical material with good temporal stability. Mol. Cryst. Liq. Cryst. Sci. Technol. Sect.
A. 1996, 280, 17. 165 Zentz, F.; Valla, A.; Guilou, R. L.; Labia, R.; Mathot, A.-G.; Sirot, D. Synthesis and
antimicrobial activities of N-substituted imides. Il Farmaco 2002, 57, 421–426. 166 Schwartz, A. L.; Lerner, L.M. J. Org. Chem. 1974, 39, 21–23. 167 (a) Bergson, J. J. Am. Chem. Soc. 1954, 76, 2835–2836. (b) Bergson, J. J. Am. Chem.
Soc. 1954, 76, 4060–4069. (c) Bergson, J.; Swidler, R. J. Am. Chem. Soc. 1953, 75,
1721–1726. 168 Walker, M. A. A High Yielding Synthesis of N-Alkylated Maleimides Using a Novel
Modification of the Mitsunobu Reaction. J. Org. Chem. 1995, 60, 5352–5355 169 Tetrahedron Lett. 1994, 35 (5), 665–668.

371
170 Aponte, M. A.; Butler, G. B. Copolymers Containing Alternating Sequences of
Nucleic Acid-Base Pairs. I. Monomer Synthesis. J. Polym. Sci., Polym. Chem. Ed. 1984,
22, 2841–2858. 171 Narita, M.; Teramoto, T.; Okawara, M. Bull. Chem. Soc. Jpn. 1971, 44, 1084. 172 Rickborn, B. Organic Reactions 1998, 52, 1–393. 173 Prill, E. D. Maleimide and N-substituted derivatives. U.S. Patent 2,524,136, 1950. 174 Clevenger, R. C.; Turnbull, K. D. Synthesis of N-Alkylated Maleimides. Synth.
Commun. 2000, 30 (8), 1379–1388. 175 Kwart, H.; Burchuk, I. J. Am. Chem. Soc. 1952, 74, 3094–3097. 176 Kayser M. Can. J. Chem. 1982, 60, 1199–1206. (NMR and m.p.) 177 Zawadowski, T.; Grabowska, M. Roczniki Chemii 1973, 47, 189–191. 178 Zawadowski, T. Acta Poloniae Pharmaceutica 1995, 52, 125–128. 179 Performed with Dr. Zhikuan Lu. 180 Simon, J.; André, J.-J. Molecular Semiconductors. Springer: Berlin, 1985. 181 Chemical Encyclopedia. Eds. Knuniants, I. L.; Zefirof, N. S. Big Russian
Encyclopedia: Moscow; Vol. 4, 55 182 Kittel, C. Introduction into Solid State Physics 8 Ed. 2005, 208. 183 Fritzsche, H. J. J. Phys. Chem. Solids 1958, 6, 69. 184 Seeger, K. Semiconductor Physics. 9-th Ed. Springer: Berlin, 2004. 185 The term “organic semiconductor” was used for the first time in 1948: (a) Vartanyan,
A. T. Zhur. Phys. Khim. 1948, 22, 769. (b) Eley, D. D. Nature 1948, 162, 819. 186 Heilmeier, G.H.; Warfield, G.; Harrison, S. E. J. Appl. Phys. 1963, 34, 2278. 187 Duke, C.; Schein, L. Phys. Today, 1980, 33, 42. 188 Simon, J.; Bassoul, P. Molecular Semiconductors: Properties and Applications. In:
Design of Molecular Materials, Chapter 6; Wiley and Sons: New York, 2000. 189 Schein L.B., Brown D.W. Mobilities In Organic Molecular Crystals. Mol. Cryst. Liq.
Cryst., 1982, 87, 1–12.

372
190 Karl, N. Defect Control in Semiconductors, Ed. Sumino, K. Elsevier Scientific:
Amsterdam, 1990; p 1725. 191 Warta, W.; Stehle, R.; Karl, N. Appl. Phys. A 1985, 36, 163. 192 Friedel, G. The mesomorphic states of matter. Ann. Phys. 1922, 18, 273–474. 193 Goodby, J. W.; Gray, G. W. Guide to the Nomenclature and Classification of Liquid
Crystals. In: Handbook of Liquid Crystals, Vol. 1, Chapter 2; Eds. Demus, D.; Goodby, J.
W.; Gray, G. W.; Spiess, H.-W.; Vill, V. Wiley-VCH: Weinheim, 1998. 194 Lehmann, O. Liquid Crystals. J. Pysique 1910, 7, 713–35. 195 Friedel, G.; Grandjean, F. The Anistropic Liquids of Lehmann. Bull. Soc. Franc. Min.
1911, 33, 192–239. 196 Collings, P.J.; Hird, M. Introduction to Liquid Crystals. Taylor & Francis: Bristol, PA,
1997. ISBN 0748406433. 197 de Gennes, P. G.; Prost, J. The Physics of Liquid Crystals. Clarendon Press: Oxford,
1993. ISBN: 0198520247. 198 Wegewijs, B. R.; Siebbeles, L. D. A.; Boden, N.; Bushby, R. J.; Movaghar, B.;
Lozman, O. R.; Liu, Q.; Pecchia, A.; Mason, L. A. Charge-carrier mobilities in binary
mixtures of discotic triphenylene derivatives as a function of temperature. Phys. Rev. B
2002, 65 (24), 245112/1–245112/8. 199 Bushby, R. J.; Lozman, O. R. Photoconducting liquid crystals. Curr. Opin. Solid State
Materials Sci. 2003, 6 (6), 569–578. 200 van de Craats, A. M.; Warman, J. M.; Fechtenkötter, A.; Brand, J. D.; Harbison, M.
A.; Müllen, K. Adv. Mater., 1999, 11, 1469. 201 Lever, L.; Kelsall, R.; Bushby, R. A band transport model for highly ordered discotic
mesophases. J. Comput. Electronics 2005, 4 (1/2), 101–104. 202 Lever, L. J.; Kelsall, R. W.; Bushby, R. J. Band transport model for discotic liquid
crystals. Phys. Rev. B 2005, 72 (3), 035130/1–035130/11. 203 Bushby, R. J.; Boden, O. R. Curr. Opin. Colloid Interface Sci. 2002, 7, 343. 204 Meier, H. Organic Semiconductors. Chemie-Verlag: Weinheim, 1974.

373
205 Burland, D. M. Phys. Rev. Lett. 1974, 33, 833. 206 Sakagami, S.; Nakamizo, M. Mesomorphic properties of 2-(4-n-
alkoxybenzylideneamino)anthracenes. Bull. Chem. Soc. Jpn. 1977, 50 (4), 1009–1010. 207 Mery, S.; Haristoy, D.; Nicoud, J.-F.; Guillon, D.; Monobe, H.; Shimizu, Y. Liquid
crystals containing a 2,6-disubstituted anthracene core — mesomorphism, charge
transport and photochemical properties. J. Mater. Chem. 2003, 13 (7), 1622–1630. 208 Gimenez, R.; Pinol, M.; Serrano, J. L. Luminescent Liquid Crystals Derived from
9,10-Bis(Phenylethynyl)anthracene. Chem. Mater. 2004, 16 (7), 1377–1383. 209 Hanna, J.; Kogo, K.; Kafuku, K. Fluorescent liquid crystalline charge transfer
materials. Eur. Pat. Appl. 1999, 67 pp. EP 915144 A1. 210 Substituted anthracenes for liquid crystal, light-emitting or semiconducting materials
and devices. Goulding, Mark John; Thompson, Marcus; Duffy, Warren; Heeney, Martin;
Mcculloch, Iain. (Merck Patent GmbH, Germany). PCT Int. Appl. (2005), 70 pp. WO
2004-EP7089 211 Norvez, S.; Tournilhac, F.-G.; Bassoul, P.; Herson, P. Mesomorphism and Polar
Distortion in 1,4,5,8-Tetrasubstituted Anthraquinones and Anthracenes. Chem. Mater.
2001, 13, 2552–2561. 212 Norvez, S. J. Org. Chem. 1993, 2414–2418. 213 Billard, J.; Dubois, J. C.; Vaucher, C.; Levelut, A. M. Structures of the two
discophases of rufigallol hexa-n-octanoate. Mol. Cryst. Liq. Cryst. 1981, 66 (1-4), 435–
442. 214 Carfagna, C.; Roviello, A.; Sirigu, A. Disk-like mesogens: synthesis and
characterization of a series of rufigallol hexa-n-alkanoates. Mol. Cryst. Liq. Cryst. 1985,
122 (1-4), 151–160. 215 Carfagna, C.; Iannelli, P.; Roviello, A.; Sirigu, A. Discotic mesomorphism of
rufigallol hexa-n-alkoxylates. Liq. Cryst. 1987, 2 (5), 611–616. 216 Billard, J.; Luz, Z.; Poupko, R.; Zimmermann, H. The mesophases of octa-
alkanoyloxy-9,10-anthraquinone. Liq. Cryst. 1994, 16 (2), 333–342.

374
217 Bender, D.; Muellen, K.. Novel alkylanthracenes synthesis, reductive alkylation, and
reductive polymerization. Chem. Ber. 1988, 121 (6), 1187–1197. 218 Pozzo, J.-L.; Clavier, G. M.; Colomes, M.; Bouas-Laurent, H. Different Synthetic
Routes towards Efficient Organogelators: 2,3-Substituted Anthracenes. Tetrahedron
1997, 53 (18), 6377–6390. 219 Haworth, R. D.; Mavin, C. R. The Structure of Diisoeugenol. J. Chem. Soc. 1931,
1363–1366. 220 Robinson, G. M. A Reaction of Homopiperonyl and of Homoveratryl Alcohols. J.
Chem. Soc. 1915, 267–276. 221 (a) Johnson, J. R.; Jobling, W. H.; Bodamer, G. W. J. Am. Chem. Soc. 1941, 63, 131–
135. (b) McDonald. E.; Suksamarn, A.; Wylie, R. D. J. Chem. Soc. Perkin Trans. 1 1979,
1893–1900. 222 (a) Harig, M.; Neumann, B. Eur. J. Org. Chem. 2004, 11, 2381–2397. (b) Leblois, D.;
Danielle, P.; Piessard, S.; Baut, G. L.; Kumar, P.; Brion J.-D. Eur. J. Med. Chem. Chim.
Ther. 1987, 22, 229. (c) Maekawa, N. Bull. Chem. Soc. Jpn. 1959, 32, 1311–1315. (d)
Perkin J. Chem. Soc. 1902, 81, 1032. (e) Perkin, W. H.; Robinson G. M. J. Chem. Soc.
1910, 97, 1139. 223 Hanson, L. Acta Chem. Scand. 1989, 43, 304–306. 224 Mono-adduct of such reaction is known: Anderson, D. R.; Koch, T. H. J. Org. Chem.
1978, 43, 2726–2728. 225 Vanzetti, B. L.; Oliverio, A. Some derivatives of veratrole and of methylvanillin. II.
2,3,6,7-Tetramethoxyanthraquinone. Gazz. Chim. Ital. 1930, 60, 620–632. 226 Shklyaev, Y. V.; Nifontov, Y. V. Three-component synthesis of 3,4-dihydroiso-
quinoline derivatives. Russ. Chem. Bull., Int. Ed. 2002, 51 (5), 844–849. 227 Miao, Q.; Nguyen, T.-Q.; Someya, T.; Blanchet, G. B.; Nuckolls, C.; J. Am. Chem.
Soc. 2003, 125 (34), 10284–10287. 228 (a) Criegee, R. Oxidation with quadrivalent lead salts. Justus Liebigs Ann. Chem.
1930, 481, 263–286. (b) Criegee, R. Chem. Ber. 1931, 64B, 260–266.

375
229 Kitamura, M.; Shen, B.; Liu, Y.; Zheng, H.; Takahashi, T. Aromatization of Highly
Alkyl-substituted Dihydroanthracenes Using n-BuLi/TMEDA/MeI. Chem. Lett. 2001,
646–647. 230 (a) Arcoleo, A.; Paternostro, M. P. Condensation of phenyl ethers with aliphatic
aldehydes. XXI. Reaction of veratrole with monobromo- and tribromoacetaldehydes.
Ann. Chim. (Rome) 1968, 58 (3), 290-7. (b) Arcoleo, A.; Natoli, M. C. Condensation of
phenolic ethers with aliphatic aldehydes. XIX. Structure of a compound derived from the
condensation of veratrole with acetaldehyde. Ann. Chim. (Rome) 1967, 57 (6), 716–722. 231 Chung, Y. J. Org. Chem. 1989, 54 (5), 1018–1032. 232 Boldt, P. Chem. Ber. 1967, 100, 1270–1280. 233 Arcoleo, A. Ann. Chim. (Rome) 1967, 57, 716–720. 234 Huang, W.; Zhou, X.; Kanno, K.-I.; Takahashi, T. Pd-Catalyzed Reactions of o-
Diiodoarenes with Alkynes for Aromatic Ring Extension. Org. Lett. 2004, 6 (14), 2429–
2431. 235 Marks, V.; Gottlieb, H. E.; Melman, A.; Byk, G.; Cohen, S.; Biali, S. E. Polyethylated
Aromatic Rings: Conformation and Rotational Barriers of 1,2,3,4,5,6,7,8-
Octaethylanthracene, 1,2,3,4,6,7,8-Heptaethylfluorene, and 1,2,3,4,5,6,7,8-
Octaethylfluorene. J. Org. Chem. 2001, 66 (20), 6711–6718. 236 Zeis, R.; Celine, C.; Siegrist, T.; Schlockermann, C.; Chi, X.; Kloc, C. Field Effect
Studies on Rubrene and Impurities of Rubrene. Chem. Mater. 2006, 18, 244–248. 237 Podzorov, V.; Menard, E.; Borissov, A.; Kiryukhin, V.; Rogers, J. A.; Gershenson, M.
E. Phys. Rev. Lett. 2004, 93, 086602. 238 (a) Laatsch, H. Liebigs Ann. Chem. 1980, 140. (b) Fieser, L. F.; Tishler, M.; Wendler,
N. L. Extensions of the Vitamin K1 Synthesis. J. Am. Chem. Soc. 1940, 62, 2861–2866./ 239 Wilds, A. L. Reduction with Aluminum Alkoxides. Organic Reactions 1944, 178–
223. 240 Hinshaw, J. C. Convenient synthesis of 2,3,6,7-tetramethylanthracene. Org. Prep.
Proc. Int. 1972, 4 (5), 211–213.

376
241 Coffey, S.; Boyd, V. The reduction of anthraquinone and other polycyclic quinones
with aluminum alkoxides (Meerwein-Pondorff reagent). J. Chem. Soc. 1954, 2468–2470. 242 Gaylord, N. G.; Stepan, V. Reduction of methylated and anthraquinones to methylated
anthracenes with aluminum tris(cyclohexyl oxide). Coll. Czech. Chem. Commun. 1974,
39 (7), 1700–1710. 243 Afzali, A.; Dimitrakopoulos, C. D.; Breen, T.L. High-Performance, Solution-
Processed Organic Thin Film Transistors from a Novel Pentacene Precursor. J. Am.
Chem. Soc. 2002, 124, 8812–8813. 244 Nelson, S. F.; Lin, Y. Y.; Gundlach, D. J.; Jackson, T. N. Appl. Phys. Lett. 1998, 72,
1854. 245 Yamabe, T,; Tonala, K.; Oheki, K. Solid State Commun. 1982, 44, 823. 246 Kivelson, S.; Chapman, O. L. Phys. Rev. B. 1983, 28, 7236. 247 Aihara, J.-I. Reduced HOMO-LUMO Gap as an Index of Kinetic Stability for
Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. A 1999, 103, 7487–7495. 248 Clar, E. Polycyclic Hydrocarbons Academic Press: London, 1964; Vols. 1, 2. 249 Harvey, R. G. Polycyclic Aromatic Hydrocarbons. Wiley-VCH: New York, 1997. 250 Aihara, J.-I. Why are some polycyclic aromatic hydrocarbons extremely reactive?
Phys. Chem. Chem. Phys. 1999, 1, 3193–3197. 251 Marschalk, C. Researches in the acene series (anthracene and its linear benzologs).
Bull. Soc. Chim. Fr. 1954, 877–892. 252 Schleyer, P. von R.; Manoharan, M.; Jiao, H.; Stahl, F. The Acenes: Is There a
Relationship between Aromatic Stabilization and Reactivity? Org. Lett. 2001, 3 (23),
3643–3646. 253 Berg, O.; Chronister, E. L.; Yamashita, T.; Scott, G. W.; Sweet, R. M.; Calabrese, J. s-
Dipentacene: Structure, Spectroscopy, and Temperature- and Pressure-Dependent
Photochemistry. J. Phys. Chem. A 1999, 103, 2451–2459. 254 Clar, E. The Aromatic Sextet. Wiley: London, 1972. 255 Herwig, P. T.; Müllen, K. Adv. Mater. 1999, 11 (6), 480–483.

377
256 Ried, W.; Anthöfer, F. Einfache Synthese für Pentacen-6,13-chinon. Angew. Chem.
1953, 601. 257 Mills, W. H.; Mills, M. J. Chem. Soc.1912, 2194. 258 (a) Brukner, V.; Karczag (Wilhelms), A.; Körmendy, K.; Mészáros, M.; Tomasz, J.
Einfache und Ausgiebige Synthese des Pentacens. Acta Chim. Acad. Sci. Hung. 1960,
443–448. (b) Brukner, V.; Karczag (Wilhelms), A.; Körmendy, K.; Mészáros, M.;
Tomasz, J. Einfache Synthese des Pentacens. Tetrahedron Lett. 1960, (1), 5–6. (c)
Brukner, V.; Tomasz, J. Weitere Vereinfachung der Pentacensynthese. Acta Chim. Acad.
Sci. Hung. 1961, 405–408. 259 Bigelow, L. A.; Reynolds, H. H. Quinizarin. Org. Syn. Coll. Vol. 1, 476. 260 Marchand, A. P.; Reddy, G. M. Mild and Highly Selective Ultrasound-Promoted
Zinc/Acetic Acid Reduction of C=C Bonds in α,β-Unsaturated γ-Dicarbonyl
Compounds. Synthesis 1991, 198–200. 261 Blaszczak, L. C.; McMurry, J. E. Reduction of Enedicarbonyl Compounds with
Titanous Ion. J. Org. Chem. 1974, 39 (2), 258. 262 (a) Serpaud, B.; Lepage, Y. Condensation of o-dialdehydes with 1,4-diphenols and γ-
diketones. Bull. Soc. Chim. Fr. 1977, (5-6, Pt. 2), 539–542. (b) Lepage, L.; Lepage, Y.
Synthesis of o-dibenzoyl compounds by unstable diene trapping. C. R. Seances Acad. Sci.
C (Paris) 1975, 280 (12), 847–849. (c) Verine, A.; Lepage, Y. Synthesis of polycyclic
quinones by carbanions. Bull. Soc. Chim. Fr. 1973, (3, Pt. 2), 1154–1159. (d) Lepage, Y.;
Verine, A. Synthesis of polycyclic quinones through carbanions. C. R. Seances Acad. Sci.
C (Paris) 1972, 274 (17), 1534–1536. 263 Roberson, L.B.; Kowalik, J.; Tolbert, L.M.; Kloc, C.; Zeis, R.; Chi, X.; Fleming, R.;
Wilkins, C. Pentacene Disproportionation during Sublimation for Field-Effect
Transistors. J. Am. Chem. Soc. 2005, 127, 3069–3075. 264 Campbell, R. B.; Robertson, J. Monteath; Trotter, J. The crystal structure of hexacene,
and a revision of the crystallographic data for tetracene and pentacene. Acta Cryst. 1962,
15, 289–290.

378
265 Holmes, D.; Kumaraswamy, S.; Matzger, A. J.; Vollhardt, K. P. C. On the nature of
nonplanarity in the [N]phenylenes. Chem. Eur. J. 1999, 11, 3399–3412. 266 Swartz, C. R.; Parkin, S. R.; Bullock, J. E.; Anthony, J. E.; Mayer, A. C.; Malliaras, G.
G. Synthesis and Characterization of Electron-Deficient Pentacenes. Org. Lett. 2005, 7
(15), 3163–3166. 267 (a) Cava, M. P. J. Org. Chem. 1980, 516. (b) Cava, M. P.; Deana, A. A.; Muth, K.
Condensed Cyclobutane Aromatic Compounds. VIII. The Mechanism of Formation of
1,2-Dibromobenzocyclobutene; A New Diels-Alder Synthesis. J. Am. Chem. Soc. 1959,
81, 6458–6460. (c) Cava, M. P. J. Am. Chem. Soc. 1958, 6149. (d) Cava, M. P. J. Am.
Chem. Soc. 1957, 1701. 268 (a) Martín, N.; Seoane, C.; Hanack, M. Org. Prep. Proced. Int. 1991, 23, 237. 269 Segura, J. L.; Martín, N. o-Quinodimethanes: Efficient Intermediates in Organic
Synthesis. Chem. Rev. 1999, 99, 3199–3246. 270 Cambridge Crystallographic Data Centre Publication CCDC-114447, ccdc.cam.ac.uk. 271 Rice, J. E. J. Org. Chem. 1988, 1775–1779. 272 Flood, D. T. Fluorobenzene. Org. Syn. Coll. Vol. 2, 295. 273 Stephenson, E. F. M. o-Xylylene Dibromide. Org. Syn. Coll. Vol. 4, 984. 274 Smith, M. B.; March J. March’s Advanced Organic Chemistry. Fifth Ed. Wiley and
Sons: New York, 2001. pp 911–914. 275 Wenner, W. Bis(bromomethyl) compounds. J. Org. Chem. 1952, 523–528. 276 Smith, M. B. Organic Synthesis. 2-nd Ed. McGraw Hill: New York, 2002. pp 204–
206. 277 Farooq, O. Oxidation of aromatic 1,2-dimethanols by activated dimethyl sulfoxide.
Synthesis 1994, (10), 1035–1036. 278 Jarvis, W. F.; Dittmer, D.C. J. Org. Chem. 1988, 53, 5750–5756. 279 Hoey, M. D.; Dittmer, D.C. J. Org. Chem. 1991, 56, 1947–1948.

379
280 Performed in collaboration with Dr. Kihong Park, Electronic Materials and Devices
Research Center, KIST, P.O. Box 131, Cheongryang, Seoul 130-650, Korea. +82-2-958-
5292. 281 Smith, J. G.; Dibble, P. W.; Sandborn, R. E.; J. Org. Chem. 1986, 51 (20), 3762–3768. 282 (a) Reetz, M. T.; Toellner, K.; Tetrahedron Lett. 1995, 36 (52), 9461–9464. (b)
Mikami, K.; Ohmura, H.; Org. Lett. 2002, 4 (20), 3355–3358. 283 LiqCryst Database, ver.4.3. 284 Allen, C. F. H.; Bell, A. Action of Grignard Reagents on Certain Pentacenequinones,
6,13-Diphenylpentacene. J. Am. Chem. Soc. 1942, 64, 1253–1260. 285 Weygand, F.; Kinkel, K. G.; Tietjen, D. The preparation of aromatic o-dialdehydes
from o-dialcohols via the cyclic selenous acid esters. Chem. Ber. 1950, 83 (4), 394–399. 286 Bhattacharjee, D.; Popp, F. D. The oxidation of a series of phthalyl alcohols. J.
Heterocycl. Chem. 1980, 17 (2), 315–320. 287 Meziane, M. A. A.; Royer, S.; Bazureau, J. P. A practical one-pot synthesis of ethyl
isoquinoline-3-carboxylate by domino reactions: a potential entry to constrained
nonproteogenic amino acid derivatives. Tetrahedron Lett. 2001, 42 (6), 1017–1020. 288 Harig, M.; Neumann, B.; Stammler, H.-G.; Kuck, D. Eur. J. Org. Chem. 2004, 11,
2381–2397. 289 Meziane, M. A. A. A.; Bazureau, J. P. Molecules 2002, 7 (2), 252–263. 290 Sato,T. J. Chem. Soc. Perkin Trans. 1 1973; 891–895. 291 Abou-Teim, O.; Jansen, R. B.; McOmie, J. F. W.; Perry, D. H. J. Chem. Soc. Perkin
Trans. 1 1980, 1841–1846. 292 Snieckus, V. directed Ortho Metallation. Tertiary Amide and O-Carbamate Directors
in Synthetic Strategies for Polysubstituted Armatics. Chem. Rev. 1990, 90 (6), 879–933. 293 Narasimhan, N. S.; Mali, R. S. Heteroatom Directed Aromatic Lithiation Reactions for
the Synthesis of Condensed Heterocyclic Compounds. In: Topics in Curr. Chem.
Springer-Verlag: Berlin, 1987, 138, 63–147.

380
294 Narasimhan, N. S.; Mali, R. S. Synthesis of Heterocyclic Compounds Involving
Aromatic Lithiation Reactions in the Key Step. Synthesis 1983, 957–986. 295 Meier, H.; Fetten, M. A new synthetic route to tribenzo[a,e,i][12]annulenes.
Tetrahedron Lett. 2000, 41, 1535–1538. 296 Gschwend, H. W.; Rodriguez, H. R. Heteroatom-facilitated lithiations. Organic
Reactions 1979, 26, 1–360. 297 Winkle, M. R.; Ronald, R. C. Regioselective metalation reactions of some substituted
(methoxymethoxy)arenes. J. Org. Chem. 1982, 47 (11), 2101–2108. 298 Ziegler, F. E.; Fowler, K. W. Substitution reactions of specifically ortho-metalated
piperonal cyclohexylimine. J. Org. Chem. 1976, 41 (9), 1564–1566. 299 Comins, D. L.; Brown, J. D. Ortho metalation directed by a-amino alkoxides. J. Org.
Chem. 1984, 49 (6), 1078–1083. 300 Napolitano, E.; Giannone, E.; Fiaschi, R.; Marsili, A. Influence of alkoxyalkyl
substituents in the regioselective lithiation of the benzene ring. J. Org. Chem. 1983, 48
(21), 3653–3657. 301 Plaumann, H. P.; Keay, B. A.; Rodrigo, R. The regiospecific lithiation of aromatic
acetals. Tetrahedron Lett. 1979, 51, 4921–4924. 302 Wakefield, B. J. The Chemistry of Organolithium Compounds. Pergamon: New York,
1974; 335 pp. 303 De Costa, B. R.; Radesca, L.; Di Paolo, L.; Bowen, W. D. Synthesis, characterization,
and biological evaluation of a novel class of N-(arylethyl)-N-alkyl-2-(1-
pyrrolidinyl)ethylamines: structural requirements and binding affinity at the s receptor. J.
Med. Chem. 1992, 35 (1), 38–47. 304 (a) Fornasier, R.; Scrimin, P.; Tecilla, P.; Tonellato, U. Bolaform and classical
cationic metallomicelles as catalysts of the cleavage of p-nitrophenyl picolinate. J. Am.
Chem. Soc. 1989, 111 (1), 224–229. (b) Hall, L. A. R.; Stephens, V. C.; Burckhalter, J. H.
2-Dimethylaminoethyl chloride hydrochloride. Org Syn. 1951, 31, 37–39 (Coll. Vol. 4,
333).

381
305 Einhorn, J.; Luche, J. L. Ultrasound in organic synthesis. 10. Selective ortho-lithiation
of the Bouveault reaction intermediate. Tetrahedron Lett. 1986, 27 (16), 1793–1796. 306 Posner, G. H.; Canella, K. A. Phenoxide-directed ortho lithiation. J. Am. Chem. Soc.
1985, 107 (8), 2571–2573. 307 Lieberman, S. V. Connor, R. p-Nitrobenzaldehyde. Org. Syn. 1943, Coll. Vol. 2,
p.441. 308 Bauer, V. J.; Duffy, B. J.; Hoffman, D.; Klioze, S. S.; Kosley, R. W., Jr.; McFadden,
A. R.; Martin, L. L.; Ong, H. H.; Geyer, H. M., III. Synthesis of spiro[isobenzofuran-
1(3H),4'-piperidines] as potential central nervous system agents. J. Med. Chem. 1976, 19
(11), 1315–1324. 309 Hartford, W. H.; Darrin, M. The chemistry of chromyl compounds. Chem. Rev. 1958,
2, 1–61. 310 Ataei, S. M. A convenient method for one step oxidative decarboxylation by KMnO4
in non aqueous media. J. Chem. Res. Synopses 2000, (3), 148–149. 311 by Dr. Lionel Sanguinet. 312 Wood, J. H.; Perry, M. A.; Tung, C. C. Bis-chloromethylation of aromatic
compounds. J. Am. Chem. Soc. 1950, 72, 2989–2991. 313 Lee, W. Y.; Park, C. H.; Kim, E. H. Orthocyclophanes. 4. Functionalization of
p1n]Orthocyclophanes on the Aromatic Rings. J. Org. Chem. 1994, 59, 4495–4500. 314 Martin, N.; Behnisch, R.; Hanack, M. Syntheses and electrochemical properties of
tetracyano-p-quinodimethane derivatives containing fused aromatic rings. J. Org. Chem.
1989, 54 (11), 2563–2568. 315 Payne, M. M.; Delcamp, J. H.; Parkin, S. R.; Anthony, J. E. Robust, Soluble
Pentacene Ethers. Org. Lett. 2004, 6 (10), 1609–1612. 316 Clar, E.; John, Fr. Polynuclear aromatic hydrocarbons and their derivatives. VII. A
new class of deeply colored radical hydrocarbons and the supposed pentacene of E.
Philippi. Chem. Ber. 1930, 63B, 2967–2977.

382
317 Sparfel, D.; Gobert, F.; Rigaudy, J. Thermal transformations of meso-acenic
photooxides. VI. Pentacenic photooxides. Tetrahedron 1980, 36 (15), 2225–2235. 318 (a) Uno, H.; Yamashita, Y.; Kikuchi, M.; Watanabe, H.; Yamada, H.; Okujima, T.;
Ogawa, T.; Ono, N. Photo precursor for pentacene. Tetrahedron Lett. 2005, 46 (12),
1981–1983. (b) Yamada, H.; Yamashita, Y.; Kikuchi, M.; Watanabe, H.; Okujima, T.;
Uno, H.; Ogawa, T.; Ohara, K.; Ono, N. Photochemical synthesis of pentacene and its
derivatives. Chem. Eur. J. 2005, 11 (21), 6212–6220. 319 Afzali, A.; Dimitrakopoulos, C. D.; Breen, T. L. High-Performance, Solution-
Processed Organic Thin Film Transistors from a Novel Pentacene Precursor. J. Am.
Chem. Soc. 2002, 124 (30), 8812–8813. 320 Afzali, A.; Kagan, C. R.; Traub, G. P. N-sulfinylcarbamate-pentacene adduct: A novel
pentacene precursor soluble in alcohols. Synthetic Metals 2005, 155 (3), 490–494. 321 Mott, N. F.; Davis, E. A. Electronic Processes in Non-Crystalline Materials, 2nd Ed.,
Clarendon Press: Oxford, 1979. 322 Holstein, T. Studies of Polaron Motion. Annals of Physics 2000, 281 (1/2), 725–773. 323 D. Emin; A.M. Kriman Transient small-polaron hopping motion. Phys. Rev. B, 1986,
34, 7278. 324 Anthony, J. E.; Eaton, D. L.; Parkin, S. R. A Road Map to Stable, Soluble, Easily
Crystallizable Pentacene Derivatives. Org. Lett. 2002, 4 (1), 15–18. 325 Emsley, J. The Elements. 2nd Ed. Clarendon Press: Oxford, 1991. 326 Periodic Table v. 2.5. http://nautilus.fis.uc.pt/st2.5/scenes-e/elem/e05392.html 327 Schwartz, L.; Melvin; I.; Henry G.; Hornig, J. F.; Photoconductivity in p-
diiodobenzene. Mol. Cryst. 1967, 2 (4), 379–384. 328 Ellman, B. Ab-Initio Study of the Electronic Structure of the Crystalline High-
Mobility Organic Semiconductor 1,4-Diiodobenzene. Submitted for publication. 329 Ellman, B.; Nene, S.; Semyonov, A. N.; Twieg, R. J.. High Mobility, Low Dispersion
Hole Transport in 1,4-Diiodobenzene. Adv. Mater. 2006, 18, accepted for publication.

383
330 Suzuki, H.; Kondo, A.; Ogawa, T. Preparation of Aromatic Iodides from Bromides via
the Reverse Halogen Exchange. Chem. Lett. 1985, 411–412. 331 Suzuki, H.; Kondo, A.; Inouye, M.; Ogawa, T. An alternative synthetic method for
polycyclic aromatic iodides. Synthesis 1986, 121–122. 332 Vonk, W. F. M.; Louw, R. Vapor phase chemistry of arenes. V. Vapor phase
iodination of benzene derivatives. Formation of aryl iodides from bromides based on
halogen exchange with iodobenzene. Rec. Trav. Chim. Pays-Bas 1977, 96 (2), 59–60. 333 Abraham, M. H.; Grellier, P. L.; Priscilla L. Heterolytic cleavage of main group
metal-carbon bonds. In: The Chemistry of Metal–Carbon Bond, Eds. Hartley, F. R.; Patai,
S. 5 vols. Wiley: New York, 1984–1990; Vol. 2, 1985, p 72. 334 Taylor, E. C.; Kienzle, F.; McKillop, A. 2-Iodo-p-xylene. Org. Syn. 1976, 55, 70–73;
Coll. Vol. 6, 709. 335 Takagi, K.; Hayama, N.; Inokawa, S. The in Situ-generated Nickel(0)-catalyzed
Reaction of Aryl Halides with Potassium Iodide and Zinc Powder. Bull. Chem. Soc. Jpn.
1980, 53 (12), 3691–3695. 336 Gan, Z.; Roy, R. Sialoside clusters as potential ligands for siglecs (sialoadhesins).
Can. J. Chem. 2002, 80 (8), 908–916. (1,3,5-Triiodobenzene from 1,3,5-tribromobenzene
via KI/Ni/DMF halogen exchange reaction.) 337 Larock, R. C. Organomercury Compounds in Organic Synthesis. Springer: New York,
1985. 338 Merkushev, E. B. Advances in the synthesis of iodoaromatic compounds. Synthesis
1988, 12, 923–937. 339 Chaikovski, V. K.; Jharlova, T. S.; Filimonov, V.D.; Saryucheva, T.A. Superactive
iodination reagent on a base of iodine chloride and silver sulfate. Synthesis 1999, 748. 340 (a) Miller, L. L.; Kujawa, E. P.; Campbell, C. B. Iodination with Electrolytically
Generated Iodine(I). J. Am. Chem. Soc. 1970, 92 (9), 2821–2825. (b) Miller, L. L.;
Watkins, B. F. Scope and Mechanism of Aromatic Iodination with Electrochemically
Generated Iodine(I). J. Am. Chem. Soc. 1976, 98 (6), 1515–1519.

384
341 Lines, R.; Parker, V. D. Electrophilic Aromatic Substitution by Positive Iodine
Species. Iodination of Deactivated Aromatic Compounds. Acta Chem. Scand. B 1980, 34
(1), 47–51. 342 Slezak, F. B.; Bluestone, H.; Magee, T. A.; Wotiz, J. H. Preparation of substituted
glycolurils and their N-chlorinated derivatives. J. Org. Chem. 1962, 27, 2181–2183. 343 Nematollahi, J.; Ketcham, R. Imidazoimidazoles. I. Reaction of ureas with glyoxal.
Tetrahydroimidazo[4,5-d]imidazole-2,5-diones. J. Org. Chem. 1963, 28 (9), 2378–2380. 344 Slezak, F. B.; Hirsch, A.; Rosen, I. Halogenation of glycoluril and diureidopentane. J.
Org. Chem. 1960, 25, 660–661. 345 Yagovkin, A. Yu.; Bakibaev, A. A.; Bystritskii, E. I. Successful synthesis of 2,4,6,8-
tetraiodo-2,4,6,8-tetraazabicyclo[3.3.0]octane-3,7-dione. Khimiya Geterotsiklicheskikh
Soedinenii 1995, (12), 1695–1696. 346 Chaikovski, V. K.; Filimonov, V. D.; Yagovkin, A. Yu.; Ogorodnikov, V. D. 2,4,6,8-
Tetraiodo-2,4,6,8-tetraazabicyclo[3.3.0]octane-3,7-dione as a mild and convenient
reagent for iodination of aromatic compounds. Russ. Chem. Bull. 2001, 50 (12), 2411–
2415. 347 Chaikovski, V. K.; Filimonov, V. D.; Yagovkin, A. Y.; Kharlova, T. S. 2,4,6,8-
Tetraiodoglycoluril in sulfuric acid as a new powerful reagent for iodination of
deactivated arenes. Tetrahedron Lett. 2000, 41 (47), 9101–9104. 348 Kajigaeshi, S.; Kakinami, T.; Yamasaki, H.; Fujisaki, S.; Kondo, M.; Okamoto, T.
Iodination of Phenols by Use of Benzyltrimethylammonium Dichloroiodate (1–). Chem.
Lett. 1987, 2109–2112. 349 Barluenga, J.; Campos, P. J.; Gonzalez, J. M.; Asensio, G. A simple and general route
to aryl iodides from arenes. J. Chem. Soc. Perkin Trans. 1 1984, (11), 2623–2624. 350 Barluenga, J.; Rodriguez, M. A.; Campos, P. J. Electrophilic additions of positive
iodine to alkynes through an iodonium mechanism. J. Org. Chem. 1990, 55 (10), 3104–
3106.

385
351 Barluenga, J.; Gonzalez, J. M.; Garcia-Martin, M. A.; Campos, P. J.; Asensio, G.
Acid-mediated reaction of bis(pyridine)iodonium(I) tetrafluoroborate with aromatic
compounds. A selective and general iodination method. J. Org. Chem. 1993, 58 (8),
2058–2060. 352 Merkushev, E. B.; Sedov, A. M.; Simakhina, N. D. Simple method for the synthesis of
iodomethylbenzenes. Zhurnal Organicheskoi Khimii 1978, 14 (5), 1115–1116. 353 Britton, D.; Gleason, W. B. Diiododurene: four centrosymmetric molecules in general
positions. Acta Cryst. C 2003, 59 (8), o439–o442. 354 Suzuki, H.; Nakamura, K.; Goto, R. The direct iodination of polyalkylbenzenes
bearing bulky groups. Bull. Chem. Soc. Jpn. 1966, 39 (1), 128–131. 355 Deacon, G. B.; Farquharson, G. J. Permercurated arenes. III. Syntheses of
periodoarenes and perchloroarenes by iodo- and chloro-demercuration of some
permercurated arenes. Aust. J. Chem. 1977, 30 (8), 1701–1713. 356 Bugueno-Hoffmann, R. Spectrochim. Acta A 1992, 48 (4), 509–517. 357 Suzuki, H; Goto, R. A convenient synthesis of certain aromatic polyiodo compounds.
Bull. Chem. Soc. Jpn. 1963, 36, 389–391. 358 Mattern, D. L. Direct Aromatic Periodination. J. Org. Chem. 1984, 49, 3051–3053. 359 Barluenga, J.; Gonzalez, J. M.; Garcia-Martin, M. A.; Campos, P. J. Polyiodination on
benzene at room temperature: a regioselective synthesis of derivatives. Tetrahedron Lett.
1993, 34 (24), 3893–3896. 360 (a) Khotsyanova, T. L.; Smirnova, V. I. Crystal and molecular structures of
hexaiodobenzene. Kristallografiya 1968, 13 (5), 787–790. (b) Steer, R J.; Watkins, S. F.;
Woodward, P. Crystal and molecular structure of hexaiodobenzene. J. Chem. Soc. C
1970, (2), 403–408. 361 Nakayama, A.; Fujihisa, H.; Takemura, K.; Aoki, K.; Carlon, R. P. Structural study on
pressure-induced metallization of C6I6. Synth. Metals 2001, 120 (1-3), 767–768.

386
362 Iwasaki, E.; Shimizu, K.; Amaya, K.; Nakayama, A.; Aoki, K.; Carlon, R. P.
Metallization and superconductivity in hexaiodobenzene under high pressure. Synth.
Metals 2001, 120 (1-3), 1003-1004. 363 Nakayama, A.; Fujihisa, H.; Aoki, K. Crystal and molecular structures of C6I6 under
high pressure. Koatsuryoku no Kagaku to Gijutsu 2000, 10 (3), 214–220. 364 Tateyama, Y.; Ohno, T. First-principles study on molecular dissociation under
metallization pressure in aromatic monomolecular crystals with iodine atoms. J. Phys.
Condensed Matter 2002, 14 (44), 10429–10432. 365 (a) Satyamurthy, N.; Barrio, J. R. Cation exchange resin (hydrogen form) assisted
decomposition of 1-aryl-3,3-dialkyltriazenes. A mild and efficient method for the
synthesis of aryl iodides. J. Org. Chem. 1983, 48 (23), 4394. (b) Barrio, J. R.;
Satyamurthy, N.; Ku, H.; Phelps, M. E. The acid decomposition of 1-aryl-3,3-
dialkyltriazenes. Mechanistic changes as a function of aromatic substitution, nucleophile
strength, and solvent. J. Chem. Soc. Chem. Commun. 1983, (8), 443–444. (c) Foster, N.
I.; Heindel, N. D.; Burns, H. D.; Muhr, W. Aryl iodides from anilines via triazene
intermediates. Synthesis 1980, (7), 572–573. 366 Wheeler, H. L.; Liddle, L. M. Researches on Halogen Amino Acids. Iodine
Derivatives of o-Toluidine. 3-Iodoaminobenzoic Acids. American Chemical Journal
1910, 42, 498–505. 367 Gore, R.; Suzuki, H. Jacobsen reaction of polyhalobenzenes and monoaikylpoly-
halobenzenes. Nippon Kagaku Zasshi 1963, 84 (2), 167–173. 368 Liu, Y. X.; Knobler, C. B.; Trueblood, K. N.; Helgeson, R. 3,4,5-Triiodotoluene,
C7H5I3. Acta Cryst. C 1985, 41 (6), 959–961. 369 Friedman, L.; Logullo, F. M. Synthesis of o-dihalobenzenes from benzenediazonium-
2-carboxylate. Angew. Chem. 1965, 5 (77), 217–218. 370 Janczewski, M.; Prajer, L. Synthesis and properties of naphthalenesulfonic acids.
Replacement of the sulfonic acid group with halogens. Roczniki Chemii 1954, 28, 681–
682.

387
371 Pan, Y.; Peng, Z. A convinient synthetic approach to 1,5-diiodonaphthalene
derivatives. Tetrahedron Lett. 2000, 41, 4537–4540. 372 Cakmak, O.; Demitras, I.; Balaydin, H. T. Selective bromination of 1-
bromonaphthalene: efficient synthesis of bromonaphthalene derivatives. Tetrahedron
2002, 58, 5603–5609. 373 Kajanus, J.; van Berlekom, S. B.; Albinsson, B.; Mårtensson, J. Synthesis of
Bis(phenylethynyl)arylene-Linked Diporphyrins Designed for Studies of Intramolecular
Energy Transfer. Synthesis 1999, (7), 1155–1162. 374 Novak, I.; Jiang, H.; Kovac, B. Intramolecular Interactions in Diiodonaphthalenes. J.
Phys. Chem. A 2003, 107 (4), 480–484. 375 Hellberg, J.; Allared, F.; Pelcman, M. Facile synthesis of 2,3-diiodonaphthalene and
2-bromo-3-iodonaphthalene. Synth. Commun. 2003, 33 (15), 2751–2756. 376 Seeboth, H. Bucherer reaction and the preparative use of its intermediate products.
Angew. Chem., Int. Ed. Eng. 1967, 6 (4), 307–317. 377 Boden, N.; Bushby, R. J.; Clements, J. Mechanism of quasi-one-dimensional
electronic conductivity in discotic liquid crystals. J. Chem. Phys. 1993, 98 (7), 5920–
5931. 378 (a) Adam, D.; Haarer, D.; Closs, F.; Frey, T.; Funhoff, D.; Siemensmeyer, K.;
Schuhmacher, P.; Ringsdorf, H. Discotic liquid crystals — A new class of fast
photoconductors. Ber. Bunsen-Ges. 1993, 97 (10), 1366–1370. (b) Adam, D.; Closs, F.;
Frey, T.; Funhoff, D.; Haarer, D.; Ringsdorf, H.; Schuhmacher, P.; Siemensmeyer, K.
Transient photoconductivity in a discotic liquid crystal. Phys. Rev. Lett. 1993, 70 (4),
457–460. 379 Marquardt, F. H. 2,3,6,7,10,11-Hexamethoxytriphenylene. J. Chem. Soc. 1965, 1517–
1518. 380 Matheson, I. M.; Musgrave, O. C.; Webster, C. J. Oxidation of veratrole by quinones.
Chem. Commun. 1965, (13), 278–279.

388
381 Boden, N.; Borner, R. C.; Bushby, R. J.; Cammidge, A. N.; Jesudason, M. V. The
synthesis of triphenylene-based discotic mesogens: new and improved routes. Liq. Cryst.
1993, 15 (6), 851–858. 382 Cooke, G.; Sage, V.; Richomme, T. Synthesis of Hexa-alkyloxytriphenylenes Using
FeCl3 Supported on Alumina. Synth. Commun. 1999, 29 (10), 1767–1771. 383 (a) Kumar, S.; Manickam, M. Oxidative trimerization of o-dialkoxybenzenes to
hexaalkoxytriphenylenes: molibdenum (V) chloride as novel reagent. Chem. Commun.
1997, 1615–1616. (b) Waldvogel, S. R. The Reaction Pattern of the MoCl5-Mediated
Oxidative Aryl-aryl Coupling. Synlett 2002, (4), 622–624. 384 Kumar, S.; Varshney, S. K. Vanadium oxychloride, a novel reagent for the oxidative
trimerization of o-dialkoxybenzenes to hexaalkoxytriphenylenes. Liq. Cryst. 1999, 26
(12), 1841–1843. 385 Kumar, S.; Varshney, S. K. Synthesis of Triphenylene and Dibenzopyrene
Derivatives: Vanadium Oxychloride a Novel Reagent. Synthesis 2001, (2), 305–311. 386 Chapuzet, J.-M.; Simonet-Gueguen, N. A versatile anodic source of triphenylenes
possessing at least one ionophoric site. Tetrahedron Lett. 1991, 32 (50), 7405–7408. 387 Chapuzet, J.-M.; Simonet, J. The anodic trimerization of aromatic orthodiethers: new
development. Tetrahedron 1991, 47 (4-5), 791–798. 388 Dietrich, M.; Heinze, J. On the determination of redox potentials of highly reactive
aromatic mono- and multications. J. Am. Chem. Soc. 1990, 112 (13), 5142–5145. 389 Bechgaard, K.; Parker, V. D. Mono-, di-, and trications of hexamethoxytriphenylene.
Novel anodic trimerization. J. Am. Chem. Soc. 1972, 94 (13), 4749–4750. 390 Ronlan, A.; Aalstad, B.; Parker, V. D. Unsymmetrical anodic coupling of veratrole
with various anisole derivatives. Products and mechanisms. Acta Chem. Scand. B 1982,
36 (5), 317–325. 391 Le Berre, V.; Angely, L.; Simonet-Gueguen, N.; Simonet, J. Anodic trimerization. A
facile one-step synthesis of tris(15-crown-5)triphenylene. J. Chem. Soc. Chem. Commun.
1987, (13), 984–986.

389
392 Private communication from Dr. Owen Roger Lozman <[email protected]>,
currently at “Avecia Inkjet Limited”, UK. 393 Bushby, R. J.; Boden, N.; Kilner, C. A.; Lozman, O. R.; Lu, Z.; Liu, Q.; Thornton-
Pett, M. A. Helical geometry and liquid crystalline properties of 2,3,6,7,10,11-
hexaalkoxy-1-nitrotriphenylenes. J. Mater. Chem. 2003, 13 (3), 470–474. 394 Kumar, S.; Manickam, M.; Varshney, S. K.; Rao, D. S. S.; Prasad, S. K. Novel
heptasubstituted discotic liquid crystals. J. Mater. Chem. 2000, 10, 2483–2489. 395 Demas, J. N.; Crosby, G. A. The Measurement of Photoluminescence Quantum
Yields. A Review. J. Phys. Chem. 1971, 75 (8), 991–1024. 396 Parker, C. A.; Rees, W. T. Correction of fluorescence spectra and measurement of
fluorescence quantum efficiency. Analyst (London) 1960, 85, 587–600. 397 Optical Radiation Measurements. Volume 3. Measurement of Photoluminescence. Ed.
Mielentz, K. D. 398 Standards in Fluorescence Spectroscopy. Ed. Miller, J. N. QD459.S72x 399 Eaton, D. F. Reference Materials for Fluorescent Measurement. Pure and Appl. Chem.
1988, 60 (7), 1107–1114. 400 Madge, D.; Wong, R.; Seybold, P. G. Fluorescence Quantum Yields and Their
Relation to Lifetimes of Rhodamine 6G and Fluorescein in Nine Solvents: Improved
Absolute Standards for Quantum Yields. Photochem. Photobiol. 2002, 75 (4), 327–334. 401 Karstens, T.; Kobs, K. Rhodamine B and Rhodamine 101 as Reference Substances for
Fluorescence Quantum Yield Measurement. J.Phys.Chem. 1980, 84, 1871–1872. 402 Langhals, H.; Karolin, J.; Johansson, L. B-Å. Spectroscopic properties of new and
convinient standards for measuring fluorescence quantum yields. J. Chem. Soc., Faraday
Trans. 1998, 94, 2919–2922. 403 Peterman, E. J. G.; Brasselet, S.; M erner, W. E. The Fluorescence Dynamics of
Single Molecules of Green Fluorescent Protein. J. Phys. Chem. A 1999, 103, 10553-
10560. 404 Dr. Katherine A. Wilets, private communication.

390
405 Cournoyer, M. E.; Dare, J. H. The use of alternatie solvent purification techniques.
Chemical Health and Safety 2003, 10 (4), 15–18. 406 Vogel, A. I. J. Chem. Soc. 1948, 631. 407 Bowman, N. S. J. Am. Chem. Soc. 1965, 87, 4477–4481. 408 Azeotropic Data. Volume III. Advances in Chemistry Series. 1973, 116.
QD1.A355no116 — includes all previous data from volumes 6 and 35. 409 Frei, U.; Kirchmayr, R. Process for the preparation of tert-alkyl esters. U.S. Patent
4,904,814, 1990 410 Chmyr, I. M.; Bukeikhanov, N. R.; Suvorov, B. V. Synthesis of p-fluorobenzonitrile
by the oxidative ammonolysis of p-fluorotoluene on vanadium-titanium oxide catalysts.
Izvestiya Akademii Nauk Kazakhskoi SSR, Seriya Khimicheskaya 1977, 27 (1), 40–41. 411 Herbst, R. M.; Wilson, K. R. Apparent acidic dissociation of some 5-aryltetrazoles.
1957, 22, 1142–1145. 412 Gillis, D. J. Org. Chem. 1971, 36, 518. 413 Ibata, T.; Isogami, Y.; Toyoda, Bull. Chem. Soc. Jpn. 1991, 64 (1), 42–49. 414 Beach, S. F.; Hepworth, J. D.; Sawyer, J.; Hallas, G.; Marsden, R. J. Chem. Soc.
Perkin Trans. 2 1984; 2, 217–222. 415 Allbright J. Med. Chem. 1983, 26, 1406. 416 Barbero, M.; Degani, I.; Dughera, S.; Fochi, R. Synthesis 2003; 5, 742–750. 417 Hellerman J. Am. Chem. Soc. 1946, 68, 1890–1891. 418 (a) Soula, G. Tris(polyoxaalkyl)amines (trident), a new class of solid-liquid phase-
transfer catalysts. J. Org. Chem. 1985, 50 (20), 3717–3721 (b) Loupy, A.; Philippon, N.;
Pigeon, P.; Sansoulet, J.; Galons, H. Solid-liquid phase transfer catalysis without solvent:
further improvement in SNAr reactions. Synthetic Communications 1990, 20 (18), 2855–
2864. 419 Kim, S. Y.; Semyonov, A. N.; Twieg, R. J.; Horwich, A. L.; Frydman, J.; Moerner, W.
E. Probing the Sequence of Conformationally-Induced Polarity Changes in the Molecular

391
Chaperonin GroEL with Fluorescence Spectroscopy J. Phys. Chem. 2005, 109 (51),
24517-24525. 420 Kalgutkar, A. S. J. Med. Chem. 1996, 39, 1692–1703. 421 Tawney, P.O. J. Org. Chem. 1961, 26, 15–21. 422 (a) Beyer, U. Monatsch.Chem. 1997, 128, 91. (b) Zentz F. Il Farmaco 2002, 57, 421–
426. 423 Germann, F. J. Am. Chem. Soc. 1927, 49, 307–312 424 Krojidlo, M.; Barth, T.; Blaha, K.; Jost, K. Coll. Czech. Chem. Commun. 1976, 41 (7),
1954–1958. 425 Boucherle, A.; Carraz, G.; Revol, A. M.; Dodu, J. N-Substituted maleimides. Bull.
Soc. Chim. Fr. 1960, 500–503. 426 Choi, D. H.; Song, S.; Jahng, W. S.; Kim, N. Mol. Cryst. Liq. Cryst. Sci. Technol.
Sect. A 1996, 280, 17–26. 427 Zawadowski, T.; Grabowska, M.; Zbikowski, B. N-Substituted derivatives of exo-7-
oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboximide. Roczniki Chemii 1973, 47 (1), 189–191. 428 Robinson, J. J. Chem. Soc. 1915, 267–276. 429 Arcoleo, A.; Oliverio, A. Ann. Chim (Rome, Italy) 1957, 47, 415–432. 430 Arcoleo, A.; Natoli, M. C.; Marino, M. L. Condensation of phenol ethers with
aliphatic aldehydes. XXX. Veratrole and isobutyraldehyde: isolation of a
dibenzo[b,f]thiepin S,S-dioxide derivative. Ann. Chim (Rome, Italy) 1970, 60 (4), 323–
334. 431 Coltart, D. M.; Charlton, J. L. Can. J. Chem. 1996, 74, 88–94. 432 Chung, Y. J. Org. Chem. 1989, 54 (5), 1018-1032. 433 Boldt, P. Chem. Ber. 1967, 100, 1270-1280. 434 Lepage Y. Synthesis 1980, 689. 435 Bhattacharjee, D.; Popp, F. D. J. Heterocycl. Chem. 1980; 17, 315–320. 436 McOmie, J. F. W.; Perry, D. H. Synthesis 1973, 416–417.

392
437 Gordon, A. J.; Ford, R. A. The Chemist’s Companion: : a handbook of practical data,
techniques, and references. Wiley: New York, 1972, 537 pp. 438 Mills, W.H.; Mills, M. J. Chem. Soc. 1912, 101, 2194. 439 Hua, D. H.; Tamura, M.; Huang, X.; Stephany, H. A.; Helfrich, B. A.; Perchellet, E.
M.; Sperfslage, B. J.; Perchellet, J.-P.; Jiang, S.; Kyle, D. E.; Chiang, P. K.J. Org. Chem.
2002, 67 (9), 2907–2912. 440 Dufraisse, C.; Rigaudy, J.; Basselier, J. J.; Nguyen-Kim-Cuong. „Abnormal” fixation
of O in three 1,4-dimethoxyanthracene photooxides, dissociable in the cold.
Luminescence during the dissociation of the photooxides. Compt. Rend. 1965, 260 (19),
5031–5036. 441 Newman, M. S.; Wiseman, E. H. J. Org. Chem. 1961, 26, 3208–3211. 442 Rice, J. E.; Czech, A.; Hussain, N.; La Voie, E. J. J. Org. Chem. 1988, 53, 1775. 443 Mattern, C. J. Org. Chem. 1984, 49, 3051. 444 Jan, C. Can. J. Chem. 2002, 80 (8), 908.

393
Appendix A
Instructions to run a GC-MS experiment
on the Thermo Electron Trace GC 2000/Polaris Q MS.

394
The compound being injected into the Trace GC must be proven (by usual GC,
TGA) to be volatile and contain NO non-volatile or thermally decomposable residues.
That is, NO reaction mixtures or crude products may be injected. Spec sensitivity is 10
picogram (!) of decafluorobenzophenone. Both volume and amount/concentration of the
sample matter, for the injector liner cannot accept more vapor volume than equivalent to
3 μl of liquid. If you need to inject more than 3 μl, change the injector liner. By default,
the split liner is set up.
Method 1. Prepare Solution #1 by dissolving 1 mg in 10 ml. Take an aliquot of 1
ml and dilute to 10 ml of Solution #2. Take an aliquot of 1 ml of Solution #2 and dilute to
10 ml of Solution #3. Inject 1 μl of Solution #3. If you get no signal on GC-gram, inject 1
μl of Solution #2.
Method 2. Prepare Solution A by dissolving 1 mg in 10 ml. Take an aliquot of 1
μl and dilute to 1 ml of Solution B. Inject 1 μl of Solution B. If you get no signal on GC-
gram, take an aliquot of 10 μl of Solution A, dilute to 1 ml of Solution C and inject 1 μl
thereof.
Method 3. Prepare Solution Z by dissolving 1 μg (0.001 mg) in 1 ml. Inject 1 μl
of Solution Z.
Inject no more than 1–100 ng (nanogram) = 10–9 g = 10–6 mg
Inject no more than 1 μl (microliter) = 10–3 ml
1 ng/μl = 1 μg/ml = 1 mg/liter
100 ng/μl = 0.1 mg/ml = 1 mg/10 ml

395
Column max = 350°C
Injector nominal = 225°C
Transfer line nominal = 300°C
MS Source nominal = 200°C
‘Alex-Stand-by.meth’:
Injector = OFF
Gas Saver Flow = 10 ml/min
RUNNING GC-MS
1. Double click on Xcalibur. This opens the "Roadmap-Home page".
Xcalibur Icon.
2. Click on Instrument Setup icon in the Roadmap-Home Page.
This opens "Untitled-Instrument Setup". File Open Open your method file
(*.meth) from D:\Methods\Your name\

396
Xcalibur Home Page.
Instrument Setup.
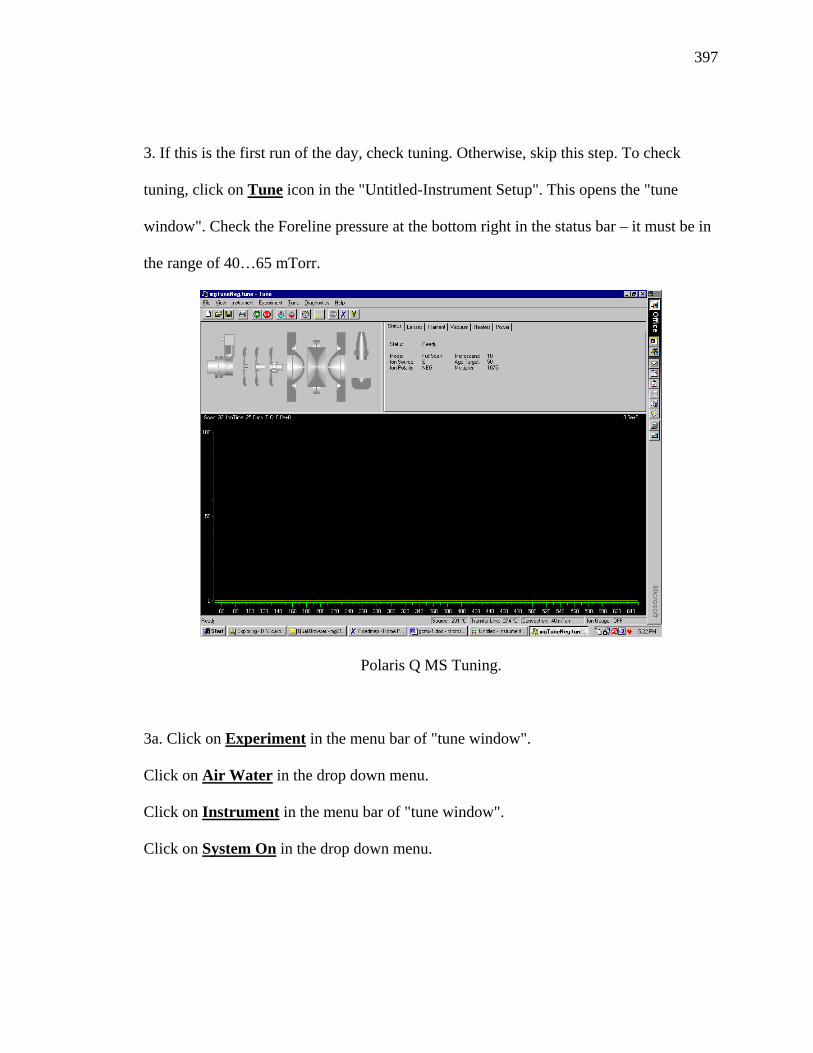
397
3. If this is the first run of the day, check tuning. Otherwise, skip this step. To check
tuning, click on Tune icon in the "Untitled-Instrument Setup". This opens the "tune
window". Check the Foreline pressure at the bottom right in the status bar – it must be in
the range of 40…65 mTorr.
Polaris Q MS Tuning.
3a. Click on Experiment in the menu bar of "tune window".
Click on Air Water in the drop down menu.
Click on Instrument in the menu bar of "tune window".
Click on System On in the drop down menu.

398
If the instrument is set for CI mode, click on “red flask” button to
switch on CI reagent gas (or from menu Instrument CI Reagent Gas). A dialog
window appears where the check-box “Reagent Gas On” must be checked and the Flow
set to 1.5 ml/min (range: 0.5 to 2.5 ml/min).
CI Reagent Gas Control.
Click on On green button in the button bar of "tune window"
(or from menu Instrument Filament/Dynode ON).
In EI mode, if the MS is working properly a spectrum with m/z= 14, 16, 18, 29
will appear. The water and oxygen peaks must NOT be the most abundant. (If you
observe high foreline pressure or you think the MS is not working properly and/or the air-
water spectrum does not look right, please let Mahinda Gangoda (2-3843 & 330-687-
4157) know about it.)

399
Click on OFF red button in the button bar of "tune window".
Close the "tune window".
Close "Untitled -Instrument setup" window. The "Roadmap-Home page" is
visible.
4a. If you do NOT use the autosampler.
Click on Sequence Setup icon in the "Roadmap-Home Page". This opens the
"Untitled[Open]-Sequence Setup" window.
Press Ctrl + O to open a sequence.
Double click on the name of the sequence belongs to your research group.
This will bring this sequence to the #1 row of the "Untitled[Open]-Sequence Setup".
Make necessary changes in #1 row. a) Double click File Name cell (Column #1)
and in the dialog window type a file name to save your data to. The file name should be
at D:\Data\Your_Name directory and end up with a *.raw extension. b) Double click on
Inst Method cell and select method file to run. c) Press Ctrl+S to save the sequence.
Unsaved sequence WILL give you an error message later on.
Click on green icon (7th from right) in the “Sequence Setup” to initialize GC.
A “Run Sequence” window opens. Click on OK in this window. Wait several seconds
until file names appear in the status window on the left and then

400
Sequence Window.
Click green icon (4th from right) in the “Sequence Setup” to initialize MS.
Check on the status window to see “GCQ/Polaris MS” is “Waiting for Contact Closure”

401
Select Rows to Run.
Then wait till the “TRACE GC 2000” turns (status window) to “Ready for Run”
(On GC key pad, the “READY TO INJECT” will turn green)
Inject only 1μL of 10-100 ng/μL (10-100 μg/ml) sample and simultaneously press
“START” (the blue round key on GC key pad) to start the run.
Click on “Real Time Plot View” (3rd icon from left) in the “Sequence Setup” window to
see the chromatogram (TIC).
The Real Time Plot View (Chromatogram) will appear after some delay time preset in the
method file (3-5 min). Wait till the run is completed (about 20 min.)

402
4b. If you DO use the autosampler.
Proceed same as 4a, but click on green icon “Run samples” (8th from right) in the
“Sequence Setup” and DO NOT click green icon “Start Analysis” (4th from right).
DATA PROCESSING
Go to “Roadmap-Home Page” by clicking on the 1st icon from left on the “Real Time
Plot” window.
Click on Qual Browser icon in “Roadmap-Home Page”
Press Ctrl + O to open the window “Open Raw File”
Double click and open your data file.
This will show the chromatogram (top) and a mass spectrum (bottom)
Click on a chromatographic peak while the mass spectral window is active (green pin on
upper right corner) to see the Mass Spectrum for a given peak.
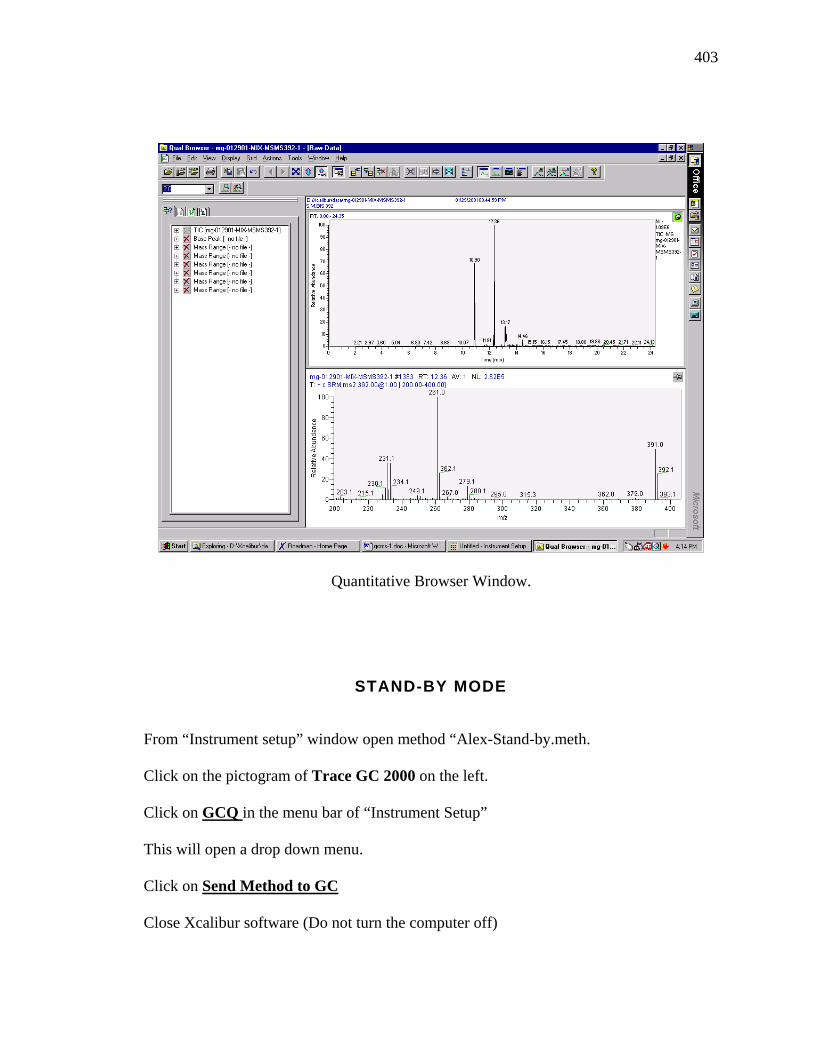
403
Quantitative Browser Window.
STAND-BY MODE
From “Instrument setup” window open method “Alex-Stand-by.meth.
Click on the pictogram of Trace GC 2000 on the left.
Click on GCQ in the menu bar of “Instrument Setup”
This will open a drop down menu.
Click on Send Method to GC
Close Xcalibur software (Do not turn the computer off)

404
Appendix B
NMR EXPERIMENTS
INSTRUCTIONS and SUMMARY

405
Bruker Avance 400 MHz
033 SRL; Phone 2-2677 host: BH041304; MAC: 00-11-0A-00-79-B8; IP: 131.123.233.48
HP XW4100 P4 2.8 GHz Bus 533MHz L1/L2/L3 20/1024/0 kB RAM 512 MB DDR 333
Topspin 1.3 for Win requirements:
CPU > 1GHz; RAM > 512 Mb
Video RAM > 64 Mb (non-shared)
Extra ethernet card
NTFS partition (for program and data)
NMRData 131.123.235.4:5505
D:\NMR\Data\Username\NMR\ExpNo\ProcNo
Run: Topspin 1.3
ej / ij to eject / inject sample tube;
rpar mg* read parameters from file;
atma auto tune/wobble;
lockdisp show lock display;
lock lock the solvent;
gradshimau perform auto shimming;
zz = zg + ftp start acquisition;
tr trace FID to hard drive;
fp Fourier process FID;
apk auto phase correctiom;
abs auto base line correction;
pp peak picking;
xwinplot = plot plot spectrum
GCOSY
Run 1H. Note rg = x value. Rpar mg-H-COSY. Enter manually rg x. atma zg xfb Adjust
intensity and save intensity. Plot: chose 2D exp first, then 1D for projections.
HMQC
Run 1H. Note rg = x value. Rpar mg-C13-1H-HMQC. Enter manually rg x. atma (tunes
for both nuclei). ns = 8. zg xfb Adjust intensity and save intensity. If separate
independent 13C is available, may plot it as y projection.

406
Sample Preparation
The 5-mm tube should be filled with a deuterated solvent to a minimum depth of
5.0…5.5 cm (about 0.60…0.75 ml). Lesser depths will make shimming the magnet
homogeneity difficult. Greater depths are O.K., except for variable temperature
experiments. The amount (concentration) of sample required for a proton spectrum
ranges from less than 1 mg/ml to about 20 mg/ml (mw=400). Too much sample can
result in a loss of resolution or a distorted spectrum. This includes not just the sample of
interest, but any proton source such as protonated buffers, residual protonated solvents,
and water. About 5mg/ml is a sufficient maximum concentration for 1H. For 13C the
higher the concentration the better. The solution should be free from any solid, such as
undissolved solute, or dust. Filter the solution through a Pasteur pipette with a tiny cotton
plug, if necessary.
At the Instrument
The Bruker AMX 300 NMR instrument may be controlled and the data may be
processed via: 1) Spectrometer Control Module (SCM) – a keypad with a black rotating
knob on the right to the computer keyboard; 2) typing commands in the command line of
uxnmr program; 3) clicking icon-buttons in Graphical User Interface (GUI) of uxnmr
program. The mouse has three buttons thus there may be left, right, and middle clicks (L-
, R-, M-click). L-Click is default.
Press orange button, and then Lift on SCM – the spinner with a tube lifts up. Insert
the spinner into the wood block. Insert your tube into the spinner all the way down.
Carefully remove the spinner from the wood block so that the tube retains its position in

407
the spinner. If the tube is inserted too deep into the spinner it will break inside the
magnet when loaded. Place the spinner on top of NMR (air should be coming out!).
Press Lift on SCM – а the tube is lowered into the magnet. Press Spin and AutoLock.
Log your name in the sign-up sheet, login into the unix shell (get your group’s login
and password from Mahinda) and type ‘uxnmr’ in the shell command prompt. Wait until
GUI is loaded. Type edc and in the appeared window enter your name in the NAME
field, “1” for 1H, “2” for 13C in EXPNO, “1” for PROCNO and hit “Save”. Raw data –
FIDs – are identified at the experiment number (EXPNO) level. Do not save more than
two files as the disk space is limited. Lack of free disk space will preclude higher-
order experiments from running. Type rpar HCDCl3H13C to read parameters from
file HCDCl3H13C, then hit “copy all”. For 13C experiment type rpar and chose
MGZGDCCHC13 file from the list.
Shimming
If necessary, reset the shims to the standard best values by typing rsh test-test or rsh
<filename>, where <filename> is the appropriate shim file. L-Click the Lock dg icon in
GUI to bring the lock level to display. Optimize Z1 and Z2 as described below. Be sure
the Fine button is illuminated on SCM. The goal is to maximize the lock level by
adjusting the shim values (LockGain is not a shim value; use LockGain to keep the lock
display in the middle of the display range). Adjust the shim values slowly since there is a
delay in the lock response. The procedure for shimming is as follows. Press Z1 and bring
the lock level as high as you can rotate the black knob on SCM. If the level goes off
scale, press LockGain and bring it back to screen. Repeat Z1 optimization until the lock

408
level reaches maximum and does not rise anymore with variation in Z1. Then press Z2
and repeat optimization rotating the knob very slowly as lock is very sensitive to Z2
variation. Return to Z1 and shim it again since these two values affect each other. Press
Standby to “lock” all the shim values from occasional change. Do not adjust Z3 or
Z4!! The proper value of Z4 requires hours of shimming.
Running experiment
Type rga (1H ONLY) to start receiver gain acquisition. After “rga:finished” has
appeared in the status line, type “rg” to see the value (1…8•103). Skip this command for
13C experiment. Type zg – this command clears all previous FIDs and starts scans. The
acquisition may be stopped at any time hereafter typing halt. It will stop acquisition and
save all FIDs acquired hitherto to the disk.
Processing
During the acquisition, all FIDs are temporarily stored in the acquisition processor’s
memory until ether halt or tr command is entered. Command tr traces all hitherto
collected FIDs from the acquisition processor’s memory to hard drive and do not
interrupt further acquisition. To see your spectrum during acquisition type tr followed by
fp (1H) or efp (13C) = em+ft+pk = exponential multiplication (em) followed by Fourier
transformation (ft) followed by last phase correction (fk). Then apply Automatic phase
correction (only once) by typing apk. To see any improvement in the signal since last tr
command, type tr followed by fp again. After the acquisition has been stopped (halted)
one need not to type tr, fp alone is sufficient.

409
The following commands are useful for 1D NMR:
em - exponential multiplication on the FID, uses the parameter LB. This improves signal
to noise at the expense of resolution.
lb - this controls the degree of broadening added and affects your signal-to-noise. To see
its effect, simply change its value and re-Fourier Transform with ef.
gm - gaussian multiplication on the fid
ft - Fourier transform. ef - combines em and ft.
gf - combines gm and ft.
pk - phase correct, applies the last phase correction to the spectrum. This process is
useful when you have phased a preliminary spectrum, (with only a few scans) and wish
to apply the same phase correction to the final spectrum.
efp - combines em, ft , and pk.
abs- automatic baseline correction
apk – automatic phase correction
Expansions
The easiest way to do horizontal expansions on the screen is to click on the left mouse
button which converts the pointer to a cursor. Position the cursor on the left edge of the
region you want to expand and click the middle mouse button. Move the cursor to the
right edge and click the middle mouse button again. Vertical expansion is performed via
L-click on ∧2 or M-click on ∨2 to increase or decrease the intensity correspondingly by
a factor of 2.

410
Calibration
Click Calibrate. Move the cursor to the reference peak (usually TMS) and click the
middle mouse button. Type in the chemical shift (sharp zero). L-Click Return.
Integration
Click Integrate. To define the integrals, convert the pointer to a cursor (click on left
mouse button), and click the middle mouse button on either side of region to be
integrated. To phase or reference an integral, they must first be selected. To select an
integral, double-click inside it using the left mouse button. An * will appear next to the
integral. To phase an integral, select it, place the pointer on slope or bias, and while
depressing the left mouse button, move the mouse to phase it. A properly defined
integral should extend beyond the apparent ends of the peak (if there is no other adjacent
peak). A properly phased integral should be horizontal before and after the peak. Good
integration requires careful attention. L-Click on Write+Return to save the integration.
Title
To add a title, type setti – this recalls vi, unix text editor. Use the following commands.
To begin type text, enter in append mode depressing “a”. To exit append mode, hit
“Esc”. To delete text, exit from append mode and press “x”. To finish: “Esc”,
Shift+”Z”+”Z”, then L-click on Clear icon. Other commands: dd – delete current line; J
– join lines; u – undo; i – insert mode; a – append mode; r – replace mode; Esc – back
from mode; ZZ – store and exit; :q! – discard and exit; :w – store.

411
Plotting
L-click on Edplot icon – editing and printing window appears. Click “Fix-wind”, chose
IntegLables and PeaksLables to display. Click Sto+Plot to print. DO NOT use
Sto+Plots – this will not print anything for you until flplot command is entered.
Finishing
Remove the tube from the magnet: Press Spin (the green light on the key goes off),
orange button, then Lift. Insert blank tube into the spinner and load the spinner with a
blank tube into the magnet, do not spin. Type “exit” two times to exit the program and to
log off from unix shell.
Commands Summary
uxnmr Unix NMR program; uxnmr –r restores default parameters.
edc Edits current file set
rpar HCDCl3H13C Reads parameters from file HCDCl3H13C, chose MGZGDCCHC13
file for 13C
ii initializes interface
Lock dg Displays lock signal window
Auto lock Automatic lock should be on and steady green (not blinking).
Lock gain Press and rotate the black knob to bring the lock value, to the top of the screen
rsh test-test Reads shim parameters from file.
Fine Should be “on”, meaning the knob changes the value slowly
Z1, Z2 Press “Z1” and bring the lock gain level as high as you can. If the level goes off
scale, press Lock gain and bring it back to screen. Repeat “Z1” optimization until the

412
level reaches maximum and does not rise anymore with variation in Z1. Then press “Z2”
and repeat optimization rotating the knob very slowly as lock is very sensitive to Z2
variation.
Stand-by Press to “lock” all the values from occasional change
lb Line broadening, more lb increases sensitivity, but lowers resolution: 2 for 1H, 0 for
13C
rga (1H ONLY) Receiver gain acquisition. rg to see value (1…8•103). Skip this
command for 13C experiment.
zg Clears all previous FIDs and starts scans
tr Traces collected FIDs to hard drive
fp (1H) efp (13C) Exponential multiplication followed by Fourier transformation
followed by phase correction
apk Automatic phase correction (type only once)
halt Terminates data acquisition and stores all FIDs to disk.
Integ Integrate the peaks and press “Write+Return”
Edplot Editing and printing window
eda Edits acquisition parameters
edg Edits graphics parameters
edp Edits processing parameters
edsp Set frequencies (also ased)
d1 1…10 for 1H, 1…5 for 13C
ns Number of scans to be completed

413
ds Number of dummy scans (to bring the spin system to an equilibrium)
lock = AutoLock. Waits 2 min to find lock.
ro = Spin. Waits 15 sec to reach set point. ro yes 15 yes will rotate with 15Hz and
modulation to suppress solvent side bands.
ej, ij – Eject and inject the sample.
lo = LockPower (0–60 dB). 45 for CDCl3.
lopo Sets lo and magnetic field wrt SOLVENT parameter.
lg = LockGain (22–140 dB).
wsh, rsh, delsh, vish, lsh, setsh – Write, read, delete, views, prints, and displays a graph
of shim file.
tune <file> –Auto shims using shim gradient file, editable with edtune <file>.
vi commands:
dd – delete current line; J – join lines; u – undo; i – insert mode; a – append mode; r –
replace mode; Esc – back from mode; ZZ – store and exit; :q! – discard and exit; :w –
store;
In .profile: UXNMR_REQMSG=NO to suppress requests for printing.
In .uxnmrrc type those uxnmr commands one would like to execute at startup (one in a
line): param no; plunit ppm; pldigit 3
To insert a part of a spectrum into a plot: get_w12 Sto+Plots fplot (flush plot)
Edpul <file> Edits and prints (“list”) specified pulse sequence file
Edcpul Edits current pulse sequence file (defined in PULPROG with eda)
1 u = 1 μs (millisecond, 10–6 sec); 1 ms = 1 microsec (10–3 sec); 1 s = 1 second

414
Lock_opt then →← to change grid.
COSY
Run usual 1H NMR in say u/user/alex/1/1. d1=1 s
Include all peaks in the sweep (expansion); include large TMS peak, but exclude small TMS peak.
Left click SW–SFO1 (accepts the display limits of your expansion for the sweep) and
note the values of SW, O1, and rg. Change delay time to de 10 μs; check aq time. Run
new spec with these parameters.
cre 2 or re 2
rpar mgcosy-CDCl3 (for all solvents)
Change (enter) the noted parameters: SW, O1, and rg:
rg = enter value
SW= enter value
1 SW = enter same value
O1=enter value
de=10
d1=200
ns=16-32
ds=4 (dummy scans to set the equilibrium in the spin system)
expt – experiment time
td – time domain size for 1D experiment
1 td – the number of cross spectra; the higher the value the higher 2D resolution
p1 p1
d1
deaq

415
rser – 1st increment
xfb – to see the spectrum (no tr)
thres Click middle button and move mouse to change intensity of threshold
DefPlot: “y” to change intensities; levels<7; contours “y”
edg edproj1 ed change 1D filename; PF1CY – length of the most intense peak.
Save.
edg edproj2 … (same)
Type plot to print. To expand: click Zoom strip → select the region: L-click, M-click2 →
DefPlot.
in co ob – operate only in intensity
Limits: F1LO and F1HI (vertical); F2LO and F2HI (horizontal).
The horisontal resolution is always higher since td > 1 td.
After you are done, delete your 2D file!!! Data → Delete → Delete 2D data → chose
your file and click “Execute”.
19F
Run usual 1H NMR, shim very well.
Remove (physically) 1H attenuators (4-2-6 dB) and place BNC connector directly to the
decoupler. Switch BNCs (together with 300 Low Pass Filter 0–31P Rejects 1H) on top of
the decoupler between 1H and 19F (X).
Tune the probe as follows. Find RED and BLACK probes in front of the decoupler
probe. For RED: push up, then turn CW until stops, then turn 21¼ turns CCW. For
BLACK: push up, then turn CW until stops, then turn 8¼ turns CCW.

416
Rpar mg-F19-CDCl3; rg=max is setup in the parameter file, so do NOT type rga.
However, if the FID signal is too strong (cut-off on top and bottom), manually reduce rg.
Set lb=1; de=10μsec. Run a spec with ns=1, use efp to transform and locate your signal.
Enlarge it and then click on SW-SFO1. Check the parameters above and collect spec
with large number of ns in the region of interest.
Return the 1H attenuators, switch back BNC connectors and tune the probes back to 1H:
For RED: push up, then turn CW until stops, then turn 9 ½ –9 ¾ turns CCW. For
BLACK: push up, then turn CW until stops, then turn 10 ½ turns CCW.
Running samples in unusual solvents
Put pure solvent in the tube. Call for Lock_dg. Press Field on SCM, look for resonance
turning the knob, or set to a known value (905 for CF3COOD). Lock power ~35…50
depending on solvent saturation limit. Set the sweep width wide: sw 2e4 Hz. Each time
the sw changed, change de back to 10 μsec: de 10. Set the reference to zero: sr 0. Take a
scan (ns=1). Find solvent reference peak and calibrate it. Check TMS signal to
correspond. Change sweep to a practical width (check de!) and run sample in the new
solvent. Save parameter file if desired.

417
Appendix C
M. Braun Solvent Purification System (SPS)
User ’s Instructions

418
A. Principles
The solvents are nitrogen-pushed through a column filled with either activated
alumina or (for hydrocarbons) alumina and active (reduced) copper. In the latter case the
solvent gets deoxygenated as well. The columns are color-coded: if the lower color band
on the top of a cylinder is white – it’s alumina packed; if green – it’s copper packed. The
active copper catalyst is compatible with hydrocarbons only and incompatible with THF,
CH2Cl2, and Et2O. The SPS does not purify solvents from non-volatile impurities and you
get the same grade of solvent as you put into the supply reservoir, yet anhydrous. Thus,
fill the supply reservoir with clean solvents only.
B. Operation
1. Start with a flask you intend to run a reaction in. Calculate the volume of
anhydrous solvent you will need. Fill the flask with acetone in that volume and make a
mark on the flask. Dry the flask in an oven or with a heat-gun. Cool it down under
nitrogen and stopper it with a matched stopper.
2. At SPS you’ll find it in Stand-by mode (Pic. 1), with nitrogen and solvent
supplies closed, vacuum pump operating, and solvent outlets capped and vacuumized.
Turn all SPS manifold valves to OFF positions (Pic. 2).

419
Pic. 1. Stand-by Mode.
Pic. 2. Manifold valves in OFF positions.
3. Check that all blue valves on all the connected solvent supply reservoirs are
closed (Pic. 3). Open main nitrogen valve and check the pressure to be less than 5-7 psi

420
(Pic. 4). Open the blue valve on the solvent supply reservoirs you need to use (Pic. 3).
Double check that all manifold valves are closed (Pic. 2).
Pic. 3. Blue valves on all solvent supply reservoirs, but the one in use, are closed.
Pic. 4. Nitrogen pressure less than 5-7 psi.

421
For exemplary purposes, let’s now consider dispensing of dry THF.
While holding the storage cap by hand:
4. Turn the Main Manifold Valve to “Nitrogen” position.
5. Slowly turn THF’s Nitrogen valve to “Nitrogen/Vacuum” position. (Pic. 5).
Pic. 5. Removing the storage cap from THF’s tap.

422
6. The cap will be filled with nitrogen. Hold the white Teflon adapter with one
hand and remove the cap with another hand. A stream of nitrogen should be heard
flowing from the tap.
7. Place your flask onto the THF’s tap and hold it with your hand throughout the
rest of operations.
8. Turn the Main Manifold Valve to “Vacuum” position. Wait for 15-30 seconds
to evacuate your flask.
9. Turn the Main Manifold Valve to “Nitrogen” position for 2-3 seconds to refill
the flask with nitrogen.
10. Repeat steps 8 and 9 three to five times.
11. Turn the Main Manifold Valve to “Vacuum” position. Wait for 30-50 seconds
to completely evacuate your flask for the final time.
12. Turn THF’s Nitrogen valve to “Fill Collection Vessel” position. (Pic. 6).

423
Pic. 6. Turn THF’s Nitrogen valve to “Fill Collection Vessel” position.
13. Slowly turn THF’s Solvent valve to “To Collection Vessel” position (Pic. 7).
Fill the flask with solvent to the necessary volume or mark. Do not fill more than 2/3 of a
flask.

424
Pic. 7. Fill the flask with solvent.
14. Turn THF’s Solvent valve to “OFF” position.
15. Turn the Main Manifold Valve to “Nitrogen” position.
16. Very slowly turn THF’s Nitrogen valve towards “Nitrogen/Vacuum” position.
(Pic. 8). Fill the flask with nitrogen. If you turn the Nitrogen valve too quickly or fully to
“Nitrogen/Vacuum” position, the blow or fast stream of nitrogen may blow the solvent
out of the flask and/or the flask itself from the tap.

425
Pic. 8. Slowly filling the void of the flask with nitrogen.
17. With the nitrogen flowing into the flask, detach the flask from the tap and
recap it with a stopper as quickly as possible. Turn THF’s Nitrogen valve completely to
“Nitrogen/Vacuum” position to flush residual solvent off.
18. Replace the storage cap on the THF’s tap.
19. Turn the Main Manifold Valve to “Vacuum” position.
20. Turn THF’s Nitrogen valve to “Nitrogen/Vacuum” position (Pic. 9).

426
Pic. 9. Recapping THF’s tap.
C. Stand-by mode
1. Close all blue valves on solvent supply reservoirs (Pic. 3).
2. Turn all “Nitrogen” valves to “Nitrogen/Vacuum” position (Pic. 1).
3. Turn the Main Manifold Valve to “Vacuum” position. (Pic. 1).
4. Close the main nitrogen valve.
The system now is in stand-by mode.




















